WO2004058835A1 - Polymers substantially free of long chain branching - Google Patents
Polymers substantially free of long chain branching Download PDFInfo
- Publication number
- WO2004058835A1 WO2004058835A1 PCT/US2003/040341 US0340341W WO2004058835A1 WO 2004058835 A1 WO2004058835 A1 WO 2004058835A1 US 0340341 W US0340341 W US 0340341W WO 2004058835 A1 WO2004058835 A1 WO 2004058835A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- butene
- copolymer
- group
- tetrafluoro
- chloride
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F210/00—Copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F210/04—Monomers containing three or four carbon atoms
- C08F210/08—Butenes
- C08F210/10—Isobutene
- C08F210/12—Isobutene with conjugated diolefins, e.g. butyl rubber
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F10/00—Homopolymers and copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/04—Polymerisation in solution
- C08F2/06—Organic solvent
Definitions
- the invention relates to new polymerization processes including diluents including hydrofluorocarbons and their use to produce novel polymers substantially free of long chain branching.
- the invention relates to copolymers of an isoolefin, preferably isobutylene, and a multiolefin, preferably a conjugated diene, more preferably isoprene, substantially free of long chain branching.
- Isoolefin polymers are prepared in carbocationic polymerization processes.
- butyl rubber which is a copolymer of isobutylene with a small amount of isoprene.
- Butyl rubber is made by low temperature cationic polymerization that generally requires that the isobutylene have a purity of >99.5 wt% and the isoprene have a purity of >98.0 wt% to prepare high molecular weight butyl rubber.
- the carbocationic polymerization of isobutylene and its copolymerization with comonomers like isoprene is mechanistically complex. See, e.g., Organic Chemistry, SIXTH EDITION, Morrison and Boyd, Prentice-Hall, 1084-1085, Englewood Cliffs, New Jersey 1992, and K. Matyjaszewski, ed, Cationic Polymerizations, Marcel Dekker, Inc., New York, 1996.
- the catalyst system is typically composed of two components: an initiator and a Lewis acid. Examples of Lewis acids include A1C1 3 and BF 3 .
- initiators include Br ⁇ nsted acids such as HCI, RCOOH (wherein R is an alkyl group), and H 2 O.
- initiation step isobutylene reacts with the Lewis acid/initiator pair to produce a carbenium ion.
- additional monomer units add to the formed carbenium ion in what is generally called the propagation step.
- steps typically take place in a diluent or solvent. Temperature, diluent polarity, and counterions affect the chemistry of propagation. Of these, the diluent is typically considered important.
- the slurry polymerization process in methyl chloride offers a number of additional advantages in that a polymer concentration of approximately 26% to 37% by volume in the reaction mixture can be achieved, as opposed to the concentration of only about 8% to 12% in solution polymerization.
- An acceptable relatively low viscosity of the polymerization mass is obtained enabling the heat of polymerization to be removed more effectively by surface heat exchange.
- Slurry polymerization processes in methyl chloride are used in the production of high molecular weight polyisobutylene and isobutylene-isoprene butyl rubber polymers. Likewise polymerizations of isobutylene and para-methylstyrene are also conducted using methyl chloride.
- the commercial reactors typically used to make these rubbers are well mixed vessels of greater than 10 to 30 liters in volume ' with a high circulation rate provided by a pump impeller.
- the polymerization and the pump both generate heat and, in order to keep the slurry cold, the reaction system needs to have the ability to remove the heat.
- An example of such a continuous flow stirred tank reactor (“CFSTR") is found in U.S. Patent No. 5,417,930, incorporated by reference, hereinafter referred to in general as a "reactor” or "butyl reactor”.
- U.S. Patent No. 2,534,698 discloses, ter alia, a polymerization process comprising the steps in combination of dispersing a mixture of isobutylene and a polyolefin having 4 to 14 carbon atoms per molecule, into a body of a fluorine substituted aliphatic hydrocarbon containing material without substantial solution therein, in the proportion of from one-half part to 10 parts of fluorine substituted aliphatic hydrocarbon having from one to five carbon atoms per molecule which is liquid at the polymerization temperature and polymerizing the dispersed mixture of isobutylene and polyolefin having four to fourteen carbon atoms per molecule at temperatures between -20°C and -164°C by the application thereto a Friedel-Crafts catalyst.
- '698 teaches
- U.S. Patent No. 2,548,415 discloses, inter alia, a continuous polymerization process for the preparation of a copolymer, the steps comprising continuously delivering to a polymerization reactors a stream consisting of a major proportion of isobutylene and a minor proportion isoprene; diluting the mixture with from 1/2 volume to 10 volumes of ethylidene difluoride; copolymerizing the mixture of isobutylene isoprene by the continuous addition to the reaction mixture of a liquid stream of previously prepared polymerization catalyst consisting of boron trifluoride in solution in ethylidene difluoride, maintaining the temperature between -40°C and -103°C throughout the entire copolymerization reaction .
- 2,644,809 teaches, inter alia, a polymerization process comprising the steps in combination of mixing together a major proportion of a monoolefin having 4 to 8, inclusive, carbon atoms per molecule, with a minor proportion of a multiolefin having from 4 to 14, inclusive, carbon atoms per molecule, and polymerizing the resulting mixture with a dissolved Friedel-Crafts catalyst, in the presence of from 1 to 10 volumes (computed upon the mixed olefins) of a liquid selected from the group consisting of dichlorodifluoromethane, dichloromethane, trichloromonofluormethane, dichloromonofluormethane, dichlorotetrafluorethane, and mixtures thereof, the monoolefin and multiolefin being dissolved in said liquid, and carrying out the polymerization at a temperature between -20oC and the freezing point of the liquid.
- CFC chlorofluorocarbons
- Hydrofluorocarbons are chemicals that are currently used as environmentally friendly refrigerants because they have a very low (even zero) ozone depletion potential. Their low ozone depletion potential is thought to be related to the lack of chlorine. The HFC's also typically have low flammability particularly as compared to hydrocarbons and chlorinated hydrocarbons.
- the invention provides for new polymerization processes comprising diluents comprising hydrofluorocarbons and their use to produce novel polymers substantially free of long chain branching or having no long chain brandling.
- the invention provides copolymers substantially free of long chain branching with new and/or improved rheological properties over a broader range of copolymer compositions than has been possible.
- the invention provides for copolymers of an isoolefin, preferably isobutylene, and a multiolefin, preferably a conjugated diene, more preferably isoprene, substantially free of long chain branching. Additionally, the invention provides for copolymers of an isoolefin, preferably isobutylene, and a multiolefin, preferably a conjugated diene, more preferably isoprene, having no long chain branching.
- the invention provides for a copolymer produced by the process comprising contacting an isoolefin, preferably isobutylene, a multiolefin, preferably a conjugated diene, more preferably isoprene, one or more Lewis acid(s), one or more initiator(s), and a diluent comprising one or more hydrofluorocarbon(s) (HFC's); wherein the copolymer is substantially free of long chain branching or has no long chain branching.
- the multiolefin, or conjugated diene, or isoprene, when present, content is from greater than 0.5 mol%.
- the multiolefin, or conjugated diene, or isoprene, when present, content is from greater than 1.0 mol%.
- the multiolefin, or conjugated diene, or isoprene, when present, content is from greater than 2.5 mol%.
- the multiolefin, or conjugated diene, or isoprene, when present, content is from greater than 5.0 mol%.
- the copolymer may be halogenated to form a halogenated copolymer.
- the halogenated copolymer is halogenated with chlorine or bromine.
- the halogen content is greater than 0.5 wt% based upon the weight of the halogenated copolymer.
- the halogen content is from
- the copolymer may have a
- Mw from greater than 50,000.
- the copolymer may have a
- Mw of from greater than 100,000.
- the copolymer may have a
- Mw from greater than 500,000.
- the copolymer may have a
- Mw of from greater than 1,000,000.
- the copolymer may have a
- MWD of from greater than 2.
- the copolymer may have a
- the copolymer may have a
- Mooney viscosity of at least 20 ⁇ 5 (ML 1 + 8 at 125°C, ASTM D 1646).
- the copolymer may have a
- the copolymer may have a g'vis.a v g. from greater than or equal to 0.978 as determined by triple detection SEC (described herein).
- the copolymer may have a g'vis.a v g. from greater than or equal to 0.980 as determined by triple detection SEC (described herein).
- the copolymer may have a g'vis.a v g. from greater than or equal to 0.990 as determined by triple detection SEC (described herein).
- the copolymer may have a g'vis.a v g. from greater than or equal to 0.995 as determined by triple detection SEC (described herein).
- the copolymer may have no long chain branching.
- This invention also relates to a polymerization process comprising contacting one or more monomers, one or more Lewis acids and one or more initiators in the presence of a diluent comprising one or more hydrofluorocarbons (HFC's) in a reactor under polymerization conditions.
- a diluent comprising one or more hydrofluorocarbons (HFC's)
- this invention relates to a process to produce polymers of monomer(s) comprising contacting, in a reactor, the monomer(s) and a Lewis acid in the presence of a hydrofluorocarbon diluent, wherein the Lewis acid is not a compound represented by formula MX 3 , where M is a group 13 metal, X is a halogen.
- the invention provides a polymerization medium suitable to polymerize one or more monomer(s) to form a polymer, the polymerization medium comprising one or more Lewis acid(s), one or more initiator(s), and a diluent comprising one or more hydrofluorocarbon(s) (HFC's).
- the invention provides a polymerization medium suitable to polymerize one or more monomer(s) to form a polymer, the polymerization medium comprising one or more Lewis acid(s) and a diluent comprising one or more hydrofluorocarbon(s) (HFC); wherein the one or more Lewis acid(s) is not a compound represented by formula MX 3 , where M is a group 13 metal and X is a halogen.
- Figure 1 is a graph of the relationship between dielectric constant and temperature.
- Figure 2 is a drawing of diluent mass uptake as a function of volume fraction of hydrofluorocarbon in methyl chloride.
- Figure 3 is a plot of peak molecular weight (M p ) versus monomer conversion of certain inventive polymers as described herein.
- Figure 4 is a graph of the relationship between the log of the intrinsic viscosity, the g' value and the log of molecular weight for polyisobutylene prepared at -95°C (Example 149). Solid lines indicate the relationship for the linear standard. Dotted lines indicate the relationship of the intrinsic viscosity calculated for the sample.
- Figure 5 is a graph of the relationship between the log of the intrinsic viscosity, the g' value and the log of molecular weight for an isobutylene/isoprene copolymer prepared in methyl chloride at -95 °C (Example 150). Solid lines indicate the relationship for the linear standard. Dotted lines indicate the relationship of the intrinsic viscosity calculated for the sample.
- Figure 6 is a graph of the relationship between the log of the intrinsic viscosity, the g' value and the log of molecular weight for an isobutylene/isoprene copolymer prepared in CH 2 FCF 3 at -95°C (Example 151). Solid lines indicate the relationship for the linear standard. Dotted lines indicate the relationship of the intrinsic viscosity calculated for the sample.
- catalyst system refers to and includes any Lewis acid(s) or other metal complex(es) (described herein) used to catalyze the polymerization of the olefinic monomers of the invention, as well as at least one initiator, and optionally other minor catalyst component(s).
- the invention provides a polymerization medium suitable to polymerize one or more monomer(s) to form a polymer, the polymerization medium comprising one or more Lewis acid(s), one or more initiator(s), and a diluent comprising one or more hydrofluorocarbon(s) (HFC's).
- a polymerization medium suitable to polymerize one or more monomer(s) to form a polymer, the polymerization medium comprising one or more Lewis acid(s), one or more initiator(s), and a diluent comprising one or more hydrofluorocarbon(s) (HFC's).
- the invention provides a polymerization medium suitable to polymerize one or more monomer(s) to form a polymer, the polymerization medium comprising one or more Lewis acid(s) and a diluent comprising one or more hydrofluorocarbon(s) (HFC); wherein the one or more Lewis acid(s) is not a compound represented by formula MX 3 , where M is a group 13 metal and X is a halogen.
- suitable to polymerize monomers to form a polymer relates to the selection of polymerization conditions and components, well within the ability of those skilled in the art necessary to obtain the production of a desired polymer in light of process parameters and component properties described herein. There are numerous permutations of the polymerization process and variations in the polymerization components available to produce the desired polymer attributes.
- such polymers include polyisobutylene homopolymers, isobutylene-isoprene (butyl rubber) copolymers, isobutylene and para-methylstyrene copolymers, and star-branched butyl rubber terpolymers.
- Diluent means a diluting or dissolving agent.
- Diluent is specifically defined to include chemicals that can act as solvents for the Lewis Acid, other metal complexes as herein described, initiators, monomers or other additives.
- the diluent does not alter the general nature of the components of the polymerization medium, i.e., the components of the catalyst system, monomers, etc. However, it is recognized that interactions between the diluent and reactants may occur. In preferred embodiments, the diluent does not react with the catalyst system components, monomers, etc. to any appreciable extent. Additionally, the term diluent includes mixtures of at least two or more diluents.
- a reactor is any container(s) in which a chemical reaction occurs.
- Slurry refers to a volume of diluent comprising monomers that have precipitated from the diluent, monomers, Lewis acid, and initiator.
- the slurry concentration is the volume percent of the partially or completely precipitated polymers based on the total volume of the slurry.
- Polymer may be used to refer to homopolymers, copolymers, interpolymers, terpolymers, etc.
- a copolymer may refer to a polymer comprising at least two monomers, optionally with other monomers.
- a polymer is referred to as comprising a monomer
- the monomer is present in the polymer in the polymerized form of the monomer or in the derivative form the monomer.
- catalyst components are described as comprising neutral stable forms of the components, it is well understood by one skilled in the art, that the ionic form of the component is the form that reacts with the monomers to produce polymers.
- Isoolefin refers to any olefm monomer having two substitutions on the same carbon.
- Multiolefin refers to any monomer having two double bonds.
- the multiolefin is any monomer comprising two conjugated double bonds.
- Elastomer or elastomeric composition refers to any polymer or composition of polymers consistent with the ASTM D1566 definition. The terms may be used interchangeably with the term "rubber(s)", as used herein.
- Alkyl refers to a paraffinic hydrocarbon group which may be derived from an alkane by dropping one or more hydrogens from the formula, such as, for example, a methyl group (CH 3 ), or an ethyl group (CH 3 CH 2 ), etc.
- Aryl refers to a hydrocarbon group that forms a ring structure characteristic of aromatic compounds such as, for example, benzene, naphthalene, phenanthrene, anthracene, etc., and typically possess alternate double bonding ("unsaturation") within its structure.
- An aryl group is thus a group derived from an aromatic compound by dropping one or more hydrogens from the formula such as, for example, phenyl, or C 6 H 5 .
- Substituted refers to at least one hydrogen group by at least one substituent selected from, for example, halogen (chlorine, bromine, fluorine, or iodine), amino, nitro, sulfoxy (sulfonate or alkyl sulfonate), thiol, alkylthiol, and hydroxy; alkyl, straight or branched chain having 1 to 20 carbon atoms which includes methyl, ethyl, propyl, tert-butyl, isopropyl, isobutyl, etc.; alkoxy, straight or branched chain alkoxy having 1 to 20 carbon atoms, and includes, for example, methoxy, ethoxy, propoxy, isopropoxy, butoxy, isobutoxy, secondary butoxy, tertiary butoxy, pentyloxy, isopentyloxy, hexyloxy, heptryloxy, octyloxy, nonyloxy, and
- this invention relates to the use of hydrofmorocarbon(s) or blends of hydrofluorocarbon(s) with hydrocarbon(s) and/or chlorinated hydrocarbon(s) to produce a polymer slurry which is less prone to fouling (i.e., also observed more glass like, less sticky particles in the reaction vessel with reduced adherence to the walls of the vessel or to the stirring impeller as well as reduced particle to particle agglomeration).
- this invention relates to the use of hydrofluorocarbon diluent(s) or HFC diluent blends with hydrocarbons and/or chlorinated hydrocarbon blends to polymerize and copolymerize isoolefins with dienes and/or alkylstyrenes to produce isoolefin homopolymers and copolymers with significantly reduced reactor fouling.
- this invention relates to the use of hydrofluorocarbon diluent(s) or diluent blends with hydrocarbons and/or chlorinated hydrocarbon blends to polymerize and copolymerize isoolefins with dienes to produce isoolefin copolymers with significantly reduced reactor fouling and hence longer run life for the reactors, as compared to conventional systems.
- the hydrofluorocarbons are used in a tubular reactor to obtain reduced polymer accumulation on the heat transfer tubes and/or reduce polymer accumulation on the impeller and thus obtain longer run life.
- the hydrofluorocarbons are used in a tubular reactor at higher temperatures to produce polymers at much greater run lengths (such as greater than 15 hours, preferably greater than 20 hours, preferably greater than 30 hours, more preferably greater than 48 hours than possible with other halogenated hydrocarbons.
- hydrofluorocarbons are used in an autorefrigerated boiling-pool reactor where heat is removed by evaporation of the diluent and monomers mixture to reduce reactor wall fouling, and agitator/impeller fouling among other things.
- hydrofluorocarbons are used in a polymerization process to obtain higher molecular weights at the same temperature than when other halogenated hydrocarbons are used.
- this invention relates to the discovery of new polymerization systems using diluents containing hydrofluorocarbons. These diluents effectively dissolve the selected catalyst system and monomers but are relatively poor solvents for the polymer product. Polymerization systems using these diluents are less prone to fouling due to the agglomeration of polymer particles to each other and their depositing on polymerization hardware.
- this invention further relates to the use of these diluents in polymerization systems for the preparation of high molecular weight polymers and copolymers at equivalent to or higher than to those polymerization temperatures using solely chlorinated hydrocarbon diluents such as methyl chloride.
- this invention relates to the discovery of new polymerization systems using fluorinated aliphatic hydrocarbons capable of dissolving the catalyst system. These polymerization systems are also beneficial for isoolefin slurry polymerization and production of a polymer slurry that is less prone to fouling, while permitting dissolution of monomer, comonomer and the commercially preferred alkylaluminum halide catalysts.
- this invention further relates to the use of these diluents for the preparation of high molecular weight polymers and copolymers at higher polymerization temperatures as compared to polymerization systems using solely chlorinated hydrocarbon diluents such as methyl chloride.
- this invention relates to the preparation of isoolefmic homopolymers and copolymers, especially the polymerization reactions required to produce the isobutylene-isoprene form of butyl rubber and isobutylene-p-alkylstyrene copolymers. More particularly, the invention relates to a method of polymerizing and copolymerizing isoolefins in a slurry polymerization process using hydrofluorocarbon diluents or blends of hydrofluorocarbons, and chlorinated hydrocarbon diluents, like methyl chloride.
- the polymerization systems of the present invention provide for copolymerizing an isomonoolefin having from 4 to 7 carbon atoms and para-alkylstyrene monomers.
- the system produces copolymers containing between about 80 and 99.5 wt. % of the isoolefin such as isobutylene and between about 0.5 and 20 wt. % of the para-alkylstyrene such as para-methylstyrene.
- the copolymers are comprised between about 10 and 99.5 wt. % of the isoolefin, or isobutylene, and about 0.5 and 90 wt. % of the para-alkylstyrene, such as para-methylstyrene.
- this invention relates to a process to produce polymers of cationically polymerizable monomer(s) comprising contacting, in a reactor, the monomer(s), a Lewis acid, and an initiator, in the presence of an HFC diluent at a temperature of 0°C or lower, preferably -10°C or lower, preferably -20°C or lower, preferably -30°C or lower, preferably -40°C or lower, preferably -50°C or lower, preferably -60°C or lower, preferably -70°C or lower, preferably -80°C or lower, preferably -90°C or lower, preferably -100°C or lower, preferably from 0°C to the freezing point of the polymerization medium, such as the diluent and monomer mixture.
- Monomers which may be polymerized by this system include any hydrocarbon monomer that is polymerizable using this invention.
- Preferred monomers include one or more of olefins, alpha-olefins, disubstituted olefins, isoolefins, conjugated dienes, non-conjugated dienes, styrenics and/or substituted styrenics and vinyl ethers.
- the styrenic may be substituted (on the ring) with an alkyl, aryl, halide or alkoxide group.
- the monomer contains 2 to 20 carbon atoms, more preferably 2 to 9, even more preferably 3 to 9 carbon atoms.
- olefins examples include styrene, para-alkylstyrene, para- methylstyrene, alpha-methyl styrene, divinylbenzene, diisopropenylbenzene, isobutylene, 2-methyl-l-butene, 3 -methyl- 1-butene, 2-methyl-2-pentene, isoprene, butadiene, 2,3-dimethyl-l,3-butadiene, ⁇ -pinene, myrcene, 6,6-dimethyl-fulvene, hexadiene, cyclopentadiene, piperylene, methyl vinyl ether, ethyl vinyl ether, and isobutyl vinyl ether and the like.
- Monomer may also be combinations of two or more monomers.
- Styrenic block copolymers may also be used a monomers.
- Preferred block copolymers include copolymers of styrenics, such as styrene, para-methylstyrene, alpha-methylstyrene, and C 4 to C 3 o diolefins, such as isoprene, butadiene, and the like.
- Particularly preferred monomer combinations include 1) isobutylene and para-methyl styrene 2) isobutylene and isoprene, as well as homopolymers of isobutylene.
- preferred monomers include those that are cationically polymerizable as described in Cationic Polymerization of Olefins, A Critical Inventory, Joseph Kennedy, Wiley Interscience, New York 1975.
- Monomers include any monomer that is cationically polymerizable, such as those monomers that are capable of stabilizing a cation or propagating center because the monomer contains an electron donating group.
- cationic catalysis please see Cationic Polymerization of Olefins, A Critical Inventory, Joseph Kennedy, Wiley Interscience, New York 1975.
- the monomers may be present in the polymerization medium in an amount ranging from 75 wt% to 0.01 wt% in one embodiment, alternatively 60 wt% to 0.1 wt%, alternatively from 40 wt% to 0.2 wt%, alternatively 30 to 0.5 wt%, alternatively 20wt% to 0.8 wt%, alternatively and from 15 wt% to 1 wt% in another embodiment.
- Preferred polymers include homopolymers of any of the monomers listed in this Section.
- Examples of homopolymers include polyisobutylene, polypara-methylstyrene, polyisoprene, polystyrene, polyalpha-methylstyrene, polyvinyl ethers (such as polymethylvinylether, polyethylvinylether).
- Preferred polymers also include copolymers of 1) isobutylene and an alkylstyrene; and 2) isobutylene and isoprene.
- butyl polymers are prepared by reacting a comonomer mixture, the mixture having at least (1) a C 4 to C 6 isoolefin monomer component such as isobutene with (2) a multiolefin, or conjugated diene monomer component.
- the isoolefin is in a range from 70 to 99.5 wt% by weight of the total comonomer mixture in one embodiment, 85 to 99.5 wt% in another embodiment. In yet another embodiment the isoolefin is in the range of 92 to 99.5 wt%.
- the conjugated diene component in one embodiment is present in the comonomer mixture from 30 to 0.5 wt% in one embodiment, and from 15 to 0.5 wt% in another embodiment. In yet another embodiment, from 8 to 0.5 wt% of the comonomer mixture is conjugated diene.
- the C to C 6 isoolefin may be one or more of isobutene, 2-methyl-l-butene, 3 -methyl- 1-butene, 2-methyl-2-butene, and 4-methyl-l-pentene.
- the multiolefin may be a C 4 to C ⁇ 4 conjugated diene such as isoprene, butadiene, 2,3-dimethyl-l,3-butadiene, ⁇ -pinene, myrcene, 6,6- dimethyl-fulvene, hexadiene, cyclopentadiene and piperylene.
- One embodiment of the butyl rubber polymer of the invention is obtained by reacting 85 to 99.5 wt% of isobutylene with 15 to 0.5 wt% isoprene, or by reacting 95 to 99.5 wt% isobutylene with 5.0 wt% to 0.5 wt% isoprene in yet another embodiment.
- the following table illustrates how the above-referenced wt % would be expressed as mol%.
- This invention further relates to terpolymers and tetrapolymers comprising any combination of the monomers listed above.
- Preferred terpolymers and tetrapolymers include polymers comprising isobutylene, isoprene and divinylbenzene, polymers comprising isobutylene, para-alkylstyrene (preferably paramethyl styrene) and isoprene, polymers comprising cyclopentadiene, isobutylene, and paraalkyl styrene (preferably paramethyl styrene), polymers of isobutylene cyclopentadiene and isoprene, polymers comprising cyclopentadiene, isobutylene, and methyl cyclopentadiene, polymers comprising isobutylene, paramethylstyrene and cyclopentadiene.
- the Lewis acid (also referred to as the co-initiator or catalyst) may be any Lewis acid based on metals from Group 4, 5, 13, 14 and 15 of the Periodic Table of the Elements, including boron, aluminum, gallium, indium, titanium, zirconium, tin, vanadium, arsenic, antimony, and bismuth.
- the metals are aluminum, boron and titanium, with aluminum being desirable.
- Illustrative examples include A1C1 3 , (alkyl)AlCl 2 , (C 2 H 5 ) 2 A1C1 and (C 2 H 5 ) 3 A1 2 C1 3 , BF 3 , SnCl 4 , TiCl 4 .
- BF 3 is not the chosen Lewis acid.
- the Group 4, 5 and 14 Lewis acids have the general formula MX 4 ; wherein M is Group 4, 5, or 14 metal; and X is a halogen independently selected from the group consisting of fluorine, chlorine, bromine, and iodine, preferably chlorine. X may also be a psuedohalogen.
- pseudohalogen is defined to be an azide, an isocyanate, a thiocyanate, an isothiocyanate or a cyanide.
- Non-limiting examples include titanium tetrachloride, titanium tetrabromide, vanadium tetrachloride, tin tetrachloride and zirconium tetrachloride.
- the Group 4, 5, or 14 Lewis acids may also contain more than one type of halogen.
- Non-limiting examples include titanium bromide trichloride, titanium dibromide dichloride, vanadium bromide trichloride, and tin chloride trifluoride.
- Group 4, 5 and 14 Lewis acids useful in this invention may also have the general formula MR devisX4-n ; wherein M is Group 4, 5, or 14 metal; wherein R is a monovalent hydrocarbon radical selected from the group consisting of Ci to C1 2 alkyl, aryl, arylalkyl, alkylaryl and cycloalkyl radicals; and n is an integer from 0 to 4; X is a halogen independently selected from the group consisting of fluorine, chlorine, bromine, and iodine, preferably chlorine. X may also be a psuedohalogen.
- pseudohalogen is defined to be an azide, an isocyanate, a thiocyanate, an isothiocyanate or a cyanide.
- arylalkyl refers to a radical containing both aliphatic and aromatic structures, the radical being at an alkyl position.
- alkylaryl refers to a radical containing both aliphatic and aromatic structures, the radical being at an aryl position.
- Non-limiting examples of these Lewis acids include benzyltitanium trichloride, dibenzyltitanium dichloride, benzylzirconium trichloride, dibenzylzirconium dibromide, methyltitanium trichloride, dimethyltitanium difluoride, dimethyltin dichloride and phenylvanadium trichloride.
- Group 4, 5 and 14 Lewis acids useful in this invention may also have the general formula M(RO) n R' m X4-(m+n); wherein M is Group 4, 5, or 14 metal, wherein RO is a monovalent hydrocarboxy radical selected from the group consisting of Ci to C 3 o alkoxy, aryloxy, arylalkoxy, alkylaryloxy radicals; R' is a monovalent hydrocarbon radical selected from the group consisting of Ci to C ⁇ 2 alkyl, aryl, arylalkyl, alkylaryl and cycloalkyl radicals as defined above; n is an integer from 0 to 4 and m is an integer from 0 to 4 such that the sum of n and m is not more than 4; X is a halogen independently selected from the group consisting of fluorine, chlorine, bromine, and iodine, preferably chlorine.
- X may also be a psuedohalogen.
- pseudohalogen is defined to be an azide, an isocyanate, a thiocyanate, an isothiocyanate or a cyanide.
- alkoxy and aryloxy are structural equivalents to alkoxides and phenoxides respectively.
- arylalkoxy refers to a radical containing both aliphatic and aromatic structures, the radical being at an alkoxy position.
- alkylaryl refers to a radical containing both aliphatic and aromatic structures, the radical being at an aryloxy position.
- Non-limiting examples of these Lewis acids include methoxytitanium trichloride, n- butoxytitanium trichloride, di(iso ⁇ ropoxy)titanium dichloride, phenoxytitanium tribromide, phenylmethoxyzirconium trifluoride, methyl methoxytitanium dichloride, methyl methoxytin dichloride and benzyl isopropoxyvanadium dichloride.
- X may also be a psuedohalogen.
- pseudohalogen is defined to be an azide, an isocyanate, a thiocyanate, an isothiocyanate or a cyanide.
- arylalkylacyloxy refers to a radical containing both aliphatic and aromatic structures, the radical being at an alkyacyloxy position.
- alkylarylacyloxy refers to a radical containing both aliphatic and aromatic structures, the radical being at an arylacyloxy position.
- Non-limiting examples of these Lewis acids include acetoxytitanium trichloride, benzoylzirconium tribromide, benzoyloxytitanium trifluoride, isopropoyloxytin trichloride, methyl acetoxytitanium dichloride and benzyl benzoyloxyvanadium chloride.
- Group 5 Lewis acids useful in this invention may also have the general formula MOX 3 ; wherein M is a Group 5 metal; wherein X is a halogen independently selected from the group consisting of fluorine, chlorine, bromine, and iodine, preferably chlorine.
- a non-limiting example is vanadium oxytrichloride.
- the Group 13 Lewis acids useful in this invention have the general formula MX 3 ; wherein M is a Group 13 metal and X is a halogen independently selected from the group consisting of fluorine, chlorine, bromine, and iodine, preferably chlorine. X may also be a psuedohalogen.
- pseudohalogen is defined to be an azide, an isocyanate, a thiocyanate, an isothiocyanate or a cyanide.
- Non-limiting examples include aluminum trichloride, boron trifluoride, gallium trichloride, and indium trifluoride.
- Group 13 Lewis acids useful in this invention may also have the general formula: MR n X 3 . n wherein M is a Group 13 metal; R is a monovalent hydrocarbon radical selected from the group consisting of Ci to C ⁇ 2 alkyl, aryl, arylalkyl, alkylaryl and cycloalkyl radicals; and n is an number from 0 to 3; X is a halogen independently selected from the group consisting of fluorine, chlorine, bromine, and iodine, preferably chlorine. X may also be a psuedohalogen.
- pseudohalogen is defined to be an azide, an isocyanate, a thiocyanate, an isothiocyanate or a cyanide.
- arylalkyl refers to a radical containing both aliphatic and aromatic structures, the radical being at an alkyl position.
- alkylaryl refers to a radical containing both aliphatic and aromatic structures, the radical being at an aryl position.
- Non-limiting examples of these Lewis acids include ethylaluminum dichloride, methylaluminum dichloride, benzylaluminum dichloride, isobutylgallium dichloride, diethylaluminum chloride, dimethylaluminum chloride, ethylaluminum sesquichloride, methylaluminum sesquichloride, trimethylaluminum and triethylaluminum.
- Group 13 Lewis acids useful in this invention may also have the general formula M(RO) n R' m X3-(m+n); wherein M is a Group 13 metal; wherein RO is a monovalent hydrocarboxy radical selected from the group consisting of C ⁇ to C 3 o alkoxy, aryloxy, arylalkoxy, alkylaryloxy radicals; R' is a monovalent hydrocarbon radical selected from the group consisting of Ci to 2 alkyl, aryl, arylalkyl, alkylaryl and cycloalkyl radicals as defined above; n is a number from 0 to 3 and m is an number from 0 to 3 such that the sum of n and m is not more than 3; X is a halogen independently selected from the group consisting of fluorine, chlorine, bromine, and iodine, preferably chlorine.
- X may also be a psuedohalogen.
- pseudohalogen is defined to be an azide, an isocyanate, a thiocyanate, an isothiocyanate or a cyanide.
- alkoxy and aryloxy are structural equivalents to alkoxides and phenoxides respectively.
- arylalkoxy refers to a radical containing both aliphatic and aromatic structures, the radical being at an alkoxy position.
- alkylaryl refers to a radical containing both aliphatic and aromatic structures, the radical being at an aryloxy position.
- Non-limiting examples of these Lewis acids include methoxyaluminum dichloride, ethoxyaluminum dichloride, 2,6-di-tert-butylphenoxyaluminum dichloride, methoxy methylaluminum chloride, 2,6-di-tert-butylphenoxy methylaluminum chloride, isopropoxygallium dichloride and phenoxy methylindium fluoride.
- X may also be a psuedohalogen.
- pseudohalogen is defined to be an azide, an isocyanate, a thiocyanate, an isothiocyanate or a cyanide.
- arylalkylacyloxy refers to a radical containing both aliphatic and aromatic structures, the radical being at an alkyacyloxy position.
- alkylarylacyloxy refers to a radical containing both aliphatic and aromatic structures, the radical being at an arylacyloxy position.
- Non-limiting examples of these Lewis acids include acetoxyaluminum dichloride, benzoyloxyaluminum dibromide, benzoyloxygallium difluoride, methyl acetoxyaluminum chloride, and isopropoyloxyindium trichloride.
- the Group 15 Lewis acids have the general formula MX y , wherein
- M is a Group 15 metal and X is a halogen independently selected from the group consisting of fluorine, chlorine, bromine, and iodine, preferably chlorine and y is 3, 4 or 5. X may also be a psuedohalogen.
- pseudohalogen is defined to be an azide, an isocyanate, a thiocyanate, an isothiocyanate or a cyanide.
- Non-limiting examples include antimony hexachloride, antimony hexafluoride, and arsenic pentafluoride.
- the Group 15 Lewis acids may also contain more than one type of halogen.
- Non- limiting examples include antimony chloride pentafluoride, arsenic trifluoride, bismuth trichloride and arsenic fluoride tetrachloride.
- Group 15 Lewis acids useful in this invention may also have the general formula MR n X y - n; wherein M is a Group 15 metal; wherein R is a monovalent hydrocarbon radical selected from the group consisting of Ci to C ⁇ 2 alkyl, aryl, arylalkyl, alkylaryl and cycloalkyl radicals; and n is an integer from 0 to 4; v is 3, 4 or 5 such that n is less than ⁇ ; X is a halogen independently selected from the group consisting of fluorine, chlorine, bromine, and iodine, preferably chlorine. X may also be a pseudohalogen.
- halogen is defined to be an azide, an isocyanate, a thiocyanate, an isothiocyanate or a cyanide.
- arylalkyl refers to a radical containing both aliphatic and aromatic structures, the radical being at an alkyl position.
- alkylaryl refers to a radical containing both aliphatic and aromatic structures, the radical being at an aryl position.
- Lewis acids include tetraphenylantimony chloride and triphenylantimony dichloride.
- Group 15 Lewis acids useful in this invention may also have the general formula M(RO) n R' m X y - (m+n ), wherein M is a Group 15 metal, wherein RO is a monovalent hydrocarboxy radical selected from the group consisting of to C 3 o alkoxy, aryloxy, arylalkoxy, alkylaryloxy radicals; R' is a monovalent hydrocarbon radical selected from the group consisting of Ci to 2 alkyl, aryl, arylalkyl, alkylaryl and cycloalkyl radicals as defined above; n is an integer from 0 to 4 and m is an integer from 0 to 4 and y is 3, 4 or 5 such that the sum of n and m is less than y; X is a halogen independently selected from the group consisting of fluorine, chlorine, bromine, and iodine, preferably chlorine.
- X may also be a psuedohalogen.
- pseudohalogen is defined to be an azide, an isocyanate, a thiocyanate, an isothiocyanate or a cyanide.
- alkoxy and aryloxy are structural equivalents to alkoxides and phenoxides respectively.
- arylalkoxy refers to a radical containing both aliphatic and aromatic structures, the radical being at an alkoxy position.
- alkylaryl refers to a radical containing both aliphatic and aromatic structures, the radical being at an aryloxy position.
- Non-limiting examples of these Lewis acids include tetrachloromethoxyantimony, dimethoxytrichloroantimony, dichloromethoxyarsine, chlorodimethoxyarsine, and difluoromethoxyarsine.
- X may also be a psuedohalogen.
- pseudohalogen is defined to be an azide, an isocyanate, a thiocyanate, an isothiocyanate or a cyanide.
- arylalkylacyloxy refers to a radical containing both aliphatic and aromatic structures, the radical being at an alkyacyloxy position.
- alkylarylacyloxy refers to a radical containing both aliphatic and aromatic structures, the radical being at an arylacyloxy position.
- Lewis acids include acetatotetrachloroantimony, (benzoato) tetrachloroantimony, and bismuth acetate chloride.
- Lewis acids may be any of those useful in cationic polymerization of isobutylene copolymers including: aluminum trichloride, aluminum tribromide, ethylaluminum dichloride, ethylaluminum sesquichloride, diethylaluminum chloride, methylaluminum dichloride, methylaluminum sesquichloride, dimethylaluminum chloride, boron trifluoride, titanium tetrachloride, etc. with ethylaluminum dichloride and ethylaluminum sesquichloride being preferred.
- Lewis acids such as methylaluminoxane (MAO) and specifically designed weakly coordinating Lewis acids such as B(C F 5 ) 3 are also suitable Lewis acids within the context of the invention.
- Initiators useful in this invention are those initiators which are capable of being complexed in a suitable diluent with the chosen Lewis acid to yield a complex which rapidly reacts with the olefm thereby forming a propagating polymer chain.
- Illustrative examples include Br ⁇ nsted acids such as H 2 O, HCI, RCOOH (wherein R is an alkyl group), and alkyl halides, such as (CH 3 ) 3 CC1, C 6 H 5 C(CH 3 ) 2 C1 and (2-Chloro-2,4,4-trimethylpentane).
- transition metal complexes such as metallocenes and other such materials that can act as single site catalyst systems, such as when activated with weakly coordinating Lewis acids or Lewis acid salts have been used to initiate isobutylene polymerization.
- the reactor and the catalyst system are substantially free of water.
- Substantially free of water is defined as less than 30 ppm (based upon total weight of the catalyst system), preferably less than 20 ppm, preferably less than 10 ppm, preferably less than 5 ppm, preferably less than 1 ppm.
- water is selected as an initiator, it is added to the catalyst system to be present at greater than 30 ppm, preferably greater than 40 ppm, and even more preferably greater than 50 ppm (based upon total weight of the catalyst system).
- the initiator comprises one or more of a hydrogen halide, a carboxylic acid, a carboxylic acid halide, a sulfonic acid, an alcohol, a phenol, a tertiary alkyl halide, a tertiary aralkyl halide, a tertiary alkyl ester, a tertiary aralkyl ester, a tertiary alkyl ether, a tertiary aralkyl ether, alkyl halide, aryl halide, alkylaryl halide, or arylalkylacid halide.
- Preferred hydrogen halide initiators include hydrogen chloride, hydrogen bromide and hydrogen iodide.
- a particularly preferred hydrogen halide is hydrogen chloride.
- carboxylic acids included both aliphatic and aromatic carboxylic acids.
- carboxylic acids useful in this invention include acetic acid, propanoic acid, butanoic acid; cinnamic acid, benzoic acid, 1- chloroacetic acid, dichloroacetic acid, trichloroacetic acid, trifluoroacetic acid, p- chlorobenzoic acid, and p-fluorobenzoic acid.
- Particularly preferred carboxylic acids include trichloroacetic acid, trifluoroacteic acid, and p-fluorobenzoic acid.
- Carboxylic acid halides useful in this invention are similar in structure to carboxylic acids with the substitution of a halide for the OH of the acid.
- the halide may be fluoride, chloride, bromide, or iodide, with the chloride being preferred.
- Preparation of acid halides from the parent carboxylic acids are known in the prior art and one skilled in the art should be familiar with these procedures.
- Carboxylic acid halides useful in this invention include acetyl chloride, acetyl bromide, cinnamyl chloride, benzoyl chloride, benzoyl bromide, trichloroacetyl chloride, trifluoroacetylchloride, trifluoroacetyl chloride and p- fluorobenzoylchloride.
- Particularly preferred acid halides include acetyl chloride, acetyl bromide, trichloroacetyl chloride, trifluoroacetyl chloride and p- fluorobenzoyl chloride.
- Sulfonic acids useful as initiators in this invention include both aliphatic and aromatic sulfonic acids.
- preferred sulfonic acids include methanesulfonic acid, trifluoromethanesulfonic acid, trichloromethanesulfonic acid and p-toluenesulfonic acid.
- Sulfonic acid halides useful in this invention are similar in structure to sulfonic acids with the substitution of a halide for the OH of the parent acid.
- the halide may be fluoride, chloride, bromide or iodide, with the chloride being preferred.
- Preparation of the sulfonic acid halides from the parent sulfonic acids are known in the prior art and one skilled in the art should be familiar with these procedures.
- Preferred sulfonic acid halides useful in this invention include methanesulfonyl chloride, methanesulfonyl bromide, trichloromethanesulfonyl chloride, trifluoromethanesulfonyl chloride and p-toluenesulfonyl chloride.
- Alcohols useful in this invention include methanol, ethanol, propanol, 2-propanol, 2-methylpropan-2-ol, cyclohexanol, and benzyl alcohol.
- Phenols useful in this invention include phenol; 2-methylphenol; 2,6- dimethylphenol; p-chlorophenol; p-fluorophenol; 2,3,4,5,6-pentafluorophenol; and 2-hydroxynaphthalene.
- Preferred tertiary alkyl and aralkyl initiators include tertiary compounds represented by the formula below:
- X is a halogen, pseudohalogen, ether, or ester, or a mixture thereof, preferably a halogen, preferably chloride and Rl, R2 and R3 are independently any linear, cyclic or branched chain alkyls, aryls or arylalkyls, preferably containing 1 to 15 carbon atoms and more preferably 1 to 8 carbon atoms, n is the number of initiator sites and is a number greater than or equal to 1 , preferably between 1 to 30, more preferably n is a number from 1 to 6.
- the arylalkyls may be substituted or unsubstituted.
- arylalkyl is defined to mean a compound containing both aromatic and aliphatic structures.
- Preferred examples of initiators include 2-chloro-2,4,4- trimethylpentane ; 2-bromo-2,4,4-trimethylpentane; 2-chloro-2-methylpropane; 2- bromo-2-methylpropane; 2-chloro-2,4,4,6,6-pentamethylheptane; 2-bromo- 2,4,4,6,6-pentamethylheptane; 1 -chloro- 1-methylethylbenzene; 1- chloroadamantane; 1-chloroethylbenzene; 1, 4-bis(l-chloro-l-methylethyl) benzene; 5-tert-butyl-l,3-bis( 1 -chloro- 1-methylethyl) benzene; 2-acetoxy-2,4,4- trimethylpentane ; 2-benzoyloxy
- Another preferred initiator is a polymeric halide, one of Rl, R2 or
- R3 is an olefin polymer and the remaining R groups are defined as above.
- Preferred olefin polymers include polyisobutylene, polypropylene, and polyvinylchloride.
- the polymeric initiator may have halogenated tertiary carbon positioned at the chain end or along or within the backbone of the polymer.
- the product may contain polymers which have a comb like structure and/or side chain branching depending on the number and placement of the halogen atoms in the olefin polymer.
- the use of a chain end tertiary polymer halide initiator provides a method for producing a product which may contain block copolymers.
- Particularly preferred initiators may be any of those useful in cationic polymerization of isobutylene copolymers including: hydrogen chloride, 2-chloro-2,4,4-trimethylpentane, 2-chloro-2-methylpropane, 1 -chloro- 1 - methylethylbenzene, and methanol.
- Catalyst system compositions useful in this invention typically comprise (1) an initiator and (2) a Lewis acid coinitiator or other metal complex described herein.
- the Lewis acid coinitiator is present anywhere from about 0.1 moles times the moles of initiator present to about 200 times the moles of initiator present.
- the Lewis acid coinitiator is present at anywhere from about 0.8 times the moles of initiator present to about 20 times the moles of initiator present.
- the initiator is present at anywhere from about 0.1 moles per liter to about 10 "6 moles per liter. It is of course understood that greater or lesser amounts of initiator are still within the scope of this invention.
- the amount of the catalyst employed will depend on desired molecular weight and molecular weight distribution of the polymer being produced. Typically the range will be from about lxlO "6 moles per liter to 3 x 10 "2 moles per liter and most preferably from 10 "4 to 10 "3 moles per liter.
- Catalyst systems useful in this invention may further comprise a catalyst composition comprising of a reactive cation and a weakly-coordinating anion ("WC anion" or “WCA” or “NCA”).
- the catalyst composition comprising the WC anion will include a reactive cation and in certain instances are novel catalyst systems.
- a weakly-coordinating anion is defined as an anion which either does not coordinate to the cation or which is weakly coordinated to the cation and when the anion is functioning as the stabilizing anion in this invention the WCA does not transfer an anionic fragment or substituent to the cation thus creating a neutral by-product or other neutral compound.
- Preferred examples of such weakly-coordinating anions include: alkyltris(pentafluorophenyl) boron
- the cation is any cation that can add to an olefin to create a carbocation.
- the anion may be combined with the cation by any method known to those of ordinary skill in the art.
- the WC anion is introduced into the diluent as a compound containing both the anion and the cation in the form of the active catalyst system.
- a composition containing the WC anion fragment is first treated to produce the anion in the presence of the cation or reactive cation source, i.e. the anion is activated.
- the WC anion may be activated without the presence of the cation or cation source which is subsequently introduced.
- a composition containing the anion and a composition containing the cation are combined and allowed to react to form a by-product, the anion and the cation.
- Any metal or metalloid compound capable of forming an anionic complex which is incapable of transferring a substituent or fragment to the cation to neutralize the cation to produce a neutral molecule may be used as the WC anion.
- any metal or metalloid capable of forming a coordination complex which is stable in water may also be used or contained in a composition comprising the anion.
- Suitable metals include, but are not limited to aluminum, gold, platinum and the like.
- Suitable metalloids include, but are not limited to, boron, phosphorus, silicon and the like.
- WC anions may be represented by the following general formula: wherein:
- M' is a metal or metalloid
- Ql to Qn are ' independently, bridged or unbridged hydride radicals, dialkylamido radicals, alkoxide and aryloxide radicals, hydrocarbyl and substituted-hydrocarbyl radicals, halocarbyl and substituted-halocarbyl radicals and hydrocarbyl and halocarbyl-substituted organometalloid radicals and any one, but not more than one of ⁇ to Q n may be a halide radical; m is an integer representing the formal valence charge of M; n is the total number of ligands q, and d is an integer greater than or equal to 1.
- Cp2 rMe2 may be combined with a composition comprising the anion (WCA ⁇ R + ) where R + acts with a Me group to leave the C 2Zr + Me WCA" catalyst system.
- Preferred WC anions comprising boron may be represented by the following general formula:
- B is a boron in a valence state of 3 ;
- Ax ⁇ and A ⁇ 2 are the same or different aromatic or substituted-aromatic hydrocarbon radicals containing from about 6 to about 20 carbon atoms and may be linked to each other through a stable bridging group;
- X3 and X4 are, independently, hydride radicals, hydrocarbyl and substituted-hydrocarbyl radicals, halocarbyl and substituted-halocarbyl radicals, hydrocarbyl- and halocarbyl-substituted organometalloid radicals, disubstituted pnictogen radicals, substituted chalcogen radicals and halide radicals, with the proviso that X3 and X4 will not be halide at the same time.
- a and Ar2 may, independently, be any aromatic of substituted-aromatic hydrocarbon radical.
- Suitable aromatic radicals include, but are not limited to, phenyl, naphthyl and anthracenyl radicals.
- Suitable substituents on the substituted-aromatic hydrocarbon radicals include, but are not necessarily limited to, hydrocarbyl radicals, organometalloid radicals, alkoxy and aryloxy radicals, fluorocarbyl and fluorohydrocarbyl radicals and the like such as those useful as X3 and X4.
- the substituent may be ortho, meta or para, relative to the carbon atoms bonded to the boron atom.
- each may be the same or a different aromatic or substituted- aromatic radical as are Aq and Ar2, or the same may be a straight or branched alkyl, alkenyl or alkynyl radical, a cyclic hydrocarbon radical or an alkyl- substituted cyclic hydrocarbon radical.
- Arj and Ar2 could be linked to either X3 or X4.
- X3 and X4 may also be linked to each other through a suitable bridging group.
- boron components which may be used as WC anions are: tetravalent boron compounds such as tetra(phenyl)boron, tetra(p-tolyl)boron, tetra(o-tolyl)boron, tetra(pentafluorophenyl)boron, tetra(o,p-dimethylphenyl)boron, tetra(m,m- dimethylphenyl)boron, (p-tri-fluoromethylphenyl)boron and the like.
- a particularly preferred WC anion comprising boron may be represented by the following general formula:
- Polymeric Q substituents on the most preferred anion offer the advantage of providing a highly soluble ion-exchange activator component and final catalyst.
- Soluble catalysts and/or precursors are often preferred over insoluble waxes, oils, or solids because they can be diluted to a desired concentration and can be transferred easily using simple equipment in commercial processes.
- WC anions containing a plurality of boron atoms may be represented by the following general formulae: or
- X, X', X", X6, X7 and X8 are, independently, hydride radicals, halide radicals, hydrocarbyl radicals, substituted-hydrocarbyl radicals, halocarbyl radicals, substituted-halocarbyl radicals, or hydrocarbyl- or halocarbyl-substituted organometalloid radicals;
- Examples of preferred WC anions of this invention comprising a plurality of boron atoms include:
- a borane or carborane anion satisfying the general formula: wherein: ax is either 0 or 1 ; ex is either 1 or 2; ax + ex 2; bx is an integer ranging from about 10 to 12; or
- WC anions include: carboranes such as dodecaborate, decachlorodecaborate, dodecachlorododecaborate, 1-carbadecaborate, 1-carbadecaborate, 1- trimethylsilyl- 1 -carbadecaborate;
- Borane and carborane complexes and salts of borane and carborane anions such as decaborane(14), 7,8-dicarbadecaborane(13), 2,7- dicarbaundecaborane(13), undecahydrido-7,8-dimethyl-7,8-dicarbaundecaborane, 6-carbadecaborate(12), 7-carbaundecaborate, 7,8-dicarbaudecaborate; and
- Metallaborane anions such as bis(nonahydrido-l,3- dicarbanonaborato)cobaltate(III), bis(undecahydrido-7,8-dicarbaundecaborato) ferrate(III), bis(undecahy drido-7 , 8 -dicarbaundecaborato) cobaltate(III), bis(undecahydrido-7,8-dicarbaunaborato) nickelate(III), bis(nonahydrido-7,8- dimethyl-7,8-dicarbaundecaborato)ferrate(III), bis(tribromooctahydrido-7,8- dicarbaundecaborato)cobaltate(III), bis(undecahydridodicarbadodecaborato) cobaltate(III) and bis(undecahy drido-7 -carbaundecaborato) cobaltate(III).
- the WC anion compositions most preferred for forming the catalyst system used in this process are those containing a trisperfluorophenyl boron, tetrapentafluorphenyl boron anion and / or two or more trispentafluorophenyl boron anion groups covalently bond to a central atomic molecular or polymeric complex or particle.
- the WC anion is combined with one or more cations that are selected from different classes of cations and cation sources.
- Some preferred classes are:
- Rj, R2 and R3 are hydrogen, alkyl, aryl, aralkyl groups or derivatives thereof, preferably C ⁇ to C30 alkyl, aryl, aralkyl groups or derivatives thereof; (C) substituted silylium; preferably those represented by the formula:
- R , R2 and R3 are hydrogen, alkyl, aryl, aralkyl groups or derivatives thereof, preferably Cj to C30 alkyl, aryl, aralkyl groups or derivatives thereof;
- compositions capable of generating a proton (D) compositions capable of generating a proton
- Ri , R2 and R3 are hydrogen, alkyl, aryl, aralkyl groups or derivatives thereof, preferably Ci to C30 alkyl, aryl, aralkyl groups or derivatives thereof, and R* is Ge, Sn or Pb.
- Preferred cyclopentadienyl transition metal derivatives include transition metals that are a mono-, bis- or tris- cyclopentadienyl derivative of a group 4, 5 or 6 transition metal, preferably a mono-cyclopentadienyl (Mono-Cp) or bis-cyclopentadienyl (Bis-Cp) group 4 transition metal compositions, particularly a zirconium, titanium or hafnium compositions.
- cyclopentadienyl derivatives that may be combined with weakly-coordinating anions are represented by the following formulae:
- (A-Cp) is either (Cp)(Cp*) or Cp-A'-Cp*;
- Cp and Cp* are the same or different cyclopentadienyl rings substituted with from zero to five substituent groups S, each substituent group S being, independently, a radical group which is a hydrocarbyl, substituted-hydrocarbyl, halocarbyl, substituted-halocarbyl, hydrocarbyl-substituted organometalloid, halocarbyl-substituted organometalloid, disubstituted boron, disubstituted pnictogen, substituted chalcogen or halogen radicals, or Cp and Cp* are cyclopentadienyl rings in which any two adjacent S groups are joined forming a C4 to C20 ring to give a saturated or unsaturated polycyclic cyclopentadienyl ligand;
- R is a substituent on one of the cyclopentadienyl radicals which is also bonded to the metal atom;
- A' is a bridging group, which group may serve to restrict rotation of the Cp and Cp* rings or (C5H5_y_ x S x ) and JR'( z _i_y) groups;
- M is a group 4, 5, or 6 transition metal; y is O or 1;
- (C5H5_y_ x S x ) is a cyclopentadienyl ring substituted with from zero to five
- JR'( z _l_y) is a heteroatom ligand in which J is a group 15 element with a coordination number of three or a group 16 element with a coordination number of 2, preferably nitrogen, phosphorus, oxygen or sulfur;
- R" is a hydrocarbyl group, preferably an alkyl group
- X and X are independently a hydride radical, hydrocarbyl radical, substituted hydrocarbyl radical, halocarbyl radical, substituted halocarbyl radical, and hydrocarbyl- and halocarbyl-substituted organometalloid radical, substituted pnictogen radical, or substituted chalcogen radicals; and
- L is an olefin, diolefin or aryne ligand, or a neutral Lewis base.
- Preferred examples include substances that are represented by the formula:
- R ⁇ , R2 and R3 are independently hydrogen, or a linear, branched or cyclic aromatic or aliphatic groups, preferably a Cj to C20 aromatic or aliphatics group, provided that only one of R ⁇ , R2 or R3 may be hydrogen. In a preferred embodiment none of Rj, R2 or R3 are H.
- Preferred aromatics include phenyl, toluyl, xylyl, biphenyl and the like.
- Preferred aliphatics include methyl, ethyl, propyl, butyl, pentyl, hexyl, octyl, nonyl, decyl, dodecyl, 3-methylpentyl, 3,5,5- trimethylhexyl and the like.
- R , R2 and R3 are phenyl groups
- the addition of an aliphatic or aromatic alcohol significantly enhances the polymerization of isobutylene.
- substituted silylium compositions preferably trisubstituted silylium compositions are combined with WCA's to polymerize monomers.
- Preferred silylium cations are those represented by the formula:
- Ri, R and R 3 are independently hydrogen, or a linear, branched or cyclic aromatic or aliphatic group, with the proviso that only one of i, R 2 and R 3 may be hydrogen.
- none of Ri, R 2 and R 3 are H.
- Ri R 2 and R 3 are, independently, a Ci to C 2 o aromatic or aliphatic group. More preferably, Ri, R 2 and R 3 are independently a Ci to C 8 alkyl group. Examples of useful aromatic groups may be selected from the group consisting of phenyl, tolyl, xylyl and biphenyl.
- Non-limiting examples of useful aliphatic groups may be selected from the group consisting of methyl, ethyl, propyl, butyl, pentyl, hexyl, octyl, nonyl, decyl, dodecyl, 3-methylpentyl and 3,5,5-trimethylhexyl.
- a particularly preferred group of reactive substituted silylium cations may be selected from the group consisting of trimethylsilylium, triethylsilylium and benzyldimethylsilylium.
- a fourth source for the cation is any compound that will produce a proton when combined with the weakly-coordinating anion or a composition containing a weakly-coordinating anion.
- Protons may be generated from the reaction of a stable carbocation salt which contains a weakly-coordinating, non- nucleophilic anion with water, alcohol or phenol present to produce the proton and the corresponding by-product, (ether in the case of an alcohol or phenol and alcohol in the case of water).
- Such reaction may be preferred in the event that the reaction of the carbocation salt is faster with the protonated additive as compared with its reaction with the olefin.
- Other proton generating reactants include thiols, carboxylic acids, and the like.
- Another method to generate a proton comprises combining a group
- a Lewis base that does not interfere with polymerization, such as an olefin. It has been observed that when a Lewis base, such as isobutylene, is present with the group 1 or 2 metal and the water, a proton is generated.
- the weakly-coordinating anion is also present in the "wet" diluent such that active catalyst is generated when the group 1 or 2 metal is added.
- Cp transition metal cations can be combined into an active catalyst in at least two ways.
- a first method is to combine a compound comprising the CpTm + with a second compound comprising the WCA" which then react to form by-product and the active "weakly-coordinating" pair.
- the CpTm + compound may also be directly combined with the WCA" to form the active catalyst system.
- WCA is combined with the cation/cation source in ratios of 1 to 1, however ratios of 100 to 1 (CpTm + to WCA) also work in the practice of this invention.
- Active cationic catalysts can be prepared by reacting a transition metal compound with some neutral Lewis acids, such as B(C F )3 n , which upon reaction with a hydrolyzable ligand (X) of the transition metal compound forms an anion, such as ([B(CgF5)3(X)]”), which stabilizes the cationic transition metal species generated by the reaction.
- B(C F )3 n some neutral Lewis acids
- X hydrolyzable ligand
- a novel aspect of this invention is the active carbocationic catalyst complex which is formed and which can be represented by the formulae:
- each G is independently hydrogen or an aromatic or aliphatic group, preferably a Cj to CJOO aliphatic group, and g is an integer representing the number of monomer units incorporated into the growing polymer chain, g is preferably a number greater than or equal to 1, preferably a number from 1 to about 150,000.
- WCA is any weakly-coordinating anion as described above. All other symbols are as defined above.
- this invention also provides active catalyst compositions which can be represented by the formulae:
- each G is independently a aliphatic or aromatic group, preferably a Ci to
- WCA is any weakly-coordinating anion as described above. All other symbols are as defined above.
- Generation of trisubstituted carbocations and silylium cations may be performed before use in the polymerization or in situ. Pre-formation and isolation of the cation or the stable cation salts may be accomplished by reacting the alkali or alkaline earth metal salt of the weakly-coordinating anion with the corresponding halogen of the potential carbocation or silylium similarly to methods known in the art. Formation of the substituted carbocations or silylium in situ occurs in a similar manner to stable salts, but within the vessel and at the desired temperature of polymerization. The advantage of the latter procedure is that it is capable of producing carbocations or silylium cations otherwise too unstable to be handled by the first method. The cation or the precursor to the cation is typically used in 1 to 1 ratios with the WCA, however ratios of 1 to 100
- a novel aspect of this invention is the active carbocationic catalyst complex which is formed and which can be represented by the formulae:
- each G is independently hydrogen or a hydrocarbyl group, preferably a C ⁇ to CjoO aliphatic group, and g is a n integer representing the number of monomer units incorporated into the growing polymer chain, g is preferably a number greater than or equal to 1, preferably a number from 1 to about 150,000.
- WCA is any weakly-coordinating anion as described above. All other symbols are as defined above.
- each G is independently hydrogen or an aliphatic or aromatic group, preferably a C to C ⁇ QO aliphatic group, and g is a n integer representing the number of monomer units incorporated into the growing polymer chain, g is preferably a number greater than or equal to 1, preferably a number from 1 to about 150,000.
- WCA is any weakly-coordinating anion as described above. All other symbols are as defined above.
- compositions of germanium, tin or lead may be used in combination with the WCA's described herein.
- Preferred compositions include those which are represented by the formula:
- R — R* R _ wherein Rj, R2 and R3 are hydrogen, alkyl, aryl, aralkyl groups or derivatives thereof, preferably Cj to C30 alkyl, aryl, aralkyl groups or derivatives thereof, and R* is Ge, Sn or Pb.
- the R groups are a C to C1 alkyl, preferably methyl, ethyl, propyl, or butyl.
- Hydrofluorocarbons are preferably used as diluents in the present invention, alone or in combination with other hydrofluorocarbons or in combination with other diluents.
- hydrofluorocarbons (“HFC's” or “HFC”) are defined to be saturated or unsaturated compounds consisting essentially of hydrogen, carbon and fluorine, provided that at least one carbon, at least one hydrogen and at least one fluorine are present.
- the diluent comprises hydrofluorocarbons represented by the formula: C x H y F z wherein x is an integer from 1 to 40, alternatively from 1 to 30, alternatively from 1 to 20, alternatively from 1 to 10, alternatively from 1 to 6, alternatively from 2 to 20 alternatively from 3 to 10, alternatively from 3 to 6, most preferably from 1 to 3, wherein y and z are integers and at least one.
- Illustrative examples include fluoromethane; difluoromethane; trifluoromethane; fluoroethane; 1,1-difluoroethane; 1,2-difluoroethane; 1,1,1- trifluoroethane; 1,1,2-trifluoroethane; 1,1,1,2-tetrafluoroethane; 1,1,2,2- tetrafluoroethane; 1,1,1,2,2-pentafluoroethane; 1-fluoropropane; 2-fluoro ⁇ ropane; 1,1-difluoropropane; 1,2-difluoropropane; 1,3-difluoropropane; 2,2- difluoropropane; 1,1,1-trifluoropropane; 1,1,2-trifluoropropane; 1,1,3- trifluoropropane; 1,2,2-trifluoropropane; 1,2,3-trifluoropropane; 1,1,1,2- tetra
- HFC's include difluoromethane, trifluoromethane, 1,1-difluoroethane, 1,1,1- trifluoroethane, fluoromethane, and 1,1,1,2-tetrafluoroethane.
- unsaturated hydrofluorocarbons include vinyl fluoride; 1,1-difluoroethene; 1,2-difluoroethene; 1,1,2-trifluoroethene; 1- fluoropropene, 1,1-difluoropropene; 1,2-difluoropropene; 1,3-difluoropropene; 2,3-difluoropropene; 3,3-difluoropropene; 1,1,2-trifluoropropene; 1,1,3- trifluoropropene; 1,2,3-trifluoropropene; 1,3,3-trifluoropropene; 2,3,3- trifluoropropene; 3,3,3-trifluoropropene; 1 -fluoro- 1-butene; 2-fluoro-l-butene; 3- fluoro-1-butene; 4-fluoro- 1-butene; 1,1-difluoro- 1-buten
- the diluent comprises non-perfluorinated compounds or the diluent is a non-perfluorinated diluent.
- Perfluorinated compounds being those compounds consisting of carbon and fluorine.
- the blend may comprise perfluorinated compound, preferably, the catalyst, monomer, and diluent are present in a single phase or the aforementioned components are miscible with the diluent as described in further detail below.
- the blend may also comprise chlorofluorocarbons (CFC's), or those compounds consisting of chlorine, fluorine, and carbon.
- suitable diluents include hydrofluorocarbons with a dielectric constant of greater than 10 at -85°C, preferably greater than 15, more preferably greater than 20, more preferably greater than 25, more preferably 40 or more.
- dielectric constant may be less than 10, or by adding larger amounts of initiator or transfer agent when the dielectric constant is above 10.
- a purpose-built instrument such as the Brookhaven Instrument Corporation BIC-870 may be used to measure dielectric constant of diluents directly.
- a comparison of the dielectric constants ( ⁇ ) of a few selected diluents at -85°C is provided below and graphically depicted in Figure 1.
- one or more HFC's are used in combination with another diluent or mixtures of diluents.
- additional diluents include hydrocarbons, especially hexanes and heptanes, halogenated hydrocarbons, especially chlorinated hydrocarbons and the like.
- Specific examples include but are not limited to propane, isobutane, pentane, methycyclopentane, isohexane, 2- methylpentane, 3-methylpentane, 2-methylbutane, 2,2-dimethylbutane, 2,3- dimethylbutane, 2-methylhexane, 3-methylhexane, 3-ethylpentane, 2,2- dimethylpentane, 2,3-dimethylpentane, 2,4-dimethylpentane, 3, 3 -dimethyl pentane, 2-methylheptane, 3-ethylhexane, 2,5-dimethylhexane, 2,24,- trimethylpentane, octane, heptane, butane, ethane, methane, nonane, decane, dodecane, undecane, hexane, methyl cyclohexane, cyclopropane, cyclobutane
- non-reactive olefins may be used as diluents in combination with HFC's.
- examples include, but are not limited to, ethylene, propylene, and the like.
- the HFC is used in combination with a chlorinated hydrocarbon such as methyl chloride. Additional embodiments include using the HFC in combination with hexanes or methyl chloride and hexanes.
- the HFC's are used in combination with one or more gases inert to the polymerization such as carbon dioxide, nitrogen, hydrogen, argon, neon, helium, krypton, zenon, and/or other inert gases that are preferably liquid at entry to the reactor.
- gases include carbon dioxide and/or nitrogen.
- the HFC's are used in combination with one or more nitrated alkanes, including Ci to C 4 o nitrated linear, cyclic or branched alkanes.
- Preferred nitrated alkanes include, but are not limited to, nitromethane, nitroethane, nitropropane, nitrobutane, nitropentane, nitrohexane, nitroheptane, nitrooctane, nitrodecane, nitrononane, nitrododecane, nitroundecane, nitrocyclomethane, nitrocycloethane, nitrocyclopropane, nitrocyclobutane, nitrocyclopentane, nitrocyclohexane, nitrocycloheptane, nitrocyclooctane, nitrocyclodecane, nitrocyclononane, nitrocyclododecane, nitrocycloundecane,
- the HFC is typically present at 1 to 100 volume % based upon the total volume of the diluents, alternatively between 5 and 100 volume %, alternatively between 10 and 100 volume %, alternatively between 15 and 100 volume %, alternatively between 20 and 100 volume %, alternatively between 25 and 100 volume %, alternatively between 30 and 100 volume %, alternatively between 35 and 100 volume %, alternatively between 40 and 100 volume %, alternatively between 45 and 100 volume %, alternatively between 50 and 100 volume %, alternatively between 55 and 100 volume %, alternatively between 60 and 100 volume %, alternatively between 65 and 100 volume %, alternatively between 70 and 100 volume %, alternatively between 75 and 100 volume %, alternatively between 80 and 100 volume %, alternatively between 85 and 100 volume %, alternatively between 90 and 100 volume %, alternatively between 95 and 100 volume %, alternatively between 97 and 100 volume %, alternatively between 98 and 100 volume %, and alternatively between 99 and 100 volume %.
- the HFC is blended with one or more chlorinated hydrocarbons.
- the HFC is selected from the group consisting of difluoromethane, trifluoromethane, 1,1-difraoroethane, 1,1,1- trifluoroethane, and 1,1,1,2-tetrafluoroethane and mixtures thereof.
- the diluent or diluent mixture is selected based upon its solubility in the polymer. Certain diluents are soluble in the polymer. Preferred diluents have little to no solubility in the polymer. Solubility in the polymer is measured by forming the polymer into a film of thickness between 50 and 100 microns, then soaking it in diluent (enough to cover the film) for 4 hours at -75°C. The film is removed from the diluent, exposed to room temperature for 90 seconds to evaporate excess diluent from the surface of the film, and weighed. The mass uptake is defined as the percentage increase in the film weight after soaking.
- the diluent or diluent mixture is chosen so that the polymer has a mass uptake of less than 4 wt%, preferably less than 3 wt%, preferably less than 2 wt%, preferably less than 1 wt%, more preferably less than 0.5 wt%.
- the diluent or diluent mixture is selected such that the difference between the measured glass transition temperature Tg of the polymer with less than 0.1 wt% of any diluent, unreacted monomers and additives is within 15°C of the Tg of the polymer measured after it has been formed into a film of thickness between 50 and 100 microns, that has been soaked in diluent (enough to cover the film) for 4 hours at -75°C.
- the glass transition temperature is determined by differential scanning calorimetry (DSC). Techniques are well described in the literature, for example, B.
- the Tg values are within 12°C of each other, preferably within 11°C of each other, preferably within 10°C of each other, preferably within 9°C of each other, preferably within 8°C of each other, preferably within 7°C of each other, preferably within 6°C of each other, preferably within 5°C of each other, preferably within 4°C of each other, preferably within 3°C of each other, preferably within 3°C of each other, preferably within 2°C of each other, preferably within 1°C of each other.
- the invention may be practiced in continuous and batch processes.
- the invention may be practiced in a plug flow reactor and/or stirred tank reactors.
- this invention may be practiced in "butyl reactors.”
- Illustrative examples include any reactor selected from the group consisting of a continuous flow stirred tank reactor, a plug flow reactor, a moving belt or drum reactor, a jet or nozzle reactor, a tubular reactor, and an autorefrigerated boiling- pool reactor.
- heat can be removed by use of heat transfer surfaces, such as in a tubular reactor where a coolant is on one side of the tube and the polymerizing mixture is on the other side. Heat may also be removed by evaporating the polymerizing mixture, such as may be found in an autorefrigerated boiling pool type reactor.
- a plug flow reactor where a portion of the polymerizing mixture is evaporated as the mixture proceeds tlirough the reactor.
- heat is removed in a plug flow reactor through surface heat transfer using coolant on the other side of a heat transfer surface.
- Another type of reactor is a jet or nozzle reactor. These reactors have a short residence time where the monomer, diluent and catalyst system are combined in the jet or nozzle and the polymerization occurs as the mixture passes tlirough the nozzle at high velocity.
- Preferred reactors include continuous flow stirred tank reactors, whether operating in batch or continuous mode, and whether operating in a tank with an agitator or in a tube type reactor. Preferred reactors also include reactors where the polymerization occurs on one side of a heat transfer surface and the coolant is present on the other side.
- An example is a reactor where tubes containing coolant run inside the reactor polymerization zone. Another example would be where the polymerization occurs inside a tube and the coolant is present on the outside of the tube in a shell.
- This invention may also be practiced in batch reactors where the monomers, diluent, and catalyst are charged to the reactor and then polymerization proceeds to completion (such as by quenching) and the polymer is then recovered.
- the invention is practiced using a slurry polymerization process.
- other polymerization methods are contemplated such as a solution polymerization process.
- the polymerization processes of the invention may be cationic polymerization processes.
- the polymerization is carried out where the catalyst, monomer, and diluent are present in a single phase.
- the polymerization is carried-out in a continuous polymerization process in which the catalyst, monomer(s), and diluent are present as a single phase.
- the monomers, catalyst(s), and initiator(s) are all miscible in the diluent or diluent mixture, i.e., constitute a single phase, while the polymer precipitates from the diluent with good separation from the diluent.
- reduced or no polymer "swelling" is exhibited as indicated by little or no Tg suppression of the polymer and/or little or no diluent mass uptake as shown in Figure 2.
- polymerization in the diluents of the present invention provides for high polymer concentration to be handled at low viscosity with good heat transfer, reduced reactor fouling, homogeneous polymerization and/or the convenience of subsequent reactions to be run directly on the resulting polymer mixture.
- the reacted monomers within the reactor form part of a slurry.
- the concentration of the solids in the slurry is equal to or greater than 10 vol%.
- the concentration of solids in the slurry is present in the reactor equal to or greater than 25 vol%.
- the concentration of solids in the slurry is less than or equal to 75 vol%.
- the concentration of solids in slurry is present in the reactor from 1 to 70 vol%.
- the concentration of solids in slurry is present in the reactor from 5 to 70 vol%.
- the concentration of solids in slurry concentration is present in the reactor from 10 to 70 vol%.
- the concentration of solids in slurry concentration is present in the reactor from 15 to 70 vol%. In yet another embodiment, the concentration of solids in slurry concentration is present in the reactor from 20 to 70 vol%. In yet another embodiment, the concentration of solids in slurry concentration is present in the reactor from 25 to 70 vol%. In yet another embodiment, the concentration of solids in slurry concentration is present in the reactor from 30 to 70 vol%. In yet another embodiment, the concentration of solids in slurry concentration is present in the reactor from 40 to 70 vol%.
- a continuous flow stirred tank-type reactor may be used.
- the reactor is generally fitted with an efficient agitation means, such as a turbo- mixer or impeller(s), an external cooling jacket and/or internal cooling tubes and/or coils, or other means of removing the heat of polymerization to maintain the desired reaction temperature, inlet means (such as inlet pipes) for monomers, diluents and catalysts (combined or separately), temperature sensing means, and an effluent overflow or outflow pipe which withdraws polymer, diluent and unreacted monomers among other things, to a holding drum or quench tank.
- the reactor is purged of air and moisture.
- the reactors are preferably designed to deliver good mixing of the catalyst and monomers within the reactor, good turbulence across or within the heat transfer tubes or coils, and enough fluid flow throughout the reaction volume to avoid excessive polymer accumulation or separation from the diluent.
- the reactor pump impeller can be of the up- pumping variety or the down-pumping variety.
- the reactor will contain sufficient amounts of the catalyst system of the present invention effective to catalyze the polymerization of the monomer containing feed-stream such that a sufficient amount of polymer having desired characteristics is produced.
- the feed-stream in one embodiment contains a total monomer concentration greater than 5 wt% (based on the total weight of the monomers, diluent, and catalyst system), preferably greater than 15 wt%, greater than 30 wt% in another embodiment. In yet another embodiment, the feed-stream will contain from 5 wt% to 50 wt% monomer concentration based on the total weight of monomer, diluent, and catalyst system.
- the feed-stream is substantially free from silica cation producing species.
- substantially free of silica cation producing species it is meant that there is no more than 0.0005 wt% based on the total weight of the monomers of these silica cation producing species in the feed stream.
- Typical examples of silica cation producing species are halo-alkyl silica compounds having the formula RjR 2 R 3 SiX or R ⁇ R 2 SiX 2 , etc., wherein "R” is an alkyl and "X" is a halogen.
- the reaction conditions will be such that desirable temperature, pressure and residence time are effective to maintain the reaction medium in the liquid state and to produce the desired polymers having the desired characteristics.
- the monomer feed-stream is typically substantially free of any impurity which is adversely reactive with the catalyst under the polymerization conditions.
- the monomer feed preferably should be substantially free of bases (such as caustic), sulfur-containing compounds (such as H 2 S, COS, and organo-mercaptans, e.g., methyl mercaptan, ethyl mercaptan), nitrogen-containing bases, oxygen containing bases such as alcohols and the like.
- bases such as caustic
- sulfur-containing compounds such as H 2 S, COS, and organo-mercaptans, e.g., methyl mercaptan, ethyl mercaptan
- nitrogen-containing bases such as alcohols and the like.
- monomer feed may be less pure, typically not less than 95% based on total olefmic content, more preferably not less than 98%, not less than 99%.
- the impurities are present at less than 10,000 ppm (by weight), preferably less that 500 pp
- reaction time, temperature, concentration, the nature of the reactants, and similar factors determine product molecular weights.
- the polymerization reaction temperature is conveniently selected based on the target polymer molecular weight and the monomer to be polymerized as well as standard process variable and economic considerations, e.g., rate, temperature control, etc.
- the temperature for the polymerization is less than 0°C, preferably between -10°C and the freezing point of the slurry in one embodiment, and from -25 °C to -120°C in another embodiment.
- the polymerization temperature is from -40°C to -100°C, and from -70°C to - 100°C in yet another embodiment.
- the temperature range is from -80°C to -100°C. Consequently, different reaction conditions will produce products of different molecular weights. Synthesis of the desired reaction product may be achieved, therefore, through monitoring the course of the reaction by the examination of samples taken periodically during the reaction; a technique widely employed in the art.
- the polymerization temperature is within 10°C above the freezing point of the diluent, preferably within 8°C above the freezing point of the diluent, preferably within 6°C above the freezing point of the diluent, preferably within 4°C above the freezing point of the diluent, preferably within 2°C above the freezing point of the diluent, preferably within 1°C above the freezing point of the diluent.
- the phrase "within X°C above the freezing point of the diluent" it means the freezing point of the diluent plus X ° C. For example if the freezing point of the diluent is -98°C, then 10°C above the freezing point of the diluent is -88°C.
- the reaction pressure will be from above 0 to 14,000 kPa in one embodiment (where 0 kPa is a total vacuum), from 7 kPa to 12,000 kPa in another embodiment, from 100 kPa to 2000 kPa in another embodiment, from 200 kPa to 1500 kPa in another embodiment, from 200 kPa to 1200 kPa in another embodiment, and from 200 kPa to 1000 kPa in yet another embodiment, from 7 kPa to 100 kPa in another embodiment, from 20 kPa to 70 kPa in another embodiment, from 40 kPa to 60 kPa in yet another embodiment, from 1000 kPa to 14,000 kPa in another embodiment, from 3000 kPa to 10,000 kPa in another embodiment, and from 3,000 kPa to 6,000 kPa in yet another embodiment.
- the order of contacting the monomer feed-stream, catalyst, initiator, and diluent may vary from one embodiment to another.
- the initiator and Lewis acid are pre- contacted by mixing together in the selected diluent for a prescribed amount of time ranging from 0.01 second to 10 hours, and then is injected into the continuous reactor through a catalyst nozzle or injection apparatus.
- Lewis acid and the initiator are added to the reactor separately.
- the initiator is blended with the feed monomers before injection to the reactor. Desirably, the monomer is not contacted with the Lewis acid, or the Lewis acid combined with the initiator before the monomers enter the reactor.
- the initiator and Lewis acid are allowed to pre-contact by mixing together in the selected diluent at temperatures between -40°C and the freezing point temperature of the diluent, with a contact time between 0.01 seconds and several hours, and between 0.1 seconds and 5 minutes, preferably less than 3 minutes, preferably between 0.2 seconds and 1 minute before injection into the reactor.
- the initiator and Lewis acid are allowed to pre-contact by mixing together in the selected diluent at temperatures between 80 and -150°C, typically between -40°C and -98°C.
- the overall residence time in the reactor can vary, depending upon, e.g., catalyst activity and concentration, monomer concentration, feed injection rate, production rate, reaction temperature, and desired molecular weight, and generally will be between about a few seconds and five hours, and typically between about 10 and 60 minutes.
- Variables influencing residence time include the monomer and diluent feed injection rates and the overall reactor volume.
- the catalyst (Lewis acid) to monomer ratio utilized will be those conventional in this art for carbocationic polymerization processes.
- the monomer to catalyst mole ratios will typically be from 500 to 10000, and in the range of 2000 to 6500 in another embodiment.
- the ratio of Lewis acid to initiator is from 0.5 to 10, or from 0.75 to 8.
- the overall concentration of the initiator in the reactor is typically from 5 to 300 ppm or 10 to 250 ppm.
- the concentration of the initiator in the catalyst feed stream is typically from 50 to 3000 ppm in one embodiment.
- Another way to describe the amount of initiator in the reactor is by its amount relative to the polymer. In one embodiment, there is from 0.25 to 20 moles polymer/mole initiator, and from 0.5 to 12 mole polymer/mole initiator in another embodiment.
- the reactor will contain sufficient amounts of the catalyst system of the present invention to catalyze the polymerization of the monomer containing feed-stream such that a sufficient amount of polymer having desired characteristics is produced.
- the feed-stream in one embodiment contains a total monomer concentration greater than 20 wt% (based on the total weight of the monomers, diluent, and catalyst system), greater than 25 wt% in another embodiment.
- the feed-stream will contain from 30 wt% to 50 wt% monomer concentration based on the total weight of monomer, diluent, and catalyst system.
- Catalyst efficiency (based on Lewis acid) in the reactor is maintained between 10,000 pounds of polymer per pound of catalyst and 300 pounds of polymer per pound of catalyst and desirably in the range of 4000 pounds of polymer per pound of catalyst to 1000 pounds of polymer per pound of catalyst by controlling the molar ratio of Lewis acid to initiator.
- the polymerization of cationically polymerizable monomers comprises several steps.
- a reactor having a pump impeller capable of up-pumping or down-pumping is provided.
- the pump impeller is typically driven by an electric motor with a measurable amperage.
- the reactor typically is equipped with parallel vertical reaction tubes within a jacket containing liquid ethylene.
- the total internal volume, including the tubes, is greater than 30 to 50 liters, thus capable of large scale volume polymerization reactions.
- the reactor typically uses liquid ethylene to draw the heat of the polymerization reaction away from the forming slurry.
- the pump impeller keeps a constant flow of slurry, diluent, catalyst system and unreacted monomers through the reaction tubes.
- a feed-stream of the cationically polymerizable monomer(s) (such as isoprene and isobutylene) in a polar diluent is charged into the reactor, the feed-stream containing less than 0.0005 wt% of cation producing silica compounds, and typically free of aromatic monomers.
- the catalyst system is then charged into the reactor, the catalyst system having a Lewis acid and an initiator present in a molar ratio of from 0.50 to 10.0.
- the feed- stream of monomers and catalyst system are allowed to contact one another, the reaction thus forming a slurry of polymer (such as butyl rubber), wherein the solids in the slurry has a concentration of from 20 vol% to 50 vol%.
- the thus formed polymer (such as butyl rubber) is allowed to exit the reactor through an outlet or outflow line while simultaneously allowing the feed-stream charging to continue, thus constituting the continuous slurry polymerization.
- the present invention improves this process in a number of ways, e.g., by ultimately reducing the amount of polymer accumulation on the reactor walls, heat transfer surfaces, agitators and/or impeller(s), and in the outflow line or exit port, as measured by pressure inconsistencies or "jumps.”
- the resultant polymer from one embodiment of the invention is a polyisobutylene/isoprene polymer (butyl rubber) that has a molecular weight distribution of from about 2 to 5, and an unsaturation of from 0.5 to 2.5 mole per 100 mole of monomer.
- This product may be subjected to subsequent halogenation to afford a halogenated butyl rubber.
- this invention relates to:
- a polymerization process comprising contacting one or more monomers, one or more Lewis acids and one or more initiators in the presence of a diluent comprising one or more hydrofluorocarbons (HFC's):
- the polymers of the invention provide chemical and physical characteristics that make them highly useful in wide variety of applications.
- the low degree of permeability to gases accounts for the largest uses of these polymers, namely inner tubes and tire innerliners. These same properties are also of importance in air cushions, pneumatic springs, air bellows, accumulator bags, and pharmaceutical closures.
- the thermal stability of the polymers of the invention make them ideal for rubber tire-curing bladders, high temperature service hoses, and conveyor belts for hot material handling.
- the polymers exhibit high damping and have uniquely broad damping and shock absorption ranges in both temperature and frequency. They are useful in molded rubber parts and find wide applications in automobile suspension bumpers, auto exhaust hangers, and body mounts.
- the polymers of the instant invention are also useful in tire sidewalls and tread compounds. In sidewalls, the polymer characteristics impart good ozone resistance, crack cut growth, and appearance.
- the polymers of the invention may also be blended. Properly formulated blends with high diene rubbers that exhibit phase co-continuity yield excellent sidewalls. Improvements in wet, snow, and ice skid resistances and in dry traction without compromises in abrasion resistance and rolling resistance for high performance tires can be accomplished by using the polymers of the instant invention.
- Blends of the polymers of the invention with thermoplastic resins are used for toughening of these compounds.
- High-density polyethylene and isotactic polypropylene are often modified with 5 to 30 wt % of polyisobutylene.
- the instant polymers provide for a highly elastic compound that is processable in thermoplastic molding equipment.
- the polymers of the instant invention may also be blended with polyamides to produce other industrial applications.
- the polymers of the instant invention may also be used as adhesives, caulks, sealants, and glazing compounds. They are also useful as plasticizers in rubber formulations with butyl, SBR, and natural rubber. In linear low density polyethylene (LLDPE) blends, they induce cling to stretch-wrap films. They are also widely employed in lubricants as dispersants and in potting and electrical cable filling materials.
- LLDPE linear low density polyethylene
- the polymers of the invention make them also useful in chewing-gum, as well as in medical applications such as pharmaceutical stoppers, and the arts for paint rollers.
- the polymerizations were performed glass reaction vessels, equipped with a teflon turbine impeller on a glass stir shaft driven by an external electrically driven stirrer. The size and design of the glass vessels is noted for each set of examples.
- the head of the reactor included ports for the stir shaft, thermocouple and addition of initiator/coinitiator solutions.
- the reactor was cooled to the desired reaction temperature, listed in the Tables, by immersing the assembled reactor into a pentane or isohexane bath in the dry box. The temperature of the stirred hydrocarbon bath was controlled to ⁇ 2°C. All apparatus in liquid contact with the reaction medium were dried at 120°C and cooled in a nitrogen atmosphere before use.
- Isobutylene (Matheson or ExxonMobil) and methyl chloride (Air Products) were dried by passing the gas through three stainless steel columns containing barium oxide and were condensed and collected as liquids in the dry box.
- methyl chloride was dried by passing the gas through stainless steel columns containing silica gel and molecular sieves. Both materials were condensed and collected as liquids in the dry box.
- Isoprene Aldrich
- p-Methylstyrene (Aldrich) was dried over calcium hydride and distilled under vacuum.
- TMPC1 (2-chloro-2,4,4,-trimethylpentane) was prepared from 2,4,4-trimethylpentene-l and a 2.0 mol/L solution of HCI in diethyl ether. The TMPC1 was distilled before use.
- the HCI (Aldrich, 99% pure) stock solution was prepared by dissolving a desirable amount of HCI gas in dry MeCl to achieve 2-3 % concentration by weight.
- the hydrofluorocarbons that were collected as clear, colorless liquids at -95 °C were used as received. Hydrofluorocarbons that remained cloudy or had visible insoluble precipitates at -95°C were distilled before use.
- Propane (Aldrich) used as received, was condensed and used as a liquid.
- Alkylaluminum dichlorides (Aldrich) were used as hydrocarbon solutions. These solutions were either purchased or prepared from the neat alkylaluminum dichloride.
- the slurry copolymerizations were performed by dissolving monomer and comonomer into the liquefied hydrofluorocarbon at polymerization temperature and stirred at a pre-determined stirring speed between 800 to 1000 ⁇ m.
- the use of a processor controlled electric stirring motor allowed control of the stirring speed to within 5 ⁇ m.
- the initiator/coinitiator solutions were prepared either in the hydrofluorocarbon or, for convenience of small-scale experiments, in a small volume of methyl chloride.
- the initiator/coinitiator solutions were prepared by dissolving the initiator into the diluent (specified in each of the examples) and adding, with mixing, a 1.0 M solution of the alkylaluminum halide.
- the initiator/coinitiator solution was used immediately.
- the initiator/coinitiator solution was added dropwise to the polymerization using a chilled glass Pasteur pipette or, optionally, a jacketed dropping funnel for examples using the 500 ml glass reaction vessels.
- a second or third initiator/coinitiator addition is specified in the examples, we refer to the preparation and addition of a second or third batch of freshly prepared initiator/coinitiator solution identical in volume and concentrations to the first batch.
- the physical behavior of the rubber particles and the state of fouling was determined at the end of the addition of each catalyst batch by stopping and removing the stir shaft and probing the particles with a chilled spatula. Stirring was begun again and the reaction quenched with the addition of greater than 100 microliters of methanol. Conversion is reported as weight percent of the monomers converted to polymer.
- Polymer molecular weights can be determined on other SEC instruments using different calibration and run protocols.
- SEC also know as GPC or gel permeation chromatography
- GPC gel permeation chromatography
- Comonomer inco ⁇ oration was determined by 1H-NMR spectrometry. NMR measurements were obtained at a field strength corresponding to 400 MHz or 500 MHz. 1H-NMR spectra were recorded at room temperature on a Bruker Avance NMR spectrometer system using CDC1 3 solutions of the polymers. All chemical shifts were referenced to TMS.
- triple detection SEC size exclusion chromatography
- Triple detection SEC was performed on a Waters (Milford, Massachusetts) 150C chromatograph operated at 40°C equipped a Precision Detectors (Bellingham, Massachusetts) PD2040 light scattering detector, a Viscotek (Houston, Texas) Model 15 OR viscometry detector and a Waters differential refractive index detector (integral with the 150C). The detectors were connected in series with the light scattering detector being first, the viscometry detector second and the differential refractive index detector third. Tetrahydrofuran was used as the eluent (0.5 ml/min.) with a set of three Polymer Laboratories, Ltd.
- the value of g' is defined to be less than or equal to one and greater than or equal to zero.
- g' is equal or nearly equal to one, the polymer is considered to be linear.
- g' is significantly less than one, the sample is long chain branched.
- a g' is calculated for each data slice of the chromatographic curve.
- a viscosity average g' or g' v is. av g. s calculated across the entire molecular weight distribution.
- the scaling factor g'vis.a v g. is calculated from the average intrinsic viscosity of the sample.
- a polymer that is substantially free of long chain branching is defined to be a polymer for which g'vi s .avg. is determined to be greater than or equal to 0.978, alternatively, greater than or equal to 0.980, alternatively, greater than or equal to 0.985, alternatively, greater than or equal to 0.990, alternatively, greater than or equal to 0.995, alternatively, greater than or equal to 0.998, alternatively, greater than or equal to 0.999, as determined by triple detection SEC as described herein.
- Table 1 lists the results of polymerizations conducted at -90 to -95
- initiator/coinitiator solutions were prepared in 1.3 ml of methyl chloride using 1.6 microliters of TMPCl and 11.5 microliters of a 1.0 M hexane solution of methylaluminum dichloride (MADC).
- MADC methylaluminum dichloride
- b Three initiator/coinitiator batches added to the reactor
- c Two initiator/coinitiator batches added to the reactor
- d ethylaluminum chloride (EADC) used in place of MADC
- Examples 13 and 14 are comparative examples. A 100 ml glass mini- resin kettle was used for these examples. TMPCl (2-chloro-2,4,4- trimethylpentane) was used as an initiator in these examples.
- Table 3 lists the results of polymerizations conducted at -95 °C in hydrofluorocarbon / methyl chloride blends.
- a 100 ml glass mini-resin kettle was used for these examples.
- TMPCl (2-chloro-2,4,4-trimethylpentane) was used as an initiator in these examples.
- a polymerization was conducted with methoxyaluminum dichloride at -95°C.
- the initiator/coinitiator solution was prepared by dissolving 0.93 microliters of anhydrous methanol into 2.6 ml of liquid 1,1,1,2- tetrafluoroethane at -35°C. To this solution was added 23 microliters of a 1.0 mol/L solution of ethylaluminum dichloride in pentane. This solution was stirred for 10 minutes. A second solution was prepared in the same way. To each solution, 3.2 microliters of 2-chloro-2,4,4-trimethylpentane was added with stirring and cooled -95°C. Both solutions were added dropwise to the polymerization solution with a chilled pipette. . A 100 ml glass mini-resin kettle was used for this example.
- Table 6 lists the results of a polymerization conducted at -95°C conducted in an 85/15 (V/V) blend of 1,1, 1,2 -tetrafluoroethane and 1,1- difluoroethane. This run was made with 30 ml of diluent, 5.4 ml of isobutylene, 0.26 ml of isoprene and used a initiator/coinitiator solution prepared in 2.6 ml methyl chloride using 3.2 microliters of TMPCl and 32.0 microliters of a 1.0 M hexane solution of methylaluminum dichloride (MADC). A 100 ml glass mini- resin kettle was used for this example.
- Table 7 lists the results of polymerizations that were conducted at -
- initiator/coinitiator solutions were prepared in 2.6 ml of methyl chloride using 3.2 microliters of TMPCl and 23.0 microliters of a 1.0 M hexane solution of ethylaluminum dichloride (EADC).
- b 32.0 microliters of a 1.0 M hexane solution of ethylaluminum dichloride was used instead of the amount noted in (a) above, c: 1.6 microliters of 2-chloro-2-methylpropane used in place of the TMPCl used in (a).
- Table 8 lists the results of polymerizations that were conducted at -
- Examples 36 and 37 are comparative examples.
- a three-neck 500 ml glass reactor was used for these examples. Prior to each polymerization, 300 ml of monomer feed containing 10 wt% of monomers were charged into the chilled reactor. The initiator/coinitiator molar ratio was controlled at 1/3 and the concentration was set at 0.1 wt% EADC in MeCl. The initiator/coinitiator solution was added dropwise to the polymerization mixture and the rate of addition is controlled in such as way so that the reactor temperature raise did not exceed 4 C. The amount of initiator/coinitiator solution added in each depended on the desired monomer conversion target.
- the examples in Table 8 demonstrate the production of high molecular weight butyl rubber using an EADC/HC1 initiator system in CHF 2 CF 3 and CH 3 CHF 2 diluents.
- the molecular weight of the butyl polymers made in CHF 2 CF 3 and CH 3 CHF 2 were significantly higher than polymers made in MeCl at similar monomer conversion under similar conditions.
- the polydispersity (Mw/Mn) of the butyl polymers made in both CH 2 FCF 3 and CH 3 CHF 2 was narrower and closer to the most probable polydispersity of 2.0 than the polymers made in MeCl under similar experimental conditions.
- the isoprene inco ⁇ oration in the copolymers made in MeCl falls between CH 2 FCF 3 and CH 3 CHF 2 .
- the polymer slurry particles made in both CH 2 FCF 3 and CH 3 CHF 2 appeared to be significantly less sticky during handling than the polymer slurry particles made in MeCl under similar conditions.
- Table 9 lists the results of copolymerizations of isobutylene and p- methylstyrene that were conducted at -95 °C in hydrofluorocarbons and methyl chloride for comparison.
- Examples 41 and 42 are comparative examples.
- a three-neck 500 ml glass reactor was used for these examples. Prior to each polymerization, 300 ml of monomer feed containing 10 wt% of monomers were charged into the chilled reactor. The initiator/coinitiator molar ratio was controlled at 1/3 and the concentration was set at 0.1 wt% EADC in MeCl.
- the initiator/coinitiator solution was added dropwise to the polymerization mixture and the rate of addition is controlled in such as way so that the reactor temperature raise did not exceed 4 C.
- the amount of initiator/coinitiator solution added in each depended on the desired monomer conversion target.
- Table 10 lists the results of copolymerizations of isobutylene/p- methylstyrene and isobutylene/isoprene that were conducted at -95°C in an 80/20 mixture (by volume) of CH 2 FCF 3 and CH 3 CHF 2 .
- a three-neck 500 ml glass reactor was used for these examples. Prior to each polymerization, 300 ml of monomer feed containing 10 wt% of monomers were charged into the chilled reactor. The initiator/coinitiator molar ratio was controlled at 1/3 and the concentration was set at 0.1 wt% EADC in MeCl.
- the initiator/coinitiator solution was added dropwise to the polymerization mixture and the rate of addition is controlled in such as way so that the reactor temperature raise did not exceed 4 C.
- the amount of initiator/coinitiator solution added in each depended on the desired monomer conversion target.
- Table 10 demonstrates the production of high molecular weight isobutylene-isoprene copolymers and isobutylene-pMS copolymers using an EADC/HC1 initiator system in a mixture of CH 2 FCF 3 and CH 3 CHF 2 as the polymerization diluent.
- the polymer slurry particles in the CH 2 FCF 3 /C 3 CHF 2 mixture demonstrate the same non-stickiness appearance as in a pure CH 2 FCF 3 or CH 3 CHF 2 diluent described above.
- Examples 48 - 117 exemplify copolymerization of isobutylene with other comonomers. The copolymerizations have been run at two temperatures and in four diluents.
- a stock solution of ethylaluminum dichloride (EADC) and hydrogen chloride (HCI) was prepared in methyl chloride by adding 0.320 ml of a 1.0 mol/L HCI solution in 1,1,1,2-tetrafluoroethane and 0.960 ml of a 1.0 mol/L ethylaluminum dichloride solution in hexane to 100 ml of methyl chloride. Polymerizations were run by adding, dropwise, 1.5 ml of this stock EADC/HC1 solution to the stirred monomer solutions. Polymerizations were terminated with the addition of 0.2 ml of methanol.
- EADC ethylaluminum dichloride
- HCI hydrogen chloride
- Isobutylene was copolymerized with either p-t-butylstyrene (t-BuS) (0.36 ml per run) or indene (Ind) (0.23 ml per run) as indicated in Table 12.
- a stock solution of ethylaluminum dichloride (EADC) and hydrogen chloride (HCI) was prepared in methyl chloride by adding 0.320 ml of a 1.0 mol/L HCI solution in 1,1,1,2- tetrafluoroethane and 0.960 ml of a 1.0 mol/L ethylaluminum dichloride solution in hexane to 100 ml of methyl chloride. Polymerizations were run by adding, dropwise, 1.5 ml of this stock EADC/HC1 solution to the stirred monomer solutions except for examples 72, 73, 74, 81, 82, 83 and 87.
- EADC ethylaluminum dichloride
- HCI hydrogen chloride
- Isobutylene was copolymerized with one of the following comonomers or comonomer pairs as indicated in Table 13: isoprene (IP) (0.20 ml per run), p- methylstyrene (pMS) (0.26 ml per run), p-t-butylstyrene (t-BuS) (0.36 ml per run), indene (Ind) (0.23 ml per run), ⁇ -pinene ( ⁇ P) (0.31 ml per run) or a 50/50 mol/mol blend (IP/pMS) of isoprene (0.10 ml) and p-methylstyrene (0.13 ml) per run as indicated in Table 20.
- IP isoprene
- pMS p- methylstyrene
- t-BuS p-butylstyrene
- IP indene
- ⁇ P ⁇ -pinene
- a stock solution of ethylaluminum dichloride (EADC) and hydrogen chloride (HCI) was prepared in methyl chloride by adding 0.320 ml of a 1.0 mol/L HCI solution in 1,1,1,2-tetrafluoroethane and 0.960 ml of a 1.0 mol/L ethylaluminum dichloride solution in hexane to 100 ml of methyl chloride. Polymerizations were run by adding, dropwise, 1.5 ml of this stock EADC/HC1 solution to the stirred monomer solutions, except for examples 98, 99, 100, 101, 102, and 103. For examples 98, 99 and 100, 3.0 ml of EADC/HC1 solution was used.
- Table 14 lists the results of copolymerization of isobutylene and butadiene (0.15 ml per ran) that were conducted at -95°C in methyl chloride (as a comparative, examples 107 and 108), 1,1,1,2-tetrafluoroethane, 1,1-difluoroethane or a 20 wt.% blend (CH 3 CHF 2 /CH 2 FCF 3 ) of 1,1-difluoroethane in 1,1,1,2- tetrafluroethane.
- a stock solution of ethylaluminum dichloride (EADC) and hydrogen chloride (HCI) was prepared in methyl chloride by adding 0.320 ml of a 1.0 mol/L HCI solution in 1,1,1,2-tetrafluoroethane and 0.960 ml of a 1.0 mol/L ethylaluminum dichloride solution in hexane to 100 ml of methyl chloride. Polymerizations were run by the adding, dropwise, 1.5 ml of this stock EADC/HCl solution to the stirred monomer solutions. Polymerizations were terminated with the addition of 0.2 ml of methanol.
- EADC ethylaluminum dichloride
- HCI hydrogen chloride
- Examples 118, 119, 120, 123, 124, 125, and 126 are comparative examples with Examples 118 and 119 being examples of this invention.
- a separate stock solution of ethylaluminum dichloride and hydrogen chloride was used for Examples 125 and 126.
- This solution was prepared from the addition of 2.0 ml of a 0.16 mol/L HCI solution in 1,1,1,2-tetrafluoroethane and 0.960 ml of a 1.0 mol/L ethylaluminum dichloride solution in hexane to 100 ml of methyl chloride.
- the final mol/L concentrations of ethylaluminum dichloride and hydrogen chloride in the stock solution is the same for both preparations. Polymerizations were terminated with the addition of 0.2 ml of methanol.
- Examples 127 - 136 are comparative examples with Examples 137 - 141 being examples of this invention.
- This solution was prepared from the addition of 0.034 ml of a 0.93 mol/L HCI solution in 1,1,1,2- tetrafluoroethane and 0.0960 ml of a 1.0 mol/L ethylaluminum dichloride solution in hexane to 10 ml of methyl chloride.
- the final mol/L concentrations of ethylaluminum dichloride and hydrogen chloride in the stock solution are the same for both preparations.
- a separate stock solution of methylaluminum dichloride (MADC) / 2-chloro-2,4,4-trimethylpentane (TMPCl) was used for Examples 132 and 133.
- the MADC/TMPC1 solution was prepared from the addition of 6.6 microliters of TMPCl and 0.0960 ml of a 1.0 mol/L methylaluminum dichloride solution in hexane to 10 ml of methyl chloride.
- the total volume of the stock solution added to the polymerization for each example is listed in Table 16.
- Table 17 lists the results of copolymerizations of isobutylene isoprene that were conducted at -95 °C CH 2 FCF 3 .
- the isobutylene/isoprene feed ratio was changed for each example.
- a three-neck 500 ml glass reactor was used for these examples. Prior to each polymerization, 300 ml of monomer feed containing 10 wt% of monomers were charged into the chilled reactor.
- the initiator/coinitiator molar ratio was controlled at 1/3 and the concentration was set at 0.1 wt% EADC in MeCl.
- the initiator/coinitiator solution was added dropwise to the polymerization mixture and the rate of addition is controlled in such as way o so that the reactor temperature raise did not exceed 4 C.
- the amount of initiator/coinitiator solution added in each depended on the desired monomer conversion target. Table 17
- the initiator/coinitiator molar ratio was controlled at 1/3 and the concentration was set at 0.1 wt% EADC in MeCl.
- the initiator/coinitiator solution was added dropwise to the polymerization mixture and the rate of addition is controlled in such as way so that the reactor temperature raise did not exceed 4 C.
- the amount of initiator/coinitiator solution added in each depended on the desired monomer conversion target.
- the data for these polymerizations are shown in Figure 3 as a plot of peak molecular weight (M p ) versus the monomer conversion in wt%. The expected decline of molecular weight with increasing conversion is observed.
- a copolymerization of isobutylene and cyclopentadiene was conducted at -93 °C in CH 2 FCF 3 .
- a molar ratio of 97/3 of isobutylene / cyclopentadiene was used for this polymerization at 10.8 wt% monomers in the feed.
- Cyclopentadiene was freshly cracked for this experiment.
- the initiator/coinitiator solution was prepared by dissolving 200 microliters of a 1.0 mol/L solution of hydrogen chloride in CH 2 FCF 3 into 50 ml of pre-chilled CH 2 FCF 3 .
- the mixture was cooled to -90°C by immersing the test tube into the cold bath. Reported observations were made after the completed heating/cooling cycle. If after this first heat/cool cycle, the catalyst did not dissolve, 0.5 ml of methyl chloride was added. The heat/cool cycle was repeated. Subsequent additions of 0.5 ml of methyl chloride were made following each heat/cool cycle until the EADC was observed to dissolve or a 50/50 V/N blend was achieved. The following observations were made and recorded.
- ethylaluminum dichloride ethylaluminum dichloride
- Pentane refers to normal pentane.
- ULB hexanes refers to an ultra low benzene grade of hexanes, an isomeric mixture, which contains less than 5 ppm benzene.
- the final 1,1,1,2-tetrafluoroethane (HFC- 134a) solution was prepared by adding the room temperature EADC stock solution, volume listed in the table, to the liquid HFC-134a in a test tube held at -35°C. In all cases, an initial solution is obtained which is clear and colorless.
- the resulting solution was then cooled by immersing the test tube into the dry box cold bath thermostated at -95 °C.
- the cloud point was determined by monitoring the temperature of the liquid with a thermometer and visually determining the onset of cloudiness.
- the solutions again became clear by allowing the solutions to warm to a temperature above the cloud point.
- the behavior of the solution around the cloud point was observed for several minutes by repeatedly cooling and warming the solution. This phenomenon was found to be reproducible.
- Examples 149 to 151 were conducted at -95°C in 500 ml glass resin kettles and were based on 300 ml of diluent.
- a linear polyisobutylene was prepared with the chain transfer agent, 2,2,4-trimethyl-l- pentene, to obtain a linear standard with a M w comparable to the polymers prepared in Examples 150 and 151.
- Examples 150 and 151 are copolymerizations of isobutylene and isoprene.
- Example 150 was prepared in methyl chloride and is provided as a comparative example.
- Example 151 was prepared in CH 2 FCF 3 and is an example of this invention. All three samples were analyzed by triple detection SEC. The plots of [ ⁇ ] (left y-axis) and g' (right y-axis) against M w for each sample are given in Figures 4, 5, and 6.
- a homopolymerization of isobutylene was conducted at -95 °C in the presence of a chain transfer agent, 2,2,4-trimethyl-l-pentene, and in CH 2 FCF 3 as a diluent.
- the polymerization was conducted with 300 ml of CH 2 FCF 3 , 56 ml of isobutylene (both collected as liquids at -95°C), and 59 microliters of 2,2,4- trimethyl-1-pentene. This solution was stirred at 1000 ⁇ m before and during the polymerization.
- an initiator/coinitiator solution was prepared in 50 ml of CH 2 FCF 3 at -85°C by adding, in order, 0.0738 ml of a 1.0 mol/L hydrogen chloride solution in CH 2 FCF 3 and 0.2213 ml of a 1.0 mol/L solution of ethylaluminum dichloride in hexane. This solution was stirred and immediately added to a chilled addition funnel. The initiator/coinitiator solution was then slowly added to the stirred monomer solution to commence polymerization. The initiator/coinitiator solution was added such that 25 ml of this solution was added. By adding the catalyst slowly the reaction temperature was maintained below -92°C.
- a copolymerization of isobutylene and isoprene was conducted in methyl chloride at -95°C.
- the monomer solution was prepared by adding 56 ml of liquid isobutylene, collected at -95 °C, 3.0 ml of isoprene to 300 ml of methyl chloride, collected as a liquid at -95°C and 1.5 ml of 2-methylbutane (internal GC standard). This solution was stirred at 1000 ⁇ before and during the polymerization.
- an initiator/coinitiator solution was prepared in 35 ml of methyl chloride at -95°C by adding, in order, 0.171 ml of a 1.2 mol/L hydrogen chloride solution in methyl chloride and 0.619 ml of a 1.0 mol/L solution of ethylaluminum dichloride in hexane. This solution was stirred and immediately added to a chilled addition funnel. The initiator/coinitiator solution was then slowly added to the stirred monomer solution to commence polymerization. The initiator/coinitiator solution was added such that 20.5 ml of this solution was added over 30 minutes. By adding the catalyst in this manner the reaction temperature was maintained below -91°C.
- the reaction was then quenched with the addition of 1-2 ml of methanol.
- the polymer was isolated by weathering off unused monomers and diluent.
- the polymer was dried in vacuo at 45°C to a constant weight. Yield: 30 g or 78 wt%.
- the copolymer contained 2.5 mol% of isoprene and exhibited a M w of 568k with a M w /M n of 2.6.
- the plot of [ ⁇ ] (left y- axis) and g' (right y-axis) against M w for Example 150 is given in Figure 5.
- the monomer solution was prepared by adding 95 ml of liquid isobutylene, collected at -95°C, 2.4 ml of isoprene to 300 ml of methyl chloride, collected as a liquid at -95 °C and 1.5 ml of 2-methylbutane (internal GC standard). This solution was stirred at 1000 ⁇ m before and during the polymerization.
- an initiator/coinitiator solution was prepared in 50 ml of CH 2 FCF 3 at -85°C by adding, in order, 0.349 ml of a 0.432 mol/L hydrogen chloride solution in methyl chloride and 0.443 ml of a 1.0 mol/L solution of ethylaluminum dichloride in hexane. This solution was stirred and immediately added to a chilled addition funnel. The initiator/coinitiator solution was then slowly added to the stirred monomer solution to commence polymerization. The initiator/coinitiator solution was added such that 30 ml of this solution was added over 75 minutes. By adding the catalyst in this manner the reaction temperature was maintained below -90°C.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
- Polymerization Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Polymerisation Methods In General (AREA)
- Polyesters Or Polycarbonates (AREA)
- Macromolecular Compounds Obtained By Forming Nitrogen-Containing Linkages In General (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Polymers With Sulfur, Phosphorus Or Metals In The Main Chain (AREA)
- Saccharide Compounds (AREA)
- Devices For Conveying Motion By Means Of Endless Flexible Members (AREA)
- Polyamides (AREA)
Abstract
Description

















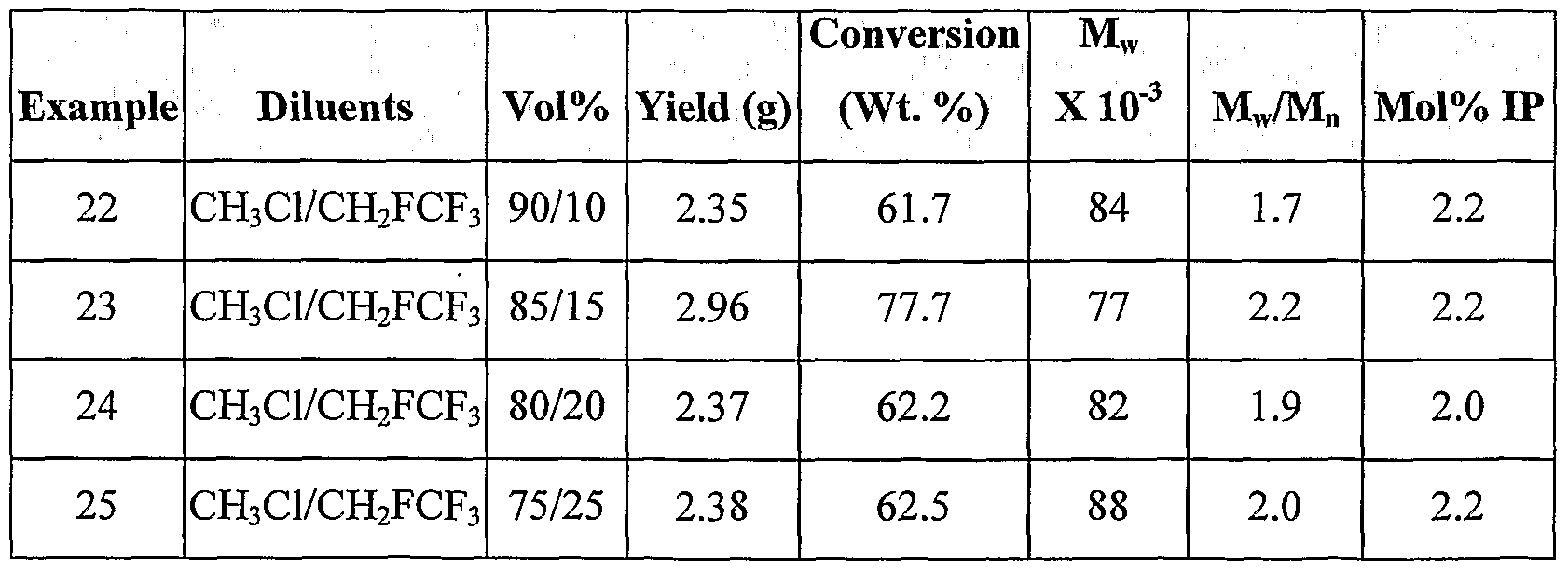
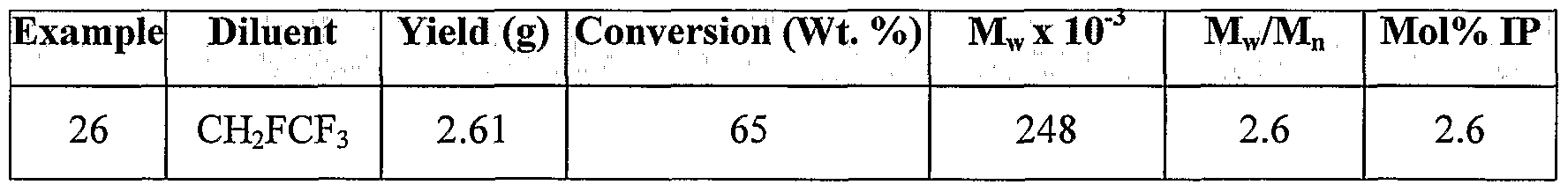


























Claims
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/538,965 US7582715B2 (en) | 2002-12-20 | 2003-12-19 | Polymers substantially free of long chain branching |
| EP03808474A EP1578822B1 (en) | 2002-12-20 | 2003-12-19 | Polymers substantially free of long chain branching |
| DE60336680T DE60336680D1 (en) | 2002-12-20 | 2003-12-19 | FAR LONG CHAIN BRANCH-FREE POLYMERS |
| AT03808474T ATE504604T1 (en) | 2002-12-20 | 2003-12-19 | LARGELY LONG CHAIN BRANCH-FREE POLYMERS |
| AU2003303476A AU2003303476A1 (en) | 2002-12-20 | 2003-12-19 | Polymers substantially free of long chain branching |
| CA2510864A CA2510864C (en) | 2002-12-20 | 2003-12-19 | Polymers substantially free of long chain branching |
| JP2005510005A JP5193424B2 (en) | 2002-12-20 | 2003-12-19 | Polymer substantially free of long chain molecules |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US43506102P | 2002-12-20 | 2002-12-20 | |
| US60/435,061 | 2002-12-20 | ||
| US46418703P | 2003-04-21 | 2003-04-21 | |
| US60/464,187 | 2003-04-21 | ||
| US47908103P | 2003-06-17 | 2003-06-17 | |
| US60/479,081 | 2003-06-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004058835A1 true WO2004058835A1 (en) | 2004-07-15 |
Family
ID=32686080
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2003/040341 WO2004058835A1 (en) | 2002-12-20 | 2003-12-19 | Polymers substantially free of long chain branching |
| PCT/US2003/040852 WO2004058825A2 (en) | 2002-12-20 | 2003-12-19 | Polymers with new sequence distributions |
| PCT/US2003/040858 WO2004058829A1 (en) | 2002-12-20 | 2003-12-19 | Polymerization processes |
| PCT/US2003/041221 WO2004067577A2 (en) | 2002-12-20 | 2003-12-19 | Polymerization processes |
Family Applications After (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2003/040852 WO2004058825A2 (en) | 2002-12-20 | 2003-12-19 | Polymers with new sequence distributions |
| PCT/US2003/040858 WO2004058829A1 (en) | 2002-12-20 | 2003-12-19 | Polymerization processes |
| PCT/US2003/041221 WO2004067577A2 (en) | 2002-12-20 | 2003-12-19 | Polymerization processes |
Country Status (9)
| Country | Link |
|---|---|
| US (6) | US7214750B2 (en) |
| EP (5) | EP1572763B1 (en) |
| JP (7) | JP4426533B2 (en) |
| AT (4) | ATE471950T1 (en) |
| AU (5) | AU2003303313A1 (en) |
| CA (5) | CA2510546C (en) |
| DE (4) | DE60333123D1 (en) |
| RU (1) | RU2341538C2 (en) |
| WO (4) | WO2004058835A1 (en) |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006002033A1 (en) * | 2004-06-15 | 2006-01-05 | Exxonmobil Chemical Patents Inc. | Elastomeric compositions, air barriers, and processes for making the same |
| WO2006062572A1 (en) | 2004-12-03 | 2006-06-15 | Exxonmobil Chemical Patents Inc. | Modified layered fillers and their use to produce nanocomposite compositions |
| WO2006071312A2 (en) | 2004-12-29 | 2006-07-06 | Exxonmobil Chemical Patents Inc. | Select elastomeric blends and their use in articles |
| JP2009522398A (en) * | 2005-12-28 | 2009-06-11 | エクソンモービル・ケミカル・パテンツ・インク | Halogenation method |
| US7723447B2 (en) | 2002-12-20 | 2010-05-25 | Exxonmobil Chemical Patents Inc. | Polymerization processes |
| US7767743B2 (en) | 2006-03-10 | 2010-08-03 | Exxonmobil Chemical Patents Inc. | Processable branched isoolefin-alkylstyrene elastomers |
| US7956138B2 (en) | 2007-05-16 | 2011-06-07 | Exxonmobil Chemical Patents Inc. | Catalyst system for olefin polymerization and polymers produced therefrom |
| EP2502742A2 (en) | 2005-10-27 | 2012-09-26 | ExxonMobil Chemical Patents Inc. | Construction comprising tie layer for pneumatic tire |
| WO2012145005A1 (en) | 2011-04-21 | 2012-10-26 | The Yokohama Rubber Co., Ltd. | Thermoplastic elastomer composition |
| WO2013052206A1 (en) | 2011-10-05 | 2013-04-11 | Exxonmobil Chemical Patents Inc. | Tire curing bladders |
| EP2930192A1 (en) | 2014-04-11 | 2015-10-14 | LANXESS Deutschland GmbH | Initiator system for the production of synthetic rubbers |
| EP3137542A4 (en) * | 2014-04-30 | 2018-06-27 | Arlanxeo Singapore Pte. Ltd. | Highly unsaturated multi-modal polyisoolefin composition and process for preparation thereof |
| WO2021011906A1 (en) | 2019-07-17 | 2021-01-21 | Exxonmobil Chemical Patents Inc. | TIRES COMPRISING RUBBER COMPOUNDS THAT COMPRISE PROPYLENE-α-OLEFIN-DIENE POLYMERS |
| WO2021262838A1 (en) | 2020-06-26 | 2021-12-30 | Exxonmobil Chemical Patents Inc. | Copolymers composed of ethylene, a-olefin, non-conjugated diene, and substituted styrene and articles therefrom |
| WO2021262842A1 (en) | 2020-06-26 | 2021-12-30 | Exxonmobil Chemical Patents Inc. | COPOLYMERS OF ETHYLENE, α-OLEFIN, NON-CONJUGATED DIENE, AND ARYL-SUBSTITUTED CYCLOALKENE, METHODS TO PRODUCE, BLENDS, AND ARTICLES THEREFROM |
Families Citing this family (90)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7714043B2 (en) | 2000-10-18 | 2010-05-11 | Exxonmobil Chemical Patents Inc. | Tire innerliners having improved cold temperature properties |
| US7838708B2 (en) | 2001-06-20 | 2010-11-23 | Grt, Inc. | Hydrocarbon conversion process improvements |
| US7425601B2 (en) * | 2002-12-20 | 2008-09-16 | Exxonmobil Chemical Patents Inc. | Polymers with new sequence distributions |
| AU2003297458A1 (en) * | 2002-12-20 | 2004-07-22 | Exxonmobil Chemical Patents Inc. | Polymerization processes |
| RU2341538C2 (en) * | 2002-12-20 | 2008-12-20 | Эксонмобил Кемикэл Пейтентс Инк. | Methods of polymerisation |
| US20050171393A1 (en) | 2003-07-15 | 2005-08-04 | Lorkovic Ivan M. | Hydrocarbon synthesis |
| CA2532367C (en) | 2003-07-15 | 2013-04-23 | Grt, Inc. | Hydrocarbon synthesis |
| US9359481B2 (en) | 2003-11-26 | 2016-06-07 | Owens Corning Intellectual Capital, Llc | Thermoplastic foams and method of forming them using nano-graphite |
| US7241398B2 (en) * | 2004-01-14 | 2007-07-10 | E.I. Du Pont De Nemours And Company | 1,1,1,3,3,-pentafluorobutane refrigerant or heat transfer fluid compositions comprising hydrofluorocarbon and uses thereof |
| US20080275284A1 (en) | 2004-04-16 | 2008-11-06 | Marathon Oil Company | Process for converting gaseous alkanes to liquid hydrocarbons |
| US7244867B2 (en) | 2004-04-16 | 2007-07-17 | Marathon Oil Company | Process for converting gaseous alkanes to liquid hydrocarbons |
| US20060100469A1 (en) | 2004-04-16 | 2006-05-11 | Waycuilis John J | Process for converting gaseous alkanes to olefins and liquid hydrocarbons |
| US8173851B2 (en) | 2004-04-16 | 2012-05-08 | Marathon Gtf Technology, Ltd. | Processes for converting gaseous alkanes to liquid hydrocarbons |
| US8642822B2 (en) | 2004-04-16 | 2014-02-04 | Marathon Gtf Technology, Ltd. | Processes for converting gaseous alkanes to liquid hydrocarbons using microchannel reactor |
| US7674941B2 (en) | 2004-04-16 | 2010-03-09 | Marathon Gtf Technology, Ltd. | Processes for converting gaseous alkanes to liquid hydrocarbons |
| CA2566855A1 (en) | 2004-05-20 | 2005-12-01 | Robert O. Hagerty | Gas olefin polymerization process |
| US7683140B2 (en) | 2004-05-20 | 2010-03-23 | Univation Technologies, Llc | Method for determining temperature value indicative of resin stickiness from data generated by polymerization reaction monitoring |
| US7781547B2 (en) * | 2004-06-25 | 2010-08-24 | Exxonmobil Chemical Patents Inc. | Reactor systems for use in polymerization processes |
| US7758562B2 (en) * | 2004-09-09 | 2010-07-20 | Plc Medical Systems, Inc. | Patient hydration system with a redundant monitoring of hydration fluid infusion |
| US7906600B2 (en) | 2004-12-29 | 2011-03-15 | Exxonmobil Chemical Patents Inc. | Processable filled, curable halogenated isoolefin elastomers |
| CA2505565C (en) * | 2005-04-28 | 2008-09-16 | Camco Inc. | Apparatus and method for controlling a clothes dryer |
| US9187608B2 (en) | 2005-09-08 | 2015-11-17 | Owens Corning Intellectual Capital, Llc | Polystyrene foam containing a modifier-free nanoclay and having improved fire protection performance |
| EP1772773B1 (en) * | 2005-10-06 | 2011-06-29 | STMicroelectronics Srl | Method for realizing a multispacer structure, use of said structure as a mould and method for producing circuital architectures using said mould |
| US8287752B2 (en) * | 2005-11-01 | 2012-10-16 | E I Du Pont De Nemours And Company | Fire extinguishing and fire suppression compositions comprising unsaturated fluorocarbons |
| KR101433781B1 (en) | 2006-02-03 | 2014-08-25 | 리액션 35, 엘엘씨 | System and method for forming hydrocarbons |
| US7883568B2 (en) | 2006-02-03 | 2011-02-08 | Grt, Inc. | Separation of light gases from halogens |
| CN103529169B (en) * | 2006-05-31 | 2016-03-02 | 埃克森美孚化学专利公司 | Isotope analysis is for measuring by the purposes of the standby aromatic hydrocarbon of methane |
| US8148450B2 (en) * | 2006-06-23 | 2012-04-03 | Exxonmobil Chemical Patents Inc. | Process to produce a hydrocarbon rubber cement utilizing a hydrofluorocarbon diluent |
| DE602007004616D1 (en) * | 2006-07-03 | 2010-03-18 | Dow Corning | CHEMICAL CURING OF ALL-IN-ONE WARM-EDGE DISTANCE HOLDER AND SEAL |
| WO2008044640A1 (en) * | 2006-10-11 | 2008-04-17 | Yasuhara Chemical Co., Ltd. | β-PINENE POLYMER AND PROCESS FOR PRODUCTION THEREOF |
| US7771667B2 (en) * | 2007-02-09 | 2010-08-10 | Exxonmobil Chemical Patents Inc. | Polymerization quench method and system |
| US7893176B2 (en) * | 2007-03-23 | 2011-02-22 | Exxonmobil Chemical Patents Inc. | Polydispersity-controlled isoolefin polymerization with polymorphogenates |
| US7402636B1 (en) | 2007-03-23 | 2008-07-22 | Exxonmobil Chemical Patents Inc | Method and apparatus for decreasing polymer deposition |
| US7981991B2 (en) * | 2007-04-20 | 2011-07-19 | Exxonmobil Chemical Patents Inc. | Separation of polymer slurries |
| AU2008256606A1 (en) | 2007-05-24 | 2008-12-04 | Grt, Inc. | Zone reactor incorporating reversible hydrogen halide capture and release |
| WO2008154612A1 (en) * | 2007-06-12 | 2008-12-18 | E.I. Du Pont De Nemours And Company | Azeotropic and azeotrope-like compositions of e-1,1,1,4,4,4-hexafluoro-2-butene |
| DE102007028238A1 (en) * | 2007-06-20 | 2008-12-24 | Osram Opto Semiconductors Gmbh | Use of a metal complex as p-dopant for an organic semiconductive matrix material, organic semiconductor material and organic light-emitting diode |
| US8282810B2 (en) | 2008-06-13 | 2012-10-09 | Marathon Gtf Technology, Ltd. | Bromine-based method and system for converting gaseous alkanes to liquid hydrocarbons using electrolysis for bromine recovery |
| US8415517B2 (en) | 2008-07-18 | 2013-04-09 | Grt, Inc. | Continuous process for converting natural gas to liquid hydrocarbons |
| HUE045444T2 (en) | 2009-03-04 | 2019-12-30 | Regenera Pharma Ltd | Compositions of polymeric myrcene |
| WO2010100650A2 (en) | 2009-03-04 | 2010-09-10 | Regenera Pharma Ltd. | Therapeutic uses of mastic gum fractions |
| US20100273964A1 (en) * | 2009-04-22 | 2010-10-28 | Stewart Lewis | Heterogeneous lewis acid catalysts for cationic polymerizations |
| US20110144216A1 (en) * | 2009-12-16 | 2011-06-16 | Honeywell International Inc. | Compositions and uses of cis-1,1,1,4,4,4-hexafluoro-2-butene |
| US8198495B2 (en) | 2010-03-02 | 2012-06-12 | Marathon Gtf Technology, Ltd. | Processes and systems for the staged synthesis of alkyl bromides |
| US8367884B2 (en) | 2010-03-02 | 2013-02-05 | Marathon Gtf Technology, Ltd. | Processes and systems for the staged synthesis of alkyl bromides |
| US9079990B2 (en) | 2010-06-01 | 2015-07-14 | Exxonmobil Chemical Patents Inc. | Methods of production of alkylstyrene/isoolefin polymers |
| US8815050B2 (en) | 2011-03-22 | 2014-08-26 | Marathon Gtf Technology, Ltd. | Processes and systems for drying liquid bromine |
| US9598655B2 (en) * | 2011-06-08 | 2017-03-21 | Tpc Group Llc | Adducts of low molecular weight PIB with low polydispersity and high vinylidene content |
| US8436220B2 (en) | 2011-06-10 | 2013-05-07 | Marathon Gtf Technology, Ltd. | Processes and systems for demethanization of brominated hydrocarbons |
| US8829256B2 (en) | 2011-06-30 | 2014-09-09 | Gtc Technology Us, Llc | Processes and systems for fractionation of brominated hydrocarbons in the conversion of natural gas to liquid hydrocarbons |
| US8802908B2 (en) | 2011-10-21 | 2014-08-12 | Marathon Gtf Technology, Ltd. | Processes and systems for separate, parallel methane and higher alkanes' bromination |
| US9193641B2 (en) | 2011-12-16 | 2015-11-24 | Gtc Technology Us, Llc | Processes and systems for conversion of alkyl bromides to higher molecular weight hydrocarbons in circulating catalyst reactor-regenerator systems |
| US8592538B2 (en) | 2011-12-20 | 2013-11-26 | Honeywell International Inc. | Azeotropes of methyl chloride with fluorocarbons |
| US9963521B2 (en) * | 2012-02-17 | 2018-05-08 | Basf Se | Process for preparing higher molecular weight polyisobutylene |
| US8927666B2 (en) | 2012-11-08 | 2015-01-06 | Honeywell International Inc. | Polymerization of monomers using fluorinated propylene solvents |
| US8987399B2 (en) * | 2012-11-08 | 2015-03-24 | Honeywell International Inc. | Azeotropes of isobutylene with fluoro-olefins |
| FR3000093B1 (en) * | 2012-12-26 | 2015-07-17 | Arkema France | AZEOTROPIC OR QUASI-AZEOTROPIC COMPOSITION OF CHLOROMETHANE |
| JP6159574B2 (en) * | 2013-05-16 | 2017-07-05 | 住友ゴム工業株式会社 | Branched conjugated diene copolymer, hydrogenated branched conjugated diene copolymer, rubber composition, and pneumatic tire |
| JP6164927B2 (en) * | 2013-05-16 | 2017-07-19 | 住友ゴム工業株式会社 | Pneumatic tire |
| JP6328773B2 (en) * | 2013-12-23 | 2018-05-23 | アランセオ・シンガポール・プライヴェート・リミテッド | High purity halogenated rubber |
| EP3087139B1 (en) | 2013-12-23 | 2022-04-13 | Arlanxeo Singapore Pte. Ltd. | Ultra pure rubber |
| US10647842B2 (en) * | 2013-12-23 | 2020-05-12 | Arlanxeo Singapore Pte. Ltd. | Anti-agglomerants for elastomeric ethylene/A-olefin copolymers |
| EP3572442B1 (en) | 2013-12-23 | 2021-04-07 | BASF South East Asia Pte. Ltd. | Novel anti-agglomerants for polyisobutylene production |
| EP3087140B1 (en) * | 2013-12-23 | 2019-05-22 | Arlanxeo Singapore Pte. Ltd. | Novel anti-agglomerants for the rubber industry |
| JP6523297B2 (en) | 2013-12-23 | 2019-05-29 | アランセオ・シンガポール・プライヴェート・リミテッド | Rubber containing adjustable levels of metal-containing antiflocculant |
| CA2947046A1 (en) * | 2014-04-30 | 2015-11-05 | Arlanxeo Singapore Pte. Ltd. | Copolymer having high multiolefin content |
| EP2940048A1 (en) | 2014-04-30 | 2015-11-04 | Lanxess Inc. | Copolymer having low isoprenoid content |
| CN106574013B (en) | 2014-04-30 | 2020-03-24 | 阿朗新科新加坡私人有限公司 | Copolymers with low isoprenoid content |
| PL3137517T3 (en) | 2014-04-30 | 2020-11-30 | Arlanxeo Singapore Pte. Ltd. | Copolymer having low cyclic oligomer content |
| KR102351740B1 (en) * | 2014-04-30 | 2022-01-14 | 아란세오 싱가포르 프라이빗 리미티드 | 2,3,3,3-tetrafluoro-1-propene as diluent for the preparation of novel butyl rubbers |
| KR20170002487A (en) * | 2014-04-30 | 2017-01-06 | 아란세오 싱가포르 프라이빗 리미티드 | Butyl rubber with new sequence distribution |
| EP2940046A1 (en) | 2014-04-30 | 2015-11-04 | Lanxess Inc. | Hydrofluorinated Olefins (HFO's) as diluents for Butyl rubber production |
| EP2940050A1 (en) | 2014-04-30 | 2015-11-04 | Lanxess Inc. | Copolymer having low cyclic oligomer content |
| KR102351739B1 (en) * | 2014-04-30 | 2022-01-14 | 아란세오 싱가포르 프라이빗 리미티드 | Hydrofluorinated olefins (hfo's) as diluents for butyl rubber production |
| EP2940047A1 (en) | 2014-04-30 | 2015-11-04 | Lanxess Inc. | Copolymer having high multiolefin content |
| JP2017516891A (en) * | 2014-04-30 | 2017-06-22 | アランセオ・シンガポール・プライヴェート・リミテッド | Diluent for the production of butyl rubber |
| PL3822294T3 (en) | 2014-06-30 | 2024-02-19 | Basf South East Asia Pte. Ltd. | Novel anti-agglomerants for polyisobutylene production |
| WO2016000073A1 (en) | 2014-06-30 | 2016-01-07 | Lanxess Inc. | Novel anti-agglomerants for the rubber industry |
| US9896525B2 (en) | 2015-04-29 | 2018-02-20 | Exxonmobil Chemical Patents Inc. | Homogeneous polymerization process using evaporative cooling |
| CN104844748B (en) * | 2015-05-12 | 2017-04-05 | 清华大学 | A kind of preparation method of butyl rubber |
| US9771442B2 (en) * | 2015-05-13 | 2017-09-26 | University Of Massachusetts | Polymerization initiating system and method to produce highly reactive olefin functional polymers |
| CN108472618B (en) | 2016-01-29 | 2020-04-28 | 埃克森美孚化学专利公司 | Systems and methods for producing functionalized olefin-based polymers |
| DE102016221346A1 (en) | 2016-10-28 | 2018-05-03 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e.V. | Process for the preparation of a polymer by nitroxyl-controlled polymerization and polymer |
| WO2019011814A1 (en) | 2017-07-13 | 2019-01-17 | Arlanxeo Deutschland Gmbh | Process for the production of isobutene polymers with improved temperature control |
| EP3444283B1 (en) * | 2017-08-17 | 2020-02-26 | Basf Se | Process for preparing high-reactivity isobutene homo- or copolymers |
| MY197010A (en) | 2017-10-14 | 2023-05-18 | Tpc Group Llc | Isobutylene copolymers, method for making isobutylene copolymers and isobutylene copolymer products |
| CN110652954A (en) * | 2019-09-19 | 2020-01-07 | 贾影丽 | Cosmetics cauldron formula reaction unit |
| JP7441321B2 (en) * | 2020-09-18 | 2024-02-29 | エルジー・ケム・リミテッド | Catalyst composition and method for producing isobutene-isoprene copolymer using the same |
| CN113861323B (en) * | 2021-11-11 | 2022-11-01 | 中国科学院长春应用化学研究所 | High-strength high-stress-at-definite-elongation bionic rubber and preparation method and application thereof |
| CN113896819B (en) * | 2021-11-11 | 2022-10-28 | 中国科学院长春应用化学研究所 | Ketone compound modified polybutadiene rubber, preparation method thereof and vulcanized rubber |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2534698A (en) * | 1944-11-29 | 1950-12-19 | Standard Oil Dev Co | Polymerization of olefins in fluorinated diluent |
| US2548415A (en) * | 1948-12-08 | 1951-04-10 | Standard Oil Dev Co | Boron fluoride in ethylidene fluoride for low-temperature polymerization |
| US4107417A (en) * | 1971-11-26 | 1978-08-15 | Snamprogetti, S.P.A. | Process for the production of polymers and copolymers of isobutylene |
Family Cites Families (110)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2356128A (en) † | 1939-10-20 | 1944-08-22 | Jasco Inc | Mixed olefinic polymerization process and product |
| US2494585A (en) * | 1944-09-30 | 1950-01-17 | Standard Oil Dev Co | Olefinic polymerization in the presence of fluorine compounds |
| US2474571A (en) | 1946-03-28 | 1949-06-28 | Standard Oil Dev Co | Low-temperature polymerization process |
| US2607764A (en) | 1947-11-28 | 1952-08-19 | Standard Oil Dev Co | Vulcanizable copolymers of isobutylene and 10-70 per cent combined butadiene |
| US2603626A (en) | 1949-03-19 | 1952-07-15 | Standard Oil Dev Co | Low-temperature polymerization process employing p2o5 treated alkyl halide catalyst solvent |
| US2644809A (en) | 1950-01-11 | 1953-07-07 | Standard Oil Dev Co | Friedel-crafts isobutylene polymerizations in chlorofluoroalkane solution |
| US3269972A (en) | 1960-10-05 | 1966-08-30 | Exxon Research Engineering Co | Butyl rubber compositions comprising methyl hydroxy stearate |
| BE613858A (en) | 1961-02-13 | 1900-01-01 | ||
| US3331822A (en) | 1963-08-20 | 1967-07-18 | Thiokol Chemical Corp | Production of polytetrafluoroethylene molding powders by the aqueous suspension polymerization of tetrafluoroethylene with small amounts of perfluoroolefins containing from 3 to 4 carbon atoms |
| DK105619C (en) | 1964-02-07 | 1966-10-17 | Henning Kaaber | Process by suspension or emulsion polymerization of ethylenically unsaturated compounds in a closed container. |
| US3470143A (en) * | 1964-03-26 | 1969-09-30 | Dart Ind Inc | Highly fluorinated hydrocarbons used as diluents in the polymerization of ethylenically unsaturated monomers |
| GB1146188A (en) * | 1965-07-30 | 1969-03-19 | Montedison Spa | Polymerization of halogen containing olefins |
| US3444117A (en) † | 1966-04-06 | 1969-05-13 | Exxon Research Engineering Co | Polymer blend |
| GB1200753A (en) | 1967-07-12 | 1970-08-05 | Ici Ltd | Polymerisation of tetrafluoroethylene |
| US3616371A (en) | 1967-08-05 | 1971-10-26 | Asahi Glass Co Ltd | Process for the production of homopolymer of vinylidene fluoride or copolymers thereof in the presence of a fluorine-containing hydrocarbon solvent |
| US3528954A (en) | 1967-10-30 | 1970-09-15 | Du Pont | Process for homopolymerization of tetrafluoroethylene and copolymerization of same with fluoro co-monomers in the solvent 1,1,2 - trichloro - 1,2,2 - trifluoroethane |
| US3642742A (en) | 1969-04-22 | 1972-02-15 | Du Pont | Tough stable tetrafluoroethylene-fluoroalkyl perfluorovinyl ether copolymers |
| US3787379A (en) | 1971-10-05 | 1974-01-22 | Pennwalt Corp | Copolymers of vinyl fluoride and hexafluoropropene |
| CH582719A5 (en) | 1973-09-12 | 1976-12-15 | Ciba Geigy Ag | |
| DE2504514A1 (en) | 1974-02-12 | 1975-08-14 | Ciba Geigy Ag | FLUORINE CARBONIC ACID AMIDES AND THEIR POLYMERIZATION PRODUCTS |
| US4100225A (en) | 1974-06-20 | 1978-07-11 | Ciba-Geigy Corporation | Stable polymer compositions containing perfluoroalkyl groups and process for making |
| US4042634A (en) | 1976-03-15 | 1977-08-16 | E. I. Du Pont De Nemours And Company | Fluorocarbon separation process |
| US4123602A (en) | 1976-05-24 | 1978-10-31 | Asahi Glass Company, Ltd. | Terpolymers of tetrafluoroethylene, ethylene and perfluoroalkyl vinyl monomer and process for producing the same |
| JPS5950162B2 (en) | 1977-05-20 | 1984-12-06 | 旭硝子株式会社 | Improved ethylene-tetrafluoroethylene copolymer and method for producing the same |
| US4194073A (en) | 1978-01-04 | 1980-03-18 | Phillips Petroleum Company | Olefin polymerization |
| DE2843157A1 (en) | 1978-10-04 | 1980-04-17 | Bayer Ag | METHOD FOR PRODUCING ACRYLNITRILE POLYMERISATS IN HALOGENATED ALIPHATIC HYDROCARBONS |
| US4248988A (en) | 1979-10-18 | 1981-02-03 | The Firestone Tire & Rubber Company | Process for the preparation of cyclized polymeric dienes |
| US4381387A (en) | 1980-06-28 | 1983-04-26 | Hoechst Aktiengesellschaft | Quaterpolymers of the tetrafluoroethylene/ethylene type |
| DE3024450A1 (en) | 1980-06-28 | 1982-01-28 | Hoechst Ag, 6000 Frankfurt | METHOD FOR PRODUCING AQUEOUS, COLLOIDAL DISPERSIONS OF TYPE TETRAFLUORETHYLENE ETHYLENE COPOLYMERS |
| DE3139721A1 (en) | 1981-10-06 | 1983-04-21 | Wacker-Chemie GmbH, 8000 München | METHOD FOR PRODUCING AQUEOUS COPOLYMER DISPERSIONS AND USE THEREOF |
| JPS58189210A (en) | 1982-04-30 | 1983-11-04 | Daikin Ind Ltd | Production of tetrafluoroethylene/fluorinated alkyl vinyl ether copolymer |
| US4501865A (en) * | 1982-07-08 | 1985-02-26 | Bayer Aktiengesellschaft | Polymerization process |
| US4435553A (en) | 1982-09-20 | 1984-03-06 | The Goodyear Tire & Rubber Company | Process for nonaqueous dispersion polymerization of butadiene in the presence of carbonylated polymeric dispersing agents |
| US4452960A (en) | 1982-09-20 | 1984-06-05 | The Goodyear Tire & Rubber Company | Process for nonaqueous dispersion polymerization of butadiene in the presence of polymeric dispersing agents |
| US4508881A (en) | 1982-09-20 | 1985-04-02 | The Goodyear Tire & Rubber Company | Process for nonaqueous dispersion polymerization of butadiene in the presence of polymeric dispersing agents |
| US4424324A (en) | 1982-12-09 | 1984-01-03 | The Goodyear Tire & Rubber Company | Process for nonaqueous dispersion polymerization of butadiene |
| US4736004A (en) | 1983-04-05 | 1988-04-05 | Green Cross Corporation | Persistent perfluoroalkyl free radicals useful as polymerization catalyst |
| US4626608A (en) | 1983-04-05 | 1986-12-02 | Green Cross Corporation | Persistent perfluoroalkyl free radicals useful as polymerization catalyst |
| US4535136A (en) | 1984-04-23 | 1985-08-13 | E. I. Du Pont De Nemours And Company | Fluoroolefin polymerization process using acyl hypofluorite catalyst |
| US4588796A (en) | 1984-04-23 | 1986-05-13 | E. I. Du Pont De Nemours And Company | Fluoroolefin polymerization process using fluoroxy compound solution as initiator |
| GB8515254D0 (en) | 1985-06-17 | 1985-07-17 | Enichem Elastomers | Butyl rubber |
| US5162445A (en) | 1988-05-27 | 1992-11-10 | Exxon Chemical Patents Inc. | Para-alkylstyrene/isoolefin copolymers and functionalized copolymers thereof |
| US5032656A (en) | 1987-11-23 | 1991-07-16 | Allied-Signal Inc. | Fluorinated copolymer and barrier films |
| IT1223409B (en) * | 1987-12-10 | 1990-09-19 | Ausimont Spa | FLUORINATED POLYMERS AND COPOLYMERS CONTAINING PERFLUOROPOLYETHER BLOCKS |
| US4948844A (en) | 1988-04-16 | 1990-08-14 | Tokuyama Soda Kabushiki Kaisha | Process for preparation of perfluorinated copolymer |
| CA1338546C (en) * | 1988-05-27 | 1996-08-20 | Kenneth William Powers | Para-alkylstyrene/isoolefin copolymers |
| US4950724A (en) | 1988-09-27 | 1990-08-21 | The Dow Chemical Company | Suspension polymerization of vinyl aromatic monomers to polymer having high syndiotacticity |
| SU1627243A1 (en) * | 1989-02-10 | 1991-02-15 | Советско-итальянское научно-исследовательское общество "Синион" | Apparatus for polymerization |
| US4983528A (en) † | 1989-06-06 | 1991-01-08 | Polysar Limited | Measurement of unsaturation level in butyl and EPDM rubbers |
| US5105047A (en) | 1989-08-02 | 1992-04-14 | E. I. Du Pont De Nemours And Company | Catalysis using blends of perfluorinated ion-exchange polymers with perfluorinated diluents |
| AU656624B2 (en) | 1991-07-29 | 1995-02-09 | Exxon Chemical Patents Inc. | Polymerization reactor |
| US5286804A (en) * | 1991-09-17 | 1994-02-15 | Exxon Chemical Patents Inc. | Halogenation of star-branched butyl rubber with improved neutralization |
| US5182342A (en) * | 1992-02-28 | 1993-01-26 | E. I. Du Pont De Nemours And Company | Hydrofluorocarbon solvents for fluoromonomer polymerization |
| US5807977A (en) | 1992-07-10 | 1998-09-15 | Aerojet General Corporation | Polymers and prepolymers from mono-substituted fluorinated oxetane monomers |
| US5310870A (en) * | 1992-08-13 | 1994-05-10 | E. I. Du Pont De Nemours And Company | Fluoroalkene/hydrofluorocarbon telomers and their synthesis |
| US5281680A (en) | 1993-01-14 | 1994-01-25 | E. I. Du Pont De Nemours And Company | Polymerization of fluorinated copolymers |
| US5506316A (en) * | 1993-02-19 | 1996-04-09 | Exxon Chemical Patents Inc. | Carbocationic catalysts and process for using said catalysts |
| DE69421021T2 (en) | 1993-03-17 | 2000-01-20 | Asahi Glass Co. Ltd., Tokio/Tokyo | METHOD FOR PRODUCING A FLUORINE POLYMER |
| JP3272475B2 (en) * | 1993-04-19 | 2002-04-08 | 旭硝子株式会社 | Method for producing ethylene-tetrafluoroethylene copolymer |
| JP2736938B2 (en) * | 1993-06-24 | 1998-04-08 | ザ ダウ ケミカル カンパニー | Titanium (II) or zirconium (II) complex and addition polymerization catalyst comprising the same |
| US5990251A (en) | 1993-07-13 | 1999-11-23 | Bp Chemicals Limited | Process for polymerising olefin with a Ziegler-Natta catalyst |
| JP2985600B2 (en) * | 1993-09-24 | 1999-12-06 | ダイキン工業株式会社 | Method for producing low molecular weight polytetrafluoroethylene |
| JPH07118596A (en) | 1993-10-25 | 1995-05-09 | Daikin Ind Ltd | Water and oil repellent composition and its production |
| BE1008739A3 (en) | 1994-01-12 | 1996-07-02 | Kanegafuchi Chemical Ind | Method of preparation of polymer isobutylene. |
| US5459212A (en) | 1994-05-26 | 1995-10-17 | E. I. Du Pont De Nemours And Company | Carbon based initiators for polymerization and telomerization of vinyl monomers |
| JP3500152B2 (en) * | 1994-06-09 | 2004-02-23 | 鐘淵化学工業株式会社 | Method for producing isobutene-based polymer |
| JPH10502690A (en) | 1994-07-08 | 1998-03-10 | ミネソタ マイニング アンド マニュファクチャリング カンパニー | Hyperbaric reaction |
| IT1274711B (en) | 1994-08-04 | 1997-07-24 | Ausimont Spa | PROCESS FOR THE PREPARATION OF FLUOROPOLYMERS CONTAINING HYDROGEN THROUGH (CO) SUSPENSION POLYMERIZATION |
| US5448001A (en) * | 1994-10-07 | 1995-09-05 | Queen's University At Kingston | Polymerization of iso-butylene |
| US5811591A (en) * | 1994-11-22 | 1998-09-22 | Nippon Shokubai Co., Ltd. | Process of producing hydroxyalkanal |
| DE69632693T2 (en) | 1995-02-06 | 2005-06-16 | E.I. Du Pont De Nemours And Co., Wilmington | Hexafluoropropene containing amorphous copolymers |
| US5478905A (en) | 1995-02-06 | 1995-12-26 | E. I. Du Pont De Nemours And Company | Amorphous tetrafluoroethylene/hexafluoropropylene copolymers |
| US6133389A (en) | 1995-02-06 | 2000-10-17 | E. I. Du Pont De Nemours And Company | Amorphous tetrafluoroethylene-hexafluoropropylene copolymers |
| US5618894A (en) | 1995-03-10 | 1997-04-08 | The University Of North Carolina | Nonaqueous polymerization of fluoromonomers |
| US5674957A (en) | 1995-03-10 | 1997-10-07 | The University Of North Carolina At Chapel Hill | Nonaqueous polymerization of fluoromonomers |
| US5552500A (en) | 1995-04-24 | 1996-09-03 | E. I. Du Pont De Nemours And Company | Fluoroalkene/hydrochlorofluorocarbon telomers and their synthesis |
| EP0828767B1 (en) | 1995-05-26 | 2003-07-30 | Igen, Inc. | Molecularly imprinted beaded polymers and stabilized suspension polymerization of the same in perfluorocarbon liquids |
| JPH08333408A (en) | 1995-06-07 | 1996-12-17 | Asahi Glass Co Ltd | Production of fluorocopolymer |
| IT1276070B1 (en) | 1995-10-31 | 1997-10-24 | Siac It Additivi Carburanti | PROCESS FOR THE PREPARATION OF ETHYLENE-BASED POLYMERS WITH LOW MOLECULAR WEIGHT |
| CA2174795C (en) * | 1996-04-23 | 2009-11-03 | Gabor Kaszas | Improved polymer bromination process |
| RU2097122C1 (en) * | 1996-05-14 | 1997-11-27 | Акционерное общество Научно-производственное предприятие "Ярсинтез" | Polymerizer |
| US5900251A (en) * | 1996-06-10 | 1999-05-04 | Breath Asure, Inc. | Internal breath freshener and digestive aid |
| EP0911347B1 (en) * | 1997-10-15 | 2006-08-23 | E.I. Du Pont De Nemours And Company | Copolymers of maleic anhydride or acid and fluorinated olefins |
| JP3818344B2 (en) | 1997-11-20 | 2006-09-06 | 旭硝子株式会社 | Process for producing fluorine-containing aliphatic ring structure-containing polymer |
| IT1298257B1 (en) | 1998-02-17 | 1999-12-20 | Ausimont Spa | perfluorodioxols |
| DE19812755A1 (en) * | 1998-03-23 | 1999-09-30 | Bayer Ag | Process for the production of partially fluorinated fluoropolymers |
| ITMI981506A1 (en) * | 1998-06-30 | 1999-12-30 | Ausimont Spa | AMORPHOUS FLUOROPOLYMER MANUFACTURES |
| CN1309670A (en) * | 1998-07-17 | 2001-08-22 | 拜尔公司 | Process for polymerizing cationically polymerizable olefin |
| TWI230712B (en) | 1998-09-15 | 2005-04-11 | Novartis Ag | Polymers |
| CN1249105C (en) * | 1998-10-05 | 2006-04-05 | 普罗米鲁斯有限责任公司 | Catalyst and methods for polymerizing cycloolefins |
| US6306989B1 (en) | 1999-09-30 | 2001-10-23 | E. I. Du Pont De Nemours And Company | Situ fluoropolymer polymerization into porous substrates |
| JP2003524675A (en) | 1998-11-30 | 2003-08-19 | イー・アイ・デュポン・ドウ・ヌムール・アンド・カンパニー | Fluoromonomer polymerization |
| KR100334163B1 (en) * | 1998-12-04 | 2002-10-25 | 삼성종합화학주식회사 | Olefin Polymerization or Copolymerization Method |
| US6337373B1 (en) | 1999-07-30 | 2002-01-08 | Montell Technology Company Bv | Polyolefin graft copolymers made with fluorinated monomers |
| IT1313597B1 (en) | 1999-08-04 | 2002-09-09 | Ausimont Spa | FLUOROPOLYEREOUS LUBRICANTS CONTAINING SULPHONYLFLUORIDE GROUPS |
| US6410982B1 (en) * | 1999-11-12 | 2002-06-25 | Intel Corporation | Heatpipesink having integrated heat pipe and heat sink |
| EP1237950B1 (en) * | 1999-11-15 | 2005-05-04 | ExxonMobil Chemical Patents Inc. | Production of isobutylene copolymers |
| US6538083B2 (en) * | 1999-12-21 | 2003-03-25 | E. I. Du Pont De Nemours And Company | Chain transfer agents in fluoroolefin polymerization |
| DE60143635D1 (en) * | 2000-02-15 | 2011-01-27 | Asahi Glass Co Ltd | Block polymer, process for producing polymer and solid polymer electrolyte fuel cell |
| US6372838B1 (en) * | 2000-06-28 | 2002-04-16 | 3M Innovative Properties Company | Fine latex and seed method of making |
| KR100818967B1 (en) * | 2000-09-21 | 2008-04-04 | 롬 앤드 하스 캄파니 | Aqueous nanocomposite dispersions: processes, compositions, and uses thereof |
| GB0026046D0 (en) * | 2000-10-24 | 2000-12-13 | Wilde Peter F | Improvements to polymerisation processes |
| DE10061727A1 (en) * | 2000-12-12 | 2002-06-13 | Basf Ag | Process for the preparation of polyisobutenes |
| CA2436558C (en) * | 2000-12-20 | 2009-11-17 | Exxonmobil Chemical Patents Inc. | Process for polymerizing cationically polymerizable monomers |
| DE10125583A1 (en) * | 2001-05-25 | 2002-11-28 | Basf Ag | Production of homo- or co-polymers of isobutene involves continuous cationic polymerisation in special tubular flow reactor with several bends in alternating directions |
| US7723447B2 (en) * | 2002-12-20 | 2010-05-25 | Exxonmobil Chemical Patents Inc. | Polymerization processes |
| AU2003297458A1 (en) * | 2002-12-20 | 2004-07-22 | Exxonmobil Chemical Patents Inc. | Polymerization processes |
| US7425601B2 (en) * | 2002-12-20 | 2008-09-16 | Exxonmobil Chemical Patents Inc. | Polymers with new sequence distributions |
| RU2341538C2 (en) * | 2002-12-20 | 2008-12-20 | Эксонмобил Кемикэл Пейтентс Инк. | Methods of polymerisation |
| CN101010377B (en) * | 2004-06-15 | 2010-11-24 | 埃克森美孚化学专利公司 | Elastomeric compositions, air barriers, and processes for making the same |
-
2003
- 2003-12-19 RU RU2005122163/04A patent/RU2341538C2/en not_active IP Right Cessation
- 2003-12-19 JP JP2005515535A patent/JP4426533B2/en not_active Expired - Fee Related
- 2003-12-19 WO PCT/US2003/040341 patent/WO2004058835A1/en active Application Filing
- 2003-12-19 JP JP2005510041A patent/JP4441487B2/en not_active Expired - Fee Related
- 2003-12-19 AU AU2003303313A patent/AU2003303313A1/en not_active Abandoned
- 2003-12-19 US US10/539,015 patent/US7214750B2/en not_active Expired - Lifetime
- 2003-12-19 AT AT03815302T patent/ATE471950T1/en not_active IP Right Cessation
- 2003-12-19 CA CA2510546A patent/CA2510546C/en not_active Expired - Fee Related
- 2003-12-19 WO PCT/US2003/040852 patent/WO2004058825A2/en active Application Filing
- 2003-12-19 AT AT03814305T patent/ATE469923T1/en not_active IP Right Cessation
- 2003-12-19 US US10/538,965 patent/US7582715B2/en not_active Expired - Fee Related
- 2003-12-19 CA CA2510864A patent/CA2510864C/en not_active Expired - Fee Related
- 2003-12-19 EP EP03814280A patent/EP1572763B1/en not_active Revoked
- 2003-12-19 AU AU2003303476A patent/AU2003303476A1/en not_active Abandoned
- 2003-12-19 CA CA2510862A patent/CA2510862C/en not_active Expired - Fee Related
- 2003-12-19 AU AU2003301223A patent/AU2003301223A1/en not_active Abandoned
- 2003-12-19 EP EP03814305.3A patent/EP1572766B2/en not_active Expired - Lifetime
- 2003-12-19 AU AU2003303342A patent/AU2003303342A1/en not_active Abandoned
- 2003-12-19 DE DE60333123T patent/DE60333123D1/en not_active Expired - Lifetime
- 2003-12-19 JP JP2005510005A patent/JP5193424B2/en not_active Expired - Fee Related
- 2003-12-19 EP EP03815302A patent/EP1572751B1/en not_active Expired - Lifetime
- 2003-12-19 CA CA2510847A patent/CA2510847C/en not_active Expired - Fee Related
- 2003-12-19 WO PCT/US2003/040858 patent/WO2004058829A1/en active Application Filing
- 2003-12-19 DE DE60336680T patent/DE60336680D1/en not_active Expired - Lifetime
- 2003-12-19 JP JP2005510040A patent/JP4690195B2/en not_active Expired - Fee Related
- 2003-12-19 AU AU2003301205A patent/AU2003301205A1/en not_active Abandoned
- 2003-12-19 US US10/539,013 patent/US7423100B2/en active Active
- 2003-12-19 WO PCT/US2003/041221 patent/WO2004067577A2/en active Application Filing
- 2003-12-19 AT AT03808474T patent/ATE504604T1/en not_active IP Right Cessation
- 2003-12-19 CA CA2509267A patent/CA2509267C/en not_active Expired - Fee Related
- 2003-12-19 DE DE60332758T patent/DE60332758D1/en not_active Expired - Lifetime
- 2003-12-19 EP EP03808474A patent/EP1578822B1/en not_active Expired - Lifetime
- 2003-12-19 DE DE60332866T patent/DE60332866D1/en not_active Expired - Lifetime
- 2003-12-19 AT AT03814280T patent/ATE469178T1/en not_active IP Right Cessation
- 2003-12-19 US US10/538,860 patent/US7332554B2/en not_active Expired - Lifetime
- 2003-12-19 EP EP03814282A patent/EP1572753B1/en not_active Expired - Lifetime
- 2003-12-19 JP JP2005510046A patent/JP5322253B2/en not_active Expired - Fee Related
-
2004
- 2004-12-10 US US11/010,092 patent/US7332555B2/en not_active Expired - Lifetime
- 2004-12-10 US US11/009,660 patent/US7491773B2/en not_active Expired - Lifetime
-
2010
- 2010-12-16 JP JP2010280330A patent/JP2011080084A/en not_active Withdrawn
-
2011
- 2011-02-04 JP JP2011022750A patent/JP2011122168A/en active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2534698A (en) * | 1944-11-29 | 1950-12-19 | Standard Oil Dev Co | Polymerization of olefins in fluorinated diluent |
| US2548415A (en) * | 1948-12-08 | 1951-04-10 | Standard Oil Dev Co | Boron fluoride in ethylidene fluoride for low-temperature polymerization |
| US4107417A (en) * | 1971-11-26 | 1978-08-15 | Snamprogetti, S.P.A. | Process for the production of polymers and copolymers of isobutylene |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7723447B2 (en) | 2002-12-20 | 2010-05-25 | Exxonmobil Chemical Patents Inc. | Polymerization processes |
| WO2006002033A1 (en) * | 2004-06-15 | 2006-01-05 | Exxonmobil Chemical Patents Inc. | Elastomeric compositions, air barriers, and processes for making the same |
| WO2006062572A1 (en) | 2004-12-03 | 2006-06-15 | Exxonmobil Chemical Patents Inc. | Modified layered fillers and their use to produce nanocomposite compositions |
| WO2006071312A2 (en) | 2004-12-29 | 2006-07-06 | Exxonmobil Chemical Patents Inc. | Select elastomeric blends and their use in articles |
| EP2502742A2 (en) | 2005-10-27 | 2012-09-26 | ExxonMobil Chemical Patents Inc. | Construction comprising tie layer for pneumatic tire |
| JP2009522398A (en) * | 2005-12-28 | 2009-06-11 | エクソンモービル・ケミカル・パテンツ・インク | Halogenation method |
| US7767743B2 (en) | 2006-03-10 | 2010-08-03 | Exxonmobil Chemical Patents Inc. | Processable branched isoolefin-alkylstyrene elastomers |
| US7956138B2 (en) | 2007-05-16 | 2011-06-07 | Exxonmobil Chemical Patents Inc. | Catalyst system for olefin polymerization and polymers produced therefrom |
| WO2012145005A1 (en) | 2011-04-21 | 2012-10-26 | The Yokohama Rubber Co., Ltd. | Thermoplastic elastomer composition |
| WO2013052206A1 (en) | 2011-10-05 | 2013-04-11 | Exxonmobil Chemical Patents Inc. | Tire curing bladders |
| EP2930192A1 (en) | 2014-04-11 | 2015-10-14 | LANXESS Deutschland GmbH | Initiator system for the production of synthetic rubbers |
| EP3137542A4 (en) * | 2014-04-30 | 2018-06-27 | Arlanxeo Singapore Pte. Ltd. | Highly unsaturated multi-modal polyisoolefin composition and process for preparation thereof |
| WO2021011906A1 (en) | 2019-07-17 | 2021-01-21 | Exxonmobil Chemical Patents Inc. | TIRES COMPRISING RUBBER COMPOUNDS THAT COMPRISE PROPYLENE-α-OLEFIN-DIENE POLYMERS |
| WO2021262838A1 (en) | 2020-06-26 | 2021-12-30 | Exxonmobil Chemical Patents Inc. | Copolymers composed of ethylene, a-olefin, non-conjugated diene, and substituted styrene and articles therefrom |
| WO2021262842A1 (en) | 2020-06-26 | 2021-12-30 | Exxonmobil Chemical Patents Inc. | COPOLYMERS OF ETHYLENE, α-OLEFIN, NON-CONJUGATED DIENE, AND ARYL-SUBSTITUTED CYCLOALKENE, METHODS TO PRODUCE, BLENDS, AND ARTICLES THEREFROM |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1578822B1 (en) | Polymers substantially free of long chain branching | |
| CA2510860C (en) | Polymerization process utilizing hydrofluorocarbons as diluents | |
| US7723447B2 (en) | Polymerization processes | |
| US7425601B2 (en) | Polymers with new sequence distributions | |
| WO2004058836A1 (en) | Polymers with new sequence distributions | |
| EP1578806B1 (en) | Polymerization processes |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2435/DELNP/2005 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 2006094847 Country of ref document: US Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10538965 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2510864 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005510005 Country of ref document: JP Ref document number: 20038A68675 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003808474 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2005122164 Country of ref document: RU Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003808474 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 10538965 Country of ref document: US |