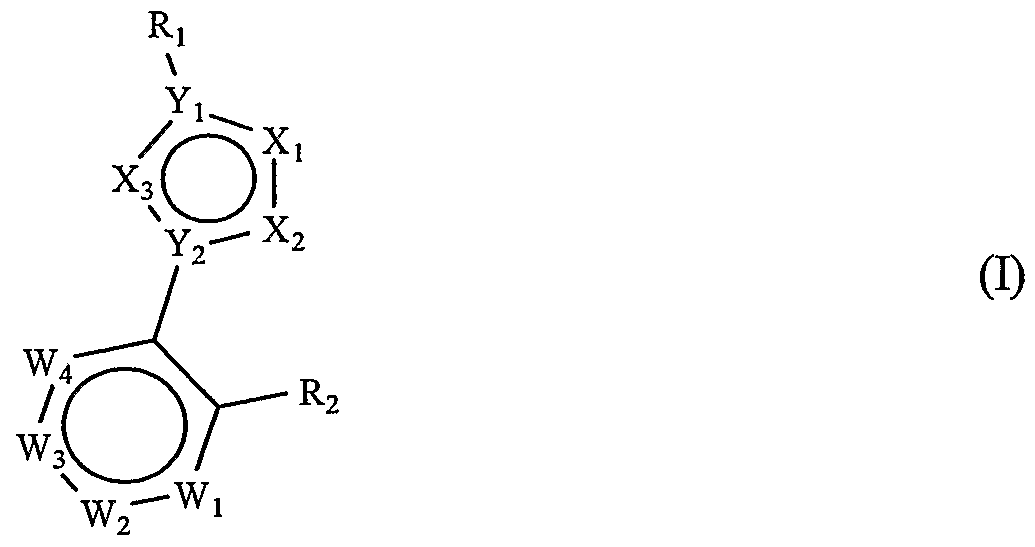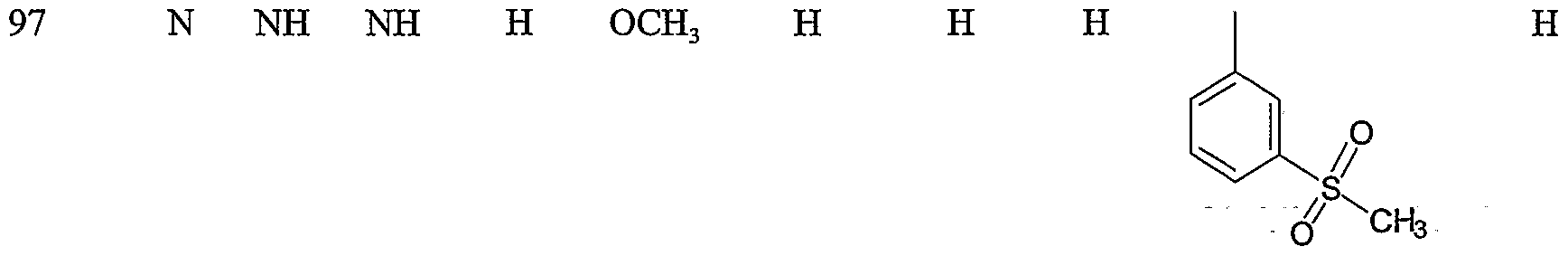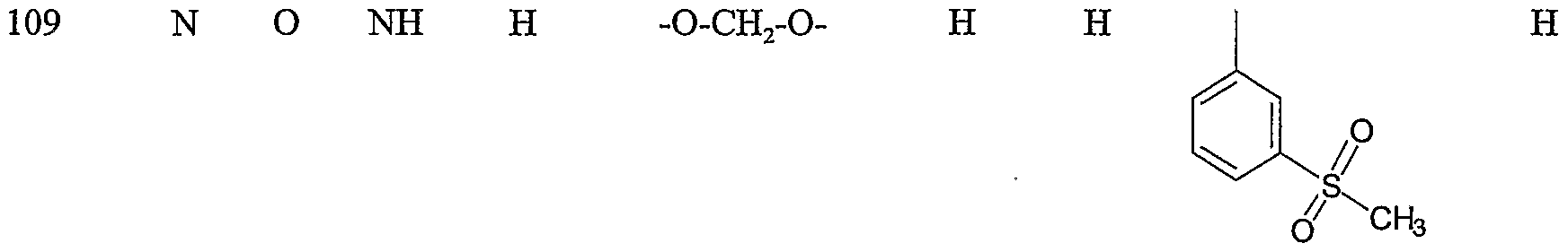A TI-A GIOGENIC COMPOUNDS AND THEIR USE IN CANCER TREATMENT
This application claims priority to U.S. Proviosnal Application No. 60/432,007, filed
December 10, 2002 and to U.S. Patent Application No.(awaited), filed December 4, 2003.
FIELD OF THE INVENTION
The present invention relates to novel five-membered ring heterocycles that inhibit the tyrosine kinase activity of VEGF receptors and are useful in the treatment of cancers, processes for making the heterocycles, pharmaceutical compositions and dosage forms that contain them, methods of treating angiogenic dependent diseases in mammals using them, and methods of manufacturing the compositions and dosage forms.
BACKGROUND OF THE INVENTION
Angiogenesis is the process by which new blood vessels develop in an organism. During angiogenesis, capillary endothelial cells proliferate and migrate f om pre-existing blood vessels into other tissues where they assemble into tubular structures and join to form tubular assemblies and closed-circuit vascular systems. These vascular systems undergo maturation to form new capillary vessels connected to the vascular network.
Angiogenesis is important to normal physiological processes like embryonic development, follicular growth, and wound healing. Angiogenesis also is necessary to the progression of some pathological conditions like neoplastic diseases (cancers) and non-neoplastic diseases that involve abnormal neovascularization like some inflammatory diseases and vascular proliferative diseases like atherosclerosis, restenosis and neovascular glaucoma (Folkman, J. and Klagsbrun, M. Støeflee 235:442-447 (1987)). In neoplastic diseases, the vascularization provides nourishment to the neoplasm and enables the migration of tumor cells through the vasculature. Inflammatory diseases whose progression is accompanied by blood vessel proliferation include rheumatoid arthritis, psoriasis, diabetic retinopathy and age-related macular degeneration. These and other
pathological conditions that are aggravated by persistent or uncontrolled angiogenesis are known as angiogenic dependent diseases.
One means of influencing the progression of angiogenic dependent diseases is to restrict the blood supply to diseased cells. Occlusion of the blood vessels that supply diseased cells by invasive procedures requires that the site(s) of the diseased cells be identified. Invasive procedures are generally limited to treatment at a single site, or a small number of sites. Direct mechanical restriction of blood supply often is ineffective at killing the diseased cells because collateral blood vessels develop, often quite rapidly, restoring the blood supply to the diseased cells. Other approaches to restricting blood supply to tumor cells have focused on the modulation of factors that are involved in the regulation of angiogenesis at the cellular level. See e.g. U.S. Patent No. 6,365,157. Angiogenesis, like many other biological mechanisms requiring regulation of cellular proliferation, is dependent on the phosphorylation of proteins by protein tyrosine kinases. Overexpression or aberrations in the pathways coupled to these kinases can produce pathological outcomes (like uncontrolled angiogenesis and tumor growth). Inhibitors, modulators or regulators of kinase activity have the potential to be therapeutically relevant agents in the treatment of tumor growth and other angiogenic dependent diseases.
Vascular endothelial growth factor ("VEGF") has been implicated in the regulation of angiogenesis in vivo (Klagsbrun, M. and D'Amore, P. (1991) Annual Rev. Physiol. 53:
217-239). VEGF is a homodimeric glycoprotein consisting of two 23 kD subunits. Four different monomeric isoforms of VEGF resulting from alternative splicing of mRNA have been identified. These include two membrane bound forms (VEGF206 and VEGFlg9) and two soluble forms (VEGF165 and VEGF121). VEGF165 is the most abundant isoform in all human tissues except placenta.
VEGF is a potent endothelial-cell specific mitogen (ED50 2-10 pM), specifically promoting proliferation of endothelial cells. VEGF is expressed in embryonic tissues (Breier et al., Development (Camb.) 114:521 (1992)), macrophages, and proliferating epidermal keratinocytes during wound healing (Brown et al., J. Exp. Med., 176:1375 (1992)), and may be responsible for tissue edema associated with inflammation (Ferrara et
al, Endocr. Rev. 13:18 (1992)). In situ hybridization studies have demonstrated high levels of VEGF expression in a number of human tumor lines including glioblastoma multiforme, hemangioblastoma, other central nervous system neoplasms and ALDS-associated Kaposi's sarcoma (Plate, K. et al. (1992) Nature 359: 845-848; Plate, K. et al. (1993) Cancer Res. 53: 5822-5827; Berkman, R. et al. (1993) J Clin. Invest. 91:
153-159; Nakamura, S. et al. (1992) AIDS Weekly, 13 (1)). A high level of VEGF expression also has been found in atherosclerotic lesions, plaques and inflammatory cells. VEGF mediates its biological effect through high affinity VEGF receptors which are selectively expressed on endothelial cells during, for example, embryogenesis (Millauer, B., et al. (1993). Cell 72: 835-846) and tumor formation. These receptors comprise a tyrosine kinase cytosolic domain that initiates the signaling pathway involved , in cell growth.
VEGF receptors are class HI receptor-type tyrosine kinases characterized by having several, typically 5 or 7, immunoglobulin-like loops in their amino-terminal extracellular receptor ligand-binding domains (Kaipainen et al, J. Exp. Med., 178:2077-88 (1993)).
The other two regions include a transmembrane region and a carboxy-terminal intracellular catalytic domain interrupted by an insertion of hydrophilic interkinase sequences of variable lengths, called the kinase insert domain (Terman et al., Oncogene, 6:1677-83 (1991)). VEGF receptors include fms-Vke tyrosine kinase receptor (flt-1), or VEGFR-1, sequenced by Shibuya et al., Oncogene, 5:519-24 (1990), kinase insert domain-containing receptor/fetal liver kinase (KDR/flk-1), or VEGFR-2, described in WO 92/14248, filed February 20, 1992, and Terman et al., Oncogene, 6:1677-83 (1991) and sequenced by Matthews et al., Proc. Natl. Acad. Sci. USA, 88:9026-30 (1991), although other receptors can also bind VEGF. Another tyrosine kinase receptor, VEGFR-3 (flt-4), binds the VEGF homologues VEGF-C and VEGF-D and is important in the development of lymphatic vessels.
It is generally believed that KDR/VEGFR-2 is the main VEGF signal transducer that results in endothelial cell proliferation, migration, differentiation, tube formation, increase of vascular permeability, and maintenance of vascular integrity. VEGFR-1 possesses a much weaker kinase activity, and is unable to generate a mitogenic response
when stimulated by VEGF, although it binds to VEGF with an affinity that is approximately 10-fold higher than KDR. VEGFR-1 has also been implicated in VEGF and placenta growth factor (P1GF) induced migration of monocytes and macrophages and production of tissue factor. When VEGF is expressed by a tumor mass, endothelial cells adjacent to the
VEGF+ tumor cells will up-regulate expression of VEGF receptor molecules, e.g., VEGFR-1 and VEGFR-2. High levels of VEGFR-2 are expressed by endothelial cells that infiltrate gliomas (Plate, K. et al., (1992) Nature 359: 845-848), and are specifically upregulated by VEGF produced by human glioblastomas (Plate, K. et al. (1993) Cancer Res. 53: 5822-5827). The finding of high levels of VEGR-2 expression in glioblastoma associated endothelial cells (GAEC) suggests that receptor activity is induced during tumor formation, since VEGFR-2 transcripts are barely detectable in normal brain endothelial cells, indicating generation of a paracrine VEGF/VEGFR loop. This upregulation is confined to the vascular endothelial cells in close proximity to the tumor. Blocking VEGF activity with neutralizing anti-VEGF monoclonal antibodies (mAbs) results in inhibition of the growth of human tumor xenografts in nude mice (Kim, K. et al. (1993) Nature 362: 841-844), suggesting a direct role for VEGF in tumor-related angiogenesis.
Accordingly, VEGFR antagonists have been developed to treat vascularized tumors and other angiogenic associated diseases. These have included neutralizing antibodies that block signaling by VEGF receptors expressed on vascular endothelial cells to reduce tumor growth by blocking angiogenesis through an endothelial-dependent paracrine loop. See, e.g., U.S. Patent No. 6,365,157 and International Publications Nos. WO 00/44777, WO 01/54723, WO 01/74296, WO 01/90192, "Bispecific Antibodies That Bind to VEGF Receptors" (Zhu, International PCT application filed June 26, 2002), and "Method of Treating Atherosclerosis and Other Inflammatory Diseases" (Carmeliet et al.; International
PCT application filed Jun. 20, 2002).
VEGF receptors also have been found on some non-endothelial cells, such as tumor cells producing VEGF, wherein an endothelial-independent autocrine loop is generated to support tumor growth. It has been demonstrated that a VEGF/human VEGFR-2 autocrine loop mediates leukemic cell survival and migration in vivo. Dias et al., "Autocrine
stimulation of VEGFR-2 activates human leukemic cell growth and migration," J. Clin. Invest. 106:511-521 (2000); Witte et al., "Treatment of non-solid mammalian tumors with vascular endothelial growth factor receptor antagonists;" and International Publication No. WO 01/74296. Similarly, VEGF production and VEGFR expression also have been reported for some solid tumor cell lines in vitro. (See Tohoku, Sato, J. Exp. Med., 185(3):
173-84 (1998); Nippon, Sanka Fujinka Gakkai Zasshi,:47(2): 133-40 (1995); and Ferrer, FA, Urology, 54(3):567-72 (1999)). It has further been demonstrated that VEGFR-1 Mabs inhibit an autocrine VEGFR/human VEGFR-1 loop in breast carcinoma cells. Wu, et al., "Monoclonal antibodys against VEGFR1 inhibits fltl -positive DU4475 human breast tumor growth by a dual mechanism involving anti-angiogenic and tumor cell growth inhibitory activities," AACR_NCI_EORTC International Conference on Molecular Targets and Cancer Therapeutics, October 29-November 2, 2001, Abstract #7; and Carmeliet et al. (International PCT application filed June 20, 2002).
In view of the foregoing, it will be appreciated that inhibitors of VEGF receptor tyrosine kinase activity can interrupt the paracrine and/or autocrine VEGF/ VEGFR loop.
Such inhibitors hold the potential to disrupt pathological angiogenisis and influence the progression of angiogenic dependent diseases.
Accordingly, there is a need for inhibitors of VEGF receptor tyrosine kinase activity. However, the road from the discovery that a compound is an inhibitor of VEGF receptor tyrosine kinases to the development of effective therapeutic agents against pathogenic vascularization is uncertain. Many factors impinge upon the outcome whether a compound shown to be effective in in vitro experiments will be an effective therapeutic agent against cancer. Such factors include side effects caused by administration of the compound in dosages necessary for anti-angiogenic effectiveness, the cost of producing or obtaining the compound, the method by which the compound must be administered and many others.
It would be highly desirable to have available new candidate therapeutic compounds with VEGF receptor tyrosine kinase activity, that are storage stable and preparable by multi-step chemical synthesis from commercially available or readily accessible starting materials.
SUMMARY OF THE INVENTION
Accordingly, it is an object of the present invention to provide active agents capable of inhibiting the tyrosine kinase activity of vascular endothelial cells, in particular vascular endothelial cells adjacent to cells affected by a disease that needs neo vascularization in order to advance to a life threatening stage. It is a further object of the invention to provide agents that are useful for the treatment of cancer. Novel small molecules have been discovered which have an inhibitory effect on the tyrosine kinase activity of VEGFR-2 receptors and utility in cancer treatment. These compounds share a 1,3-disubstituted five-membered heterocyclic ring scaffold represented by Formula (I):
wherein :
Xj and X2 are atoms or radicals independently selected from the group consisting of oxygen, sulfur, nitrogen, radicals of formula C-R3 and radicals of formula N-R3, with the proviso that at least one of Xj and X2 is oxygen, sulfur, nitrogen or a diradical of formula N-R3, wherein:
R3 is an atom or radical selected from the group consisting of: 1) hydrogen, 2) halogen,
3) alkyl, optionally substituted with one or more substituents selected
4) alkenyl, optionally substituted with one or more substituents selected from R4;
5) alkynyl, optionally substituted with one or more substituents selected from R4; wherein R4 is an atom or radical selected from the group consisting of: a) halogen; b) alkyl, which may be optionally substituted with one or more halogen, hydroxy or lower alkoxy; c) alkenyl, which may be optionally substituted with one or more halogen, hydroxy or lower alkoxy; d) nitro; e) cyano; f) oxo; g) vinyl; h) styryl; i) a group of formula -C(O)R5, -CO2R5, -OR5, -SR5, -SOR5, -
SO2R5, -NRjRg, -NCO2R5, or -OCO2R5 where R5 and R6 are atoms or radicals independently selected from the group consisting of hydrogen, lower alkyl, aralkyl, aryl and heteroaryl; X3 is selected from the group consisting of oxygen, sulfur, nitrogen and diradicals of formula N-R3,
Yj and Y2 are atoms independently selected from the group consisting of nitrogen and carbon,
Rj is a radical selected from the group consisting of:
wherein Wj is nitrogen or C-R^,,, W
2 is nitrogen or C-R^, W
3 is nitrogen or C-R
w3, W
4 is nitrogen or C-R^, W
5 is nitrogen or C-R^, W
6 is nitrogen or
and W
9 is nitrogen or C-R^ , each R^, R
w2, R
w3, R
w4, R
w5, R^, R^, R^ and R^ being an atom or radical independently selected from the group consisting of: 1) hydrogen, 2) halogen,
3) nitro,
4) cyano,
5) alkyl, optionally substituted with one or more substituents selected from R4, 6) alkenyl, optionally substituted with one or more substituents selected from R4,
7) alkynyl, optionally substituted with one or more substituents selected from R4,
8) aralkyl, optionally substituted with one or more substituents selected from R4,
9) phenyl, optionally substituted with 1 to 5 substituents selected from R4,
10) a group of the formula -CO2R5, -COR5, -OR5, -SR5, -SOR5, -SO2R6, -NR5R6, 11) pyridyl, optionally substituted with 1 to 4 substituents selected from
R ,
12) pyrazinyl, optionally substituted with 1 to 3 substituents selected from R4,
13) pyrimidinyl, optionally substituted with 1 to 3 substituents selected from R4,
14) indazolyl, optionally substituted with 1 to 5 substituents selected
15) tetrazolyl, and
14) heterocyclic radicals of formulae:
(R4)a (R4)b (R4)b
wherein X4 is O, S, or N-R4, a is 0 to 3, b is 0 to 2, c is 0 or l, or together, one or more of the combinations R^ and R^, Rw2 and Rw3, Rw3 and R^, R,v5 and Rw6, Rw6 and Rw7, R^ and R^, and Rw8 and R^ form a fused 5- or 6- membered carbocyclic ring or heterocyclic ring having one or two heteroatoms selected from nitrogen, oxygen and sulfur, Z is selected from the group consisting of oxygen, sulfur, -S(O)-, -S(O)2-, -
CR
5R
6-, -CR
5R
6O-, CR
5R
6NR
3- and -NR
3- R
2 is a radical of formula -OR
7a, -SR
7a, -S(O)R
7a,
-CR
5R
6-OR
7a wherein R
7a is a radical selected from the group consisting of: 1) -CH
2-R
8,
2) -CH2CH2-R8,
3) -CH2CH2CH2-R8, and
4) -R83 wherein R8 is a cyclic radical selected from the group consisting of: a) aromatic carbocyclic radicals, optionally substituted with 1 to 5 substituents wherein the substituents are selected from
R4 or two adjacent substituents can form a 5, 6 or 7 membered fused carbocyclic or heterocyclic ring optionally substituted with one or more substituents selected from R4, and b) aromatic and non-aromatic heterocyclicyl radicals, optionally substituted with 1 to 5 substituents wherein the substituents are either selected from R4 or two adjacent substituents can form a 5, 6 or 7 membered fused carbocyclic or heterocyclic
ring optionally substituted with one or more substituents selected from R4, or R2 is a radical selected from the group consisting of -CR5R6NR7aR7b, and -N ^R,,, and: R7a and R^ are both hydrogen,
R7a and R^ together form a 6 membered ring heterocycle, optionally substituted with one or more substituents selected from the group R4, or R-Tb is hydrogen or a radical selected from R4 and R7a is a radical selected from the group consisting of: 1) -CH2-R8,
2) -CH2CH2-R8,
3) -CH2CH2CH2-R8, and
4) -R* wherein R8 is a cyclic radical selected from the group consisting of: a) aromatic carbocyclic radicals, optionally substituted with 1 to 5 substituents wherein the substituents are selected from R4 or two adjacent substituents can form a 5, 6 or 7 membered fused carbocyclic or heterocyclic ring optionally substituted with one or more substituents selected from R4, and b) aromatic and non-aromatic heterocyclicyl radicals, optionally substituted with 1 to 5 substituents wherein the substituents are either selected from R4 or two adjacent substituents can form a 5, 6 or 7 membered fused carbocyclic or heterocyclic ring optionally substituted with one or more substituents selected from R4. Processes for preparing the active agents are also provided. The active agents may be incorporated into pharmaceutical compositions and dosage forms in accordance with further teachings of this disclosure. The active agents, compositions and dosage forms
may be used independently or together to treat angiogenic dependent diseases according to the method aspect of the invention.
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
Definitions
Unless otherwise indicated, chemical terms used in this disclosure have the meaning attributed to them by those skilled in the art of medicinal and organic chemistry and biological terms have the meaning attributed to them by those skilled in the art of molecular and cell biology. The following express definitions are consonant with those understandings.
A "hydrocarbyl" radical means a fragment of a molecule that contains carbon and hydrogen. As used in this disclosure, the term is intended to include fragments that contain, in addition to carbon and hydrogen, any number of heteroatoms. Heteroatoms may be pendant, such as the carbonyl oxygen of acetone or the fluorine atoms of 2,2- difluoropropane. Heteroatoms also may be incorporated into a hydrocarbyl fragment, such as the nitrogen of N,N-dimethylaminomethyl, the oxygen atom of diethyl ether or a polyethyleneglycol fragment. The term "alkyl" as used herein refers to a saturated hydrocarbyl radical that may be unsubstituted or substituted with one or more substituents. The alkyl radical may be straight, branched or cyclic. The term "lower alkyl" is reserved for alkyl radicals containing from 1 to 6 carbon atoms. Linear and branched lower alkyl substituents include methyl, ethyl, propyl, isopropyl, butyl and t-butyl, and the like. Cyclic lower alkyl radicals include cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
The term "alkenyl" as used herein refers to a straight, branched or cyclic non- aromatic hydrocarbyl radical containing at least one carbon to carbon double bond. The term "lower alkenyl" as used herein is reserved for an alkenyl radical containing from 2 to 6 carbon atoms. Lower alkenyl groups include ethenyl, propenyl, butenyl cyclohexenyl, and the like.
The term "alkynyl" as used herein refers to a straight, branched or cyclic hydrocarbyl radical containing at least one carbon to carbon triple bond. The term "lower alkynyl" as used herein is reserved for an alkynyl radical containing from 2 to 6 carbon atoms. Lower alkynyl groups include ethynyl, propynyl and butynyl, and the like. Alkyl, alkenyl and alkyl radicals typically have between 1 and 40 carbon atoms.
The term "aryl" as used herein refers to carbocyclic and heterocyclic radicals that are "aromatic," i.e. they possess cyclic π electron systems that have a stabilizing electromc character called resonance stabilization. Fieser & Fieser, Organic Chemistry 525-526 (3rd ed. 1956), Streitweiser, A.; Heathcock, CH. Introduction to organic Chemistry 643 (2nd ed. 1981 ). Aryl radicals can be distinguished by those skilled in the art from other radicals having cyclic π electron systems by applying the Huckel rule which states that resonance stabilization will occur when the number of electrons that are delocalized in the π electron system is 4n+2 where n is an integer. Aryl radicals include unsubstituted or substituted 6-membered aromatic rings, such as, phenyl, substituted phenyl and like, as well bicyclic rings, such as naphthyl, and polycyclic rings, such as anthracenyl, and heterocyclic radicals, such as pyridinyl as well as, imidazolyl, oxazolyl, thiazolyl, indazolyl and the like (wherein the Huckel rule is satisfied by the contribution by a heteroatom of a pair of electrons to the aromatic stabilization).
The terms "heterocycle," "heteroaryl" and "heterocyclic" mean a stable 5- to 6-membered mono- or 7- to 10-membered fused bicyclic ring system, any ring of which may be saturated or unsaturated, aromatic or non-aromatic, provided that from one to three ring atoms are selected from the heteroatoms N, O and S while the other ring atoms are carbon. The nitrogen and sulfur heteroatoms may optionally be oxidized, and the nitrogen heteroatom may optionally be quaternized. The term "heterocycles" includes the abovementioned heteroaryls, as well as reduced (e.g. dihydro and tetrahydro) analogs thereof. Heterocycles include any bicyclic group in which any of the above-defined rings is fused to a benzene ring. The heterocyclic ring may be attached at any heteroatom or carbon atom, which results in the creation of a stable structure. Examples of such heterocyclic components include piperidinyl, piperazinyl, 2-oxopiperazinyl, 2-oxopiperidinyl, 2-oxopyrrolodinyl, 2-oxoazepinyl, azepinyl, pyrrolyl, 4-piperidonyl, pyrrolidinyl, pyrazolyl, pyrazolidinyl, imidazolyl, imidazolinyl, imidazolidinyl, pyridinyl, pyrazinyl, pyrimidinyl, pyridazinyl, oxazolyl, oxazolidinyl, isoxazolyl, isoxazolidinyl, mo holinyl, thiazolyl, thiazolidinyl, isothiazolyl, - quinuclidinyl, isothiazolidinyl, indolyl, quinolinyl, isoquinolinyl, benzimidazolyl, thiadiazoyl, benzopyranyl, benzothiazolyl, benzoxazolyl, furyl, tefrahydrofuryl,
tefrahydropyranyl, thiophenyl, imidazopyridinyl, tetrazolyl, triazinyl, thienyl, benzothienyl, thiamorpholinyl sulfoxide, thiamorpholinyl sulfone, and oxadiazolyl.
The term "alkoxy" refers to a radical containing a straight, branched or cyclic hydrocarbyl radical attached via an oxygen atom. Substituents on the hydrocarbyl group may include for example, a phenyl ring, in which the alkoxy may be for example, a benzyloxy group. Examples of alkoxy groups are methoxy, ethoxy, propoxy, isopropoxy, butoxy, isobutoxy, tertiary butoxy, pentoxy, isopentoxy, hexoxy, isohexoxy allyloxy, propargyloxy, vinyloxy, and the like.
The term "aralkyl" as used herein contemplates a lower alkyl group which has as a substituent an aryl group.
The term "halo" or "halogen" as used herein is intended to include the halogen atoms fluorine, chlorine, bromine and iodine.
The chemical nomenclature used in this disclosure follows the system for naming compounds of the International Union of Pure and Applied Chemistry (IUPAC). The IUPAC system assigns a number to every atom in a compound. This disclosure also uses relative numbering where it is the clearest way to describe the invention to those skilled in the art. The absolute numbering assigned by the IUPAC system is to be distinguished from relative numbering commonly used to express bonding relationships between functional groups. Thus, it will be appreciated that the phenyl groups of 3,5-diphenyl-l,2,4-triazole are in a 1,3 relationship to each other because they are separated by three atoms including those to which they are attached. Whether atom numbering is absolute or relative will be clear to one skilled in the art from the context in which it is used.
When one or more chiral centers are present in the compounds of the present invention, the individual isomers and mixtures thereof (e.g., racemates, etc.) are intended to be encompassed by the formulae depicted herein.
1.3-Disubstituted Five-membered Ring Heterocyclic VEGF Tyrosine Kinase Inhibitors
The present invention provides novel compounds that inhibit tyrosine kinase activity of vascular endothelial growth factor receptors. The present invention is not intended to be limited to any particular mechanism of VEGF receptor kinase inhibition.
The compounds of the present invention are certain appropriately 1,3-disubstituted five- membered ring heterocycles that are encompassed by Formula (I).
X, and X2 are atoms or radicals independently selected from the group consisting of oxygen, sulfur, nitrogen, radicals of formula C-R3 and radicals of formula N-R3, with the proviso that at least one of Xj and X2 is oxygen, sulfur, nitrogen or a diradical of formula N-R3, wherein:
R3 is an atom or radical selected from the group consisting of:
1) hydrogen,
2) halogen,
3) alkyl, optionally substituted with one or more substituents selected from R4;
4) alkenyl, optionally substituted with one or more substituents selected from R4;
5) alkynyl, optionally substituted with one or more substituents selected from R4; wherein R4 is an atom or radical selected from the group consisting of: a) halogen; b) alkyl, which may be optionally substituted with one or more halogen, hydroxy or lower alkoxy; c) alkenyl, which may be optionally substituted with one or more halogen, hydroxy or lower alkoxy;
d) nitro; e) cyano; f) oxo; g) vinyl; h) styryl; i) a group of formula -C(O)R5, -CO2R5, -OR5, -SR5, -SOR5, -
SO2R5, -NR5R6, -NCO2R5, or -OCO2R5 where R5 and R6 are atoms or radicals independently selected from the group consisting of hydrogen, lower alkyl, aralkyl, aryl and heteroaryl;
X3 is selected from the group consisting of oxygen, sulfur, nitrogen and diradicals of formula N-R3,
Yj and Y2 are atoms independently selected from the group consisting of nifrogen and carbon, Rλ is a radical selected from the group consisting of:
wherein Wj is nitrogen or C-R^, W2 is nifrogen or C-R^, W3 is nitrogen or C-R^, W4 is nifrogen or C-R^, W5 is nifrogen or C-R^, W6 is nitrogen or C-Rw6, W7 is nitrogen or C-R^, W1 is nitrogen or C-R^ and W9 is nitrogen or C-R^ , each R^, R^, R^, R^, Rw5, Rw6, R^, R^ and Rw9 being an atom or radical independently selected from the group consisting of:
1) hydrogen,
2) halogen, 3) nitro,
4) cyano,
5) alkyl, optionally substituted with one or more substituents selected from R4, 6) alkenyl, optionally substituted with one or more substituents selected from R4, 7) alkynyl, optionally substituted with one or more substituents selected from R4,
8) aralkyl, optionally substituted with one or more substituents selected from R4,
9) phenyl, optionally substituted with 1 to 5 substituents selected from R4,
10) a group of the formula -CO2R5, -COR5, -OR5, -SR5, -SOR5, -SO2R6, -NR5R6,
11) pyridyl, optionally substituted with 1 to 4 substituents selected from R4, 12) pyrazinyl, optionally substituted with 1 to 3 substituents selected from R4,
13) pyrimidinyl, optionally substituted with 1 to 3 substituents selected
14) indazolyl, optionally substituted with 1 to 5 substituents selected from R
4,
15) tetrazolyl, and
14) heterocyclic radicals of formulae:
wherein X4 is O, S, or N-R4, a is 0 to 3, b is 0 to 2, c is 0 or 1, or together, one or more of the combinations R^ and Rw2, R^ and Rw3, Rw3 and R^, Rw5 and Rw6, Rw6 and R^, R^ and ^, and Rw8 and R^ form a
fused 5- or 6- membered carbocyclic ring or heterocyclic ring having one or two heteroatoms selected from nitrogen, oxygen and sulfur, Z is selected from the group consisting of oxygen, sulfur, -S(O)-, -S(O)2-, - CR5R6-, -CR5R6O-, CR5R6NR3- and -NR3- R2 is a radical of formula -OR7a, -SR7a, -S(O)R7a, -CR5R6R7a, -CR5R6-OR7a wherein R7a is a radical selected from the group consisting of:
1) -CH2-R85
2) -CH2CH2-R8,
3) -CH
2CH
2CH
2-R
8, and
wherein R
8 is a cyclic radical selected from the group consisting of: a) aromatic carbocyclic radicals, optionally substituted with 1 to 5 substituents wherein the substituents are selected from R
4 or two adjacent substituents can form a 5, 6 or 7 membered fused carbocyclic or heterocyclic ring optionally substituted with one or more substituents selected from R
4, and b) aromatic and non-aromatic heterocyclicyl radicals, optionally substituted with 1 to 5 substituents wherein the substituents are either selected from R
4 or two adjacent substituents can form a 5, 6 or 7 membered fused carbocyclic or heterocyclic ring optionally substituted with one or more substituents selected from R
4, or R
2 is a radical selected from the group consisting of -CR
5R
6-NR
7aR
7b, and -NR^R^ and:
R7a and R^ are both hydrogen,
R7a and R^ together form a 6 membered ring heterocycle, optionally substituted with one or more substituents selected from the group R4, or R-Tb is hydrogen or a radical selected from R4 and R7a is a radical selected from the group consisting of:
1) -CH2-R8,
2) -CH2CH2-R8,
3) -CH2CH2CH2-R8, and
4) -R
8, wherein R
8 is a cyclic radical selected from the group consisting of: a) aromatic carbocyclic radicals, optionally substituted with 1 to 5 substituents wherein the substituents are selected from R
4 or two adjacent substituents can form a 5, 6 or 7 membered fused carbocyclic or heterocyclic ring optionally substituted with one or more substituents selected from R
4, and b) aromatic and non-aromatic heterocyclicyl radicals, optionally substituted with 1 to 5 substituents wherein the substituents are either selected from R
4 or two adjacent substituents can form a 5, 6 or 7 membered fused carbocyclic or heterocyclic ring optionally substituted with one or more substituents selected from R
4. Generally, substituent R
2 is preferably a radical of formula -NH-R
8, -NHCH
2-R
8, - NHCH
2CH
2-R
8 or -NHCH
2CH
2CH
2-R
8, wherein R
8 is an optionally substituted phenyl radical, optionally substituted bicyclic aromatic carbocyclic radical, optionally substituted polycyclic aromatic carbocyclic radical, optionally substituted monocyclic aromatic heterocyclic radical, optionally substituted bicyclic aromatic heterocyclic radical or optionally substituted polycyclic aromatic heterocyclic radical. Preferred R
8 substituents are phenyl, optionally substituted with 1 to 5 substituents selected from R
4; pyridinyl, optionally substituted with 1 to 4 substituents selected from R
4; pyrazinyl, optionally substituted with 1 to 3 substituents selected from R
4; pyrimidinyl, optionally substituted with 1 to 3 substituents selected from R
4; indazolyl, optionally substituted with 1 to 5 subsitutents selected from R
4; tetrazolyl;or an aromatic heterocyclic radical of formula:
wherein X, R4 and a-c are as previously defined. An especially preferred Z substituent is NH.
Compounds of Formula (I) were previously unknown in any state of purity and are not a known component of any naturally occurring composition. Thus, the present invention is directed to these compounds in all states of purity. The compounds of this invention exist in different physical states as well, such as dissolved states and condensed states. Condensed states include salts and solvates of the compound (particular salts suitable for pharmaceutical use are described below). The present invention also relates to "prodrugs" of compounds of Formula (1). A "pro-drug" is a compound that is administered to a patient and is converted within the patient's body into a second compound that exerts a therapeutic effect. The present invention encompasses pro-drugs that are transformed in vivo to yield the Formula (I) compound or a protein conjugate of it. Pro-drugs are thoroughly discussed in T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems Vol. 14 of the A. C. S. Symposium Series, and in Edward B. Roche, ed.,
Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987.
The VEGF receptor antagonists of the invention are readily prepared by general methodologies described in compendia and freatises on heterocycles and organic synthesis methods. In particular, there may be mentioned Comprehensive Heterocyclic Chemistry
(Alan Katritzky and Charles Rees eds., Pergamon Press: Oxford 1984); Heterocyclic Compounds (John Wiley & Sons: New York 1981) and Methoden der Organishchen Chemie (Houben-Weyl: Stuttgart 1974). Any heterocyclic ring systems encompassed by Formula (I) that are inaccessible because they are unstable can be readily determined by those skilled in the art by consulting standard reference works in the field such as those referenced immediately above.
1.2.4-Triazole VEGF Tyrosine Kinase Inhibitors
One preferred class of 1,3-disubstituted (relative numbering) five-membered ring heterocycles of Formula (I) are 3,5-disubstiruted-l,2,4-triazoles. Triazoles are 5- membered ring heterocycles containing three nitrogen atoms. Triazoles exist in two distinct classes-l,2,3-triazoles and l,2,4-triazoles~differentiated by the positions of the nitrogen atoms in the aromatic ring. Both classes have aromatic character. 1,2,3- Triazoles characteristically have a annular nifrogen atom bonded to two other annular nifrogen atoms. The 1,2,4-triazoles characteristically have only one direct bond between annular nitrogen atoms. The triazoles that have been discovered to have VEGF receptor kinase inhibitory activity are 1,2,4-triazoles. Accordingly, one preferred class of 1,2,4- triazoles of the present invention are those that are encompassed by Formula (la):
wherein Rl5 R2, R3, W,, W2, W3 and W4 are as previously defined with respect to Formula
(I). i preferred 1,2,4-triazoles of the present invention, R3 is hydrogen. In yet more preferred 1,2,4-triazoles of the present invention, R2 is -NH-R7a, R3 is hydrogen, W2 is C-
R
w2, W
3, W
4, W
5 and W
9 are CH, W
7 is C-R^ and W
8 is C-R^. Accordingly, more preferred 1,2,4-triazoles of the present invention are those encompassed by Formula (la'):
wherein W]5 W6, R^, R^, Rw8 and R7a are as previously defined with respect to Formula
(I)-
In a second preferred group of 1,2,4-triazoles of the present invention, the triazole ring and one of the aryl rings are connected through a linking group Z. In addition, R2 is a radical of formula -NR^R^. Accordingly, this second preferred class of 1,2,4-frazoles is encompassed by Formula (la"):
wherein W]3 R^-R^, R7a, ^ and Z are as previously defined with respect to Formula (I). Yet more preferred 1,2,4-triazoles of Formula (la") are those wherein Z is -NH-, -CH2O-, or -CH2-.
1,2,4-Triazoles whose annular nitrogens are unsubstituted as they are in Formulae (la') and (la") are likely to exist as a rapidly equilibrating mixture of three isomers, a
phenomenon called protofropy. In fact, it has been stated that the parent compound 1,2,4- triazole is best represented as a proton associated with a resonance stabilized triazole anion. Ainsworth et al. J. Med. Pharm. Chem. 1962, 5, 383. Representations of the three isomers, or tautomers, may be generated by attaching the hydrogen atom whose point of attachment is unspecified to each of the annular nitrogen atoms and adjusting the positions of the two double bonds to obtain proper valency. In the general case, these tautomers are not separable at ambient temperature. All three interconverting tautomers are intended to be covered by Formulae (I), (la') and (la"), it being understood by those skilled in the art that, when protofropy occurs, one tautomer does not exist free of the other tautomers under conditions in which the compounds of the present invention will be used, e.g. in a pharmaceutical composition or the bloodstream of a mammal.
1,2,4-Triazoles are readily prepared by methods described in Comprehensive Heterocyclic Chemistry Part 4a (Alan Katritzky and Charles Rees eds., Pergamon Press: Oxford 1984); Heterocyclic Compounds Vol. 37 (John Wiley & Sons: New York 1981) stndMethoden der Organishchen Chemie (Houben-Weyl: Stuttgart 1974).
1,2,4-Triazoles can be prepared from acyclic starting materials by reacting a substituted or unsubstituted hydrazine with certain compounds containing a (-C(=O)-NH-) or -C(=NH)-NH- fragment. Methodologies for making 1,2,4-triazoles from acyclic starting materials include (1) condensation of a hydrazine (R-NHNH2) with an imide (R'-C(O)NHC(O)-R"), known as the Einhorn-Brunner reaction. Einhorn et al. Liebigs
Ann. Chem. 1905, 343, 229; Briinner £er. Dtsch Chem. Ges. 1914, 472671; Brunner /?. Chem. 1915, 3(5, 509. Additional methodologies include (2) condensation of a hydrazide (a.k.a. acylhydrazine) (R-C(O)NHNH2) with a primary amide (R'C(O)NH2), known as the Pellizzari reaction (3) condensation of an amidrazone (RC(=NH)NR'-NH2) with a carboxylic acid (R"COOH) and (4) cyclization of an acylamidrazone (RC(O)-NR'-
N=C(R")NH2).
Conversion of these compounds to 1,2,4-triazoles is generally performed in a polar organic solvent, like a lower alcohol or THF, under neutral or mildly basic conditions at a concentration of about lmM to about 2 M. Depending upon the choice of solvent, concenfration and nature of the particular starting materials, the reaction may proceed at
reduced, ambient or elevated temperatures. Generally, reaction conditions of about IM in refluxing anhydrous ethanol may be tried as a starting point for optimization of any particular ring closing reaction. Ethanol may be replaced with a high boiling solvent if the reaction does not proceed at a practicable rate. The Pellizzari reaction suffers in comparison to some of the other methods because high reaction temperatures are frequently required.
In a preferred process, the 1,2,4-triazoles of this invention are prepared by contacting a hydrazide of Formula (II)
or salt thereof, with an amidine of Formula (IH)
or salt thereof under conditions effective to condense the hydrazide group of (II) and amidine group of (in) into a 1,2,4-triazole ring. The ring closure is effected under basic conditions which can be established using a strong base, with alkali metal hydroxides and alkoxides being preferred. Other bases that may suitable include sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, trialkylamines, such as trimethylamine and triethylamine, dialkylanilines, such as N,N-diethylaniline, pyridines, such as 2,4,6-trimethylpyridine, pyridine, and dimethylaminopyridine, DBU, or DABCO, The molar amount of the base to be used is 1 to 20, preferably 1 to 5, times the amount of limiting reagent. Compounds (TJ) and (III) can be contacted in a melt (if at least one is a liquid at the reaction temperature) or in an inert diluent. Diluents include water and organic
compounds that are liquid at ambient temperature like certain lower alcohols, such as methanol, ethanol, 1-propanol, 2-propanol, 1-butanol, 2-butanol and zjo-butanol; aliphatic and aromatic hydrocarbons, such as hexane, pefroleum ether, benzene, toluene, and xylene; halogenated hydrocarbons, such as dichloromethane, chloroform and Freons; ethers, such as diethyl ether, methyl t-butyl ether, 1,2-dimethoxyethane, dioxane and tetrahydrofuran;
N,N-dialkyl amides, such as dimethylformamide and dimethylacetamide; ketones, such as acetone and methyl ethyl ketone; nitriles, such as acetonitrile and acrylonitrile; and sulfoxides, such as dimethylsulfoxide. Preferably, compounds (H) and (III) are contacted in a lower alcohol diluent, with a preferred lower alcohol being ethanol. Conveniently, the ring closure is conducted in a lower alcohol using an alkali metal salt of the corresponding alkoxide anion or different alkoxide anion as base. Although any concenfration that allows the ring closure to go substantially to completion is suitable, a concentration of from about 100 mM to about 2 M, more particularly about 1 M, is recommended. If the reaction does not go to completion in a practicable period of time at the boiling temperature of the alcohol in the recommended concentration ranges then the reaction mixture can be concentrated and the residue taken up in a higher boiling solvent, like chlorobenzene.
Alternatively, it may be desirable for functional group compatibility to prepare a hydrazide of Formula (IV)
or salt thereof, and an amidine of Formula (V)
or salt thereof and react them under generally the same conditions used when reacting (H) and (m).
The hydrazides and amidines, in turn, may be prepared conventionally by known synthetic pathways. Scheme I depicts a process whereby commercially available and readily accessible nitriles may be converted into an amidine (HI) salt that can be used in the making of the 1,2,4-triazoles of this invention. In an anhydrous lower alcohol solvent (e.g. ethanol) an appropriately substituted nitrile undergoes addition by a molecule of solvent when treated with anhydrous hydrochloric acid to form an imidate. The imidate may be conveniently isolated as an acid addition salt with the acid catalyst. The imidate salt may be carried forward in the next step of the process. Treatment of the imidate or imidate salt with ammonia transforms the imidate group into an amidine group.
Scheme I
NH NH aN II ll
R^ rT Rf "OEt - HX NH3 Rf NH2 • HX
HOEt
(HT)
Ort o-alkylamino substituted benzoic acid hydrazides can be prepared by condensation of an ester of anthranilic acid and a lower alcohol with an aldehyde followed by reduction of the so-formed imine. The resulting ort/zo-methylamino substituted benzoic acid is then contacted with hydrazine to yield hydrazide (H), as shown in Scheme H
Scheme H
(II)
To form the intermediate imine, the aldehyde and anthranilic acid lower alcohol ester are contacted under mildly acidic conditions. Preferred acid catalysts are acetic acid, methanesulfonic acid, trifluoroacetic acid, pivalic acid, hydrochloric acid, hydrobromic acid, with the most preferred acid being acetic acid. After formation of the imine, optionally with isolation, the imine group is reduced with a suitable reducing agent.
Suitable reducing agents include sodium cyanoborohydride, sodium borohydride, lithium aluminum hydride, DIBAL, diborane and the like and catalytic reducing agents such as Raney nickel, and palladium on carbon. The most preferred reducing agent is sodium cyanoborohydride. Additional reducing agents that are known to reduce imines to amines can be found in Larock R.C. Comprehensive Organic Transformations 2nd ed. (Wiley-
VCH: New York 1999). The addition-elimination and reduction can both be performed in lower alcohols, as wells as ethers and halogenated organic solvents that will solubilize the reactants and reagents that are adapted for use in homogenous liquid phase reactions. Hydrazide (II) may be prepared by reacting the carboxylic acid group of the reduced product with a hydrazine. Unsubstituted hydrazine is conveniently available as a solid hydrate (Aid. Cat. Nos. 22,581-9 and 20,794-2) and in solutions in THF and water (Aid. Cat. Nos. 43,363-2 and 30,940-0). Substituted hydrazines may be readily prepared from NH2NH2 by methods well known to those skilled in the art.
Alternatively, it maybe possible with some starting materials to prepare the 1,2,4- triazole in a reasonable yield in a one pot process by heating an appropriately substituted nitrile with a benzene hydrazide. See, Weidinger, H.; Kranz, J. Berichte 1963, 96, 1064. However, its lack of general applicability makes it a less preferred process.
Generally, the ring-forming condensation proceeds cleanly and in good yield without protection when ortAo-secondary amines are present on (V). However, compounds (II)-(V) may bear functionality that inhibits one or more reactions used in making the compounds of the present invention. Compounds (II)-(V) may be derivatized to a form that modifies interfering chemical functionality using well established techniques of functional group protection. For instance, if necessary, hydroxy groups, carboxylic acids, and primary and secondary amine groups on substituents R^ may be protected as desired as silyl ethers, esters, carbamates, carbonates and the like using techniques well known to the skilled artisan. The selection, application and removal of protective groups are the subjects of many reference works such as Green & Wuts, Protective Groups in Organic Synthesis (John Wiley & Sons: New York 1999) and McOmie, Protective Groups in Organic Chemistry (Plenum Press: London 1973). The use of protection strategies to make compounds of other heterocyclic classes discussed below also may be
advantageous.
1.3.4-Oxadiazole VEGF Tyrosine Kinase Inhibitors
A second preferred class of 1,3-disubstituted five-membered ring heterocycles of Formula (I) is 2,5-disubstituted oxadiazoles. Oxadiazoles are 5-membered ring heterocycles containing two double bonds, one oxygen atom and two nitrogen atoms. Oxadiazoles are potentially capable of existing in three distinct classes, 1,2,3-, 1,2,4-, 1,3,4- and 1,2,5-oxadiazoles. However, few unfused 1,2,3-oxadiazoles are stable. Especially preferred oxadiazoles of the present invention are 1,3,4-triazoles. Accordingly, one preferred class of 1 ,3-disubstituted five-membered ring heterocycles of the present invention are those that are encompassed by Formula (lb)
wherein Rl5 R2, W1? W2, W3 and W4 are as previously defined with respect to Formula (I). In yet more preferred 1,3,4-oxadiazoles of the present invention, Rj is a substituted or unsubstituted aniline group (-NH-Aryl) and R2 is a radical of formula -NR^ ^. Accordingly, yet more preferred 1,3,4-oxadiazoles of the present invention are those that are encompassed by Formula (lb')
Yet more preferably, Wj is nitrogen and W2, W3 and W4 are each CH.
Methods of preparing 1,3,4-oxadiazoles are described in Comprehensive Heterocyclic Chemistry Part 6, pp. 440-445 (Alan Katritzky and Charles Rees eds.,
Pergamon Press: Oxford 1984); Heterocyclic Compounds "Five- And Six-Membered Compounds with Nitrogen And Oxygen" (Richard H. Wiley ed., John Wiley & Sons: New York 1962), Katritzky, A.R.; Pozharskii, A.F. Handbook of Heterocyclic Chemistry 2nd edition pp. 591-596 (Pergamon: NY 2000); Gilchrist, T.L. Heterocyclic Chemistry 3rd edition, p. 336 (Longman Publ.: Singapore 1997); Joule, J.A. et al. Heterocyclic
Chemistry 3rd edition p.445 (Chapman & Hall: London 1995). These methods include cyclization-dehydration of diacylhydrazines (RC(O)-NHNH-C(O)-R). Each of the treatises referenced in this paragraph is hereby incorporated by reference in its entirety and, in particular, for the discussion of methods of preparing 1,3,4-oxadiazoles contained on the cited pages.
In accordance with a particularly preferred process for preparing 1,3,4-oxadiazoles of Formula (lb'), a hydrazide of Formula (II) (wherein R2=NR7aR7b), previously defined, is contacted with an isothiocyanate of Formula (VI):
to give an intermediate of Formula (VH).
This addition reaction is generally performed in an organic solvent, like dichloromethane, under neutral conditions and at ambient or elevated temperature at a concenfration of about ImM to about 2 M. The intermediate is then cyclized by treating it with a coupling agent like dicyclohexylcarbodiimide.
Isothiocyanate starting materials of Formula (VI) are either commercially available or can be conveniently prepared using well-establish techniques of functional group interconversion starting from the corresponding unsubstituted benzene derivative by electrophilic aromatic substituted with, e.g., ammonium thiocyanate; the corresponding nitro derivative by reaction with carbon disulfide; or the corresponding trialkylsilylamine, or amide by treatment with carbon disulfide and many other techniques described in Comprehensive Organic Functional Group Transformations Vol. 5 pp. 1021-1045 (Katritzky, A.R. et al. editors, Elsevier Science Ltd: Oxford 1995) which is hereby incorporated by reference in its entirety and, in particular, for its description of methods of making isothiocyanates contained in the cited pages of the reference.
Oxazole VEGF Tyrosine Kinase Inhibitors
A third preferred class of 1,3-disubstituted five-membered ring heterocycles of Formula (I) are oxazoles. Oxazoles contain two double bonds, one oxygen atom and one nitrogen atom that is not adjacent to the oxygen atom in the five-membered heterocyclic ring. One preferred class of 1,3-disubstituted oxazoles are 2,5-disubstituted oxazoles (absolute numbering) encompassed by Formula (lc):
wherein either Xj is nitrogen and X2 is C-R3 or Xj is C-R3 and X2 is nifrogen, and Rl5 R2, Wlt W2, W3 and W4 are as previously defined with respect to Formula (I). hi preferred 2,5-disubstituted oxazoles of the present invention, Rj is optionally substituted phenyl, Wl5 W2, W3 and W4 are each CH and R2 is -NH-R7a. Especially preferred 2,5-disubstituted oxazoles are those encompassed by Formula (lc'):
wherein Rw5, Rvι6 and Rw7 are as previously defined with respect to Formula (I) and X, and
X2 are as previously defined with respect to Formula (lc).
Methods of preparing oxazoles are described in Comprehensive Heterocyclic Chemistry Part 6, pp. 216-223 (Alan Katritzky and Charles Rees eds., Pergamon Press:
Oxford 1984); Heterocyclic Compounds "Oxazoles Part A" Vol. 60 pp. 4-127 (David C.
Palmer ed., John Wiley & Sons: New York 2003), Katritzky, A.R.; Pozharskϋ, A.F.
Handbook of Heterocyclic Chemistry 2nd edition pp. 591-596 (Pergamon: NY 2000);
Gilchrist, T.L. Heterocyclic Chemistry 3rd edition, pp. 320-322 (Longman Publ.: Singapore 1997); Joule, J.A. et al. Heterocyclic Chemistry 3rd edition pp.386-392
(Chapman & Hall: London 1995). These methods include cyclization-dehydration of α- acylaminocarbonyl compounds (R'C(O)C(R")-NH-C(O)-R'") known as the Robinson- Gabriel synthesis and the condensation of aldehyde cyanohydrins and aromatic aldehydes in dry ether in the presence of dry hydrochloric acid known as the Fischer Oxazole synthesis. R. H. Wiley, Chem. Rev. 1945 37, 410; J. W. Cornforth, R. H. Cornforth, J. Chem. Soc. 1949, 1028; J. W. Cornforth, Heterocyclic Compounds 1957, 5, 309; T. Onaka, Tetrahedron Letters 1971, 4391. Each of the freatises and articles referenced in this paragraph is hereby incorporated by reference in its entirety and, in particular, for the discussion of methods of preparing oxazoles contained on the cited pages.
Imidazole VEGF Tyrosine Kinase Inhibitors
A fourth preferred class of 1,3-disubstituted five-membered ring heterocycles of Formula (I) are 2,4-disubstituted imidazoles. Imidazoles contain two double bonds and two non-adjacent nifrogen atoms. in the five-membered heterocyclic ring. Accordingly, one preferred class of 2,4-disubstituted imidazoles of the present invention are those that are encompassed by Formula (Id):
wherein either X, is nitrogen and X
2 is -CR
3 or X
t is -CR
3 and X
2 is nitrogen and R„ R
2,
W
l9 W
2, W
3 and W
4 are as defined with respect to Formula (I). In preferred 2,4- disubstituted imidazoles of the present invention, X
! is CR
3 (more preferably CH or CH
3), and X
2 is nifrogen, R
j is optionally substituted phenyl, R
2 is -NH
2, and W
l5 W
2, W
3 and W
4 are each CH. Yet more preferred imidazoles of the present invention are those encompassed by Formula (Id').
Methods of preparing imidazoles are described in Comprehensive Heterocyclic Chemistry Part 5, pp. 457-496 (Alan Katritzky and Charles Rees eds., Pergamon Press: Oxford 1984); Heterocyclic Compounds "Imidazole And Derivatives Part I" pp.33-43 (Klaus Hofinann ed., Interscience Publishers: New York 1953); Gilchrist, T.L. Heterocyclic Chemistry 3rd edition, pp. 299-300 (Longman Publ.: Singapore 1997); Joule, J.A. et al. Heterocyclic Chemistry 3rd edition pp. 384-387 (Chapman & Hall: London 1995). Each of the treatises referenced in this paragraph is hereby incorporated by reference in its entirety and, in particular, for the discussion of methods of preparing imidazoles contained on the cited pages.
In a preferred process, the imidazoles of this invention are prepared by contacting an amidine of Formula (V) with an -halo carboxyl compound of Formula (VIII) :
wherein substituent "hal" is bromine or iodine, or by contacting an amidine of Formula (HI) with an α-halo ketone of Formula (IX):
When R2 is NH2, R2 is an interfering functionality. We overcame this problem by
using a 2-nitro analog of the (2-aminoaryl) α-halo ketone (DC) (NO2 in place of R2) and then reducing the nitro group with palladium-on-carbon. Accordingly, imidazoles of Formula (Id') may be prepared by a variation of the reaction expressed in Scheme HI.
Scheme UJ
The cyclization reaction is generally performed in a polar aprotic solvent like DMF or THF, under basic conditions at a concentration of about ImM to about 2 M. If the starting halide is a bromide, it can be activated by addition of sodium iodide to the reaction mixture. In addition, the reaction mixture may be refluxed to accelerate the reaction. After the cyclization, the nitro group is reduced using conditions well known in the art for reducing aryl nitro groups to amines. Such conditions are summarized and citations to primary references are given in Larock, R.C. Comprehensive Organic Transformations 2nd ed. pp. 823-827 (Wiley- VCH: New York 1999), which pages and the primary references to which they refer are hereby incorporated by reference for their teachings how to reduce nitroarenes to aminoarenes.
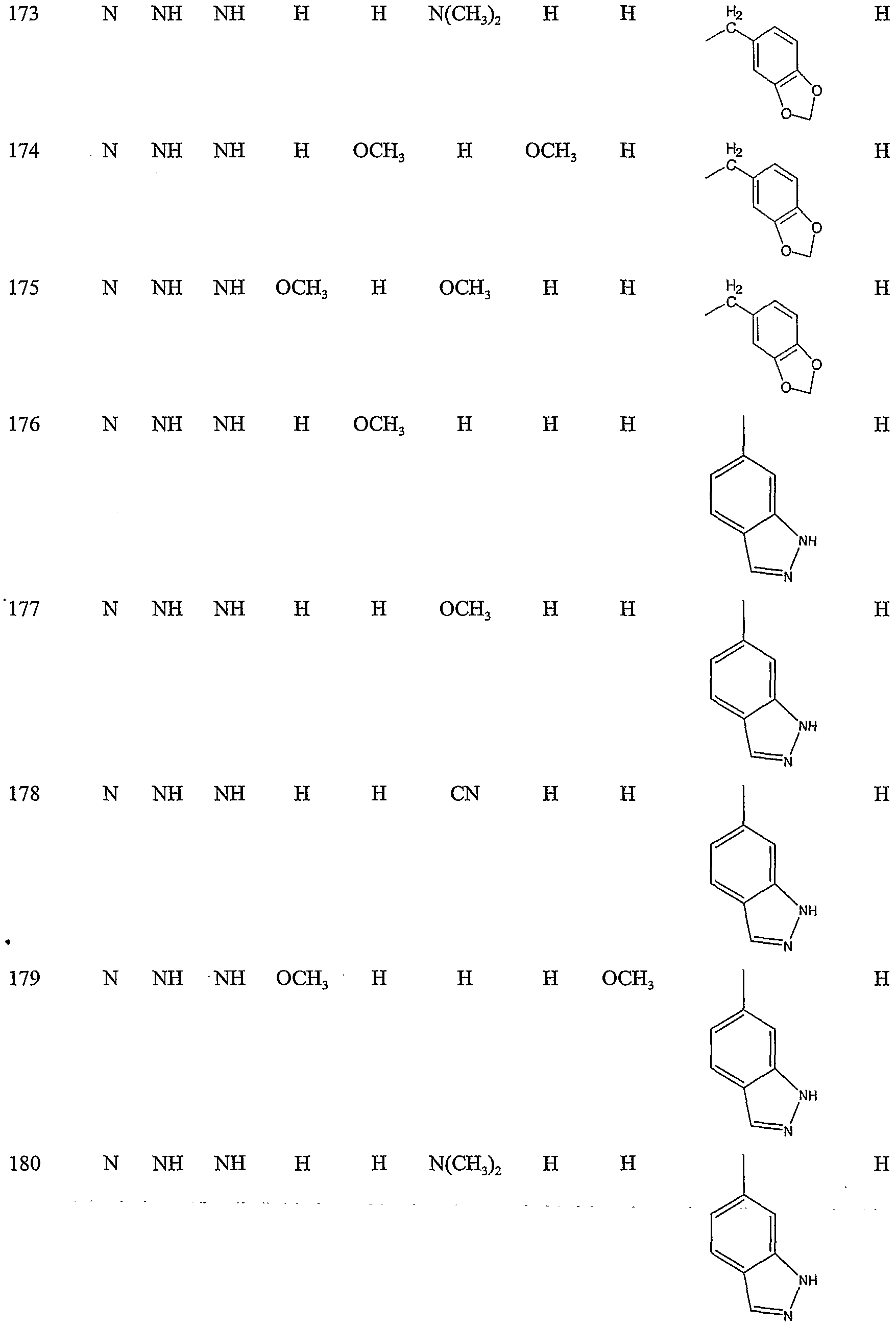
Specific representative compounds of the present invention are set forth in Tables 1, 2, 3 and 4, below
Table 1
Cmpd. No. W, W
fi ^v2 Rw7 R
*
15 CH CH H H (CH2)3N(CH3)2
16 CH CH H (CH
2)
3N(CH
3)
2 H
Cmpd. No. W, W
fi Rw2 R- 7 Rφ? R*
Cmpd. No. Wi W
fi R 7 RwS R 2.a_
45 N CH H H CF, H2 H2
H2
Cmpd. No. W, W
Λ Rw2 Rw? Rws R 2.a_
48 N N H H H H2
H2
Cmpd. No. W, W
Λ Rwa Rw7 is R*
Cmpd. No. W, W
fi Rw2 RwS R 2.a_
Table 2
Compound No. X, X,
76 N N
77 N C
78 C N
Table 3
Cmpd. No. W, X
3 Rw5 Rw( w7 Rγv8 Rw9 R,. T,
89 N NH NH H H N(CH3)2 H H H
94 N NH NH H -0-CH,-0- H H H
96 N NH NH H OCH3 H H H H2 H2 H
H2
Cmpd. No. , X
3 t w6 wi w
9 R
7, R
•7b
102 N NH NH H -0-CH,-0- H H H
103 N O NH H -0-CH,CH,-0- H H H
104 N O NH H -0-CH,CH,-0- H H H
105 N O NH H -0-CH,CH,-0- H H H
106 N O NH H -0-CH,CH,-0- H H H
107 N O NH H -0-CH,CH,-0- H H H
Cmpd. No. , X
3 R- 5 : Rwl Rw9 R
7, R
7,
108 N O NH H -0-CH,CH,-0- H H H
110 N O NH H -0-CH,CH,-0- H H H
111 N O NH H -0-CH2CH2-0- H H H2 H
N
HN-^
125 N O NH H OCH 3, O uωCHl3, H H H
Cmpd. No. W, X
3 Rγv5 Rw6 RΪV w8 9 R
7, Rτι
135 N O NH H -CH, 2CωHι 2,CωHι,2"- H H H
148 N O NH H -0-CH2-0- H H H H2
Cmpd. No. W, X
3 Rγv5 Rw6 Rw8 R
7 Rn
Cmpd. No. W, X
3 V: Rw6
lv7 RwS Rw9 R
7, R
T,
Cmpd. No. W, X
3 Rw5 w6 Rw7 RwS Rw9 R
7, Rτι
208 N NH NH H -0-CH2-0- H H -CH2CH2N(CH3)CH2CH2-
209 N NH CH2 H OCH3 H H H -CH2CH2N(CH3)CH2CH2-
210 N O NH H -0-CH2CH2-0- H H -CH2CH2N(CH3)CH2CH2-
211 CH O NH H OCH3 H H H H H
212 CH NH NH OCH3 OCH3 H H H H H
213 CH NH NH H H N(CH,), H H H H
Table 4
Cmpd. No. X! , Rw5 Rw7 R ■7a R7i
214 N N H OCH, H -CH2CH2N(CH3)CH2CH2-
215 N N H CF, H -CH2CH2N(CH3)CH2CH2
216 N N H -0-CH,-0- -CH2CH2N(CH3)CH2CH2.
217 CH CH H CF, H H H
Cmpd. No. X! W, R-W5 Rw6 w7 RT, R71
218 N CH H OCF, H H H
219 N CH Cl H H H H
220 N CH H OCH, H H H
221 N CH H H Br H H
222 N CH H Br H H H
223 C-CH, CH H C CFF3, H H H H
224 N CH H H H O OCCFF3, H H
225 N CH H HH CCFF33 H H
226 N CH 13 H CF^ H H
Pharmaceutical Compositions and Dosage Forms Useful in The Treatment Of Angiogenic
Dependent Diseases
Other aspects of the present invention are pharmaceutical compositions and dosage forms containing at least one compound of Formula (I) or a pharmaceutically acceptable salt, hydrate or pro-drug thereof, in combination with a pharmaceutically acceptable carrier.
As used herein, the terms "pharmaceutically acceptable salts" and "hydrates" refer to those salts and hydrated forms of the compound that would favorably affect the physical or pharmacokinetic properties of the compound, such as solubility, palatability, absorption, distribution, metabolism and excretion. Other factors, more practical in nature, which those skilled in the art may take into account in the selection include the cost of the raw materials, ease of crystallization, yield, stability, solubility, hygroscopicity and flowability of the resulting bulk drug.
When a compound of the present invention is present as a salt or hydrate that is not pharmaceutically acceptable, that compound can be converted in certain circumstances to a salt or hydrate form that is pharmaceutically acceptable in accordance with the present invention. When the compound is negatively charged, it is balanced by a counterion, such as, an alkali metal cation such as sodium or potassium. Other suitable counterions include calcium, magnesium, zinc, ammonium, or alkylammonium cations, such as teframethylammonium, tetrabutylammonium, choline, triethymydroammonium, meglumine, triethanol-hydroammoniiim, and the like. An appropriate number of counterions are associated with the molecule to maintain overall charge neutrality.
Likewise, when the compound is positively charged, e.g., protonated, an appropriate number of negatively charged counterions are present to maintain overall charge neutrality. These pharmaceutically acceptable salts are within the scope of the present invention.
Pharmaceutically acceptable salts of bases may be prepared by the addition of an appropriate acid. Thus, the compounds of the present invention can be used in the form of salts derived from inorganic or organic acids. Examples include acetate, adipate, alginate, aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, citrate, camphorate, camphorsulfonate, digluconate, dodecylsulfate, ethanesulfonate, fumarate, glucoheptanoate, glycerophosphate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxy-ethanesulfonate, lactate, maleate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, pamoate, pectinate, persulfate, 3-phenylpropionate, pivalate, propionate, succinate, tartrate and undecanoate.
If the compound has an acidic proton, a salt may be formed by the addition of base to form a pharmaceutically acceptable base addition salt. Base salts include ammonium salts, alkali metal salts such as sodium and potassium salts, alkaline earth metal salts such as calcium and magnesium salts, salts with organic bases such as dicyclohexylamine salts, N-methyl-D-glucamine, and salts with amino acids such as arginine, lysine, and so forth.
The basic nitrogen-containing groups may be quaternized with such agents as lower alkyl halides, such as methyl, ethyl, propyl, and butyl chloride, bromides and iodides; dialkyl sulfates like dimethyl, diethyl, dibutyl; and diamyl sulfates, long chain halides such
as decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides, aralkyl halides like benzyl and phenethyl bromides and others.
The compounds of the present invention may be employed in solid or liquid form including, for example, amorphous powder or crystalline form, in solution or in suspension. They may be administered in numerous different ways, such as orally, parenterally (intravenously or intramuscularly), topically, transdermally or by inhalation. The choice of carrier and the content of active compound in the carrier are generally determined in accordance with the solubility and chemical properties of the desired product, the particular mode of administration and well established pharmaceutical practice. The carrier may be either solid or liquid.
Examples of liquid carriers include syrup, peanut oil, olive oil, water and the like. For parenteral administration, emulsions, suspensions or solutions of the compounds according to the invention in vegetable oil, for example sesame oil, groundnut oil or olive oil, or aqueous-organic solutions such as water and propylene glycol, injectable organic esters such as ethyl oleate, as well as sterile aqueous solutions of the pharmaceutically acceptable salts, are used. Injectable forms must be fluid to the extent they can be easily syringed, and proper fluidity can be maintained, for example, by the use of a coating such as lecithin, by the maintenance of the required particle size in the case of dispersion and by the use of surfactants. Prolonged absorption of the injectable compositions can be brought about by use of agents delaying absorption, for example, aluminum monostearate and gelatin.
The active compound may be orally administered with an ingestible solid carrier. Compounds of the invention maybe enclosed in hard or soft shell gelatin capsules, or compressed into tablets, or incorporated directly with the food of the diet, or may be used in the form of ingestible tablets, buccal tablets, troches, capsules, elixirs, suspensions, syrups, wafers, and the like. Examples of oral solid dosage forms include tablets, capsules, troches, lozenges and the like. Examples of oral liquid dosage forms include solutions, suspensions, syrups, emulsions, soft gelatin capsules and the like. Carriers for oral use (solid or liquid) may include time delay materials known in the art, such as glyceryl monostearate or glyceryl distearate alone or with a wax. To prepare a capsule, it may be
advantageous to use lactose and a liquid carrier, such as high molecular weight polyethylene glycols.
Compositions and dosage forms prepared in accordance with the present invention optionally also may contain lactose, sodium citrate, calcium carbonate, dicalcium phosphate and disintegrating agents such as starch, alginic acids and certain complex silica gels combined with lubricants such as magnesium stearate, sodium lauryl sulfate and talc may be used for preparing tablets, troches, pills, capsules and the like.
Various other materials may be present as coatings or to otherwise modify the physical form of the dosage unit. For instance, tablets, pills, or capsules may be coated with shellac, sugar or both. When aqueous suspensions are used they may contain emulsifying agents or agents which facilitate suspension. Diluents such as sucrose, ethanol, polyols such as polyethylene glycol, propylene glycol and glycerol, and chloroform or mixtures thereof also may be used, hi addition, the active compound may be incorporated into sustained-release preparations and formulations. The solutions of the salts of the products according to the invention are especially useful for administration by intramuscular or subcutaneous injection. Solutions of the active compound as a free base or pharmacologically acceptable salt can be prepared in water suitably mixed with a surfactant such as hydroxypropyl-cellulose. Dispersions also can be prepared in glycerol, liquid polyethylene glycols, and mixtures thereof and in oils. The aqueous solutions, also including solutions of the salts in pure distilled water, may be used for intravenous administration with the proviso that their pH is suitably adjusted, that they are judiciously buffered and rendered isotonic with a sufficient quantity of glucose or sodium chloride and that they are sterilized by heating, irradiation, microfiltration, and/or by various antibacterial and antifungal agents, for example, parabens, chlorobutanol, phenol, sorbic acid, thimerosal, and the like.
Examples of injectable dosage forms include sterile injectable liquids, e.g., solutions, emulsions and suspensions. Sterile injectable solutions are prepared by incorporating the active compound in the required amount in the appropriate solvent with various of the other ingredients enumerated above, as required, followed by filtered sterilization. Generally, dispersions are prepared by incorporating the various sterilized
active ingredient into a sterile vehicle which contains the basic dispersion medium and the required other ingredients from those enumerated above, hi the case of sterile powders for the preparation of sterile injectable solutions, methods of preparation may include vacuum drying and a freeze-dry technique that yields a powder of the active ingredient plus any additional desired ingredient from previously sterile-filtered solution thereof.
Examples of injectable solids include powders that are reconstituted, dissolved or suspended in a liquid prior to injection, hi injectable compositions, the carrier typically includes sterile water, saline or another injectable liquid, e.g., peanut oil for intramuscular injections. Also, various buffering agents, preservatives and the like can be included within the compositions of the present invention.
Solid dosage forms include dosage forms for rectal administration, which include suppositories formulated in accordance with known methods and containing at least one compound of the present invention. Examples of solid carriers include lactose, sucrose, talc, gelatin, agar, pectin, acacia, magnesium stearate, stearic acid and the like. Topical administration, in the form of gels (water or alcohol based), creams or ointments, for example, containing compounds of the invention maybe used. Topical applications may be formulated in carriers such as hydrophobic or hydrophilic bases to form ointments, creams, lotions, in aqueous, oleaginous or alcoholic liquids to form paints or in dry diluents to form powders. Such topical formulations can be used for example, to treat ocular diseases as well as inflammatory diseases such as rheumatoid arthritis, psoriasis, contact dermatitis, delayed hypersensitivity reactions and the like.
Compounds of the invention also may be incorporated in a gel or matrix base for application in a patch, which would allow a controlled release of compound through fransdermal barrier. For administration by inhalation, compounds of the invention may be dissolved or suspended in a suitable carrier for use in a nebulizer or a suspension or solution aerosol, or may be absorbed or adsorbed onto a suitable solid carrier for use in a dry powder inhaler. Compositions according to the invention also may be formulated in a manner that resists rapid clearance from the vascular (arterial or venous) wall by convection and/or diffusion, thereby increasing the residence time of the compound at the desired site of
action. A periadventitial depot comprising a compound according to the invention may be used for sustained release. One such useful depot for administering a compound according to the invention may be a copolymer matrix, such as ethylene- vinyl acetate, or a polyvinyl alcohol gel surrounded by a Silastic shell. Alternatively, a compound according to the invention may be delivered locally from a silicone polymer implanted in the adventitia.
An alternative approach for minimizing washout of a compound according to the invention during percutaneous, transvascular delivery comprises the use of nondiffusible, drug-eluting microparticles. The microparticles may be made from any of a variety of synthetic polymers, such as polylactide, or natural substances, including proteins or polysaccharides. Such microparticles enable strategic manipulation of variables including total dose of drug and kinetics of its release. Microparticles can be injected efficiently into the arterial or venous wall through a porous balloon catheter or a balloon over stent, and are retained in the vascular wall and the periadventitial tissue for at least about two weeks. Formulations and methodologies for local, intravascular site-specific delivery of therapeutic agents are discussed in Reissen et al. (J. Am. Coll. Cardiol. 1994; 23:
1234-1244). A channeled balloon catheter (such as "channelled balloon angioplasty catheter", Mansfield Medical, Boston Scientific Corp., Watertown, Mass.) may be used. This catheter includes a conventional balloon covered with a layer of 24 perforated channels that are perfused via an independent lumen through an additional infusion orifice. Various types of balloon catheters, such as double balloon, porous balloon, microporous balloon, channel balloon, balloon over stent and hydrogel catheters, all of which maybe used to practice the invention, are disclosed in Reissen et al. (1994).
Another embodiment of the invention provides a compound according to the invention to be administered by means of perfusion balloons. These perfusion balloons, which make it possible to maintain a blood flow and thus to decrease the risks of ischaemia of the myocardium, on inflation of the balloon, also enable the compound to be delivered locally at normal pressure for a relatively long time, more than twenty minutes, which may be necessary for its optimal action.
A composition according to the invention also may comprise a hydrogel which is prepared from any biocompatible or non-cytotoxic (homo or hetero) polymer, such as a
hydrophilic polyacrylic acid polymer that can act as a drug absorbing sponge. Such polymers have been described, for example, in application WO 93/08845. Certain of them, such as, in particular, those obtained from ethylene and/or propylene oxide are commercially available. Another aspect of the present invention relates to a pharmaceutical composition including a compound according to the invention and poloxamer, such as Poloxamer 407, which is a non-toxic, biocompatible polyol, commercially available (e.g., from BASF, Parsippany, N.J.). A poloxamer impregnated with a compound according to the invention may be deposited for example, directly on the surface of the tissue to be treated, for example during a surgical intervention. Poloxamer possesses essentially the same advantages as hydrogel while having a lower viscosity. The use of a channel balloon catheter with a poloxamer impregnated with a compound according to the invention may be advantageous in that it may keep the balloon inflated for a longer period of time, while retaining the properties of facilitated sliding, and of site-specificity of the poloxamer. The composition also may be administered to a patient via a stent device. In this embodiment, the composition is a polymeric material in which the compound of the invention is incorporated, which composition is applied to at least one surface of the stent device.
Polymeric materials suitable for incorporating the compound of the invention include polymers having relatively low processing temperatures such as polycaprolactone, poly(ethylene-co-vinyl acetate) or poly(vinyl acetate or silicone gum rubber and polymers having similar relatively low processing temperatures. Other suitable polymers include non-degradable polymers capable of carrying and delivering therapeutic drugs such as latexes, urethanes, polysiloxanes, styrene-ethylene/butylene-styrene block copolymers (SEBS) and biodegradable, bioabsorbable polymers capable of carrying and delivering therapeutic drugs, such as poly-DL-lactic acid (DL-PLA), and poly-L-lactic acid (L-PLA), polyorthoesters, polyiminocarbonates, aliphatic polycarbonates, and polyphosphazenes.
The compounds of the present invention also may be formulated for use in conjunction with other therapeutically active compounds or in connection with the application of therapeutic techniques to address pharmacological conditions, which may be
ameliorated through the application of a compound according to the present invention. The percentage of active ingredient in the compositions of the invention maybe varied. Several unit dosage forms may be administered at about the same time. A suitable dose employed may be determined by a physician or qualified medical professional, and depends upon various factors including the desired therapeutic effect, the nature of the illness being treated, the route of administration, the duration of the treatment, and the condition of the patient, such as age, weight, general state of health and other characteristics, which can influence the efficacy of the compound according to the invention. In adults, doses are generally from about 0.001 to about 50, preferably about 0.001 to about 5, mg/kg body weight per day by inhalation; from about 0.01 to about 100, preferably 0.1 to 70, more preferably 0.5 to 10, mg/kg body weight per day by oral administration; from about 0.1 to about 150 mg applied externally; and from about 0.001 to about 10, preferably 0.01 to 10, mg/kg body weight per day by intravenous or
Method of the Invention.
The compounds of the present invention inhibit or regulate phosphorylation events. Tyrosine kinase inhibition may be determined by measuring the autophosphorylation level of recombinant kinase receptor, and/or phosphorylation of natural or synthetic substrates. Phosphorylation can be detected, for example, using an antibody specific for phosphotyrosine in an ELISA assay or on a western blot. Some assays for tyrosine kinase activity are described in Panek et al., J. Pharmacol. Exp. Thera., 283: 1433-44 (1997) and Batley et al., Life Sci., 62: 143-50 (1998). Detailed descriptions of conventional assays, such as those employed in phosphorylation assays, can be obtained from numerous publications, including Sambrook, J. et al, Molecular Cloning: A Laboratory Manual, 2nd ed. (Cold Spring Harbor Laboratory Press 198(. All references mentioned herein are incorporated in their entirety. hi addition, methods of detecting expression of proteins whose expression is regulated by tyrosine kinase activity can be used. These methods include immunohistochemisfry (IHC) for detection of protein expression, fluorescence in situ hybridization (FISH) for detection of gene amplification, competitive radioligand binding
assays, solid matrix blotting techniques, such as Northern and Southern blots, reverse franscriptase polymerase chain reaction (RT-PCR) and ELISA. See, e.g., Grandis et al., Cancer, 78:1284-1292. (1996); Shimizu et al., Japan J. Cancer Res., 85:567-571 (1994); Sauter et al., Am. J. Path., 148:1047-1053 (1996); Collins, Glia, 15:289-296 (1995); Radinsky et al., Clin. Cancer Res., 1:19-31 (1995); Petrides et al., Cancer Res.,
50:3934-3939 (1990); Hoffmann et al, Anticancer Res., 17:4419-4426 (1997); Wikstrand et al., Cancer Res., 55:3140-3148 (1995).
In vivo assays also can be utilized. For example, VEGF receptor tyrosine kinase inhibition can be observed by mitogenic assays using HUVEC cells (ATCC) stimulated with VEGF in the presence and absence of inhibitor. Another method involves testing for inhibition of growth of VEGF-expressing tumor cells, using for example, human tumor cells injected into a mouse. (See, U.S. Patent No. 6,365,157 to Rockwell et al.)
Also included within the scope of the present invention are methods of inhibiting VEGF receptor tyrosine kinases, especially KDR, and/or freating or preventing VEGF receptor kinase-dependent diseases and conditions in mammals using the VEGF receptor kinase inhibitors of Formula I. The VEGF receptor is usually bound to a cell, such as an endothelial or tumor cell. Alternatively, the VEGF receptor may be free from the cell. According to the treatment method of the present invention, an effective anti- angiogenic amount of one or more compounds of Formula (I), or pro-drug thereof, is administered to a mammal in need of such freatment. The diseases which may be freated or prevented by the present methods include, for example, those in which pathogenic angiogenesis or tumor growth is stimulated through a VEGF/VEGFR paracrine and/or autocrine loop. For example, paracrine VEGFR stimulation of vascular endothelium is associated with angiogenic diseases and vascularization of tumors. VEGF receptors are also found on tumor cells, indicating the presence of an autocrine and/or paracrine loop in these cells. Thus, the method is also useful for neutralizing VEGF receptors on such cells, thereby inhibiting autocrine and/or paracrine stimulation and inhibiting tumor growth.
The compounds and compositions according to the invention may be administered as frequently as necessary as determined by a skilled practitioner in order to obtain the desired therapeutic effect. Some patients may respond rapidly to a higher or lower dose
and may find much weaker maintenance doses adequate. For other patients, it may be necessary to have long-term treatments at the rate of 1 to 4 doses per day, in accordance with the physiological requirements of each particular patient. Generally, the active product may be administered orally 1 to 4 times per day. For other patients, it may be necessary to prescribe not more than one or two doses per day.
Thus, the method is effective for treating subjects with tumors and neoplasms, particularly where disease development involves angiogenesis. Tumors and neoplasms include, for example, malignant tumors and neoplasms, such as blastomas, carcinomas or sarcomas, and highly vascular tumors and neoplasms. Cancers that may be treated by the methods of the present invention include, for example, cancers of the brain, genitourinary tract, lymphatic system, stomach, renal, colon, larynx, lung and bone. Non-limiting examples further include epidermoid tumors, squamous tumors, such as head and neck tumors, colorectal tumors, prostate tumors, breast tumors, lung tumors, including lung adenocarcinoma and small cell and non-small cell lung tumors, pancreatic tumors, thyroid tumors, ovarian tumors, and liver tumors. The method is also useful for freatment of vascularized skin cancers, including squamous cell carcinoma, basal cell carcinoma, and skin cancers that can be freated by suppressing the growth of malignant keratinocytes, such as human malignant keratinocytes. Other cancers that can be treated include Kaposi's sarcoma, CNS neoplasms (neuroblastomas, capillary hemangioblastomas, meningiomas and cerebral metastases), melanoma, gastrointestinal and renal carcinomas and sarcomas, rhabdomyosarcoma, glioblastoma, including glioblastoma multiforme, histiocytic lymphoma, and leiomyosarcoma.
The freatment method of this invention includes freatment or prevention of non- neoplastic angiogenic dependent diseases like inflammatory diseases and other diseases characterized by paracrine stimulation through VEGF receptors. Examples of non-neoplastic angiogenic diseases include diseases characterized by retinal vascularization, such as neovascular glaucoma, proliferative retinopathy, including diabetic retinopathy, retinopathy of prematurity (retrolental fibroplastic), macular degeneration, and corneal graft rejection. Examples of inflammatory diseases that may be freated include, but are not limited to, atherosclerosis, rheumatoid arthritis (RA), insulin-dependent
diabetes mellitus, multiple sclerosis, myasthenia gravis, Chron's disease, autoimmune nephritis, primary biliary cirrhosis, psoriasis, acute pancreatitis, allograph rejection, allergic inflammation, contact dermatitis and delayed hypersensitivity reactions, inflammatory-bowel disease, septic shock, osteoporosis, osteoarthritis, and cognition defects induced by neuronal inflammation. The compounds of this invention are not the first 1,2,4-triazoles found to have anti-inflammatory properties. In U.S. Patent No. 4,020,064, it is reported that 1,2,4-triazole compounds having certain acetamidyl substituents on N-4 are useful as anti-inflammatory agents. Other non-limiting examples of angiogenic diseases are hemangiomas, angiofibromas, Osier- Weber syndrome, restinosis, and fungal, parasitic and viral infections, including cytomegaloviral infections.
The freatment method of the present invention also includes treatment of tumors that express VEGF receptors (e.g. KDR), for example through inhibition of an autocrine VEGF/VEGFR loop, wherein one or more of the Formula I compounds are administered in an amount effective to reduce tumor growth or size. The method can be used to treat a solid or liquid tumor that is not vascularized, or is not yet substantially vascularized.
Examples of solid tumors which may be accordingly treated include breast carcinoma, lung carcinoma, colorectal carcinoma, pancreatic carcinoma, glioma and lymphoma. Examples of liquid tumors include leukemia, multiple myeloma and lymphoma. Some examples of leukemias include acute myelogenous leukemia (AML), chronic myelogenous leukemia (CML), acute lymphocytic leukemia (ALL), chronic lymphocytic leukemia (CLL), erythrocytic leukemia or monocytic leukemia. Some examples of lymphomas include Hodgkin's and non-Hodgkin's lymphoma.
Moreover, included within the scope of the present invention is use of the present inventive compounds in vivo and in vitro for investigative, diagnostic, or prophylactic methods, which are well known in the art.
In the methods of the present invention, an effective anti-angiogenic amount of one or more of the Formula (I) compounds, including salts and solvates, is administered to a mammal in need. The term "administering" as used herein means delivering the compounds of the present invention to a mammal by any method that may achieve the result sought. They may be administered, for example, orally, parenterally (intravenously
or intramuscularly), topically, transdermally or by inhalation. The term "mammal" as used herein is intended to include, but is not limited to, humans, laboratory animals, domestic pets and farm animals. "Effective anti-angiogenic amount" means an amount of compound of the present invention that when administered to a mammal influences the progression of an angiogenic dependent disease. The influence is mediated through a slowing or arrest of pathogenic neovascularization around diseased cells. Accordingly, the influence may be a prevention or arrest of neovascularization that leads to the death of diseased cells and eradication of the disease from the body. However, the influence will be therapeutically beneficial even when not so far-reaching. Thus, as the term is used in this disclosure, "influencing the progression of the angiogenic disease" includes causing the progression of the disease to be slowed by partial or complete restriction of the blood supply to diseased cells, resulting in an increase in life expectancy of the mammal afflicted with the disease.
Having thus described the present invention with reference to certain preferred embodiments, the invention will now be further illustrated by the following non-limiting examples.
EXAMPLES
Example 1 Preparation of Ethyl Benzimidate Hydrochloride (A)
Dry HCl (g) was bubbled through a solution of benzonitrile in absolute ethanol for 4 h. A white precipitate formed and was collected by filtration, washed with absolute ethanol and dried under high vacuum to give the title compound in nearly quantitative yield.
Example 2 Preparation of Benzamidine Hydrochloride (B)
A slight molar excess of dry ammonia gas was bubbled through a solution of ethyl benzimidate hydrochloride in absolute ethanol. The mixture was stirred at room temperature for 3 h. Ammonium chloride produced from the excess ammonia was removed by filtration and the filtrate was collected and concentrated. The residue was recrystallized from ethanol, filtered and dried under high vacuum to give the title compound in nearly quantitative yield.
Example 3 Preparation of Ethyl 2- (4-Pyridinyl)methylamino]benzoate (C)
A mixture of methyl anthranilate (7.5 g) and 4-pyridinylcarboxaldehyde (8.6 g) in MeOH (300 mL) and acetic acid (3 mL) was stirred at room temperature for 12 h. Sodium cyanoborohydride (NaBH3CN) (6.9 g) was added to the reaction mixture and the resulting solution was stirred at r.t. for 12 h. The reaction mixture was then concentrated and the residue was dissolved in ethyl acetate and washed with saturated NaHCO3 and brine. The organic layer was dried with MgSO4, filtered, concentrated and purified by flash chromatography over SiO2 (1:1 ethyl acetate/hexane) to give the title compound (10.2 g, 50%).
Example 4 Preparation of 2-[(4-Pyridinyl)methylamino']benzoic Acid Hydrazide (D
A mixture of Ethyl 2-[(4-pyridinyl)methylamino]benzoate (10 g) in hydrazine (50 ml) was refluxed. After 2 h the excess hydrazine was removed and the remaining mixture was dissolved in dichloromethane, washed with brine and dried with MgSO4 and concentrated. The crude residue was purified by flash chromatography to give the title compound as a white solid (9.2 g, 97%).
Example 5 Preparation of 2-[2-f4-Pyridinyl methylaminolphenyl-4-phenyl-1.2.4-triazole (i)
A solution of sodium methylate (0.32 g, 6 mmol) in anhydrous ethanol (6 ml) was added to a solution of the benzamidine hydrochloride B prepared in Example 2 (0.7 g, 4.5 mmol) in anhydrous ethanol (7 ml) at room temperature. The milky slurry was stirred at room temperature for 45 min. and filtered. 2-[(4-Pyridinyl)methylamino]benzoic acid hydrazide D prepared in Example 4 (0.73 g, 3 mmol) was then added to the ethanol filtrate and the resulting yellow solution was refluxed for 2 hours. The ethanol was evaporated off and the residue was re-dissolved in chlorobenzene, the resulting solution was refluxed overnight. The mixture was cooled to room temperature and evaporated under vacuum.
The solid residue was washed with water and recrystallized from ethanol to give the title compound (0.9 g, 91%) as a white solid. Its structure was confirmed by NMR and elemental analysis. Η NMR (300 MHz, OMSO-d6) δ (ppm) 14.8 (br. s, IH), 8.7 (m, 3H), 8.2 (m, 3H), 7.5 (m, 5H), 7.3 (t, IH), 6.7 (m, 2H), 4.6 (d, 2H); 13C NMR (75 MHz, DMSO- d6) δ (ppm) 150.1, 149.4, 146.4, 131.2, 130.0, 129.2, 128.0, 126.4, 122.6, 115.0, 111.8,
45.7; MS 328.2 (M+H+); Formula C20H17N5 + 0.25H2O, Calculated C 72.39, Found C 72.82, Calculated H 5.32, FoundH 5.22, Calculated N 21.09, Found N 21.07.
Other 1,2,4-triazoles of this invention can be made by procedures analogous to those of Examples 1-5.
Example 6 Preparation of Compound ( 14)
To a solution of 2-(3,5-dimethoxyphenylamino)nicotinic acid hydrazide(1.0g, 3.48 mmol) in dichloromethane (20 mL) was added 4-trifluoromethoxy phenyl isothiocyanate (0.64 mL, 3.83 mmol). The reaction mixture was refluxed for 2 hours under argon. The reaction mixture was allowed to cool and a solid precipitated from the solution, filtered, the solid was washed with ether to provide a yellow solid 1.489 g. Yield: 93%. !H NMR (DMSO-^) δ 10.95 (s, IH), 10.71 (s, IH), 9.93-9.96 (m, 2H), 8.45-8.48 (m, IH), 8.25- 8.28 (m, IH), 7.60-7.66 (m, 2H), 7.35-7.42 (m, 2H), 6.90-7.05 (m, 3H), 6.20 (s, IH), 3.80 (s, 6H). MS m/z: 508.
To this yellow solid (1.45 g, 2.86 mmol) in toluene (20 mL) was added 1,3- dicyclohexylcarbodiimide (0.885 g, 4.3 mmol). Then, the reaction mixture was refluxed for 5 hours. The reaction mixture was diluted with ethyl acetate (50 mL), washed with aqueous sodium bicarbonate (30 mL) and brine (2x30 mL). The organic layer was separated and dried over anhydrous sodium sulfate. The dried solution was filtered and evaporated to give a solid. This solid was washed with warm methanol to get a white solid (1.1 g, 81.5% yield). Η NMR DMSO-d6) δ 11.10 (s, IH), 10.06 (s, IH), 8.42-8.44 (m, IH), 8.06-8.09 (m, 1 H), 7.76-7.80 (m, 2H), 7.43-7.46 (d, 2H), 7.04-7.08 (m, 3H), 6.25- 6.26 (t, IH), 3.81 (t, 6H). Ms m/z: 474.
Other 1,3,4-oxadiazoles of this invention can be made by procedures analogous to those of Example 6
Example 7 Preparation of 2-f3.5-dimethoxyphenylamino nicotinic acid hydrazide (E)
2-Chloropyridine-3 -carboxylic acid (25 g) was refluxed in 200 mL of benzene and 150 mL of SOC12. The solution was concentrated and chased with toluene. The residue obtained was refluxed in 100 mL of ethanol for 20 minutes. The solvents were removed under vacuum to give the pure product, 2-chloro-nicotinic acid ethyl ester. Light yellow oil; yield 72%; Η NMR (CDC13) δ 1.42 (t, J = 6.6 Hz, 3H), 4.43(q, J = 6.8Hz, 2H), 7.37 (br s, IH), 8.18 (d, J = 6.6Hz, IH), 8.54 (s, IH); 13C NMR δ 13.8, 61.8, 122.0, 126.9, 140.0, 149.5, 151.4, 164.2 _ 2-Chloro-nicotinic acid ethyl ester (2 mmol, 0.343g) and 3,5-dimethoxyaniline
(2mmol, 0.306g) were dissolved in ethylene glycol (lOmL) and heated to 160 °C with
stirring. The reaction mixture was maintained at this temperature for 6h. HCl gas evolved during the course of the reaction. On cooling, the reaction mixture was poured into water (10 mL) and extracted with ether (4 x 100 mL). The ethereal layer was dried over MgSO4, evaporated and the residue was distilled at 162-165 °C/0.5 mm Hg. The product,2-(3,5- dimethoxyphenylamino)nicotinic acid ethyl ester, was used in the next step without further purification. Yellow oil, yield 63%; Η NMR (CDC13) δ 1.24 (t, J= 7.1 Hz, 3H), 3.78 (s, 6H), 4.11 (t, J= 7.1 Hz, 2H), 6.17 (s, IH), 6.68 (t, J= 6.0 Hz, IH), 6.97 (s, 2H), 8.20 (d, J = 7.7 Hz, IH), 8.36 (s, IH), 10.24 (s, IH); ,3C NMR δ 20.7, 55.0, 60.1, 94.8, 98.7, 103.2, 107.1, 113.1, 139.9, 141.2, 152.7, 155.8, 160.7, 167.2, 170.9. 2-(3,5-Dimethoxyphenylamino)nicotinic acid ethyl ester (1.94 mmol, 560mg) and
85% hydrazine hydrate (1.18 mL) and 2-propanol (2 mL) were refluxed for 3 h. On cooling, the red solution deposited a yellow solid that was filtered off and washed with a little 2-propanol. Compound E appeared as a yellow solid, 84% yield: *H NMR (DMSO- d6) δ 1.22 (br s, 2H), 4.00 (s, 6H), 6.18 (t, J= 2.2 Hz, IH), 6.66-6.70 (m, IH), 6.94 (s, 2H), 7.64 (dd, J= 1.8 Hz, 7.7 Hz, IH), 7.70 (br s, IH), 8.33 (dd, J= 1.5 Hz, 4.8 Hz), 10.1
(br s, IH); 13C NMR δ 55.3, 94.8, 98.7, 100.2, 103.4, 100.4, 109.5, 113.1, 135.1, 141.5, 151.8, 160.9, 169.1.
Example 8 S.S-Dimethoxyphenvπ-^-Methylisothiourea Hvdroiodide (F)
4-Fluorobenzoyl chloride (1.03 g, 6.5 mmol) was added dropwise to a vigorously stirred hot solution of anhydrous ammonium thiocyanate (0.61 g, 7.8 mmol) in dry acetone (20 mL). The reaction mixture was refluxed for 5 min. Then, a solution of 3,5- dimethoxyaniline (1.0 g, 6.5 mmol) in dry acetone (10 mL) was added dropwise. The
reaction mixture was heated for 1 h. The solvent was evaporated and water (50 mL) was added to the residue. The precipitate was collected and recrystallized from ethyl alcohol to give the product, N-(3,5-dimethoxyphenyl)-N'-(4-fluorobenzoyl)thiourea. White needles (ethanol), Yield 69%; Η NMR (DMSO) δ 3.76 (s, 6H), 6.43 (br s, IH), 6.99 (br s, 2H), 7.35-7.41 (m, 2H), 8.04-8.09 (m, 2H), 11.62 (s, IH) ; 13C NMR (DMSO) δ . Anal. Calcd for C16H15FN2O3S: C, 57.47; H, 4.52; N, 8.38. Found: C, 57.49; H, 4.43; N, 8.26.
N-(3,5-Dimethoxyphenyl)-N'-(4-fluorobenzoyl)thiourea (1.5 g, 4.4 mmol) was heated to reflux with 5% aqueous NaOH (10 mL) for 15 min. The cooled reaction mixture was freated with concenfrated HCl until acidic to precipitate both 4-fluorobenzoic acid and N-(3,5-dimethoxyphenyl)thiourea. The mixture was then made basic (pH 9) with concentrated NH4OH to dissolve the 4-fluorobenzoic acid. The product was filtered and recrystallized from 95% ethyl alcohol to give the pure product, N-(3,5- dimethoxyphenyl)thiourea . White prisms, yield 75%; Η NMR (DMSO) δ 3.72 (s, 6H), 6.27 (br s, IH), 6.62 (br s, 2H), 7.53 (br s, 2H), 9.66 (s, IH); 13C NMR (DMSO) δ 55.2, 96.4, 100.8, 140.6, 160.4, 180.7. Anal. Calcd for C9H12N2O2S: C, 50.92; H, 5.70; N,
13.20. Found: C, 50.88; H, 5.66; N, 12.96.
A solution of N-(3,5-dimethoxyphenyl)thiourea (0.53 g, 2.5 mmol) in freshly distilled dry methanol (10 mL) was treated with CH3I (0.36 g, 2.5 mmol). The solution was refluxed for 2h, cooled, and evaporated to dryness under vacuum. The crystalline product was washed with several portion of ethyl ether and dried to give pure Compound F which appeared as white microcrystals; yield 92%; JH NMR (DMSO) δ 2.70 (s, 3H), 3.78 (s, 6H), 6.53-6.56 (m, 3H), 9.30 (br s, 2H); 13C NMR (DMSO) δ 55.6, 100.1, 103.7, 136.5, 161.1, 169.1.
Example 9
Preparation of 3.5-Dimethoxyphenyl -(3-["5- 3.5-dimethoxy-phenylamino -2H-
|"1.2.41triazol-3-yl]-ρyridin-2-yll amine (154)
A solution of 2-(3,5-dimethoxyphenylamino)nicotinic acid hydrazide (E) (lmmol) and- (3,5-dimethoxyphenyl)-S-methylisothiourea hydroiodide (F) (lmmol) in 1 mL of pyridine were refluxed for 6 h. The cooled mixture was poured into crushed ice and extracted with ether. The solvent was removed and the crude product was recrystallized from ethyl acetate (and two drops of ethanol) to give the pure product. Brown solid; yield 25%; Η NMR (OMSO-d6, 100 °C) δ 3.75 (s, 3H), 3.76 (s, 3H), 6.11 (br s, IH), 6.18 (t, J= 2.2 Hz, IH), 6.81 (d, J= 2.2 Hz, 2H), 6.89-6.93 (m, IH), 7.07 (d, J= 1.8 Hz, 2H), 8.28-8.29 (m, 2H), 9.22 (br s, IH), 10.7 (br s, IH).
Other 1,2,4-triazoles of the present invention can be prepared by procedures analogous to those of Examples 7-9.
Example 10 Preparation of 3-(Trifluoromethyl)phenacyl Bromide (G)
A solution of 3-(trifluoromethyl)acetophenone (25 g, 0.133 mol) in diethyl ether (20 ml) was cooled to 0°C. Aluminium chloride (0.15 g) was added in one portion, with stirring, followed by dropwise addition of bromine (6.85 ml, 0.133 mol), and the reaction mixture was allowed to reach room temperature overnight. Solvent was removed under vacuum and the crude product purified by distillation (94-97°C, 1mm Hg). The title compound was obtained as a colorless liquid (20.2g, 57%).
Example 11
Preparation of 2-Nitrobenzamidine hydrochloride (H)
A solution of 2-nitrobenzonitrile (12.3 g, 0.083 mol) in THF (60 ml) was added dropwise with stirring to a cooled (0°C) solution of LiHMDS (IM in THF; 99.6 ml, 0.0996 mol) in ether (60 ml) and the reaction mixture allowed to warm to room temperature overnight. The reaction was quenched at 0°C with ethereal HCl (-150 ml). A brown semi- solid formed and the solvent was decanted off. The crude product was triturated with ether
(~200 ml) and collected by filtration. After washing with ether (~1L) the title compound was obtained as a tan solid (~20 g, >100%, i.e. inorganic salts present).
Example 12 Preparation of 2-[5-(3-trifluoromethylphenyl -lH-imidazol-2-yl]nitrobenzene (I)
A mixture of compound H (13.78 g, 68.4 mmol), DMF (300 ml), compound G (18.27 g, 68.4 mmol), sodium iodide (~1 g) and triethylamine (19 ml, 136.8 mmol) was heated at 120°C for 48 hrs. Solvent was removed under vacuum and the residue dissolved in ethyl acetate (150 ml) and washed with water (3 x 200 ml) and then brine (200 ml), dried (MgSO4) and the solvent removed under vacuum. The crude product obtained was purified by 'dry flash' chromatography [Methanol/dichloromethane (1:99) - (3:97)]. Solvent was removed under vacuum and the residue dissolved in ethanol (~100 ml) and heated with activated charcoal for a few minutes then filtered through Celite. Solvent was removed under vacuum and the residue was purified by 'flash' chromatography [Methanol/ dichloromethane (1 :99) - (3 :97)] . The title compound was obtained as an orange/yellow glassy solid (7 g, 25% over 2 steps).
Example 13 Preparation of 2-|"5-(3-trifluoromethylphenyl -lH-imidazol-2-yl]aniline (217)
A mixture of compound H (6.25 g, 0.0188 mol), ethanol (350 ml) and palladium on carbon (5% wt) (1.25 g) was stirred under an atmosphere of hydrogen at room temperature overnight. The catalyst was filtered off through Celite and washed with ethanol (200 ml). Solvent was removed under vacuum to give an orange solid. This was washed with dichloromethane/petrol [200 ml, (1:1)] and the solid that remained was collected by filtration. The title compound (1.1 g, 20%) was obtained as an off-white solid after recrystallisation from dichloromethane/pefrol/methanol. MS m/z: 304 (M+H+)
Other imidazoles of the present invention can be prepared by procedures analogous to those of Examples 10-13.
Example 14
Preparation of 2-Aminobenzamidoxime (J)
2-Aminobenzonitrile hydrochloride (50 g, 0.324 mol) was suspended in anhydrous methanol in a 2 L three necked flask. One mole equivalent of sodium methoxide (17.5 g, 0.324 mol) was added portionwise to the mixture (during the addition the temperature rose to 35°C). The reaction mixture was then stirred for 30 minutes at room temperature and a second equivalent of sodium methoxide (17.5 g, 0.324 mol) was added portionwise. After
stirring for 30 minutes at room temperature, hydroxylamine hydrochloride (22.5 g, 0.324 mol) was added and the mixture heated under reflux for 20 hours. The mixture was concenfrated and the residue partitioned between EtOAc (500 ml) and 2N sodium hydroxide (500 ml). The basic phase was separated and extracted with EtOAc (4 x 500 ml). The combined organic extract was washed with brine (4 x 500 ml) and dried over magnesium sulphate. After filtration and evaporation, a brown oil was obtained. Dry flash chromatography on silica gel [dichloromethane/ethanol (95:5)] followed by precipitation from dichloromethane/petroleum ether afforded the 2-aminobenzamidoxime (J) as a pale brown solid (31 g, 63%).
Example 15
Preparation of 2-Aminobenzamidine Dihydrochloride (K)
Following generally the method described in J. Med. Chem. 2003, 46, 913-916, a suspension of 2-aminobenzamidoxime (J) (31 g, 0.205 mol) and wet Raney nickel (50% slurry in water) (ca. 15 g) in EtOH (350 ml) was stirred under 3 atmospheres of hydrogen at 60°C for 16 hours. The catalyst was removed by filtration through a pad of celite and the cake was washed with EtOH (300 ml). The solvent was evaporated to give a brown oil, 2- aminobenzamidine (250 MHz !H NMR in CDC13 consistent with structure). This material was dissolved in ethanol (200 ml) and 2N HCl in ether (230 ml, 0.46 mol) was added dropwise (during the addition the temperature rose to 35°C). The resulting precipitate was collected by filfration and dried under vacuum to give 2-aminobenzamidine dihydrochloride (K) as an off-white powder (37 g, 87%).
Example 16 General Procedure for Preparing 2-(5-Aryl-7H-[1.2.41-triazol-3-yl anilines ("218-222. 224-
226)
In a dry 100 ml three necked flask, 2-aminobenzamidine dihydrochloride (K) (12.0-16.9 mmol) was suspended in dry EtOH (18-20 ml) under nitrogen. A freshly prepared solution of sodium methoxide (2.3 eq.) in EtOH (30-35 ml) was added to the reaction mixture. The brown slurry was stirred at room temperature for 1 hour and filtered. To the ethanol filtrate was added the appropriate arylcarboxyhc acid hydrazide (1 eq.) and the reaction mixture heated under reflux for 3-6 hours. The solvent was evaporated under vacuum and the brown residue was dissolved in chlorobenzene (35-45 ml). The flask was equipped with a Dean-stark trap and the reaction mixture was heated under reflux overnight. After cooling to room temperature, the solvent was evaporated under vacuum. The brown residue was purified by dry flash chromatography on silica gel
[dichloromethane/methanol (99:1) - (95:5)], followed by trituration from dichloromethane/pefroleum ether and recrystalhsation from the appropriate solvent to give the corresponding triazole.
Although most of the arylcarboxyhc acid hydrazides used to prepare compounds 218-222 and 224-226 are commercially available, the 3-trifluoromethoxy-benzene-l- carbohydrazide was prepared in our laboratory. It was prepared as follows. 3- (Trifluoromethoxy)-methylbenzoate (5.0g, 22.7 mmol) was dissolved in dry MeOH (20mL) under nifrogen. Hydrazine monohydrate (2.27g, 45.4 mmol, 2 eq.) was added dropwise and the mixture was heated under reflux for 3 hours. The reaction mixture was then concenfrated under vacuum to give a solid. Water was removed by azeofroping with
EtOH (x2), to leave an off-white solid (5.4 g, quantitative).
Example 17 Preparation of 2-(2-Nifroberιzyl)-4- 3-trifluoromethylbenzyD-5-methylimidazole (L
l-(3-Trifluoromethyl(benzyl))-l,2-propandione (4.80 g, 22.2 mmol) was dissolved in acetic acid (250mL) under nitrogen. 2-Nitrobenzaldehyde (3.69 g, 24.4 mmol, 1.1 eq.) and ammonium acetate (34.21 g, 444 mmol, 20 eq.) were added and the resulting mixture was heated at 100°C overnight. TLC analysis (EtOAc/t-Hexane, 3:7) showed no starting material remained so the resulting solution was concentrated to approx. 50mL and the remaining acetic acid was neutralized with NaHCO
3 (solid and saturated solution). Ethyl acetate (300mL) was added and the phases separated. The aqueous layer was back- extracted with EtOAc (2x 250mL). The combined organic layers were washed with brine (250mL), dried (MgSO
4) and concentrated under vacuum to give a dark oil. Flash column chromatography [Acetone/z-Hexane (2:8)] provided the nifro-imidazole L as a dark orange oil (4.8g, 62%).
Example 18 Preparation of 2-(4-methyl-5 -(3 -trifluoromethylphenyl - 1 H-imidazol-2-yl aniline (223)
2-(2-Nitrobenzyl)-4(3-trifluoromethylbenzyl)-5-methylimidazole (M) (2.5 g, 7.2 mmol) was dissolved in EtOH (60mL) and water (lOniL). Iron powder (3.63 g, 9 eq.) and ammonium chloride (0.77 g, 14.4 mmol, 2 eq.) were added and the resulting suspension was heated under reflux for 3 hours. More ammonium chloride (0.77 g, 2 eq.) was added
and the heating was continued overnight. The reaction mixture was then concentrated and the residue partitioned between dichloromethane (75mL) and saturated NaHCO3 (aq.) (50mL). The aqueous layer was back-extracted with dichloromethane (2x 50mL). The combined organic layers were washed with water (75mL), dried (MgSO4), filtered and concenfrated under vacuum to give an orange solid. This material was recrystallised twice [dichloromethane/z'-Hexane (1:1)] to give compound 223 as a pale orange crystalline solid (1.25g, 55%). MS m z: 318 (M+H+)
Analytical Data for Selected Compounds
2- 2-("4-Pyridinyl methylaminolphenyl-4-r3-bromophenylV1.2.4-triazole ('2): 'H NMR (300 MHz, DMSO-cQ δ (ppm) 14.8 (br. s, IH), 8.7 (m, 3H), 8 (m, 9H), 6.7 (q, 2H), 4.7 (d, 2H); 13C NMR (75 MHz, DMSO- 6) δ (ppm) 150.1, 149.3, 146.4, 131.8, 131.4, 128.6, 127.6, 125.3, 122.5, 116.0, 112.0, 45.8; MS 406.1 (M+); Formula C20H16BrN5, Calculated C 59.13, H 3.97, N 17.23; Found C 58.95, H 3.94, N 17.11.
2-[2-(4-Pyridinyl methylamino]phenyl-4-r4-trifluoromethylphenyl)-1.2,4-triazole r3 : *H NMR (300 MHz, OMSO-d6) δ (ppm) 14.8 (br. s, IH), 8.8 (br. s, 2H), 8.6 (d, 2H), 8.3 (d, 2H), 7.9 (m, 3H), 7.4 (d, 2H), 7.2 (t, IH), 6.7 (m, 2H), 4.6 (d, 2H); 13C NMR (75 MHz, DMSO-rf6) δ (ppm) 150.1, 149.4, 146.5, 131.9, 127.8, 127.0, 126.2, 122.7, 122.5, 116.0,
112.0, 45.7; MS 396.2 (M+H+); Formula C21H16F3N5, Calculated C 63.79, H 4.08, N 17.7; Found C 63.85, H 4.18, N 17.57.
2-[2-r4-Pyridinyl methylamino]phenyl-4-r3-chlorophenyl -1.2.4-triazole (4 : Η NMR (300 MHz, DMSO- 6) δ (ppm) 14.8 (br. s, IH), 8.8 (m, 3H), 8.0 (m, 3H), 7.4 (m, 6H), 6.8
(q, 2H), 4.5 (d, 2H); 13C NMR (75 MHz, DMSO- 6) δ (ppm) 150.1, 149.3, 146.4, 134.0,
132.9, 131.2, 129.5, 127.8, 125.9, 124.9, 122.5, 116.0, 111.9, 45.8; MS 362.2 (M+H1");
Formula C20H16C1N5, Calculated C 66.39, H 4.46, N 19.35; Found C 66.32, H 4.48, N
19.31.
2-[2-('4-Pyridinvnmethylaminolphenyl-4-r4-biphenyl)- 1.2.4-triazole (5) : !H NMR (300 MHz, OMSO-d6) δ (ppm) 14.8 (br. s, IH), 8.8 (br. s, IH), 8.6 (m, IH), 8.3 (m, 3H), 7.9 (m, 6H), 7.3 (m, 6H), 6.7 (m, 2H), 4.6 (d, 2H); 13C NMR (75 MHz, OMSO-d6) δ (ppm) 160.2, 155.5, 150.1, 149.4, 146.4, 141.1, 139.9, 133.5, 131.8, 130.6, 129.4, 128.7, 128.4, 127.4, 122.5, 116.0, 112.1, 109.2, 45.7; MS 404.2 (M+H+); Formula C26H21N5 + 0.25H2O:
Calculated C 76.55, H 5.31, N 17.15; Found C 76.67, H 5.20, N 16.25.
2-[2-(4-Pyridinyl methylamino]phenyl-4-(3-benzoxyphenyl)-1.2.4-triazole (6): 'H NMR (300 MHz, DMSO- 6) δ (ppm) 14.6 (br. s, IH), 8 (m, 17H), 6.7 (m, 2H), 5.3 (s, 2H), 4.6 (d, 2H); 13C NMR (75 MHz, DMSO- 6) δ (ppm) 159.0, 150.1, 149.4, 146.4, 137.3, 130.3,
128.8, 128.2, 128.1, 127.6, 122.5, 118.9, 116.0, 112.5, 112.1, 69.7, 45.7; MS 434.2 (M+H+); Formula C27H23N5O: Calculated C 74.81, H 5.35, N 16.15; Found C 74.79, H 5.20, N 15.88.
2-[2-(4-Pyridinyl methylamino]phenyl-4-(4-benzoxyphenyl - 1.2.4-triazole (7) : 'HNMR
(300 MHz, DMSO- 6) δ (ppm) 14.6 (br. s, IH), 8 (m, 17H), 6.6 (m, 2H), 5.3 (s, 2H), 4.6 (d, 2H); 13C NMR (75 MHz, DMSO-tf6) δ (ppm) 159.9, 149.6, 149.0, 145.9, 136.7, 128.4, 127.8, 127.7, 115.5, 115.2, 115.0, 111.5, 111.0, 69.3, 45.3; MS 434.2 (M+H+); Formula C27H23N5O: Calculated C 74.81, H 5.35, N 16.15; Found C 74.78, H 5.34, N 16.12.
2-[2-(4-Pyridinyl methylamino-5-chloro]phenyl-4-(3-trifluoromethylphenyl -1.2.4-triazole (8): Η NMR (300 MHz, OMSO-d6) δ (ppm) 14.8 (br. s, IH), 8 (m, 17H), 6.7 (d, IH), 4.7 (d, 2H); 13C NMR (75 MHz, DMSO- 6) δ (ppm) 150.1, 148.8, 145.2, 130.9, 130.6, 130.4, 130.3, 129.9, 129.5, 127.2, 126.6, 126.2, 122.5, 122.4, 119.6, 113.7, 45.8; MS 430.1 (M+H+); Formula C21H15ClF3N5O, Calculated C 58.68, H 5.52, N 16.29; Found C 58.38, H
5.61, N 16.06.
2-[3-[2-(4-Pyridinyl)methylamino]pyridinyl]-4-r3-trifluoromethylphenyl -l,2.4-triazole (9): Η NMR (300 MHz, OMSO-d6) δ (ppm) 14.6 (br. s, IH), 8.4 (m, 7H), 7.8 (m, 2H), 7.2 (d, 2H), 6.7 (m, 2H), 4.6 (d, 2H); 13C NMR (75 MHz, DMSO- 6) δ (ppm) 153.6,
148.6, 148.5, 134.2, 129.2, 129.0, 128.9, 128.5, 128.1, 125.2, 124.8, 121.2, 121.0, 110.9, 42.3; MS 397.3 (M+H+); Formula C20H15F3N6: Calculated C 60.61, H 3.81, N 21.19; Found C 60.57, H 3.87, N 21.12.
2-[2-(4-Pyridinyl)methylamino]phenyl-4-(3-phenoxyphenyl -1.2.4-triazole (10 : 'H NMR (300 MHz, OMSO-d6) δ (ppm) 14.6 (br. s, IH), 8 (m, 16H), 6.6 (m, 2H), 4.6 (d, 2H); 13C NMR (75 MHz, DMSO- 6) δ (ppm) 157.7, 156.9, 150.0, 149.2, 146.4, 131.0, 130.5, 124.2, 122.4, 121.4, 119.4, 116.0, 111.8, 45.6; MS 420.2 (M+H ); Formula C26H21N5O: Calculated C 74.44, H 5.05, N 16.704; Found C 74.25, H 4.92, N 16.54.
2- r4-Pyridinyl methylamino]phenyl-4-(4-phenoxyphenyl -1.2.4-triazole (ll : Η NMR (300 MHz, DMSO-GQ δ (ppm) 14.6 (br. s, IH), 8 (m, 16H), 6.6 (m, 2H), 4.6 (d, 2H); 13C NMR (75 MHz, DMSO- 6) δ (ppm) 158.4, 156.4, 150.1, 149.4, 145.4, 130.5, 128.7, 128.4, 127.6, 126.6, 124.3, 122.6, 119.5, 115.9, 111.8, 45.7; MS 420.2 (M+H+); Formula C26H21N5O: Calculated C 74.44, H 5.05, N 16.70; Found C 74.22, H 4.94, N 16.45.
2- (4-Pyridinyl methylamino]phenyl-4-(4-ethvnylphenylV1.2.4-triazole (12): 'H NMR (300 MHz, DMSO- 6) δ (ppm) 14.6 (br. s, IH), 8 (m, 12H), 6.6 (m, 2H), 4.7 (d, 2H), 4.4 (s, IH); 13C NMR (75 MHz, DMSO- 6) δ (ppm) 150.1, 149.3, 146.4, 132.6, 131.5, 127.9, 126.5, 122.7, 122.5, 116.0, 111.9, 88.6, 82.5, 55.4, 45.7; MS 352.2 (M+H+); Formula
C22H17N5 + 0.25H2O: Calculated C 74.25, H 4.96, N 19.67. Found C 74.23, H 5.09, N 19.14.
2-[(4-Pyridinyl)methylamino1phenyl-4-(,3-biphenyl -1.2.4-triazole (13): *H NMR (300 MHz, DMSO-tf6) δ (ppm) 14.6 (br. s, IH), 8 (m, 16H), 6.6 (m, 2H), 4.6 (d, 2H); 13C
NMR (75 MHz, ΩMSO-d6) δ (ppm) 149.1, 148.3, 145.3, 140.2, 139.0, 130.1, 129.3, 128.9, 128.4, 127.3, 127.1, 127.0, 126.1, 124.6, 123.5, 121.5, 114.1, 110.8, 110.4, 45.7; MS 404.2 (M+H4); Formula C26H21N5: Calculated C 77.40, H 5.25, N 17.36. Found C 77.07, H 5.34, N 17.24.
2-[(4-PyridinvDmethylaminolphenyl-4-[3-(2-phenylethenyl phenyl]-1.2.4-triazole (14 : !H NMR (300 MHz, DMSO- 6) δ (ppm) 14.6 (br. s, IH), 8 (m, 18H), 6.6 (m, 2H), 4.6 (d, 2H); 13C NMR (75 MHz, OMSO-d6) δ (ppm) 150.1, 149.3, 146.4, 138.0, 137.8, 133.7, 130.6, 129.7, 129.1, 128.7, 128.2, 127.4, 127.0, 126.9, 124.7, 122.6, 116.0, 111.9, 45.8; MS 430.2 (M+H+); Formula C26H21N5O: Calculated C 78.30, H 5.40, N 16.31. Found C
78.30, H 5.40, N 16.16.
2-[(4-Pyridinyl methylamino]phenyl-4-[3-[(NN-dimethylaminomethyl)phenyl -l,2,4- triazole (15): Η ΝMR (300 MHz, CDC13) δ (ppm) 8.6 (m, 3H), 8.0 (d, IH), 7.9 (m, 3H), 7.4 (m, 3H), 7.3 (m, 3H), 6.8 (t, IH), 6.6 (d, IH), 4.6 (s, 2H), 2.7 (t, 2H), 2.4 (m, 2H), 2.3
(s, 6H), 1.9 (m, IH); 13C MR (75 MHz, CDC13) δ (ppm) 157.7, 157.2, 148.5, 148.3, 145.0, 141.2, 129.6, 128.5, 128.3, 127.5, 126.7, 125.1, 122.9, 121.2, 115.0, 110.6, 110.1, 57.8, 45.3, 43.9, 32.2, 27.5; MS 413.3 (M+H+); C25H28N6: Calculated: C, 69,75 %; H, 6.56 %; N, 19.51 %; Found: C, 69.33 %; H, 6.69 %; N, 19.41 %.
2-[(4-Pyridinyl methylamino]phenyl-4-[4-[(N.N-dimethylaminopropyl)phenyl -1.2,4- triazole (16): Η ΝMR (300 MHz, CDC13) δ (ppm) 8.6 (m, 3H), 7.9 (m, 3H), 7.4 (d, 2H), 7.2 (m, 4H), 6.8 (t, IH), 6.6 (d, IH), 4.6 (s, 2H), 2.7 (t, 2H), 2.4 (m, 2H), 2.3 (s, 6H), 1.9 (m, IH); 13C ΝMR (75 MHz, CDC13) δ (ppm) 158.7, 158.1, 149.5, 149.3, 146.0, 143.4, 130.5, 128.5, 127.7, 127.1, 126.3, 122.2, 116.0, 111.6, 111.1, 58.8, 46.2, 45.0, 33.2, 28.5;
MS 413.3 (M+H4); Formula C25H28N6: Calculated: C 69.75 %, H 6.56 %, N 19.51 %; Found: C 69.86 %, H 6.52 %, N, 19.39 %.
2- [2-(4-Pyridinyl methylamino]phenyl-4-(3 -trifluoromethylphenylV 1 ,2.4-triazole (18): !H NMR (300 MHz, DMSO- 6) δ (ppm) 14.6 (br. s, IH), 8 (m, 13H), 4.6 (d, 2H); MS 396.2
(M+H÷).
Compound (19): Η NMR (DMSO-d6) δ (ppm) 8.89 (s, IH), 8.15-8.35 (m, 4H), 7.98-8.02 (m, 2H), 7.00 (s, IH), 7.10 (s, 2H), 6.68-6.78 (m, IH), 6.00 (s, 2H), 4.67-4.69 (d, 2H), 3.28 (s, 3H); MS m/z: 450 (M+H")
Compound (20): Η NMR (DMSO-eQ δ (ppm) 14.44 (s, IH), 8.16 -8.45 (m, 2H), 7.65- 7.75 (m, 2H), 6.68-7.15 (m, 5H), 6.06 (s, 2H), 4.65-4,67 (d, 2H), 3.83-3.90 (d, 6H); MS m/z: 432 (M+H+)
Compound (21): Η NMR (OMSO-d6) δ (ppm) 14.72 (s, IH), 8.18-8.50 (m, 3H), 7.44-7.65
(m, 3H), 7.08 (s, IH), 6.72-6.77 (m, IH), 6.60-6.61 (d, 2H), 6.41-6.42 (d, IH), 4.70-4.71 (d, 2H), 3.71-3.81 (m, 9H); MS m/z: 418 (M+H+)
Compound (22): IH NMR (DMSO-d6) δ (ppm) 14.21 (s, IH), 8.52-8.56 (m, 2H), 7.98- 8.02 (m, IH), 7.55-7.64(m, 2H), 7.34-7.38 (m, 2H), 7.22-7.26 (m, IH), 7.06-7.10 (m, IH),
4.62-4.64 (d, 2H), 3.72-3.88 (m, 6H); MS m/z: 388 (M+H+)
Compound (23): 1H NMR (DMSO-^) δ (ppm) 8.55-8.61 (m, 3H), 7.90-7.98 (m, IH), 7.55-7.64 (m, 2H), 7.35-7.40 (m, 2H), 7.16-7.24 (m, IH), 6,64-6.72 (m, 2H), 6.10 (s, 2H), 4.60-4.62 (d, 2H); MS m/z: 372 (M+H+)
Compound (24): Η NMR (DMSO-c δ (ppm) 14.63 (s, IH), 8,53-8.59 (m, 3H), 8.08-8.24 (m, 2H), 7.91-7.94 (m, IH), 7.54-7.60 (m, IH), 7.40-7.42 (m, 2H), 6.65-6.75 (m, 2H), 4.63-4.65 (d, 2H); MS m z: 380 (M+H+)
Compound (25): Η NMR (DMSO-^) δ (ppm) 14.60 (s, IH), 8.61-8.72 (m, 3H), 7.90-8.04 (m, 3H), 7.20-7.71 (m, 5H), 6.62-6.84 (m, 2H), 4.64-4.68 (m, 2H); MS m/z: 346 (M+lT)
Compound (26): !H NMR (DMSO-d6) δ (ppm) 14.71(s, IH), 8.55-8.68 (m, 3H), 8.30-8.38 (m, 2H), 7.83-7.94 (m, 3H), 7.40-7.44 (m, 2H), 7.14-7.26 (m, IH), 6.63-6.78 (m, 2H),
4.64-4.66 (d, 2H); MS m z: 396 (M+H+)
Compound (27): Η NMR (DMSOO 10.59 (s, IH), 8.50-8.53 (m, 2H), 7.97-8.00 (m, IH), 7.62-7.64 (m, IH), 7.24-7.36 (m, 3H), 6.88-6.89 (d, 2H), 6.67-6.68 (m, 2H), 6.20 (s, IH), 4.66-4.68 (d, 2H), 3.76 (s, 6H); MS m z 404 (M+H+)
Compound (28): Η NMR (DMSO-d6) δ (ppm) 8.68-8.74 (m, IH), 8.50-8.56 (m, 2H), 8.30-8.35 (m, 2H), 8.04-8.08 (m, 2H), 7.91-7.96 (m, IH), 7.41-7.43 (m, 2H), 7.20-7.28 (m, IH), 6.62-6.71 (m, 2H), 4.60-4.62 (d, 2H), 3.28 (s, 3H); MS m/z: 406 (M+H4)
Compound (29): Η NMR (DMSO- 5) δ (ppm) 12.84(s, IH), 9.33 (s, IH), 8.82-8.86 (m,
IH), 8.40-8.44 (m, IH), 7.98-8.08 (s, IH), 7.30-7.82 (m, 6H), 6.92-6.99 (m, IH), 6.74-6.78 (m, IH), 4.37-4.39(d, 2H); MS m/z: 318 (M+H )
Compound (30): 'H NMR (DMSO- 6) δ (ppm) 8.48-8.52 (m, 2H), 7.40-8.02 (m, 7H), 6.70-6.94 (m, 2H), 4.35-4.42 (d, 2H); MS m/z: 385 (M+H+)
Compound (31): Η NMR (methanol-^) δ (ppm) 8.18-8.22 (m, IH), 7.85-7.97 (m, 3H), 6.90-7.00 (m, 2H), 6.54-6.60 (m, IH), 3.75-3.78 (m, 3H), 3.30-3.50 (m, 6H), 2.20-2.25 (m, 2H), 1.85-1.90 (m, 4H); MS m/z: 393 (M+H4)
Compound (32): Η NMR (OMSO-d6) δ (ppm) 12.69 (s, IH), 7.96-7.98 (m, 2H), 7.82-7.89 9m, IH), 7.67 (s, IH), 7.40-7.48 (m, 2H), 7.25-7.30 (m, IH), 7.02-7.10 (m, 2H), 6.86-6.89 (m, IH), 6.60-6.68 (m, IH), 4.26-4.27 (d, 2H), 3.78 (s, 3H); MS m z: 347 (M+H+)
Compound (33): Η NMR (CDC13) δ (ppm) 8.45-8.52 (m, IH), 8.04-8.09 (m, 2H), 7.81-
7.98 (m, 4H), 6.22-6.27 (m, IH), 3.35-3.68 (m, 6H), 2.95 (s, 3H), 2.37-2.43 (m, 2H), 1.90- 2.02 (m, 4H); MS m/z: 441(M+H+)
Compound (34): ΗNMR(CDC13) δ (ppm) 8.40 (s, IH), 8.10-8.16 (m, IH), 8.03-8.06 (m, IH), 7.50-7.55 (m, 2H), 6.65-6.71 (m, IH), 6.40-6.48 (m, IH), 5.90-5.92 (m, 2H), 3.32-
3.58 (m, 6H), 2.27-2.35 (m, 2H), 1.84-1.92 (m, 4H); MS m/z: 407 (M+H+)
Compound (35): Η NMR(methanol-^) δ (ppm) 8.27-8.34 (m, 4H), 8.01-8.13 (m, 2H), 7.52-7.67 (m, IH), 3.30-3.49 (m, 6H), 2.20-2.28 (m, 6H), 2.17-2.26 (m, 2H), 1.78-1.93 (m, 4H); MS m/z: 431 (M+IF)
Compound (36): MS m/z: 393 (M+H+)
Compound (37): Η NMR (DMSO-d6) δ (ppm) 10.11 (s, IH), 8.28-8.39 (m, 2H), 8.12 (s, IH), 7.87-7.91 (m, IH), 7.25-7.68 (m, 6H), 6.93-7.02 (m, 3H), 6.02 (s, 2H); MS m z: 437 (M+H+)
Compound (38): MS m z: 423 (M+H+)
Compound (39): MS m/z: 365 (M+H+)
Compound (40): Η NMR (DMSO-c δ (ppm) 14.38-14.39 (d, IH), 12.59 (s, IH), 7.65- 7.75 (m, 3H), 7.31-7.45 (m, 3H), 6.45-6.76 (m, 3H), 5.95 (s, 2H), 4.14 (s, 2H); MS m/z: 361 (M+H4)
Compound (41): Η NMR (OMSO-d6) δ (ppm) 9.04-9.08 (s, IH), 8.46-8.49 (m, IH), 8.14-
8.22 (m, 2H), 7.78-7.81 (m, IH), 7.34-7.40 (m, IH), 4.18 (m, IH), 2.32-2.36 (m, 6H), 2.16-2.22 (m, IH), 1.98-2.06 (m, 4H), 1.55-1.80 (m, 2H); MS m/z: 336 (M+H*)
Compound (42): MS m z: 407 (M+H+)
Compound (43): Η NMR(methanol-< δ (ppm) 9.21 (s, IH), 7.98-8.07 (m, 2H), 8.45-8.52 (m, 2H), 7.40-7.48 (m, IH), 6.63-6.72 (m, IH), 3.35-3.52 (m, 6H), 2,18-2.25 (m, 2H), 1.80-1.92 (m, 4H); MS m/z: 364 (M+H+)
Compound (44): Η NMR (methanol-^) δ (ppm) 8.15-8.22 (m, 3H), 7.96-7.99 (m, IH),
7.58-7.63 (m, 2H), 6.91-6.98 (m, IH), 4.08 (s, IH), 2.71-2.79 (m, 2H), 2.21-2.43 (m, 8H), 1.78-2.00 (m, 2H); MS m/z: 403(M+H+)
Compound (45): MS m z: 431 (M+H+)
Compound (46 : Η NMR (OMSO-d6) δ (ppm) 14.84 (s, IH), 10.98 (s, IH), 8.30-8.62 (m, 3H), 8.07 (s, IH), 7.50-7.75 (m, 4H), 7.00-7.16 (m, 2H), 6.26 (s, 2H), 3.25 (s, 3H); MS m/z: 436 (M+H4)
Compound (47): Η NMR (DMSO-c δ (ppm) 15.20 (s, IH), 11.22 (s, IH), 8.42-8.58 (m,
5H), 8.10-8.14 (m, IH), 7.85-7.96 (m, 2H), 7.55-7.70 (m, 2H), 7.12-7.17 (m, IH), 3.24 (s, 3H); MS m/z: 460 (M+H+)
Compound (48): Η NMR (methanol-^) δ (ppm) 9.32 ( s, IH), 8.52-8.64 (m, 2H), 8.26- 8.29 (m, IH), 8.11-8.16 (m, IH), 7.59-7.66 (m, IH), 6.70-6.78 (m, IH), 3.68-3.76 (m, 2H),
2.90-3.00 (m, 6H), 1.92-2.08 (m, 6H); MS m/z: 350(M+H+)
Compound (49): Η NMR (OMSO-d6) δ (ppm) 8.35-8.50 (m, 3H), 8.11-8.15 (m, IH), 7.48-7.74 (m, 5H), 7.00-7.14 (m, 2H), 3.89 (s, 3H), 3.23 (s, 3H); MS m/z: 422 (M+H )
Compound (50): Η NMR (OMSO-d6) δ (ppm) 8.75 (s, IH), 8.27-8.30 (m, IH), 8.16-8.18 (m, IH), 7.46-7.64 (m, 2H), 7.41-7.43 (m, IH), 7.00-7.07 (m, 2H), 6.87-6.93 (m, 2H), 6.71-6.75 (m, IH), 6.00 (s, 2H), 4.65-4.67 (d, 2H), 3.81 (s, 3H); MS m/z: 402 (M+H )
Compound (51): Η NMR (DMSO-d6) δ (ppm) 8.68 (s, IH), 8.27 -8.29 (m, IH), 8.17-8.19
(m, IH), 7.17-7.18 (d, 2H), 7.00-7.01 (s, IH), 6.90-6.94 (m, 2H), 6.71-6.75 (m, IH), 6.55 (s, IH), 6.00 (s, 2H), 4.64-4.66 (d, 2H), 3.79 (s, 6H); MS m/z: 432 (M+H )
Compound (52): Η NMR (DMSO-^) δ (ppm) 8.87 (s, IH), 8.10-8.25 (m, 5H), 7.50-7.60 (m, IH), 6.70-6.95 (m, 4H), 6.00 (s, 2H), 4.68-4.71 (d, 2H); MS m/z: 424 (M+H+)
Compound (53): Η NMR (DMSO-^) δ (ppm) 14.80 (s, br, IH), 9.10 (s, br, IH), 8.31- 8.38 (m, 2H), 7.35-7.85 (m, 4H), 6.70-6,95 (m, 3H), 6.00 (s, 2H), 4.65-4.70 (d, 2H); MS m/z: 390 (M+H4)
Compound (54): Η NMR (DMSO- 6) δ (ppm) 8.80 (s, IH), 8.15-8.38 (m, 4H), 7.70-7.85 (m, 2H), 7.00 (s, IH), 6.85-6.90 (m, 2H), 6.72-6.78 (m, IH), 6.00 (s, 2H), 4.65-4.67 (d, 2H); MS m/z: 440 (M+H )
Compound (55): Η NMR (DMSO-<Q 14.85 (s, br, IH), 8.80 (s, br, IH), 8.15-8.25 (m,
4H), 7.85-7.92 (m, 2H), 7.00 (s, IH), 7.11 (s, 2H), 6.68-6.72 (m, IH), 6.00 (s, 2H), 4.65- 4.68 (d, 2H); MS m/z: 440 (M+H )
Compound (56): Η NMR (DMSO- 6) δ (ppm) 14.71 (s, IH), 8.65 (s, IH), 8.48-8.59 (m, IH), 8.10-8.30 (m, 2H), 7.78-7.82 (m, IH), 7.50-7.65 (m, 2H), 7.30-7.45 (m, 2H), 7.02-
7.08 (m, IH), 6,70-6.80 (m, IH), 4.80-4.82 (m, 2H), 3.82 (s, 3H); MS m/z: 359 (M+H+)
Compound (57): Η NMR (DMSO- ) δl4.78 (s, br, IH), 8.85 (s, br, IH), 8.60 (s, IH), 8.50 (d, IH), 8.05-8.30 (m, 3H), 7.75-7.80 (m, IH), 7.50-7.60 (m, IH), 7.30-7.40(m, IH), 6.72-6.80 (m, IH), 4.75-4.82 (d, 2H); MS m/z: 381 (M+H4)
Compound (58): Η NMR (DMSO-^) δ (ppm) 14.76 (s, IH), 8,88 (s, IH), 8.65 (s, IH), 8.48 (s, IH), 8.19-8.26 (m, 3H), 7.72-7.94 (m, 3H), 7.50-7.55 (m, 2H), 6.75-6.80 (m, IH), 4.82-4.84 (d, 2H); MS m/z: 347 (M+H4)
Compound (59): Η NMR (DMSO-c δ (ppm) 14.90 (s, IH), 8.91 (s, IH), 8.68 (s, IH), 8.45-8.48 (d, IH), 8.15-8.30 (m, 4H), 7.72-7.95 (m, 3H), 7.35-7.40 (m, IH), 6.70-6.79 (m, IH), 4.80-4.85 (d, 2H); MS m z: 397 (M+H4)
Compound (60): Η NMR (DMSO-cQ δ (ppm) 14.72 (s, IH), 8.66 (s, IH), 8.47-8.48 (m,
IH), 8.17-8.31 (m, 2H), 7.81-7.83 (d, IH), 7.35-7.39 (m, IH), 7.20-7.21 (d, 2H), 6.63-6.78 (m, 2H), 4.80-4.82 (m, 2H), 3.80 (s, 6H); MS m/z: 389 (M+H4)
Compound (61): Η NMR (DMSO-c δ (ppm) 14.50 (s, IH), 8.65-8.66 (m, IH), 8.47-8.49 (m, IH), 8.16-8.18 (m, IH), 7.80-7.83 (m, IH), 7.61-7.65 (m, 2H), 7.35-7.40 (m, IH),
7.10-7.12 (d, IH), 6.73-6.77 (m, IH), 4.79-4.81 (d, 2H), 3.81-3.83 (d, 2H); MS m/z: 389 (M+H4)
Compound (62): Η NMR (OMSO-d6) δ (ppm) 14.90 (s, IH), 8.91 (s, IH), 8.66 (s, IH), 8.02-8.35 (m, 6H), 7.81-7.84 (m, IH), 7.32-7.39 (m IH), 6.75-6.80 (m, IH), 4.82-4.84 (d,
2H), 3.28-3.33 (d, 3H); MS m/z: 407 (M+H4)
Compound (63): Η NMR DMSO-d6) 14.50 (s, IH), 8.17-8.57 (m, 3H), 7.57-7.64 (m, 2H), 7.10 (s, IH), 6.41-6.61 (m, 4H), 4.74-4.75 (d, 2H), 3.75-3.87 (m, 12 H); MS m/z: 448 (M+H4)
Compound (64): Η NMR (DMSO-d6) 8.11-8.42 (m, 7H), 7.85-7.90 (m, IH), 6.55-6.60 (m, IH), 6.38-6.40 (m, IH), 4.78-4.81 (d, 2H), 3.72-3.74 (m, 9H); MS m/z: 466 (M+H4)
Compound (65): Η NMR (DMSO- 6) δ (ppm) 8.34-8.58 (m, 2H), 7.70-7.73 (m, 2H),
7.13-7.23 (m, IH), 6.85-7.10 (m, 4H), 6.19 (s, IH), 3.74-3.88 (m, 9H); MS m/z: 404 (M+H4)
Compound (66): Η NMR DMSO-d6) δ (ppm) 8.50-8.58 (m, 3H), 8.32-8.36 (m, IH), 8.24-8.27 (m IH), 7.78-7.96 (m, 2H), 6.79-6.84 (m, IH), 3.75-3.79 (m, 2H), 3.15-3.23 (m,
6H), 1.91-2.12 (m, 6H); MS m/z: 417(M+H4)
Compound (67): Η NMR DMSO-d6)δ 8.49 (m, IH), 8.29-8.33 (m, IH), 8.15-8.22 (m, IH), 7.81-7.88 (m, 2H), 7.45-7.55 (m, IH), 7.10-7.14 (m, IH), 6.72-6.78 (m, IH), 3.91 (s, 3H), 3.72-3.79 (m, 2H), 3.05-3.24 (m, 6H), 1.97-2.08 (m, 6H); MS m/z: 377(M+H4)
Compound (68): Η NMR (methanol-^) δ (ppm) 8.14-8.18 (m, IH), 7.95-7.97 (m, IH), 7.45-7.50 (m, IH), 7.51 (s, IH), 6.82-6.86 (m, IH), 6.55-6.59 (m, IH), 5.93 (s, 2H), 3.40- 3.54 (m, 2H), 2.52-2.68 (m, 6H), 1.68-1.95 (m, 6H); MS m/z: 39304+^)
Compound (69): Η NMR (methanol-^) δ (ppm) 8.25-8.28 ( , IH), 8.05-8.08 (m,lH), 7.20-7.24 (m, 2H), 6.57-6.73 (m, 2H), 3.91 (s, 6H), 3.55-3.63 (m, 2H), 2.74-2.93 (m, 6H), 1.82-2.08 (m, 6H); m z: 409(M+H4)
Compound (70): Η NMR (methanol-^) δ (ppm) 8.44-8.48 (m, IH), 8.04-8.10 (m, IH),
7.63-7.69 (m, 2H), 7.45-7.52 (m, IH), 6.98-6.12 (m, 2H), 4.32 (s, IH), 3.88 (s, 3H), 2.82- 2.91 (m, 2H), 2.64-2.68 (m, 4H), 2.38-2.46 (m, 4H), 1.95-2.15 (m, 2H); MS m/z: 365(M+H4)
Compound (71): Η NMR (OMSO-d6) δ (ppm) 11.27 (s, IH), 8.50-8.54 (m, IH), 8.34-8.37
(m, 2H), 8.00-8.06 (m, IH), 7.78-7.87 (m, 2H), 7.42-7.60 (m, 3H), 7.38 (s, 2H), 6,99-7.13 (m, 2H), 3.89 (s, 3H); MS m/z: 423 (M+H4)
Compound (72): 'H NMR fDMSOO δ (ppm) 15.30 (s, IH), 11.18 (s, IH), 8.38-8.51 (m, 5H), 7.82-7.98 (m, 3H), 7.30-7.58 (m, 4H), 7.00-7.07 (m, IH); MS m/z: 461(M+H4)
Compound (73): Η NMR (DMSO-d6) δ (ppm) 9.35 (s, IH), 8.68-8.71 (m, IH), 8.40-8.47 (m, 2H), 8.30-8.33 (m, IH), 7.59-7.66 (m, IH), 7.11 (s, 2H), 6.93-6.99 9m, IH), 6.22 (s, IH), 3.79 (s, 6H); MS m z: 375(M+H4)
Compound (74): Η NMR (OMSO-d6) δ (ppm) 14.98 (s, br, IH), 11.92 (s, br, IH), 8.60 (s, IH), 8.10-8.24 (m, 2H), 7.45-7.58 (m, 3H), 7.08-7.13 (m, IH), 6.82-6.88 (m, IH), 6.65- 6.70 (m, IH), 5.93 (s, 2H), 3.84-3.88 (m, 2H), 2.85-2.91 (m, 2H); MS m/z: 376(M+H4)
Compound (75): Η NMR (DMSO-^) δ (ppm) 14.40 (s, br, IH), 1.57 (s, br, IH), 8.55 (s,
IH), 8.15-8.22 (m, 2H), 7.42-7.65 (m, 4H), 7.07-7.10 (m, IH), 6.66-6.68 (m, IH), 6.65- 6.74 (m, IH), 3.92 (s, 3H), 3.78-3.86 (m, IH), 2.85-2.92 (m, 2H); MS m z: 362(M+H4)
Compound (76): MS m z: 397 (M+H4)
Compound (77): MS m/z: 396 (M+H4)
Compound (78): MS m/z: 396 (M+H4)
Compound (79): MS m/z: 433 (M+H4)
Compound (80): MS m/z: 388 (M+H4)
Compound (81): MS m/z: 388 (M+H4)
Compound (82): Η NMR (DMSO ) δ (ppm) 8.47-8.49 (m, 2H), 8.22-8.25 (m, IH), 7.95-7.98 (m, IH), 7.25-7.34 (m, 4H), 6.60-6.95 (m, 5H), 6.56-6.60 (d, 2H), 4.14-4.22 (d, 2H), 3.68-3.72 (m, 3H); MS m/z: 373 (M+H4)
Compound (83): MS m/z: 408 (M+H4)
Compound (84): MS m/z: 439 (M+H4)
Compound (85): MS m/z: 423 (M+H4)
Compound (86): MS m/z: 377 (M+H4)
Compound (87): Η NMR (DMSO-^) δ (ppm) 14.38 (s, IH), 10.79 (s, IH), 8.27-8.37 (m, 2H), 7.23-7.28 (m, IH), 6.82-7.00 (m, 6H), 6.14-6.16 (m, IH), 4.19 (s, IH), 3.70-3.74 (m, 9H); MS m/z: 418 (M+H4)
Compound (88): MS m/z: 410 (M+H4)
Compound (89): MS m/z: 404 (M+H4)
Compound (90): MS m z: 391 (M+H4)
Compound (91 : MS m/z: 422 (M+H4)
Compound (92 : MS m/z: 377 (M+H4)
Compound (93 : MS m/z: 417 (M+H4)
Compound (94 : MS m/z: 408 (M+H4)
Compound (95 : MS m z: 394 (M+H4)
Compound (96 : MS m/z: 394 (M+H4)
Compound (97 : MS m/z: 437 (M+H4)
Compound (98 : MS m/z: 451 (M+H4)
Compound (99): MS m/z: 380 (M+H4)
Compound (100): Η NMR(CDC13) δ (ppm) 12,35 (s, IH), 8.02-8.20 (m, 3H), 7.10-7.18 (m, IH), 6.65-6.80 (m, 3H), 6.45-6.52 (m, IH), 4.07-4.08 (d 2H), 3.72 (s, 3H), 3.42-3.52
(m, 2H), 3.25-3.38 (m, 4H), 2.15-2.22 (m, 2H), 1.80-1.90 (m, 4H); MS m/z: 407(M+H4)
Compound (101): Η NMR(CDC13) δ (ppm) 7.70-7.75 (m, IH), 7.39 9s, 2H), 7.02-7.25 (m, 2H), 6.55-6.68 (m, 5H), 4.25 (s, 2H), 3.96 9s, 2H), 3.54 (s, 3H); MS m z: 361 (M+H4)
Compound (102): MS m/z: 431 (M+H4)
Compound (103): MS m/z: 466 (M+H4)
Compound (104): MS m/z: 467 (M+H4)
Compound (105): MS m/z: 392 (M+H4)
Compound (106): MS m/z: 437 (M+H4)
Compound (107): MS m/z: 409 (M+H4)
Compound (108): MS m/z: 403 (M+H4)
Compound (109): MS m/z: 452 (M+H4)
Compound (110): MS m/z: 448 (M+H4)
Compound (111): MS m/z: 406 (M+H4)
Compound (112): MS m/z: 453 (M+H )
Compound (113): MS m/z: 488 (M+H4)
Compound (114): £ee Example 6
Compound (115): Η NMR (DMSO-d6) δ (ppm) 11.40 (s, IH), 10.0 (s, IH), 8.40-8.43 (m, IH), 8.01-8.04 (m, IH), 7.01-7.06 (m, 2H), 6.81-6.95 (m, 4H), 6.23-6.25 (t, IH), 3.80 (s, 6H); MS m/z: 426 (M+H4). Calculated for C21H17F2N5O3: 425.13
Compound (116): Η NMR (DMSO-d6) δ (ppm) 11.34 (s, IH), 10.02 (s, IH), 8.43-8.45
(m, IH), 8.06-8.16 (m, 2H), 7.87-7.90 (m, IH), 7.64-7.70 (t, IH), 7.43-7.46 (d, IH), 7.05- 7.10 (m, 3H), 6.25-6.27 (t, IH), 3.81 (s, 6H); MS m z: 458 (M+H4). Calculated for C22H18F3N5O3: 457.13
Compound (117): Η NMR (DMSO-c^) δ (ppm) 10.93 (s, IH), 10.07 (s, IH), 8.41-8.44 (m, IH), 8.04-8.08 (m, IH), 7.04-7.54 (m, 11H), 6.74-6.77 (m, IH), 6.24-6.26 (t, IH), 5.16 (s, 2H), 3.80-3.84 (d, 6H); MS m/z: 496 (M+H4). Calculated for C22H19F3N5O5: 495.19
Compound (118): Η NMR (DMSO-^) δ (ppm) 10.76 (s, IH), 10.07 (s, IH), 8.40-8.43
(m, IH), 8.02-8.05 (m, IH), 7.33-7.34 (d, IH), 6.94-7.11 (m, 5H), 6.24-6.26 (t, IH), 6.05 (s, 2H), 3.77-3.80 (d, 6H); MS m/z: 434 (M+H4). Calculated for C22H19N5O5: 433.14
Compound (119): Η NMR (OMSO-d6) δ (ppm) 10.68 (s, IH), 10.04 (s, IH), 8.40-8.42-t, IH), 8.03-8.06 (t, IH), 7.35-7.36 (d, IH), 7.00-7.19 (m, 5H), 6.25 (s, IH), 3.78-3.82 (m,
12H); MS m/z: 450 (M+H4). Calculated for C23H23N5O5: 449.17
Compound (120): Η NMR (DMSO-d6) δlθ.86 (s, IH), 10.05 (s, IH), 8..35-8.37 (m, IH), 8.00-8.02 (m, IH), 7.65-7.68 (d, 2H), 7,34-7.40 (t, 2H), 6.96-7.12 (m, 8H), 6.19-6.20 (t, IH), 3.72-3.76 (d, 6H); MS m/z: 482 (M+H4). Calculated for C27H21N5O4: 481.18
Compound (121): Η NMR (DMSO-d6) δll.05 (s, IH), 8.28-8.30 (s, IH), 8.11-8.16 (t, IH), 7.92-7.96 (m, IH), 7.73-7.76 (d, 2H), 7.17-7.29 (m, IH), 7.00 (s, IH), 6.91 (s, 2H), 6.80-6.84 (m, IH), 6.02 (s, 2H), 4.70-4.72 (d, 2H); MS m z: 471 (M+H4). Calculated for C27H17F3N4O4: 470.12
Compound (122): Η NMR (DMSO-d6) δ (ppm) 11.22 (s, IH), 8.28-8.31 (m, IH), 8.15 (s, 2H), 7.91-7.95 (m, IH), 7.81-7.84 (d, IH), 7.64-7.67 (t, IH), 7.40-7.42 (d, IH), 6.80-7.00 (m, 4H), 6.02 (s, 2H), 4.71-4.73 (d, 2H); MS m/z: 456 (M+H4). Calculated for C23H17F3N4O3: 455.12
Compound (123): Η NMR (DMSO-^) δ (ppm) 10.83 (s, IH), 8.27-8.30 (m, IH), 8.12- 8.16 (m, IH), 7.90-7.94 (m, IH), 7.17-7.52 (m, 9H), 7.00 (s, IH), 6.90 (d, 3H), 6.71-6.84 (m, 2H), 6.02 (s, IH), 5.14 (s, IH), 4.71-4.73 (d, 2H); MS m z: 494 (M+H4). Calculated for C29H24N4O4: 492.18
Compound (124): Η NMR (OMSO-d6) δ (ppm) 10.81 (s, IH), 8.27-8.29 (m, IH), 8.13- 8.17 (t, IH), 7.92-7.95 (m, IH), 7.66-7.69 (m, 2H), 7.38-7.43 (t, 2H), 6.79-7.16 (m, 9H), 6.02 (s, 2H), 4.70-4.72 (m, 2H); MS m/z: 480(M+H4). Calculated for C27H21N5O4: 479.16
Compound (125): Η NMR (DMSO-d6) δlθ.60 (s, IH), 8.26-8.29 (m, IH), 8.15-8.17 (t,
IH), 7.89-7.93 (m, IH), 7.38-7.39 (d, IH), 6.79-7.12 (m, 6H), 6.02 (s, 2H), 4.70-4.72 (d, 2H), 3.76-3.80 (d, 6H); MS m/z: 448(M+H4). Calculated for C23H21N5O5: 447.15
Compound (126): Η NMR (DMSO-d6) δ (ppm) 10.62 (s, IH), 8.23-8.25 (m, IH), 8.09 (t, IH), 7.84-7.88 (m, IH), 7.27-7.28 (d, IH), 6.75-7.03 (m, 6H), 5.99-6.00 9d, 2H), 4.66-4.68
(d, 2H); MS m/z: 432(M+H4). Calculated for C22H17N5O5: 431.12
Compound (127): Η NMR DMSO-d6) δ (ppm) 11.03 (s, IH), 8.24-8.26 ( , IH), 8.13- 8.17 (t, IH), 7.90-7.93 (m, IH), 7.70-7.73 (d, 2H), 7.78-7.40 (d, 2H), 6.78-6.82 (m, IH), 6.40-6.54 (d, 2H), 6.54-6.55 (d, IH), 6.40 (s, IH), 4.71-4.73 (d, 2H), 3.72 (s, 6H); MS m/z:
488(M+H4). Calculated for C23H20F3N5O4: 487.15
Compound (128): Η NMR (OMSO-d6) δll.20 (s, IH), 8.11-8.27 (m, 3H), 7.90-7.93 (m, IH), 7.79-7.82 (d, IH), 7.58-7.64 (t, IH), 7.37-7.39 (d, IH), 6.77-6.82 (m, IH), 6.40-6.54 (d, 2H), 6.39-6.40 (d, IH), 4.71-4.73 (d, 2H), 3.72 (s, 6H); MS m z: 472(M+H4).
Calculated for C23H20F3N5O3: 471.15
Compound (129): Η NMR (OMSO-d6) δ (ppm) 10.80 (s, IH), 8.14-8.25 (m, 2H), 7.90- 7.93 (m, IH), 7.62-7.90 (m, 2H), 6.77-7.40 (m, 7H), 6.54-6.55 (d, 2H), 6.39-6.40 (t, 2H),
3.71 (s, 6H); MS m/z: 496(M+H4). Calculated for C28H25F3N5O4: 495.19
Compound (130): Η NMR (OMSO-d6) δ (ppm) 10.80 (s, IH), 8.15-8.25 (m, 2H), 7.88- 7.91 (d, IH), 7.07-7.48 (m, 9H), 6.40-6.81 (m, 5H), 5.11 (s, 2H), 4.71-4.73 (d, 2H), 3.71 (s, 6H); MS m/z: 510(M+H4). Calculated for C29H27F3N5O4: 509.21
Compound (131): Η NMR DMSO-d6) δ (ppm) 10.64 (s, IH), 8.12-8.24 (m, 2H), 7.86- 7.89 (m, IH), 7.28-7.29 (d, IH), 6.76-7.04 (m, 3H), 6.54-6.55 (d, 2H), 6.39-6.41 (t, IH), 6.00 (s, 2H), 4.70-4.72 (d, 2H), 3.72 (s, 6H); MS m z: 448(M+H4). Calculated for
C23H21F3N5O5: 447.15
Compound (132): Η NMR DMSO-d6) δ (ppm) 10.57 (s, IH), 8.15-8.24 (m, 2H), 7.87- 7,90 (m, IH), 7.35-7,36 (m, IH), 7.06-7.10 (m, IH), 6.94-6.97 (d, IH), 6.76-6.81 (m, IH), 6.54-6.55 (d, 2H), 6.39-6.40 (t, IH), 4.71-4.73 (d, 2H), 3.72-3.77 (m, 12H); MS m/z: 464 (M+H4). Calculated for C24H25F3N5O5: 463.19
Compound (133): Η NMR (DMSO-cQ δ (ppm) 11.05 (s, IH), 8.14-8.26 (m, 2H), 7.89- 7.92 (d, IH), 7.37-7.57 (m, 3H), 7.07-7.10 (d, IH), 6.80-6.88 (m, 2H), 6.54 (s, 2H), 6.40 (s, IH), 3.72 (s, 6H); MS m z: 422 (M+H4). Calculated for C22H20FN5O3: 421.42
Compound (134): Η NMR (OMSO-d6) δ (ppm) 11.25 (s, IH), 8.13-8.27 (m, 2H), 7.72-
7.94 (m, 5H), 6.80-6.82 (m, IH), 6.54-6.55 (d, 2H), 6.40-6.41 (t, IH), 4.71-4.73 (d, 2H), 3.72 (s, 6H); MS m z: 472 (M+H4). Calculated for C23H20F3N5O3: 471.15
Compound (135): Η NMR (DMSO-^) δ (ppm) 10.62 (s, IH), 8.14-8.25 (m, 2H), 7.87- 7.90 (m, IH), 7.52 (s, IH), 7.06-7.35 (m, 3H), 6.76-6.80 (m, IH), 6.54-6.55 (d, 2H), 6.40
(s, IH), 4.70-4.72 (d, 2H), 3.72 (s, 6H), 2.78-2.88 (m, 4H), 1.96-2.06 (m, 2H); MS m/z: 444 (M+H4). Calculated for C25H25F3N5O3: 443.20
Compound (136): Η NMR (OMSO-d6) δ (ppm) 10.29 (s, IH), 8.23-8.25 (m, IH), 8.14- 8.18 (t, IH), 7.88-7.91 (m, IH), 7.07-7.33 (m, 3H), 6.77-6.81 (m, IH), 6.54-6.62 (m, 3H),
6.39 (s, IH), 4.71-4.73 (d, 2H), 3.71-3.76 (d, 9H) ; MS m/z: 420(M+H4). Calculated for C22H21N5O4: 419.16
Compound (137): Η NMR MSO-^) δ (ppm) 8.61 (s, IH), 8.45-8.46 (d, IH), 8.22-8.30 (m, 2H), 7.89-7.91 (m, IH), 7.76-7.79 (m, IH), 7.13-7.36 (m, 4H), 6.72-6.83 (m, IH),
6.59-6.61 (m, IH), 4.82-4.84 (d. 2H), 3.76 (s. 3H); MS m/z: 375(M+H4). Calculated for C20H18N6O2- 374.40
Compound (138): Η NMR (DMSO-cQ δ (ppm) 10.60 (s, IH), 8.10-8.15 (m, IH), 7.88- 7.92 (t, IH), 7.61-7.70 (m, IH), 6.41-7.14 (m, 8H), 5.85 (s, 2H), 4.49-4.51 (d, 2H), 3.62 (s,
3H); MS m/z: 418(M+H4). Calculated for C22H19N5O4: 417.14
Compound (139): Η NMR DMSO-d6) δ (ppm) 10.95 (s, IH), 9.94 (s, IH), 8.42-8.44 (m, IH), 8.06-8.11 (m, IH), 7.62-7.63 (d, IH), 6.91-7.14 (m, 5H), 6.31-6.33 (t, IH), 6.12 (s, 2H), 3.86 (s, 6H); MS m/z: 434 (M+H4). Calculated for C22H19N5O5: 433.14
Compound (140): Η NMR (T>MSO-d6) δ (ppm) 10.89 (s, IH), 10.20 (s, IH), 8.33-8.38 (m, IH), 8.01-8.04 (m, IH), 7.22-7.33 (m, 2H), 7.10-7.22 (m, IH), 7.01-7.09 (m, 3H), 6.62-6.69 (m, IH), 6.21-6.25 (m, IH), 3.78-3.87 (m, 9H); MS m/z: 420 (M+H4) Calculated for C22H21N5O4: 419.43
Compound (141): Η NMR (DMSO- d6) δ (ppm) 10.90 (s, IH), 9.91 (s, IH), 8.38-8.42 (m, IH), 7.98-8.02 (m, IH), 7.57-7.70 (m, IH), 6.94-7.38 (m, 6H), 6.66-6.70 (m, IH), 6.03 (s, 2H), 3.83 (s, 3H); MS m/z: 404 (M+H4). Calculated for C21H17N5O4: 403.13
Compound (142): Η NMR (DMSO-^ δ (ppm) 10.30 (s, IH), 8.23-8.25 (m, IH), 8.14- 8.18 ( t, IH), 7.07-7.33 (m, 3H), 6.62-6.81 (m, IH), 6.40-6.59 (m, 3H), 6.39-6.40 (t, IH), 4.71-4.73 (m, 2H), 3.67-3.76 (m, 9H); MS m/z: 434 (M+H4). Calculated for C23H23N5O4: 433.18
Compound (143): Η NMR (OMSO-d6) δ (ppm) 8.61 (s, IH), 8.45-8.46 (d, IH), 8.23-8.30 (m, 2H), 7.89-7.91 (m, IH), 7.76-7.79 (m, IH), 7.13-7.34 (m, 4H), 6.78-6.82 (m, IH), 6.59-6.61 (d, IH), 4.82-4.84 (d, 2H), 3.76 (s, 3H); MS m/z: 375 (M+H4). Calculated for
C20H18N6O2: 374.15
Compound (144): Η NMR (OMSO-d6) δ (ppm) 8.62 (s, IH), 8.42-8.45 (t, IH), 88.10- 8.23 (m, 2H), 7.76-7.79 (d, IH), 7.60-7.62 (m, IH), 7.23-7.37 (m, 2H), 6.78-6.87 (m, 3H), 6.12-6.18 (m, IH), 4.82-4.84 (d, 2H), 3.78 (s, 6H); MS m/z: 405 (M+H4). Calculated for C21H20N6O3: 404.16
Compound (145): Η NMR OMSO-d6) δ (ppm) 10.76 (s, IH), 8.22-8.25 (m, IH), 8.13- 8.17 (t, IH), 7.87-7.90 (m, IH), 7.06-7.09 (d, IH), 6.77-6.86 (m, 3H), 6.53-6.54 (d, 2H), 6.38-6.40 (t, IH), 6.19-6.21 (t, IH), 4.71-4.73 (d, 2H), 3.68-3.78 (m, 12H); MS m/z: 464 (M+H4). Calculated for C24H25N5O5: 463.19
Compound (146): Η NMR (OMSO-d6) δ (ppm) 10.60 (s, IH), 8.51-8.53 (d, 2H), 8.00- 8.01 (t, IH), 7.62-7.64 (d, IH), 7.24-7.37 9m, 3H), 6.88-6.89 (d, 2H), 6.68-6.78 (m, 2H), 6.20 (s, IH), 4.66-4.68 (d, 2H), 3.76 (s, 3H); MS m/z: 448 (M+H4). Calculated for C23H21N5O5: 447.15
Compound (147): Η NMR DMSO-d6) δ (ppm) 8.62 (s, IH), 8.46-8.47 9d, IH), 8.23-8.29 (m, 2H), 7.68-7.92 (m, 6H), 7.34-7.38 (t, IH), 6.78-6.82 (t, IH), 4.82-4.84 9d, 2H); MS m/z: 413 (M+H4). Calculated for C20H15F3N5O: 412.13
Compound (148): Η NMR (DMSO-^) δ (ppm) 10.69 (br, s, IH), 8.61-8.62 (d, IH), 8.45-
8.47 9m, IH), 8.21-8.28 (m, 2H), 7.86-7.89 (m, IH), 7.76-7.79 (d, IH), 7.31-7.38 (m, 2H), 7.02-7.06 (m, IH), 6.89-6.92 (m, IH), 6.77-6.81 (m, IH), 6.00 (s, 2H), 4.81-4.83 (d, 2H); MS m/z: 389 (M+H4). Calculated for C20H16N6O3 : 388.13
Compound (149): Η NMR (DMSO-d6) δ (ppm) 8.61 (s, IH), 8.45-8.47 (m, IH), 8.21-8.30
9m, 2H), 7.90-7.92 (m, IH), 7.76-7.79 (m, IH), 7.63-7.67 (m, 2H), 7.33-7.40 (m, 3H), 6.96-7.12 (m, 4H), 6.78-6.82 (m, IH), 4.81-4.83 (d, 2H); MS m/z: 437 (M+H4). Calculated for C25H20N6O2: 436.16 . „
Compound (150): Η NMR (DMSO-d6) δ (ppm) 8.62 (s, IH), 8.44-8.46 (m, IH), 8.21- 8.31 (m, 2H), 7.76-7.90 (m, 2H), 7.32-7.40 (m, 2H), 7.06-7.10 (m, IH), 6.93-6.96 (m, IH), 6.77-6.93 (m, IH), 4.81-4.83 (d, 2H), 3.67-3.77 (m, 6H); MS m/z: 405 (M+H4). Calculated for C21H20N6O3: 404.16
Compound (151): Η NMR (methanol-^) δ (ppm) 7,98-8.01 (m, 2H), 6.85-6.98 (m, 5H), 6.55-6.72 (m, 4H), 5.80 (s, 2H), 5.03-5.10 (d, 2H), 4.50 (s, 2H); MS m/z: 419 (M+H4)
Compound (152): Η NMR (DMSO- 5) δ (ppm) 8.60 (s, IH), 8.40-8.44 (m, IH), 8.21-8.29 (m, 2H), 8.05-8.10 (m, IH), 7.80-7.86 (m, IH), 7.08-7.19 (m, 4H), 6.77-6.83(m, IH), 5.50
(s, 2H), 4.80-4.86 (m, 2H); MS m/z: 378 (M+H4)
Compound (153): Η NMR (CDC13) δ (ppm) 8.57-8.60 (d, 2H), 8.10-8.16 (t, IH), 7.84- 7.87 (d, IH), 7.28-7.32 (m, 3H), 7.02-7.04 (m, 5H), 6.76-6.84 (m, IH), 6.58-6.64 (m, IH), 5.32 (s, 2H), 4.82-4.89 (d, 2H); MS m/z: 376 (M+H4)
Compound (154): See Example 9.
Compound (155): Η NMR (DMSO ) 11.00 (s, IH), 9.50 (s, IH), 8.28-8.32 (m, 2H), 7.10-7.28 (m, 5H), 6.92-6.97 (m, IH), 6.47-6.50 (m, IH), 6.17-6.18 (t, IH), 3.747-3.749
(d, 9H); MS m/z: 419 (M+H4). Calculated for C22H22N6O3: 418.18
Compound (156): Η NMR (methanol-^) δ (ppm) 8.25-8.35 (br s, IH), 8.19 (s, IH), 7.41- 7.45 (d, 2H), 6.83-6.96 (m, 5H), 6.16 (s, IH), 3.3-3.80 (m, 9H); MS m/z: 419 (M+H4). Calculated for C22H22N6O3: 418.18
Compound (157): Η NMR (OMSO-d6) δ (ppm) 10.30 (s, IH), 8.32-8.46 (m, 2H), 7.78- 7.93 (m, 4H), 7.00-7.77 (m, 3H), 6.25-6.27 (d, IH, J=0.069), 3.80-3.82 (d, 6H, J=0.033Hz); MS m/z: 414 (M+H4). Calculated for C22HI9N7O2: 413.16
Compound (158): Η NMR (DMSO-fi δ (ppm) 10.50 (s, IH), 9.66 (s, IH), 9.59 (s, IH), 8.14-8.32 (m, 2H), 7.48-7.55 9m, 2H), 7.16 (s, IH), 6.78-7.09 (m, 5H), 6.08 (IH), 3.65 (s, 6H); MS m/z: 389(M+H4). Calculated for C22H21N5O2: 328.14
Compound (159): Η NMR (CDC13) δ (ppm) 10.34 (s, IH), 8,27-8.29 (m, 2H), 7.77-7.78
(d, IH, J=0.03Hz), 7.36 (s, IH), 7.00-7.01 (d, 2H, J=0.03), 6.73-6.82 (m, 2H), 6,45-6.49 (m, IH), 6.14-6.15 (t, IH, J=045Hz), 3.77-3.83 (m, 12H); MS m/z: 449 (M+H4). Calculated for C23H24N6O4: 448.19
Compound (160): Η NMR (CDC13) δ (ppm) 10.56 (s, IH), 8.43-8.50 (m, 2H), 7.45-7.49
(d, 3H), 6.94-6.98 (m, 4H), 6.36-6.38 (m, IH), 3.98-4.03 (t, 6H), 3.19 (br s, 6H); MS m/z: 432 (M+H4). Calculated for C23H25N7O2: 431.21
Compound (161): ΗNMR (CDC13) δ (ppm) 10.19 (s, IH), 8.22-8.24 (m, IH), 8.11 (br s, IH), 6.60-7.39 (m, 10H), 5.91 (s, 2H), 3.81-3.85 (t, 3H); MS m/z: 403 (M+H4).
Calculated for C21H18N6O3: 402.41
Compound (162): Η NMR (CDC13) δ (ppm) 10.07 (s, IH), 8.09-8.16 (m, 2H), 7.31-7.35 (m, IH), 7.22-7.30 (m, 3H), 6.88-6.95 (m, 3H), 6,62-6.75 (m, 3H), 5.84-5.86 (d, 2H), 3.78 (s, 3H); MS m/z: 403 (M+H4). Calculated for C21H18N6O3: 402.41
Compound (163): Η NMR (DMSO-<Q δ (ppm) 10.77 (br s, IH), 10.18 (s, IH), 8.30-8.32 (d, 2H, J=0.06Hz), 7.78 (s, 4H), 7.61-7.62 (d, IH, J=0.021), 1.73-7.16 (t, IH, J=0.028), 6.94-6.97 (m, 2H), 6.06-6.05 (d, 2H, J=0.036); MS m/z: 398 (M+H4). Calculated for C21H15N7O2: 397.13
Compound (164): Η NMR (DMSO- 6) δ (ppm) 10.49 (s, IH), 8.88 (s, IH), 8.25-8.40 (m, 2H), 7,95 (s, IH), 7.64 (s, IH), 6.91-7.13 (m, 4H), 6.49-6.58 (m, IH), 6.03-6.06 (d, 2H), 3.69-3.89 (m, 6H); MS m/z: 433 (M+H4). Calculated for C22H20N6O4: 432.15
Compound (165): Η NMR (methanol-^) δ (ppm) 8.32 (s, IH), 8.09 (s, IH), 7.34 (s, IH), 6.88-7.00 (m, IH), 6.70-6.80 (m, 3H), 6.50-6.70 (m, 2H), 5.90 (d, 2H), 3.71-3.89 (m, 6H); MS m/z: 433 (M+H4). Calculated for C22H20N6O4: 432.15
Compound (166): Η NMR (methanol-^) δ (ppm) 8.09-8.21 (m, 2H), 7.30-7.38 (m, IH),
6.95-6.99 (m, IH), 6.69-6.82 (m, 5H), 6.09 (s, IH), 5.88 (s, 2H), 3.70-3.74 (m, 6H); MS m/z: 433 (M+H4). Calculated for C22H20N6O4: 432.15
Compound (167): Η NMR (DMSO-d6) δ (ppm) 13.28 (s, IH), 11.07 (s, IH), 9.27 (s, IH), 8.23-8.34 (m, 2H), 6.79-7.60 (m, 8H), 6.03 (s, 2H), 2.71 (s, 6H); MS m/z: 416 (M+H4).
Calculated for C22H2,N7O2: 415.18
Compound (168): Η NMR (CDC13) δ (ppm) 10.41 (s, IH), 8.19-8.29 (m, 2H), 7.77-7.80 (m, IH), 7.03-7.04 (d, 3H), 6.72-6.76 (m, IH), 6.51-6.55 (m, 2H), 6.14-6.16 (m, IH), 3.64- 3.90 (m, 12H); MS m/z: 449 (M+H4). Calculated for C23H24N6O4: 448.19
Compound (169): Η NMR (DMSO-^) δ (ppm) 9.64 (s, IH), 9.29 (s, IH), 8.91 (s, IH), 8.14-8.33 (m, 2H), 6.73-7.28 (m, 7H), 6.44-6.56 (m, IH), 6.01 (s, 2H), 4.70 (s, 2H), 3.75- 3.78 (d, 3H, J=0.078Hz); MS m/z: 417 (M+H4). Calculated for C22H20N6O3: 416.16
Compound (170): Η NMR (DMSO-^) δ (ppm) 9.37 (s, IH), 8.91-9.02 (d, IH, J=0.33Hz), 8.15-8.36 (m, 3H), 7.45-7.48 (d, 2H), 6.65-6.98 (m, 6H), 6.02 (s, 2H), 4.69 (s, 2H), 3.76 (s, 3H); MS m z: 417 (M+H ). Calculated for C22H20N6O3' 416.16
Compound (171): Η NMR (OMSO-d6) δ (ppm) 13.78 (s, IH), 9.78 (s, IH), 8.46 (s, IH),
7.90-7.97 (m, 2H), 7.31-7.48 (m, 4H), 6.50-6.83 (m, 4H), 5.83 (s, 2H), 4.43 (s, 2H); MS m/z: 412 (M+H4). Calculated for C22H17N7O2: 411.14
Compound (172): Η NMR (OMSO-d6) δ (ppm) 7.70-7.78 (m, 2H), 7.45 (s, IH), 7.30- 7.35(m, IH), 7.0-7.06 (m, 2H), 6.60-6.68 (m, 2H), 5.61-6.12 (m, 3H), 560-5.65(m, IH),
5.06 (s, 2H), 3.77 (s, 2H), 2.93-3.02 (d, 6H); MS m/z: 447 (M+H4). Calculated for C23H22N6O4: 446.17
Compound (173): Η NMR (methanol-^) δ (ppm) 8.16-8.19 (m, IH), 8.01-8.03 (m, IH), 7.20-7.23 (m, 2H), 6.62-6.84 (m, 6H), 5.87 (s, 2H), 4.58(s, 2H), 2.83-2.86 (m, 6H); MS m/z: 430(M+H4). Calculated for C23H23N7O2: 429.19
Compound (174): Η NMR (methanol-^) δ (ppm) 8.02-8.15 (m, 2H), 6.69-6.81 (m, 2H), 6.60-6.67 (m, 4H), 6.04 (s, IH), 5.45-5.87 (m, 2H), 4.57 (s, 2H), 3.69-3.73 (d, 6H); MS m/z: 447 (M+H4). Calculated for C23H22N6O4: 446.17
Compound (175): Η NMR (DMSO-rf ) δ (ppm) 12.20 (s, IH), 8.04-8.23 (m, 4H), 8.81- 7.90 (m, IH), 6.82-6.97 (d, 3H), 6.67-6.78 (m, 2H), 6.44-6.48 (d, IH, J=0.24Hz), 6.01 (s, 2H), 5.55-4.59 (m, 2H), 3.67-3.88 (m, 6H); MS m/z: 447 (M+H4). Calculated for C23H22N6O4: 446.17
Compound (176): Η NMR (Acetone-d6) δ (ppm) 8.78-8.95 (m, 2H), 8.30-8.60 (m, 2H), 7,95 (s, IH), 7.65-7.70 (m, IH), 7.44-7.48 (m, IH), 7.10-7.38 (m, 3H), 6.91-6.98 (m, IH), 6.55-6.65 (m, IH), 3.90 (s, 3H); MS m/z: 399(M+H4). Calculated for C21HI8N8O: 398.16
Compound (177): Η NMR (methanol-G?,) δ (ppm) 8.51 (s, IH), 8.23-8.50 (m, 2H), 7.91- 7.94 (s, IH), 7.61-7.65 (m, IH), 7.40-7.46 (m, 2H), 6.85-7.08 (m, 4H), 3.65 (s, 3H); MS m/z: 399(M+tr). Calculated for C21H18N8O: 398.16
Compound (178): Η NMR (DMSO- 6) δ (ppm) 14.26 (s, IH), 12.96 (s, IH), 11.24 (s,
IH), 10.17 (s, IH), 8.35-8.72 (m, 3H), 7.58-8.14 (m, 6H), 5.05-7.22 (m, 2H); MS m/z: 394(M+H4). Calculated for C21H15N9: 393.15
Compound (179): Η NMR (methanol-^) δ (ppm) 8.45 (s, IH), 8.35-8.37 (d, IH, J=0.057Hz), 8.23-8.26 (m, IH), 7.88-7.91 (m, 2H), 7.61-7.64 ( , IH), 7.04-7.08 (m, IH),
6.87-6.92 (m, 2H), 5.50-6.54 (m, IH), 3.88(s, 3H), 3.77 (s, 3H); MS m/z: 429 (M+H4).
Calculated for C22H20N8O2: 428.15
Compound (180): Η NMR (methanol-^) δ (ppm) 8.55 (s, IH), 8.32-8.38 (m, IH), 8.23- 8.25 (m, IH), 7.90-7.93 (t, IH), 7.60-7.63 (m, IH), 7.34-7.38 (m, 2H), 7.05-7.08 (m, IH),
6.85-6.90 (m, 3H), 2.87-2.94 (m, 6H); MS m/z: 412 (M+H ). Calculated for C22H21N9: 411.19
Compound (181): Η NMR (methanol-^) δ (ppm) 8.496 (s, IH), 8.33-8.35 (m, IH), 8.22- 8.25 (m, IH), 7.906-7.908 (d, IH), 7.779-7.808 (d, IH), 7.607-7.636 (d, IH), 7.01-7.06 (m,
IH), 6.85-6.89 (m, IH), 6.55-6.65 (m, 2H), 3.81-3.89 (m, 6H); MS m/z: 429 (M+H4). Calculated for C22H20N8O2: 428.17
Compound (182): Η NMR (DMSO- d6) δ (ppm) 8.28 (s, IH), 8.16-8.17 (d, IH), 7.71-7.74 (d, 2H), 7.26-7.32 (t, 2H), 6.96-7.01 (t, IH), 6.76-6.85 (m, 3H), 6.13 (s, IH), 3.69-3.78 (m,
6H); MS m/z: 389 (M+H4). Calculated for C21H20N6O2: 388.16
Compound (183): Η NMR (methanol-^) δ (ppm) 8.36 (s, IH), 8.32-8.33 (m, IH), 7.51 (s, IH), 7.16-7.25 (m, 4H), 7.03-7.07 (m, 1H)„ 6.85-6.90 (m, IH), 6.52-6.60 (m, 2H), 3.78- 3.80 (d, 6H); MS m/z: 389 (M+H4). Calculated for C21H20N6O2: 388.16
Compound (184): Η NMR (DMSO-^) δ (ppm) 13.55 (s, IH), 11.0 (s, IH), 9.6 (s, IH), 8.25-8.44 (m, 2H), 7.00-7.91 (m, 11H ); MS m/z: 329(M+H4). Calculated for C19H16N6: 328.14
Compound (185): Η NMR (DMSO-c δ (ppm) 8.34 (s, IH), 7.89 (s, IH), 6.96-7.40 (m, 9H), 6.60 (s, IH), 3.74-3.80(m, 3H); MS m/z: 359(M+H4). Calculated for C20H18N6O: -358.15
Compound (186): Η NMR (DMSO- 6) δ (ppm) 8.13 (s, IH), 6.65-6.88 (m, 4H), 6.51 (s, 2H), 6.35 (s, 2H), 6.05 (s, IH), 4.75 (s, 2H), 3,75 (m, 12H); MS m/z: 463(M+H4). Calculated for C24H26N6O4: 462.20
Compound (187): Η NMR (DMSO-^) δ (ppm) 13.35 (s, IH), 9.45(s, IH), 8.12 (s, 2H),
7.06-7.25 (m, 2H), 6.70-6.74 (m, IH), 6.37-6.51 (m, 4H), 4.70-4.72 (d, 2H), 3.62-3.72 (m, 9H); MS m/z: 433 (M+H4). Calculated for C23H24N6O3: 432.19
Compound (188): Η NMR (DMSO-d6) δ (ppm) 13.25 (s, IH), 9.32 (s, IH), 8.92-8.99 (d, IH), 8.08-8.39 (m, 2H), 7.40-7.45 (m, 2H), 6.38 -6.86 (m, 6H), 4.66-4.73 (m, 2H), 3.68-
3.71 (m, 9H); MS m/z: 433 (M+H4). Calculated for C23H24N6O3: 432.19
Compound (189): Η NMR (OMSO-d6) δ (ppm) 12.17 (s, IH), 8.09-8.66 (m, 4H), 7.85- 7.98 (m, IH), 6.27-6.70 (m, 6H), 4.67-4,70 (m, 2H), 3.65-3.90 (m, 12H); MS m/z: 463 (M+H4). Calculated for C24H26N6O4: 462.20
Compound (190): Η NMR DMSO-d6) δ (ppm) 11.76 (s, IH), 10.02 (s, IH), 8.74 (s, IH), 8.14-8.18 (m, 2H), 7.61 (m, 4H), 6.72-6.77 (m, IH), 6.38-6.52 (m, 3H), 4.70-4.71 (d, 2H), 3.70 (s, 6H); MS m/z: 428 (M+H4). Calculated for C23H21N7O2: 427.18
Compound (191): Η NMR (DMSO- fi) δ (ppm) 8.72 (s, IH), 8.25-8.28 (m, 2H), 8.09-8.16 (m, IH), 7.99-8.00 (d, IH), 7.79-7.80 (t, IH), 6.87-6.96 (m, IH), 6.70-6.75 (m, IH), 6.35- 6.52 (m, 4H), 4.69-4.71 (d, 2H), 3.69-3.83 (m, 12H); MS m/z: 463 (M+H4). Calculated for C24H26N6O4: 462.20
Compound (192): Η NMR (DMSO- d6) δ (ppm) 9.11 (s, IH), 8.42 (s, IH), 8.10-8.20 (d, 2H), 7.31-7.434 (d, 2H), 6.38-6.67 (m, 6H), 4.69 (s, 2H), 3.69 (s, 6H), 2.81 (s, 6H); MS m/z: 446 (M+H4). Calculated for C24H27N7O2: 445.22
Compound (193): Η NMR (DMSO- d6) δ (ppm) 8.58 (s, IH), 8.42-8.45 (m, IH), 8.10-8.25 (m, 2H), 7.67-7.75 (m, IH), 7.30-7.40 (m, 2H), 6.70-6.82 (m, 2H), 6.00-6.05 (d, IH), 4.76- 4.84 (d, 2H), 3.70-3.75 (m, 6H) ; MS m/z: 404 (M+H4). Calculated for C21H21N7O2: 403.18
Compound (194): Η NMR (methanol-d,) δ (ppm) 8.45 (s, IH), 8.26-8.28 (m, IH), 8.07- 8.08 (m, IH), 7.93-7.95 (m, IH), 7.72-7.74 (d, IH), 7.21-7.26 (m, IH), 6.99-7.08 (m, 2H), 6.83-6.86 (m, IH), 6.56-6.61 (m, IH), 6.37-6.40 (m, IH), 4.69 9s, 2H), 3.65 (s, 3H); MS m/z: 374 (M+H4). Calculated for C20H18N7O: 373.18
Compound (195): Η NMR (DMSO-^) δ (ppm) 9.19 (s, IH), 8.60-8.61 (d, IH), 8.43-8.48 (m, IH), 8.12-8.20 (m, 2H), 7.74-7.77 (m, IH), 7.33-7.44 (m, 3H), 6.70-6.85 (m, 3H) 4.77- 4.79 (d, 2H), 3.71 (s, 3H); MS m/z: 374 (M+H4). Calculated for C20Hι8N7O: 373.18
Compound (196): Η NMR (DMSO- 6) δ (ppm) 8.59 (s, IH), 8.44-8.46 (m, 2H), 8.21 (br, s, IH), 8.09-8.10 (m, IH), 7.85-7.88 (m, IH), 7.73-7.88 9m, IH), 7.31-7.36 (m, IH), 6.63- 6.73 (m, 2H), 6.44-6.69 (m, IH), 4.76-4.78 (m, 2H), 3.67-3.84 (m, 6H); MS m/z: 404 (M+H4). Calculated for C21H21N7O2: 403.18
Compound (197): Η NMR (DMSO- 6) δ (ppm) 8.10-8.12 (m, 2H), 7.93-7.94 (m, 2H),
7.71-7.74 (m, IH), 7.30-7.36 (m, IH), 6.92-6.95 (m, IH), 6.71-6.76 (m, IH), 6.43-6.47 (m, IH), 4,78-4.80 (d, 2H), 3.71-3.83 (m, 6H); MS m/z: 404 (M+H4). Calculated for C21H21N7O2: 403.18
Compound (198): Η NMR (DMSO-^) δ (ppm) 10.06 (s, IH), 8.77-8.79 (d, IH), 8.45-
8.47 (m, IH), 8.17-8.26 (m, 2H), 7.66-7.84 (m, 5H), 7.32-7.37 (m, IH), 6.74-6.79 (m, IH), 4.79-4.81 (d, 2H); MS m/z: 369 (M+H4). Calculated for C20H16N8: 368.15
Compound (199): Η NMR (DMSO-d6) δ (ppm) 9.02 (s, IH), 8.60-8.61 (d, IH), 8.46-8.47 (m, IH), 8.10-8.19 (m, 2H), 7.74-7.77 (d, IH), 7.30-7.37 (m, 3H), 6.66-6.74 (m, 3H), 4.77- 4.79 (d, 2H), 2.82 (s, 6H); MS m/z: 387 (M+H4). Calculated for C21H22N8: 386.20
Compound (200): Η NMR (DMSO-c δ (ppm) 10.89 (s, IH), 10.20 (s, IH), 8.33-8.38
(m, IH), 8.01-8.04 (m, IH), 7.22-7.33 (m, 2H), 7.10-7.22 (m, IH), 7.01-7.09 (m, 3H), 6.62-6.69 (m, IH), 6.21-6.25 (m, IH), 3.78-3.87 (m, 9H); MS m/z: 420 (M+H4) Calculated for C22H21N5O4: 419.43
Compound (201): Η NMR (methanol-d,) δ (ppm) 8.40-8.45 (m, 2H), 7.74-7.80 (m, IH),
7.42-7.45 (d, 2H), 7.08-7.35 (m, 3H), 6.94-6.98 (m, 3H), 6.95-6.98 (m, IH), 4.59 (s, 2H), 3.75 (s, 3H); MS m/z: 373 (M+H4). Calculated for C21H20N6O: 372.17
Compound (202): Η NMR (DMSO- 5) δ (ppm) 8.55-9.35 (m, 3H), 7.80-8.20 (m, 2H), 7.20-7.55 (m, 5H), 6.61-6.96 (m, 4H), 4.79 (s, 2H), 3.80 (s, 3H); MS m/z: 373 (M+H4).
Calculated for C21H20N6O: 372.17
Compound (203): Η NMR (DMSO-c δ (ppm) 11.39 (s, IH), 8.00-8.57 (m, 3H), 7.68- 7.98 (m, 6H), 7.36-7.41 (m, 3H), 6.74-6.83 (m, 2H), 4.70-4.72 (d, 2H); MS m/z: 369 (M+H4). Calculated for C21H17N7: 367.15
Compound (204): Η NMR (DMSO-cQ δ (ppm) 10.69 (s, IH), 7.75 (s, IH ), 7.60-7.62 (d, IH), 7.23-7.32 (m, 5H), 7.11-7.13 (d, IH), 6.92-6.95 (d, 2H), 6.82-6.85 (d, IH), 6.71-6.76 (t, IH), 6.58-6.61 (d, IH), 4.46-4.48 (d, 2H), 3.74-3.76 (d, 6H); MS m/z: 403 (M+H4). Calculated for C23H22N4O3: 402.17
Compound (205): Η NMR (DMSO- 6) δ (ppm) 10.71 (s, IH), 7.82-7.88 (t, IH), 7.60-7.65 (m, IH), 7.10-7.46 (m, 8H), 6.70-6.85 (m, 2H), 6.55-6.62 (m, IH), 5.54-4.58 (m, 2H), 3.78 (s, 3H); MS m/z: 391 (M+H4). Calculated for C22H19FN4O2: 390.15
Compound (206): Η NMR (OMSO-d6) δ (ppm) 7.91-7.94 (t, IH), 7.60-7.62 (d, IH), 7.10-7.41 (m, 8H), 6,67-6.77 (m, 2H), 6.55-6.58 (m, IH), 4.57-4.59 (m, IH), 4,57-4.59 (d, 2H), 3.76 (s, 3H); MS m z: 407 (M+H4). Calculated for C22H19ClN4O2: 406.12
Compound (207): !H NMR (OMSO-d6) δ (ppm) 8.24-8.26 (m, IH), 8.08-8.12 (t, IH),
7.86-7.89 (m, IH), 6.77-7.00 (m, 6H), 6.19-6.20 (t, IH), 6.00 (s, 2H), 3.74 (s, 6H); MS m/z: 404 (M+H4). Calculated for C22H21N5O3: 403.16
Compound (208): MS m/z: 380 (M+H )
Compound (209): Η NMR (CDC13) δ (ppm) 8.32-8.40 (m, 2H), 7.10-7.26 (m, 2H), 6,70- 6.89 (m, 3H), 4.09-4.12 (m, 2H), 3.83 (s, 3H), 3.10-3.17 (m, 4H), 2.54-2.60 (m, 4H), 2.27 (s, 3H); MS m/z: 365 (M+H4)
Compound (210): MS m/z: 395 (M+H4)
Compound (211): Η NMR (OMSO-d6) δ (ppm) 10.63 (s, IH), 7.54-7.55 (d, IH), 7.12- 7.52 (m, 4H), 6.87-6.90 (d, IH), 6.59-6.70 (m, 4H), 3.77 (s, 3H); MS m/z: 283 (M+H ). Calculated for C15Hι4N402: 282.11
Compound (212): Η NMR (methanol-^) δ (ppm) 7.83-7.84 (d, IH), 7.70-7.73 (d, IH), 7.12-7.18 (m, IH), 6.81-6.88 (m, 2H), 6.70-6.73 (m, IH), 6.41-6.45 (m, IH), 3.77-3.87 (d, 6H); MS m/z: 312 (M+H4). Calculated for C16H17N5O2: 311.14
Compound (213): Η NMR (methanol- 4) δ (ppm) 7.71-7.74 (d, IH), 7.30-7.33 (d, 2H),
7.09-7.15 (m, IH), 6.79-6.84 (m, 3H), 6.66-6.71 (m, IH), 2.83-2.85 (d, 6H); MS m/z: 295 (M+H ). Calculated for C16H18N6: 294.16
Compound (214): Η NMR (methanol-^) δ (ppm) 8.28-8.29 (m, IH), 8.10-8.12 (m, IH), 7.84-7.97 (m, 2H), 7.95-7.05 (m, 3H), 3.74-3.79 (m, 4H), 2.52-2.54 (m, 4H), 2.24 (s, 3H); MS m/z: 351 (M+H4)
Compound (215): Η NMR (methanol-c δ (ppm) 8.15-8.22 (m, 3H), 7.82-7.90 (m, IH),
7.50-7.63 (m, 3H), 6.90-6.94 (m, IH), 3.25-3.45 (m, 4H), 2.43-2.48 (m, 4H), 2.15-2.20 (m, 3H); MS m/z: 389(M+H4)
Compound (216): Η NMR (CDC13) δ (ppm) 8.33-8.36 (m, IH), 8.06-8.09 (m, IH), 7.71- 7.74 (m, IH), 7.63 (s, IH), 6.83-6.89 (m, 2H), 6.03 (s, 2H), 3.28-3.32 (m, 4H), 2.46-2.50
(m, 4H), 2.34 (s, 3H); MS m/z: 365 (M+H4)
2-[5-(3-1rifluoromethylphenyl)-lH-imidazol-2-yl]aniline (2l7): See Example 13.
2-(5-(3-Trifluoromethoxyphenyl)-lH- 1.2.41-triazol-3-yl)aniline (218): MS m/z: 321
(M+H4) 2.65g (49%). This material was recrystallised from methanol/water.
2-(5-(2-Chlorophenyl)-lH-[1.2.41-triazol-3-yl)aniline (219): MS m/z: 272 (M+H4) 0.965g (21%). This material was recrystallised from ethanol/water.
2-(5-(3-Methoxyphenyl)-lH-[l .2.4]-triazol-3-yl)aniline (220): MS m/z: 267 (M+H4) 1.2g (28%). This material was recrystallised from methanol.
2-(5-(4-bromophenyl)-lH-[l .2.4]-triazol-3-yl)aniline (221): MS m/z: 316 (M+H4) 1.08g (24%). This material was recrystallised from methanol.
2-(5-(3-bromophenyl)-lH-[1.2.4]-triazol-3-yl)aniline (222): MS m/z: 316 (M+H4) - 1.97g (37%). This material was recrystallised from methanol.
2-(5-(4-Trifluoromethoxyphenyl)-lH-[ 1.2.41-triazol-3-yl)aniline (224): MS m/z: 321 (M+H4); 1.15g (25%). This material was recrystallised from ethanol/water.
2-(5-(4-Trifluoromethylphenyl)-lH-ri.2.41-triazol-3-yl)aniline (225): MS m/z: 305 (M+H+); 1. Ig (37%). This material was recrystallised from methanol.
2-(5-(3-Trifluoromethylρhenyl)- lH-[ 1.2.41-triazol-3-yl)aniline (226): MS m z: 305 (M+H ); l.lg (30%). This material was recrystallised from methanol.
Example 19
Homogeneous Time-Resolved Fluorescence (HTRF) Assay for VEGFR-2 (KDR) Kinase Inhibition
Representative compounds of the invention were tested for their inhibition of VEGFR (KDR) tyrosine kinase. The percent inhibition and IC50 values of selected compounds were determined. These results are summarized in Table 5.
Table 5
KDR Enzymatic Assay Cmpd. No. % Inhibition IC50 (μM)
1 91
2 67
3 85
4 69
5 27
6 41
7 18
8 34
9 60
11 71
12 55
13 34
14 26
Cmpd. No. % Inhibition IC50(μM)
15 83
16 90
17 19
18 70
19 77 4
20 52
21 8
22 61 3.6
23 75 2.3
10 24 70 1.3
25 74 1.8
26 86
27 72 0.52
28 69 2.3
15 34 66
42 66
50 31
51 48 15
52 53 11
20 53 52 11
54 20
55 64 4.5
56 50
58 40
25 60 69 4.1
61 4.1
62 79 1.8
64 10
76 62
30 77 40
78 7
79 17
87 18
88 39
35 89 67
90 78
91 71
93 26
102 65
Cmpd. No. % Inhibition IC50 (μM)
117 10
118 81
119 11
120 3
121 6
125 12
126 12
127 6
128 4
10 129 8
130 4
132 8
133 5
134 5
15 135 5
136 3
137 24
138 43
139 7
20 140 4
141 9
142 3
143 24
144 25
25 145 3
146 85
147 0.27
148 30
149 33
30 150 245
151 90
152 8
155 19
156 39
35 157 18
159 25
160 28
161 42
162 11
40 163 14
Cmpd. No. % Inhibition ICso (μM)
164 51
165 51
166 72
167 47.5
168 46
169 55
170 63
171 55
172 61
10 173 58
174 59
175 72
176 82
178 35
15 179 70
180 84
181 84
182 9
183 29
20 185 73
186 18
187 12
188 18
189 41
25 190 14
191 41
192 23
193 79
194 81
30 195 85
196 81
200 -2
201 74
202 81
35 203 42
204 14
208 - 2 •
212 54
216 20
40
VEGFR tyrosine kinase inhibition is determined by measuring the phosphorylation level of poly-Glu-Ala-Tyr-biotin (pGAT-biotin) peptide in a Homogeneous Time- Resolved Fluorescence (HTRF) assay. Into a black 96-well Costar plate is added 2 μl/well of 25x compound in 100% DMSO (final compound concentration in the 50-μl kinase reaction is typically 1 nM to 10 μM). Next, 38 μl of reaction buffer (25 mM Hepes pH 7.5, 5 mM MgC12, 5 mM MnC12, 2 mM DTT, 1 mg/ml BSA) containing 0.5 pmoles polyGAT-biotin and 3-4 ng KDR enzyme is added to each well. After 5-10 min preincubation, the kinase reaction is initiated by the addition of 10 μl of 10 μM ATP in reaction buffer, after which the plate is incubated at room temperature for 45 min. The reaction is stopped by the addition of 50 μl of KF buffer (50 mM Hepes pH 7.5, 0.5 M KF,
1 mg/ml BSA) containing 100 mM EDTA and 0.36 μg/ml PY20K (Eu-cryptate labeled anti-phosphotyrosine antibody, CIS bio international). After 30 min, 100 μl of 5 nM SV- XL (modified- APC-labeled Streptavidin, CIS bio international) in KF buffer is added, and after an addition 2-hr incubation at room temperature, the plate is read in a RUBYstar HTRF Reader.
Example 20 Cell-based assay for VEGFR-2 (KDR) Kinase Inhibition Representative compounds of the invention were tested using the ELISA Cell-based assay for KDR inhibition. The results are reported in Table 6.
Table 6
Cell-Based Phosphorylation
Cmpd. No. % Inhibition IC50 (μM)
1 7 788 14
2 34
3 38
4 50 30
5 10
6 30
7 58 30
9 47
Cmpd. No. % Inhibition IC50(μM)
10 36
11 0
12 70 5
13 63 30
14 15
15 75 10
16 91 10
17 76 30
18 77 1
10 76 53 10
77 15
78 0
156 70
157 57
15 159 67
161 68
164 69 20
165 60
166 68
20 167 62
168 71
169 63
170 67
171 67
25 172 70 4.7
173 68
174 68 5.6
175 64
176 66 2.6
30 179 68
180 59
181 66 1.1
201 64
202 60
35 203 19
205 9
207 193
212 65
Transfection of 293 cells with DNA expressing FGFR1/KDR chimera: DNA for transfection was diluted to a final concentration of 5 μg/ml DNA in 1XBBS, 125 mM CaCl2 and incubated at room temperature for 30 min. 293 cells were seeded in 15 cm tissue culture plates using 2 xlO7 cells per plate and incubated for 4 hrs, followed by dropwise addition of 3 ml of DNA solution. The plates were incubated overnight.
The next morning, the cells were trypsinized, collected by centrifugation, resuspended (4 x 105 cell/ml), divided into wells of 48 well tissue culture plates (1 ml/well) and incubated overnight. Compounds of the invention were added to individual wells to a final concentration of 10-30 μM and incubated for 2 hours. Generally, 10 mM stock solution were diluted 1/300-1/1000, yielding a final DMSO concentration of
0.1-0.3%. Cells were lysed by resuspension in Lysis buffer (150 mM NaCI, 50 mM Hepes pH 7.5, 0.5% Trition X-100, 10 mM NaPPi, 50 mM NaF, 1 mM NajVOJ and rocked for lh at 4°C. Cells were lysed by adding in 100 μl lysis buffer per well.
ELISA for Detection of Tyrosine-phosphorylated Chimeric Receptor: 96 well ELISA plates were coated using 100 μl/well of 10 μg/ml αFGFRl, and incubated overnight at 4°C. αFGFRl is prepared in a buffer made with 16 ml 0.2M Na2CO3 and 34 ml 0.2M NaHCO3 and the pH adjusted to 9.6. Concurrent with lysis of the transfected cells, αFGFRl coated ELISA plates are washed three times with PBS+0.1% Tween-20, blocked by addition of 200 μl/well of 3% BSA in PBS and incubated for lh. Blocking solution is removed from the wells. 80 μl of lysate is then transferred to the coated and blocked wells and incubated for lh at 4°C. The plates are washed three times with PBS+0.1% Tween-20.
To detect bound phosphorylated chimeric receptor, 100 μl of anti-phosphotyrosine antibodies (RC20:HRPO, Transduction Laboratories) were added per well (final concentration 0.5 μg/ml in PBS) and incubated for lh. The plates were washed six times with PBS+0.1% Tween-20. Enzymatic activity of HRP was detected by adding 50 μl/well of equal amounts of the Kirkegaard & Perry Laboratories (KPL) Substrate A and Substrate B. (KPL cat. #54-61-0). The reaction was stopped by the addition of 50 μl/well 0. IN H2SO4 and absorbance is detected at 450nm
The above examples are intended to be illustrative only, hi particular, the invention is not intended to be limited to the methods, protocols, conditions and the like specifically recited herein, insofar as those skilled in the art would be able to substitute other conditions, methods, amounts, materials, etc. based on the present disclosure to arrive at compounds within the scope of this disclosure. While the present invention is described with respect to particular examples and preferred embodiments, the present invention is not limited to these examples and embodiments. In particular, the compounds of the present invention are not limited to the exemplary species' recited herein. Moreover, the methods of the present invention are not limited to treating only the exemplified diseases and conditions, but rather any disease or condition that may be treated by regulation of kinases. Additionally, the methods of synthesis of the present invention are not limited to the methods exemplified in the example. The methods of the present invention include methods of making any of the compounds set forth in the present invention that those skilled would be able to make in view of the present disclosure, and are not limited to the exemplified method. For example, methods encompassed by the present invention may involve the use of a different starting material depending on the desired final compound, different amounts of various ingredients, or substitution of different ingredients such as other reactants or catalysts that would be suitable depending on the starting material and result to be achieved.