WO2004050011A2 - High enantiomeric purity dexanabinol for pharmaceutical copositions - Google Patents
High enantiomeric purity dexanabinol for pharmaceutical copositions Download PDFInfo
- Publication number
- WO2004050011A2 WO2004050011A2 PCT/IL2003/001023 IL0301023W WO2004050011A2 WO 2004050011 A2 WO2004050011 A2 WO 2004050011A2 IL 0301023 W IL0301023 W IL 0301023W WO 2004050011 A2 WO2004050011 A2 WO 2004050011A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- dexanabinol
- compound
- enantiomer
- pharmaceutical composition
- over
- Prior art date
Links
- 0 CCCCCCC(C)(C)c1cc(OC(C)(C)c2cc*(CO)cc2-2)c-2c(O)c1 Chemical compound CCCCCCC(C)(C)c1cc(OC(C)(C)c2cc*(CO)cc2-2)c-2c(O)c1 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D311/78—Ring systems having three or more relevant rings
- C07D311/80—Dibenzopyrans; Hydrogenated dibenzopyrans
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/35—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom
- A61K31/352—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom condensed with carbocyclic rings, e.g. methantheline
- A61K31/353—3,4-Dihydrobenzopyrans, e.g. chroman, catechin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/10—Alcohols; Phenols; Salts thereof, e.g. glycerol; Polyethylene glycols [PEG]; Poloxamers; PEG/POE alkyl ethers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/16—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing nitrogen, e.g. nitro-, nitroso-, azo-compounds, nitriles, cyanates
- A61K47/18—Amines; Amides; Ureas; Quaternary ammonium compounds; Amino acids; Oligopeptides having up to five amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/44—Oils, fats or waxes according to two or more groups of A61K47/02-A61K47/42; Natural or modified natural oils, fats or waxes, e.g. castor oil, polyethoxylated castor oil, montan wax, lignite, shellac, rosin, beeswax or lanolin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4858—Organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/08—Drugs for disorders of the alimentary tract or the digestive system for nausea, cinetosis or vertigo; Antiemetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
Definitions
- the present invention relates to a synthetic cannabinoid, dexanabinol, of high enantiomeric purity, to pharmaceutical grade compositions comprising it, and uses thereof.
- Stereoisomers are compounds made up of the same atoms bonded by the same sequence of bonds but having different three-dimensional structures, which are not interchangeable. These three-dimensional structures are called configurations, e.g. R and S.
- Optically active compounds which have one chiral atom or more, exist as two or more isomers, called enantiomers.
- Enantiomers are mirror images of one another and have identical physical properties, except for the fact that they rotate the plane of polarized light in opposite directions, (+) clockwise for the dextro isomer and (-) counterclockwise for the levo isomer. Likewise, they have identical chemical properties except when interacting with stereospecif ⁇ c compounds.
- ⁇ 9 -tetrahydrocannabinol ⁇ 9 -THC
- the absolute configuration of ⁇ 9 -THC was established by Mechoulam e al. in 1967 and found to be of (-)-(3R,4R) stereochemistry. It was later found that the psychotropic activity of cannabinoids resides in the natural (3R,4R) series, while the opposite enantiomeric synthetic series (3S,4S) was free of these undesirable effects.
- the group of Mechoulam and coworkers also achieved the synthesis of THC (for review see Mechoulam R. and Hanus L., Chem.
- the chirality of the starting material, ⁇ -pinene determines the chirality of the final compound.
- (+)- ⁇ -pinene will yield (1S,5R) myrtenol, and corresponding derivatives, down to classical cannabinoid analogs of the (3S,4S) configuration.
- (-)- ⁇ -pinene will yield (1R,5S) myrtenol, and corresponding derivatives, down to classical cannabinoid analogs of the (3R,4R) configuration, as shown in scheme 2.
- the terpenic ring was the basis for the numbering system, and the chiral centers of THC type cannabinoids were designated at carbon atoms 3 and 4.
- THC that was previously described as ⁇ ! -THC was later renamed ⁇ 9 - THC, similarly ⁇ 6 -THC was renamed ⁇ 8 -THC, and the chiral centers are at carbons 6a and 10a.
- cannabinoids trigger additional physiological reactions, the cardiovascular effects harboring some of the more significant consequences.
- cardiovascular effects In humans, the most consistent cardiovascular effects of ⁇ 9 -THC are peripheral vasodilatation and tachycardia. These effects manifest themselves as an increase in cardiac output, increased peripheral blood flow and variable changes in blood pressure. It has been postulated that cannabinoids induce a CNS mediated increase in sympathetic and parasympathetic nerve activity, which would result in abnormal cardiovascular outputs. More recent evidence implicates peripheral site of actions, such as receptors located on sympathetic nerve terminals, receptors located in vascular tissues or in heart muscle, or a combination of all of the above.
- HU-210 is several fold more potent than ⁇ 9 -THC, in reducing psychomotor function, interfering with cognitive functions, inducing endocrine alterations, interfering or suppressing immune function, altering neurochemical development, and impairing emotional response due to anxiogenic activity.
- HU-210 has also been found to inhibit sexual behavior, to induce dependence and to have anorexic effects.
- dexanabinol ⁇ 6 -tetrahydrocannabinol, was disclosed in US 4,876,276 and subsequently assigned the trivial chemical name dexanabinol (CAS number: 112-924-45-5).
- potential therapeutic applications of dexanabinol included known attributes of marijuana itself such as anti-emesis, analgesia, and anti-glaucoma, as disclosed in US Patent No. 4,876,276.
- dexanabinol and its derivatives especially neuroprotective properties. It was later established that novel synthetic compounds could block the NMDA receptor, as disclosed in US Patent Nos. 5,284,867, 5,521,215 and 6,096,740.
- the capacity of dexanabinol and some of its analogues to block glutamate neurotoxicity has therapeutic implications for treating acute injuries to the central nervous system, including mechanical trauma, prolonged seizures, deprivation of glucose supply, and compromised blood supply (e.g. cardiac arrest or stroke), as well as chronic degenerative disorders characterized by neuronal loss (e.g. Alzheimer's disease, Huntingdon's chorea, and Parkinson's disease), and poisoning affecting the central nervous system (e.g. strychnine, picrotoxin and organophosphorous poisoning).
- neuronal loss e.g. Alzheimer's disease, Huntingdon's chorea, and Parkinson's disease
- poisoning affecting the central nervous system e.g. strychnine, picrotoxin and organophospho
- Dexanabinol and its analogues appear to share anti-oxidative, immunomodulatory and anti-inflammatory properties in addition to their capacity to block the NMDA receptor, as disclosed in US Patent Nos. 5,932,610, 6,331,560 and 6,545,041.
- the convergence of such diverse and crucial therapeutic activities in the dexanabinol molecule made it an excellent candidate for prevention or treatment of a variety of clinical conditions.
- the neuroprotective effects of dexanabinol are being assessed in clinical trials. One trial is being conducted to determine the efficacy of dexanabinol in patients suffering from traumatic brain injuries (TBI), while in another trial dexanabinol is administered during surgical procedures to assess its preventive or amelioratory effect on post-operative cognitive impairment.
- TBI traumatic brain injuries
- THC-type compounds allows for stereochemical purification through recrystallization at two steps, for the 4-oxo-myrtenyl-pivalate and for the final compound. This observation made possible the synthesis on a laboratory scale of the enantiomers in e.e. of 99.8%, as determined by HPLC analysis. Small-scale preparation of HU-211 opened the way to the study of its properties in numerous in vitro and in vivo systems. This research led to the discovery of HU-211 multifaceted therapeutic characteristics which have been above described.
- the quantitative criterion of the minimum acceptable degree of optical purity of an intended therapeutic enantiomer is dictated by the pharmacological potency of the contamination. The higher the psychotropic activity of the enantiomer, the stricter the requirement for optical purity.
- the enantiomeric pair HU-210 and HU-211 is an extreme case in point and the highly potent psychotropic effects of HU-210 require that HU-211 should be of very high enantiomeric purity.
- therapeutic dosages for humans have been shown to range from tens to hundreds of milligrams per subject, requiring that for pharmaceutical use HU-211 must actually be of enantiomeric purity even higher than any reported previously.
- the present invention now provides enantiomerically pure dexanabinol for use as an active ingredient in pharmaceutical compositions for clinical applications.
- the present invention encompasses a compound of formula (I):
- this compound or its pharmaceutically acceptable salt, ester or solvate is in enantiomeric excess of at least 99.92% over the (3R,4R) enantiomer. More preferably, the compound of formula (I) or its pharmaceutically acceptable salt, ester or solvate, is in enantiomeric excess of at least 99.95%) over the (3R,4R) enantiomer.
- the compound of formula (I) or its pharmaceutically acceptable salt, ester or solvate is in enantiomeric excess of at least 99.91% over the (3R,4R) enantiomer.
- the present invention provides a compound of formula (I) as above defined, wherein the absolute enantiomeric amount of the (3S,4S) enantiomer, or a pharmaceutically acceptable salt, ester or solvate thereof, is at least 99.95%) and the (3R,4R) enantiomer is 0.05%) or less.
- the compound of formula (I) or its pharmaceutically acceptable salt, ester or solvate is present in absolute enantiomeric amount of at least 99.96% whereas the (3R,4R) enantiomer is 0.04% or less. More preferably, the compound of formula (I) or its pharmaceutically acceptable salt, ester or solvate, is present in absolute enantiomeric amount of at least 99.97% whereas the (3R,4R) enantiomer is 0.03% or less. Most preferably, the compound of formula (I) or its pharmaceutically acceptable salt, ester or solvate, is present in absolute enantiomeric amount of at least 99.98% whereas the (3R,4R) enantiomer is 0.02%> or less.
- the present invention further encompasses pharmaceutical compositions comprising as an active ingredient dexanabinol, a compound of formula (I):
- this active ingredient is in enantiomeric excess of at least 99.92% over the (3R,4R) enantiomer. More preferably, this active ingredient is in enantiomeric excess of at least 99.95%> over the (3R,4R) enantiomer. Most preferably, this active ingredient is in enantiomeric excess of at least 99.97%) over the (3R,4R) enantiomer.
- the present invention further encompasses pharmaceutical compositions comprising as an active ingredient dexanabinol, a compound of formula (I) as above defined, wherein the absolute enantiomeric amount of the (3S,4S) enantiomer, or a pharmaceutically acceptable salt, ester or solvate of this compound, is at least 99.95%> and the (3R,4R) enantiomer is 0.05%> or less.
- the compound of formula (I) or its pharmaceutically acceptable salt, ester or solvate is present in absolute enantiomeric amount of at least 99.96% whereas the (3R,4R) enantiomer is 0.04%) or less.
- the compound of formula (I) or its pharmaceutically acceptable salt, ester or solvate is present in absolute enantiomeric amount of at least 99.97%) whereas the (3R,4R) enantiomer is 0.03% or less.
- the compound of formula (I) or its pharmaceutically acceptable salt, ester or solvate is present in absolute enantiomeric amount of at least 99.98% whereas the (3R,4R) enantiomer is 0.02% or less.
- the present invention also relates to pharmaceutical compositions comprising as an active ingredient enantiomerically pure dexanabinol, having the (3S,4S) configuration and being in enantiomeric excess of at least 99.90% over the (3R,4R) enantiomer, or a pharmaceutically acceptable salt, ester or solvate of the compound as above defined, and further comprising a pharmaceutically acceptable diluent, carrier or excipient necessary to produce a physiologically acceptable and stable formulation.
- the enantiomerically pure dexanabinol, or its pharmaceutically acceptable salt, ester or solvate is in enantiomeric excess of at least 99.92% over the (3R,4R) enantiomer.
- the enantiomerically pure dexanabinol, or its pharmaceutically acceptable salt, ester or solvate is in enantiomeric excess of at least 99.95%> over the (3R,4R) enantiomer.
- the enantiomerically pure dexanabinol, or its pharmaceutically acceptable salt, ester or solvate is in enantiomeric excess of at least 99.97%) over the (3R,4R).
- the present invention also relates to pharmaceutical compositions comprising as an active ingredient enantiomerically pure dexanabinol, or a pharmaceutically acceptable salt, ester or solvate of the compound as above defined, having the (3S,4S) configuration and being present in absolute enantiomeric amount of at least 99.95%), and further comprising a pharmaceutically acceptable diluent, carrier or excipient necessary to produce a physiologically acceptable and stable formulation.
- the enantiomerically pure dexanabinol, or its pharmaceutically acceptable salt, ester or solvate is present in absolute enantiomeric amount of at least 99.96% whereas the (3R,4R) enantiomer is 0.04%> or less.
- the enantiomerically pure dexanabinol, or its pharmaceutically acceptable salt, ester or solvate is present in absolute enantiomeric amount of at least 99.97% whereas the (3R,4R) enantiomer is 0.03% or less.
- the enantiomerically pure dexanabinol, or its pharmaceutically acceptable salt, ester or solvate is present in absolute enantiomeric amount of at least 99.98% whereas the (3R,4R) enantiomer is 0.02%> or less.
- the pharmaceutical compositions can be administered by any conventional and appropriate route including oral, parenteral, intravenous, intramuscular, subcutaneous, transdermal, intrathecal, rectal or intranasal.
- the pharmaceutical compositions Prior to their use as medicaments for preventing, alleviating or treating an individual in need thereof, the pharmaceutical compositions may be formulated in unit dosage form.
- the selected dosage of active ingredient depends upon the desired therapeutic effect, the route of administration and the duration of treatment desired.
- a further embodiment of the present invention provides a method of preventing, alleviating or treating a patient for indications including but not limited to acute neurological disorders, chronic degenerative diseases, CNS poisoning, cognitive impairment, inflammatory diseases or disorders, autoimmune diseases or disorders, pain, emesis, glaucoma and wasting syndromes, by administering to said patient a prophylactically and/or therapeutically effective amount of one of the enantiomerically pure dexanabinol compounds described herein or a pharmaceutical composition that contains such compounds as above defined wherein enantiomerically pure dexanabinol is in enantiomeric excess of at least 99.90% over the (3R,4R) enantiomer.
- the enantiomerically pure dexanabinol, or its pharmaceutically acceptable salt, ester or solvate is in enantiomeric excess of at least 99.92% over the (3R,4R) enantiomer. More preferably, the enantiomerically pure dexanabinol, or its pharmaceutically acceptable salt, ester or solvate, is in enantiomeric excess of at least 99.95% over the (3R,4R) enantiomer. Most preferably, the enantiomerically pure dexanabinol, or its pharmaceutically acceptable salt, ester or solvate, is in enantiomeric excess of at least 99.91% over the (3R.4R) enantiomer.
- a further embodiment of the present invention provides a method of preventing, alleviating or treating a patient for indications including but not limited to acute neurological disorders, chronic degenerative diseases, CNS poisoning, cognitive impairment, inflammatory diseases or disorders, autoimmune diseases or disorders, pain, emesis, glaucoma and wasting syndromes, by administering to said patient a prophylactically and/or therapeutically effective amount of one of the enantiomerically pure dexanabinol compounds described herein or a pharmaceutical composition that contains such compounds as above defined wherein enantiomerically pure dexanabinol is present in absolute enantiomeric amount of at least 99.95% whereas the (3R,4R) enantiomer is 0.05% or less.
- the enantiomerically pure dexanabinol, or its pharmaceutically acceptable salt, ester or solvate is present in absolute enantiomeric amount of at least 99.96% whereas the (3R,4R) enantiomer is 0.04% or less. More preferably, the enantiomerically pure dexanabinol, or its pharmaceutically acceptable salt, ester or solvate, is present in absolute enantiomeric amount of at least 99.97% whereas the (3R,4R) enantiomer is 0.03% or less.
- the enantiomerically pure dexanabinol, or its pharmaceutically acceptable salt, ester or solvate is present in absolute enantiomeric amount of at least 99.98% whereas the (3R,4R) is 0.02% or less.
- a further embodiment of the present invention provides use for the manufacture of a medicament for preventing, alleviating or treating acute neurological disorders, chronic degenerative diseases, CNS poisoning, cognitive impairment, inflammatory diseases or disorders, autoimmune diseases or disorders, pain, emesis, glaucoma and wasting syndromes, of one of the enantiomerically pure dexanabinol compounds described herein wherein enantiomerically pure dexanabinol is in enantiomeric excess of at least 99.90%) over the (3R,4R) enantiomer.
- the enantiomerically pure dexanabinol, or its pharmaceutically acceptable salt, ester or solvate is in enantiomeric excess of at least 99.92% over the (3R,4R) enantiomer. More preferably, the enantiomerically pure dexanabinol, or its pharmaceutically acceptable salt, ester or solvate, is in enantiomeric excess of at least 99.95% over the (3R,4R) enantiomer. Most preferably, the enantiomerically pure dexanabinol, or its pharmaceutically acceptable salt, ester or solvate, is in enantiomeric excess of at least 99.97% over the (3R,4R) enantiomer.
- a further embodiment of the present invention provides use for the manufacture of a medicament for preventing, alleviating or treating acute neurological disorders, chronic degenerative diseases, CNS poisoning, cognitive impairment, inflammatory diseases or disorders, autoimmune diseases or disorders, pain, emesis, glaucoma and wasting syndromes, of one of the enantiomerically pure dexanabinol compounds described herein wherein enantiomerically pure dexanabinol is present in absolute enantiomeric amount of at least 99.95%> whereas the (3R,4R) enantiomer is 0.05%» or less.
- the enantiomerically pure dexanabinol, or its pharmaceutically acceptable salt, ester or solvate is present in absolute enantiomeric amount of at least 99.96% whereas the (3R,4R) enantiomer is 0.04%> or less. More preferably, the enantiomerically pure dexanabinol, or its pharmaceutically acceptable salt, ester or solvate, is present in absolute enantiomeric amount of at least 99.97%) whereas the (3R,4R) enantiomer is 0.03%> or less.
- the enantiomerically pure dexanabinol, or its pharmaceutically acceptable salt, ester or solvate is present in absolute enantiomeric amount of at least 99.98% whereas the (3R,4R) is 0.02% or less.
- Figure 1 shows expanded HPLC chromatograms of four pharmaceutical grade, large scale, batches of enantiomerically pure dexanabinol.
- Figure 2 shows the profile of dexanabinol plasma concentration along time, following single or multiple injections of specified doses in the various species tested.
- Figure 3 shows the profile of dexanabinol concentrations in plasma and brain of rats inj ected with 4 mg/kg of the drug.
- the present invention provides ultrapure dexanabinol characterized by an enantiomeric excess of at least 99.90%, preferably 99.92%, more preferably 99.95% and most preferably 99.97%, for use as an active pharmaceutical ingredient in compositions for clinical applications.
- the enantiomerically pure dexanabinol of the present invention is further characterized by an absolute enantiomeric amount of at least 99.95%, preferably 99.96%, more preferably 99.91% and most preferably 99.98%.
- the respective absolute enantiomeric amount of HU-210 is 0.05% or less, preferably 0.04% or less, more preferably 0.03%) or less and most preferably 0.02% or less.
- enantiomerically pure when referring to compound of formula (I), is found in the composition in greater proportion in relation to its mirror image.
- the proportion between two enantiomers can be expressed either by the enantiomeric excess or by the absolute proportion of each enantiomer.
- enantiomeric excess represents the percent excess of one enantiomer over the other and is calculated using the following equation:
- Percent e.e. 100*([enantiomer l]-[enantiomer 2])/([enantiomer l]+[enantiomer 2]).
- the formula used to calculate the enantiomeric excess of dexanabinol over HU-210 is 100*([HU-211]-[HU-210])/([HU-211]+[HU-210]), wherein the concentration of the enantiomers is determined by HPLC and expressed as percent weight by weight.
- absolute enantiomeric amount represents the percent of each enantiomer and is calculated using the following equation:
- Absolute enantiomeric amount 100* [enantiomer l]/([enantiomer l]+[enantiomer 2]), wherein the concentration of the enantiomers is determined by HPLC and expressed as percent weight by weight.
- the enantiomeric purity of the active ingredient is determined by types of tests known in the art, for example chiral HPLC methods and reverse phase HPLC.
- the present invention required development of novel modified chiral HPLC methods (adapted from Levin S. et al., Journal of Chromatography A. 654: 53-64, 1993) exemplified hereinbelow, in conjunction with RP-HPLC.
- Analytical methods previously disclosed in the art were not validatable and did not provide reliable and reproducible results.
- the scaled up synthetic procedures according to the present invention generally adhere to the synthetic schemes used previously, with modifications to enable good manufacturing practice.
- the improvements implemented were required to obtain pharmaceutical grade dexanabinol reproducibly and with the required elevated standard of enantiomeric purity.
- Patent No. 4,876,276, are evident to persons skilled in the art and include scale-up ability, improved yield, simplified process, reduced use of toxic chemicals or dangerous reagents all leading to a safer and more cost effective production.
- the synthetic process for the preparation of dexanabinol combines two approaches for obtaining the desired enantiomer; first the utilization of enantiomerically enriched starting material, namely (+)- ⁇ -pinene, in a stereoselective multistep synthesis and then the separation of the partially resolved racemic mixture into their enantiomeric constituent using crystallization.
- enantiomerically enriched starting material namely (+)- ⁇ -pinene
- the conditions to achieve enantiomeric separation depend whether the object of the process is to maximize recovery and/or purity and they include parameters such as the composition of the solvent, the concentration, and the temperature. These parameters can be determined by one skilled in the art of recrystallization using the particular compound of which separation of the enantiomers is desired. Such additional solvents or mixtures of solvents for purifying the (3S,4S) enantiomer are embraced in the invention herein.
- the purity can be increased, if necessary, by repeating the final crystallization step with acetonitrile. Additional round(s) of recrystallization is a standard procedure and expected necessity if the initial purity is not adequate and does not fall within the specifications defined by the intended use of the product. Such additional means for purifying the (3S,4S) enantiomer are embraced in the invention herein.
- Dexanabinol is capable of further forming pharmaceutically acceptable salts and esters.
- “Pharmaceutically acceptable salts and esters” means any salt and ester that is pharmaceutically acceptable and has the desired pharmacological properties. Such salts include salts that may be derived from an inorganic or organic acid, or an inorganic or organic base, including amino acids, which is not toxic or otherwise unacceptable.
- the present invention also includes within its scope solvates of dexanabinol and salts thereof, for example, hydrates.
- prodrug represents compounds which are rapidly transformed in vivo to dexanabinol, for example by hydrolysis in the blood. All of these pharmaceutical forms are intended to be included within the scope of the present invention.
- Water-soluble derivatives of dexanabinol were synthesized and investigated over the years. They can be used as prodrugs, or active analogs depending on their hydrolytic and enzymatic stability and on their intrinsic activity.
- the two hydroxyl groups present in the dexanabinol molecule were targeted for modifications and various polar combinations or combinations bearing a permanent charge were synthesized as esters at the allylic or phenolic hydroxyls. Modifications included glycinate and N-substituted glycinates, esters of amino acids containing tertiary or quaternary heterocyclic nitrogen, phosphates, and hemiesters of dicarboxylic acids.
- prophylactically effective is intended to qualify the amount of compound which will achieve the goal of prevention, reduction or eradication of the risk of occurrence of the disorder, while avoiding adverse side effects.
- therapeutically effective is intended to qualify the amount of compound that will achieve, with no adverse effects, alleviation, diminished progression or treatment of the disorder, once the disorder cannot be further delayed and the patients are no longer asymptomatic.
- compositions of the present invention are prophylactic as well as therapeutic.
- the "individual" or “patient” for purposes of treatment includes any human or mammalian subject affected by any of the diseases where the treatment has beneficial therapeutic impact.
- compositions according to the present invention will be useful for treating indications having an inflammatory or autoimmune mechanism involved in their etiology or pathogenesis.
- diseases or disorders are exemplified by multiple sclerosis, amyotrophic lateral sclerosis, systemic lupus erythematosis, myasthenia gravis, diabetes mellitus type I, sarcoidosis; skeletal and connective tissue disorders including arthritis, rheumatoid arthritis, osteoarthritis and rheumatoid diseases; ocular inflammation related disorders; skin related disorders including psoriasis, pemphigus and related syndromes, delayed-type hypersensitivity and contact dermatitis; respiratory diseases including cystic fibrosis, chronic bronchitis, emphysema, chronic obstructive pulmonary disease, asthma, allergic rhinitis or lung inflammation, idiopathic
- compositions according to the present invention will be useful in treating acute neurological disorders, resulting either from ischemic or traumatic damage, including but not limited to stroke, head trauma and spinal cord injury.
- the composition of the present invention may also be effective in preventing or treating certain chronic degenerative diseases that are characterized by gradual selective neuronal loss such as Parkinson's disease, Alzheimer's disease, AIDS dementia, Huntington's chorea, and prion-associated neurodegeneration.
- compositions may further be effective in prevention or diminution of cognitive impairment for instance post-operative, disease induced, virally induced, therapy induced or neonatal cognitive impairment and of CNS poisoning, for instance by strychnine, picrotoxin or organophosphorous compounds.
- compositions according to the present invention will be useful in treating pain including peripheral, neuropathic and referred pain.
- compositions of the present invention will also be effective in relieving emesis and treating glaucoma, retinal eye diseases and cachexia due to acquired immunodeficiency syndrome, neoplasia or other wasting diseases.
- the present invention provides a compound of formula (I):
- the compound of formula (I), dexanabinol, or a pharmaceutically acceptable salt, ester or solvate of said compound is in enantiomeric excess of at least 99.92% over the (3R,4R) enantiomer.
- the compound of formula (I), dexanabinol, or a pharmaceutically acceptable salt, ester or solvate of said compound is in enantiomeric excess of at least 99.95% over the (3R,4R) enantiomer.
- the compound of formula (I), dexanabinol, or a pharmaceutically acceptable salt, ester or solvate of said compound is in enantiomeric excess of at least 15 99.97% over the (3R,4R) enantiomer.
- the present invention provides a compound of formula (I) as above defined, wherein the absolute enantiomeric amount of the (3S,4S) enantiomer is at least 99.95% and the (3R,4R) enantiomer is 0.05%> or less.
- the compound of formula (I), dexanabinol, or a pharmaceutically 20 acceptable salt, ester or solvate of said compound is present in absolute enantiomeric amount of at least 99.96% and the (3R,4R) enantiomer is 0.04% or less.
- the compound of formula (I), dexanabinol, or a pharmaceutically acceptable salt, ester or solvate of said compound is present in absolute enantiomeric amount of at least 99.97% and the (3R,4R) enantiomer is 0.03% or less.
- the compound of formula (I), dexanabinol, or a pharmaceutically acceptable salt, ester or solvate of said compound is present in absolute enantiomeric amount of at least 99.98% and the (3R,4R) enantiomer is 0.02% or less.
- compositions comprising as an active ingredient dexanabinol, a compound of formula (I):
- the active ingredient of the above-defined pharmaceutical composition, dexanabinol, or a pharmaceutically acceptable salt, ester or solvate of said compound is in enantiomeric excess of at least 99.92% over the (3R,4R) enantiomer.
- the active ingredient of the above-defined pharmaceutical composition, dexanabinol, or a pharmaceutically acceptable salt, ester or solvate of said compound is in enantiomeric excess of at least 99.95%» over the (3R,4R) enantiomer.
- the active ingredient of the above-defined pharmaceutical composition, dexanabinol, or a pharmaceutically acceptable salt, ester or solvate of said compound is in enantiomeric excess of at least 99.97% over the (3R,4R) enantiomer.
- the present invention provides pharmaceutical compositions comprising as an active ingredient a compound of formula (I) as above defined, wherein the absolute enantiomeric amount of the (3S,4S) enantiomer is at least 99.95% and the (3R,4R) enantiomer is 0.05% or less.
- the active ingredient of the above-defined pharmaceutical composition is present in absolute enantiomeric amount of at least 99.96% and the (3R,4R) enantiomer is 0.04% or less.
- the active ingredient of the above-defined pharmaceutical composition is present in absolute enantiomeric amount of at least 99.97% and the (3R,4R) enantiomer is 0.03%> or less.
- the active ingredient of the above-defined pharmaceutical composition is present in absolute enantiomeric amount of at least 99.98% and the (3R,4R) enantiomer is 0.02%o or less.
- the present invention also provides pharmaceutical compositions comprising as an active ingredient an enantiomerically pure compound of formula (I) having the (3S,4S) configuration and being in enantiomeric excess of at least 99.90%), preferably 99.92%, more preferably 99.95%> and most preferably 99.97%, over the (3R,4R) enantiomer, further comprising a pharmaceutically acceptable diluent or carrier.
- the present invention also provides pharmaceutical compositions comprising as an active ingredient an enantiomerically pure compound of formula (I) having the (3S,4S) configuration and being present in absolute enantiomeric amount of at least 99.95%, preferably 99.96%, more preferably 99.97%) and most preferably 99.98%, further comprising a pharmaceutically acceptable diluent or carrier.
- the pharmaceutical compositions contain in addition to the active ingredient conventional pharmaceutically acceptable carriers, diluents and excipients necessary to produce a physiologically acceptable and stable formulation.
- Some compounds of the present invention are characteristically hydrophobic and practically insoluble in water with high lipophilicity, as expressed by their high octanol/water partition coefficient expressed as log P values, and formulation strategies to prepare acceptable dosage forms will be applied. Enabling therapeutically effective and convenient administration of the compounds of the present invention is an integral part of this invention.
- Solid compositions for oral administration such as tablets, pills, capsules, softgels or the like may be prepared by mixing the active ingredient with conventional, pharmaceutically acceptable ingredients such as corn starch, lactose, sucrose, mannitol, sorbitol, talc, polyvinylpyrrolidone, polyethyleneglycol, cyclodextrins, dextrans, glycerol, polyglycolized glycerides, tocopheryl polyethyleneglycol succinate, sodium lauryl sulfate, polyethoxylated castor oils, non-ionic surfactants, stearic acid, magnesium stearate, dicalcium phosphate and gums as pharmaceutically acceptable diluents.
- conventional, pharmaceutically acceptable ingredients such as corn starch, lactose, sucrose, mannitol, sorbitol, talc, polyvinylpyrrolidone, polyethyleneglycol, cyclodextrins, dextrans, gly
- the tablets or pills can be coated or otherwise compounded with pharmaceutically acceptable materials known in the art, such as microcrystalline cellulose and cellulose derivatives such as hydroxypropylmethylcellulose (HPMC), to provide a dosage form affording prolonged action or sustained release.
- pharmaceutically acceptable materials known in the art, such as microcrystalline cellulose and cellulose derivatives such as hydroxypropylmethylcellulose (HPMC), to provide a dosage form affording prolonged action or sustained release.
- Other solid compositions can be prepared as suppositories, for rectal administration.
- Liquid forms may be prepared for oral administration or for injection, the term including but not limited to subcutaneous, transdermal, intravenous, intrathecal, intralesional, adjacent to or into tumors, and other parenteral routes of administration.
- the liquid compositions include aqueous solutions, with or without organic cosolvents, aqueous or oil suspensions including but not limited to cyclodextrins as suspending agent, flavored emulsions with edible oils, triglycerides and phospholipids, as well as elixirs and similar pharmaceutical vehicles.
- aqueous or oil suspensions including but not limited to cyclodextrins as suspending agent, flavored emulsions with edible oils, triglycerides and phospholipids, as well as elixirs and similar pharmaceutical vehicles.
- the compositions of the present invention may be formed as aerosols, for intranasal and like administration.
- Topical pharmaceutical compositions of the present invention may be formulated as solution, lotion, gel, cream, ointment, emulsion or adhesive film with pharmaceutically acceptable excipients including but not limited to propylene glycol, phospholipids, monoglycerides, diglycerides, triglycerides, polysorbates, surfactants, hydrogels, petrolatum or other such excipients as are known in the art.
- pharmaceutically acceptable excipients including but not limited to propylene glycol, phospholipids, monoglycerides, diglycerides, triglycerides, polysorbates, surfactants, hydrogels, petrolatum or other such excipients as are known in the art.
- the pharmaceutical compositions Prior to their use as medicaments, the pharmaceutical compositions will generally be formulated in unit dosage.
- the active dose for humans is generally in the range of from 0.05 mg to about 50 mg per kg body weight, in a regimen of 1-4 times a day.
- the preferred range of dosage is from 0.1 mg to about 20 mg per kg body weight.
- dosages would be determined by the attending physician, according to the disease to be treated, its severity, the method and frequency of administration, the patient's age, weight, gender and medical condition, contraindications and the like.
- the dosage will generally be lower if the compounds are administered locally rather than systematically, and for prevention or chronic treatment rather than for acute therapy.
- a further aspect of the present invention provides a method of preventing, alleviating or treating a patient for indications as above described, by administering to said patient a prophylactically and/or therapeutically effective amount of a pharmaceutical composition comprising as an active ingredient enantiomerically pure dexanabinol, having the (3S,4S) configuration and being in enantiomeric excess of at least 99.90%), preferably 99.92%, more preferably 99.95% and most preferably 99.97%, over the (3R,4R) enantiomer, or a pharmaceutically acceptable salt, ester or solvate of said compound as above defined.
- a further aspect of the present invention provides a method of preventing, alleviating or treating a patient for indications as above described, by administering to said patient a prophylactically and/or therapeutically effective amount of a pharmaceutical composition comprising as an active ingredient enantiomerically pure dexanabinol, having the (3S,4S) configuration and being present in absolute enantiomeric amount of at least 99.95%, preferably 99.96%, more preferably 99.97% and most preferably 99.98%, or a pharmaceutically acceptable salt, ester or solvate of said compound as above defined.
- a further aspect of the present invention relates to the use for the manufacture of a medicament for preventing, alleviating or treating indications as above described, of enantiomerically pure dexanabinol, having the (3S,4S) configuration and being in enantiomeric excess of at least 99.90%), preferably 99.92%, more preferably 99.95% and most preferably 99.98%, over the (3R,4R) enantiomer, or a pharmaceutically acceptable salt, ester or solvate of said compound as above defined.
- a further aspect of the present invention relates to the use for the manufacture of a medicament for preventing, alleviating or treating indications as above described, of enantiomerically pure dexanabinol, having the (3S,4S) configuration and being present in absolute enantiomeric amount of at least 99.95%), preferably 99.96%, more preferably 99.97% and most preferably 99.98%, or a pharmaceutically acceptable salt, ester or solvate of said compound as above defined.
- the 5-(r,l'-dimethylheptyl)resorcinol (12) was obtained by a 5-step synthesis which started from 2-octanone (6) and 2,6-dimethoxyphenol (8).
- (6) was transformed to 2-methyl-2-octanol (7) (Grignard reaction), which then alkylated 8 in methansulfonic acid to give (r, -dimethylheptyl)-2,6-dimethoxyphenol (9).
- the (r,r-dimethylheptyl)-2,6-dimethoxyphenyl diethylphosphite (10) was obtained.
- Step 3 Oxidation of (+) myrtenyl pivalate with sodium chromate
- the resulting solution was passed through 0.33 parts of silica gel 60-230 mesh using an eluent of 3.5 volumes of methylene chloride. After removing the solvent under reduced pressure at 50-100 Torr at a final temperature of 80°C, the residue was distilled at 120-165°C and 0.1-0.15 Torr. The distillate was diluted with two volumes of n-pentane and kept at -20°C for 40 hours. The resulting crystals of 4-oxomyrtenyl pivalate (4) were filtered, rinsed with cold pentane and dried in a clean, well- ventilated hood. A second crop of material can be obtained from the mother liquors, by removing the solvent and distilling the residue in vacuum and crystallisation from pentane.
- Step 4 Reduction of 4-oxomyrtenyl pivalate with sodium borohydride
- Step 5 Grignard synthesis of 1 ' J '-dimethylheptanol from 2-octanone
- Step 7 Esterification of 4-f ,r-dimethylheptyl -2.6-dimethoxyphenol with diethyl phosphite
- Step 8 Reduction of 4-( '-dimethylheptyl -2.6-dimethoxyphenyl diethylphosphate with lithium/ammonia
- Step 9 Demethylation of l-(r '-dimethylheptyl -3.5-dimethoxybenzene with boron tribromide
- Step 10 Coupling of 4-hydroxy myrtenyl pivalate with 5-( '-dimethylheptyl - resorcinol
- 5-( '-dimethylheptyl - resorcinol) To a mixture of 1.1 molar equivalents of 5 and 1.0 molar equivalent of 12 in 24 volumes of methylene chloride were added four molar equivalents of boron trifluoride etherate, at (-15)-(-10)°C over one hour.
- the reaction mixture was maintained at the above temperature for 2.5 hours, then treated with another four molar equivalents of boron trifluoride etherate over one hour and stirred at the same temperature for another 2.5 hours.
- the reaction mixture was poured onto 0.5 parts of crushed ice containing 29 molar equivalents of sodium bicarbonate and left overnight at 20-25°C.
- the combined extracts were washed 3 times with 0.25 volumes (each) of water and 3 times with 0.3 volume (each) of 5%> aqueous solution of sodium bicarbonate and then dried over 0.5 parts of anhydrous sodium sulphate.
- the solvents were removed in vacuum at 50 Torr and 40°C and the residue was recrystallized from 6 volumes of acetonitrile brought to temperature near reflux at 70-81.6°C.
- the white crystals of dexanabinol (14) were filtered, rinsed with cold acetonitrile (2-8°C) and dried in a vacuum oven at 60°C for three hours.
- the resulting dexanabinol was recrystallized from 28 parts 1:1.2 wate ⁇ ethanol, filtered, and dried to constant weight at 65-75°C and 1-5 Torr.
- the active pharmaceutical ingredient following crystallization from acetonitrile is superior to that recovered from any previously published procedure, both in terms of enantiomeric purity and overall yield.
- the large scale synthetic process differs from the process described in Example 1 at specific steps and the modifications are as follows.
- the changes include modifications in distillation conditions or in solvents.
- step 2 the crude myrtenyl pivalate previously used for the subsequent step without further purification, was now further distilled under high vacuum at 2 Torr up to 180°C. Under such conditions, the distillate contained at least 80% myrtenyl pivalate (3) with 53%> yield.
- step 3 the crude 4-oxomyrtenyl pivalate is further distilled at higher temperature up to 190°C under high vacuum at 1 Torr, instead of previous 120-165°C and 0.1-0.15 Torr.
- step 4 the mixture of 4-oxomyrtenyl pivalate with sodium borohydride was extracted with 2.5 volumes of dichloromethane (DCM) instead of previous hexane.
- DCM dichloromethane
- the solvent methanol/DCM was removed under reduced pressure at 50-100 Torr and temperature below 70°C.
- 1 volume of DCM was added to afford the 4-hyroxymyrtenyl pivalate (5) in DCM solution in yield of about 84.5%.
- Step 5 Grignard synthesis of 1 'J' -dimethylheptanol from 2-octanone
- a 1 liter reactor under N 2 atmosphere was filled with 468.3 g methyl magnesium chloride 23% solution in tetrahydrofuran (THF) (1.2 eq.) and 122 ml of THF. Then 153.85 g of 2-octanone (6) (1.2 mole) were added at 20-25°C during 90 minutes. The reaction mixture was then stirred for 24 hours at room temperature, while monitoring the reaction progress by gaz chromatography. The reaction mixture was then transferred to a second 1 liter reactor containing 154 ml of water, while keeping the temperature under 20°C. The reaction mixture was then passed through frit glass in order to eliminate mineral salts of magnesium.
- THF tetrahydrofuran
- Step 6 Alkylation of 2,6-dimethoxyphenol with 1 ' , 1 ' -dimethylheptanol
- Step 7 Esterification of 4-( V .1 ' -dimethylheptyl -2.6-dimethoxyphenol with diethylchlorophosphate
- Step 8 Reduction of 4-( 1 ' ⁇ ' -dimethylheptyl)-2.6-dimethoxyphenyl diethylphosphate with lithium/ammonia
- reaction mixture was then heated up to reflux to remove under atmospheric pressure water and part of the toluene to obtain a 224.5 g of a toluene solution containing about 31%) of the product (11) (0.262 mole), about 87% yield.
- Step 9 Demethylation of l-(r '-dimethylheptyl)-3,5-dimethoxybenzene with boron tribromide 0 A solution of 11 in 3 volumes of toluene was added dropwise to a stirred solution of
- Step 10 Coupling of 4-hvdroxy myrtenyl pivalate with 5-fr '-dimethylheptyl - resorcinol
- the reaction mixture was cooled to (-15)-(-20)°C under stirring and while keeping the temperature below -14°C 42.6 g of boron trifluoride etherate were added.
- the resulting brownish solution was maintained at -15°C for at least 1 hr.
- a previously prepared solution of 15.15 g of sodium bicarbonate in 288 ml of water was added while letting the temperature rise up to 20°C. Then the two phases were separated. The organic phase was washed again with sodium bicarbonate solution and again phases were separated.
- a 2 liters reactor was filled with 780 g of 12% solution of 13 (0.2 mole) and cooled down to 0-(-5)°C. Then 359 g of LiAlH 1M solution in THF were added and the reaction mixture was stirred at that temperature for 1 hour. Then 195 ml of ethyl acetate were added and while stirring vigorously 1200 ml of water were added. The reaction mixture was warmed to 25°C and 75 g of hydrochloric acid 37%> were added. Then the two phases were separated. Adding 270 ml of 5% solution of sodium bicarbonate neutralized the organic phase, and then the aqueous phase was eliminated. The organic phase was washed with 200 ml of water and the water phase was eliminated.
- the solvents from the organic phase were removed under vacuum 50 Torr at 40-50°C.
- the residue was recrystallized from 6 volumes of acetonitrile brought to temperature of about 90°C to remove residual solvents.
- the reaction mixture was allowed to cool until the beginning of the precipitation. The temperature was maintained for 1 hour at 0-5°C and the white crystals of dexanabinol (14) were filtered, rinsed with cold acetonitrile (2-8°C) and dried in a vacuum oven at 60°C for three hours.
- the resulting dexanabinol was recrystallized from ethano heptane 3:5, filtered, and dried to constant weight at 65-75°C and 1-5 Torr.
- the pivotal crystallization step is performed with acetonitrile, which is removed by recrystallization from ethanol :heptane instead of previously used water:ethanol.
- acetonitrile is removed by recrystallization from ethanol :heptane instead of previously used water:ethanol.
- the enantiomeric purity can be further increased, if necessary, by repeating the crystallization step with acetonitrile
- Example 2 The main advantages of the process of Example 2 over Example 1 lie in the utilization of solvents appropriate to industrial large-scale synthesis and in the adaptation or elimination of certain isolation and purification steps enabling a simplified continuous process.
- the new process has allowed the preparation of batches of kilogram quantities, to suit commercial production of the drug.
- HU-211 and HU-210 reference material were prepared by additional crystallization steps and chromatographic separations. Compounds that serve as reference undergo thorough analyses, which includes, on top of the assay listed in Table 1, nuclear magnetic resonance ( ⁇ MR), Mass spectra (MS) and element analysis. Per definition these ultrapure compounds will be referred to as 100%.
- the reference material for HU-211 was prepared in-house, while the reference material for HU- 210 was purchased from Tocris.
- the mobile phase is composed of 96% volume/volume (v/v) of n-hexane and 4%) v/v of isopropanol, each HPLC grade and previously filtered through a 0.45 ⁇ m nylon membrane, the mixture was degassed using a sonication bath for a few seconds.
- the HPLC is performed on a chemically modified amylose-based chiral column ChiralPak AD-H, 250x4.6 mm, 5 ⁇ m particle size (Daicel Ltd).
- the chiral stationary phase is a tris(3,5-dimethylphenylcarbamate) derivative of amylose immobilized on macroporous silica gel.
- the flow rate is 1 ml per minute, the chromatography is performed at ambient temperature of about 25°C and the detection is performed at 215 nm.
- the controls or samples are injected at a volume of 40 ⁇ l and a run is performed for 50 minutes.
- the HPLC mobile phase is injected first as a blank, then the 50 ⁇ g/ml standard of HU-210 mixed with HU-211 to determine the retention time for each enantiomer and confirm the separation of the peaks and thus the efficiency of the analytical method.
- HU-210 elutes after HU-211 with a typical relative retention time of about 1.4.
- Quantitation of HU-210 is linear at least within the range of 0.0025 up to 0.12%) w/w of dexanabinol.
- the detection and quantitation limits of HU-210 are respectively 0.00125 and 0.0025%) w/w of dexanabinol.
- the method is highly repeatable as measured by low relative standard deviation (RSD) when the same sample is injected six times (system repeatability RSD ⁇ 2%), when six replicates are injected (method repeatability RSD ⁇ 7%) and when 6 replicates are tested on two HPLC systems (intermediate precision ⁇ 5%>). This method allows to determine the level of HU-210 in the dexanabinol drug substance sample with accuracy and thus the level of enantiomeric purity of HU-211, as expressed as enantiomeric excess over HU-210, with confidence.
- the adaptations brought to the method of Levin et al. include: the use of a single shorter wavelength of detection, namely 215 nm instead of the previous double simultaneous detection at 220 and 270 nm; the utilization of smaller particles, ⁇ 5 ⁇ m instead of 10 ⁇ m; modification of the sample loading conditions with an increase in injection volume, namely 40 ⁇ l instead of 20 ⁇ l; and, in sample concentration with 5 mg/ml instead of previous 0.1 mg/ml. These modifications together lead to a significant improvement of over 30-fold in the lower limit for reliable quantitation of the (3R,4R) enantiomer in term of concentration.
- HU-210 can be detected at a concentration of 0.125 ⁇ g/ml (corresponding to an amount as low as 5 ng per sample), instead of the previous estimate of 3.9 ⁇ g/ml.
- concentration of 0.125 ⁇ g/ml corresponding to an amount as low as 5 ng per sample
- the lower limit for detection of HU-210 achieved by the present method allows confident determination of higher 5 enantiomeric excess than previously possible.
- 10 210 is 5 ng, which allows determination of enantiomeric excess above 99.99%).
- the amount of HU-211 in dexanabinol drug substance is assayed by reversed phase (RP)-HPLC.
- the HPLC column used is a Hypersil BDS RP-18 3 ⁇ m, 150x4.6 mm, maintained at 30°C.
- the mobile phase is composed of 60% acetonitrile and 40%) 10 mM ammonium acetate buffer pH 5.2.
- the injection volume is 15 ⁇ l, the flow rate
- %HU-211 (Ru/Rs)x(W s /Ns)x(Nu/Wu)xl00, wherein RQ and Rs are the peak responses of the unknown sample and standard respectively, Wu and Ws are the weights (in mg) and Nu and Ns are the volumes (in ml) of the unknown sample and standard respectively.
- Dexanabinol Formulation of dexanabinol of high enantiomeric purity for clinical use
- Dexanabinol is an extremely lipophilic compound with a computed Log P of 7.69
- Dexanabinol drug substance is formulated as a 5% w/v concentrate in a cosolvent vehicle composed of CREMOPHOR EL ® (polyoxyl 35 castor oil; 65% w/v) and absolute ethanol (26.5% w/v).
- the dexanabinol cosolvent concentrate also contains 0.01% w/v edetic acid and 0.5%> w/v Vitamin E (DL- ⁇ -tocopherol) as antioxidants.
- This parenteral 5% cosolvent solution is a clear, slightly yellow, sterile and pyrogen-free concentrate of dexanabinol for injection which must be diluted prior to intravenous infusion 1/20 to 1/100 with sterile 0.9%> sodium chloride solution for injection.
- the drug product is preservative-free and sterilization is achieved via a sterile filtration and aseptic processing technology.
- the quantitative composition of the 5% dexanabinol parenteral cosolvent concentrate is given in Table 3.
- the dexanabinol drug substance is manufactured as previously described in Example 1 or 2 and according to the specifications in Example 3, specifically in enantiomeric excess of at least 99.90%) and of absolute enantiomeric amount of at least 99.95%). All the inactive
- the parenteral concentrate formulation has to be diluted prior administration.
- the above-described clinical formulation of dexanabinol of high enantiomeric purity was diluted with sterile 0.9%) sodium chloride solution for injection at a ratio of 1 :5 up to 1 :500.
- the ready for injection diluted drug concentrate were stable at all dilution ratios for up to 24 hours as determined by HPLC analysis performed on filtrates collected at predetermined time points along the duration of the study. Table 3. Composition of dexanabinol parenteral concentrate.
- non-aqueous vehicles include surface-active agents such as TWEEN ® 80 and CREMOPHOR EL ® .
- Surfactant agents are usually incorporated into parenteral preparations to provide an increase in drug solubility through micellization and to prevent drug precipitation upon dilution.
- the vehicle of choice should provide for adequate stability, have an acceptable safety profile and allow for drug administration within the shortest period of time leading to the highest possible plasma concentration Cmax thereby providing for the maximum achievable therapeutic drug concentrations in the target organ with minimal administration risks.
- the goal of this study was to find a suitable cosolvent formulation for a concentrate of dexanabinol of high enantiomeric purity to be diluted with sterile saline solution before injection.
- the compositions of the cosolvent concentrate formulations tested are presented in Table 4. All formulations contained 1%) dexanabinol and compositions of FDA- approved cosolvent vehicles. The concentrations of the various ingredients are expressed as % weight/weight.
- ® CREMOPHOR EL :ethanol as described in Example 4 were investigated in rats, rabbits, and monkeys following intravenous administration of single doses, and 14 and 28 days of repeated dosing. Human pharmacokinetics was studied during Phase I and Phase II clinical studies. Dexanabinol used in the pharmacokinetic studies was formulated as drug concentrates of 50 and 100 mg/ml and diluted with sterile 0.9% NaCl solution prior to intravenous (i.v.) administration to the desired final doses. Determination of dexanabinol concentrations in plasma and brain extracts was carried out using a validated Gas Chromatography-Mass Spectra (GC-MS) assay following solid phase extraction of the drug and derivatization. The limit of quantitation of the assay is 0.1 ng/ml. Pharmacokinetic parameters were estimated by a non-compartmental method using
- the maximum plasma concentration (Cmax), when the drug is administered by infusion, is the concentration at the end of infusion.
- the area under the curve from time of dosing through the last time point (AUC 2 ) was calculated by the linear trapezoid method.
- the AUC extrapolated to infinity (AUCoo) was calculated from the following equation:
- AUC ⁇ AUC z + C z / ⁇ where C z is the concentration at the last time point predicted by the linear regression.
- AUCoo was normalized for dose (mg/kg) and presented as AUCoo/Dose.
- Mean residence time (MRT) when the drug is administered by infusion is described by the following equation:
- MRT (AUMC/AUC) where AUMC is the area under the first moment curve and TI is the length of infusion.
- V ss MRT x
- %F [AUC orai/Dose ora ⁇ ] / [AUC iv/Dose iv]
- Example 4 is generally well tolerated following single and/or multiple i.v. doses in rats, rabbits and monkeys.
- the NOAEL was 15 mg/kg/day in rats and 25 mg/kg/day in rabbits. In a 28-day study in monkeys the NOAEL was 25 mg/kg/day. Cmax and AUC observed following the last dose at the NOAEL in the multiple dose toxicity studies and the ratios of these values to those observed in the Phase I and II studies for the 150 mg dose are shown in Table 7. Exposure levels as exhibited by the AUCoo associated with the NOAEL in the 14-day studies and 28-day study far exceed those observed in the clinical studies.
- the NOAELs compared above are based upon 2 weeks and 4 weeks of daily dosing whereas the anticipated clinical regimen consists of a single dose. It is, therefore, reasonable to assume that the NOAELs defined in the multiple-dose animal studies represent an even greater multiple of the human dose if cumulative exposure is considered.
- the plasma concentration versus time profiles following the final administration at the NOAEL dose levels in the repeat dose studies in animals are shown in Figure 2 along with the profile obtained in humans from the Phase I and Phase II studies, that will be described below.
- the pharmacokinetic profile in all species demonstrated an initial rapid decrease in plasma related concentrations, a common characteristic of highly lipophilic compounds, followed by a slower decline. Plasma concentrations were still detectable, but low, 24 hours after injection, suggesting there might be some accumulation in the repeated dose studies. While there was some evidence for accumulation in the plasma with repeated dosing, the extent of accumulation was minimal.
- the target organ for dexanabinol therapeutic intervention in patients suffering from TBI being the brain, the monitoring of dexanabinol level in the brain was included in the rat study.
- Sprague Dawley rats of each sex received a bolus intravenous injection of 4 mg/kg of dexanabinol of high enantiomeric purity in the CREMOPHOR EL ® : ethanol clinical formulation.
- the animals were divided into eight sub-groups of 6 animals, 3 male and 3 female, assigned to a single bleeding time point. The eight bleeding time points were 5, 15, 30 minutes and 1, 2, 4, 8 and 24 hours after injection. Following bleeding the animals were euthanized and their brain were removed for analysis of brain dexanabinol concentrations.
- the divergences between genders were more pronounced in plasma, reaching 2-3 fold differences for some pharmacokinetic parameters, than in brain, where the differences are not statistically significant for most time points.
- the results for male and female were averaged in order to compare the levels of dexanabinol in plasma versus brain, following single injection of 4 mg/kg dexanabinol. The results are depicted in Figure 3. Unlike plasma concentrations, which peaked at the earliest measured time point and rapidly decline in the initial phase, the brain concentrations equilibrated with plasma concentration about 30 minutes after injection.
- dexanabinol of high enantiomeric purity was then administered to human subjects. According to standard regulatory procedures dexanabinol was first tested in healthy subjects during two Phase I studies, and once its safety was confirmed in humans it was administered to traumatic brain injury patients during a Phase II clinical study.
- Plasma concentrations were approximately 1.8 ⁇ g/ml after a 48 mg dose (0.62 mg/kg), 2.9 ⁇ g/ml at 100 mg (1.29 mg/kg), and 4.6 ⁇ g/ml at 200 mg (2.59 mg/kg).
- the total areas under the plasma concentration curve (AUCoo) differed significantly for each dose group and were related to the dose in a linear fashion.
- Total plasma clearance (CL) values of dexanabinol and Vss values increased with the dose, and the AUC ⁇ values normalized for dose (AUCoo/Dose) decreased with the dose.
- a second human Phase I study involving 24 healthy male volunteers was carried out to compare the pharmacokinetics of dexanabinol following a single i.v. dose of 48 mg or 5 150 mg.
- the subjects were divided into two groups of 12 subjects each. Each group was premedicated with ATOSIL ® 25 mg (Promethazine HI blocker), and ZANTAC ® 50 mg (Ranitidine H2 blocker) intravenously, followed 15 minutes later by a single short intravenous infusion of dexanabinol lasting 15 minutes.
- Intravenous administration of dexanabinol generated high initial plasma levels of the drug (as reflected by values obtained at the end of the drug infusion) that were dose- related.
- Maximum plasma concentrations (C max ) were 1.23 ⁇ g/ml after the 48 mg dose (0.63 mg/kg), and 5 ⁇ g/ml at 150 mg (2.05 mg/kg). In both cases, the drug levels fell 5 rapidly as a function of time with 30 min values being about 11%> of the end of infusion levels.
- the total areas under the plasma concentration curve (AUC) differed significantly for each dose group and increased proportionally to the dose.
- Total plasma clearance (CL) values of dexanabinol were similar for both dose groups and averaged 12 ml/min/kg across the two dose groups.
- dexanabinol While pharmacologically, dexanabinol bears little resemblance to naturally occurring cannabinoids, its pharmacokinetic properties are similar to those of ⁇ - THC and related materials. These properties include rapid initial distribution, long terminal elimination half-life, a rapid total plasma clearance and a large volume of distribution. Altogether, these parameters ensure extensive uptake of the drug into tissues, including the brain and central nervous system, and rapid manifestation of biological action. Beside gathering the pharmacokinetic parameters above described, the two phase I studies allowed to determine that using dexanabinol of high enantiomeric purity in human subjects was safe, well tolerated and no psychomimetic side effects were detected.
- dexanabinol is a very lipophilic compound (log P of 7.44), it will cross the blood-brain barrier easily by diffusion, thereby, brain and plasma concentrations will tend to equilibrate fairly quickly. Therefore, higher blood levels will translate into higher brain levels more readily than if some active transport process was involved or if the process was slow and required a long duration of high plasma concentrations.
- dexanabinol is a non-competitive antagonist of the NMDA receptor the faster the administration, the quicker the receptors are saturated and the sooner the pharmacological effect is established.
- a Phase II, double masked, multi-center study was conducted to evaluate the safety and tolerance of dexanabinol of high enantiomeric purity following a single intravenous administration in patients with severe head trauma (Knoller N. et al., Crit. Care Med. 30: 548-54, 2002). Treatment was administered within 6 hours of injury, based on the therapeutic window observed in relevant animal models. Additional objectives of the study were to evaluate the long-term outcome of the patients and to determine the optimal dose for Phase III studies. Medical information was collected en route or upon arrival at the hospital to determine a patient's suitability for enrollment and a randomized patient number was assigned. Written informed consent was obtained from relatives, all eligible patients being in coma.
- Antihistamines (promethazine hydrochloride PHENERGAN ® 25 mg and cimetidine 50 mg) were administered by intravenous bolus injection 15 minutes prior to study drug administration.
- Dexanabinol was manufactured and formulated as previously described (50 mg/ml in CREMOPHOR EL ® :ethanol clinical formulation) and diluted into 100 ml saline prior to injection. Solutions of dexanabinol were infused intravenously using a peristaltic pump (I vac) at a rate of 6 ml/min (or approximately 15 min/dose). The total doses of dexanabinol scheduled to be administered were 48, 150, or 200 mg per patient.
- I vac peristaltic pump
- Pharmacokinetic parameters are generally dose proportional. Cmax is somewhat lower, and the dose-dependent pharmacokinetic parameters clearance (CL) and volume of distribution at steady-state (V ss ) are somewhat higher, at the 150 mg dose level than would be expected based on the values obtained at the higher and lower doses. This is most likely the result of under-dosing of the 150 mg group. Under-dosing would also lead to underestimation of the dose-normalized AUC value (AUC/D) which indeed is lower for the 150 mg dose than for the low and high doses. Simulations of the dosing solution preparation indicated that the 150 mg group was under-dosed by approximately 20% while the 48 and 200 mg groups were within 10% of the target dose.
- the route of delivery chosen in the clinical studies was the intravenous route, which is appropriate for rapid drug delivery to the systemic circulation and to target organs in hospital setting in case of acute indications such as TBI. While the preferred route of delivery described for the CREMOPHOR EL ® :ethanol clinical formulation is intravenous (i.v.), it is possible to use this formulation for intraperitoneal (i.p.), intramuscular (i.m.), subcutaneous (s.c), intra cerebro ventricular (i.c.v.), intrathecal and per os (p.o.) administration. For chronic indications other routes can also be used for the delivery of dexanabinol of high enantiomeric purity. These additional routes of administration will also demonstrate the feasibility of further formulations to efficiently deliver dexanabinol.
- Dexanabinol of high enantiomeric purity (lot # AC9001HU) filled in hard gelatin capsules have shown good oral bioavailability.
- a pharmacokinetic study for oral bioavailability of dexanabinol in large animals using the minipig model (the best animal model for oral absorption of drugs) was carried out.
- Dexanabinol plasma levels were determined using a validated GC-MS assay up to 48 hours following administration.
- the neuroprotectant drug dexanabinol was shown to have additional potent anti- inflammatory activity in several animal models.
- Dexanabinol has demonstrated beneficial effect in a urine model of inflammatory bowel disease (IBD).
- IBD inflammatory bowel disease
- GI chronic gastrointestinal
- the rectal route is preferred for drug administration to avoid adverse effects and additional GI disturbances and to affect locally the seat of disease.
- the composition of dexanabinol rectal formulation is shown in Table 12. Table 12. Composition of dexanabinol rectal formulation.
- PEG 1000 is an excipient extensively used in rectal preparations and suppository bases.
- Xanthan gum is a high molecular weight, high viscosity polysaccharide particularly suitable for controlled release applications.
- the unique solution properties of xantham gum provide many attractive features to pharmaceutical formulations to suspend and stabilize dispersions of solids and immiscible liquids in aqueous systems.
- Xantham gum provides excellent suspension and thickening properties at very low concentrations. It hydrates well in both acid and alkaline media and the viscosity is relatively unaffected by pH.
- KELTROL ® TF dissolves in cold water at moderate concentrations to produce solutions of high viscosity. The high viscosity is often useful for providing bioadhesion to mucosal surfaces.
- Bioadhesion to mucous membranes may also be improved by combining xantham gum with polyols.
- the combination of xantham gum with PEG 1000 provides an excellent vehicle for bioadhesion and sustained release of the active ingredient dexanabinol increasing its activity and prolonging its residence time in the site of action in the proximity of GI mucosal surfaces.
- the PEG 1000 vehicle (USP/NF grade, Spectrum Quality Products, Inc.) was melted in a water bath at ⁇ 50°C. Dexanabinol of high enantiomeric purity was added to the melted PEG 1000 and mixed at ⁇ 50°C for about 2 hours until complete dispersion.
- the xantham gum KELTROL ® TF, Monsanto Pharmaceutical Ingredients was then added and the mixture was shaken for another 1 hr at 50°C until a homogeneous dispersion is obtained.
- the final product melts between 36-38°C, therefore it can be molded to obtain suppositories or it can be administered to animals rectally as an enema by melting it at 40°C to get a fluid and using a rectal catheter.
- rectal dexanabinol 10 mg/kg had the best effect (more than 50%> reduction of score) compared to its vehicle (p ⁇ 0.05). Dexanabinol 10 mg/kg reduced also the clinical disease severity compared to the vehicle.
- the rectal dexanabinol dose of 10 mg/kg was pharmacologically equivalent to 20 mg/kg i.p. dose and 80 mg/kg oral dose.
- the results of the present work demonstrate the beneficial effects of dexanabinol enema formulation in IBD murine model.
- Dexanabinol was formulated either in submicron emulsion (SME) or in hydroxypropyl-cyclodextrin (HPCD).
- the solutions of dexanabinol in HPCD were prepared as follows. First, a weighted amount of dexanabinol was dissolved in a minimum amount of absolute ethanol. The drug containing ethanol solution is then added dropwise to the HPCD powder, which is subsequently dried at 48-80 ° C until ethanol evaporates. Water is then added and mixed with the dried powder to give final dexanabinol concentrations of 0.1 to 2 mg/ml and HPCD concentrations of 5 to 45%>. Complete dissolution is obtained by sonication and heating. The homogenous solutions are then filtered through 0.2-0.45 ⁇ m sterile disposable filter unit.
- Submicron emulsions are made of homogenous oily droplets in a size range of 50-80 nm emulsified in aqueous solution.
- Various ocular drugs including timolol, pilocarpine and indomethacin were successfully formulated in SME and were advantageous over the standard formulations both in terms of irritation and bioavailability.
- the composition of dexanabinol topical formulation is shown in Table 13.
- a total volume of about 100 ml (100 g w/w%) of dexanabinol in SME was prepared.
- the oil phase stock was composed of Medium-chain triglyceride (MCT) Oil, LIPOID E- 80 ® and DL- ⁇ -Tocopherol Succinate and dexanabinol of high enantiomeric purity.
- MCT Medium-chain triglyceride
- LIPOID E- 80 ® LIPOID E- 80 ®
- DL- ⁇ -Tocopherol Succinate dexanabinol of high enantiomeric purity.
- the lipids and oil were weighed in a 250 ml beaker and mixed at 40-45°C using a magnetic stirrer for 15 min until a homogenous and almost clear solution was obtained.
- Dexanabinol was then dissolved in the oil phase by stirring at room temperature (RT).
- the water phase was prepared as follows.
- Phase (5 g) was heated to 40-45°C and added to the beaker containing the water phase (preheated to 40-45°C). The mixture was gently stirred for 10-15 minutes at room temperature.
- the droplet size of the emulsion obtained after Polytron step was lowered to the submicron (nanosize) range by submitting the emulsion to high shear homogenization using the Gaulin Microlab 70 or Emulsiflex High Pressure Homogenizers at 800 bar pressure. A total of 3 to 6 cycles were performed to obtain homogeneous SME preparation with a mean droplet diameter in the range of 50- 100 nm.
- the pH of the resultant SME was adjusted to 7.4 by adding small amounts of 1 N HC1 or 1 N NaOH solutions using a calibrated pH meter.
- the osmolalities of all SME obtained were around 300 mOsm. If the value obtained is below 300 ⁇ 30 mOsm it must be adjusted by adding Glycerol.
- the SME formulations were sterilized by filtration through a 0.2 ⁇ m sterile disposable filter unit (cellulose acetate, 0.5 liter volume, Corning, England), using vacuum supplied by water pump.
- the SME formulations were packaged under aseptic conditions in 5 ml plastic droppers in laminar flow hood using sterile (by gamma irradiation) low density polyethylene (LDPE) eye drop bottles, insert and caps. This dexanabinol SME formulation was then tested in normotensive rabbits.
- LDPE low density polyethylene
- New Zealand White albino rabbits weighing 2-2.5 kg were acclimatized in our animal facility for at least a week prior to IOP measurements.
- Baseline IOP was individualized for each animal and time point prior to drug testing. IOP was measured by a Digilab Pneumatonometer, Model 3 OR. ⁇ IOP was calculated by subtracting the baseline IOP from IOP value measured after drug application at a corresponding time point. Maximal ⁇ IOP and area under the ⁇ IOP x hours curve (AUC) were calculated and averaged for each group. Data of IOP, AUC, blood pressure and toxicity were analyzed by the Wilcoxon-Rank test.
- Average values of maximal ⁇ IOP after topical instillation of dexanabinol of high enantiomeric purity varied between 7.0 mmHg to 1.6 mmHg in several experiments with a range of dosages and formulations.
- IOP was reduced in dexanabinol treated groups compared to blank- vehicle and saline groups in virtually all experiments.
- the blank SME vehicle was devoid of any significant IOP lowering activity as compared to saline treatment.
- Ocular toxicity was tested as follows. Two groups of five animals each received topical instillation of dexanabinol in SME and blank SME for five days four times daily. The animals were examined daily with a slit lamp for ocular discharge, conjunctival hyperemia, corneal fluorescein staining and iris hyperemia. Results were scored on a 0.0- 4.0 scale in 0.5 steps. Topical treatment with dexanabinol of high enantiomeric purity resulted in mild conjunctival injection and discharge that did not significantly differ from SME-blank treated animals. Corneal staining, corneal opacities and iris hyperemia did not occur in any of the animals.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Immunology (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Ophthalmology & Optometry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Hospice & Palliative Care (AREA)
- Addiction (AREA)
- Rheumatology (AREA)
- Transplantation (AREA)
- Otolaryngology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Pyrane Compounds (AREA)
Abstract
Description



















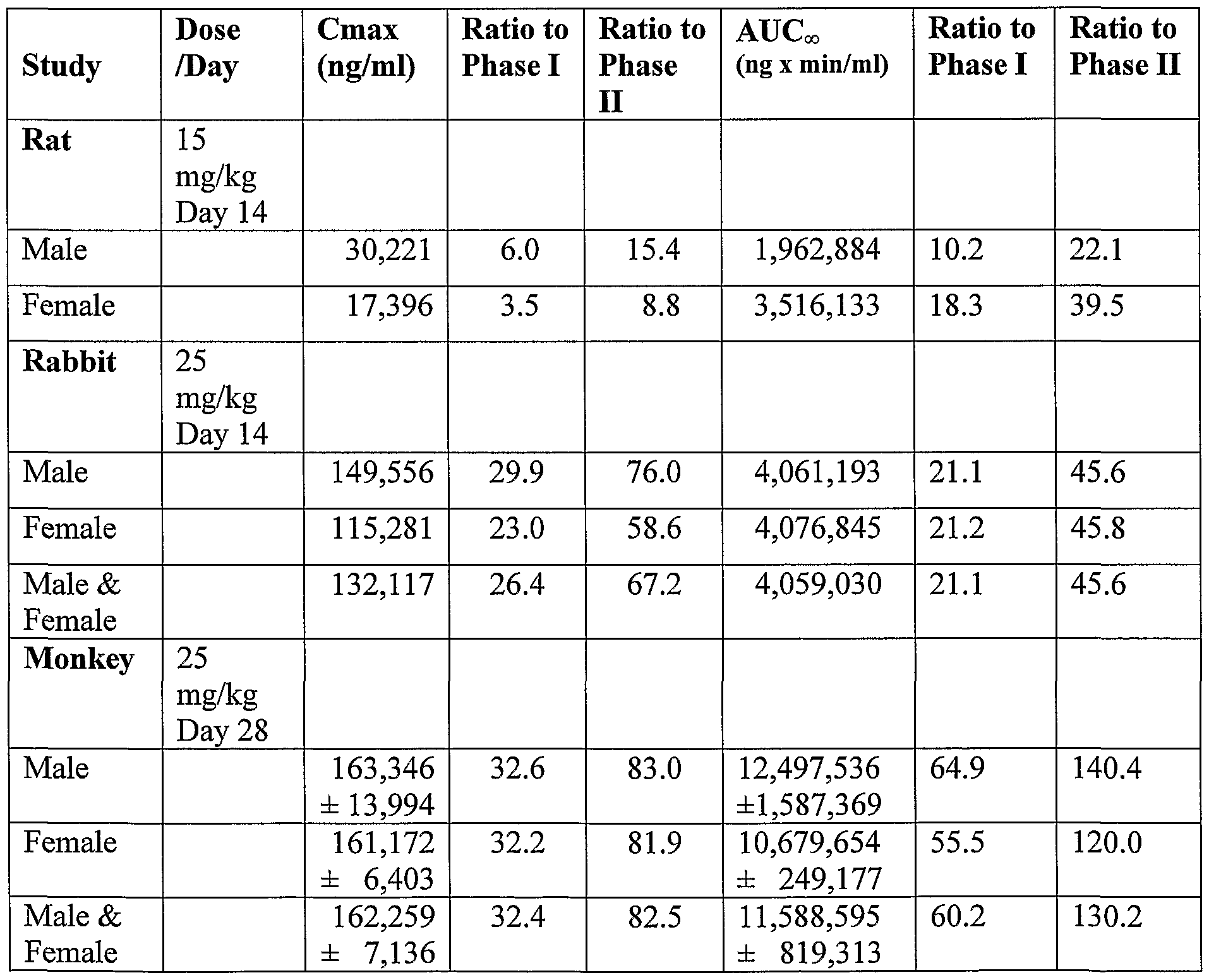




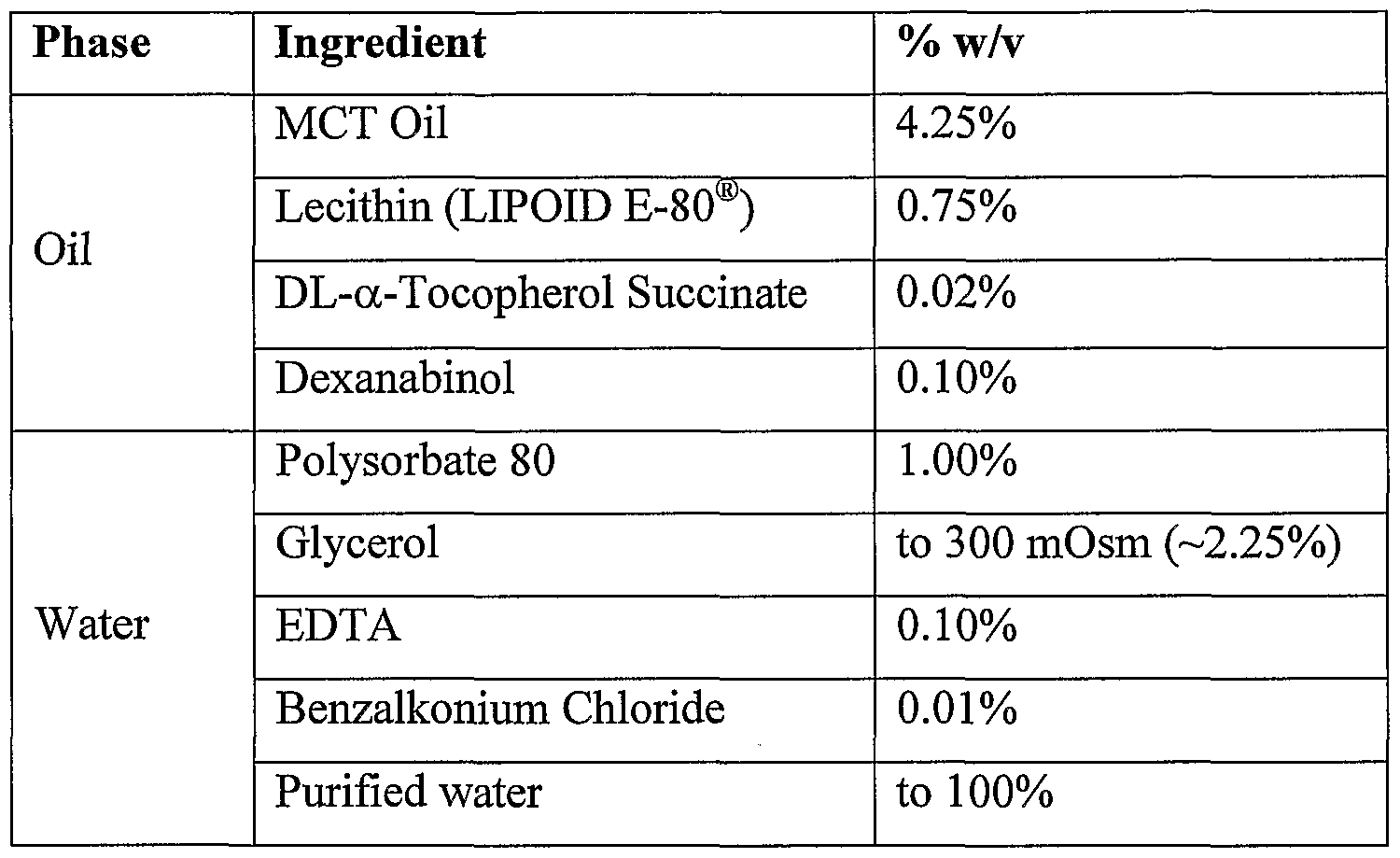
Claims


Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002507815A CA2507815A1 (en) | 2002-12-04 | 2003-12-03 | High enantiomeric purity dexanabinol for pharmaceutical compositions |
| AU2003302578A AU2003302578A1 (en) | 2002-12-04 | 2003-12-03 | High enantiomeric purity dexanabinol for pharmaceutical compositions |
| JP2004570714A JP2006509038A (en) | 2002-12-04 | 2003-12-03 | Dexanabinol with high enantiomeric purity for pharmaceutical compositions |
| EP03812258A EP1567513A2 (en) | 2002-12-04 | 2003-12-03 | High enantiomeric purity dexanabinol for pharmaceutical copositions |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IL153277 | 2002-12-04 | ||
| IL15327702A IL153277A0 (en) | 2002-12-04 | 2002-12-04 | High enantiomeric purity dexanabinol for pharmaceutical compositions |
| US10/644,687 US20040110827A1 (en) | 2002-12-04 | 2003-08-19 | High enantiomeric purity dexanabinol for pharmaceutical compositions |
| US10/644,687 | 2003-08-19 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2004050011A2 true WO2004050011A2 (en) | 2004-06-17 |
| WO2004050011A3 WO2004050011A3 (en) | 2004-07-29 |
Family
ID=32472232
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IL2003/001023 WO2004050011A2 (en) | 2002-12-04 | 2003-12-03 | High enantiomeric purity dexanabinol for pharmaceutical copositions |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20070060636A1 (en) |
| EP (1) | EP1567513A2 (en) |
| JP (1) | JP2006509038A (en) |
| AU (1) | AU2003302578A1 (en) |
| CA (1) | CA2507815A1 (en) |
| WO (1) | WO2004050011A2 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10588974B2 (en) | 2016-04-22 | 2020-03-17 | Receptor Holdings, Inc. | Fast-acting plant-based medicinal compounds and nutritional supplements |
| US11246852B2 (en) | 2016-12-02 | 2022-02-15 | Receptor Holdings, Inc. | Fast-acting plant-based medicinal compounds and nutritional supplements |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9572834B2 (en) * | 2011-04-26 | 2017-02-21 | William Marsh Rice University | Use of carbon nanomaterials with antioxidant properties to treat oxidative stress |
| FR2989169B1 (en) * | 2012-04-05 | 2015-01-23 | Commissariat Energie Atomique | METHOD AND DEVICE FOR ESTIMATING MOLECULAR PARAMETERS IN A CHROMATOGRAPHIC PROCESSED SAMPLE |
| GB201207305D0 (en) * | 2012-04-26 | 2012-06-13 | E Therapeutics Plc | Therapy |
| CN103058823A (en) * | 2012-12-26 | 2013-04-24 | 淮安万邦香料工业有限公司 | Preparation method of 2-dimethyl-2-octanol |
| BR112015019180A8 (en) | 2013-02-12 | 2018-01-30 | Corbus Pharmaceuticals Inc | ultrapure tetrahydrocannabinol-11-oic acids |
| US20190315669A1 (en) | 2016-11-13 | 2019-10-17 | Yissum Research Development Company Of The Hebrew University Of Jerusalem Ltd. | Process for the preparation of hu-910 and crystalline structure thereof |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6096740A (en) * | 1990-11-06 | 2000-08-01 | Ramot University Authority For Applied Research And Industrial Development Ltd. | Dexanabinol derivatives and their use as neuroprotective pharmaceutical compositions |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL80411A (en) * | 1986-10-24 | 1991-08-16 | Raphael Mechoulam | Preparation of dibenzopyranol derivatives and pharmaceutical compositions containing them |
| US5284867A (en) * | 1989-11-07 | 1994-02-08 | Yissum Research Development Company Of The Hebrew University In Jerusalem | NMDA-blocking pharmaceutical compositions |
| US5521215A (en) * | 1989-11-07 | 1996-05-28 | Ramot University Authority For Applied Research And Industrial Development Ltd. | NMDA-blocking pharmaceuticals |
| IL115245A (en) * | 1995-09-11 | 2002-12-01 | Yissum Res Dev Co | Tumor necrosis factor inhibiting pharmaceuticals |
-
2003
- 2003-12-03 AU AU2003302578A patent/AU2003302578A1/en not_active Abandoned
- 2003-12-03 CA CA002507815A patent/CA2507815A1/en not_active Abandoned
- 2003-12-03 JP JP2004570714A patent/JP2006509038A/en not_active Abandoned
- 2003-12-03 WO PCT/IL2003/001023 patent/WO2004050011A2/en active Application Filing
- 2003-12-03 EP EP03812258A patent/EP1567513A2/en not_active Withdrawn
-
2006
- 2006-08-14 US US11/503,250 patent/US20070060636A1/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6096740A (en) * | 1990-11-06 | 2000-08-01 | Ramot University Authority For Applied Research And Industrial Development Ltd. | Dexanabinol derivatives and their use as neuroprotective pharmaceutical compositions |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10588974B2 (en) | 2016-04-22 | 2020-03-17 | Receptor Holdings, Inc. | Fast-acting plant-based medicinal compounds and nutritional supplements |
| US11129897B2 (en) | 2016-04-22 | 2021-09-28 | Receptor Holdings, Inc. | Fast-acting plant-based medicinal compounds and nutritional supplements |
| US11246852B2 (en) | 2016-12-02 | 2022-02-15 | Receptor Holdings, Inc. | Fast-acting plant-based medicinal compounds and nutritional supplements |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1567513A2 (en) | 2005-08-31 |
| CA2507815A1 (en) | 2004-06-17 |
| WO2004050011A3 (en) | 2004-07-29 |
| JP2006509038A (en) | 2006-03-16 |
| US20070060636A1 (en) | 2007-03-15 |
| AU2003302578A1 (en) | 2004-06-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9630941B2 (en) | Compositions containing delta-9-THC-amino acid esters and process of preparation | |
| US20070060636A1 (en) | High enantiomeric purity dexanabinol for pharmaceutical compositions | |
| US5284867A (en) | NMDA-blocking pharmaceutical compositions | |
| US5434295A (en) | Neuroprotective pharmaceutical compositions of 4-phenylpinene derivatives and certain novel 4-phenylpinene compounds | |
| US6682758B1 (en) | Water-insoluble drug delivery system | |
| EP2579845B1 (en) | Ophthalmic compositions for the administration of liposoluble acitve ingredients | |
| JP2745436B2 (en) | Phenylalkanoic acid derivative, method for producing the same, and method for separating optical isomers thereof | |
| KR20060135819A (en) | Management of ophthalmologic disorders, including macular degeneration | |
| US20040110827A1 (en) | High enantiomeric purity dexanabinol for pharmaceutical compositions | |
| US7226932B2 (en) | Self-emulsifying drug delivery system | |
| SA94150314B1 (en) | pharmaceutical emulsion | |
| CN103827113A (en) | Novel fluoroergoline analogs | |
| JP5345744B1 (en) | Retinal disease prevention, amelioration, or treatment | |
| CA2900982A1 (en) | Ultrapure tetrahydrocannabinol-11-oic acids | |
| US7084157B2 (en) | Ophthalmic pharmaceutical compositions and methods for treating ocular inflammation | |
| KR20220118493A (en) | Cannabinoid Oral Formulations | |
| EP1140017B1 (en) | Water-insoluble drug delivery system | |
| US20070099988A1 (en) | Anti-emetic uses of (3r, 4r)-delta8-tetrahydrocannabinol-11-oic acids | |
| EP0277352B1 (en) | Synergistic mixture of azelastine and theophylline or of azelastine and beta-mimetics | |
| JPWO2002020540A1 (en) | Adenosine derivatives and uses thereof | |
| EP1916002B1 (en) | Method for prevention of degradation of thermally unstable substance | |
| CA1166643A (en) | Salt of ¬(1-benzyl-1h-indazol-3-yl)oxy| acetic acid (or bendazac) with the lysine | |
| JP5092266B2 (en) | Ophthalmic agent | |
| IL92238A (en) | Nmda blocking composition containing tetrahydro-cannabinol derivatives some novel tetrahydro-cannabinol derivatives and process for their preparation | |
| JPH0368515A (en) | Antiallergic drug |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2003302578 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 168819 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2507815 Country of ref document: CA Ref document number: 2003812258 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004570714 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1416/CHENP/2005 Country of ref document: IN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003812258 Country of ref document: EP |