WO2004041279A1 - Gamma-aminoamide modulators of chemokine receptor activity - Google Patents
Gamma-aminoamide modulators of chemokine receptor activity Download PDFInfo
- Publication number
- WO2004041279A1 WO2004041279A1 PCT/US2003/034009 US0334009W WO2004041279A1 WO 2004041279 A1 WO2004041279 A1 WO 2004041279A1 US 0334009 W US0334009 W US 0334009W WO 2004041279 A1 WO2004041279 A1 WO 2004041279A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mmol
- phenyl
- substituents
- heterocycle
- trifluoromethyl
- Prior art date
Links
- 102000009410 Chemokine receptor Human genes 0.000 title claims abstract description 39
- 108050000299 Chemokine receptor Proteins 0.000 title claims abstract description 39
- 230000000694 effects Effects 0.000 title claims abstract description 18
- 150000001875 compounds Chemical class 0.000 claims abstract description 199
- 125000001424 substituent group Chemical group 0.000 claims description 93
- -1 where the lH-indene Chemical compound 0.000 claims description 78
- 125000000623 heterocyclic group Chemical group 0.000 claims description 74
- 238000000034 method Methods 0.000 claims description 59
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 56
- 125000005843 halogen group Chemical group 0.000 claims description 52
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 52
- 239000001257 hydrogen Substances 0.000 claims description 38
- 229910052739 hydrogen Inorganic materials 0.000 claims description 38
- 150000003839 salts Chemical class 0.000 claims description 34
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 28
- 125000000217 alkyl group Chemical group 0.000 claims description 28
- 229910052757 nitrogen Inorganic materials 0.000 claims description 28
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 27
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 25
- 201000010099 disease Diseases 0.000 claims description 16
- 230000002757 inflammatory effect Effects 0.000 claims description 15
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 13
- 239000008194 pharmaceutical composition Substances 0.000 claims description 13
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 12
- 125000002098 pyridazinyl group Chemical group 0.000 claims description 10
- 125000004076 pyridyl group Chemical group 0.000 claims description 10
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 9
- HBEDSQVIWPRPAY-UHFFFAOYSA-N 2,3-dihydrobenzofuran Chemical compound C1=CC=C2OCCC2=C1 HBEDSQVIWPRPAY-UHFFFAOYSA-N 0.000 claims description 8
- 208000035475 disorder Diseases 0.000 claims description 8
- 230000004957 immunoregulator effect Effects 0.000 claims description 7
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 7
- 150000001204 N-oxides Chemical class 0.000 claims description 6
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 6
- 150000002431 hydrogen Chemical class 0.000 claims description 6
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 6
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims description 6
- 241000124008 Mammalia Species 0.000 claims description 5
- 125000001153 fluoro group Chemical group F* 0.000 claims description 5
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 5
- KVRZARWOKBNZMM-UHFFFAOYSA-N 1,3-dihydro-2-benzothiophene Chemical compound C1=CC=C2CSCC2=C1 KVRZARWOKBNZMM-UHFFFAOYSA-N 0.000 claims description 4
- YJUFGFXVASPYFQ-UHFFFAOYSA-N 2,3-dihydro-1-benzothiophene Chemical compound C1=CC=C2SCCC2=C1 YJUFGFXVASPYFQ-UHFFFAOYSA-N 0.000 claims description 4
- 125000002541 furyl group Chemical group 0.000 claims description 4
- 125000002883 imidazolyl group Chemical group 0.000 claims description 4
- PQNFLJBBNBOBRQ-UHFFFAOYSA-N indane Chemical compound C1=CC=C2CCCC2=C1 PQNFLJBBNBOBRQ-UHFFFAOYSA-N 0.000 claims description 4
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 claims description 4
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 4
- 125000001715 oxadiazolyl group Chemical group 0.000 claims description 4
- 125000002971 oxazolyl group Chemical group 0.000 claims description 4
- SFLGSKRGOWRGBR-UHFFFAOYSA-N phthalane Chemical compound C1=CC=C2COCC2=C1 SFLGSKRGOWRGBR-UHFFFAOYSA-N 0.000 claims description 4
- 125000003373 pyrazinyl group Chemical group 0.000 claims description 4
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 4
- 125000000168 pyrrolyl group Chemical group 0.000 claims description 4
- 125000001113 thiadiazolyl group Chemical group 0.000 claims description 4
- 125000000335 thiazolyl group Chemical group 0.000 claims description 4
- 125000001544 thienyl group Chemical group 0.000 claims description 4
- 125000001425 triazolyl group Chemical group 0.000 claims description 4
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 claims description 3
- 125000005940 1,4-dioxanyl group Chemical group 0.000 claims description 2
- 125000006288 3,5-difluorobenzyl group Chemical group [H]C1=C(F)C([H])=C(C([H])=C1F)C([H])([H])* 0.000 claims description 2
- 125000003852 3-chlorobenzyl group Chemical group [H]C1=C([H])C(=C([H])C(Cl)=C1[H])C([H])([H])* 0.000 claims description 2
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 claims description 2
- 125000006281 4-bromobenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1Br)C([H])([H])* 0.000 claims description 2
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 claims description 2
- 125000002393 azetidinyl group Chemical group 0.000 claims description 2
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 claims description 2
- 125000004601 benzofurazanyl group Chemical group N1=C2C(=NO1)C(=CC=C2)* 0.000 claims description 2
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 claims description 2
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 claims description 2
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 claims description 2
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 claims description 2
- 125000004623 carbolinyl group Chemical group 0.000 claims description 2
- 229910052799 carbon Inorganic materials 0.000 claims description 2
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 claims description 2
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 2
- 125000000723 dihydrobenzofuranyl group Chemical group O1C(CC2=C1C=CC=C2)* 0.000 claims description 2
- 125000005436 dihydrobenzothiophenyl group Chemical group S1C(CC2=C1C=CC=C2)* 0.000 claims description 2
- 125000005435 dihydrobenzoxazolyl group Chemical group O1C(NC2=C1C=CC=C2)* 0.000 claims description 2
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 claims description 2
- 125000005047 dihydroimidazolyl group Chemical group N1(CNC=C1)* 0.000 claims description 2
- 125000001070 dihydroindolyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 claims description 2
- 125000005049 dihydrooxadiazolyl group Chemical group O1N(NC=C1)* 0.000 claims description 2
- 125000005050 dihydrooxazolyl group Chemical group O1C(NC=C1)* 0.000 claims description 2
- 125000005051 dihydropyrazinyl group Chemical group N1(CC=NC=C1)* 0.000 claims description 2
- 125000005052 dihydropyrazolyl group Chemical group N1(NCC=C1)* 0.000 claims description 2
- 125000004655 dihydropyridinyl group Chemical group N1(CC=CC=C1)* 0.000 claims description 2
- 125000005053 dihydropyrimidinyl group Chemical group N1(CN=CC=C1)* 0.000 claims description 2
- 125000005054 dihydropyrrolyl group Chemical group [H]C1=C([H])C([H])([H])C([H])([H])N1* 0.000 claims description 2
- 125000005044 dihydroquinolinyl group Chemical group N1(CC=CC2=CC=CC=C12)* 0.000 claims description 2
- 125000005056 dihydrothiazolyl group Chemical group S1C(NC=C1)* 0.000 claims description 2
- 125000005057 dihydrothienyl group Chemical group S1C(CC=C1)* 0.000 claims description 2
- 125000005058 dihydrotriazolyl group Chemical group N1(NNC=C1)* 0.000 claims description 2
- 125000004634 hexahydroazepinyl group Chemical group N1(CCCCCC1)* 0.000 claims description 2
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 claims description 2
- 125000001041 indolyl group Chemical group 0.000 claims description 2
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 claims description 2
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 claims description 2
- 125000005956 isoquinolyl group Chemical group 0.000 claims description 2
- 125000001786 isothiazolyl group Chemical group 0.000 claims description 2
- 125000000842 isoxazolyl group Chemical group 0.000 claims description 2
- 125000002757 morpholinyl group Chemical group 0.000 claims description 2
- 125000003566 oxetanyl group Chemical group 0.000 claims description 2
- 125000004193 piperazinyl group Chemical group 0.000 claims description 2
- 125000003386 piperidinyl group Chemical group 0.000 claims description 2
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 claims description 2
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 2
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 claims description 2
- 125000005493 quinolyl group Chemical group 0.000 claims description 2
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 claims description 2
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 claims description 2
- 125000001412 tetrahydropyranyl group Chemical group 0.000 claims description 2
- 125000005958 tetrahydrothienyl group Chemical group 0.000 claims description 2
- 125000003831 tetrazolyl group Chemical group 0.000 claims description 2
- 125000004568 thiomorpholinyl group Chemical group 0.000 claims description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims 24
- 125000006704 (C5-C6) cycloalkyl group Chemical group 0.000 claims 2
- 102100031151 C-C chemokine receptor type 2 Human genes 0.000 abstract description 15
- 101710149815 C-C chemokine receptor type 2 Proteins 0.000 abstract description 15
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 370
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 316
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 264
- 239000000243 solution Substances 0.000 description 249
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 178
- 239000000543 intermediate Substances 0.000 description 175
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 159
- 239000000203 mixture Substances 0.000 description 155
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 142
- 239000011541 reaction mixture Substances 0.000 description 138
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 128
- 235000019439 ethyl acetate Nutrition 0.000 description 124
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 104
- 239000012267 brine Substances 0.000 description 95
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 95
- 239000000047 product Substances 0.000 description 86
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 82
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 75
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 74
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 68
- 239000002904 solvent Substances 0.000 description 68
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 66
- 238000000746 purification Methods 0.000 description 65
- 239000012044 organic layer Substances 0.000 description 63
- 239000012043 crude product Substances 0.000 description 59
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 58
- 239000002253 acid Substances 0.000 description 58
- 238000006243 chemical reaction Methods 0.000 description 53
- 238000012746 preparative thin layer chromatography Methods 0.000 description 51
- 239000007787 solid Substances 0.000 description 50
- 238000005160 1H NMR spectroscopy Methods 0.000 description 46
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 45
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 45
- 238000003756 stirring Methods 0.000 description 45
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 44
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 42
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 40
- 238000005481 NMR spectroscopy Methods 0.000 description 40
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 39
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 39
- 239000010410 layer Substances 0.000 description 37
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 36
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 35
- 238000002360 preparation method Methods 0.000 description 34
- 238000003820 Medium-pressure liquid chromatography Methods 0.000 description 30
- 230000002829 reductive effect Effects 0.000 description 29
- 239000000377 silicon dioxide Substances 0.000 description 29
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 29
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 27
- 150000002148 esters Chemical class 0.000 description 27
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 27
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 27
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 26
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 24
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 23
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 22
- 150000001299 aldehydes Chemical class 0.000 description 22
- 210000004027 cell Anatomy 0.000 description 22
- 239000012047 saturated solution Substances 0.000 description 22
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 21
- CBENFWSGALASAD-UHFFFAOYSA-N Ozone Chemical compound [O-][O+]=O CBENFWSGALASAD-UHFFFAOYSA-N 0.000 description 20
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 20
- 238000001819 mass spectrum Methods 0.000 description 20
- 238000003786 synthesis reaction Methods 0.000 description 20
- 239000002585 base Substances 0.000 description 19
- 238000003818 flash chromatography Methods 0.000 description 19
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 19
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 18
- 239000003153 chemical reaction reagent Substances 0.000 description 18
- 239000000706 filtrate Substances 0.000 description 18
- 239000003921 oil Substances 0.000 description 18
- 235000019198 oils Nutrition 0.000 description 18
- 239000000725 suspension Substances 0.000 description 18
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 17
- BHELZAPQIKSEDF-UHFFFAOYSA-N allyl bromide Chemical compound BrCC=C BHELZAPQIKSEDF-UHFFFAOYSA-N 0.000 description 17
- 150000001412 amines Chemical class 0.000 description 17
- 230000015572 biosynthetic process Effects 0.000 description 17
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 17
- 235000017557 sodium bicarbonate Nutrition 0.000 description 17
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 16
- 150000001336 alkenes Chemical class 0.000 description 16
- 235000019441 ethanol Nutrition 0.000 description 16
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 16
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 15
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 15
- ULGZDMOVFRHVEP-RWJQBGPGSA-N Erythromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 ULGZDMOVFRHVEP-RWJQBGPGSA-N 0.000 description 15
- 239000004480 active ingredient Substances 0.000 description 15
- 150000001408 amides Chemical class 0.000 description 15
- 235000011114 ammonium hydroxide Nutrition 0.000 description 15
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 15
- 238000005406 washing Methods 0.000 description 15
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 14
- QMMFVYPAHWMCMS-UHFFFAOYSA-N Dimethyl sulfide Chemical compound CSC QMMFVYPAHWMCMS-UHFFFAOYSA-N 0.000 description 14
- TUPUHSXMDIWJQT-UHFFFAOYSA-N [3-(trifluoromethoxy)phenyl]methanamine Chemical compound NCC1=CC=CC(OC(F)(F)F)=C1 TUPUHSXMDIWJQT-UHFFFAOYSA-N 0.000 description 14
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 14
- 102100021943 C-C motif chemokine 2 Human genes 0.000 description 13
- 101710155857 C-C motif chemokine 2 Proteins 0.000 description 13
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 13
- OKJHHGNJFKHGRZ-UHFFFAOYSA-N [3,5-bis(trifluoromethyl)phenyl]methanamine;hydrochloride Chemical compound Cl.NCC1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 OKJHHGNJFKHGRZ-UHFFFAOYSA-N 0.000 description 13
- 230000002378 acidificating effect Effects 0.000 description 13
- 238000007792 addition Methods 0.000 description 13
- 239000000284 extract Substances 0.000 description 13
- 239000000463 material Substances 0.000 description 13
- 239000012074 organic phase Substances 0.000 description 13
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 12
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 12
- 238000005859 coupling reaction Methods 0.000 description 12
- 210000001616 monocyte Anatomy 0.000 description 12
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical compound N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 12
- 102000019034 Chemokines Human genes 0.000 description 11
- 108010012236 Chemokines Proteins 0.000 description 11
- 230000008878 coupling Effects 0.000 description 11
- 238000010168 coupling process Methods 0.000 description 11
- 239000002808 molecular sieve Substances 0.000 description 11
- 239000011347 resin Substances 0.000 description 11
- 229920005989 resin Polymers 0.000 description 11
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 11
- 239000007858 starting material Substances 0.000 description 11
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 11
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 10
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 10
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 10
- 239000003112 inhibitor Substances 0.000 description 10
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 10
- 229910000489 osmium tetroxide Inorganic materials 0.000 description 10
- 229910000104 sodium hydride Inorganic materials 0.000 description 10
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 9
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 9
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 9
- 239000007832 Na2SO4 Substances 0.000 description 9
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 9
- 208000026935 allergic disease Diseases 0.000 description 9
- 239000007864 aqueous solution Substances 0.000 description 9
- 238000001035 drying Methods 0.000 description 9
- 230000006870 function Effects 0.000 description 9
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 9
- 238000010992 reflux Methods 0.000 description 9
- 229910052938 sodium sulfate Inorganic materials 0.000 description 9
- 235000011152 sodium sulphate Nutrition 0.000 description 9
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 9
- 238000001914 filtration Methods 0.000 description 8
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 8
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 8
- 230000004048 modification Effects 0.000 description 8
- 238000012986 modification Methods 0.000 description 8
- 102000005962 receptors Human genes 0.000 description 8
- 108020003175 receptors Proteins 0.000 description 8
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 8
- PYOKUURKVVELLB-UHFFFAOYSA-N trimethyl orthoformate Chemical compound COC(OC)OC PYOKUURKVVELLB-UHFFFAOYSA-N 0.000 description 8
- UTBULQCHEUWJNV-UHFFFAOYSA-N 4-phenylpiperidine Chemical class C1CNCCC1C1=CC=CC=C1 UTBULQCHEUWJNV-UHFFFAOYSA-N 0.000 description 7
- 241000282412 Homo Species 0.000 description 7
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 7
- DHVHORCFFOSRBP-UHFFFAOYSA-N [3,5-bis(trifluoromethyl)phenyl]methanamine Chemical compound NCC1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 DHVHORCFFOSRBP-UHFFFAOYSA-N 0.000 description 7
- 239000005557 antagonist Substances 0.000 description 7
- 230000005595 deprotonation Effects 0.000 description 7
- 238000010537 deprotonation reaction Methods 0.000 description 7
- 239000003814 drug Substances 0.000 description 7
- 239000003480 eluent Substances 0.000 description 7
- 239000012467 final product Substances 0.000 description 7
- 208000015181 infectious disease Diseases 0.000 description 7
- 239000004615 ingredient Substances 0.000 description 7
- 239000012285 osmium tetroxide Substances 0.000 description 7
- 239000000546 pharmaceutical excipient Substances 0.000 description 7
- 239000012312 sodium hydride Substances 0.000 description 7
- 229940086542 triethylamine Drugs 0.000 description 7
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 6
- NRPFNQUDKRYCNX-UHFFFAOYSA-N 4-methoxyphenylacetic acid Chemical compound COC1=CC=C(CC(O)=O)C=C1 NRPFNQUDKRYCNX-UHFFFAOYSA-N 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 6
- 241000282414 Homo sapiens Species 0.000 description 6
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 6
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 6
- 235000019270 ammonium chloride Nutrition 0.000 description 6
- 208000006673 asthma Diseases 0.000 description 6
- 239000003054 catalyst Substances 0.000 description 6
- 229940079593 drug Drugs 0.000 description 6
- 239000000796 flavoring agent Substances 0.000 description 6
- 231100000252 nontoxic Toxicity 0.000 description 6
- 230000003000 nontoxic effect Effects 0.000 description 6
- 230000036961 partial effect Effects 0.000 description 6
- 230000007170 pathology Effects 0.000 description 6
- IUBQJLUDMLPAGT-UHFFFAOYSA-N potassium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([K])[Si](C)(C)C IUBQJLUDMLPAGT-UHFFFAOYSA-N 0.000 description 6
- 125000006239 protecting group Chemical group 0.000 description 6
- 238000006268 reductive amination reaction Methods 0.000 description 6
- 239000003765 sweetening agent Substances 0.000 description 6
- 239000003826 tablet Substances 0.000 description 6
- 238000004809 thin layer chromatography Methods 0.000 description 6
- RAIPHJJURHTUIC-UHFFFAOYSA-N 1,3-thiazol-2-amine Chemical group NC1=NC=CS1 RAIPHJJURHTUIC-UHFFFAOYSA-N 0.000 description 5
- FPIRBHDGWMWJEP-UHFFFAOYSA-N 1-hydroxy-7-azabenzotriazole Chemical compound C1=CN=C2N(O)N=NC2=C1 FPIRBHDGWMWJEP-UHFFFAOYSA-N 0.000 description 5
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 5
- 201000001320 Atherosclerosis Diseases 0.000 description 5
- 208000023275 Autoimmune disease Diseases 0.000 description 5
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- AYCPARAPKDAOEN-LJQANCHMSA-N N-[(1S)-2-(dimethylamino)-1-phenylethyl]-6,6-dimethyl-3-[(2-methyl-4-thieno[3,2-d]pyrimidinyl)amino]-1,4-dihydropyrrolo[3,4-c]pyrazole-5-carboxamide Chemical compound C1([C@H](NC(=O)N2C(C=3NN=C(NC=4C=5SC=CC=5N=C(C)N=4)C=3C2)(C)C)CN(C)C)=CC=CC=C1 AYCPARAPKDAOEN-LJQANCHMSA-N 0.000 description 5
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 5
- 150000001241 acetals Chemical class 0.000 description 5
- 235000001014 amino acid Nutrition 0.000 description 5
- 150000001413 amino acids Chemical class 0.000 description 5
- 239000007900 aqueous suspension Substances 0.000 description 5
- 229910052786 argon Inorganic materials 0.000 description 5
- 230000001363 autoimmune Effects 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- 150000001735 carboxylic acids Chemical class 0.000 description 5
- 239000007859 condensation product Substances 0.000 description 5
- 238000001816 cooling Methods 0.000 description 5
- 238000002425 crystallisation Methods 0.000 description 5
- 230000008025 crystallization Effects 0.000 description 5
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 5
- 235000014113 dietary fatty acids Nutrition 0.000 description 5
- 229940043279 diisopropylamine Drugs 0.000 description 5
- 150000002009 diols Chemical class 0.000 description 5
- 125000004494 ethyl ester group Chemical group 0.000 description 5
- 239000000194 fatty acid Substances 0.000 description 5
- 229930195729 fatty acid Natural products 0.000 description 5
- 150000004665 fatty acids Chemical class 0.000 description 5
- 235000003599 food sweetener Nutrition 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 239000012458 free base Substances 0.000 description 5
- 238000004128 high performance liquid chromatography Methods 0.000 description 5
- 230000004054 inflammatory process Effects 0.000 description 5
- 210000000265 leukocyte Anatomy 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- 239000012299 nitrogen atmosphere Substances 0.000 description 5
- WURFKUQACINBSI-UHFFFAOYSA-M ozonide Chemical compound [O]O[O-] WURFKUQACINBSI-UHFFFAOYSA-M 0.000 description 5
- 230000007115 recruitment Effects 0.000 description 5
- 239000000741 silica gel Substances 0.000 description 5
- 229910002027 silica gel Inorganic materials 0.000 description 5
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 5
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 4
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 4
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Chemical compound CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 4
- 102100032366 C-C motif chemokine 7 Human genes 0.000 description 4
- 101710155834 C-C motif chemokine 7 Proteins 0.000 description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 4
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 4
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 4
- 206010061218 Inflammation Diseases 0.000 description 4
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical compound O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- 238000010976 amide bond formation reaction Methods 0.000 description 4
- 230000008485 antagonism Effects 0.000 description 4
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 4
- 230000005587 bubbling Effects 0.000 description 4
- 230000003197 catalytic effect Effects 0.000 description 4
- 239000003795 chemical substances by application Substances 0.000 description 4
- 230000035605 chemotaxis Effects 0.000 description 4
- OROGSEYTTFOCAN-DNJOTXNNSA-N codeine Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC OROGSEYTTFOCAN-DNJOTXNNSA-N 0.000 description 4
- 239000003086 colorant Substances 0.000 description 4
- QTMDXZNDVAMKGV-UHFFFAOYSA-L copper(ii) bromide Chemical compound [Cu+2].[Br-].[Br-] QTMDXZNDVAMKGV-UHFFFAOYSA-L 0.000 description 4
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 4
- 229910001873 dinitrogen Inorganic materials 0.000 description 4
- 239000002270 dispersing agent Substances 0.000 description 4
- 239000000839 emulsion Substances 0.000 description 4
- 210000003979 eosinophil Anatomy 0.000 description 4
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 4
- 238000001704 evaporation Methods 0.000 description 4
- 230000008020 evaporation Effects 0.000 description 4
- 235000013355 food flavoring agent Nutrition 0.000 description 4
- 239000007789 gas Substances 0.000 description 4
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 4
- CGIGDMFJXJATDK-UHFFFAOYSA-N indomethacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 CGIGDMFJXJATDK-UHFFFAOYSA-N 0.000 description 4
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 4
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 4
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 4
- 210000004698 lymphocyte Anatomy 0.000 description 4
- 210000002540 macrophage Anatomy 0.000 description 4
- 235000019341 magnesium sulphate Nutrition 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- 239000003960 organic solvent Substances 0.000 description 4
- 230000003647 oxidation Effects 0.000 description 4
- 238000007254 oxidation reaction Methods 0.000 description 4
- HVAMZGADVCBITI-UHFFFAOYSA-N pent-4-enoic acid Chemical compound OC(=O)CCC=C HVAMZGADVCBITI-UHFFFAOYSA-N 0.000 description 4
- 239000003755 preservative agent Substances 0.000 description 4
- 230000002265 prevention Effects 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 238000006722 reduction reaction Methods 0.000 description 4
- 239000012508 resin bead Substances 0.000 description 4
- 230000004044 response Effects 0.000 description 4
- 239000002002 slurry Substances 0.000 description 4
- 239000012279 sodium borohydride Substances 0.000 description 4
- 229910000033 sodium borohydride Inorganic materials 0.000 description 4
- 241000894007 species Species 0.000 description 4
- 239000000375 suspending agent Substances 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- 239000000080 wetting agent Substances 0.000 description 4
- RQEUFEKYXDPUSK-SSDOTTSWSA-N (1R)-1-phenylethanamine Chemical compound C[C@@H](N)C1=CC=CC=C1 RQEUFEKYXDPUSK-SSDOTTSWSA-N 0.000 description 3
- RQEUFEKYXDPUSK-ZETCQYMHSA-N (1S)-1-phenylethanamine Chemical compound C[C@H](N)C1=CC=CC=C1 RQEUFEKYXDPUSK-ZETCQYMHSA-N 0.000 description 3
- FUSFWUFSEJXMRQ-UHFFFAOYSA-N 2-bromo-1,1-dimethoxyethane Chemical compound COC(CBr)OC FUSFWUFSEJXMRQ-UHFFFAOYSA-N 0.000 description 3
- ROLMZTIHUMKEAI-UHFFFAOYSA-N 4,5-difluoro-2-hydroxybenzonitrile Chemical compound OC1=CC(F)=C(F)C=C1C#N ROLMZTIHUMKEAI-UHFFFAOYSA-N 0.000 description 3
- 208000030507 AIDS Diseases 0.000 description 3
- 206010003645 Atopy Diseases 0.000 description 3
- 241000283690 Bos taurus Species 0.000 description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 3
- 102100034871 C-C motif chemokine 8 Human genes 0.000 description 3
- 101710155833 C-C motif chemokine 8 Proteins 0.000 description 3
- 102000001902 CC Chemokines Human genes 0.000 description 3
- 108010040471 CC Chemokines Proteins 0.000 description 3
- 102000004497 CCR2 Receptors Human genes 0.000 description 3
- 108010017312 CCR2 Receptors Proteins 0.000 description 3
- 102100023688 Eotaxin Human genes 0.000 description 3
- 101710139422 Eotaxin Proteins 0.000 description 3
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 3
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 3
- 206010062016 Immunosuppression Diseases 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- LFTLOKWAGJYHHR-UHFFFAOYSA-N N-methylmorpholine N-oxide Chemical compound CN1(=O)CCOCC1 LFTLOKWAGJYHHR-UHFFFAOYSA-N 0.000 description 3
- 241001494479 Pecora Species 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- 206010039085 Rhinitis allergic Diseases 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- 210000001744 T-lymphocyte Anatomy 0.000 description 3
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric Acid Chemical compound [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 3
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical group C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 3
- 230000008484 agonism Effects 0.000 description 3
- 230000029936 alkylation Effects 0.000 description 3
- 238000005804 alkylation reaction Methods 0.000 description 3
- 201000010105 allergic rhinitis Diseases 0.000 description 3
- 229950003476 aminothiazole Drugs 0.000 description 3
- 150000008064 anhydrides Chemical class 0.000 description 3
- 239000000739 antihistaminic agent Substances 0.000 description 3
- 239000008346 aqueous phase Substances 0.000 description 3
- 125000003118 aryl group Chemical group 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 201000008937 atopic dermatitis Diseases 0.000 description 3
- 208000010668 atopic eczema Diseases 0.000 description 3
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 3
- 235000013877 carbamide Nutrition 0.000 description 3
- 235000011089 carbon dioxide Nutrition 0.000 description 3
- 239000002975 chemoattractant Substances 0.000 description 3
- 235000008504 concentrate Nutrition 0.000 description 3
- 239000012141 concentrate Substances 0.000 description 3
- 235000018417 cysteine Nutrition 0.000 description 3
- 239000002274 desiccant Substances 0.000 description 3
- 150000005690 diesters Chemical class 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- TUJCBXWUOMQYHG-UHFFFAOYSA-N ethyl 2-[2-[(2-methylpropan-2-yl)oxycarbonylamino]-1,3-thiazol-4-yl]pent-4-enoate Chemical compound CCOC(=O)C(CC=C)C1=CSC(NC(=O)OC(C)(C)C)=N1 TUJCBXWUOMQYHG-UHFFFAOYSA-N 0.000 description 3
- 238000003810 ethyl acetate extraction Methods 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- OROGSEYTTFOCAN-UHFFFAOYSA-N hydrocodone Natural products C1C(N(CCC234)C)C2C=CC(O)C3OC2=C4C1=CC=C2OC OROGSEYTTFOCAN-UHFFFAOYSA-N 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 238000006460 hydrolysis reaction Methods 0.000 description 3
- UPZJLQCUYUTZIE-UHFFFAOYSA-N hydron;4-phenylpiperidine;chloride Chemical compound Cl.C1CNCCC1C1=CC=CC=C1 UPZJLQCUYUTZIE-UHFFFAOYSA-N 0.000 description 3
- 230000001506 immunosuppresive effect Effects 0.000 description 3
- 230000008595 infiltration Effects 0.000 description 3
- 238000001764 infiltration Methods 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- UEXQBEVWFZKHNB-UHFFFAOYSA-N intermediate 29 Natural products C1=CC(N)=CC=C1NC1=NC=CC=N1 UEXQBEVWFZKHNB-UHFFFAOYSA-N 0.000 description 3
- 150000002576 ketones Chemical class 0.000 description 3
- 230000003902 lesion Effects 0.000 description 3
- 229940057995 liquid paraffin Drugs 0.000 description 3
- AEUKDPKXTPNBNY-XEYRWQBLSA-N mcp 2 Chemical compound C([C@@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CS)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CS)NC(=O)[C@H](C)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)[C@@H](N)C(C)C)C(C)C)C1=CC=CC=C1 AEUKDPKXTPNBNY-XEYRWQBLSA-N 0.000 description 3
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 3
- 150000004702 methyl esters Chemical group 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- 150000002825 nitriles Chemical class 0.000 description 3
- 239000004006 olive oil Substances 0.000 description 3
- 235000008390 olive oil Nutrition 0.000 description 3
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 3
- 229960003424 phenylacetic acid Drugs 0.000 description 3
- 239000003279 phenylacetic acid Substances 0.000 description 3
- 230000003389 potentiating effect Effects 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 description 3
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 3
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 3
- 230000000699 topical effect Effects 0.000 description 3
- 150000003672 ureas Chemical class 0.000 description 3
- 230000002792 vascular Effects 0.000 description 3
- 239000011701 zinc Substances 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- SHBIVWMKPUPDGJ-UHFFFAOYSA-N 1,1,1,4,4,4-hexafluoro-2-phenylbutan-2-ol Chemical compound FC(F)(F)CC(O)(C(F)(F)F)C1=CC=CC=C1 SHBIVWMKPUPDGJ-UHFFFAOYSA-N 0.000 description 2
- ZORQXIQZAOLNGE-UHFFFAOYSA-N 1,1-difluorocyclohexane Chemical compound FC1(F)CCCCC1 ZORQXIQZAOLNGE-UHFFFAOYSA-N 0.000 description 2
- LQQKDSXCDXHLLF-UHFFFAOYSA-N 1,3-dibromopropan-2-one Chemical compound BrCC(=O)CBr LQQKDSXCDXHLLF-UHFFFAOYSA-N 0.000 description 2
- HLVFKOKELQSXIQ-UHFFFAOYSA-N 1-bromo-2-methylpropane Chemical compound CC(C)CBr HLVFKOKELQSXIQ-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 2
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Chemical compound C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 2
- JVSFQJZRHXAUGT-UHFFFAOYSA-N 2,2-dimethylpropanoyl chloride Chemical compound CC(C)(C)C(Cl)=O JVSFQJZRHXAUGT-UHFFFAOYSA-N 0.000 description 2
- CNIIGCLFLJGOGP-UHFFFAOYSA-N 2-(1-naphthalenylmethyl)-4,5-dihydro-1H-imidazole Chemical compound C=1C=CC2=CC=CC=C2C=1CC1=NCCN1 CNIIGCLFLJGOGP-UHFFFAOYSA-N 0.000 description 2
- MGKPFALCNDRSQD-UHFFFAOYSA-N 2-(4-fluorophenyl)acetic acid Chemical compound OC(=O)CC1=CC=C(F)C=C1 MGKPFALCNDRSQD-UHFFFAOYSA-N 0.000 description 2
- MFYSUUPKMDJYPF-UHFFFAOYSA-N 2-[(4-methyl-2-nitrophenyl)diazenyl]-3-oxo-n-phenylbutanamide Chemical compound C=1C=CC=CC=1NC(=O)C(C(=O)C)N=NC1=CC=C(C)C=C1[N+]([O-])=O MFYSUUPKMDJYPF-UHFFFAOYSA-N 0.000 description 2
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- IZHVBANLECCAGF-UHFFFAOYSA-N 2-hydroxy-3-(octadecanoyloxy)propyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)COC(=O)CCCCCCCCCCCCCCCCC IZHVBANLECCAGF-UHFFFAOYSA-N 0.000 description 2
- YOETUEMZNOLGDB-UHFFFAOYSA-N 2-methylpropyl carbonochloridate Chemical compound CC(C)COC(Cl)=O YOETUEMZNOLGDB-UHFFFAOYSA-N 0.000 description 2
- RCNOGGGBSSVMAS-UHFFFAOYSA-N 2-thiophen-3-ylacetic acid Chemical compound OC(=O)CC=1C=CSC=1 RCNOGGGBSSVMAS-UHFFFAOYSA-N 0.000 description 2
- LEGPZHPSIPPYIO-UHFFFAOYSA-N 3-Methoxyphenylacetic acid Chemical compound COC1=CC=CC(CC(O)=O)=C1 LEGPZHPSIPPYIO-UHFFFAOYSA-N 0.000 description 2
- USEGQJLHQSTGHW-UHFFFAOYSA-N 3-bromo-2-methylprop-1-ene Chemical compound CC(=C)CBr USEGQJLHQSTGHW-UHFFFAOYSA-N 0.000 description 2
- KVCQTKNUUQOELD-UHFFFAOYSA-N 4-amino-n-[1-(3-chloro-2-fluoroanilino)-6-methylisoquinolin-5-yl]thieno[3,2-d]pyrimidine-7-carboxamide Chemical compound N=1C=CC2=C(NC(=O)C=3C4=NC=NC(N)=C4SC=3)C(C)=CC=C2C=1NC1=CC=CC(Cl)=C1F KVCQTKNUUQOELD-UHFFFAOYSA-N 0.000 description 2
- XVMSFILGAMDHEY-UHFFFAOYSA-N 6-(4-aminophenyl)sulfonylpyridin-3-amine Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=N1 XVMSFILGAMDHEY-UHFFFAOYSA-N 0.000 description 2
- 206010001513 AIDS related complex Diseases 0.000 description 2
- 244000215068 Acacia senegal Species 0.000 description 2
- 235000006491 Acacia senegal Nutrition 0.000 description 2
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 2
- 235000003911 Arachis Nutrition 0.000 description 2
- 244000105624 Arachis hypogaea Species 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 2
- 102100024167 C-C chemokine receptor type 3 Human genes 0.000 description 2
- 102100035875 C-C chemokine receptor type 5 Human genes 0.000 description 2
- 102100028990 C-X-C chemokine receptor type 3 Human genes 0.000 description 2
- 101710082514 C-X-C chemokine receptor type 3 Proteins 0.000 description 2
- 101150041968 CDC13 gene Proteins 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- 206010010744 Conjunctivitis allergic Diseases 0.000 description 2
- 229910021590 Copper(II) bromide Inorganic materials 0.000 description 2
- 102000010907 Cyclooxygenase 2 Human genes 0.000 description 2
- 108010037462 Cyclooxygenase 2 Proteins 0.000 description 2
- 102000004127 Cytokines Human genes 0.000 description 2
- 108090000695 Cytokines Proteins 0.000 description 2
- 201000004624 Dermatitis Diseases 0.000 description 2
- 206010012434 Dermatitis allergic Diseases 0.000 description 2
- YXHKONLOYHBTNS-UHFFFAOYSA-N Diazomethane Chemical compound C=[N+]=[N-] YXHKONLOYHBTNS-UHFFFAOYSA-N 0.000 description 2
- 241000283086 Equidae Species 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- 241000282326 Felis catus Species 0.000 description 2
- 229920000084 Gum arabic Polymers 0.000 description 2
- 239000007995 HEPES buffer Substances 0.000 description 2
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 2
- 239000012981 Hank's balanced salt solution Substances 0.000 description 2
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 2
- 102000014150 Interferons Human genes 0.000 description 2
- 108010050904 Interferons Proteins 0.000 description 2
- 208000029523 Interstitial Lung disease Diseases 0.000 description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 208000019695 Migraine disease Diseases 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 description 2
- 241000244206 Nematoda Species 0.000 description 2
- JAUOIFJMECXRGI-UHFFFAOYSA-N Neoclaritin Chemical compound C=1C(Cl)=CC=C2C=1CCC1=CC=CN=C1C2=C1CCNCC1 JAUOIFJMECXRGI-UHFFFAOYSA-N 0.000 description 2
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 2
- MITFXPHMIHQXPI-UHFFFAOYSA-N Oraflex Chemical compound N=1C2=CC(C(C(O)=O)C)=CC=C2OC=1C1=CC=C(Cl)C=C1 MITFXPHMIHQXPI-UHFFFAOYSA-N 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical compound ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 description 2
- 201000004681 Psoriasis Diseases 0.000 description 2
- 201000001263 Psoriatic Arthritis Diseases 0.000 description 2
- 208000036824 Psoriatic arthropathy Diseases 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- 206010040070 Septic Shock Diseases 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 2
- GUGOEEXESWIERI-UHFFFAOYSA-N Terfenadine Chemical compound C1=CC(C(C)(C)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 GUGOEEXESWIERI-UHFFFAOYSA-N 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 2
- ZZHLYYDVIOPZBE-UHFFFAOYSA-N Trimeprazine Chemical compound C1=CC=C2N(CC(CN(C)C)C)C3=CC=CC=C3SC2=C1 ZZHLYYDVIOPZBE-UHFFFAOYSA-N 0.000 description 2
- 208000027418 Wounds and injury Diseases 0.000 description 2
- HUCJFAOMUPXHDK-UHFFFAOYSA-N Xylometazoline Chemical compound CC1=CC(C(C)(C)C)=CC(C)=C1CC1=NCCN1 HUCJFAOMUPXHDK-UHFFFAOYSA-N 0.000 description 2
- 235000010489 acacia gum Nutrition 0.000 description 2
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 2
- 239000012346 acetyl chloride Substances 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 239000000556 agonist Substances 0.000 description 2
- 150000001350 alkyl halides Chemical class 0.000 description 2
- 201000009961 allergic asthma Diseases 0.000 description 2
- 208000002205 allergic conjunctivitis Diseases 0.000 description 2
- 230000009285 allergic inflammation Effects 0.000 description 2
- NMPVEAUIHMEAQP-UHFFFAOYSA-N alpha-bromo-acetaldehyde Natural products BrCC=O NMPVEAUIHMEAQP-UHFFFAOYSA-N 0.000 description 2
- 238000010640 amide synthesis reaction Methods 0.000 description 2
- 150000001450 anions Chemical class 0.000 description 2
- 239000002260 anti-inflammatory agent Substances 0.000 description 2
- 208000024998 atopic conjunctivitis Diseases 0.000 description 2
- 150000001540 azides Chemical class 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 229910000085 borane Inorganic materials 0.000 description 2
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 235000010216 calcium carbonate Nutrition 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 229910000389 calcium phosphate Inorganic materials 0.000 description 2
- 235000011010 calcium phosphates Nutrition 0.000 description 2
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 2
- 229960004126 codeine Drugs 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- 150000001945 cysteines Chemical class 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- 238000012217 deletion Methods 0.000 description 2
- 230000037430 deletion Effects 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- 238000001212 derivatisation Methods 0.000 description 2
- 201000001981 dermatomyositis Diseases 0.000 description 2
- 229960001271 desloratadine Drugs 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- SHQNGLYXRFCPGZ-UHFFFAOYSA-N ethyl 2-(2-amino-1,3-thiazol-4-yl)acetate Chemical compound CCOC(=O)CC1=CSC(N)=N1 SHQNGLYXRFCPGZ-UHFFFAOYSA-N 0.000 description 2
- NHOIFNKDYIKJAN-UHFFFAOYSA-N ethyl 2-[2-[(2-methylpropan-2-yl)oxycarbonylamino]-1,3-thiazol-4-yl]-4-oxobutanoate Chemical compound CCOC(=O)C(CC=O)C1=CSC(NC(=O)OC(C)(C)C)=N1 NHOIFNKDYIKJAN-UHFFFAOYSA-N 0.000 description 2
- BGXPHMCLUATZDO-UHFFFAOYSA-N ethyl 2-methyl-2-[2-[(2-methylpropan-2-yl)oxycarbonylamino]-1,3-thiazol-4-yl]pent-4-enoate Chemical compound CCOC(=O)C(C)(CC=C)C1=CSC(NC(=O)OC(C)(C)C)=N1 BGXPHMCLUATZDO-UHFFFAOYSA-N 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 2
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 2
- 238000005984 hydrogenation reaction Methods 0.000 description 2
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 description 2
- 230000009610 hypersensitivity Effects 0.000 description 2
- 229960001680 ibuprofen Drugs 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- 229960003444 immunosuppressant agent Drugs 0.000 description 2
- 239000003018 immunosuppressive agent Substances 0.000 description 2
- 229960000905 indomethacin Drugs 0.000 description 2
- 208000027866 inflammatory disease Diseases 0.000 description 2
- 230000028709 inflammatory response Effects 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 208000014674 injury Diseases 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 229940079322 interferon Drugs 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- 235000010445 lecithin Nutrition 0.000 description 2
- 239000000787 lecithin Substances 0.000 description 2
- 229940067606 lecithin Drugs 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- AHCNXVCAVUYIOU-UHFFFAOYSA-M lithium hydroperoxide Chemical compound [Li+].[O-]O AHCNXVCAVUYIOU-UHFFFAOYSA-M 0.000 description 2
- UBJFKNSINUCEAL-UHFFFAOYSA-N lithium;2-methylpropane Chemical compound [Li+].C[C-](C)C UBJFKNSINUCEAL-UHFFFAOYSA-N 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 2
- CYEBJEDOHLIWNP-UHFFFAOYSA-N methanethioamide Chemical compound NC=S CYEBJEDOHLIWNP-UHFFFAOYSA-N 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- RWIKCBHOVNDESJ-NSCUHMNNSA-N methyl (e)-4-bromobut-2-enoate Chemical compound COC(=O)\C=C\CBr RWIKCBHOVNDESJ-NSCUHMNNSA-N 0.000 description 2
- RPUSRLKKXPQSGP-UHFFFAOYSA-N methyl 3-phenylpropanoate Chemical compound COC(=O)CCC1=CC=CC=C1 RPUSRLKKXPQSGP-UHFFFAOYSA-N 0.000 description 2
- HAMGRBXTJNITHG-UHFFFAOYSA-N methyl isocyanate Chemical compound CN=C=O HAMGRBXTJNITHG-UHFFFAOYSA-N 0.000 description 2
- 230000005012 migration Effects 0.000 description 2
- 238000013508 migration Methods 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 2
- BQJCRHHNABKAKU-KBQPJGBKSA-N morphine Chemical compound O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O BQJCRHHNABKAKU-KBQPJGBKSA-N 0.000 description 2
- 229960002009 naproxen Drugs 0.000 description 2
- CMWTZPSULFXXJA-VIFPVBQESA-M naproxen(1-) Chemical compound C1=C([C@H](C)C([O-])=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-M 0.000 description 2
- LQNUZADURLCDLV-UHFFFAOYSA-N nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC=C1 LQNUZADURLCDLV-UHFFFAOYSA-N 0.000 description 2
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 2
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 2
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 2
- 239000000346 nonvolatile oil Substances 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- WYWIFABBXFUGLM-UHFFFAOYSA-N oxymetazoline Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C)=C1CC1=NCCN1 WYWIFABBXFUGLM-UHFFFAOYSA-N 0.000 description 2
- 230000001575 pathological effect Effects 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- CPJSUEIXXCENMM-UHFFFAOYSA-N phenacetin Chemical compound CCOC1=CC=C(NC(C)=O)C=C1 CPJSUEIXXCENMM-UHFFFAOYSA-N 0.000 description 2
- XKJCHHZQLQNZHY-UHFFFAOYSA-N phthalimide Chemical compound C1=CC=C2C(=O)NC(=O)C2=C1 XKJCHHZQLQNZHY-UHFFFAOYSA-N 0.000 description 2
- KNCYXPMJDCCGSJ-UHFFFAOYSA-N piperidine-2,6-dione Chemical class O=C1CCCC(=O)N1 KNCYXPMJDCCGSJ-UHFFFAOYSA-N 0.000 description 2
- 229960002702 piroxicam Drugs 0.000 description 2
- QYSPLQLAKJAUJT-UHFFFAOYSA-N piroxicam Chemical compound OC=1C2=CC=CC=C2S(=O)(=O)N(C)C=1C(=O)NC1=CC=CC=N1 QYSPLQLAKJAUJT-UHFFFAOYSA-N 0.000 description 2
- 208000005987 polymyositis Diseases 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 230000001624 sedative effect Effects 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- WBHQBSYUUJJSRZ-UHFFFAOYSA-M sodium bisulfate Chemical compound [Na+].OS([O-])(=O)=O WBHQBSYUUJJSRZ-UHFFFAOYSA-M 0.000 description 2
- 229910000342 sodium bisulfate Inorganic materials 0.000 description 2
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 2
- 238000000638 solvent extraction Methods 0.000 description 2
- 235000011069 sorbitan monooleate Nutrition 0.000 description 2
- 239000001593 sorbitan monooleate Substances 0.000 description 2
- 229940035049 sorbitan monooleate Drugs 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 230000003637 steroidlike Effects 0.000 description 2
- 239000012258 stirred mixture Substances 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 229940124530 sulfonamide Drugs 0.000 description 2
- 150000003456 sulfonamides Chemical class 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 2
- 229960001967 tacrolimus Drugs 0.000 description 2
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 2
- 208000004441 taeniasis Diseases 0.000 description 2
- IOGXOCVLYRDXLW-UHFFFAOYSA-N tert-butyl nitrite Chemical compound CC(C)(C)ON=O IOGXOCVLYRDXLW-UHFFFAOYSA-N 0.000 description 2
- ZGQQQFWPVOPRSG-UHFFFAOYSA-N tert-butyl pent-4-enoate Chemical compound CC(C)(C)OC(=O)CCC=C ZGQQQFWPVOPRSG-UHFFFAOYSA-N 0.000 description 2
- RQCNHUCCQJMSRG-UHFFFAOYSA-N tert-butyl piperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCCCC1 RQCNHUCCQJMSRG-UHFFFAOYSA-N 0.000 description 2
- ZFXYFBGIUFBOJW-UHFFFAOYSA-N theophylline Chemical compound O=C1N(C)C(=O)N(C)C2=C1NC=N2 ZFXYFBGIUFBOJW-UHFFFAOYSA-N 0.000 description 2
- QNMBSXGYAQZCTN-UHFFFAOYSA-N thiophen-3-ylboronic acid Chemical compound OB(O)C=1C=CSC=1 QNMBSXGYAQZCTN-UHFFFAOYSA-N 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- BPLUKJNHPBNVQL-UHFFFAOYSA-N triphenylarsine Chemical compound C1=CC=CC=C1[As](C=1C=CC=CC=1)C1=CC=CC=C1 BPLUKJNHPBNVQL-UHFFFAOYSA-N 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 230000009278 visceral effect Effects 0.000 description 2
- 238000010792 warming Methods 0.000 description 2
- XWTYSIMOBUGWOL-UHFFFAOYSA-N (+-)-Terbutaline Chemical compound CC(C)(C)NCC(O)C1=CC(O)=CC(O)=C1 XWTYSIMOBUGWOL-UHFFFAOYSA-N 0.000 description 1
- LUZOFMGZMUZSSK-LRDDRELGSA-N (-)-indolactam V Chemical compound C1[C@@H](CO)NC(=O)[C@H](C(C)C)N(C)C2=CC=CC3=C2C1=CN3 LUZOFMGZMUZSSK-LRDDRELGSA-N 0.000 description 1
- VCGRFBXVSFAGGA-UHFFFAOYSA-N (1,1-dioxo-1,4-thiazinan-4-yl)-[6-[[3-(4-fluorophenyl)-5-methyl-1,2-oxazol-4-yl]methoxy]pyridin-3-yl]methanone Chemical compound CC=1ON=C(C=2C=CC(F)=CC=2)C=1COC(N=C1)=CC=C1C(=O)N1CCS(=O)(=O)CC1 VCGRFBXVSFAGGA-UHFFFAOYSA-N 0.000 description 1
- UKSZBOKPHAQOMP-SVLSSHOZSA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 UKSZBOKPHAQOMP-SVLSSHOZSA-N 0.000 description 1
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 1
- XUFXOAAUWZOOIT-SXARVLRPSA-N (2R,3R,4R,5S,6R)-5-[[(2R,3R,4R,5S,6R)-5-[[(2R,3R,4S,5S,6R)-3,4-dihydroxy-6-methyl-5-[[(1S,4R,5S,6S)-4,5,6-trihydroxy-3-(hydroxymethyl)-1-cyclohex-2-enyl]amino]-2-oxanyl]oxy]-3,4-dihydroxy-6-(hydroxymethyl)-2-oxanyl]oxy]-6-(hydroxymethyl)oxane-2,3,4-triol Chemical compound O([C@H]1O[C@H](CO)[C@H]([C@@H]([C@H]1O)O)O[C@H]1O[C@@H]([C@H]([C@H](O)[C@H]1O)N[C@@H]1[C@@H]([C@@H](O)[C@H](O)C(CO)=C1)O)C)[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O XUFXOAAUWZOOIT-SXARVLRPSA-N 0.000 description 1
- RJMIEHBSYVWVIN-LLVKDONJSA-N (2r)-2-[4-(3-oxo-1h-isoindol-2-yl)phenyl]propanoic acid Chemical compound C1=CC([C@H](C(O)=O)C)=CC=C1N1C(=O)C2=CC=CC=C2C1 RJMIEHBSYVWVIN-LLVKDONJSA-N 0.000 description 1
- ZEYYDOLCHFETHQ-JOCHJYFZSA-N (2r)-2-cyclopentyl-2-[4-(quinolin-2-ylmethoxy)phenyl]acetic acid Chemical compound C1([C@@H](C(=O)O)C=2C=CC(OCC=3N=C4C=CC=CC4=CC=3)=CC=2)CCCC1 ZEYYDOLCHFETHQ-JOCHJYFZSA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- RDJGLLICXDHJDY-NSHDSACASA-N (2s)-2-(3-phenoxyphenyl)propanoic acid Chemical compound OC(=O)[C@@H](C)C1=CC=CC(OC=2C=CC=CC=2)=C1 RDJGLLICXDHJDY-NSHDSACASA-N 0.000 description 1
- GUHPRPJDBZHYCJ-SECBINFHSA-N (2s)-2-(5-benzoylthiophen-2-yl)propanoic acid Chemical compound S1C([C@H](C(O)=O)C)=CC=C1C(=O)C1=CC=CC=C1 GUHPRPJDBZHYCJ-SECBINFHSA-N 0.000 description 1
- MDKGKXOCJGEUJW-VIFPVBQESA-N (2s)-2-[4-(thiophene-2-carbonyl)phenyl]propanoic acid Chemical compound C1=CC([C@@H](C(O)=O)C)=CC=C1C(=O)C1=CC=CS1 MDKGKXOCJGEUJW-VIFPVBQESA-N 0.000 description 1
- PZIFPMYXXCAOCC-JWQCQUIFSA-N (2s,3r)-3-(2-carboxyethylsulfanyl)-2-hydroxy-3-[2-(8-phenyloctyl)phenyl]propanoic acid Chemical compound OC(=O)CCS[C@@H]([C@@H](O)C(O)=O)C1=CC=CC=C1CCCCCCCCC1=CC=CC=C1 PZIFPMYXXCAOCC-JWQCQUIFSA-N 0.000 description 1
- ICIJWOWQUHHETJ-UHFFFAOYSA-N (3,5-dichlorophenyl)methanamine Chemical group NCC1=CC(Cl)=CC(Cl)=C1 ICIJWOWQUHHETJ-UHFFFAOYSA-N 0.000 description 1
- YGZJTYCCONJJGZ-UHFFFAOYSA-N (3,5-dimethoxyphenyl)methanamine Chemical group COC1=CC(CN)=CC(OC)=C1 YGZJTYCCONJJGZ-UHFFFAOYSA-N 0.000 description 1
- LQLOGZQVKUNBRX-UHFFFAOYSA-N (3-iodophenyl)methanamine Chemical group NCC1=CC=CC(I)=C1 LQLOGZQVKUNBRX-UHFFFAOYSA-N 0.000 description 1
- ZRTGHKVPFXNDHE-UHFFFAOYSA-N (3-methyl-1,2-thiazol-5-yl)azanium;chloride Chemical compound [Cl-].CC=1C=C([NH3+])SN=1 ZRTGHKVPFXNDHE-UHFFFAOYSA-N 0.000 description 1
- CIUYJYRQKYGNQP-UHFFFAOYSA-N (3-nitrophenyl)methanamine Chemical group NCC1=CC=CC([N+]([O-])=O)=C1 CIUYJYRQKYGNQP-UHFFFAOYSA-N 0.000 description 1
- WKHABRRJMGVELW-UHFFFAOYSA-N (3-phenylphenyl)methanamine Chemical group NCC1=CC=CC(C=2C=CC=CC=2)=C1 WKHABRRJMGVELW-UHFFFAOYSA-N 0.000 description 1
- ZGGHKIMDNBDHJB-NRFPMOEYSA-M (3R,5S)-fluvastatin sodium Chemical compound [Na+].C12=CC=CC=C2N(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 ZGGHKIMDNBDHJB-NRFPMOEYSA-M 0.000 description 1
- OQANPHBRHBJGNZ-FYJGNVAPSA-N (3e)-6-oxo-3-[[4-(pyridin-2-ylsulfamoyl)phenyl]hydrazinylidene]cyclohexa-1,4-diene-1-carboxylic acid Chemical compound C1=CC(=O)C(C(=O)O)=C\C1=N\NC1=CC=C(S(=O)(=O)NC=2N=CC=CC=2)C=C1 OQANPHBRHBJGNZ-FYJGNVAPSA-N 0.000 description 1
- GBBJBUGPGFNISJ-YDQXZVTASA-N (4as,7r,8as)-9,9-dimethyltetrahydro-4h-4a,7-methanobenzo[c][1,2]oxazireno[2,3-b]isothiazole 3,3-dioxide Chemical compound C1S(=O)(=O)N2O[C@@]32C[C@@H]2C(C)(C)[C@]13CC2 GBBJBUGPGFNISJ-YDQXZVTASA-N 0.000 description 1
- OJOFMLDBXPDXLQ-SECBINFHSA-N (4r)-4-benzyl-1,3-oxazolidin-2-one Chemical compound C1OC(=O)N[C@@H]1CC1=CC=CC=C1 OJOFMLDBXPDXLQ-SECBINFHSA-N 0.000 description 1
- DLBSGOZFSFUDKO-SECBINFHSA-N (5r)-5-benzyl-1,3-oxazolidin-2-one Chemical compound O1C(=O)NC[C@H]1CC1=CC=CC=C1 DLBSGOZFSFUDKO-SECBINFHSA-N 0.000 description 1
- KFTQAKJFNJSGBS-ZDUSSCGKSA-N (5s)-5-benzyl-3-(2-cyclopropylacetyl)-1,3-oxazolidin-2-one Chemical compound O([C@@H](CC=1C=CC=CC=1)C1)C(=O)N1C(=O)CC1CC1 KFTQAKJFNJSGBS-ZDUSSCGKSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- DYLIWHYUXAJDOJ-OWOJBTEDSA-N (e)-4-(6-aminopurin-9-yl)but-2-en-1-ol Chemical compound NC1=NC=NC2=C1N=CN2C\C=C\CO DYLIWHYUXAJDOJ-OWOJBTEDSA-N 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- IYCKMNAVTMOAKD-UHFFFAOYSA-N 1,2-thiazol-3-amine Chemical compound NC=1C=CSN=1 IYCKMNAVTMOAKD-UHFFFAOYSA-N 0.000 description 1
- ZWXDAANEJMSCEX-UHFFFAOYSA-N 1-(3-anilinophenyl)ethanone Chemical compound CC(=O)C1=CC=CC(NC=2C=CC=CC=2)=C1 ZWXDAANEJMSCEX-UHFFFAOYSA-N 0.000 description 1
- JWOHBPPVVDQMKB-UHFFFAOYSA-N 1-[(2-methylpropan-2-yl)oxycarbonyl]piperidine-4-carboxylic acid Chemical compound CC(C)(C)OC(=O)N1CCC(C(O)=O)CC1 JWOHBPPVVDQMKB-UHFFFAOYSA-N 0.000 description 1
- PFVWEAYXWZFSSK-UHFFFAOYSA-N 1-[3,5-bis(trifluoromethyl)phenyl]ethanamine Chemical group CC(N)C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 PFVWEAYXWZFSSK-UHFFFAOYSA-N 0.000 description 1
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- CFJMRBQWBDQYMK-UHFFFAOYSA-N 1-phenyl-1-cyclopentanecarboxylic acid 2-[2-(diethylamino)ethoxy]ethyl ester Chemical compound C=1C=CC=CC=1C1(C(=O)OCCOCCN(CC)CC)CCCC1 CFJMRBQWBDQYMK-UHFFFAOYSA-N 0.000 description 1
- RQEUFEKYXDPUSK-UHFFFAOYSA-N 1-phenylethylamine Chemical compound CC(N)C1=CC=CC=C1 RQEUFEKYXDPUSK-UHFFFAOYSA-N 0.000 description 1
- GQODOIOBGTVQEF-UHFFFAOYSA-N 1-phenylpiperidine;hydrochloride Chemical compound Cl.C1CCCCN1C1=CC=CC=C1 GQODOIOBGTVQEF-UHFFFAOYSA-N 0.000 description 1
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 1
- RBZRMBCLZMEYEH-UHFFFAOYSA-N 1h-pyrazol-1-ium-1-carboximidamide;chloride Chemical compound Cl.NC(=N)N1C=CC=N1 RBZRMBCLZMEYEH-UHFFFAOYSA-N 0.000 description 1
- XXMFJKNOJSDQBM-UHFFFAOYSA-N 2,2,2-trifluoroacetic acid;hydrate Chemical compound [OH3+].[O-]C(=O)C(F)(F)F XXMFJKNOJSDQBM-UHFFFAOYSA-N 0.000 description 1
- KLIVRBFRQSOGQI-UHFFFAOYSA-N 2-(11-oxo-6h-benzo[c][1]benzothiepin-3-yl)acetic acid Chemical compound S1CC2=CC=CC=C2C(=O)C2=CC=C(CC(=O)O)C=C12 KLIVRBFRQSOGQI-UHFFFAOYSA-N 0.000 description 1
- XRFOEVFLFSRHLN-UHFFFAOYSA-N 2-(2-bromo-1,3-thiazol-4-yl)acetic acid Chemical compound OC(=O)CC1=CSC(Br)=N1 XRFOEVFLFSRHLN-UHFFFAOYSA-N 0.000 description 1
- MYQXHLQMZLTSDB-UHFFFAOYSA-N 2-(2-ethyl-2,3-dihydro-1-benzofuran-5-yl)acetic acid Chemical compound OC(=O)CC1=CC=C2OC(CC)CC2=C1 MYQXHLQMZLTSDB-UHFFFAOYSA-N 0.000 description 1
- KYNNBXCGXUOREX-UHFFFAOYSA-N 2-(3-bromophenyl)acetic acid Chemical compound OC(=O)CC1=CC=CC(Br)=C1 KYNNBXCGXUOREX-UHFFFAOYSA-N 0.000 description 1
- GADITLVVLBWWOW-UHFFFAOYSA-N 2-(4-fluorophenyl)pent-2-enoic acid Chemical compound CCC=C(C(O)=O)C1=CC=C(F)C=C1 GADITLVVLBWWOW-UHFFFAOYSA-N 0.000 description 1
- IZXIZTKNFFYFOF-UHFFFAOYSA-N 2-Oxazolidone Chemical class O=C1NCCO1 IZXIZTKNFFYFOF-UHFFFAOYSA-N 0.000 description 1
- DCXHLPGLBYHNMU-UHFFFAOYSA-N 2-[1-(4-azidobenzoyl)-5-methoxy-2-methylindol-3-yl]acetic acid Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(N=[N+]=[N-])C=C1 DCXHLPGLBYHNMU-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- APBSKHYXXKHJFK-UHFFFAOYSA-N 2-[2-(4-chlorophenyl)-1,3-thiazol-4-yl]acetic acid Chemical compound OC(=O)CC1=CSC(C=2C=CC(Cl)=CC=2)=N1 APBSKHYXXKHJFK-UHFFFAOYSA-N 0.000 description 1
- CGDDYPZNCJERSX-UHFFFAOYSA-N 2-[3,3,3-trifluoro-2-phenyl-2-(trifluoromethyl)propyl]isoindole-1,3-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1CC(C(F)(F)F)(C(F)(F)F)C1=CC=CC=C1 CGDDYPZNCJERSX-UHFFFAOYSA-N 0.000 description 1
- TYCOFFBAZNSQOJ-UHFFFAOYSA-N 2-[4-(3-fluorophenyl)phenyl]propanoic acid Chemical compound C1=CC(C(C(O)=O)C)=CC=C1C1=CC=CC(F)=C1 TYCOFFBAZNSQOJ-UHFFFAOYSA-N 0.000 description 1
- JIEKMACRVQTPRC-UHFFFAOYSA-N 2-[4-(4-chlorophenyl)-2-phenyl-5-thiazolyl]acetic acid Chemical compound OC(=O)CC=1SC(C=2C=CC=CC=2)=NC=1C1=CC=C(Cl)C=C1 JIEKMACRVQTPRC-UHFFFAOYSA-N 0.000 description 1
- WGDADRBTCPGSDG-UHFFFAOYSA-N 2-[[4,5-bis(4-chlorophenyl)-1,3-oxazol-2-yl]sulfanyl]propanoic acid Chemical compound O1C(SC(C)C(O)=O)=NC(C=2C=CC(Cl)=CC=2)=C1C1=CC=C(Cl)C=C1 WGDADRBTCPGSDG-UHFFFAOYSA-N 0.000 description 1
- 125000005273 2-acetoxybenzoic acid group Chemical group 0.000 description 1
- XKSAJZSJKURQRX-UHFFFAOYSA-N 2-acetyloxy-5-(4-fluorophenyl)benzoic acid Chemical compound C1=C(C(O)=O)C(OC(=O)C)=CC=C1C1=CC=C(F)C=C1 XKSAJZSJKURQRX-UHFFFAOYSA-N 0.000 description 1
- SPCKHVPPRJWQRZ-UHFFFAOYSA-N 2-benzhydryloxy-n,n-dimethylethanamine;2-hydroxypropane-1,2,3-tricarboxylic acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O.C=1C=CC=CC=1C(OCCN(C)C)C1=CC=CC=C1 SPCKHVPPRJWQRZ-UHFFFAOYSA-N 0.000 description 1
- KVVDRQDTODKIJD-UHFFFAOYSA-N 2-cyclopropylacetic acid Chemical compound OC(=O)CC1CC1 KVVDRQDTODKIJD-UHFFFAOYSA-N 0.000 description 1
- RSGUVKAALVOJNZ-UHFFFAOYSA-N 2-phenyl-n,n-bis(trifluoromethyl)ethanamine Chemical compound FC(F)(F)N(C(F)(F)F)CCC1=CC=CC=C1 RSGUVKAALVOJNZ-UHFFFAOYSA-N 0.000 description 1
- ILYSAKHOYBPSPC-UHFFFAOYSA-N 2-phenylbenzoic acid Chemical class OC(=O)C1=CC=CC=C1C1=CC=CC=C1 ILYSAKHOYBPSPC-UHFFFAOYSA-N 0.000 description 1
- SMJRBWINMFUUDS-UHFFFAOYSA-N 2-thienylacetic acid Chemical compound OC(=O)CC1=CC=CS1 SMJRBWINMFUUDS-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- MPJPPDINXOVGRK-UHFFFAOYSA-N 2h-pyridin-1-ylmethanamine Chemical group NCN1CC=CC=C1 MPJPPDINXOVGRK-UHFFFAOYSA-N 0.000 description 1
- LDWLIXZSDPXYDR-UHFFFAOYSA-N 3,5-bis(trifluoromethyl)benzaldehyde Chemical compound FC(F)(F)C1=CC(C=O)=CC(C(F)(F)F)=C1 LDWLIXZSDPXYDR-UHFFFAOYSA-N 0.000 description 1
- HJTLKVYOWNTDPF-UHFFFAOYSA-N 3-bromo-5-(trifluoromethyl)aniline Chemical compound NC1=CC(Br)=CC(C(F)(F)F)=C1 HJTLKVYOWNTDPF-UHFFFAOYSA-N 0.000 description 1
- KAKZYGLQCHMNGS-UHFFFAOYSA-N 3-phenylpiperidine-2,6-dione Chemical compound O=C1NC(=O)CCC1C1=CC=CC=C1 KAKZYGLQCHMNGS-UHFFFAOYSA-N 0.000 description 1
- YVWANCNXGXRHKE-UHFFFAOYSA-N 4,4-dimethoxy-2-thiophen-3-ylbutanoic acid Chemical compound COC(OC)CC(C(O)=O)C=1C=CSC=1 YVWANCNXGXRHKE-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- AFYALJSDFPSAAZ-UHFFFAOYSA-N 4-(4-fluorophenyl)piperidine Chemical compound C1=CC(F)=CC=C1C1CCNCC1 AFYALJSDFPSAAZ-UHFFFAOYSA-N 0.000 description 1
- QBLLOOKBLTTXHB-UHFFFAOYSA-N 4-fluoropiperidine Chemical compound FC1CCNCC1 QBLLOOKBLTTXHB-UHFFFAOYSA-N 0.000 description 1
- SYCHUQUJURZQMO-UHFFFAOYSA-N 4-hydroxy-2-methyl-1,1-dioxo-n-(1,3-thiazol-2-yl)-1$l^{6},2-benzothiazine-3-carboxamide Chemical compound OC=1C2=CC=CC=C2S(=O)(=O)N(C)C=1C(=O)NC1=NC=CS1 SYCHUQUJURZQMO-UHFFFAOYSA-N 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-M 4-hydroxybenzoate Chemical compound OC1=CC=C(C([O-])=O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-M 0.000 description 1
- PJJGZPJJTHBVMX-UHFFFAOYSA-N 5,7-Dihydroxyisoflavone Chemical compound C=1C(O)=CC(O)=C(C2=O)C=1OC=C2C1=CC=CC=C1 PJJGZPJJTHBVMX-UHFFFAOYSA-N 0.000 description 1
- LSLYOANBFKQKPT-DIFFPNOSSA-N 5-[(1r)-1-hydroxy-2-[[(2r)-1-(4-hydroxyphenyl)propan-2-yl]amino]ethyl]benzene-1,3-diol Chemical compound C([C@@H](C)NC[C@H](O)C=1C=C(O)C=C(O)C=1)C1=CC=C(O)C=C1 LSLYOANBFKQKPT-DIFFPNOSSA-N 0.000 description 1
- BSYNRYMUTXBXSQ-FOQJRBATSA-N 59096-14-9 Chemical compound CC(=O)OC1=CC=CC=C1[14C](O)=O BSYNRYMUTXBXSQ-FOQJRBATSA-N 0.000 description 1
- IXJCHVMUTFCRBH-SDUHDBOFSA-N 7-[(1r,2s,3e,5z)-10-(4-acetyl-3-hydroxy-2-propylphenoxy)-1-hydroxy-1-[3-(trifluoromethyl)phenyl]deca-3,5-dien-2-yl]sulfanyl-4-oxochromene-2-carboxylic acid Chemical compound CCCC1=C(O)C(C(C)=O)=CC=C1OCCCC\C=C/C=C/[C@@H]([C@H](O)C=1C=C(C=CC=1)C(F)(F)F)SC1=CC=C2C(=O)C=C(C(O)=O)OC2=C1 IXJCHVMUTFCRBH-SDUHDBOFSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 208000032484 Accidental exposure to product Diseases 0.000 description 1
- 206010027654 Allergic conditions Diseases 0.000 description 1
- 206010058284 Allergy to arthropod sting Diseases 0.000 description 1
- 229940077274 Alpha glucosidase inhibitor Drugs 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 206010002198 Anaphylactic reaction Diseases 0.000 description 1
- 241001147672 Ancylostoma caninum Species 0.000 description 1
- 241001465677 Ancylostomatoidea Species 0.000 description 1
- 241000244023 Anisakis Species 0.000 description 1
- 206010002556 Ankylosing Spondylitis Diseases 0.000 description 1
- 102000001381 Arachidonate 5-Lipoxygenase Human genes 0.000 description 1
- 108010093579 Arachidonate 5-lipoxygenase Proteins 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 description 1
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 description 1
- 229930003347 Atropine Natural products 0.000 description 1
- 241000271566 Aves Species 0.000 description 1
- 208000009137 Behcet syndrome Diseases 0.000 description 1
- ZBJJDYGJCNTNTH-UHFFFAOYSA-N Betahistine mesilate Chemical compound CS(O)(=O)=O.CS(O)(=O)=O.CNCCC1=CC=CC=N1 ZBJJDYGJCNTNTH-UHFFFAOYSA-N 0.000 description 1
- 229940123208 Biguanide Drugs 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- 102100031172 C-C chemokine receptor type 1 Human genes 0.000 description 1
- 101710149862 C-C chemokine receptor type 3 Proteins 0.000 description 1
- 102100037853 C-C chemokine receptor type 4 Human genes 0.000 description 1
- 101710149870 C-C chemokine receptor type 5 Proteins 0.000 description 1
- 102100023702 C-C motif chemokine 13 Human genes 0.000 description 1
- 101710112613 C-C motif chemokine 13 Proteins 0.000 description 1
- 102100036849 C-C motif chemokine 24 Human genes 0.000 description 1
- 102100031650 C-X-C chemokine receptor type 4 Human genes 0.000 description 1
- 101710082513 C-X-C chemokine receptor type 4 Proteins 0.000 description 1
- 108010039171 CC cytokine receptor-4 Proteins 0.000 description 1
- 108050006947 CXC Chemokine Proteins 0.000 description 1
- 102000019388 CXC chemokine Human genes 0.000 description 1
- 101150093802 CXCL1 gene Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 241000282465 Canis Species 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 229930186147 Cephalosporin Natural products 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 241000242722 Cestoda Species 0.000 description 1
- ZKLPARSLTMPFCP-UHFFFAOYSA-N Cetirizine Chemical compound C1CN(CCOCC(=O)O)CCN1C(C=1C=CC(Cl)=CC=1)C1=CC=CC=C1 ZKLPARSLTMPFCP-UHFFFAOYSA-N 0.000 description 1
- 108010083647 Chemokine CCL24 Proteins 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 229920001268 Cholestyramine Polymers 0.000 description 1
- OIRAEJWYWSAQNG-UHFFFAOYSA-N Clidanac Chemical compound ClC=1C=C2C(C(=O)O)CCC2=CC=1C1CCCCC1 OIRAEJWYWSAQNG-UHFFFAOYSA-N 0.000 description 1
- 206010009344 Clonorchiasis Diseases 0.000 description 1
- 229920002911 Colestipol Polymers 0.000 description 1
- 206010009900 Colitis ulcerative Diseases 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- JPVYNHNXODAKFH-UHFFFAOYSA-N Cu2+ Chemical compound [Cu+2] JPVYNHNXODAKFH-UHFFFAOYSA-N 0.000 description 1
- 238000006969 Curtius rearrangement reaction Methods 0.000 description 1
- 206010011686 Cutaneous vasculitis Diseases 0.000 description 1
- NOTFZGFABLVTIG-UHFFFAOYSA-N Cyclohexylethyl acetate Chemical compound CC(=O)OCCC1CCCCC1 NOTFZGFABLVTIG-UHFFFAOYSA-N 0.000 description 1
- 229940093444 Cyclooxygenase 2 inhibitor Drugs 0.000 description 1
- 229940122204 Cyclooxygenase inhibitor Drugs 0.000 description 1
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 1
- 229930105110 Cyclosporin A Natural products 0.000 description 1
- 108010036949 Cyclosporine Proteins 0.000 description 1
- 201000000077 Cysticercosis Diseases 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 206010012438 Dermatitis atopic Diseases 0.000 description 1
- 206010012442 Dermatitis contact Diseases 0.000 description 1
- YIIMEMSDCNDGTB-UHFFFAOYSA-N Dimethylcarbamoyl chloride Chemical compound CN(C)C(Cl)=O YIIMEMSDCNDGTB-UHFFFAOYSA-N 0.000 description 1
- 206010013700 Drug hypersensitivity Diseases 0.000 description 1
- 206010014096 Echinococciasis Diseases 0.000 description 1
- 208000009366 Echinococcosis Diseases 0.000 description 1
- 206010014824 Endotoxic shock Diseases 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 206010014954 Eosinophilic fasciitis Diseases 0.000 description 1
- 241000283073 Equus caballus Species 0.000 description 1
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 1
- 241000282324 Felis Species 0.000 description 1
- RBBWCVQDXDFISW-UHFFFAOYSA-N Feprazone Chemical compound O=C1C(CC=C(C)C)C(=O)N(C=2C=CC=CC=2)N1C1=CC=CC=C1 RBBWCVQDXDFISW-UHFFFAOYSA-N 0.000 description 1
- 201000006353 Filariasis Diseases 0.000 description 1
- 108091006027 G proteins Proteins 0.000 description 1
- 102000030782 GTP binding Human genes 0.000 description 1
- 108091000058 GTP-Binding Proteins 0.000 description 1
- 241000287828 Gallus gallus Species 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- HEMJJKBWTPKOJG-UHFFFAOYSA-N Gemfibrozil Chemical compound CC1=CC=C(C)C(OCCCC(C)(C)C(O)=O)=C1 HEMJJKBWTPKOJG-UHFFFAOYSA-N 0.000 description 1
- 206010018364 Glomerulonephritis Diseases 0.000 description 1
- 244000068988 Glycine max Species 0.000 description 1
- 235000010469 Glycine max Nutrition 0.000 description 1
- 208000009329 Graft vs Host Disease Diseases 0.000 description 1
- 206010018691 Granuloma Diseases 0.000 description 1
- 208000030836 Hashimoto thyroiditis Diseases 0.000 description 1
- 208000006968 Helminthiasis Diseases 0.000 description 1
- 208000002250 Hematologic Neoplasms Diseases 0.000 description 1
- 229940122957 Histamine H2 receptor antagonist Drugs 0.000 description 1
- 101000777564 Homo sapiens C-C chemokine receptor type 1 Proteins 0.000 description 1
- 101000980744 Homo sapiens C-C chemokine receptor type 3 Proteins 0.000 description 1
- 101000946926 Homo sapiens C-C chemokine receptor type 5 Proteins 0.000 description 1
- 101000947174 Homo sapiens C-X-C chemokine receptor type 1 Proteins 0.000 description 1
- 101000973997 Homo sapiens Nucleosome assembly protein 1-like 4 Proteins 0.000 description 1
- 101000947178 Homo sapiens Platelet basic protein Proteins 0.000 description 1
- 241000725303 Human immunodeficiency virus Species 0.000 description 1
- RKUNBYITZUJHSG-UHFFFAOYSA-N Hyosciamin-hydrochlorid Natural products CN1C(C2)CCC1CC2OC(=O)C(CO)C1=CC=CC=C1 RKUNBYITZUJHSG-UHFFFAOYSA-N 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 201000009794 Idiopathic Pulmonary Fibrosis Diseases 0.000 description 1
- 208000016300 Idiopathic chronic eosinophilic pneumonia Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010021460 Immunodeficiency syndromes Diseases 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- 206010069803 Injury associated with device Diseases 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 102000003996 Interferon-beta Human genes 0.000 description 1
- 108090000467 Interferon-beta Proteins 0.000 description 1
- 108010002352 Interleukin-1 Proteins 0.000 description 1
- 102000000589 Interleukin-1 Human genes 0.000 description 1
- 108090001007 Interleukin-8 Proteins 0.000 description 1
- 108010063738 Interleukins Proteins 0.000 description 1
- 102000015696 Interleukins Human genes 0.000 description 1
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- HUYWAWARQUIQLE-UHFFFAOYSA-N Isoetharine Chemical compound CC(C)NC(CC)C(O)C1=CC=C(O)C(O)=C1 HUYWAWARQUIQLE-UHFFFAOYSA-N 0.000 description 1
- PWWVAXIEGOYWEE-UHFFFAOYSA-N Isophenergan Chemical compound C1=CC=C2N(CC(C)N(C)C)C3=CC=CC=C3SC2=C1 PWWVAXIEGOYWEE-UHFFFAOYSA-N 0.000 description 1
- UETNIIAIRMUTSM-UHFFFAOYSA-N Jacareubin Natural products CC1(C)OC2=CC3Oc4c(O)c(O)ccc4C(=O)C3C(=C2C=C1)O UETNIIAIRMUTSM-UHFFFAOYSA-N 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- 229930182816 L-glutamine Natural products 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 208000034624 Leukocytoclastic Cutaneous Vasculitis Diseases 0.000 description 1
- 208000032514 Leukocytoclastic vasculitis Diseases 0.000 description 1
- 239000000867 Lipoxygenase Inhibitor Substances 0.000 description 1
- 229940122142 Lipoxygenase inhibitor Drugs 0.000 description 1
- 241000186779 Listeria monocytogenes Species 0.000 description 1
- 201000009324 Loeffler syndrome Diseases 0.000 description 1
- 208000019693 Lung disease Diseases 0.000 description 1
- 102000009571 Macrophage Inflammatory Proteins Human genes 0.000 description 1
- 108010009474 Macrophage Inflammatory Proteins Proteins 0.000 description 1
- SBDNJUWAMKYJOX-UHFFFAOYSA-N Meclofenamic Acid Chemical compound CC1=CC=C(Cl)C(NC=2C(=CC=CC=2)C(O)=O)=C1Cl SBDNJUWAMKYJOX-UHFFFAOYSA-N 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- FQISKWAFAHGMGT-SGJOWKDISA-M Methylprednisolone sodium succinate Chemical compound [Na+].C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)COC(=O)CCC([O-])=O)CC[C@H]21 FQISKWAFAHGMGT-SGJOWKDISA-M 0.000 description 1
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 1
- 102000014962 Monocyte Chemoattractant Proteins Human genes 0.000 description 1
- 108010064136 Monocyte Chemoattractant Proteins Proteins 0.000 description 1
- UCHDWCPVSPXUMX-TZIWLTJVSA-N Montelukast Chemical compound CC(C)(O)C1=CC=CC=C1CC[C@H](C=1C=C(\C=C\C=2N=C3C=C(Cl)C=CC3=CC=2)C=CC=1)SCC1(CC(O)=O)CC1 UCHDWCPVSPXUMX-TZIWLTJVSA-N 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 101100000659 Mus musculus Ackr1 gene Proteins 0.000 description 1
- 241000872931 Myoporum sandwicense Species 0.000 description 1
- 201000002481 Myositis Diseases 0.000 description 1
- IJHNSHDBIRRJRN-UHFFFAOYSA-N N,N-dimethyl-3-phenyl-3-(2-pyridinyl)-1-propanamine Chemical compound C=1C=CC=NC=1C(CCN(C)C)C1=CC=CC=C1 IJHNSHDBIRRJRN-UHFFFAOYSA-N 0.000 description 1
- PRQROPMIIGLWRP-UHFFFAOYSA-N N-formyl-methionyl-leucyl-phenylalanin Chemical compound CSCCC(NC=O)C(=O)NC(CC(C)C)C(=O)NC(C(O)=O)CC1=CC=CC=C1 PRQROPMIIGLWRP-UHFFFAOYSA-N 0.000 description 1
- 241001274216 Naso Species 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 102000005348 Neuraminidase Human genes 0.000 description 1
- 108010006232 Neuraminidase Proteins 0.000 description 1
- JZFPYUNJRRFVQU-UHFFFAOYSA-N Niflumic acid Chemical compound OC(=O)C1=CC=CN=C1NC1=CC=CC(C(F)(F)F)=C1 JZFPYUNJRRFVQU-UHFFFAOYSA-N 0.000 description 1
- IOVCWXUNBOPUCH-UHFFFAOYSA-N Nitrous acid Chemical compound ON=O IOVCWXUNBOPUCH-UHFFFAOYSA-N 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- HVAUUPRFYPCOCA-AREMUKBSSA-O PAF Chemical compound CCCCCCCCCCCCCCCCOC[C@@H](OC(C)=O)COP(O)(=O)OCC[N+](C)(C)C HVAUUPRFYPCOCA-AREMUKBSSA-O 0.000 description 1
- 208000030852 Parasitic disease Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- BELBBZDIHDAJOR-UHFFFAOYSA-N Phenolsulfonephthalein Chemical compound C1=CC(O)=CC=C1C1(C=2C=CC(O)=CC=2)C2=CC=CC=C2S(=O)(=O)O1 BELBBZDIHDAJOR-UHFFFAOYSA-N 0.000 description 1
- 102000004861 Phosphoric Diester Hydrolases Human genes 0.000 description 1
- 108090001050 Phosphoric Diester Hydrolases Proteins 0.000 description 1
- VQDBNKDJNJQRDG-UHFFFAOYSA-N Pirbuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=N1 VQDBNKDJNJQRDG-UHFFFAOYSA-N 0.000 description 1
- 102100036154 Platelet basic protein Human genes 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- TVQZAMVBTVNYLA-UHFFFAOYSA-N Pranoprofen Chemical compound C1=CC=C2CC3=CC(C(C(O)=O)C)=CC=C3OC2=N1 TVQZAMVBTVNYLA-UHFFFAOYSA-N 0.000 description 1
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- GOOHAUXETOMSMM-GSVOUGTGSA-N R-propylene oxide Chemical compound C[C@@H]1CO1 GOOHAUXETOMSMM-GSVOUGTGSA-N 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 206010063837 Reperfusion injury Diseases 0.000 description 1
- GOOHAUXETOMSMM-VKHMYHEASA-N S-propylene oxide Chemical compound C[C@H]1CO1 GOOHAUXETOMSMM-VKHMYHEASA-N 0.000 description 1
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 1
- 206010039710 Scleroderma Diseases 0.000 description 1
- 208000021386 Sjogren Syndrome Diseases 0.000 description 1
- 208000006045 Spondylarthropathies Diseases 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 206010042254 Strongyloidiasis Diseases 0.000 description 1
- 229940100389 Sulfonylurea Drugs 0.000 description 1
- 201000009594 Systemic Scleroderma Diseases 0.000 description 1
- 206010042953 Systemic sclerosis Diseases 0.000 description 1
- 241000244031 Toxocara Species 0.000 description 1
- 206010052779 Transplant rejections Diseases 0.000 description 1
- 241000242541 Trematoda Species 0.000 description 1
- 241000869417 Trematodes Species 0.000 description 1
- 206010044608 Trichiniasis Diseases 0.000 description 1
- UFLGIAIHIAPJJC-UHFFFAOYSA-N Tripelennamine Chemical compound C=1C=CC=NC=1N(CCN(C)C)CC1=CC=CC=C1 UFLGIAIHIAPJJC-UHFFFAOYSA-N 0.000 description 1
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 1
- 201000006704 Ulcerative Colitis Diseases 0.000 description 1
- 208000024780 Urticaria Diseases 0.000 description 1
- 206010047115 Vasculitis Diseases 0.000 description 1
- 206010047124 Vasculitis necrotising Diseases 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- YEEZWCHGZNKEEK-UHFFFAOYSA-N Zafirlukast Chemical compound COC1=CC(C(=O)NS(=O)(=O)C=2C(=CC=CC=2)C)=CC=C1CC(C1=C2)=CN(C)C1=CC=C2NC(=O)OC1CCCC1 YEEZWCHGZNKEEK-UHFFFAOYSA-N 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- WERKSKAQRVDLDW-ANOHMWSOSA-N [(2s,3r,4r,5r)-2,3,4,5,6-pentahydroxyhexyl] (z)-octadec-9-enoate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO WERKSKAQRVDLDW-ANOHMWSOSA-N 0.000 description 1
- BJTWPJOGDWRYDD-UHFFFAOYSA-N [3,5-bis(trifluoromethyl)phenyl]methanol Chemical group OCC1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 BJTWPJOGDWRYDD-UHFFFAOYSA-N 0.000 description 1
- YKNZTUQUXUXTLE-UHFFFAOYSA-N [3-(trifluoromethyl)phenyl]methanamine Chemical group NCC1=CC=CC(C(F)(F)F)=C1 YKNZTUQUXUXTLE-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 229960002632 acarbose Drugs 0.000 description 1
- XUFXOAAUWZOOIT-UHFFFAOYSA-N acarviostatin I01 Natural products OC1C(O)C(NC2C(C(O)C(O)C(CO)=C2)O)C(C)OC1OC(C(C1O)O)C(CO)OC1OC1C(CO)OC(O)C(O)C1O XUFXOAAUWZOOIT-UHFFFAOYSA-N 0.000 description 1
- 229960004892 acemetacin Drugs 0.000 description 1
- FSQKKOOTNAMONP-UHFFFAOYSA-N acemetacin Chemical compound CC1=C(CC(=O)OCC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 FSQKKOOTNAMONP-UHFFFAOYSA-N 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- XPOLVIIHTDKJRY-UHFFFAOYSA-N acetic acid;methanimidamide Chemical compound NC=N.CC(O)=O XPOLVIIHTDKJRY-UHFFFAOYSA-N 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 239000003929 acidic solution Substances 0.000 description 1
- 239000011149 active material Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- NDAUXUAQIAJITI-UHFFFAOYSA-N albuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-UHFFFAOYSA-N 0.000 description 1
- 229960005142 alclofenac Drugs 0.000 description 1
- ARHWPKZXBHOEEE-UHFFFAOYSA-N alclofenac Chemical compound OC(=O)CC1=CC=C(OCC=C)C(Cl)=C1 ARHWPKZXBHOEEE-UHFFFAOYSA-N 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 229960003790 alimemazine Drugs 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- 208000002029 allergic contact dermatitis Diseases 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 238000005937 allylation reaction Methods 0.000 description 1
- 229960004663 alminoprofen Drugs 0.000 description 1
- FPHLBGOJWPEVME-UHFFFAOYSA-N alminoprofen Chemical compound OC(=O)C(C)C1=CC=C(NCC(C)=C)C=C1 FPHLBGOJWPEVME-UHFFFAOYSA-N 0.000 description 1
- 239000003888 alpha glucosidase inhibitor Substances 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 229940024548 aluminum oxide Drugs 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 230000000202 analgesic effect Effects 0.000 description 1
- 229940051880 analgesics and antipyretics pyrazolones Drugs 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 208000003455 anaphylaxis Diseases 0.000 description 1
- 239000000730 antalgic agent Substances 0.000 description 1
- 229960002469 antazoline Drugs 0.000 description 1
- REYFJDPCWQRWAA-UHFFFAOYSA-N antazoline Chemical compound N=1CCNC=1CN(C=1C=CC=CC=1)CC1=CC=CC=C1 REYFJDPCWQRWAA-UHFFFAOYSA-N 0.000 description 1
- 230000001088 anti-asthma Effects 0.000 description 1
- 230000001387 anti-histamine Effects 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 239000000924 antiasthmatic agent Substances 0.000 description 1
- 239000003529 anticholesteremic agent Substances 0.000 description 1
- 229940127226 anticholesterol agent Drugs 0.000 description 1
- 229940125708 antidiabetic agent Drugs 0.000 description 1
- 239000003472 antidiabetic agent Substances 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 229940125715 antihistaminic agent Drugs 0.000 description 1
- 229940111133 antiinflammatory and antirheumatic drug oxicams Drugs 0.000 description 1
- 229940111131 antiinflammatory and antirheumatic product propionic acid derivative Drugs 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000006286 aqueous extract Substances 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- 201000009361 ascariasis Diseases 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- GXDALQBWZGODGZ-UHFFFAOYSA-N astemizole Chemical compound C1=CC(OC)=CC=C1CCN1CCC(NC=2N(C3=CC=CC=C3N=2)CC=2C=CC(F)=CC=2)CC1 GXDALQBWZGODGZ-UHFFFAOYSA-N 0.000 description 1
- 230000000923 atherogenic effect Effects 0.000 description 1
- 230000003143 atherosclerotic effect Effects 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 229960005370 atorvastatin Drugs 0.000 description 1
- RKUNBYITZUJHSG-SPUOUPEWSA-N atropine Chemical compound O([C@H]1C[C@H]2CC[C@@H](C1)N2C)C(=O)C(CO)C1=CC=CC=C1 RKUNBYITZUJHSG-SPUOUPEWSA-N 0.000 description 1
- 229960000396 atropine Drugs 0.000 description 1
- 230000002238 attenuated effect Effects 0.000 description 1
- WOIIIUDZSOLAIW-NSHDSACASA-N azapropazone Chemical compound C1=C(C)C=C2N3C(=O)[C@H](CC=C)C(=O)N3C(N(C)C)=NC2=C1 WOIIIUDZSOLAIW-NSHDSACASA-N 0.000 description 1
- 229960001671 azapropazone Drugs 0.000 description 1
- 229960000383 azatadine Drugs 0.000 description 1
- SEBMTIQKRHYNIT-UHFFFAOYSA-N azatadine Chemical compound C1CN(C)CCC1=C1C2=NC=CC=C2CCC2=CC=CC=C21 SEBMTIQKRHYNIT-UHFFFAOYSA-N 0.000 description 1
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 1
- 229960002170 azathioprine Drugs 0.000 description 1
- 210000003651 basophil Anatomy 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 210000003323 beak Anatomy 0.000 description 1
- NBMKJKDGKREAPL-DVTGEIKXSA-N beclomethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(Cl)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O NBMKJKDGKREAPL-DVTGEIKXSA-N 0.000 description 1
- 229940092705 beclomethasone Drugs 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Substances 0.000 description 1
- 229960005430 benoxaprofen Drugs 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 150000003939 benzylamines Chemical class 0.000 description 1
- 229940124748 beta 2 agonist Drugs 0.000 description 1
- 229960002537 betamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-DVTGEIKXSA-N betamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-DVTGEIKXSA-N 0.000 description 1
- 150000004283 biguanides Chemical class 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 239000012148 binding buffer Substances 0.000 description 1
- 229960004620 bitolterol Drugs 0.000 description 1
- FZGVEKPRDOIXJY-UHFFFAOYSA-N bitolterol Chemical compound C1=CC(C)=CC=C1C(=O)OC1=CC=C(C(O)CNC(C)(C)C)C=C1OC(=O)C1=CC=C(C)C=C1 FZGVEKPRDOIXJY-UHFFFAOYSA-N 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 150000001649 bromium compounds Chemical class 0.000 description 1
- 229960000725 brompheniramine Drugs 0.000 description 1
- ZDIGNSYAACHWNL-UHFFFAOYSA-N brompheniramine Chemical compound C=1C=CC=NC=1C(CCN(C)C)C1=CC=C(Br)C=C1 ZDIGNSYAACHWNL-UHFFFAOYSA-N 0.000 description 1
- IJTPQQVCKPZIMV-UHFFFAOYSA-N bucloxic acid Chemical compound ClC1=CC(C(=O)CCC(=O)O)=CC=C1C1CCCCC1 IJTPQQVCKPZIMV-UHFFFAOYSA-N 0.000 description 1
- 229950005608 bucloxic acid Drugs 0.000 description 1
- 235000019437 butane-1,3-diol Nutrition 0.000 description 1
- HNCWWKMHPKJIHO-UHFFFAOYSA-N butyl n-(1-hydroxyethyl)carbamate Chemical compound CCCCOC(=O)NC(C)O HNCWWKMHPKJIHO-UHFFFAOYSA-N 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- OFAIGZWCDGNZGT-UHFFFAOYSA-N caramiphen Chemical compound C=1C=CC=CC=1C1(C(=O)OCCN(CC)CC)CCCC1 OFAIGZWCDGNZGT-UHFFFAOYSA-N 0.000 description 1
- 229960004160 caramiphen Drugs 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 229960003184 carprofen Drugs 0.000 description 1
- IVUMCTKHWDRRMH-UHFFFAOYSA-N carprofen Chemical compound C1=CC(Cl)=C[C]2C3=CC=C(C(C(O)=O)C)C=C3N=C21 IVUMCTKHWDRRMH-UHFFFAOYSA-N 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 229940124587 cephalosporin Drugs 0.000 description 1
- 150000001780 cephalosporins Chemical class 0.000 description 1
- 229960001803 cetirizine Drugs 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 230000010002 chemokinesis Effects 0.000 description 1
- 230000003399 chemotactic effect Effects 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 235000013330 chicken meat Nutrition 0.000 description 1
- 238000004296 chiral HPLC Methods 0.000 description 1
- SOYKEARSMXGVTM-UHFFFAOYSA-N chlorphenamine Chemical compound C=1C=CC=NC=1C(CCN(C)C)C1=CC=C(Cl)C=C1 SOYKEARSMXGVTM-UHFFFAOYSA-N 0.000 description 1
- 229960003291 chlorphenamine Drugs 0.000 description 1
- 230000001906 cholesterol absorption Effects 0.000 description 1
- 238000013375 chromatographic separation Methods 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 201000009323 chronic eosinophilic pneumonia Diseases 0.000 description 1
- 229960001265 ciclosporin Drugs 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 229960002881 clemastine Drugs 0.000 description 1
- YNNUSGIPVFPVBX-NHCUHLMSSA-N clemastine Chemical compound CN1CCC[C@@H]1CCO[C@@](C)(C=1C=CC(Cl)=CC=1)C1=CC=CC=C1 YNNUSGIPVFPVBX-NHCUHLMSSA-N 0.000 description 1
- 229950010886 clidanac Drugs 0.000 description 1
- KNHUKKLJHYUCFP-UHFFFAOYSA-N clofibrate Chemical compound CCOC(=O)C(C)(C)OC1=CC=C(Cl)C=C1 KNHUKKLJHYUCFP-UHFFFAOYSA-N 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- GMRWGQCZJGVHKL-UHFFFAOYSA-N colestipol Chemical compound ClCC1CO1.NCCNCCNCCNCCN GMRWGQCZJGVHKL-UHFFFAOYSA-N 0.000 description 1
- 229960002604 colestipol Drugs 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 230000006957 competitive inhibition Effects 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 239000003246 corticosteroid Substances 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 229960000265 cromoglicic acid Drugs 0.000 description 1
- 239000013058 crude material Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 239000003255 cyclooxygenase 2 inhibitor Substances 0.000 description 1
- YMGUBTXCNDTFJI-UHFFFAOYSA-N cyclopropanecarboxylic acid Chemical compound OC(=O)C1CC1 YMGUBTXCNDTFJI-UHFFFAOYSA-N 0.000 description 1
- 150000001942 cyclopropanes Chemical class 0.000 description 1
- 229930182912 cyclosporin Natural products 0.000 description 1
- 229960001140 cyproheptadine Drugs 0.000 description 1
- JJCFRYNCJDLXIK-UHFFFAOYSA-N cyproheptadine Chemical compound C1CN(C)CCC1=C1C2=CC=CC=C2C=CC2=CC=CC=C21 JJCFRYNCJDLXIK-UHFFFAOYSA-N 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 239000000850 decongestant Substances 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 1
- SOYKEARSMXGVTM-HNNXBMFYSA-N dexchlorpheniramine Chemical compound C1([C@H](CCN(C)C)C=2N=CC=CC=2)=CC=C(Cl)C=C1 SOYKEARSMXGVTM-HNNXBMFYSA-N 0.000 description 1
- 229960001882 dexchlorpheniramine Drugs 0.000 description 1
- 150000004985 diamines Chemical class 0.000 description 1
- 229960001259 diclofenac Drugs 0.000 description 1
- DCOPUUMXTXDBNB-UHFFFAOYSA-N diclofenac Chemical compound OC(=O)CC1=CC=CC=C1NC1=C(Cl)C=CC=C1Cl DCOPUUMXTXDBNB-UHFFFAOYSA-N 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- HUPFGZXOMWLGNK-UHFFFAOYSA-N diflunisal Chemical compound C1=C(O)C(C(=O)O)=CC(C=2C(=CC(F)=CC=2)F)=C1 HUPFGZXOMWLGNK-UHFFFAOYSA-N 0.000 description 1
- 229960000616 diflunisal Drugs 0.000 description 1
- XYYVYLMBEZUESM-UHFFFAOYSA-N dihydrocodeine Natural products C1C(N(CCC234)C)C2C=CC(=O)C3OC2=C4C1=CC=C2OC XYYVYLMBEZUESM-UHFFFAOYSA-N 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 229960001760 dimethyl sulfoxide Drugs 0.000 description 1
- AMTWCFIAVKBGOD-UHFFFAOYSA-N dioxosilane;methoxy-dimethyl-trimethylsilyloxysilane Chemical compound O=[Si]=O.CO[Si](C)(C)O[Si](C)(C)C AMTWCFIAVKBGOD-UHFFFAOYSA-N 0.000 description 1
- 229960000520 diphenhydramine Drugs 0.000 description 1
- OWQUZNMMYNAXSL-UHFFFAOYSA-N diphenylpyraline Chemical compound C1CN(C)CCC1OC(C=1C=CC=CC=1)C1=CC=CC=C1 OWQUZNMMYNAXSL-UHFFFAOYSA-N 0.000 description 1
- 229960000879 diphenylpyraline Drugs 0.000 description 1
- VLARUOGDXDTHEH-UHFFFAOYSA-L disodium cromoglycate Chemical compound [Na+].[Na+].O1C(C([O-])=O)=CC(=O)C2=C1C=CC=C2OCC(O)COC1=CC=CC2=C1C(=O)C=C(C([O-])=O)O2 VLARUOGDXDTHEH-UHFFFAOYSA-L 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 239000002934 diuretic Substances 0.000 description 1
- 230000001882 diuretic effect Effects 0.000 description 1
- DLNKOYKMWOXYQA-UHFFFAOYSA-N dl-pseudophenylpropanolamine Natural products CC(N)C(O)C1=CC=CC=C1 DLNKOYKMWOXYQA-UHFFFAOYSA-N 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 238000002651 drug therapy Methods 0.000 description 1
- 239000000428 dust Substances 0.000 description 1
- 239000012039 electrophile Substances 0.000 description 1
- 239000003974 emollient agent Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 206010014881 enterobiasis Diseases 0.000 description 1
- 201000001564 eosinophilic gastroenteritis Diseases 0.000 description 1
- 210000003743 erythrocyte Anatomy 0.000 description 1
- 239000012259 ether extract Substances 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- RPWXYCRIAGBAGY-UHFFFAOYSA-N ethyl 2-pyridin-3-ylacetate Chemical compound CCOC(=O)CC1=CC=CN=C1 RPWXYCRIAGBAGY-UHFFFAOYSA-N 0.000 description 1
- UREBWPXBXRYXRJ-UHFFFAOYSA-N ethyl acetate;methanol Chemical compound OC.CCOC(C)=O UREBWPXBXRYXRJ-UHFFFAOYSA-N 0.000 description 1
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 230000028023 exocytosis Effects 0.000 description 1
- 201000001155 extrinsic allergic alveolitis Diseases 0.000 description 1
- OLNTVTPDXPETLC-XPWALMASSA-N ezetimibe Chemical compound N1([C@@H]([C@H](C1=O)CC[C@H](O)C=1C=CC(F)=CC=1)C=1C=CC(O)=CC=1)C1=CC=C(F)C=C1 OLNTVTPDXPETLC-XPWALMASSA-N 0.000 description 1
- ZWJINEZUASEZBH-UHFFFAOYSA-N fenamic acid Chemical class OC(=O)C1=CC=CC=C1NC1=CC=CC=C1 ZWJINEZUASEZBH-UHFFFAOYSA-N 0.000 description 1
- ZPAKPRAICRBAOD-UHFFFAOYSA-N fenbufen Chemical compound C1=CC(C(=O)CCC(=O)O)=CC=C1C1=CC=CC=C1 ZPAKPRAICRBAOD-UHFFFAOYSA-N 0.000 description 1
- 229960001395 fenbufen Drugs 0.000 description 1
- 229950006236 fenclofenac Drugs 0.000 description 1
- IDKAXRLETRCXKS-UHFFFAOYSA-N fenclofenac Chemical compound OC(=O)CC1=CC=CC=C1OC1=CC=C(Cl)C=C1Cl IDKAXRLETRCXKS-UHFFFAOYSA-N 0.000 description 1
- 229950011481 fenclozic acid Drugs 0.000 description 1
- 229960002297 fenofibrate Drugs 0.000 description 1
- YMTINGFKWWXKFG-UHFFFAOYSA-N fenofibrate Chemical compound C1=CC(OC(C)(C)C(=O)OC(C)C)=CC=C1C(=O)C1=CC=C(Cl)C=C1 YMTINGFKWWXKFG-UHFFFAOYSA-N 0.000 description 1
- MQOBSOSZFYZQOK-UHFFFAOYSA-N fenofibric acid Chemical class C1=CC(OC(C)(C)C(O)=O)=CC=C1C(=O)C1=CC=C(Cl)C=C1 MQOBSOSZFYZQOK-UHFFFAOYSA-N 0.000 description 1
- 229960001419 fenoprofen Drugs 0.000 description 1
- 229960001022 fenoterol Drugs 0.000 description 1
- 229960002428 fentanyl Drugs 0.000 description 1
- IVLVTNPOHDFFCJ-UHFFFAOYSA-N fentanyl citrate Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O.C=1C=CC=CC=1N(C(=O)CC)C(CC1)CCN1CCC1=CC=CC=C1 IVLVTNPOHDFFCJ-UHFFFAOYSA-N 0.000 description 1
- 229960002679 fentiazac Drugs 0.000 description 1
- 229960000489 feprazone Drugs 0.000 description 1
- RWTNPBWLLIMQHL-UHFFFAOYSA-N fexofenadine Chemical compound C1=CC(C(C)(C(O)=O)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 RWTNPBWLLIMQHL-UHFFFAOYSA-N 0.000 description 1
- 229960003592 fexofenadine Drugs 0.000 description 1
- LPEPZBJOKDYZAD-UHFFFAOYSA-N flufenamic acid Chemical compound OC(=O)C1=CC=CC=C1NC1=CC=CC(C(F)(F)F)=C1 LPEPZBJOKDYZAD-UHFFFAOYSA-N 0.000 description 1
- 229960004369 flufenamic acid Drugs 0.000 description 1
- 229950007979 flufenisal Drugs 0.000 description 1
- 229950001284 fluprofen Drugs 0.000 description 1
- 229960002390 flurbiprofen Drugs 0.000 description 1
- SYTBZMRGLBWNTM-UHFFFAOYSA-N flurbiprofen Chemical compound FC1=CC(C(C(O)=O)C)=CC=C1C1=CC=CC=C1 SYTBZMRGLBWNTM-UHFFFAOYSA-N 0.000 description 1
- 229960003765 fluvastatin Drugs 0.000 description 1
- 210000000497 foam cell Anatomy 0.000 description 1
- 238000005187 foaming Methods 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 229950010931 furofenac Drugs 0.000 description 1
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 229960003627 gemfibrozil Drugs 0.000 description 1
- 229940074045 glyceryl distearate Drugs 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- 208000024908 graft versus host disease Diseases 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 description 1
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-UHFFFAOYSA-N hexane-1,2,3,4,5,6-hexol Chemical compound OCC(O)C(O)C(O)C(O)CO FBPFZTCFMRRESA-UHFFFAOYSA-N 0.000 description 1
- 235000009200 high fat diet Nutrition 0.000 description 1
- 229960001340 histamine Drugs 0.000 description 1
- 239000003485 histamine H2 receptor antagonist Substances 0.000 description 1
- 230000006801 homologous recombination Effects 0.000 description 1
- 238000002744 homologous recombination Methods 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- LLPOLZWFYMWNKH-CMKMFDCUSA-N hydrocodone Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)CC(=O)[C@@H]1OC1=C2C3=CC=C1OC LLPOLZWFYMWNKH-CMKMFDCUSA-N 0.000 description 1
- 229960000240 hydrocodone Drugs 0.000 description 1
- 229960000890 hydrocortisone Drugs 0.000 description 1
- USZLCYNVCCDPLQ-UHFFFAOYSA-N hydron;n-methoxymethanamine;chloride Chemical compound Cl.CNOC USZLCYNVCCDPLQ-UHFFFAOYSA-N 0.000 description 1
- UWYVPFMHMJIBHE-OWOJBTEDSA-N hydroxymaleic acid group Chemical group O/C(/C(=O)O)=C/C(=O)O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- ZQDWXGKKHFNSQK-UHFFFAOYSA-N hydroxyzine Chemical compound C1CN(CCOCCO)CCN1C(C=1C=CC(Cl)=CC=1)C1=CC=CC=C1 ZQDWXGKKHFNSQK-UHFFFAOYSA-N 0.000 description 1
- 229960000930 hydroxyzine Drugs 0.000 description 1
- 230000000260 hypercholesteremic effect Effects 0.000 description 1
- 208000022098 hypersensitivity pneumonitis Diseases 0.000 description 1
- 201000006362 hypersensitivity vasculitis Diseases 0.000 description 1
- 229950009183 ibufenac Drugs 0.000 description 1
- CYWFCPPBTWOZSF-UHFFFAOYSA-N ibufenac Chemical compound CC(C)CC1=CC=C(CC(O)=O)C=C1 CYWFCPPBTWOZSF-UHFFFAOYSA-N 0.000 description 1
- 150000003949 imides Chemical class 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 230000000495 immunoinflammatory effect Effects 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 229960004187 indoprofen Drugs 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 208000030603 inherited susceptibility to asthma Diseases 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 229940102213 injectable suspension Drugs 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 229960001388 interferon-beta Drugs 0.000 description 1
- 208000036971 interstitial lung disease 2 Diseases 0.000 description 1
- 238000003402 intramolecular cyclocondensation reaction Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 229960001361 ipratropium bromide Drugs 0.000 description 1
- KEWHKYJURDBRMN-ZEODDXGYSA-M ipratropium bromide hydrate Chemical compound O.[Br-].O([C@H]1C[C@H]2CC[C@@H](C1)[N@@+]2(C)C(C)C)C(=O)C(CO)C1=CC=CC=C1 KEWHKYJURDBRMN-ZEODDXGYSA-M 0.000 description 1
- 229950000831 iralukast Drugs 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 229960001268 isoetarine Drugs 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 229950011455 isoxepac Drugs 0.000 description 1
- QFGMXJOBTNZHEL-UHFFFAOYSA-N isoxepac Chemical compound O1CC2=CC=CC=C2C(=O)C2=CC(CC(=O)O)=CC=C21 QFGMXJOBTNZHEL-UHFFFAOYSA-N 0.000 description 1
- YYUAYBYLJSNDCX-UHFFFAOYSA-N isoxicam Chemical compound OC=1C2=CC=CC=C2S(=O)(=O)N(C)C=1C(=O)NC=1C=C(C)ON=1 YYUAYBYLJSNDCX-UHFFFAOYSA-N 0.000 description 1
- 229950002252 isoxicam Drugs 0.000 description 1
- 235000015110 jellies Nutrition 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- DKYWVDODHFEZIM-UHFFFAOYSA-N ketoprofen Chemical compound OC(=O)C(C)C1=CC=CC(C(=O)C=2C=CC=CC=2)=C1 DKYWVDODHFEZIM-UHFFFAOYSA-N 0.000 description 1
- 229960000991 ketoprofen Drugs 0.000 description 1
- 229960004752 ketorolac Drugs 0.000 description 1
- OZWKMVRBQXNZKK-UHFFFAOYSA-N ketorolac Chemical compound OC(=O)C1CCN2C1=CC=C2C(=O)C1=CC=CC=C1 OZWKMVRBQXNZKK-UHFFFAOYSA-N 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- VNYSSYRCGWBHLG-AMOLWHMGSA-N leukotriene B4 Chemical compound CCCCC\C=C/C[C@@H](O)\C=C\C=C\C=C/[C@@H](O)CCCC(O)=O VNYSSYRCGWBHLG-AMOLWHMGSA-N 0.000 description 1
- 239000003199 leukotriene receptor blocking agent Substances 0.000 description 1
- 150000002617 leukotrienes Chemical class 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 229910003002 lithium salt Inorganic materials 0.000 description 1
- 159000000002 lithium salts Chemical class 0.000 description 1
- MLTFKRDFAJSIEX-UHFFFAOYSA-N lithium;1,3-oxazolidin-2-one Chemical compound [Li].O=C1NCCO1 MLTFKRDFAJSIEX-UHFFFAOYSA-N 0.000 description 1
- JCCNYMKQOSZNPW-UHFFFAOYSA-N loratadine Chemical compound C1CN(C(=O)OCC)CCC1=C1C2=NC=CC=C2CCC2=CC(Cl)=CC=C21 JCCNYMKQOSZNPW-UHFFFAOYSA-N 0.000 description 1
- 229960003088 loratadine Drugs 0.000 description 1
- 229960004844 lovastatin Drugs 0.000 description 1
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 1
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 229960000816 magnesium hydroxide Drugs 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 1
- CCERQOYLJJULMD-UHFFFAOYSA-M magnesium;carbanide;chloride Chemical compound [CH3-].[Mg+2].[Cl-] CCERQOYLJJULMD-UHFFFAOYSA-M 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 229960003803 meclofenamic acid Drugs 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 229960003464 mefenamic acid Drugs 0.000 description 1
- 229960000582 mepyramine Drugs 0.000 description 1
- YECBIJXISLIIDS-UHFFFAOYSA-N mepyramine Chemical compound C1=CC(OC)=CC=C1CN(CCN(C)C)C1=CC=CC=N1 YECBIJXISLIIDS-UHFFFAOYSA-N 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- KBOPZPXVLCULAV-UHFFFAOYSA-N mesalamine Chemical compound NC1=CC=C(O)C(C(O)=O)=C1 KBOPZPXVLCULAV-UHFFFAOYSA-N 0.000 description 1
- 229960004963 mesalazine Drugs 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- LMOINURANNBYCM-UHFFFAOYSA-N metaproterenol Chemical compound CC(C)NCC(O)C1=CC(O)=CC(O)=C1 LMOINURANNBYCM-UHFFFAOYSA-N 0.000 description 1
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 1
- 229960003105 metformin Drugs 0.000 description 1
- HTMIBDQKFHUPSX-UHFFFAOYSA-N methdilazine Chemical compound C1N(C)CCC1CN1C2=CC=CC=C2SC2=CC=CC=C21 HTMIBDQKFHUPSX-UHFFFAOYSA-N 0.000 description 1
- 229960004056 methdilazine Drugs 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- KWETUWOIEQLFLM-ILDUYXDCSA-N methyl (2s)-3-methyl-2-(oxolan-2-ylmethylcarbamoylamino)pentanoate Chemical compound CCC(C)[C@@H](C(=O)OC)NC(=O)NCC1CCCO1 KWETUWOIEQLFLM-ILDUYXDCSA-N 0.000 description 1
- HFLMYYLFSNEOOT-UHFFFAOYSA-N methyl 4-chloro-3-oxobutanoate Chemical compound COC(=O)CC(=O)CCl HFLMYYLFSNEOOT-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 229960004584 methylprednisolone Drugs 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- OJGQFYYLKNCIJD-UHFFFAOYSA-N miroprofen Chemical compound C1=CC(C(C(O)=O)C)=CC=C1C1=CN(C=CC=C2)C2=N1 OJGQFYYLKNCIJD-UHFFFAOYSA-N 0.000 description 1
- 229950006616 miroprofen Drugs 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 230000007193 modulation by symbiont of host erythrocyte aggregation Effects 0.000 description 1
- 229960005285 mofebutazone Drugs 0.000 description 1
- REOJLIXKJWXUGB-UHFFFAOYSA-N mofebutazone Chemical compound O=C1C(CCCC)C(=O)NN1C1=CC=CC=C1 REOJLIXKJWXUGB-UHFFFAOYSA-N 0.000 description 1
- 239000003068 molecular probe Substances 0.000 description 1
- 230000023185 monocyte chemotactic protein-1 production Effects 0.000 description 1
- ATHHXGZTWNVVOU-UHFFFAOYSA-N monomethyl-formamide Natural products CNC=O ATHHXGZTWNVVOU-UHFFFAOYSA-N 0.000 description 1
- 229960005127 montelukast Drugs 0.000 description 1
- 229960005181 morphine Drugs 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 206010028417 myasthenia gravis Diseases 0.000 description 1
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 1
- CQGGFCOCFOTAHO-UHFFFAOYSA-N n-[4-(2-nitroimidazol-1-yl)butyl]acridin-9-amine;hydrochloride Chemical compound Cl.[O-][N+](=O)C1=NC=CN1CCCCNC1=C(C=CC=C2)C2=NC2=CC=CC=C12 CQGGFCOCFOTAHO-UHFFFAOYSA-N 0.000 description 1
- GVUWDIFTNXLBBB-UHFFFAOYSA-N n-benzyl-1,1,1-trifluoro-n-(trifluoromethyl)methanamine Chemical compound FC(F)(F)N(C(F)(F)F)CC1=CC=CC=C1 GVUWDIFTNXLBBB-UHFFFAOYSA-N 0.000 description 1
- UPPGACROUYOUAA-UHFFFAOYSA-N n-benzyl-1,1,1-trifluoromethanamine;hydrochloride Chemical compound Cl.FC(F)(F)NCC1=CC=CC=C1 UPPGACROUYOUAA-UHFFFAOYSA-N 0.000 description 1
- CJYQZTZSYREQBD-UHFFFAOYSA-N n-fluorobenzenesulfonamide Chemical compound FNS(=O)(=O)C1=CC=CC=C1 CJYQZTZSYREQBD-UHFFFAOYSA-N 0.000 description 1
- 229960005016 naphazoline Drugs 0.000 description 1
- NVSYANRBXPURRQ-UHFFFAOYSA-N naphthalen-1-ylmethanamine Chemical group C1=CC=C2C(CN)=CC=CC2=C1 NVSYANRBXPURRQ-UHFFFAOYSA-N 0.000 description 1
- 230000017074 necrotic cell death Effects 0.000 description 1
- 230000002956 necrotizing effect Effects 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- 229960003512 nicotinic acid Drugs 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 229960000916 niflumic acid Drugs 0.000 description 1
- 150000002828 nitro derivatives Chemical class 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- 239000003402 opiate agonist Substances 0.000 description 1
- 229960002657 orciprenaline Drugs 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 229960002739 oxaprozin Drugs 0.000 description 1
- OFPXSFXSNFPTHF-UHFFFAOYSA-N oxaprozin Chemical compound O1C(CCC(=O)O)=NC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 OFPXSFXSNFPTHF-UHFFFAOYSA-N 0.000 description 1
- 150000002924 oxiranes Chemical class 0.000 description 1
- MUMZUERVLWJKNR-UHFFFAOYSA-N oxoplatinum Chemical compound [Pt]=O MUMZUERVLWJKNR-UHFFFAOYSA-N 0.000 description 1
- 229960001528 oxymetazoline Drugs 0.000 description 1
- 229960000649 oxyphenbutazone Drugs 0.000 description 1
- HFHZKZSRXITVMK-UHFFFAOYSA-N oxyphenbutazone Chemical compound O=C1C(CCCC)C(=O)N(C=2C=CC=CC=2)N1C1=CC=C(O)C=C1 HFHZKZSRXITVMK-UHFFFAOYSA-N 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- 229940124641 pain reliever Drugs 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- UQPUONNXJVWHRM-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 UQPUONNXJVWHRM-UHFFFAOYSA-N 0.000 description 1
- 229960005489 paracetamol Drugs 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 208000014837 parasitic helminthiasis infectious disease Diseases 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 230000008529 pathological progression Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 229960003436 pentoxyverine Drugs 0.000 description 1
- 125000001151 peptidyl group Chemical group 0.000 description 1
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 1
- 239000008024 pharmaceutical diluent Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 229960003893 phenacetin Drugs 0.000 description 1
- 229960001190 pheniramine Drugs 0.000 description 1
- 229960003531 phenolsulfonphthalein Drugs 0.000 description 1
- DGTNSSLYPYDJGL-UHFFFAOYSA-N phenyl isocyanate Chemical compound O=C=NC1=CC=CC=C1 DGTNSSLYPYDJGL-UHFFFAOYSA-N 0.000 description 1
- 229960002895 phenylbutazone Drugs 0.000 description 1
- VYMDGNCVAMGZFE-UHFFFAOYSA-N phenylbutazonum Chemical compound O=C1C(CCCC)C(=O)N(C=2C=CC=CC=2)N1C1=CC=CC=C1 VYMDGNCVAMGZFE-UHFFFAOYSA-N 0.000 description 1
- 229960001802 phenylephrine Drugs 0.000 description 1
- SONNWYBIRXJNDC-VIFPVBQESA-N phenylephrine Chemical compound CNC[C@H](O)C1=CC=CC(O)=C1 SONNWYBIRXJNDC-VIFPVBQESA-N 0.000 description 1
- 229960000395 phenylpropanolamine Drugs 0.000 description 1
- DLNKOYKMWOXYQA-APPZFPTMSA-N phenylpropanolamine Chemical compound C[C@@H](N)[C@H](O)C1=CC=CC=C1 DLNKOYKMWOXYQA-APPZFPTMSA-N 0.000 description 1
- 229960005095 pioglitazone Drugs 0.000 description 1
- 150000004885 piperazines Chemical class 0.000 description 1
- 150000003053 piperidines Chemical class 0.000 description 1
- 229960005414 pirbuterol Drugs 0.000 description 1
- 229960000851 pirprofen Drugs 0.000 description 1
- PIDSZXPFGCURGN-UHFFFAOYSA-N pirprofen Chemical compound ClC1=CC(C(C(O)=O)C)=CC=C1N1CC=CC1 PIDSZXPFGCURGN-UHFFFAOYSA-N 0.000 description 1
- 229940096701 plain lipid modifying drug hmg coa reductase inhibitors Drugs 0.000 description 1
- 230000007505 plaque formation Effects 0.000 description 1
- 229910003446 platinum oxide Inorganic materials 0.000 description 1
- 229950011515 pobilukast Drugs 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 239000004810 polytetrafluoroethylene Substances 0.000 description 1
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- UAJUXJSXCLUTNU-UHFFFAOYSA-N pranlukast Chemical compound C=1C=C(OCCCCC=2C=CC=CC=2)C=CC=1C(=O)NC(C=1)=CC=C(C(C=2)=O)C=1OC=2C=1N=NNN=1 UAJUXJSXCLUTNU-UHFFFAOYSA-N 0.000 description 1
- 229960004583 pranlukast Drugs 0.000 description 1
- 229960003101 pranoprofen Drugs 0.000 description 1
- 229960002965 pravastatin Drugs 0.000 description 1
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 description 1
- 238000011533 pre-incubation Methods 0.000 description 1
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 1
- 229960004618 prednisone Drugs 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 238000009117 preventive therapy Methods 0.000 description 1
- 150000003138 primary alcohols Chemical class 0.000 description 1
- FYPMFJGVHOHGLL-UHFFFAOYSA-N probucol Chemical compound C=1C(C(C)(C)C)=C(O)C(C(C)(C)C)=CC=1SC(C)(C)SC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 FYPMFJGVHOHGLL-UHFFFAOYSA-N 0.000 description 1
- 229960003912 probucol Drugs 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 230000000770 proinflammatory effect Effects 0.000 description 1
- 229960003910 promethazine Drugs 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- 150000005599 propionic acid derivatives Chemical class 0.000 description 1
- 229960000786 propylhexedrine Drugs 0.000 description 1
- JCRIVQIOJSSCQD-UHFFFAOYSA-N propylhexedrine Chemical compound CNC(C)CC1CCCCC1 JCRIVQIOJSSCQD-UHFFFAOYSA-N 0.000 description 1
- 239000002599 prostaglandin synthase inhibitor Substances 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 229950010450 pseudophedrine Drugs 0.000 description 1
- 201000009732 pulmonary eosinophilia Diseases 0.000 description 1
- JEXVQSWXXUJEMA-UHFFFAOYSA-N pyrazol-3-one Chemical class O=C1C=CN=N1 JEXVQSWXXUJEMA-UHFFFAOYSA-N 0.000 description 1
- HDOUGSFASVGDCS-UHFFFAOYSA-N pyridin-3-ylmethanamine Chemical group NCC1=CC=CN=C1 HDOUGSFASVGDCS-UHFFFAOYSA-N 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 230000033300 receptor internalization Effects 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 238000005057 refrigeration Methods 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 206010039083 rhinitis Diseases 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 229960000672 rosuvastatin Drugs 0.000 description 1
- BPRHUIZQVSMCRT-VEUZHWNKSA-N rosuvastatin Chemical compound CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC(O)=O BPRHUIZQVSMCRT-VEUZHWNKSA-N 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 229960002052 salbutamol Drugs 0.000 description 1
- 150000003873 salicylate salts Chemical class 0.000 description 1
- 201000004409 schistosomiasis Diseases 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 238000007423 screening assay Methods 0.000 description 1
- 230000036303 septic shock Effects 0.000 description 1
- 239000003352 sequestering agent Substances 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 229940083037 simethicone Drugs 0.000 description 1
- 229960002855 simvastatin Drugs 0.000 description 1
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 1
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 1
- 229960002930 sirolimus Drugs 0.000 description 1
- 208000017520 skin disease Diseases 0.000 description 1
- 210000000329 smooth muscle myocyte Anatomy 0.000 description 1
- 235000015424 sodium Nutrition 0.000 description 1
- 229940083542 sodium Drugs 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 229910001467 sodium calcium phosphate Inorganic materials 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 235000010288 sodium nitrite Nutrition 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 201000005671 spondyloarthropathy Diseases 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 229950005175 sudoxicam Drugs 0.000 description 1
- GGCSSNBKKAUURC-UHFFFAOYSA-N sufentanil Chemical group C1CN(CCC=2SC=CC=2)CCC1(COC)N(C(=O)CC)C1=CC=CC=C1 GGCSSNBKKAUURC-UHFFFAOYSA-N 0.000 description 1
- 229960001940 sulfasalazine Drugs 0.000 description 1
- NCEXYHBECQHGNR-UHFFFAOYSA-N sulfasalazine Natural products C1=C(O)C(C(=O)O)=CC(N=NC=2C=CC(=CC=2)S(=O)(=O)NC=2N=CC=CC=2)=C1 NCEXYHBECQHGNR-UHFFFAOYSA-N 0.000 description 1
- 229960000894 sulindac Drugs 0.000 description 1
- MLKXDPUZXIRXEP-MFOYZWKCSA-N sulindac Chemical compound CC1=C(CC(O)=O)C2=CC(F)=CC=C2\C1=C/C1=CC=C(S(C)=O)C=C1 MLKXDPUZXIRXEP-MFOYZWKCSA-N 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 229960004492 suprofen Drugs 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- LXIKEPCNDFVJKC-QXMHVHEDSA-N tenidap Chemical compound C12=CC(Cl)=CC=C2N(C(=O)N)C(=O)\C1=C(/O)C1=CC=CS1 LXIKEPCNDFVJKC-QXMHVHEDSA-N 0.000 description 1
- 229960003676 tenidap Drugs 0.000 description 1
- 229960000195 terbutaline Drugs 0.000 description 1
- 229960000351 terfenadine Drugs 0.000 description 1
- ZPZQBIKXAJZZGK-UHFFFAOYSA-N tert-butyl 3-amino-1,2-thiazole-4-carboxylate Chemical compound CC(C)(C)OC(=O)C1=CSN=C1N ZPZQBIKXAJZZGK-UHFFFAOYSA-N 0.000 description 1
- IYRULVWLVRRRIS-UHFFFAOYSA-N tert-butyl 3-amino-2-hydroxypropanoate Chemical class CC(C)(C)OC(=O)C(O)CN IYRULVWLVRRRIS-UHFFFAOYSA-N 0.000 description 1
- QAMKZPZMTWAMLU-UHFFFAOYSA-N tert-butyl 4-(2-aminoanilino)piperidine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC1NC1=CC=CC=C1N QAMKZPZMTWAMLU-UHFFFAOYSA-N 0.000 description 1
- WINCJOWZQAVIPN-UHFFFAOYSA-N tert-butyl 4-(2-ethoxy-2-oxoethyl)-2-[(2-methylpropan-2-yl)oxycarbonylimino]-1,3-thiazole-3-carboxylate Chemical compound CCOC(=O)CC1=CSC(=NC(=O)OC(C)(C)C)N1C(=O)OC(C)(C)C WINCJOWZQAVIPN-UHFFFAOYSA-N 0.000 description 1
- XBSYPKCFRQMSOB-UHFFFAOYSA-N tert-butyl 5-amino-3-methyl-3h-1,2-thiazole-2-carboxylate;hydrochloride Chemical compound Cl.CC1C=C(N)SN1C(=O)OC(C)(C)C XBSYPKCFRQMSOB-UHFFFAOYSA-N 0.000 description 1
- RUPAXCPQAAOIPB-UHFFFAOYSA-N tert-butyl formate Chemical compound CC(C)(C)OC=O RUPAXCPQAAOIPB-UHFFFAOYSA-N 0.000 description 1
- 239000012414 tert-butyl nitrite Substances 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 229960000278 theophylline Drugs 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- CWERGRDVMFNCDR-UHFFFAOYSA-M thioglycolate(1-) Chemical compound [O-]C(=O)CS CWERGRDVMFNCDR-UHFFFAOYSA-M 0.000 description 1
- 229960001312 tiaprofenic acid Drugs 0.000 description 1
- 229950002345 tiopinac Drugs 0.000 description 1
- 229950006150 tioxaprofen Drugs 0.000 description 1
- YEZNLOUZAIOMLT-UHFFFAOYSA-N tolfenamic acid Chemical compound CC1=C(Cl)C=CC=C1NC1=CC=CC=C1C(O)=O YEZNLOUZAIOMLT-UHFFFAOYSA-N 0.000 description 1
- 229960002905 tolfenamic acid Drugs 0.000 description 1
- 229960001017 tolmetin Drugs 0.000 description 1
- UPSPUYADGBWSHF-UHFFFAOYSA-N tolmetin Chemical compound C1=CC(C)=CC=C1C(=O)C1=CC=C(CC(O)=O)N1C UPSPUYADGBWSHF-UHFFFAOYSA-N 0.000 description 1
- LLPOLZWFYMWNKH-UHFFFAOYSA-N trans-dihydrocodeinone Natural products C1C(N(CCC234)C)C2CCC(=O)C3OC2=C4C1=CC=C2OC LLPOLZWFYMWNKH-UHFFFAOYSA-N 0.000 description 1
- 238000011830 transgenic mouse model Methods 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- CFQJBWKKHCMCGJ-UHFFFAOYSA-N tributyl(pyridin-3-yl)stannane Chemical compound CCCC[Sn](CCCC)(CCCC)C1=CC=CN=C1 CFQJBWKKHCMCGJ-UHFFFAOYSA-N 0.000 description 1
- 208000003982 trichinellosis Diseases 0.000 description 1
- 201000007588 trichinosis Diseases 0.000 description 1
- 208000009920 trichuriasis Diseases 0.000 description 1
- 229960003223 tripelennamine Drugs 0.000 description 1
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 1
- 229960001128 triprolidine Drugs 0.000 description 1
- CBEQULMOCCWAQT-WOJGMQOQSA-N triprolidine Chemical compound C1=CC(C)=CC=C1C(\C=1N=CC=CC=1)=C/CN1CCCC1 CBEQULMOCCWAQT-WOJGMQOQSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- GXPHKUHSUJUWKP-UHFFFAOYSA-N troglitazone Chemical compound C1CC=2C(C)=C(O)C(C)=C(C)C=2OC1(C)COC(C=C1)=CC=C1CC1SC(=O)NC1=O GXPHKUHSUJUWKP-UHFFFAOYSA-N 0.000 description 1
- 229960001641 troglitazone Drugs 0.000 description 1
- GXPHKUHSUJUWKP-NTKDMRAZSA-N troglitazone Natural products C([C@@]1(OC=2C(C)=C(C(=C(C)C=2CC1)O)C)C)OC(C=C1)=CC=C1C[C@H]1SC(=O)NC1=O GXPHKUHSUJUWKP-NTKDMRAZSA-N 0.000 description 1
- 241001529453 unidentified herpesvirus Species 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 238000005292 vacuum distillation Methods 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 239000003039 volatile agent Substances 0.000 description 1
- 238000002424 x-ray crystallography Methods 0.000 description 1
- 229960000833 xylometazoline Drugs 0.000 description 1
- 229960004764 zafirlukast Drugs 0.000 description 1
- 229950007802 zidometacin Drugs 0.000 description 1
- MWLSOWXNZPKENC-SSDOTTSWSA-N zileuton Chemical compound C1=CC=C2SC([C@H](N(O)C(N)=O)C)=CC2=C1 MWLSOWXNZPKENC-SSDOTTSWSA-N 0.000 description 1
- 229960005332 zileuton Drugs 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- GTLDTDOJJJZVBW-UHFFFAOYSA-N zinc cyanide Chemical compound [Zn+2].N#[C-].N#[C-] GTLDTDOJJJZVBW-UHFFFAOYSA-N 0.000 description 1
- 229960003414 zomepirac Drugs 0.000 description 1
- ZXVNMYWKKDOREA-UHFFFAOYSA-N zomepirac Chemical compound C1=C(CC(O)=O)N(C)C(C(=O)C=2C=CC(Cl)=CC=2)=C1C ZXVNMYWKKDOREA-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/06—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with radicals, containing only hydrogen and carbon atoms, attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/438—The ring being spiro-condensed with carbocyclic or heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/14—Decongestants or antiallergics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/10—Anthelmintics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P39/00—General protective or antinoxious agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/14—Radicals substituted by nitrogen atoms, not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/10—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with radicals containing only carbon and hydrogen atoms attached to ring carbon atoms
- C07D211/14—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with radicals containing only carbon and hydrogen atoms attached to ring carbon atoms with hydrocarbon or substituted hydrocarbon radicals attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/20—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by singly bound oxygen or sulphur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/40—Oxygen atoms
- C07D211/44—Oxygen atoms attached in position 4
- C07D211/52—Oxygen atoms attached in position 4 having an aryl radical as the second substituent in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/60—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D211/62—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/60—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D211/62—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals attached in position 4
- C07D211/64—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals attached in position 4 having an aryl radical as the second substituent in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D221/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00
- C07D221/02—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00 condensed with carbocyclic rings or ring systems
- C07D221/20—Spiro-condensed ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D451/00—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof
- C07D451/02—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing not further condensed 8-azabicyclo [3.2.1] octane or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane; Cyclic acetals thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
Definitions
- chemokines are a family of small (70-120 amino acids), proinflammatory cytokines, with potent chemotactic activities. Chemokines are chemotactic cytokines that are released by a wide variety of cells to attract various cells, such as monocytes, macrophages, T cells, eosinophils, basophils and neutrophils to sites of inflammation (reviewed in Schall, Cvtokine, 3, 165-183 (1991) and Murphy, Rev. Immun., 12, 593-633 (1994)). These molecules were originally defined by four conserved cysteines and divided into two subfamilies based on the arrangement of the first cysteine pair.
- CXC-chemokine family which includes IL-8, GRO , NAP-2 and IP- 10
- these two cysteines are separated by a single amino acid
- CC-chemokine family which includes RANTES, MCP-1, MCP-2, MCP-3, MlP-l ⁇ , MTP-l ⁇ and eotaxin
- the chemokines are secreted by a wide variety of cell types and bind to specific
- GPCRs G-protein coupled receptors
- chemokine receptors form a sub-family of GPCRs, which, at present, consists of fifteen characterized members and a number of orphans.
- chemokine receptors are more selectively expressed on subsets of leukocytes.
- generation of specific chemokines provides a mechanism for recruitment of particular leukocyte subsets.
- chemokine receptors On binding their cognate ligands, chemokine receptors transduce an intracellular signal though the associated trimeric G protein, resulting in a rapid increase in intracellular calcium concentration.
- CCR-1 or "CKR-1" or "CC-CKR- 1" [MEP-l ⁇ , MlP-l ⁇ , MCP-3, RANTES] (Ben-Barruch, et al., J. Biol.
- the ⁇ -chemokines include eotaxin, -VHP ("macrophage inflammatory protein"), MCP ("monocyte chemoattractant protein”) and RANTES ("regulation-upon-activation, normal T expressed and secreted”) among other chemokines.
- Chemokine receptors such as CCR-1, CCR-2, CCR-2A, CCR-2B, CCR-3, CCR- 4, CCR-5, CXCR-3, CXCR-4, have been implicated as being important mediators of inflammatory and immunoregulatory disorders and diseases, including asthma, rhinitis and allergic diseases, as well as autoimmune pathologies such as rheumatoid arthritis and atherosclerosis.
- Humans who are homozygous for the 32-basepair deletion in the CCR-5 gene appear to have less susceptibility to rheumatoid arthritis (Gomez, et al., Arthritis & Rheumatism, 42, 989-992 (1999)).
- chemokines are potent chemoattractants for monocytes and macrophages. The best characterized of these is MCP-1 (monocyte chemoattractant protein-1), whose primary receptor is CCR2.
- MCP-1 is produced in a variety of cell types in response to inflammatory stimuli in various species, including rodents and humans, and stimulates chemotaxis in monocytes and a subset of lymphocytes. In particular, MCP-1 production correlates with monocyte and macrophage infiltration at inflammatory sites. Deletion of either MCP-1 or CCR2 by homologous recombination in mice results in marked attenuation of monocyte recruitment in response to thioglycollate injection and Listeria monocytogenes infection (Lu et al., J. Exp. Med.. 187, 601-608 (1998); Kurihara et al. J. Exp. Med., 186, 1757-1762 (1997); Boring et al. J. Clin.
- agents which modulate chemokine receptors such as the CCR-2 receptor would be useful in such disorders and diseases.
- the recruitment of monocytes to inflammatory lesions in the vascular wall is a major component of the pathogenesis of atherogenic plaque formation.
- MCP-1 is produced and secreted by endothelial cells and intimal smooth muscle cells after injury to the vascular wall in hypercholesterolemic conditions.
- Monocytes recruited to the site of injury infiltrate the vascular wall and differentiate to foam cells in response to the released MCP-1.
- CCR2 antagonists may inhibit atherosclerotic lesion formation and pathological progression by impairing monocyte recruitment and differentiation in the arterial wall.
- the present invention is further directed to compounds which are modulators of chemokine receptor activity and are useful in the prevention or treatment of certain inflammatory and immunoregulatory disorders and diseases, allergic diseases, atopic conditions including allergic rhinitis, dermatitis, conjunctivitis, and asthma, as well as autoimmune pathologies such as rheumatoid arthritis and atherosclerosis.
- the invention is also directed to pharmaceutical compositions comprising these compounds and the use of these compounds and compositions in the prevention or treatment of such diseases in which chemokine receptors are involved.
- the present invention is directed to compounds of the formula I:
- W is selected from the group consisting of:
- X is selected from the group consisting of:
- R*0 is independently selected from: hydrogen, C ⁇ _6 alkyl, benzyl, phenyl, and Ci-6 alkyl-C3_6 cycloalkyl, which is unsubstituted or substituted with 1-3 substituents where the substituents are independently selected from: halo, Ci-3alkyl, C 1 -3 alkoxy and trifluoromethyl ; or where R 10 and R 2 may be joined together to form a 5- or 6-membered ring,
- Y is selected from: a single bond, -O-, -S-, -SO-, -SO2-, and -NR10-, and where the phenyl, heterocycle, alkyl and the cycloalkyl are unsubstituted or substituted with 1-7 substituents where the substituents are independently selected from:
- R9 is independently selected from: hydrogen, Ci_6 alkyl, C5-.6 cycloalkyl, benzyl or phenyl, which is unsubstituted or substituted with 1-3 substituents where the substituents are independently selected from: halo, C1-.3a.kyl, Ci_3alkoxy and trifluoromethyl,
- R3 is selected from: hydrogen, (C ⁇ -6 a lkyl)-phenyl, (Co- 6 alkyl)-heterocycle, C ⁇ - 6 alkyl, CF 3 , C 3 - cycloalkyl, - NR9R10, -CO2R 9 , -NR9-SO2-R 10 , -NR 9 CONR 9 R 10 , and -CONR 9 R 10 , where the alkyl is unsubstituted or substituted with 1-5 substituents where the substituents are independently selected from:
- R4 is selected from:
- R3 and R ⁇ may be joined together to form a ring which is selected from:
- R 5 , R 6 , R7 a nd R 8 are independently selected from
- n is an integer selected from 0, 1, 2 and 3;
- More preferred compounds of the present invention include those of formula lb:
- Rl, R2, and R ⁇ are defined herein, and wherein Rl5 and R ⁇ are independently selected from:
- More preferred compounds of the present invention also include those of formula Ic:
- More preferred compounds of the present invention include those of formula Id:
- Rl, R ⁇ , and R ⁇ are defined herein, and where Z is a heterocycle selected from the group consisting of: benzoimidazolyl, benzofuranyl, benzofurazanyl, benzopyrazolyl, benzotriazolyl, benzothiophenyl, benzoxazolyl, carbazolyl, carbolinyl, cinnolinyl, furanyl, imidazolyl, indolinyl, indolyl, indolazinyl, indazolyl, isobenzofuranyl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, naphthpyridinyl, oxadiazolyl, oxazolyl, oxetanyl, pyranyl, pyrazinyl, pyrazolyl, pyridazinyl, pyridopyridinyl, pyridazinyl,
- X is -CONH-.
- Rl is selected from: -Co- 6 alkyl-phenyl, C 0 . 6 alkyl-heterocycle, -Ci-6alkyl, -C ⁇ -6alkyl-O-Ci-6alkyl-, -C ⁇ -
- R 9 is independently selected from: hydrogen, Ci-6 alkyl, C5-.6 cycloalkyl, benzyl or phenyl, which is unsubstituted or substituted with 1-3 substituents where the substituents are independently selected from: halo, C ⁇ _3alkyl, C1-.3a.koxy and trifluoromethyl, (h) -CN,
- Rl is selected from: (1) -Ci_6alkyl, which is unsubstituted or substituted with 1-6 substituents where the substituents are independently selected from: (a) halo, (b) hydroxy,
- phenyl which is unsubstituted or substituted with 1-5 substituents where the substituents are independently selected from:
- R is selected from:
- R is selected from: -(C ⁇ -4alkyl)-phenyl and -(C ⁇ -4alkyl)-heterocycle, where heterocycle is selected from: furanyl, imidazolyl, oxadiazolyl, oxazolyl, pyrazolyl, pyrazinyl, pyridyl, pyridazinyl, pyrimidyl, pyrrolyl, thiadiazolyl, thiazolyl, thienyl, and triazolyl, and
- R ⁇ is selected from:
- heterocycle is selected from: pyridyl, pyridazinyl, pyrimidyl, and N-oxides thereof, and where the phenyl or heterocycle is unsubstituted or substituted with 1-3 substituents where the substituents are independently selected from: (a) halo,
- R2 is selected from:
- R? is phenyl or heterocycle, where the phenyl or heterocycle is unsubstituted or substituted with 1-5 substituents where the substituents are independently selected from:
- R3 is phenyl or heterocycle, where the phenyl or heterocycle is unsubstituted or substituted with 1-3 substituents where the substituents are independently selected from: (a) halo,
- R ⁇ is phenyl, para-fluorophenyl, 3-carboxyphenyl, 3-pyridyl, 3,5-pyrimidyl, 1-benzimidazole, 3-indole, 1- indazole, 1 -pyrrole, imidazoyl, diazoyl, triazoyl or tetrazoyl.
- R4 is selected from:
- R5 and R6 are independently selected from:
- RU is hydrogen
- Rl2 is hydrogen
- Especially preferred compounds of the present invention include those of the formula:
- Especially preferred compounds of the present invention include those of the formula:
- R ⁇ and R2 are defined herein; and pharmaceutically acceptable salts and individual diastereomers thereof.
- Especially preferred compounds of the present invention include those of the formula:
- R! and R are defined herein; and pharmaceutically acceptable salts and individual diastereomers thereof.
- Representative compounds of the present invention include those presented in the Examples and pharmaceutically acceptable salts and individual diastereomers thereof.
- the compounds of the instant invention have at least one asymmetric center at the position ⁇ -to the amide carbonyl. Additional asymmetric centers may be present depending upon the nature of the various substituents on the molecule. Each such asymmetric center will independently produce two optical isomers and it is intended that all of the possible optical isomers and diastereomers in mixtures and as pure or partially purified compounds are included within the ambit of this invention.
- the absolute configurations of the more preferred compounds of this invention are of the configuration shown below, where the position ⁇ -to the amide carbonyl
- Ci -.3 alkyl is defined to identify the group as having 1, 2 or 3 carbons in a linear or branched arrangement, such that C1-.3 alkyl specifically includes methyl, ethyl, n-propyl, and iso-propyl.
- pharmaceutically acceptable refers to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- pharmaceutically acceptable salts refer to derivatives wherein the parent compound is modified by making acid or base salts thereof. Examples of pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like.
- the pharmaceutically acceptable salts include the conventional non-toxic salts or the quaternary ammonium salts of the parent compound formed, for example, from non-toxic inorganic or organic acids.
- such conventional non-toxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like; and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, sulfanilic, 2-acetoxybenzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, isethionic, and the like.
- the pharmaceutically acceptable salts of the present invention can be prepared from the parent compound which contains a basic or acidic moiety by conventional chemical methods. Generally, such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, nonaqueous media such as ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are preferred. Suitable salts are found, e.g. in Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, PA, 1985, p. 1418. Specific compounds within the present invention include a compound which selected from the group consisting of: the title compounds of the Examples; and pharmaceutically acceptable salts thereof and individual diastereomers thereof.
- the subject compounds are useful in a method of modulating chemokine receptor activity in a patient in need of such modulation comprising the administration of an effective amount of the compound.
- the present invention is directed to the use of the foregoing compounds as modulators of chemokine receptor activity.
- these compounds are useful as modulators of the chemokine receptors, in particular CCR-2.
- Receptor affinity in a CCR-2 binding assay was determined by measuring inhibition of 125 I-MCP-1 to the endogenous CCR-2 receptor on various cell types including monocytes, THP-1 cells, or after heterologous expression of the cloned receptor in eukaryotic cells.
- the cells were suspended in binding buffer (50 mM HEPES, pH 7.2, 5 mM MgCl 2 , 1 M CaCl 2 , and 0.50% BSA) with and added to test compound or DMSO and 125 I-MCP-1 at room temperature for 1 h to allow binding.
- the cells were then collected on GFB filters, washed with 25 mM HEPES buffer containing 500 mM NaCl and cell bound 125 I-MCP-1 was quantified.
- chemotaxis assay was performed using T cell depleted PBMC isolated from venous whole or leukophoresed blood and purified by Ficoll-Hypaque centrifugation followed by rosetting with neuraminidase-treated sheep erythrocytes. Once isolated, the cells were washed with HBSS containing 0.1 mg/ml BSA and suspended at lxlO 7 cells/ml. Cells were fluorescently labeled in the dark with 2 ⁇ M Calcien-AM (Molecular Probes), for 30 min at 37° C.
- the compounds of the following examples had activity in binding to the CCR-2 receptor in the aforementioned assays, generally with an IC50 °f l ess than about 1 ⁇ M. Such a result is indicative of the intrinsic activity of the compounds in use as modulators of chemokine receptor activity.
- Mammalian chemokine receptors provide a target for interfering with or promoting eosinophil and or lymphocyte function in a mammal, such as a human.
- Compounds which inhibit or promote chemokine receptor function are particularly useful for modulating eosinophil and/or lymphocyte function for therapeutic purposes.
- compounds which inhibit or promote chemokine receptor function would be useful in treating, preventing, ameliorating, controlling or reducing the risk of a wide variety of inflammatory and immunoregulatory disorders and diseases, allergic diseases, atopic conditions including allergic rhinitis, dermatitis, conjunctivitis, and asthma, as well as autoimmune pathologies such as rheumatoid arthritis and atherosclerosis.
- an instant compound which inhibits one or more functions of a mammalian chemokine receptor e.g., a human chemokine receptor
- one or more inflammatory processes such as leukocyte emigration, chemotaxis, exocytosis (e.g., of enzymes, histamine) or inflammatory mediator release, is inhibited.
- mammals including, but not limited to, cows, sheep, goats, horses, dogs, cats, guinea pigs, rats or other bovine, ovine, equine, canine, feline, rodent or murine species can be treated.
- the method can also be practiced in other species, such as avian species (e.g., chickens).
- the disease or condition is one in which the actions of lymphocytes are to be inhibited or promoted, in order to modulate the inflammatory response.
- Diseases or conditions of humans or other species which can be treated with inhibitors of chemokine receptor function include, but are not limited to: inflammatory or allergic diseases and conditions, including respiratory allergic diseases such as asthma, particularly bronchial asthma, allergic rhinitis, hypersensitivity lung diseases, hypersensitivity pneumonitis, eosinophilic pneumonias (e.g., Loeffler's syndrome, chronic eosinophilic pneumonia), delayed-type hypersentitivity, interstitial lung diseases (ILD) (e.g., idiopathic pulmonary fibrosis, or ELD associated with rheumatoid arthritis, systemic lupus erythematosus, ankylosing spondylitis, systemic sclerosis, Sjogren's syndrome, polymyositis or dermatomyositis); systemic anaphylaxis or hypersensitivity responses, drug allergies (e.g., to penicillin, cephalosporins), insect sting allergies; autoimmune diseases, such as r
- Other diseases or conditions in which undesirable inflammatory responses are to be inhibited can be treated, including, but not limited to, reperfusion injury, atherosclerosis, certain hematologic malignancies, cytokine-induced toxicity (e.g., septic shock, endotoxic shock), polymyositis, dermatomyositis.
- Immunosuppression such as that in individuals with immunodeficiency syndromes such as AIDS or other viral infections, individuals undergoing radiation therapy, chemotherapy, therapy for autoimmune disease or drug therapy (e.g., corticosteroid therapy), which causes immunosuppression; immunosuppression due to congenital deficiency in receptor function or other causes; and infections diseases, such as parasitic diseases, including, but not limited to helminth infections, such as nematodes (round worms), (Trichuriasis, Enterobiasis, Ascariasis, Hookworm, Strongyloidiasis, Trichinosis, filariasis), trematodes (flukes) (Schistosomiasis, Clonorchiasis), cestodes (tape worms) (Echinococcosis, Taeniasis saginata, Cysticercosis),
- helminth infections such as nematodes (round worms), (Trichuriasis, Enterobia
- treatment of the aforementioned inflammatory, allergic and autoimmune diseases can also be contemplated for promoters of chemokine receptor function if one contemplates the delivery of sufficient compound to cause the loss of receptor expression on cells through the induction of chemokine receptor internalization or delivery of compound in a manner that results in the misdirection of the migration of cells.
- the compounds of the present invention are accordingly useful in treating, preventing, ameliorating, controlling or reducing the risk of a wide variety of inflammatory and immunoregulatory disorders and diseases, allergic conditions, atopic conditions, as well as autoimmune pathologies.
- the present invention is directed to the use of the subject compounds for treating, preventing, ameliorating, controlling or reducing the risk of autoimmune diseases, such as rheumatoid arthritis or psoriatic arthritis.
- the instant invention may be used to evaluate putative specific agonists or antagonists of chemokine receptors, including CCR-2. Accordingly, the present invention is directed to the use of these compounds in the preparation and execution of screening assays for compounds which modulate the activity of chemokine receptors.
- the compounds of this invention are useful for isolating receptor mutants, which are excellent screening tools for more potent compounds.
- the compounds of this invention are useful in establishing or determining the binding site of other compounds to chemokine receptors, e.g., by competitive inhibition.
- the compounds of the instant invention are also useful for the evaluation of putative specific modulators of the chemokine receptors, including CCR-2.
- the present invention is further directed to a method for the manufacture of a medicament for modulating chemokine receptor activity in humans and animals comprising combining a compound of the present invention with a pharmaceutical carrier or diluent.
- the present invention is further directed to the use of the present compounds in treating, preventing, ameliorating, controlling or reducing the risk of infection by a retrovirus, in particular, herpes virus or the human immunodeficiency virus (FflV) and the treatment of, and delaying of the onset of consequent pathological conditions such as AIDS.
- a retrovirus in particular, herpes virus or the human immunodeficiency virus (FflV)
- FflV human immunodeficiency virus
- Treating ADDS or preventing or treating infection by HTV is defined as including, but not limited to, treating a wide range of states of H1N infection: AIDS, ARC (AIDS related complex), both symptomatic and asymptomatic, and actual or potential exposure to HTV.
- the compounds of this invention are useful in treating infection by H1N after suspected past exposure to HTV by, e.g., blood transfusion, organ transplant, exchange of body fluids, bites, accidental needle stick, or exposure to patient blood during surgery.
- a subject compound may be used in a method of inhibiting the binding of a chemokine to a chemokine receptor, such as CCR-2, of a target cell, which comprises contacting the target cell with an amount of the compound which is effective at inhibiting the binding of the chemokine to the chemokine receptor.
- the subject treated in the methods above is a mammal, preferably a human being, male or female, in whom modulation of chemokine receptor activity is desired.
- “Modulation” as used herein is intended to encompass antagonism, agonism, partial antagonism, inverse agonism and/or partial agonism. In a preferred aspect of the present invention, modulation refers to antagonism of chemokine receptor activity.
- therapeutically effective amount means the amount of the subject compound that will elicit the biological or medical response of a tissue, system, animal or human that is being sought by the researcher, veterinarian, medical doctor or other clinician.
- composition as used herein is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- pharmaceutically acceptable it is meant the carrier, diluent or excipient must be compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
- administration of and or “administering a” compound should be understood to mean providing a compound of the invention to the individual in need of treatment.
- treatment refers both to the treatment and to the prevention or prophylactic therapy of the aforementioned conditions.
- Combined therapy to modulate chemokine receptor activity for thereby treating, preventing, ameliorating, controlling or reducing the risk of inflammatory and immunoregulatory disorders and diseases, including asthma and allergic diseases, as well as autoimmune pathologies such as rheumatoid arthritis and atherosclerosis, and those pathologies noted above is illustrated by the combination of the compounds of this invention and other compounds which are known for such utilities.
- the present compounds may be used in conjunction with an antiinflammatory or analgesic agent such as an opiate agonist, a lipoxygenase inhibitor, such as an inhibitor of 5- lipoxygenase, a cyclooxygenase inhibitor, such as a cyclooxygenase-2 inhibitor, an interleukin inhibitor, such as an interleukin- 1 inhibitor, an NMD A antagonist, an inhibitor of nitric oxide or an inhibitor of the synthesis of nitric oxide, a non-steroidal antiinflammatory agent, or a cytokine-suppressing antiinflammatory agent, for example with a compound such as acetaminophen, aspirin, codeine, usinel, fentanyl, ibuprofen, indomethacin, ketorolac, morphine, naproxen, phenacetin, piroxicam, a steroidal analgesic, sufentany
- analgesic agent such as an opiate agonist, a lipoxy
- the instant compounds may be administered with a pain reliever; a potentiator such as caffeine, an H2-antagonist, simethicone, aluminum or magnesium hydroxide; a decongestant such as phenylephrine, phenylpropanolamine, pseudophedrine, oxymetazoline, ephinephrine, naphazoline, xylometazoline, propylhexedrine, or levo-desoxy-ephedrine; an antiitussive such as codeine, hydrocodone, caramiphen, carbetapentane, or dextramethorphan; a diuretic; and a sedating or non-sedating antihistamine.
- a pain reliever such as caffeine, an H2-antagonist, simethicone, aluminum or magnesium hydroxide
- a decongestant such as phenylephrine, phenylpropanolamine, pseudophedrine, oxymetazoline, ephinep
- compounds of the present invention may be used in combination with other drugs that are used in the treatment/prevention/suppression or amelioration of the diseases or conditions for which compounds of the pressent invention are useful.
- Such other drugs may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with a compound of the present invention.
- a pharmaceutical composition containing such other drugs in addition to the compound of the present invention is preferred.
- the pharmaceutical compositions of the present invention include those that also contain one or more other active ingredients, in addition to a compound of the present invention.
- Examples of other active ingredients that may be combined with a compound of the present invention, either administered separately or in the same pharmaceutical compositions, include, but are not limited to: (a) NLA-4 antagonists such as those described in US 5,510,332, WO95/15973, WO96/01644, WO96/06108, WO96/20216, WO96/22966, WO96/31206, WO96/40781, WO97/03094, WO97/02289, WO 98/42656, WO98/53814, WO98/53817, WO98/53818, WO98/54207, and WO98/58902; (b) steroids such as beclomethasone, methylprednisolone, betamethasone, prednisone, dexamethasone, and hydrocortisone; (c) immunosuppressants such as cyclosporin, tacrolimus, rapamycin and other FK-506 type immunosuppressants; (d) antihistamines
- the weight ratio of the compound of the present invention to the second active ingredient may be varied and will depend upon the effective dose of each ingredient. Generally, an effective dose of each will be used. Thus, for example, when a compound of the present invention is combined with an NS AID the weight ratio of the compound of the present invention to the NSAED will generally range from about 1000:1 to about 1:1000, preferably about 200:1 to about 1 :200. Combinations of a compound of the present invention and other active ingredients will generally also be within the aforementioned range, but in each case, an effective dose of each active ingredient should be used. In such combinations the compound of the present invention and other active agents may be administered separately or in conjunction. In addition, the administration of one element may be prior to, concurrent to, or subsequent to the administration of other agent(s). The compounds of the present invention may be administered by oral, parenteral
- intramuscular, intraperitoneal, intravenous, ICV, intracisternal injection or infusion, subcutaneous injection, or implant by inhalation spray, nasal, vaginal, rectal, sublingual, or topical routes of administration and may be formulated, alone or together, in suitable dosage unit formulations containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles appropriate for each route of administration.
- suitable dosage unit formulations containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles appropriate for each route of administration.
- the compounds of the invention are effective for use in humans.
- compositions for the administration of the compounds of this invention may conveniently be presented in dosage unit form and may be prepared by any of the methods well known in the art of pharmacy. All methods include the step of bringing the active ingredient into association with the carrier which constitutes one or more accessory ingredients.
- the pharmaceutical compositions are prepared by uniformly and intimately bringing the active ingredient into association with a liquid carrier or a finely divided solid carrier or both, and then, if necessary, shaping the product into the desired formulation.
- the active object compound is included in an amount sufficient to produce the desired effect upon the process or condition of diseases.
- composition is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- the pharmaceutical compositions containing the active ingredient may be in a form suitable for oral use, for example, as tablets, troches, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsions, hard or soft capsules, or syrups or elixirs.
- compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations.
- Tablets contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients which are suitable for the manufacture of tablets.
- excipients may be for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, for example starch, gelatin or acacia, and lubricating agents, for example magnesium stearate, stearic acid or talc.
- the tablets may be uncoated or they may be coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- a time delay material such as glyceryl monostearate or glyceryl distearate may be employed. They may also be coated by the techniques described in the U.S. Patents 4,256,108; 4,166,452; and 4,265,874 to form osmotic therapeutic tablets for control release.
- Formulations for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example peanut oil, liquid paraffin, or olive oil.
- Aqueous suspensions contain the active materials in admixture with excipients suitable for the manufacture of aqueous suspensions.
- excipients are suspending agents, for example sodium carboxymethylcellulose, methylcellulose, hydroxy- propylmethylcellulose, sodium alginate, polyvinyl-pyrrolidone, gum tragacanth and gum acacia; dispersing or wetting agents may be a naturally-occurring phosphatide, for example lecithin, or condensation products of an alkylene oxide with fatty acids, for example polyoxyethylene stearate, or condensation products of ethylene oxide with long chain aliphatic alcohols, for example heptadecaethy ⁇ ene-oxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate, or condensation products of ethylene oxide with partial esters derived from fatty acids and hexitol anhydrides, for example polyethylene sorbitan monooleate.
- dispersing or wetting agents may be a naturally-occurring phosphatide, for example
- the aqueous suspensions may also contain one or more preservatives, for example ethyl, or n-propyl, p-hydroxybenzoate, one or more coloring agents, one or more flavoring agents, and one or more sweetening agents, such as sucrose or saccharin.
- Oily suspensions may be formulated by suspending the active ingredient in a vegetable oil, for example arachis oil, olive oil, sesame oil or coconut oil, or in a mineral oil such as liquid paraffin.
- the oily suspensions may contain a thickening agent, for example beeswax, hard paraffin or cetyl alcohol. Sweetening agents such as those set forth above, and flavoring agents may be added to provide a palatable oral preparation.
- compositions may be preserved by the addition of an anti-oxidant such as ascorbic acid.
- Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives. Suitable dispersing or wetting agents and suspending agents are exemplified by those already mentioned above. Additional excipients, for example sweetening, flavoring and coloring agents, may also be present.
- the pharmaceutical compositions of the invention may also be in the form of oil-in-water emulsions.
- the oily phase may be a vegetable oil, for example olive oil or arachis oil, or a mineral oil, for example liquid paraffin or mixtures of these.
- Suitable emulsifying agents may be naturally- occurring gums, for example gum acacia or gum tragacanth, naturally- occurring phosphatides, for example soy bean, lecithin, and esters or partial esters derived from fatty acids and hexitol anhydrides, for example sorbitan monooleate, and condensation products of the said partial esters with ethylene oxide, for example polyoxyethylene sorbitan monooleate.
- the emulsions may also contain sweetening and flavoring agents. Syrups and elixirs may be formulated with sweetening agents, for example glycerol, propylene glycol, sorbitol or sucrose. Such formulations may also contain a demulcent, a preservative and flavoring and coloring agents.
- the pharmaceutical compositions may be in the form of a sterile injectable aqueous or oleagenous suspension.
- This suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents which have been mentioned above.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally-acceptable diluent or solvent, for example as a solution in 1,3-butane diol.
- the acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid find use in the preparation of injectables.
- the compounds of the present invention may also be administered in the form of suppositories for rectal administration of the drug.
- These compositions can be prepared by mixing the drug with a suitable non-irritating excipient which is solid at ordinary temperatures but liquid at the rectal temperature and will therefore melt in the rectum to release the drug.
- a suitable non-irritating excipient which is solid at ordinary temperatures but liquid at the rectal temperature and will therefore melt in the rectum to release the drug.
- Such materials are cocoa butter and polyethylene glycols.
- creams, ointments, jellies, solutions or suspensions, etc., containing the compounds of the present invention are employed.
- composition and method of the present invention may further comprise other therapeutically active compounds as noted herein which are usually applied in the treatment of these pathological conditions.
- an appropriate dosage level will generally be about 0.01 to 500 mg per kg patient body weight per day which can be administered in single or multiple doses.
- the dosage level will be about 0.1 to about 250 mg/kg per day; more preferably about 0.5 to about 100 mg/kg per day.
- a suitable dosage level may be about 0.01 to 250 mg/kg per day, about 0.05 to 100 mg kg per day, or about 0.1 to 50 mg/kg per day.
- the dosage may be 0.05 to 0.5, 0.5 to 5 or 5 to 50 mg/kg per day.
- the compositions are preferably provided in the form of tablets containing 1.0 to 1000 milligrams of the active ingredient, preferably 2.0 to 500, more preferably 3.0 to 200, particularly 1, 5, 10, 15, 20, 25, 30, 50, 75, 100, 125, 150, 175, 200, 250, 300, 400, 500, 600, 750, 800, 900, and 1000 milligrams of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated.
- the compounds may be administered on a regimen of 1 to 4 times per day, preferably once or twice per day.
- ester 1-2 (where R is an alkyl group).
- Conversion of ester 1-2 to the carboxylic acid l-2a can be achieved by a number of conditions depending on the nature of the ester. For example, methyl or ethyl esters can be readily saponified with sodium hydroxide, or lithium hydroxide; tert-butyl ester can be removed by treatment with TFA.
- Coupling of the acid l-2a with amine 1-3 to give amide 1-4 can be accomplished by the standard amide bond formation conditions using a coupling reagent such as DCC, EDC and a catalyst such as DMAP, HOBT or HO AT.
- Oxidation of the olefin 1-4 to the aldehyde 1-5 can be carried out under numerous conditions, such as with ozone followed by treatment with methyl sulfide or triphenylphosphine, or with osmium tetroxide and sodium periodate (see March J. "Advanced Organic Chemistry", 4th ed., John Wiley & Sons, New York, pp. 1167-1171 (1992)). Reductive amination with amine 1-6 in the presence of a borohydride such as sodium triacetoxyborohydride or sodium cyanoborohydride then provides the compound of formula I.
- a borohydride such as sodium triacetoxyborohydride or sodium cyanoborohydride
- compounds of formula I may be prepared in one pot by reductive amination of the ozonide without converting it to the ketone.
- R 1 is not hydrogen
- a chiral center is generated by alkylation with allyl bromide.
- the resulting acid l-2a can be resolved in a variety of ways, including by crystallization with optically pure amines such as ⁇ -methylbenzylamine.
- the resolved acid can then be used to prepare I as a single enantiomer, or as a single diastereomer when other chiral centers are present.
- aldehydes 1-5 as intermediates can also be achieved as depicted in Scheme 1A.
- Alkylation of 1-1 with commercially available bromoacetaldehyde dimethyl acetal can be achieved with a strong base such as sodium, lithium or potassium hexamethyldisilazide, lithium diisopropylamide, and the like, to give 1-7.
- Conversion of ester 1- 7 to the carboxylic acid l-7a can be achieved by a number of conditions depending on the nature of the ester.
- methyl or ethyl esters can be readily saponified with potassium hydroxide, sodium hydroxide, or lithium hydroxide.
- Coupling of the acid l-7a with amine 1-3 to give amide 1-8 can be accomplished by the standard amide bond formation conditions using a coupling reagent such as DCC, EDC and a catalyst such as DMAP, HOBT or HO AT. These compounds can be then converted to the compound of formula I according to Scheme 1.
- a coupling reagent such as DCC, EDC and a catalyst such as DMAP, HOBT or HO AT.
- N-acyloxazolidinone 1-11 to the optically pure carboxylic acid l-2a can be achieved by a number of conditions including treatment with lithium hydroperoxide, or bases such as potassium hydroxide, sodium hydroxide, or lithium hydroxide.
- Scalemic l-2a can be then converted to isomerically pure compounds of formula I according to Scheme 1.
- Coupling of the acid 2-2a with amine 1-3 to give the compound of the formula I can be accomplished by the standard amide bond formation conditions using a coupling reagent such as DCC, EDC and a catalyst such as DMAP, HOBT or HO AT.
- a coupling reagent such as DCC, EDC and a catalyst such as DMAP, HOBT or HO AT.
- Scheme 3 shows one general example involving 1-lb, where R 1 is 4-(2-aminothiazole).
- R 1 is 4-(2-aminothiazole).
- Compound 1-lb is commercially available, where R 10 is ethyl. Protection of 1-lb with a t-butoxycarbonyl group can be accomplished with BOC O to give 3-lb.
- Deprotonation with a strong base such as n- butyl lithium, followed by treatment with allyl bromide provides l-2b, which can be carried to compounds of formula I in the usual way as described in Scheme 2.
- the approach shown in Scheme 2b can be applied to a variety of closely related aminoheterocycles, for example 3-(5- aminoisothiazole) and 5-(2-aminothiazole).
- esters 1-2 as intermediates can also be achieved as depicted in Scheme 4.
- Commercially available acid 4-1 can be protected as an ester by various means, depending upon the nature of R 17 (i.e., methyl, ethyl, benzyl, t-butyl).
- the t-butyl ester can be prepared using t-butanol, magnesium sulfate, and sulfuric acid according to the method of Wright, et al (Wright, S.W., Hageman, D.L., Wright, A.S., McClure, L.D. Tetrahedron Lett. 1997, 3 ⁇ S(42), 7345).
- Deprotonation of 4-2 with a strong base such as LDA, followed by treatment with an electrophile such as an alkyl halide, a ketone, an aldehyde, an epoxide, or an ⁇ , ⁇ -unsaturated ester, nitrile or nitro compound can provide 1-2, which in turn may be used to prepare compounds of the formula I as described in Schemes 1 and 2.
- compounds l-2c can be prepared by alkylation of the enolate of 4-2 with ⁇ -bromo- ⁇ , ⁇ -unsaturated ester or nitrile 4-3.
- the trans cyclopropanes are obtained and the diastereoselectivity of this reaction to give erythro or threo products can be controlled by solvent choice according to the work of Prempree, et al. (J. Org. Chem. 1983, 48, 3553, and Tetrahedron Lett. 1985, 26, 1723).
- Compounds l-2c may be used to prepare compounds of the formula I as described in Schemes 1 and 2.
- R 11 is something other than hydrogen, for example methyl, hydroxy, and fluoro.
- deprotonation of 1-2 with a strong base such as potassium hexamethylsilazide, followed by treatment with camphorsulfonyl oxaziridine or N-fluorobenzenesulfonamide provides hydroxy and fluoro compounds l-2e and l-2f, respectively.
- Oxidation of the olefin 5-2 to the ketone followed by reduction to the alcohol 5-3 can be carried out under numerous conditions, such as with ozone followed by treatment with sodium borohydride, or with osmium tetroxide and sodium periodate (see March J. "Advanced Organic Chemistry", 4th ed., John Wiley & Sons, New York, pp. 1167-1171 (1992)), again followed by sodium borohydride.
- Mitsunobo reaction Mitsunobo reaction (Mitsunobu, O. Synthesis 1981, 1) of 5-3 with glutarimides 5-4 (glutarimide is commercially available, substituted analogs are known from the literature) gives intermediates 5-5.
- Reduction of the imide 5-5 can be accomplished with BH »DMS to provide 5-6.
- Hydrolysis of the ester 5-6 can be achieved by a number of conditions depending on the nature of the ester. For example, methyl or ethyl esters can be readily saponified with sodium hydroxide, or lithium hydroxide; tert-butyl ester can be removed by treatment with TFA.
- Coupling of the acid 5-6a with amine 1-3 to give the compound of the formula la can be accomplished by the standard amide bond formation conditions using a coupling reagent such as DCC, EDC and a catalyst such as DMAP, HOBT or HO AT.
- Oxidation of the olefin 6-2 to the aldehyde 6-3 can be carried out under numerous conditions, such as with ozone followed by treatment with methyl sulfide or triphenylphosphine, or with osmium tetroxide and sodium periodate (see March J. "Advanced Organic Chemistry", 4th ed., John Wiley & Sons, New York, pp. 1167-1171 (1992)). Reductive amination with amine 1-6 in the presence of a borohydride such as sodium triacetoxyborohydride or sodium cyanoborohydride then provides the compound of formula lb.
- a borohydride such as sodium triacetoxyborohydride or sodium cyanoborohydride
- heterocycles can be modified in a variety of ways as shown in Scheme 7, 7A and 7B, as well as by other means.
- Compounds Id prepared as described in Schemes 2 and 3, can be deprotected with TFA to give compounds of the formula Ie.
- Coversion of Ie to the corresponding amides If, Ureas Ig, carbamates Ih, and guanidmes Ii, can be accomplished using under a variety of conditions, including those outlined in Scheme 7.
- Scheme 7a gives two examples.
- Deprotection with, for example, TFA gives diamines 7-2, which in turn can be cyclized in a number of different ways, including by treatment with phosgene in the presence of a base such as diisopropylethylamine to provide compounds of the formula Ij.
- Related compounds of the formula Ik can be prepared starting from compounds Ie.
- Amide coupling under a variety of conditions affords 7-3. Rearrangement is accomplished by treatment with NaH and DMF to provide 7-4. Cyclization can be achieved by standard amide coupling with, for example, EDC and catalytic DMAP, to give the target compounds of the formula Ik.
- Scheme 7b shows how the thiazole core may be linked to various aryl and heteroaryl groups.
- Intermediate 1-lb can be converted to the corresponding 2-halothiazole 7-5 by, for example, treatment with t-butylONO and CuBr 2 .
- Elaboration to compounds of the formula 7-6 is accomplished using protocols described in Schemes 1 and 2.
- Compounds 7-6 can readily be coupled to aryl and heteroarylboronic acids and stannanes under palladium catalyzed conditions, to give compounds of the formula U.
- Substituted cyclopropyl compounds may be modified in a variety of ways as shown in part in Scheme 8.
- Compounds l-2c (prepared as described in Scheme 4A) can be elaborated to compounds 8-1 using the methods shown in Schemes 1 or 2. These, in turn can be hydrolyzed to the corresponding carboxylic acids Im.
- the acids can be converted to carbamates by a Curtius rearrangement and then further converted to amines, amides, ureas, sulfonamides, heterocycles, etc, using standard techniques known to those skilled in the art.
- the acids can be reduced to the corresponding primary alcohols which, themselves may be carried on to amines, amides, carbamates, ureas, sulfonamides, and heterocycles, using standard techniques known to those skilled in the art.
- the carboxylic acids and nitriles 8-1 can themselves be converted to heterocycles.
- Substituted cyclopropyl compounds of the formulas 8- 1, and Im-r have at least three stereocenters, allowing for 8 possible isomers. These can be separated a various stages of the syntheses by a number of means including, preparative thin layer chromatography, flash chromatography, HPLC, and HPLC using columns with chiral packing components.
- the subject compounds may be prepared by modification of the procedures disclosed in the Examples as appropriate. Starting materials are made by known procedures or as illustrated. The following examples are provided for the purpose of further illustration only and are not intended to be limitations on the disclosed invention. The following are representative procedures for the preparation of the compounds used in the following Examples or which can be substituted for the compounds used in the following Examples which may not be commercially available.
- the enantiomeric purity of the acid prepared above was determined by derivatization with L-Trp-OMe (amide formation).
- the methyl ester signal for the resolved acid (one singlet, 3.59 ppm)) was compared with that of the racemic acid (two singlets, 3.59 and 3.66 ppm) and determined to be >95% de, hence the acid is >95% ee.
- the procedure for derivatization is as follows:
- 4-(4-Fluorophenyl piperidine hydrochloride is now commercially available from Arch Corporation (catalog # AR01507). It can also be prepared in quantitative yield from commercially available 4-(4-fluorophenyl)-l,2,3,6-tetrahvdropyridine hydrochloride (20 g. 94 mmol) by hydrogenation using Pd/C catalyst (2g) in 200 mL of ethanol at 40 psi H? pressure for 2 days using a Parr apparatus.
- the aqueous layer was extracted again with DCM (200 mL), and the organic layers were combined and washed with IN HCl (250 mL), saturated NaHCO 3 solution (250 mL), and brine (250 mL). The organic layer was dried over MgSO , filtered, and concentrated to give 22.8 g of crude bis- mesylate, which was used immediately. If not used immediately the bis-mesylate underwent decomposition.
- N-Boc-isonipecotic acid (8.04 g, 35.1 mmol) was combined with N,O-dimethylhydroxylamine hydrochloride (5.13 g, 52.6 mmol), EDC (10.1 g, 52.6 mmol) and DIEA (9.2 mL, 53 mmol) in DCM (100 mL). Then N,N-dimethylaminopyridine (-200 mg) was added and the reaction mixture was permitted to stir at rt for 2h. The reaction mixture was diluted with more DCM and washed with 2 N HCl solution, saturated NaHCO 3 solution, and brine.
- the crude aldehyde intermediate 1 (107 mg, 0.255 mmol), prepared as described above, was combined with 4-phenylpiperidine (49.3 mg, 0.306 mmol), NaB(OAc) 3 H (108 mg, 0.510 mmol) and 4 °A molecular sieves (250 mg) in DCE (5 mL) and stirred overnight.
- the reaction mixture was filtered through celite, diluted with ethyl acetate, and washed with saturated NaHCO 3 solution, followed by brine.
- the organic layer was dried over MgSO 4 , filtered, and concentrated.
- Ozone was bubbled through a cooled (-78 °C) solution of the olefin from Step B (4.0 g, 15 mmol) in methanol until a green/blue color persisted. Nitrogen was bubbled through the solution until the blue color has disappeared. Then sodium borohydride (576 mg, 15.2 mmol) was added and the mixture was stirred for 1.5 h. The reaction mixture was quenched with water and concentrated to remove the methanol.
- Step D To a cooled (0 °C) solution of the alcohol prepared as described in Step B (1.19 g, 4.46 mmol), 4-phenyl glutarimide (1.04 g, 5.35 mmol), and triphenylphosphine (1.40 g, 5.35 mmol) in THF (20 mL) was added a solution of diisopropyl azodicarboxylate (1.05 mL, 5.35 mmol). The reaction mixture was permitted to warm to rt and stir for 3 h. The reaction mixture was concentrated, redissolved in ethyl acetate, and washed with saturated NaHCO 3 solution, then brine, dried over anhydrous MgSO 4 , filtered, and concentrated. Purification by flash chromatography (silica, 50% ethyl acetate/hexane), followed by MPLC (silica, 75% ethyl acetate/hexane) gave 968 mg of the desired product. Step D:
- the organic layer was separated, washed with brine (1 x 50 mL), dried with anhydrous sodium sulfate and the solvent was evaporated in vacuo to yield 11.50 g (87 %) of the crude product.
- the crude residue was purified by flash column (gradient eluant 20% ethyl acetate/hexane to 60% ethyl acetate/hexane) to yield 9.0 g (68%) of the racemic desired product.
- the desired S isomer was obtained through crystallization with (R) (+) -alpha methyl benzylamine (2.55 ml, 20.11 mmol, 0.5 eq) in ether (80 ml) cooled by refrigeration overnight.
- step A A mixture of the acid (described in step A, 1.0 g, 4.46 mmol), 3,5- bis(trifluoromethyl)benzylamine hydrochloride (1.25 g, 4.46 mmol), HOBt (602 mg, 4.46 mmol), N,N-diisopropyl ethylamine (776 ⁇ l, 4.46 mmol) in dichloromethane (20 mL) was treated with l-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDC, 1.71 g, 8.92 mmol) and stirred at room temperature overnight.
- EDC l-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride
- Step B The intermediate in Step B (1.0 g, 2.23 mmol) was treated with solution of 90% trifluoroacetic acid/water (20 ml) for 10 minutes.
- the reaction mixture was diluted with water (30 mL) and extracted with ether (3x 40 ml).
- the organic layer was washed with saturated sodium bicarbonate (3 x 50 mL), brine (1 x 50 mL), dried over anhydrous sodium sulfate and the solvent was evaporated in vacuo to yield 512 mg (57 %) of the crude product. No further purification was done and the material was stored under a blanket of nitrogen in the freezer (- 16°C).
- a solution of 4-methoxyphenylacetic acid (5.0 g, 30.088 mmol) in tetrahydrofuran (50 mL) was added drop-wise onto a suspension of sodium hydride (1.60 g, 40.20 mmol, 60 %) in THF (50 mL), and then heated to reflux for 2 hrs.
- the suspension of the salt was cooled to 10 °C and a solution of lithium diisopropylamid (generated from 4.64 mL, 33.09 mmol) of diisopropylamine and 21.0 mL of nBuLi (1.6 M, hexanes)) in 50 mL of tetrahydrofuran was added via canula.
- the olefin (980 mg, 2.27 mmol) was ozonized at -78 °C in dichloromethane until permanent blue color indicated complete consumption of the olefin.
- the excess ozone was purged with a stream of nitrogen, and the ozonide was reduced with dimethylsulfide (1.6 mL, 122.7 mmol).
- the cooling bath was removed, and the reaction mixture was gradually warmed up to room temperature.
- the solvent and excess of DMS was concentrated, and the resulting residue was used in the next reaction without purification.
- Example 6 was synthesized starting from aldehyde Intermediate 13 and 4-spiroindenylpiperidine hydrochloride according to a procedure described for preparation of Example 1, except that 1.2 equivalents of DIEA was added and the solvent was DCM instead of DCE. MS: for C 33 H 32 N 2 O 2 F 6 [M + H] + calculated: 603.24, found 603.20.
- Example 7 was synthesized starting from acid Intermediate 13 and 4-phenylpiperidine hydrochloride according to the procedure described for preparation of Example 6. MS: for C 31 H 32 N 2 O 2 F 6 [M + H] + calculated: 579.24, found 579.00.
- Example 7 A solution of Example 7 (100 mg, as hydrochloride, 0.163 mmol) was dissolved in dry dichloromethane (10 mL), and treated at 0°C with neat boron tribromide (980 ⁇ L, 0.975 mmol). After stirring at 0°C for 30 minutes the temperature was allowed warm up to ambient, and stirring was continued for another 3 hrs. The reaction mixture was cooled again to 0°C and quenched with ammonium hydroxide (4.0 mL, 30 % aq. Solution). After 30 minutes of stirring the product was extracted with dichloromethane (3 x 20 mL). After drying (anhydrous sodium sulfate) the solvent was removed in vacuo to leave 93 mg (100 %) of the desired phenol. MS: for C 3 oH 3 oN 2 O 2 F 6 [M+H] + calculated: 565.22, found 565.00. EXAMPLE 9
- Example 9 was prepared using the synthetic sequence described for preparation of Example 7 except that 3-methoxyphenylacetic acid was used instead of 4-methoxyphenylacetic acid. MS: for C 31 H 32 N 2 O 2 F 6 [M+H] + calculated: 579.24, found 579.2.
- Example 10 was prepared using the synthetic sequence described for preparation of Example 6 except that 3-methoxyphenylacetic acid was used instead of 4-methoxyphenylacetic acid. MS: for C 33 H 32 N 2 O 2 F 6 [M+H] + calculated: 603.24, found 646.5.
- Example 11 (640 mg, 0.93 mmol) and TFA (7 mL) were combined and stirred for half hour before concentrating in vacuo. The crude product was redissolved in DCM and salted out with 4N HCl in dioxane to yield Example 12 (600mg, 99 + %).
- Example 12 A variety of N-substituted derivatives of Example 12 were synthesized. These compounds were made by simple coupling reactions known to those skilled in the art. Table 6 below summarizes these compounds. TABLE 6
- 13-F 13-D (52 mg, 0.135 mmol) was first dissolved in a solution of THF (ImL), MeOH (ImL), and H 2 O (ImL). The mixture was cooled to 0 °C before LiOH (23 mg, 0.538 mmol) was added. The solution was stirred at 0 °C for 1 hour before raised to room temperature and stirred overnight. The reaction mixture was concentrated in vacuo and the residue was redissolved in a mininum amount of H 2 O. After the pH was adjusted to 7.0 with 2N HCl, the aqueous layer was extracted with DCM (5x). The combined organic layers were concentrated in vacuo to yield the crude 13- F (38.7 mg, 77.4%), which was used on next step.
- Example 14 (187 mg, 0.297 mmol) was first dissolved in a solution of THF (2mL), MeOH (2mL), and H 2 O (2mL). The mixture was cooled to 0 °C before LiOH (25 mg, 0.594 mmol) was added. The solution was stirred at 0 °C for 10 minutes before warming to room temperature and stirring for another 4 hours. TLC showed starting material so leq of NaOH was added to push the reaction to completion. The reaction mixture was concentrated in vacuo and redissolved in DCM. A few drops of 4N HCl in dioxane was added and the mixture was concentrated and purified by preparative TLC (80/20 ethyl acetate/hexanes) to yield Example 15 (20mg, 10.9%). LC-MS: MW calculated 616.59, found 617.4.
- Example 15 The product from Example 15 (18 mg, 0.029 mmol), ethylamine 2M (15 ⁇ L, 0.029 mmol), HO At (4 mg, 0.029 mmol), EDC (9 mg, 0.044 mmol), and DCM were stirred overnight before washing with IN NaOH. The organic layer was dried over MgSO and concentrated in vacuo. The crude product was purified by preparative TLC (silica, 100% ethyl acetate) to yield Example 16. LC-MS: MW calculated 643.26, found 644.4.
- reaction mixture was diluted with dichloromethane (20 mL), washed with water (2 x 20 mL), brine (1 x 20 mL), dried over anhydrous sodium sulfate and the solvent was evaporated. Purification was accomplished by preparative TLC (eluant: 40% ethyl acetate/hexane) to yield 295 mg (90 %) of the desired product.
- the solution was dried with magnesium sulfate, the drying agent was filtered off, and to the filtrate was added spiroindenylpiperidine hydrochloride (68 mg, 0.307 mmol), diisopropylethylamine (42 ⁇ L, 0.307 mmol), crushed 4 A molecular sieves (50 mg) and the resulting mixture was treated with sodium triacetoxyborohydride (325 g, 1.54 mmol). After stirring at ambient temperature for 24 hours, the sieves were filtered off, the filtrate was washed with a saturated solution of sodium bicarbonate (1 x 10 mL), water (3 x 5 mL) and brine (1 x 10 mL).
- Lithium diisopropyl amide (LDA) was freshly prepared by treating a solution of diisopropylamine (3.75 ml, 26.8 mmol) in dry THF (10 ml) under nitrogen cooled to -78°C with 2.5 M 72-butyl lithium (10.72 ml, 26.79 mmol) added slowly via syringe. To this solution was added methyl hydrocinnamate (4.0 g, 24 mmol) in dry THF (50 ml) via syringe dropwise over a 30 minute period.
- LDA Lithium diisopropyl amide
- 4-(4-FormyI-3-methoxyphenoxy)butyryl AM resin was swelled in dichloroethane (20 ml) for 30 minutes after which the sovent was drain. A fresh cocktail of 5% trimethyl orthoformate in dichloroethane (15 ml) was added to the resin. To this suspension was then added 3,5-bis(trifluoromethyl)benzyl amine (1.00 g, 4.05 mmol) and sodium triacetoxyborohydride (858 mg, 4.05 mmol) and the resulting mixture was spun on a mechanical rotary for 15 hours, releasing pressure every 15 minutes for the first 5 hours.
- the solvent was drained and the resin beads washed with methanol (2 x 10 ml), dichloroethane (3 x 10 ml), dimethyl formamide (5 x 10 ml), dichloromethane (3 x 10 ml) and ether (3 x 10 ml).
- the resin was first dried by flowing nitrogen through the container; then under high vacuum overnight.
- the solvent was drained and the resin beads washed with methanol (2 x 10 ml), dichloroethane (3 x 10 ml), dimethyl formamide (5 x 10 ml), dichloromethane (3 x 10 ml) and ether (3 x 10 ml).
- the resin was first dried by flowing nitrogen through the container; then under high vacuum overnight.
- the prepared resin (Intermediate 16, 50 mg, 0.027 mmol) was swelled in dichloroethane (2 ml) for 15 minutes after which the solvent was drain. A fresh cocktail of 5% trimethyl orthoformate in dichloroethane (2 ml) was added to the resin. To this suspension was then added spiroindenylpiperidine hydrochloride (30 mg, 0.14 mmol), diisopropylethylamine (24 ⁇ L, 0.14 mmol), and sodium triacetoxyborohydride (57 g, 0.27 mmol) and the resulting mixture was spun on a mechanical rotary for 15 hours, releasing pressure every 15 minutes for the first 3 hours. The solvent was drained and the resin beads washed with methanol (3 2 ml), dichloroethane (5 x 2 ml), dimethyl formamide (5 x 2ml), dichloromethane (10 x 2 ml).
- the resin was then immediately treated with a pre-mixed cocktail of 25% trifluoroacetic acid in dichloromethane (2 ml) for 2 hours.
- the beads became dark red after treatment with the acidic solution.
- the solvent was collected along with the dichloromethane washings (3 x 2 ml) and concentrated to dryness under reduced pressure to afford 2.5 mg (31%) of the desired final product with an HPLC purity analysis of 91 % .
- Lithium diisopropyl amide (LDA) was freshly prepared by treating a solution of diisopropylamine (280 ⁇ l, 2.0 mmol) in dry THF (4 ml) under nitrogen cooled to -78°C with 2.5 M n-butyl lithium (791 ⁇ l, 2.00 mmol) added slowly via syringe. To this solution was added product from Step B, Intermediate 17 (520 mg, 2.00 mmol) in dry THF (16 ml) via syringe dropwise over a 15 minute period.
- LDA Lithium diisopropyl amide
- reaction mixture was diluted with dichloromethane (5 mL), washed with water (2 x 5 mL), brine (1 5 mL), dried over anhydrous sodium sulfate and the solvent was evaporated.. Purification was done by preparative TLC (eluant: 5% methanol / 95% ethyl acetate) to yield 14.1 mg (40 %) of the desired final product.
- the reaction mixture was diluted with water, and the non-acidic components were extracted with diethyl ether.
- the pH of the aqueous solution was set to acidic (20 g of citric acid) and extracted with diethyl ether again.
- the combined (acidic) extracts were dried with anhydrous magnesium sulfate and evaporated to dryness.
- the crude product was further purified by distillation, which yielded 4.7447 g of recovered starting cyclopropanecacetic acid (b.p.: 75°C/0.3 mmHg) and 6.1538 g (32 %) of the desired product, b.p.: 110°C/0.3 mmHg.
- Procedure B A solution of the olefin Intermediate 19 (603 mg, 1.65 mmol) in dichloromethane (50 mL) was cooled to -78°C and a stream of ozone was passed through until the permanent blue color indicated complete consumption of the olefin. The excess of ozone was purged with nitrogen, and the solution of the ozonide was allowed to warm up to room temperature. The solution was dried with anhydrous magnesium sulfate and filtered.
- Example 21 was synthesized according to the procedure described for preparation of Example 20, except that 4-spiroindenylpiperidine was used instead of 3-methyl-4-spiroindenylpiperidine in Step D, Intermediate 18.
- Example 22 was synthesized according to the procedure described for preparation of Example 20, except that 4-(4-fluorophenyl)piperidine was used instead of 3-methyl-4- spiroindenylpiperidine in Step D, Intermediate 18.
- Example 23 was synthesized according to the procedure described for preparation of Example 20, except that 4-phenylpiperidine was used instead of 3-methyl-4-spiroindenylpiperidine in Step D, intermediate 18.
- Example 24 was synthesized according to a procedure analogous to that described for preparation of Example 20.
- Example 25 was synthesized according to a procedure analogous to that described for preparation of Example 20.
- Lithium diisopropyl amide (LDA) was freshly prepared by treating a solution of diisopropylamine (3.24 ml, 23.1 mmol) in dry THF (40 ml) under nitrogen cooled to -78°C with 2.5 M n-butyl lithium (9.244 ml, 23.11 mmol) added slowly via syringe. To this solution was added ethyl cyclohexylacetate (3.77 g, 21.0 mmol) in dry THF (60 ml) via syringe dropwise over a 30 minute period.
- LDA Lithium diisopropyl amide
- the solution was dried with magnesium sulfate, the drying agent was filtered off, and to the filtrate was added spiroindenylpiperidine hydrochloride (419 mg, 1.80 mmol), diisopropylethylamine (328 ⁇ L, 1.80 mmol), crushed 4 A molecular sieves (250 mg) and the resulting mixture was treated with sodium triacetoxyborohydride (2.00 g, 9.46 mmol). After stirring at ambient temperature for 24 hours, the sieves were filtered off, the filtrate was washed with a saturated solution of sodium bicarbonate (1 x 100 mL), water (3 x 50 mL) and brine (1 x 100 mL).
- Lithium diisopropyl amide (LDA) was freshly prepared by treating a solution of diisopropylamine (1.80 ml, 12.8 mmol) in dry THF (20 ml) under nitrogen cooled to -78°C with 2.5 M n-butyl lithium (5.13 ml, 12.8 mmol) added slowly via syringe. To this solution was added the product from Step A of Intermediate 21 (3.0 g, 12 mmol) in dry THF (80 ml) via syringe dropwise over a 30 minute period.
- LDA Lithium diisopropyl amide
- the solution was dried with magnesium sulfate, the drying agent was filtered off, and to the filtrate was added spiroindenylpiperidine hydrochloride (745 mg, 3.36 mmol), diisopropylethylamine (585 ⁇ L, 3.36 mmol), crushed 4 A molecular sieves (500 mg) and the resulting mixture was treated with sodium triacetoxyborohydride (3.56 g, 16.8 mmol). After stirring at ambient temperature for 24 hours, the sieves were filtered off, the filtrate was washed with a saturated solution of sodium bicarbonate (1 x 100 mL), water (3 x 50 mL) and brine (1 x 100 mL).
- the pH of the aqueous solution was set to 3, (HCl, 2 N) and the crude acid was extracted into diethyl ether (3 x 50 mL). The combined organic extracts were dried with anhydrous sodium sulfate, and the solvent was evaporated to dryness to leave 1.12 g (27 %) of the crude acid, used in the next step without additional purification.
- This racemic acid could be resolved into its respective enantiomers using (R)- and/or (S)- ⁇ - phenylethylamine salts via crystallization from ethyl acetate.
- the acid, which ultimately led to the more active enantiomer of the racemate shown under Example 30, was obtained by crystallizations using the salts derived form the (S)- amine.
- Example 35 was prepared in a similar manner to Example 34, substituting 3- trifluoromethylbenzylamine for 3-trifluoromethoxybenzylamine. MS: for C 29 H 29 F 3 N 2 OS [M + H] + calculated: 511.20, found 511.4.
- Example 36 was prepared in a similar manner to Example 34, substituting 3-iodobenzylamine for 3-trifluoromethoxybenzylamine. MS: for C 28 H 29 IN 2 S [M + H] + calculated: 569.10, found 569.3.
- Example 37 was prepared in a similar manner to Example 34, substituting 3-phenylbenzylamine for 3-trifluoromethoxybenzylamine. MS: for C 3 H 3 N 2 OS [M + H] + calculated: 519.24, found 519.50.
- Example 38 was prepared in a similar manner to Example 34, substituting 1- aminomethylnaphthalene for 3-trifluoromethoxybenzylamine. MS: for C 32 H 32 N 2 OS [M + H] 4 calculated: 493.22, found 493.50.
- Example 39 was prepared in a similar manner to Example 34, substituting 3,5- dichlorobenzylamine for 3-trifluoromethoxybenzylamine. MS: for C 28 H 28 Cl 2 N 2 OS [M + H] + calculated: 511.13, found 511.3.
- Example 40 was prepared in a similar manner to Example 34, substituting 3-nitrobenzylamine for 3-trifluoromethoxybenzylamine. MS: for C 28 H 29 N 3 O 3 S [M + H] + calculated: 488.19, found 488.3.
- Example 41 was prepared in a similar manner to Example 34, substituting 3,5- bistrifluoromethyl- ⁇ -methylbenzylamine for 3-trifluoromethoxybenzylamine. MS: for C 3 ⁇ H 30 N 2 OSF 6 [M + Hf calculated: 593.20, found 593.4.
- Example 42 was prepared in a similar manner to Example 34, substituting 3,5- dimethoxybenzylamine for 3-trifluoromethoxybenzylamine. MS: for C 3 oH 34 N 2 O 3 S [M + H] + calculated: 503.23, found 503.5.
- Example 43 was prepared in a similar manner to Example 34, substituting 3,5- bistrifluoromethylbenzylalcohol for 3-trifluoromethoxybenzylamine. MS: for C 3 oH 7 NO 2 SF [M + H] + calculated: 580.17, found 580.4.
- Example 44 was prepared in a similar manner to Example 34, substituting 3- aminomethylpyri dine for 3-trifluoromethoxybenzylamine. MS: for C 2 H 2 N 3 OS [M + H] + calculated: 444.20, found 444.40.
- Example 45 was prepared in a similar manner to Example 34, substituting 1- aminomethylpyridine for 3-trifluoromethoxybenzylamine. MS: for C 27 H 2 N 3 OS [M + H] 4 calculated: 444.20, found 444.40.
- Example 47 was prepared in a similar manner to Example 46, substituting 4-phenylpiperidine hydrochloride for and 4-spiroindenylpiperidine hydrochloride.
- Example 48 was prepared in a similar manner to Example 34, substituting Intermediate 25 with Intermediate 28 and 3-trifluoromethoxybenzylamine with 3,5-bistrifluoromethylbenzylamine.
- Example 49 was prepared in a similar manner to Example 34, substituting Intermediate 25 with Intermediate 29 and 3-trifluoromethoxybenzylamine with 3,5-bistrifluoromethylbenzylamine.
- Example 50 was synthesized starting from acid Intermediate 31 and 3,5- bistrifluoromethylbenzylamine according to a procedure described for preparation of Example 34. MS: for C 29 H 29 N 3 OF 6 [M + H] + calculated: 550.22, found 550.4.
- Example 51 was synthesized starting from acid Intermediate 32 and 3,5- bistrifluoromethylbenzylamine according to a procedure described for preparation of Example 34. MS: for C 31 H 29 N 3 OF 6 [M + H] + calculated: 574.22, found 574.3. INTERMEDIATE 33
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Pulmonology (AREA)
- Diabetes (AREA)
- Physical Education & Sports Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Dermatology (AREA)
- Virology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Neurology (AREA)
- Oncology (AREA)
- Rheumatology (AREA)
- Endocrinology (AREA)
- Communicable Diseases (AREA)
- Urology & Nephrology (AREA)
- Cardiology (AREA)
- Hematology (AREA)
- Heart & Thoracic Surgery (AREA)
- Toxicology (AREA)
- AIDS & HIV (AREA)
- Transplantation (AREA)
- Neurosurgery (AREA)
- Ophthalmology & Optometry (AREA)
- Pain & Pain Management (AREA)
- Molecular Biology (AREA)
Abstract
Description
























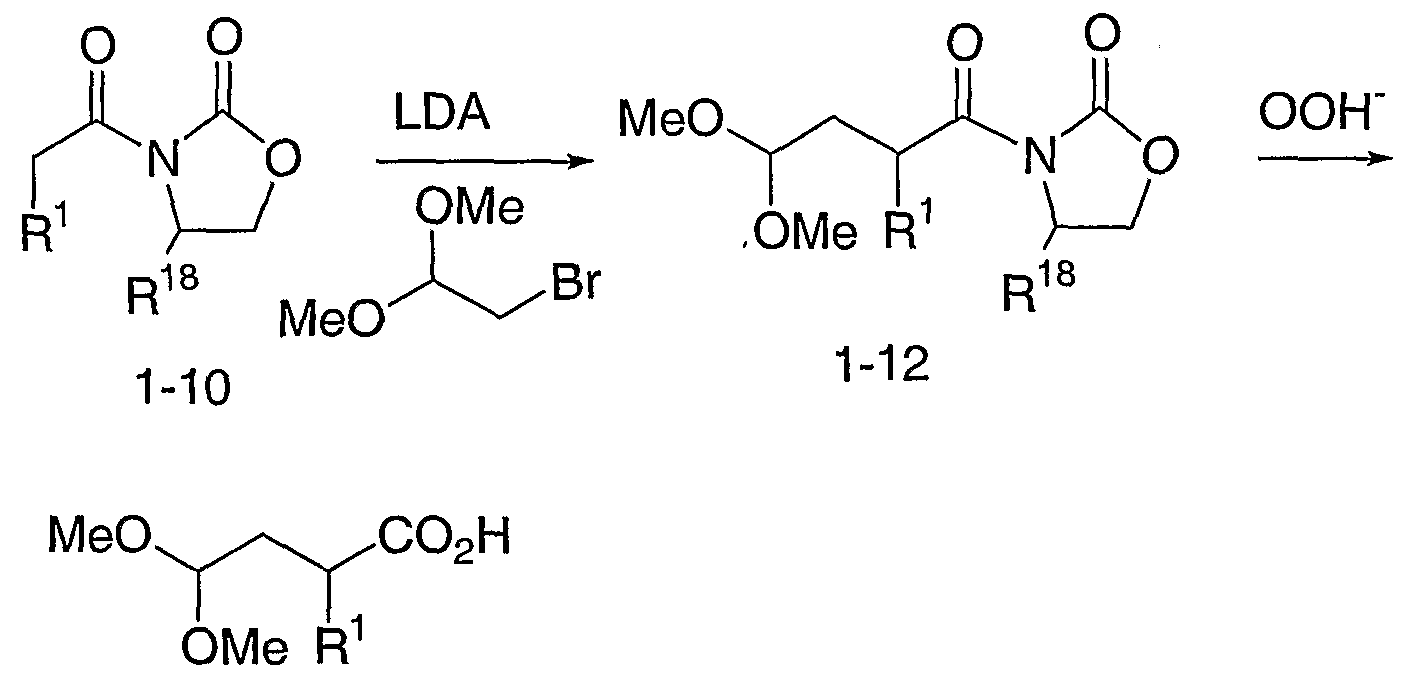










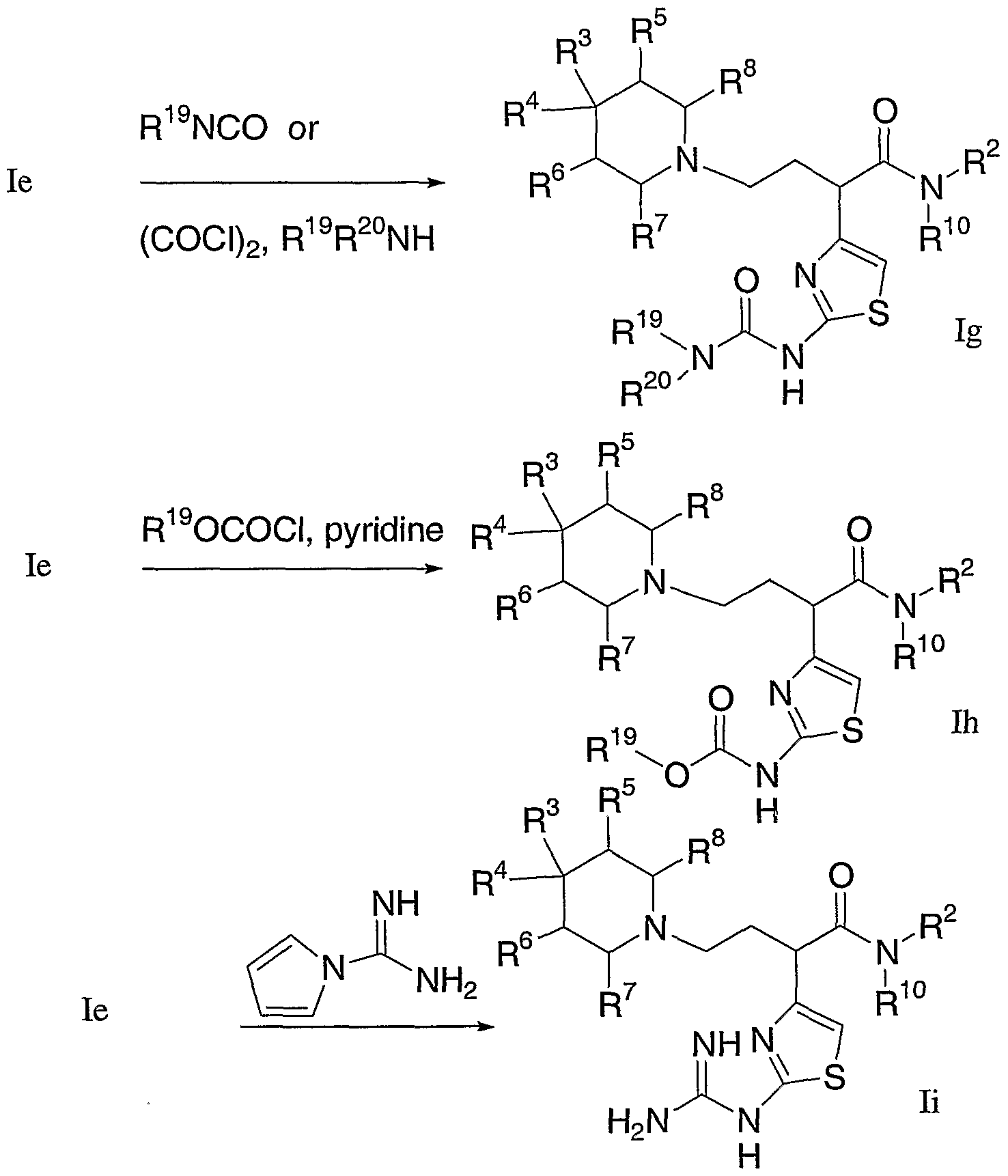

























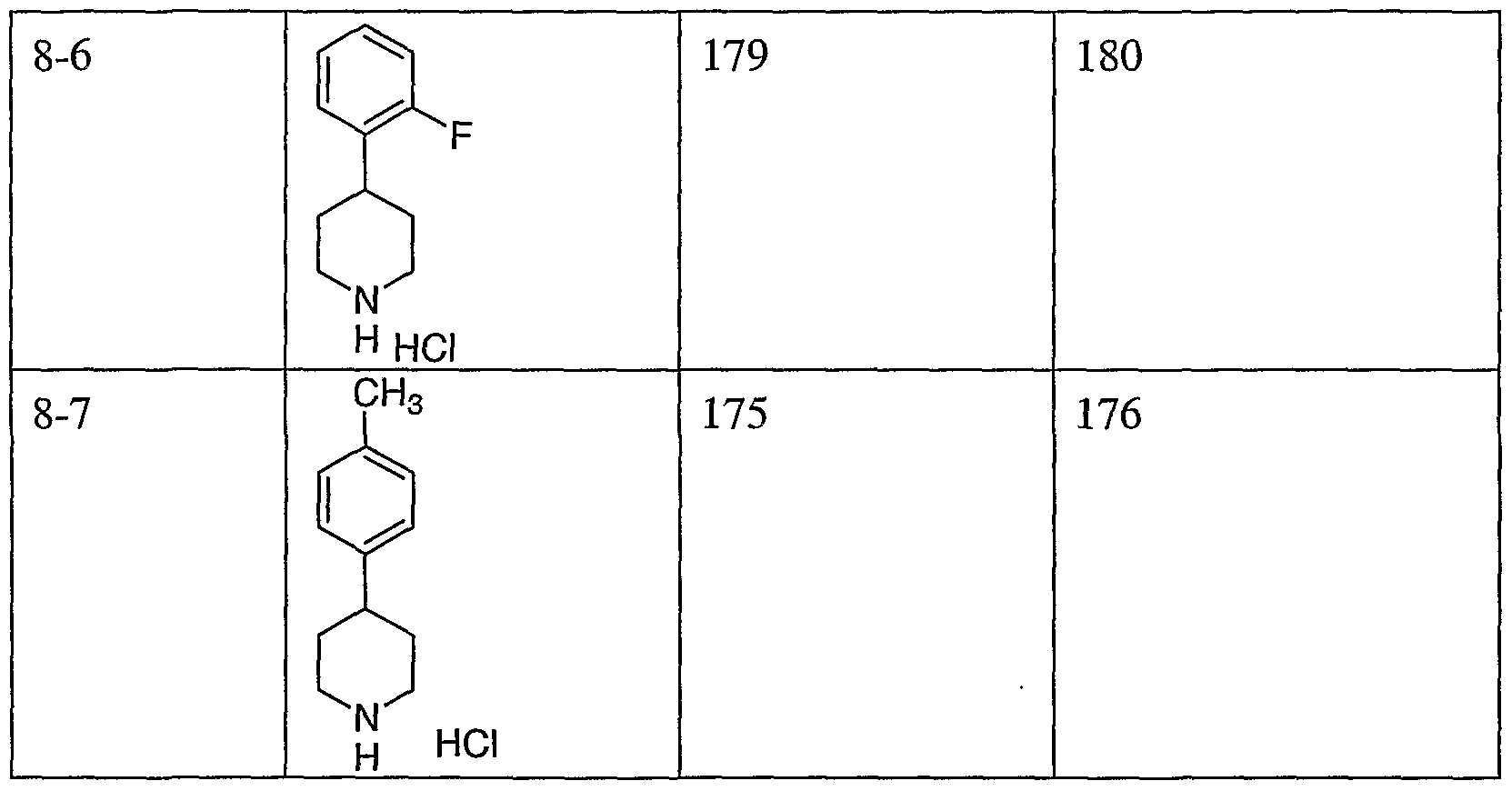










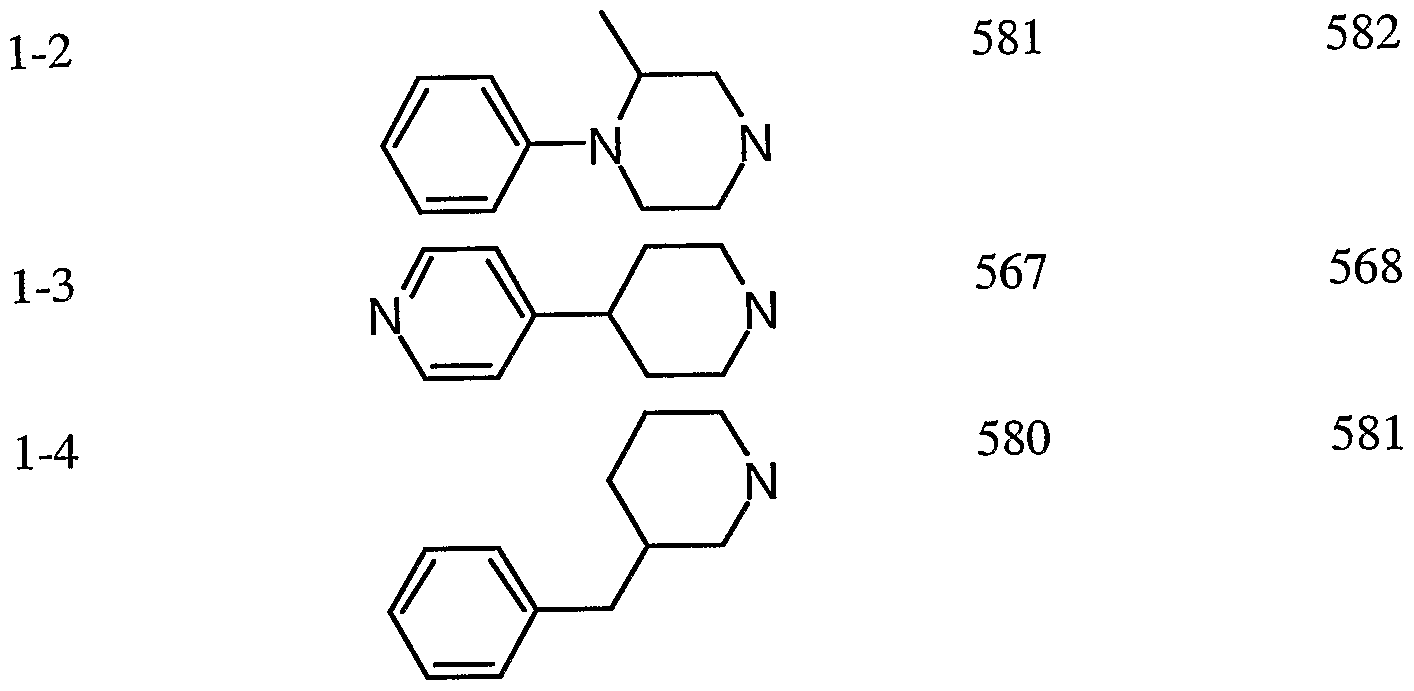
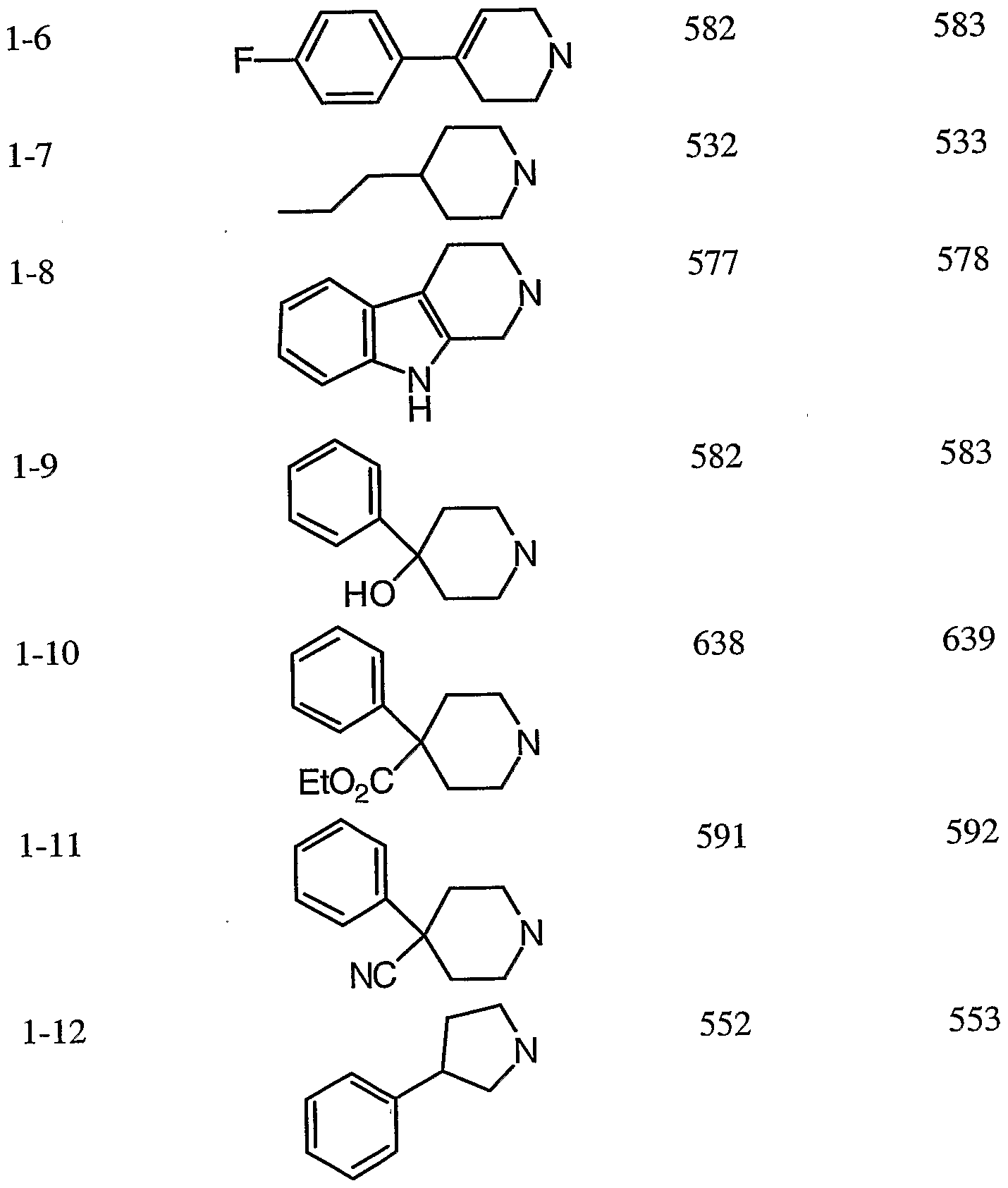








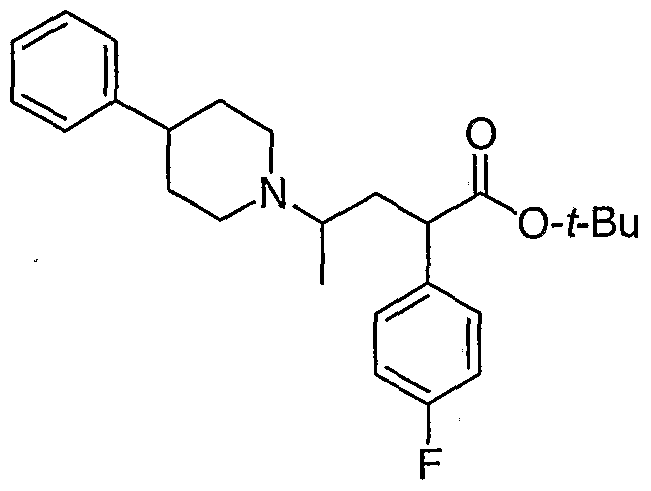





















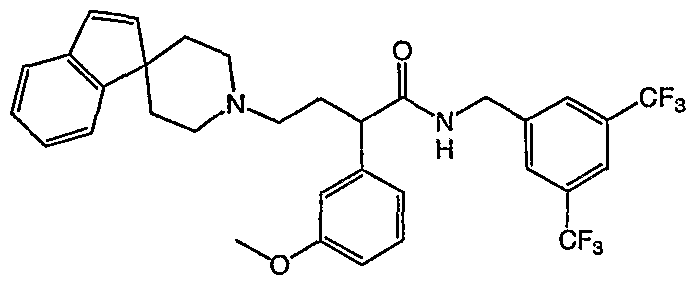









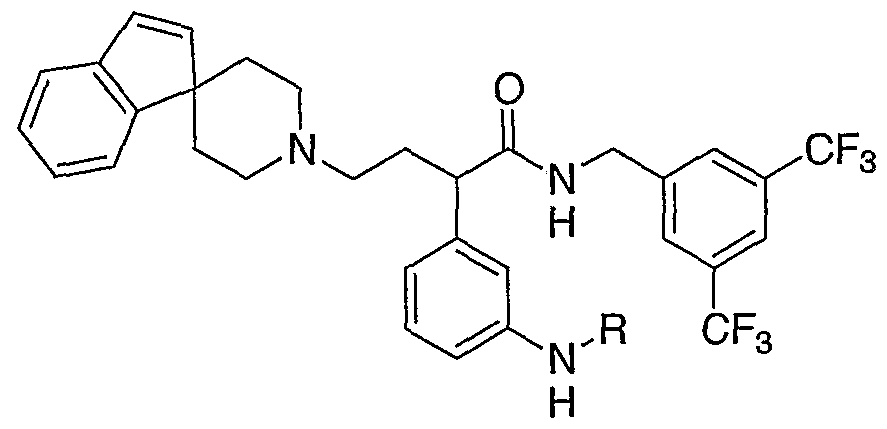

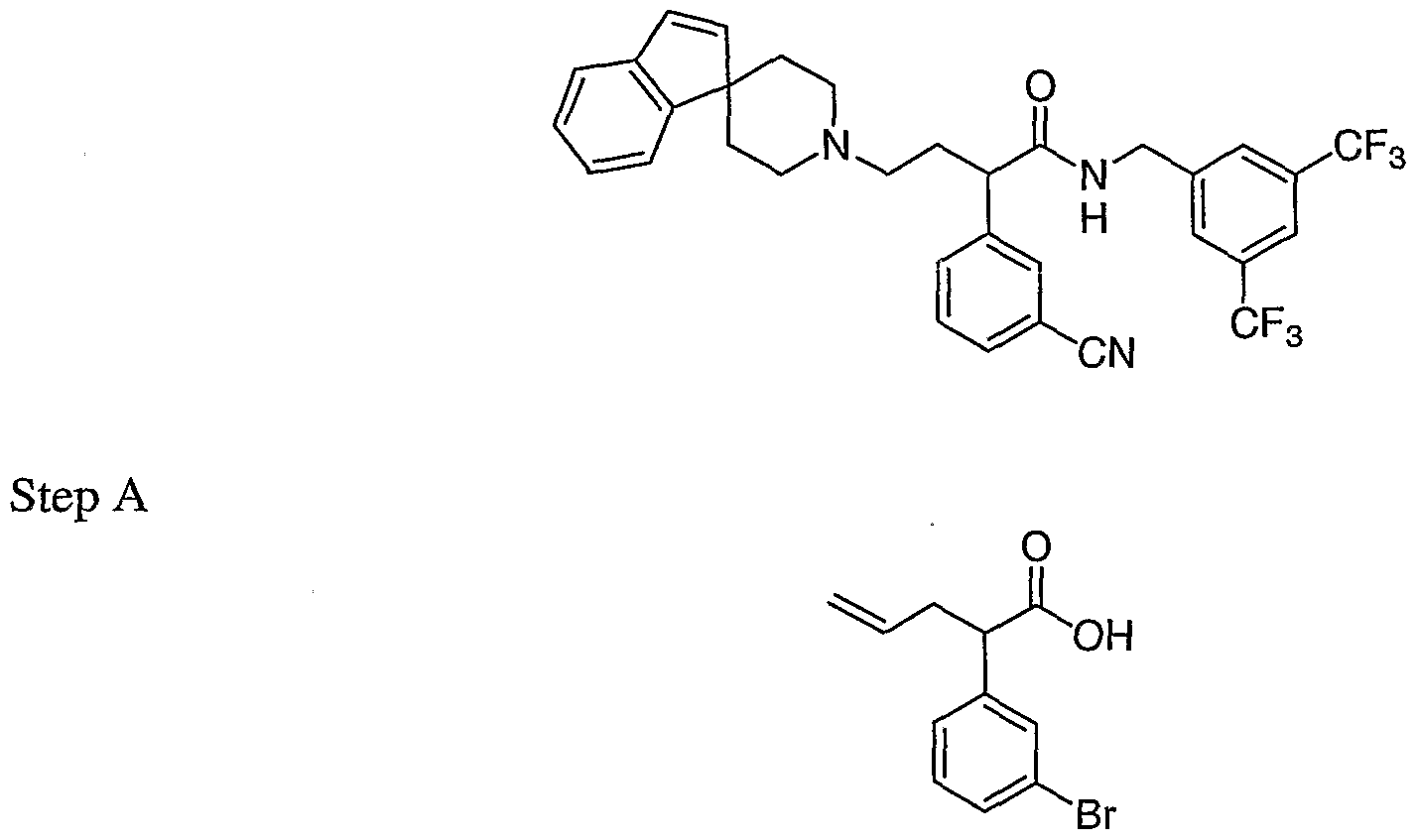














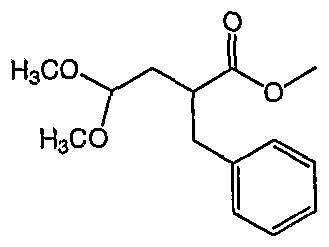
































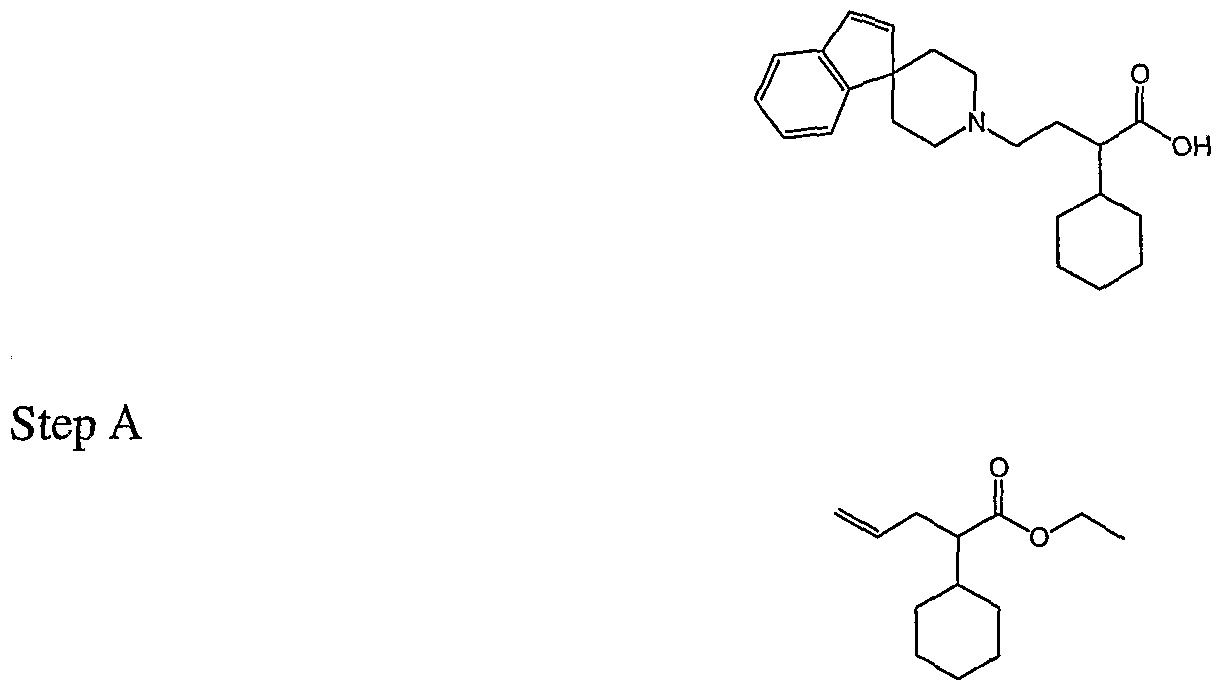















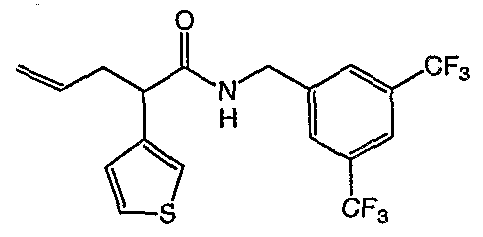





















































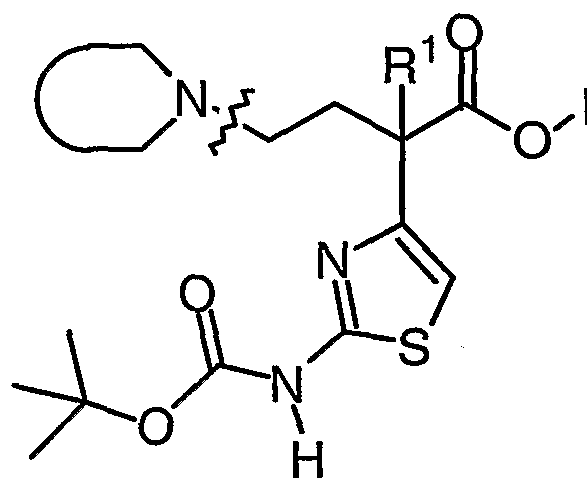









































































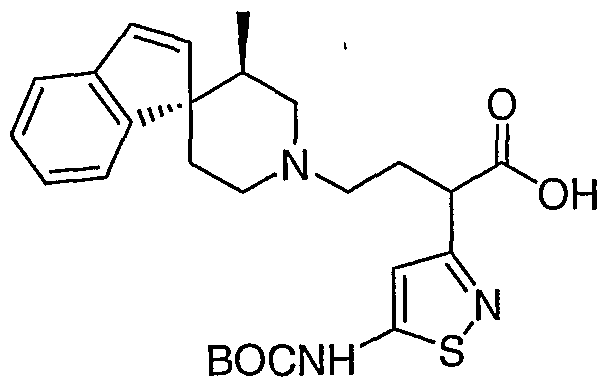





































Claims
















Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP03779303A EP1558250A4 (en) | 2002-10-30 | 2003-10-24 | Gamma-aminoamide modulators of chemokine receptor activity |
| JP2004550131A JP2006511500A (en) | 2002-10-30 | 2003-10-24 | γ-Aminoamide-based chemokine receptor activity modulator |
| AU2003284984A AU2003284984B2 (en) | 2002-10-30 | 2003-10-24 | Gamma-aminoamide modulators of chemokine receptor activity |
| CA002503720A CA2503720A1 (en) | 2002-10-30 | 2003-10-24 | Gamma-aminoamide modulators of chemokine receptor activity |
| US10/528,329 US7247725B2 (en) | 2002-10-30 | 2003-10-24 | Gamma-aminoamide modulators of chemokine receptor activity |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US42226802P | 2002-10-30 | 2002-10-30 | |
| US60/422,268 | 2002-10-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004041279A1 true WO2004041279A1 (en) | 2004-05-21 |
Family
ID=32312486
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2003/034009 WO2004041279A1 (en) | 2002-10-30 | 2003-10-24 | Gamma-aminoamide modulators of chemokine receptor activity |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US7247725B2 (en) |
| EP (1) | EP1558250A4 (en) |
| JP (1) | JP2006511500A (en) |
| AU (1) | AU2003284984B2 (en) |
| CA (1) | CA2503720A1 (en) |
| WO (1) | WO2004041279A1 (en) |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005058884A3 (en) * | 2003-12-15 | 2005-09-09 | Japan Tobacco Inc | Cyclopropane compounds and pharmaceutical use thereof |
| WO2006060919A1 (en) | 2004-12-09 | 2006-06-15 | Virochem Pharma Inc. | Novel spirotropane compounds and methods for the modulation of chemokine receptor activity |
| WO2007037742A1 (en) * | 2005-09-29 | 2007-04-05 | Astrazeneca Ab | Novel azetidine compounds useful in the treatment of functional gastrointestinal disorders, ibs and functional dyspepsia |
| WO2007037743A1 (en) * | 2005-09-29 | 2007-04-05 | Astrazeneca Ab | Novel azetidine compounds useful in the treatment of functional gastrointestinal disorders, ibs and functional dyspepsia |
| WO2007049771A1 (en) * | 2005-10-28 | 2007-05-03 | Ono Pharmaceutical Co., Ltd. | Compound containing basic group and use thereof |
| US7279486B2 (en) | 2004-03-29 | 2007-10-09 | Pfizer Inc. | Alpha aryl or heteroaryl methyl beta piperidino propanoic acid compounds as ORL1-receptor antagonists |
| US7307086B2 (en) | 2004-05-11 | 2007-12-11 | Incyte Corporation | 3-(4-heteroarylcyclohexylamino)cyclopentanecarboxamides as modulators of chemokine receptors |
| US7449467B2 (en) | 2004-06-28 | 2008-11-11 | Pfizer Inc. | 3-aminocyclopentanecarboxamides as modulators of chemokine receptors |
| US7449475B2 (en) | 2002-07-08 | 2008-11-11 | Astrazeneca Ab | Tricyclic spiropiperidines or spiropyrrolidines |
| JP2008540333A (en) * | 2005-04-30 | 2008-11-20 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Novel piperidine substituted indoles |
| JP2008543858A (en) * | 2005-06-15 | 2008-12-04 | ジェンザイム・コーポレーション | Chemokine receptor binding compounds |
| US7498338B2 (en) | 2003-11-20 | 2009-03-03 | Astrazeneca Ab | Compounds |
| US7524856B2 (en) | 2003-12-22 | 2009-04-28 | Astrazeneca Ab | Tricyclic spiroderivatives as modulators of chemokine receptor activity |
| US7576089B2 (en) | 2003-12-18 | 2009-08-18 | Incyte Corporation | 3-cycloalkylaminopyrrolidine derivatives as modulators of chemokine receptors |
| US7618970B2 (en) | 2004-06-28 | 2009-11-17 | Incyte Corporation | 3-aminocyclopentanecarboxamides as modulators of chemokine receptors |
| EP2166848A1 (en) * | 2007-06-20 | 2010-03-31 | Glaxo Group Limited | Spiroindolines as modulators of chemokine receptors |
| EP2190286A1 (en) * | 2007-08-14 | 2010-06-02 | Glaxo Group Limited | Spiroindenes and spiroindanes as modulators of chemokine receptors |
| US7834021B2 (en) | 2002-11-27 | 2010-11-16 | Incyte Corporation | 3-aminopyrrolidine derivatives as modulators of chemokine receptors |
| US8076349B2 (en) | 2004-12-09 | 2011-12-13 | Virochem Pharma Inc. | Spirotropane compounds and methods for the modulation of chemokine receptor activity |
| CN111559992A (en) * | 2020-05-29 | 2020-08-21 | 华中科技大学 | Preparation method of 2-aryl-gamma-aminobutyric acid derivative |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2503844A1 (en) * | 2002-10-30 | 2004-05-21 | Merck & Co., Inc. | Piperidinyl-alpha-aminoamide modulators of chemokine receptor activity |
| US7491737B2 (en) * | 2002-10-30 | 2009-02-17 | Merck & Co., Inc. | Heterarylpiperidine modulators of chemokine receptor activity |
| JP2006507301A (en) * | 2002-10-30 | 2006-03-02 | メルク エンド カムパニー インコーポレーテッド | Piperidinylcyclopentylarylbenzylamide modulators of chemokine receptor activity |
| US7262196B2 (en) * | 2003-02-11 | 2007-08-28 | Prosidion Limited | Tri(cyclo) substituted amide glucokinase activator compounds |
| JP4736043B2 (en) | 2003-03-14 | 2011-07-27 | 小野薬品工業株式会社 | Nitrogen-containing heterocyclic derivatives and drugs containing them as active ingredients |
| ES2457041T3 (en) * | 2004-09-13 | 2014-04-24 | Ono Pharmaceutical Co., Ltd. | N-4-piperidylurea derivatives and medicines containing them as active ingredient |
| US8003642B2 (en) * | 2006-03-10 | 2011-08-23 | Ono Pharmaceutical Co., Ltd. | Nitrogenated heterocyclic derivative, and pharmaceutical agent comprising the derivative as active ingredient |
| CN101565369B (en) * | 2009-05-22 | 2012-10-03 | 江苏开元医药化工有限公司 | Method for preparing 3-bromine-5-trifluoromethylbenzoic acid |
| US8592395B2 (en) * | 2009-07-24 | 2013-11-26 | Glaxosmithkline Llc | Therapeutic compounds |
| CN111606920A (en) * | 2020-05-29 | 2020-09-01 | 凯美克(上海)医药科技有限公司 | Synthesis method of 7-bromothieno [2,3-B ] pyrazine |
Citations (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4166452A (en) | 1976-05-03 | 1979-09-04 | Generales Constantine D J Jr | Apparatus for testing human responses to stimuli |
| US4256108A (en) | 1977-04-07 | 1981-03-17 | Alza Corporation | Microporous-semipermeable laminated osmotic system |
| US4265874A (en) | 1980-04-25 | 1981-05-05 | Alza Corporation | Method of delivering drug with aid of effervescent activity generated in environment of use |
| US5403842A (en) | 1992-02-25 | 1995-04-04 | Recordati S.A., Chemical And Pharmaceutical Company | Benzopyran and benzothiopyran derivatives |
| WO1995015973A1 (en) | 1993-12-06 | 1995-06-15 | Cytel Corporation | Cs-1 peptidomimetics, compositions and methods of using the same |
| WO1995028931A1 (en) | 1994-04-26 | 1995-11-02 | Merck & Co., Inc. | Spiro-substituted azacycles as neurokinin-3 antagonists |
| WO1996001644A1 (en) | 1994-07-11 | 1996-01-25 | Athena Neurosciences, Inc. | Inhibitors of leukocyte adhesion |
| WO1996006108A2 (en) | 1994-08-25 | 1996-02-29 | Cytel Corporation | Cyclic cs-1 peptidomimetics, compositions and methods of using the same |
| US5510332A (en) | 1994-07-07 | 1996-04-23 | Texas Biotechnology Corporation | Process to inhibit binding of the integrin α4 62 1 to VCAM-1 or fibronectin and linear peptides therefor |
| WO1996020216A1 (en) | 1994-12-24 | 1996-07-04 | Zeneca Limited | Fibronectin adhesion inhibitors |
| WO1996022966A1 (en) | 1995-01-23 | 1996-08-01 | Biogen, Inc. | Cell adhesion inhibitors |
| WO1996031206A2 (en) | 1995-04-07 | 1996-10-10 | Warner-Lambert Company | Flavones and coumarins as agents for the treatment of atherosclerosis |
| WO1996040781A1 (en) | 1995-06-07 | 1996-12-19 | Tanabe Seiyaku Co., Ltd. | CYCLIC PEPTIDE INHIBITORS OF β1 AND β2 INTEGRIN-MEDIATED ADHESION |
| WO1997002289A1 (en) | 1995-07-06 | 1997-01-23 | Zeneca Limited | Peptide inhibitors of fibronectine |
| WO1997003094A1 (en) | 1995-07-11 | 1997-01-30 | Biogen, Inc. | Cell adhesion inhibitors |
| WO1998042656A1 (en) | 1997-03-21 | 1998-10-01 | Cytel Corporation | Novel compounds |
| WO1998053818A1 (en) | 1997-05-29 | 1998-12-03 | Merck & Co., Inc. | Sulfonamides as cell adhesion inhibitors |
| WO1998054207A1 (en) | 1997-05-30 | 1998-12-03 | Celltech Therapeutics Limited | Anti-inflammatory tyrosine derivatives |
| WO1998053817A1 (en) | 1997-05-29 | 1998-12-03 | Merck & Co., Inc. | Biarylalkanoic acids as cell adhesion inhibitors |
| WO1998053814A1 (en) | 1997-05-29 | 1998-12-03 | Merck & Co., Inc. | Heterocyclic amide compounds as cell adhesion inhibitors |
| WO1998058902A1 (en) | 1997-06-23 | 1998-12-30 | Tanabe Seiyaku Co., Ltd. | INHIBITORS OF α4β1MEDIATED CELL ADHESION |
| US6117880A (en) * | 1997-10-30 | 2000-09-12 | Merck & Co., Inc. | Somatostatin agonists |
| WO2002066446A1 (en) | 2001-02-16 | 2002-08-29 | Aventis Pharmaceuticals Inc. | Heterocyclic substituted carbonyl derivatives and their use as dopamine d3 receptor ligands |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2937180A (en) * | 1958-12-22 | 1960-05-17 | Janssen Paul Adriaan Jan | Alkanoic acid amides |
| JPS5849550B2 (en) * | 1974-02-07 | 1983-11-05 | 住友化学工業株式会社 | Synchronous aniline usage information |
| US4166119A (en) * | 1978-04-14 | 1979-08-28 | American Hoechst Corporation | Analgesic and tranquilizing spiro[dihydrobenzofuran]piperidines and pyrrolidines |
| JPH07100688B2 (en) * | 1986-02-12 | 1995-11-01 | 大日本製薬株式会社 | Cyclic amine derivative |
| NZ243065A (en) * | 1991-06-13 | 1995-07-26 | Lundbeck & Co As H | Piperidine derivatives and pharmaceutical compositions |
| HRP930210A2 (en) * | 1992-02-25 | 1995-06-30 | Recordati Chem Pharm | Heterobicyclic compounds and pharmaceutical preparations containing them |
| WO1994017045A1 (en) * | 1993-01-28 | 1994-08-04 | Merck & Co., Inc. | Spiro-substituted azacycles as tachykinin receptor antagonists |
| GB9310713D0 (en) * | 1993-05-24 | 1993-07-07 | Zeneca Ltd | Aryl substituted heterocycles |
| AU680020B2 (en) * | 1993-06-07 | 1997-07-17 | Merck & Co., Inc. | Spiro-substituted azacycles as neurokinin antagonists |
| EP1003514A4 (en) * | 1997-07-25 | 2000-10-11 | Merck & Co Inc | Cyclic amine modulators of chemokine receptor activity |
| US6620438B2 (en) * | 2001-03-08 | 2003-09-16 | Boehringer Ingelheim Pharma Kg | Pharmaceutical compositions based on anticholinergics and NK1-receptor antagonists |
| AUPR201600A0 (en) * | 2000-12-11 | 2001-01-11 | Fujisawa Pharmaceutical Co., Ltd. | Quinazolinone derivative |
| NZ527680A (en) * | 2001-03-21 | 2005-07-29 | Pharmacopeia Drug Discovery | Aryl and biaryl compounds having MCH modulatory activity |
| WO2002088089A1 (en) * | 2001-04-19 | 2002-11-07 | Banyu Pharmaceutical Co., Ltd. | Spiropiperidine derivatives, nociceptin receptor antagonists containing the same as the active ingredient and medicinal compositions |
| US20030078278A1 (en) * | 2001-06-26 | 2003-04-24 | Pfizer Inc. | Spiropiperidine compounds as ligands for ORL-1 receptor |
| UA77814C2 (en) * | 2002-07-03 | 2007-01-15 | Lundbeck & Co As H | Spirocyclic piperidines as antagonists of mch1 and use thereof |
| CA2503844A1 (en) * | 2002-10-30 | 2004-05-21 | Merck & Co., Inc. | Piperidinyl-alpha-aminoamide modulators of chemokine receptor activity |
-
2003
- 2003-10-24 AU AU2003284984A patent/AU2003284984B2/en not_active Ceased
- 2003-10-24 EP EP03779303A patent/EP1558250A4/en not_active Withdrawn
- 2003-10-24 US US10/528,329 patent/US7247725B2/en not_active Expired - Fee Related
- 2003-10-24 CA CA002503720A patent/CA2503720A1/en not_active Abandoned
- 2003-10-24 WO PCT/US2003/034009 patent/WO2004041279A1/en active Application Filing
- 2003-10-24 JP JP2004550131A patent/JP2006511500A/en active Pending
Patent Citations (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4166452A (en) | 1976-05-03 | 1979-09-04 | Generales Constantine D J Jr | Apparatus for testing human responses to stimuli |
| US4256108A (en) | 1977-04-07 | 1981-03-17 | Alza Corporation | Microporous-semipermeable laminated osmotic system |
| US4265874A (en) | 1980-04-25 | 1981-05-05 | Alza Corporation | Method of delivering drug with aid of effervescent activity generated in environment of use |
| US5403842A (en) | 1992-02-25 | 1995-04-04 | Recordati S.A., Chemical And Pharmaceutical Company | Benzopyran and benzothiopyran derivatives |
| WO1995015973A1 (en) | 1993-12-06 | 1995-06-15 | Cytel Corporation | Cs-1 peptidomimetics, compositions and methods of using the same |
| WO1995028931A1 (en) | 1994-04-26 | 1995-11-02 | Merck & Co., Inc. | Spiro-substituted azacycles as neurokinin-3 antagonists |
| US5510332A (en) | 1994-07-07 | 1996-04-23 | Texas Biotechnology Corporation | Process to inhibit binding of the integrin α4 62 1 to VCAM-1 or fibronectin and linear peptides therefor |
| WO1996001644A1 (en) | 1994-07-11 | 1996-01-25 | Athena Neurosciences, Inc. | Inhibitors of leukocyte adhesion |
| WO1996006108A2 (en) | 1994-08-25 | 1996-02-29 | Cytel Corporation | Cyclic cs-1 peptidomimetics, compositions and methods of using the same |
| WO1996020216A1 (en) | 1994-12-24 | 1996-07-04 | Zeneca Limited | Fibronectin adhesion inhibitors |
| WO1996022966A1 (en) | 1995-01-23 | 1996-08-01 | Biogen, Inc. | Cell adhesion inhibitors |
| WO1996031206A2 (en) | 1995-04-07 | 1996-10-10 | Warner-Lambert Company | Flavones and coumarins as agents for the treatment of atherosclerosis |
| WO1996040781A1 (en) | 1995-06-07 | 1996-12-19 | Tanabe Seiyaku Co., Ltd. | CYCLIC PEPTIDE INHIBITORS OF β1 AND β2 INTEGRIN-MEDIATED ADHESION |
| WO1997002289A1 (en) | 1995-07-06 | 1997-01-23 | Zeneca Limited | Peptide inhibitors of fibronectine |
| WO1997003094A1 (en) | 1995-07-11 | 1997-01-30 | Biogen, Inc. | Cell adhesion inhibitors |
| WO1998042656A1 (en) | 1997-03-21 | 1998-10-01 | Cytel Corporation | Novel compounds |
| WO1998053818A1 (en) | 1997-05-29 | 1998-12-03 | Merck & Co., Inc. | Sulfonamides as cell adhesion inhibitors |
| WO1998053817A1 (en) | 1997-05-29 | 1998-12-03 | Merck & Co., Inc. | Biarylalkanoic acids as cell adhesion inhibitors |
| WO1998053814A1 (en) | 1997-05-29 | 1998-12-03 | Merck & Co., Inc. | Heterocyclic amide compounds as cell adhesion inhibitors |
| WO1998054207A1 (en) | 1997-05-30 | 1998-12-03 | Celltech Therapeutics Limited | Anti-inflammatory tyrosine derivatives |
| WO1998058902A1 (en) | 1997-06-23 | 1998-12-30 | Tanabe Seiyaku Co., Ltd. | INHIBITORS OF α4β1MEDIATED CELL ADHESION |
| US6117880A (en) * | 1997-10-30 | 2000-09-12 | Merck & Co., Inc. | Somatostatin agonists |
| WO2002066446A1 (en) | 2001-02-16 | 2002-08-29 | Aventis Pharmaceuticals Inc. | Heterocyclic substituted carbonyl derivatives and their use as dopamine d3 receptor ligands |
Non-Patent Citations (28)
| Title |
|---|
| BEN-BARRUCH ET AL., J. BIOL. CHEM., vol. 270, 1995, pages 22123 - 22128 |
| BEOTE ET AL., CELL, vol. 72, 1993, pages 415 - 425 |
| BORING ET AL., J. CLIN. INVEST., vol. 100, 1997, pages 2552 - 2561 |
| CHAUDHUN ET AL., J. BIOL. CHEM., vol. 269, 1994, pages 7835 - 7838 |
| EVANS, D.A.; ENNIS, M.D.; MATHRE, D.J., J. AM. CHEM. SOC., vol. 104, 1982, pages 1737 |
| GOMEZ ET AL., ARTHRITIS & RHEUMATISM, vol. 42, 1999, pages 989 - 992 |
| GOSLING ET AL., J. CLIN. INVEST., vol. 103, 1999, pages 773 - 778 |
| GREENE, T; WUTS, P. G. M.: "Protective Groups in Organic Synthesis", 1991, JOHN WILEY & SONS |
| GREENE, T; WUTS, P. G. M.: "Protective Groups in Organic Synthesis", 1991, JOHN WILEY & SONS, INC. |
| HORUK, TRENDS PHARM. SCI., vol. 15, 1994, pages 159 - 165 |
| J. ORG. CHEM., vol. 48, 1983, pages 3553 |
| KITA, H. ET AL., J. EXP. MED., vol. 183, 1996, pages 2421 - 2426 |
| KURIHARA ET AL., J. EXP. MED., vol. 186, 1997, pages 1757 - 1762 |
| KUZIEL ET AL., PROC. NATL. ACAD. SCI., vol. 94, 1997, pages 12053 - 12058 |
| LU ET AL., J. EXP. MED., vol. 187, 1998, pages 601 - 608 |
| LUSTGER, A.D., NEW ENGLAND J. MED., vol. 338, no. 7, 1998, pages 426 - 445 |
| MARCH J.: "Advanced Organic Chemistry", 1992, JOHN WILEY & SONS, pages: 1167 - 1171 |
| MITSUNOBU, O., SYNTHESIS, 1981, pages 1 |
| MURPHY, REV. IMMUN., vol. 12, 1994, pages 593 - 633 |
| NATURE, vol. 394, 1998, pages 894 - 897 |
| ROLLINS ET AL., BLOOD, vol. 90, 1997, pages 908 - 928 |
| SANSON ET AL., BIOCHEMISTRY, vol. 35, 1996, pages 3362 - 3367 |
| SCHALL, CYTOKINE, vol. 3, 1991, pages 165 - 183 |
| See also references of EP1558250A4 * |
| TETRAHEDRON LETT., vol. 26, 1985, pages 1723 |
| VAN RIPER ET AL., J. EXP. MED, vol. 177, 1993, pages 851 - 856 |
| WARMINGTON ET AL., AM J. PATH., vol. 154, 1999, pages 1407 - 1416 |
| WRIGHT, S.W. ET AL., TETRAHEDRON LETT., vol. 38, no. 42, 1997, pages 7345 |
Cited By (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7449475B2 (en) | 2002-07-08 | 2008-11-11 | Astrazeneca Ab | Tricyclic spiropiperidines or spiropyrrolidines |
| US7985730B2 (en) | 2002-11-27 | 2011-07-26 | Incyte Corporation | 3-aminopyrrolidine derivatives as modulators of chemokine receptors |
| US8362003B2 (en) | 2002-11-27 | 2013-01-29 | Incyte Corporation | 3-aminopyrrolidine derivatives as modulators of chemokine receptors |
| US8729063B2 (en) | 2002-11-27 | 2014-05-20 | Incyte Corporation | 3-aminopyrrolidine derivatives as modulators of chemokine receptors |
| US7834021B2 (en) | 2002-11-27 | 2010-11-16 | Incyte Corporation | 3-aminopyrrolidine derivatives as modulators of chemokine receptors |
| US7498338B2 (en) | 2003-11-20 | 2009-03-03 | Astrazeneca Ab | Compounds |
| WO2005058884A3 (en) * | 2003-12-15 | 2005-09-09 | Japan Tobacco Inc | Cyclopropane compounds and pharmaceutical use thereof |
| US7351825B2 (en) | 2003-12-15 | 2008-04-01 | Japan Tobacco Inc. | Cyclopropane compounds and pharmaceutical use thereof |
| US7576089B2 (en) | 2003-12-18 | 2009-08-18 | Incyte Corporation | 3-cycloalkylaminopyrrolidine derivatives as modulators of chemokine receptors |
| US7524856B2 (en) | 2003-12-22 | 2009-04-28 | Astrazeneca Ab | Tricyclic spiroderivatives as modulators of chemokine receptor activity |
| US7279486B2 (en) | 2004-03-29 | 2007-10-09 | Pfizer Inc. | Alpha aryl or heteroaryl methyl beta piperidino propanoic acid compounds as ORL1-receptor antagonists |
| US7307086B2 (en) | 2004-05-11 | 2007-12-11 | Incyte Corporation | 3-(4-heteroarylcyclohexylamino)cyclopentanecarboxamides as modulators of chemokine receptors |
| US8563582B2 (en) | 2004-06-28 | 2013-10-22 | Incyte Corporation | 3-aminocyclopentanecarboxamides as modulators of chemokine receptors |
| US7618970B2 (en) | 2004-06-28 | 2009-11-17 | Incyte Corporation | 3-aminocyclopentanecarboxamides as modulators of chemokine receptors |
| US8470827B2 (en) | 2004-06-28 | 2013-06-25 | Incyte Corporation | 3-aminocyclopentanecarboxamides as modulators of chemokine receptors |
| US7449467B2 (en) | 2004-06-28 | 2008-11-11 | Pfizer Inc. | 3-aminocyclopentanecarboxamides as modulators of chemokine receptors |
| EP1831222A4 (en) * | 2004-12-09 | 2009-10-21 | Virochem Pharma Inc | Novel spirotropane compounds and methods for the modulation of chemokine receptor activity |
| EP1831222A1 (en) * | 2004-12-09 | 2007-09-12 | Virochem Pharma Inc. | Novel spirotropane compounds and methods for the modulation of chemokine receptor activity |
| US7960403B2 (en) | 2004-12-09 | 2011-06-14 | Virochem Pharma Inc. | Spirotropane compounds and methods for the modulation of chemokine receptor activity |
| WO2006060919A1 (en) | 2004-12-09 | 2006-06-15 | Virochem Pharma Inc. | Novel spirotropane compounds and methods for the modulation of chemokine receptor activity |
| US8076349B2 (en) | 2004-12-09 | 2011-12-13 | Virochem Pharma Inc. | Spirotropane compounds and methods for the modulation of chemokine receptor activity |
| JP2008540333A (en) * | 2005-04-30 | 2008-11-20 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Novel piperidine substituted indoles |
| JP2008543858A (en) * | 2005-06-15 | 2008-12-04 | ジェンザイム・コーポレーション | Chemokine receptor binding compounds |
| US8288370B2 (en) | 2005-09-29 | 2012-10-16 | Albireo Ab | Substituted azetidine compounds of formula (I) useful in the treatment of functional gastrointestinal disorders, IBS, and functional dyspepsia |
| WO2007037743A1 (en) * | 2005-09-29 | 2007-04-05 | Astrazeneca Ab | Novel azetidine compounds useful in the treatment of functional gastrointestinal disorders, ibs and functional dyspepsia |
| WO2007037742A1 (en) * | 2005-09-29 | 2007-04-05 | Astrazeneca Ab | Novel azetidine compounds useful in the treatment of functional gastrointestinal disorders, ibs and functional dyspepsia |
| US8318931B2 (en) | 2005-10-28 | 2012-11-27 | Ono Pharmaceutical Co., Ltd. | Chemokine receptor antagonists and use thereof |
| EP2657235A1 (en) * | 2005-10-28 | 2013-10-30 | Ono Pharmaceutical Co., Ltd. | Compound containing basic group and use thereof |
| US8614323B2 (en) | 2005-10-28 | 2013-12-24 | Ono Pharmaceutical Co., Ltd. | Chemokine receptor antagonists and use thereof |
| WO2007049771A1 (en) * | 2005-10-28 | 2007-05-03 | Ono Pharmaceutical Co., Ltd. | Compound containing basic group and use thereof |
| US8871210B2 (en) | 2005-10-28 | 2014-10-28 | Ono Pharmaceutical Co., Ltd. | Chemokine receptor antagonists and use thereof |
| EP2166848A4 (en) * | 2007-06-20 | 2011-08-03 | Glaxo Group Ltd | Spiroindolines as modulators of chemokine receptors |
| EP2166848A1 (en) * | 2007-06-20 | 2010-03-31 | Glaxo Group Limited | Spiroindolines as modulators of chemokine receptors |
| EP2190286A4 (en) * | 2007-08-14 | 2011-06-01 | Glaxo Group Ltd | Spiroindenes and spiroindanes as modulators of chemokine receptors |
| EP2190286A1 (en) * | 2007-08-14 | 2010-06-02 | Glaxo Group Limited | Spiroindenes and spiroindanes as modulators of chemokine receptors |
| CN111559992A (en) * | 2020-05-29 | 2020-08-21 | 华中科技大学 | Preparation method of 2-aryl-gamma-aminobutyric acid derivative |
| CN111559992B (en) * | 2020-05-29 | 2022-04-08 | 华中科技大学 | Preparation method of 2-aryl-gamma-aminobutyric acid derivative |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2003284984A1 (en) | 2004-06-07 |
| EP1558250A1 (en) | 2005-08-03 |
| EP1558250A4 (en) | 2006-11-02 |
| CA2503720A1 (en) | 2004-05-21 |
| US7247725B2 (en) | 2007-07-24 |
| US20050261325A1 (en) | 2005-11-24 |
| JP2006511500A (en) | 2006-04-06 |
| AU2003284984B2 (en) | 2008-10-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2003284984B2 (en) | Gamma-aminoamide modulators of chemokine receptor activity | |
| US6545023B2 (en) | Cyclopentyl modulators of chemokine receptor activity | |
| AU2003284165B2 (en) | Piperidinyl-alpha-aminoamide modulators of chemokine receptor activity | |
| US7514431B2 (en) | Piperidinyl cyclopentyl aryl benzylamide modulators of chemokine receptor activity | |
| AU2005214319B2 (en) | Amino heterocyclic modulators of chemokine receptor activity | |
| EP1615699B1 (en) | Benzoxazinyl-amidocyclopentyl-heterocyclic modulators of chemokine receptors | |
| AU2001283345A1 (en) | Cyclopentyl modulators of chemokine receptor activity | |
| WO2004041161A2 (en) | Tetrahydropyranyl cyclopentyl benzylamide modulators of chemokine receptor activity | |
| EP1558599A2 (en) | Heteroarylpiperidine modulators of chemokine receptor activity | |
| WO2004082616A2 (en) | Tetrahydropyranyl cyclopentyl heterocylic amide modulators of chemokine receptor activity | |
| AU2004313486A1 (en) | Alkylamino, arylamino, and sulfonamido cyclopentyl amide modulators of chemokine receptor activity | |
| US20070299104A1 (en) | Tetrahydropyranyl Cyclopentyl 1-Substituted and 1,1-Disubstituted Tetrahydroisoquinoline Modulators of Chemokine Receptor Activity | |
| US20070238723A1 (en) | Benzoxazinyl-amidocyclopentyl-heterocyclic modulators of chemokine receptors | |
| WO2005014537A2 (en) | Tetrahydropyran heterocyclic cyclopentyl heteroaryl modulators of chemokine receptor activity |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2003284984 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10528329 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003779303 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2503720 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004550131 Country of ref document: JP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003779303 Country of ref document: EP |