MODIFIED PNA MOLECULES
The present invention concerns novel drugs for use in combating various diseases. More particular the invention concerns peptide nucleic acid (PNA) drugs, which are optionally modified in order to obtain novel PNA molecules with cell-specific delivery.
BACKGROUND OF THE INVENTION
Antisense agents offer a novel strategy in combating diseases, as well as opportuni- ties to employ new chemical classes in the drug design.
Oligonucleotides can interact with native DNA and RNA in several ways. One of these is duplex formation between an oligonucleotide and a single stranded nucleic acid. Another is triplex formation between an oligonucleotide and double stranded DNA to form a triplex structure.
Results from basic research have been encouraging, and antisense oligonucleotide drug formulations against viral and disease causing human genes are progressing through clinical trials. Efficient antisense inhibition of bacterial genes also has wide applications.
Peptide nucleic acids (PNA) are compounds that in certain respects are similar to oligonucleotides and their analogs and thus may mimic DNA and RNA. In PNA, the deoxyribose backbone of oligonucleotides has been replaced by a pseudo-peptide backbone (Nielsen et al. 1991 (1)). Each subunit, or monomer, has a naturally occurring or non-naturally occurring nucleobase attached to this backbone. One such backbone is constructed of repeating units of N-(2-aminoethyl)glycine linked through amide bonds. PNA hybridises with complementary nucleic acids through Watson and Crick base pairing and helix formation (Egholm et al. 1993 (2)). The Pseudo- peptide backbone provides superior hybridization properties (Egholm et al. 1993 (2)), resistance to enzymatic degradation (Demidov et al. 1994 (3)) and access to a variety of chemical modifications (Nielsen and Haaima 1997 (4), WO 94/25472, WO98/03542).
PNA binds both DNA and RNA to form PNA/ DNA or PNA/RNA duplexes. The resulting PNA/DNA or PNA/RNA duplexes are bound with greater affinity than corresponding DNA/DNA or DNA/RNA duplexes as determined by the melting point temperature (Tm). This high thermal stability might be attributed to the lack of charge repulsion due to the neutral backbone in PNA. In addition to increased affinity, PNA has also been shown to bind to DNA with increased specificity. When a PNA/DNA duplex mismatch is melted relative to the DNA/DNA duplex, there is seen an 8°C to 20°C drop in the melting point temperature.
Furthermore, homopyrimidine PNA oligomers form extremely stable PNA2-DNA (RNA) triplexes with sequence complementary targets in DNA or RNA oligomers. Finally, PNA's may bind to double stranded DNA or RNA by helix invasion.
An advantage of PNA compared to oligonucleotides is that the PNA polyamide backbone (having appropriate nucleobases or other side chain groups attached thereto) is not recognised by either nucleases or proteases and are thus not cleaved. As a result, PNA's are resistant to degradation by enzymes unlike nucleic acids and peptides.
For antigene and antisense application, target bound PNA can cause steric hindrance of DNA and RNA polymerases, reverse transcription, telomerase and of the ribosomes (Hanvey et al. 1992 (5), Knudsen et al. 1996 (6), Good and Nielsen 1998 (7,8)), by targeting, among others, DNA, mRNA, rRNA, or tRNA.
A general difficulty when using antisense agents is cell uptakeand targeting of specific organs. A variety of strategies to improve uptake can be envisioned and there are reports of improved uptake into eukaryotic cells using lipids (Lewis et al. 1996 (9)), encapsulation (Meyer et al. 1998 (10)) and carrier strategies (Nyce and Metzger 1997 (11), Pooga et al, 1998 (12)).
WO 99/05302 discloses a PNA conjugate consisting of PNA and the transporter peptide transportan, which peptide may be used for transport cross a lipid mem-
brane and for delivery of the PNA into interactive contact with intracellular polynucleotides.
US-A-5 777 078 discloses a pore-forming compound, which comprises a delivery agent recognising the target cell and being linked to a pore-forming agent, such as a bacterial exotoxin. The compound is administered together with a drug such as PNA.
WO 96/11205 discloses PNA conjugates, wherein a conjugated moiety may be placed on terminal or non-terminal parts of the backbone of PNA in order to func- tionalise the PNA. The conjugated moieties may be reporter enzymes or molecules, steroids, carbohydrate, terpenes, peptides, proteins, etc. It is suggested that the conjugates among other properties may possess improved transfer properties for crossing cellular membranes.
WO 01/27261 discloses conjugates of cationic peptides and aeg-PNA (cf. Figure 4).
WO 98/52614 discloses a method of enhancing transport over biological membranes. According to this publication, biological active agents such as PNA may be conjugated to a transporter polymer in order to enhance the transmembrane transport. The transporter polymer consists of 6-25 subunits; at least 50% of which contain a guanidino or amidino sidechain moiety and wherein at least 6 contiguous sub- units contain guanidino and/or amidino sidechains. A preferred transporter polymer is a polypeptide containing nine arginine subunits ((Arg)9).
However, the present methods of transport of PNA oligomers across biological membranes lack efficiency and specificity. Only little information is available on the pharmacokinetic behaviour of PNA oligomers, e.g. the dynamic and kinetic mechanisms of exogenous absorption, biotransformation, distribution, release, transport, uptake, and elimination of PNA oligomers as a function of dosage and extent and rate of metabolic processes. However, data indicates that PNA oligomers are fairly quickly excreted in the urine according to McMahon et al (2002 (13)), being a rather hydrophilic compound, which rarely binds to proteins like albumin in serum.
It could be of significant medicinal interest to functionalise the PNAs, in order to control the bio-distribution of the molecule. Functionalisation of the PNA backbone may dramatically change the physico-chemical properties of the PNA, and it is plausible that such changes would significantly influence its pharmacokinetic behaviour.
Zhang et al (2001 (14)) describes a method by which uptake of PNA oligomers in a liver cell was promoted by modifying the terminal ends of PNA with lactose. Lactose, being recognized by the hepatic asialoglycoprotein receptor, provided an efficient entry of lactose modified PNAs into HepG2 cells.
Biessen et al (2002, (15)) presents work in which the parenchymal liver cell uptake of antisense PNA drug was improved by targeting to the asialoglycoprotein receptor of the liver cell, a glycoconjuga ted antisense PNA. The PNA was conjugated with N- acetyl-galactos-aminyls at the 5' end through lysine side chain linkers.
Prior art shows an increased organ and cell specificity of PNAs by end glycosylation of the PNA. However, incorporation of the saccharides into the PNA backbone would improve the medicinal chemistry opportunities, the biostability and biodistribu- tion of the drug, resulting in lower dosage and reduced side effects.
SUMMARY OF THE INVENTION
The present invention relates to glycosylated peptide nucleic acid (PNA) monomers. More particular, the invention concerns the incorporation of glycosylated monomers into an antisense PNA oligomer, in order to improve the cell and/or organ-specific uptake of PNAs and thereby the pharmacokinetic behavior.
It has been found that by integrating saccharides within PNA monomers and by subsequent incorporation of at least one modified PNA monomer in an antisense PNA oligomer, an enhanced efficacy and organ-specificity is observed, without major influence on the DNA or RNA hybridisation potency of the PNA. The important feature of the modified PNA molecules is the incorporation of at least one glycosy-
lated PNA monomer in the PNA oligomer chain. This results in versatility in synthesis and design in terms of character, position and number of saccharides, which is crucial for biological activity and also high bio-stability.
Thus, the present invention concerns a PNA monomer of formula (I):
wherein B is a naturally-occurring nucleobase preferably A, T, G, or C, or a non- naturally-occurring nucleobase;
(Pr) is hydrogen or a protection group;
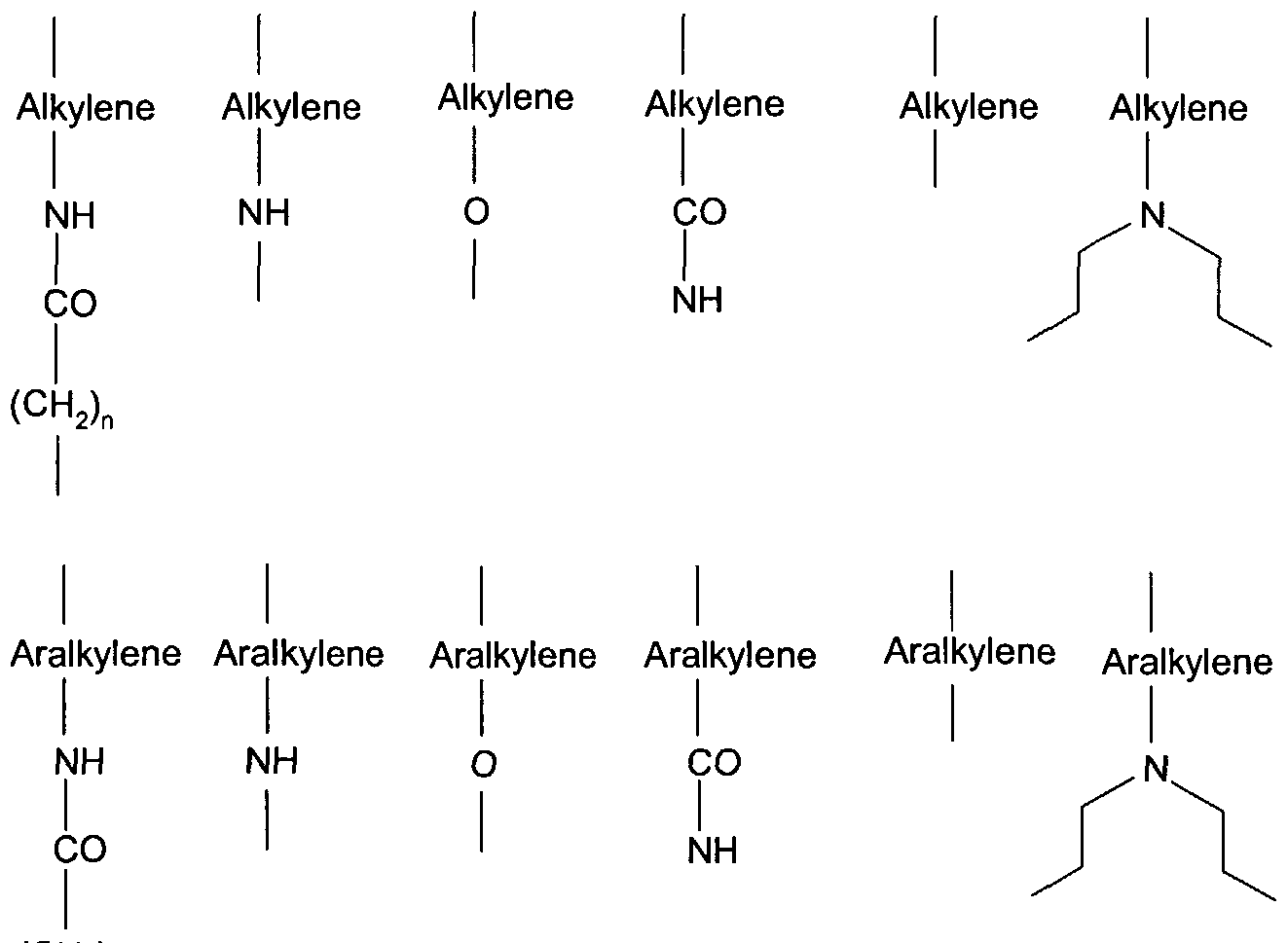
R1, R2 and R3 are, independently, hydrogen, an amino acid side chain, or an C2-6- alkyl, aryl, aralkyl, heteroaryl, hydroxy, C^-alkoxy, d-6-alkylthio, hydroxy- or alkoxy- or alkylthio-substituted C^-alkyl, -NR4R5, (wherein R4and R5 independently are hy- drogen, d-e-alkyl, hydroxy- or alkoxy- or alkylthio-substituted Ci-e-alkyl), or Z1-Z2, wherein Z is a bond or one of the radicals of formula (II):
Alkylene Alkylene Alkylene Alkylene Alkylene Alkylene
(CH2)π
wherein n is form 0 to 8; and Z2 is alfa- or beta forms of a monosaccharide, a disaccharide, a polysaccharide, or one of the radicals of formula (III):
provided that at least one of R1, R2, or R3 is Z1-Z2.
Enclosed is also a compound according to claim 1, wherein Z1 is one of the radicals of formula (IV):
(
Enclosed is further a compound according to claim 1 or 3, wherein Z1-Z2 comprises ligands of formula (V):
Enclosed is also a peptide nucleic acid oligomer with from 4 to 50 monomers selected from the group consisting of PNA monomers and at least one monomer of claim 1, said PNA oligomer conjugated either directly or through a linking moiety to hydrogen or a reporter enzyme, a reporter molecule, a steroid, a carbohydrate, a terpene, a peptide, a protein, an aromatic lipophilic molecule, a non aromatic lipo- philic molecule, a phosphortipid, an intercalator, a cell receptor binding molecule, a
crosslinking agent, a water soluble vitamin, a lipid soluble vitamin, an RNA/ DNA cleaving complex, a metal chelator, a porphyrin, an alkylator, or a polymeric compound selected from polymeric amines, polymeric glycols and polyethers.
The present invention further comprises a peptide nucleic acid molecule comprising a peptide nucleic acid oligomer with from 4 to 50 aeg-PNA monomers and one or more conjugates bound to said peptide nucleic acid either directly or through one or more linking moieties, wherein said conjugate is a reporter enzyme, a reporter molecule, a steroid, a carbohydrate, a terpene, a peptide, a protein, an aromatic lipophilic molecule, a non aromatic lipophilic molecule, a phosphortipid, an intercalator, a cell receptor binding molecule, a crosslinking agent, a water soluble vitamin, a lipid soluble vitamin, an RNA/ DNA cleaving complex, a metal chelator, a porphyrin, an alkylator, or a polymeric compound selected from polymeric amines, polymeric glycols and polyethers.
In a preferred embodiment the glycoside residues (sugar residues) are chosen from monosaccharides having a high intrinsic affinity for the asialoglycoprotein receptor, including β-D-galactosyl, 2-acetamido-2-deoxy-galactopyranosyl, 1-phenyl-β-D- galactosyl, 1-propyl-β-D-galactosyl or 1-butyl-β-D-galactosyl.
In another preferred embodiment of the invention the linking moiety is an amino acid sequence of from 1 to 10 positively charged amino acids or amino acid analogues.
By the term "positively charged amino acids or amino acid analogues" is to be un- derstood any natural or non-natural occurring amino acid or amino acid analogue which have a positive charge at physiological pH.
Among the positively charged amino acids and amino acid analogs may be mentioned lysine (Lys, K), arginine (Arg, R), diamino butyric acid (DAB) and ornithine (Orn). The skilled person will be aware of further positively charged amino acids and amino acid analogs.
In one aspect of the invention, the modified PNA molecules are used in the manu-
facture of medicaments for the treatment or prevention of a disease selected from bacterial and viral infections, cardiac or vascular diseases, metabolic diseases or immunological disorders or for disinfecting non-living objects.
In a further aspect, the invention concerns a composition for treating or preventing disease selected from bacterial and viral infections, cardiac or vascular diseases, metabolic diseases or immunological disorders or for disinfecting non-living objects.
In another aspect, the invention concerns the treatment or prevention of disease se- lected from bacterial and viral infections, cardiac or vascular diseases, metabolic diseases or immunological disorders or for disinfecting non-living objects.
Preferred targeting is organ related diseases e.g. liver diseases such as hepatitis and liver cancer, known for a person skilled in the art.
Liver cancer is the fifth most common cancer worldwide. More than 400,000 cases were reported in 1990. Hepatocellular carcinoma (HCC) accounts for 80% of all liver cancer. Liver cancer can result from both viral infection and chemical exposure.
Known risk factors include hepatitis B and C virus infection and exposure to aflatoxin 1. It is not known whether distinct routes to liver cancer affect the same or different cellular pathways. No mutational model has yet been developed for liver cancer as it has been for other cancers such as colon cancer.
According to the invention one of numerous described high affinity ligands for the αvβ3 integrin receptor is used for PNA delivery to angiogenic blood vessels in the treatment of diseases that depend on angiogenisis and vascular remodelling, including cancer, see for example Hood JD et al. Science (2002) 296(5577):2404-7.
In yet a further aspect, the present invention concerns a method of identifying spe- cific advantageous antisense PNA sequences, which may be used in the modified PNA molecule according to the invention.
DETAILED DESCRIPTION OF THE INVENTION
Antisense PNAs can inhibit bacterial gene expression with gene and sequence specificity (Good and Nielsen 1998a,b (12, 13) and WO 99/13893). The approach may prove practical as a tool for functional genomics and as a source for novel antimicrobial drugs. However, improvements on standard PNA are required to increase antisense potencies. The major limit to activity appears to be cellular entry and cellular specificity. Cell membranes effectively exclude the entry of large molecular weight foreign compounds, and previous results for in vitro and cellular assays seem to show that the cell barrier restricts antisense effects. Accordingly, the present invention concerns strategies to improve the activity and specifidity of antisense potencies.
Without being bound by theory, it is believed that glycosylation of PNA oligomers lead to an improved cell specific PNA uptake. It is believed that the glycosylated peptides are recognised by receptors in cell membranes such as the hepatic asialoglycoprotein receptor thereby taken up through the glucose pathway, allowing the modified PNA molecule to cross the cell wall, reaching structures inside the cell, such as the genome, mRNA's, the ribosome, etc.
According to the invention, PNA molecules modified with saccharides enable specific and efficient inhibition of genes with nanomolar concentrations. Antisense potencies in this concentration are consistent with practical applications of the technology. It is believed that the present invention for the first time demonstrates that pep- tides with a certain pattern of glycosylation can be used as carriers to deliver PNAs across cell membranes. Further, the present invention has made it possible to administer PNA in an efficient concentration, which is also acceptable to the patient.
The terms "C^-alky!" as used herein, represent a branched or straight alkyl group having from one to six carbon atoms. Typical C^-alkyl groups include, but are not limited to, methyl, ethyl, n-propyl, iso-propyl, butyl, iso-butyl, sec-butyl, tert-butyl, pentyl, iso-pentyl, hexyl, iso-hexyl and the like.
The number of modified PNA monomers in the PNA oligomer may be chosen between 1 and full modification. It appears that at least 2 monomers, according to claim 1 , are preferable to obtain the advantageous effect.
The modified PNA molecule according to the present invention comprises a PNA oligomer of a sequence, which is complementary to at least one target nucleotide sequence in the target cell. The target may be a nucleotide sequence of any RNA, which is essential for the growth, and/or reproduction of the cell.
The binding of a PNA strand to a DNA or RNA strand can occur in one of two orientations, anti-parallel or parallel. As used in the present invention, the term complementary as applied to PNA does not in itself specify the orientation parallel or anti- parallel. It is significant that the most stable orientation of PNA/DNA and PNA/RNA is anti-parallel. In a preferred embodiment, PNA targeted to single strand RNA is complementary in an anti-parallel orientation.
The ability of PNAs to act as an antisense drug may be measured in many ways, which should be clear to the skilled person. To illustrate one way of preparing glycosylated PNA monomers with subsequent measuring of cell specificity, the following procedure may be used. However, the present invention is not limited hereto.
Preparation of O-glycosyla ted PNAs
The initially employed strategy is outlined in Scheme 1. As ortogonal protection group for the synthesis of O-glycosylated PNAs, Fmoc, tetf-butyl and allyl were ap- plied for amine, hydroxyl and carboxylic acid protection, respectively.
Scheme 1
(9-10) (-11.-12)
M2/M3
R=H) 1, 3,5,7,9,11,13,1s R=methyl) 2,4,6,8,10,12,14,2s
Initially, the carboxyl group of Fmoc-serine(t-Bu)-OH and Fmoc-threonine(t-Bu)-OH were allylated using allyl bromide in presence of DIPEA (18) to obtain high yields of 1-2. Fmoc-deprotection by piperidinolys treatment gave the free amine of 3-4 (19). The PNA backbone 5-6 were prepared using 2-Boc-aminoacetaldehyde (20) by reductive amination in presence of sodium cyanoborohydride. Subsequently, thymine- 1-yl acetic acid (21 ) was condensed to the PNA backbone to give 7-8. Boc and tert- butyl group removal by TFA resulted in the intermediates 9-10. The ethereal solution was neutralised by addition of excess solid sodium carbonate. The free amine was re-protected with Fmoc using Fmoc-O-succinimide to obtain 11-12.
Galactose donors were prepared by the method shown in scheme 2. Commercially available β-D-galactose pentaacetate was converted stereoselectively to the 1-O- deacetylated form M1 by treatment with ammonia in a THF-methanol solution. The reaction was monitored by TLC, in order to avoid undesired further deacetylations, and M1 was obtained in quantitative yield in the α-form (16). Galactosyl trichloroace-
timidate derivatives M2 & M3 were prepared by the method of Schmidt et. al. (17). In situ deprotonation of M1 under basic condition (K2CO3), reaction with trichloroace- tonit le followed by separation on silica gel column, gave α and β anomers in good yields.
Scheme 2
A) ammonia in THF-methanol (7:3 v/v), 1.5 h, rt, (_. 100%).
B) K2C03, CCI3CN, mol. sieves. 4A°, DCM, over night, rt, purification by silicagel chromatography (hexane-EtOAc 2:1 v/v) → α: 36%, β: 48%
The reaction of both α- and β-galactosyl trichloroacetimidates with 11 gave 13 as a 1 :1 anomeric mixture, and reaction with 12 gave 14 predominantly as the β-form. The β-glycosidic linkage in 13 and 14 were verified by the 1H NMR triplets at δ 5.05 ppm and 5.08 ppm respectively (J1ι2 = 7.7 Hz). Finally, removal of the allyl group was performed in a high yield by treatment with N-ethyl aniline in the presence of catalytic amount of tetrakis (Ph3P) Pd to give 1s-2s.
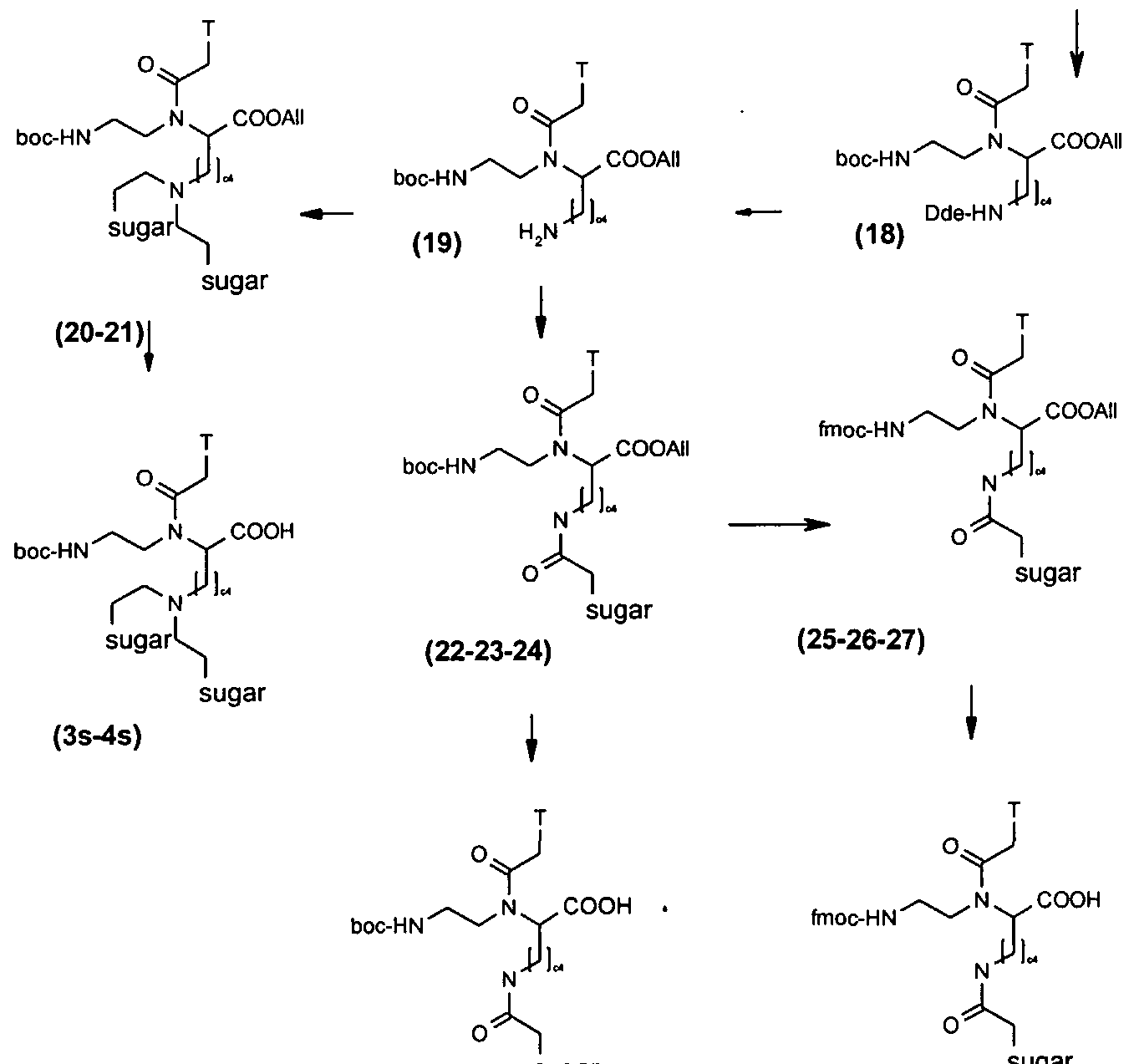
Preparation of lysine based N-C-glycosyla ted PNAs
Scheme 3
fmoc-HN ._ COOH fmoc-HN . ^ C COOOOAAIIII H,N. ^COOAII . l lu/s. . N- ..COOAII
Dde-HN I → Dde-HN J — ■ Dde-HN X Dde-HN X
(15) (16) (17)
(5s) (6s-7s-8s)
Λ/-Fmoc-lysine-Λ/'-(Dde)-OH was allylated using allyl bromide in presence of DIPEA and heating (18) resulting in 15. Following Fmoc deprotection with 20% piperidine in DCM, the free amine of 16 was achieved. It has been reported that the protection group Dde is able to migrate from the side chain of lysine to an unprotected amine group of another lysine residue (22). Therefore, fresh 16 was used for the synthesis of the PNA backbone 17. Subsequently, thymine-1-yl acetic acid (21 ) was condensed to the PNA backbone to yield 18. The Dde protection group was removed by
2% hydrazine in DMF to give 19. Allyl alcohol was added the deprotection solution to prevent reduction of the allyl group by hydrazine (23).
Scheme 4
a) galactose b) mannose c) fucose
A) Allyl trimethylsilane B) OsO4, KIO4, dioxane-water (8:2 v/v), 12h, rt, (80%).
C) 5% aqueous NaH2PO4, 1 M aqueous KMnO4, t-BuOH, 15 min, rt, (85%).
The sugar derivatives M6 were prepared by standard literature methods (Scheme 4). Peracetylated galactose, mannose and fucose were converted to the corre- sponding α-C-allyl-derivatives M4 (24) and only α-epimers were isolated. M4 were then oxidated to (α-D-)-acetaldehydes M5 (25) using potassium periodate in the presence of OsO as catalyst. Further oxidation of the galactose and mannose derivatives by KMnO4 gave the corresponding acids M6 (26). Attachment of 2 eq of M5a or M5c (galactose, fucose) derivatives to the free amino group of 19, and sub- sequent reduction by NaCNBH3 gave good yields of 20-21. The reaction of M6a-c with 19 in the presence of DCC and DhbtOH gave 22-24. These products were converted to the Fmoc protected derivatives in two steps. They were first treated with 5% TES in TFA in order to remove the Boc group. The ethereal solution of the intermediates were neutralised with excess solid sodium carbonate, and the free amine re-protected with Fmoc using Fmoc-O-succinimide to obtain 25-27. Removal of the allyl protection group from 20-22 and 25-27 yielded 3s-7s.
Preparation of N-glycosylated PNAs
Scheme 5 fmoc-HN COOH fmoc-HN COO-Allyl x COO-fBu — - (28) I cOO-fBu
H fmoc N-~^_- NTCOO-AHyl H2NγCOO-Allyl
H (,3A0). COO-fBu fm "oc-amino- , C-ΛOO.-f »oBu acetaldehyde
(29)
Thymine Thymine o - O^ fmoc N-^NγCOO-Allyl fmoc N-^N ^COO-Allyl
H H
COO-tBU
(31) 02)
M7a-e
yl
Λ/-Fmoc-L-asp(OtBu)-OH was allylated to give 28. The fmoc group was removed and the free amine 29 reacted with fmoc-aminoacetaldehyde (27) to give 30. Fmoc- aminoacetaldehyde was prepared by oxidation of fmoc-amino-2,3-propandiol (28) with potassium-m-periodate in dioxane-water (8:2 v/v). Thymine-1-ylacetic acid was attached to the backbone 30 to give 31. tButyl group on the side chain carboxylic acid was removed with 5% TES in TFA to give 32 in a moderate yield. All glyco- sylamines (scheme 6) were prepared by standard literature methods (29) via glyco- syl-azides (30) and were obtained as β-epimers.
Scheme 6
M7a-e
M7a) R=Br, R'=OAc, galactose M7b) R=Br, R'=OAc, mannose M7c) R=Br, R'=OAc, fucose M7d) R=CI, R'=NHAc, glucosamine M7e) R=CI, R'=NHAc, galactosamine
Attachment of these sugar-amine compounds to the free carboxyl group of 32 was accomplished by the reaction of 2 eq of amine in presence of DCC and DhbtOH to give 33-37. Final removal of the allyl groups was performed in a high yield whereby 9s-13s were obtained.
Preparation of C-galactosyla ted PNA
Scheme 7
(38) (39) (40)
M5a
(43) i <42> (41)
(44) (14s)
The C-galactosylated PNA monomer was prepared from phosphonate 38 (Scheme 7). Alkaline hydrolysis of 38 in methanol afforded carboxylic acid 39, which was subsequently converted to the t-Butyl ester 40 in 90% yield by treatment with EEDQ in t- butanol and chloroforn for 24 h. Condensation of galactosyl-aldehyde derivative M5 with phosphorylglycine t-butyl ester 40 in the presence of N,N,N
',N
'- tetramethyl- guanidine as base gave 41 a mixture of E/Z isomers (5:95). As already pointed out by U. Schmidt and co-workers (31 ), the use of a strong base such as DBU or TMG favours the formation of the Z form. The E/Z ratio of 41 was measured to approximately 1 :20 by proton NMR spectroscopy. Hydrogenation of 41 reduced the double bond and removed Cbz simultaneously, whereby intermediate 42 was obtained. This intermediate was subsequently treated with Fmoc-aminoacetaldehyde yielding 43 as a separable mixture of stereoisomers (25:75 according to TLC). The major diasteromer was readily isolated by flash chromatography. Attachment of thymine-1- yl acetic acid to the modified PNA backbone afforded 44. Finally, the t-butyl group was removed by TFA scavenger and 14s was obtained in a moderate yield.
Solid Phase Synthesis
Procedure a) O-Glycosylated PNA
A well-characterised decamer PNA was applied as an antibacterial agent (32). The thymine-based PNA monomers of the decamer were replaced with O-galactosylated PNA monomers (1s-2s). The high lability of O-glycosidic bonds necessitated the use of a mild method for solid phase synthesis of O-glycosylated PNA oligomers. The Fmoc strategy was applied, since it has been adapted to O-glycosyl peptide solid phase synthesis (33). The stability of the o-galactosylated PNA to acidic cleavage conditions was tested on several resins. The Fmoc-Sieber-TG resin was found suitable due to its susceptibility to mild cleavage conditions (2% TFA in DCM). The β- elimination of the sugar moiety during synthesis and deprotection steps was also investigated. The HATU-collidine was found the most suitable coupling reagent. HATU was neutralised with collidine prior to acid pre-activation. The deprotection was carried out by anhydrous 50% morpholine in DMF at the minimum time required for de-protection (10+5 min). Coupling completion was investigated by Kaiser test and capping was avoided. As a cleavage mixture, 5% water, 30% TFA in DCM was
sufficient to cleave the BHOC groups. Finally, a methanolic hydrazine solution (pH 9.5) was applied for the post-cleavage de-acetylation of the sugar-residue.
Procedure b) Lysine-based -C, N, C-glycosylated PNA Initially, the Boc-strategy for the solid phase synthesis of PNA oligomers containing lysine based c-glycosylated-PNA monomers was applied. In the case of oligomers containing one residue of s5, following cleavage with TFMSA, a side product consisting of oligomer minus one acetyl-group was detected by MALDI, and HPLC. Although the final deacetylation of both the product and the side product gave the tar- get oligomer, it was decided to switch to the fmoc strategy, in order to avoid the use of strong acidic cleavage condition in boc strategy. For this purpose 22-24 was converted to the corresponding fmoc protected derivatives 5s-8s. C-and N-glycosylated PNA monomers 9s-14s were also prepared as fmoc protected derivatives. C- and N-glycosylated PNA monomers was also prepared as fmoc protected derivatives. PNA oligomers were synthesized on Fmoc-PAL-PEG resin. HATU-DIPEA was used as the coupling reagents and 20% piperidine in DMF at minimum required time for deprotections (3+2 min). Coupling yields were detected by Kaiser test and capping steps were avoided. A cleavage reagent of 5% TES in TFA was used and acetyl groups were removed with methanolic hydrazine (pH 10) solution following cleav- age.
Several oligomers were synthesised and melting point temperature (Tm) values were determined by standard methods. All of glycosylated PNAs showed reasonable binding affinity.
c) Glycosylated PNAs having cysteine at the N-terminal
In order to develop the chemically conjugates of glycosylated PNA with biologically interesting compounds such as peptides or labelling compounds, it was necessary to optimise a method for the solid phase synthesis of the conjugate. For this pur- pose, cysteine was introduced at the N-terminal end of the glycosylated PNA as a S- tri tyl-protected derivative. Deprotection of sugar hydroxyl groups were performed on the resin prior to cleavage.
Eight decamers were prepared with incorporated glycosylated PNA monomers. The monomers were introduced in two or three residues shown as T* in Table 1.
Table 1
Cys-C-T***-C-A-T**-A-C-T*-C-T-NH2
In-vivo imaging of glycosylated PNAs
Positron Emission Tomography (PET), a high-resolution sensitive and non-invasive imaging technique for the labelling of oligonucleotides (34) containing a single phos- phorotioate monoester with an electrophilic moiety such as 2-bromo-N-substitu ted acetamides was applied. N-(4-halobenzyl)-2-bromoacetamide was designed as a radiochemically feasible reagent, the benzyl function offering the opportunity to act as the carrier of a radioactive halogen such as fluorine-18, the most widely used positron emitter (T1 2 = 109,8 min.). Cysteine has a high and selective reactivity towards N-(4-fluorobenzyl)-2-bromoacetate, due to its nucleophilic thiol function. Based hereupon, eight glycosylated PNA decamers (shown in table 1 ) were pre- pared.
a) Synthesis of non-radioactive references
PNA monomers were conjugated with N-(4-fluorobenzyl)-2-bromoacetamide in a mixture of acetonitrile and phosphate buffer (0,1 M aq., pH 8.75) for 20 minutes at 60°C (scheme 8). The conjugated PNAs were purified by semi-preparative reverse phase HPLC and characterised by mass spectroscopy analysis (MALDI-TOF).
b) Radiochemistry
N-(4-[18F]fluorobenzyl)-2-bromoacetamide was synthesised in three steps using a robot. Typically, 60-90 mCi of pure N-(4-[18F]fluorobenzyl)-2-bromoacetamide was obtained in 85-95 min. starting from a 550-650 mCi of a cyclotron [18F]F- production batch (scheme 9).
Scheme 9
100 W, 1 mm _5'C, 2 mm
The HPLC-collected fraction containing N-(4-[18F]fluorobenzyl)-2-bromoacetamide was concentrated to dryness at 80°C under a nitrogen stream, before diluted with 0.4 ml acetonitrile. A solution of 1.3 mg PNA in 0.5 ml phosphate buffer (0.1 M, pH 8.75) and 0.1 ml of acetonitrile was rapidly added. The reactor was placed in a heat- ing block and heated without stirring under a slight flow of nitrogen at 60°C for 20 minutes. Before total dryness, 1 ml distilled water was added to the reaction mixture, and the suspension was subjected to the HPLC purification for separation of labelled PNA[18F], unreacted PNA and unreacted N-(4-[18F]fluorobenzyl)-2-bromoacetamide. Labelled PNAs co-eluted with authentic synthesized unlabelled reference com- pounds. The HPLC fraction containing the labelled PNA was concentrated and formulated by transferring into a volume of 1-2 ml of serum.
c) In-vivo PET imaging
Two male and two female Spargue-Dawley rats (200 g) were injected with 40 mCi labelled PNA in the tail vein placed in a Siemens ECAT EXACT HR+ camera under anesthesia and whole body images acquired in 3D mode for 2 hours.
d) Ex-vivo study of bio-distribution
Following imaging, the animals were sacrificed and the kidneys, liver, spleen, heart, lungs, brain, muscles, blood and adrenals immediately collected. Aliquots of these organs were weighed and radioactivity was counted on a radiocounter. Radioactivity was expressed as percentage of injected dose per gram of organ (%ID/g) and reported as the mean ± standard deviation.
Example of PNA oligomers containing glycosylated monomers used for pharmacokinetic analyses by 18F-isotope PET scanning is shown in Figure 1 :
Organ distribution of PNA oligomers in rats is shown in Figure 2. The N-acetyl- galacotsamine PNA shown in Figure 2 is preferentially targeting the liver. Similar results were obtained in baboons.
Pharmacokinetics of PNA oligomers of the type shown in figure 1 analysed by PET scanning. The N-acetyl-galactosamine PNA shown in Fig. 3 is preferentially accumulated in the liver. Similar results were obtained baboons.
e) Study of metabolism
Plasma supematants were centrifuged in microfilter/10000 fixed Eppendorf tubes and subjected to analytical RP-HPLC. Urine samples were subjected to the HPLC directly. In some cases the samples were co-injected with unlabelled PNA.
Pharmaceutical Compositions
The PNA drugs of the present invention are used in the manufacture of medicaments for the treatment or prevention of bacterial, viral, protozoal, and fungal infections, cancer, metabolic diseases, cardiovascular diseases, autoimmune and immu- nological disorders, or for disinfecting non-living objects, such as surgery tools, hospital inventory, dental tools, slaughterhouse inventory and tool, dairy inventory and tools, barbers and beauticians tools and the like.
Within the present invention, the compounds of the invention may be prepared in the form of pharmaceutically acceptable salts, especially acid-addition salts, including salts of organic acid, fumaric acid, acetic acid, propionic acid, glycolic acid, lactic acid, pyruvic acid, oxalic acid, succinic acid, malic acid, tartaric acid, citric acid, ben- zoic acid, salicylic acid, and the like. Suitable inorganic acid-addition salts include salts of hydrochloric, hydrobromic, sulphuric- and phosphoric acids and the like. Fur- ther examples of pharmaceutically acceptable inorganic or organic acid addition salts include the pharmaceutically acceptable salts listed in Journal of Pharmaceutical Science, Berge et al 1977 (19), which are known to the skilled artisan. Also intended as pharmaceutically acceptable acid-addition salts are the hydrates, which the present compounds are able to form. The acid-addition salts may be obtained as the direct drugs of compound synthesis. In the alternative, the free base may be dissolved in a suitable solvent containing the appropriate acid, and the salt isolated by evaporating the solvent or otherwise separating the salt and solvent.
The compounds of this invention may form solvates with standard low molecular weight solvents using methods known to the skilled artisan.
In one aspect, the invention concerns the manufacture of a composition for treating or preventing bacterial, viral, protozoal, and fungal infections, cancer, metabolic diseases, cardiovascular diseases, autoimmune and immunological disorders, or disinfecting non-living objects, such as surgery tools, hospital inventory, dental tools, slaughterhouse inventory and tool, dairy inventory and tools, barbers and beauti- cians tools and the like.
Typical compositions include a compound of the invention or a pharmaceutically acceptable acid-addition salt thereof, associated with a pharmaceutically acceptable excipient which may be a carrier or a diluent or be diluted by a carrier, or enclosed within a carrier, which can be in the form of a capsule, sachet, paper or other container. In making the compositions, conventional techniques for the preparation of pharmaceutical compositions may be used. For example, the active compound will usually be mixed with a carrier, or diluted by a carrier, or enclosed within a carrier, which may be in the form of an ampoule, capsule, sachet, paper, or other container. When the carrier serves as a diluent, it may be solid, semi-solid, or liquid material, which acts as a vehicle, excipient, or medium for the active compound. The active compound can be adsorbed on a granular solid container for example in a sachet. Some examples of suitable carriers are water, salt solutions, alcohol's, polyethylene glycol's, polyhydroxyethoxyla ted castor oil, peanut oil, olive oil, glycine, gelatin, lac- tose, terra alba, sucrose, glucose, cyclodextrine, amylose, magnesium stearate, talc, gelatin, agar, pectin, acacia, stearic acid or lower alkyl ethers of cellulose, silicic acid, fatty acids, fatty acid amines, fatty acid monoglycerides and diglycerides, pen- taerythritol fatty acid esters, polyoxyethylene, hydroxymethylcellulose and polyvi- nylpyrrolidone. Similarly, the carrier or diluent may include any sustained release material known in the art, such as glyceryl monostearate or glyceryl distearate, alone or mixed with a wax. The formulations may also include wetting agents, emulsifying and suspending agents, preserving agents, sweetening agents, thickeners or flavoring agents. The formulations of the invention may be formulated so as to pro-
vide quick, sustained, or delayed release of the active ingredient after administration to the patient by employing procedures well known in the art.
The pharmaceutical compositions can be sterilized and mixed, if desired, with auxil- iary agents, emulsifiers, salt for influencing osmotic pressure, buffers and/or coloring substances and the like, which do not dele teriously react with the active compounds.
For therapeutic or prophylactic treatment, the PNA drug of the invention can be formulated in a pharmaceutical composition, which may include one or more active in- gredients such as antimicrobial agents, anti-inflammatory agents, anaesthetics, and the like in addition to PNA.
The pharmaceutical composition may be administered in a number of ways depending on whether local or systemic treatment is desired, and on the area to be treated. Administration may be done topically (including ophthalmically, vaginally, rectally, in tranasally), orally, by inhalation, or parenterally, for example by intravenous drip or subcutaneous, intraperitoneal or intramuscular injection.
Formulations for topical administration may include ointments, lotions, creams, gels, drops, suppositories, sprays, liquids and powders. Conventional pharmaceutical carriers, aqueous, powder or oily bases, thickeners and the like may be necessary or desirable. Coated condoms may also be useful.
Compositions for oral administration include powders or granules, suspensions or solutions in water or non-aqueous media, capsules, sachets, or tablets. Thickeners, flavourings, diluents, emulsifiers, dispersing aids or binders may be desirable.
If a solid carrier is used for oral administration, the preparation may be tabletted placed in a hard gelatin capsule in powder or pellet form or it can be in the form of a troche or lozenge. If a liquid carrier is used, the preparation may be in the form of a suspension or solution in water or a non-aqueous media, a syrup, emulsion or soft gelatin capsules. Thickeners, flavorings, diluents, emulsifiers, dispersing aids or binders may be added.
Formulations for parenteral administration may include sterile aqueous solutions, which may also contain buffers, diluents and other suitable additives.
For nasal administration, the preparation may contain a compound of the invention dissolved or suspended in a liquid carrier, in particular an aqueous carrier, for aerosol application. The carrier may contain additives such as solubilising agents, e.g. propylene glycol, surfactants, absorption enhancers such as lecithin (phosphatidyl- choline) or cyclodextrine, or preservatives such as parabenes. For parenteral application, particularly suitable are injectable solutions or suspensions, preferably aqueous solutions with the active compound dissolved in polyhy- droxylated castor oil.
Tablets, dragees, or capsules having talc and/or a carbohydrate carrier or binder or the like are particularly suitable for oral application. Preferable carriers for tablets, dragees, or capsules include lactose, cornstarch, and/or potato starch. A syrup or elixir can be used in cases where a sweetened vehicle can be employed.
In yet another aspect, the invention concerns the treatment or prevention of bacte- rial, viral, protozoal, and fungal infections, cancer, metabolic diseases, cardiovascular diseases, autoimmune and immunological disorders, or treatment of non-living objects.
Dosing is dependent on severity and responsiveness of the condition to be treated, but will normally be one or more doses per day, with course of treatment lasting from several days to several months or until a cure is effected or a diminution of disease state is achieved. Persons of ordinary skill can easily determine optimum dosages, dosing methodologies and repetition rates.
Usually, dosage forms suitable for oral, nasal, pulmonal or transdermal administration comprise from about 0.01 mg to about 500 mg, preferably from about 0.01 mg to about 100 mg of the compounds of the invention admixed with a pharmaceutically acceptable carrier or diluent.
Treatments of this type can be practiced on a variety of organisms ranging from unicellular prokaryotic and eukaryotic organisms to multicellular eukaryotic organisms. Any organism that utilises DNA-RNA transcription or RNA-protein translation as a fundamental part of its hereditary, metabolic or cellular control is susceptible to therapeutic and/or prophylactic treatment in accordance with the invention. Seemingly diverse organisms such as bacteria, yeast, protozoa, algae, all plants and all higher animal forms, including warm-blooded animals, can be treated. Further, since each cell of multicellular eukaryotes can be treated since they include both DNA- RNA transcription and RNA-protein translation as integral parts of their cellular activity. Furthermore, many of the organelles, (e.g. mitochondria and chloroplasts) of eukaryotic cells also include transcription and translation mechanisms. Thus, single cells, cellular populations or organelles can also be included within the definition of organisms that can be treated with therapeutic or diagnostic PNA drug. As used herein, therapeutics is meant to include the eradication of a disease state, by killing an organism or by control of erratic or harmful cellular growth or expression.
EXPERIMENTAL
The following examples are merely illustrative of the present invention and should not be considered limiting of the scope of the invention in any way.
Abbreviations
The following abbreviations related to reagents are used in the experimental part:
Reagents and solvents were obtained from commercial sources and used without further purification, unless indicated. NMR spectra were recorded in CDCI3 and CD3OH on Varian 400, 300 MHz and Brucher 250 MHz unity spectrometers, FAB mass spectra on a JEOL HX 110/110 mass spectrometer, MALDI-TOF mass spectra on a Cratos Compact Maldi II spectrometer. The Microanalysis Department of HC0 Institute, University of Copenhagen, performed elementary analyses. Flash chromatography was carried out using Silica Gel 60 (Merck particle size 0.040- 0.063mm). The oligomers were analysed on a Delta Pak Cι8 column (5μM, 3.9x150mm) and were purified on a Delta Pak C18 column (15μM, 19x300mm). A gradient composed of A (0.1 %TFA in water) and B (0.1% TFA in 10% H2O/90% acetonitrile) was used for analytical and preparative HPLC. Analytical: Time 0, 0% B. Time 35 min, 50% B (Flow, 1ml/min). Preparative: Time 0, 15% B. Time 45 min, 40% B (Flow, 8ml/min).
EXAMPLE 1
Procedure (a): allylation of N-protected amino acids
5 mmol of Λ/-protected aminoacid was dissolved in a mixture of 10 ml acetonitrile and 12 ml allylbromide. (2.1 eq) N-N-diisopropylethylamine was added and the reac- tion mixture was stirred for 4 h at 40°C. Subsequently 200 ml ethylacetate was added and the solution was extracted with half saturated KHSO4 (2 x 50 ml) and half saturated NaHCO3 (2 x 50 ml) and 100 ml brine respectively. Organic phase was dried over MgSO4 and volatile were removed under vacuum. The remaining oil was used for the next step (Yield = 87%-95%).
EXAMPLE 2
Procedure (b): preparation of N-boc/fmoc protected PNA backbone
12 mmol amine and 10 mmol boc/fmoc-aminoacetaldehyde were stirred in 40 ml methanol for 10 min. 0.8 ml (13 mmol) Acetic acid and 0.6 g (10 mmol) natrium cyanoborohydrate was added sequentially. The reaction mixture was stirred for further 1 h at room temperature. Vola tiles was removed under vacuum and the remaining solid dissolved in 100 ml ethyacetate and extracted with NaHCO3 saturated solution and brine. Organic phase was dried over magnesium sulphate and evaporated to dryness under vacuum. The residue was purified on silica gel column eluting hex- ane-ethylacetate 1 :1. (Yield = 50% - 80%).
EXAMPLE 3
Procedure (c): coupling of thymine-1-yl-acetic acid to the PNA backbone 17 mmol Thymine-1-ylacetic acid and 18 mmol DhbtOH were dissolved in 50 ml dry DMF. 20 mmol DCC was added and the solution stirred for 20 minutes. 8.5 mmol backbone was added in 20 ml dry DMF to the reaction mixture and stirred for further 6 h at room temperature. Volatiles were removed under vacuum and the remaining dissolved in 200 ml ethylacetate. Insoluble DCU was filtered off and the filtrate ex- tracted with NaHCO3 saturated solution (2 x 100 ml) and brine (100 ml). After drying over magnesium sulphate, the organic phase was evaporated to dryness. The residue was purified on a silica gel column eluting the mixture of ethylacetate-methanol 10:0 to 10:1 (Yield = 63%-87%).
EXAMPLE 4
Procedure (d): boc de-protection and subsequent fmoc protection
3 mmol boc-protected compound was added to 20 ml solution of 5% triethylsilane in trifluoroacetic acid at 0°C and stirred until TLC did not show any starting material left. 50 ml Toluene was added and volatile removed under vacuum. Further 3 x 100 ml DCM was added and evaporated in order to removal of TFA. The remaining was dissolved in 50 ml diethylether and 5 g natrium carbonate was added as well powdered solid. The suspension was stirred for 30 min and then evaporated. The solid residue was suspended in 50 ml Acetonitrile and 3.2 mmol fmoc-O-Su was added
and stirred for further 2h. The solution was evaporated under vacuum and the crude was purified on silica gel column eluting ethylacetate-methanol 10:0 to 10:1 (Yield = 88%-94%).
EXAMPLE 5
Procedure (e): N-glycosylation
2 mmol 32, 2.2 mmol DhbtOH, and 3 mmol DCC were mixed in 10 ml DMF and stirred for 30 min under Nitrogen. A solution 2 mmol Sugar-amine in 10 ml DMF was added and the reaction mixture was stirred overnight. Volatile was removed under vacuum and the residue dissolved in 200 ml ethylacetate. Insoluble DCU was filtered off and the filtrate extracted with NaHCO3 saturated solution (2 x 100ml) and brine (100 ml). After drying over magnesium sulphate the organic phase was evaporated to dryness. The residue subjected to silica gel column eluting the mixture of ethylacetate-methanol 10:0 to 10:1 (Yields = 60%-77%).
EXAMPLE 6
Procedure (f): removing of allyl group
0.1 mmol of ester was dissolved in 2 ml THF. 23 mg (0.02 mmol, 0.2 eq)
Tetrakis(triphenylphosphine)Pd(0) was added. 10 eq N-ethyl-aniline was added drop wise to the reaction mixture and stirred at room temperature. The reaction was checked by TLC (ethylacetate-methanole 10:1 ). After complete conversion of starting material, the reaction mixture was poured dropwise in a 20 ml vigorously stirring solution of diethylether-n-hexane 1 :1. White precipitate was collected by filtration and washed with n-hexane (Yield = 80%-95%).
EXAMPLE 7
Threonine(t-Bu)-Allyl (4)
To 4.4 g (10 mmol) 2 [prepared by procedure (a)] was added 50 ml of a solution of
20% piperidine in DCM. After 30 min, 100 ml toluene was added and volatile were removed under vacuum. The residue was purified with a short silica gel column eluting ethylacetate-methanole 10:0 to 10:1. 1.6 g (7.5 mmol) titled compound was obtained as colourless oil (Yield = 75%). [α] obtained as colourless oil in 83% yield.
[α]D 22 = -1.53 (c = 1 , methanol); MS (FAB) m/z 216 (M+H); 1H-NMR, (CDCI3): δ 5.94 (9 line m, 1H, -CH=CH2), 5.3 (dd, 1H, Jtrans = 17.03, 1.37 Hz), 5.2 (dd, 1H, Jcis= 10.5, 1.1 Hz), 4.64-4.46 (dddt, 2H, J = 33.08, 10.7, 5.8, 1.1 , O-CH2-CH=), 4 (m, 1 H, βCH), 3.26 (d, 1 H, J=3.3 Hz, αCH), 1.65 (s, 2H, -NH2), 1.2 (d, 3H, J=6.3 Hz, βC-CH3), 1.09 (s, 9H, boc); 13C-NMR (CDCI3): δ 174.51 (COO-Allyl), 132.09 (-CH=CH2), 118.87 (- CH=CH2), 73.77 [-O-C(Me)3], 68.63, 65.67, 60.84, 28.68 [-COO-C(CH3)3], 20.94 (βC-CH3).
C.H.N analyse for CnH21NO3. Y2 H2O: calc. C 60.11 , H 9.86, N 6.37; found C 59.87, H 9.95, N 6.34.
EXAMPLE 8 Λ/-(2-Boc-aminoethyl)-Serine(t-Bu)-Allyl (5)
Preparation: Procedure (b)
[α]D 22= -10.26 (c=1 , methanol); MS (FAB) m/z 345(M+H); 1H-NMR, (CDCI3): δ 5.96- 5.89 (8 line m, 1 H, -CH=CH2), 5.26 (dd, 1 H, Jtra_s=17.2, 1.46 Hz), 5.17 (dd, 1 H, Jcis=10.4, 1.28 Hz), 5.05 (br.s, boc-HN-), 4.57-4.55 (m, 2H, O-CH2-CH=), 3.52 (ABq, 2H, J=4.95Hz, βCH), 3.36 (t, J=4.76 Hz, 1 H, αCH), 3.2-3.1 (m, 2H), 2.8 (m, 1H), 2.6 (m, 1 H), 1.37 (s, 9H, t-butyl), 1.08 (s, 9H, boc); 13C-NMR (CDCI3): δ 172.50 (COO- Allyl), 155.9 (-NH-COO), 131.74 (-CH=CH2), 118.42 (-CH=CH2), 118.27, 73.13 [-O- C(Me)3], 65.263 (-CH2-CH=), 62.66 (β carbon), 61.22 (α carbon), 47.18, 40.03, 28.25 [-COO-C(CH3)3], 27.17 [-O-C(CH3)3].
C.H.N analyse for C17H32N2O5. V_ H2O: calc. C 57.77, H 9.41 , N 7.93; found C 58.11 , H 9.20, N 8.14.
EXAMPLE 9 ιV-(2-Boc-aminoethyl)-Threonine(t-Bu)-Allyl (6)
Preparation: Procedure (b)
(Yield = 67%) [α]D 22= -3.39 (c=1 , methanol); MS (FAB) m/z 359 (M+H); 1H-NMR, (CDCI3): δ 5.90-5.83 (8 line m, 1H, -CH=CH2), 5.26 (dt, 1 H, Jtra_s=17, 1.4 Hz), 5.18 (dt, 1H, Jcis=10, 1.1 Hz), 5.05 (br.s, boc-HN-), 4.60-4.47 (dddt, 2H, J=33.3, 13.2, 5.8, 1.3 HZ, O-CH2-CH=), 3.9 (q, 1 H, PCH), 3.14-3.08 (dm, 2H), 3.05 (d, 1 H, J=3.5, αCH), 2.8 (m, 1 H), 2.7 (m, 1 H), 1.37 (s, 9H, t-butyl), 1.18 (d, 3H, J=6.2 Hz, βC-CH3), 1.06 (s, 9H, boc); 13C-NMR (CDCI3): δ 172.91 (COO-Allyl), 155.88 (-NH-COO), 131.65 (-
CH=CH2), 118.54 (-CH=CH2), 78.81 [-COO-C(Me)3], 73.66 [-O-C(Me)3], 68.14 (β carbon), 66.37 (α carbon), 65.270 (-CH2-CH=), 47.51 , 39.88, 28.23 [-COO-C(CH3)3], 28.19 [-O-C(CH3)3], 20.49 (PC-CH3).
C.H.N analyse for C18H34N2O5: calc. C 60.31 , H 9.56, N 7.81 ; found C 60.16, H 9.98, N 7.75.
EXAMPLE 10 Λ/-(2-Boc-aminoethyl)-Λ/-(Thymine-1-ylacetyl)Serine(t-Bu)-Allyl (7)
Preparation: Procedure (c) Mp = 70-72; MS (FAB) m/z 216 (M+H); 1 H-NMR, (CDCI3): (major rotamer) δ 9.08 (s, 1 H, Thymine aromatic), 6.83 (s, 1 H, Thymine-NH-), 5.87-5.77 (8 line m, 1 H, - CH=CH2), 5.6 (br.s, 1 H, boc-HN-), 5.25 (dd, 1 H, Jtra_s=17.2, 1.28 Hz), 5.17 (dd, 1 H, Jcιs=10.2, 0.91 Hz), 4.6-4.4 (dm, 2H, -O-CH2-CH=), 4.55 (s, 2H, -CO-CH2-Thymine), 4.26 (br.d, 1H, J=5.3), 3.93 (t, 1 H), 3.77 (dd, 1 H, J=10, 3.1 Hz), 3.6-3.4 (dm, 2H), 3.35 (m, 2H), 1.83 (s, 3H, Thymine-CH3), 1.37 (s, 9H, t-butyl), 1.1 (s, 9H, boc); 13C- NMR (CDCI3): δ 168.93, 167.15, 164.07, 155.85 (-NH-COO), 150.72, 140.72, 131.36 (-CH=CH2), 118.84 (-CH=CH2), 111.59, 110.43, 79.62 [-COO-C(Me)3], 73.84 [-O- C(Me)3], 66.06 (-CH2-CH=), 60.79, 60.24, 59,50, 38.84, 33.66, [28.30, 28.10, -COO- C(CH3)3], 27.15 [-O-C(CH3)3], 12.25 (Thymine-CH3).
EXAMPLE 11
Λ/-(2-Boc-aminoethyl)-Λ/-(Thymine-1-ylacetyl)Threonine(t-Bu)-Allyl (8) Preparation: Procedure (c) Mp = 71-73; Yield = 76%; MS (FAB) m/z 525(M+H); 1H-NMR, (CDCI3): (major ro- tamer) δ 8.59 (s, 1 H, Thymine aromatic), 6.9 (s, 1 H, Thymine-NH-), 5.95-5.85 (8 line m, 1 H, -CH=CH2), 5.5 (br.s, 1 H, boc-HN-), 5.3 (dd, 1 H, Jtra_s=17.1 , 1.4 Hz), 5.25 (d, 1 H, Jcιs=10.2 Hz), 4.66 (s, 2H, -CO-CH2-Thymine), 4.52-4.41 (overlapping m, 3H, - O-CH2-CH=, αCH), 4.1 (q, 1 H), 3.8 (dt, 1 H, J=15.3, 6 Hz), 3.6 (m, 1 H), 3.5-3.4 (m, 2H), 1.89 (s, 3H, Thymine-CH3), 1.44 (s, 9H, t-butyl), 1.28 (d, 3H, J=6Hz, -βC-CH3). 1.1 (s, 9H, boc); 13C-NMR (CDCI3): (major rotamer) δ 169.12, 167.95, 163.78, 155.78 (-NH-COO), 150.59, 140.73, 131.20 (-CH=CH2), 119.41 (-CH=CH2), 110.43, 79.61 [-COO-C(Me)3], 74.55 [-O-C(Me)3], 66.14 (-CH2-CH=), 64.39, 39.16, 33.79,
[28.69, 28.60, 28.40, -COO-C(CH3)3], [25.53, 24.87, -O-C(CH3)3], 21.71 (βC-CH3), 12.37 (Thymine-CH3).
EXAMPLE 12 Λ/-Fmoc-aminoethyl-Λ-(Thymine-1-ylacetyl)Serine(OH)-Allyl (11)
Preparation: Procedure (d)
Mp = 101-103; [α]D 22= -5.36 (c=0.25, methanol); MS (FAB) m/z 577(M+H); 1H-NMR, (CDCI3): δ 9.63 (s, 1 H,Thymine aromatic), 7.56-7.07 (8H, Florenyl aromatic protons), 6.59 (s, 1 H, Thymine-NH-), 5.9 (s, 1 H, fmoc-HN-), 5.8-5.6 (m, 1H, -CH=CH2), 5.12 (d, 1 H, J,rans=1 .2 Hz), 5.05 (dd, 1 H, Jcis=10.4, 0.91 Hz), 4.6-4.4 (m, 3H, O-CH2-CH=, αCH), 4.24 (s, 2H, -CO-CH2-Thymine), 4-3.8 (m, 4H), 3.4-3.2 (m, 4H), 2.73 (br.s, 1 H), 1.7 (s, 3H, Thymine-CH3); 13C-NMR (CDCI3): δ 168.50, 168.06, 167.94, 167.07, 164.05, 162.58, 156.59, 151.33, 143.58 and 143.39 (florenyl), 140.98, 131.14 (- CH=CH2), [(130.69, 127.54, 126.88, 124.82, 124.36, 119.75 (florenyl)], 118.95 (- CH=CH2), 110.90, 66.75, 66.17, 63.43 (-CH2-CH=), 59.47, 49.24, 48.81 , 47.10, 39.45, 31.54, 12.30 (Thymine-CH3).
C.H.N analyse for C30H32N4O8. V_ H2O: calc. C 61.35, H 5.68, N 9.57; found. C 61.39, H 5.53, N 9.36.
EXAMPLE 13
Λ/-Fmoc-Λ/-(Thymine-1-ylacetyl)Threonine(OH)-Allyl (12)
Preparation: Procedure (d)
Mp = 99-101 ; Yield = 91 %; [α]D 22= -0.93 (c=0.75, methanol); MS (FAB) m/z 591
(M+H); 1H-NMR, (CDCI3): δ 8.9 (s, 1H,Thymine aromatic), 7.6-7.1 (8H, Florenyl aromatic protons), 6.6 (s, 1 H, Thymine-NH-), 5.8-5.6 (overlapping m, 2H, -CH=CH2, fmoc-HN-) 5.12 (dt, 1 H, Jtrans=16.1 , 8.8 Hz), 5.07 (dd, 1 H, Jcis = 10.3, 1 Hz), 4.5 (overlapping m, 3H), 4.42-4.26 (m, 2H, O-CH2-CH=), 4.2 (t, 1 H, J=7.4 Hz) 4.03 (d, 2H, J=6 Hz), 3.65 (br.d, 1 H, J=5.3 Hz), 3.53-3.47 (m, 1 H), 3.4-3.24 (m, 3H), 2.8 (br.s, 1 H), 1.68 (s, 3H, Thymine-CH3), 1.14 (d, 3H, J=5.86 Hz, -βC-CH3); 13C-NMR (CDCI3): δ 168.67, 167.85, 163.99, 156.60, 151.10, [143.67, 143.50, 141.15, 140.98 (florenyl)], 131.22 (-CH=CH2), 127.64, 126.96, 124.88, 124.36, 119.87, 119.25 (- CH=CH2), 110.75, 67.50, 66.61 , 66.19, 65.16, 49.55, 48.86, 47.06, 39.41 , 21.30 (βC- CH3), 12.30 (Thymine-CH3).
C.H.N analyse for C31H34N4O8. 1/2 H2O: calc. C 62.09, H 5.88, N 9.34; found C 62.10, H 5.86, N 9.22.
EXAMPLE 14 Λ -Fmoc-Λ-(Thymine-1-ylacetyl)Serine(2,3,4,5-tetra-0-acetyl-α-D-Galactose-1- yl)-Allyl (13)
Preparation: 0.7g (1.2 mmol) 11 and 0.75 g (1.5 mmol) galactose trichloroacetamide were dissolved in 5 ml ethylacetate. 0.5 g molecular sieve was suspended and 1.2 ml (9.5 mmol) borontriflate in 1 ml ethylacetate was added at 0°C. Ice bath was re- moved and the reaction mixture was stirred under nitrogen over night. 50 ml ethylacetate was added and the reaction mixture was filtered over celite. The filtrate was extracted with an ice cold half saturated NaHCO3 aqueous solution (2 x 25 ml), and brine (50 ml), dried over magnesium sulphate and evaporated to dryness. The residue was purified on silica gel column eluting hexane-ethylacetate 1 :1. 0.4 g (0.9 mmol) α anomer was obtained as white crystalline.(yield = 37%)
Mp = 115-118; [α]D 22= -24.17 (c=0.5, methanol); MS (FAB) m/z 907(M+H); 1H-NMR, (CDCI3): δ 8.75 (s, 1 H, Thymine aromatic), 7.68-7.21 (m, 8H, Florenyl aromatic protons), 6.8 (s, 1H, Thymine-NH-), 5.9-5.7 (overlapping m, 2H, -CH=CH2> fmoc-HN-), 5.3(d, 1 H, J=2.9 Hz, sugar CH), 5.25 (d.d, 1 H, Jtrans = 17.2, 1.26 Hz), 5.15 (d, 1 H, Hz), 5.05 (t, 1 H, Jι,2=7.7 Hz, J2,3=10.4 Hz, sugar 2CH), 4.95 (dd, 1 H, J=10.4, 3.2 Hz, sugar 3CH), 4.7-4.3 (m, 6H), 4.25, 3.95 (m, 6H), 3.8 (t, 2H, J=6.4, sugar 5CH), 3.6-3.3 (m, 3H), 2.9 (br.s, 1 H), 2.1-1.9 (4s, 12H, sugar acetyl protons), 1.8 (s, 3H, Thymine-CH3); 13C-NMR (CDCI3): δ 169.37, 169.09, 168.96, 168.80, 166.84, 166.61 , 163.09, 155.81 (-NH-COO), 149.67, 142.76, 140.27, 130.37 (- CH=CH2), 126.73, 126.06, 123.91 , 118.98, 118.24 (-CH=CH2), 109.26, 100.03 (sugar C1 ), 70.09, 69.57, 67.75, 66.38, 65.93, 65.54, 65.43, 60.22, 60.09, 47.48, 47.02, 46.29, 38.60, 19.78-19.54 (sugar-CO-CH3 groups), 11.28 (Thymine-CH3). HRMS (M+Na)+, calculated (found) for C44H50N4Oι7Na are 929.3069 (929.3087).
EXAMPLE 15
Λ -Fmoc-Λ/-(Thymine-1-ylacetyl)Threonine(2,3,4,5-tetra-0-acetyl-α-D-Galactose- 1-yl)-Allyl (14)
Preparation: The Procedure is the same as described above for the synthesis of 13
Yield = 77%, mp = 106-108; [α]D 22= -27.38 (c=0.5, methanol); MS (FAB) m/z 921(M+H); 1H-NMR, (CDCI3): δ 1H-NMR, (CDCI3): δ 8.54 (s, 1 H,Thymine aromatic), 7.78-7.76 (8H, Florenyl aromatic protons), 6.95 (1 H, Thymine-NH-), 5.92-5.82 (m, 1 H, -CH=CH2), 5.81 (s, 1 H, fmoc-HN-), 5.38 (d, 1 H, J=2.5 Hz, sugar 4CH), 5.3 (d, 1 H, Jtrans=17.4 Hz), 5.2 (d, 1 H, Jcιs=10.4 Hz), 5.08 (t, 1 H, J1 ι2=7.7 Hz, J2,3=10.4 Hz, sugar CH ), 5.03 (dd, 1H, J=10.4, 3.3 Hz, sugar 3CH), 4.9 (d, 1 H, J=16.3), 4.7-4.5 (overlapping s ddd and s, 4H, J=49.6, 13.2, 5.86 Hz, -O-CH2-CH and CO-CH2- Thymine), 4.44 (overlapping, 2H), 4.35 (d, 2H, J=16.3 Hz), 4.2(t, 2H, J=7 Hz), 4.07 (dd, 1 H, J=22.9, 6.41 Hz, sugar 6CH), 3.88 (dd, 1 H, J5,6=6.23Hz, J5,6 =6.59, sugar 5CH), 3.6 (overlapping s, 2H), 3.43 (br.s, 1 H), 2.18-2 (3s, 12H, sugar acetyl protons), 1.88 (s, 3H, Thymine-CH3), 1.27 (d, 3H, J=5.86, βC-CH3); 13C-NMR (CDCI3): δ 170.11 , 169.89, 167.97, 163.65, 156.64(-NH-COO), 150.35, 143.53, 141.21 , 141.05, 131.31(-CH=CH2), 127.51 , 126.82, 124.78, 119.80, 118.78 (-CH=CH2), 110.07, 98.73 (sugar C1 ), 73.34, 71.53, 70.70, 70.48, 70.01 , 68.80, 66.66, 66.55, 66.17, 65.65, 64.10, 62.79, 60.84, 60.31 , 48.48, 47.19, 39.70, [21.05, 20.83, 20.67, 20.55 (sugar-CO-CH3 groups)], 17.38, 14.20 (βC-CH3), 12.29 (Thymine-CH3). C.H.N analyse for C45H52N4O17. H2O: calc. C 57.56, H 5.80, N 5.97; found C 57.51 , H 5.52, N 5.82.
EXAMPLE 16
Lysine(Dde)-Allyl (16)
Preparation: 5.7g (10 mmol) ester 15 [prepared by Procedure (a)] was added to a solution of 20% piperidine in DCM and stirred for 30 min. 100 ml toluene was added and volatile were removed under vacuum. The residue was purified on a short sili- cagel column eluting ethylacetate-methanole 10:0 to 10:1. 2.5 g (7 mmol) Titled compound was obtained as slightly yellow oil which was used subsequently for the next step (Yield = 71%).
MS (FAB) m/z 351 (M+H); 1 H-NMR, (CDCI3): δ 13.34(s, 1 H, -HN-Dde), 5.9-5.8 (ddt, 1 H, J=5.57, 10.55, 10.43 Hz, -CH=CH2), 5.27(dd, 1 H, Jtrans=17.3, 1.47 Hz), 5.22(dd, 1 H, JCis=10.5, 1.17 Hz), 4.56 (dt, 5.86, 1.17Hz, O-CH2-CH=), 3.4 (dd, 1 H, J=7.5, 5 Hz, αCH), 3.3 (ABq, 2H, JAB=12.3, -CH2-NH-Dde), 2.49 (s, 3H, -HN-C-CH3), 2.3 (s, 4H, 2x-CH2- of Dde), 1.77 (s, 2H, -NH2), 1.74-1.38 (overlapping m, 6H), 0.97 (s, 6H, 2x-CH3 of Dde);
13C-NMR (CDCI3): δ [198.59, 196.83 (-CO- of Dde)], 175.19 (=C(CH3)-NH), 173.14 (COO-Allyl), 131.60 (-CH=CH2), 118.51 (-CH=CH2), 107.56 (=C= of Dde), 65.33 (O- CH2-CH=), 60.10 (-CH2- of Dde), 53.92, 42.96, 33.98, 29.83, 28.59, 28.02, 22.87, 17.65 (CH3 of Dde).
EXAMPLE 17
Λ/-(2-Boc-aminoethyl)- Lysine(Dde)-Allyl (17)
Preparation: Procedure (b), yield= %65
MS (FAB) /z 494 (M+H); 1H-NMR, (CDCI3): δ 13.45 (s, 1 H, -HN-Dde), 5.98-5.85 (10 line m, -CH=CH2), 5.33 (dd, 1 H, Jtrans=17.29, 1.47 Hz), 5.28 (dd, 1 H, Jcis=10.55, 1.17 Hz), 4.56 (dd, 5.86, 1.17Hz, O-CH2-CH=), 3.4 (overlapping m, 3H, αCH, -CH2- NH-Dde), 3.2 (m, 2H), 2.8 (m, 1H), 2.7 (m, 1 H), 2.55 (s, 3H, -HN-C-CH3), 2.36 (s, 4H, 2x-CH2- of Dde), 1.8-1.5 (2 x m, 6H), 1.44 (s, 9H, boc), 1.03 (s, 6H, 2x-CH3 of Dde); 13C-NMR (CDCI3): δ [198.79, 197.21 (-CO- of Dde)], 174.68 (=C(CH3)-NH), 173.68 (COO-Allyl), 156.37 [-NH-COO-C(Me)3], 132.00 (-CH=CH2), 119.27(- CH=CH2), 108.08(=C= of Dde), 79.47 [COO-C(Me)3], 65.83 (O-CH2-CH=), 60.99 (- CH2- of Dde), 53.07, 47.83, 43.40, 40.55, 32.94, 30.35, [28.96, 28.68, 28.52, - COO-C(CH3)3], 23.35, 87, 18.18 (CH3 of Dde). C.H.N analyse for C26H43N3O6. H2O: calc. C 61.03, H 8.86, N 8.21 ; found C 61.12, H 8.78, N 8.13.
EXAMPLE 18 Λ/-(2-Boc-aminoethyl)-Λ/-(Thymine-1-ylacetyl)-Lysine-N'-(Dde) O-Allyl (18)
Preparation: Procedure (c), light yellow crystalline. Yield = 87% Mp = 84-86; MS (FAB) m/z 660 (M+H); 1H-NMR, (CDCI3): δ 13.4 (s, 1 H, -HN-Dde), 9.56 (s, 1 H, aromatic proton of Thymine), 6.99 (S, 1 H, -NH- of Thymine), 5.97-5.84 (m, 1H, -CH=CH2), 5.66 (s, 1 H, boc-NH-), 5.35 (dt,1 H, Jtrans=17.28, 1.46 Hz), 5.25 (dd, 1 H, Jcis=10.55, 1.17 Hz), 4.7-4.4 (overlapping m and s, 4H, O-CH2-CH=, CO- CHz-Thymine), 4.2 (t, 1 H), 3.7 (m, 1 H), 3.44-3.27 (overlapping m, 5H), 2.55 (s, 3H, - HN-C-CH3), 2.37 (s, 4H, 2 x-CH2- of Dde), 2.05 (m, 4H), 1.89 (s, 3H, CH3 of Thymine)1.7 (m, 2H), 1.45 (s, 9H, boc), 1.03 (s, 6H, 2x-CH3 of Dde); 13C-NMR (CDCI3): δ = 197.81 (-CO- of Dde), 173.54, 170.33, 167.50, 163.94, 155.89 (-NH- COO), 150.74, 140.91 , 131.26 (-CH=CH2), 119.26 (-CH=CH2), 110.41 , 107.80 (=C=
of Dde), 79.85 [-COO-C(Me)3], 66.25 (-CH2-CH=), 60.27 (-CH2- of Dde), 52.69, 48.30, 47.66, 42.93, 39.11 , 30.01 , [28.31 , 28.13, 28.01 , -COO-C(CH3)3], 23.36, 17.89 (CH3 of Dde), 12.29 (CH3 of Thymine).
C.H.N analyse for C33H49N5O9: calc. C 60.08, H 7.49, N 10.61 ; found C 59.70, H 7.49, N 10.50.
EXAMPLE 19 Λ -(2-Boc-aminoethyl)-Λ/-(Thymine-1-ylacetyl)-Lysine-0-Allyl (19)
Preparation: 1.4 g (2.1 mmol) 18 was dissolved in a solution of 18 ml allyl alcohol and 2 ml hydrazin hydrate and stirred for 15 min. 200 ml DMF was added and the reaction mixture was evaporated under high vacuum. 50 ml Water was added to the remaining oil and extracted with diethylether (3 x 50 ml). The water phase was freeze dried and 0.8 g (1.6 mmol) titled compound was obtained as slightly yellow crystalline, which was used for the next step without further purification (yield = 76%). A sample of product was purified by HPLC and collected as TFA salt.
Mp = 81-83; MS (FAB) m/z 496 (M+H); 1 H-NMR, (CD3OD): δ 7.34 (S, aromatic proton of Thymine), 6.03-5.94 (12 line m, 1 H, -CH=CH2), 5.37(ddd,1 H, Jtrans=17.21 , 3.11 , 1.46 Hz), 5.27 (dd, 1 H, Jcιs=10.44, 2.56, 1.28 Hz), 4.8-4.6 (ddt, 2H, J=32, 5.67, 1.28 Hz, O-CH2-CH=), 4.78 (s, 2H, CO-CH2-Thymine), 4.37 (dd, 1 H, J=8.8, 5.86 Hz, αCH), 3.7 (m, 1 H, boc-HN-CH2-CHH'-), 3.4 [overlapping m and d (J=0.74), 3H, boc- HN-CH2-CHH'-, -NH2), 3.23 (m, 1 H), 3.03 (m, 1H), 3.97 (t, 2H, j=7.51 , -CH2-NH2), 2.2-2 (dm, 2H, βCH2), 1.9 (s, 3H, CH3 of Thymine) 1.7 (m, 2H), 1.49 [overlapping s and m, 11H, boc(9H), -CH2-CH2-NH2]; 13C-NMR (CD3OD): (major rotamer) δ 171.79, 169.81 , 166.98, 158.50 (-NH-COO-t-butyl), 153.12, 143.74, 133.44 (- CH=CH2), 119.03 (-CH=CH2), 116.38, 111.11 , 80.74 [-COO-C(Me)3], 67.21 (-CH2- CH=), 61.94, 50.21 , 40.61, 29.61, 28.81, 28.37, 27.83, 24.30, 12.32 (CH3 of Thymine).
C.H.N analyse for C23H37N5O7. 2 CF3COOH: calc. C 44.82, H 5.43, N 9.68; found C 44.61 , H 5.66, N 9.92.
EXAMPLE 20
Λ/-boc-Λ/-(Thymine-1-ylacetyl)Lysine-[Λ/-Λ/-bis(2,3,4,5-tetra-0-acetyl Galactose- 1-yl)-ethyl]-0-Allyl (20)
Preparation: 300mg (0.6 mmol) 19 was dissolved in 10 ml methanol, 500 mg (1.33 mmol, 2.2 eq) (M5) was added and stirred for 10 min. 80 μml (1.3 mmol) Acetic acid and 0.6g (10 mmol) natrium cyanoborohydrate was added sequentially. After 15 min the same amounts of acetic acid and NaCNBH3 were added and the reaction mixture was stirred for further 30 min at room temperature. Volatile was removed under vacuum and remaining dissolved in 100 ml ethylacetate and extracted with NaHCO3 (2 x 50ml) saturated solution and 50 ml brine respectively. Organic phase was dried over magnesium sulphate and evaporated to dryness under vacuum. The residue was purified on silica gel column eluting ethylacetate-methanol 10:0 to 10:0.5. 440 mg (0.36 mmol) titled compound was obtained as white crystalline. Yield = 60%. Mp = 106-108; MS (FAB) m/z 1212 (M+H); 1H-NMR, (CDCI3): resolved signals: δ 7 (s, 1 H, aromatic proton of Thymine), 5.9 (m, 1 H, -CH=CH2), 5.5 (br.s, 1 H, -NH-boc), 5.37 (d, 2H, J=2.74 Hz, 2x4CH-sugar), 5.3 (dd, 1 H, Jtrans=17.2, 1.28 Hz), 5.2 (overlapping, 5H, -CH=CHH\ 2x3CH- and 2x2CH- of sugar), 4.64 (s, 2H), 4.58 (d, 2H, J=5.86 Hz, O-CH2-CH=), 4.29 (br.d, 2H, J=10.07), 4.15 (br., 2H), 4 (br.d 2H, J=8.4), 2.2-2 (4s, 24H, CO-CH3 of sugar), 1.8 (s, 3H, CH3 of Thymine), 1.4 (overlapping s, 11 H, 9H of boc, 2H of lysine side chain); 13C-NMR (CDCI3): δ 170.75, 169.97, 169.65, 169.59, 167.58, 164.13, 156.00, 151.06, 141.38, 131.24, 119.06, 110.31 , 79.65, 68.20, 67.44, 66.65, 66.18, 60.75, 60.18, 50.01 , 39.12, 28.25, 20.66, 20.61 , 20.48, 12.00. HRMS (M+H)+, calcd (found) for C55H82N5O25 are 1212.5299 (1212.5360).
EXAMPLE 21
Λ/-boc-/V-(Thymine-1 -ylacetyl)Lysine-[Λ-Λ/-bis(2,3,4-tri-0-acetyl Fucose-1 -yl)- ethyl]-0-Allyl (21)
Preparation: The Procedure is the same as described above for the synthesis of 20. yield = 63%.
Mp = ; MS (FAB) m/z 1096 (M+H); 1H-NMR, (CDCI3): resolved signals: δ 7 (s, 1 H, aromatic proton of Thymine), 5.9 (m, 1 H, -CH=CH2), 5.6 (br.s, 1 H, -NH-boc), 5.3 (s, 1 H), 5.2 (overlapping, 4H, -CH=CHH', sugar), 4.58 (s, 2H, O-CH2-CH=), 4.2 (br.s,
1 H), 4 (dd, 2H, J=7.3, 0.8 Hz), 3.4-3.2 (br. 2H), 2.9 (br.s, 1 H), 2.8 (br.s, 1 H), 2 (3s, 18H, CO-CH3 of sugar), 1.8 (s, 3H, CH3 of Thymine), 1.4 (s, 9H, boc), 1.2 (dd, 2H, J=7.3, 1.2 of lysine side chain), 1.1 (d, 6H, J=3.8, methyl group of fucose); 13C-NMR (CDCI3): δ 170.87, 170.45, 170.17, 169.95, 167.21 , 164.46, 155.90, 151.11 , 141.38, 131.18, 130.89, 119.34, 118.79, 1 10.26, 80.08, 72.51 , 70.39, 69.75, 69.50, 68.07, 67.86, 66.05, 60.80, 60.08, 53.51 , 50.20, 48.69, 48.01 , 38.89, 36.44, 31.28, 28.09, 23.63, 21.65, 20.73, 20.60, 20.48, 20.38, 16.00, 15.47, 13.87, 11.94. HRMS (M+H)+, calcd (found) for C5ιH78N5O2ι are 1096.5189(1096.5192).
EXAMPLE 22
/V-boc-Λ -(Thymine-1-ylacetyl)Lysine-[Λ -(2,3,4,5-tetra-0-acetyl Galactose-1-yl)- ace tyl]-0-Allyl (22)
1.03 g (2.64 mmol, 1.1 eq) (M6), 470 mg (2.88 mmol, 1.2 eq) DhbtOH, and 740mg (3.6 mmol, 1.5 eq) DCC were mixed in 20 ml DMF and stirred for 30 min under Ni- trogen. A solution of 1.2 g (2.4 mmol) 19 in 10 ml DMF was added and the reaction mixture was stirred for further 3h. Volatiles were removed under vacuum and the residue dissolved in 200ml ethylacetate. Insoluble DCU was filtered off and the filtrate was extracted with NaHCO3 saturated solution (2 x 100 ml) and brine (100 ml) respectively. Organic phase was dried over magnesium sulphate and evaporated to dryness under vacuum. The remaining was purified on sillica gel column eluting ethylacetate. 1.1 g (1.2 mmol) title compound was obtained as white crystalline. Yield = 61 %.
Mp = 82-84; MS (FAB) m/z 868 (M+H); 1 H-NMR, (CDCI3): resolved signals: δ 9.39 (aromatic proton of Thymine), 6.94 (s, 1 H, -NH- of Thymine), 6.64 (s, 1H, -NH-CO- sugar), 5.92-5.85 (m, H, -CH=CH2), 5.42 (overlapping d, 2H, J=2.75, 4CH-sugar, - NH-boc), 5.32 (dd, 1 H, Jtrans= 7.2, 1.46 Hz), 5.25 (overlapping, 3H, -CH=CHH', 3CH- and 2CH- of sugar), 4.7 (m, 1 H), 4.6 (d, 2H, J=5.86), 4.5 (m, 1 H), 4.3-4.1 (m, 5H), 3.6 (m, 1 H), 3.35 (m, 3H), 3.2 (br., 2H), 2.6 (m, 1 H), 2.45 (m, 1 H), 2.15-2 (3s, 12H, CO-CH3 of sugar), 1.9 (s, 3H, CH3 of Thymine), 1.6 (br., 2H), 1.4 (overlapping s, 11 H, 9H of boc, 2H of lysine side chain); 13C-NMR (CDCI3): δ 170.56, 169.85, 169.77, 169.44, 167.47, 163.93, 155.82, 151.40, 140.89, 131.23 (-CH=CH2), 119.09 (-CH=CH2), 110.93, 79.86 [-O-C(Me)3], 69.23, 68.87, 68.23, 67.60, 66.82, 66.12,
60.85, 59.64, 48.88, 47.13, 38.99, 38.59, 34.74, 28.28, 28.04, 27.58, 22.79, 20.61 , 20.51 , 12.20(CH3 of Thymine)
C.H.N analyse for C39H57N5O17.1/2 H2O: calc. C 53.42, H 6.67, N 7.99; found C 53.25, H 6.71 , N 7.44.
EXAMPLE 23
Λ/-boc- V-(Thymine-1-ylacetyl)Lysine-[Λ/-(2,3,4,5-tetra-0-acetyl-Mannose-1-yl)- acetyl]-0-Allyl (23)
Preparation: The Procedure is the same as described above for the synthesis of 22. Yield = 58%.
Mp = ; MS (FAB) m/z 868 (M+H); 1 H-NMR, (CDCI3): resolved signals: δ 9.35 (aromatic proton of Thymine), 6.94 (s, 1H, -NH- of Thymine), 6.78 (s, 1H, -NH-CO- sugar), 5.8 (m, 1 H, -CH=CH2), 5.45 (s, 1 H, -NH-boc), 5.35 (dd, 1 H, Jtrans=17, 1.2 Hz), 5.25 (m, 2H of sugar), 4.6 (d, 2H, J=5.5), 4.5 (s, 1H), 4.3 (dd, 1H, J=12.1 , 5.7 Hz), 4.2 (dd, 1H, J= 12.1 , 8.2), 4.1 (m, 1H), 3.6 (m, 1H), 3.35 (m, 5H), 2.6 (m, 2H), 2.15-2 (4s, 12H, CO-CH3 of sugar), 1.9 (s, 3H, CH3 of Thymine), 1.6 (br., 2H), 1.4 (overlapping s, 11 H, 9H of boc, 2H of lysine side chain); 13C-NMR (CDCI3): δ 170.73, 170.19, 169.97, 169.48, 169.28, 167.36, 163.82, 156.56, 151.39, 143.55, 141.16, 131.21 (-CH=CH2), 127.70, 127.01 , 124.81 , 119.93, 119.21 (-CH=CH2), 110.87, 71.56, 70.33, 69.35, 68.11 , 67.26, 66.67, 66.24, 61.73, 59.90, 47.06, 39.51 , 38.79, 36.95, 28.14, 27.62, 22.85, 20.66, 12.18 (CH3 of Thymine).
EXAMPLE 24
/V-boc-Λ/-(Thymine-1-ylacetyl)Lysine-[Λ/-(2,3,4,5-tetra-0-acetyl fucose-1-yl)- acetyl]-0-Allyl (24)
Preparation: The Procedure is the same as described above for the synthesis of 22. Yield = 65%.
Mp= ; MS (FAB) m/z 810 (M+H); 1H-NMR, (CDCI3): resolved signals: δ 9.64 (aromatic proton of Thymine), 6.94 (s, 1 H, -NH- of Thymine), 6.88 (s, 1 H, -NH-CO- sugar), 6-5.8 (m, H, -CH=CH2), 5.5 (s, 1 H, -NH-boc), 5.37 (d, 1 H, J=3.8) 5.3 (overlapping, 2H, -CH=CHH'), 5.16 (m, 1H of sugar), 4.7 (d, 1H, J=15.5), 4.65 (m, 1H), 4.6 (d, 2H, J=5.8), 4.3 (m, 1 H), 4.1 (m, 1 H), 3.9 (m, 1 H), 3.8 (m, 1 H), 3.5-3.1 (m, 5H), 2.9 (dd, 1 H, J=23.1 , 2.3Hz), 2.7 (m, 1 H), 2.4-2.2 (m, 2H), 2.15-2 (3s, 9H, CO-
CH3 of sugar), 1.9 (s, 3H, CH3 of Thymine), 1.4 (s, 9H of boc), 1.2 (m, 2H, of lysine side chain); 13C-NMR (CDCI3): δ 170.42, 169.95, 169.74, 169.68, 169.55, 167.13, 163.86, 162.37, 155.81 , 151.53, 140.81 , 131.25 (-CH=CH2), 119.08 (-CH=CH2), 111.01 , 79.83 [-O-C(Me)3], 69.36, 68.43, 68.23, 67.95, 66.16, 61.64, 60.24, 48.95, 38.83, 37.41 , 36.34, 35.68, 33.78, 28.72, 28.34, 27.33, 27.46, 24.80, 22.67, 20.91 , 20.69, 20.62, 14.63, 14.05, 12.30 (CH3 of Thymine).
EXAMPLE 25
Λ/-Fmoc-Λ/-(Thymine-1-ylacetyl)Lisine-[/V-(2,3,4,5-tetra-0-acetyl Galactose-1-yl)- acetyl]-0-Allyl (25)
Preparation: procedure (d). Yield = 91%.
Mp = 93-95; MS (FAB) m/z 990 (M+H); 1H-NMR, (CDCI3): δ 9.33 (aromatic proton of Thymine), 7.6-7.1 (8H, florenyl aromatics), 6.64 (s, 1 H, -NH- of Thymine), 6.51 (s, 1 H, -NH-CO-sugar), 5.8-5.6 (overlapping m, 2H, -CH=CH2, -NH-fmoc), 5.27 (s, 1 H, 4CH-sugar), 5.15 (d, 1 H, Jtrans=16.66 Hz), 5.07 (overlapping, 3H, -CH=CHH', 3CH and 2CH of sugar), 4.55 (br.d, 1 H, J=3.84), 4.45 (d, 2H, J=5.86 Hz, O-CH2-CH=), 4.28 (overlapping s, 3H), 4.2 (d, 1H, J=7.14), 4.1-4 (m, 5H), 3.42 (m, 1 H), 3.29-3.26 (overlapping s, 3H), 3.04 (br.s, 2H), 2.46 (dd, 1 H, J=14.83, 9.15 Hz), 2.26 (m, 1 H), 2-1.8 (4s, 12H, -CO-CH3 of sugar), 1.68 (s, 3H, CH3 of Thymine), 1.4 (m, 1 H), 1.2 (m, 3H); 13C-NMR (CDCI3): δ 170.56, 170.46, 169.80, 169.72, 169.41 , 169.36, 167.51 , 163.98, 156.48, 151.29, 143.47, 143.44, 141.07, 131.18, 127.60, 127.49, 126.89, 124.71, 119.83, 119.03, 110.59, 77.41 , 76.90, 76.39, 69.21 , 68.75, 68.17, 67.64, 67.52, 66.73, 66.48, 66.06, 60.77, 59.54, 48.68, 46.97, 39.52, 38.62, 34.66, 28.16, 27.69, 22.87, 20.52, 20.43, 12.08. HRMS (M+H)+, calcd (found) for C49H6oN5O17 are 990.3984(990.3940).
EXAMPLE 26
Λ/-Fmoc-Λ/-(Thymine-1-ylacetyl)Lysine-[Λ-(2,3,4,5-tetra-0-acetyl mannose-1-yl)- acetyl]-0-Allyl (26) Preparation: procedure (d). Yield = 88%.
Mp= ; MS (FAB) m/z 990 (M+H); 1H-NMR, (CDCI3): δ 9.24 (aromatic proton of Thymine), 7.7-7.2 (8H, florenyl aromatics), 6.71 (overlapping-s, 2H, -NH- of Thymine, -NH-CO-sugar), 5.8 (overlapping-m, 2H, -CH=CH2, -NH-fmoc), 5.28 (s,
1 H, sugar), 5.2(m, 2H, -CH=CHH'), 5.06 (t, 2H, J=6.7), 4.55 (d, 2H, J=4.1 ), 4.4 (3, 2H, O-CH2-CH=), 4.3 (m, 1 H), 4.2 (m, 1 H), 4.1 (m, 1 H), 3.6 (m, 1 H), 3.4-3.1 (m, 3H), 2.5 (m, 2H), 2 (s, 12H, -CO-CH3 of sugar), 1.78 (s, 3H, CH3 of Thymine), 1.5 (m, 1 H), 1.2 (m, 3H); 13C-NMR (CDCI3): δ 170.72, 169.54, 165.72, 163.81 , 151.40, 143.57, 141.17, 136.86, 131.22, 127.70, 127.01 , 124.89, 119.93, 119.22, 110.10, 81.47, 67.27, 66.65, 66.24, 61.74, 47.07, 20.65, 20.43, 12.18. HRMS (M+H)+, calcd (found) for C49H60N5O17 are 990.3984(990.3987).
EXAMPLE 27 Λ -Fmoc-Λ-(Thymine-1-ylacetyl)Lysine-[Λ/-(2,3,4-tri-0-acetyl fucose-1-yl)- acetyl]-0-Allyl (27)
Preparation: procedure (d). Yield = 89% 3
Mp= ; MS (FAB) m/z 932 (M+H); 1 H-NMR, (CDCI3): δ 9.85 (aromatic proton of
Thymine), 7.7-7.2 (8H, florenyl aromatics), 7 (s, 1 H, -NH- of Thymine), 6.8 (s, 1 H, - NH-CO-sugar), 6 (s, 1 H, -NH-fmoc), 5.9 (m, 1 H, -CH=CH2), 5.28 (s, 1H, sugar), 5.2 (m, 2H, -CH=CHH'), 5.1 (s, 1 H), 4.6 (overlapping-s, 3H), 4.4 (m, 2H), 4.25 (br.s, 1 H), 4.2(m, 1 H), 2 (s, 9H, -CO-CH3 of sugar), 1.8 (s, 3H, CH3 of Thymine), 1.5-1.2 (m, 4H of Lysine side chain); 13C-NMR (CDCI3): δ 170.99, 170.46, 170.16, 169.98, 169.79, 169.62, 167.11 , 163.90, 156.50, 151.60, 143.65, 143.31 , 141.11 , 131.22, 130.90, 127.65, 127.02, 126.96, 124.87, 119.88, 119.13, 110.87, 69.05, 68.19, 67.70, 66.60, 66.20, 61.55, 60.23, 48.77, 46.99, 39.32, 37.69, 35.20, 28.77, 27.46, 22.74, 20.89, 20.61 , 20.56, 20.47, 14.72, 14.02, 12.15. HRMS (M+H)+, calcd (found) for C47H58N5O15 are 932.3929(932.3945).
EXAMPLE 28
Λ.-(2-Fmoc-aminoethyl)- Asp(tBu)-Allyl (30)
To 4.5 g (10 mmol) 28 [prepared by procedure (a)] was added 50 ml of a solution of 20% piperidine in DCM. After 30 min, 100 ml toluene was added and volatile were removed under vacuum. The residue was purified with a short silica gel column eluting ethylacetate-methanole 10:0 to 10:1. 1.8 g (7.9 mmol) 29 was obtained as slightly yellow oil. This oil was used for procedure (b). 1.7 g (3.4 mmol) title compound was obtained as colourless oil. Overall yield = 34%.
[α]D 22= (c=1 , methanol); MS (FAB) m/z 495 (M+H); 1H-NMR, (CDCI3): δ 7.7-7.2 (8H, Florenyl aromatic protons), 5.96-5.84 (16 line m, 1 H, -CH=CH2), 5.56 (br.s, 1 H, fmoc-HN-), 5.35 (dt, 1 H, Jtrans=15.9, 1.46 Hz), 5.25 (dt, 1 H, Jcis=10.5, 1.28Hz), 4.6 (dd, 2H, J=5.9, 1.1 Hz, O-CH2-CH=), 4.4 (d, 2H, J=7 Hz), 4.2 (t, 1H, J=7 Hz), 3.6(t, 1 H, J=7.3 Hz, αCH), 3.3 (m, 2H, βCH), 2.9 (m, 1 H), 2.7 (m, 2H), 2.6 (m, 1 H), 2.3 (br.s, 1 H), 1.4 (s, 9H, f-butyl) 13C-NMR (CDCI3): δ 173.02 (COO-Allyl), 169.89, 156.37, 143.84, 141.06, 131.47(-CH=CH2), 127.41 , 126.81 , 124.95, 119,71 , 118.68(-CH=CH2), 81.17[-O-C(Me)3], 66.5 (-CH2-CH=), 65.62 (β carbon), 57.12 (α carbon), 47.05, 40.34, 38.97, 27.85 [-COO-C(CH3)3]. HRMS (M+H)+, calcd (found) for C28H35N2O6 are 495.2495(495.2480).
EXAMPLE 29 Λ/-(2-fmoc-aminoethyl)- V-(Thymine-1-ylacetyl)-Asp-(tBu)-0-Allyl (31)
Preparation: procedure (c). Yield = 81%. Mp= ; [α]D 22= (c=0.25, methanol); MS (FAB) m/z 661 (M+H); 1 H-NMR, (CDCI3): δ 9.15 (s, 1 H,Thymine aromatic), 7.75-7.26 (8H, Florenyl aromatic protons), 6.75 (s, 1 H, Thymine-NH-), 5.9 (s, 1 H, fmoc-HN-), 5.8 (m, 1 H, -CH=CH2), 5.3 (d, 1 H, Jtrans=17.3 Hz), 5.2 (d, 1 H, JCis=10.5 Hz), 4.6 (m, 3H, O-CH2-CH=, αCH), 4.45 (s, 2H, -CO-CH2-Thymine), 4.2-4.1 (m, 3H), 3.4-3.4 (m, 2H), 3.4-3.2 (m, 2H), 2.9-2.7(m, 2H), 1.8 (s, 3H, Thymine-CH3), 1.4 (s, 9H, -COOtBu); 13C-NMR (CDCI3): δ 170.49, 169.36, 167.05, 164.06, 156.52, 150.62, 143,63, 143.52, 141.09, 140.85, 131.10(- CH=CH2), [(130.71 , 127.57, 126.92, 124.83, 119.79(florenyl)], 119.24(-CH=CH2), 110.32, 82.27, 81.41, 66.84, 66.51, 60.24(-CH2-CH=), 58.08, 48.85, 47.82, 47.10, 39.15, 35.41 , 27.85, 14.04, 12.19(Thymine-CH3). HRMS (M+H)+, calcd (found) for C35H41N4O9 are 661.2874(661.2881).
EXAMPLE 30 Λ/-(2-fmoc-aminoethyl)-/V-(Thymine-1-ylacetyl)-Asp-0-ANyl (32)
Preparation: To 3.3 g (5 mmol) 31 was added a solution of 95% TFA 5% TES. The reaction mixture was stirred until total conversion of starting material according to TLC. TFA solution was co-evaporated with DCM under vacuum. Titled compound was obtained as slightly yellow crystalline which was used for the next step without further purification (Yield = 62%); Mp = [α]D 22= (c=0.25, methanol); MS (FAB)=605
m/z (M+H); 1 H-NMR, (DMSO): δ 11.3 (s, 1 H,Thymine aromatic), 7.91-7.22 (8H, Florenyl aromatic protons, 1H, Thymine-NH-), 5.8 (m, 1H, -CH=CH2), 5.3 (dd, 1H, Jtrans=17.3, 1.76 Hz), 5.1 (dd, 1 H, Jcιs=10.55, 1 .47 Hz), 4.6 (d, 2H, J=7 Hz, O-CH2- CH=), 4.5 (s, 2H, -CO-CH2-Thymine), 4.4 (t, 1 H, J=6.4, αCH), 4.3 (d, 2H, J=6.8, florenyl-CH2-O-), 4.2 (t, 1 H, J=6.8, florenyl=CH-CH2-O-), 3.5 (m, 2H), 3.4 (m, 2H), 3.2 (dd, 2H, J=17, 7.3 Hz), 1.7 (s, 3H, Thymine-CH3); 13C-NMR (CD3OD): δ 174.50, 170.90, 169.54, 166.94, 158.90, 152.80, 145.30, 143,64, 142.57, 142.16, 133.25, 131.09 (-CH=CH2), [(130.44, 128.83, 128.21 , 127.13, 126.20, 121.00(florenyl)], 118.96 (-CH=CH2), 110.89, 67.80, 67.38, 61.59, 59.96(-CH2-CH=), 40.47, 35.32, 14.53, 12.34 (Thymine-CH3).
HRMS (M+H)+, calcd (found) for C3ιH33N4O9 are 605.2224(605.2248).
EXAMPLE 31 fmoc-Λ/-(Thymine-1-ylacetyl)Asp-[0-(2,3,4,5-tetra-0-acetyl Galactose-1-yl)- amide]-0-Allyl (33)
Preparation: procedure (e). Yield = 64%.
Mp = ; [α]D 22= (c=0.5, methanol); MS (FAB) m/z 934 (M+H); 1H-NMR, (CDCI3): δ 9.5 (s, 1 H,Thymiήe aromatic), 7.7-7.2 (m, 8H, Florenyl aromatic protons), 6.6 (s, 1 H, Thymine-NH-), 5.9 (overlapping m, 2H, -CH=CH2, fmoc-HN-), 5.3 (d, 1 H, J=1.6Hz), 5.25-5.15 (m, 3H), 5.05 (d, 2H, J=5.3 Hz), 4.6 (m, 2H), 4.4 (m, 2H), 4.1 (m, 1 H), 4 (m, 3H), 3.6-3.2 (m, 5H), 2.8 (m, 1 H), 2.1-1.9 (4s, 12H, sugar acetyl protons), 1.8 (s, 3H, Thymine-CH3); 13C-NMR (CDCI3): δ 171.21 , 170.22, 169.84, 169.76, 169.58, 166.56, 164.17, 156.81 , 156.57, 151.39, 143.68, 143.45, 141.17, 140.94, 131.20(- CH=CH2), 127.71 , 127.04, 124.89, 119.92, 119.16 (-CH=CH2), 110.60, 80.39, 78.26, 72.13, 71.36, 67.86, 67.19, 66.81 , 66.55, 61.44, 61.06, 57.81 , 49.22, 48.92, 48.77, 47.16, 39.19, 36.00, 33.69, 25.47, 24.79, 20.71 , 20.61 , 20.57, 20.46 (sugar- CO-CH3 groups), 12.26 (Thymine-CH3).
C.H.N analyse for C45H51N5O17, H2O: calc. C 56.78, H 5.61 , N 7.36; found C 56.87, H 5.94, N 7.57.
EXAMPLE 32
Fmoc-/V-(Thymine-1-ylacetyl)Asp-[0-(2,3,4,5-tetra-0-acetyl mannose-1-yl)- amide]-0-Allyl (34)
Preparation: procedure (e). Yield = 60%. Mp= ; [α]D 22= (c=0.5, methanol); MS (FAB) m/z 934(M+H); 1H-NMR, (CDCI3): δ 9.5 (s, 1 H,Thymine aromatic), 7.7-7.2 (m, 8H, Florenyl aromatic protons), 6.7 (s, 1 H, Thymine-NH-), 6 (br.s, 1H, fmoc-HN-), 5.9 (m, 1H, -CH=CH2), 5.5 (d, 1H, J=8.8Hz), 5.4 (d, 1 H, , J=8.8 Hz), 5.3-5 (m, 5H), 4.6 (m, 2H), 4.4 (m, 2H), 4.2 (m, 3H), 4.1 (m, 2H), 3.8-3.4 (m, 5H), 3.1 (m, 1 H), 2.9 (m, 1 H), 2.2-1.9 (4s, 12H, sugar acetyl pro- tons), 1.8 (s, 3H, Thymine-CH3); 3C-NMR (CDCI3). Mp = ; [α]D 22= (c=0.5, methanol); MS (FAB) m/z 934 (M+H); 1H-NMR, (CDCI3 13C-NMR (CDCI3): δ 171.13, 170.95, 170.49, 170.30, 169.93, 169.85, 164.62, 157.14, 156.95, 151.54, 144.08, 141.49, 131.57(-CH=CH2), 128.04, 127.35, 125.24, 120.27, 119.56 (-CH=CH2), 110.77, 74.43, 71.83, 69.87, 67.00, 66.87, 66.05, 65.62, 62.61, 58.53, 49.56, 49.34, 47.44, 39.47, 34.13, 25.83, 25.16, 21.21, 21.01 , 20.95, 20.84 (sugar-CO-CH3 groups), 12.05 (Thymine-CH3). HRMS (M+H)+, calcd (found) for C45H52N5O17 is 934.3358 (934.3342).
EXAMPLE 33 Fmoc-Λ/-(Thymine-1-ylacetyl)Asp-[0-(2,3,4-tri-0-acetyl fucose-1-yl)-amide]-0- Allyl (35)
Preparation: procedure (e). Yield = 77%.
Mp = ; [α]D 22= (c=0.5, methanol); MS (FAB) m/z 876 (M+H); 1 H-NMR, (CDCI3): δ 9.1
(s, 1 H,Thymine aromatic), 7.7-7.2 (m, 8H, Florenyl aromatic protons), 6.7 (s, 1 H, Thymine-NH-), 5.9 (br.s, 1 H, fmoc-HN-), 5.8 (m, 1 H, -CH=CH
2), 5.3(s, 1 H), 5.25 (s, 2H), 5.2 (m, 2H), 5 (m, 2H), 4.6 (m, 2H), 4.4 (m, 2H), 4.2-4 (m, 2H), 3.8 (d, 1 H, J=6.4 Hz), 3.7 (q, 1 H, J=6.4 Hz),3.4 (m, 2H), 3 (m, 1 H), 2.9 (m, 1 H), 2.2-2 (3s, 9H, sugar acetyl protons), 1.9 (s, 3H, Thymine-CH
3), 1.1 (s, 3H, fucose-CH
3). HRMS (M+H)
+, calcd (found) for
is 876.3303(876.3304).
EXAMPLE 34
Fmoc-Λ/-(Thymine-1-ylacetyl)Asp-[0-(2,3,4,5-tetra-0-acetyl glucosamine-1-yl)- amide]-0-Allyl (36)
Preparation: procedure (e). Yield = 73%. Mp = ; [α]D 22= (c=0.5, methanol); MS (FAB) m/z 933 (M+H); 1H-NMR, (CDCI3): δ 10 (s, 1H,Thymine aromatic), 7.7-7.2 (m, 8H, Florenyl aromatic protons), 6.8 (s, 1 H, Thymine-NH-), 6 (br.s, 1H, fmoc-HN-), 5.8 (m, 1H, -CH=CH2) , 5.4-5 (m, 5H), 4.7 (m, 1 H), 4.6 (m, 2H), 4.4 (m, 2H), 4.2 (m, 3H), 4.1 (m, 3H), 3.8-3.2 (m, 6H), 2.1-2 (4s, 12H, sugar acetyl protons), 1.9 (s, 3H, Thymine-CH3); 13C-NMR (CDCI3): δ 171.78, 171.08, 170.97, 170.71 , 170.51 , 169.42, 169.23, 166.94, 164.52, 156.47, 151.12, 143.58, 141.06, 131.05, 130.79 (-CH=CH2), 127.60, 126.92, 124.82, 119.84, 119.51 , 119.17 (-CH=CH2), 110.40, 79.39, 73.07, 67.89, 66.48, 61.68, 57.94, 52.75, 48.96, 48.28, 47.00, 39.14, 35.51 , 33.67, 24.73, 22.74, 20.54, 20.43, (sugar-CO-CH3 groups), 12.11 (Thymine-CH3). C.H.N analyse for C45H52N6O16, 3/2 H2O: calc. C 56.30, H 5.78, N 8.75; found C 56.43, H 5.70, N 8.72.
EXAMPLE 35
Fmoc-/V-(Thymine-1-ylacetyl)Asp-[0-(2,3,4,5-tetra-0-acetyl galactosamine-1- yl)-amide]-0-Allyl (37)
Preparation: procedure (e). Yield = 60%.
Mp = ; [α]D 22= (c=0.5, methanol); MS (FAB) m/z 933 (M+H); 1H-NMR, (CDCI3): δ 10.2 (s, 1H,Thymine aromatic), 7.9-7 (m, 8H, Florenyl aromatic protons), 6.8 (s, 1 H, Thymine-NH-), 6.3 (m, 1 H), 6.1 (br.s, 1 H, fmoc-HN-), 5.8 (m, 1 H, -CH=CH2) , 5.4-5.1 (m, 4H), 4.7 (m, 2H), 4.4 (m, 2H), 4.2 (m, 5H), 3.6-3.3 (m, 4H), 2.8 (m, 4H), 2.1-2 (m, 12H, sugar acetyl protons), 1.8 (s, 3H, Thymine-CH3); 13C-NMR (CDCI3): δ 171.78, 171.08, 170.97, 170.71 , 170.51 , 169.42, 169.23, 13C-NMR (CDCI3): δ 172.32, 170.82, 170.54, 170.33, 170.04, 169.55, 169.37, 167.03, 164.53, 156.62, 151.35, 143.55, 141.04, 131.09, 127.59, 126.91, 124.80, 119.82, 119.07(-CH=CH2), 110.41 , 79.57, 71.83, 70.46, 67.91 , 67.37, 66.86, 66.70, 66.45, 61.77, 61.31 , 58.59, 49.20, 48.98, 47.96, 47.01 , 39.18, 23.04, 22.95, 22.77, 20.52 (sugar-CO-CH3 groups), 12.04 (Thymine-CH3). HRMS (M+H)+, calcd (found) for C45H53N6O16 is 933.3518(933.3549).
EXAMPLE 36
4-C-(2,3,4,6-tera-0-acetyl-galactose-1-yl)-2-Z-amino-2-butenoic acid t-butyl ester (41) Preparation: 2.2 g (5.8 mmol, 1.1 eq) 40 was dissolved in 20 ml dry THF. 0.8 ml ( mmol, 1.1 eq) tetramethylguanidine was added at (-78°C) and stirred for 5 min. 1.9 g (5.27 mmol, 1 eq) 2,3,4,5 tetraacetyl galactose-1yl-acetaldehyde was added in 10 ml dry THF solution. The reaction mixture was allowed to reach the room temperature. Volatile was evaporated and the residue was purified on silica gel column eluting hexane-ethylacetate 1 :1 solution. 2.2 g (3.5 mmol) titled compound was obtained as colourless oil. Yield = 66%.
Mp: ; [α]D 22= (c=0.5, methanol); MS (FAB) m/z 622 (M+H); 1H-NMR, (CDCI3): δ 7.4 (m, 4H of cbz aromatic protons), 6.5 (overlapping, 2H, cbz-ring-4CH-, cbz-HN-), 5.4 (t, 1 H, J=3 Hz), 5.2 (dd, 1 H, J=9.1 , 4.8 Hz), 5.18 (dd, 1 H, J=9.1 , 3.3 Hz), 5.15 (d, 2H, J=1.3 Hz), 4.35 (m, 1 H), 4.45 (m, 1 H), 4.1-4 (m, 2H), 2.6 (m, 1 H), 2.4 (m, 1 H), 2.18 (d, 1 H, J=8.4), 2.15-2 (4s, 12H, sugar acetyl protons), 1.4 (s, 9H, t-Bu); 13C- NMR (CDCI3): δ 170.42, 169.86, 169.68, 163.08, 153.86, 135.81 , 129.60, 128.43, 128.16, 128.06, 82.06, 74.04, 71.02, 68.91 , 68.51, 67.72, 67.55, 67.32, 67.25, 61.18, 27.87, 25.82, 20.62, 20.54. HRMS (M+H)+, calcd (found) for C3oH40NOi3 is 622.2500(622.2495).
EXAMPLE 37
4-C-(2,3,4,6-tera-0-acetyl-galactose-1-yl)-2(2-fmoc-aminoethyl)-aminobutanoic acid t-butyl ester (43) Preparation: 2.2 g (3.5 mmol) 41 was dissolved in 10 ml methanole. 50 mg Pd/C catalyst was added and the mixture hydrogenated at 1 atm for 3 hours. The catalyst was filtered off over celite and methanol was removed under vacuum. The remaining oil 42 was directly used for procedure (b). 2.1g (2.78 mmol) titled compound was obtained as white solid. Yield = 82%. Mp = ; [α]D 22= (c=0.5, methanol); MS (FAB) m/z 755 (M+H); 1H-NMR, (CDCI3): δ 7.7- 7.2 (m, 8H, fmoc aromatic protons), 5.5 (overlapping s, 2H), 5.2 (m, 2H), 4.4 (d, 2H, 7 Hz), 4.2 (s, 2H), 4.1 (m, 2H), 3.3-3 (m, 4H), 2.8 (m, 1 H), 2.6 (m, 1 H), 2.2-2 (4s, 12H, sugar acetyl protons), 1.4(s, 9H, t-Bu); CDCI3 13C-NMR (CDCI3): δ 174.13,
170.53, 170.47, 170.40, 170.01, 169.93, 169.86, 169.76, 169.71, 16963, 156.37, 143.79, 143.69, 141.07, 127.45, 126.82, 124.90, 119.75, 81.43, 71.60, 68.26, 68.20, 68.00, 67.80, 67.71 , 67.58, 67.44, 67.35, 66.51 , 61.42, 61.24, 61.07, 61.00, 60.19, 47.33, 47.05, 28.79, 27.89, 27.47, 21.75, 20.86, 20.59, 20.53, 20.46 (sugar-CO-CH3 groups), 14.01.
HRMS (M+H)+, calcd (found) for C39H51N2Oι3 is 755.3391 (755.3371 ).
EXAMPLE 38
4-C-(2,3,4,6-tera-0-acetyl-galactose-1-yl)-2[N,N(2-fmoc-aminoethyl)-(thymine-1- yl-methyl-carbonyl)-aminobutanoic acid t-butyl ester (44)
Preparation: procedure (c). Yield = 66%.
Mp: ; [α]D 22= (c=0.5, methanol); MS (FAB) m/z 951 (M+H); 1H-NMR, (CDCI3): δ 9.2 (s, 1 H, thymine aromatic), 7.8-7.3 (m, 8H, fmoc aromatic), 6.9 (s, 1 H, thymine-NH-), 5.8 (s, 1 H, fmoc-HN-), 5.4 (s, 1 H), 5.35 (dd, 1H, J=10.2, 3.3 Hz), 5.25 (dd, 1 H, J=10.2, 5.7 Hz), 5.2 (d, 1 H, J=3 Hz), 4.4 (m, 3H), 4.2-4 (m, 5H), 3.6-3.3 (m, 4H), 2.2-2 (4s, 12H, sugar acetyl protons), 1.8 (s, 3H, thymine-methyl), 1.4 (s, 9H, t-Bu); 13C-NMR (CDCI3): δ 170.46, 169.94, 169.84, 169.07, 168.19, 166.85, 163.99, 156.44, 150.82, 143.54, 141.08, 127.59, 126.89, 124.78, 119.83, 110.23, 82.49, 72.31 , 71.83, 67.49, 66.93, 66.61 , 61.81 , 61.29, 60.20, 59.30, 48.87, 48.40, 47.89, 46.96, 46.10, 39.98, 39.38, 33.72, 27.75, 25.41 , 24.88, 22.60, 21.16, 20.86, 20.68, 20.55, 14.01 , 12.16. HRMS (M+H)\ calcd (found) for C46H57N4O16 is 921.3770(921.3782).
EXAMPLE 39 Λ/-Fmoc-/V-(Thymine-1-ylacetyl)Serine(2,3,4,5-tetra-0-acetyl-α-D-Galactose-1- yl)-COOH (1s)
Preparation: procedure (f). Yield = 86%.
Mp = 124-127; [α]D 22= (c=0.5, methanol); MS (FAB) m/z 867 (M+H); H-NMR, (CD3OD): characteristic signals: δ = 7.8-7.3 [m, 9H, (8H, Florenyl aromatic protons, 1 H, Thymine aromatic)], 5.4 (s, 1 H, sugar 4CH), 5.15 (m, 2H, sugar 3CH and 2CH), 4.65 (overlapping s, 3H, sugar 1CH, -CO-CH2-Thymine), 2.2-2 (4s, 12H, sugar acetyl protons), 1.8 (s, 3H, Thymine-CH3); 13C-NMR (CD3OD): δ 170.20, 170.13, 170.05, 169.75, 165.15, 143.56, 143.46, 141.98, 140.73, 133.95, 128.16, 127.99, 126.92,
126.36, 126.30, 124.40, 124.30, 119.06, 108.83, 100.26 (sugar C1 ), 70.33, 70.13, 68.53, 68.44, 66.90, 65.86, 60.58, 47.95, 38.61 , 18.88, 18.68, 18.59, 10.36. HRMS (M+H)+, calcd (found) for C41H47N4O17 is 867.2936 (867.2924).
EXAMPLE 40
Λ/-Fmoc-Λ/-(Thymine-1-ylacetyl)Threonine(2,3,4,5-tetra-0-acetyl-α-D-Galactose- 1-yl)-COOH (2s)
Preparation: procedure (f). Yield = 90%.
Mp = 128-131 ; [α]D 22= (c=0.5, methanol); MS (FAB) m/z 881 (M+H); 1H-NMR, (CD3OD): characteristic signals: δ = 7.84-7.32 []m, 9H, (8H, Florenyl aromatic protons, 1 H, Thymine aromatic), 5.41 (d, 1H, J=3.3 Hz, sugar 4CH), 5.2 (dd, 1 H, j=10.44, 3.3 Hz, sugar 3CH), 5.1 (dd, 1 H, Jι,2=7.5, J2,3=10.5, sugar 2CH), 2.18-2 (4s, 12H, sugar acetyl protons), 1.9 (s, 3H, Thymine-CH3), 1.36 (d, 3H, J=5.86 Hz, βC- CH3); 13C-NMR (CD3OD): δ = 172.06, 171.51 , 167.09, 145.38, 143.86, 142.61, 128.87, 128.26, 126.26, 121.03, 116.69, 110.78, 100.40(sugar C1 ), 74.76, 72.31 , 71.97, 70.66, 68.80, 67.78, 62.69, 62.49, 20.93, 20.73, 20.62, 12.41. HRMS (M+H)+, calcd (found) for C42H49N4O17 is 881.3043 (881.3073).
EXAMPLE 41 /V-boc-ιV-(Thymine-1-ylacetyl)Lisine-[Λ-Λ -bis(2,3,4,5-tetra-0-acetyl Galactose- 1-yl)-ethyl]-OH (3s)
Preparation: procedure (f). Yield = 80%.
Mp = 118-120; [α]D 22= (c=0.5, methanol); MS (FAB) m/z 1172 (M+H); 1H-NMR,
(CD3OH): resolved signals: δ 7.4 (s, 1 H, aromatic proton of Thymine), 5.45 (d, 2H, J=2.74 Hz, 2 x 4CH-sugar), 5.31 (dd, 2H, J=9.16, 3.3 Hz, 2 x 3CH-sugar), 5.25 (m, 2H, 2 x 2CH-sugar), 4.38 (m, 4H), 4.25 (m, 2H), 4.15 (m, 3H), 3.95 (dd, 1 H, J=12.82, 3.66Hz), 3.65 (dt, 1 H, J=2.38, 12.45 Hz), 3.5 (m, 2H), 3.15 (d, 2H, J=13.7 Hz), 2.9 (br, 2H), 2.8(m, 2H), 2.2-2 (4s, 24H, CO-CH3 of sugar), 1.9 (s, 3H, CH3 of Thymine), 1.8-1.6 (br. 4H), 1.47 (overlapping s, 11 H, 9H of boc, 2H of lysine side chain); 13C- NMR (CD3OH): δ 172.41 , 172.23, 171.91 , 171.72, 171.69, 171.50, 170.39, 169.75, 167.08, 158.28, 153.16, 144.45, 144.16, 110.77, 110.57, 80.42, 71.31 , 71.24, 70.29, 69.86, 69.78, 69.37, 69.20, 69.07, 66.30, 65.76, 62.88, 62.70, 54.96, 51.67, 50.87,
45.10, 40.60, 39.83, 32.81 , 31.39, 30.56, 28.95, 28.88, 26.81 , 26.26, 25.95, 25.66,
23.46, 23.01 , 20.99, 20.79, 20.68, 12.43.
HRMS (M+H)+, calcd (found) for C52H78N5O25 is 1172.4986 (1172.5027).
EXAMPLE 42
Λ-boc-/V-(Thymine-1-ylacetyl)Lisine-[Λ/-Λ/-bis(2,3,4-tri-0-acetyl fucose-1-yl)- ethyl]-OH (4s)
Preparation: procedure (f). Yield = 88%.
Mp = ; [α]
D 22= (c=0.5, methanol); MS (FAB) m/z 1056 (M+H);
13C-NMR (CD
3OH): δ 172.19, 171.65, 166.98, 162.58, 158.30, 153.14, 144.9, 133.86, 133.18, 133.05, 130.66, 130.13, 128.83, 128.35, 123.36, 114.36, 110.83, 108.98, 80.44, 80.08, 71.82, 71.51, 69.82, 69.34, 67.49, 62.40, 54.33, 51.85, 51.32, 50.66, 45.12, 40.60, 39.81 , 30.22, 28.97, 28.90, 25.24, 22.13, 21.02, 20.83, 20.67, 16.43, 12.49, 11.99. HRMS (M+H)
+, calcd (found) for
is 1056.4876(1056.4886).
Λ -boc-/V-(Thymine-1-ylacetyl)Lisine-[Λ-(2,3,4,5-tetra-0-acetyl Galactose-1-yl)- acetyl]-OH (5s)
Preparation: procedure (f). Yield = 95%.
Mp = 115-118; [α]D 22= (c=0.5, methanol); MS (FAB) m/z 828 (M+H); 1H-NMR, (CD3OD): resolved signals: δ 7.37 (s, 1 H, aromatic proton of Thymine), 5.44 (d, 1 H, J=2.56 Hz, 4CH-sugar), 5.3 (overlapping, 2H, 3CH- and 2CH-sugar), 4.7 (m, 2H), 4.5(m, 1 H), 4.25 (m, 1 H), 4.1 (m, 3H), 3.5 (m, 1 H), 3.25 (m, 2H), 2.7 (m, 1 H), 2.5 (dd, 1 H, J=5.3, 2.2 Hz), 2.2-2 (4s, 12H, CO-CH3 of sugar), 1.9 (s, 3H, CH3 of Thymine), 1.6 (br. 2H), 1.4 (overlapping s, 11 H, 9H of boc, 2H of lysine side chain); 13C-NMR (CD3OD): δ 173.13, 172.16, 170.41-169.52 (sugar-O-CO-Me), 167.69, 164.97 (-CO-CH2-Thymine), 156.48 (-NH-COO-t-butyl), 151.46, 141.90, 109.37, 78.68 [-O-C(Me)3], 74.39 (sugar C5), 69.32-66.93 (sugar C1-4), 60.52 (sugar C6), 48.68 (lysine, α carbon), 40.93, 38.31, 37.08, 33.06, 29.22-25.82 [-COO-C(CH3)3], 22.94-18.65 (sugar-O-CO-CH3), 10.34 (Thymine-CH3). HRMS (M+H)+, calcd (found) for C36H5 N5Oι7 is 828.3515 (828.3515).
EXAMPLE 43
Λ/-fmoc-/V-(Thymine-1-ylacetyl)Lisine-[Λ/-(2,3,4,5-tetra-0-acetyl Galactose-1-yl)- acetyl]-OH (6s)
Preparation: procedure (f). Yield = 83%. Mp = 130-132; [α]D 22= (c=0.5, methanol); MS (FAB) m/z 951 (M+H); 1H-NMR, (CD3OH): resolved signals: δ 7.8-7.3 (9H, aromatic proton of Thymine, 8H of florenyl aromatics), 5.43 (d, 1 H, J=2.4 Hz, 4CH-sugar), 5.3 (overlapping, 2H, 3CH- and 2CH- sugar), 4.72 (m, 3H), 4.45 (d, 1 H, J=6.6), 4.4 (m, 2H), 4.3-4.2 (m, 3H), 4.2-4.1 (m, 3H), 3.6 (m, 1 H), 3.45 (m, 2H), 3.2 (m, 3H), 2.7 (dd, 1 H, J=14.65, 9.34 Hz), 2.5 (dd, 1 H, J=14.65, 5.31 Hz), 2.2-2 (3s, 12H, CO-CH3 of sugar), 1.9 (s, 3H, CH3 of Thymine), 1.6 (br. 3H), 1.2 (m, 1 H); 13C-NMR (CD3OH): δ 174.33, 172.23, 172.20, 171.89, 171.56, 171.41 , 170.43, 169.78, 166.98, 158.89, 153.02, 145.02, 143.91, 142.67, 128.88, 128.23, 126.22, 121.04, 110.92, 71.28, 70.14, 69.41 , 69.15, 69.00, 67.81 , 62.52, 61.65, 50.20, 40.80, 40.30, 35.08, 30.12, 29.78, 24.96, 20.83, 20.76, 20.72, 20.62, 12.37(Thymine-CH3).
HRMS (M+H)\ calcd (found) for C46H56N5O17 is 950.3671 (950.3666).
EXAMPLE 44
Λ -fmoc-Λ/-(Thymine-1-ylacetyl)Lysine-[Λ/-(2,3,4,5-tetra-0-acetyl mannose-1-yl)- acetyl]-OHI (7s)
Preparation: procedure (f). Yield = 92%.
Mp = ; [α]D 22= (c=0.5, methanol); MS (FAB) m/z 951 (M+H); 13C-NMR (CDCI3): δ 193.55, 178.08, 172.71 , 172.46, 171.75, 171.56, 171.44, 166.88, 153.09, 145.27, 144.01 , 142.58, 130.33, 128.89, 128.24, 126.23, 121.04, 116.26, 110.95, 73.18, 72.40, 72.30, 71.40, 70.19, 68.28, 67.88, 63.44, 62.41 , 40.15, 37.31, 30.17, 25.05, 20.87, 20.71 (acetyl groups on sugar), 12.40 (CH3 of Thymine)
EXAMPLE 45
Λ/-fmoc-Λ/-(Thymine-1-ylacetyl)Lysine-[Λ/-(2,3,4-ttri-0-acetyl fucose-1-yl)- ace tyl]-OHl (8s)
Preparation: procedure (f). Yield = 86%.
Mp = ; [α]D 22= (c=0.5, methanol); MS (FAB) m/z 892 (M+H); 1 H-NMR, (CD3OD): resolved protons δ 7.8-7.2 (m, 8H, Florenyl aromatic protons), 6.9 (s, 1 H, Thymine-
NH-), 5.3 (overlapping s, 2H), 4.7 (s, 2H, -CO-CH2-Thymine), 4.44 (d, 1 H, J=6.6Hz), 4.38 (m, 1 H), 4.25 (m, 1 H), 4.15 (m, 1 H), 3.6-3.2 (multiplets, 5H), 2.7 (m, 1 H), 2.5 (m, 1H), 2.2-2 (3s, 9H, sugar acetyl protons), 1.85 (s, 3H, Thymine-CH3), 1.6-1.3 (m, 4H, lysine side chain protons), 1.1 (d, 3H, J=4.2, fucose-CH3); 13C-NMR (CD3OD): δ 174.67, 172.51 , 172.24, 171.51 , 170.32, 169.68, 166.93, 158.86, 152.99, 145.29, 143.93, 142.62, 133.19, 131.77, 130.48, 129.92, 128.89, 128.24, 126.22, 121.83, 121.05, 117.01 , 110.89, 72.044, 71.53, 69.88, 69.00, 67.81, 61.93, 42.08, 40.75, 40.12, 34.79, 30.15, 29.75, 26.77, 26.09, 24.91 , 20.80, 20.59, 16.50, 12.38. HRMS (M+H)+, calcd (found) for C44H54N5Oi5 is 892.3616(892.3635).
EXAMPLE 46 fmoc-Λ-(Thymine-1-ylacetyl)Asp-[0-(2,3,4,5-tetra-0-acetyl Galactose-1-yl)- amide]-OH (9s)
Preparation: procedure (f). Yield = 94%. Mp = ; [α]D 22= (c=0.5, methanol); MS (FAB) m/z 894 (M+H); 1H-NMR, (CD3OD): resolved protons δ 7.8-7.3 (m, 8H, Florenyl aromatic protons), 7.2 (s, 1 H, Thymine- NH-), 5.45 (s, 1 H, sugar 4CH), 5.37 (d, 1 H, J=8.5 Hz, 2CH), 4.24(m, 2H), 5.23 (d, 1 H, J=13.4 Hz), 4.48 (t, 1 H, J=6 Hz), 4.42 (d, 1 H, J=3.3 Hz, -CO-CH2-Thymine), 4.3-4.1 (m, 3H), 2.2-2 (4s, 12H, sugar acetyl protons), 1.8 (s, 3H, Thymine-CH3); 3C-NMR (CD3OD): δ 173.62, 172.93, 172.00, 171.79, 171.53, 169.34, 166.98, 158.89, 152.90, 145.33, 143.68, 142.64, 133.85, 133.21, 130.14, 128.89, 128.27, 126.29, 121.06, 110.93, 79.30, 73.41 , 73.09, 69.81 , 69.03, 68.36, 62.72, 59.72, 40.46, 37.15, 34.80, 32.77, 30.19, 26.79, 26.10, 24.91 , 23.73, 20.94, 20.70, 20.64, 14.51 , 12.38. HRMS (M+H)+, calcd (found) for C42H48N5O17 is 894.3045 (894.3072).
EXAMPLE 47
Fmoc-Λ/-(Thymine-1-ylacetyl)Asp-[0-(2,3,4,5-tetra-0-acetyl mannose-1-yl)- amide]-OH (10s) Preparation: procedure (f). Yield = 95%.
Mp = ; [α]D 2 = (c=0.5, methanol); MS (FAB) m/z 894 (M+H); 1H-NMR, (DMSO): resolved protons δ 11.2 (s, 1H, thymine aromatic), 8.7 (d, 1 H, J=8.8 Hz, Thymine-NH-) 7.8-7.3 (m, 8H, Florenyl aromatic protons), 5.6 (d, 1H, J=8.8 Hz), 5.37 (dd, 1 H,
J=10.2, 3.8 Hz), 5.15 (d, 1H, J=3.3 Hz), 5 (t, 1 H, J=10 Hz), 4.6 (m, 2H), 4.3 (d, 1 H, J=6 Hz), 4.2 (m, 2H), 4 (d, 1 H, J=11 Hz, 2.2-2 (4s, 12H, sugar acetyl protons), 1.8 (s, 3H, Thymine-CH3); (CDCI3 13C-NMR (CDCI3): δ 170.13, 169.62, 143.91 , 140.76, 127.67, 127.13, 125.17, 120.15, 108.05, 72.60, 70.88, 65.58, 47.84, 46.75, 33.39, 24.53, 21.03, 20.61 , (sugar-CO-CH3 groups), 11.99 (Thymine-CH3). HRMS (M+H)+, calcd (found) for C42H48N5O17 is 894.3045 (893.3065).
EXAMPLE 48
Fmoc-Λ/-(Thymine-1-ylacetyl)Asp-[0-(2,3,4-tri-0-acetyl fucose-1-yl)-amide]-OW (11s)
Preparation: procedure (f). Yield = 88%.
Mp = ; [α]D 22= (c=0.5, methanol); MS (FAB) 836 m/z (M+H); 7.8-7.3 (m, 8H, Florenyl aromatic protons), 7.2 (s, 1 H, Thymine-NH-), 5.3 (overlapping s, 2H), 5.2 (d, 1 H, J=3 Hz), 4.66 (s, 1 H), 4.4 (m, 2H), 4.25 (m, 1 H), 4 (m, 1 H), 3.5 (m, 3H), 3.15 (m, 2H), 2.2-2 (singles, 9H, sugar acetyl protons), 1.8 (s, 3H, Thymine-CH3), 1.1 (s, 3H, fucose-CH3); 13C-NMR (CD3OD): δ 172.30, 171.81 , 170.68, 170.30, 169.96, 167.74, 165.39, 157.35, 151.41 , 143.84, 143.74, 142.15, 141.04, 134.35, 132.27, 131.60, 131.49, 128.52, 128.40, 127.30, 126.68, 124.77, 124.07, 119.44, 109.36, 86.67, 77.61, 71.92, 71.46, 71.33, 70.51 , 68.57, 68.26, 68.12, 67.93, 66.40, 58.96, 39.00, 35.92, 33.26, 25.26, 24.57, 19.27, 19.11 , 19.05, 15.05, 10.90. HRMS (M+H)+, calcd (found) for C40H46N5Oι5 is 836.2990 (836.2980).
EXAMPLE 49
Fmoc-Λ/-(Thymine-1-ylacetyl)Asp-[0-(2,3,4,5-tetra-acetyl glucosamine-1-yl)- amide]-OH (12s)
Preparation: procedure (f). Yield = 90%.
Mp = ; [α]D 22= (c=0.5, methanol); MS (FAB) m/z 893 (M+H); 1H-NMR, (CD3OD): δ 7.8-7.3 (m, 8H, Florenyl aromatic protons), 7.2 (d, 1 H, J=9Hz, Thymine-NH-), 5.3 (m, 2H), 5.1 (d, 1 H, J=9.7Hz), 4.7 (m, 1 H), 4.4 (m, 2H), 4.2 (m, 2H), 4.1 (m, 1 H), 3.9 (m, 1 H), 3.6-2.8 (multiplets, 5H), 2.1-1.9 (4s, 12H, sugar acetyl protons), 1.8 (s, 3H, Thymine-CHs); 13C-NMR (CD3OD): δ 173.76, 172.94, 172.43, 172.05, 171.40, 170.14, 169.26, 166.94, 158.85, 153.13, 152.87, 145.32, 143.72, 142.59, 130.13, 129.93, 128.88, 128.26, 126.24, 121.05, 111.17, 110.92, 79.53, 74.85, 74.62, 69.97,
69.82, 67.86, 63.34, 59.82, 54.28, 40.46, 37.16, 23.09, 23.01 , 22.87, 22.74, 20.72,
12.39.
HRMS (M+H)+, calcd (found) for C42H49N5O16 is 893.3205 (893.3232).
EXAMPLE 50
Fmoc-Λ/-(Thymine-1-ylacetyl)Asp-[0-(2,3,4,5-tetra-acetyl galactosamine-1-yl)- amide]-OH (13s)
Preparation: procedure (f). Yield = 91%.
Mp = ; [α]D 22= (c=0.5, methanol); MS (FAB) m/z 893 (M+H); 1H-NMR, (CD3OD): δ 7.8-7.3 (m, 8H, Florenyl aromatic protons), 7.2 (s, 1 H, Thymine-NH-), 5.4 (s, 1 H), 5.3 (d, 1 H, J=9.5 Hz), 5.2 (dd, 1 H, J=11 , 3.3 Hz), 5.1 (m, 1 H), 4.7 (s, 1 H), 4.4 (m, 2H), 4.3-4.1 (m, 4H), 3.8 (m, 1 H), 3.6 (m, 1 H), 3.5-2.8 (multiplets, 5H), 2.2-1.9 (4s, 12H, sugar acetyl protons), 1.85 (s, 3H, Thymine-CH3); 13C-NMR (CD3OD): δ 174.06, 173.57, 172.94, 172.18, 172.05, 171.81 , 171.74, 169.28, 166.98, 158.88, 153.14, 152.9, 145.34, 143.75, 142.62, 135.10, 134.97, 133.05, 131.43, 130.22, 129.95, 128.87, 128.24, 126.24, 121.03, 110.89, 80.06, 73.36, 72.71 , 68.91 , 68.23, 67.89, 62.86, 59.86, 40.46, 37.19, 26.55, 23.12, 20.70, 12.39. HRMS (M+H)\ calcd (found) for C42H49N6O16 is 893.3205 (893.3245).
EXAMPLE 51
4-C-(2,3,4,6-tera-0-acetyl-galactose-1-yl)-2[N,N(2-fmoc-aminoethyl)-(thymine-1- yl-methyl-carbonyl)-aminobutanoic acid (14s)
920 mg (1 mmol) 44 was added to 10 ml solution of 5% triethylsilane in trifluoroace- tic acid at 0°C and stirred until TLC did not show any starting material left. 50 ml DCM was added and volatile removed under vacuum. Further 3 x 100 ml DCM was added and evaporated in order to removal of TFA. The product was precipitated with adding diethylether. 830 mg (0.94 mmol) titled compound was obtained as white powder (Yield = 94%). Mp= ; [α]D 22= (c=0.5, methanol); MS (FAB) m/z 865 (M+H); 1H-NMR, (CD3OH): δ 7.8-7.3 (m, 8H, fmoc aromatic), 7.2 (s, 1 H, thymine-NH-), 5.4 (overlapping s, 2H), 5.3 (m, 1 H), 4.7(s, 1 H), 4.6 (m, 1 H), 4.5(d, 2H, J=6.6 Hz), 4.2 (overlapping s, 2H), 4.1 (overlapping s, 2H), 3.6 (s, 1 H), 3.5 (overlapping s, 2H), 3.3 (s, 1 H), 2.2-2 (4s, 12H, sugar acetyl protons), 1.85 (s, 3H, thymine-methyl), 13C-NMR (CD3OD): δ
172.75, 171.22, 170.86, 170.75, 170.63, 170.52, 168.56, 165.80, 157.63, 151.63, 144.09, 142.89, 142.70, 141.43, 127.66, 127.01 , 125.00, 119.82, 109.43, 72.16, 68.62, 68.45, 68.26, 68.08, 39.52, 33.60, 31.59, 25.58, 24.91 , 24.76, 22.55, 21.73, 19.60, 19.43, 13.31 , 11.16. HRMS (M+H)+, calcd (found) for C42H49N4O16 is 865.3140 (865.3144).
EXAMPLE 52
Solid Phase Synthesis of Type IV PNA monomers
Boo- -AsvOH A B
F
N -Boc-Diaminopropionic acid
A) To a solution of Boc-L-Asn-OH (15.0 g, 60.3 mmol) in EtOAc (75 mL), CH3CN (75 mL) and water (30 mL) at 10°C was added PIDA (24.9 g, 77.3 mmol, 1.3 eq.). The mixture was stirred for 30 min, heated to 20 C and stirred for further 2.5 h. The mixture was heated to 70°C until completely dissolved and then slowly cooled to 20 C and filtered. The remanence was washed with EtOAc (2 x 50 mL) and dried. Yield 8.9 g (68%). 1H NMR (DMSO-d6 + TFA) δ 8.06 (bs, 3NH), 7.36 (d, 1 NH, J = 8.7 Hz),
4.40-4.25 (m, 1 H), 3.35-3.25 (m, 1 H), 3.15-3.00 (m, 1 H), 1.51 (s, 9H). MS: [M+H]+: expected: 205.1 ; observed: 205.1.
N -Boc-l -Z-Diaminopropionic acid B) To a solution of Nα-Boc-Diaminopropionic acid (8.90 g, 43.6 mmol) in water (270 ml) was added NaHCO3 (7.5 g) and subsequently a solution of benzyl chloroformate (6.85 mL, 48.0 mmol, 1.1 eq.) in Et2O (30 mL) was added dropwise with vigorous stirring. Another quantity of NaHCO3 (7.5 g) was added, and the mixture was stirred vigorously for 3 h. The mixture was washed with Et2O (2 x 200 mL), cooled to 0 C, acidified with solid citric acid to pH 3 and extracted with EtOAc (2 x 200 mL). The combined organic phases were washed with 10% aq. citric acid (100 mL), dried (Na2SO4) and concentrated in vacuo Xo a foam. Yield 12.55 g (85%). 1H NMR (ace- tone- 6) δ 11.22 (bs, 1 H), 7.40-7.20 (m, 5H), 6.58 (bs, 1 NH), 6.23 (d, 1 NH, J = 7.7 Hz), 5.07 (s, 2H), 4.35-4.25 (m, 1 H), 3.65-3.60 (m, 1 H), 3.55-3.50 (m, 1 H), 1.39 (s, 9H). 3C-NMR (acetone-de) δ 171.76, 157.22, 155.89, 137.70, 128.69, 128.11, 78.91 , 66.22, 54.56, 42.57, 28.03. MS: expected: 338.4; observed: 338.2.
(2-tert-Butoxycarbonylamino-2-methylcarbamoyl-ethyl)-carbamic acid benzyl ester
C) To a solution of N^Boc-N^-Z-Diaminopropionic acid (11.5 g; 34 mmol), HOBt (7,8 g, 51 mmol, 1 ,5 eq.) and methylamine (8M in ethanol, 6.4 mL, 1.5 eq.) in DCM:DMF
(10:1 ; 110 mL) at 0°C was added DCC (8.4 g, 40.8 mmol, 1.2 eq.) and the mixture was stirred for 2 h while warming to 20°C. 10% aq. NaHCO3 (30 mL) was added and the mixture was stirred for further 30 min. The mixture was filtered and the phases separated. The organic phase was extracted with aq. KHSO4 (0.5M, 30 mL) and brine (30 mL), dried (MgSO4) and concentrated in vacuo to a white solid. Yield 7.5 g (63%). MS: [M+H] expected: 352.4; observed: 351.9.
(2-Amino-1-methylcarbamoyl-ethyl)-carbamic acid tert-butyl ester
D) To a solution of (2-tert-Butoxycarbonylamino-2-methylcarbamoyl-ethyl)-carbamic acid benzyl ester (7.0 g, 19.9 mmol) in methanol (100 mL) was added Pd/C (310 mg). The mixture was hydrogenated for 1 h at 1 atm, filtered (celite) and concentrated in vacuo to a foam. Yield 4.3 g (100%). MS: [M+H] expected: 218.30; observed: 218.07.
(2-tert-Butoxycarbonylamino-2-methylcarbamoyl-ethylamino)-acetic acid ethyl ester E) To a stirred mixture of (2-Amino-1-methylcarbamoyl-ethyl)-carbamic acid tert- butyl ester (4.2 g, 19.4 mmol) and triethylamine (3.5 mL, 25.2 mmol, 1.3 eq.) in THF (80 mL) was added a solution of ethyl bromoacetate (2.37 mL, 21.3 mmol, 1.1 eq.) in THF (20 mL) dropwise over 15 min. The mixture was stirred for 16 h, filtered and concentrated in vacuo to an oil, which was purified by flash chromatography (silica, 7.5% methanol in dichloromethane). Yield: 3.5 g (59%). MS: [M+H] expected: 304.36; observed: 304.04.
[[2-(6-Benzyloxycarbonylamino-purin-9-yl)-acetyl]-(2-tert-butoxycarbonylamino-2- methylcarbamoyl-ethyl)-amino]-acetic acid ethyl ester
F) To a stirred suspension of (2-tert-Butoxycarbonylamino-2-methylcarbamoyl- ethylamino)-acetic acid ethyl ester (3.27 g, 10 mmol) and DHBt-OH (1.2 eq.) in DMF (50 mL) was added a solution of (6-benzyloxy-carbonylamino-purin-9-yl)-acetic acid (3.0 g, 10 mmol, 1.0 eq.) in DMF (25 mL). The mixture was cooled to 0°C and DCC (2.26 g, 11 mmol, 1.1 eq.) was added. The mixture was stirred for 1 h at 0°C and then at 20°C for 16 h. DCU was removed by filtration and washed with DMF (20 mL). The filtrate was concentrated in vacuo to 25% volume and DCM (100 mL) was added. The solution was washed with 0.5M KHSO4 (2 x 50 mL) and 5% aq. Na- HCO3 (2 x 50 mL) and activated carbon (1 g) was added. The mixture was stirred for 1 h, filtered and concentrated in vacuo to an oil, which was purified by flash chromatography (silica, 5% methanol in dichloromethane). Yield: 2.7 g (44%) MS: [M+H] expected: 613.27; observed: 613.10.
[[2-(6-Benzyloxycarbonylamino-purin-9-yl)-acetyl]-(2-tert-butoxycarbonylamino-2- methylcarbamoyl-ethyl)-amino]-acetic acid
G) To a mixture of [[2-(6-Benzyloxycarbonylamino-purin-9-yl)-acetyl]-(2-tert- butoxycarbonylamino-2-methylcarbamoyl-ethyl)-amino]-acetic acid ethyl ester (2.6 g, 4.25 mmol) in THF (50 mL) was added a mixture of LiOH (1.25 eq.) in water (7 mL). The mixture was stirred for 2 h and then concentrated in vacuo to 15% vol. pH was adjusted to -2.5 with 1 M aq. HCl and ethyl acetate (20 mL) was added. The
phases were separated and the organic phase was concentrated in vacuo to a foam. Yield: 2.47 g (99%). MS: [M+H] expected: 585.23; observed: 585.3.
EXAMPLE 53
Compounds of the invention were prepared according to the following reaction schemes.
First, a carbohydrate-linker building block is synthesized according to Scheme 1 :
1) TFA
Scheme 1
Then, a peptide nucleic acid molecule of the invention is synthesized according to Scheme 2:
Deprotection of Bz groups
Scheme 2
wherein B is a naturally-occurring nucleobase preferably A, T, G, or C and n is an integer of from 3 to 49 and wherein the nucleobases are selected in order to bind to the target DNA or target RNA.
EXAMPLE 54
Compounds of the invention were prepared as described in Example 53 based upon the following carbohydrate-linker building block:
resulting in a compound of the following formula:
wherein B is a naturally-occurring nucleobase preferably A, T, G, or C and n is an integer of from 3 to 49 and wherein the nucleobases are selected in order to bind to the target DNA or target RNA.
EXAMPLE 55
Compounds of the invention were prepared as described in Example 53 based upon the following carbohydrate-linker building block:
resulting in a compound of the following formula:
wherein B is a naturally-occurring nucleobase preferably A, T, G, or C and n is an integer of from 3 to 49 and wherein the nucleobases are selected in order to bind to the target DNA or target RNA.
EXAMPLE 56 Preparation of GalNAc(OBz)3-0-(CH2)4-COOH (G)
GalNAc(OAc)4 (A)
D-Galactosamine, HCl (0,185mol; 40g), DMAP (0,018mol; 2,26g) and acetic anhy- dride (2,4mol; 240mL) were mixed in pyridine (400 mL) at RT. After 16h the reaction mixture was evaporated to an oil, and ethyl acetate (200 mL) was added. The mixture was stirred and after 2 h filtered through a glass filter, and the white product
washed with one portion of ethyl acetate. The filtrate evaporated again, and by adding ethyl acetate more product was isolated. The white crystals were dried in vacuum oven at 25°C, 10mbar. Yield 59g (83%)
Intermediate (B)
A (0,051 mol; 20g) was dissolved in dry 1 ,2-dichloroethane (250mL) in a dry flask. Trimethylsilyl trifluoromethanesulfonate (0,056mol; 10mL) was added and reacted by heating to 50°C for 2 h. The reaction was performed in N2-atmosphere. Triethylamine was added to the reaction until pH 8-9 and the resulting mixture was filtered through a short silica-gel column (2 cm), evaporated and further purified on a silica- gel column using ethyl acetat + 0,1% triethylamine as eluent. Yield: 16,6g (99%)
GalNAc(OAc)3-0-(CH2)5-OTr (C)
B (0,050mol; 16,6g) and 5-Trityloxy-pentan-1-ol (0,060mol, 20,8g) were dissolved in dichloroethane (200mL) under N2. Molecular sieve 4A (10g) was added to the reaction. After 30 min trimethylsilyl trifluoromethanesulfonate (0,005mol; 1mL) was added to the yellow mixture. After stirring at room temperature for 20 h, the mixture was added triethylamine (20mL) and filtered through a short silica-gel column (2 cm), The filter was washed 5 times with a mixture of ethyl acetate + 0,1% triethyl- amine (5 * 100mL) and the combined organic phases were evaporated. The resulting mixture was purified on a silica-gel column using ethyl acetate/ toluene (6:4) as eluent. Fractions identified as C were collected and evaporated. Yield: 17,4g (50%)
GalNAc(OBz)3-0-(CH2)5-OTr (D) C (0,025mol; 17,2g) was suspended in methanol (220 mL) under N2 and potassium tert-butoxide (0,002mol; 0,2g) was added. After 1 h the reaction was evaporated to dryness and subsequently treated three times with pyridine (3 * 30mL) followed by evaporation. The resulting oil was dissolved in pyridine (200mL) under N2 and ben- zoic anhydride (0,075mol; 20g) and a catalytic amount of 4-dimethylaminopyridine (0,5g) was added. After stirring at room temperature for 16 h, the mixture was evaporated to dryness, dissolved in ethyl acetate (200mL) and extracted with half- saturated NaHCO3 (2 * 100mL) and water (2 * 200 mL)* DMEDA (20mL) was added to the organic layer, and this was extracted with half-saturated KHSO4 (6 * 100mL),
brine (200mL) and dried over MgSO4. Filtering and evaporation gave the product D. Yield: 16,4g (75%)
GalNAc(OBz)3-0-(CH2) -OH (E) D (0,018mol; 16g) was dissolved in a mixture of methanol/ formic acid (100mL/
120mL). The mixture was stirred at room temperature for 1 h and slowly added over dry K2CO3 (approx. 150g). The mixture was stirred in 3 h, and extracted between ethyl acetate (300mL) and water (200mL). The organic layer was drying over MgSO . Filtering and evaporation gave a crude product, which was further purified on a silica-gel column using toluene/ ethyl acetate (6:4) as eluent. Yield: 8,4g (75%)
GalNAc(OBz)3-0-(CH2)4-CHO (F)
Oxalylchloride (0,028mol; 2,47mL) was dissolved in DCM (100mL) and cooled on acetone/ dryice bath to -60°C. DMSO (30mL) in DCM (2M) was added and reacted in 5 min. A solution of E (0,014mol; 8,4g) in DCM (100mL) was added dropwise (10 min). After stirring cold for 40 min triethyl amine (17,3mL) was added and coolbath removed. To the resulting mixture was added ethyl acetate (300mL) and extracted with water (200mL), 10% NaHCO3 in water (2 * 150mL) and finally water (200mL). The organic phase was evaporated to dryness, and subsequently treated three times with toluene followed by evaporation. Yield: 6,8g (99%)
GalNAc(OBz)3-0-(CH2)4-COOH (G)
F (0,011 mol; 6,8g) was dissolved in a mixture of NaH2PO4 (0,073mol; 8,72g), Na- CIO2 (90%)(0,078mol; 8,83g), 2-Methyl-2-butene (0,715mol; 75mL) in acetonitrile/ water (1 :1 ; 400mL). After stirring at room tempeature for 1 h, the mixture was added ethyl acetate (50mL) and water (30mL), followed by separation of the organic layer. The aq. phase was extracted with ethyl acetate (3 * 50mL). The combined organic phases were dried over MgSO4, and finally filtering and evaporation gave the product G. LC-MS (electrospray): found m/z 633,2; theoretical m/z 633,2. Purity (RP-HPLC, 251 nm): 97%. Yield: 6,92g (99%).
EXAMPLE 57
Preparation of GalNAc(OBz)3-0-(CH2)4-CONH-Lys(GalNAc(OBz)3-0-(CH2)4-
CONH-)-Gly-OH (K).
Boc-Lys(Boc)-Gly-OBn (H)
Boc-Lys(Boc)-OSu (11 ,57mmol; 5,0g) was dissolved in DCM (50mL) and H-Gly- OBzl, Ts (12,4mmol; 4,17g) was dissolved in DCM, deprotonated with DIEA (12,4mmol; 2,0mL) and added to the reaction. The mixture was stirred for 1 h, extracted with 50% sat. aq. NaHCO3 (50mL) and 50% sat. aq. KHSO4 (50mL). The organic phase was dried over MgSO4 and concentrated in vacuo to an oil. Yield: 5,5g (99%).
GalNAc(OBz)3-0-(CH2)4-CONH-Lys(GalNAc(OBz)3-0-(CH2)4-CONH-)-Gly-OBn (J)
HOAt (1 ,32mmol; 180mg), EDC (1 ,32mmol; 254mg), DIEA (1 ,32mmol; 230μL) and G (1 ,2mmol; 543mg) were dissolved in dry DCM (4mL). Reaction time 3 h.
H (0,551 mmol; 272mg) was stirred in DCM/ trifluoroacetic acid (1:1 ; 6mL) for 1 h and then concentrated in vacuo and coevaporeted with DCM (5mL), toluene (5mL) and DCM (2 * 5mL). The resulting oil was dissolved in DCM/ DMF (5:1 ; 6mL). pH adjust to 8 with NMM and added to the mixture of the activated acid. Reaction time 20 h. Ethyl acetate (8mL) and water (5mL) was added to the reaction, followed by separation of the organic layer. The organic layer was extracted with 50% sat. aq. KHSO4 (2 * 5mL), 50% sat. aq. NaHCO3 (2 * 5mL), water (2 * 5mL) and brine (5mL). The organic phase was dried over MgSO4, filtered and evaporated to give the crude product, which was purified on a silica-gel column using DCM/ methanol (15:1 ) as eluent. Yield: 420mg (50%).
GalNAc(OBz)3-0-(CH2)4-CONH-Lys(GalNAc(OBz)3-0-(CH2)4-CONH-)-Gly-OH (K)
J (0,275mmol; 420mg) was dissolved in ethanol/ THF (1 :1 ; 75mL) under N2, 10% Pd/C (50mg) was added and the mixture was hydrogenated with H2 gas. After 2 h the reaction mixture was filtered through Hyflo®, which was rinsed afterwards with 1 portion of ethyl acetate (20mL). The combined organic phases were evaporated to dryness, and subsequently treated three times with toluene followed by evaporation. LC-MS (electrospray): found m/z 1434,3; theoretical m/z 1433,6. Purity (RP-HPLC,
210 nm): 97%. Yield: 395mg (99%).
EXAMPLE 58
Preparation of GalNAc(OBz)3-0-(CH2)4-CONH-Lys(GalNAc(OBz)3-0-(CH2)4- CONH)-Lys(GalNAc(OBz)3-0-(CH2)4-CONH)-Gly-OH (S)
Boc-Lys(Z)-OMe (L)
Boc-Lys(Z)-OH (0,026mol; 10,0g) and NaHCO3 (0,079mol; 6,6g) were dissolved in DMF (75mL) in a dark flask (light sensitive!). Methyl iodide (0,029mol; 1 ,8mL) was added to the reaction. After stirring at room temperature for 16 h, ethyl acetate (400mL) was added and the mixture was extracted with water (2 * 100mL), brine (100mL), dried over MgSO4 and filtered through a short silica-gel column. The filtrate was concentrated in vacuo to a yellow oil. Yield: 10,2g (99%).
Boc-Lys-OMe (M)
L (0,028mol; 11 ,6g) was dissolved in a mixture of ethanol/ THF (1 :1 ; 100mL) under N2. 10 % Pd/C (0,6g) was added and the mixture was hydrogenated with H2 gas. After 1 h the reaction mixture was filtered through Hyflo®, which was rinsed afterwards with 2 portions of ethyl acetate (2 * 25mL). The combined organic phases were evaporated to dryness, and subsequently treated three times with toluene followed by evaporation. Yield: 7,9g colourless oil (99%).
Boc-Lys(Boc-Lys(Boc))-OMe (N)
M (0,007mol; 1 ,85g) was dissolved in dry DCM (20mL) under N2. After cooling to 0°C on ice bath Boc-Lys(Boc)-OSu (0,007mol; 3,2g) was added to the reaction and the ice bath was removed. The mixture was stirred for 5 h. DMEDA (2mL) was added. After stirring for 15 min the reaction mixture was extracted with water (20mL), 20% KHSO4 (20mL), water (20mL) and brine (20mL). The organic layer was dried over MgSO4, filtered and evaporated. The resulting mixture was purified on a silica-gel column using DCM/ methanol (19:1 ) as eluent. Yield: 2,7g white crystals (65%).
Boc-Lys(Boc-Lys(Boc))-OH (P)
N (0,004mol; 2,7g) was dissolved in THF (200mL) and 1N LiOH (15mL) was added. After stirring for 3 h water (100mL) was added, and pH adjusted with 2N HCl to pH 3. The mixture was extracted with ethyl acetate (3 * 100mL) and the combined or- ganic phases were extracted with brine (150mL), dried over MgSO4, filtered and evaporated to dryness. Yield: 2,65g colourless oil (99%).
Boc-Lys(Boc-Lys(Boc))-Gly-OBn (Q)
P (0,0045mol; 2,6g) was dissolved in DCM (50mL) and cooled to -10°C. H-Gly-OBzl, Ts (0,0054mol; 1,82g) was deprotonated with DIEA and added to the reaction. HOBt (0,0054; 0,73g), EDC (0,0054mol; 1,03g) were added and the pH was adjusted with DIEA to pH 7-8. After 2 h reaction pH was adjusted again. After 20 h reaction 10% aq. NaHCO3 (30mL) was added. After stirring for 30 min the reaction was extracted with water (2 * 50mL), 50% sat. aq. KHSO4 (2 * 50mL), water (2 * 50 mL) and brine (50mL). The organic phase was evaporated, and the resulting oil was purified on a silica-gel column using DCM/ methanol (19:1 ) as eluent. Yield: 2,2g colourless oil (68%).
GalNAc(OBz)3-0-(CH2)4-CONH-Lys(GalNAc(OBz)3-0-(CH2)4-CONH)- Lys(GalNAc(OBz)3-0-(CH2)4-CONH)-Gly-OBn (R)
HOAt (I .Ommol; 0,14g), EDC (1,1mmol; 0,21g), DIEA (1,2mmol; 210μL) and G (1 ,0mmol; 0,64g) were dissolved in dry DCM (10mL). Reaction time 3 h. In another flask Q (0,305mmol; 0,22g) was dissolved in 5mL DCM and trifluoroacetic acid (5mL) was added. After 2 h reaction the mixture was evaporated and pH adjust to 8- 9 with DIEA. The deprotected compound Q was dissolved in DCM (5mL) and DMF (1 mL) and added to the mixture of the activated acid. Reaction time 20 h. Ethyl acetate (20mL) and water (25mL) was added to the reaction, followed by separation of the organic layer. The organic layer was extracted with 50% sat. aq. KHSO4 (2 * 20mL), 50% sat. aq. NaHCO3 (2 * 20 mL), water (2 * 20mL) and brine (20mL). The organic phase was dried over MgSO4, filtered and evaporated to give the crude product, which was purified on a silica-gel column using DCM/ methanol (15:1 ) as eluent. Yield: 76mg colourless oil (50%).
GalNAc(OBz)3-0-(CH2)4-CONH-Lys(GalNAc(OBz)3-0-(CH2)4-CONH)- Lys(GalNAc(OBz)3-0-(CH2)4-CONH)-Gly-OH (S)
R (0,003mmol; 76mg) was dissolved in ethanol/ THF (1 :1 ; 10mL) under N2, 10% Pd/C (25mg) was added and the mixture was hydrogenated with H2 gas. After 2 h the reaction mixture was filtered through Hyflo®, which was rinsed afterwards with 1 portion of ethyl acetate (4mL). The combined organic phases were evaporated to dryness, and subsequently treated three times with toluene followed by evaporation. LC-MS (electrospray): found m/z 2176,9; theoretical m/z 2177,3. Purity (RP-HPLC, 254 nm): 97%. Yield: 60 mg colourless oil (82%).
EXAMPLE 59
Preparation of GalNAc(OBz)3-0-(CH2)4-CONH -Lys(GalNAc(OBz)3-0-(CH2)4- CONH -Lys(GalNAc(OBz)3-0-(CH2)4-CONH -Lys(GalNAc(OBz)3-0-(CH2)4- CONH)))-Gly-OH (Z)
Boc-Lys(Boc-Lys(Z))-OMe (T)
M (18,05mmol; 4,7g) was dissolved in dry DCM/ dry DMF (10:1 ; 100mL) under N2. After cooling to 0°C on ice bath Boc-Lys(Z)-OH (18,05mmol; 6,9g), HOBt (18,05mmol; 2,4g), EDC (19,85mmol; 3,8g) and DIPEA (27mmol; 4,7mL) was added to the reaction and the ice bath was removed. The mixture was stirred for 20 h. The reaction mixture was extracted with water (100mL), 20% sat. aq. KHSO4 (100mL), water (100mL) and brine (100mL). The organic layer was dried over MgSO4, filtered and evaporated. The resulting oil was precipitated with DCM/ Petroleum ether (40- 60°C). Yield: 6,7g white crystals (60%).
Boc-Lys(Boc-Lys(H))-OMe (U)
T (10,44mmol; 6,5g) was dissolved in a mixture of ethanol/ THF (1 :1 ; 100mL) under N2. 10 % Pd/C (0,5g) was added and the mixture was hydrogenated with H2 gas. After 2 h the reaction mixture was filtered through Hyflo®, which was rinsed afterwards with 2 portions of ethyl acetate (2 * 50mL). The combined organic phases were evaporated to dryness, and subsequently treated three times with toluene followed by evaporation. Yield: 5,1 g colourless oil (99%).
Boc-Lys(Boc-Lys(Boc-Lys(Boc)))-OMe (V)
U (10,64mmol; 5,1g) was dissolved in dry DCM (20mL) under N2. After cooling to 0°C on ice bath Boc-Lys(Boc)-OSu (10,64; 4,7g) was added to the reaction and the ice bath was removed. The mixture was stirred for 20 h. The reaction mixture was extracted with water (20mL), 20% KHSO4 (20mL), water (20mL) and brine (20mL). The organic layer was dried over MgSO4, filtered and evaporated. The resulting mixture was purified on a silica-gel column using DCM/ methanol (15:1 ) as eluent. The resulting oil was dissolved in hot ethyl acetate and precipitated with hexane. Yield: 4,7g white crystals (55%).
Boc-Lys(Boc-Lys(Boc-Lys(Boc)))-OH (W)
V (4,89mmol; 4,0g) was dissolved in THF (60mL) and 1 N LiOH (7,3mL) was added. After stirring for 6 h pH adjusted with 2N HCl to pH 4. The mixture was evaporated to an oil, dissolved in DCM (50mL) and extracted with water (50mL), brine (150mL), dried over MgSO4, filtered and evaporated to an oil. Yield: 3,95g colourless oil (99%).
Boc-Lys(Boc-Lys(Boc-Lys(Boc)))-Gly-OBn (X) W (4,89mmol; 4g) was dissolved in DCM/ DMF (5:1 ; 50mL) and cooled to -10°C. H- Gly-OBzl, p-TsOH (4,89mmol; 1 ,7g) was dissolved in DCM, deprotonated with DIEA (12,25mmol; 2,1mL) and added to the reaction. HOBt (4,89mmol; 0,66g), EDC (5,38mmol; 1,03g) were added and the pH was adjusted with DIEA to pH 7-8. After 2 h reaction pH was adjusted again. After 20 h reaction 10% aq. NaHCO3 (30mL) was added. After stirring for 30 min the reaction was extracted with water (2 * 50mL), 50% sat. aq. KHSO4 (2 * 50mL), water (2 * 50 mL) and brine (50mL). The organic phase was evaporated, and the resulting oil was dissolved in refluxing ethyl acetate. The mixture was placed on an ice bath and the compound precipitate after 20min. Yield: 4,1g white crystals (87%).
GalNAc(OBz)3-0-(CH2)4-CONH -Lys(GalNAc(OBz)3-0-(CH2)4-CONH - Lys(GalNAc(OBz)3-0-(CH2)4-CONH -Lys(GalNAc(OBz)3-0-(CH2)4-CONH)))-Gly- OBn (Y)
HOAt (6,94mmol; 0,88g), EDC (7,57mmol; 1 ,31g), DIEA (9,47mmol; 1 ,31mL) and G (6,313mmol; 4,0g) were dissolved in dry DCM (50mL). Reaction time 3 h. In another flask X (1 ,05mmol; 1 ,0g) was dissolved in DCM (10mL) and trifluoroacetic acid (10mL) was added. After 2 h reaction the mixture was evaporated and pH adjust to 8-9 with DIEA. The deprotected compound X was dissolved in DCM (50mL) and DMF (20mL) and added to the mixture of the activated acid. Reaction time 20 h. Ethyl acetate (200mL) and water (100mL) was added to the reaction, followed by separation of the organic layer. The organic layer was extracted with 50% sat. aq. KHSO4 (50mL), 50% sat. aq. NaHCO3 (50mL), water (50mL) and brine (50mL). The organic phase was dried over MgSO4, filtered and evaporated to give the crude product, which was purified on a silica-gel column using DCM/ methanol (15:1 ) as eluent. Yield: 1 ,5g (40%).
GalNAc(OBz)3-0-(CH2)4-CONH -Lys(GalNAc(OBz)3-0-(CH2)4-CONH -
Lys(GalNAc(OBz)3-0-(CH2)4-CONH -Lys(GalNAc(OBz)3-0-(CH2)4-CONH)))-Gly-
OH (Z) Y (0,498mmol; 1 ,5g) was dissolved in ethanol/ THF (3: 1 ; 10OmL) under N2, 10% Pd/C (150mg) was added and the mixture was hydrogenated with H2 gas (balloon). After 20 h the reaction mixture was filtered through Hyflo®, which was rinsed afterwards with 1 portion of ethyl acetate (100mL). The combined organic phases were evaporated to dryness, and subsequently treated three times with toluene followed by evaporation. LC-MS (electrospray): found m/z 2921 ,1 ; theoretical m/z 2920,2. Purity (RP-HPLC, 254 nm): 91%. Yield: 1,2g (82%).
EXAMPLE 60 Preparation of (GalNAc(OH)3)2-Lys-Gly-CATCACTGGCAGACCCTG-NH2
In a 10-mL round-bottomed flask an aeg-PNA of the sequence H- CATCACTGGCAGACCCTG-NH2 (85,42 mg; 0,0176 mmoles) and K (50,60 mg;
0,0353 mmoles, 2,00 eq,) were dissolved in DMSO (7,7 mL). A 0.125M solution of HOBt in DMSO (296,3 μL; 0,0370 mmoles, 2,10 eq.) was added and pH (5-6) adjusted to 7-8 by addition of 10% NMM in DMSO (500 μL). A 0.125M solution of DIC in DMSO (564,4 μL; 0,0706 mmoles, 4,00 eq.) was finally added and the reaction mixture was stirred at room temperature for 45 h.
Deprotection of the benzoyl groups was performed by addition of 1 % NaOH in MeOH (1 ,47 mL) to the reaction mixture until the pH was raised to 10-12. The de- benzoylation reaction was completed after 2h 30min. and the crude construct was precipitated by pouring the mixture over cold diethyl ether (90 mL). The suspension thus obtained was centrifuged, the ether discarded and the pellet washed thoroughly two times with ether (2 x 90 mL). The resulting pellet was dissolved in water (11 mL) and the pH adjusted to 1-2 with TFA to facilitate dissolution of the solid. The crude solution was purified by RP-HPLC and after freeze-drying 21 mg of the pure compound was obtained as a white powder. LC-MS (electrospray): found m/z 5631 ,8; theoretical m/z 5632,5. Purity (RP-HPLC, 210 nm): 97%. Total yield: 21 ,1 %
EXAMPLE 61
Preparation of (GalNAc(OH)3)3Lys2Gly-GTGGATGATACCTGGATC-NH2
In a 25-mL round-bottomed flask an aeg-PNA of the sequence GTGGATGA- TACCTGGATC-NH2 (200,62 mg; 0,04052 mmoles) was dissolved in DMSO (4 mL). A solution of S (140,92 mg; 0,06469 mmoles, 1 ,60 eq) in DMSO (1 mL) was added followed by a 0,125M solution of HOBt in DMSO (543 μL; 0,06787 mmoles, 1 ,68 eq). The pH («3) was raised to 7-8 by addition of 10% NMM in DMSO (1 ,1 mL). Then, a 0,125M solution of DIC in DMSO (2,07 mL; 0,2588 mmoles, 6.40 eq) was added together with DMSO (1 ,29 mL) to reach a final concentration of 20 mg PNA/ mL. The reaction mixture was stirred at room temperature for 21 h 30 min. To perform debenzoylation, 1%NaOH in MeOH was added over the mixture until de pH was raised to 10-12 (3,2 mL). After a few seconds the solution turned opalescent and afterwards a fine white powder precipitated. Deprotection of the benzoyl groups was completed after 2 h. The crude was precipitated in cold diethyl ether (160 mL), pelleted by centrifugation and further washed with ether (200 mL). The crude com-
pound was isolated by centrifugation, dissolved in water (6mL) and pH adjusted to
2-3.
The crude solution was purified by RP-HPLC and after freeze-drying, 140,7 mg of pure compound was obtained as a white powder. LC-MS (electrospray): found m/z
6173,8; theoretical m/z 6174,1. Purity (RP-HPLC, 210 nm): 96%.
Total yield: 56,2%
EXAMPLE 62
Preparation of (GalNAc(OH)3)4Lys3Gly-GTGGATGATACCTGGATC-NH2
In a 25-mL round-bottomed flask, an aeg-PNA of the sequence H- GTGGATGATACCTGGATC-NH2 (350,42 mg; 0,0708 mmoles) was dissolved in DMSO (5 mL). A solution of Z (333,50 mg; 0,1141 mmoles, 1 ,61 eq) in DMSO (2 mL) was added followed by a 0,125M solution of HOBt in DMSO (951 μL; 0,1188 mmoles, 1 ,68 eq). By addition of 10% NMM in DMSO (1,8 mL), the pH («3) was raised to 7-8 and then a 0.125M solution of DIC in DMSO (3,62 mL; 0,4525 mmoles, 6.39 eq) was added together with DMSO (4,13 mL) to reach a final concentration of 20 mg PNA/mL. The reaction mixture was stirred at room temperature for 21 h 30'. Debenzoylation was performed by addition of 1% NaOH in MeOH (7,5mL) until the pH was raised to 10-12. A fine white suspension was obtained, and after 2h 40min. the reaction was completed. The mixture was poured over cold diethyl ether (160 mL), the compound separated by centrifugation, washed thoroughly with ether (200 mL) and the suspension further centrifuged. The crude pellet was dissolved in water (12 mL) and the pH adjusted to 2-3. After purification (RP-HPLC) and freeze-drying, 200,08 mg of compound was obtained as a white powder. LC-MS (electrospray): found m/z 6604,5; theoretical m/z 6605,6. Purity (RP-HPLC, 210 nm): 96%. Total yield: 42,8%.
EXAMPLE 63
Preparation of Folate-CCTCTTACCTCAGTTACA-NH2
An aeg-PNA of the sequence H-CCTCTTACCTCAGTTACA-NH2 (10,00 mg;
0,0021 mmoles) was dissolved in DMSO (1 mL). A 0,13M solution of folic acid in DMSO (161 ,4 μL; 0,0210 mmoles) was added together with a 0,125M solution of HOBt in DMSO (167,8 μL; 0,0210 mmoles). The pH of the mixture (5-6) was adjusted to 7-8 by addition of 10% NMM in DMSO (60 μL), and then a 0,125M solution of DIC in DMSO (167,8 μL; 0,0210 mmoles) was added.
The mixture was stirred at room temperature for 21 h 30 min. The conjugate and excess of folic acid were precipitated by pouring the reaction mixture over cold diethyl ether (10 mL) and pelleted by centrifugation. The pellet was washed twice with ether (2 x10 mL) and then dissolved in a mixture of DMSO (0,5 mL) and 0,1% TFA in wa- ter (3,5mL).
After purification (RP-HPLC) and freeze-drying, 5,00 mg of compound was obtained as a yellow powder. LC-MS (electrospray): found m/z 5188,1 ; theoretical m/z 5189,1. Purity (RP-HPLC, 210 nm): 94% Total yield: 45,9%
EXAMPLE 64
Preparation of PNA and PNA Conjugates
The PNA was synthesized on MBHA-resin in a sequential manner. The oligomers were synthesized from Boc-protected PNA-monomers using a standard SPPS protocol. The products were cleaved from the resin using trifluoromethane sulphonic acid. Crude deprotected PNAs were purified by preparative HPLC and subsequently lyophilized.
Two 18mer PNA oligomers, PNA-I and PNA-II, were synthesized, cleaved, purified and lyophilized. The compounds were isolated in 1 g quantities.
GalNAc constructs
GalNAc(OBz)
3-O-pentanoic acid (A, Scheme I) was prepared in five steps according to known procedures. The synthesis, which starts from inexpensive D- galactosamine, is high yielding and scalable.
A
Scheme
A was used in the preparation of di-, tri- and tetr a-antennary ligands (Sceme l-IV). Solid-phase synthesis of similar di- and tri-antennary lysine-based cluster galactosides has previously been reported. However, attempts to apply the described pro- cedures resulted in very poor yields and, consequently, the synthetic protocol was redesigned for solution phase synthesis. In contrast to the original solid phase procedure the developed procedure is robust and scalable.
Achiral glycine was incorporated as a C-terminal linker in the cluster galactosides to avoid racemization in the subsequent condensation with the PNA oligomer.
The di-an tennary ligand (C, Scheme II) was synthesized by initial coupling of pre- activated Boc-Lys(Boc)-OSu to H-Gly-OBn giving scaffold Boc-Lys(Boc)-Gly-OBn (B). Cleavage of the Boc groups and coupling with IBC activated GalNAc(OBz)3-O- pentanoic acid (A) gave the di-substituted construct, which was hydrogenated to the desired product (C).
Scheme II
In the synthesis of the tri- and tetra-antennary GalNAc-constructs three orthogonal protecting groups were necessary and, hence, the synthesis was performed in a slightly different manner. Boc-Lys(Z)-OH was initially methylated and the Z-group was removed by hydrogenation giving Boc-Lys-OMe (D). Coupling with pre- activated Boc-Lys(Boc)-OSu gave Boc-Lys(Boc-Lys(Boc))-OMe (E). Hydrolysis of the methyl ester and subsequent coupling with H-Gly-OBn gave Boc-Lys(Boc- Lys(Boc))-Gly-OBn (F). Removal of the Boc groups and coupling with IBC activated GalNAc(OBz)3-O-pentanoic acid (A) gave the tri-substituted construct, which was hydrogenated yielding the desired product (G).
Scheme III
The tetra-antennary GalNAc-construct (H, Scheme) was synthesized in a manner similar to the tri-antennary GalNAc-construct (G)
H
Scheme IV
The GalNAc monomer (A) was synthesized in 15 g scale, and the three ligands were synthesized in 2-5 g quantities.
Ligand-PNA Conjugation
Previously, transporter-PNA constructs have been synthesized in a sequential manner, e.g. KFF-PNA, or by different cross-linking methods involving thiols, e.g. disul- fides or maleimides. These methodologies are not compatible with the GalNAc-PNA chemistry for a number of reasons. The sequential synthesis involves coupling of the GalNAc-construct to PNA on resin. It is not feasible because of the subsequent cleavage conditions, which will also cleave the glycoside bonds. Disulfides might be attractive for screening purposes, but the poor biological and chemical stability of the disulfide bond impedes the use in a ligand-S-S-PNA drug. Incorporation of maleimides, e.g. using SMCC, is unattractive because of the resulting introduction of the bulky linker between the ligand and the PNA. Furthermore, the maleimide coupling results in two diastereomeres.
As a consequence of the limitations in the known cross-linking methods the development of an alternative procedure was initiated. It was attempted to perform a solu- tion phase segment condensation involving an amid bond formation between the C- terminal of the GalNAc-construct and the N-terminal of the PNA.
There are numerous factors that complicate such a condensation. The poor solubility of PNA in common organic solvents limits the choice of solvent to DMSO. The degree of activation on the carboxylic acid was limited by the demand of high selectivity between the N-terminal amine and the amines on the nucleobases. Furthermore, a quantitative and clean de-protection of the benzoyl protecting groups on the GalNAc-moieties was mandatory. Finally, the product should be isolated from the DMSO and dissolved in an aqueous buffer for purification.
A clean and high yielding one-pot segment condensation/de-protection was developed. The synthesis was performed at pH 7-8 in DMSO using DIC/HOBt activation. The subsequent hydrolysis of the Benzoyl protecting groups was achieved by addi-
tion of NaOH in methanol. Finally, the product was precipitated from the reaction mixture in diethyl ether.
The method is versatile and does not limit the choice of linker. It allows the incorporation of any ligand with a free carboxylic acid, e.g. protected peptides, bile acids and folic acid. Furthermore the formed amid bond is chemically and enzymatically stable.
Four constructs were synthesized in 100 mg scale:
• Construct la, based upon PNA-I and a ligand of formula C
• Construct lb, based upon PNA-I and a ligand of formula G
• Construct lla, based upon PNA-II and a ligand of formula G
• Construct lib, based upon PNA-II and a ligand of formula H.
PNA-I is an aeg-PNA of the sequence H-CATCACTGGCAGACCCTG-NH2 PNA-II is an aeg-PNA of the sequence H-GTGGATGATACCTGGATC-NH2
Synthesized compounds were purified on an automated preparative HPLC system and subsequently lyophilised. Identity and purity were determined using analytical HPLC and mass spectrometry. All compounds had an HPLC purity >95% at 210 nm.
EXAMPLE 65
In vivo testing of PNA and PNA Conjugates
Identification of PNAs provoking exon skipping of pre-mRNA expressed in mouse liver
Classical antisense molecules recruit RNaseH activity, which leads to degradation of the targeted mRNA, which in turn leads to reduced synthesis of the protein encoded by the targeted mRNA. This can be visualized by Northern blot, quantitative RT- PCR and Western blot analysis. However there is always a risk that such down regulations are due to unspecific effects on gene expression or translation not re-
lated to the desired antisense activity on the intended target. For this reason the lack of effect of scrambled or mismatch oligonucleotides is frequently reported in parallel. However such compounds are poor controls since alterations in the nucleotide sequence is likely to affect the compounds overall pharmaco-kinetic properties.
We therefore sought to obtain compounds with an inbuilt antisense control. We chose to target the splice pattern of pre-mRNA. There are numerous examples in the literature of non-RNaseH-recrui ting oligonucleotides capable of changing the splice pattern of pre-mRNA by hybridising to and thereby blocking a splice site. Such effects are easily visualized by RT-PCR analysis. It is very unlikely that splice variations specific for the targeted exon could occur by unspecific effects not related to a direct antisense interaction at the intended target.
Hnf4 The Hnf4 gene, encoding the Hepatocyte Nuclear Factor 4α (HNF4 α), was chosen as a model target for PNA liver-uptake studies for a number of reasons:
1. It is expressed in hepatocytes carrying the asialoglycoprotein (ASGP) receptors 2. it encodes a transcription factor of which both mRNA and protein are expected to have a short half-life
3. it is constitutively expressed, enabling simple experimental testing
PNAs were designed as 18-mers, spanning the intron/exon or exon/intron boundary with 9 bases overlapping either side. The PNA sequence was checked for possible problematic characteristics (self-complementarity, high purine content and long N- terminal purine stretches). Possible problems were corrected by choosing adjacent 18-mer sequences (still overlapping the splice site, but non-symmetrical) free of unwanted characteristics. A total of 13 PNAs were designed targeting different splice sites of Hnf4 pre-mRNA. The PNAs were evaluated in vitro using a BNL CL.2 mouse liver cell line. PNA intracellular delivery was achieved by annealing the PNA to a partly complementary DNA-oligo followed by co-transfection using a polyethylen- imine transfection reagent.
PNA effect on the splice pattern of Hnf4 mRNA was evaluated by RT-PCR on RNA extracted 20 hrs. after PNA-treatment of the cells.
Among active PNAs, PNA-II (H-GTGGATGATACCTGGATC-NH2) and PNA-I (H- CATCACTGGCAGACCCTG-NH2- were selected for in vivo studies.
PNA-II (targeting the 3'-end of exon 7) causes skipping exclusively of Hnf4 exon 7, observed as a new shorter RT-PCR product obtained with PCR primers external to exon 7. Minute hardly detectable levels of this splice variant does occur naturally, but the ratio between the normal and the rare splice variant is strongly shifted following PNA-II treatment.
PNA-II binds to Hnf4 pre-mRNA with all 18 bases and with 7 bases to correct spliced Hnf4 mRNA.
PNA-I (targeting the 5'-end of exon 9) causes exclusive skipping of Hnf4 exon 9. Again, very low levels of this splice variant occurs naturally, but the ratio between the normal and the rare splice variant, was strongly shifted following PNA-I treatment. PNA-I binds to Hnf4 pre-mRNA with all 18-bases and with coincidently with no less than 17 bases to correct spliced Hnf4 mRNA. As a consequence the PNA is capable of binding to Hnf4 mRNA and inhibits reverse transcriptase. Thus, traditional RT-PCR could not be used for evaluating the effect on splicing. However this problem was solved with a specially designed PCR triple primer mix (see "Analysis of liver samples" below).
Exon 7 and exon 9 skipping both yield in frame deletions in the reading frame of hnf4 mRNA. As a result a shorter HNF4 protein was synthesized which could be detected by Western blot analysis. This feature is an additional internal control of antisense activity and made PNA-II and PNA-I particularly interesting candidates for GalNAc modification.
Animal studies
Animals and housing
NMRI female mice (approx. weight 25 to 30 g at the day of the study) were housed in transparent macrolone cages and light cycles of 12 hours light and 12 hours darkness were used. The room temperature was 21°C ± 1°C and the relative humid- ity 50±10%. The animals were fed ad libitum using a complete rodent diet and had free access to clean drinking water. On the day of study the animals were randomly allocated to groups of two animals each for the different treatments.
Experimental design Animals were dosed subcutaneously (s.c). For dosing, 400 μM solutions (as determined by spectrometry from a theoretically calculated coefficient of extinction) of the respective test compounds in 0.625 mM AcOH, 5 % glucose in water, were used. At each dosing, the animal received 0.3 ml of the respective PA. Each dose corresponded to approximately 4.8 μmol PNA/kg equivalent to approximately 25 mg/kg. For multiple dosings, 6-hour intervals were used between same day dosings and 18- hour intervals before next day dosing.
Sampling
At the indicated number of days after the first dosing, the animals were anaesthe- tized by carbon di-oxide/oxygen and euthanized by partial decapitation and exsan- guination. Immediately hereafter, the abdominal cavity was opened and the liver excised. From the liver, the caudate lobe was isolated (approximate weight 0.1 g) and transferred to 1 ml of RNA preservation solution (RNAIater). Liver tissue for Western blot analysis was immersed in a protease inhibitor solution (ProPrep), homogenized and frozen for later analysis. The remaining liver tissue was frozen and stored at - 18°C for later compound analysis (not presented here).
From selected animals, different other tissues were also excised for RT-PCR analysis for Hnf4: a 3 mm section from the middle of the sagitally cut left kidney, a 3 mm transverse section from the middle of colon descendens, a 3 mm transverse section from the middle of jejunum, and a 3 mm transverse section from the pancreas. These tissues were preserved by immersion in 1 ml of RNA preservation solution (RNAIater).
Analysis of tissue samples
RT-PCR
Approximately 15-20 mg the stabilized liver sample was transferred into 1 ,0 ml RTL lysis buffer and homogenized. Total RNA was purified according to the RNeasy mini kit protocol and the concentration determined by OD260 measurement. cDNA was synthesized from 1 μg of RNA using random decamer primers in a total vol. of 20 μl. Briefly, RNA was preincubated at 70°C for 4 min prior to addition of M- MLV reverse transcriptase, followed by 60 min incubation at 42°C in the presence of 0,5 mM of each dNTP.
The PCR reaction was carried out in 35 cycles using SuperTaq polymerase in presence of 0,4 μM primers and 0,2 mM of each dNTP.
For PNA-II a single set of primers could be used as this sequence does not inhibit reverse transcription of Hnf4 mRNA: caatgaatatgcctgcctcaa (forward) attcagatcccgagccactt (reverse)
For PNA-I, RT-PCR analysis was complicated by the fact that PNA-I is a highly potent inhibitor of reverse transcription of Hnf4 mRNA between exon9 and exonδ due to extensive base complementarity to the exonδ/ 9 junction. This effect is specific for normally spliced Hnf4 mRNA. PNA-I does not affect reverse transcription of exon9 skipped Hnf4 mRNA. It has been found that PNA contaminates the RNA extractions from liver tissue. As a consequence cDNA formation of normally spliced mRNA is selectively inhibited at exon9 and the result of the ensuing PCR reaction (with prim- ers surrounding exon 9) is therefore artificially skewed in favour of the exonθ- skipped cDNA.
Exon 9 skip caused by PNA-I was therefore evaluated using a modified RT-PCR strategy based on two independent PCR reactions that specifically amplify either normally spliced or exon9 skipped Hnf4 mRNA. The two reactions were performed simultaneously in the same tube using a triple primer set. The forward primer in exon 10 is common to the two reactions. Two different reverse primers were designed to hybridise specifically with the exon8/10 junction (in exon 9 skipped mRNA)
and specifically with exon 9 respectively (in the normally spliced mRNA). The two reverse primers were in addition such designed that they give rise to slightly differently sized PCR products that can be separated and distinguished on an agarose gel. To validate this concept, PCR products specific for normally spliced mRNA and exon9 skipped mRNA were generated and mixed in 100:1 , 10:1 , 1 :1 , 1 :10 and 1 :100 relative concentrations. Subsequent PCR with the triple primer set yielded two products of the expected sizes that accurately reported the relative concentration of template (as long as the templates were sufficiently diluted). The technique was subsequently successfully used to evaluate the efficacy of PNA-I. The triple primer set used for RT-PCR of mRNA from PNA-l-treated animals were: Exon10 forward primer: ggtccctcgtgtcacatctt Exon9 reverse primer: cctcacctgatgcaagaaca Exon8/10 reverse primer: tgcttctcggagccactc
PCR products were loaded onto 2% agarose gels (containing EtBr) along side a DNA mass ruler. TBE running buffer was used for electrophoresis.
Western blot
Approximately 150 mg liver from mice treated with two different PNAs (PNA-I and PA5233, 2x4.8 μmol/kg/day for four days, PD-03-025) was homogenized in 4 ml detergent solution supplemented with proteinase inhibitors (Pro-Prep Solution from iN- tRON Biotechnology). The samples were kept at -20°C until analysis. The total protein concentrations in liver samples were measured by using a detergent compatible protein assay based on bicinchoninic acid (BCA) for calorimetric detection and quantification (Pierce).
About 70 μg total proteins from each sample was run on a 10% NuPage Bis-Tris gel (Invitrogen) and blotted onto a PVDF membrane according to manufacturers instructions (Invitrogen). The membrane was incubated with 0.25 μg/ml anti-HNF4α rabbit IgG targeting the C-terminal amino acid residues 455-465 of the human HNF4α pro- tein (Active Motif) followed by incubation with secondary antibodies and a chro- mogenic substrate from Invitrogen.
Gelshift assay
We have used a gel mobility shift assay (13) to quantify the amount of PNA in various murine organs. The principle in the gel-shift assay is that PNA-DNA hybrids migrate more slowly on a polyacrylamide gel than the DNA oligo alone, and that the amount of PNA can be quantified relative to a standard curve. The standard curve was constructed by mixing different amounts (pg) of PNA with 1 ng 33p labeled DNA oligo complementary to the PNA. For optimal annealing, the mixtures were heated to 95°C followed by a slow decrease in temperature. Organs from mice treated with PNA (and vehicle) were homogenized in 1.5 v/w 1 mM Tris-HCI, pH 8.0. The samples were boiled 5 min. followed by centrifugation. The supematants were diluted appropriately and incubated with 1 ng 33p labeled DNA oligo as described above. Glycerol was added to a final concentration of 10% and the samples were run on a 20% polyacrylamide TBE gel and blotted onto a nylon membrane. The result was visualized and quantified using an Instant Imager (an example is shown in figure 10).
Results
Initially we administered PNA to the mice either intravenously, intraperitoneally or subcutanously. Based on published results of in vivo activity of 4xlysin conjugated PNA (36), we chose a high dosis of 4.8 μM PNA/kg administered twice in a day (~50mg/kg/ day). We obtained no antisense effects in the liver with IV administration, while both SC and IP administration gave initial promising results. We carried out all the following experiments with SC administration.
Animals were sacrificed 18 hours after the last dose and liver biopsies were sam- pled for RT-PCR analysis. Virtually no antisense activity was observed with IV administration and only weak activities were registered upon SC and IP administration - at best the RT-PCR product of exon skipped mRNA corresponded to -1% of the total Hnf4 mRNA (data not shown). However this limited success was anticipated. Although the uptake of GalNAc-PNA in hepatocytes might be rapid and effective, entrapment of PNA within vesicles after the asialoglycoprotein receptor-mediated endocytosis is expected. Release of PNA to the nucleus, the site of pre-mRNA splicing, is dependent on the rupture of the vesicles - a process that could take considerable time. We therefore studied the kinetics of PNA antisense activity in the liver
upon a single day of SC administration. Our preliminary results are displayed in Figure 5. Animals were dosed subcutaneously twice in one day (4.8 μmol/kg/dosing) with the indicated PNA. Liver samples were gathered from animals sacrificed the indicated number of days after the PNA administration. RT-PCR analysis was per- formed on these samples. Antisense effect is visualized by a shifted ratio between PCR products corresponding to normal mRNA (359 bp) and exon 7 skipped mRNA (203 bp). Very low levels of exon 7 skipped mRNA could be detected in animals sacrificed 18 hours after PNA administration (not shown), but significant levels were observed after three days (-10%) and antisense activity increased radically 5 days after administration. At this time between 50-60% (estimated roughly on the relative level of EtBr staining) of the Hnf4 mRNA was lacking exon 7. This level of antisense activity remained unchanged until day 10 and in an ongoing study the activity appears to be only slightly receding at day 20 (data not shown). The number of animals in this pilot study are very limited and as seen in Figure 5, some animal to ani- mal variation is apparent. However the trend of a relatively slow occurrence of antisense activity peaking at around 5 days after PNA administration is clear.
The total lack of activity of unmodified PNA (Figure 5) demonstrated that the Gal- NAc modification dramatically improved the hepatic uptake of PNA. In addition the slow antisense kinetics was an expected feature of receptor-mediated uptake. To obtain further evidence of this uptake mechanism we looked for signs of antisense activity in tissues collected from other organs known to express Hnf4. Animals were dosed subcutaneously with the indicated PNA twice in one day (4.8 μmol/kg/dosing) and sacrificed 7 days after dosing. Samples from various organs were collected and subjected to RT-PCR analysis. Antisense effect is visualized (figure 6) by a shifted ratio between PCR products corresponding to normal mRNA (359 bp) and exon 7 skipped mRNA (203 bp). As can be seen in Figure 6 the antisense activity is restricted to the liver. This strongly suggests that the hepatocellular PNA uptake is indeed receptor-mediated since none of the cells in the other analysed tissues ex- press the asialoglycoprotein receptor. The lack of activity in the kidney is particularly significant, since the most of the PNA is known to accumulate rapidly in the kidney where it is then effectively secreted in the urine (37). Our preliminary data suggests that this is also true for GalNAc modified PNA (data not shown).
The long-lived antisense activity suggests that once highly stable PNA has been released to the cytosol and nucleus it is not readily excreted out of the cell. We therefore reasoned that it should be possible to accumulate PNA in hepatocytes by re- peated dosing over several days and thereby enhance the antisense activity. Figure 7 shows the results of 1 day and 4 days dosing of 2xGalNAc-PNA-l (2x4.8 μmol/kg/day). The mice were sacrificed 5 days after the last PNA administration and liver samples were subjected to RT-PCR analysis. Antisense effect is visualized by a shifted ratio between PCR products corresponding to normal mRNA (349 bp) and exon 9 skipped mRNA (254 bp). The result of a single days dosing regime is seen in lane 8 and 9. At this point only minute levels of exon 9 skipping (-1%) was observed with this PNA. Lane 6 and 7 shows the dramatic impact of 4 days of repeated dosing. Using a 33P-end labelled exon 10 forward primer in the PCR reaction we were able to quantify that the exon 9 skipped mRNA made up around 75% of the total Hnf4 mRNA (figure 8) - proof that GalNAc PNA indeed accumulates upon repeated dosing. It seems likely that we could achieve near complete down regulation of Hnf4 gene expression upon prolonged dosing. Again the unmodified PNA had no effect at all.
Exon 9 skipping is predicted to delete 51 amino acids from the HNF4α protein. If
75% of the Hnf4 mRNA is missing exon 9 this should be roughly reflected at the protein level. Figure 9 shows a Western blot analysis with HNF4α antibodies on protein extracts from the same liver samples analysed by RT-PCR in Figure 7. The figure shows the appearance of a novel slightly shorter protein at the expense of the nor- mal 51 KD HNF4α protein in the GalNac-PNA treated animals only. The proportion of the two different HNF4α proteins appears to reflect pretty accurately the 75% exon 9 skipped Hnf4 mRNA.
GalNAc modified PNA accumulates in the liver
GalNAc modification of PNA is supposed to ensure enhanced uptake selectively in hepatocytes and the above mentioned antisense activity clearly suggests that this is the case. In order to quantify PNA in tissues from PNA treated mice we imple- mented a slightly modified version of a published gel mobility shift assay (13), which we found to be very sensitive and accurate. The detection limit was in the range of 25pg PNA/g tissue - as shown in figure 10.
Table 1 Effect of GalNAc ligands on PNA delivery to the liver
Animals were dosed subcutaneously with the indicated PNA twice in one day or twice daily (4.8 μmol/kg/dosing) for four days as indicated. The mice were sacrificed 5 days after the last PNA administration and tissue samples were analysed for PNA content by gel mobility shift assay.
1 day 4 days 4 days
PNA μg PNA/g tissue μg PNA/g tissue μg PNA/g tissue
Liver Liver Kidney
PNA-II - 1 50
1xGalNAc-PNA-ll 2 9 22
2xGalNAc-PNA-ll 150 580 50
3xGalNAc-PNA-ll 240 700 60
4xGalNAc-PNA-ll 250 700 23
Table 1 summarizes a typical result of PNA distribution analysis. Tissue distribution was studied in animals sacrificed 5 days after last administration to ensure that the detected PNA was actually taken up by the tissue and not simply circulating in blood vessels. The ligand with a single GalNAc moiety had little or no effect on tissue distribution whereas the ligands with two or more GalNAc moieties caused significant accumulation in the liver. PNA conjugated with the bivalent GalNAc ligand accumulated in 100 fold higher concentration in the liver than unmodified PNA after a single day dose regime and 600 fold in the 4 day dose regime. Tri- and tetravalent ligands appeared only modestly more efficient at directing PNA to the liver than the bivalent ligand. GalNAc modification had no effect on distribution to any of the other tested organs and tissues - data not shown (muscle, colon, kidney and brain). The amounts of PNA found in muscle and colon was roughly half of that in the kidney, whereas we were unable to detect any PNA in the brain. PNA distribution to the brain upon IV administration has previously been described (13). However we find no evidence of this (at least not after SC administration) and conclude that PNA seems incapable of penetrating the blood-brain barrier.
The amounts of PNA accumulated in the liver are quite significant. Simple calculations based on the distribution data in table 1 show that after a four day dose regime at least 10% of the administered 2xGalNAc modied PNA is retained by the liver (the livers weighed on average 1 ,1g). In comparison only 0.02% of the dosed unmodified PNA is found in the liver. If the GalNAc modification ensures hepatic uptake via the asialoglycoprotein receptor, then majority of the PNA is likely to be trapped in vesicles upon endocytosis. In order to investigate the intracellular distribution of 2xGalNAc-PNA in hepatocytes we treated a mouse with a fluorescein tagged Gal- NAc modified PNA. The animal was dosed subcutaneously twice in one day (4.8 μmol/kg/dose) and sacrificed 5 days later. Histological examination of liver samples by fluorescense microscopy clearly show PNA accumulation in foci that is compatible with vesicular accumulation (see fig. 11 ).
REFERENCES
I . Nielsen, P.E., Egholm, M., Berg, R.H. and Buchardt, O., Science (1991 ) 254, 1497-1500. 2. Egholm, M., Buchardt, O., Christensen, L., Behrens, O, Freier, S. M., Driver, D , Berg, R.H., Kim, S.K., Norden, B., Nielsen, P.E., Nature (1993) 365, 566- 568.
3. Demidov, V., Potaman, V.N., Frank-Kamenetskii, M.D., Egholm, M., Buchardt, O. Sδnnichsen, H. S., Nielsen, P.E., Biochem. Pharmacol. (1994) 48, 1310- 1313.
4. Nielsen, P.E. and Haaima, G., Chemical Society Reviews (1997) 73-78.
5. Hanvey J.C., Peffer N.J., Bisi J.E., Thomson S.A., Cadilla R., Josey J.A., Ricca D.J., Hassman C.F., Bonham M.A., Au K.G., Science (1992) 258 (5087),1481-5.
6. Knudsen, H. and Nielsen, P.E., Nucleic Acids Res. (1996) 24, 494-500. 7. Good, L. and Nielsen, P.E., Proc. Natl. Acad. Sci. USA (1998) 95, 2073-2076.
8. Good, L. and Nielsen, P.E., Nature Biotechnology (1998) 16, 355-358.
9. Lewis, L.G. et al. Proc. Natl. Acad. Sci. USA (1996) 93, 3176-81.
10. Meyer, O. et al. J. Biol. Chem. (1998) 273, 15621-7.
I I . Nyce, J.W. and Metzger, W.J. Nature (1997) 385 721-725. 12. Pooga, M. et al, Nature Biotechnology (1998) 16, 857-61.
13. McMahon, B.M.; Mays, D.; Lipsky, J.; Stewart, J. A.; Fauq, A.; Richelson, E. Antisense & Nucleic Acid Drug Development 2002, 12, 65-70.
14. Zhang, X.; Simmons, C. G.; Corey, D. R.; Bioorganic & Medicinal Chemistry Letters 2001, 11 , 1269-1272. 15. Biessen, E. A. L.; Sliedregt-Bol, K.; Hoen, P.C.T.; Prince, P. Bilt, E. Valentijn, A. R. P. M.; Meeuwenoord, N. J.; Princen, H.; Bijsterbosch, M. K.; Marel, G. A., Boom, J. H., Berkel, T. J. C. Bioconjugate Chem. 2002, 13, 295-302.
16. Litt. 5
17. Lift. 6 18. Litt. 1
19. Litt. 2
20. Litt. 3
21. Litt.4
22. Litt. 7
23. Litt. 8
24. Litt. 9
25. Litt. 10 26. Litt. 11
27. Litt. 12
28. Litt. 13
29. Litt. 14
30. Litt. 15 31. Litt. 16
32. Ref.
33. Ref.
34. Litt. 17
35. Berge, S. M., Bighley L.D., Monkhouse D.C., Pharmaceutical Science (1977) 66, 1-19.
36. Sazani P et al. Nat Biotechnol (2002) 20 (12):1228-33
37. McMahon BM et al. Antisense Nucleic Acid Drug Dev (2002) 12(2):65-70