WO2004013135A1 - 2-phenylpyridin-4-yl derivatives as alk5 inhibitors - Google Patents
2-phenylpyridin-4-yl derivatives as alk5 inhibitors Download PDFInfo
- Publication number
- WO2004013135A1 WO2004013135A1 PCT/EP2003/008496 EP0308496W WO2004013135A1 WO 2004013135 A1 WO2004013135 A1 WO 2004013135A1 EP 0308496 W EP0308496 W EP 0308496W WO 2004013135 A1 WO2004013135 A1 WO 2004013135A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pyridin
- alkyl
- hydrogen
- compounds
- formula
- Prior art date
Links
- -1 2-phenylpyridin-4-yl Chemical class 0.000 title claims description 34
- 239000003112 inhibitor Substances 0.000 title abstract description 6
- 238000011282 treatment Methods 0.000 claims abstract description 24
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 16
- 201000010099 disease Diseases 0.000 claims abstract description 10
- 239000003814 drug Substances 0.000 claims abstract description 8
- 230000001404 mediated effect Effects 0.000 claims abstract description 6
- 150000001875 compounds Chemical class 0.000 claims description 360
- 229910052739 hydrogen Inorganic materials 0.000 claims description 72
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 65
- 239000001257 hydrogen Substances 0.000 claims description 59
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 claims description 53
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 46
- 125000005843 halogen group Chemical group 0.000 claims description 41
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 38
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 claims description 35
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 claims description 34
- 229910052757 nitrogen Inorganic materials 0.000 claims description 31
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 30
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 27
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 claims description 22
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 20
- 229920006395 saturated elastomer Polymers 0.000 claims description 18
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 15
- 125000001153 fluoro group Chemical group F* 0.000 claims description 12
- 229910052717 sulfur Inorganic materials 0.000 claims description 12
- 150000003839 salts Chemical class 0.000 claims description 11
- 239000012453 solvate Substances 0.000 claims description 11
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 10
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 10
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 claims description 9
- 125000001246 bromo group Chemical group Br* 0.000 claims description 9
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 9
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims description 9
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 9
- GAMYYCRTACQSBR-UHFFFAOYSA-N 4-azabenzimidazole Chemical compound C1=CC=C2NC=NC2=N1 GAMYYCRTACQSBR-UHFFFAOYSA-N 0.000 claims description 8
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 claims description 8
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 claims description 8
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 claims description 8
- 125000005842 heteroatom Chemical group 0.000 claims description 8
- 125000000623 heterocyclic group Chemical group 0.000 claims description 8
- 208000017169 kidney disease Diseases 0.000 claims description 8
- 229910052760 oxygen Inorganic materials 0.000 claims description 8
- 229910052701 rubidium Inorganic materials 0.000 claims description 8
- 125000001424 substituent group Chemical group 0.000 claims description 8
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 7
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 7
- 150000003852 triazoles Chemical class 0.000 claims description 7
- 201000001320 Atherosclerosis Diseases 0.000 claims description 6
- 241000711549 Hepacivirus C Species 0.000 claims description 6
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 claims description 6
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 claims description 6
- 125000003545 alkoxy group Chemical group 0.000 claims description 6
- 125000004429 atom Chemical group 0.000 claims description 6
- 208000035475 disorder Diseases 0.000 claims description 6
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 claims description 6
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 6
- 125000004193 piperazinyl group Chemical group 0.000 claims description 6
- ROSDSFDQCJNGOL-UHFFFAOYSA-N protonated dimethyl amine Natural products CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 claims description 6
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 5
- 206010016654 Fibrosis Diseases 0.000 claims description 5
- 206010023421 Kidney fibrosis Diseases 0.000 claims description 5
- 241000124008 Mammalia Species 0.000 claims description 5
- 208000025865 Ulcer Diseases 0.000 claims description 5
- 208000027418 Wounds and injury Diseases 0.000 claims description 5
- 125000003118 aryl group Chemical group 0.000 claims description 5
- 125000004432 carbon atom Chemical group C* 0.000 claims description 5
- 208000020832 chronic kidney disease Diseases 0.000 claims description 5
- 230000004761 fibrosis Effects 0.000 claims description 5
- 238000004519 manufacturing process Methods 0.000 claims description 5
- 239000008194 pharmaceutical composition Substances 0.000 claims description 5
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 5
- 231100000397 ulcer Toxicity 0.000 claims description 5
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 claims description 4
- 208000007342 Diabetic Nephropathies Diseases 0.000 claims description 4
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 4
- 229910003827 NRaRb Inorganic materials 0.000 claims description 4
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 claims description 4
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 claims description 4
- 206010052428 Wound Diseases 0.000 claims description 4
- 230000001154 acute effect Effects 0.000 claims description 4
- 229910052799 carbon Inorganic materials 0.000 claims description 4
- 208000033679 diabetic kidney disease Diseases 0.000 claims description 4
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical compound C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 claims description 4
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 claims description 4
- 238000011321 prophylaxis Methods 0.000 claims description 4
- 208000037803 restenosis Diseases 0.000 claims description 4
- VLLMWSRANPNYQX-UHFFFAOYSA-N thiadiazole Chemical compound C1=CSN=N1.C1=CSN=N1 VLLMWSRANPNYQX-UHFFFAOYSA-N 0.000 claims description 4
- 229930192474 thiophene Natural products 0.000 claims description 4
- 230000029663 wound healing Effects 0.000 claims description 4
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 claims description 3
- BAXOFTOLAUCFNW-UHFFFAOYSA-N 1H-indazole Chemical compound C1=CC=C2C=NNC2=C1 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 claims description 3
- IMSODMZESSGVBE-UHFFFAOYSA-N 2-Oxazoline Chemical compound C1CN=CO1 IMSODMZESSGVBE-UHFFFAOYSA-N 0.000 claims description 3
- VHMICKWLTGFITH-UHFFFAOYSA-N 2H-isoindole Chemical compound C1=CC=CC2=CNC=C21 VHMICKWLTGFITH-UHFFFAOYSA-N 0.000 claims description 3
- MGADZUXDNSDTHW-UHFFFAOYSA-N 2H-pyran Chemical compound C1OC=CC=C1 MGADZUXDNSDTHW-UHFFFAOYSA-N 0.000 claims description 3
- 208000024827 Alzheimer disease Diseases 0.000 claims description 3
- 208000008439 Biliary Liver Cirrhosis Diseases 0.000 claims description 3
- 208000033222 Biliary cirrhosis primary Diseases 0.000 claims description 3
- 206010007559 Cardiac failure congestive Diseases 0.000 claims description 3
- 201000009273 Endometriosis Diseases 0.000 claims description 3
- 206010019280 Heart failures Diseases 0.000 claims description 3
- 208000018565 Hemochromatosis Diseases 0.000 claims description 3
- 241000700721 Hepatitis B virus Species 0.000 claims description 3
- 208000014919 IgG4-related retroperitoneal fibrosis Diseases 0.000 claims description 3
- 208000002260 Keloid Diseases 0.000 claims description 3
- 206010027398 Mesenteric fibrosis Diseases 0.000 claims description 3
- 208000022873 Ocular disease Diseases 0.000 claims description 3
- 208000001132 Osteoporosis Diseases 0.000 claims description 3
- WYNCHZVNFNFDNH-UHFFFAOYSA-N Oxazolidine Chemical compound C1COCN1 WYNCHZVNFNFDNH-UHFFFAOYSA-N 0.000 claims description 3
- 208000012654 Primary biliary cholangitis Diseases 0.000 claims description 3
- 206010038979 Retroperitoneal fibrosis Diseases 0.000 claims description 3
- JTBSSXWFYSSVJP-UHFFFAOYSA-N [S].C1=CSC=N1 Chemical compound [S].C1=CSC=N1 JTBSSXWFYSSVJP-UHFFFAOYSA-N 0.000 claims description 3
- 206010003246 arthritis Diseases 0.000 claims description 3
- 208000019425 cirrhosis of liver Diseases 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 208000006454 hepatitis Diseases 0.000 claims description 3
- 231100000283 hepatitis Toxicity 0.000 claims description 3
- 230000001771 impaired effect Effects 0.000 claims description 3
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 claims description 3
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 claims description 3
- 210000001117 keloid Anatomy 0.000 claims description 3
- 230000007658 neurological function Effects 0.000 claims description 3
- 208000005069 pulmonary fibrosis Diseases 0.000 claims description 3
- JWVCLYRUEFBMGU-UHFFFAOYSA-N quinazoline Chemical compound N1=CN=CC2=CC=CC=C21 JWVCLYRUEFBMGU-UHFFFAOYSA-N 0.000 claims description 3
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 2
- RFRXIWQYSOIBDI-UHFFFAOYSA-N benzarone Chemical compound CCC=1OC2=CC=CC=C2C=1C(=O)C1=CC=C(O)C=C1 RFRXIWQYSOIBDI-UHFFFAOYSA-N 0.000 claims description 2
- 239000003085 diluting agent Substances 0.000 claims description 2
- 238000000034 method Methods 0.000 abstract description 36
- 102000004887 Transforming Growth Factor beta Human genes 0.000 abstract description 28
- 108090001012 Transforming Growth Factor beta Proteins 0.000 abstract description 28
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 abstract description 28
- 238000002360 preparation method Methods 0.000 abstract description 11
- 108091000080 Phosphotransferase Proteins 0.000 abstract description 6
- 102000020233 phosphotransferase Human genes 0.000 abstract description 6
- 108010009583 Transforming Growth Factors Proteins 0.000 abstract description 4
- 102000009618 Transforming Growth Factors Human genes 0.000 abstract description 4
- 230000019491 signal transduction Effects 0.000 abstract description 3
- 230000037361 pathway Effects 0.000 abstract description 2
- 230000026731 phosphorylation Effects 0.000 abstract description 2
- 238000006366 phosphorylation reaction Methods 0.000 abstract description 2
- 230000002265 prevention Effects 0.000 abstract description 2
- 239000000543 intermediate Substances 0.000 description 197
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 148
- 239000000243 solution Substances 0.000 description 124
- 239000000203 mixture Substances 0.000 description 116
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 94
- 238000006243 chemical reaction Methods 0.000 description 89
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 77
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 73
- 239000007787 solid Substances 0.000 description 71
- 230000002829 reductive effect Effects 0.000 description 69
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 55
- 239000002904 solvent Substances 0.000 description 55
- 229910052938 sodium sulfate Inorganic materials 0.000 description 54
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 53
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 52
- 238000000668 atmospheric pressure chemical ionisation mass spectrometry Methods 0.000 description 52
- 239000012074 organic phase Substances 0.000 description 52
- 239000007832 Na2SO4 Substances 0.000 description 51
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 48
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 46
- 238000004587 chromatography analysis Methods 0.000 description 46
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 45
- 239000000741 silica gel Substances 0.000 description 43
- 229910002027 silica gel Inorganic materials 0.000 description 43
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 38
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 37
- 238000010992 reflux Methods 0.000 description 37
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 35
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 34
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 32
- 239000011347 resin Substances 0.000 description 32
- 229920005989 resin Polymers 0.000 description 32
- 238000005160 1H NMR spectroscopy Methods 0.000 description 29
- 239000011541 reaction mixture Substances 0.000 description 29
- 239000003921 oil Substances 0.000 description 26
- 235000019198 oils Nutrition 0.000 description 26
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 24
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 24
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 24
- 235000019439 ethyl acetate Nutrition 0.000 description 23
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 22
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 21
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 20
- 229910000029 sodium carbonate Inorganic materials 0.000 description 19
- 239000000047 product Substances 0.000 description 18
- 101800002279 Transforming growth factor beta-1 Proteins 0.000 description 17
- 210000004027 cell Anatomy 0.000 description 17
- 238000000605 extraction Methods 0.000 description 17
- 150000002431 hydrogen Chemical group 0.000 description 17
- 239000007858 starting material Substances 0.000 description 17
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 16
- 102000046299 Transforming Growth Factor beta1 Human genes 0.000 description 16
- 230000000694 effects Effects 0.000 description 16
- 229910052763 palladium Inorganic materials 0.000 description 16
- 238000002531 positive electrospray ionisation time-of-flight mass spectrometry Methods 0.000 description 16
- 229910000027 potassium carbonate Inorganic materials 0.000 description 16
- 108020003175 receptors Proteins 0.000 description 15
- 102000005962 receptors Human genes 0.000 description 15
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 14
- 239000011734 sodium Substances 0.000 description 14
- 229940086542 triethylamine Drugs 0.000 description 14
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 13
- 239000000843 powder Substances 0.000 description 13
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 12
- 229960000583 acetic acid Drugs 0.000 description 12
- 229910052786 argon Inorganic materials 0.000 description 12
- 239000006071 cream Substances 0.000 description 12
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 12
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 11
- 238000001816 cooling Methods 0.000 description 11
- 235000019441 ethanol Nutrition 0.000 description 11
- 238000001665 trituration Methods 0.000 description 11
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 10
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 10
- 239000008346 aqueous phase Substances 0.000 description 10
- 239000013078 crystal Substances 0.000 description 10
- 239000000706 filtrate Substances 0.000 description 10
- 239000012044 organic layer Substances 0.000 description 10
- 238000000746 purification Methods 0.000 description 10
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 10
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 9
- LSZMVESSGLHDJE-UHFFFAOYSA-N 2-bromo-4-methylpyridine Chemical compound CC1=CC=NC(Br)=C1 LSZMVESSGLHDJE-UHFFFAOYSA-N 0.000 description 9
- JMTMSDXUXJISAY-UHFFFAOYSA-N 2H-benzotriazol-4-ol Chemical compound OC1=CC=CC2=C1N=NN2 JMTMSDXUXJISAY-UHFFFAOYSA-N 0.000 description 9
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 9
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 9
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 9
- 238000003556 assay Methods 0.000 description 9
- 229910052796 boron Inorganic materials 0.000 description 9
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- 0 Cc1cccc(-c2n[n]c(*)c2-c2ccnc(Br)c2)n1 Chemical compound Cc1cccc(-c2n[n]c(*)c2-c2ccnc(Br)c2)n1 0.000 description 8
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 8
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 8
- 229910052794 bromium Inorganic materials 0.000 description 8
- 239000003054 catalyst Substances 0.000 description 8
- 238000001704 evaporation Methods 0.000 description 8
- 230000008020 evaporation Effects 0.000 description 8
- 239000002244 precipitate Substances 0.000 description 8
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 8
- 108090000765 processed proteins & peptides Proteins 0.000 description 8
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 8
- 229910000104 sodium hydride Inorganic materials 0.000 description 8
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 8
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 8
- VXWBQOJISHAKKM-UHFFFAOYSA-N (4-formylphenyl)boronic acid Chemical compound OB(O)C1=CC=C(C=O)C=C1 VXWBQOJISHAKKM-UHFFFAOYSA-N 0.000 description 7
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 7
- 108060001084 Luciferase Proteins 0.000 description 7
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 7
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical group C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 7
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 7
- 238000002425 crystallisation Methods 0.000 description 7
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 7
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 7
- 239000012312 sodium hydride Substances 0.000 description 7
- 239000003981 vehicle Substances 0.000 description 7
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 6
- ZNEWDLYYLKDPRH-UHFFFAOYSA-N 4-[4-[5-(6-methylpyridin-2-yl)-2-propan-2-yl-1h-imidazol-4-yl]pyridin-2-yl]-n-(oxan-4-yl)benzamide Chemical compound N1C(C(C)C)=NC(C=2C=C(N=CC=2)C=2C=CC(=CC=2)C(=O)NC2CCOCC2)=C1C1=CC=CC(C)=N1 ZNEWDLYYLKDPRH-UHFFFAOYSA-N 0.000 description 6
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 6
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 6
- 239000005089 Luciferase Substances 0.000 description 6
- YNHIGQDRGKUECZ-UHFFFAOYSA-L bis(triphenylphosphine)palladium(ii) dichloride Chemical compound [Cl-].[Cl-].[Pd+2].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-L 0.000 description 6
- 229910000024 caesium carbonate Inorganic materials 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- 239000000460 chlorine Substances 0.000 description 6
- 229910052801 chlorine Inorganic materials 0.000 description 6
- 239000010779 crude oil Substances 0.000 description 6
- 239000012043 crude product Substances 0.000 description 6
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 6
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 5
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 5
- 150000001408 amides Chemical class 0.000 description 5
- 210000002744 extracellular matrix Anatomy 0.000 description 5
- 238000001914 filtration Methods 0.000 description 5
- 239000012442 inert solvent Substances 0.000 description 5
- 230000005764 inhibitory process Effects 0.000 description 5
- 239000003446 ligand Substances 0.000 description 5
- JUJWROOIHBZHMG-UHFFFAOYSA-O pyridinium Chemical compound C1=CC=[NH+]C=C1 JUJWROOIHBZHMG-UHFFFAOYSA-O 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 4
- ICSNLGPSRYBMBD-UHFFFAOYSA-N 2-aminopyridine Chemical compound NC1=CC=CC=N1 ICSNLGPSRYBMBD-UHFFFAOYSA-N 0.000 description 4
- JUIKUQOUMZUFQT-UHFFFAOYSA-N 2-bromoacetamide Chemical compound NC(=O)CBr JUIKUQOUMZUFQT-UHFFFAOYSA-N 0.000 description 4
- BICZJRAGTCRORZ-UHFFFAOYSA-N 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(O)C=C1 BICZJRAGTCRORZ-UHFFFAOYSA-N 0.000 description 4
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 4
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 4
- 108090000331 Firefly luciferases Proteins 0.000 description 4
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 4
- ZSXGLVDWWRXATF-UHFFFAOYSA-N N,N-dimethylformamide dimethyl acetal Chemical compound COC(OC)N(C)C ZSXGLVDWWRXATF-UHFFFAOYSA-N 0.000 description 4
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 4
- 241000700159 Rattus Species 0.000 description 4
- 238000006069 Suzuki reaction reaction Methods 0.000 description 4
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 4
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 4
- UHOVQNZJYSORNB-UHFFFAOYSA-N benzene Substances C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 4
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 4
- 230000001413 cellular effect Effects 0.000 description 4
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 4
- 238000003776 cleavage reaction Methods 0.000 description 4
- 230000008878 coupling Effects 0.000 description 4
- 238000010168 coupling process Methods 0.000 description 4
- 238000005859 coupling reaction Methods 0.000 description 4
- 230000002401 inhibitory effect Effects 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 239000011159 matrix material Substances 0.000 description 4
- 239000012286 potassium permanganate Substances 0.000 description 4
- 239000012047 saturated solution Substances 0.000 description 4
- 230000007017 scission Effects 0.000 description 4
- 235000010288 sodium nitrite Nutrition 0.000 description 4
- 235000011152 sodium sulphate Nutrition 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- PYOKUURKVVELLB-UHFFFAOYSA-N trimethyl orthoformate Chemical compound COC(OC)OC PYOKUURKVVELLB-UHFFFAOYSA-N 0.000 description 4
- SEDZOYHHAIAQIW-UHFFFAOYSA-N trimethylsilyl azide Chemical compound C[Si](C)(C)N=[N+]=[N-] SEDZOYHHAIAQIW-UHFFFAOYSA-N 0.000 description 4
- COIQUVGFTILYGA-UHFFFAOYSA-N (4-hydroxyphenyl)boronic acid Chemical compound OB(O)C1=CC=C(O)C=C1 COIQUVGFTILYGA-UHFFFAOYSA-N 0.000 description 3
- VDUKDQTYMWUSAC-UHFFFAOYSA-N (4-methylsulfonylphenyl)boronic acid Chemical compound CS(=O)(=O)C1=CC=C(B(O)O)C=C1 VDUKDQTYMWUSAC-UHFFFAOYSA-N 0.000 description 3
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 3
- IYDKBQIEOBXLTP-UHFFFAOYSA-N 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(C(O)=O)C=C1 IYDKBQIEOBXLTP-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- 244000166102 Eucalyptus leucoxylon Species 0.000 description 3
- 235000004694 Eucalyptus leucoxylon Nutrition 0.000 description 3
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 3
- 101000712674 Homo sapiens TGF-beta receptor type-1 Proteins 0.000 description 3
- 238000005481 NMR spectroscopy Methods 0.000 description 3
- 235000019502 Orange oil Nutrition 0.000 description 3
- 206010051246 Photodermatosis Diseases 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 208000007107 Stomach Ulcer Diseases 0.000 description 3
- 102100033456 TGF-beta receptor type-1 Human genes 0.000 description 3
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 3
- 150000001412 amines Chemical class 0.000 description 3
- 230000003143 atherosclerotic effect Effects 0.000 description 3
- 230000009286 beneficial effect Effects 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 150000001642 boronic acid derivatives Chemical class 0.000 description 3
- 239000003638 chemical reducing agent Substances 0.000 description 3
- 230000001684 chronic effect Effects 0.000 description 3
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 3
- 239000003480 eluent Substances 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 229910052736 halogen Inorganic materials 0.000 description 3
- 150000002367 halogens Chemical class 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine hydrate Chemical compound O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 3
- 125000002883 imidazolyl group Chemical group 0.000 description 3
- 239000010410 layer Substances 0.000 description 3
- 210000002540 macrophage Anatomy 0.000 description 3
- REPVNSJSTLRQEQ-UHFFFAOYSA-N n,n-dimethylacetamide;n,n-dimethylformamide Chemical compound CN(C)C=O.CN(C)C(C)=O REPVNSJSTLRQEQ-UHFFFAOYSA-N 0.000 description 3
- 230000003472 neutralizing effect Effects 0.000 description 3
- 229910052759 nickel Inorganic materials 0.000 description 3
- 239000010502 orange oil Substances 0.000 description 3
- AHVQYHFYQWKUKB-UHFFFAOYSA-N oxan-4-amine Chemical compound NC1CCOCC1 AHVQYHFYQWKUKB-UHFFFAOYSA-N 0.000 description 3
- 230000008845 photoaging Effects 0.000 description 3
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 229940075993 receptor modulator Drugs 0.000 description 3
- 230000011664 signaling Effects 0.000 description 3
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 235000010356 sorbitol Nutrition 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 239000006188 syrup Substances 0.000 description 3
- 235000020357 syrup Nutrition 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 3
- BDZBKCUKTQZUTL-UHFFFAOYSA-N triethyl phosphite Chemical compound CCOP(OCC)OCC BDZBKCUKTQZUTL-UHFFFAOYSA-N 0.000 description 3
- JBWKIWSBJXDJDT-UHFFFAOYSA-N triphenylmethyl chloride Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(Cl)C1=CC=CC=C1 JBWKIWSBJXDJDT-UHFFFAOYSA-N 0.000 description 3
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 2
- UKSZBOKPHAQOMP-SVLSSHOZSA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 UKSZBOKPHAQOMP-SVLSSHOZSA-N 0.000 description 2
- QBLFZIBJXUQVRF-UHFFFAOYSA-N (4-bromophenyl)boronic acid Chemical compound OB(O)C1=CC=C(Br)C=C1 QBLFZIBJXUQVRF-UHFFFAOYSA-N 0.000 description 2
- CVBNDDGEVPNUNA-UHFFFAOYSA-N (4-ethylsulfonylphenyl)boronic acid Chemical compound CCS(=O)(=O)C1=CC=C(B(O)O)C=C1 CVBNDDGEVPNUNA-UHFFFAOYSA-N 0.000 description 2
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 description 2
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 2
- RAIPHJJURHTUIC-UHFFFAOYSA-N 1,3-thiazol-2-amine Chemical compound NC1=NC=CS1 RAIPHJJURHTUIC-UHFFFAOYSA-N 0.000 description 2
- FSNGFFWICFYWQC-UHFFFAOYSA-N 1-(2-chloroethyl)pyrrolidine;hydron;chloride Chemical compound Cl.ClCCN1CCCC1 FSNGFFWICFYWQC-UHFFFAOYSA-N 0.000 description 2
- FJJYHTVHBVXEEQ-UHFFFAOYSA-N 2,2-dimethylpropanal Chemical compound CC(C)(C)C=O FJJYHTVHBVXEEQ-UHFFFAOYSA-N 0.000 description 2
- SJAHYONZGTUULM-UHFFFAOYSA-N 2-(4-ethylsulfonylphenyl)pyridine-4-carbaldehyde Chemical compound C1=CC(S(=O)(=O)CC)=CC=C1C1=CC(C=O)=CC=N1 SJAHYONZGTUULM-UHFFFAOYSA-N 0.000 description 2
- VHMOZRYEXQYKHZ-UHFFFAOYSA-N 2-bromo-4-(2-phenyl-5-pyridin-2-yl-1h-imidazol-4-yl)pyridine Chemical compound C1=NC(Br)=CC(C2=C(N=C(N2)C=2C=CC=CC=2)C=2N=CC=CC=2)=C1 VHMOZRYEXQYKHZ-UHFFFAOYSA-N 0.000 description 2
- OOGALSDKTFQSKW-UHFFFAOYSA-N 2-bromo-n-methoxy-n-methylpyridine-4-carboxamide Chemical compound CON(C)C(=O)C1=CC=NC(Br)=C1 OOGALSDKTFQSKW-UHFFFAOYSA-N 0.000 description 2
- YBTKGKVQEXAYEM-UHFFFAOYSA-N 2-bromopyridine-4-carboxylic acid Chemical compound OC(=O)C1=CC=NC(Br)=C1 YBTKGKVQEXAYEM-UHFFFAOYSA-N 0.000 description 2
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 2
- UCFSYHMCKWNKAH-UHFFFAOYSA-N 4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound CC1(C)OBOC1(C)C UCFSYHMCKWNKAH-UHFFFAOYSA-N 0.000 description 2
- ZANPJXNYBVVNSD-UHFFFAOYSA-N 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(N)C=C1 ZANPJXNYBVVNSD-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- 239000005541 ACE inhibitor Substances 0.000 description 2
- 108010059616 Activins Proteins 0.000 description 2
- 102000005606 Activins Human genes 0.000 description 2
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical class N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 2
- 239000005695 Ammonium acetate Chemical class 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 102000004127 Cytokines Human genes 0.000 description 2
- 108090000695 Cytokines Proteins 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- 206010056340 Diabetic ulcer Diseases 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 241000254158 Lampyridae Species 0.000 description 2
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 2
- 240000007472 Leucaena leucocephala Species 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- 102000012335 Plasminogen Activator Inhibitor 1 Human genes 0.000 description 2
- 108010022233 Plasminogen Activator Inhibitor 1 Proteins 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 2
- 239000007983 Tris buffer Substances 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 239000000488 activin Substances 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 125000003342 alkenyl group Chemical group 0.000 description 2
- 229950003476 aminothiazole Drugs 0.000 description 2
- 235000019257 ammonium acetate Nutrition 0.000 description 2
- 229940043376 ammonium acetate Drugs 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 239000002333 angiotensin II receptor antagonist Substances 0.000 description 2
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 2
- 239000012131 assay buffer Substances 0.000 description 2
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 2
- 230000004221 bone function Effects 0.000 description 2
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 235000011089 carbon dioxide Nutrition 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 238000006555 catalytic reaction Methods 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 235000012000 cholesterol Nutrition 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 208000000718 duodenal ulcer Diseases 0.000 description 2
- FQYYIPZPELSLDK-UHFFFAOYSA-N ethyl pyridine-2-carboxylate Chemical compound CCOC(=O)C1=CC=CC=N1 FQYYIPZPELSLDK-UHFFFAOYSA-N 0.000 description 2
- 230000003176 fibrotic effect Effects 0.000 description 2
- 238000011049 filling Methods 0.000 description 2
- 238000003818 flash chromatography Methods 0.000 description 2
- 238000002875 fluorescence polarization Methods 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 125000002541 furyl group Chemical group 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 230000014509 gene expression Effects 0.000 description 2
- 230000035876 healing Effects 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 208000027866 inflammatory disease Diseases 0.000 description 2
- UEXQBEVWFZKHNB-UHFFFAOYSA-N intermediate 29 Natural products C1=CC(N)=CC=C1NC1=NC=CC=N1 UEXQBEVWFZKHNB-UHFFFAOYSA-N 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- 230000003902 lesion Effects 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 2
- 239000006210 lotion Substances 0.000 description 2
- 238000003670 luciferase enzyme activity assay Methods 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 230000005012 migration Effects 0.000 description 2
- 238000013508 migration Methods 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- CDXVUROVRIFQMV-UHFFFAOYSA-N oxo(diphenoxy)phosphanium Chemical compound C=1C=CC=CC=1O[P+](=O)OC1=CC=CC=C1 CDXVUROVRIFQMV-UHFFFAOYSA-N 0.000 description 2
- UQPUONNXJVWHRM-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 UQPUONNXJVWHRM-UHFFFAOYSA-N 0.000 description 2
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 150000003217 pyrazoles Chemical class 0.000 description 2
- PYWVYCXTNDRMGF-UHFFFAOYSA-N rhodamine B Chemical compound [Cl-].C=12C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C2C=1C1=CC=CC=C1C(O)=O PYWVYCXTNDRMGF-UHFFFAOYSA-N 0.000 description 2
- 230000036573 scar formation Effects 0.000 description 2
- 230000037390 scarring Effects 0.000 description 2
- 235000004400 serine Nutrition 0.000 description 2
- XGVXKJKTISMIOW-ZDUSSCGKSA-N simurosertib Chemical compound N1N=CC(C=2SC=3C(=O)NC(=NC=3C=2)[C@H]2N3CCC(CC3)C2)=C1C XGVXKJKTISMIOW-ZDUSSCGKSA-N 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 2
- CWMFRHBXRUITQE-UHFFFAOYSA-N trimethylsilylacetylene Chemical group C[Si](C)(C)C#C CWMFRHBXRUITQE-UHFFFAOYSA-N 0.000 description 2
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 239000008215 water for injection Substances 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- 230000037314 wound repair Effects 0.000 description 2
- UBWXUGDQUBIEIZ-UHFFFAOYSA-N (13-methyl-3-oxo-2,6,7,8,9,10,11,12,14,15,16,17-dodecahydro-1h-cyclopenta[a]phenanthren-17-yl) 3-phenylpropanoate Chemical compound CC12CCC(C3CCC(=O)C=C3CC3)C3C1CCC2OC(=O)CCC1=CC=CC=C1 UBWXUGDQUBIEIZ-UHFFFAOYSA-N 0.000 description 1
- XFQNWPYGEGCIMF-HCUGAJCMSA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].[Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 XFQNWPYGEGCIMF-HCUGAJCMSA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 1
- PQCXFUXRTRESBD-UHFFFAOYSA-N (4-methoxycarbonylphenyl)boronic acid Chemical compound COC(=O)C1=CC=C(B(O)O)C=C1 PQCXFUXRTRESBD-UHFFFAOYSA-N 0.000 description 1
- VOAAEKKFGLPLLU-UHFFFAOYSA-N (4-methoxyphenyl)boronic acid Chemical compound COC1=CC=C(B(O)O)C=C1 VOAAEKKFGLPLLU-UHFFFAOYSA-N 0.000 description 1
- ALSTYHKOOCGGFT-KTKRTIGZSA-N (9Z)-octadecen-1-ol Chemical compound CCCCCCCC\C=C/CCCCCCCCO ALSTYHKOOCGGFT-KTKRTIGZSA-N 0.000 description 1
- ZORQXIQZAOLNGE-UHFFFAOYSA-N 1,1-difluorocyclohexane Chemical compound FC1(F)CCCCC1 ZORQXIQZAOLNGE-UHFFFAOYSA-N 0.000 description 1
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 1
- CLEVZBPYLTYZHC-UHFFFAOYSA-N 1-(4-bromophenyl)-4-ethylpiperazine Chemical compound C1CN(CC)CCN1C1=CC=C(Br)C=C1 CLEVZBPYLTYZHC-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 1
- CWRJPBXIVVHQOC-UHFFFAOYSA-N 1-[2-(4-iodophenoxy)ethyl]pyrrolidine Chemical compound C1=CC(I)=CC=C1OCCN1CCCC1 CWRJPBXIVVHQOC-UHFFFAOYSA-N 0.000 description 1
- QUFYPJILXRCKSB-UHFFFAOYSA-N 1-ethyl-4-phenylpiperazine Chemical compound C1CN(CC)CCN1C1=CC=CC=C1 QUFYPJILXRCKSB-UHFFFAOYSA-N 0.000 description 1
- WGCYRFWNGRMRJA-UHFFFAOYSA-N 1-ethylpiperazine Chemical compound CCN1CCNCC1 WGCYRFWNGRMRJA-UHFFFAOYSA-N 0.000 description 1
- WZTRQGJMMHMFGH-UHFFFAOYSA-N 1-methyl-imidazole-4-carboxylic acid Chemical compound CN1C=NC(C(O)=O)=C1 WZTRQGJMMHMFGH-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- MVRIZYXWRMPEMI-UHFFFAOYSA-N 2-(2-bromopyridin-4-yl)-1-(3-chloro-4-fluorophenyl)ethanone Chemical compound C1=C(Cl)C(F)=CC=C1C(=O)CC1=CC=NC(Br)=C1 MVRIZYXWRMPEMI-UHFFFAOYSA-N 0.000 description 1
- ZNEQPLGDZLUBDY-UHFFFAOYSA-N 2-(2-bromopyridin-4-yl)-1-(3-chlorophenyl)ethanone Chemical compound ClC1=CC=CC(C(=O)CC=2C=C(Br)N=CC=2)=C1 ZNEQPLGDZLUBDY-UHFFFAOYSA-N 0.000 description 1
- ZLGHROSJIVPXMQ-UHFFFAOYSA-N 2-(2-bromopyridin-4-yl)-1-(6-fluoropyridin-2-yl)ethanone Chemical compound FC1=CC=CC(C(=O)CC=2C=C(Br)N=CC=2)=N1 ZLGHROSJIVPXMQ-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- KZMKVPDVYKCZLG-UHFFFAOYSA-N 2-bromo-2-(2-bromopyridin-4-yl)-1-pyridin-2-ylethanone Chemical compound C=1C=NC(Br)=CC=1C(Br)C(=O)C1=CC=CC=N1 KZMKVPDVYKCZLG-UHFFFAOYSA-N 0.000 description 1
- BWUAMQFOSCMITC-UHFFFAOYSA-N 2-bromo-4-[2-tert-butyl-5-(6-methylpyridin-2-yl)-1h-imidazol-4-yl]pyridine Chemical compound CC1=CC=CC(C2=C(NC(=N2)C(C)(C)C)C=2C=C(Br)N=CC=2)=N1 BWUAMQFOSCMITC-UHFFFAOYSA-N 0.000 description 1
- GKBTUXZBWAQUET-UHFFFAOYSA-N 2-bromo-4-[5-(6-methylpyridin-2-yl)-1h-pyrazol-4-yl]pyridine Chemical compound CC1=CC=CC(C=2C(=CNN=2)C=2C=C(Br)N=CC=2)=N1 GKBTUXZBWAQUET-UHFFFAOYSA-N 0.000 description 1
- YMQRPXBBBOXHNZ-UHFFFAOYSA-N 2-chloro-1-morpholin-4-ylethanone Chemical compound ClCC(=O)N1CCOCC1 YMQRPXBBBOXHNZ-UHFFFAOYSA-N 0.000 description 1
- KJKIPRQNFDUULB-UHFFFAOYSA-N 2-chloro-4-iodopyridine Chemical compound ClC1=CC(I)=CC=N1 KJKIPRQNFDUULB-UHFFFAOYSA-N 0.000 description 1
- UFPOSTQMFOYHJI-UHFFFAOYSA-N 2-chloropyridine-4-carbaldehyde Chemical compound ClC1=CC(C=O)=CC=N1 UFPOSTQMFOYHJI-UHFFFAOYSA-N 0.000 description 1
- LQLJZSJKRYTKTP-UHFFFAOYSA-N 2-dimethylaminoethyl chloride hydrochloride Chemical compound Cl.CN(C)CCCl LQLJZSJKRYTKTP-UHFFFAOYSA-N 0.000 description 1
- UDMNVTJFUISBFD-UHFFFAOYSA-N 2-fluoro-6-methylpyridine Chemical compound CC1=CC=CC(F)=N1 UDMNVTJFUISBFD-UHFFFAOYSA-N 0.000 description 1
- YFBIAMYFNHEBPR-UHFFFAOYSA-N 2-fluoroacetyl isothiocyanate Chemical compound FCC(=O)N=C=S YFBIAMYFNHEBPR-UHFFFAOYSA-N 0.000 description 1
- LXBGSDVWAMZHDD-UHFFFAOYSA-N 2-methyl-1h-imidazole Chemical compound CC1=NC=CN1 LXBGSDVWAMZHDD-UHFFFAOYSA-N 0.000 description 1
- ICGYBJNONRDZOP-UHFFFAOYSA-N 2-morpholin-4-ium-4-ylacetic acid;chloride Chemical compound Cl.OC(=O)CN1CCOCC1 ICGYBJNONRDZOP-UHFFFAOYSA-N 0.000 description 1
- LGCYVLDNGBSOOW-UHFFFAOYSA-N 2H-benzotriazol-4-ol 1-hydroxybenzotriazole Chemical compound OC1=CC=CC2=C1N=NN2.C1=CC=C2N(O)N=NC2=C1 LGCYVLDNGBSOOW-UHFFFAOYSA-N 0.000 description 1
- CPVDUQPNEZJPSD-UHFFFAOYSA-N 3,4-difluorobenzoic acid;ethyl 3,4-difluorobenzoate Chemical compound OC(=O)C1=CC=C(F)C(F)=C1.CCOC(=O)C1=CC=C(F)C(F)=C1 CPVDUQPNEZJPSD-UHFFFAOYSA-N 0.000 description 1
- UMCMPZBLKLEWAF-BCTGSCMUSA-N 3-[(3-cholamidopropyl)dimethylammonio]propane-1-sulfonate Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCCC[N+](C)(C)CCCS([O-])(=O)=O)C)[C@@]2(C)[C@@H](O)C1 UMCMPZBLKLEWAF-BCTGSCMUSA-N 0.000 description 1
- PKTSBFXIHLYGEY-UHFFFAOYSA-N 3-chloro-4-fluorobenzoic acid Chemical compound OC(=O)C1=CC=C(F)C(Cl)=C1 PKTSBFXIHLYGEY-UHFFFAOYSA-N 0.000 description 1
- FAXDZWQIWUSWJH-UHFFFAOYSA-N 3-methoxypropan-1-amine Chemical compound COCCCN FAXDZWQIWUSWJH-UHFFFAOYSA-N 0.000 description 1
- LZPWAYBEOJRFAX-UHFFFAOYSA-N 4,4,5,5-tetramethyl-1,3,2$l^{2}-dioxaborolane Chemical compound CC1(C)O[B]OC1(C)C LZPWAYBEOJRFAX-UHFFFAOYSA-N 0.000 description 1
- UJTKZWNRUPTHSB-UHFFFAOYSA-N 4-(4-bromophenyl)morpholine Chemical compound C1=CC(Br)=CC=C1N1CCOCC1 UJTKZWNRUPTHSB-UHFFFAOYSA-N 0.000 description 1
- QVAQFEBMXMVWJD-UHFFFAOYSA-N 4-(chloromethyl)-1-methylimidazole Chemical compound CN1C=NC(CCl)=C1 QVAQFEBMXMVWJD-UHFFFAOYSA-N 0.000 description 1
- BLBDTBCGPHPIJK-UHFFFAOYSA-N 4-Amino-2-chloropyridine Chemical compound NC1=CC=NC(Cl)=C1 BLBDTBCGPHPIJK-UHFFFAOYSA-N 0.000 description 1
- KNSAMIHAIQCBHB-UHFFFAOYSA-N 4-[4-[4-[3-(6-methylpyridin-2-yl)-1-tritylpyrazol-4-yl]pyridin-2-yl]phenyl]morpholine Chemical compound CC1=CC=CC(C=2C(=CN(N=2)C(C=2C=CC=CC=2)(C=2C=CC=CC=2)C=2C=CC=CC=2)C=2C=C(N=CC=2)C=2C=CC(=CC=2)N2CCOCC2)=N1 KNSAMIHAIQCBHB-UHFFFAOYSA-N 0.000 description 1
- SIAVMDKGVRXFAX-UHFFFAOYSA-N 4-carboxyphenylboronic acid Chemical compound OB(O)C1=CC=C(C(O)=O)C=C1 SIAVMDKGVRXFAX-UHFFFAOYSA-N 0.000 description 1
- VSMDINRNYYEDRN-UHFFFAOYSA-N 4-iodophenol Chemical compound OC1=CC=C(I)C=C1 VSMDINRNYYEDRN-UHFFFAOYSA-N 0.000 description 1
- FHQRDEDZJIFJAL-UHFFFAOYSA-N 4-phenylmorpholine Chemical compound C1COCCN1C1=CC=CC=C1 FHQRDEDZJIFJAL-UHFFFAOYSA-N 0.000 description 1
- ZHEXKZSHMBMFEK-UHFFFAOYSA-N 5-(2-bromopyridin-4-yl)-4-(6-methylpyridin-2-yl)-1,3-thiazol-2-amine Chemical compound CC1=CC=CC(C2=C(SC(N)=N2)C=2C=C(Br)N=CC=2)=N1 ZHEXKZSHMBMFEK-UHFFFAOYSA-N 0.000 description 1
- VNHBYKHXBCYPBJ-UHFFFAOYSA-N 5-ethynylimidazo[1,2-a]pyridine Chemical compound C#CC1=CC=CC2=NC=CN12 VNHBYKHXBCYPBJ-UHFFFAOYSA-N 0.000 description 1
- DIEMCUFYSOEIDU-UHFFFAOYSA-N 6-fluoropyridine-2-carboxylic acid Chemical compound OC(=O)C1=CC=CC(F)=N1 DIEMCUFYSOEIDU-UHFFFAOYSA-N 0.000 description 1
- MAQAGRJURDEYDQ-UHFFFAOYSA-N 6-methylpyridine Chemical compound CC1=C=CC=C[N]1 MAQAGRJURDEYDQ-UHFFFAOYSA-N 0.000 description 1
- AHISYUZBWDSPQL-UHFFFAOYSA-N 6-methylpyridine-2-carbaldehyde Chemical compound CC1=CC=CC(C=O)=N1 AHISYUZBWDSPQL-UHFFFAOYSA-N 0.000 description 1
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 1
- 235000019489 Almond oil Nutrition 0.000 description 1
- 108010064733 Angiotensins Proteins 0.000 description 1
- 102000015427 Angiotensins Human genes 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N Benzoic acid Natural products OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 102000007350 Bone Morphogenetic Proteins Human genes 0.000 description 1
- 108010007726 Bone Morphogenetic Proteins Proteins 0.000 description 1
- 101000856746 Bos taurus Cytochrome c oxidase subunit 7A1, mitochondrial Proteins 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- CCGRXCIJCYWWOY-UHFFFAOYSA-N C(COCC1)N1c(cc1)ccc1-c1cc(-c2c(-c3ncccc3)nc3[n]2cccc3)ccn1 Chemical compound C(COCC1)N1c(cc1)ccc1-c1cc(-c2c(-c3ncccc3)nc3[n]2cccc3)ccn1 CCGRXCIJCYWWOY-UHFFFAOYSA-N 0.000 description 1
- HJNVDARWHYFCEY-UHFFFAOYSA-N CC(C)(C)c1nc(-c2c(C)cnc(-c(cc3)ccc3OCC(N)=O)c2)c(-c2nc(C)ccc2)[nH]1 Chemical compound CC(C)(C)c1nc(-c2c(C)cnc(-c(cc3)ccc3OCC(N)=O)c2)c(-c2nc(C)ccc2)[nH]1 HJNVDARWHYFCEY-UHFFFAOYSA-N 0.000 description 1
- WQHRMZMXTJBGKJ-UHFFFAOYSA-N CC(C=CC1)=NC1c1c(-c2ccnc(-c(cc3)ccc3S(C)(=O)=O)c2)nn[nH]1 Chemical compound CC(C=CC1)=NC1c1c(-c2ccnc(-c(cc3)ccc3S(C)(=O)=O)c2)nn[nH]1 WQHRMZMXTJBGKJ-UHFFFAOYSA-N 0.000 description 1
- NXGLRKJZZLZWOO-UHFFFAOYSA-N CC1(C)OS(c(cc2)ccc2C(N2CCOCC2)=O)OC1(C)C Chemical compound CC1(C)OS(c(cc2)ccc2C(N2CCOCC2)=O)OC1(C)C NXGLRKJZZLZWOO-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- YSOAMSPADJLAHI-UHFFFAOYSA-N Cc1cc2nc(-c3nc(C)ccc3)c(-c3ccnc(-c(cc4)ccc4OCC(N)=O)c3)[n]2cc1 Chemical compound Cc1cc2nc(-c3nc(C)ccc3)c(-c3ccnc(-c(cc4)ccc4OCC(N)=O)c3)[n]2cc1 YSOAMSPADJLAHI-UHFFFAOYSA-N 0.000 description 1
- XICZBKYSRPYSOL-UHFFFAOYSA-N Cc1cccc(-c2c(-c3ccnc(-c(cc4)ccc4N)c3)[n](cccc3)c3n2)n1 Chemical compound Cc1cccc(-c2c(-c3ccnc(-c(cc4)ccc4N)c3)[n](cccc3)c3n2)n1 XICZBKYSRPYSOL-UHFFFAOYSA-N 0.000 description 1
- MVKWEFIEDUOTEQ-UHFFFAOYSA-N Cc1cccc(C(Cc2cc(-c(cc3)ccc3C(NC3CCOCC3)=O)ncc2)=O)n1 Chemical compound Cc1cccc(C(Cc2cc(-c(cc3)ccc3C(NC3CCOCC3)=O)ncc2)=O)n1 MVKWEFIEDUOTEQ-UHFFFAOYSA-N 0.000 description 1
- CWORZGVWEFJASB-UHFFFAOYSA-N Cc1nc(C#Cc2ccnc(Cl)c2)ccc1 Chemical compound Cc1nc(C#Cc2ccnc(Cl)c2)ccc1 CWORZGVWEFJASB-UHFFFAOYSA-N 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- BZKFMUIJRXWWQK-UHFFFAOYSA-N Cyclopentenone Chemical compound O=C1CCC=C1 BZKFMUIJRXWWQK-UHFFFAOYSA-N 0.000 description 1
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 1
- 239000004150 EU approved colour Substances 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- 241000400611 Eucalyptus deanei Species 0.000 description 1
- 102000016359 Fibronectins Human genes 0.000 description 1
- 108010067306 Fibronectins Proteins 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 206010018366 Glomerulonephritis acute Diseases 0.000 description 1
- 206010018367 Glomerulonephritis chronic Diseases 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- VWVYILCFSYNJHF-UHFFFAOYSA-N Goe 6976 Chemical compound C1=CC=C2N(CCC#N)C3=C4N(C)C5=CC=CC=C5C4=C(C(=O)NC4)C4=C3C2=C1 VWVYILCFSYNJHF-UHFFFAOYSA-N 0.000 description 1
- 206010070737 HIV associated nephropathy Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- 108010004250 Inhibins Proteins 0.000 description 1
- 102000002746 Inhibins Human genes 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 235000019759 Maize starch Nutrition 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- 208000034827 Neointima Diseases 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 229920001774 Perfluoroether Polymers 0.000 description 1
- 206010034650 Peritoneal adhesions Diseases 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 208000027032 Renal vascular disease Diseases 0.000 description 1
- 241000242739 Renilla Species 0.000 description 1
- 108010052090 Renilla Luciferases Proteins 0.000 description 1
- 108700008625 Reporter Genes Proteins 0.000 description 1
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 102000007374 Smad Proteins Human genes 0.000 description 1
- 108010007945 Smad Proteins Proteins 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 239000004141 Sodium laurylsulphate Substances 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 108091005735 TGF-beta receptors Proteins 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 102000016715 Transforming Growth Factor beta Receptors Human genes 0.000 description 1
- 102000011117 Transforming Growth Factor beta2 Human genes 0.000 description 1
- 102000004060 Transforming Growth Factor-beta Type II Receptor Human genes 0.000 description 1
- 108010082684 Transforming Growth Factor-beta Type II Receptor Proteins 0.000 description 1
- 101800000304 Transforming growth factor beta-2 Proteins 0.000 description 1
- KOOADCGQJDGAGA-UHFFFAOYSA-N [amino(dimethyl)silyl]methane Chemical compound C[Si](C)(C)N KOOADCGQJDGAGA-UHFFFAOYSA-N 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- SPEUIVXLLWOEMJ-UHFFFAOYSA-N acetaldehyde dimethyl acetal Natural products COC(C)OC SPEUIVXLLWOEMJ-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 239000011149 active material Substances 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 1
- 239000008168 almond oil Substances 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 230000003444 anaesthetic effect Effects 0.000 description 1
- 238000002399 angioplasty Methods 0.000 description 1
- 229940126317 angiotensin II receptor antagonist Drugs 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 150000001543 aryl boronic acids Chemical class 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 125000002785 azepinyl group Chemical group 0.000 description 1
- 150000001540 azides Chemical class 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- KXDAEFPNCMNJSK-UHFFFAOYSA-N benzene carboxamide Natural products NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000003310 benzodiazepinyl group Chemical group N1N=C(C=CC2=C1C=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004622 benzoxazinyl group Chemical group O1NC(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 229940112869 bone morphogenetic protein Drugs 0.000 description 1
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 210000000748 cardiovascular system Anatomy 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000001364 causal effect Effects 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 230000009137 competitive binding Effects 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 239000012050 conventional carrier Substances 0.000 description 1
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 239000013058 crude material Substances 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 125000002576 diazepinyl group Chemical group N1N=C(C=CC=C1)* 0.000 description 1
- GLUUGHFHXGJENI-UHFFFAOYSA-N diethylenediamine Natural products C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 1
- 125000004609 dihydroquinazolinyl group Chemical group N1(CN=CC2=CC=CC=C12)* 0.000 description 1
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 1
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- 125000000532 dioxanyl group Chemical group 0.000 description 1
- 125000005879 dioxolanyl group Chemical group 0.000 description 1
- FVWDBVACVTXVJN-UHFFFAOYSA-L dipotassium;propan-2-one;carbonate Chemical compound [K+].[K+].CC(C)=O.[O-]C([O-])=O FVWDBVACVTXVJN-UHFFFAOYSA-L 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- NLHWCTNYFFIPJT-UHFFFAOYSA-N disodium bis(trimethylsilyl)azanide Chemical compound [Na+].[Na+].C[Si](C)(C)[N-][Si](C)(C)C.C[Si](C)(C)[N-][Si](C)(C)C NLHWCTNYFFIPJT-UHFFFAOYSA-N 0.000 description 1
- UZZWBUYVTBPQIV-UHFFFAOYSA-N dme dimethoxyethane Chemical compound COCCOC.COCCOC UZZWBUYVTBPQIV-UHFFFAOYSA-N 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 239000003221 ear drop Substances 0.000 description 1
- 229940047652 ear drops Drugs 0.000 description 1
- 239000008157 edible vegetable oil Substances 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 239000003974 emollient agent Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- LHWWETDBWVTKJO-UHFFFAOYSA-N et3n triethylamine Chemical compound CCN(CC)CC.CCN(CC)CC LHWWETDBWVTKJO-UHFFFAOYSA-N 0.000 description 1
- OCLXJTCGWSSVOE-UHFFFAOYSA-N ethanol etoh Chemical compound CCO.CCO OCLXJTCGWSSVOE-UHFFFAOYSA-N 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- NYURFOITSAPYLU-UHFFFAOYSA-N ethyl 3-chloro-4-fluorobenzoate Chemical compound CCOC(=O)C1=CC=C(F)C(Cl)=C1 NYURFOITSAPYLU-UHFFFAOYSA-N 0.000 description 1
- FMSXHQBTRLUYHP-UHFFFAOYSA-N ethyl 6-methylpyridine-2-carboxylate Chemical compound CCOC(=O)C1=CC=CC(C)=N1 FMSXHQBTRLUYHP-UHFFFAOYSA-N 0.000 description 1
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 239000003885 eye ointment Substances 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 230000000893 fibroproliferative effect Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 238000002290 gas chromatography-mass spectrometry Methods 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000003193 general anesthetic agent Substances 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 206010061989 glomerulosclerosis Diseases 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- USZLCYNVCCDPLQ-UHFFFAOYSA-N hydron;n-methoxymethanamine;chloride Chemical compound Cl.CNOC USZLCYNVCCDPLQ-UHFFFAOYSA-N 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 150000002460 imidazoles Chemical class 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 150000005232 imidazopyridines Chemical class 0.000 description 1
- 125000004857 imidazopyridinyl group Chemical group N1C(=NC2=C1C=CC=N2)* 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000000893 inhibin Substances 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 102000006495 integrins Human genes 0.000 description 1
- 108010044426 integrins Proteins 0.000 description 1
- 229940079865 intestinal antiinfectives imidazole derivative Drugs 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- 208000019423 liver disease Diseases 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- BCVXHSPFUWZLGQ-UHFFFAOYSA-N mecn acetonitrile Chemical compound CC#N.CC#N BCVXHSPFUWZLGQ-UHFFFAOYSA-N 0.000 description 1
- 230000028161 membrane depolarization Effects 0.000 description 1
- COTNUBDHGSIOTA-UHFFFAOYSA-N meoh methanol Chemical compound OC.OC COTNUBDHGSIOTA-UHFFFAOYSA-N 0.000 description 1
- 210000003584 mesangial cell Anatomy 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- XRDRKVPNHIWTBX-UHFFFAOYSA-N methyl 3-chlorobenzoate Chemical compound COC(=O)C1=CC=CC(Cl)=C1 XRDRKVPNHIWTBX-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 238000000491 multivariate analysis Methods 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000008692 neointimal formation Effects 0.000 description 1
- 239000002687 nonaqueous vehicle Substances 0.000 description 1
- 239000003883 ointment base Substances 0.000 description 1
- XMLQWXUVTXCDDL-UHFFFAOYSA-N oleyl alcohol Natural products CCCCCCC=CCCCCCCCCCCO XMLQWXUVTXCDDL-UHFFFAOYSA-N 0.000 description 1
- 229940055577 oleyl alcohol Drugs 0.000 description 1
- 229940006093 opthalmologic coloring agent diagnostic Drugs 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 125000005489 p-toluenesulfonic acid group Chemical class 0.000 description 1
- 239000003182 parenteral nutrition solution Substances 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 125000005010 perfluoroalkyl group Chemical group 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 239000008024 pharmaceutical diluent Substances 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 125000005544 phthalimido group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 230000010287 polarization Effects 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- QDQZNQGEYULIKI-UHFFFAOYSA-N propan-2-yl 6-fluoropyridine-2-carboxylate Chemical compound CC(C)OC(=O)C1=CC=CC(F)=N1 QDQZNQGEYULIKI-UHFFFAOYSA-N 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 1
- 235000018102 proteins Nutrition 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 239000003586 protic polar solvent Substances 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 238000006268 reductive amination reaction Methods 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 208000015670 renal artery disease Diseases 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 125000006413 ring segment Chemical group 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000003355 serines Chemical group 0.000 description 1
- 125000003607 serino group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(O[H])([H])[H] 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 210000000329 smooth muscle myocyte Anatomy 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- WGRULTCAYDOGQK-UHFFFAOYSA-M sodium;sodium;hydroxide Chemical compound [OH-].[Na].[Na+] WGRULTCAYDOGQK-UHFFFAOYSA-M 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 238000010532 solid phase synthesis reaction Methods 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000001593 sorbitan monooleate Substances 0.000 description 1
- 235000011069 sorbitan monooleate Nutrition 0.000 description 1
- 229940035049 sorbitan monooleate Drugs 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 235000011044 succinic acid Nutrition 0.000 description 1
- 150000003444 succinic acids Chemical class 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- IIACRCGMVDHOTQ-UHFFFAOYSA-N sulfamic acid group Chemical class S(N)(O)(=O)=O IIACRCGMVDHOTQ-UHFFFAOYSA-N 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- 230000006103 sulfonylation Effects 0.000 description 1
- 238000005694 sulfonylation reaction Methods 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000002511 suppository base Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 229940042055 systemic antimycotics triazole derivative Drugs 0.000 description 1
- 239000002278 tabletting lubricant Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000005207 tetraalkylammonium group Chemical group 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- PHCBRBWANGJMHS-UHFFFAOYSA-J tetrasodium;disulfate Chemical compound [Na+].[Na+].[Na+].[Na+].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O PHCBRBWANGJMHS-UHFFFAOYSA-J 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000005308 thiazepinyl group Chemical group S1N=C(C=CC=C1)* 0.000 description 1
- 125000002769 thiazolinyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000005505 thiomorpholino group Chemical group 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 239000012049 topical pharmaceutical composition Substances 0.000 description 1
- 231100000167 toxic agent Toxicity 0.000 description 1
- 239000003440 toxic substance Substances 0.000 description 1
- 230000002110 toxicologic effect Effects 0.000 description 1
- 231100000759 toxicological effect Toxicity 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 238000011830 transgenic mouse model Methods 0.000 description 1
- 229960001727 tretinoin Drugs 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000009827 uniform distribution Methods 0.000 description 1
- 231100000216 vascular lesion Toxicity 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/56—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Definitions
- This invention relates to novel 2-phenylpyridin-4-yl heterocyclyl derivatives which are inhibitors of the transforming growth factor, (“TGF”)- ⁇ signalling pathway, in particular, the phosphorylation of smad2 or smad3 by the TGF- ⁇ type I or activin-like kinase (“ALK”)-5 receptor, methods for their preparation and their use in medicine, specifically in the treatment and prevention of a disease state mediated by this pathway.
- TGF transforming growth factor
- ALK activin-like kinase
- TGF- ⁇ 1 is the prototypic member of a family of cytokines including the TGF- ⁇ s, activins, inhibins, bone morphogenetic proteins and M ⁇ llerian-inhibiting substance, that signal through a family of single transmembrane serine/threonine kinase receptors. These receptors can be divided into two classes, the type I or activin like kinase (ALK) receptors and type II receptors.
- ALK activin like kinase
- the ALK receptors are distinguished from the type II receptors in that the ALK receptors (a) lack the serine/threonine rich intracellular tail, (b) possess serine/threonine kinase domains that are very homologous between type I receptors, and (c) share a common sequence motif called the GS domain, consisting of a region rich in glycine and serine residues.
- the GS domain is at the amino terminal end of the intracellular kinase domain and is critical for activation by the type II receptor.
- the type II receptor phosphorylates the GS domain of the type I receptor for TGF- ⁇ , ALK5, in the presence of TGF- ⁇ .
- the ALK5 in turn, phosphorylates the cytoplasmic proteins smad2 and smad3 at two carboxy terminal serines.
- the phosphorylated smad proteins translocate into the nucleus and activate genes that contribute to the production of extracellular matrix. Therefore, preferred compounds of this invention are selective in that they inhibit the type I receptor and thus matrix production.
- the invention provides a compound of formula (I), a pharmaceutically acceptable salt, solvate or derivative thereof:
- A is furan, dioxolane, thiophene, pyrrole, imidazole, pyrrolidine, pyran, pyridine, pyrimidine, morpholine, piperidine, oxazole, isoxazole, oxazoline, oxazolidine, thiazole, isothiazole, thiadiazole, benzofuran, indole, isoindole, indazole, imidazopyridine, quinazoline, quinoline, isoquinoline, pyrazole or triazole;
- X is N or CH;
- R 1 is hydrogen, C ⁇ -6 alkyl, C 1-6 alkenyl, C 1-6 alkoxy, halo, cyano, perfluoro C -6 alkyl, perfluoroC 1-6 alkoxy, -NR 5 R 6 , -(CH 2 ) n NR 5 R 6 , -O(CH 2 ) n OR 7 , -O(CH 2 ) n -Het, -O(CH 2 ) n NR 5 R 6 , -CONR 5 R 6 , -CO(CH 2 ) n NR 5 R 6 , -SO 2 R 7 , -SO 2 NR 5 R 6 ,
- R 2 is hydrogen, C 1-6 alkyl, halo, cyano or perfluoroC 1 . 6 alkyl;
- R 3 is hydrogen or halo;
- R 4 is hydrogen, halo, phenyl, C 1-6 alkyl or -NR 5 R 6 ;
- R 5 and R 6 are independently selected from hydrogen; Het; C 3-6 cycloalkyl optionally substituted by C 1-6 alkyl; or by C 1-6 alkyl optionally substituted by Het, alkoxy, cyano or -NR a R b (where R a and R b which may the same or different are hydrogen or C 1-6 alkyl, or R a and R b together with the nitrogen atom to which they are attached may form a 4,5 or 6-membered saturated ring); or R 5 and
- R 6 together with the nitrogen atom to which they are attached form a 3, 4, 5, 6 or 7-membered saturated or unsaturated ring which may contain one or more heteroatoms selected from N, S or O, and wherein the ring may be further substituted by one or more substituents selected from halo (such as fluoro, chloro, bromo), cyano, -CF 3 , hydroxy, -OCF 3 , C 1-6 alkyl and C 1-6 alkoxy;
- halo such as fluoro, chloro, bromo
- cyano, -CF 3 cyano, -CF 3 , hydroxy, -OCF 3 , C 1-6 alkyl and C 1-6 alkoxy
- R 7 is selected from hydrogen and C 1-6 alkyl
- Het is a 5 or 6-membered C-linked heterocyclyl group which may be saturated, unsaturated or aromatic, which may contain one or more heteroatoms selected from N, S or O and which may be substituted by C 1-6 alkyl; and n is 1-4; with the provisos that: a) when A is thiazole (wherein the thiazole sulfur is on the same side as the 4-pyridyl moiety); X is N; R 1 is hydrogen, C 1-6 alkyl, C 1-6 alkoxy, halo, cyano, perfluoroC 1-6 alkyl or perfluoroC ⁇ alkoxy; R 2 is hydrogen, C 1-6 alkyl, halo, cyano or perfluoroC 1-6 alkyl; and R 3 is hydrogen or halo; then R 4 is not NH 2 ; and b) when X is N, A is pyrazole (where the ring containing X is attached to the pyrazole ring at carbon atom next
- C 1-6 alkyl refers to a straight or branched chain saturated aliphatic hydrocarbon radical of 1 to 6 carbon atoms, unless the chain length is limited thereto, including, but not limited to methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, isobutyl, tert-butyl, pentyl and hexyl.
- alkenyl as a group or part of a group refers to a straight or branched chain mono- or poly-unsaturated aliphatic hydrocarbon radical containing the specified number(s) of carbon atoms.
- References to “alkenyl” groups include groups which may be in the E- or Z-form or mixtures thereof.
- alkoxy refers to an alkyl ether radical, wherein the term “alkyl” is defined above.
- alkoxy groups in particular include methoxy, ethoxy, n-propoxy, iso-propoxy, n-butoxy, iso-butoxy, sec-butoxy and tert- butoxy.
- perfluoroalkyl as used herein includes compounds such as trifluoromethyl.
- perfluoroalkoxy as used herein includes compounds such as trifluoromethoxy.
- halo or halogen are used interchangeably herein to mean radicals derived from the elements chlorine, fluorine, iodine and bromine.
- heterocyclyl as used herein includes cyclic groups containing 5 to 7 ring- atoms up to 4 of which may be hetero-atoms such as nitrogen, oxygen and sulfur, and may be saturated, unsaturated or aromatic.
- heterocyclyl groups are furyl, thienyl, pyrrolyl, pyrrolinyl, pyrrolidinyl, imidazolyl, dioxolanyl, oxazolyl, thiazolyl, imidazolyl, imidazolinyl, imidazolidinyl, pyrazolyl, pyrazolinyl, pyrazolidinyl, isoxazolyl, isothiazolyl, oxadiazolyl, triazolyl, thiadiazolyl, pyranyl, pyridyl, piperidinyl, dioxanyl, morpholino, dithianyl, thiomorpholino, pyridazinyl, pyr
- heterocyclyl includes fused heterocyclyl groups, for example benzimidazolyl, benzoxazolyl, imidazopyridinyl, benzoxazinyl, benzothiazinyl, oxazolopyridinyl, benzofuranyl, quinolinyl, quinazolinyl, quinoxalinyl, dihydroquinazolinyl, benzothiazolyl, phthalimido, benzofuranyl, benzodiazepinyl, indolyl and isoindolyl.
- A is furan, thiophene, pyrrole, imidazole, pyridine, pyrimidine, oxazole, isoxazole, thiazole, isothiazole, thiadiazole, imidazopyridine, pyrazole or triazole; each of which is optionally substituted by one or more of the substituents R 4 .
- A is triazole, imidazopyridine, thiazole, imidazole or pyrazole; each of which is optionally substituted by one or more of the substituents R 4 .
- A is imidazopyridine, thiazole or imidazole; each of which is optionally substituted by one R 4 substitutent.
- A is imidazole optionally substituted by one R 4 substitutent.
- X is N.
- R 1 is C 1-6 alkyF, C 1-6 alkoxy, halo, cyano, perfluoroC 1-6 alkoxy, -NR 5 R 6 , -(CH 2 ) n NR 5 R 6 , -O(CH 2 ) n OR 7 , -O(CH 2 ) n -Het, -O(CH 2 ) n NR 5 R 6 , -CONR 5 R 6 , -SO 2 R 7 , -NR 5 SO 2 R 7 , -NR 5 COR 7 -O(CH 2 ) n CONR 5 R 6 , -NR 5 CO(CH 2 ) n NR 5 R 6 or -C(O)R 7 .
- R 1 is C 1-6 alkoxy, halo, perfluoroC 1-6 alkoxy, -NR 5 R 6 , -(CH 2 )nNR 5 R 6 , -O(CH 2 ) n OR 7 , -O(CH 2 ) n -Het, -O(CH 2 ) n NR 5 R 6 , -CONR 5 R 6 , -SO 2 R 7 or -O(CH 2 ) n CONR 5 R 6 .
- R 2 is hydrogen, C 1-6 alkyl or fluoro. More preferably R 2 is hydrogen or methyl. More preferably still, R 2 is methyl. Preferably R 3 is hydrogen.
- R 2 is methyl. More preferably when X is N and R 2 is methyl, R 3 is hydrogen.
- R 4 is hydrogen, phenyl, C 1-6 alkyl or halo. More preferably tert-butyl, isopropyl or methyl.
- R 5 and R 6 are independently selected from hydrogen; Het (preferably tetrahydropyranyl); C 3 ⁇ cycloalkyl optionally substituted by C ⁇ -6 alkyl; or by C 1-6 alkyl optionally substituted by Het (preferably furyl), alkoxy, cyano or-NR a R b (where R a and R b which may the same or different are hydrogen or C 1-6 alkyl, or R a and R b together with the nitrogen atom to which they are attached may form a 4, 5 or 6- membered saturated ring); or R 5 and R 6 together with the atom to which they are attached form a morpholine, piperidine, pyrrolidine or piperazine ring, each of which may be substituted by halo (such as fluoro, chloro, bromo), cyano, -CF 3 , hydroxy, -OCF 3 , C 1-4 alkyl or C 1- alkoxy.
- halo such as fluoro, chloro, brom
- R 5 and R 6 are independently selected from hydrogen, Het
- R 5 and R 6 together with the atom to which they are attached form a morpholine, piperidine, pyrrolidine or piperazine ring, each of which may be substituted by halo (such as fluoro, chloro, bromo), cyano, -CF 3 , hydroxy, -OCF 3 , C 1-4 alkyl or C 1-4 alkoxy.
- halo such as fluoro, chloro, bromo
- A is imidazole;
- X is N;
- R 1 is C 1-6 alkyl, C 1-6 alkoxy, halo, cyano, perfluoroC 1-6 alkoxy, -NR 5 R 6 , -(CH 2 ) n NR 5 R 6 ,
- -(CH 2 ) n OR 7 -O(CH 2 )n-Het (preferably imidazolyl), -O(CH 2 ) n NR 5 R 6 , -CONR 5 R 6 , -SO 2 R 7 , -NR 5 SO 2 R 7 , -R 5 COR 7 , -O(CH 2 ) n CONR 5 R 6 , -NR 5 CO(CH 2 ) n NR 5 R 6 or . -C(O)R 7 ;
- R 2 is hydrogen, C ⁇ alkyl or fluoro
- R 3 is hydrogen or halo
- R 4 is hydrogen, phenyl, C 1-6 alkyl or halo
- R 5 and R 6 are independently selected from hydrogen, Het (preferably tetrahydropyranyl) or C 1-6 alkyl; or R 5 and R 6 together with the atom to which they are attached form a morpholine, piperidine, pyrrolidine or piperazine ring, each of which may be substituted by halo (such as fluoro, chloro, bromo), cyano, -CF 3 , hydroxy, -OCF 3 , C 1-4 alkyl or C 1-4 alkoxy; R 7 is selected from hydrogen and C 1-6 alkyl;
- Het is a 5 or 6-membered C-linked heterocyclyl group which may be saturated, unsaturated or aromatic, which may contain one or more heteroatoms selected from N, S or O and which may be substituted by C 1-6 alkyl; and n is 1-4.
- Compounds of formula (I) which are of special interest as agents useful in the treatment or prophylaxis of disorders characterised by the overexpression of TGF- ⁇ are selected from the list: 4- ⁇ 2-tert-Butyl-5-[6-methyl]-pyridin-2-yl-1H-imidazol-4-yl ⁇ -2-(4-methanesulfonyl- phenyl)-pyridine (Example 84); 4- ⁇ 4-[4-(2-tetf-Butyl-5- ⁇ 6-methyl ⁇ -pyridin-2-yl-1H-imidazol-4-yl)-pyridin-2-yl]-phenyl ⁇ - morpholine (Example 86);
- substituted means substituted by one or more defined groups.
- groups may be selected from a number of alternative groups, the selected groups may be the same or different.
- the term independently means that where more than one substituent is selected from a number of possible substituents, those substituents may be the same or different.
- pharmaceutically acceptable derivative means any pharmaceutically acceptable salt, solvate, ester or amide, or salt or solvate of such ester or amide, of the compound of formula (I), or any other compound which upon administration to the recipient is capable of providing (directly or indirectly) the a compound of formula (I) or an active metabolite or residue thereof, e.g., a prodrug.
- Preferred pharmaceutically acceptable derivatives according to the invention are any pharmaceutically acceptable salts, solvates or prodrugs.
- Suitable pharmaceutically acceptable salts of the compounds of formula (I) include acid salts, for example sodium, potassium, calcium, magnesium and tetraalkylammonium and the like, or mono- or di- basic salts with the appropriate acid for example organic carboxylic acids such as acetic, lactic, tartaric, malic, isethionic, lactobionic and succinic acids; organic sulfonic acids such as methanesulfonic, ethanesulfonic, benzenesulfonic and p-toluenesulfonic acids and inorganic acids such as hydrochloric, sulfuric, phosphoric and sulfamic acids and the like.
- organic carboxylic acids such as acetic, lactic, tartaric, malic, isethionic, lactobionic and succinic acids
- organic sulfonic acids such as methanesulfonic, ethanesulfonic, benzenesulfonic and p-toluen
- Some of the compounds of this invention may be crystallised or recrystallised from solvents such as aqueous and organic solvents. In such cases solvates may be formed.
- This invention includes within its scope stoichiometric solvates including hydrates as well as compounds containing variable amounts of water that may be produced by processes such as lyophilisation.
- compounds, their pharmaceutically acceptable salts, their solvates and polymorphs, defined in any aspect of the invention are referred to as "compounds of the invention".
- the compounds of the invention may exist in one or more tautomeric forms. All tautomers and mixtures thereof are included in the scope of the present invention.
- Compounds of the invention may exist in the form of optical isomers, e.g. diastereoisomers and mixtures of isomers in all ratios, e.g. racemic mixtures.
- the invention includes all such forms, in particular the pure isomeric forms.
- the different isomeric forms may be separated or resolved one from the other by conventional methods, or any given isomer may be obtained by conventional synthetic methods or by stereospecific or asymmetric syntheses.
- the compounds of the invention are intended for use in pharmaceutical compositions it will readily be understood that they are each preferably provided in substantially pure form, for example at least 60% pure, more suitably at least 75% pure and preferably at least 85%, especially at least 98% pure (% are on a weight for weight basis). Impure preparations of the compounds may be used for preparing the more pure forms used in the pharmaceutical compositions; these less pure preparations of the compounds should contain at least 1%, more suitably at least 5% and preferably from 10 to 59% of a compound of the invention.
- Preferred catalysts include tetrakis(triphenlyphosphine) palladium(O), palladium(ll) acetate, dichlorobis(triphenylphosphine) palladium(ll), tris(dibenzylideneacetone) dipalladium(O) and dichlorobis(triphenylphosphine) nickel.
- Compounds of formula (IVA) may be deprotected under acidic conditions (preferably hydrochloric acid) to give compounds of formula (lAa).
- Compounds of formula (lAb) may be prepared according to reaction scheme 2A from compounds of formula (VA), by reacting compounds of formula (VA) with dimethylformamide dimethyl acetal and acetic acid in a solvent such as DMF at room temperature, followed by treatment with hydrazine.
- Compounds of formula (VA) may be prepared using Suzuki coupling methodology (see reaction scheme 1A) from compounds of formula (VIA) according to reaction scheme 3A.
- Compounds of formula (VIA) may in turn be prepared in two steps from 2-bromo-4-pyridinecarboxylic acid.
- Compounds of formula (IVAc), i.e. compounds of formula (IVA) (see reaction scheme 1A) where R 1 is -CH2NR 5 R 6 , may be prepared according to reaction scheme 6A by reacting compounds of formula (IXA) with R 5 R 6 NH in the presence of a reducing agent, preferably sodium triacetoxyborohydride in acetic acid, in a solvent such as dichloroethane at room temperature.
- a reducing agent preferably sodium triacetoxyborohydride in acetic acid
- Compounds of formula (IIA) may be prepared according to reaction scheme 7A. Firstly, 2-bromo-4-methy!pyridine may be coupled to compounds of formula (XA) to give compounds of formula (XIA). Preferred reaction conditions comprise treatment with a base such as sodium bis(trimethylsilyl)amide or potassium bis(trimethyisilyl)amide in tetrahydrofuran at a range of temperature from -70°C to 0°C. Compounds of formula (XIA) may then be reacted with dimethylformamide dimethyl acetal and acetic acid in a solvent such as DMF at room temperature followed by treatment with hydrazine to give compounds of formula (XIIA) where R 2 is hydrogen.
- a base such as sodium bis(trimethylsilyl)amide or potassium bis(trimethyisilyl)amide in tetrahydrofuran at a range of temperature from -70°C to 0°C.
- Compounds of formula (IB) may be prepared from compounds of formula (IIB) by treatment with an azide source according to reaction scheme 1 B.
- Preferred reaction conditions comprise treating compounds of formula (IIB) with trimethylsilylazide at elevated temperature in a suitable solvent such as dimethylformamide.
- Compounds of formula (IIB) may be prepared by reacting compounds of formula (IIIB) (where Y is a leaving group such as halogen preferably chlorine) with boronic acid derivatives of formula (lVB) according to reaction scheme 2B.
- Preferred conditions are those developed by Miyaura et al (Chem.Rev. 1995, 95: 2457), typically comprising reaction inert solvent in the presence of a base and a palladium or nickel catalyst at a temperature of between room temperature and 130°C for a period between 30 minutes and 48 hours.
- Suitable bases include sodium carbonate, potassium carbonate, potassium hydroxide, sodium hydroxide.
- Suitable catalysts include tetrakis(triphenlyphosphine) palladium(O), palladium(ll) acetate, dichlorobis(triphenylphosphine) palladium(ll), tris(dibenylideneacetone) dipalladium(O) and dichlorobis(triphenylphosphine) nickel.
- Compounds of formula (IIIB) may be prepared by Sonagashira coupling of compounds of formula (VB) (where preferably Y is chlorine and Z is iodine) with compounds of formula (VIB) according to reaction scheme 3.
- Preferred reaction conditions comprise reaction in an inert solvent in the presence of a base and a palladium catalyst at a temperature of between room temperature and 80°C, for a period of between 30 minutes and 48 hours.
- Suitable bases include TMEDA or triethyl amine.
- Suitable palladium catalysts include tetrakisftriphenlyphosphine) palladium(O) and dichlorobis(triphenylphosphine) palladium(ll).
- Compounds of formula (VIB) may be prepared according to reaction scheme 4B where Y 1 in compounds of formula (VIIB) is a leaving group, preferably bromine.
- Preferred reaction conditions for the preparation of compounds of formula (VIIIB) comprise treating compounds of formula (VIIB) with trimethylsilylacetylene in the presence of TMEDA and copper iodide under palladium catalysis in an inert solvent such as tetrahydrofuran at elevated temperature.
- the trimethylsilyl group may be removed by treating compounds of formula (VIIIB) with a base such as potassium carbonate in a protic solvent such as methanol.
- Compounds of formula (IIBa), i.e. compounds of formula (IIB) where R 1 is -O(CH 2 ) 2 NR 5 R 6 , may be prepared from compounds of formula (IIIB) (where Y is preferably chlorine) according to reaction scheme 5B.
- Compounds of formula (IIIB) may be reacted with compounds of formula (IXB) to give compounds of formula (IIBa) in one step.
- compounds of formula (IIIB) may firstly be reacted with 4-hydroxy-phenyl boronic acid, followed by alkylation with R 5 R 6 N(CH 2 ) 2 CI in the presence of a base such as potassium carbonate or sodium hydride in a solvent such as dimethylformamide.
- Compounds of formula (IC) may be prepared from compounds of formula (IIC) according to reaction scheme 1C, by reacting compounds of formula (IIC) with compounds of formula (IIIC).
- Preferred reaction conditions comprise boron coupling of compounds of formula (IIIC) where Y is -B(OH)2 or 4,4,5,5-tetramethyl-[1 ,3,2]- dioxaborolan-2-yl cyclic derivative, with a compound of formula (IIC) in the presence of a suitable palladium catalysis (preferably Pd(PPh 3 ) 4 ) and a suitable base (preferably sodium carbonate) in an inert solvent (preferably 1 ,2-dimethoxyethane) at elevated temperature.
- a suitable palladium catalysis preferably Pd(PPh 3 ) 4
- a suitable base preferably sodium carbonate
- Compounds of formula (ICa), i.e. compounds of formula (IC) where R 1 is -CH2NR 5 R 6 , may be prepared by reductive amination of compounds of formula (IVC) according to reaction scheme 2C.
- Preferred reaction conditions comprise reacting (IVC) with HNR 5 R 6 in the presence of NaHB(OAc) 3 , in a suitable solvent (preferably dichloromethane) at room temperature.
- Compounds of formula (ICb), i.e. compounds of formula (IC) where R 1 is -NR 5 R 6 may be prepared according to reaction scheme 3C by reacting compounds of formula (ICc), i.e. compounds of formula (IC) where R 1 is bromine, with HNR 5 R 6 in the presence of a catalyst system preferably tris(dibenzylideneacetone)dipalladium(0) and 2,2'-bis(diphenylphosphino)-1 , 1 '- binaphthyl (Binap) in potassium tert-butoxide in a suitable solvent such as toluene at elevated temperature.
- a catalyst system preferably tris(dibenzylideneacetone)dipalladium(0) and 2,2'-bis(diphenylphosphino)-1 , 1 '- binaphthyl (Binap) in potassium tert-butoxide in a suitable solvent such as toluene at elevated temperature.
- Compounds of formula (ICd), i.e. compounds of formula (IC) where R is -OCH2CH2NR 5 R 6 , may be prepared according to reaction scheme 4C by reacting compounds of formula (VC) with 1 ,2-dibromoethane in the presence of a base preferably potassium carbonate in a suitable solvent, such as acetone, at elevated temperature. Treatment with HNR 5 R 6 in a suitable solvent such as tetrahydrofuran at elevated temperature gives (ICd).
- Compounds of formula (ICe), i.e. compounds of general formula (IC) where R 1 is -CONR 5 R 6 , may be prepared according to reaction scheme 5C.
- Compounds of formula (VIC) (where R is methyl or ethyl) are firstly saponified by heating with sodium hydroxide in methanol, followed by conversion of the resulting carboxylic acid to amide (ICe).
- Preferred reaction conditions comprise treating the intermediate carboxylic acid with HNR 5 R 6 in the presence of HOBT, EDCI and a suitable base such as triethylamine in a suitable solvent such as dimethylformamide at room temperature.
- Compounds of formula (ICg), i.e. compounds of general formula (IC) where R 1 is -NHSO2CF3, may be prepared in two steps according to reaction scheme 6C. Firstly the acetyl group is removed from compounds of formula (ICh) by treatment with sodium hydroxide in methanol at elevated temperature. The resulting amine is then treated with CF3SO2CI preferably in the presence of a base such as triethylamine in a suitable solvent such as dichloromethane at room temperature.
- a base such as triethylamine
- a suitable solvent such as dichloromethane
- compounds of formula (IC) may also be prepared by introducing R 1 before formation of the imidazopyridine.
- compounds of formula (ICi) i.e. compounds of formula (IC) where R 1 is morpholine, X is N and R 3 is H may be prepared according to reaction scheme 7C.
- Compounds of formula (IIC) may be prepared in two steps according to reaction scheme 8C.
- Compounds of formula (VI IC) are firstly reacted with a suitable polymer-supported bromine reagent, such as polymer-supported pyridinium perbromide, in a suitable solvent such as dichloromethane at room temperature.
- a suitable solvent such as dichloromethane at room temperature.
- Treatment with a compound of formula (VIIIC) in a suitable solvent such as ethanol at elevated temperature gives compounds of formula (IIC).
- Compounds of formula (VIIC) may be prepared according to reaction scheme 9C by reacting 2-bromo-4-methylpyridine with compounds of formula (IXC) in the presence of a suitable base such as sodium bis(trimethylsilyl)amide in a suitable solvent such as tetrahydrofuran at -78 °C to -30°C.
- a suitable base such as sodium bis(trimethylsilyl)amide
- a suitable solvent such as tetrahydrofuran at -78 °C to -30°C.
- Compounds of formula (ID) may be prepared according to Scheme 1D.
- Compounds of formula (IID) may be treated with sodium nitrite in HCI to give compounds of formula (MID).
- Compounds of formula (HID) may then be condensed with a suitably substituted aldehyde and ammonium acetate followed by treatment with triethylphosphite to give compounds of formula (IVD) according to the method outlined in US Pat. 5,656,644.
- Boronic acid coupling gives compounds of formula (ID).
- Preferred coupling conditions are those developed by Miyaura et al (Chem.Rev.
- Suitable bases include sodium carbonate, potassium carbonate, potassium hydroxide, sodium hydroxide.
- Suitable catalysts include tetrakis(triphenlyphosphine) palladium(O), palladium(ll) acetate, dichlorobis(triphenylphosphine) palladium(ll), tris(dibenylideneacetone) dipalladium(O) and dichlorobis(triphenylphosphine) nickel.
- Compounds of formula (IDb), i.e. compounds of formula (ID) where X is N, R 1 is - NR 5 R 6 and R 3 is hydrogen, may be prepared according to reaction scheme 3D by reacting compounds of formula (VID) with HNR 5 R 6 in the presence of a catalyst system preferably tris(dibenzylidene acetone)dipalladium(O) and 2,2'- bis(diphenylphosphino)-1,1 '-binaphthyl (Binap) in potassium tert-butoxide in a suitable solvent such as toluene at elevated temperature.
- a catalyst system preferably tris(dibenzylidene acetone)dipalladium(O) and 2,2'- bis(diphenylphosphino)-1,1 '-binaphthyl (Binap) in potassium tert-butoxide in a suitable solvent such as toluene at elevated temperature.
- Preferred reaction conditions comprise treating the intermediate carboxylic acid with HNR 5 R 6 in the presence of hydroxybenzotriazole (HOBT), 1-[3-(Dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDCI) and a suitable base such as triethylamine in a suitable solvent such as dimethylformamide at room temperature.
- HBT hydroxybenzotriazole
- EDCI 1-[3-(Dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride
- a suitable base such as triethylamine
- Compounds of formula (lEa), i.e. compounds of general formula (IE) where A is S, B is N and R 4 is NH 2 , may be prepared by reacting compounds of formula (ME) with a suitable polymer-supported bromine reagent, such as polymer-supported pyridinium perbromide, followed by treatment with thiourea in a suitable solvent such as ethanol, preferably at elevated temperatures (see reaction scheme 1 E).
- a suitable polymer-supported bromine reagent such as polymer-supported pyridinium perbromide
- Compounds of formula (HE) may be prepared by reacting compounds of formula (IVE) with compounds of formula (VE) where Y is a boron containing moiety such as -B(OH) 2 or 4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl according to reaction scheme 3E.
- Preferred conditions comprise reaction with a suitable catalyst such as tetrakis(triphenylphosphine) palladium (0), in the presence of a suitable base such as sodium carbonate in a suitable solvent such as DME at elevated temperature.
- compounds of formula (HE) may be prepared by reacting compounds of formula (VIE) with compounds of formula (VIIE) according to reaction scheme 4E.
- Preferred reaction conditions comprise reacting (VIE) with sodium bis-
- VIE VIE
- VIIE VIIE
- HE HE
- HIE Compounds of formula (HIE) may be prepared according to reaction scheme 5E by reacting compounds of formula (VIIIE) with compounds of formula (VE) (where Y is as defined for reaction scheme 3E) using analogous reaction conditions to those of reaction scheme 3E.
- compounds of formula (HIE) may be prepared according to reaction scheme 6E by reacting compounds of formula (IXE) with compounds of formula (XE) in the presence of a suitable base such as cesium carbonate in a suitable solvent such as tetrahydrofuran and isopropanol at room temperature.
- a suitable base such as cesium carbonate
- a suitable solvent such as tetrahydrofuran and isopropanol
- Compounds of formula (IXE) may be prepared in two steps according to reaction scheme 8E.
- Preferred reaction conditions for the first step are analogous to those described for reaction scheme 3E.
- Preferred reaction conditions for the second step comprise reacting compounds of formula (XIE) with aniline and diphenylphosphite in a suitable solvent such as isopropanol at room temperature.
- Compounds of general formula (IE) may also be prepared using solid supported chemistry.
- Compounds of formula (lEc), i.e. compounds of general formula (I) where A is S, B is N, R is -OR (where R is for example -(CH 2 ) n -Het or -CH 2 CONR 5 R 6 ) and R 4 is NH 2 , may be prepared from solid supported compounds of formula (XllE) by reaction with RX (where X is a suitable leaving group such as chlorine) followed by cleavage under acidic conditions from the solid support, according to reaction scheme 9E.
- Preferred conditions comprise treating (XllE) with RX under basic conditions such as potassium carbonate in a suitable solvent such as DMSO at elevated temperature.
- Preferred cleavage conditions are trifluoroacetic acid in a suitable solvent such as dichloromethane at room temperature.
- Compounds of formula (XVE) may be prepared from solid-phase synthesis according to reaction scheme 13E.
- Compounds of formula (XVIE) may be prepared by treating compounds of formula (IVE) (see scheme 3E) with a suitable polymer-supported bromine reagent, such as polymer-supported pyridinium perbromide.
- a suitable polymer-supported bromine reagent such as polymer-supported pyridinium perbromide.
- Treatment of a resin bound thiourea with a dioxane solution of compounds of formula (XVI) gives the compounds (XV) using general conditions described in the literature (Kearney P.C, J. Org. Chem., (1998), 63, 196).
- the compounds of the invention may be prepared singly or as compound libraries comprising at least 2, for example 5 to 1,000 compounds, and more preferably 10 to 100 compounds.
- Libraries of compounds of the invention may be prepared by a combinatorial 'split and mix' approach or by multiple parallel synthesis using either solution phase or solid phase chemistry, by procedures known to those skilled in the art.
- a compound library comprising at least 2 compounds of the invention.
- TGF- ⁇ 1 Activation of the TGF- ⁇ 1 axis and expansion of extracellular matrix are early and persistent contributors to the development and progression of chronic renal disease and vascular disease. Border W.A., et al, N. Engl. J. Med., 1994; 331(19), 1286-92. Further, TGF- ⁇ 1 plays a role in the formation of fibronectin and plasminogen activator inhibitor-1 , components of sclerotic deposits, through the action of smad3 phosphorylation by the TGF- ⁇ 1 receptor ALK5. Zhang Y., et al, Nature, 1998; 394(6696), 909-13; Usui T., et al, Invest. Ophthalmol. Vis. Sci., 1998; 39(11), 1981-9.
- TGF- ⁇ 1 has been implicated in many renal fibrotic disorders. Border W.A., et al, N. Engl. J. Med., 1994; 331(19), 1286-92. TGF- ⁇ 1 is elevated in acute and chronic glomerulonephritis Yoshioka K., et al, Lab.
- TGF- ⁇ 1 normal glomeruli, mesangial cells and non-renal cells can be induced to produce extracellular-matrix protein and inhibit protease activity by exogenous TGF- ⁇ 1 in vitro.
- TGF- ⁇ 1 and its receptors are increased in injured blood vessels and are indicated in neointima formation following balloon angioplasty Saltis J., et al, Clin. Exp. Pharmacol. Physiol., 1996; 23(3), 193-200.
- TGF- ⁇ 1 is a potent stimulator of smooth muscle cell ("SMC") migration in vitro and migration of SMC in the arterial wall is a contributing factor in the pathogenesis of atherosclerosis and restenos ⁇ s.
- SMC smooth muscle cell
- TGF- ⁇ receptor ALK5 correlated with total cholesterol (P ⁇ 0.001) Blann A.D., et al, Atherosclerosis, 1996; 120(1-2), 221-6.
- SMC derived from human atherosclerotic lesions have an increased ALK5/TGF- ⁇ type II receptor ratio. Because TGF- ⁇ 1 is over-expressed in fibroproliferative vascular lesions, receptor- variant cells would be allowed to grow in a slow, but uncontrolled fashion, while overproducing extracellular matrix components McCaffrey T.A., et al, Jr., J. Clin. Invest., 1995; 96(6), 2667-75.
- TGF- ⁇ 1 was immunolocalized to non-foamy macrophages in atherosclerotic lesions where active matrix synthesis occurs, suggesting that non-foamy macrophages may participate in modulating matrix gene expression in atherosclerotic remodelling via a TGF- ⁇ -dependent mechanism. Therefore, inhibiting the action of TGF- ⁇ 1 on ALK5 is also indicated in atherosclerosis and restenosis. TGF- ⁇ is also indicated in wound repair. Neutralizing antibodies to TGF- ⁇ 1 have been used in a number of models to illustrate that inhibition of TGF- ⁇ 1 signalling is beneficial in restoring function after injury by limiting excessive scar formation during the healing process.
- TGF- ⁇ 1 and TGF- ⁇ 2 reduced scar formation and improved the cytoarchitecture of the neodermis by reducing the number of monocytes and macrophages as well as decreasing dermal fibronectin and collagen deposition in rats Shah M., J. Cell. Sci., 1995, 108, 985- 1002.
- TGF- ⁇ antibodies also improve healing of corneal wounds in rabbits Moller-Pedersen T., Curr. Eye Res., 1998, 17, 736-747, and accelerate wound healing of gastric ulcers in the rat, Ernst H., Gut, 1996, 39, 172-175.
- TGF- ⁇ is also implicated in peritoneal adhesions Saed G.M., et al, Wound Repair Regeneration, 1999 Nov-Dec, 7(6), 504-510. Therefore, inhibitors of ALK5 would be beneficial in preventing peritoneal and sub-dermal fibrotic adhesions following surgical procedures.
- TGF- ⁇ is also implicated in photoaging of the skin (see Fisher GJ. Kang SW. Varani J. Bata-Csorgo Z. Wan YS. Data S. Voorhees JJ. , Mechanisms of photoaging and chronological skin ageing, Archives of Dermatology, 138(11): 1462-1470, 2002 Nov. and Schwartz E. Sapadin AN. Kligman LH. "Ultraviolet B radiation increases steady state mRNA levels for cytokines and integrins in hairless mouse skin- modulation by topical tretinoin", Archives if Dermatological Research, 290(3): 137-144, 1998 Mar.)
- the invention provides the use of a compound defined in the first aspect in the preparation of a medicament for treating or preventing a disease or condition mediated by ALK-5 inhibition.
- the disease or condition mediated by ALK-5 inhibition is selected from the list: chronic renal disease, acute renal disease, wound healing, arthritis, osteoporosis, kidney disease, congestive heart failure, ulcers (including diabetic ulcers, chronic ulcers, gastric ulcers, and duodenal ulcers), ocular disorders, corneal wounds, diabetic nephropathy, impaired neurological function, Alzheimer's disease, atherosclerosis, peritoneal and sub-dermal adhesion, any disease wherein fibrosis is a major component, including, but not limited to kidney fibrosis, lung fibrosis and liver fibrosis, for example, hepatitis B virus (HBV), hepatitis C virus (HCV), alcohol- induced hepatitis, haemochromatosis, primary biliary cirrhosis, restenosis, retroperitoneal fibrosis, mesenteric fibrosis, endometriosis, keloids, cancer, abnormal bone function, inflammatory disorders,
- the disease or condition mediated by ALK-5 inhibition is fibrosis.
- fibrosis Preferably kidney fibrosis.
- references herein to treatment extend to prophylaxis as well as the treatment of established conditions.
- Compounds of the invention may be administered in combination with other therapeutic agents, for example antiviral agents for liver diseases, or in combination with ACE inhibitors or angiotensin II receptor antagonists for kidney diseases.
- other therapeutic agents for example antiviral agents for liver diseases, or in combination with ACE inhibitors or angiotensin II receptor antagonists for kidney diseases.
- the compounds of the invention may be administered in conventional dosage forms prepared by combining a compound of the invention with standard pharmaceutical carriers or diluents according to conventional procedures well known in the art.
- compositions of the invention may be formulated for - administration by any route, and include those in a form adapted for oral, topical or parenteral administration to mammals including humans.
- compositions may be formulated for administration by any route.
- the compositions may be in the form of tablets, capsules, powders, granules, lozenges, creams or liquid preparations, such as oral or sterile parenteral solutions or suspensions.
- topical formulations of the present invention may be presented as, for instance, ointments, creams or lotions, eye ointments and eye or ear drops, impregnated dressings and aerosols, and may contain appropriate conventional additives such as preservatives, solvents to assist drug penetration and emollients in ointments and creams.
- the formulations may also contain compatible conventional carriers, such as cream or ointment bases and ethanol or oleyl alcohol for lotions.
- suitable conventional carriers such as cream or ointment bases and ethanol or oleyl alcohol for lotions.
- Such carriers may be present as from about 1% up to about 98% of the formulation. More usually they will form up to about 80% of the formulation.
- Tablets and capsules for oral administration may be in unit dose presentation form, and may contain conventional excipients such as binding agents, for example syrup, acacia, gelatin, sorbitol, tragacanth, or polyvinylpyrrolidone; fillers, for example lactose, sugar, maize-starch, calcium phosphate, sorbitol or glycine; tabletting lubricants, for example magnesium stearate, talc, polyethylene glycol or silica; disintegrants, for example potato starch; or acceptable wetting agents such as sodium lauryl sulphate.
- the tablets may be coated according to methods well known in normal pharmaceutical practice.
- Oral liquid preparations may be in the form of, for example, aqueous or oily suspensions, solutions, emulsions, syrups or elixirs, or may be presented as a dry product for reconstitution with water or other suitable vehicle before use.
- Such liquid preparations may contain conventional additives, such as suspending agents, for example sorbitol, methyl cellulose, glucose syrup, gelatin, hydroxyethyl cellulose, carboxymethyl cellulose, aluminium stearate gel or hydrogenated edible fats, emulsifying agents, for example lecithin, sorbitan monooleate, or acacia; non-aqueous vehicles (which may include edible oils), for example almond oil, oily esters such as glycerine, propylene glycol, or ethyl alcohol; preservatives, for example methyl or propyl p-hydroxybenzoate or sorbic acid, and, if desired, conventional flavouring or colouring agents.
- suspending agents for example sorbitol, methyl cellulose, glucose syrup, gelatin, hydroxyethyl cellulose, carboxymethyl cellulose, aluminium stearate gel or hydrogenated edible fats, emulsifying agents, for example lecithin, sorbitan monooleate, or
- Suppositories will contain conventional suppository bases, e.g. cocoa-butter or other glyceride.
- fluid unit dosage forms are prepared utilising the compound and a sterile vehicle, water being preferred.
- the compound depending on the vehicle and concentration used, can be either suspended or dissolved in the vehicle.
- the compound can be dissolved in water for injection and filter sterilised before filling into a suitable vial or ampoule and sealing.
- agents such as a local anaesthetic, preservative and buffering agents can be dissolved in the vehicle.
- the composition can be frozen after filling into the vial and the water removed under vacuum. The dry lyophilised powder is then sealed in the vial and an accompanying vial of water for injection may be supplied to reconstitute the liquid prior to use.
- Parenteral suspensions are prepared in substantially the same manner except that the compound is suspended in the vehicle instead of being dissolved and sterilisation cannot be accomplished by filtration.
- the compound can be sterilised by exposure to ethylene oxide before suspending in the sterile vehicle.
- a surfactant or wetting agent is included in the composition to facilitate uniform distribution of the compound.
- compositions may contain from 0.1% by weight, preferably from 10-60% by weight, of the active material, depending on the method of administration. Where the compositions comprise dosage units, each unit will preferably contain from 50-500 mg of the active ingredient.
- the dosage as employed for adult human treatment will preferably range from 100 to 3000 mg per day, for instance 1500 mg per day depending on the route and frequency of administration. Such a dosage corresponds to 1.5 to 50 mg/kg per day. Suitably the dosage is from 5 to 20 mg/kg per day.
- the optimal quantity and spacing of individual dosages of a compound of the invention will be determined by the nature and extent of the condition being treated, the form, route and site of administration, and the particular mammal being treated, and that such optimums can be determined by conventional techniques. It will also be appreciated by one of skill in the art that the optimal course of treatment, i.e., the number of doses of a compound of the invention given per day for a defined number of days, can be ascertained by those skilled in the art using conventional course of treatment determination tests.
- composition comprising a compound of the invention and a pharmaceutically acceptable carrier or diluent
- a disorder selected from chronic renal disease, acute renal disease, wound healing, arthritis, osteoporosis, kidney disease, congestive heart failure, ulcers (including diabetic ulcers, chronic ulcers, gastric ulcers, and duodenal ulcers), ocular disorders, corneal wounds, diabetic nephropathy, impaired neurological function, Alzheimer's disease, atherosclerosis, peritoneal and sub-dermal adhesion, any disease wherein fibrosis is a major component, including, but not limited to kidney fibrosis, lung fibrosis and liver fibrosis, for example, hepatitis B virus (HBV), hepatitis C virus (HCV), alcohol-induced hepatitis, haemochromatosis, primary biliary cirrhosis, restenosis, retroperitoneal fibrosis, mesenteric fibrosis, endometriosis, keloids, cancer, abnormal bone function, inflammatory disorders
- HBV hepatitis B virus
- HCV
- a combination of a compound of the invention with an ACE inhibitor or an angiotensin II receptor antagonist iv) a combination of a compound of the invention with an ACE inhibitor or an angiotensin II receptor antagonist.
- the invention provides a compound of formula (I), a pharmaceutically acceptable salt, solvate or derivative thereof, wherein
- X is N or CH
- A is selected from the list: furan, dioxolane, thiophene, pyrrole, imidazole, pyrrolidine, pyran, pyridine, pyrimidine, morpholine, piperidine, oxazole, isoxazole, oxazoline, oxazolidine, thiazole, isothiazole, thiadiazole, benzof ⁇ ran, indole, isoindole, indazole, imidazopyridine, quinazoline, quinoline, isoquinoline and triazole;
- R 1 is selected from H, C -6 alkyl, C 1-6 alkenyl, C ⁇ alkoxy, halo, cyano, perfluoro C. ⁇ alkyl, perfluoroC 1-6 alkoxy, -NR 5 R 6 , -(CH 2 ) n NR 5 R 6 , -O(CH 2 ) n OR 5 , -O(CH 2 ) n NR 5 R 6 , -CONR 5 R 6 , -CO(CH 2 ) n NR 5 R 6 , -SO 2 R 5 , -SO 2 NR 5 R 6 ,
- R 2 is selected from H, C ⁇ alkyl, halo, CN or perfluoroC 1-6 alkyl;
- R 3 is selected from H or halo
- R 4 is selected from H, halo, C 1-6 alkyl or -NR 5 R 6 ;
- R 5 and R 6 are independently selected from H or C ⁇ alkyl; or R 5 R 6 together with the atom to which they are attached form a 3, 4, 5, 6 or 7-membered saturated or unsaturated ring which may contain one or more heteroatoms selected from N, S or O, and wherein the ring may be further substituted by one or more substituents selected from halo (such as fluoro, chloro, bromo), -CN, -CF 3 , - OH, -OCF 3 , C 1-6 alkyl and C 1-6 alkoxy; and n is 1-4; with the provisos that : a) when A is thiazole (wherein the thiazole sulfur is on the same side as the 4-pyridyl moiety);
- X is N;
- R 1 is hydrogen, C 1-6 alkyl, C 1-6 alkoxy, halo,
- Step 1 Rink Argopore resin (12g, 0.58 mmol/g substitution) was placed into a peptide vessel and washed with CH 2 CI 2 (3x100mL). The resin was then treated for 10min with a solution of piperidine 20% in DMF (3x40mL). After washing with DMF (3x100mL) and CH 2 CI 2 (3x100mL), the resin was treated with a solution of Fmoc- NCS (0.2M) in CH 2 CI 2 (170mL) under argon at room temperature for 1h.
- Step 2 To a solution of intermediate 39 (8.5g, 29mmol) in dioxane (145mL) was added under argon polymer-supported pyridinium perbromide (1.8mmol/g, 16g). The suspension was shaken under argon at room temperature overnight.
- Step 3 The product from step 1 was stirred with the product from step 2 (0.18M) in dioxane (175mL) for 4h at room temperature under argon. The resin was washed with dioxane (3x100mL). A second exposure with the product from step 2 (0.18M in dioxane, 175mL) was performed. The resin was washed with DMF (3x100mL), EtOH (3x100mL), CH 2 CI 2 (3x100mL) and dried under a stream of nitrogen overnight.
- Intermediate 129 was prepared in analogous fashion to intermediate 128 starting from intermediate 40. After step 3, 2 mg of the obtained resin were cleaved with a solution of TFA 20% in CH 2 CI 2 to give the title compound which was characterised by LC-MS (purity>96%); [APCI MS] m/z 347/ 349/ 350 (MH+).
- Example 8 4-[3-(6-methylpyridin-2-yl)-1 H-pyrazol-4-yl]-2-f4-(pyrrolidin-1- ylmethvPphenv ⁇ pyridine
- Step 1 To a solution of intermediate 96 (0.633g, 1mmoI) in toluene (10ml) were added morpholine (0.348g, 4mmol, 4eq), Pd 2 (dba) 3 (0.045g, 0.049mmol, 0.05eq), binap (0.062g, 0.1 mmol, 0.1 eq) and t-BuOK (0.134g, 1.4mmol, 1.4eq) and the reaction mixture was refluxed for 5 hours. The mixture was then poured into ice and extracted with EtOAc. The organic phase was washed with water and dried over Na 2 SO 4 .
- Step 2 4-(4- ⁇ 4-[3-(6-Methyl-2-pyridinyI)-1-(triphenylmethyl)-1 H-pyrazol-4-yl]-2- pyridinyl ⁇ phenyl)morpholine was treated with a mixture of MeOH/HCI 1N (3 :2, 50ml) under reflux for 2 hours. The reaction mixture was poured into water and extracted with CH 2 CI 2 . The aqueous phase was basified with NaOH (1N) and extracted with CH 2 CI 2 .
- Example 75 7-methyl-2-(6-methyl-pyridin-2-yl)-3-(2-r4-((1-methyl-imidazol-4- yl)methyloxy)phenyl1-pyridin-4-yl)-imidazoM,2-alpyridine
- Example 77 7-methyl-2-(6-methyl-pyridin-2-yl)-3-l2- r4(aminocarbonylmethyloxy)phenvn-pyridin-4-yl -imidazo[1,2-alpyridine
- Example 78 8-methyl-2-(6-methyl-pyridin-2-yl)-3-(2-r4- (aminocarbonylmethylo ⁇ yl)phenvn-pyridin-4-yl)-imidazo[1,2-alpyridine
- Example 80 7-methyl-2-(6-methyl-pyridin-2-yl)-3-(2-r4-(morpholin-4-yl)phenv ⁇ - ⁇ pyridin-4-yl)-imidazo[1,2-alPyridine
- intermediate 120 (0,9 ⁇ g, 2. ⁇ 6mmol) in a mixture of DME (30ml) and water (1 ⁇ ml) were added intermediate 33 (0.93g, 2.81 mmol), tetrakis(triphenylphosphine) palladium(O) (0.1g, 0.086mmol) and Na 2 CO 3 (solution 2M, 5ml) and the mixture was heated under reflux overnight and then poured into water. After extraction with CH CI 2 , the organic phase was dried over Na 2 SO 4 , and concentrated under reduced pressure. The residue was recrystallised from EtOAc to afford the title compound as yellow crystals (0.77g, 56.36%); m.p. 174°C ; TOF MS ES + exact mass calculated for C 3 oH 33 N 5 O 2 : 496.2712(MH+). Found : 496.2662 (MH+).
- Example 106 4-(2-tert-Butyl-5-f6-methyl)-pyridin-2-yl-1H-imidazol-4-vn-2-r4-(1- methyl-1H-imidazol-4-ylmethoxy)-phenyll-pyridine
- intermediate 126 (0.49g, 1.27mmol) in DMF (20ml) was added portionwise sodium hydride (60% in mineral oil, 0.162g, 3.81 mmol) and the mixture was stirred at room temperature for 10 minutes.
- Intermediate 22 (0.3g, 1.8mmol) was ⁇ then added and the mixture was stirred for 18 hours at room temperature and then poured into water. After extraction with EtOAc, the organic phase was washed with a solution of NaOH (1 N) and water, dried over Na 2 SO 4 and concentrated under reduced pressure.
- Example 108 4-(2-tert-Butyl- ⁇ -f6-methyl -pyridin-2-yl-1 H-imidazol-4-yl)-2-f4-(2- Pyrrolidin-1-yl-ethoxy)-phenvn-pyridine
- example 83 (0.26g , 0.67mmol) in CH 2 CI 2 (40ml) was added boron tribromide (2.1ml, 2.1 mmol, 3.2eq, solution 1 in CH 2 CI 2 ). The mixture was stirred at room temperature overnight. The reaction mixture was evaporated and neutralised with NaOH (1 N), the resulting mixture was warmed up to 60°C and stirred for 1 hour. After cooling to room temperature, the mixture was extracted with CH 2 CI . The aqueous phase was acidified with HCI (1 N) and extracted with CH 2 CI 2 .
- Example 110 4- ⁇ 2-Phenyl-5-[6-methvn-pyridin-2-yl-1 H-imidazol-4-yl)-2-(4- methanesulfonyl-phenvD-pyridine
- Step 1 Intermediate 128 supported on resin (1g) was weighed out into a peptide vessel. Then 4-formylphenylboronic acid (870mg, 5.8mmol, 10eq), Pd(PPh 3 ) (134 mg, 0.16mmol, 0.2eq), and sodium carbonate (615mg, 5.8mmol, 2M) were added and suspended in toluene/EtOH (8:2, 20mL). The reaction vessel was purged with argon for 5 min, and the mixture was stirred at 90°C for 16h. The resin was washed with DMF (3x1 OmL), water (3x1 OmL), EtOH (3x1 OmL) and CH 2 CI 2 (3x1 OmL).
- Step 2 The product from step 1 was placed into a peptide vessel with a solution of NHR 5 R 6 ( ⁇ . ⁇ mmol, 10eq) in trimethylorthoformate (5.4mL). Then a solution of sodium cyanoborohydride (0.2M) in THF (5.4mL) with acetic acid (110 ⁇ L) was added. The reaction vessel was purged with argon for 5 min and the mixture was stirred at 60°C for 16h. The resin was washed with DMF (3x1 OmL), EtOH (3x1 OmL) and CH 2 CI 2 (3x1 OmL). The resin was treated with a solution of 20% TFA in CH 2 CI 2 and the solvent was removed under reduced pressure. Purification of the residue by HPLC chromatography (water/ acetonitrile gradient) gave the products of formula (lEh) shown in Table 22.
- Step 1 Intermediate 128 or intermediate 129 supported on resin (1g) were weighed out into a peptide vessel. Then 4-hydroxyphenylboronic acid (800mg, 5.8mmol, 10eq), Pd(PPh 3 ) 4 (134 mg, 0.16mmol, 0.2eq), and sodium carbonate (615mg, ⁇ . ⁇ mmol, 2M) were added and suspended in toluene/EtOH (8:2, 20mL). The reaction vessel was purged with argon for 5 min, and the mixture was stirred at 90°C for 16h. The resin was washed with DMF (3x1 OmL), water (3x1 OmL), EtOH (3x1 OmL) and CH 2 CI 2 (3x10mL).
- Step 2 The product from step 1 was placed into a peptide vessel with a solution of R-CI ( ⁇ . ⁇ mmol, 10eq) in DMSO (10mL). Then a solution of potassium carbonate (802mg, ⁇ . ⁇ mmol, 10eq) in DMSO ( ⁇ mL) was added. The reaction vessel was purged with argon for ⁇ min and the mixture was stirred at 90°C for 16h. The resin was washed with DMF (3x1 OmL), EtOH (3x1 OmL) and CH 2 Ci 2 (3x1 OmL). The resin was treated with a solution of 20% TFA in CH 2 CI 2 and the solvent was removed under reduced pressure. Purification of the residue by HPLC chromatography (water/ acetonitrile gradient) gave the products of formula (lEj) shown in Table 23.
- Step 1 Intermediate 129 supported on resin (1g) was weighed out into a peptide vessel. Then 4-methoxycarbonylphenylboronic acid (1.05g, ⁇ . ⁇ mmol, 10eq), Pd(PPh 3 ) (0.134 g, 0.16mmol, 0.2eq), and a aqueous solution of sodium carbonate (0.615g, ⁇ . ⁇ mmol, 2M) were added and suspended in toluene/EtOH ( ⁇ :2, 20mL). The reaction vessel was purged with argon for ⁇ min, and the mixture was stirred at 90°C for 16h.
- the biological activity of the compounds of the invention may be assessed using the following assays: 106
- the potential for compounds of the invention to inhibit TGF- ⁇ signalling may be demonstrated, for example, using the following in vitro assay.
- the assay was performed in HepG2 cells stably transfected with the PAI-1 promoter (known to be a strong TGF- ⁇ responsive promoter) linked to a luciferase (firefly) reporter gene.
- the compounds were selected on their ability to inhibit luciferase activity in cells exposed to TGF- ⁇ .
- cells were transfected with a second luciferase (Renilla) gene which was not driven by a TGF- ⁇ responsive promoter and was used as a toxicity control.
- 96 well microplates were seeded, using a multidrop apparatus, with the stably transfected cell line at a concentration of 35000 cells per well in 200 ⁇ l of serum- containing medium. These plates were placed in a cell incubator. 16 to 24 hours later (Day 2), cell-incubation procedure was launched. Cells were incubated with TGF- ⁇ and a candidate compound at concentrations in the range 50 nM to 10 ⁇ M (final concentration of DMSO 1%). The final concentration of TGF- ⁇ (rhTGF ⁇ -1) used in the test was 1 ng/mL. Cells were incubated with a candidate compound 15-30 mins prior to the addition of TGF- ⁇ . The final volume of the test reaction was 150 ⁇ l. Each well contained only one candidate compound and its effect on the PAI-1 promoter was monitored.
- Columns 11 and 12 were employed as controls. Column 11 contained ⁇ wells in which the cells were incubated in the presence of TGF- ⁇ , without a candidate compound. Column 11 was used to determine the 'reference TGF- ⁇ induced firefly luciferase value' against which values measured in the test wells (to quantify inhibitory activity) were compared. In wells A12 to D12, cells were grown in medium without TGF- ⁇ . The firefly luciferase values obtained from these positions are representative of the 'basal firefly luciferase activity'. In wells E12 to H12, cells were incubated in the presence of TGF- ⁇ and 600 ⁇ M CPO (Cyclopentenone, Sigma), a cell toxic compound. The toxicity was revealed by decreased firefly and renilla luciferase activities (around 60 % of those obtained in column 11).
- luciferase quantification procedure was launched. The following reactions were performed using reagents obtained from a Dual Luciferase Assay Kit (Promega). Cells were washed and lysed with the addition of 10 ⁇ l of passive lysis buffer (Promega). Following agitation (15 to 30 mins), luciferase activities of the plates were read in a dual-injector luminometer (BMG lumistar). For this purpose, 50 ⁇ l of luciferase assay reagent and 60 ⁇ l of 'Stop & Glo' buffer were injected sequentially to quantify the activities of both luciferases. Data obtained from the measurements were processed and analysed using suitable software.
- the mean Luciferase activity value obtained in wells A11 to H11 (Column 11 , TGF- ⁇ only) was considered to represent 100% and values obtained in wells A12 to D12 (cells in medium alone) gave a basal level (0%).
- a concentration response curve was constructed from which an IC 50 value was determined graphically.
- Kinase inhibitor compounds conjugated to fluorophores can be used as fluorescent ligands to monitor ATP competitive binding of other compounds to a given kinase.
- This protocol details the use of a rhodamine green-labelled ligand for assays using recombinant GST-ALK5 (residues 198-603).
- Assay buffer components 62.5 mM Hepes pH 7.5 (Sigma H-4034), 1 mM DTT (Sigma D-0632), 12.5 mM MgCI 2 (Sigma M-9272), 1.26 mM CHAPS (Sigma C-3023).
- ALK5 was added to assay buffer containing the above components and 1 nM of the rhodamine green-labelled ligand so that the final ALK5 concentration was 10 nM based on active site titration of the enzyme.
- the enzyme/ligand reagent 39 ⁇ l was added to each well of the previously prepared assay plates.
- a control compound (1 ⁇ l) was added to column 12, rows E-H for the low control values.
- the plates were read immediately on a LJL Acquest fluorescence reader (Molecular Devices, serial number AQ1048) with excitation, emission, and dichroic filters of 485nm, 530 nm, and 505 nm, respectively.
- the fluorescence polarization for each well was calculated by the Acquest reader and then imported into curve fitting software for construction of concentration response curves.
- the normalized response was determined relative to the high controls (1 ⁇ l DMSO in column 12, rows A-D) and the low controls (1 ⁇ l of control compound in column 12, rows E-H). An IC 50 value was then calculated for each compound
- Example 86 4- ⁇ 4-[4-(2-tett-Butyl-5- ⁇ 6-methyl ⁇ -pyridin-2-yl-1H-imidazol-4-yl)-pyridin-2-yl]-phenyl ⁇ - morpholine (Example 86) showed an ALK5 receptor modulator activity of 34 nM and TGF- ⁇ cellular activity of 183 nM.
- N-(tetrahydropyran-4-yl)-4-(4- ⁇ 2-isopropyl- ⁇ -[6-methyl-pyridin-2-yl]-1 H-imidazol-4-yl ⁇ - pyridin-2-yl)-benzamide (Example 96) showed an ALK ⁇ receptor modulator activity of 26 nM and TGF- ⁇ cellular activity of ⁇ 14 nM.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Biomedical Technology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Cardiology (AREA)
- Diabetes (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Endocrinology (AREA)
- Heart & Thoracic Surgery (AREA)
- Hospice & Palliative Care (AREA)
- Urology & Nephrology (AREA)
- Emergency Medicine (AREA)
- Immunology (AREA)
- Pain & Pain Management (AREA)
- Vascular Medicine (AREA)
- Psychiatry (AREA)
- Reproductive Health (AREA)
- Dermatology (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Ophthalmology & Optometry (AREA)
- Pulmonology (AREA)
- Gastroenterology & Hepatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description



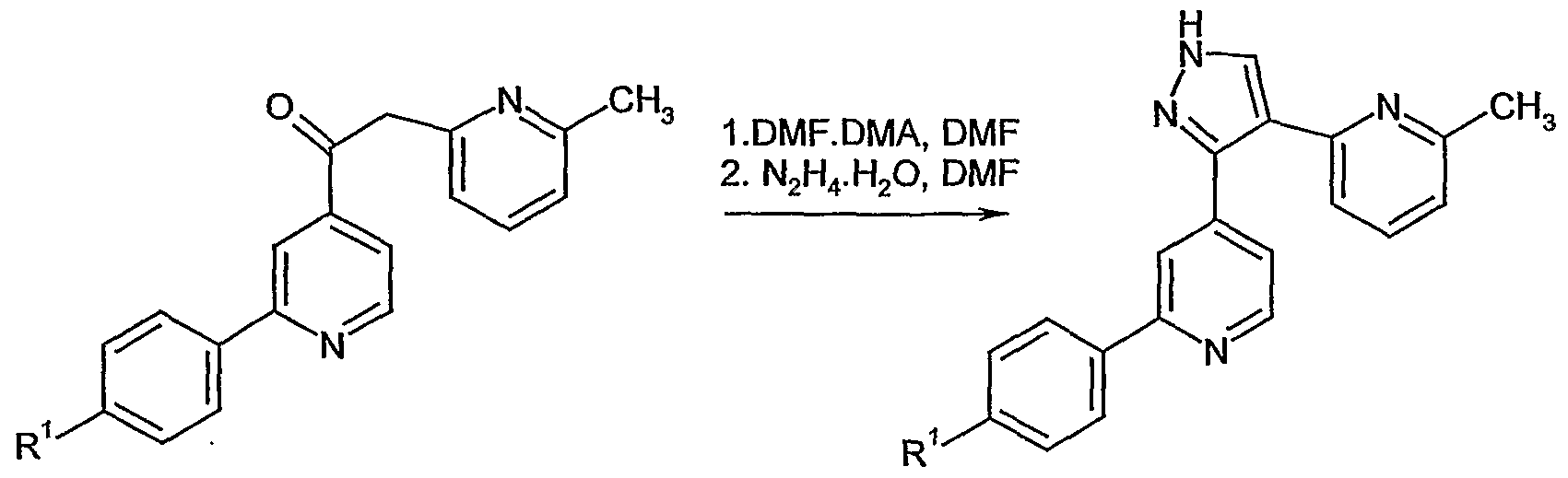










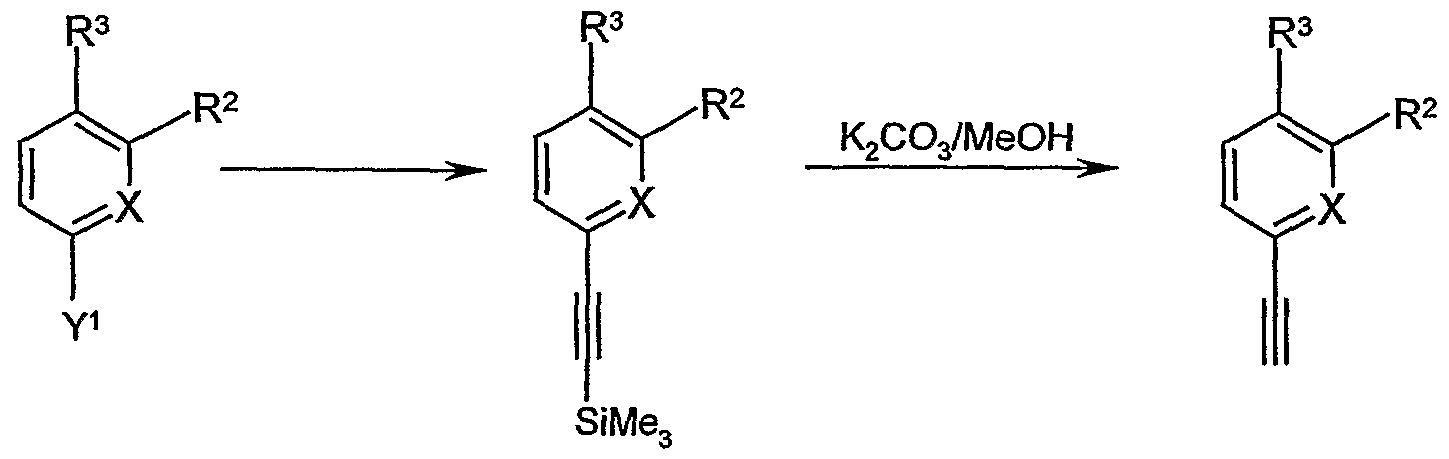









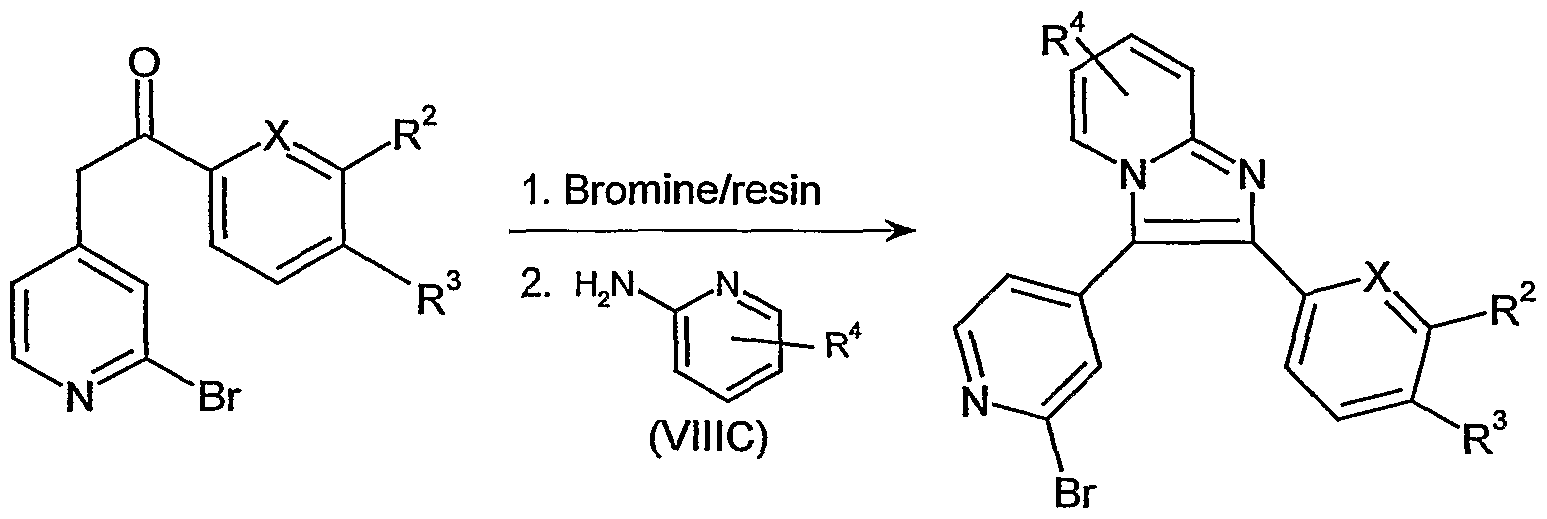

















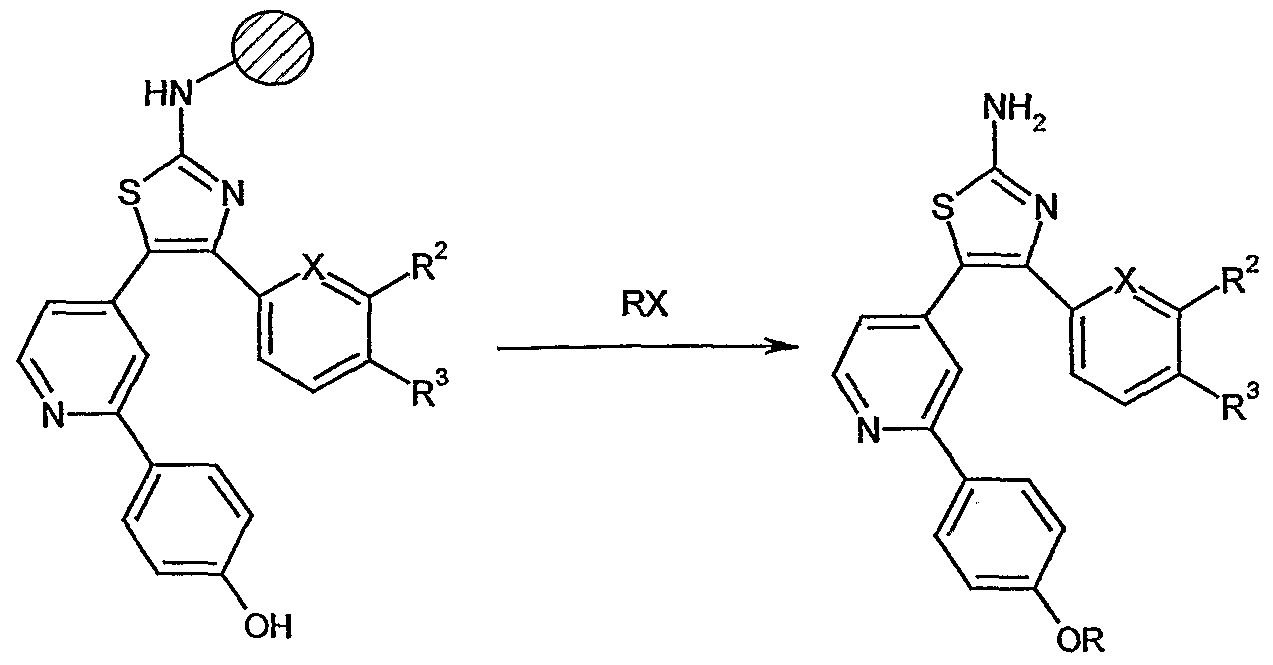
















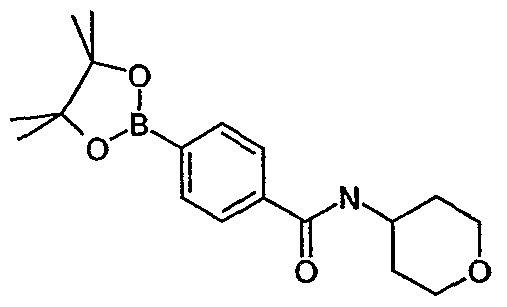






























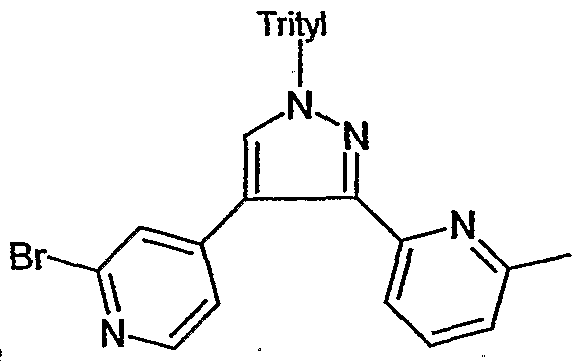









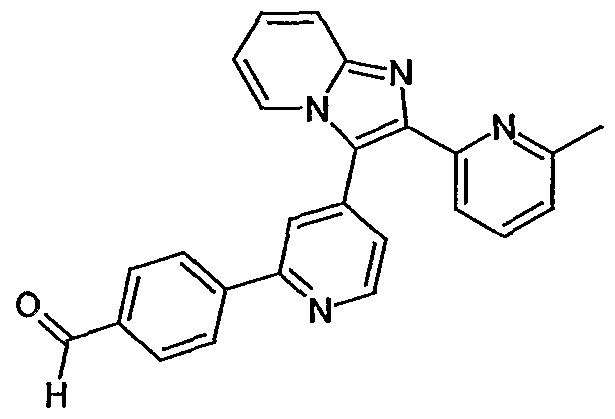













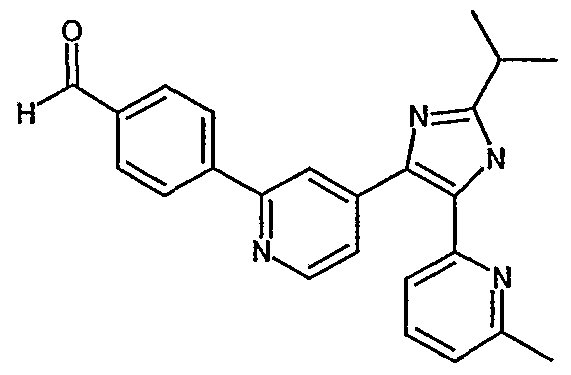

















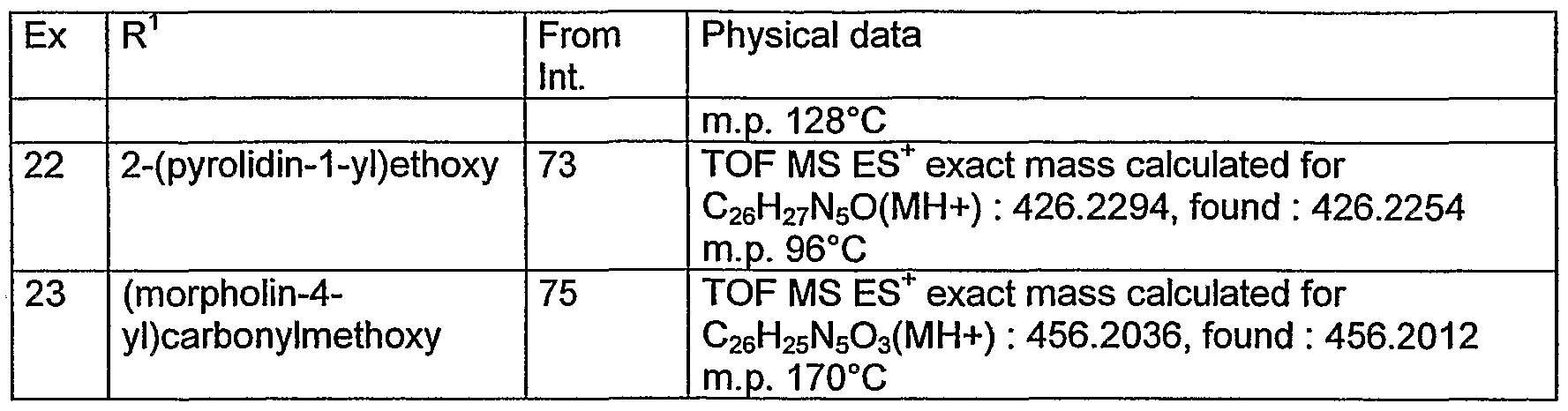








































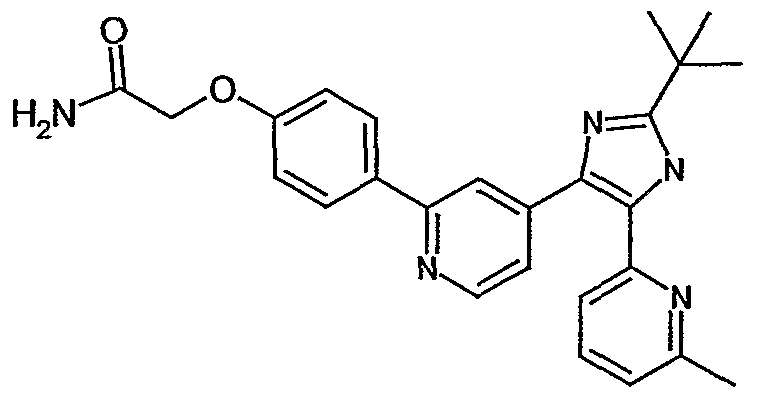







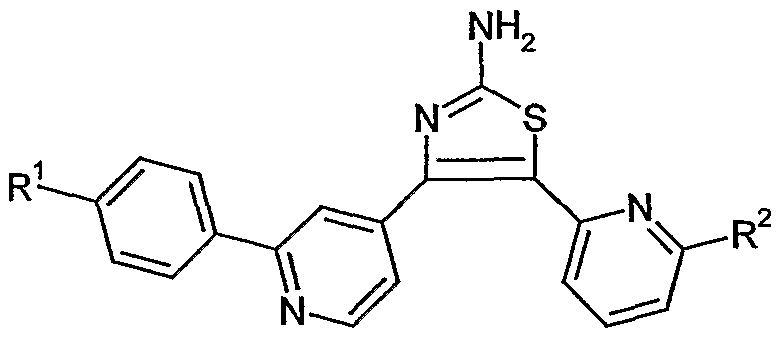









Claims

Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/522,969 US20050245520A1 (en) | 2002-07-31 | 2003-07-29 | 2-Phenylpyridin-4-yl derivatives as alk5 inhibitors |
| JP2004525405A JP2005539000A (en) | 2002-07-31 | 2003-07-29 | 2-Phenylpyridin-4-yl derivatives as ALK5 inhibitors |
| AU2003260345A AU2003260345A1 (en) | 2002-07-31 | 2003-07-29 | 2-phenylpyridin-4-yl derivatives as alk5 inhibitors |
| EP03766385A EP1539748A1 (en) | 2002-07-31 | 2003-07-29 | 2-phenylpyridin-4-yl derivatives as alk5 inhibitors |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0217751.7 | 2002-07-31 | ||
| GB0217751A GB0217751D0 (en) | 2002-07-31 | 2002-07-31 | Compounds |
| GB0314698A GB0314698D0 (en) | 2003-06-24 | 2003-06-24 | Compounds |
| GB0314698.2 | 2003-06-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004013135A1 true WO2004013135A1 (en) | 2004-02-12 |
Family
ID=31497253
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2003/008496 WO2004013135A1 (en) | 2002-07-31 | 2003-07-29 | 2-phenylpyridin-4-yl derivatives as alk5 inhibitors |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20050245520A1 (en) |
| EP (1) | EP1539748A1 (en) |
| JP (1) | JP2005539000A (en) |
| AR (1) | AR040726A1 (en) |
| AU (1) | AU2003260345A1 (en) |
| TW (1) | TW200417547A (en) |
| WO (1) | WO2004013135A1 (en) |
Cited By (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004078110A2 (en) * | 2003-03-04 | 2004-09-16 | Pfizer Products Inc. | Fused imizadoles as transforming growth factor (tgf) inhibitors |
| EP1546112A2 (en) * | 2002-09-06 | 2005-06-29 | Biogen Idec MA Inc. | Imidazolopyridines and methods of making and using the same |
| US6958354B2 (en) | 2002-09-18 | 2005-10-25 | Pfizer Inc. | Pyrazole compounds as transforming growth factor (TGF) inhibitors |
| US7030125B2 (en) | 2002-09-18 | 2006-04-18 | Pfizer, Inc. | Isothiazole and isoxazole compounds as transforming growth factor (TGF) inhibitors |
| US7053095B2 (en) | 2002-09-18 | 2006-05-30 | Pfizer Inc. | Triazole compounds as transforming growth factor (TGF) inhibitors |
| JP2006517592A (en) * | 2003-02-12 | 2006-07-27 | バイオジェン・アイデック・エムエイ・インコーポレイテッド | Pyrazole and methods of making and using them |
| US7153872B2 (en) | 2002-09-18 | 2006-12-26 | Pfizer Inc. | Imidazole compounds as transforming growth factor (TGF) inhibitors |
| WO2007059359A2 (en) * | 2005-11-21 | 2007-05-24 | Biogen Idec Ma Inc. | Substituted pyrazalones |
| US7273936B2 (en) | 2002-09-18 | 2007-09-25 | Pfizer Inc. | Oxazole and thiazole compounds as transforming growth factor (TGF) inhibitors |
| JPWO2005085241A1 (en) * | 2004-03-05 | 2008-01-17 | 大正製薬株式会社 | Thiazole derivative |
| EP1928237A2 (en) * | 2005-09-02 | 2008-06-11 | Abbott Laboratories | Novel imidazo based heterocycles |
| WO2008094574A2 (en) * | 2007-01-30 | 2008-08-07 | Biogen Idec Ma Inc. | Furanone compounds and methods of making and using the same |
| JP2008543971A (en) * | 2005-06-27 | 2008-12-04 | エグゼリクシス, インコーポレイテッド | LXR modulator based on imidazole |
| WO2009087212A2 (en) * | 2008-01-11 | 2009-07-16 | Novartis Ag | Pyridine derivatives |
| WO2009087224A1 (en) | 2008-01-11 | 2009-07-16 | Novartis Ag | Pyrimidines as kinase inhibitors |
| US7973069B2 (en) | 2004-07-14 | 2011-07-05 | Ptc Therapeutics, Inc. | Methods for treating hepatitis C |
| WO2012003405A1 (en) * | 2010-06-30 | 2012-01-05 | Ironwood Pharmaceuticals, Inc. | Sgc stimulators |
| EP2409708A1 (en) * | 2010-07-21 | 2012-01-25 | B & G Partners, LLC | Compounds and methods for selectively targeting tumor-associated mucins |
| JP2013530192A (en) * | 2010-07-05 | 2013-07-25 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツング | Bipyridyl derivatives useful for the treatment of kinase-induced diseases |
| US8507472B2 (en) | 2010-04-07 | 2013-08-13 | Bayer Cropscience Ag | Bicyclic pyridinylpyrazoles |
| WO2014024077A1 (en) * | 2012-08-07 | 2014-02-13 | Aurigene Discovery Technologies Limited | 5-(1H-PYRAZOL-4-YL)-1H-PYRROLO[2,3-b]PYRIDINE DERIVATIVES AS KINASE INHIBITORS |
| KR20140072220A (en) * | 2009-12-18 | 2014-06-12 | 미쓰비시 타나베 파마 코퍼레이션 | Novel antiplatelet agent |
| CN104066731A (en) * | 2011-12-27 | 2014-09-24 | 铁木医药有限公司 | 2 - benzyl, 3 - (pyrimidin- 2 -yl) substituted pyrazoles useful as sgc stimulators |
| US9061030B2 (en) | 2010-11-09 | 2015-06-23 | Ironwood Pharmaceuticals, Inc. | sGC stimulators |
| WO2016210292A1 (en) | 2015-06-25 | 2016-12-29 | Children's Medical Center Corporation | Methods and compositions relating to hematopoietic stem cell expansion, enrichment, and maintenance |
| WO2017161001A1 (en) | 2016-03-15 | 2017-09-21 | Children's Medical Center Corporation | Methods and compositions relating to hematopoietic stem cell expansion |
| WO2018019106A1 (en) | 2016-07-29 | 2018-02-01 | 上海璎黎药业有限公司 | Nitrogenous heterocyclic aromatic compound, preparation method therefor, pharmaceutical composition thereof, and application thereof |
| WO2018086609A1 (en) * | 2016-11-14 | 2018-05-17 | 江苏恒瑞医药股份有限公司 | 3,4-bipyridyl pyrazole derivative, and preparation method therefor and medical application thereof |
| US10041047B2 (en) | 2013-03-14 | 2018-08-07 | Massachusetts Institute Of Technology | Compositions and methods for epithelial stem cell expansion and culture |
| WO2019183245A1 (en) | 2018-03-20 | 2019-09-26 | Icahn School Of Medicine At Mount Sinai | Kinase inhibitor compounds and compositions and methods of use |
| WO2019236766A1 (en) | 2018-06-06 | 2019-12-12 | Ideaya Biosciences, Inc. | Methods of culturing and/or expanding stem cells and/or lineage committed progenitor cells using lactam compounds |
| US10568883B2 (en) | 2014-09-03 | 2020-02-25 | Massachusetts Institute Of Technology | Compositions, systems, and methods for generating inner ear hair cells for treatment of hearing loss |
| WO2020142485A1 (en) | 2018-12-31 | 2020-07-09 | Icahn School Of Medicine At Mount Sinai | Kinase inhibitor compounds and compositions and methods of use |
| US11021687B2 (en) | 2016-01-08 | 2021-06-01 | The Brigham And Women's Hospital, Inc. | Production of differentiated enteroendocrine cells and insulin producing cells |
| US11033546B2 (en) | 2016-03-02 | 2021-06-15 | Frequency Therapeutics, Inc. | Solubilized compositions for controlled proliferation of stem cells / generating inner ear hair cells using a GSK3 inhibitor: I |
| US11066419B2 (en) | 2016-12-30 | 2021-07-20 | Frequency Therapeutics, Inc. | 1H-pyrrole-2,5-dione compounds and methods of using same |
| WO2021167703A1 (en) * | 2020-02-19 | 2021-08-26 | Nammi Therapeutics, Inc. | Formulated and/or co-formulated liposome compositions containing tgfb antagonist prodrugs useful in the treatment of cancer and methods thereof |
| US11160868B2 (en) | 2016-03-02 | 2021-11-02 | Frequency Therapeutics, Inc. | Thermoreversible compositions for administration of therapeutic agents |
| US11162071B2 (en) | 2018-08-17 | 2021-11-02 | Frequency Therapeutics, Inc. | Compositions and methods for generating hair cells by upregulating JAG-1 |
| US11260130B2 (en) | 2016-03-02 | 2022-03-01 | Frequency Therapeutics, Inc. | Solubilized compositions for controlled proliferation of stem cells / generating inner ear hair cells using a GSK3 inhibitor: IV |
| US11447490B2 (en) | 2018-01-24 | 2022-09-20 | Shanghai Yingli Pharmaceutical Co., Ltd | Aromatic heterocyclic substituted olefin compound, preparation method for same, pharmaceutical composition of same, and applications thereof |
| US11584745B2 (en) | 2018-01-24 | 2023-02-21 | Shanghai Yingli Pharmaceutical Co., Ltd | Aromatic heterocyclic compound, intermediate thereof, preparation method therefor, and pharmaceutical composition and use thereof |
| US11617745B2 (en) | 2018-08-17 | 2023-04-04 | Frequency Therapeutics, Inc. | Compositions and methods for generating hair cells by downregulating FOXO |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0217783D0 (en) * | 2002-07-31 | 2002-09-11 | Glaxo Group Ltd | Compounds |
| TW200639163A (en) * | 2005-02-04 | 2006-11-16 | Genentech Inc | RAF inhibitor compounds and methods |
| EP2044056B1 (en) * | 2006-07-14 | 2012-08-22 | Novartis AG | Pyrimidine derivatives as alk-5 inhibitors |
| EA201000615A1 (en) * | 2007-10-17 | 2010-12-30 | Новартис Аг | IMIDAZO [1,2-A] PYRIDINE DERIVATIVES APPLICABLE AS APC INHIBITORS (ACTIVIN-like KINASE) |
| US20120220581A1 (en) | 2009-10-30 | 2012-08-30 | Janssen-Cilag, S.A. | IMIDAZO[1,2-b]PYRIDAZINE DERIVATIVES AND THEIR USE AS PDE10 INHIBITORS |
| AR080754A1 (en) | 2010-03-09 | 2012-05-09 | Janssen Pharmaceutica Nv | IMIDAZO DERIVATIVES (1,2-A) PIRAZINA AND ITS USE AS PDE10 INHIBITORS |
| CA2838645C (en) | 2011-06-27 | 2020-03-10 | Janssen Pharmaceutica Nv | 1-aryl-4-methyl-[1,2,4]triazolo[4,3-a]quinoxaline derivatives |
| US9669035B2 (en) | 2012-06-26 | 2017-06-06 | Janssen Pharmaceutica Nv | Combinations comprising PDE 2 inhibitors such as 1-aryl-4-methyl-[1,2,4]triazolo-[4,3-A]]quinoxaline compounds and PDE 10 inhibitors for use in the treatment of neurological of metabolic disorders |
| ES2607184T3 (en) | 2012-07-09 | 2017-03-29 | Janssen Pharmaceutica, N.V. | Phosphodiesterase 10 enzyme inhibitors |
| WO2016025629A1 (en) | 2014-08-12 | 2016-02-18 | The Regents Of The University Of California | Molecular composition for enhancing and rejuvenating maintenance and repair of mammalian tissues |
| AU2018431586A1 (en) * | 2018-07-09 | 2021-01-21 | Synthis Therapeutics, Inc | Antibody-ALK5 inhibitor conjugates and their uses |
| WO2020256721A1 (en) * | 2019-06-19 | 2020-12-24 | Synthis, Llc | Antib0dy-alk5 inhibitor conjugates and their uses |
| CN115490687B (en) * | 2022-09-19 | 2023-10-27 | 国家烟草质量监督检验中心 | 3-benzofuranyl thioimidazo pyridine derivative, preparation method and application thereof |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002040468A1 (en) * | 2000-11-16 | 2002-05-23 | Smithkline Beecham Corporation | Compounds |
| WO2002040476A1 (en) * | 2000-11-16 | 2002-05-23 | Smithkline Beecham Corporation | Pyridyl-substituted triazoles as tgf inhibitors |
| WO2002062753A1 (en) * | 2001-02-02 | 2002-08-15 | Glaxo Group Limited | Thiazolamines and their use as tgf-beta inhibitors |
| WO2002066462A1 (en) * | 2001-02-02 | 2002-08-29 | Glaxo Group Limited | Pyrazole derivatives against tgf overexpression |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9809869D0 (en) * | 1998-05-09 | 1998-07-08 | Medical Res Council | Inhibition of protein kinases |
| GB0007405D0 (en) * | 2000-03-27 | 2000-05-17 | Smithkline Beecham Corp | Compounds |
| PE20020506A1 (en) * | 2000-08-22 | 2002-07-09 | Glaxo Group Ltd | PIRAZOLE DERIVATIVES FUSED AS PROTEIN KINASE INHIBITORS |
| GB0217783D0 (en) * | 2002-07-31 | 2002-09-11 | Glaxo Group Ltd | Compounds |
-
2003
- 2003-07-29 EP EP03766385A patent/EP1539748A1/en not_active Withdrawn
- 2003-07-29 US US10/522,969 patent/US20050245520A1/en not_active Abandoned
- 2003-07-29 WO PCT/EP2003/008496 patent/WO2004013135A1/en not_active Application Discontinuation
- 2003-07-29 AU AU2003260345A patent/AU2003260345A1/en not_active Abandoned
- 2003-07-29 JP JP2004525405A patent/JP2005539000A/en active Pending
- 2003-07-29 AR AR20030102722A patent/AR040726A1/en not_active Application Discontinuation
- 2003-07-29 TW TW092120588A patent/TW200417547A/en unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002040468A1 (en) * | 2000-11-16 | 2002-05-23 | Smithkline Beecham Corporation | Compounds |
| WO2002040476A1 (en) * | 2000-11-16 | 2002-05-23 | Smithkline Beecham Corporation | Pyridyl-substituted triazoles as tgf inhibitors |
| WO2002062753A1 (en) * | 2001-02-02 | 2002-08-15 | Glaxo Group Limited | Thiazolamines and their use as tgf-beta inhibitors |
| WO2002066462A1 (en) * | 2001-02-02 | 2002-08-29 | Glaxo Group Limited | Pyrazole derivatives against tgf overexpression |
Cited By (77)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1546112A2 (en) * | 2002-09-06 | 2005-06-29 | Biogen Idec MA Inc. | Imidazolopyridines and methods of making and using the same |
| EP1546112A4 (en) * | 2002-09-06 | 2006-06-07 | Biogen Idec Inc | Imidazolopyridines and methods of making and using the same |
| US7151110B2 (en) | 2002-09-18 | 2006-12-19 | Pfizer Inc. | Pyrazole compounds as transforming growth factor (TGF) inhibitors |
| US7638537B2 (en) | 2002-09-18 | 2009-12-29 | Pfizer Inc. | Pyrazole compounds as transforming growth factor (TGF) inhibitors |
| US7030125B2 (en) | 2002-09-18 | 2006-04-18 | Pfizer, Inc. | Isothiazole and isoxazole compounds as transforming growth factor (TGF) inhibitors |
| US7053095B2 (en) | 2002-09-18 | 2006-05-30 | Pfizer Inc. | Triazole compounds as transforming growth factor (TGF) inhibitors |
| US7635702B2 (en) | 2002-09-18 | 2009-12-22 | Pfizer Inc. | Imidazole compounds as transforming growth factor (TGF) inhibitors |
| US6958354B2 (en) | 2002-09-18 | 2005-10-25 | Pfizer Inc. | Pyrazole compounds as transforming growth factor (TGF) inhibitors |
| US7273936B2 (en) | 2002-09-18 | 2007-09-25 | Pfizer Inc. | Oxazole and thiazole compounds as transforming growth factor (TGF) inhibitors |
| US7153872B2 (en) | 2002-09-18 | 2006-12-26 | Pfizer Inc. | Imidazole compounds as transforming growth factor (TGF) inhibitors |
| JP2006517592A (en) * | 2003-02-12 | 2006-07-27 | バイオジェン・アイデック・エムエイ・インコーポレイテッド | Pyrazole and methods of making and using them |
| WO2004078110A3 (en) * | 2003-03-04 | 2004-11-11 | Pfizer Prod Inc | Fused imizadoles as transforming growth factor (tgf) inhibitors |
| WO2004078110A2 (en) * | 2003-03-04 | 2004-09-16 | Pfizer Products Inc. | Fused imizadoles as transforming growth factor (tgf) inhibitors |
| US7417041B2 (en) | 2003-03-04 | 2008-08-26 | Pfizer Inc. | Imidazopyrimidines as transforming growth factor (TGF) inhibitors |
| JP4853284B2 (en) * | 2004-03-05 | 2012-01-11 | 大正製薬株式会社 | Thiazole derivative |
| JPWO2005085241A1 (en) * | 2004-03-05 | 2008-01-17 | 大正製薬株式会社 | Thiazole derivative |
| US7973069B2 (en) | 2004-07-14 | 2011-07-05 | Ptc Therapeutics, Inc. | Methods for treating hepatitis C |
| JP2008543971A (en) * | 2005-06-27 | 2008-12-04 | エグゼリクシス, インコーポレイテッド | LXR modulator based on imidazole |
| US9000022B2 (en) | 2005-06-27 | 2015-04-07 | Exelixis Patent Company Llc | Imidazole based LXR modulators |
| US8569352B2 (en) | 2005-06-27 | 2013-10-29 | Exelixis Patent Company Llc | Imidazole based LXR modulators |
| EP1928237A2 (en) * | 2005-09-02 | 2008-06-11 | Abbott Laboratories | Novel imidazo based heterocycles |
| EP1928237A4 (en) * | 2005-09-02 | 2011-03-09 | Abbott Lab | Novel imidazo based heterocycles |
| WO2007059359A3 (en) * | 2005-11-21 | 2007-12-21 | Biogen Idec Inc | Substituted pyrazalones |
| WO2007059359A2 (en) * | 2005-11-21 | 2007-05-24 | Biogen Idec Ma Inc. | Substituted pyrazalones |
| WO2008094574A3 (en) * | 2007-01-30 | 2008-09-25 | Biogen Idec Inc | Furanone compounds and methods of making and using the same |
| WO2008094574A2 (en) * | 2007-01-30 | 2008-08-07 | Biogen Idec Ma Inc. | Furanone compounds and methods of making and using the same |
| WO2009087224A1 (en) | 2008-01-11 | 2009-07-16 | Novartis Ag | Pyrimidines as kinase inhibitors |
| WO2009087212A3 (en) * | 2008-01-11 | 2009-09-24 | Novartis Ag | Pyridine derivatives |
| WO2009087212A2 (en) * | 2008-01-11 | 2009-07-16 | Novartis Ag | Pyridine derivatives |
| US8343966B2 (en) | 2008-01-11 | 2013-01-01 | Novartis Ag | Organic compounds |
| US8431578B2 (en) | 2008-01-11 | 2013-04-30 | Novartis Ag | Organic compounds |
| KR101692126B1 (en) | 2009-12-18 | 2017-01-02 | 미쓰비시 타나베 파마 코퍼레이션 | Novel antiplatelet agent |
| KR20140072220A (en) * | 2009-12-18 | 2014-06-12 | 미쓰비시 타나베 파마 코퍼레이션 | Novel antiplatelet agent |
| US8507472B2 (en) | 2010-04-07 | 2013-08-13 | Bayer Cropscience Ag | Bicyclic pyridinylpyrazoles |
| US10189809B2 (en) | 2010-06-30 | 2019-01-29 | Ironwood Pharmaceuticals, Inc. | SGC stimulators |
| WO2012003405A1 (en) * | 2010-06-30 | 2012-01-05 | Ironwood Pharmaceuticals, Inc. | Sgc stimulators |
| US8748442B2 (en) | 2010-06-30 | 2014-06-10 | Ironwood Pharmaceuticals, Inc. | sGC stimulators |
| CN103313976A (en) * | 2010-06-30 | 2013-09-18 | 铁木医药有限公司 | SGC stimulators |
| CN107021951A (en) * | 2010-06-30 | 2017-08-08 | 铁木医药有限公司 | SGC stimulants |
| CN107021951B (en) * | 2010-06-30 | 2020-10-20 | 赛克里翁治疗有限公司 | sGC stimulators |
| EP3173407A1 (en) * | 2010-06-30 | 2017-05-31 | Ironwood Pharmaceuticals, Inc. | Sgc stimulators |
| EA026692B1 (en) * | 2010-06-30 | 2017-05-31 | Айронвуд Фармасьютикелз, Инк. | sGC STIMULATORS |
| JP2013530192A (en) * | 2010-07-05 | 2013-07-25 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツング | Bipyridyl derivatives useful for the treatment of kinase-induced diseases |
| US8871744B2 (en) | 2010-07-21 | 2014-10-28 | B & G Partyers, LLC | Compounds and methods for selectively targeting tumor-associated mucins |
| EP2409708A1 (en) * | 2010-07-21 | 2012-01-25 | B & G Partners, LLC | Compounds and methods for selectively targeting tumor-associated mucins |
| US9061030B2 (en) | 2010-11-09 | 2015-06-23 | Ironwood Pharmaceuticals, Inc. | sGC stimulators |
| CN106117194A (en) * | 2011-12-27 | 2016-11-16 | 铁木医药有限公司 | Can be used as 2 benzyls of SGC stimulant, 3 (pyrimidine 2 base) substituted pyrazoles |
| US9139564B2 (en) | 2011-12-27 | 2015-09-22 | Ironwood Pharmaceuticals, Inc. | 2-benzyl, 3-(pyrimidin-2-yl) substituted pyrazoles useful as sGC stimulators |
| CN104066731A (en) * | 2011-12-27 | 2014-09-24 | 铁木医药有限公司 | 2 - benzyl, 3 - (pyrimidin- 2 -yl) substituted pyrazoles useful as sgc stimulators |
| WO2014024077A1 (en) * | 2012-08-07 | 2014-02-13 | Aurigene Discovery Technologies Limited | 5-(1H-PYRAZOL-4-YL)-1H-PYRROLO[2,3-b]PYRIDINE DERIVATIVES AS KINASE INHIBITORS |
| US10041047B2 (en) | 2013-03-14 | 2018-08-07 | Massachusetts Institute Of Technology | Compositions and methods for epithelial stem cell expansion and culture |
| US10041046B2 (en) | 2013-03-14 | 2018-08-07 | Massachusetts Institute Of Technology | Compositions and methods for epithelial stem cell expansion and culture |
| US10954490B2 (en) | 2013-03-14 | 2021-03-23 | The Brigham And Women's Hospital, Inc. | Compositions and methods for epithelial stem cell expansion and culture |
| US11369607B2 (en) | 2014-09-03 | 2022-06-28 | The Brigham And Women's Hospital, Inc. | Compositions, systems, and methods for generating inner ear hair cells for treatment of hearing loss |
| US10568883B2 (en) | 2014-09-03 | 2020-02-25 | Massachusetts Institute Of Technology | Compositions, systems, and methods for generating inner ear hair cells for treatment of hearing loss |
| WO2016210292A1 (en) | 2015-06-25 | 2016-12-29 | Children's Medical Center Corporation | Methods and compositions relating to hematopoietic stem cell expansion, enrichment, and maintenance |
| US11021687B2 (en) | 2016-01-08 | 2021-06-01 | The Brigham And Women's Hospital, Inc. | Production of differentiated enteroendocrine cells and insulin producing cells |
| US11033546B2 (en) | 2016-03-02 | 2021-06-15 | Frequency Therapeutics, Inc. | Solubilized compositions for controlled proliferation of stem cells / generating inner ear hair cells using a GSK3 inhibitor: I |
| US11160868B2 (en) | 2016-03-02 | 2021-11-02 | Frequency Therapeutics, Inc. | Thermoreversible compositions for administration of therapeutic agents |
| US11260130B2 (en) | 2016-03-02 | 2022-03-01 | Frequency Therapeutics, Inc. | Solubilized compositions for controlled proliferation of stem cells / generating inner ear hair cells using a GSK3 inhibitor: IV |
| WO2017161001A1 (en) | 2016-03-15 | 2017-09-21 | Children's Medical Center Corporation | Methods and compositions relating to hematopoietic stem cell expansion |
| EP4049665A1 (en) | 2016-03-15 | 2022-08-31 | Children's Medical Center Corporation | Methods and compositions relating to hematopoietic stem cell expansion |
| WO2018019106A1 (en) | 2016-07-29 | 2018-02-01 | 上海璎黎药业有限公司 | Nitrogenous heterocyclic aromatic compound, preparation method therefor, pharmaceutical composition thereof, and application thereof |
| US11466003B2 (en) | 2016-07-29 | 2022-10-11 | Shanghai Yingli Pharmaceutical Co., Ltd | Nitrogenous heterocyclic aromatic compound, preparation method therefor, pharmaceutical composition thereof, and application thereof |
| CN108779100B (en) * | 2016-11-14 | 2021-11-16 | 江苏恒瑞医药股份有限公司 | 3, 4-dipyridyl pyrazole derivatives, preparation method and medical application thereof |
| US10899741B2 (en) | 2016-11-14 | 2021-01-26 | Jiangsu Hengrui Medicine Co., Ltd. | 3,4-bipyridyl pyrazole derivative, and preparation method therefor and medical application thereof |
| WO2018086609A1 (en) * | 2016-11-14 | 2018-05-17 | 江苏恒瑞医药股份有限公司 | 3,4-bipyridyl pyrazole derivative, and preparation method therefor and medical application thereof |
| CN108779100A (en) * | 2016-11-14 | 2018-11-09 | 江苏恒瑞医药股份有限公司 | 3,4- bipyridyls pyrazole derivatives, preparation method and its application in medicine |
| US11066419B2 (en) | 2016-12-30 | 2021-07-20 | Frequency Therapeutics, Inc. | 1H-pyrrole-2,5-dione compounds and methods of using same |
| US11447490B2 (en) | 2018-01-24 | 2022-09-20 | Shanghai Yingli Pharmaceutical Co., Ltd | Aromatic heterocyclic substituted olefin compound, preparation method for same, pharmaceutical composition of same, and applications thereof |
| US11584745B2 (en) | 2018-01-24 | 2023-02-21 | Shanghai Yingli Pharmaceutical Co., Ltd | Aromatic heterocyclic compound, intermediate thereof, preparation method therefor, and pharmaceutical composition and use thereof |
| WO2019183245A1 (en) | 2018-03-20 | 2019-09-26 | Icahn School Of Medicine At Mount Sinai | Kinase inhibitor compounds and compositions and methods of use |
| WO2019236766A1 (en) | 2018-06-06 | 2019-12-12 | Ideaya Biosciences, Inc. | Methods of culturing and/or expanding stem cells and/or lineage committed progenitor cells using lactam compounds |
| US11162071B2 (en) | 2018-08-17 | 2021-11-02 | Frequency Therapeutics, Inc. | Compositions and methods for generating hair cells by upregulating JAG-1 |
| US11617745B2 (en) | 2018-08-17 | 2023-04-04 | Frequency Therapeutics, Inc. | Compositions and methods for generating hair cells by downregulating FOXO |
| WO2020142485A1 (en) | 2018-12-31 | 2020-07-09 | Icahn School Of Medicine At Mount Sinai | Kinase inhibitor compounds and compositions and methods of use |
| WO2021167703A1 (en) * | 2020-02-19 | 2021-08-26 | Nammi Therapeutics, Inc. | Formulated and/or co-formulated liposome compositions containing tgfb antagonist prodrugs useful in the treatment of cancer and methods thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1539748A1 (en) | 2005-06-15 |
| TW200417547A (en) | 2004-09-16 |
| AU2003260345A1 (en) | 2004-02-23 |
| AR040726A1 (en) | 2005-04-20 |
| US20050245520A1 (en) | 2005-11-03 |
| JP2005539000A (en) | 2005-12-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2004013135A1 (en) | 2-phenylpyridin-4-yl derivatives as alk5 inhibitors | |
| KR100451523B1 (en) | N-heteroaryl-pyridine sulfonamide derivatives and their use as antagonists | |
| KR101589332B1 (en) | 2h-chromene compound and derivative thereof | |
| BR112020026748A2 (en) | CYCLINE DEPENDENT KINASE INHIBITORS | |
| EP1506191A1 (en) | Benzoxazine and benzoxazinone substituted triazoles | |
| US20060058329A1 (en) | Pyrazole inhibitors of the transforming growth factor | |
| EP1543003B1 (en) | Imidazo¬1,2-a|pyridines | |
| WO2004065392A1 (en) | Condensed pyridines and pyrimidines and their use as alk-5 receptor ligands | |
| MXPA02008082A (en) | Pyridinylimidazoles. | |
| CA3009669A1 (en) | Bruton's tyrosine kinase inhibitors | |
| EP1444232A1 (en) | Phenyl substituted triazoles and their use as selective inhibors of akl5 kinase | |
| JP2019520366A (en) | Novel pyrazole derivative as ALK5 inhibitor and use thereof | |
| WO2004111036A1 (en) | 4- (heterocyclyl- fused phenyl)- 3- (phenyl or pyrid -2- yl) pyrazoles as inhibitors of the alk-5- receptor | |
| KR20190022728A (en) | Mitochondrial inhibitors for the treatment of proliferative disorders | |
| US20060074244A1 (en) | Pyridinyl substituted (1,2,3,)triazoles as inhibitors of the tgf-beta signalling pathway | |
| US20060247233A1 (en) | Thiazoles inhibitors of the alk-5 receptor | |
| US20060004051A1 (en) | Compounds | |
| JP6670832B2 (en) | Novel dihydroquinoline pyrazolyl compounds as aldosterone synthase inhibitors | |
| JP2018188364A (en) | Heteroaromatic ring derivative | |
| TW202304884A (en) | Substituted pyrrole carboxamides, process for their preparation and their use as kinase inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 10522969 Country of ref document: US Ref document number: 2004525405 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003766385 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003766385 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2003766385 Country of ref document: EP |