NOVEL CARBOXAMIDE COMPOUNDS FOR USE IN MCH RECEPTOR RELATED DISORDERS
Field of the invention The present invention relates to novel compounds that interact with a melanin- concentrating hormone receptor, a MCH receptor. The compounds have modulating activity on the MCH receptor such as e.g. antagonistic, agonistic or allosteric activity and are useful for medicinal or cosmetic purposes such as, e.g. in the treatment or prevention of feeding disorders like obesity, metabolic syndrome, Type II diabetes, bulimia etc. or in the treatment or prevention of depression.
The invention also relates to therapeutic and/or prophylactic use of the compounds, to processes for the preparation of the novel compounds, to pharmaceutical compositions comprising the compounds, to the manufacture of such compositions and to methods for the treatment and/or prevention of MCH receptor related disorders.
Background of the invention
Melanin-concentrating hormone (MCH) is a cyclic peptide that originally was isolated from salmoid pituitaries. In the fish, the 17 amino acid peptide causes aggregation of melanin and inhibits the release of ACTH. Mammalian MCH (19 amino acids) is highly conserved between rat, mouse and human exhibiting 100% amino acid identity. In the last decades there has been increasing activity in the research in the physiologic roles of MCH. It has been reported that MCH is involved in the feeding or body weight regulation, in energy balance, in response to stress, in water balance, in energy metabolism, in the general arousal/attention state, memory and cognitive functions and in psychiatric disorders. The biological effects of MCH are believed to be mediated by specific MCH receptors, and the MCH1 and MCH2 receptors have been described. Antagonists of MCH receptor (e.g. MCH1 receptor) may be suitable for use as obesity or weight reducing agents and they are also believed to have antidepressant and/or anxiolytic properties.
The present invention provides novel compounds that have a MCH modulating activity, i.e. antagonistic, inverse agonistic/negative antagonism, allosteric modulator, partial agonist or agonistic action.
Detailed description of the invention
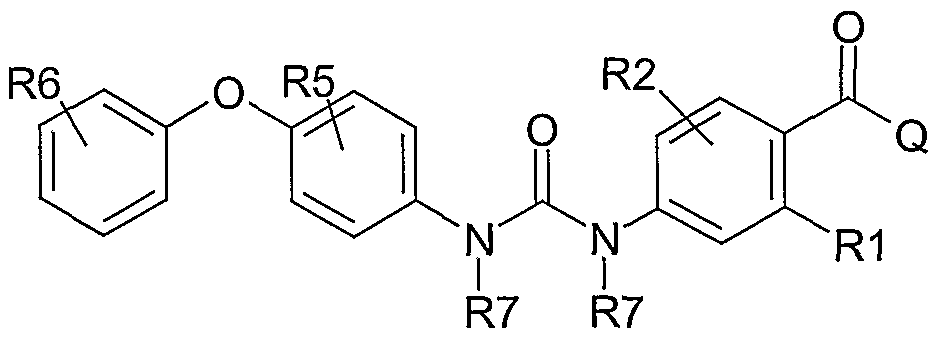
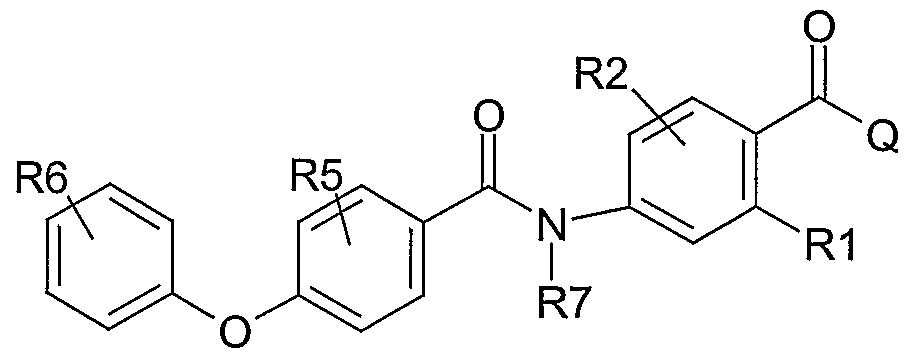
The present invention relates to a compound with the following structure (Formula I)
wherein -A- is a linker, which is selected from the group consisting of
R7
.0 I
.S
R7 R7 R7
and, wherein the linker -A- may be attached via either of the two free bonds to the A group;
B is a connecting moiety selected from the group consisting of:
o . . o
,<
R7 R7 1
which may be attached via either of the two free bonds to the Ar-, group;
and R7 is the same or different and is hydrogen or a straight or branched C C4 alkyl or alkenyl group;
Ar-i and Ar2 being the same or different aryl or heteroaryl group such as, e.g. phenyl, pyridine, pyrimidine, pyrazine, thiophene, oxazole, isothiazole, pyrazole, pyrrole, imidazole, indole, benzimidazole, quinoline, isoquinoline, furan, benzofuran, benzothiophene, benzothiazole, indazole, thiazole, isoxazole, oxadiazole, indan;
R1 and R2 are the same or different and selected from hydrogen, halogen atoms, CF3, OCF3, SCF3, SCH3, nitrile, alkyl, alkenyl, alkynyl groups; R1 and R2 could be connected to each other to form annelated rings;
R5 and R6 are the same or different selected from hydrogen, halogen atoms, alkoxy groups (AlkO-), hydroxy, alkylamino groups (AlkNH-), dialkylamino groups (Alk2N-), hydroxylalkyl groups, carboxamido groups (-CONH2, -CONHAIk, -CONAIk2), acylamido groups (-NHCO-Alk), acyl groups (-CO-Alk), -CHO, nitrile, alkyl, alkenyl or alkynyl groups, -SCH3, partially or fully fluorinated alkyl, alkoxy or thioalkoxy groups such as -CH2CF3, - CF2CF3, -CF3, -OCF3, -SCF3; -SO2NH2, -SO2NHAIk, -SO2NAIk2, -SO2Alk;
more than one R5 group, same or different, may be present on An and more than one R6 group, same or different, may be present on Ar2; when more than one R5 group is present they could be connected to each other, directly or with a suitable connecting moiety, to form rings; when more than one R6 group is present they could be connected to each other, directly or with a suitable connecting moiety, to form rings; R5 and R6 could be connected to each other, directly or with a suitable connecting moiety, to form rings
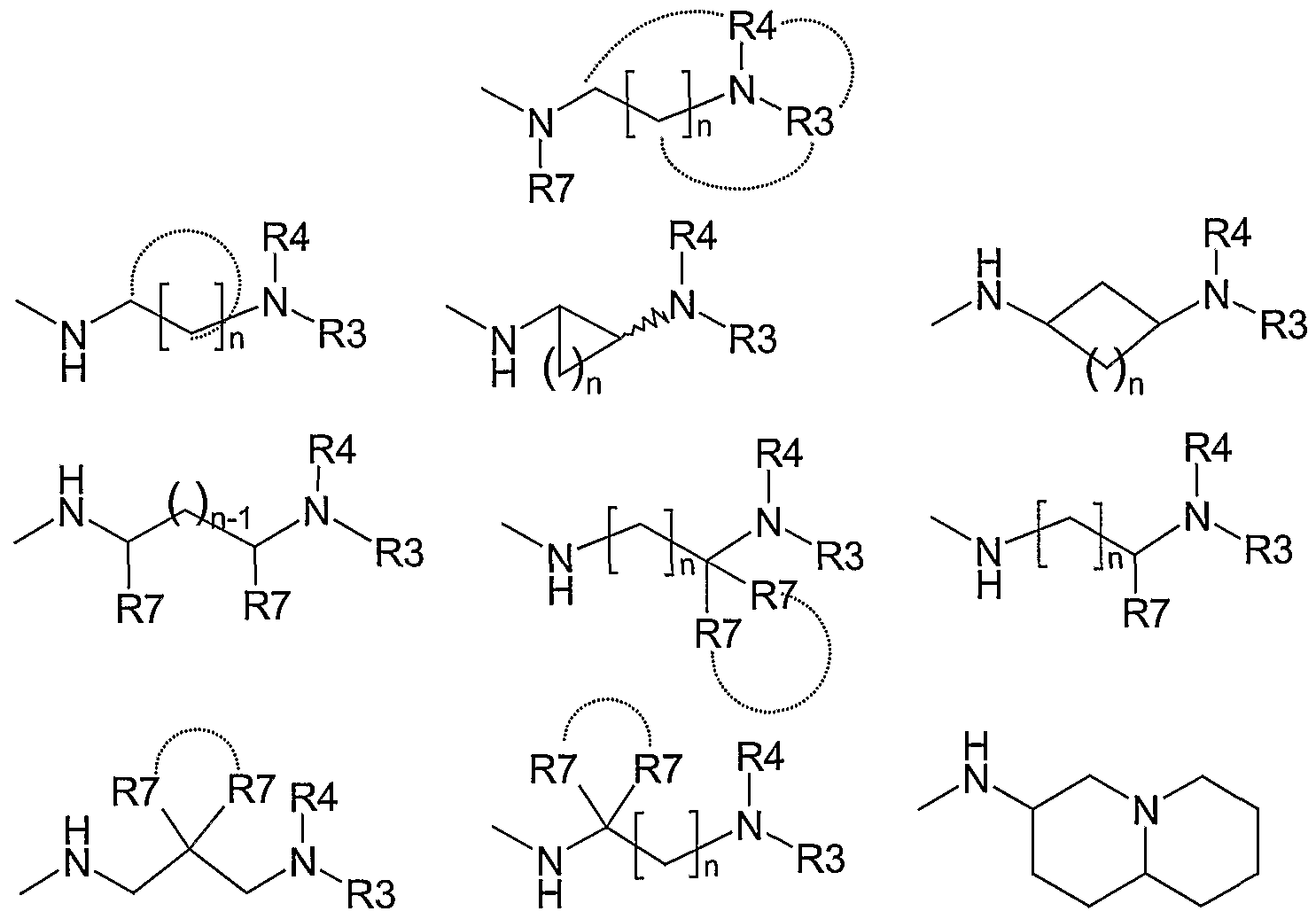
Q is selected from the group consisting of
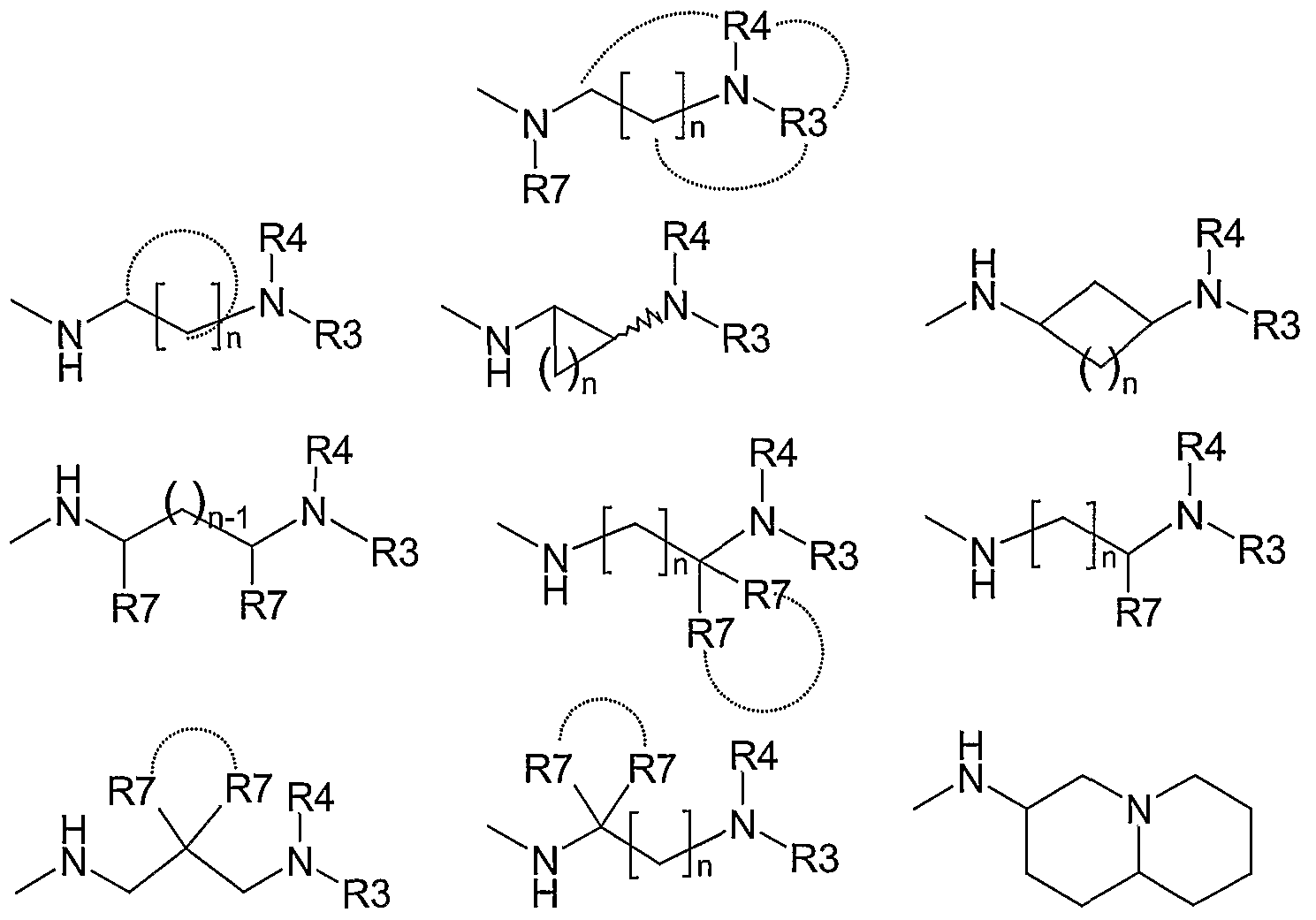
R3 and R4 are the same or different selected from straight or branched alkyl, alkenyl or alkynyl groups with 1-8 carbon atoms; cycloalkyl groups with 3-7 carbons; alkylcycloalkyl with 4-9 carbons atoms; alkylaryl groups such as benzyl, 2-ethylphenyl, 3-propylphenyl, 4- butylphenyl; alkylheterocyclyl groups such as 2-ethylpiperazine, 3-propylpiperidine; alkylheteroaryl groups; the aryl, heterocyclyl and heteroaryl groups may be substituted with substituents such as Alk-CONH-, Alk-O-, HO-, NC-, AlkNH-, Alk2N-, -CONH2, - CONHAIk, -CONAIk2, aryl, substituted aryl, benzyl, substituted benzyl groups, or fused moieties such as -O-CH2-O-, -N=CH-NH-, -O-CH=N-, -N=CH-CH=CH- ;
Alk is the same or a different alkyl, alkenyl or alkynyl group;
R3 and R4 may optionally be linked to each other, when possible, as indicated in Formula I; and oxygen or nitrogen atoms may be inserted in the chain or ring in a chemically stable position;
n is 1 , 2 or 3.
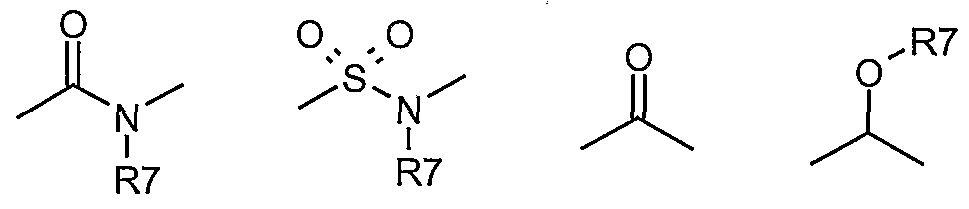
In the present context, the term "alkyl" is intended to indicate a branched or straight-chain, saturated chemical group containing 1-8 carbon atoms such as, e.g., 1-6 carbon atoms, such as methyl, ethyl, propyl, isopropyl, butyl, sec. butyl, tert. butyl, pentyl, isopentyl, hexyl, isohexyl, heptyl, octyl etc.
The term "lower alkyl" is intended to indicate an alkyl group containing 1-6 carbon atoms, unless otherwise specified. Likewise, "lower alkenyl" and "lower alkynyl" are intended to indicate alkenyl and alkynyl groups, respectively containing 2-6 carbon atoms.
The term "alkenyl" is intended to indicate an unsaturated alkyl group having one or more double bonds and 2-8 carbon atoms unless otherwise specified.
The term "alkynyl" is intended to indicate an unsaturated alkyl group having one or more triple bonds and 2-8 carbon atoms unless otherwise specified.
The term "cycloalkyl" is intended to denote a cyclic, saturated alkyl group of 3-7 carbon atoms.
The term "cycloalkenyl" is intended to denote a cyclic, unsaturated alkyl group of 5-7 carbon atoms having one or more double bonds.
The term "alkoxy" is intended to indicate the group alkyl-O-.
The term "aryl" is intended to denote an aromatic (unsaturated), typically 6-membered, ring, which may be a single ring (e.g. phenyl) or fused with other 5- or 6-membered rings (e.g. naphthyl or indole).
The term "heteroaryl" is intended to denote an aromatic (unsaturated), 5- or 6-membered, ring, which may be a single ring (e.g. pyridyl) or fused with other 5- or 6-membered rings (e.g. quinoline or indole).
The term "heterocyclyl" is intended to indicate a cyclic unsaturated (heteroalkenyl), aromatic ("heteroaryl") or saturated ("heterocycloalkyl") group comprising at least one heteroatom.
The present invention also relates to a compound with the following structure
wherein An, Ar2, A, B, R1 , R2, Q, R5, and R6 are as defined herein.
The present invention further relates to a compound with the following structure
wherein An, Ar2, A, B, R1 , R2, Q, R5, and R6 are as defined herein.
Additionally, the present invention relates to a compound with the following structure
wherein An, Ar2, A, B, R1 , R2, Q, R5, and R6 are as defined in herein.
In an embodiment of the present invention Q may be:
wherein R3, R4, R7 and n are as defined in herein.
Additionally, Q may be
wherein R3, R4 and n are as defined in claim 1.
In a specific embodiment, the invention relates to compounds wherein A is selected from the group consisting of:
wherein R7 is as defined herein.
More specifically, A may be selected from the group consisting of:
wherein R7 is as defined herein.
The compounds according to the present invention contain a B moiety, which may be selected from the group consisting of: o . .0
-°\ .s;
R7 R7
wherein R7 is as defined herein.
Furthermore, the present invention relates to compounds wherein B is selected from the group consisting of:
R7
wherein R7 is as defined herein.
Interesting compounds according to the present invention have the structure
wherein R1 , R2, R5, R6 and R7 are as defined herein.
Other interesting compounds according to the present invention have one of the structures:
wherein R1 , R2, R5, R6 and R7 are as defined herein.
In an embodiment of the present invention, Q may be:
wherein R3, R4, R7 and n are as defined herein.
wherein R3, R4, R7 and n are as defined herein.
The substitution of the carboxamide is important, and compounds according to the present invention may have a -B- moiety not placed ortho- to the -A- linker. It is also of interest for at least one of R1 and R2 to be a substituent i.e. R1 and R2 are not both hydrogen. Additionally, R1 or R2 may be hydrogen.
The present invention relates to compounds wherein An and Ar2 are the same or different aryl or heteroaryl groups such as, e.g., phenyl, pyridine, thiophene.
In an embodiment of the present invention, R1 is hydrogen and R2 is F, Cl, Br, I, CF3, OCF3, OCH3, SCF3, SCH3 or lower alkyl or alkenyl group.
In a further embodiment, R5 and R6 may be the same or different selected from hydrogen, halogen atoms, alkoxy groups (AlkO-), alkyamino groups (AlkNH-), dialkylamino groups (Alk2N-), carboxamido groups (-CONH2, -CONHAIk, CONAIk2), acylamido groups (-NHCO-Alk), nitrile, lower alkyl groups, -CF3, -OCF3, -SCF3, -SCH3.
Other compounds according to the invention have the following formulas:
Other specific embodiments appear from the appended claims and the examples herein.
Synthetic routes
Compounds of Formula I are preferably made by connecting an appropriately functionalised (A") benzamide moiety III with a suitably functionalised (A') diaryl moiety using well-known synthetic routes according to the following general scheme (Route 1):
(I)
For example, urea bonds -A- can be formed by reaction of II having A' as isocyanate with III having A" equal to NH-R7 using appropriate catalysis by base or acid. The reverse use of III having A" as isocyanate with II having A' equal to NH-R7 can also be applied. Analogously, carbamates can for example be made by reaction of II having A' as isocyanate with III having A" equal to OH or the reverse use of OH and isocyanate in A' and A".
Preparation of amide and sulphonamide bonds
in the connecting A-linkage can be made via reaction of A" in compound III being NH-R7 with activated forms, e.g. acid chlorides or active esters, of A' in compound II being COOH or SO2OH. Alternatively, the conversion can be made directly with the acids having A' as COOH using suitable coupling reagents such as dicyclohexylcarbodiimide (DCC), and promoters such as 1-hydroxybenzotriazole. The reverse use of A' and A" in II and III can be applied as well to form the linker in the opposite direction.
Formation of the connecting A-linkage to form
bonds in either direction between Ar1 and the benzamide can be made by N-, O- or S- alkylations of compound II with A' being OH, NH-R7, or SH with compound III with A" being a CH2-L wherein L being a suitable leaving group such as halogen (Cl, Br, I), tosyl or mesyl using appropriate catalysts and conditions. The alkene linkage can be made by a Wittig reaction with compound II with A' being CHO and compound III with A" being CH2- PPh3. The reverse use of A' and A" in II and III can be applied as well to form the linker in the opposite direction.
The 5-membered heterocyclic linkers
can be made according to standard cyclisation procedures using appropriate solvents, catalysts and temperatures. For example, formation of 1 ,2,4-triazole can be made from II with A' being acylhydrazide with III with A" being amide or thioamide or the reverse orientation of A' and A". 1 ,2,4-Oxadiazole can be formed from II with A' being amidoxime with III with A" being carboxylic ester or the reverse orientation of A' and A". 1 ,3,4- Oxadiazole can be formed from II with A' being acylhydrazide with III with A" being carboxylic ester or the reverse orientation of A' and A".
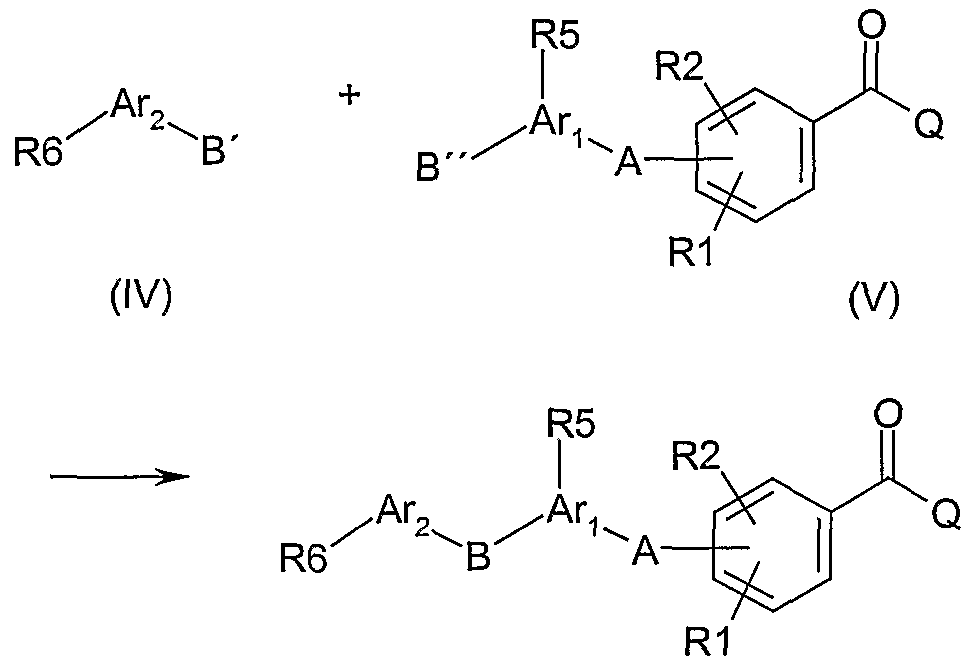
Alternatively, compounds of Formula I are made by connecting an appropriately functionalised (B
") arylated benzamide moiety V with a suitably functionalised (B') aryl moiety IV using well-known synthetic routes according to the following general scheme (Route 2):
(I)
Thus, formation of the connecting B-linkage to form
bonds in either direction between Ar1 and Ar2 can be made by N-, O- or S-alkylations of compounds IV having B' as OH, NH-R7, or SH with compounds V having B" as CH2-L, wherein L is a suitable leaving group such as halogen (Cl, Br, I), tosyl or mesyl using appropriate catalysts and conditions. The reverse use of B' and B" in IV and V can be applied as well to form the linker in the opposite direction.
Formation of the connecting B-linkage to form
R7 O .0
bonds can be made via coupling reactions of compounds IV with B' being OH, NH-R7, or SH with compound V having B" as a suitable metal-reagent capable of forming the bond using appropriate catalysts and conditions or with B" being a halogen that can be replaced under thermal or metal-catalysed conditions. The reverse use of B' and B" in IV and V can be applied as well. The -SO2- linkage may be obtained by oxidation of the corresponding -S- derivative.
Formation of the connecting B-linkage to form
can be made by Friedel-Craft chemistry utilising compounds IV having B' as e.g. CO-CI and compounds V having B" as hydrogen to form the -CO- linkage followed by reduction to -CH(OH)-, that can be alkylated to give -CH(OAIk), or complete reduction to -CH2-. The amide bond is made according to standard reactions involving compounds IV having B' as NH-R7 and activated derivatives of compound V with B" being COOH or coupling reagents and promotors. The reverse use of B' and B" in IV and V can be applied as well. The sulphonamides are made analogously from the corresponding SO2-CI derivatives and NH-R7 derivatives.
Notably, the -B- linkage is preferably introduced during the synthesis of intermediates II that are used in the coupling with III according to Route 1. In most cases the -B- linkage is made in compounds having A' groups that are compatible with the reaction conditions and that can be transformed into the required reactive moieties for subsequently forming the - A- linkage.
R5
I
.Ar, .Ar,. (II)
R6 SB' 'A'
Aromatic substituents R4, R5 and R6 are preferably introduced prior to formation of the A- or B-linkage either direct or via a masked functionality that is compatible with the subsequent synthetic steps.
Compounds of Formula I in which Q is as shown below are also obtained by connecting carboxylic acid derivatives VI with amines VII using well-known synthetic routes according to the following general scheme (Route 3):
Thus, the benzamide bond is formed by reacting a suitably activated carboxylic acid VI (e.g. acid chloride) with the corresponding amines VII in the presence of a base or using suitable coupling reagents such as DCC in presence of promoting agents and a suitable base.
Alternatively, compounds of Formula I can be made by N-alkylation of compounds of Formula I having R3 and R4 being hydrogen using well-known synthetic routes such as reductive alkylation or alkylation with alkyl halides in case the functionalisation of the molecule is compatible with this type of reactions (Route 4).
Synthetic method 1A
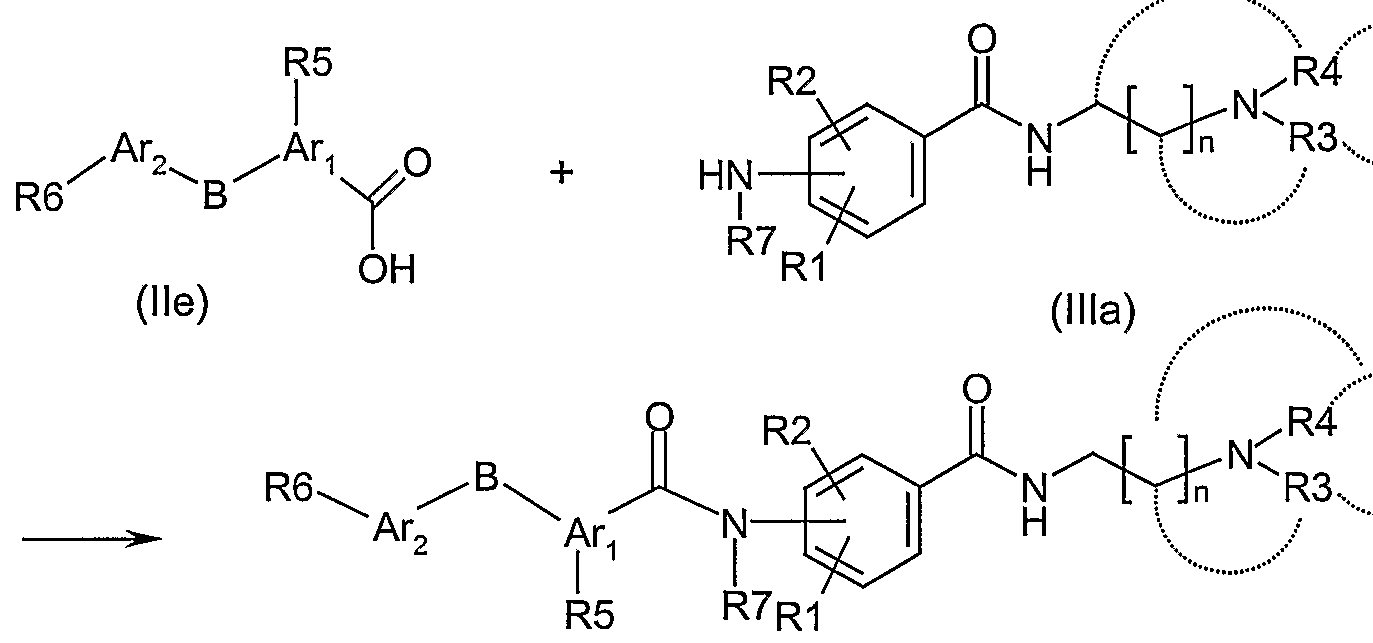
Thus, compound (lb) having NHCON-R7 as linker A with R7 defined as hydrogen or lower alkyl or alkenyl group, can be produced, for instance, by the following urea reaction.
(lb)
Compound lla and compound Ilia are reacted in an inert solvent in accordance with standard procedures. Typically, inert solvents can be ether solvents, halogenated hydrocarbon solvents, nitrile solvents and aromatic solvents. Reaction temperature is usually room temperature and the reaction time is 2 hours to 1 day.
Compound lla can be produced from the corresponding carboxylic acid. For instance, 4- phenoxyphenylisocyanate can be produced in accordance with methods such as described in "Comprehensive Organic Transformation", 2nd Edition (Wiley); R.C. Larock.
Synthetic method 1B
Compound Ic having N-AlkCON-R7 as linker A with R7 defined as hydrogen or lower alkyl or alkenyl group, can be produced, for instance, by the following urea reaction.
(|c)
Compound Ilia and 1 equivalent of compound lib are reacted in an inert solvent in the presence of an excess of a base in accordance with known procedures (e.g. WO 9205174; J.Med.Chem. 43(20), 3653-3664, 2000). Suitable inert solvents can be ether solvents, halogenated hydrocarbon solvents, nitrile solvents and aromatic solvents. As a base can be used for instance triethylamine, diisopropylethylamine and sodium carbonate. Typically, the reaction temperature is 0 °C to room temperature and the reaction time is 1 hour to 1 day.
Compound lib can be produced from the corresponding N-alkyl aromatic amine by well- known methods. For instance, N-methyl-N-4-phenoxyphenylcarbamoyl chloride can be
produced in accordance with methods such as described in J. Labelled Compd. Radiopharma 29(2), 149-155, 1991.
Synthetic method 1C Compound If having 5-membered ring urea as linker A can be produced, for instance, by the following reaction sequence.
(le)
Compound le and 1 equivalent of carbonyldiimidazole are reacted in an inert solvent at elevated temperature until the reaction is completed. Typically, the reaction is conducted at reflux in acetonitrile for less than 24 hours.
Compounds lie, Id and le can be produced following the functional group conversions described in procedures like the one in J.Med.Chem. 43(20), 3653-3664, 2000.
Synthetic method 1D
Compound li having CON-R7 as linker A with R7 defined as hydrogen or lower alkyl or alkenyl group, can be produced by the following amidation reaction.
(li)
The amide bonds are formed by reacting a suitably activated carboxylic acid lie (acid chloride, mixed anhydrides, esters with phenol bearing electron withdrawing substituents, 1-hydroxybenzotriazole, N-hydroxysuccinimide, 2-hydroxypyridine) with anilines Ilia in an inert solvent in the presence of a base. As inert solvents can be used ether solvents, amide solvents and halogenated hydrocarbon solvents. Suitable bases that can be used are triethylamine, diiisopropylethylamine, pyridine, 4-dimethylaminopyridine (DMAP) and sodium carbonate. The reaction temperature is usually between 0°C to 30°C and reaction time is 1 hour to 1 day.
The coupling can also be performed directly from lie using suitable coupling reagents such as dicyclohexylcarbodiimide (DCC), 1-(3-dimethylaminopropyl)-3-ethyl-cabodiimide (EDCI), N-ethoxycarbonyl-2-ethoxy-1 ,2-dihydroquinoline (EEDQ) preferably in presence of promoting agents capable of forming an active ester such as 1-hydroxybenzotriazole, N-hydroxysuccinimide, 2-hydroxypyridine in an inert solvent in the presence of a base. As inert solvents can be used ether solvents, amide solvents and halogenated hydrocarbon solvents. Suitable bases that can be used are triethylamine, diiisopropylethylamine, pyridine, N-ethyldiisopropylamine, and 4-methylmorpholine. The reaction temperature is usually between 0°C to 30°C and reaction time is 1 hour to 1 day.
Analogously, a sulphonamide group, as the connecting A-linkage to form
bonds can be made via the corresponding reaction of Ar-NH-R7 (Ilia) with activated forms of sulphonic acids, such sulphonyl chlorides, in the presence of base.
Synthetic method 2
Compound Ih having 1 ,2,4-oxadiazole (X=O) or 1 ,2,4-triazole (X=NH) heterocyclic rings as linker A can be produced, for instance, by the following cyclodehydratation reaction.
(lid) (lllc)
(ig)
(Ih)
Compound Ig is reacted in an inert solvent with or without the presence of a suitable base or acid (e.g. N-tetrabutyl ammonium fluoride, sodium hydride, sodium ethoxide or polyphosphoric acid) in accordance with standard methods such as described in Tetrahedron Lett. 42, 1441-1443, 2001 ; Tetrahedron Lett. 42, 1495-1498, 2001. Suitable, inert solvents can be ether solvents, amide solvents and aromatic solvents. The reaction temperature is usually room temperature to 100°C and the reaction time is 1 hour to 3 days.
Compound Ig can be produced by reacting an activated derivative of compound lid with 1 equivalent of compound lllc in an inert solvent in the presence of a base. As inert solvents
can be used ether solvents, amide solvents and halogenated hydrocarbon solvents. Suitable bases that can be used are triethylamine, diiisopropylethylamine, pyridine and sodium carbonate.
Appropriate examples of the activated derivatives of compound lid include active esters (e.g. esters with phenol bearing electron withdrawing substituents, 1-hydroxybenzotriazole, N-hydroxysuccinamide), acid chlorides, symmetrical or unsymmetrical anhydrides and orthoesters. The reaction temperature is usually between 0°C to 30°C and reaction time is 1 hour to 1 day.
Compound lllc can be produced from the corresponding amino compound Nib by well known methods such as described in "Comprehensive Organic Transformation", 2nd Edition (Wiley), R.C. Larock; In "Handbook of Heterocyclic Chemistry", 2nd Edition (Pergamon), A.R. Katritzky).
Synthetic method 3
Benzamide bonds are formed by reacting a suitably activated carboxylic acid VI (acid chloride, mixed anhydrides, esters with phenol bearing electron withdrawing substituents, 1-hydroxybenzotriazole, N-hydroxysuccinimide, 2-hydroxypyridine) with the corresponding amines VII in an inert solvent in the presence of a base. As inert solvents can be used ether solvents, amide solvents and halogenated hydrocarbon solvents. Suitable bases that can be used are triethylamine, diiisopropylethylamine, pyridine, 4-dimethylamino- pyridine (DMAP) and sodium carbonate. The reaction temperature is usually between 0°C to 30°C and reaction time is 1 hour to 1 day.
The coupling can also be performed by using suitable coupling reagents such as dicyclohexylcarbodiimide (DCC), 1-(3-dimethylaminopropyl)-3-ethyl-cabodiimide (EDCI), N-ethoxycarbonyl-2-ethoxy-1 ,2-dihydroquinoline (EEDQ) preferably in presence of promoting agents capable of forming an active ester such as 1-hydroxybenzotriazole, N- hydroxysuccinimide, 2-hydroxypyridine in an inert solvent in the presence of a base. As inert solvents can be used ether solvents, amide solvents and halogenated hydrocarbon solvents. Suitable bases that can be used are triethylamine, diiisopropylethylamine, pyridine, N-ethyldiisopropylamine, and 4-methylmorpholine. The reaction temperature is usually between 0°C to 30°C and reaction time is 1 hour to 1 day.
Synthetic method 4 Intermediates II
wherein A' being groups that are compatible with the reaction conditions and that can be transformed into the required reactive moieties for subsequently forming the -A- linkage (e.g. -CO2H, -NCO, -NAIkCOCI and -NHCOCO2Alk) can be produced by firstly connecting Ar1 to Ar2 to each other in accordance with standard methods including N-, O- and S-alkylations and metal-catalysed cross couplings. One or several aromatic substituents R5 and R6, depending on their chemical properties, can be introduced either before or after the connection of Ar1 and Ar2 to each other.
Compounds II with B = -O-, -NH-R7-, or -S- are prepared from a suitable aryl halide and the corresponding phenol, aniline or thiol by heating with for example NaH or K2CO3 as base with the presence of a copper salt in DMF, pyridine or other high boiling solvents. An example of a metal assisted preparation of diaryl ethers is the coupling of a phenol with an arylbromide in the presence of Pd(OAc)2 together with a phosphine ligand and K3PO4. For instance, 4-(4-chloro-phenoxy)benzoic acid can be produced in a two-steps synthesis from the corresponding 4-fluoro-acetophenone and 4-chlorophenol in accordance with methods such as described in Synthesis, 63-68, 1991 and Eur. J. Med. Chem., 3, 205- 214, 1984.
For compounds II with B equal to -CH2O-, the preparation is performed by heating a benzyl halide and phenol with K2CO3 or NaOMe as base. These ethers can also be prepared from suitable benzyl alcohols and phenols utilising Mitsunobu conditions (DEAD
and PPh3). Compounds II with B equal to -CH2N-R7- can be prepared from an aniline and a benzyl halide using K2CO3 as base. The corresponding thioether can be formed from a benzyl halide and thiophenol using KOH or NaOMe as bases and with for example ethanol as the solvent.
When B is equal to -CO- the compounds II can be synthesised from an arylic acid chloride either through a Friedel Craft reaction with an appropriate benzene derivative or via addition of a suitable Grignard reagent. Reduction of the same compound with NaBH4 gives the compound II with B = -CH(OH)- that can be alkylated to produce -CH(OAIk)-. Utilising hydrogenation with PtO2 as catalyst or Zn(Hg) as reducing agent yields compounds II with B = -CH2-.
Compounds II with B = -SO2- can be prepared from the corresponding sulfide by oxidation with H2O2 or KMnO4.
When B is an amide linkage compounds II can be prepared according to standard protocol from an activated carboxylic acid derivative (acid chloride, mixed anhydrides, esters with phenol bearing electron withdrawing substituents, 1-hydroxybenzotriazole, N- hydroxysuccinimide, 2-hydroxypyridine) and an amine in an inert solvent and in the presence of a base. Suitable bases that can be used are triethylamine, diiisopropylethylamine, pyridine, 4-dimethylaminopyridine (DMAP) and sodium carbonate. The coupling can also be performed by using suitable coupling reagents such as dicyclohexylcarbodiimide (DCC), 1 -(3-dimethylaminopropyl)-3-ethyl-cabodimidmide (EDCI), N-ethoxycarbonyl-2-ethoxy-1 ,2-dihydroquinoline (EEDQ) preferably in presence of promoting agents capable of forming an active ester such as 1-hydroxybenzotriazole, N-hydroxysuccinimide, and 2-hydroxypyridine.
Synthetic method 5 Intermediate 11 lb
can be prepared by reacting an activated carboxylic acid derivative VIII according to methods described above, preferably having the aniline nitrogen suitably protected (e.g. Boc, CF3CO), with the corresponding amine VII. The nitrogen may also be masked as a nitro group that subsequently is reduced to form lllb. The N-alkylated derivative Ilia may be obtained via reductive alkylation of lllb.
The carboxylic acids VIII are produced by well-known organic reactions including electrophilic substitutions or organometallic reactions such as ortho-lithiation and halogen- metal exchange followed by capture with electrophilic reagents. Alternatively, the aniline nitrogen may be introduced by a benzyne reaction.
Compounds
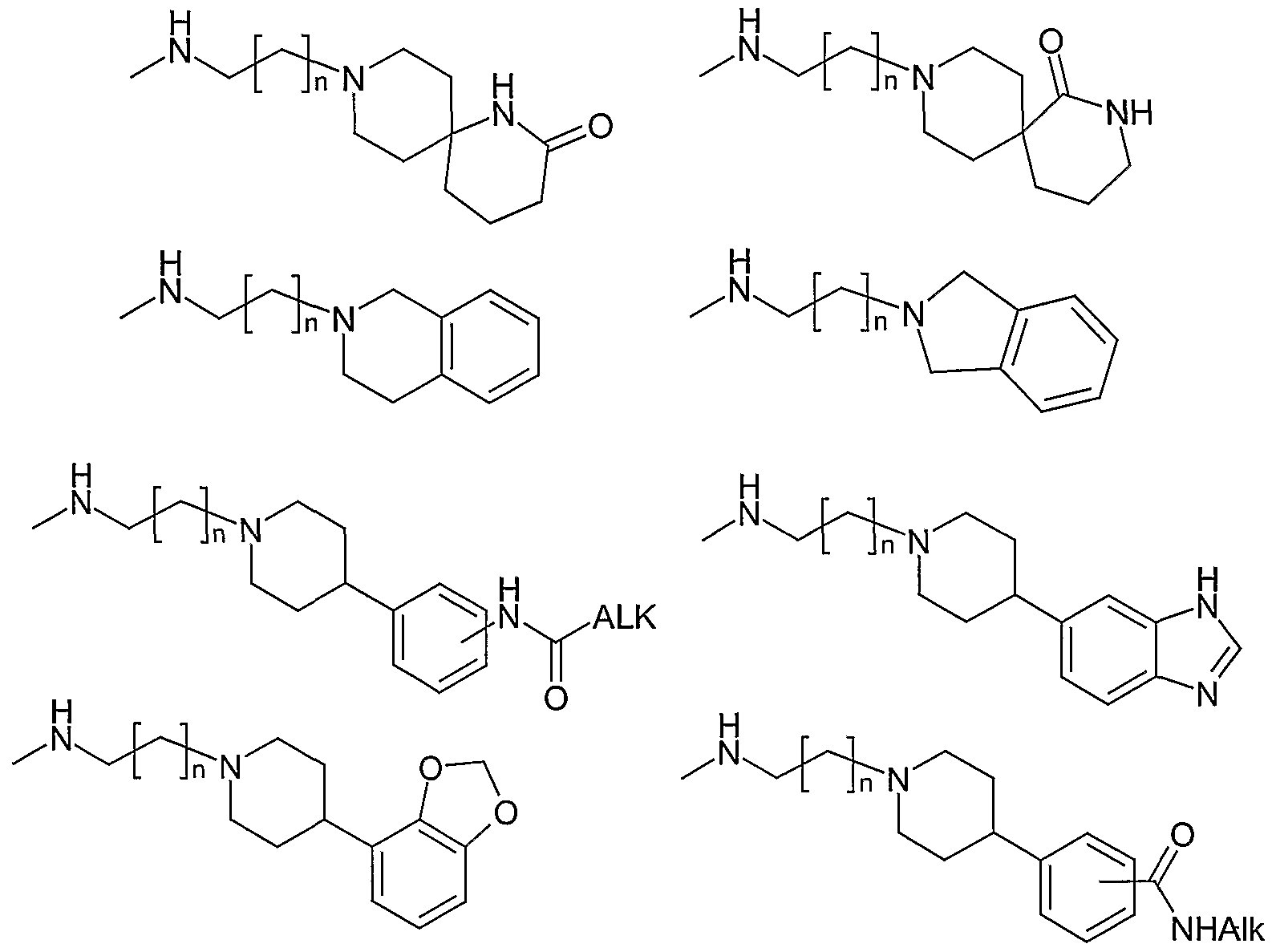
Below follows some examples of specific compounds according to the invention. In the compounds mentioned, one part of the molecule such as e.g. the amine group, the linker -A-, the linker -B-, the An or Ar2 group, the R1 , R2, R5, R6 group or the chain length is varied, while the other parts are conserved. Though not shown nor specifically mentioned, the invention also includes all compounds wherein all variations in one part of the molecule, e.g. linker -A- is combined with all variations in another of the features, e.g. variation in the An group.
4-[3-(4-Phenoxy-phenyl)-ureido]-Λ/-(3-pyrrolidin-1-yl-propyl)-benzamide, Λ/-(4-Dimethylamino-butyl)-[3-(4-phenoxy-phenyl)-ureido]-benzamide, Λ/-(3-Dimethylamino-2,2-dimethyl-propyl)-4-[3-(4-phenoxy-phenyl)-ureido]-benzamide, Λ/-(3-Dipropylamino-propyl)-4-[3-(4-phenoxy-phenyl)-ureido]-benzamide,
Variation of the linker A
Λ/-(2-Diethylamino-ethyl)-4-(4-phenoxy-benzoylamino)-benzamide, Λ/-(2-Diethylamino-ethyl)-4-[methyl-(4-phenoxy-benzoyl)-amino]-benzamide, Λ/-(2-Diethylamino-ethyl)-4-(4-phenoxy-phenylacetylamino)-benzamide, Λ/-(2-Diethylamino-ethyl)-4-(α-(4-phenoxy-phenyl)propanoylamino)-benzamide, Λ/-(2-Diethylamino-ethyl)-4-(α-(4-phenoxy-phenyl)butanoylamino)-benzamide, Λ/7-(2-Diethylamino-ethyl)- Λ/4-(4-phenoxy-phenyl)-terephthalamide, Λ/f-(2-Diethylamino-ethyl)-Λ/ -methyl- Λ/4-(4-phenoxy-phenyl)-terephthalamide, A/^-(2-Diethylamino-ethyl)-/V4-(4-phenoxy-benzyl)-terephthalamide, Λ/7-(2-Diethylamino-ethyl)-Λ/4-methyl- Λ/ -(4-phenoxy-benzyl)-terephthalamide, Λ/-(2-Diethylamino-ethyl)-4 4-phenoxy-benzenesulfonylamino)-benzamide, A/-(2-Diethylamino-ethyl)-4-| methyl-(4-phenoxy-benzenesulfonyl)-amino]-benzamide, Λ/-(2-Diethylamino-ethyl)-4- 4-phenoxy-phenylsulfamoyl)-benzamide, Λ/-(2-Diethylamino-ethyl)-4- 1 ,3-dimethyl-3-(4-phenoxy-phenyl)-ureido]-benzamide, Λ/-(2-Diethylamino-ethyl)-4-| '2-oxo-3-(4-phenoxy-phenyl)-imidazolidin-1-yl]-benzamide, Λ/-(2-Diethylamino-ethyl)-4- 3-methyl-3-(4-phenoxy-phenyl)-ureido]-benzamide, Λ/-(2-Diethylamino-ethyl)-4' '2-oxo-3-(4-phenoxy-phenyl)-tetrahydro-pyrimidin-1-yl]- benzamide,
Λ/-(2-Diethylamino-ethyl)-4 5-(4-phenoxy-phenyl)-[1 ,2,4]oxadiazol-3-yl]-benzamide, Λ/-(2-Diethylamino-ethyl)-4- 3-(4-phenoxy-phenyl)-[1 ,2,4]oxadiazol-5-yl]-benzamide, Λ/-(2-Diethylamino-ethyl)-4-| 5-(4-phenoxy-phenyl)-4r- -imidazol-2-yl]-benzamide, Λ/-(2-Diethylamino-ethyl)-4- 5-(4-phenoxy-phenyl)-1 H-[1 ,2,4]triazol-3-yl]-benzamide, Λ/-(2-Diethylamino-ethyl)-4- 5-(4-phenoxy-phenyl)-[1 ,3,4]oxadiazol-2-yl]-benzamide,
Λ/-(2-Diethylamino-ethyl)-4-[5-(4-phenoxy-phenyl)-2H-[1 ,2,4]triazol-3-yl]-benzamide, Λ/-(2-Diethylamino-ethyl)-4-[2-(4-phenoxy-phenyl)-5H-imidazol-4-yl]-benzamide, Λ/-(2-Diethylamino-ethyl)-4-[2-(4-phenoxy-phenyl)-vinyl]-benzamide, Λ/-(2-Diethylamino-ethyl)-4-(4-phenoxy-phenoxymethyl)-benzamide, Λ/-(2-Diethylamino-ethyl)-4-(4-phenoxy-benzyloxy)-benzamide, Λ/-(2-Diethylamino-ethyl)-4-(4-phenoxy-benzylamino)-benzamide, Λ/-(2-Diethylamino-ethyl)-4-[methyl-(4-phenoxy-benzyl)-amino]-benzamide, Λ/-(2-Diethylamino-ethyl)-4-[(4-phenoxy-phenylamino)-methyl]-benzamide, Λ/-(2-Diethylamino-ethyl)-4-{[methyl-(4-phenoxy-phenyl)-amino]-methyl}-benzamide, Λ/-(2-Diethylamino-ethyl)-4-(4-phenoxy-phenylsulfanylmethyl)-benzamide, Λ/-(2-Diethylamino-ethyl)-4-(4-phenoxy-benzylsulfanyl)-benzamide
Variation of the linker B
4-[3-(4-Benzyl-phenyl)-ureido]-Λ/-(2-diethylamino-ethyl)-benzamide,
Λ/-(2-Diethylamino-ethyl)-4-[3-(4-phenylsulfanyl-phenyl)-ureido]-benzamide,
4-[3-(4-Benzenesulfonyl-phenyl)-ureido]-Λ/-(2-diethylamino-ethyl)-benzamide, 4-[3-(4-Benzoyl-phenyl)-ureido]-/V-(2-diethylamino-ethyl)-benzamide,
A/-(2-Diethylamino-ethyl)-4-{3-[4-(hydroxy-phenyl-methyl)-phenyl]-ureido}-benzamide, Λ/-(2-Diethylamino-ethyl)-4-{3-[4-(methoxy-phenyl-methyl)-phenyl]-ureido}-benzamide, Λ/-(2-Diethylamino-ethyl)-4-[3-(4-phenoxymethyl-phenyl)-ureido]-benzamide, /V-(2-Diethylamino-ethyl)-4-[3-(4-phenylsulfanylmethyl-phenyl)-ureido]-benzamide, Λ/-(2-Diethylamino-ethyl)-4-[3-(4-phenylaminomethyl-phenyl)-ureido]-benzamide, Λ/-(2-Diethylamino-ethyl)-4-(3-{4-[(methyl-phenyl-amino)-methyl]-phenyl}-ureido)- benzamide,
4-[3-(4-Benzylamino-phenyl)-ureido]-Λ/-(2-diethylamino-ethyl)-benzamide, 4-{3-[4-(Benzyl-methyl-amino)-phenyl]-ureido}-Λ/-(2-diethylamino-ethyl)-benzamide, 4-[3-(4-Benzylsulfanyl-phenyl)-ureido]-V-(2-diethylamino-ethyl)-benzamide, 4-[3-(4-Benzyloxy-phenyl)-ureido]-Λ/-(2-diethylamino-ethyl)-benzamide, 4-[3-(4-Benzoylamino-phenyl)-ureido]-Λ/-(2-diethylamino-ethyl)-benzamide, 4-{3-[4-(Benzoyl-methyl-amino)-phenyl]-ureido}-Λ/-(2-diethylamino-ethyl)-benzamide, Λ/-(2-Diethylamino-ethyl)-4-[3-(4-phenylcarbamoyl-phenyl)-ureido]-benzamide,
Λ/-(2-Diethylamino-ethyl)-4-{3-[4-(methyl-phenyl-carbamoyl)-phenyl]-ureido}-benzamide,
Λ/-(2-Diethylamino-ethyl)-4-[3-(4-phenylamino-phenyl)-ureido]-benzamide,
Λ/-(2-Diethylamino-ethyl)-4-{3-[4-(methyl-phenyl-amino)-phenyl]-ureido}-benzamide.
Variation of the aromatic rings as well as their substituents
A/-(2-Diethylamino-ethyl)-4-{3-[5-(pyridin-3-yloxy)-pyridin-2-yl]-ureido}-benzamide, Λ/-(2-Diethylamino-ethyl)-4-{3-[4-(pyridin-3-yloxy)-phenyl]-ureido}-benzamide, Λ/-(2-Diethylamino-ethyl)-4-{3-[4-(pyrimidin-2-yloxy)-phenyl]-ureido}-benzamide, A/-(2-Diethylamino-ethyl)-4-[3-(2-phenoxy-pyrimidin-5-yl)-ureido]-benzamide, Λ/-(2-Diethylamino-ethyl)-4-[3-(5-phenoxy-pyrazin-2-yl)-ureido]-benzamide, Λ/-(2-Diethylamino-ethyl)-4-{3-[4-(thiophen-3-yloxy)-phenyl]-ureido}-benzamide, Λ/-(2-Diethylamino-ethyl)-4-{3-[4-(isothiazol-4-yloxy)-phenyl]-ureido}-benzamide, Λ/-(2-Diethylamino-ethyl)-4-{3-[4-(oxazol-4-yloxy)-phenyl]-ureido}-benzamide, Λ/-(2-Diethylamino-ethyl)-4-{3-[4-(1H-pyrazol-4-yloxy)-phenyl]-ureido}-benzamide, Λ/-(2-Diethylamino-ethyl)-4-[3-(5-phenoxy-thiophen-3-yl)-ureido]-benzamide, Λ/-(2-Diethylamino-ethyl)-4-[3-(2-phenoxy-oxazol-4-yl)-ureido]-benzamide, Λ/-(2-Diethylamino-ethyl)-4-[3-(4-phenoxy-oxazol-2-yl)-ureido]-benzamide, 4-{3-[4-(4-Chloro-phenoxy)-phenyl]-ureido}-A/-(2-diethylamino-ethyl)-benzamide, 4-{3-[4-(3,4-Dichloro-phenoxy)-phenyl]-ureido}-Λ/-(2-diethylamino-ethyl)-benzamide, 4-{3-[4-Fluoro-3-chloro-phenoxy)-phenyl]-ureido}-Λ/-(2-diethylamino-ethyl)-benzamide, 4-{3-[4-(4-bromo-3-trifluoromethoxy-phenoxy)-phenyl]-ureido}-Λ/-(2-diethylamino-ethyl)- benzamide,
4-{3-[4-(3,4-methylenedioxy-phenoxy)-phenyl]-ureido}-Λ/-(2-diethylamino-ethyl)- benzamide, 4-{3-[4-(4-acetamido-phenoxy)-phenyl]-ureido}-Λ/-(2-diethylamino-ethyl)-benzamide, 4-{3-[4-(3-hydroxymethyl-phenoxy)-phenyl]-ureido}-Λ/-(2-diethylamino-ethyl)-benzamide, 4-{3-[4-(4-trifluoromethyl-phenoxy)-phenyl]-ureido}-Λ/-(2-diethylamino-ethyl)-benzamide, 4-{3-(4-p-tolyloxy-phenyl)-ureido}-Λ/-(2-diethylamino-ethyl)-benzamide, Λ/-(2-Diethylamino-ethyl)-4-{3-[4-(3-fluoro-4-methoxy-phenoxy)-phenyl]-ureido}- benzamide,
A/-(2-Diethylamino-ethyl)-4-{3-[4-(4-hydroxy-phenoxy)-phenyl]-ureido}-benzamide, Λ/-(2-Diethylamino-ethyl)-4-{3-[4-(4-dimethylamino-phenoxy)-phenyl]-ureido}-benzamide, Λ/-(2-Diethylamino-ethyl)-2-methoxy-4-{3-[4-(4-methylamino-phenoxy)-phenyl]-ureido}- benzamide, 4-{3-[4-(4-Cyano-3-chloro-phenoxy)-phenyl]-ureido}-Λ/-(2-diethylamino-ethyl)-benzamide, 4-{3-[4-(4-Carbamoyl-phenoxy)-phenyl]-ureido}-A/-(2-diethylamino-ethyl)-benzamide, 4-[3-(3-Chloro-4-phenoxy-phenyl)-ureido]-A/-(2-diethylamino-ethyl)-benzamide, Λ/-(2-Diethylamino-ethyl)-4-[3-(2-fluoro-3-methoxy-4-phenoxy-phenyl)-ureido]-benzamide, Λ/-(2-Diethylamino-ethyl)-4-[3-(3-bromo-6-methoxy-4-phenoxy-phenyl)-ureido]-benzamide, Λ/-(2-Diethylamino-ethyl)-4-[3-(3-methylamino-4-phenoxy-phenyl)-ureido]-benzamide, Λ/-(2-Diethylamino-ethyl)-4-[3-(3-hydroxymethyl-4-phenoxy-phenyl)-ureido]-benzamide, Λ/-(2-Diethylamino-ethyl)-4-[3-(3-carboxamido-4-phenoxy-phenyl)-ureido]-benzamide, Λ/-(2-Diethylamino-ethyl)-4-[3-(3-(V,/V-dimethylcarboxamido)-4-phenoxy-phenyl)-ureido]- benzamide, Λ/-(2-Diethylamino-ethyl)-4-[3-(3-methyl-4-phenoxy-phenyl)-ureido]-benzamide,
Λ/-(2-Diethylamino-ethyl)-4-[3-(4-phenoxy-3-trifluoromethoxy-phenyl)-ureido]-benzamide,
A/-(2-Diethylamino-ethyl)-4-[3-(4-phenoxy-2-trifluoromethyl-phenyl)-ureido]-benzamide,
Λ/-(2-Diethylamino-ethyl)-4-{3-[4-(4-trifluoromethoxy-phenoxy)-phenyl]-ureido}-benzamide,
Substituents on the benzamide moiety
Λ/-(2-Diethylamino-ethyl)-3-fluoro-4-[3-(4-phenoxy-phenyl)-ureido]-benzamide, Λ/-(2-Diethylamino-ethyl)-2-fluoro-5-chloro-4-[3-(4-phenoxy-phenyl)-ureido]-benzamide,
3-Chloro-Λ/-(2-diethylamino-ethyl)-3-methyl-4-[3-(4-phenoxy-phenyl)-ureido]-benzamide,
3-Bromo-Λ/-(2-diethylamino-ethyl)-2-flouro-5-ethyl-4-[3-(4-phenoxy-phenyl)-ureido]- benzamide,
2-Amino-3-chloro-Λ/-(2-diethylamino-ethyl)-4-[3-(4-phenoxy-phenyl)-ureido]-benzamide, 2-Amino-3-bromo-Λ/-(2-diethylamino-ethyl)-4-[3-(4-phenoxy-phenyl)-ureido]-benzamide,
Λ/-(2-Diethylamino-ethyl)-4-[3-(4-phenoxy-phenyl)-ureido]-3-trifluoromethyl-benzamide,
Λ/-(2-Diethylamino-ethyl)-4-[3-(4-phenoxy-phenyl)-ureido]-3-trifluoromethoxy-benzamide.
Salts, complexes or solvates
The invention also relates to physiologically acceptable salts, complexes, solvates or prodrugs of the compounds of the invention.
When a compound of the invention possesses a basic functional group it can form a salt with an inorganic or organic acid.
Examples of physiologically acceptable salts of the compounds according to the invention include salts with inorganic acids, salts with organic acids, and salts with basic or acidic amino acids.
Examples of salts with inorganic acids include salts with hydrochloric acid, hydrobromic acid, hydroiodic acid, nitric acid (to form e.g. a nitrate or a nitrite), sulfuric acid (to form e.g., a H2SO3 salt, a sulfate or a H2SO5 salt) and phosphoric acid (to form e.g. a H3PO3 salt or a H3PO4 salt)
Examples of salts with organic acids include salts with formic acid, acetic acid, propionic acid, butyric acid, pentanoic acid, oxalic acid, tartaric acid, malonic acid, succinic acid, citric acid, C H8(COOH)2, C5H10(COOH)2, acrylic acid, malic acid, fumaric acid, H2CO3, lactic acid, ascorbic acid, benzoic acid, salicylic acid and phthalic acid, trifluoroacetic acid, methanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid and 3-chlorobenzoic acid.
Examples of salts with acidic amino acids include salts with aspartic acid and glutamic acid.
Optical isomers
When a compound of the invention contains optical isomers, diastereomers or other stereroisomers these are included as a compound of the invention as well as the racemate, i.e. mixture of enantiomers. Each of them can be obtained by methods known by a person skilled in the art. For example the optical isomer can be obtained using an optically active synthetic intermediate, an asymmetric synthesis or subjecting the racemic mixture of the final product or a suitable intermediate to optical resolution in accordance with known methods such as, e.g., fractional recrystallisation method, chiral column method, diastereomer method etc.
Other forms
The invention also encompasses a compound in amorphous, any polymorphous or any crystalline form.
Disorders
The compounds according to the invention can be used in medicine and modulate the activity of a MCH receptor. The compounds may be used as agents for preventing or treating diseases caused by or involving a melanin-concentrating hormone, i.e. they are useful for treating or preventing a MCH or MCH receptor related disorder or abnormality in a subject such as, e.g., an animal or a mammal such as, e.g., a human.
The compounds according to the invention may have antagonistic, inverse agonistic, agonistic or allosteric activity against a MCH receptor, normally antagonistic activity.
In the present context an agonist is defined as a compound that increases the functional activity of a MCH receptor (e.g. the signal transduction through a receptor). The term "agonist" includes partial agonist, i.e. which increases the functional activity of the receptor to a submaximal level. An inverse agonist (or negative antagonist) is defined as a compound that decreases the basal functional activity of a MCH receptor. An allosteric compound is defined as a compound that enhances or diminishes the effects of other receptor ligands.
An antagonist is defined as a compound that decreases the functional activity of a MCH receptor either by inhibiting the action of an agonist or by its own intrinsic activity.
The MCH receptors mentioned in the invention include MCH1 and MCH2 receptors. It also includes MCH receptors having at least about 80% such as, e.g. at least about 85% or at least about 90% homology to the amino acid sequences CTLITAMDAN or CTIITSLDTC.
The MCH receptors may be an animal or a mammalian or non-mammalian receptor, such as a human receptor.
Increasing or decreasing the activity of a MCH receptor such as, e.g. a MCH1 receptor alleviates a MCH-related disorder or abnormality. In specific embodiments the disorder is
a steroid or pituitary hormone disorder, an epinephrine release disorder, a gastrointestinal disorder, a cardiovascular disorder, an electrolyte balance disorder, hypertension, diabetes, a respiratory disorder, asthma, a reproductive function disorder, a muscoskeletal disorder, a neuroendocrine disorder, a cognitive disorder, a memory disorder such as, e.g., Alzheimer's disease, a sensory modulation and transmission disorder, a motor coordination disorder, a sensory integration disorder, a motor integration disorder, a dopaminergic function disorder such as, e.g. Parkinson's disease, a sensory transmission disorder, an olfaction disorder, a sympathetic innervation disorder, an affective disorder such as, e.g. depression, a stress-related disorder, a fluid-balance disorder, a urinary disorder such as, e.g., urinary incontinence, a seizure disorder, pain, psychotic behaviour such as, e.g., schizophrenia, morphine or opioid tolerance, opiate addiction or migraine.
More specifically, the compounds of the invention are useful for the treatment or prevention of feeding disorders such as, e.g., overweight, adiposity, obesity and bulimia (e.g. malignant mastocytosis, exogeneous obesity, hyperinsulinar obesity, hyperplasmic obesity, hypophyseal adposity, hypoplasmic obesity, hypophysal adiposity, hypoplasmic obesity, hypothyroid obesity, hypothalamic obesity, symptomatic obesity, infantile obesity, upper body obesity, alimentary obesity, hypogonadal obesity, systemic mastocytosis, simple obesity, central obesity etc.), hyperfagia, emotional disorders, dementia or hormonal disorders.
In the present context the term body mass index or BMI is defined as body weight (kg)/height2 (m2), and the term overweight is intended to indicate a BMI in a range from about 25 to about 29.9, whereas obesity is intended to indicate a BMI, which is at least about 30.
A compound of the invention is also useful as an agent for preventing or treating lifestyle diseases such as, e.g., diabetes, diabetic complications (e.g. retinopathy, neuropathy, nephropathy etc.), arteriosclerosis and gonitis.
The present invention further relates to a cosmetic method for reducing overweight and/or for treating of and/or preventing overweight, bulimia, bulimia nervosa, obesity and/or complications thereto, the method comprising administering to an animal such as, e.g. a human in need thereof, an effective amount of a compound according to the invention
The invention also relates to a method for the treatment and/or prophylaxis of diseases caused by a melanin-concentrating hormone, the method comprising administering to a mammal in need thereof an efficient amount of a compound according to the invention.
A mentioned above, the MCH-related disorders may be a feeding disorder. Accordingly, the invention relates to a method for the treatment and/or prophylaxis of diseases caused by feeding disorders, the method comprising administering to a mammal in need thereof an efficient amount of a compound according to the invention.
The invention also relates to a method for modifying the feeding behaviour of a mammal, the method comprising administering to a mammal in need thereof an efficient amount of a compound according to the invention.
Furthermore, the invention relates to a method for the reduction of body mass, the method comprising administering to a mammal in need thereof an efficient amount of a compound according to the invention.
Moreover, the invention relates to a method for the treatment and/or prophylaxis of Syndrome X (metabolic syndrome) or any combination of obesity, insulin resistance, dyslipidemia, impaired glucose tolerance and hypertension, the method comprising administering to a mammal in need thereof an efficient amount of a compound according to the invention.
Another aspect of the invention is a method for the treatment and/or prophylaxis of Type II diabetes or Non Insulin Dependent Diabetes Mellitus (NIDDM), the method comprising administering to a mammal in need thereof an efficient amount of a compound according to the invention.
A still further aspect of the invention is a method for the treatment and/or prophylaxis of bulimia, bulimia nervosa and/or obesity, the method comprising administering to a mammal in need thereof an efficient amount of a compound according to the invention.
Moreover, the invention relates to a method for the treatment and/or prophylaxis of depression and/or anxiety, the method comprising administering to a mammal in need thereof an efficient amount of a compound according to the invention.
Pharmaceutical compositions
The compounds for use in the methods according to the invention are normally presented in the form of a pharmaceutical or a cosmetic composition comprising the specific compound or a physiologically acceptable salt thereof together with one or more physiologically acceptable excipients.
The compounds may be administered to the animal including a mammal such as, e.g., a human by any convenient administration route such as, e.g., the oral, buccal, nasal, ocular, pulmonary, topical, transdermal, vaginal, rectal, ocular, parenteral (including inter alia subcutaneous, intramuscular, and intravenous), route in a dose that is effective for the individual purposes. A person skilled in the art will know how to chose a suitable administration route.
The pharmaceutical or cosmetic composition comprising a compound according to the invention may be in the form of a solid, semi-solid or fluid composition.
The solid composition may be in the form of tablets such as, e.g. conventional tablets, effervescent tablets, coated tablets, melt tablets or sublingual tablets, pellets, powders, granules, granulates, particulate material, solid dispersions or solid solutions.
A semi-solid form of the composition may be a chewing gum, an ointment, a cream, a liniment, a paste, a gel or a hydrogel.
The fluid form of the composition may be a solution, an emulsion including nano- emulsions, a suspension, a dispersion, a liposomal composition, a spray, a mixture, a syrup or a aerosol.
Fluid compositions, which are sterile solutions or dispersions can utilized by for example intraveneous, intramuscular, intrathecal, epidural, intraperitoneal or subcutaneous injection of infusion. The compounds may also be prepared as a sterile solid composition, which may be dissolved or dispersed before or at the time of administration using e.g. sterile water, saline or other appropriate sterile injectable medium.
Other suitable dosages forms of the pharmaceutical compositions according to the invention may be vagitories, suppositories, plasters, patches, tablets, capsules, sachets, troches, devices etc.
The dosage form may be designed to release the compound freely or in a controlled manner e.g. with respect to tablets by suitable coatings.
The pharmaceutical composition may comprise a therapeutically effective amount of a compound according to the invention.
The content of a compound of the invention in a pharmaceutical composition of the invention is e.g. from about 0.1 to about 100% w/w of the pharmaceutical composition.
The pharmaceutical or cosmetic compositions may be prepared by any of the method well known to a person skilled in pharmaceutical or cosmetic formulation.
In pharmaceutical or cosmetic compositions, the compounds are normally combined with a pharmaceutical excipient, i.e. a therapeutically inert substance or carrier.
The carrier may take a wide variety of forms depending on the desired dosage form and administration route.
The pharmaceutically or cosmetically acceptable excipients may be e.g. fillers, binders, d is integrants, diluents, glidants, solvents, emulsifying agents, suspending agents, stabilizers, enhancers, flavours, colors, pH adjusting agents, retarding agents, wetting agents, surface active agents, preservatives, antioxidants etc. Details can be found in pharmaceutical handbooks such as, e.g., Remington's Pharmaceutical Science or Pharmaceutical Excipient Handbook.
Dosage
Optimal dosages to be administered may be determined by those skilled in the art, and will vary with the particular compound in use, the strength of the composition, the route of administration, the frequency of administration, the age, weight, gender, diet and condition of the subject to be treated and the condition being treated and the advancement of the disease condition etc.
Suitable dosages may be from about 0.001 mg to about 1 g such as, e.g. from about 0.005 to about 750 mg, from about 0.01 to about 500 mg, from about 0.05 to about 500 mg, from about 0.1 to about 250 mg, from about 0.1 to about 100 mg or from about 0.5 to about 50 mg.
The amounts can be divided into one or several doses for administration daily, every second day, weekly, every two weeks, monthly or with any other suitable frequency. Normally, the administration is daily.
A compound or a pharmaceutical composition according to the invention may be used in combination with other drug substances such as agents for treating disorders like e.g. diabetes, diabetes complications, obesity, hypertension, hyperlipidemia, arteriosclerosis, arthritis, anxiety, and/or depression etc.
Experimental
Materials and methods
Transfections and Tissue Culture - The cDNA encoding the human MCH-1 receptor was cloned from a human brain cDNA library and cloned into the eukaryotic expression vector pcDNA3.1 (Invitrogen). Assays were performed on transiently transfected COS-7 cells or stably transfected CHO (Chinese Hamster Ovary) cells, expressing the human MCH-1 receptor in pcDNA3.1. Stable MCH-1 receptor transfectants of CHO cells were obtained using 5 μg plasmid cDNA and a standard calcium phosphate transfection method
(Johansen et. al., 1990; Gether et. al., 1992) with subsequent selection in 1 mg/ml G418 (Life Technology). Clones were screened by a MCH receptor radioligand binding assay (as described below). Stably transfected CHO cells were maintained in RPMI 1640 culture medium (Invitrogen), supplemented with 10 % fetal calf serum (Invitrogen), 100 U/ml penicillin, 100 μg/ml streptomycin (Life Technology), and 500 μg/ml G418 (Life
Technology). COS-7 cells were grown in Dulbecco's modified Eagle's medium (DMEM) 1885 (Invitrogen) supplemented with 10 % fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and were transiently transfected by a standard calcium phosphate transfection method (Johansen et al., 1990; Gether et. al., 1992) two days before assay.
Radioligand Binding Assay -Transiently transfected COS-7 cells or stably transfected CHO cells, expressing human MCH-1 receptor were seeded in multi-well culture plates one day before the assay. The number of cells per well was determined by the apparent expression efficiency of the cell line aiming at 5 - 10 % binding of the added radioligand. Cells were assayed by competition binding for 3 hours at room temperature using 15 pM [125I]-MCH (Amersham Pharmacia Biotech) plus variable amounts of unlabeled ligand in 0.5 ml of a 25 mM Hepes buffer, pH 7.4, supplemented with 10 mM MgCI2, 5 mM MnCI2,
10 mM NaCI, 0.1 % (w/v) bovine serum albumin (BSA), 100 μg/ml bacitracin. The assay was performed in duplicate. Nonspecific binding was determined as the binding in the presence of 1 μM MCH (Bachem). Binding data were analyzed and IC5o values determined by non-linear regression using the Prism software (GraphPad software, San Diego). Values of the dissociation and inhibition constants (Kd and Kj) were estimated from competition binding using the equations Kd=IC50- and Kj=IC5o/(1+L/Kd), respectively, where L is the concentration of radioligand.
Phosphatidylinositol assay - To assay phosphatidylinositol turnover, transiently transfected COS-7 cells or stably transfected CHO cells, expressing human MCH-1 receptor (2x105 cells/well) were incubated for 24 h with 5 μCi of [3H]-myo-inositol (Amersham Pharmacia Biotech) in 0.5 ml inositol-free culture medium. Cells were washed twice in Pl-buffer: 20 mM HEPES, pH 7.4, supplemented with 140 mM NaCI, 5 mM KCI, 1 mM MgSO4, 1 mM CaCI2, 10 mM glucose, 0.02% (w/v) bovine serum; and were incubated in 0.5 ml Pl-buffer supplemented with 10 mM LiCI at 37 °C for 45 min. Phosphatidylinositol turnover was stimulated by submaximal concentrations of MCH, i.e. 10 nM in the presence of increasing amounts of ligand. The ligand was added 5 min. before adding the agonist (MCH). Cells were extracted with 10 mM ice-cold Formic acid, and the generated [3H]-inositol phosphates were purified on Bio-Rad AG 1-X8 anion-exchange resin. Determinations were made in duplicate. PI data were analyzed and IC50 values determined by non-linear regression using the Prism software (GraphPad software, San Diego).
Scintillation Proximity Assay (SPA)- Measurement of [125|]_|\/|CHbinding was performed in duplicates by incubating membranes and beads with tracer in the presences of various concentrations of test compounds (10
"8 to 10
"4 M ) in DMSO (3 μl) at room temperature for two hours. Membranes and beads were pre-incubated for 20 min. The binding buffer contained 50 mM Tris (pH 7.4), 8 mM MgCI2, 12% glycerol, 0.1% (w/v) bovine serum albumin (BSA), and protease inhibitors (Complete protease inhibitor cocktail tablets, Roche). A final
Ci/mmol; Amersham Pharmacia Biotech) concentration of 75.000 cpm/well (33.8 nCi) was applied and PEI-treated WGA-coupled PVT SPA beads, type B from Amersham Pharmacia Biotech were used at a final concentration of 0.4 mg/well. Moreover, CHO-K1 membranes expressing the hMCHreceptor were purchased from Euroscreen (ES-370-M) and a final concentration of 2μg/well were used. Binding data were analyzed and IC50 values determined by non-linear regression using the Prism software (GraphPad software, San Diego). Values of the inhibition constant (K
j)
were estimated from competition binding using the equation
where L and K
fj are the concentration and affinity constant, respectively, of the radioligand. were analyzed by one-way ANOVA followed by post hoc Bonferroni test.
References:
Gether, U., Marray, T., Schwartz, T.W., and Johansen, T.E. (1992). Stable expression of high affinity NK-] (substance P) and NK2 (neurokinin A) receptors but low affinity NK3 (neurokinin B) receptors in transfected CHO cells. FEBS Lett., 296, 241-244.
Johansen, T.E., Schøller, M.S., Tolstoy, S. and Schwartz, T.W. (1990). Biosynthesis of peptide precursors and protease inhibitors using new constitutive and inducible eukaryotic expressions vectors. FEBS Lett., 267, 289-294.
Examples
General comments: 1H NMR data are given either in full detailed or with characteristic selected peaks. LC-MS were obtained with positive ion scanning.
Example 1 4-[3-(4-Phenoxyphenyl)ureido]benzoic acid
To a suspension of 4-aminobenzoic acid (430 mg, 3.13 mmol) in DCM (10 ml) under nitrogen atmosphere was added dropwise 4-phenoxyphenyl isocyanate (0.70 ml, 3.8 mmol). The reaction was stirred at room temperature for 3 days. The precipitate was filtered off and washed with DCM to give 866 mg (79%) of a white solid. 1H NMR (300 MHz, DMSO-d6) δ 12.58 (s, 1 H), 9.03 (s, 1 H), 8.80 (s, 1 H), 7.87 (d, J = 8.5, 1 H).
Example 2 Λ/-(3-Dimethylaminopropyl)-4-[3-(4-phenoxyphenyl)ureido]benzamide
To a solution of 4-[3-(4-phenoxyphenyl)ureido]benzoic acid (Ex 1) (50 mg, 0.14 mmol) in DCM/DMF (9:1 , 3.5 ml) was added PS-DCC resin (500 mg, 1 ,35 mmol/g, 5 equiv), HOBt (40 mg, 0.30 mmol) and N,N-dimethylaminopropylamine (18 μl, 0.14 mmol). The reaction
mixture was stirred for 3 days. SP-Trisamine (200 mg, 5 equiv) was added, and the reaction mixture was strirred for 3 more days. The resin was filtered off and washed with DCM (2x). The product was purified by filtration of the combined organic solutions through an SPE-column (1.0 g SCX, 0.3 mmol/g), washing with MeOH, and elution of the product with 5% ammonia in MeOH. Concentration gave 14.4 mg (23%) of a white solid. LC/MS [M+H]+ m/z 433.1.
Example 3-16
The following compounds were prepared according to the procedure outlined in Example 2.
Ex 14
Ex 15
Example 3
4-[3-(4-Phenoxyphenyl)-ureido]-Λ/-(2-piperidin-1-yl-ethyl)-benzamide
4-[3-(4-Phenoxyphenyl)-ureido]-benzoic acid (Ex 1) (50 mg, 0.14 mmol) and 1-(2- aminoethyl)-piperidine (20 μl, 0.14 mmol) was coupled as described in Example 2 to give 19.5 mg (28%) of the title compound. LC/MS [M+H]+ m/z 459.2.
Example 4
Λ/-(2-Morpholin-4-yl-ethyl)-4-[3-(4-phenoxy-phenyl)-ureido]-benzamide 4-[3-(4-Phenoxyphenyl)-ureido]-benzoic acid (Ex 1) (50 mg, 0.14 mmol) and 4-(2- aminoethyl)-morpholine (19 μl, 0.14 mmol) was coupled as described in Example 2 to give 17.1 mg (26%) of the title compound. LC/MS [M+H]+ m/z 461.1.
Example 5 4-[3-(4-Phenoxy-phenyl)-ureido]-W-(3-pyrrolidin-1-yl-ethyl)-benzamide
4-[3-(4-Phenoxyphenyl)-ureido]-benzoic acid (Ex 1) (50 mg, 0.14 mmol) and Λ/-(2- aminoethyl)-pyrrolidine (18 μl, 0.14 mmol) was coupled as described in Example 2 to give
16.8 mg (26%) of the title compound. LC/MS [M+H]+ m/z 445.1.
Example 6 Λ/-(2-Diethylaminoethyl)-4-[3-(4-phenoxyphenyl)-ureido]-benzamide 4-[3-(4-Phenoxyphenyl)-ureido]-benzoic acid (Ex 1) (50 mg, 0.14 mmol) and N,N- diethylethylenediamine (20 μl, 0.14 mmol) was coupled as described in Example 2 to give
17.9 mg (28%) of the title compound. LC/MS [M+H]+ m/z 447.1.
Example 7 1 -[4-(4-Methyl-piperazine-1 -carbonyl)-phenyl]-3-(4-phenoxy-phenyl)-urea
4-[3-(4-Phenoxyphenyl)-ureido]-benzoic acid (Ex 1) (50 mg, 0.14 mmol) and N- methylpiperazine (15.8 μl, 0.14 mmol) was coupled as described in Example 2 to give 20.7 mg (34%) of the title compound. LC/MS [M+H]+ m/z 431.1.
Example 8
Λ -(4-Benzyl-morpholin-2-ylmethyl)-4-[3-(4-phenoxy-phenyl)-ureido]-benzamide
4-[3-(4-Phenoxyphenyl)-ureido]-benzoic acid (Ex 1) (50 mg, 0.14 mmol) and (4-benzyl- 1 ,4-oxazinan-2-yl)methylamine (29 μl, 0.14 mmol) was coupled as described in Example 2 to give 23.7 mg (31%) of the title compound. LC/MS [M+H]+ m/z 537.2.
Example 9 Λ -(2-Dimethylamino-ethyl)-4-[3-(4-phenoxy-phenyl)-ureido]-benzamide
4-[3-(4-Phenoxyphenyl)-ureido]-benzoic acid (Ex 1) (50 mg, 0.14 mmol) and N,N- dimethylethylenediamine (16 μl, 0.14 mmol) was coupled as described in Example 2 to give 14.3 mg (24%) of the title compound. LC/MS [M+H]+ m/z 419.1.
Example 10
W-(1-Benzyl-piperidin-4-yl)-4-[3-(4-phenoxy-phenyl)-ureido]-benzamide
4-[3-(4-Phenoxyphenyl)-ureido]-benzoic acid (Ex 1) (50 mg, 0.14 mmol) and 4-amino-1- benzylpiperidine (27 μl, 0.14 mmol) was coupled as described in Example 2 to give 11.6 mg (16%) of the title compound. LC/MS [M+H]+ m/z 521.2.
Example 11
Λ -(1-Ethyl-pyrrolidin-2-ylmethyl)-4-[3-(4-phenoxy-phenyl)-ureido]-benzamide 4-[3-(4-Phenoxyphenyl)-ureido]-benzoic acid (Ex 1) (50 mg, 0.14 mmol) and 2- (aminomethyl)-l-ethylpyrrolidine (18 μl, 0.14 mmol) was coupled as described in Example 2 to give 22.5 mg (34%) of the title compound. LC/MS [M+H]+ m/z 459.2.
Example 12 Λ/-(2-Diisopropylamino-ethyl)-4-[3-(4-phenoxy-phenyl)-ureido]-benzamide
4-[3-(4-Phenoxyphenyl)-ureido]-benzoic acid (Ex 1) (50 mg, 0.14 mmol) and N,N- diisopropylethylenediamine (20 μl, 0.14 mmol) was coupled as described in Example 2 to give 26.0 mg (38%) of the title compound. 1H NMR (300 MHz, DMSO-d6) δ 8.92 (s, 1 H), 8.79 (s, 1 H), 7.77 (d, J = 8.9 Hz, 2H), 7.45-7.55 (m, 4H), 7.32-7.40 (m, 2H), 7.05-7.13 (m, 1 H), 6.93-7.02 (m, 4H), 3.12-3.30 (m, 2H), 2.91-3.04 (m, 2H), 0.98 (d, J = 6.4 Hz, 12H). LC/MS [M+H]+ m/z 575.2.
Example 13
W-[2-(4-Benzyl-piperazin-1-yl)-ethyl]-4-[3-(4-phenoxy-phenyl)-ureido]-benzamide 4-[3-(4-Phenoxyphenyl)-ureido]-benzoic acid (Ex 1) (50 mg, 0.14 mmol) and 2-(4- benzylpiperazino)ethan-1 -amine (31 μl, 0.14 mmol) was coupled as described in Example 2 to give 28.6 mg (36%) of the title compound. 1H NMR (300 MHz, DMSO-d6) δ 8.92 (s, 1 H), 8.78 (s, 1 H). LC/MS [M]+ m/z 459.2.
Example 14 Λ -(2-Dimethylamino-ethyl)-/V-methyl-4-[3-(4-phenoxy-phenyl)-ureido]-benzamide 4-[3-(4-Phenoxyphenyl)-ureido]-benzoic acid (Ex 1) (50 mg, 0.14 mmol) and N,N,N'- trimethylethylenediamine (18 μl, 0.14 mmol) was coupled as described in Example 2 to give 17.2 mg (28%) of the title compound. LC/MS [M+H]+ m/z 433.1.
Example 15
W-Methyl-Λ/-[3-(4-methyl-piperazin-1-yl)-propyl]-4-[3-(4-phenoxy-phenyl)-ureido]- benzamide 4-[3-(4-Phenoxyphenyl)-ureido]-benzoic acid (Ex 1) (50 mg, 0.14 mmol) and 1-(3- aminopropyl)-4-methylpiperazine (22.5 mg, 0.14 mmol) was coupled as described in Example 2 to give 23.6 mg (34%) of the title compound. LC/MS [M+H]+ m/z 488.2.
Example 16 W-(3-Dimethylamino-propyl)-4-[3-(4-phenoxy-phenyl)-ureido]-benzamide
4-te/τ-Butyloxycarbonylaminobenzoic acid (193 mg, 0.81 mmol), prepared from 4- aminobenzoic acid and di-fe/τ-butyl dicarbonate by standard procedures, in DCM/DMF (9:1 , 7 ml) was added PS-DCC resin (1.25 g, 1.35 mmol/g, 2 equiv), HOBt (120 mg, 0.77 mmol) and N,N-dimethylaminopropylamine (97 μl, 0.77 mmol). The reaction mixture was stirred for 3 days. SP-Trisamine (1 g, 5 equiv) was added, and the reaction mixture was stirred for 3 more days. The resin was filtered off and washed with DCM (2x). The combined organic phases were concentrated to 5 ml. TFA (1.5 ml) was added, and the reaction was stirred at room temperature for 7 days. The reaction was concentrated, added DCM (10 ml) and washed with sat. aqueous Na2CO3 (5 ml). The organic phase was concentrated to give 184.3 mg (97%) of 4-amino-Λ/-(2- dimethylaminoethyl)benzamide, containing 5 % DMF. 1H NMR (300 MHz, CDCI3) δ 8.00 (s, 1 H), 7.59 (d, J = 8.7 Hz, 2H), 6.64 (d, J = 8.7 Hz, 2H), 4.01 (s, 2H), 3.51 (dt, J = 6.2, 5.1 Hz, 2H), 2.45 (t, J = 5.9 Hz, 2H), 2.27 (s, 6H), 1.73 (p, J = 6.1 Hz, 2H).
A solution of 4-amino-Λ/-(2-dimethylaminoethyl)benzamide (57mg, 0.26 mmol) in dioxane (1 ml), under argon at 10 °C was added 4-phenoxybenzoyl chloride (149.8 mg, 0.64 mmol) in DCM (1 ml), and the reaction was stirred at room temperature for 3 days. PS- Trisamine (700 mg, 3.38 mmol/g) was added, and the stirring was continued for 12 h. The resin was filtered off, and the product was purified with an SPE-column (1.0 g SCX, 0.3 mmol/g). Concentration gave 27 mg (25%) of the title compound as a white solid. LC/MS [M+H]+ m/z 418.1.
Example 17
4-[3-(4-Phenoxy-phenyl)-ureido]-N-(2-piperidin-1-yl-ethyl)-benzamide
A solution of 4-terf-butyloxycarbonylaminobenzoic acid (193 mg, 0.81 mmol) in DCM/DMF (9:1 , 7 ml) was added PS-DCC resin (1.25 g, 1.35 mmol/g, 2 equiv), HOBt (120 mg, 0.77 mmol) and A/-(2-aminoethyl)piperidine (110 μl, 0.77 mmol). The reaction mixture was stirred for 3 days. SP-Trisamine (1 g, 5 equiv) was added, and the reaction mixture was stirred for 3 more days. The resin was filtered off and washed with DCM (2x). The combined organic phases were concentrated to 5 ml. TFA (1.5 ml) was added, and the reaction was stirred at room temperature for 7 days. The reaction was concentrated, added DCM (10 ml) and washed with sat. aq. Na2CO3 (5 ml). The organic phase was concentrated to give 61 mg (32%) of 4-amino-Λ/-(2-dimethylaminoethyl)benzamide, containing 5 % DMF.
A solution of 4-amino-Λ/-(2-dimethylaminoethyl)benzamide (57mg, 0.26 mmol) in dioxane (1 ml), under argon at 10 °C was added 4-phenoxybenzoyl chloride (149.8 mg, 0.64 mmol, prepared from 4-phenoxybenzoic acid with oxalyl chloride by standard methods) in DCM (1 ml), and the reaction was stirred at room temperature for 3 days. PS-Trisamine (700 mg, 3.38 mmol/g) was added, and the stirring was continued for 12 h. The resin was filtered off, and the product was purified with an SPE-column (1.0 g SCX, 0.3 mmol/g). Concentration gave 33 mg (29%) of the title compound as a white solid. LC/MS [M+H]+ m/z 443.1.
Example 18 4-[3-(4-Phenoxy-phenyl)-ureido]-Λ/-(2-piperidin-1-yl-ethyl)-benzamide
A solution of 4-tetτ-butyloxycarbonylaminobenzoic acid (193 mg, 0.81 mmol) in DCM/DMF (9:1 , 7 ml) was added PS-DCC resin (1.25 g, 1.35 mmol/g, 2 equiv), HOBt (120 mg, 0.77 mmol) and Λ/-(2-aminoethyl)morpholine (101 μl, 0.77 mmol). The reaction mixture was stirred for 3 days. SP-Trisamine (1 g, 5 equiv) was added, and the reaction mixture was stirred for 3 more days. The resin was filtered off and washed with DCM (2x). The combined organic phases were concentrated to 5 ml. TFA (1.5 ml) was added, and the
reaction was stirred at room temperature for 7 days. The reaction was concentrated, added DCM (10 ml) and washed with sat. aq. Na2CO3 (5 ml). The organic phase was concentrated to give 59 mg (31 %) of 4-amino-Λ/-(2-morpholin-4-yl-ethyl)benzamide, containing 5 % DMF.
A solution of 4-amino-Λ/-(2-dimethylaminoethyl)benzamide (57mg, 0.26 mmol) in dioxane (1 ml), under argon at 10 °C was added 4-phenoxybenzoyl chloride (149.8 mg, 0.64 mmol) in DCM (1 ml), and the reaction was stirred at room temperature for 3 days. PS- Trisamine (700 mg, 3.38 mmol/g) was added, and the stirring was continued for 12 h. The resin was filtered off, and the product was purified with an SPE-column (1.0 g SCX, 0.3 mmol/g). Concentration gave 14 mg (12%) of the title compound as a white solid.
Example 19 tø-(2-Diethylamino-ethyl)-4-methylamino-benzamide
A mixture of paraformaldehyde (2.87g, 31.9 mmol) and sodium methoxide (3.55 g, 65.7 mmol) in MeOH (275 ml) was added procainamide (5.00 g, 21.2 mmol). The mixture was stirred at 40 °C for 12 h. Sodium borohydride (1.88 g, 50.0 mmol) was added carefully, and the reaction was stirred at 50 °C for 24 h. The reaction was extracted with DCM (800 ml). The extract was washed with 5% NaHCO3 (40 ml), dried (MgSO4) and concentrated to give 5.0 g (94%) the title compound. LC/MS [M+H]+ 250.1.
Example 20 (General method for the preparation of compounds of Example 21-25) W-(2-Diethylaminoethyl)-4-[W-methyl-4-biphenylcarboxamido]-benzamide
A solution of Ex 19 (100 mg, 0.40 mmol) in CHCI3 (6 ml) was added DMAP (100 mg, 0.80 mmol), SP-DCC resin (1.0 g, 1.37 mmol/g, 1.4 mmol) and 4-phenylbenzoic acid (119 mg,
0.60 mmol), and the reaction was stirred for 13 days. The resin was filtered off and washed with DCM. The combined organic phases were concentrated and the title compound was isolated by chromatography on SiO2 (EtOAc/heptane/Et3N 10:5:1) to give
53 mg (31%) of the title compound. LC/MS [M+HJ+ 430.1.
Example 21-25
The following compounds were prepared according to the procedure outlined in Example 20.
Ar2-B-Ar CO
Example 26 Λ/-(2-Diethylamino-ethyl)-4-[1-methyl-3-(4-phenoxy-phenyl)-ureido]-benzamide
prepared by reaction of Ex 19 with 20% excess of 4-phenoxyphenyl isocyanate in DCM under nitrogen atmosphere.
Example 27 Λ/-(2-Diethylamino-ethyl)-4-[5-(4-phenoxy-phenyl)-[1,2,4]oxadiazol-3-yl]-benzamide
Example 136 (40 mg, 0.11 mmol), PS-cabodiimidazole (0.15 g, 1.35 mmol/g), hydroxyl benztriazole (23 mg, 0.17 mmol), and Λ/,Λ/-diethylethylenediamine (14 DL, 0.10 mmol) were placed in a flask under nitrogen atmosphere with dichloromethane (2 mL) and the reaction was stirred for three days. PS-Trisamine (0.14 g, 3.58 mmol/g) and more dichloromethane (4 mL) were added and left stirring for 2h before the resins were filtered off. The solvent was removed in vacuo. The crude product was chromatographed (silica, dichloromethane/methanol/ammoniak, 95:5+0.5%) giving 20 mg (40%) of the title product. 1H NMR (300 MHz, CD3CI): δ 1.07 (t, 6H), 2.60 (q, 4H), 7.13 (d, 4H). LCMS (an20p10); Rt = 6.35 min, (M+1) = 457
Example 28
N-(1-Ethyl-pyrrolidin-2-ylmethyl)-4-[5-(4-phenoxy-phenyl)-[1,2,4]oxadiazol-3-yl]- benzamide
The title product was synthesised using the same method as described in example 137 from example 136 (40 mg, 0.11 mmol) and 2-(4-benzylpiperazino)ethane-1-amine (27 μL, 0.10 mmol) giving 44 mg (70 %) of the product. 1H NMR (300 MHz, CD3CI): δ 2.03 (t, 6H), 2.93 (d, 2H), 3.56 (q, 4H), 7.93 (d, 2H), 7.45 (t, 2H). LCMS (an20p10); Rt = 7.40 min, M = 559
Example 29 V-[2-(4-Benzyl-piperazin-1-yl)-ethyl]-4-[5-(4-phenoxy-phenyl)-[1,2,4]oxadiazol-3-yl]- benzamide
The title product was synthesised using the same method as described in example 137 from example 136 (40 mg, 0.11 mmol) and 2-(aminomethyl)-1-ethylpyrrolidine (15 μL, 0.13 mmol) giving 43 mg (81 %) of the product.
1H NMR (300 MHz, CD
3CI): δ 7.13 (d, 4H), 7.44 (t, 2H), 8.67 (s, 1 H). LCMS (an20p10); Rt = 6.27 min, (M+1 ) = 469.
Example 30
In vitro tests of compounds according to the invention
The following compounds are prepared as described in previous examples.
4-{3-[4-(4-Chloro-phenoxy)-phenyl]-ureido}-A/-(2-diethylamino-ethyl)-benzamide Λ/-(1-Benzyl-piperidin-4-yl)-4-{3-[4-(4-chloro-phenoxy)-phenyl]-ureido}-benzamide Λ/-{3-[4-(4-Acetylamino-phenyl)-piperidin-1-yl]-propyl}-4-[3-(4-phenoxy-phenyl)-ureido]- benzamide
Λ/-{3-[4-(3-Acetylamino-phenyl)-piperidin-1-yl]-propyl}-4-[3-(4-phenoxy-phenyl)-ureido]- benzamide Λ/-(3-Morpholin-4-yl-propyl)-4-[3-(3-phenoxy-phenyl)-ureido]-benzamide 4-[1-Methyl-3-(3-phenoxy-phenyl)-ureido]-Λ/-(3-morpholin-4-yl-propyl)-benzamide 4-[1-Methyl-3-(4-phenoxy-phenyl)-ureido]-Λ/-(3-morpholin-4-yl-propyl)-benzamide Λ/-(3-Morpholin-4-yl-propyl)-4-[3-(3-phenoxy-phenyl)-ureido]-benzamide 4-[1-Methyl-3-(3-phenoxy-phenyl)-ureido]-A/-(3-morpholin-4-yl-propyl)-benzamide 4-[3-(4-Benzyloxy-phenyl)-1-methyl-ureido]-2-A/-(3-morpholin-4-yl-propyl)-benzamide 4-[3-(4-Benzyloxy-phenyl)-ureido]-Λ/-(3-morpholin-4-yl-propyl)-benzamide
4-[3-(4-Benzyl-phenyl)-1-methyl-ureido]-Λ/-(3-morpholin-4-yl-propyl)-benzamide 4-[3-(4-Benzyl-phenyl)-ureido]-Λ/-(3-morpholin-4-yl-propyl)-benzamide Λ/-(3-Morpholin-4-yl-propyl)-4-(4-phenoxy-benzoylamino)-benzamide Λ/-(3-Morpholin-4-yl-propyl)-4-(3-phenoxy-benzoylamino)-benzamide 4-(4-Benzyloxy-benzoylamino)-Λ/-(3-morpholin-4-yl-propyl)-benzamide Λ/-(3-Morpholin-4-yl-propyl)-4-(4-pyrimidin-2-yl-benzoylamino)-benzamide 4-[4-(2,5-Dimethyl-pyrrol-1-yl)-benzoylamino]-Λ/-(3-morpholin-4-yl-propyl)-benzamide 4-(4-Benzoimidazol-1-ylmethyl-benzoylamino)-/V-(3-morpholin-4-yl-propyl)-benzamide Λ/-(1-Benzyl-piperidin-4-yl)-4-[1-methyl-3-(3-phenoxy-phenyl)-ureido]-benzamide /V-(1-Benzyl-piperidin-4-yl)-4-[3-(3-phenoxy-phenyl)-ureido]-benzamide 4-[3-(4-Benzyloxy-phenyl)-ureido]-A/-(1-benzyl-piperidin-4-yl)-benzamide 4-[3-(4-Benzyloxy-phenyl)-1-methyl-ureido]-/V-(1-benzyl-piperidin-4-yl)-benzamide 4-[3-(4-Benzyl-phenyl)-1-methyl-ureido]-Λ/-(1-benzyl-piperidin-4-yl)-benzamide 4-[3-(4-Benzyl-phenyl)-ureido]-Λ/-(1-benzyl-piperidin-4-yl)-benzamide 4-(4-Benzoimidazol-1 -ylmethyl-benzoylamino)-Λ/-(1 -benzyl-piperidin-4-yl)-benzamide Λ/-(1-Benzyl-piperidin-4-yl)-4-[4-(2,5-dimethyl-pyrrol-1-yl)-benzoylamino]-benzamide Λ/-(1-Benzyl-piperidin-4-yl)-4-(4-phenoxy-benzoylamino)-benzamide /V-[2-(4-Benzyl-piperazin-1-yl)-ethyl]-4-[3-(3-phenoxy-phenyl)-ureido]-benzamide Λ/-[2-(4-Benzyl-piperazin-1-yl)-ethyl]-4-[1-methyl-3-(3-phenoxy-phenyl)-ureido]-benzamide Λ/-[2-(4-Benzyl-piperazin-1-yl)-ethyl]-4-[1-methyl-3-(4-phenoxy-phenyl)-ureido]-benzamide 4-[3-(4-Benzyloxy-phenyl)-ureido]-A/-[2-(4-benzyl-piperazin-1-yl)-ethyl]-benzamide
4-[3-(4-Benzyloxy-phenyl)-1-methyl-ureido]-Λ/-[2-(4-benzyl-piperazin-1-yl)-ethyl]- benzamide
4-[3-(4-Benzyl-phenyl)-1-methyl-ureido]-Λ/-[2-(4-benzyl-piperazin-1-yl)-ethyl]-benzamide 4-[3-(4-Benzyl-phenyl)-ureido]-Λ/-[2-(4-benzyl-piperazin-1-yl)-ethyl]- benzamide N-[2-(4-Benzyl-piperazin-1-yl)-ethyl]-4-(3-phenoxy-benzoylamino)-benzamide
4-(4-Benzyloxy-benzoylamino)-Λ/-[2-(4-benzyl-piperazin-1-yl)-ethyl]-2-methoxy-benzamide Λ/-[2-(4-Benzyl-piperazin-1-yl)-ethyl]-4-(4-pyrimidin-2-yl-benzoylamino)-benzamide A/-[2-(4-Benzyl-piperazin-1-yl)-ethyl]-4-[4-(2,5-dimethyl-pyrrol-1-yl)-benzoylamino]- benzamide 4-(4-Benzoimidazol-1-ylmethyl-benzoylamino)-/V-[2-(4-benzyl-piperazin-1-yl)-ethyl]- benzamide