WO2003082871A1 - Imidazo fused compounds - Google Patents
Imidazo fused compounds Download PDFInfo
- Publication number
- WO2003082871A1 WO2003082871A1 PCT/EP2003/003178 EP0303178W WO03082871A1 WO 2003082871 A1 WO2003082871 A1 WO 2003082871A1 EP 0303178 W EP0303178 W EP 0303178W WO 03082871 A1 WO03082871 A1 WO 03082871A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- alkyl
- optionally
- substituted
- compound according
- Prior art date
Links
- 0 ***C1C2=C(*)CCCN2C2=NC(S*)=*CC=C2C*1 Chemical compound ***C1C2=C(*)CCCN2C2=NC(S*)=*CC=C2C*1 0.000 description 5
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/14—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains three hetero rings
- C07D487/14—Ortho-condensed systems
Definitions
- the present invention relates to certain heterocylic compounds as p38 protein kinase ("p38") inhibitors.
- the present invention relates to imidazo-substituted hetero-bicyclic compounds, a process for their manufacture, pharmaceutical preparations comprising the same, and methods for using the same.
- Mitogen-activated protein kinases is a family of proline-directed serine/threonine kinases that activate their substrates by dual phosphorylation.
- the kinases are activated by a variety of signals including nutritional and osmotic stress, UV light, growth factors, endotoxin and inflammatory cytokines.
- One group of MAP kinases is the p38 kinase group which includes various isoforms (e.g., p38 ⁇ , p38 ⁇ , p38 ⁇ and p38 ⁇ ).
- the p38 kinases are responsible for phosphorylating and activating transcription factors as well as other kinases, and are themselves activated by physical and chemical stress, pro-inflammatory cytokines and bacterial lipopolysaccharide.
- TNF- ⁇ is a cytokine produced primarily by activated monocytes and macrophages. Its excessive or unregulated production has been implicated as playing a causative role in the pathogenesis of rheumatoid arthritis. More recently, inhibition of TNF production has been shown to have broad application in the treatment of inflammation, inflammatory bowel disease, Alzheimer's disease, Crohn's disease, multiple sclerosis and asthma.
- TNF has also been implicated in viral infections, such as HIV, influenza virus, and herpes virus including herpes simplex virus type-1 (HSN-1), herpes simplex virus type-2 (HSV-2), cytomegalo virus (CMN), varicella-zoster virus (VZV), Epstein-Barr virus, human herpes virus-6 (HHV-6), human herpesvirus-7 (HHV-7), human herpesvirus-8 (HHN-8), pseudorabies and rhinotracheitis, among others.
- HSN-1 herpes simplex virus type-1
- HSV-2 herpes simplex virus type-2
- CPN cytomegalo virus
- VZV varicella-zoster virus
- Epstein-Barr virus Epstein-Barr virus
- HHV-6 human herpes virus-6
- HHV-7 human herpesvirus-7
- HHN-8 human herpesvirus-8
- pseudorabies and rhinotracheitis among others.
- IL-1 is produced by activated monocytes and macrophages, and plays a role in many pathophysiological responses including rheumatoid arthritis, fever and reduction of bone resorption.
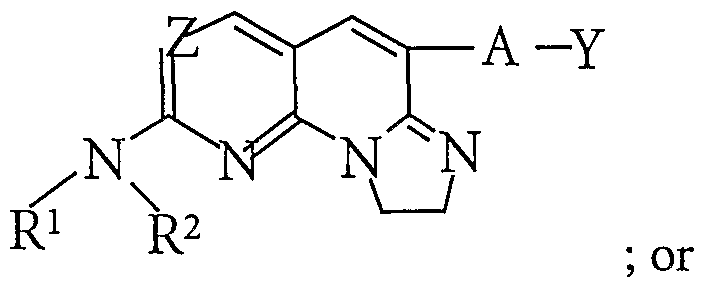
- the present invention provides compounds represented by the formula I:
- Z 1 is selected from ⁇ and CH when the bond between atoms C5 and Z 1 is a single bond, and Z 1 is C when the bond between C5 and Z 1 is a double bond;
- R 1 is hydrogen or alkyl;
- R- is alkyl, aralkyl, cycloalkyl, heteroalkyl, heterocyclyl, aryl, or heteroaryl;
- R' is hydrogen or alkyl
- R 3 is hydrogen, alkyl, aryl, heteroaryl, or cycloalkyl
- the bond between atoms C5 and Z 1 is a single or double bond
- the bond between atoms C8 and C9 is a single or double bond
- Y is an alkyl, heteroalkyl, cycloalkyl, aryl, or heteroaryl; or isomers, pharmaceutically acceptable salts, esters or prodrugs thereof.
- Y is phenyl optionally substituted with one to two groups selected from halo, hydroxy, amino, alkyl or heteroalkyl; or isomers, pharmaceutically acceptable salts, esters or prodrugs thereof; or
- R is selected from cyclopentyl, cyclohexyl, tetrahydropyranyl, piperidinyl, thianonyl, tetrahydro-l,l-dioxide-2-H- thiopyranyl, phenyl, and benzyl, wherein each of said R groups is optionally substituted with one of hydroxy, halogen, O(C ⁇ - 4 alkyl), or S(O) (C ⁇ - 4 alkyl); or R .2 is selected from C ⁇ - 4 alkyl optionally substituted with one of hydroxy, O(C ⁇ - 4 alkyl) or S(O) 2 (C 1 - 4 alkyl); or
- R is selected from cyclopentyl, cyclohexyl, tetrahydropyranyl, piperidinyl, thianonyl, tetrahydro-l,l-dioxide-2-H- thiopyranyl, phenyl, and benzyl, wherein each of said R groups is optionally- substituted with one of hydroxy, halogen, O(CH 3 ), or S(O) 2 (CH 3 ); or R 2 is selected from C ⁇ - 4 alkyl optionally substituted with one of hydroxy, O(CH 3 ) or S(O) 2 (CH 3 ); or
- R is selected from cyclopentyl, cyclohexyl, tetrahydropyranyl, piperidinyl, thianonyl, tetrahydro- l,l-dioxide-2-H-thiopyranyl, phenyl, and benzyl, wherein each of said R 2 groups is optionally-substituted with one of hydroxy, halogen, O(C ⁇ . 4 alkyl), or S(O) 2 (C ⁇ . - 4 alkyl); or R 2 is selected from C ⁇ - 4 alkyl optionally substituted with one of hydroxy, O(C ⁇ - 4 alkyl) or S(O) 2 (C ⁇ - 4 alkyl); or
- A is absent or O
- R 1 is hydrogen
- R is alkyl, hydroxyalkyl, optionally-substituted cycloalkyl, optionally-substituted heteocyclyl, optionally-substituted aryl, or optionally-substituted aralkyl; and the bond between C8 and C9 is a single or double bond; or
- R is 4-tetrahydropyranyl and Y is 4-fluorophenyl;
- R 2 is 4-fluorophenyl and Y is 4-fluorophenyl;
- R 2 is 4-hydroxycyclohexyl and Y is 2-chlorophenyl;
- R 2 is 4-tetrahydropyranyl and Y is 2-chlorophenyl;
- R 2 is 4-(N-methyl-sulfonylpiperdinyl) and Y is 2-chlorophenyl;
- R 2 is cyclopentyl and Y is 2-chlorophenyl
- R 2 is 4-(N-methyl-sulfonylpiperdinyl) and Y is 2-chlorophenyl;
- R 2 is 4-tetrahydro,l,l-dioxide-2-H-thiopyranyl and Y is 2-chlorophenyl; R 2 is isopropyl and Y is 2-chlorophenyl; or
- R 2 is 4-fluorbenzyl and Y is 2-chlorophenyl;
- the compounds of formula I and their aforementioned salts are inhibitors of protein kinases and exhibit effective activity against p38 in vivo. Therefore, the compounds can be used for the treatment of diseases mediated by the pro- inflammatory cytokines such as TNF and IL-1.
- the present invention relates to methods for the treatment of p38 mediated diseases or conditions in which a therapeutically effective amount of a compound of formula I is administered to a patient in need of such treatment.
- the present invention relates to methods for preparing the compounds described above.
- the present invention relates to methods for preparing medicaments useful for the treatment of the p38 mediated diseases and conditions.
- alkyl means a linear saturated monovalent hydrocarbon radical or a branched saturated monovalent hydrocarbon radical of one to six carbon atoms, e.g., methyl, ethyl, n-propyl, 2-propyl, tert-butyl, pentyl, and the like.
- aryl refers to a monovalent monocyclic or bicyclic aromatic hydrocarbon radical preferably phenyl which is optionally substituted independently with one or more substituents.
- the substituents are preferably selected from the group consisting of alkyl, haloalkyl, halo, hydroxy, nitro, cyano, amino, haloalkoxy, heteroalkyl, methylenedioxy, ethylenedioxy, Y-aryl, Y- heteroaryl, Y-cycloalkyl, Y-heterocyclyl, Y-OR p -Y-NR p R q , -Y-C(O)-R p , -YS(O) 0 -2R P .
- aryl includes, but is not limited to, phenyl, chlorophenyl, methoxyphenyl, 1-naphthyl, 2-naphthyl, and the derivatives thereof.
- Alkyl means a radical -R x R y where R x is an alkylene group and R is an aryl group as defined above, e.g. benzyl, phenylethylene, and the like.
- cycloalkyl refers to a saturated monovalent cyclic hydrocarbon radical of three to seven ring carbons, e.g., cyclopentyl, cyclobutyl, cyclohexyl, and the like.
- the saturated monovalent cyclic hydrocarbon radical as defined above may be optionally substituted with one, two or three substituents which are not hydrogen.
- the substituents are selected from the group consisting of alkyl, hydroxy, alkoxy, amino, monosubstituted amino, disubstituted amino, haloalkyl, halo, cyanoalkyl, oxo (i.e., carbonyl oxygen), heteroalkyl, heterocyclyl, hydroxyalkyl, and -(X) n -C(O)R' (where, X is O or NR", n is 0 or 1, R" is hydrogen, alkyl, haloalkyl, amino, monosubstituted amino, disubstituted amino, hydroxy, alkoxy, alkyl or optionally substituted phenyl, and R' is H or alkyl), and -S(O) n R' (wherein n is 0 to 2). More specifically, the term substituted cycloalkyl includes, for example, substituted cyclopentyl, substituted cyclohexyl, and the like.
- halo when referring to a substituent means fluoro, chloro, bromo, or iodo, preferably chloro.
- haloalkyl means alkyl substituted with one or more same or different halo atoms, e.g., -CH2CI, -CF3, -CH2CF3, -CH2CCI3, and the like, and further includes those alkyl groups such as perfluoroalkyl in which all alkyl hydrogen atoms are replaced by fluorine atoms.
- heteroalkyl as used herein means an alkyl radical defined above, wherein one, two or three hydrogen atoms have been replaced with a substituent independently selected from the group consisting of -OR a , -NR b R c , and S(O) n R d (where n is an integer from 0 to 2), with the understanding that the point of attachment of the heteroradical is through a carbon atom, wherein R a is hydrogen, acyl, alkyl, cycloalkyl, or cycloalkylalkyl; or R b and R c are independently of each other hydrogen, acyl, alkyl, cycloalkyl, or cycloalkylakyl, or R b and R c together with the nitrogen atom to which they are attached form heterocyclo or heteroaryl; and when n is 0, R d is hydrogen, alkyl, cycloalkyl, or cycloalkylakyl, and when n is 1 or 2, R
- Representative examples include, but are not limited to, 2- hydroxyethyl, 3-hydroxypropyl, 2-hydroxymethylethyl, 2,3-dihydroxypropyl, 1-hydroxy-methylethyl, 3-hydroxybutyl, 2,3-dihydroxybutyl, 2-hydroxy-l- methylpropyl, 2-aminoethyl, 3-aminopropyl, 2-methylsulfonylethyl, aminosulfonylmethyl, aminosulfonylethyl, methylaminosulfonylmethyl, methylaminosulfonylethyl, methylaminosulfonylpropyl, and the like.
- R a is hydrogen
- the radical -OR a is also referred to as "hydroxyalkyl" and includes, but is not limited to, 2-hydroxyethyl, 3-hydroxypropyl, 2-hydroxymethylethyl,
- Heteroaryl means a monvalent monocyclic or bicyclic radical of 5 to 12 ring atoms having at least one aromatic ring containing one, two, or three ring heteroatoms selected from N, O, or S, the remaining ring atoms being C, with the understanding that when the heteroaryl is a bicyclic system in which one of the rings is carbocyclic and/or non-aromatic, the attachment point of the heteroaryl radical will be on a heteroaryl ring.
- the heteroaryl ring is optionally substituted with one or more substituents, preferably one or two substituents, independently from each other selected from alkyl, haloalkyl, halo, nitro, cyano, amino, methylenedioxy, Y-aryl, Y-heteroaryl, Y-cycloalkyl, -Y-heterocyclyl, -Y-OR', -YNR'R", -Y-C(O)R ⁇ -Y-O-C(O)-R', -Y-S(O) 0 - 2 -R ⁇ -Y-N-SO 2 -R', -Y-SO 2 - NR'R", and -Y-N-C(O)-NR'R", where Y is absent or a C ⁇ -C 3 alkylene group and R' and R" are each independently from each other hydrogen, alkyl, haloalkyl, hydroxy, alkoxy, aryl, heteroaryl,
- heteroaryl includes, but is not limited to, pyridyl, furanyl, thienyl, thiazolyl, isothiazolyl, triazolyl, imidazolyl, isoxazolyl, pyrrolyl, pyrazolyl, pyrimidinyl, benzofuranyl, tetrahydrobenzofuranyl, isobenzofuranyl, benzo- thiazolyl, benzoiosthiazolyl, benzotriazolyl, indolyl, isoindolyl, benzoxazolyl, quinolyl, tetrahydroquinolnyl, isoquinolyl, benzimidazolyl, benzisoxazolyl or benzothienyl, imidazo[l,2-a]-pryidinyl, imidazo[2,l-b]thiazolyl, and derivatives thereof.
- “Monosubstituted amino” means a radical -NHR e where R e is alkyl, heteroalkyl, haloalkyl, cycloalkyl, cycloalkylalkyl, hydroxyalkyl, aryl, aralkyl, aralkenyl, heteroaryl, heteroaralkyl, heteroaralkenyl, heterocyclyl, or heterocyclylalkyl, e.g., methylamino, ethylamino, phenylamine, benzylamine, and the like.
- disubstituted amino refers to a radical -NR g R h wherein R s and R h are, independently of each other, alkyl, heteroalkyl, haloalkyl, cycloalkyl, cycloalkylalkyl, hydroxyalkyl, aryl, aralkyl, aralkenyl, heteroaryl, heteroaralkyl, heteroaralkenyl, heterocyclyl, or heterocyclylalkyl, or R g and R h together with the nitrogen atom to which they are attached form a heterocyclyl ring.
- Representative examples include, but are not limited to, dimethylamino, methylethylamino, di(l-methyl-ethyl)amino, piperazinyl, and the like.
- the heterocyclyl ring may be optionally substituted independently with one, two, or three substituents selected from alkyl, hydroxy, hydroxyalkyl, alkoxy, heteroalkyl, haloalkyl, and -(X) n -C(O)R (where, X is O or NR', n is 0 or 1, R is hydrogen, alkyl, haloalkyl, amino, monosubstituted amino, disubstituted amino, hydroxy, alkoxy, or optionally-substituted phenyl, and R' is H or alkyl), Y-aryl, Y-heteroaryl, Y-cycloalkyl, Y-heterocyclyl, Y-OR p , -Y- NR p R q , -Y-C(O)-R p , -YS(O)o_2R p .
- Y is absent or a C ⁇ -C 3 alkylene group
- R p and R q are each independently from each other selected from hydrogen, alkyl, haloalkyl, hydroxy, alkoxy, aryl, heteroaryl, cycloalkyl, and heterocyclyl.
- heterocyclyl includes, but is not limited to, tetrahydropyranyl, piperidinyl, piperazinyl, morpholinyl, and the derivatives thereof.
- acyl refers to the group -C(O)R r where R r is alkyl, haloalkyl, heteroalkyl, aryl, heteroaryl, aralkyl or heteroaralkyl.
- Alkoxy means a radical - OR where R is an alkyl, aryl, aralkyl, or heteroaralkyl, respectively, as defined above, e.g., methoxy, phenoxy, pyridin-2-ylmethyloxy, benzyloxy, and the like.
- R is an alkyl, aryl, aralkyl, or heteroaralkyl, respectively, as defined above, e.g., methoxy, phenoxy, pyridin-2-ylmethyloxy, benzyloxy, and the like.
- the two bonds appears as a dotted line, as in , it should be understood that unless otherwise specifically stated, this notation means the bond optionally may be a single bond or a double bond, with appropriate selections being made for adjacent atoms.
- the dotted line is a bond
- the dotted line is not a bond
- Leaving group has the meaning conventionally associated with it in synthetic organic chemistry, i.e., an atom or a group capable of being displaced by a nucleophile and includes halo (such as chloro, bromo, and iodo), alkanesulfonyloxy, arenesulfonyloxy, alkylcarbonyloxy (e.g., acetoxy), arylcarbonyloxy, mesyloxy, tosyloxy, trifluoromethanesulfonyloxy, aryloxy (e.g., 2,4-dinitrophenoxy), methoxy, N-O-dimethylhydroxylamino, and the like.
- halo such as chloro, bromo, and iodo
- alkanesulfonyloxy arenesulfonyloxy
- alkylcarbonyloxy e.g., acetoxy
- arylcarbonyloxy mesyloxy, tosyloxy
- “Pharmaceutically acceptable excipient” means an excipient that is useful in preparing a pharmaceutical composition that is generally safe, non-toxic and neither biologically nor otherwise undesirable, and includes excipients that are acceptable for veterinary use as well as human pharmaceutical use.
- a "pharmaceutically acceptable excipient” as used in the specification and claims includes both one and more than one such excipient.
- “Pharmaceutically acceptable salt” of a compound means a salt that is pharmaceutically acceptable and that possesses the desired pharmacological activity of the parent compound.
- Such salts include: (1) acid addition salts, formed with inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like; or formed with organic acids such as acetic acid, propionic acid, hexanoic acid, cyclopentanepropionic acid, glycolic acid, pyruvic acid, lactic acid, malonic acid, succinic acid, malic acid, maleic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, 3-(4-hydroxybenzoyl)benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, 1,2- ethane-disulfonic acid, 2-hydroxyethanesulfonic acid, benzenesulf
- prodrug and “prodrug” are used interchangeably herein and refer to any compound which releases an active parent drug according to Formula I in vivo when such prodrug is administered to a mammalian subject.
- Prodrugs of a compound of Formula I are prepared by modifying one or more functional group(s) present in the compound of Formula I in such a way that the modification(s) may be cleaved in vivo to release the parent compound.
- Prodrugs include compounds of Formula I wherein a hydroxy, amino, or sulfhydryl group in a compound of Formula I is bonded to any group that may be cleaved in vivo to regenerate the free hydroxyl, amino, or sulfhydryl group, respectively.
- prodrugs include, but are not limited to, esters (e.g., acetate, formate, and benzoate derivatives), carbamates (e.g., N,N-dimethylaminocarbonyl) of hydroxy functional groups in compounds of Formula I, and the like.
- esters e.g., acetate, formate, and benzoate derivatives
- carbamates e.g., N,N-dimethylaminocarbonyl
- Protecting group refers to a grouping of atoms that when attached to a reactive group in a molecule masks, reduces or prevents that reactivity. Examples of protecting groups can be found in T.W. Green and P.G. Futs, Protective Groups in Organic Chemistry, (Wiley, 2 nd ed. 1991) and Harrison and Harrison et al., Compendium of Synthetic Organic Methods, Nols. 1-8 (John Wiley and Sons, 1971-1996).
- Representative amino protecting groups include, formyl, acetyl, trifluoroacetyl, benzyl, benzyloxycarbonyl (CBZ), tert-butoxycarbonyl (Boc), trimethyl silyl (TMS), 2-trimethylsilyl-ethanesulfonyl (SES), trityl and substituted trityl groups, allyloxycarbonyl, 9-fluorenylmethyloxycarbonyl (FMOC), nitro- veratryloxycarbonyl (NVOC), and the like.
- hydroxy protecting groups include those where the hydroxy group is either acylated or alkylated such as benzyl, and trityl ethers as well as alkyl ethers, tetrahydropyranyl ethers, trialkylsilyl ethers and allyl ethers.
- Treating" or “treatment” of a disease includes: (1) preventing the disease, i.e., causing the clinical symptoms of the disease not to develop in a mammal that may be exposed to or predisposed to the disease but does not yet experience or display symptoms of the disease; (2) inhibiting the disease, i.e., arresting or reducing the development of the disease or its clinical symptoms; or (3) relieving the disease, i.e., causing regression of the disease or its clinical symptoms.
- a therapeutically effective amount means the amount of a compound that, when administered to a mammal for treating a disease, is sufficient to effect such treatment for the disease. The “therapeutically effective amount” will vary depending on the compound, the disease and its severity and the age, weight, etc., of the mammal to be treated.
- the present invention provides compounds represented by the formula:
- Preferred compounds of formula I includee those wherein A is absent or - O-, both Z and Z 1 are N, Y is an aryl, R 1 is hydrogen, and R 2 is as defined above for compounds of formula (I). More preferred compounds are compounds of formula I, wherein Z is N, Z 1 is C such that the bond between C5 and Z 1 is a double bond, A is absent or -O, Y is aryl (more preferably optionally-substituted phenyl), R is hydrogen, and R is as defined above for compounds of formula I. Still more preferred compounds are compounds of formula 1(a):
- Z is N, A or absent or -O-, Y is optionally-substituted phenyl, R 1 is hydrogen, and R 2 is as defined above.
- Most preferred compounds are those of formula 1(a) wherein Z is N, A is absent, Y is a phenyl substituted with a halo, hydroxy, amino, alkyl or heteroalkyl, R 1 is hydrogen, and R 2 is alkyl, heteroalkyl, optionally-substituted cycloalkyl, optionally-substituted heterocycyl or substituted phenyl or benzyl.
- Particularly preferred compounds are compounds of formula 1(a) wherein A is absent, Y is phenyl substituted with a halo, the dotted line is a double bond, R 1 is hydrogen, and R is alkyl (more preferably lower alkyl optionally substituted with hydroxy, methoxy or methylsulfonyl), heteroalkyl, optionally-substituted cycloalkyl (more preferably cyclopentyl or cyclohexyl optionally substituted with methoxy), optionally-substituted heterocycyl (more preferably optionally- substituted piperidinyl), or substituted phenyl or benzyl (more preferably phenyl or benzyl substituted with a halo).
- Table I lists some of the representative compounds of formula 1(a).
- a preferred compound (12) of formula 1(c) is wherein Z is N, A is absent, R is hydrogen, Y is 2-chlorophenyl, and R is 4-hydroxycyclohexyl. Mass spectra of MH+ 399.
- Representative compounds of formula 1(a) are compounds wherein the dotted line is a bond, Z is N, R 1 is hydrogen, and the values of R 2 , A, and Y are as set forth below:
- a preferred compound (13) of formula 1(a) is a compound wherein Z is N, the dotted line is not a bond, R 1 is hydrogen, R 2 is 4-hydroxycylcohexyl, and Y is 2-chlorophenyl. MH+ 396, M.P. 169.3°C to 175.8°C.
- Particularly preferred compounds of formula 1(a) are compounds (3) and (7) of Table 1, above.
- the compounds of the present invention can exist in unsolvated forms as well as solvated forms, including hydrated forms and are intended to be encompassed within the scope of the invention. Furthermore, as stated above, the present invention also includes all pharmaceutically acceptable salts of the compounds along with prodrug forms of the compounds and all stereoisomers whether in a pure chiral form or a racemic mixture of other forms of mixture.
- the compounds of formula I are capable of further forming pharmaceutically acceptable acid addition salts. All of these forms are also contemplated within the scope of the claimed invention.
- Pharmaceutically acceptable acid addition salts of the compounds of formula I include salts derived from inorganic acids such as hydrochloric, nitric, phosphoric, sulfuric, hydrobromic, hydriodic, phosphorus, and the like, as well as the salts derived from organic acids, such as aliphatic mono- and dicarboxylic acids, phenyl-substituted alkanoic acids, hydroxy alkanoic acids, alkanedioic acids, aromatic acids, aliphatic and aromatic sulfonic acids, etc.
- Such salts include sulfate, pyrosulfate, bisulfate, sulfite, bisulfite, nitrate, phosphate, monohydro- genphosphate, dihydrogenphosphate, metaphosphate, pyrophosphate, chloride, bromide, iodide, acetate, propionate, caprylate, isobutyrate, oxalate, malonate, succinate, suberate, sebacate, fumarate, maleate, mandelate, benzoate, chlorobenzoate, methylbenzoate, dinitrobenzoate, pthalate, benzenesulfonate, toluenesulfonate, phenylacetate, citrate, lactate, maleate, tartarate, methane- sulfonate, and the like.
- salts of amino acids such as arginate and the like and gluconate, galacturonate (see, for example, Berge et al., "Pharmaceutical Salts," J. of Pharmaceutical Science, 1977, 66, 1-19).
- the acid addition salts of the basic compounds can be prepared by contacting the free base form with a sufficient amount of the desired acid to produce the salt in the conventional manner.
- the free base form can be regenerated by contacting the salt form with a base and isolating the free base in the conventional manner.
- the free base forms may differ from their respective salt forms somewhat in certain physical properties such as solubility in polar solvents, but otherwise the salts are equivalent to their respective free base for the purposes of the present invention.
- Pharmaceutically acceptable base addition salts can be formed with metal ions or amines, such as alkali and alkaline earth metal ions or organic amines.
- metal ions which are used as cations include sodium, potassium, magnesium, calcium, and the like.
- suitable amines are N, N'- dibenzylethylenediamine, chlororocaine, choline, diethanolamine, ethylenediamine, N-methylglucamine, and procaine (see, for example, Berge et al., supra).
- the basic addition salts of acidic compounds can be prepared by contacting the free acid form with a sufficient amount of the desired base to produce the salt in the conventional manner.
- the free acid form can be regenerated by contacting the salt form with an acid and isolating the free acid in the conventional manner.
- the free acid forms may differ from their respective salt forms somewhat in certain physical properties such as solubility in polar solvents, but otherwise the salts are equivalent to their respective free acid for the purposes of the present invention.
- the compounds of the present invention can be prepared by a variety of methods, using procedures well-known to those of skill in the art.
- NMP l-methyl-2-pyrrolidinone.
- THF tetrahydrofuran.
- LAH lithium aluminum hydride
- DMF dimethylformamide DMPU: l,3-dimethyl-3,4,5,6-tetrahydro-2(lH)-pyrimidinone
- Scheme 1 describes a general method of synthesizing compounds of formula 1(a) (1) (compounds of formula 1(a) wherein the dotted line is not a bond) starting with a protected thiopyridine-5-carboxylate 1, wherein R p and R pa are protecting groups such as lower alkyl and benzyl; and R L is a leaving group such as a halo.
- the protecting and leaving groups are as defined on page 6, lines 30 through page 7, line 9 and page 5, lines 22-26, respectively.
- the general method of preparing compound 5 from carboxlyate 1 in the Scheme is described in WO 0129042 and WO 0129041.
- Compound 1 is treated with triethylamine and aqueous ammonium to afford compound 2.
- Compound 2 is reduced with lithium aluminum hydride or other reducing agents well known in the art to yield compound 3.
- Compound 3 is oxidized to aldehyde 4 by treating it with activated manganese oxide powder or by other oxidizing methods known in the art.
- Compound 4_ is reacted with an ester of general formula -A-Y-C(O)OR z , wherein A and Y are as defined above and R z is lower alkyl, aryl or cycloalkyl, to afford pyrimidinone 5.
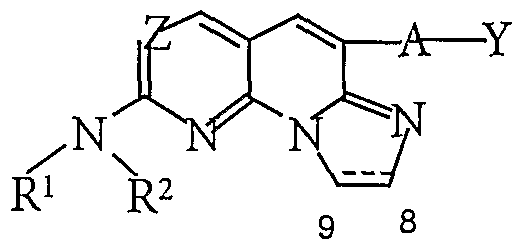
- Tricyclic sulfide 8 may be directly reacted with an amine of the general formula -NH R R to afford a compound of general formula 1(a)(1).
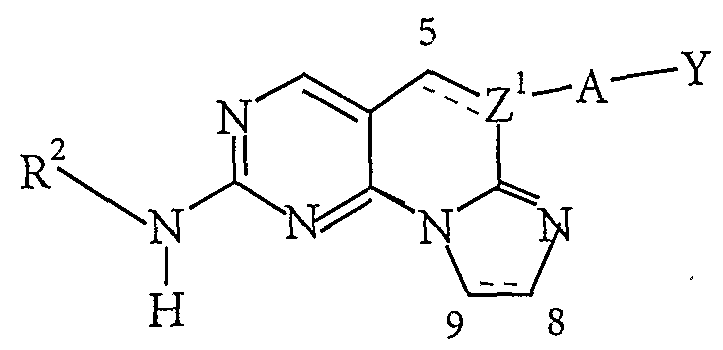
- Scheme 2 describes a general method of preparing a compound of formula 1(a)(2) (compounds of formula 1(a) wherein the dotted line is a bond), by starting with the aldehyde 4 from Scheme 1 above and heating it with a cyanide of general formula CN-CH 2 -A-Y, wherein A and Y are as defined above, and potassium carbonate in DPMU to obtain aminopyridopyrimidine 9.
- Compound 9 is treated with triethylamine and 1,2-dichloroethyl ethyl ether to afford sulfide of formula 10.
- Scheme 3 describes a general method of preparing compound of formula 1(c) starting with compound 12.
- Compound 12 is prepared according to the method described in WO 0129042 and WO 0129041.
- Compound Y2 is treated with triphenylphosphine, diethylazodicarboxylate and N- (2-hydroxyethyl)-phthalimide to obtain compound 13.
- Compound 13 is then reacted with hydrazine hydrate to obtain compound 14, which is then reacted with trimethylaluminum solution to obtain compound 15.
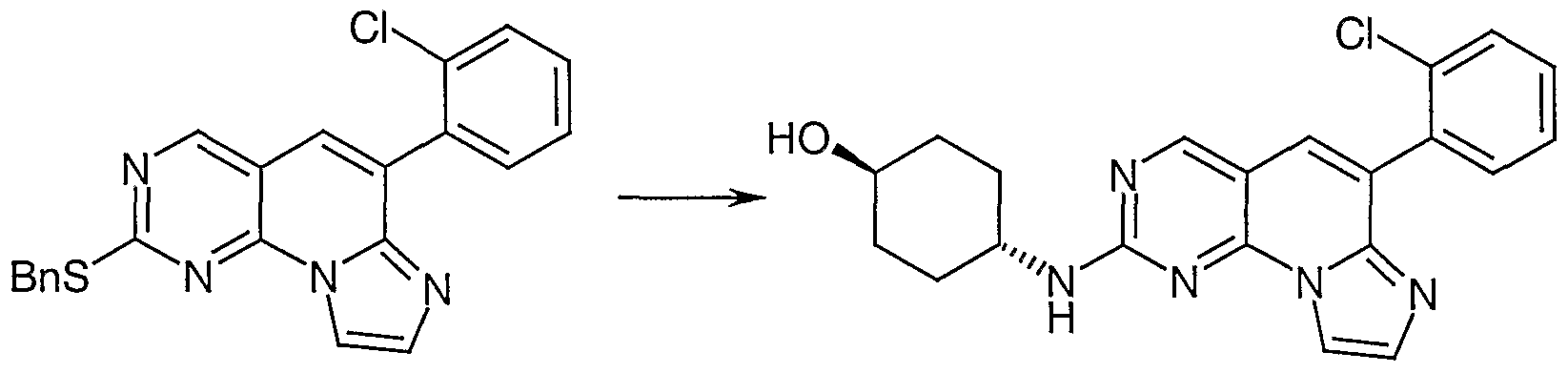
- Compound 15 is reacted with an amine of formula -H 2 NR R to obtain a compound of formula 1(c).
- compound 15 can be oxidized with 3-chloroperbenzoic acid to obtain the corresponding sulfoxide (not shown) which is then reacted with amine -NR ! R 2 to obtain the desired compound of formula I (c).
- a process of making a compound according to Claim 1, comprising: reacting a compound of formula A with a cyanide of formula CN-CH 2 -A-Y wherein A, Z and Y are as defined in Claim 1, and R p is selected from lower alkyl and benzyl, to afford a compound of formula B;
- a process of preparing a compound according to Claim 1 comprising: a) reacting a compound of formula E wherein Z, A and Y are as defined in Claim 1 and R p is lower alkyl or benzyl, with a reagent selected from triphenylphosphine, diethylazodicarboxylate, and N-(2-hydroxyethyl)-phthalimide to obtain compound F;
- a reagent selected from 3- chloroperbenzoic acid, hydrogen peroxide/formic acid, hydrogen peroxide/ methyltrioxorhenium VII, or OXONE
- the compounds of formula I and the pharmaceutically acceptable salts of basic compounds of formula I with acids can be used as medicaments, e.g. in the form of pharmaceutical preparations.
- the pharmaceutical preparations can be administered enterally, e.g. orally in the form of tablets, coated tablets, dragees, hard and soft gelatine capsules, solutions, emulsions or suspensions, nasally, e.g. in the form of nasal sprays, or rectally, e.g. in the form of suppositories. However, they may also be administered parenterally, e.g. in the form of injection solutions.
- the compounds of formula I and their aforementioned pharmaceutically acceptable salts can be processed with pharmaceutically inert, organic or inorganic carriers for the production of pharmaceutical preparations.
- Lactose, com starch or derivatives thereof, talc, stearic acid or its salts and the like can be used, for example, as such carriers for tablets, coated tablets, dragees and hard gelatine capsules.
- Suitable carriers for soft gelatine capsules are, for example, vegetable oils, waxes, fats, semi-solid and liquid polyols and the like; depending on the nature of the active ingredient no carriers are, however, usually required in the case of soft gelatine capsules.
- Suitable carriers for the production of solutions and syrups are, for example, water, polyols, sucrose, invert sugar, glucose and the like.
- Suitable carriers for suppositories are, for example, natural or hardened oils, waxes, fats, semi-liquid or liquid polyols and the like.
- the pharmaceutical preparations can also contain preservatives, solubilizers, stabilizers, wetting agents, emulsifiers, sweeteners, colorants, flavorants, salts for varying the osmotic pressure, buffers, masking agents or antioxidants. They can also contain therapeutically valuable substances other than the compounds of formula I and their aforementioned pharmaceutically acceptable salts.
- Medicaments which contain a compound of formula I or a pharmaceutically acceptable salt of a basic compound of formula I with an acid in association with a compatible pharmaceutical carrier material are also an object of the present invention, as is a process for the production of such medicaments which comprises bringing one or more of these compounds or salts and, if desired, one or more other therapeutically valuable substances into a galenical administration form together with a compatible pharmaceutical carrier.
- the compounds of formula I and their aforementioned pharmaceutically acceptable salts can be used in accordance with the invention as therapeutically active substances, especially as antiinflammatory agents or for the prevention of graft rejection following transplant surgery.
- the dosage can vary within wide limits and will, of course, be fitted to the individual requirements in each particular case. In general, in the case of administration to adults a convenient daily dosage should be about 0.1 mg/kg to about 100 mg/kg, preferably about 0.5 mg/kg to about 5 mg/kg.
- the daily dosage may be administered as a single dose or in divided doses and, in addition, the upper dosage limit referred to earlier may be exceeded when this is found to be indicated.
- the present invention provides a method of treating a cytokine-mediated disease which comprises administering an effective cytokine- interfering amount of a compound of Formula I, or a pharmaceutically acceptable salt or tautomer thereof.
- Compounds of Formula I would be useful for, but not limited to, the treatment of inflammation in a subject, and for use as antipyretics for the treatment of fever.
- Compounds of the invention would be useful to treat arthritis, including but not limited to, rheumatoid arthritis, spondyloarthropathies, gouty arthritis, osteoarthritis, systemic lupus erythematosus and juvenile arthritis, and other arthritic conditions.
- Such compounds would be useful for the treatment of pulmonary disorders or lung inflammation, including adult respiratory distress syndrome, pulmonary sarcoidosis, asthma, silicosis, and chronic pulmonary inflammatory disease.
- the compounds are also useful for the treatment of viral and bacterial infections, including sepsis, septic shock, gram negative sepsis, malaria, meningitis, cachexia secondary to infection or malignancy, cachexia secondary to acquired immune deficiency syndrome (AIDS), AIDS, ARC (AIDS related complex), pneumonia, and herpes virus.
- the compounds are also useful for the treatment of bone resorption diseases, such as osteoporosis, endotoxic shock, toxic shock syndrome, reperfusion injury, autoimmune disease including graft vs.
- cardiovascular diseases including atherosclerosis, thrombosis, congestive heart failure, and cardiac reperfusion injury, renal reperfusion injury, liver disease and nephritis, and myalgias due to infection.
- the compounds are also useful for the treatment of influenza, multiple sclerosis, cancer, diabetes, systemic lupus erthrematosis (SLE), skin-related conditions such as psoriasis, eczema, burns, dermatitis, keloid formation, and scar tissue formation.
- Compounds of the invention also would be useful to treat gastrointestinal conditions such as inflammatory bowel disease, Crohn's disease, gastritis, irritable bowel syndrome and ulcerative colitis.
- the compounds would also be useful in the treatment of ophthalmic diseases, such as retinitis, retinopathies, uveitis, ocular photophobia, and of acute injury to the eye tissue.
- Compounds of the invention also would be useful for treatment of angiogenesis, including neoplasia; metastasis; ophthalmological conditions such as corneal graft rejection, ocular neovascularization, retinal neovascularization including neovascularization following injury or infection, diabetic retinopathy, retrolental fibroplasia and neovascular glaucoma; ulcerative diseases such as gastric ulcer; pathological, but non-malignant, conditions such as hemangiomas, including infantile hemangiomas, angiofibroma of the nasopharynx and avascular necrosis of bone; diabetic nephropathy and cardiomyopathy; and disorders of the female reproductive system such as endometriosis.
- the compounds of the invention may also
- these compounds are also useful for veterinary treatment of companion animals, exotic animals and farm animals, including mammals, rodents, and the like. More preferred animals include horses, dogs, and cats.
- the present compounds may also be used in co-therapies, partially or completely, in place of other conventional antiinflammatories, such as together with steroids, cyclooxygenase-2 inhibitors, NSAIDs, DMARDS, immunosuppressive agents, 5-lipoxygenase inhibitors, LTB 4 antagonists and LTA 4 hydrolase inhibitors.
- TNF mediated disorder refers to any and all disorders and disease states in which TNF plays a role, either by control of TNF itself, or by TNF causing another monokine to be released, such as but not limited to IL-1, IL-6 or IL-8.
- TNF a disease state in which, for instance, IL-1 is a major component, and whose production or action, is exacerbated or secreted in response to TNF, would therefore be considered a disorder mediated by TNF.
- p38 mediated disorder refers to any and all disorders and disease states in which p38 plays a role, either by control of p38 itself, or by p38 causing another factor to be released, such as but not limited to IL- 1, IL-6 or IL-8.
- IL-1 is a major component, and whose production or action, is exacerbated or secreted in response to p38, would therefore be considered a disorder mediated by p38.
- TNF- ⁇ has close structural homology with TNF- ⁇ (also known as cachectin), and since each induces similar biologic responses and binds to the same cellular receptor, the synthesis of both TNF- ⁇ and TNF- ⁇ are inhibited by the compounds of the present invention and thus are herein referred to collectively as "TNF” unless specifically delineated otherwise.
- a glass sealed-tube (teflon screw cap) reactor was charged with 0.4 g (1.2 mmol) of the sulfoxide 10. This was purged with nitrogen for 2 minutes as the reaction vessel was cooled with an ice-water bath. 5 ml of isopropyl amine was added and the tube was sealed. The red solution was heated in a 64°C oil bath for 2 hours until reaction was completed. The mixture was cooled and the reaction vessel was rinsed with dichloromethane. The solvent was removed in vacuo and the sample was purified by column chromatography with silica eluting with a gradient of 5 to 10% ethyl acetate in dichloromethane affording 0.34 g (87% yield) of product.
- Example 13 The following are representative pharmaceutical formulations containing a compound of Formula (I).
- Tablet formulation The following ingredients are mixed intimately and pressed into single scored tablets.
- Capsule formulation The following ingredients are mixed intimately and loaded into a hard-shell gelatin capsule.
- Quantity per Ingredient capsule mg compound of this invention 200 lactose, spray-dried 148 magnesium stearate 2 Suspension formulation The following ingredients are mixed to form a suspension for oral administration.
- Ingredient Amount compound of this invention 1.0 g fumaric acid 0.5 g sodium chloride 2.0 g methyl paraben 0.15 g propyl paraben 0.05 g granulated sugar 25.5 g Sorbitol (70% solution) 12.85 g
- Veegum K (Vanderbilt Co.) 1.0 g flavoring 0.035 ml colorings 0.5 mg distilled water q.s. to 100 ml
- Injectable formulation The following ingredients are mixed to form an injectable formulation.
- Suppository formulation A suppository of total weight 2.5 g is prepared by mixing the compound of the invention with Witepsol® H-15 (triglycerides of saturated vegetable fatty acid; Riches-Nelson, Inc., New York), and has the following composition:
- the p-38 MAP kinase inhibitory activity of compounds of this invention in vitro was determined by measuring the transfer of the ⁇ -phosphate from ⁇ - P- ATP by p-38 kinase to Myelin Basic Protein (MBP), using the a minor modification of the method described in Ahn, N. G.; et al. J. of Biol. Chem. Vol. 266(7), 4220- 4227, (1991)
- the phosphorylated form of the recombinant p38 MAP kinase was expressed with SEK-1 and MEK-K in E. Coli (see, Khokhlatchev, A. et al. J. of Biol. Chem. Vol. 272(17), 11057-11062, (1997) and then purified by affinity chromatography using a Nickel column.
- the phosphorylated p38 MAP kinase was diluted in kinase buffer (20 mM
- the reaction was terminated by adding 0.75% phosphoric acid.
- the phosphorylated MBP was then separated from the residual ⁇ - 33 P-ATP using a phosphocellulose membrane (Millipore, Bedfrod, MA) and quantitated using a scintillation counter (Packard, Meriden, CT).
- IC 50 the concentration causing 50% inhibition of the p-38 enzyme being analyzed
- Table 2 shows representative values for inhibition of p38 using the Ex. 14 assay:
Abstract
Description


























































Claims










Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002479644A CA2479644A1 (en) | 2002-04-03 | 2003-03-27 | Imidazo fused compounds |
| MXPA04009585A MXPA04009585A (en) | 2002-04-03 | 2003-03-27 | Imidazo fused compounds. |
| AU2003215675A AU2003215675B2 (en) | 2002-04-03 | 2003-03-27 | Imidazo fused compounds |
| EP03745276A EP1492790A1 (en) | 2002-04-03 | 2003-03-27 | Imidazo fused compounds |
| JP2003580336A JP2006503802A (en) | 2002-04-03 | 2003-03-27 | Imidazo condensation compounds |
| KR1020047015609A KR100656205B1 (en) | 2002-04-03 | 2003-03-27 | Imidazo fused compounds |
| BR0308937-1A BR0308937A (en) | 2002-04-03 | 2003-03-27 | Fused imidazo compounds |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US36992902P | 2002-04-03 | 2002-04-03 | |
| US60/369,929 | 2002-04-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2003082871A1 true WO2003082871A1 (en) | 2003-10-09 |
Family
ID=28675587
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2003/003178 WO2003082871A1 (en) | 2002-04-03 | 2003-03-27 | Imidazo fused compounds |
Country Status (13)
| Country | Link |
|---|---|
| US (4) | US6949560B2 (en) |
| EP (1) | EP1492790A1 (en) |
| JP (1) | JP2006503802A (en) |
| KR (1) | KR100656205B1 (en) |
| CN (1) | CN1293078C (en) |
| AR (1) | AR039219A1 (en) |
| AU (1) | AU2003215675B2 (en) |
| BR (1) | BR0308937A (en) |
| CA (1) | CA2479644A1 (en) |
| MX (1) | MXPA04009585A (en) |
| PL (1) | PL374544A1 (en) |
| RU (1) | RU2310657C2 (en) |
| WO (1) | WO2003082871A1 (en) |
Cited By (39)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005021551A1 (en) * | 2003-08-27 | 2005-03-10 | Amgen Inc. | Substituted heterocyclic compounds and methods of use |
| JP2008506673A (en) * | 2004-07-15 | 2008-03-06 | サノフイ−アベンテイス | Pyrido-pyrimidine derivatives, their preparation, their therapeutic use for cancer treatment |
| US9266892B2 (en) | 2012-12-19 | 2016-02-23 | Incyte Holdings Corporation | Fused pyrazoles as FGFR inhibitors |
| US9388185B2 (en) | 2012-08-10 | 2016-07-12 | Incyte Holdings Corporation | Substituted pyrrolo[2,3-b]pyrazines as FGFR inhibitors |
| US9533984B2 (en) | 2013-04-19 | 2017-01-03 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US9533954B2 (en) | 2010-12-22 | 2017-01-03 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of FGFR3 |
| US9580423B2 (en) | 2015-02-20 | 2017-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9611267B2 (en) | 2012-06-13 | 2017-04-04 | Incyte Holdings Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US9708318B2 (en) | 2015-02-20 | 2017-07-18 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9890156B2 (en) | 2015-02-20 | 2018-02-13 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| WO2018153373A1 (en) * | 2017-02-27 | 2018-08-30 | 贝达药业股份有限公司 | Fgfr inhibitor and application thereof |
| CN109721600A (en) * | 2017-10-30 | 2019-05-07 | 如东凌达生物医药科技有限公司 | A kind of nitrogenous fused ring compound and its preparation method and application |
| US10342786B2 (en) | 2017-10-05 | 2019-07-09 | Fulcrum Therapeutics, Inc. | P38 kinase inhibitors reduce DUX4 and downstream gene expression for the treatment of FSHD |
| EP3476846A4 (en) * | 2016-06-28 | 2019-11-20 | Hanmi Pharmaceutical Co., Ltd. | Novel heterocyclic derivative compound and use thereof |
| US10611762B2 (en) | 2017-05-26 | 2020-04-07 | Incyte Corporation | Crystalline forms of a FGFR inhibitor and processes for preparing the same |
| US10851105B2 (en) | 2014-10-22 | 2020-12-01 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US11066404B2 (en) | 2018-10-11 | 2021-07-20 | Incyte Corporation | Dihydropyrido[2,3-d]pyrimidinone compounds as CDK2 inhibitors |
| US11091485B2 (en) | 2017-05-23 | 2021-08-17 | Shengke Pharmaceuticals (Jiangsu) Ltd. | Imidazo[1′,2′:1,6]pyrido[2,3-d]pyrimidine compound as protein kinase inhibitor |
| US11174257B2 (en) | 2018-05-04 | 2021-11-16 | Incyte Corporation | Salts of an FGFR inhibitor |
| WO2022032071A1 (en) * | 2020-08-07 | 2022-02-10 | Abm Therapeutics Corporation | Kinase inhibitors and uses thereof |
| US11291659B2 (en) | 2017-10-05 | 2022-04-05 | Fulcrum Therapeutics, Inc. | P38 kinase inhibitors reduce DUX4 and downstream gene expression for the treatment of FSHD |
| EP3845534A4 (en) * | 2018-08-27 | 2022-04-06 | Betta Pharmaceuticals Co., Ltd | Salt form and crystal form of novel azatricyclic compound and use thereof |
| US11384083B2 (en) | 2019-02-15 | 2022-07-12 | Incyte Corporation | Substituted spiro[cyclopropane-1,5′-pyrrolo[2,3-d]pyrimidin]-6′(7′h)-ones as CDK2 inhibitors |
| US11407750B2 (en) | 2019-12-04 | 2022-08-09 | Incyte Corporation | Derivatives of an FGFR inhibitor |
| US11427567B2 (en) | 2019-08-14 | 2022-08-30 | Incyte Corporation | Imidazolyl pyrimidinylamine compounds as CDK2 inhibitors |
| US11440914B2 (en) | 2019-05-01 | 2022-09-13 | Incyte Corporation | Tricyclic amine compounds as CDK2 inhibitors |
| US11447494B2 (en) | 2019-05-01 | 2022-09-20 | Incyte Corporation | Tricyclic amine compounds as CDK2 inhibitors |
| US11466004B2 (en) | 2018-05-04 | 2022-10-11 | Incyte Corporation | Solid forms of an FGFR inhibitor and processes for preparing the same |
| US11472791B2 (en) | 2019-03-05 | 2022-10-18 | Incyte Corporation | Pyrazolyl pyrimidinylamine compounds as CDK2 inhibitors |
| US11566028B2 (en) | 2019-10-16 | 2023-01-31 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11591329B2 (en) | 2019-07-09 | 2023-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11607416B2 (en) | 2019-10-14 | 2023-03-21 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11628162B2 (en) | 2019-03-08 | 2023-04-18 | Incyte Corporation | Methods of treating cancer with an FGFR inhibitor |
| US11851426B2 (en) | 2019-10-11 | 2023-12-26 | Incyte Corporation | Bicyclic amines as CDK2 inhibitors |
| US11897891B2 (en) | 2019-12-04 | 2024-02-13 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
| US11919904B2 (en) | 2019-03-29 | 2024-03-05 | Incyte Corporation | Sulfonylamide compounds as CDK2 inhibitors |
| US11939331B2 (en) | 2021-06-09 | 2024-03-26 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
| US11976073B2 (en) | 2021-12-10 | 2024-05-07 | Incyte Corporation | Bicyclic amines as CDK2 inhibitors |
| US11981671B2 (en) | 2022-06-21 | 2024-05-14 | Incyte Corporation | Bicyclic pyrazolyl amines as CDK2 inhibitors |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008114119A2 (en) * | 2007-03-20 | 2008-09-25 | Cadila Pharmaceuticals Limited | P38 inhibitors |
| EP2768506A4 (en) * | 2011-10-21 | 2015-08-19 | Glaxosmithkline Llc | Compounds and methods for enhancing innate immune responses |
| CN105481858B (en) * | 2014-10-11 | 2019-05-17 | 上海医药集团股份有限公司 | A kind of nitrogenous fused heterocyclic compound, preparation method, composition and application |
| CA3003433A1 (en) | 2015-10-27 | 2017-05-04 | Children's Hospital Medical Center | Use of mapk inhibitors to reduce loss of hematopoietic stem cells during ex vivo culture and/or genetic manipulation |
| US20190060286A1 (en) | 2016-02-29 | 2019-02-28 | University Of Florida Research Foundation, Incorpo | Chemotherapeutic Methods |
| WO2018004258A1 (en) * | 2016-06-28 | 2018-01-04 | 한미약품 주식회사 | Novel heterocyclic derivative compound and use thereof |
| ES2968252T3 (en) * | 2016-11-16 | 2024-05-08 | Impact Therapeutics Shanghai Inc | Compound 8,9-dihydroimidazole[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-ketone |
| JP7041821B2 (en) * | 2017-10-30 | 2022-03-25 | シャンハイ リンジーン バイオファーマ カンパニー リミテッド | Amino-substituted nitrogen-containing condensed ring compound, its preparation method and use |
| WO2020038458A1 (en) * | 2018-08-23 | 2020-02-27 | 如东凌达生物医药科技有限公司 | Class of fused ring triazole compound, preparation method, and use |
| CN113646314B (en) * | 2019-03-08 | 2023-12-08 | 首药控股(北京)股份有限公司 | FGFR4 kinase inhibitor and preparation method and application thereof |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001029042A1 (en) * | 1999-10-21 | 2001-04-26 | F. Hoffmann-La Roche Ag | Heteroalkylamino-substituted bicyclic nitrogen heterocycles as inhibitors of p38 protein kinase |
| WO2002018380A1 (en) * | 2000-08-31 | 2002-03-07 | F. Hoffmann-La Roche Ag | 7-oxo pyridopyrimidines as inhibitors of a cellular proliferation |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4522947A (en) * | 1982-03-29 | 1985-06-11 | Usv Pharmaceutical Corporation | Certain 1,3-oxazolo[4,5H]quinolines useful as anti-allergy agents |
| US4656281A (en) * | 1983-03-18 | 1987-04-07 | Usv Pharmaceutical Corp. | Imidazo quinoline compounds useful as anti-allergy agents |
| DE3502689A1 (en) * | 1985-01-26 | 1986-07-31 | Hoechst Ag, 6230 Frankfurt | ELECTROPHOTOGRAPHIC RECORDING MATERIAL |
| US4831040A (en) * | 1988-02-01 | 1989-05-16 | Rorer Pharmaceutical Corporation | Method of prevention and treatment of peptic ulcers |
| GB9123916D0 (en) | 1991-11-11 | 1992-01-02 | Wellcome Found | Heterocyclic compounds |
| JPH11292878A (en) | 1998-04-09 | 1999-10-26 | Yamanouchi Pharmaceut Co Ltd | Imidazonaphthylidine derivative |
| HU229604B1 (en) | 2001-02-12 | 2014-02-28 | Hoffmann La Roche | 6-substituted pyrido-pyrimidines, process for their preparation and pharmaceutical compositions containing them |
-
2003
- 2003-03-27 EP EP03745276A patent/EP1492790A1/en not_active Withdrawn
- 2003-03-27 AU AU2003215675A patent/AU2003215675B2/en not_active Ceased
- 2003-03-27 CN CNB038075369A patent/CN1293078C/en not_active Expired - Fee Related
- 2003-03-27 JP JP2003580336A patent/JP2006503802A/en active Pending
- 2003-03-27 KR KR1020047015609A patent/KR100656205B1/en not_active IP Right Cessation
- 2003-03-27 PL PL03374544A patent/PL374544A1/en not_active Application Discontinuation
- 2003-03-27 CA CA002479644A patent/CA2479644A1/en not_active Abandoned
- 2003-03-27 WO PCT/EP2003/003178 patent/WO2003082871A1/en active IP Right Grant
- 2003-03-27 BR BR0308937-1A patent/BR0308937A/en not_active IP Right Cessation
- 2003-03-27 MX MXPA04009585A patent/MXPA04009585A/en active IP Right Grant
- 2003-03-27 RU RU2004132204/04A patent/RU2310657C2/en active
- 2003-04-02 AR ARP030101141A patent/AR039219A1/en unknown
- 2003-04-03 US US10/406,364 patent/US6949560B2/en not_active Expired - Fee Related
-
2005
- 2005-05-04 US US11/122,137 patent/US7081462B2/en not_active Expired - Fee Related
-
2006
- 2006-07-13 US US11/486,193 patent/US7285559B2/en not_active Expired - Fee Related
- 2006-07-13 US US11/486,348 patent/US7285561B2/en not_active Expired - Fee Related
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001029042A1 (en) * | 1999-10-21 | 2001-04-26 | F. Hoffmann-La Roche Ag | Heteroalkylamino-substituted bicyclic nitrogen heterocycles as inhibitors of p38 protein kinase |
| WO2002018380A1 (en) * | 2000-08-31 | 2002-03-07 | F. Hoffmann-La Roche Ag | 7-oxo pyridopyrimidines as inhibitors of a cellular proliferation |
Cited By (68)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005021551A1 (en) * | 2003-08-27 | 2005-03-10 | Amgen Inc. | Substituted heterocyclic compounds and methods of use |
| JP2008506673A (en) * | 2004-07-15 | 2008-03-06 | サノフイ−アベンテイス | Pyrido-pyrimidine derivatives, their preparation, their therapeutic use for cancer treatment |
| US10213427B2 (en) | 2010-12-22 | 2019-02-26 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of FGFR3 |
| US9533954B2 (en) | 2010-12-22 | 2017-01-03 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of FGFR3 |
| US10813930B2 (en) | 2010-12-22 | 2020-10-27 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of FGFR3 |
| US10131667B2 (en) | 2012-06-13 | 2018-11-20 | Incyte Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US11840534B2 (en) | 2012-06-13 | 2023-12-12 | Incyte Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US9611267B2 (en) | 2012-06-13 | 2017-04-04 | Incyte Holdings Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US11053246B2 (en) | 2012-06-13 | 2021-07-06 | Incyte Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US9388185B2 (en) | 2012-08-10 | 2016-07-12 | Incyte Holdings Corporation | Substituted pyrrolo[2,3-b]pyrazines as FGFR inhibitors |
| US9745311B2 (en) | 2012-08-10 | 2017-08-29 | Incyte Corporation | Substituted pyrrolo[2,3-b]pyrazines as FGFR inhibitors |
| US9266892B2 (en) | 2012-12-19 | 2016-02-23 | Incyte Holdings Corporation | Fused pyrazoles as FGFR inhibitors |
| US11530214B2 (en) | 2013-04-19 | 2022-12-20 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US10040790B2 (en) | 2013-04-19 | 2018-08-07 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US10947230B2 (en) | 2013-04-19 | 2021-03-16 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US10450313B2 (en) | 2013-04-19 | 2019-10-22 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US9533984B2 (en) | 2013-04-19 | 2017-01-03 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US10851105B2 (en) | 2014-10-22 | 2020-12-01 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US11014923B2 (en) | 2015-02-20 | 2021-05-25 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10016438B2 (en) | 2015-02-20 | 2018-07-10 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9708318B2 (en) | 2015-02-20 | 2017-07-18 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10251892B2 (en) | 2015-02-20 | 2019-04-09 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US11667635B2 (en) | 2015-02-20 | 2023-06-06 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US11173162B2 (en) | 2015-02-20 | 2021-11-16 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9580423B2 (en) | 2015-02-20 | 2017-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10632126B2 (en) | 2015-02-20 | 2020-04-28 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9801889B2 (en) | 2015-02-20 | 2017-10-31 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10738048B2 (en) | 2015-02-20 | 2020-08-11 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10214528B2 (en) | 2015-02-20 | 2019-02-26 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9890156B2 (en) | 2015-02-20 | 2018-02-13 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| EP3476846A4 (en) * | 2016-06-28 | 2019-11-20 | Hanmi Pharmaceutical Co., Ltd. | Novel heterocyclic derivative compound and use thereof |
| TWI741155B (en) | 2017-02-27 | 2021-10-01 | 大陸商貝達藥業股份有限公司 | FGFR inhibitor and its application |
| US11365196B2 (en) | 2017-02-27 | 2022-06-21 | Betta Pharmaceuticals Co., Ltd. | FGFR inhibitor and application thereof |
| RU2745035C1 (en) * | 2017-02-27 | 2021-03-18 | Бетта Фармасьютикалз Ко., Лтд. | Fgfr inhibitor and its application |
| WO2018153373A1 (en) * | 2017-02-27 | 2018-08-30 | 贝达药业股份有限公司 | Fgfr inhibitor and application thereof |
| EP3587419A4 (en) * | 2017-02-27 | 2020-08-05 | Betta Pharmaceuticals Co., Ltd. | Fgfr inhibitor and application thereof |
| AU2018226315B2 (en) * | 2017-02-27 | 2021-01-28 | Betta Pharmaceuticals Co., Ltd. | FGFR inhibitor and application thereof |
| US11091485B2 (en) | 2017-05-23 | 2021-08-17 | Shengke Pharmaceuticals (Jiangsu) Ltd. | Imidazo[1′,2′:1,6]pyrido[2,3-d]pyrimidine compound as protein kinase inhibitor |
| US10611762B2 (en) | 2017-05-26 | 2020-04-07 | Incyte Corporation | Crystalline forms of a FGFR inhibitor and processes for preparing the same |
| US11472801B2 (en) | 2017-05-26 | 2022-10-18 | Incyte Corporation | Crystalline forms of a FGFR inhibitor and processes for preparing the same |
| US10537560B2 (en) | 2017-10-05 | 2020-01-21 | Fulcrum Therapeutics. Inc. | P38 kinase inhibitors reduce DUX4 and downstream gene expression for the treatment of FSHD |
| US11479770B2 (en) | 2017-10-05 | 2022-10-25 | Fulcrum Therapeutics, Inc. | Use of p38 inhibitors to reduce expression of DUX4 |
| US11291659B2 (en) | 2017-10-05 | 2022-04-05 | Fulcrum Therapeutics, Inc. | P38 kinase inhibitors reduce DUX4 and downstream gene expression for the treatment of FSHD |
| US10342786B2 (en) | 2017-10-05 | 2019-07-09 | Fulcrum Therapeutics, Inc. | P38 kinase inhibitors reduce DUX4 and downstream gene expression for the treatment of FSHD |
| CN109721600B (en) * | 2017-10-30 | 2021-04-27 | 上海凌达生物医药有限公司 | Nitrogen-containing fused ring compounds and preparation method and application thereof |
| CN109721600A (en) * | 2017-10-30 | 2019-05-07 | 如东凌达生物医药科技有限公司 | A kind of nitrogenous fused ring compound and its preparation method and application |
| US11174257B2 (en) | 2018-05-04 | 2021-11-16 | Incyte Corporation | Salts of an FGFR inhibitor |
| US11466004B2 (en) | 2018-05-04 | 2022-10-11 | Incyte Corporation | Solid forms of an FGFR inhibitor and processes for preparing the same |
| EP3845534A4 (en) * | 2018-08-27 | 2022-04-06 | Betta Pharmaceuticals Co., Ltd | Salt form and crystal form of novel azatricyclic compound and use thereof |
| US11066404B2 (en) | 2018-10-11 | 2021-07-20 | Incyte Corporation | Dihydropyrido[2,3-d]pyrimidinone compounds as CDK2 inhibitors |
| US11866432B2 (en) | 2018-10-11 | 2024-01-09 | Incyte Corporation | Dihydropyrido[2,3-d]pyrimidinone compounds as CDK2 inhibitors |
| US11384083B2 (en) | 2019-02-15 | 2022-07-12 | Incyte Corporation | Substituted spiro[cyclopropane-1,5′-pyrrolo[2,3-d]pyrimidin]-6′(7′h)-ones as CDK2 inhibitors |
| US11472791B2 (en) | 2019-03-05 | 2022-10-18 | Incyte Corporation | Pyrazolyl pyrimidinylamine compounds as CDK2 inhibitors |
| US11628162B2 (en) | 2019-03-08 | 2023-04-18 | Incyte Corporation | Methods of treating cancer with an FGFR inhibitor |
| US11919904B2 (en) | 2019-03-29 | 2024-03-05 | Incyte Corporation | Sulfonylamide compounds as CDK2 inhibitors |
| US11440914B2 (en) | 2019-05-01 | 2022-09-13 | Incyte Corporation | Tricyclic amine compounds as CDK2 inhibitors |
| US11447494B2 (en) | 2019-05-01 | 2022-09-20 | Incyte Corporation | Tricyclic amine compounds as CDK2 inhibitors |
| US11591329B2 (en) | 2019-07-09 | 2023-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11427567B2 (en) | 2019-08-14 | 2022-08-30 | Incyte Corporation | Imidazolyl pyrimidinylamine compounds as CDK2 inhibitors |
| US11851426B2 (en) | 2019-10-11 | 2023-12-26 | Incyte Corporation | Bicyclic amines as CDK2 inhibitors |
| US11607416B2 (en) | 2019-10-14 | 2023-03-21 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11566028B2 (en) | 2019-10-16 | 2023-01-31 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11897891B2 (en) | 2019-12-04 | 2024-02-13 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
| US11407750B2 (en) | 2019-12-04 | 2022-08-09 | Incyte Corporation | Derivatives of an FGFR inhibitor |
| WO2022032071A1 (en) * | 2020-08-07 | 2022-02-10 | Abm Therapeutics Corporation | Kinase inhibitors and uses thereof |
| US11939331B2 (en) | 2021-06-09 | 2024-03-26 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
| US11976073B2 (en) | 2021-12-10 | 2024-05-07 | Incyte Corporation | Bicyclic amines as CDK2 inhibitors |
| US11981671B2 (en) | 2022-06-21 | 2024-05-14 | Incyte Corporation | Bicyclic pyrazolyl amines as CDK2 inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| US20030232847A1 (en) | 2003-12-18 |
| US20050197352A1 (en) | 2005-09-08 |
| PL374544A1 (en) | 2005-10-31 |
| CN1293078C (en) | 2007-01-03 |
| US7285559B2 (en) | 2007-10-23 |
| RU2310657C2 (en) | 2007-11-20 |
| AR039219A1 (en) | 2005-02-09 |
| JP2006503802A (en) | 2006-02-02 |
| US6949560B2 (en) | 2005-09-27 |
| MXPA04009585A (en) | 2005-01-11 |
| US7285561B2 (en) | 2007-10-23 |
| KR20040099384A (en) | 2004-11-26 |
| CN1646529A (en) | 2005-07-27 |
| AU2003215675A1 (en) | 2003-10-13 |
| AU2003215675B2 (en) | 2007-07-12 |
| RU2004132204A (en) | 2005-11-20 |
| US20060252783A1 (en) | 2006-11-09 |
| CA2479644A1 (en) | 2003-10-09 |
| US20060252784A1 (en) | 2006-11-09 |
| US7081462B2 (en) | 2006-07-25 |
| KR100656205B1 (en) | 2006-12-12 |
| EP1492790A1 (en) | 2005-01-05 |
| BR0308937A (en) | 2005-01-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2003215675B2 (en) | Imidazo fused compounds | |
| AU776250B2 (en) | Heteroalkylamino-substituted bicyclic nitrogen heterocycles as inhibitors of P38 protein kinase | |
| EP1226144B1 (en) | Alkylamino substituted bicyclic nitrogen heterocycles as inhibitors of p38 protein kinase | |
| US7439247B2 (en) | Bicyclic pyridine and pyrimidine P38 kinase inhibitors | |
| EP1919918A2 (en) | P38 map kinase inhibitors and methods for using the same | |
| EP1685131B1 (en) | Hydroxyalkyl substituted pyrido-7-pyrimidin-7-ones | |
| WO2004092144A2 (en) | Quinazoline compounds useful as p38 kinase inhibitors | |
| EP1620105B1 (en) | (6-(phenoxy)-pyrido¬3,4-d|pyrimidin-2-yl)-amine derivatives as p38 kinase inhibitors for the treatment of inflammatory conditions such as rheumatoid arthritis | |
| US20080146590A1 (en) | P38 map kinase inhibitors and methods for using the same | |
| JP4028236B6 (en) | Alkylamino substituted bicyclic nitrogen heterocycles as inhibitors of p38 protein kinase |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SD SE SG SK SL TJ TM TN TR TT TZ UA UG UZ VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2479644 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003745276 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20038075369 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2004/009585 Country of ref document: MX Ref document number: 374544 Country of ref document: PL Ref document number: 1020047015609 Country of ref document: KR Ref document number: 2003215675 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003580336 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 2004132204 Country of ref document: RU Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020047015609 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003745276 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2003215675 Country of ref document: AU |