DESCRIPTION HIGH POTENCY SIRNAS FOR REDUCING THE EXPRESSION OF TARGET GENES
BACKGROUND OF THE INVENTION
The present application claims the benefit of U.S. Provisional Application Serial No. 60/353,332 filed February 1, 2002, the entire text of which is herein incorporated by reference.
1. Field of the Invention
The present invention relates to improved methods for making small interfering RNA (siRNA), improved siRNA made by such methods, their use in the modulation of gene expression in mammalian and other cell types and their use in medical therapies.
2. Description of Related Art
RNA interference (RNAi) is a phenomenon in which a double stranded RNA (dsRNA) specifically suppresses the expression of a gene bearing its complementary sequence. The phenomenon was originally discovered in Caenorhabditis elegans by Fire and Mello (Fire et al. , 1998). RNAi has since become a useful research tool for many organisms. Although the mechanism by which dsRNA suppresses gene expression is not entirely understood, experimental data provide important insights. In non-mammalian systems, it appears that longer dsRNA are processed into small, 21-23 nt dsRNAs by an enzyme containing RNase HI motifs (Bernstein et al, 2001 ; Grishok et al, 2001; Hamilton and Baulcombe, 1999; Knight and Bass, 2001; Zamore et al, 2000). It has been theorized that the RNAi nuclease complex, called RNA- induced silencing complex (RISC), helps the small dsRNAs recognize complementary mRNAs through base-pairing interactions. Following the siRNAs interaction with its substrate, the RNA is targeted for degradation, perhaps by enzymes that are present in the RISC (Montgomery et al, 1998).
Until recently, RNAi could only be used in non-mammalian cells. This is because mammalian cells have a potent antiviral response pathway that induces global changes in gene expression when the cells are challenged with long (>30 nucleotides) dsRNA molecules. This pathway has made it impossible to specifically suppress the expression of proteins in mammalian cells using the typical RNAi molecules, which are hundreds of nucleotides long.
Recently Elbashir et al (2001) published a method to bypass the antiviral response and induce gene specific silencing in mammalian cells. Several 21 nucleotide (nt) dsRNAs with 2 nt 3' overhangs were transfected into mammalian cells without inducing the antiviral response. These small dsRNAs, referred to as small interfering RNAs (siRNAs) proved capable of inducing the specific suppression of target genes. In one set of experiments, siRNAs complementary to a luciferase gene were co-transfected with a luciferase reporter plasmid into NIH3T3, COS-7, HeLaS3, and 293 cells. In all cases, the siRNAs were able to specifically reduce luciferase gene expression. In addition, the authors demonstrated that siRNAs could reduce the expression of several endogenous genes in human cells. The endogenous targets were lamin A/C, lamin B 1 , nuclear mitotic apparatus protein, and vimentin. The use of siRNAs to modulate gene expression in mammalian cells has now been repeated at least twice (Caplen et al, 2001; Hutvagner et al, 2001). This technology has great potential as a tool to study gene function in mammalian cells and may lead to the development of pharmacological agents based upon siRNA.
To realize this potential, siRNAs must be designed so that they are specific and effective in suppressing the expression of the genes of interest. Methods of selecting the target sequences, i.e. those sequences present in the gene or genes of interest to which the siRNAs will guide the degradative machinery, are directed to avoiding sequences that may interfere with the siRNA's guide function while including sequences that are specific to the gene or genes. Typically, siRNA target sequences of about 21 to 23 nucleotides in length are most effective. This length reflects the lengths of digestion products resulting from the processing of much longer RNAs as described above.
The making of siRNAs to date has been through direct chemical synthesis or through processing of longer, double stranded RNAs through exposure to Drosophila embryo lysates or through an in vitro system derived from S2 cells. Use of cell lysates or in vitro processing may further involve the subsequent isolation of the short, 21-23 nucleotide siRNAs from the lysate, etc., making the process somewhat cumbersome and expensive. Chemical synthesis proceeds by making two single stranded RNA-oligomers followed by the annealing of the two single stranded oligomers into a double stranded RNA. Methods of chemical synthesis are diverse. Non- limiting examples are provided in U.S. Pat. No.s 5,889,136; 4,415,732; 4,458,066, expressly incorporated herein by reference, and in Wincott et al (1995).
Elbashir and colleagues have published the procedure that they use to design, prepare, and transfect siRNAs for mammalian RNAi experiments. ("The siRNA user guide" August 26, 2001). Similar protocols and procedures are available in Dharmacon Technical Bulletin #003, July 2001. These guides recommend chemically synthesizing two 21-mer RNA oligomers with two deoxythymidines at the 3' terminus and 19 nucleotide complementary sequences. The two ribo-oligomers are mixed to allow them to hybridize. The products are then mixed with a transfection agent and added to cell culture at concentrations of about 100 nM. The pamphlet further recommends that the selection of the target sequence should be constrained so that they begin with AA and end with TT, so that the AA and TT overhang sequences may be fashioned from the target sequence itself. The pamphlet indicates that the symmetric 3' overhangs aid the formation of approximately equimolar ratios of sense and antisense target RNA-cleaving siRNAs.
Several further modifications to siRNA sequences have been suggested in order to alter their stability or improve their effectiveness. It is suggested that synthetic complementary 21mer RNAs having di-nucleotide overhangs (i.e. 19 complementary nucleotides + 3' non- complementary dimers) may provide the greatest level of suppression, although actual data demonstrating this advantage is lacking. These protocols primarily use a sequence of two (2'- deoxy) thymidine nucleotides as the di-nucleotide overhangs. These dinucleotide overhangs are often written as dTdT to distinguish them from the typical nucleotides incorporated into RNA. The literature has indicated that the use of dT overhangs is primarily motivated by the need to reduce the cost of the chemically synthesized RNAs. It is also suggested that the dTdT overhangs might be more stable than UU overhangs, though the data available shows only a slight (< 20%) improvement of the dTdT overhang compared to an siRNA with a UU overhang.
To date, such chemically synthesized siRNAs are found to work optimally when they are in cell culture at concentrations of 25-100 nM. Elbashir et al used concentrations of about 100 nM to achieve effective suppression of expression in mammalian cells. Unfortunately, ribo- oligomers are very expensive to chemically synthesize, making the procedure less appealing and not cost effective to many researchers and pharmaceutical companies. Furthermore, in foreseeable medical applications of siRNA, it would be desirable to achieve target gene inhibition with as little siRNA as possible. Therefore, siRNAs that are still as effective, if not more so, at lower concentrations would be significantly advantageous. There is therefore a need in the art for siRNAs that function at lower concentrations to modulate or attenuate the expression of target genes.
siRNAs have been most effective in mammalian cell culture at about 100 nM. In several instances, however, lower concentrations of chemically synthesized siRNA have been used. Caplen, et al. used chemically synthesized siRNAs at 18 nM. However, Caplen used semi- quantitative RT-PCR to monitor reduction of transcripts. The semi-quantitative nature of {he assay makes unclear how great an effect this low concentration of siRNA had on transcript levels. Hutvagner, et al. used chemically synthesized siRNAs at concentrations of 70 nM to elicit a response. Although less than 100 nM, 70 nM may still represent a substantially prohibitive concentration for some applications. Although Elbashir efa also indicated that they could use lower amounts of siRNA in the cell culture and still observe suppression, they did not provide data nor did they indicate by how much the expression was reduced at these lower levels.
WO 99/32619 and WO 01/68836 suggest that RNA for use in siRNA may be chemically or enzymatically synthesized. Both of these texts are incorporated herein in their entirety by reference. The enzymatic synthesis contemplated in these references is by a cellular RNA polymerase or a bacteriophage RNA polymerase (e.g. T3, T7, SP6) via the use and production of an expression construct as is known in the art. For example, see U.S. Pat. No. 5,795,715. The contemplated constructs provide templates that produce RNAs that contain nucleotide sequences identical to a portion of the target gene. The length of identical sequences provided by these references is at least 25 bases, and may be as many as 400 or more bases in length. An important aspect of this reference is that the authors contemplate digesting longer dsRNAs to 21-25mer lengths with the endogenous nuclease complex that converts long dsRNAs to siRNAs in vivo. They do not describe or present data for synthesizing and using in vitro transcribed 21-25mer dsRNAs. No distinction is made between the expected properties of chemical or enzymatically synthesized dsRNA in its use in RNA interference.
Similarly, WO 00/44914, incorporated herein by reference, suggests that single strands of
RNA can be produced enzymatically or by partial/total organic synthesis. Preferably, single stranded RNA is enzymatically synthesized from the PCR products of a DNA template, preferably a cloned cDNA template and the RNA product is a complete transcript of the cDNA, which may comprise hundreds of nucleotides. WO 01/36646, incorporated herein by reference, places no limitation upon the manner in which the siRNA is synthesized, providing that the RNA may be synthesized in vitro or in vivo, using manual and/or automated procedures. This reference also provides that in vitro synthesis may be chemical or enzymatic, for example using cloned RNA polymerase (e.g. T3, T7, SP6) for transcription of the endogenous DNA (or cDNA)
template, or a mixture of both. Again, no distinction in the desirable properties for use in RNA interference is made between chemically or enzymatically synthesized siRNA.
U.S. Pat. No. 5,795,715 reports the simultaneous transcription of two complementary DNA sequence strands in a single reaction mixture, wherein the two transcripts are immediately hybridized. The templates used are preferably of between 40 and 100 base pairs, and which is equipped at each end with a promoter sequence. The templates are preferably attached to a solid surface. After transcription with RNA polymerase, the resulting dsRNA fragments may be used for detecting and/or assaying nucleic acid target sequences. U.S. Pat. No. 5,795,715 was filed June 17, 1994, well before the phenomenon of RNA interference was described by Fire et al (1998). The production of siRNA was, therefore, not contemplated by these authors.
As described above, there is a need for siRNAs of increased potency, both for general research and for use as medical or veterinary therapies. siRNAs of increased potency would decrease the risk or adverse reactions or other, undesired effects of medical therapies using siRNA. Fewer molecules of siRNA of increased potency would be needed for such therapies, with concomitant benefits to patients.
SUMMARY OF THE INVENTION
The present invention is directed to compositions and methods useful in the production of double stranded RNAs (dsRNAs) of increased potency for use as small interfering RNA (siRNA) in the suppression of gene expression and the treatment of disease. The invention provides methods of synthesis and use of siRNA that result in less costly siRNA and siRNA of substantially and significantly higher potency.
SiRNAs of increased potency are those wherein the provision of fewer molecules of siRNA is effective in achieving modulation or attenuation of gene expression when compared to the number of standard siRNA molecules required to achieve the same level of modulation or attenuation of target gene expression. Standard siRNAs are those provided by typical chemical synthesis methods and incorporating the usual, unmodified nucleotides that make up the RNA polymer, i.e. adenine, cytosine, guanine, and uracil.
Potency of siRNA may be evaluated by a number of means as will be appreciated by those of skill in the art. The level of attenuation of gene expression may be compared between replicate cells or organisms treated with equal molar amounts of standard and siRNAs of increased potency designed to target the same target sequence within a target gene or genes. Additionally, response curves may be generated wherein levels of expression of a target gene in response to varying concentrations of siRNAs are measured and displayed for standard and siRNAs of increased potency delivered to replicate cells or organisms. Other means of comparison are contemplated. Generally, any means will suffice that reveals that fewer molecules of a particular siRNA are required to achieve the same level of modulation or attenuation as a standard siRNA used under equivalent conditions and targeting the same target sequence.
The use of siRNAs of increased potency therefore results in an increased modulation or attenuation of gene expression in comparison to standard siRNAs when both are used under identical conditions. Such increased attenuation may be recognized in a number of measures. Non limiting examples include altered gene expression measurable through decreased transcript abundance, decreased protein product abundance, decreased activity associated with the protein product, or an altered phenotype associated with the protein product. Such changes in expression or phenotype may be large or small yet still reveal the high potency of the presently disclosed siRNAs.
The choice of effect measured, the context of its measurement, and the metric used all interact to determine the absolute magnitude of the response. Nevertheless, the identification of siRNA of increased potency is unambiguous even though the relative effectiveness of standard and siRNAs of increased potency may be indicated by small relative changes in phenotype or expression levels.
A relative change in expression levels or phenotype may be on the order of any level greater than 1% to 100%. Thus, in one example, relative cell proliferation may be altered by about 2% at an siRNA concentration of 1 nM (FIG. 10) and will be indicative of the high potency of the siRNA provided by the present invention. In another example, the relative transcript levels of a gene may be reduced more effectively by siRNAs of increased potency of the present invention across a continuous range of concentrations (FIG. 11), resulting in relative potency ranging continuously from greater than 0% to 100%. siRNAs of increased potency may be 1%, 2,%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%,
60%, 70%, 80%, 90%, or 100% more potent. siRNAs of increased potency may also be described as 1-, 2-, 3-, 4-, 5-, 6-, 7-, 8-, 9-, 10-, 11-, 12-, 13-, 14-, 15-, 20-, 25-, 30-, 40-, 50-, 60-, 70-, 80-, 90-, 100-, 200-, and more fold more potent than standard siRNAs. Potency may thus be further defined with respect to specific processes used to determine potency. Particularly preferred embodiments for calculating increased potency include the comparison of the magnitudes or importance of the desired effects of administering siRNAs of the present invention to the effects of administering siRNAs made from standard or currently available protocols.
The inventors have made the surprising discovery that the method of synthesis of siRNA can dictate its potency. Specifically, the inventors have discovered that enzymatic synthesis of siRNAs through the methods of the present invention provides for siRNA with substantially and significantly greater potency. In one example, the optimal concentration of siRNA made by the present invention for use in the transfection of cells is unexpectedly up to 20-fold less than what is commonly used. Thus, these siRNAs of increased potency may be 20-fold more potent than standard siRNAs. Since siRNAs synthesized by the methods and compositions of the present invention are unexpectedly, and significantly more potent that those available otherwise, methods of use of these siRNAs are provided that incorporate this highly surprising and unexpected potency. These methods include methods of attenuating gene expression in host cells, organs, tissues, and whole organisms.
An advantage of this aspect of the present invention lies in providing for siRNA that may be enzymatically synthesized from a great range of template sequences. Templates are provided that contain structures specifically adapted for the efficient synthesis of siRNAs of increased potency in a variety of situations without limitation and for any target gene sequence that may be desired. These siRNAs maybe from 15 to 30 nucleotides in length, may contain dinucleotide or other overhang sequences, may contain modified nucleotides, or other modifications as desired. The methods of the present invention also provide for the use of dT, and other overhang sequences, as well as the incorporation of modified nucleotide analogs, which may also increase effectiveness of the siRNA.
Compositions of the present invention include oligonucleotide and polynucleotide templates and methods for the use of those templates in enzymatic synthesis of siRNA of increased potency, which is employed in the specific inhibition of target gene expression. The templates may comprise a polynucleotide sequence comprising a target sequence. The target
sequence may be derived from the sequence of a target gene. A target gene is a gene whose expression is targeted for interference, inhibition, attenuation, disruption, augmentation, or other modulation. Preferably, the expression is targeted for interference. Most preferably the expression is targeted for attenuation.
The inventors have also made the surprising discovery that incorporation of modified nucleotide analogs may increase the potency of siRNA. Modified nucleotide analogs may be incorporated in siRNAs through either enzymatic synthesis or chemical synthesis. Enzymatic synthesis is the use of RNA polymerases to polymerize nucleotides into single or double stranded RNA for use as siRNA through the methods and compositions presently disclosed. Chemical synthesis is the use of any other method to synthesize single or double stranded RNA for use in siRNA.
The inventors have found that modified nucleotide analogs increase the potency of siRNAs whether incorporated through enzymatic or chemical synthesis, h particular, enzymatic incorporation of modified nucleotide analogs produces even further enhancement of siRNA potency over that achieved through enzymatic synthesis alone. Modified nucleotide analogs incorporated through chemical synthesis have also been found to unexpectedly enhance siRNA potency. Increased potency of siRNA may thus be achieved through the enzymatic synthesis of siRNA, through the incorporation of modified nucleotide analogs through enzymatic synthesis, and through the incorporation of modified nucleotide analogs through chemical synthesis.
The inventors have also discovered that siRNAs wherein the duplex structure of the double stranded RNA is of reduced stability are significantly more potent. In particular, nucleotide analogs that reduce the stability of RNA duplexes enhance the potency of siRNAs.
RNA duplex stability may be measured by several methods, as will be appreciated by one of skill in the art. A typical example is the measurement of the melting temperature (TM) of the duplex in a specified set of conditions, such as, but not limited to specified salt concentrations, pH, etc. See, for example the thermal melting analysis provided in U.S. Pat. No. 6,005,087, incorporated herein by reference. Also see, generally, Sambrook (2001), and by way of additional example, Dubins, et al (2001) and Testa etal (1999). Reduced duplex stability may be on the order of a 1%, 2,%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50% or more decrease in TM or like measure in comparison to unmodified, or fully complementary, or other RNA, DNA, or RNA DNA duplexes. Reduced RNA duplex stability
may be further defined by the particular methods chosen, as will be appreciated by one of skill in the art. Such methods can include assays of siRNA potency as described above.
Reduced stability of the siRNA duplex may be achieved through a variety of techniques.
Nucleotide analogs, as described above, may be introduced such that the resultant duplex of the siRNA is of reduced stability. The following discussion is a non-limiting list of possible modifications to be made to the siRNA that may result in higher potency through reduced stability of the siRNA duplex structure.
Phosphorothioates - Phosphorothioates reduce duplex stability approximately 0.5°C to 1°C per modification. They can be substituted at one or more nucleotide positions along the length of the siRNA.
Inosine - Substitution of inosine (I) for G's at one or more positions in the siRNA will reduce duplex stability and thereby enhance siRNA potency. I:C base pairs form only two hydrogen bonds (as opposed to three in G:C base-pairs), reducing the stability of the duplex (Kawase et al 1986).
4-Thio uridine - 4-thio uridine forms only a single hydrogen bond with adenosine
(Testa et al 1999) and therefore the substitution of one or more uracils (U) in the siRNA results in duplex structures of reduced stability.
4-Ethyl cytosine - 4-ethyl cytosine forms only two hydrogen bonds with guanosine, reducing the stability of G:C base-pairs (Nguyen et al. 1997). Use of 4-ethyl cytosine at one or more positions in the siRNA is expected to reduce stability of the duplex structure.
3-Nitropyrrole nucleoside and 5-nitroindole nucleoside (5-nitroindole) - Both.of these nucleosides hybridize to all four natural nucleosides, but with lower affinity than canonical base^ pairs (Bergstrom et al.1997). Thus, substitution of an appropriate number of nucleotides of the siRNA with these nucleosides will result in reduced overall duplex stability without loss of appropriate sequence specificity. The selection of the appropriate number and position of such nucleoside substitutions are well within the skill of the ordinary artisan (Bergstrom et al.1997).
Abasic sites — There are several nucleotide linkers that do not have an associated base.
These can be introduced at one or more sites in the sense strand of the siRNA to eliminate one or more base-pairs and reduce the stability of the siRNA duplex. Nucleic acid helices with abasic sites have reduced melting temperatures, i.e. reduced duplex stabilities. (Shishkina et al 2000).
Mismatches - One or more mismatches can be introduced along the length of the siRNA duplex. The mismatched bases should be in the sense strand of the siRNA so as not to reduce the binding affinity of the anti-sense strand for the mRNA target. The net effect of such mismatches on duplex stability is the same as that achieved by the other chemical substitutions described above. That is, the stability of the duplex structure of the siRNA is reduced relative to that of a completely and absolutely complementary match between the two single strand sequences that make up the RNA duplex.
All of the discoveries described are directed to the modulation, especially the attenuation of the expression of a target gene. The target gene may include sequences encoding polypeptides or polynucleotide sequences that regulate the replication, transcription, translation or other process important to the expression of the gene. The target gene need not necessarily encode a polypeptide but may encode other cellular components, such as ribosomal RNA, splicosome RNA, transfer RNA, etc.
The target gene may exist as an endogenous gene, which occurs naturally within a cell, or an exogenous gene, which does not naturally occur within a cell. Exogenous genes may, for example, be a transgene or synthetic gene, or a gene of a pathogen, parasite, or commensal organism. Preferably, the target gene exists within a vertebrate cell, although the invention is not limited to the making of siRNA for use in vertebrate cells.
The target sequence may be the entire sequence of the target gene, or, preferably, only a portion of the target gene. Preferably, the target sequence is a contiguous subsequence of the target gene sequence and is from 15 to 30 nucleotides in length. The target sequence may be 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 or 30 nucleotides in length. The size and sequence of the target gene used as the target sequence may be selected by those of skill in the art so as to optimize the interfering effects of the product siRNAs.
Within a polynucleotide template for making siRNA, the target sequence may be operatively linked to a promoter (FIG. 1). Preferably, the promoter is a sequence sufficient to direct the transcription of the template sequence into RNA when contacted with RNA polymerase. Such promoters are well known to those of skill in the art and include, but are not limited to the T7, T3, and SP6 promoters. Polymerases may be chosen so as to maximize the benefits of enzymatic synthesis of siRNA. Such polymerases are well known to those of skill in the art and include, but are not limited to the T7, T3, SP6, polymerases and derivatives thereof.
Such a choice is within the skill of one in the art. In one embodiment, the polymerase is selected from T7, T3, and SP6. In a preferred embodiment, the polymerase is T7 RNA polymerase. Naturally, the selection of appropriate polymerase is performed in conjunction with the selection of appropriate promoter so that the polymerase functionally recognizes the promoter, leading to the synthesis of an RNA strand.
If operatively linked to a T7, T3, SP6 or similar promoter, the target sequence may be contiguous with the promoter sequence (FIG. 1). Milligan et al. (1987) reports the use of relatively short template nucleotides in the synthesis of single-stranded RNA products. In particular, Milligan et al. disclose necessary sequence constraints imposed by the use of T7, T3, or SP6 RNA polymerase promoters. Therefore, in an embodiment of the invention, if the target sequence is contiguous with the promoter sequence, the target sequence adjacent to the promoter sequence preferably begins with the dinucleotide sequence of GG or GA.
However, the present invention in part provides compositions and methods that surmount the limitations identified by Milligan et al by providing a template comprising at least one leader sequence interposed between the promoter and the template sequence (FIG. 2 and FIG. 3). Thus, in another preferred embodiment, the target sequence present in the polynucleotide template for making siRNA may be operatively linked to a promoter without being contiguous with it. A spacer or leader sequence may be interposed between the promoter sequence and the target sequence of the template. Preferably, such a leader sequence provides the dinucleotide sequences of GG or GA that aid the efficient transcription from a T7, T3, SP6 or similar promoter. Such a leader sequence may be any length that does not interfere with the effective functioning of the promoter. Such a leader sequence may be 10, 12, 14, 16, 18, 20, 22, 24, 26, 28, 30, 40, or 50 or more nucleotides in length. Preferably, the leader sequence is about 10 nt, but may be 9, 8, 7, 6, 5, 4, 3, or even 2 or 1 nt in length. In one embodiment, the leader sequence is selected from SEQ ID NO: 1 and SEQ JO NO: 2. In another embodiment, the leader sequence is that of SEQ ID NO: 1. Li another embodiment, the leader sequence is that of SEQ ID NO: 2. In yet another embodiment, the leader sequences of two templates are different sequences.
Of course, and as will be appreciated by those of skill in the art, specifying the template sequence for enzymatic synthesis is generally accomplished by listing the nucleotide sequence of the template in the 5' to 3' direction, and by convention, as the top, or sense strand of a double stranded duplex. (FIGs.1-4). However, the template sequence so specified in the context of the
present invention is not limited to the sense strand. For example, the double stranded molecule, as discussed above, can function as a template for RNA synthesis. Furthermore, the antisense strand alone can function as a template for RNA synthesis. Therefore, following convention, as used herein the description of polynucleotides will be generally from 5' to 3' and will recite the sense strand, although the invention is not limited to transcription from the sense strand as listed, but will encompass both the sense and the antisense (or bottom) strand and a single, antisense strand (FIGs. 1-4). Therefore, for example, the embodiments that comprise templates comprising overhang encoding sequences comprising the nucleotide sequence of TT, are designed so that the resultant RNA strand formed comprises an overhang of UU or the like.
In preferred embodiments, the invention provides for RNases that have activity in relation to the nucleotide sequences of the leader sequence. Thus, RNase TI preferentially digests single-stranded RNA at G residues. RNase A preferentially digests at C and U residues. If RNase TI is to be used, the last nucleotide in the leader sequence is preferred to be a G to eliminate the entire leader sequence from the siRNA. Likewise, if RNase A is to be used, the last nucleotide in the leader sequence should be a C or U. In additional, preferred embodiments, the RNase may be RNase Sa, RNase Sa2, or RNase Sa3. In a particularly preferred embodiment, the RNase is RNase Sa.
In yet another preferred embodiment, the polynucleotide template for synthesizing small interfering RNA comprises a promoter, a target sequence, and a complementary sequence to the target sequence positioned between the target sequence and the 3' terminus of the template (FIG. 4).
The templates provided by the invention may be used in the synthesis of siRNA of increased potency. The embodiments of the synthesis methods preferably comprise the transcription of the templates by RNA polymerase, annealing of the single stranded RNA products to form double stranded RNA, and RNAse digestion to remove unnecessary single stranded RNA from the double stranded RNA product (FIGS. 2, 3, and 4).
In one preferred embodiment, the synthesized RNA is such that it is partially self- complementary and so forms a stem and loop structure (FIG. 4). In alternative and preferred embodiments, two strands of RNA maybe made that are substantially complementary so that they form a duplex upon provision of appropriate conditions (FIGS. 1, 2, and 3). In yet another preferred embodiment, a single RNA strand may be synthesized that complements a template
RNA strand itself made through other means (RNA copying). Additionally, a single RNA strand may be synthesized that complements a pre-existing single stranded RNA molecule and allowed to form a duplex therewith, forming a dsRNA functional as a siRNA of increased potency. In a further preferred embodiment, the synthesized RNA is further treated with nucleases in order to digest or remove template nucleic acids or portions of the synthesized RNA. In an additional preferred embodiment, once synthesized, the RNA products may be purified or further manipulated before use as siRNA. In an alternative embodiment, the RNA products may be used directly without further manipulation.
In still more preferred embodiments, the siRNA includes modifications to either the phosphate-sugar backbone or the nucleoside. In one embodiment, the phosphodiester linkages of natural RNA are modified to include at least one of a nitrogen or sulfur hetero-atom. In another embodiment, bases may be modified to block the activity of adenosine deaminase. In some embodiments, the modified nucleotide analogs may be selected from the group of aminoallyl UTP, pseudo-UTP, 5-I-UTP, 5-I-CTP, 5-Br-UTP, alpha-S ATP, alpha-S CTP, alpha-S GTP, alpha-S UTP, 4-thio UTP, 2-thio-CTP, 2'NH2 UTP, 2'NH2 CTP, and 2' F UTP.
In preferred embodiments, modified nucleotide analogs are incorporated into the synthesized siRNA that decrease duplex stability. Modified nucleotide analogs may be incorporated through enzymatic or chemical synthesis. In one preferred embodiment, modified nucleotide analogs are incorporated through enzymatic synthesis. In another preferred embodiment modified nucleotide analogs are incorporated through chemical synthesis
siRNA can be introduced into cells in a number of ways. Preferred embodiments include micro-injection, bombardment by particles covered by the siRNA, soaking the cell or organism in a solution of the siRNA, electroporation of cell membranes in the presence of siRNA, liposome-mediated delivery of siRNA and transfection mediated by chemicals such as polyamines, calcium phosphate, viral infection, transformation, and the like. In further preferred embodiments, siRNA is introduced along with components that enhance RNA uptake by the cell, stabilize the annealed strands, or otherwise increase inhibition of the target gene. In a most preferred embodiment, cells are conveniently incubated in a solution containing the siRNA.
In further embodiments, siRNA is delivered to a cell indirectly by introducing one or more vectors that encode both single strands of a siRNA (or, in the case of a self-complementary RNA, the single self-complementary strand) into the cell. The vectors of these embodiments
contain elements of the templates described above such that the RNA is transcribed inside the cell, annealed to form siRNA and effects attenuation of the target gene expression. See WO 99/32619, WO 00/44914, WO 01/68836 (each of which is expressly incorporated herein by reference) and references therein for further examples of methods known to the art for introducing siRNA into cells.
Thus, in some embodiments of the present invention, a method for making siRNA of increased potency comprises obtaining nucleotides, and incorporating the nucleotides into siRNA such that an RNA duplex of from 15 to 30 contiguous nucleotides is formed, wherein the siRNA has a sequence that is substantially identical to at least a portion of a selected target gene. In a preferred embodiment, the siRNA is further defined as having reduced duplex stability. In an additional preferred embodiment the siRNA is further defined as comprising obtaining at least one modified nucleotide analog and incorporating the at least one modified nucleotide analog into the siRNA.
In additional, preferred embodiments, the modified nucleotide analog is selected from the group consisting of aminoallyl UTP, pseudo-UTP, 5-I-UTP, 5-I-CTP, 5-Br-UTP, alpha-S ATP, alpha-S CTP, alpha-S GTP, alpha-S UTP, 4-thio UTP, 2-thio-CTP, 2'NH2 UTP, 2'NH2 CTP, and F UTP.
In another preferred embodiemnt, the nucleotides or nucleotide analogs may be incorporated into siRNA through enzymatic synthesis. In one embodiement, the nucleotides or nucleotide analogs may be incorporated into siRNA through chemical synthesis.
In some embodiments the invention comprises a method of attenuating the expression of a target gene, the method comprising the steps of:
(a) obtaining a first polynucleotide template comprising a first promoter operatively linked to a first target sequence that has 5' and 3' ends that is substantially identical to at least a portion of the target gene;
(b) obtaining a second polynucleotide template comprising a second promoter operatively linked to a second target sequence that has 5* and 3' ends and substantially the reverse complement of the first target sequence of the first template;
(c) contacting the first template with a reaction mixture comprising an RNA polymerase and nucleotides to transcribe the first template to form a first RNA product;
(d) contacting the second template with a reaction mixture comprising an RNA polymerase and nucleotides to transcribe the second template to form a second RNA product;
(e) annealing the first and second RNA products to form a double stranded RNA product; and
(f) introducing the double stranded RNA product into the cell in an amount sufficient to attenuate expression of the target gene.
Other preferred embodiments comprise the steps of:
(a) obtaining a polynucleotide template comprising a promoter operatively linked to a first target sequence, a loop sequence, and a second target sequence having 5' and 3 '.ends and that is substantially the reverse complement of the first target sequence;
(b) contacting the template with a reaction mixture comprising an RNA polymerase and nucleotides to transcribe the template to form an RNA product; and
(c) introducing the RNA product into the cell in an amount sufficient to attenuate expression of the target gene.
In other embodiments a method of attenuating target gene expression comprises the steps of:
(a) obtaining a single-stranded polynucleotide template comprising a target sequence substantially identical to at least a portion of the target gene;
(b) contacting the template with a reaction mixture comprising an RNA replicase and nucleotides to synthesize RNA of a complementary sequence to that of the target sequence so as to form a double stranded RNA product; and
(c) introducing the double stranded RNA product into the cell in an amount sufficient to attenuate expression of the target gene.
Additional preferred embodiments comprise the steps of:
(a) obtaining a polynucleotide template comprising a promoter operatively linked to a first target sequence, a loop sequence, and a second target sequence having 5' and 3' ends and that is substantially the reverse complement of the first target sequence;
(b) enzymatically incorporating nucleotides into RNA by contacting the template with a reaction mixture comprising an RNA polymerase and nucleotides to transcribe the template to form an RNA product;
(c) annealing the RNA product to form a stem and loop siRNA product; and
(d) introducing the RNA product to a cell comprising a target gene.
Further embodiments comprise the steps of:
(a) obtaining a polynucleotide template comprising a promoter operatively linked to a first target sequence, a loop sequence, and a second target sequence having 5' and 3' ends and that is substantially the reverse complement of the first target sequence;
(b) enzymatically incorporating nucleotides into RNA by contacting the template with a reaction mixture comprising an RNA polymerase and nucleotides to transcribe the template to form an RNA product;
(c) introducing the RNA product to a cell comprising a target gene; and
(d) annealing the RNA product to form a stem and loop siRNA product within the cell.
Further preferred embodiments comprise the steps of treating the template nucleotides with an appropriate nuclease, e.g. DNase or similar enzyme, to remove template nucleic acids. Yet another preferred embodiment comprises the steps of treating the enzymatic or chemically synthesized dsRNA with RNases to remove single stranded leader sequences or other, undesired sequences.
Surprisingly and unexpectedly, siRNAs of from 15 to 30 nucleotides in length made through these embodiments of the invention are significantly more potent than siRNAs of the same length made through standard chemical synthesis or without appropriate incorporation of modified nucleotide analogs.
Any of the compositions described herein may be comprised in a kit formulated for performing the methods disclosed. In a non-limiting example the kits may comprise, in suitable container means, a promoter primer, corresponding polymerases, reagents and materials for performing the methods of the invention. In another non-limiting example, the kits may
comprise in a suitable container means, modified nucleotide analogs, reagents and materials for incorporation of the modified nucleotide analogs through chemical synthesis means. In a further example, the kits may comprise siRNAs that have incorporated in them modified nucleotide analogs that reduce dsRNA duplex stability.
The kits may comprise compositions of the present invention in suitable aliquots, whether labeled or unlabeled, as may be used to practice the methods of the invention. The components of the kits may be packaged either in aqueous media or in lyophilized form. The container means of the kits will generally include at least one vial, test tube, flask, bottle, syringe or other container means, into which a component may be placed, and preferably, in suitable aliquots. Where there is more than one component in the kit, the kit also will generally contain a second, third or other additional container into which the additional components may be separately placed. However, various combinations of components may be comprised in a vial, kits may also contain cells in which the expression of target genes is attenuated by the methods of the present invention.
Therapeutic kits of the present invention are kits comprising a promoter, a polymerase, and the reagents, agents, and materials that may be required to practice the methods of the invention, including, but not limited to those reagents necessary for transfection or transformation of cells with siRNA. Such kits may also comprise siRNA made by the methods of the present invention. Such kits will generally contain, in suitable container means, a pharmaceutically acceptable formulation of siRNA made by the methods of the present invention. Such kits may also contain cells in which the expression of target genes is attenuated by the methods of the present invention. The kit may have a single container means, and/or it may have distinct container means for each compound or each reaction mixture or step.
When the components of the kit are provided in one and/or more liquid solutions, the liquid solution is an aqueous solution, with a sterile aqueous solution being particularly preferred. The compositions may also be formulated into a syringeable composition. In which case, the container means may itself be a syringe, pipette, and/or other such like apparatus, from which the formulation may be applied to an infected area of the body, injected into an animal, and/or even applied to and/or mixed with the other components of the kit. However, the components of the kit may be provided as dried powder(s). When reagents and/or components are provided as a dry powder, the powder can be reconstituted by the addition of a suitable solvent. It is envisioned that, the solvent may also be provided in another container means.
The container means will generally include at least one vial, test tube, flask, bottle, syringe and/or other container means, into which the siRNA or cells in which the expression of target gene or genes is attenuated are placed, preferably, suitably allocated. The kits may also comprise a second container means for containing a sterile, pharmaceutically acceptable buffer and/or other diluent. The kits of the present invention also will typically include a means for containing the materials for practicing the methods of the invention, and any other reagent containers in close confinement for commercial sale. Such containers may include injection or blow-molded plastic containers into which the desired vials are retained. Irrespective of the number or type of containers, the kits of the invention may also comprise, or be packaged with, an instrument for assisting with the injection/administration and/or placement of the ultimate cells in which the expression of a target gene or genes is attenuated within the body of an animal. Such an instrument may be a syringe, pipette, forceps, and/or any such medically approved delivery vehicle.
Following long-standing patent law, the words "a" and "an," when used in conjunction with the word "comprising" in the claims or specification, denotes one or more.
BRIEF DESCRIPTION OF THE DRAWINGS
The following drawings form part of the present specification and are included to further demonstrate certain aspects of the present invention. The invention may be better understood by reference to one or more of these drawings in combination with the detailed description of specific embodiments presented herein.
FIG. 1 : Production of siRNA by transcription of templates without leader sequences.
FIG. 2: Production of siRNA by transcription of templates with leader sequences and subsequent digestion by RNase TI .
FIG. 3 : Production of siRNA by transcription of templates with leader sequences and subsequent digestion with RNase A.
FIG. 4: Production of siRNA by transcription of templates encoding hairpin structures.
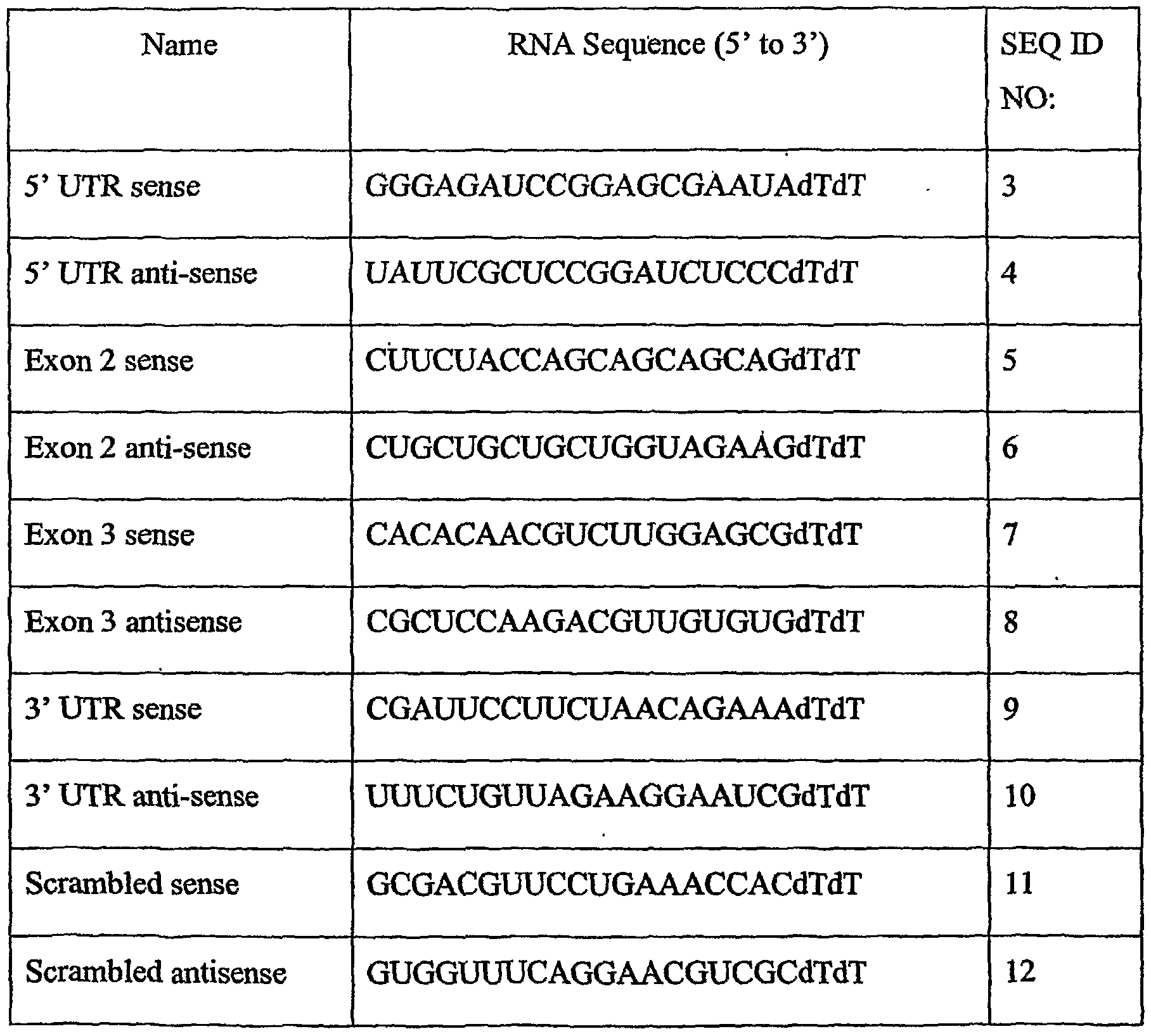
FIG. 5: c-myc mRNA transcript organization. The locations of siRNAs made are indicated by the boxes above the line representing the transcript.
FTG. 6: Relative cell proliferation (as compared to a buffer-transfected control) of HeLa cells transfected with the various siRNAs to c-myc, as well as with the optimized antisense oligonucleotide, 48 hr after transfection.
FIG. 7: Immunofluorescence experiments using an anti-myc antibody were used to measure c-myc protein levels to confirm that siRNAs were reducing expression of c-myc.
FIG. 8: The siRNA against the 3' UTR leads to a drastic reduction in c-myc protein levels.
FIG. 9: Relative reduction in GAPDH mRNA levels in siRNA transfected cells versus untreated cells.
FIG. 10: Comparison of siRNA effectiveness between enzymatically synthesized and siRNA made through standard chemical synthesis in attenuation of c-myc mediated cell proliferation.
FIG. 11: Comparison of siRNA effectiveness between enzymatically synthesized and siRNA made through standard chemical synthesis in expression of GAPDH mRNA.
FIG. 12: Comparison of siRNA effectiveness between siRNA made through standard chemical synthesis and enzymatically synthesized siRNA via RNA copying in attenuation of c-myc mediated cell proliferation.
FIG. 13: Impact of Nucleotide Analogs on siRNA Potency in enzymatically synthesized siRNA.
DETAILED DESCRIPTION OF THE INVENTION
The present invention provides a method for preparing double-stranded RNA molecules that are of increased potency when used to modulate or attenuate gene expression in cells, tissues, organs, and organisms. The method includes enzymatic polymerization of individual ribonucleotides in a sequence that effectively matches or hybridizes with the nucleotide sequence
of a target gene, thereby facilitating the specific modulation of target gene expression. Surprisingly, such enzymatic preparation of dsRNA yields siRNA that is many fold more potent in modulating gene expression than corresponding siRNA prepared through chemical synthesis.
The methods of the present invention therefore provide for much more efficient and cost- effective means for producing and using siRNA in RNA interference applications, which include a wide range of research, industrial, and medical processes, materials, and applications. Medical applications include, by way of example, anti-viral compositions and therapies, anti-tumor compositions and therapies, and compositions and therapies for inherited disorders. One example of the latter application would be the enzymatic synthesis of siRNA for use in therapies to treat autosomal dominant genetic disease such as Huntington's chorea. Additional examples of therapeutic uses include the management of transplant rejection through the treatment of tissues to be introduced into a subject with the siRNAs of the invention in order to modulate or attenuate the expression of genes promoting transplant rejection. For example, hepatocytes may be incubated with enzymatically synthesized siRNA designed to attenuate expression of genes that prompt a host immune response.
The siRNA provided by the present invention allows for the modulation and especially the attenuation of target gene expression when such a gene is present and liable to expression within a cell. Modulation of expression can be partial or complete inhibition of gene function, or even the up-regulation of other, secondary target genes or the enhancement of expression of such genes in response to the inhibition of the primary target gene. Attenuation of gene expression may include the partial or complete suppression or inhibition of gene function, transcript processing or translation of the transcript. In the context of RNA interference, modulation of gene expression is thought to proceed through a complex of proteins and RNA, specifically including small, dsRNA that may act as a "guide" RNA. The siRNA therefore is thought to be effective when its nucleotide sequence sufficiently corresponds to at least part of the nucleotide sequence of the target gene. Although the present invention is not limited by this mechanistic hypothesis, it is highly preferred that the sequence of nucleotides in the siRNA be substantially identical to at least a portion of the target gene sequence.
A target gene generally means a polynucleotide comprising a region that encodes a polypeptide, or a polynucleotide region that regulates replication, transcription or translation or other processes important tot expression of the polypeptide, or a polynucleotide comprising both a region that encodes a polypeptide and a region operably linked thereto that regulates
expression. The targeted gene can be chromosomal (genomic) or extrachromosomal. It may be endogenous to the cell, or it may be a foreign gene (a transgene). The foreign gene can be integrated into the host genome, or it may be present on an extrachromosomal genetic construct such as a lasmid or a cosmid. The targeted gene can also be derived from a pathogen, such as a virus, bacterium, fungus or protozoan, which is capable of infecting an organism or cell. Target genes may be viral and pro-viral genes that do not elicit the interferon response, such as retroviral genes. The target gene may be a protein-coding gene or a non-protein coding gene, such as a gene which codes for ribosmal RNAs, splicosomal RNA, tRNAs, etc.
Any gene being expressed in a cell can be targeted. Preferably, a target gene is one involved in or associated with the progression of cellular activities important to disease or of particular interest as a research object. Thus, by way of example, the following are classes of possible target genes that may be used in the methods of the present invention to modulate or attenuate target gene expression: developmental genes (e.g. adhesion molecules, cyclin kinase inhibitors, Wnt family members, Pax family members, Winged helix family members, Hox family members, cytokines/lymphokines and their receptors, growth or differentiation factors and their receptors, neurotransmitters and their receptors), oncogenes (e.g. ABLI, BLC1 , BCL6, CBFA1, CBL, CSFIR, ERBA, ERBB, EBRB2, ETS1, ETS1, ETV6, FGR, FOX, FYN, HCR, HRAS, JUN, KRAS, LCK, LYN, MDM2, MLL, MYB, MYC, MYCL1, MYCN, NRAS, PIM1, PML, RET, SRC, TALI, TCL3 and YES), tumor suppresser genes (e.g. APC, BRCA1, BRCA2, MADH4, MCC, NF1, NF2, RBI, TP53 and WT1), and enzymes (e.g. ACP desaturases and hycroxylases, ADP-glucose pyrophorylases, ATPases, alcohol dehycrogenases, amylases, amyloglucosidases, catalases, cellulases, cyclooxygenases, decarboxylases, dextrinases, esterases, DNA and RNA polymerases, galactosidases, glucanases, glucose oxidases, GTPases, helicases, hemicellulases, integrases, invertases, isomersases, kinases, lactases, lipases, lipoxygenases, lysozymes, pectinesterases, peroxidases, phosphatases, phospholipases, phophorylases, polygalacturonases, proteinases and peptideases, pullanases, recombinases, reverse transcriptases, topoisomerases, xylanases).
The nucleotide sequence of the siRNA is defined by the nucleotide sequence of its target gene. The siRNA contains a nucleotide sequence that is essentially identical to at least a portion of the target gene. Preferably, the siRNA contains a nucleotide sequence that is completely identical to at least a portion of the target gene. Of course, when comparing an RNA sequence to a DNA sequence, an "identical" RNA sequence will contain ribonucleotides where the DNA
sequence contains deoxyribonucleotides, and further that the RNA sequence will typically contain a uracil at positions where the DNA sequence contains thymidine.
A siRNA comprises a double stranded structure, the sequence of which is "substantially identical" to at least a portion of the target gene. "Identity," as known in the art, is the relationship between two or more polynucleotide (or polypeptide) sequences, as determined by comparing the sequences. In the art, identity also means the degree of sequence relatedness between polynucleotide sequences, as determined by the match of the order of nucleotides between such sequences. Identity can be readily calculated. See, for example: Computational Molecular Biology, Lesk, A.M., ed. Oxford University Press, New York, 1988; Biocomputing: Informatics and Genome Projects, Smith, D.W., ed., Academic Press, New York, 1993; and the methods disclosed in WO 99/32619, WO 01/68836, WO 00/44914, and WO 01/36646, specifically incorporated herein by reference. While a number of methods exist for measuring identity between two nucleotide sequences, the term is well known in the art. Methods for determining identity are typically designed to produce the greatest degree of matching of nucleotide sequence and are also typically embodied in computer programs. Such programs are readily available to those in the relevant art. For example, the GCG program package (Devereux et al), BLASTP, BLASTN, and FASTA (Atschul et al.) and CLUSTAL (Higgins etal, 1992; Thompson, etal, 1994).
One of skill in the art will appreciate that two polynucleotides of different lengths may be compared over the entire length of the longer fragment. Alternatively, small regions may be compared. Normally sequences of the same length are compared for a final estimation of their utility in the practice of the present invention. It is preferred that there be 100% sequence identity between the dsRNA for use as siRNA and at least 15 contiguous nucleotides of the target gene, although a dsRNA having 70%, 75%, 80%, 85%, 90%, or 95% or greater may also be used in the present invention. A siRNA that is essentially identical to a least a portion of the target gene may also be a dsRNA wherein one of the two complementary strands (or, in the case of a self-complementary RNA, one of the two self-complementary portions) is either identical to the sequence of that portion or the target gene or contains one or more insertions, deletions or single point mutations relative to the nucleotide sequence of that portion of the target gene. siRNA technology thus has the property of being able to tolerate sequence variations that might be expected to result from genetic mutation, strain polymorphism, or evolutionary divergence.
Alternatively, a siRNA that is "essentially identical to at least a portion of the target gene can be functionally a dsRNA wherein one of the two complementary strands (or, in the case of a self-complementary RNA, one of the two self-complementary portions) is capable of hybridizing with a portion of the target gene transcript (e.g. under conditions including 400 mM NaCl, 40 mM PIPES pH 6.4, 1 mM EDTA, 50 degrees C or 70 degrees C hybridization for 12-26 hours; followed by washing).
RNA (ribonucleic acid) is known to be the transcription product of a molecule of DNA (deoxyribonucleic acid) synthesized under the action of an enzyme, DNA-dependent RNA polymerase. There are diverse applications of the obtaining of specific RNA sequences, such as, for example, the synthesis of RNA probes or of oligoribonucleotides (Milligan et al), or the expression of genes (see, in particular, Steen et al, Fuerst, et al. and Patent Applications WO 91/05,866 and EP 0,178,863), or alternatively gene amplification as described by Kievits, et al. and Kwoh et al. or in Patent Applications WO 88/10,315 and WO 91/02,818, and U.S. Pat. No. 5,795,715, all of which are expressly incorporated herein by reference .
One of the distinctive features of most DNA-dependent RNA polymerases is that of initiating RNA synthesis according to a DNA template from a particular start site as a result of the recognition of a nucleic acid sequence, termed a promoter, which makes it possible to define the precise localization and the strand on which initiation is to be effected. Contrary to DNA- dependent DNA polymerases, polymerization by DNA-dependent RNA polymerases is not initiated from a 3'-OH end, and their natural substrate is an intact DNA double strand.
Compared to bacterial, eukaryotic or mitochondrial RNA polymerases, phage RNA polymerases are very simple enzymes. Among these, the best known are the RNA polymerases of bacteriophages T7, T3 and SP6. These enzymes are very similar to one another, and are composed of a single subunit of 98 to 100 kDa. Two other phage polymerases share these similarities: that of Klebsiella phage Kl 1 and that of phage BA14 (Diaz et al). Any DNA dependent RNA polymerase is expected to perform in conjunction with a functionally active promoter as desired in the present invention. These include, but are not limited to the above listed polymerases, active mutants thereof, E. coli RNA polymerase, and RNA polymerases I., II, and m from a variety of eukaryotic organisms.
Initiation of transcription with T7, SP6 RNA and T3 RNA Polymerases is highly specific for the T7, SP6 and T3 phage promoters, respectively. The properties and utility of these
polymerases are well known to the art. Their properties and sources are described in U.S. Pat. Nos.: (T7) 5,869,320; 4,952,496; 5,591,601; 6,114,152; (SP6) 5,026,645; (T3) 5,102,802; 5,891,681 ; 5,824,528; 5,037,745, all of which are expressly incorporated herein by reference.
Reaction conditions for use of these RNA polymerases are well known in the art, and are exemplified by those conditions provided in the examples and references. The result of contacting the appropriate template with an appropriate polymerase is the synthesis of an RNA product, which is typically single-stranded. Although under appropriate conditions, double stranded RNA may be made from a double stranded DNA template. See U.S. Pat. No. 5,795,715, incorporated herein by reference. The process of sequence specific synthesis may also be known as transcription, and the product the transcript, whether the product represents an entire, functional gene product or not.
dsRNA for use as siRNA may also be enzymatically synthesized through the use of RNA dependent RNA polymerases such as Q beta replicase, Tobacco mosaic virus replicase, brome mosaic virus replicase, potato virus replicase, etc. Reaction conditions for use of these RNA polymerases are well known in the art, and are exemplified by those conditions provided in the examples and references. Also see U.S. Pat. No. RE35,443, and U.S. Pat. No.4,786,600, both of which are incorporated herein by reference. The result of contacting the appropriate template with an appropriate polymerase is the synthesis of an RNA product, which is typically double- stranded. Employing these RNA dependent RNA polymerases therefore may utilize a single stranded RNA or single stranded DNA template. If utilizing a single stranded DNA template, the enzymatic synthesis results in a hybrid RNA/DNA duplex that is also contemplated as useful as siRNA.
The templates for enzymatic synthesis of siRNA are nucleic acids, typically, though not exclusively DNA. A nucleic acid may be made by any technique known to one of ordinary skill in the art. Non-limiting examples of synthetic nucleic acid, particularly a synthetic oligonucleotide, include a nucleic acid made by in vitro chemical synthesis using phosphotriester, phosphite or phosphoramidite chemistry and solid phase techniques such as described in EP 266,032, incorporated herein by reference, or via deoxynucleoside H- phosphonate intermediates as described by Froehler et al, 1986, and U.S. Patent Serial No. 5,705,629, each incorporated herein by reference. A non-limiting example of enzymatically produced nucleic acid include one produced by enzymes in amplification reactions such as PCR™ (see for example, U.S. Patent 4,683,202 and U.S. Patent 4,682,195, each incorporated
herein by reference), or the synthesis of oligonucleotides described in U.S. Patent No. 5,645,897, incorporated herein by reference. A non-limiting example of a biologically produced nucleic acid includes recombinant nucleic acid production in living cells (see for example, Sambrook, 2001, incorporated herein by reference).
A nucleic acid may be purified on polyacrylamide gels, cesium chloride centrifugation gradients, or by any other means known to one of ordinary skill in the art (see for example, Sambrook (2001), incorporated herein by reference).
The term "nucleic acid" will generally refer to at least one molecule or strand of DNA, RNA or a derivative or mimic thereof, comprising at least one nucleotide base, such as, for example, a naturally occurring purine or pyrimidine base found in DNA (e.g., adenine "A," guanine "G," thymine "T," and cytosine "C") or RNA (e.g. A, G, uracil "U," and C). The term "nucleic acid" encompasses the terms "oligonucleotide" and "polynucleotide." These definitions generally refer to at least one single-stranded molecule, but in specific embodiments will also encompass at least one additional strand that is partially, substantially or fully complementary to the at least one single-stranded molecule. Thus, a nucleic acid may encompass at least one double-stranded molecule or at least one triple-stranded molecule that comprises one or more complementary strand(s) or "comρlement(s)" of a particular sequence comprising a strand of the molecule.
As will be appreciated by one of skill in the art, the useful form of nucleotide or modified nucleotide to be incorporated will be dictated largely by the nature of the synthesis to be performed. Thus, for example, enzymatic synthesis typically utilizes the free form of nucleotides and nucleotide analogs, typically represented as nucleotide triphospates, or NTPs.
These forms thus include, but are not limited to aminoallyl UTP, pseudo-UTP, 5-I-UTP, 5-1-
CTP, 5-Br-UTP, alpha-S ATP, alpha-S CTP, alpha-S GTP, alpha-S UTP, 4-thio UTP, 2-thio- CTP, 2'NH2 UTP, 2'NH2 CTP, and 2' F UTP. As will also be appreciated by one of skill in the art, the useful form of nucleotide for chemical syntheses may be typically represented as aminoallyl uridine, pseudo-uridine, 5-I-uridine, 5-I-cytidine, 5-Br-uridine, alpha-S adenosine, alpha-S cytidine, alpha-S guanosine, alpha-S uridine, 4-thio uridine, 2-thio-cytidine, 2'NH2 uridine, 2'NH2 cytidine, and 2' F uridine. In the present invention, the listing of either form is non-limiting in that the choice of nucleotide form will be dictated by the nature of the synthesis to be performed. In the present invention, then, the inventors use the terms aminoallyl uridine, pseudo-uridine, 5-I-uridine, 5-I-cytidine, 5-Br-uridine, alpha-S adenosine, alpha-S cytidine,
alpha-S guanosine, alpha-S uridine, 4-thio uridine, 2-thio-cytidine, 2'NH2 uridine, 2'NH2 cytidine, and 2' F uridine generically to refer to the appropriate nucleotide or modified nucleotide, including the free phosphate (NTP) forms as well as all other useful forms of the nucleotides.
In certain embodiments, a "gene" refers to a nucleic acid that is transcribed. As used herein, a "gene segment" is a nucleic acid segment of a gene. In certain aspects, the gene includes regulatory sequences involved in transcription, or message production or composition. In particular embodiments, the gene comprises transcribed sequences that encode for a protein, polypeptide or peptide. In other particular aspects, the gene comprises a nucleic acid, and/or encodes a polypeptide or peptide-coding sequences of a gene that is defective or mutated in a hematopoietic and lympho-hematopoietic disorder. In keeping with the terminology described herein, an "isolated gene" may comprise transcribed nucleic acid(s), regulatory sequences, coding sequences, or the like, isolated substantially away from other such sequences, such as other naturally occurring genes, regulatory sequences, polypeptide or peptide encoding sequences, etc. In this respect, the term "gene" is used for simplicity to refer to a nucleic acid comprising a nucleotide sequence that is transcribed, and the complement thereof. In particular aspects, the transcribed nucleotide sequence comprises at least one functional protein, polypeptide and/or peptide encoding unit. As- will be understood by those in the art, this functional term "gene" includes both genomic sequences, RNA or cDNA sequences, or smaller engineered nucleic acid segments, including nucleic acid segments of a non-transcribed part of a gene, including but not limited to the non-transcribed promoter or enhancer regions of a gene. Smaller engineered gene nucleic acid segments may express, or may be adapted to express using nucleic acid manipulation technology, proteins, polypeptides, domains, peptides, fusion proteins, mutants and/or such like. Thus, a "truncated gene" refers to a nucleic acid sequence that is missing a stretch of contiguous nucleic acid residues.
Various nucleic acid segments may be designed based on a particular nucleic acid sequence, and may be of any length. By assigning numeric values to a sequence, for example, the first residue is 1, the second residue is 2, etc., an algorithm defining all nucleic acid segments can be created:
n to n + y
where n is an integer from 1 to the last number of the sequence and y is the length of the nucleic acid segment minus one, where n + y does not exceed the last number of the sequence.
Thus, for a 10-mer, the nucleic acid segments correspond to bases 1 to 10, 2 to 11, 3 to 12 ... and/or so on. For a 15-mer, the nucleic acid segments correspond to bases 1 to 15, 2 to 16, 3 to 17 ... and/or so on. For a 20-mer, the nucleic segments correspond to bases 1 to 20, 2 to 21, 3 to 22 ... and/or so on.
The nucleic acid(s) of the present invention, regardless of the length of the sequence itself, may be combined with other nucleic acid sequences, including but not limited to, promoters, enhancers, polyadenylation signals, restriction enzyme sites, multiple cloning sites, coding segments, and the like, to create one or more nucleic acid construct(s). The overall length may vary considerably between nucleic acid constructs. Thus, a nucleic acid segment of almost any length may be employed, with the total length preferably being limited by the ease of preparation or use in the intended protocol.
To obtain the RNA corresponding to a given template sequence through the action of an RNA polymerase, it is necessary to place the target sequence under the control of the promoter recognized by the RNA polymerase.
A "promoter" is a control sequence that is a region of a nucleic acid sequence at which initiation and rate of transcription are controlled. It may contain genetic elements at which regulatory proteins and molecules may bind, such as RNA polymerase and other transcription factors, to initiate the specific transcription a nucleic acid sequence. The phrases "operatively positioned," "operatively linked," "under control," and "under transcriptional control" mean that a promoter is in a correct functional location and/or orientation in relation to a nucleic acid sequence to control transcriptional initiation and/or expression of that sequence.
A promoter generally comprises a sequence that functions to position the start site for RNA synthesis. The best known example of this is the TATA box, but in some promoters lacking a TATA box, such as, for example, the promoter for the mammalian terminal deoxynucleotidyl transferase gene and the promoter for the SV40 late genes, a discrete element overlying the start site itself helps to fix the place of initiation. Additional promoter elements regulate the frequency of transcriptional initiation. Typically, these are located in the region 30-110 bp upstream of the start site, although a number of promoters have been shown to contain functional elements downstream of the start site as well. To bring a coding sequence "under the control of . a promoter, one positions the 5' end of the transcription initiation site of the
transcriptional reading frame "downstream" of (i.e., 3' of) the chosen promoter. The "upstream" promoter stimulates transcription of the DNA and synthesis of the RNA.
The spacing between promoter elements frequently is flexible, so that promoter function is preserved when elements are inverted or moved relative to one another. The spacing between promoter elements can be increased to 50 bp apart before activity begins to decline. Depending on the promoter, it appears that individual elements can function either cooperatively or independently to activate transcription. A promoter may or may not be used in conjunction with an "enhancer," which refers to a czs-acting regulatory sequence involved in the transcriptional activation of a nucleic acid sequence.
T7, T3, or SP6 RNA polymerases display a high fidelity to their respective promoters.
The natural promoters specific for the RNA polymerases of phages T7, T3 and SP6 are well known. Furthermore, consensus sequences of promoters are known to be functional as promoters for these polymerases. The bacteriophage promoters for T7, T3, and SP6 consist of 23 bp numbered -17 to +6, where +1 indicates the first base of the coded transcript. An important observation is that, of the +1 through +6 bases, only the base composition of +1 and +2 are critical and must be a G and purine, respectively, to yield an efficient transcription template. In addition, synthetic oligonucleotide templates only need to be double-stranded in the -17 to -1 region of the promoter, and the coding region can be all single-stranded. (See Milligan et al.) This can reduce the cost of synthetic templates, since the coding region (i.e., from +1 on) can be left single-stranded and the short oligonucleotides required to render the promoter region double-stranded can be used with multiple templates. A further discussion of consensus promoters and a source of naturally occurring bacteriophage promoters is U.S. Pat. No. 5,891,681, specifically incorporated herein by reference.
Use of a T7, T3 or SP6 cytoplasmic expression system is another possible embodiment. Eukaryotic cells can support cytoplasmic transcription from certain bacterial promoters if the appropriate bacterial polymerase is provided, either as part of the delivery complex or as an additional genetic expression construct.
When made in vitro, siRNA is formed from one or more strands of polymerized ribonucleotide. When formed of only one strand, it takes the form of a self-complementary hairpin-type or stem and loop structure that doubles back on itself to form a partial duplex. The self-duplexed portion of the RNA molecule may be referred to as the "stem" and the remaining,
connecting single stranded portion referred to as the "loop" of the stem and loop structure (FIG. 4). When made of two strands, they are substantially complementary (FIGS. 1, 2, and 3).
The cell containing the target gene may be derived from or contained in any organism (e.g. plant, animal, protozoan, virus, bacterium, or fungus). The plant may be a monocot, dicot or gynmosperm; the animal may be a vertebrate or invertebrate. Preferred microbes are those used in agriculture or by industry, and those that a pathogenic for plants or animals. Fungi include organisms in both the mold and yeast morphologies. Examples of vertebrates include fish and mammals, including cattle, goat, pig, sheep, hamster, mouse, rate and human; invertebrate animals include nematodes, insects, arachnids, and other arthropods. Preferably, the cell is a vertebrate cell. More preferably, the cell is a mammalian cell.
The cell having the target gene maybe from the germ line or somatic, totipotent or pluripotent, dividing or non-dividing, parenchyma or epithelium, immortalized or transformed, or the like. The cell can be a gamete or an embryo; if an embryo, it can be a single cell embryo or a constituent cell or cells from a multicellular embryo. The term "embryo" thus encompasses fetal tissue. The cell having the target gene may be an undifferentiated cell, such as a stem cell, or a differentiated cell, such as from a cell of an organ or tissue, including fetal tissue, or any other cell present in an organism. Cell types that are differentiated include adipocytes, fibroblasts, myocytes, cardiomyocytes, endothelium, neurons, glia, blood cells, megakaryocytes, lymphocytes, macrophages, neutrophils, eosinophils, basophils, mast cells, leukocytes, granulocytes, keratinocytes, chondrocytes, osteoblasts, osteoclasts, hepatocytes, and cells, of the endocrine or exocrine glands.
As used herein, the terms "cell," "cell line," and "cell culture" may be used interchangeably. All of these terms also include their progeny, which is any and all subsequent generations formed by cell division. It is understood that all progeny may not be identical due to deliberate or inadvertent mutations. A host cell may be "transfected" or "transformed," which refers to a process by which exogenous nucleic acid is transferred or introduced into the host cell. A transformed cell includes the primary subject cell and its progeny. As used herein, the terms "engineered" and "recombinant" cells or host cells are intended to refer to a cell into which an exogenous nucleic acid sequence, such as, for example, a small, interfering RNA or a template construct encoding such an RNA has been introduced. Therefore, recombinant cells are distinguishable from naturally occurring cells which do not contain a recombinantly introduced nucleic acid.
In certain embodiments, it is contemplated that RNAs or proteinaceous sequences may be co-expressed with other selected RNAs or proteinaceous sequences in the same host cell. Co-expression may be achieved by co-transfecting the host cell with two or more distinct recombinant vectors. Alternatively, a single recombinant vector may be constructed to include multiple distinct coding regions for RNAs, which could then be expressed in host cells transfected with the single vector.
A tissue may comprise a host cell or cells to be transformed or contacted with a nucleic acid delivery composition and/or an additional agent. The tissue may be part or separated from an organism. In certain embodiments, a tissue and its constituent cells may comprise, but is not limited to, blood (e.g., hematopoietic cells (such as human hematopoietic progenitor cells, human hematopoietic stem cells, CD34+ cells CD4+cells), lymphocytes and other blood lineage cells), bone marrow, brain, stem cells, blood vessel, liver, lung, bone, breast, cartilage, cervix, colon, cornea, embryonic, endometrium, endothelial, epithelial, esophagus, facia, fibroblast, follicular, ganglion cells, glial cells, goblet cells, kidney, lymph node, muscle, neuron, ovaries, pancreas, peripheral blood, prostate, skin, skin, small intestine, spleen, stomach, testes.
In certain embodiments, the host cell or tissue may be comprised in at least one organism. In certain embodiments, the organism may be, human, primate or murine. In other embodiments the organism maybe any eukaryote or even a rokayrote (e.g., a eubacteria, an archaea), as would be understood by one of ordinary skill in the art. One of skill in the art would further understand the conditions under which to incubate all of the above described host cells to maintain them and to permit their division to form progeny.
EXAMPLES
The following examples are included to demonstrate preferred embodiments of the invention. It should be appreciated by those of skill in the art that the techniques disclosed in the examples that follow represent techniques discovered by the inventor to function well in the practice of the invention, and thus can be considered to constitute preferred modes for its practice. However, those of skill in the art should, in light of the present disclosure, appreciate that many changes can be made in the specific embodiments which are disclosed and still obtain a like or similar result without departing from the spirit and scope of the invention.
Materials and Methodology Employed in Examples 1 - 5.
A. Cell proliferation assays.
Transfected HeLa cells were analyzed using Alamar Blue (BioSource International, Inc., CA) at 24 hr intervals. Alamar Blue is a compound, that when reduced by cellular metabolism, changes from anon-fluorescent blue color to a fluorescent red form that is easily quantified. The amount of Alamar Blue reduced is directly proportional to the cell number, providing a rapid - method for assessing cell proliferation. To perform the assay, the Alamar Blue reagent was added into the tissue culture media at a 10% final concentration. The mixture was incubated for 3-6 hr in growth conditions after which fluorescence was quantified using a Spectra Max™ GeminiXS™ (Molecular Devices, Sunnyvale, CA).
B. Immunofluorescence.
HeLa cells used for immunofluorescence were grown on chamber slides in DMEM/10% FBS and transfected with the siRNAs, buffer, or phosphorothioate oligonucleotides. 48 hr after transfection, the cells were fixed with 4% paraformaldehyde PBS. Cells were permeabihzed by exposure to 0.1% Triton X-100/PBS for 5 min and then incubated with 3% BSA in PBS for 1 hr. After incubating with a mouse anti-myc monoclonal antibody (Neomarkers) at a 1:200 dilution in PBS, the cells were washed briefly with PBS, incubated with fluorescein-conjugated goat anti- mouse IgG (Jackson ImmunoResearch Laboratories). The cells were mounted with Vectashield™ (Vector Laboratories). Images were analyzed using an Olympus BX60™ microscope and acquired with the help of a Hitachi KP-c571™ camera and Adobe® Photoshop®.
C. Transfection of HeLa Cells with siRNAs.
Hela S3 cells @ 5 x 103 cells/well were plated in complete medium w/out antibiotics. The cells were incubated overnight @ 37°C in a humidified 5% CO2 incubator. Chemically synthesized siRNA stocks were diluted into 40 ul Opti-MEM™ (Invitrogen) to indicated final concentrations in 250 ul total volume per well. Enzymatically synthesized siRNA stocks were diluted into 40 ul Opti-MEM™ to indicated final concentrations in 250 ul total volume per well.
For transfections, 1.5 ul of Oligofectamine™ (Invitrogen) was added to 6 ul of OptiMEM™ for each well being transfected. The mixture was incubated at room temperature for 5 - 10 min. The diluted Oligofectamine™ was added to prepared siRNAs, mixed gently, and incubated at RT for 15 -20 min. The cell medium was aspirated, and 200 ul fresh growth medium was added to each well. The medium was mixed gently, and then overlaid with ~50 ul of the appropriate siRNA/oligofectamine complex. The transfected cells were incubated at 37 °C in a humidified 5% CO2 incubator.
D. Direct Recovery of siRNAs.
To prepare chemically or enzymatically produced siRNAs for transfection, they must be purified from accompanying salts, proteins, and non-dsRNA nucleic acids. The hybridization, transcription, or nuclease digestion reactions used to produce siRNAs can be phenol extracted with 2X volumes of buffered phenol, extracted with 2X volumes of ether or chloroform, and precipitated by adding NH4OAC to a final concentration of 0.5 M and 2X volumes of ethanol. The siRNA is recovered by centrifuging at 13,200 RPM. The siRNA pellet is washed one time with 70% ethanol.
E. Gel Purification of siRNAs.
To gel purify siRNAs, 10 μl of 50% sucrose/0.25% bromophenol blue is added to the hybridization, transcription, or nuclease digestion reaction. The sample is loaded on a 12% polyacrylamide gel and electrophoresed at 250 volts for one hour. The siRNA is detected by UV shadowing (Sambrook 2001). The product band is excised from the gel, transferred to a microfuge tube with 400 μl of 2 mM EDTA, and incubated overnight at 37°C. The siRNA is recovered by transferring the solution to a new microfuge tube, adding NH4OAC to a final concentration of 0.5 M, adding 2X volumes of ethanol, incubating at -20°C for fifteen minutes, and centrifuging at 13,200 RPM for fifteen minutes. The siRNA pellet is washed IX with 70% ethanol and then dried.
F. Column Purification of siRNAs.
400 μl of 1.2X Binding Buffer (625 mM NaCl, 62.5% EtOH) is added to hybridization, transcription, or nuclease-digestion reaction. A glass fiber filter in a column is equilibrated with
400 μl of IX Binding Buffer (500 mM NaCl, 50% EtOH). The siRNA/binding buffer mixture is added to the pre-wet column and spun at 13,200 RPM for 2 minutes. The column is washed 2X with 500 μl of IX Binding Buffer. 100 μl of nuclease-free water pre-heated to 75°C is added to the column and incubated for two minutes. The column is spun at 13,200 RPM for 2 minutes. siRNA is in the elute.
G. RNAse digestion.
In cases where a leader sequence is used to improve the yield of siRNAs and reduce template constraints, the following nuclease digestion step is employed to remove a non- homologous leader sequence and non-hybridized RNAs. Add 50 μl of nuclease free water, 6 μl of 10X Nuclease buffer (100 mM Tri pH 7.5, 25 mM MgCl2, 1 mM CaCl2), and 10 U of RNase A or 10,000 U of RNase TI, or similar unit activity of RNase Sa. Incubate for thirty minutes at room temperature.
Leader sequence used may dictate which of the nucleases to use. Single strand specific Rnases are preferred.. Thus, RNase TI preferentially digests single-stranded RNA at G residues. RNase A preferentially digests at C and U residues. If RNase TI is to be used, the last nucleotide in the leader sequence is preferred to be a G to eliminate the entire leader sequence from the siRNA. Likewise, if RNase A is to be used, the last nucleotide in the leader sequence should be a C or U. Additionally, RNase Sa, RNase Sa2, or RNase Sa3, whose use and properties are well known to those of skill in the art, may be employed to digest the leader sequences. See Hebert et al. (1997) and Pace et al. (1998), both of which are expressly incorporated herein by reference.
H. T7 RNA Polymerase
T7 RNA Polymerase is used as a preparation that includes 200 U/ul of T7 RNA Polymerase, Inorganic Pyrophosphatase (IPP) 0.05 U/ul, Placental Ribonuclease Inhibitor (Sambrook 2001) 0.3 U/ul, Superasin™ (Ambion) 2 U/ul, and 1% chaps.
EXAMPLE 1: Model target genes.
C-myc and GAPDH were chosen to evaluate the impact of siRNA on the expression of genes in mammalian cells. The c-myc proto-oncogene can be a transcription repressor and activator and has important roles in cell death and cell cycle progression (reviewed in Ryan et al, 1996). GAPDH is the metabolic protein Glyceraldehyde-3-phosphate
Dehydrogenase. It is involved in glycolysis. Reducing the expression of either gene slows the cell division rate, which can be tracked using the Alomar Blue assay described above or by quantifying the number of healthy cells. In addition, the abundance of the mRNA and protein from each gene can be calculated using standard techniques (RT-PCR, Northern analysis, immunofluorescence, or Western analysis).
EXAMPLE 2: siRNA Target Site Selection.
Four different double-stranded 21mer siRNAs were designed and prepared for both c- myc and GAPDH. These siRNAs were tested to determine which siRNA provided the greatest effect without affecting non-target genes.
c-myc siRNA Development
The siRNAs specific to different regions of the c-myc gene are listed in Table 1 (SEQ ID NOS: 3-10) and diagrammed in FIG. 5. Also shown are the locations of the start codon (start),* stop codon (stop), coding region, 5' and 3' UTR's as well as the binding site of a well- characterized antisense oligonucleotide. The antisense oligonucleotide that we used has previously been shown to reduce c-myc expression (Kimura et al, 1995) and served as a positive control in our experiments.
1.5 nanomoles of the sense and anti-sense siRNAs were mixed in a solution comprising 100 mM KOAc, 30 mM HEPES-KOH pH 7.4, and 2 mM MgOAc. The solutions were incubated at 37oC for one minute, then at room temperature for one hour. The samples were evaluated by non-denaturing 12% PAGE to confirm that the majority of the RNA was double- stranded. The siRNAs were then kept in aliquots at -20°C until they were transfected.
HeLa cells were transfected with 100 nM of the siRNAs and the antisense oligonucleotide. Since reduction in c-myc expression levels can lead to a reduction in cell division rates, cell proliferation was monitored at 24 hr intervals following transfection. Differences in proliferation rates were first noted 48 hr after the HeLa cells had been transfected with the siRNAs. FIG. 6 depicts the relative cell proliferation (as compared to a buffer- transfected control) of HeLa cells transfected with the various siRNAs, as well as with the optimized antisense oligonucleotide, 48 hr after transfection. The data demonstrate that siRNAs to different regions of the mRNA have variable effects on cell proliferation.
Table 1.
The reduction in cell proliferation observed with the siRNA to the 3' UTR was similar to that found using the optimized antisense phosphorothioate oligonucleotide complementary to the start codon of the c-myc mRNA (Kimura etal, 1995). In contrast, siRNAs against the 5' UTR
and Exon 2 of the c-myc mRNA affected cell proliferation similarly to the scrambled siRNA sequence, which was used as a negative control (FIG. 6). All of the transfections and cell proliferation assays were reproduced in independent experiments and the differences in cell proliferation rates were shown to be statistically significant.
Immunofluorescence experiments using an anti-myc antibody were used to measure c- myc protein levels to confirm that siRNAs were reducing expression of c-myc (FIG. 7). As with the cell proliferation assay, the siRNA corresponding to the 3' UTR induced the greatest reduction in fluorescence (FIG. 7), indicating the lowest levels of protein. A representative example of the immunofluorescence data for c-myc protein levels is shown in FIG. 8. These data clearly demonstrate that the siRNA against the 3' UTR leads to a drastic reduction in c-myc protein levels. The amount of fluorescence detected for the 3' UTR sample was nearly equivalent to that observed for the "secondary antibody-only" control and no change in GAPDH protein levels was detected after transfection with any of the siRNAs complementary to the c-myc mRNA (data not shown), indicating that the antiviral response pathway was not induced. The 3' UTR specific c-myc siRNA was selected for subsequent studies.
GAPDH siRNA Development
The siRNAs specific to different regions of the GAPDH gene are listed in Table 2 (SEQ TO NOS: 14-21) The four siRNAs were prepared from DNA oligonucleotides using in vitro transcription with T7 RNA polymerase.
Table 2.
The following synthetic DNA oligomers were purchased from Integrated DNA Technologies:
Table 3.
In separate reactions, the T7 promoter primer was mixed with each of the sense and antisense templates in separate reactions and converted to transcription templates. Templates for in vitro transcription may be double-stranded over the length of the promoter sequence (Milligan et al 1987). Making the entire template double-stranded improves the transcription of siRNAs, therefore the following procedure is used to convert DNA oligonucleotides to transcription templates for siRNA synthesis.
The DNA templates were diluted to 100 μM in nuclease-free water. 2 μl of each DNA template was mixed with 2 μl of 100 μM Promoter Primer and 6 μl of Hybridization Buffer (20 mM Tris pH 7.0, 100 mM KC1, 1 mM EDTA). The oligonucleotide mixtures were heated to 70°C for five minutes, then incubate at 37°C for five minutes. 2 μl of 10X reaction Buffer (150 mM Tris pH 7.0, 850 mM KC1, 50 mM MgCl2, 50 mM (NH4)2SO ), 2 μl of 10 dNTP mix (2.5 mM dATP, 2.5 mM dCTP, 2.5 mM dGTP, and 2.5 mM dTTP), 4 μl of water, and 2 μl of 5 U/ml klenow DNA polymerase was added to each oligonucleotide mixture. The reaction was incubated at 37°C for thirty minutes.
The templates were transcribed using T7 RNA polymerase by mixing together the following: 2 μl siRNA DNA Template; 2 μl 75 mM ATP; 2 μl 75 mM CTP; 2 μl 75 mM GTP; 2 μl 75 mM UTP; 2 μl 1 OX Transcription Buffer (400 mM Tris pH 8.0, 240 mM MgCl2, 20 mM Spermidine, 100 mM DTT); 6 μl Nuclease-Free Water; and 2 μl T7 RNA Polymerase (T7 RNA Polymerase- 200 U/ul, Inorganic Pyrophosphatase (IPP) 0.05 U/ul, RNase Inhibitor 0.3 U/ul, Superasin 2 U/ul, 1% chaps).
This reaction mix was incubated for two to four hours at 37°C. The RNA products were then mixed and incubated overnight at 37°C to facilitate annealing of the complementary strands of the siRNAs. The leader sequences were removed by treatment with RNase TI and the resulting siRNAs were gel purified.
HeLa cells were transfected with 10 nM of each of the GAPDH-specific siRNAs. 48 hours after transfection, the cells were harvested and RNA was isolated using the RNAquesous kit (Ambion). Equal amounts of the RNA samples were fractionated by agarose gel electrophoresis and transferred to positively charged nylon membranes using the NorthernMax- Gly kit (Ambion). The Northern blots were probed for GAPDH, cyclophilin, and 28s rRNA using the reagents and protocols of the NorthernMax-Gly kit. The Northern blots were exposed to a phosphorimager screen and quantified using the Molecular Analyst (BioRad). The relative reduction in GAPDH mRNA levels in siRNA transfected cells versus untreated cells is provided in FIG. 9. For GAPDH, the 5' Medial siRNA provided the greatest level of gene silencing and was selected for subsequent evaluation.
EXAMPLE 3: Potency of Chemical versus Enzymatic synthesis of siRNA- c-myc.
The 3' UTR siRNA described above was produced by in vitro transcription to compare the potency of siRNAs prepared by enzymatic means to siRNAs generated by chemical synthesis. The following synthetic DNA oligomers were purchased from Integrated DNA Technologies:
Table 4.
The T7 promoter primer was mixed with the 3' UTR sense and antisense templates in separate reactions and converted to transcription templates. Templates for in vitro transcription may be double-stranded over the length of the promoter sequence (Milligan et al. 1987). Making the entire template double-stranded improves the transcription of siRNAs, therefore the following procedure is used to convert DNA oligonucleotides to transcription templates for siRNA synthesis.
The DNA templates were diluted to 100 μM in nuclease-free water. 2 μl of each DNA template was mixed with 2 μl of 100 μM Promoter Primer and 6 μl of Hybridization Buffer (20 mM Tris pH 7.0, 100 mM KC1, 1 mM EDTA). The oligonucleotide mixtures were heated to 70°C for five minutes, then incubated at 37°C for five minutes. 2 μl of 10X reaction Buffer (150 mM Tris pH 7.0, 850 mM KC1, 50 mM MgCl2, 50 mM (NIL 2SO4), 2 μl of 10 dNTP mix (2.5 mM dATP, 2.5 mM dCTP, 2.5 mM dGTP, and 2.5 mM dTTP), 4 μl of water, and 2 μl of 5 U/ml klenow DNA polymerase was added to each oligonucleotide mixture. The reaction was incubated at 37°C for thirty minutes.
The templates were transcribed using T7 RNA polymerase by mixing together the following: 2 μl siRNA DNA Template; 2 μl 75 mM ATP; 2 μl 75 M CTP; 2 μl 75 mM GTP; 2 μl 75 mM UTP; 2 μl 10X Transcription Buffer (400 mM Tris pH 8.0, 240 mM MgCl2, 20 mM Spermidine, 100 mM DTT); 6 μl Nuclease-Free Water; and 2 μl T7 RNA Polymerase (T7 RNA Polymerase- 200 U/ul, Inorganic Pyrophosphatase (IPP) 0.05 U/ul, RNase Inhibitor 0.3 U/ul, Superasin 2 U/ul, 1% chaps).
This reaction mix was incubated for two to four hours at 37°C. The RNA products were then mixed and incubated overnight at 37°C to facilitate annealing to form siRNA. The leader sequences were removed by digestion with RNase TI . The resulting siRNA was then gel purified.
Different molar amounts of in vitro transcribed and chemically synthesized siRNAs specific to the 3' UTR of the c-myc mRNA were transfected into HeLa S3 cells. The HeLa S3 cells were evaluated for proliferation using Alamar Blue (BioSource International, Inc., CA) at 72 hrs post-transfection. The in vitro transcribed siRNAs at a concentration of 10 nM reduced cell proliferation by greater than 50% (FIG. 10). In contrast, synthetic siRNA to the same target sequence at a concentration of 100 nM reduced proliferation by only 40% (FIG. 10). The experiment was done in triplicate and has been repeated many times with identical results.
The in vitro transcribed and chemically synthesized siRNAs were quantified by both the Picogreen Assay (Molecular Probes) and by measuring the absorbance of the samples at 260 nm (Sambrook 2001). Both methods confirmed the concentrations of the siRNAs, supporting our conclusion that our preparative procedure yields siRNAs that are at least ten-fold more potent than siRNAs prepared by standard methods.
To rule out the possibility that purification procedures were providing an advantage to enzymatic siRNA preparations, both the in vitro transcribed and chemically synthesized siRNAs were gel purified. Gel purification did not enhance the potency of the chemically synthesized siRNA, confirming that there is a fundamental difference between the siRNAs produced by the two methods. Three different methods to purify siRNAs prior to transfection: Phenol extraction/ethanol precipitation, gel purification, and column purification. Each of the methods yield siRNAs that are at least ten times as potent as equivalent siRNAs prepared by standard chemical synthesis.
EXAMPLE 4: Potency of Chemical versus Enzymatic synthesis of siRNA - GAPDH.
To confirm the general enhanced potency of enzymatically synthesized siRNAs and to confirm that the higher potency was due to a reduction in target mRNA concentration, siRNAs specific to GAPDH were compared for their capacity to reduce GAPDH mRNA levels in HeLa cells. Chemically synthesized and enzymatically synthesized siRNAs specific to the same target sequence in the 5' Medial Region of the GAPDH mRNA were prepared and transfected at varying concentrations into HeLa cells. The cells were harvested forty-eight hours after transfection. Total RNA from the samples was harvested, fractionated by agarose gel electrophoresis, and transferred to a membrane. The resulting Northern blot was incubated with a probe specific to the GAPDH mRNA. The relative abundance of GAPDH mRNA in the various samples was determined by imaging the probe signal on the Northern blot using a phosphorimager. Cyclophilin (a common housekeeping gene used for sample normalization) was assessed on the same Northern blots to normalize the samples.
FIG. 11 shows the GAPDH signal normalized to cyclophilin in the samples treated with varying concentrations of the two siRNAs. Consistent with our results with c-myc, the in vitro transcribed siRNAs were ten- to twenty-fold more potent than the chemically synthesized siRNA to the same target sequence. This experiment confirms that the improved potency of enzymatically synthesized siRNAs is consistent for different gene targets and that the improved potency derives from an ability to decrease the concentration of the target mRNA and not through some other cellular process.
EXAMPLE 5: SiRNA of increased potency synthesis by RNA copying (via RNA dependent RNA polymerase).
Recombinant protein P2 of the double-stranded RNA bacteriophage phi6 (P2 replicase) is an RNA polymerase that binds single-stranded RNAs and synthesizes a complementary strand to create dsRNA (Makeyev and Bamford, 2000). P2 replicase was used to convert single-stranded 21mer oligonucleotides bearing sequence identical to or complementary to c-myc mRNA into 21mer dsRNAs. These dsRNAs were transfected into HeLa cells at concentrations of 5 or 10 nM in the cell culture medium. Seventy-two hours after transfection, the cells were counted. Reduction in c-myc protein levels limits cell-division, thus more potent siRNAs result in reduced numbers of cells.
FIG. 12 shows the effect on cell growth of four different siRNAs transfected at two different concentrations. The first and second sets show cell numbers for cells transfected with chemically synthesized, single-stranded RNAs that had been converted to dsRNAs by P2 replicase. The first set was a dsRNA specific to the c-myc mRNA. The second set was a scrambled sequence bearing a nucleotide composition equivalent to the c-myc-specific siRNA. The third and fourth sets are c-myc-specific and scrambled sequence siRNAs whereby both strands were chemically synthesized. The P2 replicase-generated dsRNAs specific to c-myc are 10-20 fold more potent than the equivalent chemically synthesized siRNA (FIG. 12). The enhanced potency is sequence specific as the equivalently treated scrambled sequence has a profile equivalent to the chemically synthesized, scrambled sequence siRNA.
Surprisingly, both sense and anti-sense strand chemically synthesized RNA can be converted to siRNA of increased potency by the action of the P2 replicase (data not shown). These data indicate that the chemically synthesized anti-sense strand of the siRNA can target mRNA degradation as well as an enzymatically prepared RNA.
EXAMPLE 6: Enzymatically synthesized siRNA incorporating nucleotide analogs.
Parrish et al. (2000) found that several nucleotide analogs could be present in double- stranded RNAs without eliminating the capacity of the dsRNAs to suppress gene expression. However, Parrish et al. (2000) observed no particular benefit to using the nucleotide analogs in their studies.
Elbashir et al (2001) reported a slight improvement in the effectiveness of siRNAs that had 3' overhanging di-nucleotide deoxy-thymidines rather than ribo-uridines. Elbashir et al. suggested that the improvement might be due to improved nuclease stability of the deoxy- thymidines, though they also indicated that they saw improved yield of the siRNAs with the deoxy-thymidines which could translate to improved siRNA quality and thus an improved molecule for siRNA experiments.
A variety of nucleotide analogs were systematically tested for their capacity to improve the potency of siRNAs. Analogs of UTP, CTP, GTP, and ATP were incorporated by transcription into siRNAs specific to GAPDH. The siRNAs were prepared and transfected into HeLa cells. Forty-eight hours after transfection, the treated cells were harvested, total RNA was recovered, and the levels of GAPDH mRNA were assessed by Northern analysis. Cyclophilin (a
common housekeeping gene used for sample normalization) and 28S rRNA were also probed to normalize the samples. Many of the analogs performed equivalently to the unmodified siRNA (FIG. 13), consistent with what Parrish et al (2000) had observed in their experiments. However, several analogs provided a 2-4-fold enhancement in siRNA potency, over and above the enhancement of potency resulting from enzymatic synthesis alone.
Interestingly, replacing an oxygen with a thiol on the bridging phosphate 5 ' to any of the four nucleotides improved the potency of the GAPDH-specific siRNA (see alpha-S ATP, alpha- S CTP, alpha-S GTP, alpha-S UTP). Each of these analogs are reported to provide some protection to nucleases (Black et al. (1972) "Studies on the Toxicity and Antiviral Activity of Various Polynucleotides," Antimicrob. Agents Chemotherap. 3, 198-206), though our work suggests that the level of protection is significantly less than two-fold (data not shown). Notably, each of the alpha-thio modified NTPs provide similar enhancements in siRNA potency. If nuclease stability was important in improving siRNA potency as was suggested (Elbashir 2001), then one would expect that the modified uridine would be more beneficial because it would ensure that the single-stranded, overhanging di-nucleotide remained intact.
Further evidence that nuclease stability is not that critical to siRNA potency is the observation that the siRNAs with 2'NH2 Uridine and 2' NH2 Cytidine as well as 2'F Uridine (these analogs are known to improve RNA stability) perform no better than the unmodified siRNAs (FIG. 13). Based on these data, it is not nuclease stability but rather the stability of the siRNA duplex that has a substantial impact on tile potency of the siRNAs. Alpha-thio- nucleotides reduce the stability of the dsRNA duplex. Presumably, an early step in the gene silencing pathway is the dissociation of the double-stranded siRNA to facilitate hybridization of the siRNA to the mRNA target. Reducing the stability of the siRNA duplex would make it less difficult for the proteins involved in the gene silencing pathway to dissociate the anti-sense and sense strands of the siRNA, thus improving their potency. Analogs other than alpha-thio-NTPs that decrease dsRNA stability and thus would be expected to improve siRNA potency include 4- thio UTP, inosine triphosphate, 4-Ethyl CTP, etc.
To test this hypothesis, GAPDH-specific siRNAs with 4-thio UTP and 2-thio UTP were synthesized . The 2-thio modification stabilizes A-U base pairs whereas the 4-thio modification destabilizes A-U basepairs (Testa et al, 1999). If duplex stability truly is an important predictor of siRNA potency, then the 2-thio modified siRNA would reduce potency and the 4-thio modification would enhance potency. The data are consistent with this hypothesis (FIG. 13).
EXAMPLE 7: Chemical Synthesis and use of siRNAs with reduced duplex stability.
Standard and modified 21mer ribo-oligomers of the two sequences provided below were chemically synthesized using an Expedite Nucleic Acid Synthesis System™ (Applied Biosystems) and the associated synthesis program. Two types of phosphoramidites were used: 5 '-O-DMT-N6-phenoxyacetyl)-2 '-O-t-butyldimethylsilyl group (TBDMS)-nucleoside-3 '-O-(B- cyanoethyl-N,N-diisopropylamino) phosphoramidite (Wu et al.,1989) and 5'-O-DMT-N6- phenoxyacetyl)- 2'O-TriisoρropylsilylOxyMethyl (TOM)-3'-O-(B-cyanoethyl-N,N- diisopropylamino) phosphoramidite. For both types of amidites, we followed the same basic procedure for coupling and deprotecting as described (Wincott et al. 1995). Following the addition of the last nucleotide, ribo-oligomers generated with either phosphoramidite were cleaved from the solid support with 40% aqueous methylamine and deprotected with tetrabutylammomum fluoride (Wincott 1995).
Ribo-oligomers with phosphorothioates at defined positions were produced by incubating the elongating oligonucleotide with thiosulfonate for 30 seconds after the appropriate nucleoside was added (Iyer et al 1990). Ribo-oligomers with 4-thio-U and inosine were produced by substituting a 4-thio-U phosphoramidite for the U precursor and an Inosine phosphoramidite for the G precursor in the standard synthesis procedure.
The incorporation of these modified nucleotide analogs, because they act to effectively reduce overall duplex stability in RNA duplexes, is expected to lead to substantially enhanced potency of the resultant siRNAs.
All of the compositions and/or methods disclosed and claimed herein can be made and executed without undue experimentation in light of the present disclosure. While the compositions and methods of this invention have been described in terms of preferred embodiments, it will be apparent to those of skill in the art that variations may be applied to the compositions and/or methods and in the steps or in the sequence of steps of the method described herein without departing from the concept, spirit and scope of the invention. More specifically, it will be apparent that certain agents which are both chemically and physiologically related may be substituted for the agents described herein while the same or similar results would be achieved. All such similar substitutes and modifications apparent to those skilled in the art are deemed to be within the spirit, scope and concept of the invention as defined by the appended claims.
REFERENCES
The following references, to the extent that they provide exemplary procedural or other details supplementary to those set forth herein, are specifically incorporated herein by reference.
U.S. Patent No. 4,415,732 U.S. Patent No. 4,458,066
U.S. Patent No. 4,682,195
U.S. Patent No. 4,683,202
U.S. Patent No. 4,786,600
U.S. Patent No. 4,952,496 U.S. Patent No. 5,026,645
U.S. Patent No. 5,037,745
U.S. Patent No. 5,102,802
U.S. Patent No. 5,591,601
U.S. Patent No. 5,645,897 U.S. Patent No. 5,705,629
U.S. Patent No. 5,795,715
U.S. Patent No. 5,824,528
U.S. Patent No. 5,869,320
U.S. Patent No. 5,889,136 U.S. Patent No. 5,891,681
U.S. Patent No. 6,005,087
U.S. Patent No. 6,114,152
U.S. Patent No. RE35,443
Euro. Patent No. 178,863 Euro. Patent No. 266,032
WO 00/44914
WO 01/36646
WO 01/68836
WO 88/10,315 WO 91/02,818
WO 91/05,866
WO 99/32619
Anonymous, "The siRNA user guide." Revised August 26, 2001, [online], [retrieved on 2002- 01-31] Retrieved from Max Planck Institute for Biophysical Chemistry using Internet <URL:http://www.mpibρc.gwdg.de/abteilungen/l 00/105/siRNAuserguide.pdf . Atschul, S.F. et al, J. Molec. Biol. 215:403 (1990).
Bergstrom D.E., Zhang, P. and Johnson W.T. (1997) Nucleic Acids Res. 25:1935-1942. Bernstein, E, Caudy, AA, Hammond, SM, and Hannon, GJ (2001). Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409: 363-366. Black et al. (1972) "Studies on the Toxicity and Antiviral Activity of Various Polynucleotides," Antimicrob. Agents Chemotherap. 3, 198-206.
Caplen, NJ, Parrish, S, Imani, F, Fire, A, and Morgan, RA (2001). Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc Natl Acad Sci USA 98: 9742-9747. Devereux, J., et al, Nucleic Acid. Res. 12(1):387 (1984). Dharmacon Research. (July, 2001) siRNA Oligonucleotides for RNAi Applications: Dharmacon siACE-RNAi™ Options. Technical Bulletin #003. [online], [retrieved on 2002-01-31] using Internet
<URL:http://www .dharmacon.com/tech tech003.html>. Diaz et al, J. Mol. Biol. 229: 805-811 (1993). Dubins, DN, Lee, A, Macgbregor, RB, and Chalikian, TV (2001). On the stability of double stranded nucleic acids. J. Am. Chem. Soc. 123:9254-59. Elbashir, SM, Harborth, J, Lendeckel, W, Yalcin, A, Weber, K, and Tuschl, T (2001). Duplexes of 21 -nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411: 494-498. Fire, A, Xu, S, Montgomery, MK, Kostas, SA, Driver, SE, and Mello, CC (1998). Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391: 806-811. Froehler, etal (1986). Synthesis of DNA via deoxynucleoside H-phosphonate intermediates. Nucleic Acids Res. 14(13):5399-407. Fuerst, T. R. et al (1987) Molecular and Cellular Probes 7, 2538-2544.
Grishok, A, Pasquinelli, AE, Conte, D, Li, N, Parrish, S, Ha, I, Baillie, DL, Fire, A, Ruvkun, G, and Mello, CC (2001). Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell 106: 23-34.
Hamilton, AJ, and Baulcombe, DC (1999). A species of small antisense RNA in posttranscriptional gene silencing in plants. Science 286: 950-952. Hammond, SM, Boettcher, S, Caudy, AA, Kobayashi, R, and Harmon, GJ (2001). Argonaute2, a link between genetic and biochemical analyses of RNAi. Science 293: 1146-1150. Hebert EJ, Grimsley GR, Hartley RW, Horn G, Schell D, Garcia S, Both V, Sevcik J, and Pace
CN. (1997). Purification of ribonucleases Sa, Sa2, and Sa3 after expression in
Escherichia coli. Protein Expr. Purif. 11, 162-8. Higgins, D.G., Bleasby, A.J. and Fuchs, R. (1992) CLUSTAL V: improved software for multiple sequence alignment. Computer Applications in the Biosciences (CABIOS), 8 (2) : 189- 191. Hutvagner, G., McLachlan, J., Pasquinelli, A. E., Balint, E., Tuschl, T., and Zamore, P. D.
(2001). A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 293: 834-838. Iyer RP, Egan W, Regan JB, Beaucage SL (1990) J. Am. Chem. Soc. 112, 1253-1254. Kawase, Y., Iwai, S., Inoue, H., Miura, K. and Ohtsuka E. (1986) Nucleic Acids Res. 19:7727- 7736.
Kievits, T. et al Journal of Virological Methods 35, 273-286 (1991).
Kimura S, Maekawa T, Hirakawa K, Murakami A and Abe T (1995). Alterations of c-myc expression by antisense oligodeoxynucleotides enhance the induction of apoptosis in HL-
60 cells. Cancer Research 55: 1379-1384. Knight, SW, and Bass, BL (2001). A Role for the RNase HI Enzyme DCR-1 in RNA
Interference and Germ Line Development in C. elegans. Science 2: 2. Kwoh, D. Y. et al. Proc. Natl. Acad. Sci. USA 86, 1173-1177 (1989). Lesk, A.M., ed., (1988). Computational Molecular Biology. Oxford University Press, New
York, 1988. Makeyev EV, Bamford DH [2000] "Replicase activity of purified recombinant protein P2 of double-stranded RNA bacteriophage phi6," EMBO J 19:124-33. Milligan, J. F., Groebe, D. R., Witherell, G. W. and U enbeck, O. C. (1987) Nucleic Acids Res.
25, 8783-8798 Montgomery, MK, Xu, S and Fire, A (1998). RNA as a target of double-stranded RNA-mediated genetic interference in Caenorhabditis elegans. Proc Natl Acad Sci USA 95: 15502-
15507. Nguyen HK, Auffray P, Asseline U, Dupret D, Thuong NT (1997) Nucl. Acids Res.25, 3059-65.
Pace CN, Hebert EJ, Shaw KL, Schell D, Both V, Krajcikova D, Sevcik J, Wilson KS, Dauter Z, Hartley RW, and Grimsley GR (1998). Conformational stability and thermodynamics of folding of ribonucleases Sa, Sa2 and Sa3. J. Mol. Biol. 279, 271-86.
Parrish et al. (2000) Functional anatomy of a dsRNA trigger: differential requirement for the two trigger strands in RNA interference. Mol Cell.6(5): 1077-87.
Ryan, KM and Birnie GD (1996). Myc oncogenes: the enigmatic family. Biochem. J. 314: 713- 721.
Sambrook, J, Russell DW (2001) Molecular Cloning, Cold Spring Harbor Laboratory Press.
Shishkina, I.G. and F. Johnson, Chem Res Toxicol, 2*000, 13, 907-912. Smith, D.W., ed. (1986) Biocomputing: Informatics and Genome Projects. Academic Press, New York, 1993.
Stein, CA (2001) The experimental use of antisense oligonucleotides: a guide for the perplexed. J. Clinical Invest. 108: 641-644. .
Testa SM, Disney MD, Turner DH, Kierzek R (1999) "Thermodynamics of RNA-RNA Duplexes with 2- or 4-thiouridines," Biochemistry 38, 16655-62.
Thompson J.D., Higgins D.G., Gibson TJ. "CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice." Nucleic Acids Res. 22:4673-4680 (1994).
Wincott F, DiRenzo A, Grimm S, Tracz D, Workman C, Sweedler D, Gonzalez C, Scaringe S, Usman N (1995) "Synthesis, Deprotection, analysis and Purification of RNA and
Ribozymes," Nucl. Acids Res. 23, 2677-2684.
Wu,T., Ogilvie,K.K., and Pon, R.T. (1989) Nucl. Acids Res. 17, 3501-3517.
Zamore, PD, Tuschl, T, Sharp, PA, and Bartel, DP (2000). RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 101: 25-33.