WO2003059910A1 - Intermediates for preparing glycogen phosphorylase inhibitors - Google Patents
Intermediates for preparing glycogen phosphorylase inhibitors Download PDFInfo
- Publication number
- WO2003059910A1 WO2003059910A1 PCT/IB2003/000034 IB0300034W WO03059910A1 WO 2003059910 A1 WO2003059910 A1 WO 2003059910A1 IB 0300034 W IB0300034 W IB 0300034W WO 03059910 A1 WO03059910 A1 WO 03059910A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- acid
- mixture
- reaction
- solution
- Prior art date
Links
- 0 CC1(C)O[C@@](COC2C*)C2O1 Chemical compound CC1(C)O[C@@](COC2C*)C2O1 0.000 description 4
- DEIWANNNLCOXCI-IATFEOPMSA-N C=[O][C@H](C(C1)([C@@H]1c1ccccc1)NC(c1cc2cc(Cl)ccc2[nH]1)=O)C(N(C[C@H]1O)C[C@@H]1O)=O Chemical compound C=[O][C@H](C(C1)([C@@H]1c1ccccc1)NC(c1cc2cc(Cl)ccc2[nH]1)=O)C(N(C[C@H]1O)C[C@@H]1O)=O DEIWANNNLCOXCI-IATFEOPMSA-N 0.000 description 1
- XZYBWCDJMBCCJX-OLQVQODUSA-N CC1(C)O[C@@H](CNC2)[C@@H]2O1 Chemical compound CC1(C)O[C@@H](CNC2)[C@@H]2O1 XZYBWCDJMBCCJX-OLQVQODUSA-N 0.000 description 1
- BUOXLOFKQFXCFL-UHFFFAOYSA-N CCC1OC(C)(C)OC1CC Chemical compound CCC1OC(C)(C)OC1CC BUOXLOFKQFXCFL-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
Definitions
- the instant invention provides novel processes and intermediates useful in the preparation of certain N-(indole-2-carbonyl)- ⁇ -alaninamide compounds, which compounds are glycogen phosphorylase inhibitors useful in the treatment of diseases such as hypercholesterolemia, hyperglycemia, hyperinsulinemia, hyperlipidemia, hypertension, atherosclerosis, diabetes, diabetic cardiomyopathy, infection, tissue ischemia, myocardial ischemia, and in inhibiting tumor growth.
- diseases such as hypercholesterolemia, hyperglycemia, hyperinsulinemia, hyperlipidemia, hypertension, atherosclerosis, diabetes, diabetic cardiomyopathy, infection, tissue ischemia, myocardial ischemia, and in inhibiting tumor growth.
- Type 1 diabetes insulin dependent diabetes mellitus
- Type 2 diabetes non-insulin dependent diabetes mellitus
- Treatment of non-insulin dependent diabetes mellitus usually consists of a combination of diet, exercise, oral agents, e.g., sulfonylureas, and, in more severe cases, insulin.
- clinically available hypoglycemic agents can have other side effects that limit their use. In any event, where one of these agents may fail in an individual case, another may succeed. A continuing need for hypoglycemic agents, which may have fewer side effects or succeed where others fail, is clearly evident.
- Atherosclerosis a disease of the arteries, is recognized to be the leading cause of death in the United States and Western Europe.
- the pathological sequence leading to atherosclerotic development and occlusive heart disease is well known. The earliest stage in this sequence is the formation of "fatty streaks" in the carotid, coronary, and cerebral arteries, and in the aorta. These lesions are yellow in color due to the presence of lipid deposits found principally within smooth-muscle cells and in macrophages of the intima layer of the arteries and aorta.
- fibrous plaque which consists of accumulated intimal smooth muscle cells laden with lipid and surrounded by extra-cellular lipid, collagen, elastin, and proteoglycans. These cells, plus matrix, form a fibrous cap that covers a deeper deposit of cell debris and more extra cellular lipid, which comprises primarily free and esterified cholesterol.
- the fibrous plaque forms slowly, and is likely in time to become calcified and necrotic, advancing to the so-called “complicated lesion” which accounts for the arterial occlusion and tendency toward mural thrombosis and arterial muscle spasm that characterize advanced atherosclerosis.
- CVD cardiovascular disease
- CVD cardiovascular disease
- medical professionals have placed renewed emphasis on lowering plasma cholesterol levels, and low density lipoprotein cholesterol in particular, as an essential step in prevention of CVD.
- the upper limits of so-called "normal" cholesterol are now known to be significantly lower than heretofore appreciated.
- independent risk factors include glucose intolerance, left ventricular hypertrophy, hypertension, and being male.
- Cardiovascular disease is especially prevalent among diabetic subjects, at least in part because of the existence of multiple independent risk factors in this population. Successful treatment of hyperlipidemia in the general population, and in diabetic subjects in particular, is therefore of exceptional medical importance.
- Hypertension is a condition that occurs in the human population as a secondary symptom to various other disorders such as renal artery stenosis, pheochromocytoma, or endocrine disorders.
- hypertension is also evidenced in many patients in whom the causative agent, or disorder, is unknown. While such essential hypertension is often associated with disorders such as obesity, diabetes, and hypertriglyceridemia, the relationship between these disorders has not been elucidated. Additionally, many patients present with symptoms of high blood pressure in the complete absence of any other signs of disease, or disorder.
- hypertension can directly lead to heart failure, renal failure, and stroke, which conditions are all capable of causing short-term death.
- Hypertension also contributes to the development of atherosclerosis, and coronary disease, which conditions gradually weaken a patient and can lead, in long-term, to death.
- the precise etiology of essential hypertension is unknown, although a number of factors are believed to contribute to the onset of the disease. Among such factors are stress, uncontrolled emotions, unregulated hormone release (the renin, angiotensin, aldosterone system), excessive salt and water due to kidney malfunction, wall thickening and hypertrophy of the vasculature resulting in vascular constriction, and genetic pre-disposition.
- Hypertension has further been associated with elevated blood insulin levels, a condition known as hyperinsulinemia.
- Insulin a peptide hormone whose primary actions are to promote glucose utilization, protein synthesis, and the formation and storage of neutral lipids, also acts, inter alia, to promote vascular cell growth and increase renal sodium retention. These latter functions can be accomplished without affecting glucose levels and are known causes of hypertension.
- Peripheral vasculature growth for example, can cause constriction of peripheral capillaries; while sodium retention increases blood volume.
- the lowering of insulin levels in hyperinsulinemics can prevent abnormal vascular growth and renal sodium retention caused by high insulin levels and thereby alleviate hypertension.
- Cardiac hypertrophy is a significant risk factor in the development of sudden death, myocardial infarction, and congestive heart failure. These cardiac events are due, at least in part, to increased susceptibility to myocardial injury after ischemia and reperfusion which can occur in both out-patient and perioperative settings. There is currently an unmet medical need to prevent or minimize adverse myocardial perioperative outcomes, particularly perioperative myocardial infarction. Both cardiac and non-cardiac surgery are associated with substantial risks for myocardial infarction or death, and some 7 million patients undergoing non-cardiac surgery are considered to be at risk, with incidences of perioperative death and serious cardiac complications as high as 20-25% in some instances.
- perioperative myocardial infarction is estimated to occur in 5% and death in 1-2%.
- There is currently no commercial drug therapy in this area which reduces damage to cardiac tissue from perioperative myocardial ischemia or enhances cardiac resistance to ischemic episodes.
- Such a therapy is anticipated to be life- saving and reduce hospitalizations, enhance quality of life and reduce overall health care costs of high risk patients.
- the mechanism(s) responsible for the myocardial injury observed after ischemia and reperfusion is not fully understood, however, it has been reported (M. F. Allard, et al. Am. J.
- Hepatic glucose production is an important target for Type 2 diabetes therapy.
- the liver is the major regulator of plasma glucose levels in the post absorptive (fasted) state, and the rate of hepatic glucose production in Type 2 diabetes patients is significantly elevated relative to normal individuals.
- hepatic glucose production is abnormally high in Type 2 diabetes patients.
- Glycogenolysis is an important target for interruption of hepatic glucose production.
- the liver produces glucose by glycogenolysis (breakdown of the glucose polymer glycogen) and gluconeogenesis (synthesis of glucose from 2- and 3-carbon precursors).
- glycogenolysis breakdown of the glucose polymer glycogen
- gluconeogenesis synthesis of glucose from 2- and 3-carbon precursors.
- glycogen phosphorylase is catalyzed in liver, muscle, and brain by tissue-specific isoforms of the enzyme glycogen phosphorylase. This enzyme cleaves the glycogen macromolecule releasing glucose-1 -phosphate and a new shortened glycogen macromolecule.
- Two types of glycogen phosphorylase inhibitors have been reported to date: glucose and glucose analogs [J. L. Martin, et al., Biochemistry, 30, 10101 , (1991)], and caffeine and other purine analogs [P. J. Kasvinsky, et al., J. Biol. Chem., 253, 3343-3351 and 9102-9106 (1978)].
- glycogen phosphorylase inhibitors have been postulated to be of potential use for the treatment of Type 2 diabetes by decreasing hepatic glucose production and lowering glycemia. See, for example, T. B. Blundell, et al., Diabetologia, 35 (Suppl. 2), 569-576 (1992), and Martin et al., supra. Recently, glycogen phosphorylase inhibitors have been disclosed in, inter alia, PCT
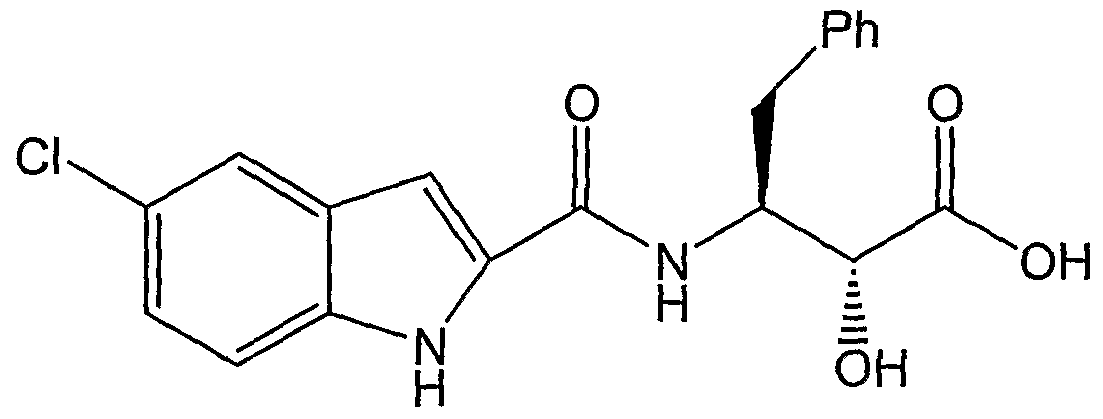
- the present invention relates to improved processes useful in the preparation of the N- (indole-2-carbonyl)- ⁇ -alaninamides disclosed in the aforementioned U.S. Patent Nos. 6,107,329, 6,277,877, and 6,297,269, including 5-chloro-N-[(1S,2R)-3-[3R,4S]-3,4-dihydroxy- 1-pyrrolidinyl]-2-hydroxy-3-oxo-1-(phenylmethyl)propyl]-1 H-indole-2-carboxamide (I); certain intermediates related thereto; and processes useful in preparing such intermediates.
- the instant invention provides novel processes and intermediates useful in the preparation of certain N-(indole-2-carbonyl)- ⁇ -alaninamide compounds, which compounds are glycogen phosphorylase inhibitors useful in the treatment of diseases such as hypercholesterolemia, hyperglycemia, hyperinsulinemia, hyperlipidemia, hypertension, atherosclerosis, diabetes, diabetic cardiomyopathy, infection, tissue ischemia, myocardial ischemia, and in inhibiting tumor growth.
- diseases such as hypercholesterolemia, hyperglycemia, hyperinsulinemia, hyperlipidemia, hypertension, atherosclerosis, diabetes, diabetic cardiomyopathy, infection, tissue ischemia, myocardial ischemia, and in inhibiting tumor growth.
- the present invention provides novel processes and intermediates useful in the preparation of certain N-(indole-2-carbonyl)- ⁇ -alaninamides. More particularly, the invention provides novel processes for preparing the compound 5-chloro-N-[(1S,2R)-3-[3R,4S]-3,4- dihydroxy-1 -pyrrolidinyl]-2-hydroxy-3-oxo-1 -(phenylmethyl)propyl]-1 H-indole-2-carboxamide (I). The invention further provides intermediates useful in the preparation of the aforementioned compound, and processes for the production of such intermediates.
- Such coupling reaction may be effected according to standard synthetic methodologies known to one of ordinary skill in the art.
- an appropriate coupling reagent such as 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), in the presence of 1-hydroxybenzotriazole (HOBT), 2-ethyloxy-1- ethyloxy-carbonyl-1,2-dihydroquinone (EEDQ), CDI/HOBT, propanephosphonic anhydride (PPA), or diethylphosphorylcyanide, and the like, in an aprotic, reaction-inert solvent, such as dichloromethane, acetonitrile, diethylether, tetrahydrofuran, optionally in the presence of a tertiary amine base, such as triethylamine or N,N'-diisopropylethylamine (Hunig's Base).
- EDC 1-(3-dimethylaminopropy
- Such coupling is typically effected at a temperature range of from about 0° C to about the reflux temperature of the solvent employed.
- the coupling reaction is effected at ambient temperature in tetrahydrofuran using EDC, and a catalytic amount of HOBT, in the presence of an organic base selected from triethylamine or Hunig's Base.
- Hunig's Base in such coupling is especially preferred.
- the 3-pyrroline starting material may be obtained from commercial sources.
- the oxidation reaction set forth in Step (b) may be effected according to synthetic methodologies known to one of ordinary skill in the art for converting olefins into c/s-diols.
- Such oxidation may be carried out using ruthenium(lll) chloride, with sodium periodate as a co- oxidant, AgO (J. Org. Chem., 6_1, 4801 (1996)), osmium tetroxide, or a catalyst with N- methylmorpholine N-oxide (NMO) in a reaction-inert, polar organic solvent such as acetonitrile, tetrahydrofuran, alkyl ethers, and the like.
- ruthenium(lll) chloride with sodium periodate as a co- oxidant, AgO (J. Org. Chem., 6_1, 4801 (1996))
- osmium tetroxide osmium tetroxide
- NMO N- methylmorpholine N-oxide
- the oxidation of (lb) to compound (I) is effected using catalytic osmium tetroxide and N-methylmorpholine N-oxide (NMO) in tetrahydrofuran (Rosenberg, et al.; J. Med. Chem., 33, 1962 (1990)).
- NMO N-methylmorpholine N-oxide
- Step (b) The product of Step (b) is then preferably isolated according to well-known methodologies known to one of ordinary skill in the art.
- the invention provides a process for preparing a compound of structural formula (I)
- Step (a) cleaving the acetonide derivative (lla) formed in Step (a) to furnish the compound of structural formula (I).
- the coupling of compound (la) with (IVi) to form the acetonide derivative (lla) can be effected according to the methods disclosed hereinabove for the preparation of compound (lb).
- the coupling is performed using EDC and HOBT in the presence of Hunig's Base.
- the HOBT may be employed catalytically, i.e., in an amount less than one equivalent.
- a range of from about 0.05 to about 0.50 equivalents may be employed in the coupling step, however, it is generally preferred that the HOBT be employed in a catalytic ratio of about 0.15 to about 0.25 molar equivalents of acid (la).
- acetonide (lla) can be employed directly in the subsequent cleavage step, it may occasionally be preferable, for reasons of improved color and purity, to isolate acetonide (lla) prior to such cleavage.
- the isolation of the less polar acetonide (lla) allows a purge of more polar impurities then, following the deprotection step, the more polar substrate (I) is isolated by crystallization, thereby allowing for a purge of less polar impurities that may be present.
- acetonide (lla) into compound (I) may be effected according to generally known methods, for example, by treatment of the isolated acetonide (lla) with a mineral acid, such as hydrochloric or hydrobromic acid, or an organic acid, such as methanesulfonic or p-toluenesulfonic acid, all in the presence of water.
- a mineral acid such as hydrochloric or hydrobromic acid
- organic acid such as methanesulfonic or p-toluenesulfonic acid
- compound (I) may also be conveniently prepared by the production, and in situ cleavage, of acetonide (lla).
- the preparation of a solution of acetonide (lla) in a suitable solvent may be effected as outlined hereinabove.
- the in situ conversion of acetonide (lla) into compound (I), described in Example 5 hereinbelow, may also be conveniently effected according to known methods, for example, by treating the solution of acetonide (lla) with an aqueous mineral acid, such as hydrochloric or hydrobromic acid, or an organic acid, such as methanesulfonic, or p-toluenesulfonic acid, also under aqueous conditions.
- Compound (I) so produced may then be isolated according to known preparative methods.
- Step (b) desolvating the ethanol solvate (Ilia) formed in Step (a) to furnish the compound of structural formula (I).
- the coupling of compound (la) to form ethanol solvate (Ilia) may be performed according to those coupling methods previously described hereinabove for the preparation of compound (lb) and acetonide (Ma).
- the coupling is effected using EDC and HOBT in the presence of a tertiary amine base, such as triethylamine, or Hunig's Base.
- Hunig's Base is especially preferred..
- the ethanol solvate (Ilia) may be desolvated to form compound (I) by dissolving (Ilia) in an aprotic solvent, such as ethyl acetate or toluene, distilling the solution to remove residual ethanol, treating the solution with water such that a concentration of water in the range of between about 1% to about 3% water is achieved, and warming the aqueous solution to reflux temperature, at which point crystallization of (I) begins.
- the addition of seed crystals to the aqueous solution prior to reflux is typically preferred.
- the reflux period may comprise from a few hours to one or more days, preferably from about eight to about twenty hours.
- the present invention provides a process for preparing a compound of structural formula (I)
- the coupling of compound (la) with c/s-3,4-dihydroxypyrrolidine free base (V) to form compound (I) may also be performed according to those coupling methods previously described hereinabove for the preparation of compound (lb), acetonide (lla), or ethanol solvate (Ilia).
- the free base of c/s-3,4-dihydroxypyrrolidine (V) may be prepared according to the several synthetic methods described in detail hereinbelow including, for example, the process disclosed in Example 18.
- the compound of structural formula (I) so prepared is then preferably isolated according to standard methodologies that are well known to one of ordinary skill in the art.
- Another aspect of the invention provides synthetic methods useful for preparing compound (V), and the acid addition salts thereof, which compound, or which acid addition salts, are intermediates useful in the preparation of compound (I). These exemplary synthetic methods are described in detail hereinbelow in Schemes 1 to 7.
- the c/s-3,4- dihydroxypyrrolidine, p-toluenesulfonate salt (Vi) may be obtained commercially.
- the invention provides a process useful in preparing compound (V), or an acid addition salt thereof, which process comprises the steps outlined hereinbelow in Scheme 1.
- the BOC protecting group of (Vb) may be subsequently removed by treatment with a suitable acid, for example, trifluoroacetic acid, methanesulfonic acid, and the like, in the presence of a reaction-inert solvent such as tetrahydrofuran, dichloromethane, or acetonitrile, to form (V).
- a suitable acid for example, trifluoroacetic acid, methanesulfonic acid, and the like
- a reaction-inert solvent such as tetrahydrofuran, dichloromethane, or acetonitrile
- compound (V) is then isolated, either in the form of the free base, or in the form of an acid addition salt thereof, wherein such acid addition salt may be prepared according to known methods.
- acid addition salts may include, for example, the hydrochloride, hydrobromide, sulfate, hydrogen sulfate, phosphate, hydrogen phosphate, dihydrogen phosphate, acetate, succinate, citrate, methanesulfonate (mesylate), and 4- methylbenzenesulfonate (p-toluenesulfonate) acid addition salts.
- Such acid addition salts may be prepared readily by reacting compound (V) with an appropriate conjugate acid.
- the desired salt is of a monobasic acid (e.g., hydrochloride, hydrobromide, tosylate, acetate, etc.)
- the hydrogen form of a dibasic acid e.g., hydrogen sulfate, succinate, etc.
- the dihydrogen form of a tribasic acid e.g., dihydrogen phosphate, citrate, etc.
- at least one molar equivalent, and usually a molar excess, of the acid is employed.
- the appropriate and stoichiometric equivalent of the acid will generally be employed.
- the free base and the acid are normally combined in a co-solvent from which the desired acid addition salt then precipitates, or can be otherwise isolated by concentration of the mother liquor, or by the precipitative effect resulting from the addition of a non-solvent.
- Especially preferred acid addition salts of compound (V) comprise the p-toluenesulfonate (Vi) and hydrochloride acid addition salts.
- the dibromo diketone starting material is reduced in the presence of a suitable reducing agent, such as sodium borohydride, in a reaction-inert solvent, such as an ether (tetrahydrofuran or methyl terf-butyl ether), or other suitable solvent(s) to provide a mixture of the syn- and anti-alcohols (Via) and (Via').
- a suitable reducing agent such as sodium borohydride
- a reaction-inert solvent such as an ether (tetrahydrofuran or methyl terf-butyl ether), or other suitable solvent(s) to provide a mixture of the syn- and anti-alcohols (Via) and (Via').
- Alcohols (Via) and (Via') are then cyclized with benzylamine in the presence of a suitable base, such as sodium bicarbonate, to yield diol (Vlb).
- a suitable base such as sodium bicarbonate
- the benzyl protecting group of (Vlb) may be subsequently removed by standard methods, such as hydrogenation using a catalyst such as palladium on carbon in a reaction- inert solvent, such as an alcohol or ether, to form compound (V), followed by acid addition salt formation, if desired.
- a catalyst such as palladium on carbon in a reaction- inert solvent, such as an alcohol or ether
- acid addition salt formation if desired.
- Yet another alternative method that may be employed in the preparation of (V), or an acid addition salt thereof, comprises the process depicted in Scheme 3.
- amide bond formations from carboxylic acids may be aided by addition of coupling agents such as dicyclohexylcarbodiimide, N,N'-carbonyldiimidazole, or ethyl-1 ,2-dihydro-2-ethoxy-1- quinolinecarboxylate (EEDQ).
- Diol (Vllb) is then reduced to diol (Vlb) through the use of known reducing reagents, such as lithium aluminum hydride, diborane, or sodium borohydride, in the presence of boron trifluoride.
- the benzyl protecting group of (Vlb) may be subsequently removed by standard methods, such as hydrogenation using a catalyst such as palladium on carbon in a suitable solvent, such as an alcohol or ether, to form compound (V), followed by acid addition salt formation, if desired.
- a catalyst such as palladium on carbon in a suitable solvent, such as an alcohol or ether
- Yet another method useful in the preparation of compound (V), or an acid addition salt thereof, comprises the steps shown in Scheme 4.
- the benzyl protecting group of (Vlb) may be subsequently removed by standard methods, such as hydrogenation using a catalyst such as palladium on carbon in a suitable solvent, such as an alcohol or ether, to form compound (V), followed by acid addition salt formation, if desired.
- a catalyst such as palladium on carbon in a suitable solvent, such as an alcohol or ether
- acid addition salt formation if desired.
- Another. method useful in the preparation of (V), or an acid addition salt thereof comprises the process shown in Scheme 5.
- (E)-1 ,4-dichloro-2-butene is di-hydroxylated to furnish diol (IXa) employing conditions known to one of ordinary skill in the art, for example, hydrogen peroxide and formic acid, or m-chloroperoxybenzoic acid and water.
- Diol (IXa) is then cyclized with benzylamine in the presence of a suitable base, such as sodium bicarbonate, to give diol (Vlb).
- a suitable base such as sodium bicarbonate
- the benzyl protecting group of (Vlb) may be subsequently removed by standard methods, such as hydrogenation using a catalyst such as palladium on carbon in a reaction- inert solvent, such as an alcohol or ether, to form compound (V), followed by acid addition salt formation, if desired.
- (Z)-1 ,4-dichloro-2-butene is di-hydroxylated to furnish diol (IXa) according to synthetic methods known to one of ordinary skill in the art.
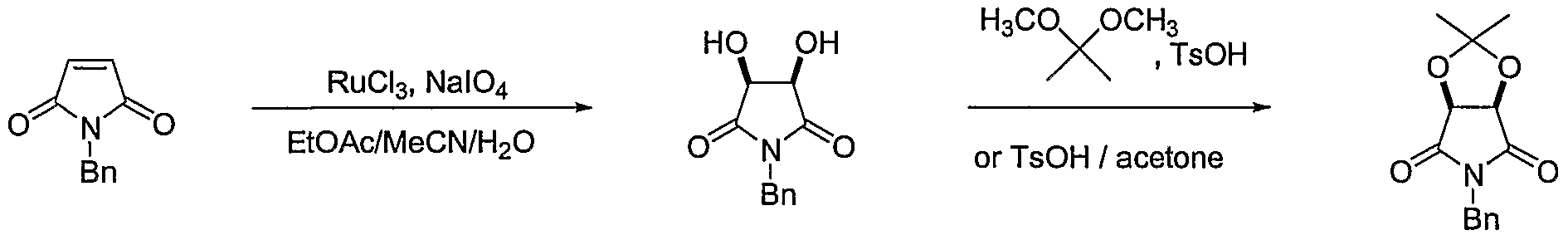
- oxidation may be effected employing a mixture of sodium periodate and a ruthenium salt in a reaction-inert, aprotic solvent such as acetontrile, or a halogenated hydrocarbon solvent such as chloroform, methylene chloride, or carbon tetrachloride.
- solvent mixtures comprising reaction-inert, aprotic solvents, for example, acetonitrile and ethyl acetate, may also be utilized.
- the oxidation reaction is effected utilizing ruthenium(lll) chloride hydrate and sodium periodate in a cooled acetonitrile/ethyl acetate solvent mixture.
- Diol (IXa) is then cyclized using benzylamine in the presence of a suitable base, such as sodium bicarbonate, to furnish compound diol (Vlb).
- a suitable base such as sodium bicarbonate
- an additive such as potassium iodide, to assist in cyclization may be employed if desired, and/or appropriate.
- the benzyl protecting group of (Vlb) may be subsequently removed by standard methods, such as hydrogenation using a catalyst such as palladium on carbon in a suitable solvent, such as an alcohol or ether, to form compound (V), followed by acid addition salt formation, if desired.
- the aminodiol starting material is protected with BOC-anhydride in the presence of an organic or Br ⁇ nsted base in an aprotic solvent.
- the BOC protected diol (Xla) is then oxidized to dialdehyde (Xlb) by methods generally known to those skilled in the art.
- diol (Xla) may be oxidized using a strong oxidant such as potassium permanganate, ruthenium tetroxide, manganese dioxide, or Jones' reagent (chromic acid and sulfuric acid in water).
- oxidation of (Xla) to (Xlb) may be effected by catalytic dehydrogenation using reagents such as copper chromite, Raney nickel, palladium acetate, copper oxide, and the like.
- reagents such as copper chromite, Raney nickel, palladium acetate, copper oxide, and the like.
- the dialdehyde (Xlb) may then be cyclized to BOC-protected diol (Vb) via pinacol coupling.
- Known methods of effecting such coupling may comprise direct electron transfer using active metals such as sodium, magnesium, or aluminum, or through the use of titanium trichloride.
- the BOC-group of (Vb) can then be removed by treatment with a suitable acid as described hereinabove.
- compound (V) is then isolated, either in the form of the free base, or in the form of an acid addition salt thereof, wherein such acid addition salt may be prepared as described hereinabove.
- Another aspect of the instant invention provides synthetic methods useful for preparing compound (IV) hereinbelow, and the acid addition salts thereof, which compound and acid addition salts, are also intermediates useful in the preparation of compound (I). Such exemplary synthetic methods are depicted in detail hereinbelow in Schemes 8 to 10. In one aspect, compound (IV), or an acid addition salt thereof, may be prepared according to the process shown in Scheme 8.
- ribose is protected by forming the acetonide derivative (Xlla) thereof.
- acetonide formation can be effected in a variety of ways, for example, according to those methods described in Greene, T.W., et al., Protective Groups in Organic Synthesis, 2 nd Edition, Wiley-lnterscience, (1991).
- the formation of protected diol (Xlla) may be performed using acetone in the presence of iodine.
- the oxidation of (Xlla) to (Xllb) may be effected using reagents including sodium periodate in methanol.
- (Xllb) may be performed according to known methods, for example, through the use of lithium aluminum hydride or sodium borohydride in the presence of acid, such as acetic acid.
- Amine (IVc) is prepared by treating (Xllb) with benzylamine in methylene chloride or similar reaction-inert solvents. The benzyl protecting group of (IVc) can be subsequently removed according to standard methods, such as hydrogenation, using a catalyst such as palladium on carbon in a suitable solvent, such as an alcohol or ether, to form compound (IV).
- compound (IV) is then isolated, either in the form of the free base, or in the form of an acid addition salt thereof, wherein such acid salt may be prepared as described hereinabove.
- acid addition salts of compound (IV) are the p- toluenesulfonate (IVi) and hydrochloride acid addition salts.
- meso-erythritrol is protected using standard methodologies to form the di-pivaloyl derivative (Xllla). Such protection is preferably effected using pivaloyl chloride in the presence of a strong organic base, such as pyridine.
- the resulting diol (Xllla) may be protected by formation of the acetonide (Xlllb) by treatment of (Xllla) with tosic acid in acetone or by treatment with 2,2-dimethoxypropane (DMP).
- the Piv- groups of (Xlllb) may be subsequently removed according to standard methods, for example those methods disclosed in Greene, T.W., et al., Protective Groups in Organic Synthesis, 2 nd Edition, Wiley- lnterscience, (1991), to form deprotected derivative (Xlllc).
- the deprotection of (Xlllb) may be effected using a strong inorganic base, such as sodium or potassium hydroxide, in an aqueous solvent, such as an alcohol.
- Mesylate activation of the diol (Xlllc) in a suitable non-reactive solvent in the presence of a base such as triethylamine, gives compound (Xllld).
- compound (IV) is then isolated, either in the form of the free base, or in the form of an acid addition salt thereof, wherein such acid salt may be prepared as described hereinabove.
- the invention provides a generally preferred process for the preparation of compound (IV), or the preferred p-toluenesulfonate acid addition salt (IVi) thereof, which process is depicted hereinbelow in Scheme 10.
- N-benzylmaleimide to diol may be performed according to synthetic methods known to one of ordinary skill in the art.
- such oxidation may be effected employing a mixture of sodium periodate and a ruthenium salt in a reaction-inert, aprotic solvent such as acetonitrile, or a halogenated hydrocarbon solvent such as chloroform, methylene chloride, or carbon tetrachloride.
- solvent mixtures comprising reaction-inert, aprotic solvents, for example, acetonitrile and ethyl acetate, may also be utilized.
- the oxidation reaction is effected utilizing ruthenium(lll) chloride hydrate and sodium periodate in a acetonitrile/ethyl acetate solvent mixture at below ambient temperature.
- acetonide (IVb) may be effected according to synthetic methodologies known to one of ordinary skill in the art.
- such protection may be performed by condensing diol (Vllb) with acetone, 2,2-dimethoxypropane, or a mixture of both, in the presence of an acid catalyst, such as sulfuric, p-toluenesulfonic, or methanesulfonic acid.
- an acid catalyst such as sulfuric, p-toluenesulfonic, or methanesulfonic acid.
- the protection reaction is effected by condensing diol (Vila) in 2,2-dimethoxypropane with a catalytic amount of methanesulfonic acid.
- the reduction of acetonide (IVb) to (IVc) may be effected according to synthetic methodologies known to one of ordinary skill in the art. For example, such reduction may be performed using a boron or aluminum hydride complex including, for example, BH 3 THF, BH 3 etherate, or Red-AI ® (sodium bis(2-methoxyethoxy)aluminum hydride; Aldrich Chemical Co., Milwaukee, Wl), in an aprotic, reaction-inert solvent, such as toluene or diethylether.
- the reduction of protected acetonide (IVb) to (IVc) is effected using Red-AI ® in toluene.
- the deprotection of (IVc) may be effected according to synthetic methodologies known to one of ordinary skill in the art. For example, such using palladium salts, or complexes, such as Pd(OH) 2 , or Pd/C in polar, protic solvents, such as methanol or ethanol, in a non-protic solvent, such as tetrahydrofuran, or in a mixture of such solvents.
- palladium salts, or complexes such as Pd(OH) 2 , or Pd/C in polar, protic solvents, such as methanol or ethanol, in a non-protic solvent, such as tetrahydrofuran, or in a mixture of such solvents.
- such deprotection may be effected under hydrogenation-transfer conditions, i.e., Pd/C with cyclohexene.
- the deprotection reaction is effected using Pd(OH) 2 /C in methanol.
- the deprotected product (IV), is then preferably isolated, in the form of the preferred p- toluenesulfonate acid addition salt (IVi) thereof, which may be either prepared as described hereinabove, or obtained commercially.
- the mixture was treated with 0.6 g (0.33 equiv.) of 1-hydroxybenzotriazole hydrate (HOBT) and the mixture was cooled to between 0 ° and 5 ° C.
- N,N-diisopropylethylamine (2.08 ml, 2.1 equiv.) was added to the mixture over 15 minutes at 0 ° to 5 ° C.
- the mixture was then treated with 1-(3-dimethylaminopropyl)-3- ethylcarbodiimide hydrochloride (EDC) (2.78 g, 1.1 equiv.) at -10 ° to -6 ° C.
- EDC 1-(3-dimethylaminopropyl)-3- ethylcarbodiimide hydrochloride
- the reaction was allowed to warm to about 20 ° C and was stirred at ambient temperature for about 24 hours.
- reaction mixture was treated with water (50 ml) and ethyl acetate (50 ml) to give a two- phase mixture.
- the layers were settled and the organic layer was separated and concentrated to furnish a solid by distillation under partial vacuum. A total of 5.1 g (92.7% yield) of the pure title product was isolated.
- the reaction solution was cooled to between 0 ° and -10 ° C and treated with 1-(3-dimethylaminopropyl)-3- ethylcarbodiimide hydrochloride (EDC) (14.2 g, 0.0741 mol), and hydroxybenzotriazole hydrate (HOBT) (10.0 g, 0.074 mol).
- EDC 1-(3-dimethylaminopropyl)-3- ethylcarbodiimide hydrochloride
- HOBT hydroxybenzotriazole hydrate
- the reaction mixture was stirred at -6 ° to -10 ° C for about 30 minutes.
- the reaction was allowed to warm to ambient temperature over about 45 minutes and stirred for about 2 hours.
- the reaction mixture was treated with 50% aqueous sodium hydroxide to give a pH of about 10, and the two-phase mixture was allowed to settle.
- the organic layer was concentrated to an oil by rotary evaporation using partial vacuum. A total of 31 g (88 % yield
- the pH was adjusted to 6.5 to 7.5 with 50% sodium hydroxide, and the solution was reduced to a small volume by atmospheric distillation at a pot temperature of about 90 ° C.
- a total of 100 ml of ethyl acetate was added, the organic layer was washed with 50 ml of water, and the organic layer was diluted with 50 ml of toluene.
- the mixture was refluxed overnight, stirred for about 10 hours at ambient temperature, and filtered.
- the residual solid was dried in vacuo at a temperature of about 45 ° C to afford 10.4 g (86.6% yield) of the title product.
- reaction mixture was then treated with c/s-3,4-dihydroxypyrrolidine, p-toluenesulfonate (Vi) (41.1 kg, 149.3 mol) and the reaction was allowed to stir for about 30 minutes at 0 ° to 5 ° C. The reaction was then warmed to ambient temperature and stirred for about 6 hours. The reaction mixture was treated with water (175 gallons), stirred for about 1 hour, and then allowed to settle. The aqueous layer was separated off and was washed twice with ethyl acetate (2 x 35 gallons).
- the ethyl acetate layers were combined and washed three times with aqueous sodium bicarbonate (2 x 23.8 kg sodium bicarbonate in 70 gallons of water and 1 x 11.9 kg sodium bicarbonate in 35 gallons of water).
- the ethyl acetate solution was combined with 20 gallons of ethyl acetate and 35 gallons of water, stirred for about 30 minutes and then allowed to settle.
- the ethyl acetate layer was separated off, treated with decolorizing charcoal (0.55 kg), and then stirred for about 15 minutes. The mixture was filtered to remove the charcoal and the solution was concentrated in vacuo to a volume of about 80 gallons.
- the ethyl acetate was displaced by distillation using ethanol (4 x 55 gallons), whereupon a thick white slurry formed at a final volume of about 110 gallons.
- the product was stirred at ambient temperature for about 18 hours.
- a total of 83.2 kg of the title compound was isolated by filtration as an ethanol-wet cake.
- the solids were filtered off, washed with ethyl acetate (8 gallons), and then blown dry under a nitrogen stream. The solid was dissolved in ethyl acetate and the solution was stirred at ambient temperature for about 11 days, whereupon a solid product gradually formed. The solid was then filtered off and vacuum dried at 30 ° to 45 ° C to give the title compound (30.9 kg, 71.6 % yield).
- the inorganic salts were removed by suction filtration, and the filter cake was washed with ethyl acetate.
- the combined filtrates were washed with water and allowed to settle.
- the aqueous layer was extracted with ethyl acetate and the product-rich organic layers were combined and washed with a solution of 8 kg of sodium chloride in 72 L of water.
- the organic extracts were concentrated by atmospheric distillation at a temperature of about 75 ° C, cooled to room temperature, and allowed to granulate for 2 to 4 hours. Hexanes (360 L) was added to the cooled (5 ° C to 15 ° C) slurry and granulation was continued for about 1 hour.
- the precipitated solids were collected by suction filtration, washed well with ethyl acetate followed by hexanes, and then dried in vacuo at a temperature of about 40 ° C to about 45 ° C to provide the title compound (42.0 kg, 71% yield) as a white solid.
- the mixture was cooled to 30 ° C and 50 ° C, 473 L of methanol was added, and the mixture was concentrated to a final volume of about 145 L again by atmospheric distillation as previously described.
- the concentrate was cooled to about room temperature and about 1 L of water was added.
- the resulting solution of the title compound was used directly in the following step.
- the resulting solution was then treated, over a time period of about 1 hour, with a solution of 34.6 kg of p-toluenesulfonic acid in 102 L of methyl ethyl ketone and the mixture was allowed to granulate for about 5 hours at 10 ° C to 20 ° C.
- the slurry was cooled to between 0 ° C and 5 ° C, and granulated for a further 2 hours.
- the precipitated product was collected by filtration, washed with cold methyl ethyl ketone, and dried in vacuo at 40 ° C to 45 ° C to furnish the title compound (44.8 kg, 74% yield) as a white crystalline solid.
- the reaction was then cooled to between 10 ° and 15 ° C, and quenched slowly with 39 L of water.
- the lower, product layer was removed and the aqueous layer was then washed with about 2 gallons of ethyl acetate.
- the organic and product layers were combined and washed three times with sodium bicarbonate solutions (one wash with a solution of 1.37 kg sodium bicarbonate in 4 gallons water, followed by two washes with a solution of 687 g sodium bicarbonate in 2 gallons water).
- the organic layer was treated with decolorizing charcoal, filtered, and the residue washed with 1 gallon of ethyl acetate.
- the filtrate was concentrated to a volume of about 2 gallons, diluted with 16 L of ethanol, and then concentrated in vacuo to a volume of about 8 L. An additional 10 L of ethanol was added, and the resulting suspension was stirred overnight. An additional 10 L of ethanol was added, and the mixture was filtered. The collected solid was washed with 3 L of ethanol, and dried in vacuo at a temperature of about 35 ° C to furnish 2.47 kg of the title compound.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Indole Compounds (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description












































Claims


Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| HU0500513A HUP0500513A3 (en) | 2002-01-18 | 2003-01-06 | Intermediates for preparing glycogen phosphorylase inhibitors |
| KR10-2004-7011084A KR20040072730A (en) | 2002-01-18 | 2003-01-06 | Intermediates for Preparing Glycogen Phosphorylase Inhibitors |
| AU2003200853A AU2003200853A1 (en) | 2002-01-18 | 2003-01-06 | Intermediates for preparing glycogen phosphorylase inhibitors |
| CA002472205A CA2472205A1 (en) | 2002-01-18 | 2003-01-06 | Intermediates for preparing glycogen phosphorylase inhibitors |
| MXPA04006937A MXPA04006937A (en) | 2002-01-18 | 2003-01-06 | Intermediates for preparing glycogen phosphorylase inhibitors. |
| EP03700024A EP1470128A1 (en) | 2002-01-18 | 2003-01-06 | Intermediates for preparing glycogen phosphorylase inhibitors |
| JP2003560013A JP2005514460A (en) | 2002-01-18 | 2003-01-06 | Intermediates for producing glycogen phosphorylase inhibitors |
| BR0307006-9A BR0307006A (en) | 2002-01-18 | 2003-01-06 | Intermediates for preparing glycogen phosphorylase inhibitors |
| IL16150703A IL161507A0 (en) | 2002-01-18 | 2003-01-06 | Intermediates for preparing glycogen phosphorylase inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US34964702P | 2002-01-18 | 2002-01-18 | |
| US60/349,647 | 2002-01-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2003059910A1 true WO2003059910A1 (en) | 2003-07-24 |
Family
ID=23373348
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2003/000034 WO2003059910A1 (en) | 2002-01-18 | 2003-01-06 | Intermediates for preparing glycogen phosphorylase inhibitors |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US6696574B2 (en) |
| EP (1) | EP1470128A1 (en) |
| JP (1) | JP2005514460A (en) |
| KR (1) | KR20040072730A (en) |
| CN (1) | CN1592749A (en) |
| AU (1) | AU2003200853A1 (en) |
| BR (1) | BR0307006A (en) |
| CA (1) | CA2472205A1 (en) |
| HU (1) | HUP0500513A3 (en) |
| IL (1) | IL161507A0 (en) |
| MX (1) | MXPA04006937A (en) |
| PL (1) | PL371408A1 (en) |
| RU (1) | RU2004121986A (en) |
| TW (1) | TW200302220A (en) |
| WO (1) | WO2003059910A1 (en) |
| ZA (1) | ZA200402621B (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007128761A2 (en) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Uses of dpp-iv inhibitors |
| CN103626845A (en) * | 2013-09-30 | 2014-03-12 | 承德医学院 | Arylpyrrole-2-formamide dipeptide derivatives serving as glycogen phosphorylase inhibitor and preparation method and medical application thereof |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006059163A1 (en) * | 2004-12-02 | 2006-06-08 | Prosidion Limited | Treatment of diabetes with glycogen phosphorylase inhibitors |
| ES2699773T3 (en) * | 2011-05-31 | 2019-02-12 | Theravance Biopharma R&D Ip Llc | Neprilysin inhibitors |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996039385A1 (en) * | 1995-06-06 | 1996-12-12 | Pfizer Inc. | Substituted n-(indole-2-carbonyl-) amides and derivatives as glycogen phosphorylase inhibitors |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE69523182T2 (en) | 1995-06-06 | 2002-02-07 | Pfizer | SUBSTITUTED N- (INDOL-2-CARBONYL) GLYCINAMIDES AND DERIVATIVES AS GLYCOGEN PHOSPHORYLASE INHIBITORS |
| US6277877B1 (en) | 2000-08-15 | 2001-08-21 | Pfizer, Inc. | Substituted n-(indole-2-carbonyl)glycinamides and derivates as glycogen phosphorylase inhibitors |
-
2003
- 2003-01-06 CA CA002472205A patent/CA2472205A1/en not_active Abandoned
- 2003-01-06 CN CNA038015455A patent/CN1592749A/en active Pending
- 2003-01-06 BR BR0307006-9A patent/BR0307006A/en not_active Application Discontinuation
- 2003-01-06 WO PCT/IB2003/000034 patent/WO2003059910A1/en not_active Application Discontinuation
- 2003-01-06 KR KR10-2004-7011084A patent/KR20040072730A/en not_active Application Discontinuation
- 2003-01-06 AU AU2003200853A patent/AU2003200853A1/en not_active Abandoned
- 2003-01-06 PL PL03371408A patent/PL371408A1/en not_active Application Discontinuation
- 2003-01-06 HU HU0500513A patent/HUP0500513A3/en unknown
- 2003-01-06 JP JP2003560013A patent/JP2005514460A/en active Pending
- 2003-01-06 MX MXPA04006937A patent/MXPA04006937A/en unknown
- 2003-01-06 EP EP03700024A patent/EP1470128A1/en not_active Withdrawn
- 2003-01-06 RU RU2004121986/04A patent/RU2004121986A/en not_active Application Discontinuation
- 2003-01-06 IL IL16150703A patent/IL161507A0/en unknown
- 2003-01-13 TW TW092100616A patent/TW200302220A/en unknown
- 2003-01-16 US US10/345,661 patent/US6696574B2/en not_active Expired - Fee Related
-
2004
- 2004-04-02 ZA ZA200402621A patent/ZA200402621B/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996039385A1 (en) * | 1995-06-06 | 1996-12-12 | Pfizer Inc. | Substituted n-(indole-2-carbonyl-) amides and derivatives as glycogen phosphorylase inhibitors |
| US6297269B1 (en) * | 1995-06-06 | 2001-10-02 | Pfizer Inc. | Substituted n-(indole-2-carbonyl-) amides and derivatives as glycogen phosphorylase inhibitors |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007128761A2 (en) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Uses of dpp-iv inhibitors |
| EP2351568A2 (en) | 2006-05-04 | 2011-08-03 | Boehringer Ingelheim International GmbH | Uses of dpp-iv inhibitors |
| CN103626845A (en) * | 2013-09-30 | 2014-03-12 | 承德医学院 | Arylpyrrole-2-formamide dipeptide derivatives serving as glycogen phosphorylase inhibitor and preparation method and medical application thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| IL161507A0 (en) | 2004-09-27 |
| US20030187051A1 (en) | 2003-10-02 |
| KR20040072730A (en) | 2004-08-18 |
| EP1470128A1 (en) | 2004-10-27 |
| US6696574B2 (en) | 2004-02-24 |
| MXPA04006937A (en) | 2004-12-06 |
| BR0307006A (en) | 2004-11-03 |
| CA2472205A1 (en) | 2003-07-24 |
| HUP0500513A3 (en) | 2006-07-28 |
| TW200302220A (en) | 2003-08-01 |
| RU2004121986A (en) | 2005-11-20 |
| PL371408A1 (en) | 2005-06-13 |
| HUP0500513A2 (en) | 2005-11-28 |
| CN1592749A (en) | 2005-03-09 |
| ZA200402621B (en) | 2005-05-27 |
| AU2003200853A1 (en) | 2003-07-30 |
| JP2005514460A (en) | 2005-05-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1309594B1 (en) | Benzimidazole derivatives, preparation and therapeutic use thereof | |
| EP0656360B1 (en) | Trifluoromethylpyrroloindole carboxylic ester derivative and process for producing the same | |
| EP0031741A1 (en) | Substituted imino-acids, process for their preparation and their use as enzyme inhibitors | |
| WO2006078006A1 (en) | Indole compound and pharmaceutical composition containing the same | |
| KR20040107520A (en) | (s)-4-amino-5-chloro-2-methoxy-n-[1-[1-(2-tetrahydrofurylcarbonyl)-4-piperidinylmethyl]-4-piperidinyl]benzamide, process for the preparation thereof, pharmaceutical composition containing the same, and intermediate therefor | |
| EP0429341A2 (en) | Heterocyclic derivatives, their preparation and pharmaceuticals containing them | |
| WO2006030847A1 (en) | Novel bicyclic pyrazole derivative | |
| EP0401981B1 (en) | Pyridazinone deriratives | |
| EP0350403A1 (en) | (Aza)naphthalenesultam derivatives, process for their preparation and drugs containing same | |
| EP1242418A1 (en) | Antidiabetic thiazolidinediones and their preparation | |
| US6696574B2 (en) | Processes and intermediates for preparing glycogen phosphorylase inhibitors | |
| EP0266102B1 (en) | Quinoxalinone derivatives | |
| FR2477149A1 (en) | NOVEL CARBOSTYRILE DERIVATIVES, PROCESS FOR PREPARING THEM AND THEIR THERAPEUTIC APPLICATION | |
| EP1383762B1 (en) | Tetrahydropyridyl-alkyl-heterocycles, method for preparing same and pharmaceutical compositions containing same | |
| KR940000785B1 (en) | Process for the preparation of carbostyril derivatives and salts thereof | |
| EP0382628A1 (en) | Aminoalkoxylphenyl derivatives, process for their preparation and compositions containing them | |
| EP1383761B1 (en) | Phenyl- and pyridyl-piperidines with tnf activity | |
| KR19990083196A (en) | Pharmaceutical Composition for Preventing Allograft Rejection | |
| CN109280028B (en) | Quinoline compound and application thereof in DPP-4 enzyme inhibitor | |
| JP2002515064A (en) | New heterocyclic compounds | |
| US6683106B2 (en) | N-(indole-2-carbonyl)-b-alaninamide crystal forms | |
| KR20240109240A (en) | Beta-adrenergic agonists and methods of their use | |
| WO1998022443A1 (en) | N-(imidazolylbutyl) benzenesulphonamide derivatives, their preparation and therapeutic application | |
| KR19990022042A (en) | 3 (2H) -pyridazinone derivatives and pharmaceutical compositions containing these compounds | |
| CZ372799A3 (en) | Piperidine compounds and pharmaceutical preparation containing thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SD SE SG SK SL TJ TM TN TR TT TZ UA UG US UZ VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2004/02621 Country of ref document: ZA Ref document number: 200402621 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 910/DELNP/2004 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003200853 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 161507 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003700024 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20038015455 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003560013 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2472205 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P-593/04 Country of ref document: YU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004121986 Country of ref document: RU Ref document number: PA/a/2004/006937 Country of ref document: MX Ref document number: 1020047011084 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003700024 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2003700024 Country of ref document: EP |