WO2003055482A1 - Amide derivatives as gk activators - Google Patents
Amide derivatives as gk activators Download PDFInfo
- Publication number
- WO2003055482A1 WO2003055482A1 PCT/DK2002/000880 DK0200880W WO03055482A1 WO 2003055482 A1 WO2003055482 A1 WO 2003055482A1 DK 0200880 W DK0200880 W DK 0200880W WO 03055482 A1 WO03055482 A1 WO 03055482A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- aryl
- compound according
- thiazol
- substituents
- Prior art date
Links
- 0 C*(C(C(c1ccc(*)cc1)NC1CCCC1)=O)C1=NC=C*1 Chemical compound C*(C(C(c1ccc(*)cc1)NC1CCCC1)=O)C1=NC=C*1 0.000 description 1
- WZMLRQKJWNKJBY-UHFFFAOYSA-N CC(C)CN(C(Nc1ncc[s]1)=O)c(cc1Cl)ccc1Cl Chemical compound CC(C)CN(C(Nc1ncc[s]1)=O)c(cc1Cl)ccc1Cl WZMLRQKJWNKJBY-UHFFFAOYSA-N 0.000 description 1
- ICXFPEDUFABSBM-UHFFFAOYSA-N CCCCC(C(Nc1ncc[s]1)=O)Oc(cc1)cc(OC)c1OC Chemical compound CCCCC(C(Nc1ncc[s]1)=O)Oc(cc1)cc(OC)c1OC ICXFPEDUFABSBM-UHFFFAOYSA-N 0.000 description 1
- RLSJRWKTKWFZJU-UHFFFAOYSA-N CCOC(Cc1c[s]c(NC(C(CC2CCCCC2)Oc(cc2)ccc2OC)=O)n1)=O Chemical compound CCOC(Cc1c[s]c(NC(C(CC2CCCCC2)Oc(cc2)ccc2OC)=O)n1)=O RLSJRWKTKWFZJU-UHFFFAOYSA-N 0.000 description 1
- FPMIASRJYBNQFX-UHFFFAOYSA-N CNC(Cc1c[s]c(NC(C(c(cc2Cl)ccc2Cl)SC2CCCC2)=O)n1)=O Chemical compound CNC(Cc1c[s]c(NC(C(c(cc2Cl)ccc2Cl)SC2CCCC2)=O)n1)=O FPMIASRJYBNQFX-UHFFFAOYSA-N 0.000 description 1
- SEBPTBFZCHJPID-UHFFFAOYSA-N COc1ccc(C(C(Nc2ncc[s]2)=O)SC2CCCC2)cc1 Chemical compound COc1ccc(C(C(Nc2ncc[s]2)=O)SC2CCCC2)cc1 SEBPTBFZCHJPID-UHFFFAOYSA-N 0.000 description 1
- LXDDQGCNOXDAHW-UHFFFAOYSA-N O=C(C(c(cc1)cc(F)c1F)SC1CCCC1)Nc1ncccc1 Chemical compound O=C(C(c(cc1)cc(F)c1F)SC1CCCC1)Nc1ncccc1 LXDDQGCNOXDAHW-UHFFFAOYSA-N 0.000 description 1
- UDDIAZJEOLFJKS-UHFFFAOYSA-N O=C(C(c(cc1)cc2c1OCO2)SC1CCCC1)Nc1ncc[s]1 Chemical compound O=C(C(c(cc1)cc2c1OCO2)SC1CCCC1)Nc1ncc[s]1 UDDIAZJEOLFJKS-UHFFFAOYSA-N 0.000 description 1
- LXKRVXFDQCSSIH-UHFFFAOYSA-N O=C(C(c(cc1)ccc1Br)Oc(cc1)ccc1F)Nc1ncc[s]1 Chemical compound O=C(C(c(cc1)ccc1Br)Oc(cc1)ccc1F)Nc1ncc[s]1 LXKRVXFDQCSSIH-UHFFFAOYSA-N 0.000 description 1
- OQCARJFHRFUTEO-UHFFFAOYSA-N O=C(C(c(cc1)ccc1Br)SC1CCCC1)Nc1ccccn1 Chemical compound O=C(C(c(cc1)ccc1Br)SC1CCCC1)Nc1ccccn1 OQCARJFHRFUTEO-UHFFFAOYSA-N 0.000 description 1
- LCMNCWWWBYYHHH-UHFFFAOYSA-N O=C(C(c(cc1)ccc1Cl)OCc(cc1)cc(Cl)c1Cl)Nc1ncccc1 Chemical compound O=C(C(c(cc1)ccc1Cl)OCc(cc1)cc(Cl)c1Cl)Nc1ncccc1 LCMNCWWWBYYHHH-UHFFFAOYSA-N 0.000 description 1
- KOXYVBJAVWQLCR-UHFFFAOYSA-N O=C(C(c(cc1)ccc1F)Oc(cc1)ccc1F)Nc1ncc[s]1 Chemical compound O=C(C(c(cc1)ccc1F)Oc(cc1)ccc1F)Nc1ncc[s]1 KOXYVBJAVWQLCR-UHFFFAOYSA-N 0.000 description 1
- BDQMTKUTOBJNSB-UHFFFAOYSA-N O=C(C(c1ccccc1)SC1CCCC1)Nc1ncccc1 Chemical compound O=C(C(c1ccccc1)SC1CCCC1)Nc1ncccc1 BDQMTKUTOBJNSB-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/38—Nitrogen atoms
- C07D277/44—Acylated amino or imino radicals
- C07D277/46—Acylated amino or imino radicals by carboxylic acids, or sulfur or nitrogen analogues thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/426—1,3-Thiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/18—Sulfonamides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4402—Non condensed pyridines; Hydrogenated derivatives thereof only substituted in position 2, e.g. pheniramine, bisacodyl
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
- A61P5/50—Drugs for disorders of the endocrine system of the pancreatic hormones for increasing or potentiating the activity of insulin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/75—Amino or imino radicals, acylated by carboxylic or carbonic acids, or by sulfur or nitrogen analogues thereof, e.g. carbamates
Definitions
- This invention relates to compounds which are activators of glucokinase (GK), which may be useful for the management, treatment, control, or adjunct treatment of diseases, where increasing glucokinase activity is beneficial.
- GK glucokinase
- Diabetes is characterised by an impaired glucose metabolism manifesting itself among other things by an elevated blood glucose level in the diabetic patients. Underlying defects lead to a classification of diabetes into two major groups: Type 1 diabetes, or insulin demanding diabetes mellitus (IDDM), which arises when patients lack 7-cells producing insulin in their pancreatic glands, and type 2 diabetes, or non-insulin dependent diabetes mellitus (NIDDM), which occurs in patients with an impaired yff-cell function besides a range of other abnormalities.
- IDDM insulin demanding diabetes mellitus
- NIDDM non-insulin dependent diabetes mellitus
- Type 1 diabetic patients are currently treated with insulin, while the majority of type 2 diabetic patients are treated either with sulphonylureas that stimulate ⁇ -ce ⁇ function or with agents that enhance the tissue sensitivity of the patients towards insulin or with insulin.
- agents applied to enhance tissue sensitivity towards insulin metformin is a representative example.
- the liver produces glucose in order to avoid hypoglycaemia.
- This glucose production is derived either from the release of glucose from glycogen stores or from gluconeogenesis, which is a de novo intracellular synthesis of glucose.
- type 2 diabetes the regulation of hepatic glucose output is poorly controlled and is increased, and may be doubled after an overnight fast.
- hepatic glucose production will be increased in type 1 diabetes, if the disease is not properly controlled by insulin treatment. Since existing forms of therapy of diabetes does not lead to sufficient glycaemic control and therefore are unsatisfactory, there is a great demand for novel therapeutic approaches.
- Atherosclerosis a disease of the arteries, is recognized to be the leading cause of death in the United States and Western Europe.
- the pathological sequence leading to atherosclerosis and occlusive heart disease is well known. The earliest stage in this sequence is the formation of "fatty streaks" in the carotid, coronary and cerebral arteries and in the aorta. These lesions are yellow in colour due to the presence of lipid deposits found principally within smooth-muscle cells and in macrophages of the intima layer of the arteries and aorta.
- fibrous plaque which consists of accumulated intimal smooth muscle cells laden with lipid and surrounded by extra-cellular lipid, collagen, elastin and proteoglycans.
- the cells plus matrix form a fibrous cap that covers a deeper deposit of cell debris and more extracellular lipid.
- the lipid is primarily free and esterified cholesterol.
- the fibrous plaque forms slowly, and is likely in time to become calcified and necrotic, advancing to the "complicated lesion” which accounts for the arterial occlusion and tendency toward mural thrombosis and arterial muscle spasm that characterize advanced atherosclerosis.
- CVD cardiovascular disease
- leaders of the medical profession have placed renewed emphasis on lowering plasma cholesterol levels, and low density lipoprotein cholesterol in particular, as an essential step in prevention of CVD.
- the upper limits of "normal” are now known to be significantly lower than heretofore appreciated.
- Independent risk factors include glucose intolerance, left ventricular hypertrophy, hypertension, and being of the male sex.
- Cardiovascular disease is especially prevalent among diabetic subjects, at least in part because of the existence of multiple independent risk factors in this population. Successful treatment of hyperlipidemia in the general population, and in diabetic subjects in particular, is therefore of exceptional medical importance.
- Hypertension is a condition, which occurs in the human population as a secondary symptom to various other disorders such as renal artery stenosis, pheochromocytoma, or endocrine disorders.
- hypertension is also evidenced in many patients in whom the causative agent or disorder is unknown. While such "essential" hypertension is often associated with disorders such as obesity, diabetes, and hypertriglyceridemia, the relationship between these disorders has not been elucidated. Additionally, many patients display the symptoms of high blood pressure in the complete absence of any other signs of disease or disorder.
- hypertension can directly lead to heart failure, renal failure, and stroke (brain haemorrhaging). These conditions are capable of causing short-term death in a patient. Hypertension can also contribute to the development of atherosclerosis and coronary disease. These conditions gradually weaken a patient and can lead to long-term death.
- the exact cause of essential hypertension is unknown, though a number of factors are believed to contribute to the onset of the disease. Among such factors are stress, uncontrolled emotions, unregulated hormone release (the renin, angiotensin aldosterone system), excessive salt and water due to kidney malfunction, wall thickening and hypertrophy of the vasculature resulting in constricted blood vessels and genetic factors.
- stress uncontrolled emotions
- unregulated hormone release the renin, angiotensin aldosterone system
- excessive salt and water due to kidney malfunction
- wall thickening and hypertrophy of the vasculature resulting in constricted blood vessels and genetic factors.
- Hypertension has been associated with elevated blood insulin levels, a condition known as hyperinsulinemia.
- Insulin a peptide hormone whose primary actions are to promote glucose utilization, protein synthesis and the formation and storage of neutral lipids, also acts to promote vascular cell growth and increase renal sodium retention, among other things. These latter functions can be accomplished without affecting glucose levels and are known causes of hypertension.
- Peripheral vasculature growth for example, can cause constriction of peripheral capillaries, while sodium retention increases blood volume.
- the lowering of insulin levels in hyperinsulinemics can prevent abnormal vascular growth and renal sodium retention caused by high insulin levels and thereby alleviates hypertension.
- Cardiac hypertrophy is a significant risk factor in the development of sudden death, myocardial infarction, and congestive heart failure. Theses cardiac events are due, at least in part, to increased susceptibility to myocardial injury after ischemia and reperfusion, which can occur in out-patient as well as perioperative settings. There is an unmet medical need to prevent or minimize adverse myocardial perioperative outcomes, particularly perioperative myocardial infarction. Both non-cardiac and cardiac surgery are associated with substantial risks for myocardial infarction or death. Some 7 million patients undergoing non-cardiac surgery are considered to be at risk, with incidences of perioperative death and serious cardiac complications as high as 20-25% in some series.
- perioperative myocardial infarction is estimated to occur in 5% and death in 1-2%.
- drug therapy is anticipated to be life- saving and reduce hospitalizations, enhance quality of life and reduce overall health care costs of high risk patients.
- Obesity is a well-known risk factor for the development of many very common diseases such as atherosclerosis, hypertension, and diabetes. The incidence of obese people and thereby also these diseases is increasing throughout the entire industrialised world. Except for exercise, diet and food restriction no convincing pharmacological treatment for reducing body weight effectively and acceptably currently exist. However, due to its indirect but important effect as a risk factor in mortal and common diseases it will be important to find treatment for obesity and/or means of appetite regulation.
- the term obesity implies an excess of adipose tissue.
- obesity is best viewed as any degree of excess adiposity that imparts a health risk.
- the cut off between normal and obese individuals can only be approximated, but the health risk imparted by the obesity is probably a continuum with increasing adiposity.
- the Framingham study demonstrated that a 20% excess over desirable weight clearly imparted a health risk (Mann GV N.Engl.J.Med 291, 226 (1974)).
- BMI body mass index
- the satiety centre may be activated by the increases in plasma glucose and/or insulin that follow a meal. Meal-induced gastric distension is another possible inhibitory factor. Additionally the hypothalamic centres are sensitive to catecholamines, and beta-adrenergic stimulation inhibits eating behaviour. Ultimately, the cerebral cortex controls eating behaviour, and impulses from the feeding centre to the cerebral cortex are only one input. Psychological, social, and genetic factors also influence food intake.
- initial weight loss is not an optimal therapeutic goal. Rather, the problem is that most obese patients eventually regain their weight.
- An effective means to establish and/or sustain weight loss is the major challenge in the treatment of obesity today.
- Glucokinase plays an essential role in blood glucose homeostasis. GK catalyses glucose phosphorylation, and is the rate-limiting reaction for glycolysis in hepatocytes and pancreatic yff-cells. In liver GK determine the rates of both glucose uptake and glycogen synthesis, and it is also thought to be essential for the regulation of various glucose-responsive genes (Girard, J.et al., Annu Rev Nutr 17, 325-352 (1997)). In ⁇ e ⁇ - cells, GK determines glucose utilization and thus is necessary for glucose-stimulated insulin secretion. GK is also expressed in a population of neurones in the hypothalamus where it might be involved in feeding behaviour and in the gut where it might contribute to the secretion of enteroincretins.
- GK has two main distinctive characteristics: its expression, which is limited to tissues that require glucose-sensing (mainly liver and pancreatic ?-cells), and its S 05 for glucose, which is much higher (8-12 mM) than that of the other members of the hexokinase family. Due to these kinetic characteristics, changes in serum glucose levels are paralleled by changes in glucose metabolism in liver which in turn regulate the balance between hepatic glucose output and glucose consumption.
- Activators of glucokinase may thus be useful for treating diseases where increasing the activity of glucokinase is beneficial.
- agents which activate glucokinase and increase glucokinase enzymatic activity would be useful for the treatment of type I diabetes and type II diabetes.
- WO 00/58293, WO 01/44216, WO/0183465, WO/0183478, WO/0185706, and WO 01/85707, to Hoffman-La Roche disclose compounds as glucokinase activators for treatment of type 2 diabetes.
- This invention provides amide derivatives which are activators of glucokinase.
- the compounds of the present invention are useful as activators of glucokinase and thus are useful for the management, treatment, control and adjunct treatment of diseases where increasing the activity of glucokinase is beneficial.
- diseases include type I diabetes and type II diabetes.
- the present invention provides compounds as described below, pharmaceutical compositions comprising the compounds, their use for increasing the activity of glucokinase, their use in preparation of a medicament for treating said diseases and conditions and the use of compounds or pharmaceutical preparations of the present invention for treating said diseases and conditions as well as methods for treating said diseases and conditions, which methods comprise administering to a subject in need thereof an effective amount of a compound according to the present invention.
- the present invention provides the use of a compound according to the present invention for increasing the activity of glucokinase.
- the present invention provides the use of a compound according to the present invention or a pharmaceutical composition according to the present invention for the treatment of hyperglycemia.
- the present invention provides the use of a compound according to the present invention for the preparation of a medicament for the treatment of hyperglycemia.
- the present invention provides the use of a compound according to the present invention or a pharmaceutical composition according to the present invention for treatment of IGT.
- the present invention provides the use of a compound according to the present invention for the preparation of a medicament for the treatment of IGT.
- the present invention provides the use of a compound according to the present invention or a pharmaceutical composition according to the present invention for the treatment of Syndrome X.
- the present invention provides the use of a compound according to the present invention for the preparation of a medicament for the treatment of Syndrome X.
- the present invention provides the use of a compound according to the present invention or a pharmaceutical composition according to the present invention for the treatment of type 2 diabetes.
- the present invention provides the use of a compound according to the present invention for the preparation of a medicament for the treatment of type 2 diabetes.
- the present invention provides the use of a compound according to the present invention or a pharmaceutical composition according to the present invention for the treatment of type 1 diabetes.
- the present invention provides the use of a compound according to the present invention for the preparation of a medicament for the treatment of type 1 diabetes.
- the present invention provides the use of a compound according to the present invention or a pharmaceutical composition according to the present invention for the treatment of dyslipidemia or hyperlipidemia.
- the present invention provides the use of a compound according to the present invention for the preparation of a medicament for the treatment of dyslipidemia or hyperlipidemia.
- the present invention provides the use of a compound according to the present invention or a pharmaceutical composition according to the present invention for the treatment of hypertension.
- the present invention provides the use of a compound according to the present invention for the preparation of a medicament for the treatment of hypertension.
- the present invention provides the use of a compound according to the present invention or a pharmaceutical composition according to the present invention for lowering of food intake.
- the present invention provides the use of a compound according to the present invention for the preparation of a medicament lowering of food intake.
- the present invention provides the use of a compound according to the present invention or a pharmaceutical composition according to the present invention for appetite regulation.
- the present invention provides the use of a compound according to the present invention for the preparation of a medicament for appetite regulation.
- the present invention provides the use of a compound according to the present invention or a pharmaceutical composition according to the present invention for the treatment or prophylaxis of obesity.
- the present invention provides the use of a compound according to the present invention for the preparation of a medicament for the treatment or prophylaxis of obesity.
- the present invention provides the use of a compound according to the present invention or a pharmaceutical composition according to the present invention for regulating feeding behaviour.
- the present invention provides the use of a compound according to the present invention for the preparation of a medicament for regulating feeding behaviour.
- the present invention provides the use of a compound according to the present invention or a pharmaceutical composition according to the present invention for enhancing the secretion of enteroincretins.
- said enteroincretin is GLP-1.
- the present invention provides the use of a compound according to the present invention for the preparation of a medicament for enhancing the secretion of enteroincretins.
- said enteroincretin is GLP-1.
- use according to the present invention as described above is for a regimen, which comprises treatment with a further antidiabetic agent, such as a further antidiabetic agent selected from insulin or an insulin analogue, a sulphonylurea, a biguanide, a meglitinide, an insulin sensitizer, a thiazolidinedione insulin sensitizer, an ⁇ -glucosidase inhibitor, a glycogen phosphorylase inhibitor, and an agent acting on the ATP-dependent potassium channel of the pancreatic yff-cells.
- a further antidiabetic agent selected from insulin or an insulin analogue, a sulphonylurea, a biguanide, a meglitinide, an insulin sensitizer, a thiazolidinedione insulin sensitizer, an ⁇ -glucosidase inhibitor, a glycogen phosphorylase inhibitor, and an agent acting on the ATP-dependent potassium channel of the pancreatic yf
- the use according to the present invention as described above is for a regimen, which comprises treatment with a further antihyperlipidemic agent, such as a further antihyperlipidemic agent selected from cholestyramine, colestipol, clofibrate, gemfibrozil, lovastatin, pravastatin, simvastatin, probucol, and dextrothyroxine.
- a further antihyperlipidemic agent selected from cholestyramine, colestipol, clofibrate, gemfibrozil, lovastatin, pravastatin, simvastatin, probucol, and dextrothyroxine.
- the use according to the present invention as described above is for a regimen, which comprises treatment with a further antiobesity agent.
- the use according to the present invention as described above is for a regimen, which comprises treatment with a further antihypertensive agent.
- halogen or "halo” means fluorine, chlorine, bromine or iodine.
- perhalomethyl means trifluoromethyl, trichloromethyl, tribromomethyl, or triiodomethyl.
- C x-y -alkyl, C x . y -alkenyl, C x-y -alkynyl, C x-y - cycloalkyl or C x-y -cycloalkyl-C x . y -alkenyl designates radical of the designated type having from x to y carbon atoms.
- alkyl refers to a straight or branched chain saturated monovalent hydrocarbon radical having from one to ten carbon atoms, for example C 1-8 -alkyl.
- Typical C 1-8 -alkyl groups include, but are not limited to e.g.
- C ⁇ -alkyl as used herein also includes secondary Cs- ⁇ -alkyl and tertiary C 4 ⁇ -alkyl.
- alkylene refers to a straight or branched chain saturated divalent hydrocarbon radical having from one to ten carbon atoms, for example C 1-8 -alkylene.
- alkylene as used herein include, but are not limited to, methylene, ethylene, and the like.
- alkenyl refers to a straight or branched chain monovalent hydrocarbon radical containing from two to ten carbon atoms and at least one carbon-carbon double bond, for example C 2-8 -alkenyl.
- Typical C 2-3 -alkenyl groups include, but are not limited to, vinyl, 1-propenyl, 2-propenyl, iso-propenyl, 1,3- butadienyl, 1-butenyl, 2-butenyl, 3-butenyl, 2-methyl-1-propenyl, 1 -pentenyl, 2-pentenyl, 3- pentenyl, 4-pentenyl, 3-methyl-2-butenyl, 1-hexenyl, 2-hexenyl, 3-hexenyl, 2,4-hexadienyl, 5- hexenyl and the like.
- alkenylene refers to a straight or branched chain divalent hydrocarbon radical having from two to ten carbon atoms and at least one carbon-carbon double bond, for example C( 2-8 )-alkenylene.
- Typical C( 2-8 )- alkenylene groups include, but are not limited to, ethene-1 ,2-diyl, propene-1 ,3-diyl, methylene-1,1-diyl, and the like.
- alkynyl refers to a straight or branched hydrocarbon group containing from 2 to the specified number of carbon atoms and at least one triple carbon-carbon bond, for example C 2-8 -alkynyl.
- Typical C 2-8 -alkynyl groups include, but are not limited to, ethynyl, 1-propynyl, 2-propynyl, 1-butynyl, 2-butynyl, 3-butynyl, 1-pentynyl, 2-pentynyl, 3-pentynyl, 4-pentynyl, 1-hexynyl, 2-hexynyl, 3-hexynyl, 5-hexynyl, 2,4-hexadiynyl and the like.
- alkynylene refers to a straight or branched chain divalent hydrocarbon radical having from two to ten carbon atoms and at least one carbon-carbon triple bond, for example C 2 . 8 -alkynylene.
- Typical C 2-8 -alkynylene groups include, but are not limited to, ethyne-1 ,2-diyl, propyne-1 ,3-diyl, and the like.
- cycloalkyl refers to a non- aromatic carbocyclic monovalent hydrocarbon radical having from three to twelve carbon atoms, and optionally with one or more degrees of unsaturation, for example Such a ring may be optionally fused to one or more benzene rings or to one or more of other cycloalkyl ring(s).
- Typical C 3 . ⁇ -cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl and the like.
- cycloalkylene refers to a non- aromatic carbocyclic divalent hydrocarbon radical having from three to twelve carbon atoms and optionally possessing one or more degrees of unsaturation, for example ene. Such a ring may be optionally fused to one or more benzene rings or to one or more of other cycloalkyl ring(s).
- Typical C 3-8 -cycloalkylene groups include, but are not limited to, cyclopropyl-1,1-diyl, cyclopropyl-1 ,2-diyl, cyclobutyl-1 ,2-diyl, cyclopentyl-1 ,3-diyl, cyclohexyl- 1 ,4-diyl, cycloheptyl-1 ,4-diyl, or cyclooctyl- 1,5-diyl, and the like.
- heterocyclic or the term “heterocyclyl” as used herein, alone or in combination, refers to a three to twelve membered heterocyclic ring having one or more degrees of unsaturation containing one or more heteroatomic substitutions selected from S, SO, SO 2 , O, or N, for example Cs ⁇ -heterocyclyl. Such a ring may be optionally fused to one or more of another "heterocyclic" ring(s) or cycloalkyl ring(s).
- Typical C 3 - 3 -heterocyclyl groups include, but are not limited to, tetrahydrofuran, 1 ,4-dioxane, 1 ,3-dioxane, piperidine, pyrrolidine, morpholine, piperazine, and the like.
- heterocyclylene refers to a three to twelve-membered heterocyclic ring diradical optionally having one or more degrees of unsaturation containing one or more heteroatoms selected from S, SO, SO 2 , O, or N. Such a ring may be optionally fused to one or more benzene rings or to one or more of another "heterocyclic" rings or cycloalkyl rings.
- heterocyclylene examples include, but are not limited to, tetrahydrofuran-2,5-diyl, morpholine-2,3-diyl, pyran-2,4-diyl, 1 ,4-dioxane-2,3-diyl, 1 ,3-dioxane-2,4-diyl, piperidine-2,4-diyl, piperidine-1 ,4-diyl, pyrrolidine-1 ,3-diyl, morpholine- 2,4-diyl, piperazine-1 ,4-dyil, and the like.
- alkoxy refers to the monovalent radical R a O-, where R a is alkyl as defined above, for example C (1 ⁇ r alkyl giving C(i-8)-alkoxy.
- Typical C ⁇ v-alkoxy groups include, but are not limited to, methoxy, ethoxy, n-propoxy, isopropoxy, butoxy, sec-butoxy, terf-butoxy, pentoxy, isopentoxy, hexoxy, isohexoxy and the like.
- alkylthio refers to a straight or branched monovalent radical comprising an alkyl group as described above linked through a divalent sulphur atom having its free valence bond from the sulphur atom, for example C 1-8 - alkylthio.
- Typical C 1-8 -alkylthio groups include, but are not limited to, methylthio, ethylthio, propylthio, butylthio, pentylthio, hexylthio and the like.
- alkoxycarbonyl refers to the monovalent radical R a OC(O)- , where R a is alkyl as described above, for example C ⁇ - 8 -alkoxycarbonyl.
- Typical C 1-8 - alkoxycarbonyl groups include, but are not limited to, methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, isopropoxycarbonyl, n-butoxycarbonyl, sec-butoxycarbonyl, tertbutoxycarbonyl, 3-methylbutoxycarbonyl, n-hexoxycarbonyl and the like.
- carbamoyl refers to NH 2 C(O)-.
- aryl refers to a carbocyclic aromatic ring radical with for instance 6 to 8 member atoms, or to an aromatic ring system radical with for instance from 12 to 18 member atoms.
- Aryl is also intended to include the partially hydrogenated derivatives of the carbocyclic systems.
- heteroaryl refers to an aromatic ring radical with for instance 5 to 7 member atoms, or to an aromatic ring system radical with for instance from 7 to 18 member atoms, containing one or more heteroatoms selected from nitrogen, oxygen, or sulfur heteroatoms, wherein N-oxides and sulfur monoxides and sulfur dioxides are permissible heteroaromatic substitutions; such as e.g.
- aryl and “heteroaryl” includes, but are not limited to phenyl, biphenyl, indene, fluorene, naphthyl (1-naphthyl, 2-naphthyl), anthracene (1-anthracenyl, 2- anthracenyl, 3-anthracenyl), thiophene (2-thienyl, 3-thienyl), furyl (2-furyl, 3-furyl), indolyl, oxadiazolyl, isoxazolyl, thiadiazolyl, oxatriazolyl, thiatriazolyl, quinazolin, fluorenyl, xanthenyl, isoindanyl, benzhydryl, acridinyl, thiazolyl, pyrrolyl (1 -pyrrolyl, 2-pyrrolyl, 3-pyrrolyl), pyrazolyl (1 -pyrazolyl, 3-pyrroly
- fused arylheterocyclyl refers to an aryl group fused to a heterocyclyl group, the two having two atoms in common, and wherein the aryl group is the point of substitution.
- fused arylheterocyclyl used herein include 4-(2,3-benzo- dioxin), 3,4-methylenedioxy-l -phenyl, and the like.
- fused heterocyclylaryl refers to a heterocyclyl group fused to an aryl group, the two having two atoms in common, and wherein the heterocyclyl group is the point of substitution.
- fused heterocyclylaryl used herein include 2- (1 ,3-benzodioxole and the like.
- fused heteroarylheterocyclyl refers to a heteroaryl group fused to an heterocyclyl group, the two having two atoms in common, and wherein the heteroaryl group is the point of substitution.
- fused heteroarylheterocyclyl examples include 1 ,2,3,4-tetrahydro-beta-carboline and the like.
- fused heterocyclylheteroaryl refers to a heterocyclyl group fused to an heteroaryl group, the two having two atoms in common, and wherein the heterocyclyl group is the point of substitution.
- fused heterocyclylheteroaryl examples include 2-[1,3]-dioxolo[4,5-c]pyridine and the like.
- fused arylcycloalkyl refers to a aryl group fused to a cycloalkyl group, the two having two atoms in common, and wherein the aryl group is the point of substitution.
- fused cycloalkylaryl examples include 5-indanyl, 6- (1,2,3,4-tetrahydronaphthyl), and the like.
- fused cycloalkylaryl refers to a cycloalkyl group fused to an aryl group, the two having two atoms in common, and wherein the cycloalkyl group is the point of substitution
- fused cycloalkylaryl used herein include 1-indanyl, 2- indanyl, 1-(1,2,3,4-tetrahydronaphthyl), and the like.
- fused heteroarylcycloalkyl refers to a heteroaryl group fused to an cycloalkyl group, the two having two atoms in common, and wherein the heteroaryl group is the point of substitution.
- fused heteroarylcycloalkyl used herein include 5-aza-6-indanyl and the like.
- fused cycloakylheteroaryl refers to a cycloalkyl group fused to an heteroaryl group, the two having two atoms in common, and wherein the cycloalkyl group is the point of substitution.
- fused cycloalkylheteroaryl used herein include 5-aza-1-indanyl and the like.
- arylene refers to carbocyclic aromatic ring diradical or to a aromatic ring system diradical.
- Examples of “arylene” include, but are not limited to, benzene-1 ,4-diyl, naphthalene-1,8-diyl, and the like.
- heteroarylene refers to a five to seven membered aromatic ring diradical, or to a aromatic ring system diradical, containing one or more heteroatoms selected from nitrogen, oxygen, or sulfur heteroatoms, wherein N- oxides and sulfur monoxides and sulfur dioxides are permissible heteroaromatic substitutions.
- heteroarylene used herein are furan-2,5-diyl, thiophene-2,4-diyl, 1 ,3,4-oxadiazole-2,5-diyl, 1 ,3,4-thiadiazole-2,5-diyl, 1 ,3-thiazole-2,4-diyl, 1 ,3-thiazole-2,5-diyl, pyridine-2,4-diyl, pyridine-2,3-diyl, pyridine-2,5-diyl, pyrimidine-2,4-diyl, quinoline-2,3-diyl, and the like.
- alkylsulfanyl refers to the group R a S-, where R a is alkyl as described above.
- alkylsulfenyl refers to the group R a S(O)-, where R a is alkyl as described above.
- alkylsulfonyl refers to the group R a SO 2 -, where R a is alkyl as described above.
- acyl refers to the group R a C(O)- , where R a is alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, or heterocyclyl as described above.
- aroyl refers to the group R a C(O)- , where R a is aryl as described above.
- heteroaroyl refers to the group R a C(O)- , where R a is heteroaryl as described above.
- acyloxy refers to the group R a C(O)O- , where R a is alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, or heterocyclyl as described above.
- aroyloxy refers to the group R a C(O)O- , where R a is aryl as described above.
- heteroaroyloxy refers to the group R a C(O)O- , where R a is heteroaryl as described above.
- R a is heteroaryl as described above.
- mercapto shall refer to the substituent -SH.
- carboxy shall refer to the substituent -COOH.
- cyano shall refer to the substituent -CN.
- aminosulfonyl shall refer to the substituent -SO 2 NH 2 .
- sulfenyl shall refer to the substituent -S(O)-.
- sulfonyl shall refer to the substituent -S(O) 2 -.
- direct bond where part of a structural variable specification, refers to the direct joining of the substituents flanking (preceding and succeeding) the variable taken as a "direct bond”.
- lower refers to an group having between one and six carbons, and may be indicated with the prefix C x ⁇ -.
- Lower alkyl may thus be indicated as C ⁇ -alkyl, while lower alkylene may be indicated as C 2 ⁇ -alkylene.
- a radical such as C x-y -cycloalkyl-C a - b -alkenyl shall designate that the radical's point of attachment is in part of the radical mentioned last.
- the term “optionally” means that the subsequently described event(s) may or may not occur, and includes both event(s) which occur and events that do not occur.
- substituted refers to substitution with the named substituent or substituents, multiple degrees of substitution being allowed unless otherwise stated.
- the terms "contain” or “containing” can refer to in-line substitutions at any position along the above defined alkyl, alkenyl, alkynyl or cycloalkyl substituents with one or more of any of O, S, SO, SO 2 , N, or N-alkyl, including, for example, -CH 2 -O-CH 2 -, -CH 2 -SO 2 -CH 2 -, -CH 2 -NH-CH 3 and so forth.
- solvate is a complex of variable stoichiometry formed by a solute (in this invention, a compound of formula (I), (II), or (III)) and a solvent.
- solvents for the purpose of the present invention may not interfere with the biological activity of the solute.
- Solvents may be, by way of example, water, ethanol, or acetic acid.
- biohydrolyzable ester is an ester of a drug substance (in this invention, a compound of formula (I), (II), or (III)) which either a) does not interfere with the biological activity of the parent substance but confers on that substance advantageous properties in vivo such as duration of action, onset of action, and the like, or b) is biologically inactive but is readily converted in vivo by the subject to the biologically active principle.
- a drug substance in this invention, a compound of formula (I), (II), or (III)
- a) does not interfere with the biological activity of the parent substance but confers on that substance advantageous properties in vivo such as duration of action, onset of action, and the like
- b) is biologically inactive but is readily converted in vivo by the subject to the biologically active principle.
- the advantage is that, for example, the biohydrolyzable ester is orally absorbed from the gut and is transformed to (I) in plasma.
- lower alkyl esters e.g., C ⁇ -C 4
- lower acyloxyalkyl esters lower alkoxyacyloxyalkyl esters
- alkoxyacyloxy esters alkyl acylamino alkyl esters
- choline esters e.g., choline esters
- biohydrolyzable amide is an amide of a drug substance (in this invention, a compound of general formula (I), (II), or (III)) which either a) does not interfere with the biological activity of the parent substance but confers on that substance advantageous properties in vivo such as duration of action, onset of action, and the like, or b) is biologically inactive but is readily converted in vivo by the subject to the biologically active principle.
- the advantage is that, for example, the biohydrolyzable amide is orally absorbed from the gut and is transformed to (I) in plasma.
- Many examples of such are known in the art and include by way of example lower alkyl amides, ⁇ -amino acid amides, alkoxyacyl amides, and alkylaminoalkylcarbonyl amides.
- prodrug includes biohydrolyzable amides and biohydrolyzable esters and also encompasses a) compounds in which the biohydrolyzable functionality in such a prodrug is encompassed in the compound of formula (I): for example, the lactam formed by a carboxylic group in R 2 and an amine in R 4 , and b) compounds which may be oxidized or reduced biologically at a given functional group to yield drug substances of formula (I).
- Examples of these functional groups include, but are not limited to, 1 ,4- dihydropyridine, N-alkylcarbonyl-1 ,4-dihydropyridine, 1 ,4-cyclohexadiene, tert-butyl, and the like.
- pharmaceutically effective amount or shall mean that amount of a drug or pharmaceutical agent that will elicit the biological or medical response of a tissue, animal or human that is being sought by a researcher or clinician. This amount can be a therapeutically effective amount.
- therapeutically effective amount shall mean that amount of a drug or pharmaceutical agent that will elicit the therapeutic response of an animal or human that is being sought.
- treatment means the management and care of a patient for the purpose of combating a disease, disorder or condition.
- the term is intended to include the full spectrum of treatments for a given disorder from which the patient is suffering, such as the delaying of the progression of the disease, disorder or condition, the alleviation or relief of symptoms and complications, and/or the cure or elimination of the disease, disorder or condition.
- the patient to be treated is preferably a mammal, in particular a human being.
- the present invention provides carboxamide or sulfonamide activator of glucokinase having a heteroatom in the alpha, beta, or gamma position relative to the carboxamide or sulfonamide, respectively.
- the present invention provides compounds of the general formula (I)
- G is -S(O 2 )-, or -C(O)-;
- A is >N-, and
- X is a direct bond, -O-, -S-, -S(O)-, -S(O 2 )-, or -N(R 6 ) -, wherein R 6 is hydrogen or alkyl, which may optionally be substituted with one or more substituents R 16 , R 17 , and R 18 , and L 1 is -(CH 2 ) n -C(R 9 )(R 10 )m-Y-, or a direct bond, wherein n is an integer of from 1 to 6,
- R 9 and R 10 independently of each other are selected from alkyl, or cycloalkyl, optionally substituted by one or more substituents R 19 , R 20 , and R 21 ; or from aryl optionally substituted by one or more substituents R 40 , R 41 ,
- m is an integer of 0 to 1 .
- Y is a direct bond, -O- or -N(R 7 )-, wherein
- R 7 is hydrogen or alkyl, which may optionally be substituted with one or more substituents R 22 , R 23 , and R 24 ; or
- X is alkylene, which may optionally be substituted with one or more substituents R 25 , R 26 , and R 27 , or a direct bond
- L 1 is -O-, or -N(R 8 )-, wherein R 8 is hydrogen or alkyl, which may optionally be substituted with one or more substituents R 28 , R 29 , and R 30 ; or
- A is >C(R 2 )-, wherein R 2 is hydrogen or alkyl, optionally substituted with one or more substituents R 31 , R 32 , and R 33 , and X is -O-, -S-, -S(O)-, -S(O 2 )-, or -N(R 6 )-, wherein
- R 6 is as defined above, and L 1 is -(CH 2 ) deliberately-Y-, or a direct bond, wherein n is an integer of from 1 to 6, and
- Y is a direct bond, O, or -N(R 7 )-, wherein R 7 is as defined above;
- X is alkylene, which may optionally be substituted with one or more substituents R 25 ,
- L 1 is -O-, or -N(R 8 )-, wherein R 8 is as defined above;
- R 1 and R 3 independently of each other are selected from alkyl, alkenyl, alkynyl, cycloalkyl, and heterocyclyl, optionally substituted with one or more substituents R 34 , R 35 , and R 36 ; or from aryl, heteroaryl, fused heterocyclylaryl, fused heteroarylheterocyclyl, fused heterocyclylheteroaryl, fused arylcycloalkyl, fused cycloalkylaryl, fused heteroarylcycloalkyl, and fused cycloalkylheteroaryl, optionally substituted with one or more substituents R 44 , R 45 , R 46 , and R 47 ; or
- R 1 and R 3 may be taken together with the atoms to which they are attached to form a cycloalkyl or heterocyclyl ring, optionally substituted with one or more substituents R 34 , R 35 , and R 36 , and optionally fused to a heteroaryl or aryl ring, optionally substituted with one or more substituents R 44 , R 45 , R 46 , and R 47 ; or, when A is >C(R 2 )-, then
- R 1 and R 2 may be taken together with the atoms to which they are attached to form a cycloalkyl or heterocyclyl ring, optionally substituted with one or more substituents R 34 , R 35 , and R 36 , and optionally fused to a heteroaryl or aryl ring, optionally substituted with one or more substituents R 44 , R 45 , R 46 , and R 47 ; or
- R 2 and R 3 may be taken together with the atoms to which they are attached to form a cycloalkyl or heterocyclyl ring, optionally substituted with one or more substituents R 34 , R 35 , and R 36 , and optionally fused to a heteroaryl or aryl ring, optionally substituted with one or more substituents R 44 , R 45 , R 46 , and R 47 ;
- R 4 is hydrogen or alkyl, optionally substituted with one or more substituents R 37 , R 38 , and R 39 ;
- R 5 is aryl or heteroaryl, optionally substituted with one or more substituents R 48 , R 49 , R 50 , and
- R 37 , R 38 , and R 39 independently of each other are selected from
- C 2-6 -alkenyl and C 2-6 -alkynyl which may optionally be substituted with one or more substituents selected from -CN, -CF 3 , -OCF 3 , -OR 52 , -NR ⁇ R 53 and d-e-alkyl; C 3 - 8 -cycloalkyl, 0 3 - ⁇ -cycloalkylthio, heterocyclyl-C 1-6 -alkyl, heterocyclyl-C 2 - ⁇ -alkenyl, heterocyclyl-C 2-6 -alkynyl, aryl, aryloxy, aryloxycarbonyl, aroyl, aryl-C ⁇ -alkoxy, aryl-C ⁇ -alky
- C ⁇ .e-alkyl, C 2-6 -alkenyl and C 2 ⁇ -alkynyl which may optionally be substituted with one or more substituents selected from -CN, -CF 3 , -OCF 3 , -OR 52 , -NR ⁇ R 53 , and
- R 40 , R 41 , R 42 , and R 43 , or two of R 44 , R 45 , R 46 , and R 47 on adjacent carbon atoms may independently be taken together to form -O-CH 2 -O-, wherein R 52 and R 53 independently of each other are hydrogen, C ⁇ -alky!, aryl-C 1- ⁇ - alkyl or aryl; or
- R 52 and R ⁇ when attached to the same nitrogen atom, together with the said nitrogen atom may form a 3 to 8 membered heterocyclic ring optionally containing one or two further heteroatoms selected from nitrogen, oxygen and sulfur, and optionally containing one or two double bonds; and R 48 , R 49 , R 50 , and R 51 independently of each other are selected from halogen, perfluoroalkyl, cyano, alkyl-Z-, aryl-Z-, aryl-alkylene-Z-, N(R 63 )(R 64 )-alkylene-Z-; and R 65 -W-alkylene-Z-; wherein Z and W independently of each other are selected from a direct bond, alkylene, -O-, -N(R 66 )-, -S-, -SO 2 -, -C(O)N(R 66 )-, -N(R 66 )C(O)-, - N(R 6 ⁇ )C(
- R 66 and R 67 independently of each other are hydrogen or alkyl;
- R 63 , R 64 , and R 65 are selected from the group consisting of hydrogen, aryl, alkyl, and aryl-alkylene-; or
- R 63 and R 64 may be taken together to form a ring having the formula -(CH 2 ) r E-(CH 2 ) k - bonded to the nitrogen atom to which R 63 and R 64 are attached, wherein j is an integer of from 1 to 4; k is an integer of from 1 to 4; and
- E is a direct bond, -CH 2 -, -O-, -S-, -S(O 2 )-, -C(O)-, -C(O)N(H)-,
- R 68 and R 69 are selected from the group consisting of hydrogen, aryl, alkyl, and aryl-alkylene-; or a pharmaceutically acceptable salt, solvate or prodrug thereof.
- the present invention provides compounds of the general formula (II)
- G is -S(O 2 )-, or -C(O)-;
- R 2 is hydrogen or alkyl, which may optionally be substituted with one or more substituents R 31 , R 32 , and R 33 ;
- X is -O-, -S-, -S(O)-, -S(O 2 )-, or -N(R 6 )-, wherein R 6 is as described above;
- L 1 is -(CH 2 ) n -Y-, or a direct bond, wherein n is an integer of from 1 to 6, Y is a direct bond, O, or -N(R 7 )-, wherein R 7 is as described above;
- R 1 and R 3 independently of each other are selected from alkyl, alkenyl, alkynyl, cycloalkyl, and heterocyclyl, optionally substituted with one or more substituents R 34 , R 35 , and R 36 ; or from aryl, heteroaryl, fused heterocyclylaryl, fused heteroarylheterocyclyl, fused heterocyclylheteroaryl, fused arylcycloalkyl, fused cycloalkylaryl, fused heteroarylcyclo
- R 1 and R 2 may be taken together with the atoms to which they are attached to form a cycloalkyl or heterocyclyl ring, optionally substituted with one or more substituents R 34 , R 35 , and R 36 , and optionally fused to a heteroaryl or aryl ring, optionally substituted with one or more substituents R 44 , R 45 , R 46 , and R 47 ;
- the present invention provides compounds of the general formula (II)
- G is -S(O 2 )-, or -C(O)-, and X is -O-, -S-, -S(O)-, -S(O 2 )-, or -N(R 6 )-, wherein
- R 6 is as described above, and L 1 is -(CH 2 ) n -Y-, or a direct bond, wherein n is an integer of from 1 to 6, and Y is a direct bond, O, or -N(R 7 )-, wherein R 7 is as described above; or X is alkylene, which may optionally be substituted with one or more substituents R 25 ,
- L 1 is -O-, or -N(R 8 )-, wherein
- R 8 is as described above;
- R 1 is selected from alkyl, alkenyl, alkynyl, cycloalkyl, and heterocyclyl, optionally substituted with one or more substituents R 34 , R 35 , and R 36 ; or from aryl, heteroaryl, fused heterocyclylaryl, fused heteroarylheterocyclyl, fused heterocyclylheteroaryl, fused arylcycloalkyl, fused cycloalkylaryl, fused heteroarylcycloalkyl, and fused cycloalkylheteroaryl, optionally substituted with one or more substituents R 44 , R 45 , R 46 , and R 47 ;
- R 2 and R 3 are taken together with the atoms to which they are attached to form a cycloalkyl or heterocyclyl ring, optionally substituted with one or more substituents R 34 , R 35 , and R 36 , and optionally fused to a heteroaryl or aryl ring, optionally substituted with one or more substituents R 44 , R 45 , R 46 , and R 47 ;
- the present invention provides compounds of the general formula (II)
- G is -S(O 2 )-, or -C(O)-;
- R 2 is hydrogen or alkyl, which may optionally be substituted with one or more substituents
- X is alkylene, which may optionally be substituted with one or more substituents R 25 , R 26 , and
- L 1 is -O-, or -N(R 8 )-, wherein R 8 is as described above;
- R 1 and R 3 independently of each other are selected from alkyl, alkenyl, alkynyl, cycloalkyl, and heterocyclyl, optionally substituted with one or more substituents R 34 , R 35 , and R 36 ; or from aryl, heteroaryl, fused heterocyclylaryl, fused heteroarylheterocyclyl, fused heterocyclylheteroaryl, fused arylcycloalkyl, fused cycloalkylaryl, fused heteroarylcycloalkyl, and fused cycloalkylheteroaryl, optionally substituted with one or more substituents R 44 , R 45 , R 46 , and R 47 ; or
- R 1 and R 2 may be taken together with the atoms to which they are attached to form a cycloalkyl or heterocyclyl ring, optionally substituted with one or more substituents R 34 , R 35 , and R 36 , and optionally fused to a heteroaryl or aryl ring, optionally substituted with one or more substituents R 44 , R 45 , R 46 , and R 47 ; or
- R 2 and R 3 may be taken together with the atoms to which they are attached to form a cycloalkyl or heterocyclyl ring, optionally substituted with one or more substituents R 34 , R 35 , and R 36 , and optionally fused to a heteroaryl or aryl ring, optionally substituted with one or more substituents R 44 , R 45 , R 46 , and R 47 ;
- R 4 is as described above; and R 5 is as described above; wherein R 25 , R 26 , R 27 , R 31 , R 32 , R 33 , R 34 , R 35 , R 36 , R 44 , R 45 , R 46 , and R 47 are as described above; or a pharmaceutically acceptable salt, solvate or prodrug thereof.
- the present invention provides compounds of the general formula (III)
- X is a direct bond, -O-, -S-, -S(O)-, -S(O 2 )-, or -N(R 6 )-, wherein R 6 is as described above, and L 1 is -(CH 2 ) n - C(R 9 )(R 10 ) m -Y-, or a direct bond, wherein n is an integer of from 1 to 6, R 9 and R 10 are as described above; m is an integer of 0 to 1 , and Y is a direct bond, -O- or -N(R 7 )-, wherein R 7 is as described above; or X is alkylene, which may optionally be substituted with one or more substituents R 25 ,
- R 8 is as described above;
- R 1 and R 3 independently of each other are selected from alkyl, alkenyl, alkynyl, cycloalkyl, and heterocyclyl, optionally substituted with one or more substituents R 34 , R 35 , and R 36 , or from aryl, heteroaryl, fused heterocyclylaryl, fused heteroarylheterocyclyl, fused heterocyclylheteroaryl, fused arylcycloalkyl, fused cycloalkylaryl, fused heteroarylcycloalkyl, and fused cycloalkylheteroaryl, optionally substituted with one or more substituents R 44 , R 45 , R 46 , and R 47 ; or
- R 1 and R 3 may be taken together with the atoms to which they are attached to form a cycloalkyl or heterocyclyl ring, optionally substituted with one or more substituents R 34 , R 35 , and R 36 , and optionally fused to a heteroaryl or aryl ring, optionally substituted with one or more substituents R 44 , R 45 , R 46 , and R 47 ;
- R 4 is as described above; and R 5 is as described above; wherein R 25 , R 26 , R 27 , R 34 , R 35 , R 38 , R 44 , R 45 , R 46 , and R 47 are as described above; or a pharmaceutically acceptable salt, solvate or prodrug thereof.
- the present invention also provides a compound according to the present invention for use as a medicament.
- the present invention provides a compound according to the present invention for treatment of hyperglycemia.
- the present invention provides a compound according to the present invention for treatment of IGT.
- the present invention provides a compound according to the present invention for treatment of Syndrome X.
- the present invention provides a compound according to the present invention for treatment of type 2 diabetes. In one embodiment, the present invention provides a compound according to the present invention for treatment of type 1 diabetes. In one embodiment, the present invention provides a compound according to the present invention for treatment of dyslipidemia or hyperlipidemia.
- the present invention provides a compound according to the present invention for treatment of hypertension. In one embodiment, the present invention provides a compound according to the present invention for the treatment or prophylaxis of obesity.
- the present invention provides a compound according to the present invention for lowering of food intake.
- the present invention provides a compound according to the present invention for appetite regulation.
- the present invention provides a compound according to the present invention for regulating feeding behaviour.
- the present invention provides a compound according to the present invention for enhancing the secretion of enteroincretins.
- said enteroincretin is GLP-1.
- the present invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising, as an active ingredient, at least one compound according to the present invention together with one or more pharmaceutically acceptable carriers or excipients.
- the present invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising, as an active ingredient, at least one compound according to the present invention together with one or more pharmaceutically acceptable carriers or excipients in unit dosage form, comprising from about 0.05 mg to about 1000 mg, preferably from about 0.1 mg to about 500 mg and especially preferred from about 0.5 mg to about 200 mg of the compound according to the present invention.
- the pharmaceutical composition according to the present invention comprises a further antidiabetic agent.
- said further antidiabetic agent is insulin or an insulin analogue.
- said further antidiabetic agent is a sulphonylurea.
- said further antidiabetic agent is a biguanide. In one embodiment, said further antidiabetic agent is a meglitinide.
- said further antidiabetic agent is an insulin sensitizer.
- said further antidiabetic agent is a thiazolidinedione insulin sensitizer.
- said further antidiabetic agent is an ⁇ -glucosidase inhibitor. In one embodiment, said further antidiabetic agent is a glycogen phosphorylase inhibitor. In one embodiment, said further antidiabetic agent is an agent acting on the ATP- dependent potassium channel of the pancreatic ?-cells.
- the pharmaceutical composition according to the present invention comprises a further antihyperlipidemic agent.
- said further antihyperlipidemic agent is cholestyramine.
- said further antihyperlipidemic agent is colestipol.
- said further antihyperlipidemic agent is clofibrate.
- said further antihyperlipidemic agent is gemfibrozil.
- said further antihyperlipidemic agent is lovastatin. In one embodiment, said further antihyperlipidemic agent is pravastatin.
- said further antihyperlipidemic agent is simvastatin.
- said further antihyperlipidemic agent is probucol.
- said further antihyperlipidemic agent is dextrothyroxine.
- the present invention also covers the individual enantiomers of the compounds represented by formula (I) above as mixtures with diastereoisomers thereof in which one or more stereocenters are inverted.
- the present compounds are activators of glucokinase and are as such useful for the activation of glucokinase.
- the present invention provides a method for activating glucokinase in a patient in need thereof, which method comprises administering to a subject in need thereof a compound of the present invention, preferably a compound of formula (I), (II), or (III), preferably a pharmacologically effective amount, more preferably a therapeutically effective amount.
- the present invention also provides a method for lowering blood glucose in a patient in need thereof, which method comprises administering to a subject in need thereof a compound of the present invention, preferably a compound of formula (I), (II), or (III), preferably a pharmacologically effective amount, more preferably a therapeutically effective amount.
- the present invention also provides a method for prevention and/or treatment of glucokinase deficiency-mediated human diseases, the method comprising administration to a human in need thereof a therapeutically effective amount of a compound of the present invention, preferably a compound of formula (I), (II), or (III).
- a subject in need thereof includes mammalian subjects, preferably humans, who either suffer from one or more of the aforesaid diseases or disease states or are at risk for such.
- this method also is comprised of a method for treating a mammalian subject prophylactically, or prior to the onset of diagnosis such disease(s) or disease state(s).
- Other embodiments of such methods will be clear from the following description.
- Compounds according to the present invention are useful for the treatment of disorders, diseases and conditions, wherein the activation of glucokinase is beneficial. Accordingly, the present compounds are useful for the treatment of hyperglycemia,
- IGT insulin resistance syndrome
- syndrome X type 1 diabetes, type 2 diabetes
- dyslipidemia dyslipoproteinemia (abnormal lipoproteins in the blood) including diabetic dyslipidemia, hyperlipidemia, hyperlipoproteinemia (excess of lipoproteins in the blood) including type I, ll-a (hypercholesterolemia), ll-b, III, IV (hypertriglyceridemia) and V (hypertriglyceridemia) hyperiipoproteinemias, and obesity.
- they may be useful for the treatment of albuminuria, cardiovascular diseases such as cardiac hypertrophy, hypertension and arteriosclerosis including atherosclerosis; gastrointestinal disorders; acute pancreatitis; and appetite regulation or energy expenditure disorders.
- the effective amount of the compound according to the present invention is in the range of from about 0.05 mg to about 2000 mg, preferably from about 0.1 mg to about 1000 mg and especially preferred from about 0.5 mg to about 500 mg per day.
- the method is used in a regimen which comprises treatment with a further antidiabetic agent, such as a further antidiabetic agent selected from insulin or an insulin analogue, a sulphonylurea, a biguanide, a meglitinide, an insulin sensitizer, a thiazolidinedione insulin sensitizer, an ⁇ -glucosidase inhibitor, a glycogen phosphorylase inhibitor, and an agent acting on the ATP- dependent potassium channel of the pancreatic ?-cells.
- a further antidiabetic agent selected from insulin or an insulin analogue, a sulphonylurea, a biguanide, a meglitinide, an insulin sensitizer, a thiazolidinedione insulin sensitizer, an ⁇ -glucosidase inhibitor, a glycogen phosphorylase inhibitor, and an agent acting on the ATP- dependent potassium channel of the pancreatic ?-cells.
- the method is used in a regimen which comprises treatment with a further antihyperlipidemic agent, such as a further antihyperlipidemic agent selected from cholestyramine, colestipol, clofibrate, gemfibrozil, lovastatin, pravastatin, simvastatin, probucol, and dextrothyroxine.
- a further antihyperlipidemic agent selected from cholestyramine, colestipol, clofibrate, gemfibrozil, lovastatin, pravastatin, simvastatin, probucol, and dextrothyroxine.
- the method is used in a regimen which comprises treatment with a further antiobesity agent. In one embodiment of a method according to the present invention, the method is used in a regimen which comprises treatment with a further antihypertensive agent.
- the invention relates to a compound according to the present invention for use as a medicament.
- the invention also relates to pharmaceutical compositions comprising, as an active ingredient, at least one compound of the present invention together with one or more pharmaceutically acceptable carriers or excipients.
- the pharmaceutical composition is preferably in unit dosage form, comprising from about 0.05 mg to about 1000 mg, preferably from about 0.1 mg to about 500 mg and especially preferred from about 0.5 mg to about 200 mg of the compound of the present invention, such as a compound with formula (I), (II), or (III).
- the present compounds are used for the preparation of a medicament for the treatment of hyperglycemia.
- hyperglycemia is to be taken as generally understood in the art, with reference for example to the Report of the Expert Committee of the Diagnosis and Classification of Diabetes Mellitus, published in Diabetes Care 20, 1183-1197, (1997), but is usually taken to mean an elevated plasma glucose level exceeding about 110 mg/dl.
- the present compounds are effective in lowering the blood glucose both in the fasting and postprandial stage.
- the present compounds are used for the preparation of a pharmaceutical composition for the treatment of IGT.
- the present compounds are used for the preparation of a pharmaceutical composition for the treatment of Syndrome X.
- the present compounds are used for the preparation of a pharmaceutical composition for the treatment of type 2 diabetes.
- Such treatment includes ia treatment for the purpose of the delaying of the progression from IGT to type 2 diabetes as well as delaying the progression from non-insulin requiring type 2 diabetes to insulin requiring type 2 diabetes.
- the present compounds are used for the preparation of a pharmaceutical composition for the treatment of type 1 diabetes.
- Such therapy is normally accompanied by insulin administration.
- the present compounds are used for the preparation of a pharmaceutical composition for the treatment of dyslipidemia and hyperlipidemia.
- the present compounds are used for the preparation of a pharmaceutical composition for the treatment of obesity.
- treatment of a patient with the present compounds are combined with diet and/or exercise.
- the present invention provides methods of activating glucokinase activity in a mammal, which methods comprise administering, to a mammal in need of activation of glucokinase activity, a therapeutically defined amount of a compound of the present invention, such as a compound of formula (I), (II), or (III), defined above, as a single or polymorphic crystalline form or forms, an amorphous form, a single enantiomer, a racemic mixture, a single stereoisomer, a mixture of stereoisomers, a single diastereoisomer, a mixture of diastereoisomers, a solvate, a pharmaceutically acceptable salt, a solvate, a prodrug, a biohydrolyzable ester, or a biohydrolyzable amide thereof.
- a compound of the present invention such as a compound of formula (I), (II), or (III), defined above, as a single or polymorphic crystalline form or forms
- the present invention provides a method of activating glucokinase, comprising the step of administering to a mammal in need thereof a pharmacologically effective amount of a compound of the present invention.
- the invention further provides a pharmaceutical composition comprising a pharmaceutically acceptable carrier and a pharmacologically effective amount of a compound of the present invention sufficient to activate glucokinase.
- a glucokinase - activating amount can be an amount that reduces or inhibits a PTPase activity in the subject.
- a pharmaceutical composition comprising a pharmaceutically acceptable carrier and a pharmacologically effective amount of a compound of the present invention sufficient to treat type I diabetes.
- Also provided by the present invention is a pharmaceutical composition
- a pharmaceutical composition comprising a pharmaceutically acceptable earner and a pharmacologically effective amount of a compound of the present invention sufficient to treat type II diabetes.
- the compounds of the present invention can be administered to any mammal in need of activation of glucokinase activity.
- mammals can include, for example, horses, cows, sheep, pigs, mice, dogs, cats, primates such as chimpanzees, gorillas, rhesus monkeys, and, most preferably humans.
- the present compounds are administered in combination with one or more further active substances in any suitable ratios.
- Such further active agents may be selected from antidiabetic agents, antihyperlipidemic agents, antiobesity agents, antihypertensive agents and agents for the treatment of complications resulting from or associated with diabetes.
- Suitable antidiabetic agents include insulin, GLP-1 (glucagon like peptide-1) derivatives such as those disclosed in WO 98/08871 (Novo Nordisk A/S), which is incorporated herein by reference, as well as orally active hypoglycemic agents.
- Suitable orally active hypoglycemic agents preferably include imidazolines, sulfonylureas, biguanides, meglitinides, oxadiazolidinediones, thiazolidinediones, insulin sensitizers, ⁇ -glucosidase inhibitors, agents acting on the ATP-dependent potassium channel of the pancreatic ?-cells eg potassium channel openers such as those disclosed in WO 97/26265, WO 99/03861 and WO 00/37474 (Novo Nordisk A/S) which are incorporated herein by reference, potassium channel openers, such as ormitiglinide, potassium channel blockers such as nateglinide or BTS-67582, glucagon antagonists such as those disclosed in WO 99/01423 and WO 00/39088 (Novo Nordisk A/S and Agouron Pharmaceuticals, Inc.), all of which are incorporated herein by reference, GLP-1 agonists such as those disclosed in WO
- the present compounds are administered in combination with insulin or insulin analogues.
- the present compounds are administered in combination with a sulphonylurea eg tolbutamide, chlorpropamide, tolazamide, glibenclamide, glipizide, glimepiride, glicazide or glyburide.
- a sulphonylurea eg tolbutamide, chlorpropamide, tolazamide, glibenclamide, glipizide, glimepiride, glicazide or glyburide.
- the present compounds are administered in combination with a biguanide eg metformin.
- the present compounds are administered in combination with a meglitinide eg repaglinide or senaglinide/nateglinide.
- a meglitinide eg repaglinide or senaglinide/nateglinide.
- the present compounds are administered in combination with a thiazolidinedione insulin sensitizer eg troglitazone, ciglitazone, pioglitazone, rosiglitazone, isaglitazone, darglitazone, englitazone, CS-011/CI- 1037 or T 174 or the compounds disclosed in WO 97/41097 (DRF-2344), WO 97/41119, WO 97/41120, WO 00/41121 and WO 98/45292 (Dr. Reddy's Research Foundation), which are incorporated herein by reference.
- a thiazolidinedione insulin sensitizer eg troglitazone, ciglitazone, pioglitazone, rosiglitazone, isaglitazone, darglitazone, englitazone, CS-011/CI- 1037 or T 174 or the compounds disclosed in WO 97/41097 (DRF-2344), WO
- the present compounds may be administered in combination with an insulin sensitizer eg such as GI 262570, YM-440, MCC- 555, JTT-501, AR-H039242, KRP-297, GW-409544, CRE-16336, AR-H049020, LY510929, MBX-102, CLX-0940, GW-501516 or the compounds disclosed in WO 99/19313 (NN622/DRF-2725), WO 00/50414, WO 00/63191 , WO 00/63192, WO 00/63193 (Dr.
- an insulin sensitizer eg such as GI 262570, YM-440, MCC- 555, JTT-501, AR-H039242, KRP-297, GW-409544, CRE-16336, AR-H049020, LY510929, MBX-102, CLX-0940, GW-501516 or the compounds disclosed in WO 99/19313 (NN622/
- the present compounds are administered in combination with an ⁇ -glucosidase inhibitor eg voglibose, emiglitate, miglitol or acarbose.
- an ⁇ -glucosidase inhibitor eg voglibose, emiglitate, miglitol or acarbose.
- the present compounds are administered in combination with a glycogen phosphorylase inhibitor eg the compounds described in WO 97/09040 (Novo Nordisk A/S).
- the present compounds are administered in combination with an agent acting on the ATP-dependent potassium channel of the pancreatic ?-cells eg tolbutamide, glibenclamide, glipizide, glicazide, BTS-67582 or repaglinide.
- an agent acting on the ATP-dependent potassium channel of the pancreatic ?-cells eg tolbutamide, glibenclamide, glipizide, glicazide, BTS-67582 or repaglinide.
- the present compounds are administered in combination with nateglinide. In one embodiment of the present invention the present compounds are administered in combination with an antihyperlipidemic agent or a antilipidemic agent eg cholestyramine, colestipol, clofibrate, gemfibrozil, lovastatin, pravastatin, simvastatin, probucol or dextrothyroxine.
- an antihyperlipidemic agent or a antilipidemic agent eg cholestyramine, colestipol, clofibrate, gemfibrozil, lovastatin, pravastatin, simvastatin, probucol or dextrothyroxine.
- the present compounds are administered in combination with more than one of the above-mentioned compounds eg in combination with metformin and a sulphonylurea such as glyburide; a sulphonylurea and acarbose; nateglinide and metformin; acarbose and metformin; a sulfonylurea, metformin and troglitazone; insulin and a sulfonylurea; insulin and metformin; insulin, metformin and a sulfonylurea; insulin and troglitazone; insulin and lovastatin; etc.
- the compounds according to the present invention may be administered in combination with one or more antiobesity agents or appetite regulating agents.
- Such agents may be selected from the group consisting of CART (cocaine amphetamine regulated transcript) agonists, NPY (neuropeptide Y) antagonists, MC3 (melanocortin 3) agonists, MC4 (melanocortin 4) agonists, orexin antagonists, TNF (tumor necrosis factor) agonists, CRF (corticotropin releasing factor) agonists, CRF BP (corticotropin releasing factor binding protein) antagonists, urocortin agonists, ⁇ Z adrenergic agonists such as CL-316243, AJ-9677, GW-0604, LY362884, LY377267 or AZ-40140, MSH (melanocyte-stimulating hormone) agonists, MCH (melanocyte-concentrating hormone) antagonists, CCK (cholecystokinin) agonists, serotonin reuptake inhibitors (fluoxetine, seroxat or citalopram
- the antiobesity agent is leptin.
- the antiobesity agent is a serotonin and norepinephrine reuptake inhibitor eg sibutramine.
- the antiobesity agent is a lipase inhibitor eg orlistat.
- the antiobesity agent is an adrenergic CNS stimulating agent eg dexamphetamine, amphetamine, phentermine, mazindol phendimetrazine, diethylpropion, fenfluramine or dexfenfluramine.
- an adrenergic CNS stimulating agent eg dexamphetamine, amphetamine, phentermine, mazindol phendimetrazine, diethylpropion, fenfluramine or dexfenfluramine.
- the present compounds may be administered in combination with one or more antihypertensive agents.
- antihypertensive agents are ⁇ -blockers such as alprenolol, atenolol, timolol, pindolol, propranolol and metoprolol, ACE (angiotensin converting enzyme) inhibitors such as benazepril, captopril, enalapril, fosinopril, lisinopril, quinapril and ramipril, calcium channel blockers such as nifedipine, felodipine, nicardipine, isradipine, nimodipine, diltiazem and verapamil, and ⁇ -blockers such as doxazosin, urapidil, prazosin and terazosin. Further reference can be made to Remington: The Science and Practice of Pharmacy, 19th Edition, Gennaro, Ed., Mack Publishing Co
- the compounds of the present invention may be administered alone or in combination with pharmaceutically acceptable carriers or excipients, in either single or multiple doses.
- the pharmaceutical compositions according to the present invention may be formulated with pharmaceutically acceptable carriers or diluents as well as any other known adjuvants and excipients in accordance with conventional techniques such as those disclosed in Remington: The Science and Practice of Pharmacy, 19 th Edition, Gennaro, Ed., Mack Publishing Co., Easton, PA, 1995.
- compositions may be specifically formulated for administration by any suitable route such as the oral, rectal, nasal, pulmonary, topical (including buccal and sublingual), transdermal, intracisternal, intraperitoneal, vaginal and parenteral (including subcutaneous, intramuscular, intrathecal, intravenous and intradermal) route, the oral route being preferred. It will be appreciated that the preferred route will depend on the general condition and age of the subject to be treated, the nature of the condition to be treated and the active ingredient chosen.
- compositions for oral administration include solid dosage forms such as hard or soft capsules, tablets, troches, dragees, pills, lozenges, powders and granules. Where appropriate, they can be prepared with coatings such as enteric coatings or they can be formulated so as to provide controlled release of the active ingredient such as sustained or prolonged release according to methods well known in the art.
- Liquid dosage forms for oral administration include solutions, emulsions, aqueous or oily suspensions, syrups and elixirs.
- compositions for parenteral administration include sterile aqueous and non-aqueous injectable solutions, dispersions, suspensions or emulsions as well as sterile powders to be reconstituted in sterile injectable solutions or dispersions prior to use. Depot injectable formulations are also contemplated as being within the scope of the present invention.
- a typical oral dosage is in the range of from about 0.001 to about 100 mg/kg body weight per day, preferably from about 0.01 to about 50 mg/kg body weight per day, and more preferred from about 0.05 to about 10 mg/kg body weight per day administered in one or more dosages such as 1 to 3 dosages.
- the exact dosage will depend upon the frequency and mode of administration, the sex, age, weight and general condition of the subject treated, the nature and severity of the condition treated and any concomitant diseases to be treated and other factors evident to those skilled in the art.
- a typical unit dosage form for oral administration one or more times per day such as 1 to 3 times per day may contain from 0.05 to about 1000 mg, preferably from about 0.1 to about 500 mg, and more preferred from about 0.5 mg to about 200 mg.
- the compounds of this invention are generally utilized as the free substance or as a pharmaceutically acceptable salt thereof.
- examples are an acid addition salt of a compound having the utility of a free base and a base addition salt of a compound having the utility of a free acid.
- pharmaceutically acceptable salts refers to non-toxic salts of the compounds of this invention which are generally prepared by reacting the free base with a suitable organic or inorganic acid or by reacting the acid with a suitable organic or inorganic base.
- salts are prepared in a conventional manner by treating a solution or suspension of the compound with a chemical equivalent of a pharmaceutically acceptable acid.
- a compound of the present invention such as a compound of formula (I), (II), or (III)
- contains a free acid such salts are prepared in a conventional manner by treating a solution or suspension of the compound with a chemical equivalent of a pharmaceutically acceptable base.
- Physiologically acceptable salts of a compound with a hydroxy group include the anion of said compound in combination with a suitable cation such as sodium or ammonium ion.
- Other salts which are not pharmaceutically acceptable may be useful in the preparation of compounds of the present invention and these form a further aspect of the present invention.
- solutions of the novel compounds of the formula (I) in sterile aqueous solution, aqueous propylene glycol or sesame or peanut oil may be employed.
- aqueous solutions should be suitably buffered if necessary and the liquid diluent first rendered isotonic with sufficient saline or glucose.
- the aqueous solutions are particularly suitable for intravenous, intramuscular, subcutaneous and intraperitoneal administration.
- the sterile aqueous media employed are all readily available by standard techniques known to those skilled in the art.
- Suitable pharmaceutical carriers include inert solid diluents or fillers, sterile aqueous solution and various organic solvents.
- solid carriers are lactose, terra alba, sucrose, cyclodextrin, talc, gelatine, agar, pectin, acacia, magnesium stearate, stearic acid and lower alkyl ethers of cellulose.
- liquid carriers are syrup, peanut oil, olive oil, phospholipids, fatty acids, fatty acid amines, polyoxyethylene and water.
- the carrier or diluent may include any sustained release material known in the art, such as glyceryl monostearate or glyceryl distearate, alone or mixed with a wax.
- the pharmaceutical compositions formed by combining the novel compounds of the present invention and the pharmaceutically acceptable carriers are then readily administered in a variety of dosage forms suitable for the disclosed routes of administration.
- the formulations may conveniently be presented in unit dosage form by methods known in the art of pharmacy.
- Formulations of the present invention suitable for oral administration may be presented as discrete units such as capsules or tablets, each containing a predetermined amount of the active ingredient, and which may include a suitable excipient.
- the orally available formulations may be in the form of a powder or granules, a solution or suspension in an aqueous or non-aqueous liquid, or an oil-in-water or water-in-oil liquid emulsion.
- compositions intended for oral use may be prepared according to any known method, and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents, and preserving agents in order to provide pharmaceutically elegant and palatable preparations.
- Tablets may contain the active ingredient in admixture with non-toxic pharmaceutically-acceptable excipients which are suitable for the manufacture of tablets.
- excipients may be for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example corn starch or alginic acid; binding agents, for example, starch, gelatin or acacia; and lubricating agents, for example magnesium stearate, stearic acid or talc.
- the tablets may be uncoated or they may be coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- a time delay material such as glyceryl monostearate or glyceryl distearate may be employed.
- Formulations for oral use may also be presented as hard gelatin capsules where the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or a soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example peanut oil, liquid paraffin, or olive oil.
- an inert solid diluent for example, calcium carbonate, calcium phosphate or kaolin
- an oil medium for example peanut oil, liquid paraffin, or olive oil.
- Aqueous suspensions may contain the active compounds in admixture with excipients suitable for the manufacture of aqueous suspensions.
- excipients are suspending agents, for example sodium carboxymethylcellulose, methylcellulose, hydroxypropylmethylcellulose, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia; dispersing or wetting agents may be a naturally-occurring phosphatide such as lecithin, or condensation products of an alkylene oxide with fatty acids, for example polyoxyethylene stearate, or condensation products of ethylene oxide with long chain aliphatic alcohols, for example, heptadecaethyl-eneoxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate, or condensation products of ethylene oxide with partial esters derived from fatty acids and hexitol anhydrides, for example polyethylene
- the aqueous suspensions may also contain one or more coloring agents, one or more flavoring agents, and one or more sweetening agents, such as sucrose or saccharin.
- Oily suspensions may be formulated by suspending the active ingredient in a vegetable oil, for example arachis oil, olive oil, sesame oil or coconut oil, or in a mineral oil such as a liquid paraffin.
- the oily suspensions may contain a thickening agent, for example beeswax, hard paraffin or cetyl alchol. Sweetening agents such as those set forth above, and flavoring agents may be added to provide a palatable oral preparation. These compositions may be preserved by the addition of an anti-oxidant such as ascorbic acid.
- Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide the active compound in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives.
- a dispersing or wetting agent e.g., talc, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, mannitol, mannitol, mannitol, mannitol, mannitol, mannitol, mannitol, mannitol, mannitol,
- the pharmaceutical compositions of the present invention may also be in the form of oil-in-water emulsions.
- the oily phase may be a vegetable oil, for example, olive oil or arachis oil, or a mineral oil, for example a liquid paraffin, or a mixture thereof.
- Suitable emulsifying agents may be naturally-occurring gums, for example gum acacia or gum tragacanth, naturally-occurring phosphatides, for example soy bean, lecithin, and esters or partial esters derived from fatty acids and hexitol anhydrides, for example sorbitan monooleate, and condensation products of said partial esters with ethylene oxide, for example polyoxyethylene sorbitan monooleate.
- the emulsions may also contain sweetening and flavoring agents.
- Syrups and elixirs may be formulated with sweetening agents, for example glycerol, propylene glycol, sorbitol or sucrose. Such formulations may also contain a demulcent, a preservative and flavoring and coloring agents.
- the pharmaceutical compositions may be in the form of a sterile injectible aqueous or oleaginous suspension. This suspension may be formulated according to the known methods using suitable dispersing or wetting agents and suspending agents described above.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally-acceptable diluent or solvent, for example as a solution in 1,3-butanediol.
- Suitable vehicles and solvents that may be employed are water, Ringer's solution, and isotonic sodium chloride solution.
- sterile, fixed oils are conveniently employed as solvent or suspending medium.
- any bland fixed oil may be employed using synthetic mono- or diglycerides.
- fatty acids such as oleic acid find use in the preparation of injectables.
- compositions may also be in the form of suppositories for rectal administration of the compounds of the present invention.
- These compositions can be prepared by mixing the drug with a suitable non-irritating excipient which is solid at ordinary temperatures but liquid at the rectal temperature and will thus melt in the rectum to release the drug.
- suitable non-irritating excipient include cocoa butter and polyethylene glycols, for example.
- topical applications For topical use, creams, ointments, jellies, solutions of suspensions, etc., containing the compounds of the present invention are contemplated.
- topical applications shall include mouth washes and gargles.
- the compounds of the present invention may also be administered in the form of liposome delivery systems, such as small unilamellar vesicles, large unilamellar vesicles, and multilamellar vesicles.
- Liposomes may be formed from a variety of phospholipids, such as cholesterol, stearylamine, or phosphatidylcholines.
- some of the compounds of the present invention may form solvates with water or common organic solvents. Such solvates are also encompassed within the scope of the present invention.
- a pharmaceutical composition comprising a compound of the present invention, or a pharmaceutically acceptable salt, solvate, or prodrug therof, and one or more pharmaceutically acceptable carriers, excipients, or diluents.
- the preparation may be tabletted, placed in a hard gelatine capsule in powder or pellet form or it can be in the form of a troche or lozenge.
- the amount of solid carrier will vary widely but will usually be from about 25 mg to about 1 g.
- the preparation may be in the form of a syrup, emulsion, soft gelatine capsule or sterile injectable liquid such as an aqueous or non-aqueous liquid suspension or solution.
- a typical tablet that may be prepared by conventional tabletting techniques may contain: Core:
- Active compound (as free compound or salt thereof) 5.0 mg
- Polacrillin potassium NF tablet disintegrant, Rohm and Haas. ** Acylated monoglyceride used as plasticizer for film coating.
- composition of the present invention may comprise a compound according to the present invention in combination with further active substances such as those described in the foregoing.
- the present invention also provides a method for the synthesis of compounds useful as intermediates in the preparation of compounds of formula (I) along with methods for the preparation of compounds of formula (I).
- the compounds can be prepared readily according to the following reaction Schemes (in which all variables are as defined before, unless so specified) using readily available starting materials, reagents and conventional synthesis procedures. In these reactions, it is also possible to make use of variants which are themselves known to those of ordinary skill in this art, but are not mentioned in greater detail.
- R f relative TLC mobility
- rt room temperature
- s.c. subcutaneous
- DMPU 1,3-dimethypropylene urea
- DMSO dimethylsulfoxide
- HMPA hexamethylphosphoric triamide
- HOBt 1-hydroxybenzotriazole
- LAH lithium aluminum hydride
- NMM N-methylmorpholine
- 4-methylmorpholine TEA triethylamine
- TTF fluoro-N.N.N'-tetramethylformamidinium hexafluorophosphate
- Scheme 1 describes the preparation of compounds of formula (I).
- L ⁇ is -(CH 2 ) n -C(R 9 )(R 10 ) m - , wherein n, m, R 9 , and R 10 are as defined for formula (I).
- the sulfonyl chloride (3) may be treated with the amine (4) in the presence of a tertiary amine base such as TEA to afford (5) where G is S(O) 2 .
- R 59 may be R 4 , or R 59 may be a linkage to a polymer support such as Wang Resin.
- Treatment of such a polymer - supported (5) with TFA in a suitable solvent such as dichloromethane affords (I) where R 4 is H.
- (1) may be treated with the amine (4) to afford (5), and subsequently (I), in like manner.
- the acid (2) may be activated by treatment with a carbodiimide reagent such as EDC, or with a coupling agent such as TFFH, in a solvent such as DCM or DMF, in the presence of (4) to afford (5).
- (5) may be converted in like manner to (I).
- the carbamyl chloride (6) may be prepared by treatment of (4) with a reagent such as phosgene, diphosgene, or triphosgene in a solvent such as DCM in the presence of a tertiary amine base such as TEA. Treatment of (8) with (6) in the presence of a tertiary amine base such as TEA affords (5), and in like manner, (I).
- the chlorosulfonamide (7) may be prepared by treatment of (4) with sulfuryl chloride in the presence of a tertiary amine base such as TEA or DIEA. (7) may be treated with (8) in a suitable solvent such as DCM in the presence of a tertiary amine base such as TEA or DIEA to afford 5, and, in like manner to the previous, (I).
- Scheme 2 describes the preparation of the compound of formula (2).
- the ester (9), where PG 2 is a carboxy protecting group, may be treated with N- bromosuccinimide in the presence of benzoyl peroxide to afford (10) where R 60 is bromide. Typically this procedure is preferable where R 1 is aryl or heteroaryl.
- (10) where R 60 is bromide may be treated with a reagent R 3 -SH, R 3 -N(R 6 )H, or R 3 -OH and a base such as sodium hydride or potassium tert-butoxide to afford (11 ) where X is S, N(R 6 ), or O, respectively.
- (11) may be deprotected with, for example, aqueous alkali where PG 2 is methyl or ethyl, to afford (2).
- L 1 is a direct bond
- treatment of (9) with base such as LDA and an oxidizing agent such as a sulfonyl-oxaziridine reagent affords (10) where R 60 is OH.
- treatment of such with a reagent R 3 -LG 1 where LG 1 is a nudeofugal group such as Br, CI, I, or sulfonate, and a base such as DBU or sodium hydride affords (11).
- treatment of (12) with two equivalents of strong base such as LDA and a reagent R 3 -S-LG 2 , where LG 2 is an arylsulfinate group or halogen, affords (2) where X is S.
- Scheme 3 describes an alternate preparation of a compund of formula (2).
- An amine (13) may be alkylated with an alkyl halide R 3 -CH 2 -LG 1 , where LG 1 is a nudeofugal group such as tosylate or bromide or iodide, in the presence of a base such as potassium carbonate in a solvent such as DMF to afford (14).
- (13) may be treated with an aldehyde R 3 -CHO in the presence of a reducing agent such as sodium cyanoborohydride to afford (14).
- the amine (14) may be treated with a reagent LG 1 -L 1 -COO- PG 2 , where LG 1 is a nudeofugal group such as tosylate, iodide, or bromide, in the presence of an organic base such as potassium carbonate, in a solvent such as DMF, to afford (15).
- LG 1 is a nudeofugal group such as tosylate, iodide, or bromide
- an organic base such as potassium carbonate
- PG 2 is a carboxyl protecting group such as allyl or methyl, or benzyl, which may be removed by hydrolysis in, for example, aqueous base to afford (2).
- the hydroxyester (16) may be treated with R 3 - LG 1 in the presence of a base such as DBU, DIEA, or sodium hydride, to afford (18) where X is O.
- (16) may be treated with methanesulfonyl chloride, toluenesulfonyl chloride, or trifluoromethanesulfonic anhydride to afford (17) where R 61 is an arylsulfonate or alkylsulfonate group.
- (17) may then be treated with R 3 -XH, where X is O, S, or N-R ⁇ , in the presence of a suitable base such as TEA, DIEA, NaH, DBU, potassium t- butoxide, or the like, to afford (18).
- (18) may be deprotected as described above to afford (2).
- Scheme 4 describes an alternate preparation of compounds of formula (I).
- the acid (2) may be treated with oxalyl chloride or thionyl chloride in a solvent such as DCM, followed by sodium azide, to afford an acyl azide intermediate.
- (2) may be treated with diphenylphosphoryl azide in the presence of a base such as DIEA, to afford an acyl azide intermediate.
- the acyl azide intermediate is heated at a temperature of from 25 to 100°C to afford the isocyanate (19), which may be treated with an amine (4) to afford (20).
- (19) may be hydrolyzed in weak aqueous acid or weak aqueous base to afford the amine (20).
- the amine (20) may be treated with an aldehyde or ketone embodying the R 6 group, in the presence of a reducing agent such as sodium triacetoxyborohydride, to afford (23). (23) may be treated with reagents (6) or (7) in manner analogous to Scheme 1 to afford (24). The amine (20) may be treated with reagents (6) or (7) in like manner to afford (22). Where R 30 is a solid support such as Wang Resin, (22) and (24) may be treated with TFA as in Scheme 1 to afford (I).
- a reducing agent such as sodium triacetoxyborohydride
- amino protecting group refers to substituents of the amino group commonly employed to block or protect the amino functionality while reacting other functional groups on the compound.
- amino-protecting groups include the formyl group, the trityl group, the phthalimido group, the trichloroacetyl group, the chloroacetyl, bromoacetyl and iodoacetyl groups, urethane-type blocking groups (PG 1 as used herein) such as benzyloxy- carbonyl, 4-phenylbenzyloxycarbonyl, 2-methylbenzyloxycarbonyl, 4-methoxybenzyloxy- carbonyl, 4-fluorobenzyloxycarbonyl, 4-chlorobenzyloxycarbonyl, 3-chlorobenzyloxycarbonyl, 2-chlorobenzyloxycarbonyl, 2,4-dichlorobenzyloxycarbonyl, 4-bro
- amino-protecting group employed is not critical so long as the derivatized amino group is stable to the condition of subsequent reaction(s) on other positions of the compound of formula (I) and can be removed at the desired point without disrupting the remainder of the molecule.
- Preferred amino-protecting groups are the allyloxycarbonyl, the t-butoxycarbonyl, 9-fluorenylmethoxycarbonyl, and the trityl groups. Similar amino-protecting groups used in the cephalosporin, penicillin and peptide art are also embraced by the above terms. Further examples of groups referred to by the above terms are described by J. W. Barton, "Protective Groups In Organic Chemistry", J. G. W.
- alcohol -protecting groups include the 2-tetrahydropyranyl group, 2- ethoxyethyl group, the trityl group, the methyl group, the ethyl group, the allyl group, the trimethylsilylethoxymethyl group, the 2,2,2-trichloroethyl group, the benzyl group, and the trialkylsilyl group, examples of such being trimethylsilyl, tert-butyldimethylsilyl, phenyldimethylsilyl, triiospropylsilyl and thexyldimethylsilyl.
- carboxyl protecting group employed is not critical so long as the derivatized alcohol group is stable to the condition of subsequent reaction(s) on other positions of the compound of the formulae and can be removed at the desired point without disrupting the remainder of the molecule.
- N-Boc aryl amino acid is coupled with heteroaromatic amine as described in procedure E.
- the N-Boc protected aryl amino acid amide (2 mmol) is treated with 4N HCl in dioxane (5 ml). The mixture is stirred at 25°C for 30 min. The solution is concentrated in vacuo and the residue is treated with TEA (5 mmol). The mixture is diluted with ethyl acetate (30 ml). The organic layer is washed with water (2x20 ml), brine (2x20 ml) and dried (Na 2 SO 4 ). Concentration in vacuo afforded the corresponding Boc-deprotected amine.
- the secondary amine (1 eq) is dissolved dry DCM (0.1-1 M) and triethylamine (1-3 eq) is added. Then triphosgene (1-3 eq) is added at -20°C. After a few hours at 25°C a primary or secondary amine (1 eq) is added to the reaction mixture. After consumption of starting material, the mixture is subjected to aqueous workup. Concentration in vacuo afforded the product urea.
- Polymer supported carbamyl chloride (1.0 g, 1.46 mmol 1 eq ) is treated with primary or secondary amine (3-10 eq) in the presence of DCE (0.02-3 M)) and diisopropylethylamine (3-20 eq). After 1-10 h, the resin is then washed with three cycles of DMF/methanol/DCM and dried in vacuo to afford the urea.
- Carboxylic acid (1 eq) in DCM (0.02-2 M) is treated with N.O- dimethylhydroxyl amine hydrochloride (1 eq) and triethylamine (1 eq), DCC, EDC, or other carbodiimide reagent (1 eq) is added. After 1-24 hr, The solution is concentrated in vacuo and the residue is removed by filtration. Alternately, the mixture may be given an aqueous workup. The filtrate is concentrate in vacuo. The product is used directly or is purified by silica gel chromatography.
- the ketone 1 eq in THF or ether (0.02 - 2 M) is treated with an aluminum hydride reagent such as LiAIH 4 (0.25 - 2 eq) at - 78°C - 25°C, followed by quench and filtration, affording the secondary alcohol.
- an aluminum hydride reagent such as LiAIH 4 (0.25 - 2 eq) at - 78°C - 25°C
- 2-(3,4-Dichlorophenoxy)hexanoic acid (0.3 g, 55%) is prepared from ethyl 2-hyd- roxyhexanoate (0.32 g, 2.0 mmol) and 3,4-dichlorophenol (0.39 g, 2.4 mmol) following general procedure A.
- a solution of this acid (69 mg, 0.25 mmol) in THF (3 ml) is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following general procedure E to obtain 2-(3,4-dichloro- phenoxy)-hexanoic acid thiazol-2-yl amide (64 mg, 72%).
- LCMS (m/z): 360 (M + H) +
- 2-(4-Fluorophenoxy)hexanoic acid (0.23 g, 52%) is prepared from ethyl 2-hydroxy- hexanoate (0.32 g, 2.0 mmol) and 4-fluorophenol (0.27 g, 2.4 mmol) following the general procedure A.
- a solution of this acid (56 mg, 0.25 mmol) in THF (3 ml) is reacted with 2- aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-(4-fluoro- phenoxy)-N-1 ,3-thiazol-2-ylhexanamide (58 mg, 76%).
- 2-(4-Methoxyphenoxy)hexanoic acid (0.23 g, 48%) is prepared from ethyl 2-hydroxy- hexanoate (0.32 g, 2.0 mmol) and 4-methoxyphenol (0.3 g, 2.4 mmol) following the general procedure A.
- a solution of this acid (60 mg, 0.25) in THF (3 ml) is reacted with 2-amino- thiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-(4-methoxy- phenoxy)-N-1,3-thiazol-2-ylhexanamide (130 mg, 82%).
- LCMS (m/z): 321 (M + H) +
- 2-(3,4-dichlorophenoxy)-4-methylpentanoic acid (0.26 g, 46%) is prepared from methyl 2-hydroxy-4-methylpentanoate (0.32 g, 2.0 mmol) and 3,4-dichlorophenol (0.39 g, 2.4 mmol) following the general procedure A.
- a solution of this acid (69 mg, 0.25 mmol) in THF (3 ml) is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-(3,4-dichlorophenoxy)-4-methyl-N-1 ,3-thiazol-2-ylpentanamide (74 mg, 82%).
- LCMS (m/z): 359 (M + H) +
- 2-(4-phenylphenoxy)hexanoic acid (0.15 g, 26%) is prepared from ethyl 2-hydroxy- hexanoate (0.32 g, 2.0 mmol) and 4-hydroxybiphenyl (0.39 g, 2.4 mmol) following the general procedure A.
- a solution of this acid (71 mg, 0.25 mmol) in THF (3 ml) is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-(1 ,1'-biphe- nyl-4-yloxy)-N-1 ,3-thiazol-2-ylhexanamide (60 mg, 66%).
- LCMS (m/z): 367 (M + H) +
- 2-(4-lsopropylphenoxy)hexanoic acid (0.23 g, 46%) is prepared from ethyl 2-hyd- roxyhexanoate (0.32 g, 2.0 mmol) and 4-isopropylphenol (0.33 g, 2.4 mmol) following the general procedure A.
- a solution of this acid (63 mg, 0.25 mmol) in THF (3 ml) is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-(4-iso- propylphenoxy)-N-1 ,3-thiazol-2-ylhexanamide (68 mg, 82%).
- LCMS (m/z): 333 (M+ H) +
- 2-(3-Methoxyphenoxy)hexanoic acid (0.37 g, 82%) is prepared from 2-bromohexanoic acid (0.39 g, 2.0 mmol) and 3-methoxyphenol (0.25 g, 2.0 mmol) following the general procedure B.
- a solution of this crude acid (60 mg, 0.25 mmol) in THF (3 ml) is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-(3-methoxyphenoxy)-N-1,3-thiazol-2-ylhexanamide (61 mg, 75%).
- 2-(2,3-Dimethoxyphenoxy)hexanoic acid (0.34 g, 64%) is prepared from 2-bromohexanoic acid (0.39 g, 2.0 mmol) and 2,3-dimethoxyphenol (0.31 g, 2.0 mmol) following the general procedure B.
- a solution of this crude acid (67 mg, 0.25 mmol) in THF (3 ml) is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-(2,3-dimethoxyphenoxy)-N-1 ,3-thiazol-2-ylhexanamide (64 mg, 73%).
- 2-(3,4-Dimethoxyphenoxy)hexanoic acid (0.34 g, 63%) is prepared from 2-bromohexanoic acid (0.39 g, 2.0 mmol) and 3,4-dimethoxyphenol (0.31 g, 2.0 mmol) following the general procedure B.
- a solution of this crude acid (67 mg, 0.25 mmol) in THF (3 ml) is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain
- 2-(3,5-Dimethoxyphenoxy)hexanoic acid (0.43 g, 81%) is prepared from 2-bromohexanoic acid (0.39 g, 2.0 mmol) and 3,5-dimethoxyphenol (0.31 g, 2.0 mmol) following the general procedure B.
- a solution of this crude acid (67 mg, 0.25 mmol) in THF (3 ml) is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-(3,5-dimethoxyphenoxy)-N-1,3-thiazol-2-ylhexanamide (77 mg, 89%).
- 2-(2-Naphthoxy)hexanoic acid (0.39 g, 75%) is prepared from 2-bromohexanoic acid (0.39 g, 2.0 mmol) and 2-naphthol (0.29 g, 2.0 mmol) following the general procedure B.
- a solution of this crude acid (65 mg, 0.25 mmol) in THF (3 ml) is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-(2-naphthyloxy)-N-1 ,3- thiazol-2-ylhexanamide (62 mg, 73%).
- 2-(2,4-Difluorophenoxy)hexanoic acid (0.34 g, 71%) is prepared from 2-bromo- hexanoic acid (0.39 g, 2.0 mmol) and 2,4-difluorophenol (0.26 g, 2.0 mmol) following the general procedure B.
- a solution of this crude acid (61 mg, 0.25 mmol) in THF (3 ml) is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-(2,4-difluorophenoxy)-N-1,3-thiazol-2-ylhexanamide (61 mg, 74%).
- 2-(3,4-Difluorophenoxy)hexanoic acid (0.4 g, 82%) is prepared from 2-bromohexanoic acid (0.39 g, 2.0 mmol) and 3,4-difluorophenol (0.26 g, 2.0 mmol) following the general procedure B.
- a solution of this crude acid (61 mg, 0.25 mmol) in THF (3 ml) is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain
- 2-(3,4-Methylenedioxyphenoxy)hexanoic acid (0.42 g, 83%) is prepared from 2- bromohexanoic acid (0.39 g, 2.0 mmol) and 3,4-methylenedioxyphenol (0.28 g, 2.0 mmol) following the general procedure B.
- a solution of this crude acid (63 mg, 0.25 mmol) in THF (3 ml) is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-(1,3-benzodioxol-5-yloxy)-N-1,3-thiazol-2-ylhexanamide (75 mg, 91%).
- 2-(4-Methylsulfonylphenoxy)hexanoic acid (0.37 g, 66%) is prepared from 2-bromohexanoic acid (0.39 g, 2.0 mmol) and 4-methylsulfonylphenol (0.35 g, 2.0 mmol) following the general procedure B.
- a solution of this crude acid (70 mg, 0.25 mmol) in THF (3 ml) is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain
- 2-(2,4,6-Trichlorophenoxy)hexanoic acid (0.48 g, 76%) is prepared from 2-bromo- hexanoic acid (0.39 g, 2.0 mmol) and 2,4,6-trichlorophenol (0.4 g, 2.0 mmol) following the general procedure B.
- a solution of this crude acid (80 mg, 0.25 mmol) in THF (3 ml) is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-(2,4,6-Trichlorophenoxy)-N-1,3-thiazol-2-ylhexanamide (77 mg, 79%).
- 2-(2,4-Dichlorophenoxy)hexanoic acid (0.4 g, 72%) is prepared from 2-bromohexanoic acid (0.39 g, 2.0 mmol) and 2,4-dichlorophenol (0.33 g, 2.0 mmol) following the general procedure B.
- a solution of this crude acid (69 mg, 0.25 mmol) in THF (3 ml) is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-(2,4-dichlorophenoxy)-N-1 ,3-thiazol-2-ylhexanamide (81 mg, 91%).
- 2-(4-Phenoxyphenoxy)hexanoic acid (0.52 g, 87%) is prepared from 2-bromohexanoic acid (0.39 g, 2.0 mmol) and 4-phenoxyphenol (0.37 g, 2.0 mmol) following the general procedure B.
- a solution of this crude acid (75 mg, 0.25 mmol) in THF (3 ml) is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-(4-phenoxyphenoxy)-N-1 ,3-thiazol-2-ylhexanamide (87 mg, 92%).
- 2-(4-Cyanophenoxy)hexanoic acid (0.35 g, 75%) is prepared from 2-bromohexanoic acid (0.39 g, 2.0 mmol) and 4-cyanophenol (0.24 g, 2.0 mmol) following the general procedure B.
- a solution of this crude acid (58 mg, 0.25 mmol) in THF (3 ml) is reacted with 2- aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-(4-cyano- phenoxy)-N-1,3-thiazol-2-ylhexanamide (67 mg, 86%).
- 2-(4-Chloro-3-trifluoromethylphenoxy)hexanoic acid (0.50 g, 82%) is prepared from 2-bromohexanoic acid (0.39 g, 2.0 mmol) and 2-chloro-5-hydroxybenzotrifluoride (0.39 g, 2.0 mmol) following the general procedure B.
- a solution of this crude acid (77 mg, 0.25 mmol) in THF (3 ml) is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-(4-chloro-3-trifluoromethylphenoxy)-N-1 ,3-thiazol-2-ylhexanamide (80 mg, 82%).
- 2-(3,4-Dichlorophenoxy)-3-cyclopentylpropionic acid (0.43 g, 72%) is prepared from 2-bromo-3-cyclopentylpropionic acid (0.44 g, 2.0 mmol) and 3,4-dichlorophenol (0.33 g, 2.0 mmol) following the general procedure B.
- a solution of this crude acid (75 mg, 0.25 mmol) in THF (3 ml) is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-(3,4-difluorophenoxy)-N-1 ,3-thiazol-2-ylhexanamide (75 mg, 78%).
- 2-(4-Methoxyphenoxy)-3-cyclopentyloxypropionic acid (0.36 g, 68%) is prepared from 2-bromo-3-cyclopentylpropionic acid (0.44 g, 2.0 mmol) and 4-methoxyphenol (0.25 g, 2.0 mmol) following the general procedure B.
- a solution of this crude acid (65 mg, 0.25 mmol) in THF (3 ml) is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-(4-methoxyphenoxy)-3-cyclopentyl-N-1 ,3-thiazol-2-ylpropionamide (73 mg, 85%).
- 2-Bromo-N-1 ,3-thiazol-2-ylheptanamide was prepared from 2-bromohexanoic acid (0.19 g, 1 mmol) and 2-aminothiazole (0.1 g, 1 mmol) as described in procedure E.
- To the reaction mixture was added indoline (0.3 g, 2.5 mmol) and heated at 80°C for 12 h.
- the reaction mixture was concentrated and purified by column chromatography (silica, 10-20% ethyl acetate in hexanes) to obtain 2-(indolin-1-yl)-N-(1 ,3-thiazol-2-yl)hexanamide (120 mg, 38%).
- 3-(4-Chlorophenyl)-N-pyridin-2-yl-3-(tetrahydro-2H-thiopyran-4-ylamino)propan- amide (206 mg, 55%) is prepared from 3-N-Boc-3-(4-chloro phenyl)propionic acid (300 mg, 1 mmol), 2-amino pyridine (225 mg, 2.4 mmol ) and 4-tetrahydrothiopyranone (127 mg, 1.1 mmol), following the general procedure F.
- 3-(4-Chlorophenyl)-3-(tetrahydro-2H-thiopyran-4-ylamino)-N-1,3-thiazol-2-ylpropan- amide (198 mg, 52%) is prepared from 3-N-Boc-3-(4-chloro phenyl)propionic acid (300 mg, 1 mmol), 2-amino thiazole (240 mg, 2.4 ) and 4-tetrahydrothiopyranone(127 mg, 1.1 mmol), following the general procedure F.
- 2-(4-Bromophenoxy)-2-(4-chlorophenyl) acetic acid (450 mg, 54%) is prepared from 4-chloromandelic acid methyl ester (402 mg, 2 mmol), 4-bromophenol (415 mg, 2.4 mmol) following general procedure A.
- a solution of this acid (104 mg, 0.25 mmol) in THF is reacted with 2-aminopyridine (56 mg, 0. 6 mmol) following the general procedure E to obtain 2-(4- bromophenoxy)-2-(4-chlorophenyl)-N-pyridin-2-ylacetamide (75 mg, 72%).
- 2-(4-Fluorophenoxy)-2-(4-chlorophenyl) acetic acid (296 mg, 53%) is prepared from 4-chloro-mandalic acid methyl ester (402 mg, 2 mmol), 4-fluorophenol (269 mg, 2.4 mmol) following the general procedure A.
- a solution of this acid (70 mg, 0.25 mmol) in THF is reacted with 2-aminothiazole (60 mg, 0.60 mmol) following the general procedure E to obtain 2-(4-chlorophenyl)-2-(4-fluorophenoxy)-N-1,3-thiazol-2-ylacetamide (71 mg, 78%).
- 2-(3,4-Dichlorophenoxy)-2-(4-chlorophenly) acetic acid (363 mg, 55%) is prepared from 4-chloro-mandalic acid methyl ester (402 mg, 2 mmol), 3,4-dichlorophenol (390 mg 2.4 mmol) following the general procedure A.
- a solution of this acid (83 mg, 0.25 mmol) in THF is reacted with 2-aminopyridine (56 mg, 0.60 mmol) following the general procedure E to obtain 2-(4-chlorophenyl)-2-(3,4-dichlorophenoxy)-N-pyridin-2-ylacetamide (82 mg, 80%).
- 2-(4-Methylphenoxy)-2-(4-bromophenyl) acetic acid (372 mg, 58%) is prepared from 4-bromomandelic acid methyl ester (490 mg, 2 mmol), 4-methylphenol (260 mg, 2.4 mmol) following the general procedure A.
- a solution of this acid (80 mg, 0.25 mmol) in THF is reacted with 2-aminopyridine (56 mg, 0.6 mmol) following the general procedure E to obtain 2-(4-bromophenyl)-2-(4-methylphenoxy)-N-pyridin-2-ylacetamide (89 mg, 90%).
- 2-(4-Fluorophenoxy)-2-(4-bromophenyl) acetic acid (472 mg, 58%) is prepared from 4-bromomandelic acid methyl ester (490 mg, 2 mmol) and 4-fluorophenol (268 mg, 2.4 mmol) following the general procedure A.
- a solution of this acid (81 mg, 0.25 mmol) in THF is reacted with 2-aminothiazole (60 mg, 6.0 mmol) following the general procedure E to obtain 2-(4-bromophenyl)-2-(4-fluorophenoxy)-N-1 ,3-thiazol-2-ylacetamide (73g, 72%).
- 2-Phenoxy-2-(4-bromophenyl) acetic acid (319 mg, 52%) is prepared from 4-bromomandelic acid methyl ester (490 mg, 2 mmol), phenol (226 mg, 2.4mmol) following the general procedure A.
- a solution of this acid (76.78 mg, 0.25 mmol) in THF is reacted with 2- aminothiazole (60mg, 0.6 mmol) following the general procedure E to obtain 2-(4-bromo- phenyl)-2-phenoxy-N-1 ,3-thiazol-2-ylacetamide (82 mg, 85%).
- LCMS (m/z): 390 (M + H) +
- 2-(4-Fluorophenoxy)-2-(4-fluorophenyl) acetic acid (317 mg, 60%) is prepared from 4-fluoromandelic acid methyl ester (368 mg, 2 mmol) and 4-fluorophenol (268 mg, 2.4 mmol) following the general procedure A.
- a solution of this acid (66 mg, 0.25 mmol) in THF is reacted with 2-aminopyridine (5 mg, 0.6 mmol) following the general procedure E to obtain 2- (4-fluorophenoxy)-2-(4-fluorophenyl)-N-pyridin-2-ylacetamide (73 mg, 76%).
- 2-(4-Methylphenoxy)-2-(4-fluorophenyl) acetic acid (264 mg, 50%) is prepared from 4-fluoromandalic acid methyl ester (368 mg, 2 mmol) and 4-methylphenol (259 mg, 2.4 mmol) following the general procedure A.
- a solution of this acid (66 mg, 0.25 mmol) in THF is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-(4-fluorophenyl)-2-(4-methylphenoxy)-N-1,3-thiazol-2-ylacetamide (69 mg, 80%).
- LCMS (m/z): 347 (M +H) +
- 2-(4-Bromophenoxy-2-(4-fluorophenyl) acetic acid (338 mg, 52%) is prepared from 4-fluoromandelic acid methyl ester (368 mg, 2 mmol) and 4-bromophenol (415mg, 2.4 mmol) following the general procedure A.
- a solution of this acid (81 mg, 0.25 mmol) in THF is reacted with 2-aminopyridine (56 mg, 0.6 mmol) following the general procedure E to obtain 2-(4-bromophenoxy)-2-(4-fluorophenyl)-N-pyridin-2-ylacetamide (86 mg, 86%).
- LCMS (m/z): 402 (M + H) +
- 2-(4-Fluorophenoxy-2-(4-trifluoromethylphenyl) acetic acid (390 mg, 62%) is prepared from 4-trifluoromethylmandelic acid methyl ester (468 mg, 2 mmol) and 4-fluorophenol (269 mg, 2.4 mmol) following the general procedure A.
- a solution of this acid (79 mg, 0.5 mmol) in THF is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-(4-fluorophenoxy)-N-1,3-thiazol-2-yl-2-[4-(trifluoromethyl)phenyl]- acetamide (87 mg, 88%).
- 2-(4-Bromophenoxy-2-(4-trifluoromethylphenyl) acetic acid (405 mg, 54%) is prepared from 4-trifluoromethylmandelic acid methyl ester (468 mg, 2 mmol) and 4-bromophenol (415 mg, 2.4 mmol) following the general procedure A.
- a solution of this acid (94 mg, 0.25 mmol) in THF is reacted with 2-aminopyridine (56 mg, 0.6 mmol) following the general procedure E to obtain 2-(4-bromophenoxy)-N-pyridin-2-yl-2-[4-(trifluoromethyl)phenyl]- acetamide (102 mg, 90%).
- 2-(3,4-Dichlorophenoxy)-2-(3,4-dichlorophenyl)acetic acid 400 mg, 55%) is prepared from 3,4-dichloromandalic acid methyl ester (468 mg, 2 mmol) and 3,4-dichlorophenol (389 mg 2.4 mmol) following the general procedure A.
- a solution of this acid (91 mg, 0.25 mmol) in THF is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-(3,4-dichlorophenoxy)-2-(3,4-dichlorophenyl)-N-1,3-thiazol-2- ylacetamide (72 mg, 65 %).
- 2-Cyclopentylthio-2-phenylacetic acid (330 mg, 70%) is prepared from 2-bromo- phenylacetic acid methyl ester (458 mg, 2 mmol) and cyclopentane thiol (245 mg, 2.4 mmol) following the general procedure C.
- a solution of this acid (59 mg, 0.25 mmol) in THF is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-cyclopentylsulfanyl-2-phenyl-N-1,3-thiazol-2-yl-acetamide (63 mg, 79%).
- LCMS (m/z): 319 (M+ 1 H) +
- 2-Cyclopentylthio-2-(4-fluorophenyl)acetic acid (345 mg, 68%) is prepared from 2- bromo-2-(4-fluorophenyl)acetic acid methyl ester (494 mg, 2 mmol) and cyclopentane thiol (245 mg, 2.4 mmol) following the general procedure C.
- a solution of this acid (64 mg, 0.25 mmol) in THF is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-cyclopentylsulfanyl-2-(4-fluorophenyl)-N-1 ,3-thiazol-2-yl-acetamide (59 mg, 70%).
- LCMS (m/z): 337 (M + H) +
- THF is reacted with 2-aminopyridine (57 mg, 0.6 mmol) following the general procedure E to obtain 2-cyclopentylsulfanyl-2-(4-fluorophenyl)-N-pyridin-2-yl-acetamide (60 mg, 72%).
- 2-Cyclopentylthio-2-(3-chlorophenyl)acetic acid (351 mg, 65%) is prepared from 2- bromo-2-(3-chlorophenyl)acetic acid methyl ester (527 mg, 2 mmol) and cyclopentane thiol (245 mg, 2.4 mmol) following the general procedure C.
- a solution of this acid (68 mg, 0.25 mmol) in THF is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-cyclopentylsulfanyl-2-(3-chlorophenyl)-N-1 ,3-thiazol-2-yl-acetamide (63 mg, 72%).
- LCMS (m/z): 353 (M + H) +
- 2-Cyclopentylthio-2-(4-chlorophenyl)acetic acid (390 mg, 72%) is prepared from 2- bromo-2-(4-chlorophenyl)acetic acid methyl ester (528 mg, 2 mmol) and cyclopentane thiol (245 mg, 2.4 mmol) following the general procedure C.
- a solution of this acid (68 mg, 0.25 mmol) in THF is reacted with 2-aminopyridine (60 mg, 0.6 mmol) following the general procedure E to obtain 2-cyclopentylsulfanyl-2-(4-chlorophenyl)-N-pyridin-2-yl-acetamide (62 mg, 72%).
- 2-Cyclopentylthio-2-(4-bromophenyl)acetic acid (441 mg, 70%) is prepared from 2- bromo-2-(4-bromophenyl)acetic acid methyl ester (616 mg, 2 mmol) and cyclopentane thiol (245 mg, 2.4 mmol) following the general procedure C.
- a solution of this acid (79 mg, 0.25 mmol) in THF is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-cyclopentylsulfanyl-2-(4-bromophenyl)-N-1 ,3-thiazol-2-yl-acetamide (71 mg, 72%).
- 2-Cyclopentylthio-2-(4-methoxyphenyl)acetic acid (319 mg, 60%) is prepared from 4-methoxymandelic acid methyl ester (392 mg, 2 mmol) and cyclopentane thiol (245 mg, 2.4 mmol) following the general procedure D.
- a solution of this acid (67 mg, 0.25 mmol) in THF is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-cyclopentylsulfanyl-2-(4-methoxyphenyl)-N-1 ,3-thiazol-2-yl-acetamide (65 mg, 75%).
- LCMS (m/z): 349 (M + H) +
- 2-Cyclopentylthio-2-(3-cyanophenyl)acetic acid (323 mg, 62%) is prepared from 3- cyanomandelic acid methyl ester (382 mg, 2 mmol) and cyclopentane thiol (245 mg, 2.4 mmol) following the general procedure D.
- a solution of this acid (65 mg, 0.25 mmol) in THF is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-cyclopentylsulfanyl-2-(3-cyanophenyl)-N-1 ,3-thiazol-2-yl-acetamide (64 mg, 74%).
- LCMS (m/z): 344 (M + H) +
- 2-Cyclopentylthio-2-(4-cyanophenyl)acetic acid (313 mg, 60%) is prepared from 4- cyanomandelic acid methyl ester (382 mg, 2 mmol) and cyclopentane thiol (245 mg, 2.4 mmol) following the general procedure D.
- a solution of this acid (65 mg, 0.25 mmol) in THF is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-cyclopentylsulfanyl-2-(4-cyanophenyl)-N-1 ,3-thiazol-2-yl-acetamide (58 mg, 68%).
- LCMS (m/z): 344 (M + H) +
- 2-Cyclopentylthio-2-(4-nitrophenyl)acetic acid (270 mg, 48%) is prepared from 2- bromo-2-(4-nitrophenyl)acetic acid methyl ester (548 mg, 2 mmol) and cyclopentane thiol (245 mg, 2.4 mmol) following the general procedure C.
- a solution of this acid (70 mg, 0.25 mmol) in THF is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-cyclopentylsulfanyl-2-(4-nitrophenyl)-N-1 ,3-thiazol-2-yl-acetamide (67 mg, 74%).
- LCMS (m/z): 364 (M+ H) +
- 2-Cyclopentylthio-2-(4-methylsulfonylphenyl)acetic acid (471 mg, 75%) is prepared from 2-bromo-2-(4-methylsulfonylphenyl)acetic acid methyl ester (614 mg, 2 mmol) and cyclopentane thiol (245 mg, 2.4 mmol) following the general procedure C.
- 2-Cyclopentylthio-2-(4-trifluoromethylphenyl)acetic acid (413 mg, 68%) is prepared from 2-bromo-2-(4-trifluoromethylphenyl)acetic acid methyl ester (594 mg, 2 mmol) and cyclopentane thiol (245 mg, 2.4 mmol) following the general procedure C.
- 2-Cyclopentylthio-2-(3-trifluoromethoxyphenyl)acetic acid (416 mg, 65%) is prepared from 2-bromo-2-(3-trifluoromethoxyphenyl)acetic acid methyl ester (626 mg, 2 mmol) and cyclopentane thiol (245 mg, 2.4 mmol) following the general procedure C.
- 2-Cyclopentylthio-2-(4-trifluoromethoxyphenyl)acetic acid (448 mg, 70%) is prepared from 2-bromo-2-(4-trifluoromethoxylphenyl)acetic acid methyl ester (626 mg, 2 mmol) and cyclopentane thiol (245 mg, 2.4 mmol) following the general procedure C.
- 2-Cyclopentylthio-2-(4-phenyl)phenylacetic acid (406 mg, 65%) is prepared from 2- bromo-biphenylacetic acid methyl ester (610 mg, 2 mmol) and cyclopentane thiol (245 mg, 2.4 mmol) following the general procedure C.
- a solution of this acid (78 mg, 0.25 mmol) in THF is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-cyclopentylsulfanyl-2-(4-phenyl)phenyl-N-1,3-thiazol-2-yl-acetamide (69 mg, 70%).
- 2-Cyclopentylthio-2-(4-phenoxyphenyl)acetic acid (459 mg, 70%) is prepared from 2-bromo-(4-phenoxyphenyl)acetic acid methyl ester (642 mg, 2 mmol) and cyclopentane thiol (245 mg, 2.4 mmol) following the general procedure C.
- a solution of this acid (82 mg, 0.25 mmol) in THF is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-cyclopentylsulfanyl-2-(4-phenoxyphenyl)-N-1 ,3-thiazol-2-yl- acetamide (81 mg, 79%).
- 2-Cyclopentylthio-2-(3,5-difluorophenyl)acetic acid (326 mg, 60%) is prepared from 2-bromo-2-(3,5-difluorophenyl)acetic acid methyl ester (530 mg, 2 mmol) and cyclopentane thiol (245 mg, 2.4 mmol) following the general procedure C.
- 2-Cyclopentylthio-2- ⁇ 3,4-(methylenedioxy)phenyl ⁇ acetic acid (336 mg, 60%) is prepared from 3,4-(methylenedioxy)mandelic acid methyl ester (420 mg, 2 mmol) and cyclopentane thiol (245 mg, 2.4 mmol) following the general procedure D.
- a solution of this acid (70 mg, 0.25 mmol) in THF is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-cyclopentylsulfanyl-2- ⁇ 3,4-(methylenedioxy)phenyl ⁇ -N- 1 ,3-thiazol-2-yl-acetamide (59 mg, 65%).
- LCMS (m/z): 363 (M + H) *
- 2-Cyclopentylthio-2-[3,5-bis(trifluoromethyl)phenyl]acetic acid (521 mg, 70%) is prepared from 2-bromo-2-[3,5-bis(trifIuoromethyl)phenyl]acetic acid methyl ester (730 mg, 2 mmol) and cyclopentane thiol (245 mg, 2.4 mmol) following the general procedure C.
- 2-Cyclopentylthio-2-(3-chloro-4-methoxyphenyl)acetic acid (421 mg, 70%) is prepared from 2-bromo-2-(3-chloro-4-methoxyphenyl)acetic acid methyl ester (588 mg, 2 mmol) and cyclopentane thiol (245 mg, 2.4 mmol) following the general procedure C.
- 2-lsopropyllthio-2-(3,4-dichlorophenyl)acetic acid (458 mg, 75%) is prepared from 2- bromo-2-(3,4-dichlorophenyl)acetic acid methyl ester (594 mg, 2 mmol) and isopropane thiol (183 mg, 2.4 mmol) following the general procedure C.
- a solution of this acid (76 mg, 0.25 mmol) in THF is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-isopropylsulfanyl-2-(3,4-dichlorophenyl)-N-1 ,3-thiazol-2-yl-acetamide
- THF is reacted with 2-aminopyridine (57 mg, 0.6 mmol) following the general procedure E to obtain 2-isopropylsulfanyl-2-(3,4-dichlorophenyl)-N-pyridin-2-yl-acetamide (64 mg, 72%).
- 2-Allylthio-2-(3,4-dichlorophenyl)acetic acid (458 mg, 75%) is prepared from 2- bromo-2-(3,4-dichlorophenyl)acetic acid methyl ester (594 mg, 2 mmol) and allyl thiol (178 mg, 2.4 mmol) following the general procedure C.
- a solution of this acid (76 mg, 0.25 mmol) in THF is reacted with 2-aminothiazole (60 mg, 0.6 mmol) following the general procedure E to obtain 2-allylsulfanyl-2-(3,4-dichlorophenyl)-N-1 ,3-thiazol-2-yl-acetamide (72 mg, 80%).
- 2-(2-Methylpropanethio)-2-(3,4-dichloro phenyl) acetic acid (457 mg, 78%) is prepared from 2-bromo-2-(3,4-dichloro phenyl) acetic acid methyl ester (594 mmol, 2 mmol) and 2-methylpropanethiol (216 mg, 2.4 mmol) following the general procedure C.
- 2-(2-Furanylmethylthio)-2-(3,4-dichloro phenyl) acetic acid (482 mg, 76%) is prepared from 2-bromo-2-(3,4-dichloro phenyl) acetic acid methyl ester (594 mg, 2 mmol) and 2-furanylmethylthiol (274 mg, 2.4 mmol) following the general procedure C.
- 2-(4-Methylphenylthio)-2-phenylacetic acid (310 mg, 60%) is prepared from a- bromophenylacetic acid methyl ester (458 mg, 2 mmol) and 4-methylthiophenol (298 mg, 2.4 mmol) following the general procedure C.
- a solution of this acid (65 mg, 0.25 mmol) in THF is reacted with 2-aminopyridine (57 mg, 0.6 mmol) following the general procedure E to obtain 2-(4-methylphenylthio)-2-phenyl-N-pyridin-2-ylacetamide (61 mg, 72%).
- LCMS (m/z): 335 (M + H) + .
- 2-(4-Chlorophenlythio)-2-(3,4-dichlorophenyl)acetic acid (542 g, 78%) is prepared from 2-bromo-2-(3,4-dichlorophenyl)acetic acid methyl ester (594 mg, 2 mmol), 4-chloro- benzenethiol (347 mg, 2.4 mmol) following the general procedure C.
- 2-(4-Fluorophenlythio)-2-(4-trifluoromethylphenyl)acetic acid (760 mg, 92%) is prepared from 2-hydroxy-2-(4-trifluoromethyl phenyl)acetic acid methyl ester (860 mg, 2.5 mmol) and 4-fluorobenzenethiol (308 mg, 2.4 mmol) following the general procedure D.
- 2-(4-Fluorophenlythio)-2-(4-fluorophenyl)acetic acid 400 mg, 75%) is prepared from 2-hydroxy-2-(4-fluorophenyl)acetic acid methyl ester (368 mg, 2 mmol) and 4-fluorobenzene- thiol (307 mg, 2.4mmol) following the general procedure D.
- a solution of this acid (66 mg, 0.25 mmol) in THF is reacted with 2-aminopyridine (56 mg, 0.6 mmol) following the general procedure E to obtain 2-(4-fluorophenyl)-2-[(4-fluorophenyl)thio]-N-pyridin-2-ylacetamide (73 mg, 82%).
- 2-(4-Fluorophenlythio)-2-(4-bromophenyl) acetic acid (593 mg, 87%) is prepared from 2-hydroxy-2-(4-bromophenyl) acetic acid methyl ester (490 mg, 2 mmol) and 4-fluoro- benzenethiol (307 mg, 2.4 mmol) following the general procedure D.
- 2-(4-Methylphenlythio)-2-(4-bromophenyl)acetic acid (559 mg, 83%) is prepared from 2-hydroxy-2-(4-bromophenyl) acetic acid methyl ester (490 mg, 2 mmol) and 4-methyl- benzenethiol (298 mg, 2.4 mmol) following the general procedure D.
- a solution of this acid (84 mg, 0.25 mmol) in THF is reacted with 2-aminopyridine (56 mg, 0.6 mmol) following the general procedure E to obtain 2-(4-bromophenyl)-2-[(4-methylphenyl)thio]-N-pyridin-2-ylacet- amide (85 mg, 82%).
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Diabetes (AREA)
- Epidemiology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Endocrinology (AREA)
- Urology & Nephrology (AREA)
- Child & Adolescent Psychology (AREA)
- Emergency Medicine (AREA)
- Vascular Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Thiazole And Isothizaole Compounds (AREA)
- Nitrogen- Or Sulfur-Containing Heterocyclic Ring Compounds With Rings Of Six Or More Members (AREA)
- Pyridine Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description










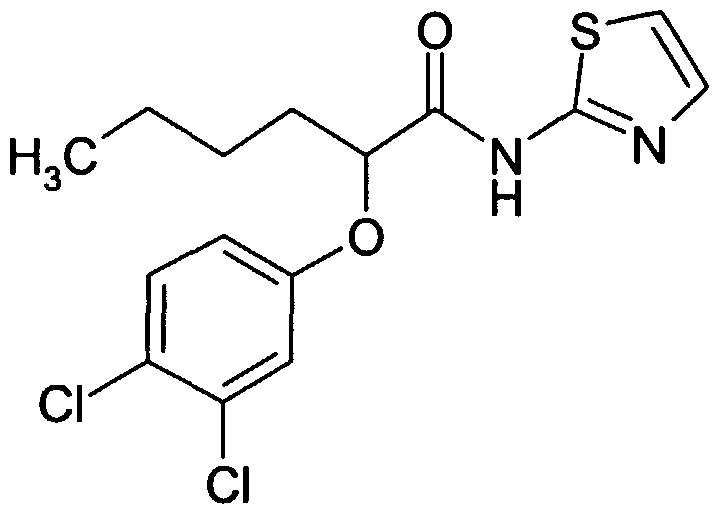
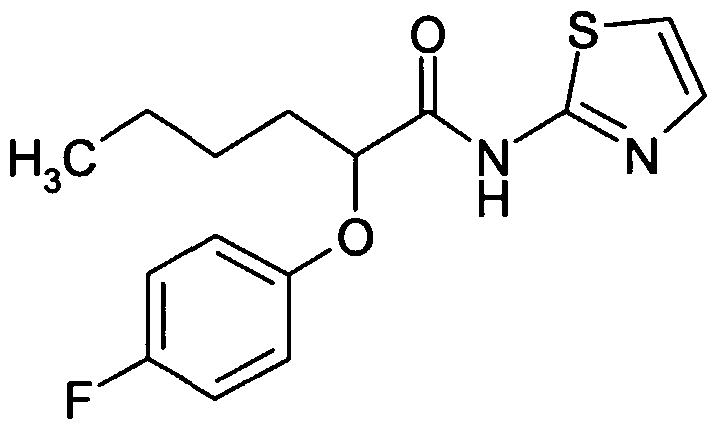
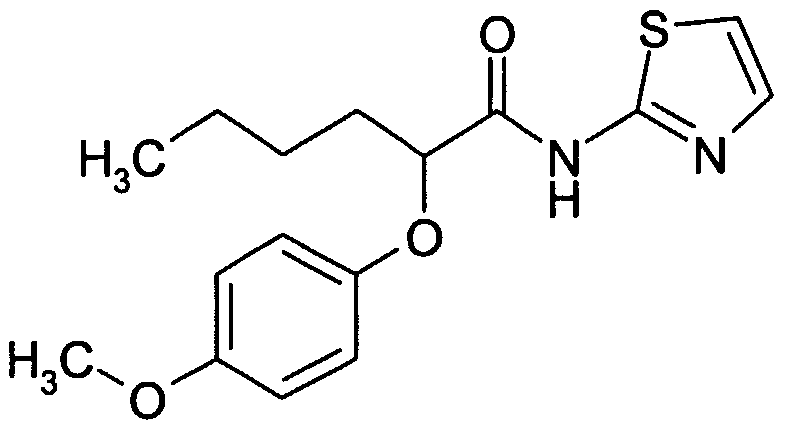










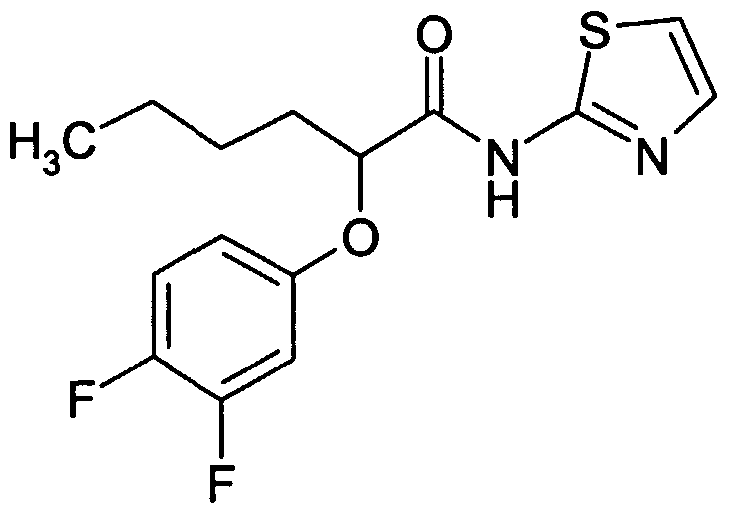







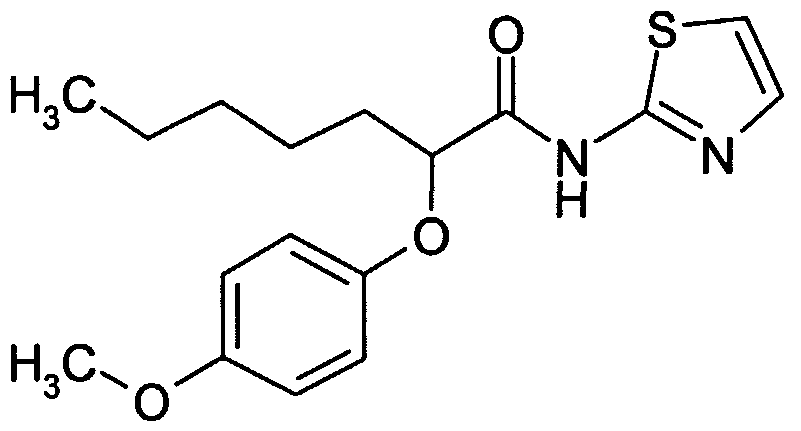



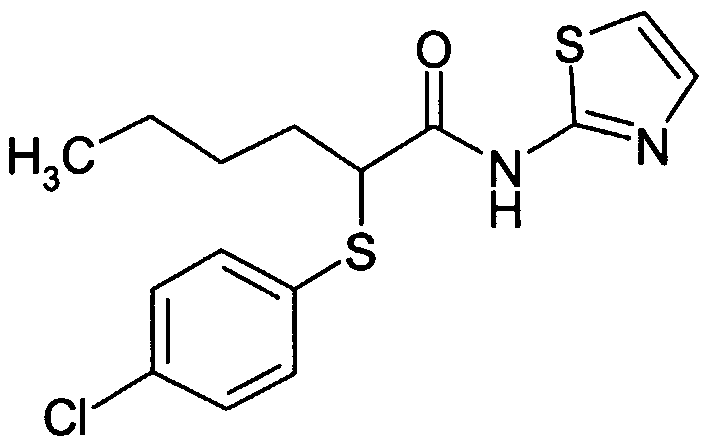
















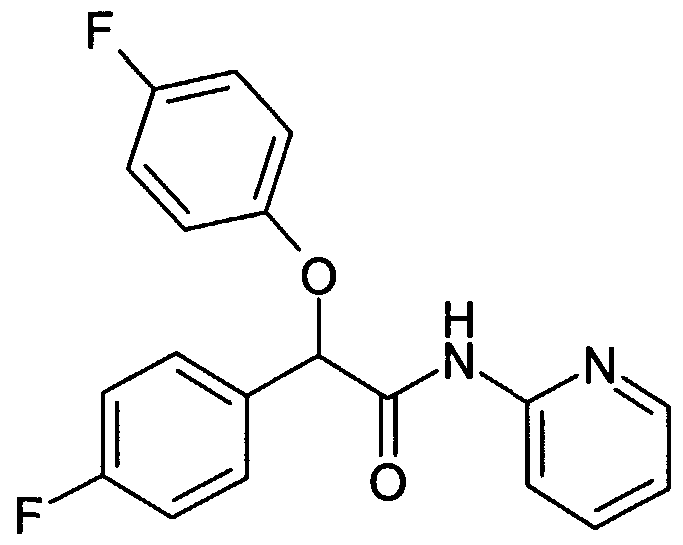






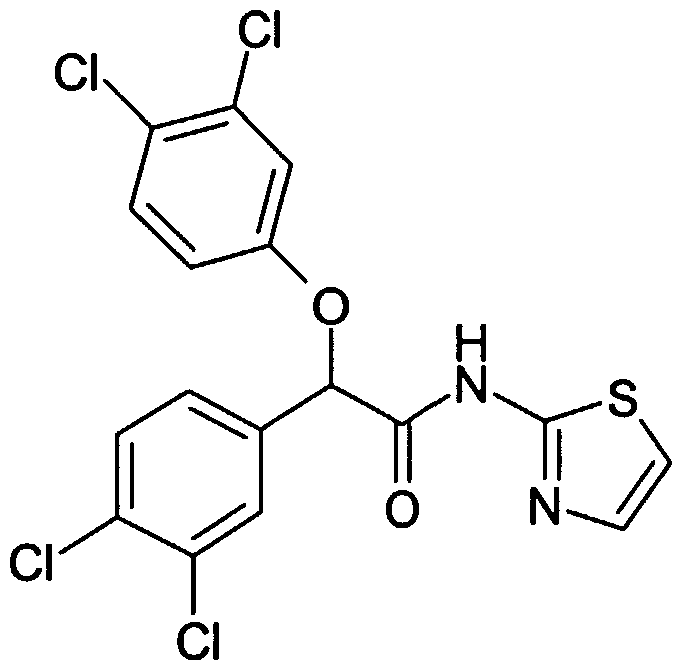


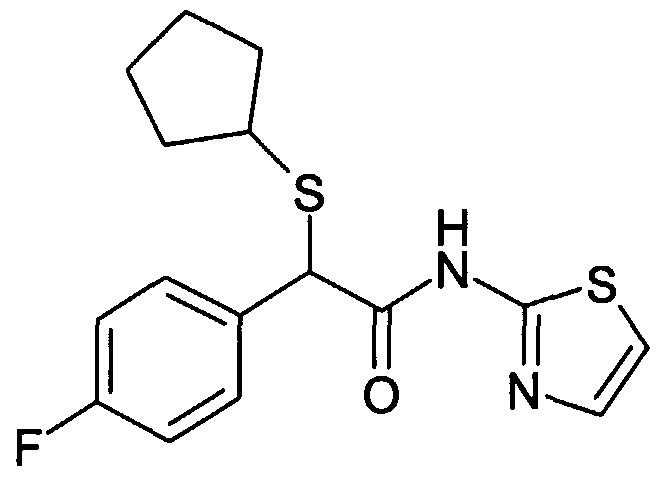

















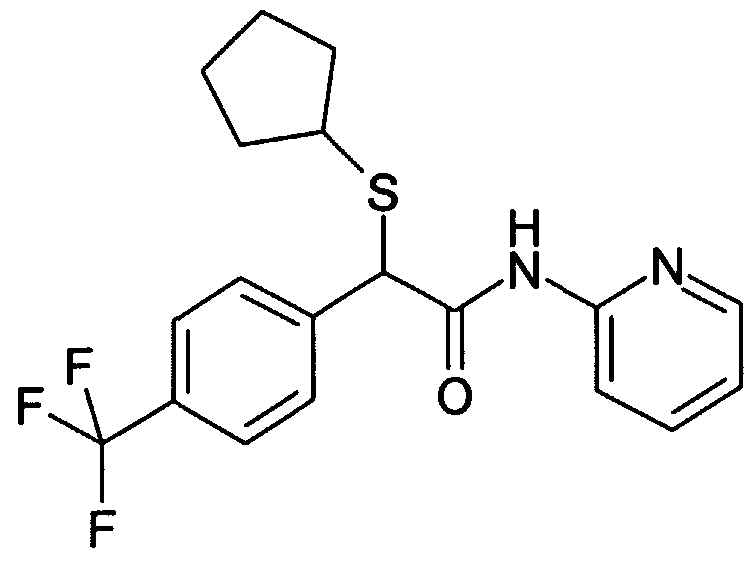
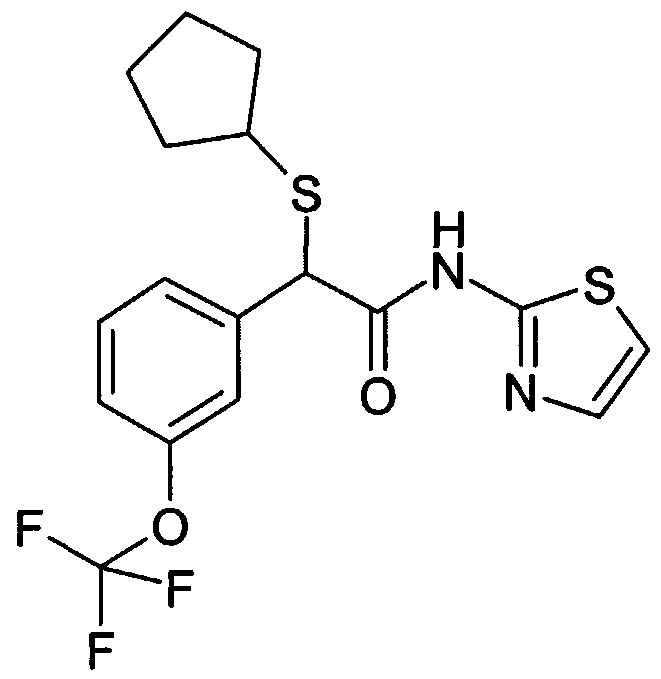



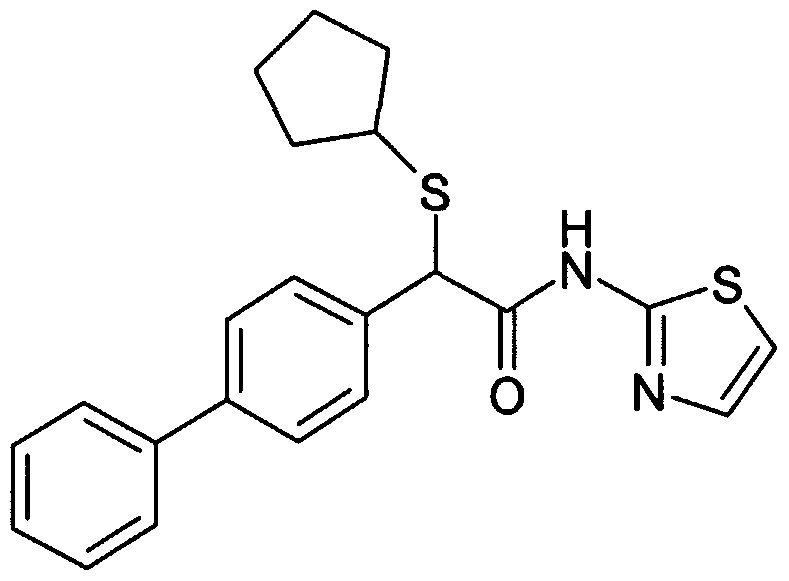











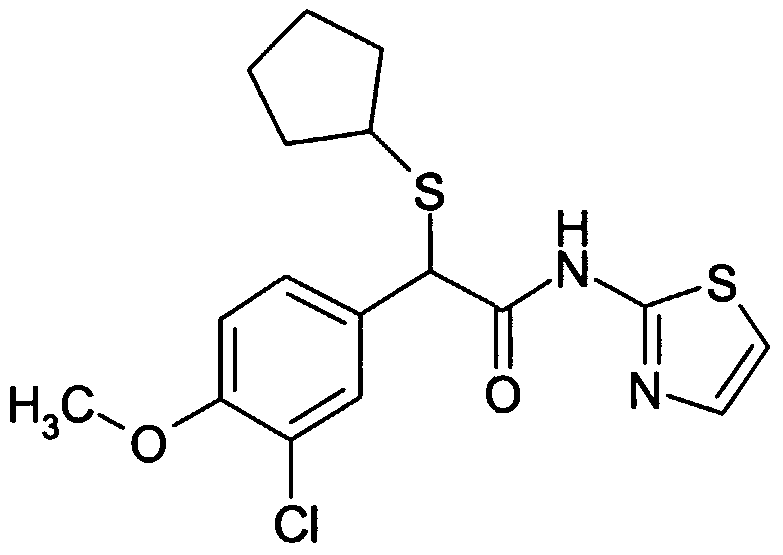





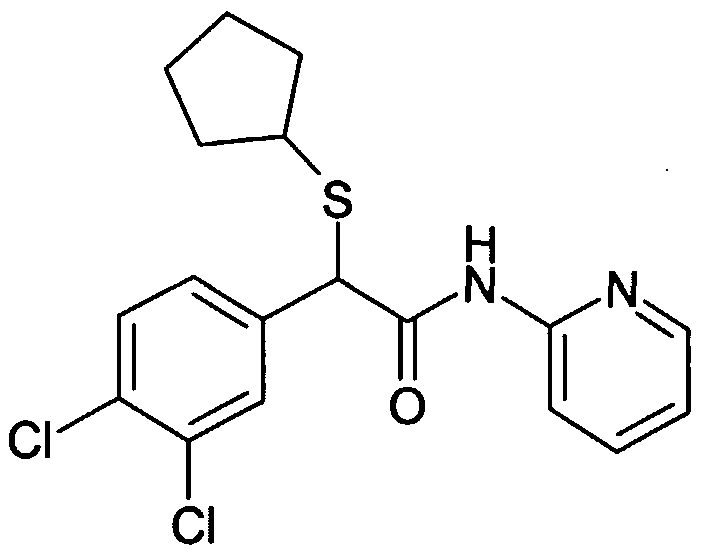













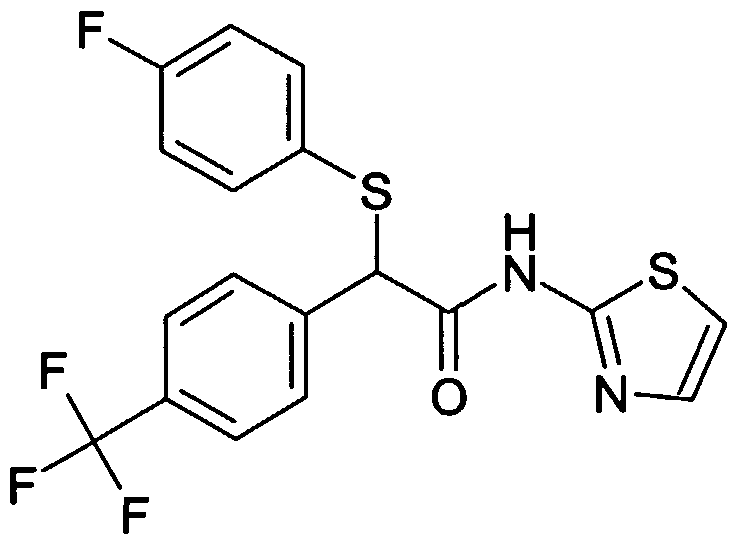












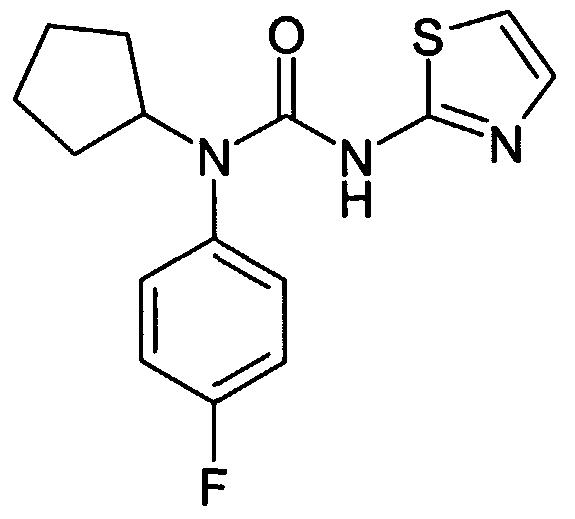











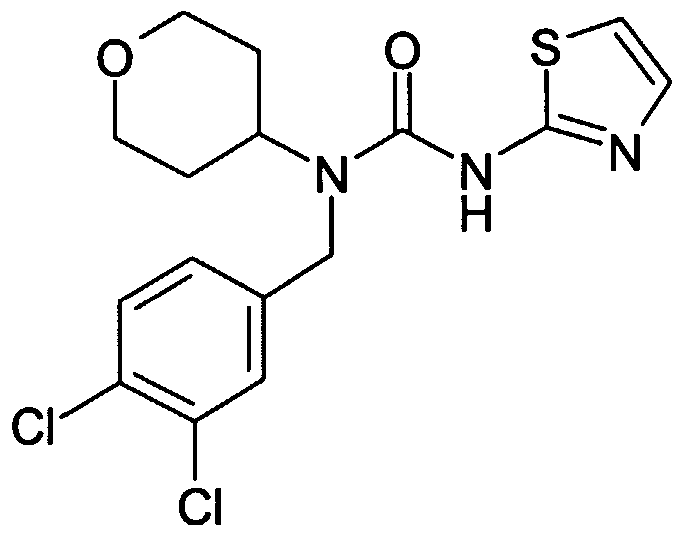












Claims







Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| UA20040604430A UA84390C2 (en) | 2001-12-21 | 2002-12-19 | Amide derivatives as glucokinase activators |
| JP2003556060A JP2005518391A (en) | 2001-12-21 | 2002-12-19 | Amide derivatives as GK activators |
| KR1020047009841A KR101018318B1 (en) | 2001-12-21 | 2002-12-19 | Amide derivatives as gk activators |
| CA002471049A CA2471049A1 (en) | 2001-12-21 | 2002-12-19 | Amide derivatives as gk activators |
| MXPA04006048A MXPA04006048A (en) | 2001-12-21 | 2002-12-19 | Amide derivatives as gk activators. |
| BR0215212-6A BR0215212A (en) | 2001-12-21 | 2002-12-19 | Carboxamide or glycoside sulfonamide activator, compound, pharmaceutical composition, and use of a compound |
| AU2002351748A AU2002351748B2 (en) | 2001-12-21 | 2002-12-19 | Amide derivatives as GK activators |
| HU0402309A HUP0402309A3 (en) | 2001-12-21 | 2002-12-19 | Amide derivatives as glucokinase activators and pharmaceutical compositions containing them |
| EP20020787463 EP1458382A1 (en) | 2001-12-21 | 2002-12-19 | Amide derivatives as gk activators |
| IL16262002A IL162620A0 (en) | 2001-12-21 | 2002-12-19 | Amide derivatives as gk activators |
| TW092100480A TWI355938B (en) | 2002-02-19 | 2003-01-10 | Amide derivatives as therapeutic agents |
| NO20043116A NO20043116L (en) | 2001-12-21 | 2004-07-20 | Amide derivatives such as GK activators |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US38618501P | 2001-12-21 | 2001-12-21 | |
| US60/386,185 | 2001-12-21 | ||
| EP02388015.6 | 2002-02-19 | ||
| EP02388015A EP1336607A1 (en) | 2002-02-19 | 2002-02-19 | Amide derivatives as glucokinase activators |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2003055482A1 true WO2003055482A1 (en) | 2003-07-10 |
Family
ID=26077624
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/DK2002/000880 WO2003055482A1 (en) | 2001-12-21 | 2002-12-19 | Amide derivatives as gk activators |
Country Status (17)
| Country | Link |
|---|---|
| US (1) | US20030171411A1 (en) |
| EP (2) | EP2305648A1 (en) |
| JP (2) | JP2005518391A (en) |
| KR (1) | KR101018318B1 (en) |
| CN (1) | CN100506807C (en) |
| AU (1) | AU2002351748B2 (en) |
| BR (1) | BR0215212A (en) |
| CA (1) | CA2471049A1 (en) |
| CZ (1) | CZ2004747A3 (en) |
| HU (1) | HUP0402309A3 (en) |
| IL (1) | IL162620A0 (en) |
| MX (1) | MXPA04006048A (en) |
| NO (1) | NO20043116L (en) |
| PL (1) | PL370989A1 (en) |
| RU (1) | RU2374236C2 (en) |
| UA (1) | UA84390C2 (en) |
| WO (1) | WO2003055482A1 (en) |
Cited By (90)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004002481A1 (en) * | 2002-06-27 | 2004-01-08 | Novo Nordisk A/S | Aryl carbonyl derivatives as therapeutic agents |
| WO2004050645A1 (en) * | 2002-10-03 | 2004-06-17 | Novartis Ag | Substituted (thiazol-2-yl) -amide or sulfonamide as glycokinase activators useful in the treatment of type 2 diabetes |
| WO2004063194A1 (en) * | 2003-01-06 | 2004-07-29 | Eli Lilly And Company | Heteroaryl compounds |
| WO2004072066A1 (en) * | 2003-02-11 | 2004-08-26 | Prosidion Limited | Tri(cyclo) substituted amide glucokinase activator compounds |
| WO2005066145A1 (en) * | 2004-01-06 | 2005-07-21 | Novo Nordisk A/S | Heteroaryl-ureas and their use as glucokinase activators |
| WO2005121110A1 (en) | 2004-06-05 | 2005-12-22 | Astrazeneca Ab | Hetroaryl benzamide derivatives for use as glk activators in the treatment of diabetes |
| WO2007006761A1 (en) * | 2005-07-08 | 2007-01-18 | Novo Nordisk A/S | Dicycloalkylcarbamoyl ureas as glucokinase activators |
| WO2007006814A1 (en) | 2005-07-14 | 2007-01-18 | Novo Nordisk A/S | Urea glucokinase activators |
| WO2007007041A1 (en) | 2005-07-09 | 2007-01-18 | Astrazeneca Ab | Heteroaryl benzamide derivatives for use as glk activators in the treatment of diabetes |
| WO2007006760A1 (en) * | 2005-07-08 | 2007-01-18 | Novo Nordisk A/S | Dicycloalkyl urea glucokinase activators |
| US7199140B2 (en) | 2001-06-26 | 2007-04-03 | Astrazeneca Ab | Vinyl phenyl derivatives as GLK activators |
| WO2007039177A2 (en) | 2005-09-29 | 2007-04-12 | Sanofi-Aventis | Phenyl- and pyridinyl- 1, 2 , 4 - oxadiazolone derivatives, processes for their preparation and their use as pharmaceuticals |
| US7214681B2 (en) | 2003-02-11 | 2007-05-08 | Prosidion Limited | Tri(cyclo) substituted amide compounds |
| WO2007053662A1 (en) * | 2005-11-01 | 2007-05-10 | Janssen Pharmaceutica N.V. | Dihydroisoindolones as allosteric modulators of glucokinase |
| WO2007053503A1 (en) * | 2005-11-01 | 2007-05-10 | Janssen Pharmaceutica N.V. | Substituted dihydroisoindolones as allosteric modulators of glucokinase |
| WO2007053657A1 (en) * | 2005-11-01 | 2007-05-10 | Janssen Pharmaceutica N.V. | Substituted pyrrolones as allosteric modulators of glucokinase |
| US7230108B2 (en) | 2002-11-19 | 2007-06-12 | Astrazeneca Ab | Quinoline derivatives as glucokinase ligands |
| WO2007053765A3 (en) * | 2005-11-01 | 2007-07-05 | Janssen Pharmaceutica Nv | Substituted cycloalkylpyrrolones as allosteric modulators of glucokinase |
| WO2007128761A2 (en) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Uses of dpp-iv inhibitors |
| US7342118B2 (en) | 2004-03-23 | 2008-03-11 | Pfizer Inc | Imidazole compounds for the treatment of neurodegenerative disorders |
| US7390908B2 (en) | 2001-08-17 | 2008-06-24 | Astrazeneca Ab | Compounds effecting glucokinase |
| WO2008084043A1 (en) * | 2007-01-09 | 2008-07-17 | Novo Nordisk A/S | Urea glucokinase activators |
| WO2008084044A1 (en) | 2007-01-11 | 2008-07-17 | Novo Nordisk A/S | Urea glucokinase activators |
| WO2008084872A1 (en) | 2007-01-10 | 2008-07-17 | Mitsubishi Tanabe Pharma Corporation | Hydrazone derivative |
| WO2008104994A2 (en) | 2007-02-28 | 2008-09-04 | Advinus Therapeutics Private Limited | 2,2,2-tri-substituted acetamide derivatives as glucokinase activators, their process and pharmaceutical application |
| EP2048137A1 (en) | 2004-02-18 | 2009-04-15 | AstraZeneca AB | Benzamide derivatives and their use as glucokinase activating agents. |
| US7541373B2 (en) | 2002-06-27 | 2009-06-02 | Novo Nordisk A/S | Aryl carbonyl derivatives as therapeutic agents |
| WO2009047798A3 (en) * | 2007-10-08 | 2009-06-04 | Advinus Therapeutics Private L | Acetamide derivatives as glucokinase activators, their process and medicinal applications |
| US7595397B2 (en) | 2005-12-15 | 2009-09-29 | Boehringer Ingelheim International Gmbh | Compounds which modulate the CB2 receptor |
| US7645789B2 (en) | 2006-04-07 | 2010-01-12 | Vertex Pharmaceuticals Incorporated | Indole derivatives as CFTR modulators |
| US7659268B2 (en) | 2005-11-08 | 2010-02-09 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| WO2010047982A1 (en) | 2008-10-22 | 2010-04-29 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| WO2010051206A1 (en) | 2008-10-31 | 2010-05-06 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| US7750020B2 (en) | 2004-04-02 | 2010-07-06 | Novartis Ag | Sulfonamide-thiazolpyridine derivatives as glucokinase activators useful the treatment of Type 2 diabetes |
| US7754739B2 (en) | 2007-05-09 | 2010-07-13 | Vertex Pharmaceuticals Incorporated | Modulators of CFTR |
| US7776905B2 (en) | 2006-04-07 | 2010-08-17 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US7781435B2 (en) | 2005-09-22 | 2010-08-24 | Pfizer Inc | Imidazole compounds for the treatment of neurological disorders |
| US7781451B2 (en) | 2004-04-02 | 2010-08-24 | Novartis Ag | Thiazolopyridine derivatives, pharmaceut ical conditions containing them and methods of treating glucokinase mediated conditions |
| US7795257B2 (en) | 2005-09-30 | 2010-09-14 | Novartis Ag | Organic compounds |
| EP2272834A1 (en) | 2006-10-26 | 2011-01-12 | AstraZeneca AB | Benzoyl amino heterocyclyl compounds as glucokinase (GLK) activators |
| WO2011008475A1 (en) * | 2009-06-30 | 2011-01-20 | Allergan, Inc. | Optionally substituted 2-(arylmethyl, aryloxy or arylthio) -n- pyridin-2 -yl-aryl acetamide or 2, 2-bis (aryl) -n-pyridin-2-yl acetamide compounds as medicaments for the treatment of eye diseases |
| WO2011009845A1 (en) | 2009-07-23 | 2011-01-27 | F. Hoffmann-La Roche Ag | Pyridone glucokinase activators |
| US7928123B2 (en) | 2006-09-25 | 2011-04-19 | Boehringer Ingelheim International Gmbh | Compounds which modulate the CB2 receptor |
| US7935715B2 (en) | 2006-07-28 | 2011-05-03 | Boehringer Ingelheim International Gmbh | Compounds which modulate the CB2 receptor |
| WO2011080755A1 (en) | 2009-12-29 | 2011-07-07 | Advinus Therapeutics Private Limited | Fused nitrogen heterocyclic compounds, process of preparation and uses thereof |
| WO2011095997A1 (en) | 2010-02-08 | 2011-08-11 | Advinus Therapeutics Private Limited | Benzamide compounds as glucokinase activators and their pharmaceutical application |
| US8008332B2 (en) | 2006-05-31 | 2011-08-30 | Takeda San Diego, Inc. | Substituted indazoles as glucokinase activators |
| WO2011106273A1 (en) | 2010-02-25 | 2011-09-01 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| US8034822B2 (en) | 2006-03-08 | 2011-10-11 | Takeda San Diego, Inc. | Glucokinase activators |
| US8039491B2 (en) | 2005-12-28 | 2011-10-18 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| WO2011130459A1 (en) | 2010-04-14 | 2011-10-20 | Bristol-Myers Squibb Company | Novel glucokinase activators and methods of using same |
| US8048899B2 (en) | 2008-09-25 | 2011-11-01 | Boehringer Ingelheim International Gmbh | Compounds which selectively modulate the CB2 receptor |
| EP2402327A1 (en) | 2010-06-29 | 2012-01-04 | Advinus Therapeutics Private Limited | Acetamide compounds as glucokinase activators, their process and medicinal applications |
| US8124617B2 (en) | 2005-09-01 | 2012-02-28 | Takeda San Diego, Inc. | Imidazopyridine compounds |
| US8124781B2 (en) | 2007-12-07 | 2012-02-28 | Vertex Pharmaceuticals Incorporated | Processes for producing cycloalkylcarboxamido-pyridine benzoic acids |
| WO2012051450A1 (en) | 2010-10-13 | 2012-04-19 | Takeda Pharmaceutical Company Limited | Method of making azaindazole derivatives |
| US8163779B2 (en) | 2006-12-20 | 2012-04-24 | Takeda San Diego, Inc. | Glucokinase activators |
| US8173645B2 (en) | 2007-03-21 | 2012-05-08 | Takeda San Diego, Inc. | Glucokinase activators |
| US8252931B2 (en) | 2005-09-30 | 2012-08-28 | Novartis Ag | Thiazolo[5,4-B]pyridine glucokinase activators |
| WO2012116145A1 (en) | 2011-02-25 | 2012-08-30 | Merck Sharp & Dohme Corp. | Novel cyclic azabenzimidazole derivatives useful as anti-diabetic agents |
| US8466282B2 (en) | 2008-06-19 | 2013-06-18 | Takeda Pharmaceutical Company Limited | Heterocyclic compound and use thereof |
| US8563730B2 (en) | 2008-05-16 | 2013-10-22 | Takeda San Diego, Inc. | Pyrazole and fused pyrazole glucokinase activators |
| WO2014022528A1 (en) | 2012-08-02 | 2014-02-06 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| US8809342B2 (en) | 2010-12-23 | 2014-08-19 | Pfizer Inc. | Glucagon receptor modulators |
| WO2014130608A1 (en) | 2013-02-22 | 2014-08-28 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| WO2014139388A1 (en) | 2013-03-14 | 2014-09-18 | Merck Sharp & Dohme Corp. | Novel indole derivatives useful as anti-diabetic agents |
| US8846936B2 (en) | 2010-07-22 | 2014-09-30 | Boehringer Ingelheim International Gmbh | Sulfonyl compounds which modulate the CB2 receptor |
| US8859591B2 (en) | 2011-02-08 | 2014-10-14 | Pfizer Inc. | Glucagon receptor modulators |
| US8865744B1 (en) | 2013-05-17 | 2014-10-21 | Boehringer Ingelheim International Gmbh | (Cyano-dimethyl-methyl)-isoxazoles and -[1,3,4]thiadiazoles |
| US8927577B2 (en) | 2011-07-22 | 2015-01-06 | Pfizer Inc. | Quinolinyl glucagon receptor modulators |
| US8987295B2 (en) | 2009-03-30 | 2015-03-24 | Transtech Pharma, Llc | Substituted azoanthracene derivatives, pharmaceutical compositions, and methods of use thereof |
| WO2015051725A1 (en) | 2013-10-08 | 2015-04-16 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| US9315454B2 (en) | 2010-01-15 | 2016-04-19 | Boehringer Ingelheim International Gmbh | Compounds which modulate the CB2 receptor |
| US9340506B2 (en) | 2007-10-08 | 2016-05-17 | Advinus Therapeutics Limited | Acetamide derivatives as glucokinase activators, their process and medicinal applications |
| US9725440B2 (en) | 2007-05-09 | 2017-08-08 | Vertex Pharmaceuticals Incorporated | Modulators of CFTR |
| US9732080B2 (en) | 2006-11-03 | 2017-08-15 | Vertex Pharmaceuticals Incorporated | Azaindole derivatives as CFTR modulators |
| US9751890B2 (en) | 2008-02-28 | 2017-09-05 | Vertex Pharmaceuticals Incorporated | Heteroaryl derivatives as CFTR modulators |
| US9840499B2 (en) | 2007-12-07 | 2017-12-12 | Vertex Pharmaceuticals Incorporated | Solid forms of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)cyclopropanecarboxamido)-3-methylpyridin-2-yl) benzoic acid |
| WO2018106518A1 (en) | 2016-12-06 | 2018-06-14 | Merck Sharp & Dohme Corp. | Antidiabetic heterocyclic compounds |
| WO2018118670A1 (en) | 2016-12-20 | 2018-06-28 | Merck Sharp & Dohme Corp. | Antidiabetic spirochroman compounds |
| US10022352B2 (en) | 2006-04-07 | 2018-07-17 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US10058546B2 (en) | 2012-07-16 | 2018-08-28 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions of (R)-1-(2,2-difluorobenzo[D][1,3]dioxo1-5-y1)-N-(1-(2,3-dihydroxypropy1)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-y1)-1H-indol-5-y1) cyclopropanecarbox-amide and administration thereof |
| US10071979B2 (en) | 2010-04-22 | 2018-09-11 | Vertex Pharmaceuticals Incorporated | Process of producing cycloalkylcarboxamido-indole compounds |
| US10076513B2 (en) | 2010-04-07 | 2018-09-18 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions of 3-(6-(1-(2,2-difluorobenzo[D][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl) benzoic acid and administration thereof |
| US10081621B2 (en) | 2010-03-25 | 2018-09-25 | Vertex Pharmaceuticals Incorporated | Solid forms of (R)-1(2,2-difluorobenzo[D][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide |
| US10206877B2 (en) | 2014-04-15 | 2019-02-19 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cystic fibrosis transmembrane conductance regulator mediated diseases |
| US10231932B2 (en) | 2013-11-12 | 2019-03-19 | Vertex Pharmaceuticals Incorporated | Process of preparing pharmaceutical compositions for the treatment of CFTR mediated diseases |
| US10302602B2 (en) | 2014-11-18 | 2019-05-28 | Vertex Pharmaceuticals Incorporated | Process of conducting high throughput testing high performance liquid chromatography |
| US11690841B2 (en) | 2017-09-14 | 2023-07-04 | Queen Mary University Of London | Glycolysis-activating agents for treatment or prevention of disease |
| US11833136B2 (en) | 2018-06-12 | 2023-12-05 | Vtv Therapeutics Llc | Therapeutic uses of glucokinase activators in combination with insulin or insulin analogs |
Families Citing this family (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2477604A1 (en) | 2002-03-13 | 2003-09-25 | Signum Biosciences, Inc. | Modulation of protein methylation and phosphoprotein phosphate |
| GB0226930D0 (en) * | 2002-11-19 | 2002-12-24 | Astrazeneca Ab | Chemical compounds |
| US7687625B2 (en) | 2003-03-25 | 2010-03-30 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7678909B1 (en) | 2003-08-13 | 2010-03-16 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| BRPI0413452A (en) | 2003-08-13 | 2006-10-17 | Takeda Pharmaceutical | compound, pharmaceutical composition, kit, article of manufacture, and methods of inhibiting dpp-iv, therapeutic, and treating a disease state, cancer, autoimmune disorders, a condition and HIV infection |
| US7169926B1 (en) | 2003-08-13 | 2007-01-30 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7790734B2 (en) | 2003-09-08 | 2010-09-07 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7732446B1 (en) | 2004-03-11 | 2010-06-08 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| AU2004318013B8 (en) | 2004-03-15 | 2011-10-06 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| WO2005118555A1 (en) | 2004-06-04 | 2005-12-15 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| WO2006019965A2 (en) | 2004-07-16 | 2006-02-23 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
| EP2805953B1 (en) * | 2004-12-21 | 2016-03-09 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7923041B2 (en) | 2005-02-03 | 2011-04-12 | Signum Biosciences, Inc. | Compositions and methods for enhancing cognitive function |
| WO2006084033A1 (en) | 2005-02-03 | 2006-08-10 | Signum Biosciences, Inc. | Compositions and methods for enhancing cognitive function |
| EP1942898B2 (en) | 2005-09-14 | 2014-05-14 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors for treating diabetes |
| JP5122462B2 (en) | 2005-09-16 | 2013-01-16 | 武田薬品工業株式会社 | Dipeptidyl peptidase inhibitor |
| WO2007061923A2 (en) * | 2005-11-18 | 2007-05-31 | Takeda San Diego, Inc. | Glucokinase activators |
| EP2295432A1 (en) * | 2006-02-10 | 2011-03-16 | TransTech Pharma Inc. | Process for the preparation of aminobenzimidazole derivatives |
| WO2007112347A1 (en) | 2006-03-28 | 2007-10-04 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8324383B2 (en) | 2006-09-13 | 2012-12-04 | Takeda Pharmaceutical Company Limited | Methods of making polymorphs of benzoate salt of 2-[[6-[(3R)-3-amino-1-piperidinyl]-3,4-dihydro-3-methyl-2,4-dioxo-1(2H)-pyrimidinyl]methyl]-benzonitrile |
| US20080107725A1 (en) * | 2006-10-13 | 2008-05-08 | Albano Antonio A | Pharmaceutical Solid Dosage Forms Comprising Amorphous Compounds Micro-Embedded in Ionic Water-Insoluble Polymers |
| JPWO2008050600A1 (en) * | 2006-10-25 | 2010-02-25 | 株式会社ニュージェン・ファーマ | Therapeutic or preventive agent for intractable diseases with molecular background of oxidative stress cell death |
| TW200838536A (en) | 2006-11-29 | 2008-10-01 | Takeda Pharmaceutical | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| US8093236B2 (en) | 2007-03-13 | 2012-01-10 | Takeda Pharmaceuticals Company Limited | Weekly administration of dipeptidyl peptidase inhibitors |
| PL2195312T3 (en) * | 2007-10-09 | 2013-04-30 | Merck Patent Gmbh | Pyridine derivatives useful as glucokinase activators |
| US8258134B2 (en) * | 2008-04-16 | 2012-09-04 | Hoffmann-La Roche Inc. | Pyridazinone glucokinase activators |
| CA2758424C (en) | 2008-04-21 | 2018-03-06 | Signum Biosciences, Inc. | Tryptamine derivatives as pp2a methylation modulators |
| US8921576B2 (en) * | 2011-10-19 | 2014-12-30 | Kowa Company, Ltd. | Spiroindoline compound, and medicinal agent comprising same |
| EP2878339A1 (en) * | 2013-12-02 | 2015-06-03 | Siena Biotech S.p.A. | SIP3 antagonists |
| WO2017004383A1 (en) * | 2015-06-30 | 2017-01-05 | Dana-Farber Cancer Institute, Inc. | Inhibitors of egfr and methods of use thereof |
| US11147788B2 (en) * | 2017-12-14 | 2021-10-19 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| CA3093546A1 (en) * | 2018-03-28 | 2019-10-03 | Hanlim Pharmaceutical Co., Ltd. | 2-cyanopyrimidin-4-yl carbamate or urea derivative or salt thereof, and pharmaceutical composition including same |
| US20230265083A1 (en) * | 2020-08-10 | 2023-08-24 | Prelude Therapeutics Incorporated | Heterocycle CDK Inhibitors And Their Use Thereof |
Citations (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2151766A1 (en) | 1971-10-14 | 1973-04-19 | Schering Ag | N-thiazol(in)y-2-phenoxyalkanoamides - with herbicidal activity |
| DE2264983A1 (en) | 1972-02-10 | 1975-10-16 | Schering Ag | N-thiazolyl phenoxy (acet-or propion) amides - - herbicides |
| US4166452A (en) | 1976-05-03 | 1979-09-04 | Generales Constantine D J Jr | Apparatus for testing human responses to stimuli |
| US4265871A (en) | 1979-05-07 | 1981-05-05 | Allied Chemical Corporation | Purification of boron-containing sulfuric acid |
| US4356108A (en) | 1979-12-20 | 1982-10-26 | The Mead Corporation | Encapsulation process |
| WO1997009040A1 (en) | 1995-09-08 | 1997-03-13 | Novo Nordisk A/S | 2-alkylpyrrolidines |
| WO1997026265A1 (en) | 1996-01-17 | 1997-07-24 | Novo Nordisk A/S | Fused 1,2,4-thiadiazine and fused 1,4-thiazine derivatives, their preparation and use |
| WO1997041120A1 (en) | 1996-07-26 | 1997-11-06 | Dr. Reddy's Research Foundation | Thiazolidinedione compounds having antidiabetic, hypolipidaemic, antihypertensive properties, process for their preparation and pharmaceutical compositions thereof |
| WO1997041119A1 (en) | 1997-05-02 | 1997-11-06 | Dr. Reddy's Research Foundation | Novel antidiabetic compounds having hypolipidaemic, antihypertensive properties, process for their preparation and pharmaceutical compositions containing them |
| WO1997041097A2 (en) | 1996-12-31 | 1997-11-06 | Dr. Reddy's Research Foundation | Novel heterocyclic compounds process for their preparation and pharmaceutical compositions containing them and their use in the treatment of diabetes and related diseases |
| WO1998008871A1 (en) | 1996-08-30 | 1998-03-05 | Novo Nordisk A/S | Glp-1 derivatives |
| WO1998045292A1 (en) | 1997-12-02 | 1998-10-15 | Dr. Reddy's Research Foundation | Thiazolidinedione and oxazolidinedione derivatives having antidiabetic, hypolipidaemic and antihypertensive properties |
| WO1999001423A1 (en) | 1997-07-01 | 1999-01-14 | Novo Nordisk A/S | Glucagon antagonists/inverse agonists |
| WO1999003861A1 (en) | 1997-07-16 | 1999-01-28 | Novo Nordisk A/S | Fused 1,2,4-thiadiazine derivatives, their preparation and use |
| WO1999019313A1 (en) | 1997-10-27 | 1999-04-22 | Dr. Reddy's Research Foundation | Novel tricyclic compounds and their use in medicine; process for their preparation and pharmaceutical compositions containing them |
| WO2000023416A1 (en) | 1998-10-21 | 2000-04-27 | Novo Nordisk A/S | New compounds, their preparation and use |
| WO2000023445A1 (en) | 1998-10-21 | 2000-04-27 | Novo Nordisk A/S | New compounds, their preparation and use |
| WO2000023425A1 (en) | 1998-10-21 | 2000-04-27 | Novo Nordisk A/S | New compounds, their preparation and use |
| WO2000023417A1 (en) | 1998-10-21 | 2000-04-27 | Novo Nordisk A/S | New compounds, their preparation and use |
| WO2000023451A1 (en) | 1998-10-21 | 2000-04-27 | Novo Nordisk A/S | New compounds, their preparation and use |
| WO2000023415A1 (en) | 1998-10-21 | 2000-04-27 | Novo Nordisk A/S | New compounds, their preparation and use |
| WO2000037474A1 (en) | 1998-12-18 | 2000-06-29 | Novo Nordisk A/S | Fused 1,2,4-thiadiazine derivatives, their preparation and use |
| WO2000039088A1 (en) | 1998-12-23 | 2000-07-06 | Novo Nordisk A/S | Glucagon antagonists/inverse agonists |
| WO2000041121A1 (en) | 1999-01-07 | 2000-07-13 | Ccrewards.Com | Method and arrangement for issuance and management of digital coupons and sales offers |
| WO2000042026A1 (en) | 1999-01-15 | 2000-07-20 | Novo Nordisk A/S | Non-peptide glp-1 agonists |
| WO2000042023A1 (en) | 1999-01-18 | 2000-07-20 | Novo Nordisk A/S | Substituted imidazoles, their preparation and use |
| WO2000050414A1 (en) | 1999-02-24 | 2000-08-31 | Dr.Reddy's Research Foundation | Novel tricyclic compounds and their use in medicine; process for their preparation and pharmaceutical composition containing them |
| WO2000058293A2 (en) | 1999-03-29 | 2000-10-05 | F. Hoffmann-La Roche Ag | Glucokinase activators |
| WO2000063191A1 (en) | 1999-04-16 | 2000-10-26 | Dr. Reddy's Research Foundation | Novel polymorphic forms of an antidiabetic agent: process for their preparation and a pharmaceutical composition containing them |
| WO2000063190A1 (en) | 1999-04-20 | 2000-10-26 | Novo Nordisk A/S | New compounds, their preparation and use |
| WO2000063153A1 (en) | 1999-04-20 | 2000-10-26 | Novo Nordisk A/S | New compounds, their preparation and use |
| WO2000063189A1 (en) | 1999-04-16 | 2000-10-26 | Novo Nordisk A/S | Crystalline r- guanidines, arginine or (l) -arginine (2s) -2- ethoxy -3-{4- [2-(10h -phenoxazin -10-yl)ethoxy]phenyl}propanoate |
| WO2000063192A1 (en) | 1999-04-16 | 2000-10-26 | Dr. Reddy's Research Foundation | Novel polymorphic forms of an antidiabetic agent: process for their preparation and pharmaceutical compositions containing them |
| WO2000063208A1 (en) | 1999-04-16 | 2000-10-26 | Novo Nordisk A/S | Substituted imidazoles, their preparation and use |
| WO2000063196A1 (en) | 1999-04-20 | 2000-10-26 | Novo Nordisk A/S | New compounds, their preparation and use |
| WO2000063209A1 (en) | 1999-04-20 | 2000-10-26 | Novo Nordisk A/S | New compounds, their preparation and use |
| WO2000064884A1 (en) | 1999-04-26 | 2000-11-02 | Novo Nordisk A/S | Piperidyl-imidazole derivatives, their preparations and therapeutic uses |
| WO2001044216A1 (en) | 1999-12-15 | 2001-06-21 | F. Hoffmann-La Roche Ag | Trans olefinic glucokinase activators |
| WO2001083465A2 (en) | 2000-05-03 | 2001-11-08 | F. Hoffmann-La Roche Ag | Alkynyl phenyl heteroaromatic glucokinase activators |
| WO2001083478A2 (en) | 2000-05-03 | 2001-11-08 | F. Hoffmann-La Roche Ag | Hydantoin-containing glucokinase activators |
| WO2001085707A1 (en) | 2000-05-08 | 2001-11-15 | F. Hoffmann-La Roche Ag | Para-amine substituted phenylamide glucokinase activators |
| WO2001085706A1 (en) | 2000-05-08 | 2001-11-15 | F. Hoffmann-La Roche Ag | Substituted phenylacetamides and their use as glucokinase activators |
| WO2002008209A1 (en) | 2000-07-20 | 2002-01-31 | F. Hoffmann-La Roche Ag | Alpha-acyl and alpha-heteroatom-substituted benzene acetamide glucokinase activators |
Family Cites Families (53)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3152136A (en) * | 1958-10-30 | 1964-10-06 | Dow Chemical Co | Dinitroaroyl-nu-pyridyl amides |
| US3067250A (en) * | 1959-01-26 | 1962-12-04 | Dow Chemical Co | 4-aryl, 1, 1-di propynyl-semicarbazides |
| US3317534A (en) * | 1963-10-30 | 1967-05-02 | Chugai Pharmaceutical Co Ltd | Benzamidopyrimidines |
| US3551442A (en) * | 1965-04-06 | 1970-12-29 | Pechiney Saint Gobain | Thiazole derivatives |
| US4175081A (en) * | 1968-02-01 | 1979-11-20 | Mobil Oil Corporation | 5-Substituted thiadiazole ureas |
| GB1266769A (en) * | 1969-08-15 | 1972-03-15 | ||
| US3682163A (en) * | 1970-09-18 | 1972-08-08 | Walter A Plummer | Snap-on orthopedic splint |
| US3887709A (en) * | 1971-09-16 | 1975-06-03 | Zdzislaw Brzozowski | 2-Pyrazoline-1-carboxamide sulfonamide derivatives useful as hypoglycemic agents |
| US3862163A (en) * | 1971-10-14 | 1975-01-21 | Schering Ag | Phenoxycarboxylic acid amides |
| US3874873A (en) * | 1972-03-27 | 1975-04-01 | Fmc Corp | Herbicidal compositions based on 1,2,3-thiadiazol-5-yl ureas |
| IL44058A (en) * | 1973-02-02 | 1978-10-31 | Ciba Geigy Ag | 3amino-1,2,4-benzotriazine 1,4-di-noxide derivatives, their preparation and compositions for the control of microorganisms containing them |
| JPS5614643B2 (en) * | 1973-07-02 | 1981-04-06 | ||
| PL106114B1 (en) * | 1976-12-31 | 1979-11-30 | Akad Medyczna | METHOD OF MAKING NEW N- (4- / 2- / PYRAZOLO-1-CARBONAMIDO / -ETHYL / -BENZENSULPHONYL) -Urea |
| DE2712630A1 (en) * | 1977-03-23 | 1978-09-28 | Bayer Ag | 1,3,4-THIADIAZOL-2-YL-UREA, METHOD OF MANUFACTURING AND USE AS FUNGICIDES |
| DE2716324A1 (en) * | 1977-04-07 | 1978-10-12 | Schering Ag | 1,2,3-THIADIAZOL-3-IN-5-YLIDEN-UREAS, METHOD FOR THE PRODUCTION OF THESE COMPOUNDS AND THEIR CONTAINING AGENTS WITH GROWTH REGULATORY EFFECT FOR PLANTS |
| JPS6033109B2 (en) * | 1977-04-28 | 1985-08-01 | 塩野義製薬株式会社 | Synthesis method of urea derivatives |
| US4241072A (en) * | 1979-01-18 | 1980-12-23 | Merck & Co., Inc. | Substituted ureas and processes for their preparation |
| DE2928485A1 (en) * | 1979-07-14 | 1981-01-29 | Bayer Ag | USE OF UREA DERIVATIVES AS A MEDICINAL PRODUCT IN THE TREATMENT OF FATTY METABOLISM DISORDERS |
| US4265874A (en) | 1980-04-25 | 1981-05-05 | Alza Corporation | Method of delivering drug with aid of effervescent activity generated in environment of use |
| US4694004A (en) * | 1984-07-09 | 1987-09-15 | Fujisawa Pharmaceutical Co., Ltd. | Semicarbazide derivatives, processes for preparation thereof and pharmaceutical composition comprising the same |
| US4808722A (en) * | 1985-10-31 | 1989-02-28 | Fmc Corporation | Pyridinylurea N-oxide compounds and agricultural uses |
| JPH05294935A (en) * | 1991-03-15 | 1993-11-09 | Green Cross Corp:The | Aminopyridine-based compound |
| US5371086A (en) * | 1991-03-15 | 1994-12-06 | The Green Cross Corporation | Aminopyridine compounds |
| KR950701621A (en) * | 1992-05-28 | 1995-04-28 | 알렌 제이. 스피겔 | Acyl coenzyme A: N-aryl and N-heteroaryl urea derivatives as inhibitors of cholesterol acyl transferase (ACAT) A: CHOLESTEROL ACYL TRANSFERASE (ACAT) |
| US5849769A (en) * | 1994-08-24 | 1998-12-15 | Medivir Ab | N-arylalkyl-N-heteroarylurea and guandine compounds and methods of treating HIV infection |
| US5556969A (en) * | 1994-12-07 | 1996-09-17 | Merck Sharp & Dohme Ltd. | Benzodiazepine derivatives |
| WO1996040629A1 (en) * | 1995-06-07 | 1996-12-19 | Sugen, Inc. | Tyrphostin-like compounds for the treatment of cell proliferative disorders or cell differentiation disorders |
| US5846990A (en) * | 1995-07-24 | 1998-12-08 | Bristol-Myers Squibb Co. | Substituted biphenyl isoxazole sulfonamides |
| JPH09124620A (en) * | 1995-10-11 | 1997-05-13 | Bristol Myers Squibb Co | Substituted biphenylsulfonamide endothelin antagonist |
| CA2197364A1 (en) * | 1996-02-15 | 1997-08-16 | Toshikazu Suzuki | Phenol compound and process for preparing the same |
| TW523506B (en) * | 1996-12-18 | 2003-03-11 | Ono Pharmaceutical Co | Sulfonamide or carbamide derivatives and drugs containing the same as active ingredients |
| US5846985A (en) * | 1997-03-05 | 1998-12-08 | Bristol-Myers Squibb Co. | Substituted biphenyl isoxazole sulfonamides |
| SE9702001D0 (en) * | 1997-05-28 | 1997-05-28 | Astra Pharma Prod | Novel compounds |
| US6268384B1 (en) * | 1997-08-29 | 2001-07-31 | Vertex Pharmaceuticals Incorporated | Compounds possessing neuronal activity |
| US6225346B1 (en) * | 1997-10-24 | 2001-05-01 | Sugen, Inc. | Tyrphostin like compounds |
| US6407124B1 (en) * | 1998-06-18 | 2002-06-18 | Bristol-Myers Squibb Company | Carbon substituted aminothiazole inhibitors of cyclin dependent kinases |
| GT199900147A (en) * | 1998-09-17 | 1999-09-06 | 1, 2, 3, 4- TETRAHIDROQUINOLINAS 2-SUBSTITUTED 4-AMINO SUBSTITUTED. | |
| GB9823873D0 (en) * | 1998-10-30 | 1998-12-30 | Pharmacia & Upjohn Spa | 2-ureido-thiazole derivatives,process for their preparation,and their use as antitumour agents |
| US6500817B1 (en) * | 1999-03-08 | 2002-12-31 | Bayer Aktiengesellschaft | Thiazolyl urea derivatives and their utilization as antiviral agents |
| US6610846B1 (en) * | 1999-03-29 | 2003-08-26 | Hoffman-La Roche Inc. | Heteroaromatic glucokinase activators |
| US6503949B1 (en) * | 1999-05-17 | 2003-01-07 | Noro Nordisk A/S | Glucagon antagonists/inverse agonists |
| US6114365A (en) * | 1999-08-12 | 2000-09-05 | Pharmacia & Upjohn S.P.A. | Arylmethyl-carbonylamino-thiazole derivatives, process for their preparation, and their use as antitumor agents |
| MXPA02012795A (en) * | 2000-06-28 | 2004-07-30 | Teva Pharma | Carvedilol. |
| US6645990B2 (en) * | 2000-08-15 | 2003-11-11 | Amgen Inc. | Thiazolyl urea compounds and methods of uses |
| BR0116850A (en) * | 2001-01-31 | 2004-02-25 | Pfizer Prod Inc | Thiazolyl-, oxazolyl-, pyrrolyl- and imidazolyl- amide derivatives useful as inhibitors of pde4 isoenzymes |
| EP1256578B1 (en) * | 2001-05-11 | 2006-01-11 | Pfizer Products Inc. | Thiazole derivatives and their use as cdk inhibitors |
| EP1408950B1 (en) * | 2001-07-11 | 2007-04-25 | Boehringer Ingelheim Pharmaceuticals Inc. | Methods of treating cytokine mediated diseases |
| US6881746B2 (en) * | 2001-12-03 | 2005-04-19 | Novo Nordick A/S | Glucagon antagonists/inverse agonists |
| DE60221627D1 (en) * | 2001-12-21 | 2007-09-20 | Virochem Pharma Inc | Thiazole derivatives and their use for the treatment or prevention of infections by flaviviruses |
| CA2744893A1 (en) * | 2002-06-27 | 2004-01-08 | Novo Nordisk A/S | Aryl carbonyl derivatives as glucokinase activators |
| TW200505894A (en) * | 2003-08-08 | 2005-02-16 | Yamanouchi Pharma Co Ltd | Tetrahydro-2H-thiopyran-4-carboxamide derivative |
| BRPI0506662B8 (en) * | 2004-01-06 | 2021-05-25 | Novo Nordisk As | glucokinase activating compounds |
| KR20080024211A (en) * | 2005-07-08 | 2008-03-17 | 노보 노르디스크 에이/에스 | Dicycloalkyl urea glucokinase activators |
-
2002
- 2002-12-19 JP JP2003556060A patent/JP2005518391A/en not_active Withdrawn
- 2002-12-19 CN CNB028275012A patent/CN100506807C/en not_active Expired - Fee Related
- 2002-12-19 RU RU2004122407/04A patent/RU2374236C2/en not_active IP Right Cessation
- 2002-12-19 AU AU2002351748A patent/AU2002351748B2/en not_active Ceased
- 2002-12-19 MX MXPA04006048A patent/MXPA04006048A/en active IP Right Grant
- 2002-12-19 US US10/323,290 patent/US20030171411A1/en not_active Abandoned
- 2002-12-19 EP EP10181515A patent/EP2305648A1/en not_active Withdrawn
- 2002-12-19 IL IL16262002A patent/IL162620A0/en unknown
- 2002-12-19 HU HU0402309A patent/HUP0402309A3/en unknown
- 2002-12-19 CA CA002471049A patent/CA2471049A1/en not_active Abandoned
- 2002-12-19 WO PCT/DK2002/000880 patent/WO2003055482A1/en active Application Filing
- 2002-12-19 UA UA20040604430A patent/UA84390C2/en unknown
- 2002-12-19 PL PL02370989A patent/PL370989A1/en not_active Application Discontinuation
- 2002-12-19 CZ CZ2004747A patent/CZ2004747A3/en unknown
- 2002-12-19 EP EP20020787463 patent/EP1458382A1/en not_active Withdrawn
- 2002-12-19 BR BR0215212-6A patent/BR0215212A/en not_active IP Right Cessation
- 2002-12-19 KR KR1020047009841A patent/KR101018318B1/en not_active IP Right Cessation
-
2004
- 2004-07-20 NO NO20043116A patent/NO20043116L/en not_active Application Discontinuation
-
2010
- 2010-08-06 JP JP2010178083A patent/JP2011006435A/en not_active Withdrawn
Patent Citations (44)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2151766A1 (en) | 1971-10-14 | 1973-04-19 | Schering Ag | N-thiazol(in)y-2-phenoxyalkanoamides - with herbicidal activity |
| DE2264983A1 (en) | 1972-02-10 | 1975-10-16 | Schering Ag | N-thiazolyl phenoxy (acet-or propion) amides - - herbicides |
| US4166452A (en) | 1976-05-03 | 1979-09-04 | Generales Constantine D J Jr | Apparatus for testing human responses to stimuli |
| US4265871A (en) | 1979-05-07 | 1981-05-05 | Allied Chemical Corporation | Purification of boron-containing sulfuric acid |
| US4356108A (en) | 1979-12-20 | 1982-10-26 | The Mead Corporation | Encapsulation process |
| WO1997009040A1 (en) | 1995-09-08 | 1997-03-13 | Novo Nordisk A/S | 2-alkylpyrrolidines |
| WO1997026265A1 (en) | 1996-01-17 | 1997-07-24 | Novo Nordisk A/S | Fused 1,2,4-thiadiazine and fused 1,4-thiazine derivatives, their preparation and use |
| WO1997041120A1 (en) | 1996-07-26 | 1997-11-06 | Dr. Reddy's Research Foundation | Thiazolidinedione compounds having antidiabetic, hypolipidaemic, antihypertensive properties, process for their preparation and pharmaceutical compositions thereof |
| WO1998008871A1 (en) | 1996-08-30 | 1998-03-05 | Novo Nordisk A/S | Glp-1 derivatives |
| WO1997041097A2 (en) | 1996-12-31 | 1997-11-06 | Dr. Reddy's Research Foundation | Novel heterocyclic compounds process for their preparation and pharmaceutical compositions containing them and their use in the treatment of diabetes and related diseases |
| WO1997041119A1 (en) | 1997-05-02 | 1997-11-06 | Dr. Reddy's Research Foundation | Novel antidiabetic compounds having hypolipidaemic, antihypertensive properties, process for their preparation and pharmaceutical compositions containing them |
| WO1999001423A1 (en) | 1997-07-01 | 1999-01-14 | Novo Nordisk A/S | Glucagon antagonists/inverse agonists |
| WO1999003861A1 (en) | 1997-07-16 | 1999-01-28 | Novo Nordisk A/S | Fused 1,2,4-thiadiazine derivatives, their preparation and use |
| WO1999019313A1 (en) | 1997-10-27 | 1999-04-22 | Dr. Reddy's Research Foundation | Novel tricyclic compounds and their use in medicine; process for their preparation and pharmaceutical compositions containing them |
| WO1998045292A1 (en) | 1997-12-02 | 1998-10-15 | Dr. Reddy's Research Foundation | Thiazolidinedione and oxazolidinedione derivatives having antidiabetic, hypolipidaemic and antihypertensive properties |
| WO2000023416A1 (en) | 1998-10-21 | 2000-04-27 | Novo Nordisk A/S | New compounds, their preparation and use |
| WO2000023445A1 (en) | 1998-10-21 | 2000-04-27 | Novo Nordisk A/S | New compounds, their preparation and use |
| WO2000023425A1 (en) | 1998-10-21 | 2000-04-27 | Novo Nordisk A/S | New compounds, their preparation and use |
| WO2000023417A1 (en) | 1998-10-21 | 2000-04-27 | Novo Nordisk A/S | New compounds, their preparation and use |
| WO2000023451A1 (en) | 1998-10-21 | 2000-04-27 | Novo Nordisk A/S | New compounds, their preparation and use |
| WO2000023415A1 (en) | 1998-10-21 | 2000-04-27 | Novo Nordisk A/S | New compounds, their preparation and use |
| WO2000037474A1 (en) | 1998-12-18 | 2000-06-29 | Novo Nordisk A/S | Fused 1,2,4-thiadiazine derivatives, their preparation and use |
| WO2000039088A1 (en) | 1998-12-23 | 2000-07-06 | Novo Nordisk A/S | Glucagon antagonists/inverse agonists |
| WO2000041121A1 (en) | 1999-01-07 | 2000-07-13 | Ccrewards.Com | Method and arrangement for issuance and management of digital coupons and sales offers |
| WO2000042026A1 (en) | 1999-01-15 | 2000-07-20 | Novo Nordisk A/S | Non-peptide glp-1 agonists |
| WO2000042023A1 (en) | 1999-01-18 | 2000-07-20 | Novo Nordisk A/S | Substituted imidazoles, their preparation and use |
| WO2000050414A1 (en) | 1999-02-24 | 2000-08-31 | Dr.Reddy's Research Foundation | Novel tricyclic compounds and their use in medicine; process for their preparation and pharmaceutical composition containing them |
| WO2000058293A2 (en) | 1999-03-29 | 2000-10-05 | F. Hoffmann-La Roche Ag | Glucokinase activators |
| WO2000063208A1 (en) | 1999-04-16 | 2000-10-26 | Novo Nordisk A/S | Substituted imidazoles, their preparation and use |
| WO2000063193A1 (en) | 1999-04-16 | 2000-10-26 | Dr. Reddy's Research Foundation | Novel polymorphic forms of an antidiabetic agent: process for their preparation and a pharmaceutical composition containing them |
| WO2000063191A1 (en) | 1999-04-16 | 2000-10-26 | Dr. Reddy's Research Foundation | Novel polymorphic forms of an antidiabetic agent: process for their preparation and a pharmaceutical composition containing them |
| WO2000063189A1 (en) | 1999-04-16 | 2000-10-26 | Novo Nordisk A/S | Crystalline r- guanidines, arginine or (l) -arginine (2s) -2- ethoxy -3-{4- [2-(10h -phenoxazin -10-yl)ethoxy]phenyl}propanoate |
| WO2000063192A1 (en) | 1999-04-16 | 2000-10-26 | Dr. Reddy's Research Foundation | Novel polymorphic forms of an antidiabetic agent: process for their preparation and pharmaceutical compositions containing them |
| WO2000063153A1 (en) | 1999-04-20 | 2000-10-26 | Novo Nordisk A/S | New compounds, their preparation and use |
| WO2000063196A1 (en) | 1999-04-20 | 2000-10-26 | Novo Nordisk A/S | New compounds, their preparation and use |
| WO2000063190A1 (en) | 1999-04-20 | 2000-10-26 | Novo Nordisk A/S | New compounds, their preparation and use |
| WO2000063209A1 (en) | 1999-04-20 | 2000-10-26 | Novo Nordisk A/S | New compounds, their preparation and use |
| WO2000064884A1 (en) | 1999-04-26 | 2000-11-02 | Novo Nordisk A/S | Piperidyl-imidazole derivatives, their preparations and therapeutic uses |
| WO2001044216A1 (en) | 1999-12-15 | 2001-06-21 | F. Hoffmann-La Roche Ag | Trans olefinic glucokinase activators |
| WO2001083465A2 (en) | 2000-05-03 | 2001-11-08 | F. Hoffmann-La Roche Ag | Alkynyl phenyl heteroaromatic glucokinase activators |
| WO2001083478A2 (en) | 2000-05-03 | 2001-11-08 | F. Hoffmann-La Roche Ag | Hydantoin-containing glucokinase activators |
| WO2001085707A1 (en) | 2000-05-08 | 2001-11-15 | F. Hoffmann-La Roche Ag | Para-amine substituted phenylamide glucokinase activators |
| WO2001085706A1 (en) | 2000-05-08 | 2001-11-15 | F. Hoffmann-La Roche Ag | Substituted phenylacetamides and their use as glucokinase activators |
| WO2002008209A1 (en) | 2000-07-20 | 2002-01-31 | F. Hoffmann-La Roche Ag | Alpha-acyl and alpha-heteroatom-substituted benzene acetamide glucokinase activators |
Non-Patent Citations (8)
| Title |
|---|
| "Expert Committee of the Diagnosis and Classification of Diabetes Mellitus", DIABETES CARE, vol. 20, 1997, pages 1183 - 1197 |
| "Remington: The Science and Practice of Pharmacy", 1995, MACK PUBLISHING CO. |
| ANN INTERN MED, vol. 103, 1985, pages 147 |
| GIRARD, J. ET AL., ANNU REV NUTR, vol. 17, 1997, pages 325 - 352 |
| J. W. BARTON: "Protective Groups In Organic Chemistry", 1973, PLENUM PRESS |
| MANN GV, N.ENGL.J.MED, vol. 291, 1974, pages 226 |
| PROC. NATL. ACAD. SCI. U.S.A., vol. 88, 1991, pages 7294 - 7297 |
| T. W. GREENE: "Protective Groups in Organic Synthesis", 1981, JOHN WILEY AND SONS |
Cited By (176)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7199140B2 (en) | 2001-06-26 | 2007-04-03 | Astrazeneca Ab | Vinyl phenyl derivatives as GLK activators |
| US7390908B2 (en) | 2001-08-17 | 2008-06-24 | Astrazeneca Ab | Compounds effecting glucokinase |
| US7384967B2 (en) | 2002-06-27 | 2008-06-10 | Novo Nordisk A/S | Aryl carbonyl derivatives as therapeutic agents |
| US7897628B2 (en) | 2002-06-27 | 2011-03-01 | Novo Nordisk A/S | Aryl carbonyl derivatives as therapeutic agents |
| US8063081B2 (en) | 2002-06-27 | 2011-11-22 | Novo Nordisk A/S | Aryl carbonyl derivatives as therapeutic agents |
| US7541373B2 (en) | 2002-06-27 | 2009-06-02 | Novo Nordisk A/S | Aryl carbonyl derivatives as therapeutic agents |
| WO2004002481A1 (en) * | 2002-06-27 | 2004-01-08 | Novo Nordisk A/S | Aryl carbonyl derivatives as therapeutic agents |
| USRE45670E1 (en) | 2002-06-27 | 2015-09-15 | Novo Nordisk A/S | Aryl carbonyl derivatives as therapeutic agents |
| EP1549626A1 (en) * | 2002-10-03 | 2005-07-06 | Novartis AG | Substituted (thiazol-2-yl) -amide or sulfonamide as glycokinase activators useful in the treatment of type 2 diabetes |
| WO2004050645A1 (en) * | 2002-10-03 | 2004-06-17 | Novartis Ag | Substituted (thiazol-2-yl) -amide or sulfonamide as glycokinase activators useful in the treatment of type 2 diabetes |
| US7812167B2 (en) | 2002-10-03 | 2010-10-12 | Novartis, Ag | Substituted (thiazol-2-yl)-amides or sulfonamides as glucokinase activators useful in the treatment of type 2 diabetes |
| US7230108B2 (en) | 2002-11-19 | 2007-06-12 | Astrazeneca Ab | Quinoline derivatives as glucokinase ligands |
| WO2004063194A1 (en) * | 2003-01-06 | 2004-07-29 | Eli Lilly And Company | Heteroaryl compounds |
| WO2004072066A1 (en) * | 2003-02-11 | 2004-08-26 | Prosidion Limited | Tri(cyclo) substituted amide glucokinase activator compounds |
| US7582632B2 (en) | 2003-02-11 | 2009-09-01 | Prosidion Limited | Tri(cyclo) substituted amide compounds |
| US7214681B2 (en) | 2003-02-11 | 2007-05-08 | Prosidion Limited | Tri(cyclo) substituted amide compounds |
| AU2005203930B2 (en) * | 2004-01-06 | 2011-07-14 | Vtv Therapeutics Llc | Heteroaryl-ureas and their use as glucokinase activators |
| JP2011201892A (en) * | 2004-01-06 | 2011-10-13 | Novo Nordisk As | Heteroaryl-urea, and use thereof as glucokinase activator |
| JP2007517812A (en) * | 2004-01-06 | 2007-07-05 | ノボ ノルディスク アクティーゼルスカブ | Heteroaryl ureas and their use as glucokinase activators |
| AU2005203930C1 (en) * | 2004-01-06 | 2012-02-23 | Vtv Therapeutics Llc | Heteroaryl-ureas and their use as glucokinase activators |
| USRE45183E1 (en) | 2004-01-06 | 2014-10-07 | Novo Nordisk A/S | Heteroaryl-ureas and their use as glucokinase activators |
| KR101126225B1 (en) | 2004-01-06 | 2012-04-12 | 노보 노르디스크 에이/에스 | Heteroaryl-ureas and their use as glucokinase activators |
| US7872139B2 (en) * | 2004-01-06 | 2011-01-18 | Novo Nordisk A/S | Heteroaryl-ureas and their use as glucokinase activators |
| WO2005066145A1 (en) * | 2004-01-06 | 2005-07-21 | Novo Nordisk A/S | Heteroaryl-ureas and their use as glucokinase activators |
| KR101196313B1 (en) * | 2004-01-06 | 2012-11-07 | 노보 노르디스크 에이/에스 | Heteroaryl-ureas and their use as glucokinase activators |
| JP4834840B2 (en) * | 2004-01-06 | 2011-12-14 | ノヴォ ノルディスク アー/エス | Heteroaryl ureas and their use as glucokinase activators |
| NO337003B1 (en) * | 2004-01-06 | 2015-12-21 | Novo Nordisk As | Compounds, combinations comprising said compound and one or more additional active compounds, pharmaceutical composition comprising said compound, as well as the compound, combination and pharmaceutical composition's use in the treatment of disease |
| RU2386622C9 (en) * | 2004-01-06 | 2021-04-21 | Ново Нордиск А/С | Heteroaromatic derivatives of urea and use thereof as glucokinase activators |
| EP2444397A1 (en) * | 2004-01-06 | 2012-04-25 | Novo Nordisk A/S | Heteroaryl-ureas and their use as glucokinase activators |
| CN102516240A (en) * | 2004-01-06 | 2012-06-27 | 诺和诺德公司 | Heteroaryl-ureas and their use as glucokinase activators |
| US7598391B2 (en) | 2004-01-06 | 2009-10-06 | Novo Nordisk A/S | Heteroaryl-ureas and their use as glucokinase activators |
| US10626111B2 (en) | 2004-01-30 | 2020-04-21 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| EP2048137A1 (en) | 2004-02-18 | 2009-04-15 | AstraZeneca AB | Benzamide derivatives and their use as glucokinase activating agents. |
| US7795447B2 (en) | 2004-03-23 | 2010-09-14 | Pfizer Inc | Imidazole compounds for the treatment of neurodegenerative disorders |
| US7951958B2 (en) | 2004-03-23 | 2011-05-31 | Pfizer Inc. | Imidazole compounds for the treatment of neurodegenerative disorders |
| US7342118B2 (en) | 2004-03-23 | 2008-03-11 | Pfizer Inc | Imidazole compounds for the treatment of neurodegenerative disorders |
| US7781451B2 (en) | 2004-04-02 | 2010-08-24 | Novartis Ag | Thiazolopyridine derivatives, pharmaceut ical conditions containing them and methods of treating glucokinase mediated conditions |
| US7750020B2 (en) | 2004-04-02 | 2010-07-06 | Novartis Ag | Sulfonamide-thiazolpyridine derivatives as glucokinase activators useful the treatment of Type 2 diabetes |
| WO2005121110A1 (en) | 2004-06-05 | 2005-12-22 | Astrazeneca Ab | Hetroaryl benzamide derivatives for use as glk activators in the treatment of diabetes |
| EP1992623A1 (en) | 2004-06-05 | 2008-11-19 | Astra Zeneca AB | Heteroaryl benzamide derivative for use as GLK activator in the treatment of diabetes |
| US7999114B2 (en) | 2005-07-08 | 2011-08-16 | Novo Nordisk A/S | Dicycloalkylcarbamoyl ureas as glucokinase activators |
| WO2007006761A1 (en) * | 2005-07-08 | 2007-01-18 | Novo Nordisk A/S | Dicycloalkylcarbamoyl ureas as glucokinase activators |
| US7582769B2 (en) | 2005-07-08 | 2009-09-01 | Novo Nordisk A/S | Dicycloalkyl urea glucokinase activators |
| WO2007006760A1 (en) * | 2005-07-08 | 2007-01-18 | Novo Nordisk A/S | Dicycloalkyl urea glucokinase activators |
| EP2301929A1 (en) | 2005-07-09 | 2011-03-30 | AstraZeneca AB (Publ) | Heteroaryl benzamide derivatives for use as GLK activators in the treatment of diabetes |
| WO2007007041A1 (en) | 2005-07-09 | 2007-01-18 | Astrazeneca Ab | Heteroaryl benzamide derivatives for use as glk activators in the treatment of diabetes |
| EP2305674A1 (en) | 2005-07-09 | 2011-04-06 | AstraZeneca AB | Heteroaryl benzamide derivatives for use as GLK activators in the treatment of diabetes |
| EP2301935A1 (en) | 2005-07-09 | 2011-03-30 | AstraZeneca AB (Publ) | Heteroaryl benzamide derivatives for use as GLK activators in the treatment of diabetes |
| US7884210B2 (en) | 2005-07-14 | 2011-02-08 | Novo Nordisk A/S | Ureido-thiazole glucokinase activators |
| US8586614B2 (en) | 2005-07-14 | 2013-11-19 | Novo Nordisk A/S | Urea glucokinase activators |
| KR101446973B1 (en) | 2005-07-14 | 2014-10-07 | 트랜스테크 파르마 엘엘씨 | Urea glucokinase activators |
| WO2007006814A1 (en) | 2005-07-14 | 2007-01-18 | Novo Nordisk A/S | Urea glucokinase activators |
| EP2377856A1 (en) | 2005-07-14 | 2011-10-19 | Novo Nordisk A/S | Urea glucokinase activators |
| US8124617B2 (en) | 2005-09-01 | 2012-02-28 | Takeda San Diego, Inc. | Imidazopyridine compounds |
| US7781435B2 (en) | 2005-09-22 | 2010-08-24 | Pfizer Inc | Imidazole compounds for the treatment of neurological disorders |
| WO2007039177A2 (en) | 2005-09-29 | 2007-04-12 | Sanofi-Aventis | Phenyl- and pyridinyl- 1, 2 , 4 - oxadiazolone derivatives, processes for their preparation and their use as pharmaceuticals |
| US7795257B2 (en) | 2005-09-30 | 2010-09-14 | Novartis Ag | Organic compounds |
| US8252931B2 (en) | 2005-09-30 | 2012-08-28 | Novartis Ag | Thiazolo[5,4-B]pyridine glucokinase activators |
| WO2007053503A1 (en) * | 2005-11-01 | 2007-05-10 | Janssen Pharmaceutica N.V. | Substituted dihydroisoindolones as allosteric modulators of glucokinase |
| WO2007053657A1 (en) * | 2005-11-01 | 2007-05-10 | Janssen Pharmaceutica N.V. | Substituted pyrrolones as allosteric modulators of glucokinase |
| WO2007053765A3 (en) * | 2005-11-01 | 2007-07-05 | Janssen Pharmaceutica Nv | Substituted cycloalkylpyrrolones as allosteric modulators of glucokinase |
| WO2007053662A1 (en) * | 2005-11-01 | 2007-05-10 | Janssen Pharmaceutica N.V. | Dihydroisoindolones as allosteric modulators of glucokinase |
| US7928246B2 (en) | 2005-11-01 | 2011-04-19 | Janssen Pharmaceutica Nv | Dihydroisoindolones as allosteric modulators of glucokinase |
| US8680122B2 (en) | 2005-11-01 | 2014-03-25 | Janssen Pharmacetica N.V. | Substituted pyrrolones as allosteric modulators of glucokinase |
| US7531671B2 (en) | 2005-11-01 | 2009-05-12 | Janssen Pharmaceutica N.V. | Dihydroisoindolones as allosteric modulators of glucokinase |
| US8796313B2 (en) | 2005-11-01 | 2014-08-05 | Janssen Pharmaceutica N.V. | Substituted dihydroisoindolones as allosteric modulators of glucokinase |
| US11084804B2 (en) | 2005-11-08 | 2021-08-10 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US7973038B2 (en) | 2005-11-08 | 2011-07-05 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US7956052B2 (en) | 2005-11-08 | 2011-06-07 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US9216969B2 (en) | 2005-11-08 | 2015-12-22 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US7659268B2 (en) | 2005-11-08 | 2010-02-09 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US7741321B2 (en) | 2005-11-08 | 2010-06-22 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US7595397B2 (en) | 2005-12-15 | 2009-09-29 | Boehringer Ingelheim International Gmbh | Compounds which modulate the CB2 receptor |
| US8039491B2 (en) | 2005-12-28 | 2011-10-18 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US8034822B2 (en) | 2006-03-08 | 2011-10-11 | Takeda San Diego, Inc. | Glucokinase activators |
| US7645789B2 (en) | 2006-04-07 | 2010-01-12 | Vertex Pharmaceuticals Incorporated | Indole derivatives as CFTR modulators |
| US10987348B2 (en) | 2006-04-07 | 2021-04-27 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US8952049B2 (en) | 2006-04-07 | 2015-02-10 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US8952050B2 (en) | 2006-04-07 | 2015-02-10 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US9758510B2 (en) | 2006-04-07 | 2017-09-12 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US11639347B2 (en) | 2006-04-07 | 2023-05-02 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US9974781B2 (en) | 2006-04-07 | 2018-05-22 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US7776905B2 (en) | 2006-04-07 | 2010-08-17 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US10022352B2 (en) | 2006-04-07 | 2018-07-17 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US8415387B2 (en) | 2006-04-07 | 2013-04-09 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US10239867B2 (en) | 2006-04-07 | 2019-03-26 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US10975061B2 (en) | 2006-04-07 | 2021-04-13 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| EP2351568A2 (en) | 2006-05-04 | 2011-08-03 | Boehringer Ingelheim International GmbH | Uses of dpp-iv inhibitors |
| WO2007128761A2 (en) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Uses of dpp-iv inhibitors |
| US8008332B2 (en) | 2006-05-31 | 2011-08-30 | Takeda San Diego, Inc. | Substituted indazoles as glucokinase activators |
| US8394843B2 (en) | 2006-05-31 | 2013-03-12 | Takeda California, Inc. | Substituted isoindoles as glucokinase activators |
| US7935715B2 (en) | 2006-07-28 | 2011-05-03 | Boehringer Ingelheim International Gmbh | Compounds which modulate the CB2 receptor |
| US8829034B2 (en) | 2006-09-25 | 2014-09-09 | Boehringer Ingerlheim International GmbH | Compounds which modulate the CB2 receptor |
| US7928123B2 (en) | 2006-09-25 | 2011-04-19 | Boehringer Ingelheim International Gmbh | Compounds which modulate the CB2 receptor |
| EP2272834A1 (en) | 2006-10-26 | 2011-01-12 | AstraZeneca AB | Benzoyl amino heterocyclyl compounds as glucokinase (GLK) activators |
| US9732080B2 (en) | 2006-11-03 | 2017-08-15 | Vertex Pharmaceuticals Incorporated | Azaindole derivatives as CFTR modulators |
| US8163779B2 (en) | 2006-12-20 | 2012-04-24 | Takeda San Diego, Inc. | Glucokinase activators |
| WO2008084043A1 (en) * | 2007-01-09 | 2008-07-17 | Novo Nordisk A/S | Urea glucokinase activators |
| WO2008084872A1 (en) | 2007-01-10 | 2008-07-17 | Mitsubishi Tanabe Pharma Corporation | Hydrazone derivative |
| US8314247B2 (en) | 2007-01-10 | 2012-11-20 | Mitsubishi Tanabe Pharma Corporation | Hydrazone derivative |
| WO2008084044A1 (en) | 2007-01-11 | 2008-07-17 | Novo Nordisk A/S | Urea glucokinase activators |
| WO2008104994A2 (en) | 2007-02-28 | 2008-09-04 | Advinus Therapeutics Private Limited | 2,2,2-tri-substituted acetamide derivatives as glucokinase activators, their process and pharmaceutical application |
| US8940900B2 (en) | 2007-02-28 | 2015-01-27 | Advinus Therapeutics Private Limited | 2,2,2-tri-substituted acetamide derivatives as glucokinase activators, their process and pharmaceutical application |
| US8173645B2 (en) | 2007-03-21 | 2012-05-08 | Takeda San Diego, Inc. | Glucokinase activators |
| US7754739B2 (en) | 2007-05-09 | 2010-07-13 | Vertex Pharmaceuticals Incorporated | Modulators of CFTR |
| US9725440B2 (en) | 2007-05-09 | 2017-08-08 | Vertex Pharmaceuticals Incorporated | Modulators of CFTR |
| WO2009047798A3 (en) * | 2007-10-08 | 2009-06-04 | Advinus Therapeutics Private L | Acetamide derivatives as glucokinase activators, their process and medicinal applications |
| JP2010540679A (en) * | 2007-10-08 | 2010-12-24 | アドビヌス・セラピューティクス・プライベート・リミテッド | Acetamide derivatives as glucokinase activators, their preparation and pharmaceutical applications |
| US9340506B2 (en) | 2007-10-08 | 2016-05-17 | Advinus Therapeutics Limited | Acetamide derivatives as glucokinase activators, their process and medicinal applications |
| US8501955B2 (en) | 2007-10-08 | 2013-08-06 | Advinus Therapeutics Private Limited | Acetamide derivatives as glucokinase activators, their process and medicinal application |
| CN101827832B (en) * | 2007-10-08 | 2014-05-07 | 阿德维纳斯治疗私人有限公司 | Acetamide derivatives as glucokinase activators, their process and medicinal applications |
| AU2008310519B2 (en) * | 2007-10-08 | 2013-05-02 | Advinus Therapeutics Private Limited | Acetamide derivatives as glucokinase activators, their process and medicinal applications |
| US8124781B2 (en) | 2007-12-07 | 2012-02-28 | Vertex Pharmaceuticals Incorporated | Processes for producing cycloalkylcarboxamido-pyridine benzoic acids |
| US12065432B2 (en) | 2007-12-07 | 2024-08-20 | Vertex Pharmaceuticals Incorporated | Solid forms of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)cyclopropanecarboxamido)-3-methylpyridin-2-yl) benzoic acid |
| US9840499B2 (en) | 2007-12-07 | 2017-12-12 | Vertex Pharmaceuticals Incorporated | Solid forms of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)cyclopropanecarboxamido)-3-methylpyridin-2-yl) benzoic acid |
| US10597384B2 (en) | 2007-12-07 | 2020-03-24 | Vertex Pharmaceuticals Incorporated | Solid forms of 3-(6-(1-(2,2-difluorobenzo[D][1,3]dioxol-5-yl)cyclopropanecarboxamido)-3-methylpyridin-2-yl) benzoic acid |
| US9776968B2 (en) | 2007-12-07 | 2017-10-03 | Vertex Pharmaceuticals Incorporated | Processes for producing cycloalkylcarboxamido-pyridine benzoic acids |
| US9751890B2 (en) | 2008-02-28 | 2017-09-05 | Vertex Pharmaceuticals Incorporated | Heteroaryl derivatives as CFTR modulators |
| US8563730B2 (en) | 2008-05-16 | 2013-10-22 | Takeda San Diego, Inc. | Pyrazole and fused pyrazole glucokinase activators |
| US9139598B2 (en) | 2008-05-16 | 2015-09-22 | Takeda California, Inc. | Glucokinase activators |
| US8466282B2 (en) | 2008-06-19 | 2013-06-18 | Takeda Pharmaceutical Company Limited | Heterocyclic compound and use thereof |
| US9221836B2 (en) | 2008-06-19 | 2015-12-29 | Takeda Pharmaceutical Company Limited | Heterocyclic compound and use thereof |
| US9045436B2 (en) | 2008-06-19 | 2015-06-02 | Takeda Pharmaceutical Company Limited | Heterocyclic compound and use thereof |
| US8664380B2 (en) | 2008-06-19 | 2014-03-04 | Takeda Pharmaceutical Company Limited | Heterocyclic compound and use thereof |
| US8048899B2 (en) | 2008-09-25 | 2011-11-01 | Boehringer Ingelheim International Gmbh | Compounds which selectively modulate the CB2 receptor |
| WO2010047982A1 (en) | 2008-10-22 | 2010-04-29 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| WO2010051206A1 (en) | 2008-10-31 | 2010-05-06 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| US8987295B2 (en) | 2009-03-30 | 2015-03-24 | Transtech Pharma, Llc | Substituted azoanthracene derivatives, pharmaceutical compositions, and methods of use thereof |
| US9175003B2 (en) | 2009-03-30 | 2015-11-03 | Vtv Therapeutics Llc | Substituted azoanthracene derivatives and intermediates for preparation thereof |
| WO2011008475A1 (en) * | 2009-06-30 | 2011-01-20 | Allergan, Inc. | Optionally substituted 2-(arylmethyl, aryloxy or arylthio) -n- pyridin-2 -yl-aryl acetamide or 2, 2-bis (aryl) -n-pyridin-2-yl acetamide compounds as medicaments for the treatment of eye diseases |
| WO2011009845A1 (en) | 2009-07-23 | 2011-01-27 | F. Hoffmann-La Roche Ag | Pyridone glucokinase activators |
| WO2011080755A1 (en) | 2009-12-29 | 2011-07-07 | Advinus Therapeutics Private Limited | Fused nitrogen heterocyclic compounds, process of preparation and uses thereof |
| US9315454B2 (en) | 2010-01-15 | 2016-04-19 | Boehringer Ingelheim International Gmbh | Compounds which modulate the CB2 receptor |
| WO2011095997A1 (en) | 2010-02-08 | 2011-08-11 | Advinus Therapeutics Private Limited | Benzamide compounds as glucokinase activators and their pharmaceutical application |
| WO2011106273A1 (en) | 2010-02-25 | 2011-09-01 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| US11578062B2 (en) | 2010-03-25 | 2023-02-14 | Vertex Pharmaceuticals Incorporated | Solid forms of (R)-1(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide |
| US10906891B2 (en) | 2010-03-25 | 2021-02-02 | Vertex Pharmaceuticals Incoporated | Solid forms of (R)-1(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide |
| US10081621B2 (en) | 2010-03-25 | 2018-09-25 | Vertex Pharmaceuticals Incorporated | Solid forms of (R)-1(2,2-difluorobenzo[D][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide |
| US10076513B2 (en) | 2010-04-07 | 2018-09-18 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions of 3-(6-(1-(2,2-difluorobenzo[D][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl) benzoic acid and administration thereof |
| US11052075B2 (en) | 2010-04-07 | 2021-07-06 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl) benzoic acid and administration thereof |
| WO2011130459A1 (en) | 2010-04-14 | 2011-10-20 | Bristol-Myers Squibb Company | Novel glucokinase activators and methods of using same |
| US10071979B2 (en) | 2010-04-22 | 2018-09-11 | Vertex Pharmaceuticals Incorporated | Process of producing cycloalkylcarboxamido-indole compounds |
| EP2402327A1 (en) | 2010-06-29 | 2012-01-04 | Advinus Therapeutics Private Limited | Acetamide compounds as glucokinase activators, their process and medicinal applications |
| US8846936B2 (en) | 2010-07-22 | 2014-09-30 | Boehringer Ingelheim International Gmbh | Sulfonyl compounds which modulate the CB2 receptor |
| WO2012051450A1 (en) | 2010-10-13 | 2012-04-19 | Takeda Pharmaceutical Company Limited | Method of making azaindazole derivatives |
| US8809342B2 (en) | 2010-12-23 | 2014-08-19 | Pfizer Inc. | Glucagon receptor modulators |
| US8933104B2 (en) | 2010-12-23 | 2015-01-13 | Pfizer Inc. | Glucagon receptor modulators |
| US9056834B2 (en) | 2010-12-23 | 2015-06-16 | Pfizer Inc. | Glucagon receptor modulators |
| US8859591B2 (en) | 2011-02-08 | 2014-10-14 | Pfizer Inc. | Glucagon receptor modulators |
| US9452999B2 (en) | 2011-02-08 | 2016-09-27 | Pfizer Inc. | Glucagon receptor modulators |
| US9073871B2 (en) | 2011-02-08 | 2015-07-07 | Pfizer Inc. | Glucagon receptor modulators |
| EP3243385A1 (en) | 2011-02-25 | 2017-11-15 | Merck Sharp & Dohme Corp. | Novel cyclic azabenzimidazole derivatives useful as anti-diabetic agents |
| WO2012116145A1 (en) | 2011-02-25 | 2012-08-30 | Merck Sharp & Dohme Corp. | Novel cyclic azabenzimidazole derivatives useful as anti-diabetic agents |
| US8927577B2 (en) | 2011-07-22 | 2015-01-06 | Pfizer Inc. | Quinolinyl glucagon receptor modulators |
| US9139538B2 (en) | 2011-07-22 | 2015-09-22 | Pfizer Inc. | Quinolinyl glucagon receptor modulators |
| US10058546B2 (en) | 2012-07-16 | 2018-08-28 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions of (R)-1-(2,2-difluorobenzo[D][1,3]dioxo1-5-y1)-N-(1-(2,3-dihydroxypropy1)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-y1)-1H-indol-5-y1) cyclopropanecarbox-amide and administration thereof |
| WO2014022528A1 (en) | 2012-08-02 | 2014-02-06 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| WO2014130608A1 (en) | 2013-02-22 | 2014-08-28 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| WO2014139388A1 (en) | 2013-03-14 | 2014-09-18 | Merck Sharp & Dohme Corp. | Novel indole derivatives useful as anti-diabetic agents |
| US10112934B2 (en) | 2013-05-17 | 2018-10-30 | Centrexion Therapeutics Corporation | (Cyano-dimethyl-methyl)-isoxazoles and -[1,3,4]thiadiazoles |
| US8865744B1 (en) | 2013-05-17 | 2014-10-21 | Boehringer Ingelheim International Gmbh | (Cyano-dimethyl-methyl)-isoxazoles and -[1,3,4]thiadiazoles |
| US11084810B2 (en) | 2013-05-17 | 2021-08-10 | Centrexion Therapeutics Corporation | (Cyano-dimethyl-methyl)-isoxazoles and -[1,3,4]thiadiazoles |
| US11725004B2 (en) | 2013-05-17 | 2023-08-15 | Centrexion Therapeutics Corporation | (Cyano-dimethyl-methyl)-isoxazoles and -[1,3,4]thiadiazoles |
| US9650370B2 (en) | 2013-05-17 | 2017-05-16 | Centrexion Therapeutics Corporation | (Cyano-dimethyl-methyl)-isoxazoles and -[1,3,4]thiadiazoles |
| US10570125B2 (en) | 2013-05-17 | 2020-02-25 | Centrexion Therapeutics Corporation | (Cyano-dimethyl-methyl)-isoxazoles and -[1,3,4]thiadiazoles |
| WO2015051725A1 (en) | 2013-10-08 | 2015-04-16 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| US10231932B2 (en) | 2013-11-12 | 2019-03-19 | Vertex Pharmaceuticals Incorporated | Process of preparing pharmaceutical compositions for the treatment of CFTR mediated diseases |
| US10206877B2 (en) | 2014-04-15 | 2019-02-19 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cystic fibrosis transmembrane conductance regulator mediated diseases |
| US10980746B2 (en) | 2014-04-15 | 2021-04-20 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cystic fibrosis transmembrane conductance regulator mediated diseases |
| US11951212B2 (en) | 2014-04-15 | 2024-04-09 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cystic fibrosis transmembrane conductance regulator mediated diseases |
| US10302602B2 (en) | 2014-11-18 | 2019-05-28 | Vertex Pharmaceuticals Incorporated | Process of conducting high throughput testing high performance liquid chromatography |
| WO2018106518A1 (en) | 2016-12-06 | 2018-06-14 | Merck Sharp & Dohme Corp. | Antidiabetic heterocyclic compounds |
| WO2018118670A1 (en) | 2016-12-20 | 2018-06-28 | Merck Sharp & Dohme Corp. | Antidiabetic spirochroman compounds |
| US11690841B2 (en) | 2017-09-14 | 2023-07-04 | Queen Mary University Of London | Glycolysis-activating agents for treatment or prevention of disease |
| US11833136B2 (en) | 2018-06-12 | 2023-12-05 | Vtv Therapeutics Llc | Therapeutic uses of glucokinase activators in combination with insulin or insulin analogs |
| US11974989B2 (en) | 2018-06-12 | 2024-05-07 | Vtv Therapeutics Llc | Therapeutic uses of glucokinase activators in combination with insulin or insulin analogs |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2002351748B2 (en) | 2009-07-09 |
| CN1658871A (en) | 2005-08-24 |
| EP2305648A1 (en) | 2011-04-06 |
| PL370989A1 (en) | 2005-06-13 |
| AU2002351748A1 (en) | 2003-07-15 |
| UA84390C2 (en) | 2008-10-27 |
| HUP0402309A2 (en) | 2005-02-28 |
| RU2374236C2 (en) | 2009-11-27 |
| RU2004122407A (en) | 2005-04-10 |
| HUP0402309A3 (en) | 2008-09-29 |
| NO20043116L (en) | 2004-09-20 |
| MXPA04006048A (en) | 2004-09-27 |
| US20030171411A1 (en) | 2003-09-11 |
| JP2005518391A (en) | 2005-06-23 |
| JP2011006435A (en) | 2011-01-13 |
| BR0215212A (en) | 2004-12-07 |
| EP1458382A1 (en) | 2004-09-22 |
| IL162620A0 (en) | 2005-11-20 |
| CA2471049A1 (en) | 2003-07-10 |
| CN100506807C (en) | 2009-07-01 |
| CZ2004747A3 (en) | 2004-11-10 |
| KR101018318B1 (en) | 2011-03-04 |
| KR20040075900A (en) | 2004-08-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2002351748B2 (en) | Amide derivatives as GK activators | |
| EP1531815B1 (en) | Glucokinase activators | |
| EP1904467B1 (en) | Urea glucokinase activators | |
| RU2340605C2 (en) | Arylcarbonyl derivatives as therapeutic agents | |
| US8148412B2 (en) | Heteroaromatic glucokinase activators | |
| EP1723128B1 (en) | Heteroaryl-ureas and their use as glucokinase activators | |
| EP1336607A1 (en) | Amide derivatives as glucokinase activators | |
| US6881746B2 (en) | Glucagon antagonists/inverse agonists | |
| US7645791B2 (en) | Salicylic anilides | |
| EP1519723A1 (en) | Novel glucagon antagonists/inverse agonists | |
| US7939690B2 (en) | Haloalkylsulfone substituted compounds useful for treating obesity and diabetes | |
| US20090062396A1 (en) | Novel Haloalkoxy-Substituted Salicylic Anilides | |
| US20070004794A1 (en) | Novel salicylic anilides | |
| CN101434585A (en) | Amide derivatives as GK activators | |
| ES2526192T3 (en) | Glucokinase activators | |
| AU2011265421A1 (en) | Urea glucokinase activators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SC SD SE SG SK SL TJ TM TN TR TT TZ UA UG UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR IE IT LU MC NL PT SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2002787463 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200404521 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002351748 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 162620 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2471049 Country of ref document: CA Ref document number: PA/a/2004/006048 Country of ref document: MX Ref document number: 1371/CHENP/2004 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003556060 Country of ref document: JP Ref document number: PV2004-747 Country of ref document: CZ Ref document number: 1020047009841 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004122407 Country of ref document: RU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20028275012 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2002787463 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2004-747 Country of ref document: CZ |