WO2003026652A1 - Lactam-containing compounds and derivatives thereof as factor xa inhibitors - Google Patents
Lactam-containing compounds and derivatives thereof as factor xa inhibitors Download PDFInfo
- Publication number
- WO2003026652A1 WO2003026652A1 PCT/US2002/029491 US0229491W WO03026652A1 WO 2003026652 A1 WO2003026652 A1 WO 2003026652A1 US 0229491 W US0229491 W US 0229491W WO 03026652 A1 WO03026652 A1 WO 03026652A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- substituted
- ring
- oxo
- amino
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 269
- 150000003951 lactams Chemical class 0.000 title abstract description 14
- 229940123583 Factor Xa inhibitor Drugs 0.000 title description 19
- 125000000623 heterocyclic group Chemical group 0.000 claims abstract description 117
- 150000003839 salts Chemical group 0.000 claims abstract description 47
- -1 methylenedioxy Chemical group 0.000 claims description 324
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 268
- 125000005842 heteroatom Chemical group 0.000 claims description 214
- 125000000217 alkyl group Chemical group 0.000 claims description 202
- 229910052757 nitrogen Inorganic materials 0.000 claims description 201
- 125000004432 carbon atom Chemical group C* 0.000 claims description 195
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 166
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 155
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 141
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 131
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 123
- DBTDEFJAFBUGPP-UHFFFAOYSA-N Methanethial Chemical compound S=C DBTDEFJAFBUGPP-UHFFFAOYSA-N 0.000 claims description 109
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 106
- 229910052731 fluorine Inorganic materials 0.000 claims description 91
- 125000004429 atom Chemical group 0.000 claims description 89
- 229910052801 chlorine Inorganic materials 0.000 claims description 89
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 claims description 85
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 claims description 75
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 64
- 229910052717 sulfur Inorganic materials 0.000 claims description 64
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 63
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 claims description 62
- 238000000034 method Methods 0.000 claims description 57
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 claims description 56
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 56
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 52
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 51
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 50
- 229920006395 saturated elastomer Polymers 0.000 claims description 50
- 229910052794 bromium Inorganic materials 0.000 claims description 40
- 208000035475 disorder Diseases 0.000 claims description 39
- 230000009424 thromboembolic effect Effects 0.000 claims description 38
- 239000003814 drug Substances 0.000 claims description 37
- CPRRHERYRRXBRZ-SRVKXCTJSA-N methyl n-[(2s)-1-[[(2s)-1-hydroxy-3-[(3s)-2-oxopyrrolidin-3-yl]propan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]carbamate Chemical compound COC(=O)N[C@@H](CC(C)C)C(=O)N[C@H](CO)C[C@@H]1CCNC1=O CPRRHERYRRXBRZ-SRVKXCTJSA-N 0.000 claims description 37
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 35
- 125000001475 halogen functional group Chemical group 0.000 claims description 34
- 125000004076 pyridyl group Chemical group 0.000 claims description 31
- 230000004927 fusion Effects 0.000 claims description 28
- VDBNYAPERZTOOF-UHFFFAOYSA-N isoquinolin-1(2H)-one Chemical compound C1=CC=C2C(=O)NC=CC2=C1 VDBNYAPERZTOOF-UHFFFAOYSA-N 0.000 claims description 28
- LISFMEBWQUVKPJ-UHFFFAOYSA-N quinolin-2-ol Chemical compound C1=CC=C2NC(=O)C=CC2=C1 LISFMEBWQUVKPJ-UHFFFAOYSA-N 0.000 claims description 28
- DENPQNAWGQXKCU-UHFFFAOYSA-N thiophene-2-carboxamide Chemical compound NC(=O)C1=CC=CS1 DENPQNAWGQXKCU-UHFFFAOYSA-N 0.000 claims description 28
- FFSJPOPLSWBGQY-UHFFFAOYSA-N triazol-4-one Chemical compound O=C1C=NN=N1 FFSJPOPLSWBGQY-UHFFFAOYSA-N 0.000 claims description 28
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 26
- 229910052740 iodine Inorganic materials 0.000 claims description 24
- 125000000043 benzamido group Chemical group [H]N([*])C(=O)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 claims description 23
- XUWHAWMETYGRKB-UHFFFAOYSA-N piperidin-2-one Chemical compound O=C1CCCCN1 XUWHAWMETYGRKB-UHFFFAOYSA-N 0.000 claims description 23
- 125000000242 4-chlorobenzoyl group Chemical group ClC1=CC=C(C(=O)*)C=C1 0.000 claims description 22
- 229910052799 carbon Inorganic materials 0.000 claims description 21
- 125000001624 naphthyl group Chemical group 0.000 claims description 21
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 claims description 20
- 125000002619 bicyclic group Chemical group 0.000 claims description 18
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 18
- 125000001544 thienyl group Chemical group 0.000 claims description 18
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 claims description 17
- 238000004519 manufacturing process Methods 0.000 claims description 17
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 16
- AOJDZKCUAATBGE-UHFFFAOYSA-N bromomethane Chemical compound Br[CH2] AOJDZKCUAATBGE-UHFFFAOYSA-N 0.000 claims description 16
- VUWZPRWSIVNGKG-UHFFFAOYSA-N fluoromethane Chemical compound F[CH2] VUWZPRWSIVNGKG-UHFFFAOYSA-N 0.000 claims description 16
- 239000008194 pharmaceutical composition Substances 0.000 claims description 16
- 125000003545 alkoxy group Chemical group 0.000 claims description 15
- 125000002950 monocyclic group Chemical group 0.000 claims description 15
- 125000003368 amide group Chemical group 0.000 claims description 14
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 13
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 claims description 13
- 229910052701 rubidium Inorganic materials 0.000 claims description 13
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 claims description 12
- 125000004179 3-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(Cl)=C1[H] 0.000 claims description 12
- 125000001072 heteroaryl group Chemical group 0.000 claims description 12
- IBBMAWULFFBRKK-UHFFFAOYSA-N picolinamide Chemical compound NC(=O)C1=CC=CC=N1 IBBMAWULFFBRKK-UHFFFAOYSA-N 0.000 claims description 11
- 125000003373 pyrazinyl group Chemical group 0.000 claims description 11
- 125000002098 pyridazinyl group Chemical group 0.000 claims description 11
- XXJGBENTLXFVFI-UHFFFAOYSA-N 1-amino-methylene Chemical compound N[CH2] XXJGBENTLXFVFI-UHFFFAOYSA-N 0.000 claims description 10
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 claims description 10
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 10
- 208000007536 Thrombosis Diseases 0.000 claims description 10
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 10
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 claims description 10
- 235000005152 nicotinamide Nutrition 0.000 claims description 10
- 239000011570 nicotinamide Substances 0.000 claims description 10
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 9
- UUFQTNFCRMXOAE-UHFFFAOYSA-N 1-methylmethylene Chemical group C[CH] UUFQTNFCRMXOAE-UHFFFAOYSA-N 0.000 claims description 9
- 229910052739 hydrogen Inorganic materials 0.000 claims description 9
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 claims description 9
- 125000002883 imidazolyl group Chemical group 0.000 claims description 9
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 claims description 9
- 125000000842 isoxazolyl group Chemical group 0.000 claims description 9
- 125000002971 oxazolyl group Chemical group 0.000 claims description 9
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 9
- 125000000168 pyrrolyl group Chemical group 0.000 claims description 9
- 125000000335 thiazolyl group Chemical group 0.000 claims description 9
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 claims description 8
- 239000008280 blood Substances 0.000 claims description 8
- 210000004369 blood Anatomy 0.000 claims description 8
- 239000003937 drug carrier Substances 0.000 claims description 8
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 8
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims description 7
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 7
- 125000003342 alkenyl group Chemical group 0.000 claims description 7
- 125000002837 carbocyclic group Chemical group 0.000 claims description 7
- 125000004005 formimidoyl group Chemical group [H]\N=C(/[H])* 0.000 claims description 7
- UBQKCCHYAOITMY-UHFFFAOYSA-N pyridin-2-ol Chemical compound OC1=CC=CC=N1 UBQKCCHYAOITMY-UHFFFAOYSA-N 0.000 claims description 7
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 claims description 6
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 claims description 6
- 206010047249 Venous thrombosis Diseases 0.000 claims description 6
- WBLIXGSTEMXDSM-UHFFFAOYSA-N chloromethane Chemical compound Cl[CH2] WBLIXGSTEMXDSM-UHFFFAOYSA-N 0.000 claims description 6
- VZWXIQHBIQLMPN-UHFFFAOYSA-N chromane Chemical compound C1=CC=C2CCCOC2=C1 VZWXIQHBIQLMPN-UHFFFAOYSA-N 0.000 claims description 6
- 230000002526 effect on cardiovascular system Effects 0.000 claims description 6
- 208000010125 myocardial infarction Diseases 0.000 claims description 6
- RGSFGYAAUTVSQA-UHFFFAOYSA-N pentamethylene Natural products C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 claims description 6
- 125000003386 piperidinyl group Chemical group 0.000 claims description 6
- YMNAJWHTELQUJU-UHFFFAOYSA-N quinoline-6-carboxamide Chemical compound N1=CC=CC2=CC(C(=O)N)=CC=C21 YMNAJWHTELQUJU-UHFFFAOYSA-N 0.000 claims description 6
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 claims description 5
- 125000004502 1,2,3-oxadiazolyl group Chemical group 0.000 claims description 5
- 125000004511 1,2,3-thiadiazolyl group Chemical group 0.000 claims description 5
- 125000001399 1,2,3-triazolyl group Chemical group N1N=NC(=C1)* 0.000 claims description 5
- 125000004504 1,2,4-oxadiazolyl group Chemical group 0.000 claims description 5
- 125000004514 1,2,4-thiadiazolyl group Chemical group 0.000 claims description 5
- 125000001376 1,2,4-triazolyl group Chemical group N1N=C(N=C1)* 0.000 claims description 5
- 125000004506 1,2,5-oxadiazolyl group Chemical group 0.000 claims description 5
- 125000001781 1,3,4-oxadiazolyl group Chemical group 0.000 claims description 5
- 125000004520 1,3,4-thiadiazolyl group Chemical group 0.000 claims description 5
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 claims description 5
- 125000004604 benzisothiazolyl group Chemical group S1N=C(C2=C1C=CC=C2)* 0.000 claims description 5
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 claims description 5
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 claims description 5
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 claims description 5
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 claims description 5
- 125000002541 furyl group Chemical group 0.000 claims description 5
- 125000001041 indolyl group Chemical group 0.000 claims description 5
- 125000005438 isoindazolyl group Chemical group 0.000 claims description 5
- 125000001786 isothiazolyl group Chemical group 0.000 claims description 5
- 125000002757 morpholinyl group Chemical group 0.000 claims description 5
- 229910052760 oxygen Inorganic materials 0.000 claims description 5
- 125000004193 piperazinyl group Chemical group 0.000 claims description 5
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 5
- 238000002560 therapeutic procedure Methods 0.000 claims description 5
- WNBQDDAKLKODPH-UHFFFAOYSA-N 1,2,4-triazolidine Chemical compound C1NCNN1 WNBQDDAKLKODPH-UHFFFAOYSA-N 0.000 claims description 4
- 125000004517 1,2,5-thiadiazolyl group Chemical group 0.000 claims description 4
- KJUGUADJHNHALS-UHFFFAOYSA-N 1H-tetrazole Chemical compound C=1N=NNN=1 KJUGUADJHNHALS-UHFFFAOYSA-N 0.000 claims description 4
- ZYHQGITXIJDDKC-UHFFFAOYSA-N 2-[2-(2-aminophenyl)ethyl]aniline Chemical group NC1=CC=CC=C1CCC1=CC=CC=C1N ZYHQGITXIJDDKC-UHFFFAOYSA-N 0.000 claims description 4
- 125000004182 2-chlorophenyl group Chemical group [H]C1=C([H])C(Cl)=C(*)C([H])=C1[H] 0.000 claims description 4
- ATVJJNGVPSKBGO-UHFFFAOYSA-N 3,4-dihydro-2h-thiopyran Chemical compound C1CSC=CC1 ATVJJNGVPSKBGO-UHFFFAOYSA-N 0.000 claims description 4
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 claims description 4
- NSPMIYGKQJPBQR-UHFFFAOYSA-N 4H-1,2,4-triazole Chemical compound C=1N=CNN=1 NSPMIYGKQJPBQR-UHFFFAOYSA-N 0.000 claims description 4
- 125000001054 5 membered carbocyclic group Chemical group 0.000 claims description 4
- 125000004008 6 membered carbocyclic group Chemical group 0.000 claims description 4
- 125000001960 7 membered carbocyclic group Chemical group 0.000 claims description 4
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 claims description 4
- 201000001320 Atherosclerosis Diseases 0.000 claims description 4
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 4
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 claims description 4
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 claims description 4
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 claims description 4
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 claims description 4
- 125000000304 alkynyl group Chemical group 0.000 claims description 4
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cyclohexene Chemical compound C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 claims description 4
- LPIQUOYDBNQMRZ-UHFFFAOYSA-N cyclopentene Chemical compound C1CC=CC1 LPIQUOYDBNQMRZ-UHFFFAOYSA-N 0.000 claims description 4
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 claims description 4
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical compound O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 claims description 4
- 125000002962 imidazol-1-yl group Chemical group [*]N1C([H])=NC([H])=C1[H] 0.000 claims description 4
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 claims description 4
- AQIAIZBHFAKICS-UHFFFAOYSA-N methylaminomethyl Chemical compound [CH2]NC AQIAIZBHFAKICS-UHFFFAOYSA-N 0.000 claims description 4
- 125000004092 methylthiomethyl group Chemical group [H]C([H])([H])SC([H])([H])* 0.000 claims description 4
- VMWJCFLUSKZZDX-UHFFFAOYSA-N n,n-dimethylmethanamine Chemical compound [CH2]N(C)C VMWJCFLUSKZZDX-UHFFFAOYSA-N 0.000 claims description 4
- 125000003261 o-tolyl group Chemical group [H]C1=C([H])C(*)=C(C([H])=C1[H])C([H])([H])[H] 0.000 claims description 4
- 125000000565 sulfonamide group Chemical group 0.000 claims description 4
- ZYXWYDDFNXBTFO-UHFFFAOYSA-N tetrazolidine Chemical compound C1NNNN1 ZYXWYDDFNXBTFO-UHFFFAOYSA-N 0.000 claims description 4
- IBBLKSWSCDAPIF-UHFFFAOYSA-N thiopyran Chemical compound S1C=CC=C=C1 IBBLKSWSCDAPIF-UHFFFAOYSA-N 0.000 claims description 4
- 125000001425 triazolyl group Chemical group 0.000 claims description 4
- 125000004198 2-fluorophenyl group Chemical group [H]C1=C([H])C(F)=C(*)C([H])=C1[H] 0.000 claims description 3
- 125000004204 2-methoxyphenyl group Chemical group [H]C1=C([H])C(*)=C(OC([H])([H])[H])C([H])=C1[H] 0.000 claims description 3
- 125000004180 3-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(F)=C1[H] 0.000 claims description 3
- 208000004476 Acute Coronary Syndrome Diseases 0.000 claims description 3
- 206010002388 Angina unstable Diseases 0.000 claims description 3
- 200000000007 Arterial disease Diseases 0.000 claims description 3
- 206010003178 Arterial thrombosis Diseases 0.000 claims description 3
- 206010008088 Cerebral artery embolism Diseases 0.000 claims description 3
- 206010008092 Cerebral artery thrombosis Diseases 0.000 claims description 3
- 206010051055 Deep vein thrombosis Diseases 0.000 claims description 3
- 208000005189 Embolism Diseases 0.000 claims description 3
- 206010014513 Embolism arterial Diseases 0.000 claims description 3
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 claims description 3
- 208000010378 Pulmonary Embolism Diseases 0.000 claims description 3
- 206010037437 Pulmonary thrombosis Diseases 0.000 claims description 3
- 208000006011 Stroke Diseases 0.000 claims description 3
- 206010042434 Sudden death Diseases 0.000 claims description 3
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 claims description 3
- 208000032109 Transient ischaemic attack Diseases 0.000 claims description 3
- 208000007814 Unstable Angina Diseases 0.000 claims description 3
- 230000002612 cardiopulmonary effect Effects 0.000 claims description 3
- 238000001631 haemodialysis Methods 0.000 claims description 3
- 125000005843 halogen group Chemical group 0.000 claims description 3
- 230000000322 hemodialysis Effects 0.000 claims description 3
- 239000007943 implant Substances 0.000 claims description 3
- 201000004332 intermediate coronary syndrome Diseases 0.000 claims description 3
- 201000010849 intracranial embolism Diseases 0.000 claims description 3
- 230000000302 ischemic effect Effects 0.000 claims description 3
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 claims description 3
- 210000003734 kidney Anatomy 0.000 claims description 3
- PVNIIMVLHYAWGP-UHFFFAOYSA-M nicotinate Chemical compound [O-]C(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-M 0.000 claims description 3
- WCPAKWJPBJAGKN-UHFFFAOYSA-N oxadiazole Chemical compound C1=CON=N1 WCPAKWJPBJAGKN-UHFFFAOYSA-N 0.000 claims description 3
- 230000002093 peripheral effect Effects 0.000 claims description 3
- 230000000306 recurrent effect Effects 0.000 claims description 3
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 claims description 3
- 201000005060 thrombophlebitis Diseases 0.000 claims description 3
- 201000010875 transient cerebral ischemia Diseases 0.000 claims description 3
- MKVBVTOSWPBSNC-UHFFFAOYSA-N 1,2,3,4-tetrahydro-1,2,4-triazine Chemical compound C1NNC=CN1 MKVBVTOSWPBSNC-UHFFFAOYSA-N 0.000 claims description 2
- OQJVXNHMUWQQEW-UHFFFAOYSA-N 1,2,3,4-tetrahydropyrazine Chemical compound C1CNC=CN1 OQJVXNHMUWQQEW-UHFFFAOYSA-N 0.000 claims description 2
- JQIZHNLEFQMDCQ-UHFFFAOYSA-N 1,2,3,4-tetrahydropyridazine Chemical compound C1CC=CNN1 JQIZHNLEFQMDCQ-UHFFFAOYSA-N 0.000 claims description 2
- OTPDWCMLUKMQNO-UHFFFAOYSA-N 1,2,3,4-tetrahydropyrimidine Chemical compound C1NCC=CN1 OTPDWCMLUKMQNO-UHFFFAOYSA-N 0.000 claims description 2
- OVFJHQBWUUTRFT-UHFFFAOYSA-N 1,2,3,4-tetrahydrotetrazine Chemical compound C1=CNNNN1 OVFJHQBWUUTRFT-UHFFFAOYSA-N 0.000 claims description 2
- FIDRAVVQGKNYQK-UHFFFAOYSA-N 1,2,3,4-tetrahydrotriazine Chemical compound C1NNNC=C1 FIDRAVVQGKNYQK-UHFFFAOYSA-N 0.000 claims description 2
- UGUHFDPGDQDVGX-UHFFFAOYSA-N 1,2,3-thiadiazole Chemical compound C1=CSN=N1 UGUHFDPGDQDVGX-UHFFFAOYSA-N 0.000 claims description 2
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical compound C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 claims description 2
- UUZJJNBYJDFQHL-UHFFFAOYSA-N 1,2,3-triazolidine Chemical compound C1CNNN1 UUZJJNBYJDFQHL-UHFFFAOYSA-N 0.000 claims description 2
- BBVIDBNAYOIXOE-UHFFFAOYSA-N 1,2,4-oxadiazole Chemical compound C=1N=CON=1 BBVIDBNAYOIXOE-UHFFFAOYSA-N 0.000 claims description 2
- BLWJJPNXUDWVCG-UHFFFAOYSA-N 1,2,4-oxadiazolidine Chemical compound C1NCON1 BLWJJPNXUDWVCG-UHFFFAOYSA-N 0.000 claims description 2
- YGTAZGSLCXNBQL-UHFFFAOYSA-N 1,2,4-thiadiazole Chemical compound C=1N=CSN=1 YGTAZGSLCXNBQL-UHFFFAOYSA-N 0.000 claims description 2
- YFCCVJQVYQHMOG-UHFFFAOYSA-N 1,2,4-thiadiazolidine Chemical compound C1NCSN1 YFCCVJQVYQHMOG-UHFFFAOYSA-N 0.000 claims description 2
- FYADHXFMURLYQI-UHFFFAOYSA-N 1,2,4-triazine Chemical compound C1=CN=NC=N1 FYADHXFMURLYQI-UHFFFAOYSA-N 0.000 claims description 2
- USSYLANTAAUELR-UHFFFAOYSA-N 1,2-dihydro-1,2,4-triazine Chemical compound N1NC=NC=C1 USSYLANTAAUELR-UHFFFAOYSA-N 0.000 claims description 2
- QYMGRIFMUQCAJW-UHFFFAOYSA-N 1,2-dihydropyrazine Chemical compound C1NC=CN=C1 QYMGRIFMUQCAJW-UHFFFAOYSA-N 0.000 claims description 2
- BKWQKVJYXODDAC-UHFFFAOYSA-N 1,2-dihydropyridazine Chemical compound N1NC=CC=C1 BKWQKVJYXODDAC-UHFFFAOYSA-N 0.000 claims description 2
- WCFAPJDPAPDDAQ-UHFFFAOYSA-N 1,2-dihydropyrimidine Chemical compound C1NC=CC=N1 WCFAPJDPAPDDAQ-UHFFFAOYSA-N 0.000 claims description 2
- CIISBYKBBMFLEZ-UHFFFAOYSA-N 1,2-oxazolidine Chemical compound C1CNOC1 CIISBYKBBMFLEZ-UHFFFAOYSA-N 0.000 claims description 2
- CZSRXHJVZUBEGW-UHFFFAOYSA-N 1,2-thiazolidine Chemical compound C1CNSC1 CZSRXHJVZUBEGW-UHFFFAOYSA-N 0.000 claims description 2
- FKASFBLJDCHBNZ-UHFFFAOYSA-N 1,3,4-oxadiazole Chemical compound C1=NN=CO1 FKASFBLJDCHBNZ-UHFFFAOYSA-N 0.000 claims description 2
- SKLDBJVFTOQKHE-UHFFFAOYSA-N 1,3,4-oxadiazolidine Chemical compound C1NNCO1 SKLDBJVFTOQKHE-UHFFFAOYSA-N 0.000 claims description 2
- MBIZXFATKUQOOA-UHFFFAOYSA-N 1,3,4-thiadiazole Chemical compound C1=NN=CS1 MBIZXFATKUQOOA-UHFFFAOYSA-N 0.000 claims description 2
- BRQBVEHLEITSPT-UHFFFAOYSA-N 1,3,4-thiadiazolidine Chemical compound C1NNCS1 BRQBVEHLEITSPT-UHFFFAOYSA-N 0.000 claims description 2
- OGYGFUAIIOPWQD-UHFFFAOYSA-N 1,3-thiazolidine Chemical compound C1CSCN1 OGYGFUAIIOPWQD-UHFFFAOYSA-N 0.000 claims description 2
- YNGDWRXWKFWCJY-UHFFFAOYSA-N 1,4-Dihydropyridine Chemical compound C1C=CNC=C1 YNGDWRXWKFWCJY-UHFFFAOYSA-N 0.000 claims description 2
- QXKKDVUEMSAYPG-UHFFFAOYSA-N 1-(4-methoxyphenyl)-6-[4-(2-oxopiperidin-1-yl)phenyl]-3-[2-(pyrrolidin-1-ylmethyl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridin-7-one Chemical compound C1=CC(OC)=CC=C1N1C(C(=O)N(CC2)C=3C=CC(=CC=3)N3C(CCCC3)=O)=C2C(C=2C(=CC=CC=2)CN2CCCC2)=N1 QXKKDVUEMSAYPG-UHFFFAOYSA-N 0.000 claims description 2
- CMUVAVDDRSOCEG-UHFFFAOYSA-N 1-[3-(aminomethyl)-4-fluorophenyl]-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide Chemical compound C1=C(F)C(CN)=CC(N2C=3C(=O)N(CCC=3C(C(N)=O)=N2)C=2C=CC(=CC=2)N2C(CCCC2)=O)=C1 CMUVAVDDRSOCEG-UHFFFAOYSA-N 0.000 claims description 2
- QWENRTYMTSOGBR-UHFFFAOYSA-N 1H-1,2,3-Triazole Chemical compound C=1C=NNN=1 QWENRTYMTSOGBR-UHFFFAOYSA-N 0.000 claims description 2
- OXBLVCZKDOZZOJ-UHFFFAOYSA-N 2,3-Dihydrothiophene Chemical compound C1CC=CS1 OXBLVCZKDOZZOJ-UHFFFAOYSA-N 0.000 claims description 2
- FJRPOHLDJUJARI-UHFFFAOYSA-N 2,3-dihydro-1,2-oxazole Chemical compound C1NOC=C1 FJRPOHLDJUJARI-UHFFFAOYSA-N 0.000 claims description 2
- YTQQIHUQLOZOJI-UHFFFAOYSA-N 2,3-dihydro-1,2-thiazole Chemical compound C1NSC=C1 YTQQIHUQLOZOJI-UHFFFAOYSA-N 0.000 claims description 2
- ZABMHLDQFJHDSC-UHFFFAOYSA-N 2,3-dihydro-1,3-oxazole Chemical compound C1NC=CO1 ZABMHLDQFJHDSC-UHFFFAOYSA-N 0.000 claims description 2
- OYJGEOAXBALSMM-UHFFFAOYSA-N 2,3-dihydro-1,3-thiazole Chemical compound C1NC=CS1 OYJGEOAXBALSMM-UHFFFAOYSA-N 0.000 claims description 2
- LWTIGYSPAXKMDG-UHFFFAOYSA-N 2,3-dihydro-1h-imidazole Chemical compound C1NC=CN1 LWTIGYSPAXKMDG-UHFFFAOYSA-N 0.000 claims description 2
- KEQTWHPMSVAFDA-UHFFFAOYSA-N 2,3-dihydro-1h-pyrazole Chemical compound C1NNC=C1 KEQTWHPMSVAFDA-UHFFFAOYSA-N 0.000 claims description 2
- JKTCBAGSMQIFNL-UHFFFAOYSA-N 2,3-dihydrofuran Chemical compound C1CC=CO1 JKTCBAGSMQIFNL-UHFFFAOYSA-N 0.000 claims description 2
- UYPSMNKRWIJATL-UHFFFAOYSA-N 2-(5-chloropyridin-2-yl)-7-methoxy-3-[4-(2-oxopiperidin-1-yl)phenyl]isoquinolin-1-one Chemical compound C=1C=C(Cl)C=NC=1N1C(=O)C2=CC(OC)=CC=C2C=C1C(C=C1)=CC=C1N1CCCCC1=O UYPSMNKRWIJATL-UHFFFAOYSA-N 0.000 claims description 2
- CTNKBHHERILYRT-UHFFFAOYSA-N 2-[(4-chlorobenzoyl)amino]-n-[4-(2-oxopiperidin-1-yl)phenyl]benzamide Chemical compound C1=CC(Cl)=CC=C1C(=O)NC1=CC=CC=C1C(=O)NC1=CC=C(N2C(CCCC2)=O)C=C1 CTNKBHHERILYRT-UHFFFAOYSA-N 0.000 claims description 2
- CECYSALGBDWNMC-UHFFFAOYSA-N 2-[7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-3-(trifluoromethyl)-4,5-dihydropyrazolo[3,4-c]pyridin-1-yl]benzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=CC=C1N1C(C(=O)N(CC2)C=3C=CC(=CC=3)N3C(CCCC3)=O)=C2C(C(F)(F)F)=N1 CECYSALGBDWNMC-UHFFFAOYSA-N 0.000 claims description 2
- JXLWCABYHOMQHE-UHFFFAOYSA-N 2-methyl-3,7-dihydropurin-6-one Chemical compound N1C(C)=NC(=O)C2=C1N=CN2 JXLWCABYHOMQHE-UHFFFAOYSA-N 0.000 claims description 2
- VSWICNJIUPRZIK-UHFFFAOYSA-N 2-piperideine Chemical compound C1CNC=CC1 VSWICNJIUPRZIK-UHFFFAOYSA-N 0.000 claims description 2
- RSEBUVRVKCANEP-UHFFFAOYSA-N 2-pyrroline Chemical compound C1CC=CN1 RSEBUVRVKCANEP-UHFFFAOYSA-N 0.000 claims description 2
- MGADZUXDNSDTHW-UHFFFAOYSA-N 2H-pyran Chemical compound C1OC=CC=C1 MGADZUXDNSDTHW-UHFFFAOYSA-N 0.000 claims description 2
- GZPHSAQLYPIAIN-UHFFFAOYSA-N 3-pyridinecarbonitrile Chemical compound N#CC1=CC=CN=C1 GZPHSAQLYPIAIN-UHFFFAOYSA-N 0.000 claims description 2
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 claims description 2
- BUDQDWGNQVEFAC-UHFFFAOYSA-N Dihydropyran Chemical compound C1COC=CC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 claims description 2
- WRYCSMQKUKOKBP-UHFFFAOYSA-N Imidazolidine Chemical compound C1CNCN1 WRYCSMQKUKOKBP-UHFFFAOYSA-N 0.000 claims description 2
- WYNCHZVNFNFDNH-UHFFFAOYSA-N Oxazolidine Chemical compound C1COCN1 WYNCHZVNFNFDNH-UHFFFAOYSA-N 0.000 claims description 2
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 claims description 2
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 claims description 2
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 claims description 2
- HXJUTPCZVOIRIF-UHFFFAOYSA-N Tetrahydrothiophene-1,1-dioxide, Natural products O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 claims description 2
- DPOPAJRDYZGTIR-UHFFFAOYSA-N Tetrazine Chemical compound C1=CN=NN=N1 DPOPAJRDYZGTIR-UHFFFAOYSA-N 0.000 claims description 2
- YPWFISCTZQNZAU-UHFFFAOYSA-N Thiane Chemical compound C1CCSCC1 YPWFISCTZQNZAU-UHFFFAOYSA-N 0.000 claims description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 claims description 2
- 125000005037 alkyl phenyl group Chemical group 0.000 claims description 2
- 125000004202 aminomethyl group Chemical group [H]N([H])C([H])([H])* 0.000 claims description 2
- DMEGYFMYUHOHGS-UHFFFAOYSA-N heptamethylene Natural products C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 claims description 2
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical compound C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 claims description 2
- LVWZTYCIRDMTEY-UHFFFAOYSA-N metamizole Chemical compound O=C1C(N(CS(O)(=O)=O)C)=C(C)N(C)N1C1=CC=CC=C1 LVWZTYCIRDMTEY-UHFFFAOYSA-N 0.000 claims description 2
- LWAWERGZGHRDHT-UHFFFAOYSA-N n'-hydroxy-3-[7-oxo-6-[4-(2-oxopyridin-1-yl)phenyl]-3-(trifluoromethyl)-4,5-dihydropyrazolo[3,4-c]pyridin-1-yl]benzenecarboximidamide Chemical compound ONC(=N)C1=CC=CC(N2C=3C(=O)N(CCC=3C(=N2)C(F)(F)F)C=2C=CC(=CC=2)N2C(C=CC=C2)=O)=C1 LWAWERGZGHRDHT-UHFFFAOYSA-N 0.000 claims description 2
- WURVWPLRXOYNOU-UHFFFAOYSA-N n'-methoxy-3-[7-oxo-6-[4-(2-oxopyridin-1-yl)phenyl]-3-(trifluoromethyl)-4,5-dihydropyrazolo[3,4-c]pyridin-1-yl]benzenecarboximidamide Chemical compound CONC(=N)C1=CC=CC(N2C=3C(=O)N(CCC=3C(=N2)C(F)(F)F)C=2C=CC(=CC=2)N2C(C=CC=C2)=O)=C1 WURVWPLRXOYNOU-UHFFFAOYSA-N 0.000 claims description 2
- 235000001968 nicotinic acid Nutrition 0.000 claims description 2
- 239000011664 nicotinic acid Substances 0.000 claims description 2
- DTHHUAXKOMWYBI-UHFFFAOYSA-N oxadiazolidine Chemical compound C1CONN1 DTHHUAXKOMWYBI-UHFFFAOYSA-N 0.000 claims description 2
- USPWKWBDZOARPV-UHFFFAOYSA-N pyrazolidine Chemical compound C1CNNC1 USPWKWBDZOARPV-UHFFFAOYSA-N 0.000 claims description 2
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 claims description 2
- RAOIDOHSFRTOEL-UHFFFAOYSA-N tetrahydrothiophene Chemical compound C1CCSC1 RAOIDOHSFRTOEL-UHFFFAOYSA-N 0.000 claims description 2
- RLTPJVKHGBFGQA-UHFFFAOYSA-N thiadiazolidine Chemical compound C1CSNN1 RLTPJVKHGBFGQA-UHFFFAOYSA-N 0.000 claims description 2
- BUGOPWGPQGYYGR-UHFFFAOYSA-N thiane 1,1-dioxide Chemical compound O=S1(=O)CCCCC1 BUGOPWGPQGYYGR-UHFFFAOYSA-N 0.000 claims description 2
- 229930192474 thiophene Natural products 0.000 claims description 2
- 125000004205 trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 claims description 2
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims description 2
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 claims 2
- RXGHULSMJIVVTA-UHFFFAOYSA-N thieno[2,3-b]pyridine-2-carboxamide Chemical compound C1=CN=C2SC(C(=O)N)=CC2=C1 RXGHULSMJIVVTA-UHFFFAOYSA-N 0.000 claims 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims 1
- GVNVAWHJIKLAGL-UHFFFAOYSA-N 2-(cyclohexen-1-yl)cyclohexan-1-one Chemical compound O=C1CCCCC1C1=CCCCC1 GVNVAWHJIKLAGL-UHFFFAOYSA-N 0.000 claims 1
- 101150065749 Churc1 gene Proteins 0.000 claims 1
- 102100025027 E3 ubiquitin-protein ligase TRIM69 Human genes 0.000 claims 1
- 101000830203 Homo sapiens E3 ubiquitin-protein ligase TRIM69 Proteins 0.000 claims 1
- 102100038239 Protein Churchill Human genes 0.000 claims 1
- 125000000031 ethylamino group Chemical group [H]C([H])([H])C([H])([H])N([H])[*] 0.000 claims 1
- HNQIVZYLYMDVSB-UHFFFAOYSA-N methanesulfonimidic acid Chemical compound CS(N)(=O)=O HNQIVZYLYMDVSB-UHFFFAOYSA-N 0.000 claims 1
- 239000003112 inhibitor Substances 0.000 abstract description 42
- 108010074860 Factor Xa Proteins 0.000 abstract description 29
- 108010036927 trypsin-like serine protease Proteins 0.000 abstract description 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 190
- 238000006243 chemical reaction Methods 0.000 description 101
- 239000000543 intermediate Substances 0.000 description 71
- 235000019439 ethyl acetate Nutrition 0.000 description 70
- 239000000203 mixture Substances 0.000 description 60
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 55
- 239000000460 chlorine Substances 0.000 description 52
- 229940093499 ethyl acetate Drugs 0.000 description 50
- 239000011734 sodium Substances 0.000 description 49
- 239000012267 brine Substances 0.000 description 46
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 46
- 238000005481 NMR spectroscopy Methods 0.000 description 42
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 42
- 239000003795 chemical substances by application Substances 0.000 description 39
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 36
- 239000000243 solution Substances 0.000 description 35
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 34
- 235000002639 sodium chloride Nutrition 0.000 description 34
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 33
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 32
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 32
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 30
- 239000000047 product Substances 0.000 description 27
- 239000007787 solid Substances 0.000 description 25
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 24
- 229940124597 therapeutic agent Drugs 0.000 description 24
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 23
- 238000011282 treatment Methods 0.000 description 21
- 239000004480 active ingredient Substances 0.000 description 19
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 19
- 238000000746 purification Methods 0.000 description 19
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 18
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 18
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 18
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 18
- 229940002612 prodrug Drugs 0.000 description 18
- 239000000651 prodrug Substances 0.000 description 18
- 239000012044 organic layer Substances 0.000 description 17
- 125000001424 substituent group Chemical group 0.000 description 17
- 108090000190 Thrombin Proteins 0.000 description 16
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 15
- 238000004949 mass spectrometry Methods 0.000 description 15
- 238000010898 silica gel chromatography Methods 0.000 description 15
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 14
- 230000005764 inhibitory process Effects 0.000 description 14
- 229960004072 thrombin Drugs 0.000 description 14
- 239000003480 eluent Substances 0.000 description 13
- 230000002401 inhibitory effect Effects 0.000 description 13
- 230000015572 biosynthetic process Effects 0.000 description 12
- 230000000694 effects Effects 0.000 description 12
- 150000002148 esters Chemical group 0.000 description 12
- 239000003146 anticoagulant agent Substances 0.000 description 11
- 201000010099 disease Diseases 0.000 description 11
- 239000010410 layer Substances 0.000 description 11
- 229940086542 triethylamine Drugs 0.000 description 11
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 10
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 10
- 230000008878 coupling Effects 0.000 description 10
- 238000010168 coupling process Methods 0.000 description 10
- 238000005859 coupling reaction Methods 0.000 description 10
- 238000004128 high performance liquid chromatography Methods 0.000 description 10
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 10
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 10
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 10
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 9
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 150000001408 amides Chemical group 0.000 description 9
- 229940127218 antiplatelet drug Drugs 0.000 description 9
- 125000003118 aryl group Chemical group 0.000 description 9
- 238000003556 assay Methods 0.000 description 9
- 229940079593 drug Drugs 0.000 description 9
- 235000019441 ethanol Nutrition 0.000 description 9
- 239000000706 filtrate Substances 0.000 description 9
- 239000007858 starting material Substances 0.000 description 9
- 239000000758 substrate Substances 0.000 description 9
- 238000003786 synthesis reaction Methods 0.000 description 9
- OKRUMSWHDWKGHA-UHFFFAOYSA-N 5-bromopentanoyl chloride Chemical compound ClC(=O)CCCCBr OKRUMSWHDWKGHA-UHFFFAOYSA-N 0.000 description 8
- 239000002253 acid Substances 0.000 description 8
- 150000001412 amines Chemical class 0.000 description 8
- 239000002552 dosage form Substances 0.000 description 8
- 238000010992 reflux Methods 0.000 description 8
- 238000003756 stirring Methods 0.000 description 8
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 7
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 7
- 241000124008 Mammalia Species 0.000 description 7
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 7
- 239000002775 capsule Substances 0.000 description 7
- 239000003527 fibrinolytic agent Substances 0.000 description 7
- 239000000463 material Substances 0.000 description 7
- 239000003921 oil Substances 0.000 description 7
- 235000019198 oils Nutrition 0.000 description 7
- 239000003826 tablet Substances 0.000 description 7
- 238000012360 testing method Methods 0.000 description 7
- 238000005160 1H NMR spectroscopy Methods 0.000 description 6
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 6
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 6
- 239000005552 B01AC04 - Clopidogrel Substances 0.000 description 6
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- 102000004190 Enzymes Human genes 0.000 description 6
- 108090000790 Enzymes Proteins 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- 229940127219 anticoagulant drug Drugs 0.000 description 6
- 239000002585 base Substances 0.000 description 6
- JFDZBHWFFUWGJE-UHFFFAOYSA-N benzonitrile Chemical compound N#CC1=CC=CC=C1 JFDZBHWFFUWGJE-UHFFFAOYSA-N 0.000 description 6
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 6
- GKTWGGQPFAXNFI-HNNXBMFYSA-N clopidogrel Chemical compound C1([C@H](N2CC=3C=CSC=3CC2)C(=O)OC)=CC=CC=C1Cl GKTWGGQPFAXNFI-HNNXBMFYSA-N 0.000 description 6
- 229960003009 clopidogrel Drugs 0.000 description 6
- 229940088598 enzyme Drugs 0.000 description 6
- CGIGDMFJXJATDK-UHFFFAOYSA-N indomethacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 CGIGDMFJXJATDK-UHFFFAOYSA-N 0.000 description 6
- 125000002346 iodo group Chemical group I* 0.000 description 6
- 125000005647 linker group Chemical group 0.000 description 6
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 6
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 6
- 238000000524 positive electrospray ionisation mass spectrometry Methods 0.000 description 6
- 229910000027 potassium carbonate Inorganic materials 0.000 description 6
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 6
- 239000011780 sodium chloride Substances 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 150000003536 tetrazoles Chemical class 0.000 description 6
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 5
- VLVCDUSVTXIWGW-UHFFFAOYSA-N 4-iodoaniline Chemical compound NC1=CC=C(I)C=C1 VLVCDUSVTXIWGW-UHFFFAOYSA-N 0.000 description 5
- 108010058207 Anistreplase Proteins 0.000 description 5
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 5
- 102000007625 Hirudins Human genes 0.000 description 5
- 108010007267 Hirudins Proteins 0.000 description 5
- 229920002472 Starch Polymers 0.000 description 5
- 229940122388 Thrombin inhibitor Drugs 0.000 description 5
- 108090000435 Urokinase-type plasminogen activator Proteins 0.000 description 5
- 102000003990 Urokinase-type plasminogen activator Human genes 0.000 description 5
- 238000002835 absorbance Methods 0.000 description 5
- 229960001138 acetylsalicylic acid Drugs 0.000 description 5
- 150000001350 alkyl halides Chemical class 0.000 description 5
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 5
- 229910000024 caesium carbonate Inorganic materials 0.000 description 5
- 238000004587 chromatography analysis Methods 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 238000004108 freeze drying Methods 0.000 description 5
- 229920000669 heparin Polymers 0.000 description 5
- WQPDUTSPKFMPDP-OUMQNGNKSA-N hirudin Chemical compound C([C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC(OS(O)(=O)=O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H]1NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@@H]2CSSC[C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@H](C(=O)N[C@H](C(NCC(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N2)=O)CSSC1)C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]1NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=2C=CC(O)=CC=2)NC(=O)[C@@H](NC(=O)[C@@H](N)C(C)C)C(C)C)[C@@H](C)O)CSSC1)C(C)C)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 WQPDUTSPKFMPDP-OUMQNGNKSA-N 0.000 description 5
- 229940006607 hirudin Drugs 0.000 description 5
- 230000007062 hydrolysis Effects 0.000 description 5
- 238000006460 hydrolysis reaction Methods 0.000 description 5
- 235000019359 magnesium stearate Nutrition 0.000 description 5
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 5
- 230000004048 modification Effects 0.000 description 5
- 238000012986 modification Methods 0.000 description 5
- 150000002825 nitriles Chemical class 0.000 description 5
- 231100000252 nontoxic Toxicity 0.000 description 5
- 230000003000 nontoxic effect Effects 0.000 description 5
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 5
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 5
- 229920000642 polymer Polymers 0.000 description 5
- 239000011541 reaction mixture Substances 0.000 description 5
- 239000002464 receptor antagonist Substances 0.000 description 5
- 229940044551 receptor antagonist Drugs 0.000 description 5
- 239000008107 starch Substances 0.000 description 5
- 235000019698 starch Nutrition 0.000 description 5
- 239000003868 thrombin inhibitor Substances 0.000 description 5
- 229960000103 thrombolytic agent Drugs 0.000 description 5
- 229960005356 urokinase Drugs 0.000 description 5
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- JZUFKLXOESDKRF-UHFFFAOYSA-N Chlorothiazide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC2=C1NCNS2(=O)=O JZUFKLXOESDKRF-UHFFFAOYSA-N 0.000 description 4
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 4
- 241000282414 Homo sapiens Species 0.000 description 4
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 4
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical class CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 4
- 102000007399 Nuclear hormone receptor Human genes 0.000 description 4
- 108020005497 Nuclear hormone receptor Proteins 0.000 description 4
- 102000012479 Serine Proteases Human genes 0.000 description 4
- 108010022999 Serine Proteases Proteins 0.000 description 4
- RRUDCFGSUDOHDG-UHFFFAOYSA-N acetohydroxamic acid Chemical compound CC(O)=NO RRUDCFGSUDOHDG-UHFFFAOYSA-N 0.000 description 4
- 229960001171 acetohydroxamic acid Drugs 0.000 description 4
- RWZYAGGXGHYGMB-UHFFFAOYSA-N anthranilic acid Chemical class NC1=CC=CC=C1C(O)=O RWZYAGGXGHYGMB-UHFFFAOYSA-N 0.000 description 4
- 229940127090 anticoagulant agent Drugs 0.000 description 4
- 230000023555 blood coagulation Effects 0.000 description 4
- 230000037396 body weight Effects 0.000 description 4
- 238000001035 drying Methods 0.000 description 4
- 230000009977 dual effect Effects 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 210000001035 gastrointestinal tract Anatomy 0.000 description 4
- 239000007903 gelatin capsule Substances 0.000 description 4
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 4
- 239000008101 lactose Substances 0.000 description 4
- 230000001404 mediated effect Effects 0.000 description 4
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- UHZYTMXLRWXGPK-UHFFFAOYSA-N phosphorus pentachloride Chemical compound ClP(Cl)(Cl)(Cl)Cl UHZYTMXLRWXGPK-UHFFFAOYSA-N 0.000 description 4
- 239000004033 plastic Substances 0.000 description 4
- 229920003023 plastic Polymers 0.000 description 4
- 125000006239 protecting group Chemical group 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 4
- 238000013268 sustained release Methods 0.000 description 4
- 239000012730 sustained-release form Substances 0.000 description 4
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 4
- 238000005406 washing Methods 0.000 description 4
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 3
- SGTNSNPWRIOYBX-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-{[2-(3,4-dimethoxyphenyl)ethyl](methyl)amino}-2-(propan-2-yl)pentanenitrile Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC=C(OC)C(OC)=C1 SGTNSNPWRIOYBX-UHFFFAOYSA-N 0.000 description 3
- 125000006306 4-iodophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1I 0.000 description 3
- CVZCYPSLFBHLRM-UHFFFAOYSA-N 5-carbamoyl-2-(3-cyano-4-fluorophenyl)pyrazole-3-carboxylic acid Chemical compound N1=C(C(=O)N)C=C(C(O)=O)N1C1=CC=C(F)C(C#N)=C1 CVZCYPSLFBHLRM-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 239000005528 B01AC05 - Ticlopidine Substances 0.000 description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 3
- 229940127291 Calcium channel antagonist Drugs 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 3
- 102000009123 Fibrin Human genes 0.000 description 3
- 108010073385 Fibrin Proteins 0.000 description 3
- BWGVNKXGVNDBDI-UHFFFAOYSA-N Fibrin monomer Chemical compound CNC(=O)CNC(=O)CN BWGVNKXGVNDBDI-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 3
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 description 3
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 3
- 108090000373 Tissue Plasminogen Activator Proteins 0.000 description 3
- 102000003978 Tissue Plasminogen Activator Human genes 0.000 description 3
- 239000000556 agonist Substances 0.000 description 3
- 229960000983 anistreplase Drugs 0.000 description 3
- 239000005557 antagonist Substances 0.000 description 3
- 239000012298 atmosphere Substances 0.000 description 3
- 239000000480 calcium channel blocker Substances 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 235000012000 cholesterol Nutrition 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000000576 coating method Methods 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- NLUNLVTVUDIHFE-UHFFFAOYSA-N cyclooctylcyclooctane Chemical compound C1CCCCCCC1C1CCCCCCC1 NLUNLVTVUDIHFE-UHFFFAOYSA-N 0.000 description 3
- 229950003499 fibrin Drugs 0.000 description 3
- 239000012065 filter cake Substances 0.000 description 3
- 238000003818 flash chromatography Methods 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 229960002897 heparin Drugs 0.000 description 3
- 239000001257 hydrogen Substances 0.000 description 3
- 229960001680 ibuprofen Drugs 0.000 description 3
- 229960000905 indomethacin Drugs 0.000 description 3
- 125000000468 ketone group Chemical group 0.000 description 3
- 229940127215 low-molecular weight heparin Drugs 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- 229960002009 naproxen Drugs 0.000 description 3
- CMWTZPSULFXXJA-VIFPVBQESA-N naproxen Chemical compound C1=C([C@H](C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-N 0.000 description 3
- 239000012299 nitrogen atmosphere Substances 0.000 description 3
- 239000000123 paper Substances 0.000 description 3
- QYSPLQLAKJAUJT-UHFFFAOYSA-N piroxicam Chemical compound OC=1C2=CC=CC=C2S(=O)(=O)N(C)C=1C(=O)NC1=CC=CC=N1 QYSPLQLAKJAUJT-UHFFFAOYSA-N 0.000 description 3
- 229960002702 piroxicam Drugs 0.000 description 3
- 239000000106 platelet aggregation inhibitor Substances 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- OSFBJERFMQCEQY-UHFFFAOYSA-N propylidene Chemical compound [CH]CC OSFBJERFMQCEQY-UHFFFAOYSA-N 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 230000002441 reversible effect Effects 0.000 description 3
- 239000000741 silica gel Substances 0.000 description 3
- 229910002027 silica gel Inorganic materials 0.000 description 3
- 239000001632 sodium acetate Substances 0.000 description 3
- 235000017281 sodium acetate Nutrition 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 235000000346 sugar Nutrition 0.000 description 3
- 239000011885 synergistic combination Substances 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 230000002537 thrombolytic effect Effects 0.000 description 3
- 229960005001 ticlopidine Drugs 0.000 description 3
- PHWBOXQYWZNQIN-UHFFFAOYSA-N ticlopidine Chemical compound ClC1=CC=CC=C1CN1CC(C=CS2)=C2CC1 PHWBOXQYWZNQIN-UHFFFAOYSA-N 0.000 description 3
- 229960000187 tissue plasminogen activator Drugs 0.000 description 3
- 229960001722 verapamil Drugs 0.000 description 3
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 3
- ZYECEUZMVSGTMR-LBDWYMBGSA-N (4s)-4-[[(2s,3s)-2-benzamido-3-methylpentanoyl]amino]-5-[[2-[[(2s)-5-(diaminomethylideneamino)-1-(4-nitroanilino)-1-oxopentan-2-yl]amino]-2-oxoethyl]amino]-5-oxopentanoic acid Chemical compound N([C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CCCN=C(N)N)C(=O)NC=1C=CC(=CC=1)[N+]([O-])=O)C(=O)C1=CC=CC=C1 ZYECEUZMVSGTMR-LBDWYMBGSA-N 0.000 description 2
- AOEWVBNOFYEGQB-UHFFFAOYSA-N 1-(trifluoromethyl)-5,6-dihydro-4H-pyrazolo[3,4-c]pyridin-7-one Chemical compound FC(F)(F)N1N=CC2=C1C(NCC2)=O AOEWVBNOFYEGQB-UHFFFAOYSA-N 0.000 description 2
- JIVPVXMEBJLZRO-CQSZACIVSA-N 2-chloro-5-[(1r)-1-hydroxy-3-oxo-2h-isoindol-1-yl]benzenesulfonamide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC([C@@]2(O)C3=CC=CC=C3C(=O)N2)=C1 JIVPVXMEBJLZRO-CQSZACIVSA-N 0.000 description 2
- XTWYTFMLZFPYCI-KQYNXXCUSA-N 5'-adenylphosphoric acid Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O XTWYTFMLZFPYCI-KQYNXXCUSA-N 0.000 description 2
- PSWCIARYGITEOY-UHFFFAOYSA-N 6-nitro-1h-indole Chemical compound [O-][N+](=O)C1=CC=C2C=CNC2=C1 PSWCIARYGITEOY-UHFFFAOYSA-N 0.000 description 2
- 239000005541 ACE inhibitor Substances 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- XTWYTFMLZFPYCI-UHFFFAOYSA-N Adenosine diphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(O)=O)C(O)C1O XTWYTFMLZFPYCI-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 229940123208 Biguanide Drugs 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Chemical compound CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- 102000016622 Dipeptidyl Peptidase 4 Human genes 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- 108010056764 Eptifibatide Proteins 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- 101000930822 Giardia intestinalis Dipeptidyl-peptidase 4 Proteins 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 229930194542 Keto Natural products 0.000 description 2
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 2
- CESYKOGBSMNBPD-UHFFFAOYSA-N Methyclothiazide Chemical compound ClC1=C(S(N)(=O)=O)C=C2S(=O)(=O)N(C)C(CCl)NC2=C1 CESYKOGBSMNBPD-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 2
- 102000003729 Neprilysin Human genes 0.000 description 2
- 108090000028 Neprilysin Proteins 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- 108010022233 Plasminogen Activator Inhibitor 1 Proteins 0.000 description 2
- 102100039418 Plasminogen activator inhibitor 1 Human genes 0.000 description 2
- 229920000954 Polyglycolide Polymers 0.000 description 2
- CYLWJCABXYDINA-UHFFFAOYSA-N Polythiazide Polymers ClC1=C(S(N)(=O)=O)C=C2S(=O)(=O)N(C)C(CSCC(F)(F)F)NC2=C1 CYLWJCABXYDINA-UHFFFAOYSA-N 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 2
- 108010023197 Streptokinase Proteins 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- 229960000446 abciximab Drugs 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 2
- 230000000996 additive effect Effects 0.000 description 2
- 230000002776 aggregation Effects 0.000 description 2
- 238000004220 aggregation Methods 0.000 description 2
- 229940083712 aldosterone antagonist Drugs 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- XSDQTOBWRPYKKA-UHFFFAOYSA-N amiloride Chemical compound NC(=N)NC(=O)C1=NC(Cl)=C(N)N=C1N XSDQTOBWRPYKKA-UHFFFAOYSA-N 0.000 description 2
- 229960002576 amiloride Drugs 0.000 description 2
- HTIQEAQVCYTUBX-UHFFFAOYSA-N amlodipine Chemical compound CCOC(=O)C1=C(COCCN)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1Cl HTIQEAQVCYTUBX-UHFFFAOYSA-N 0.000 description 2
- 229960000528 amlodipine Drugs 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 2
- 230000003288 anthiarrhythmic effect Effects 0.000 description 2
- 229940121363 anti-inflammatory agent Drugs 0.000 description 2
- 239000002260 anti-inflammatory agent Substances 0.000 description 2
- 239000000883 anti-obesity agent Substances 0.000 description 2
- 230000003262 anti-osteoporosis Effects 0.000 description 2
- 230000001028 anti-proliverative effect Effects 0.000 description 2
- 230000002785 anti-thrombosis Effects 0.000 description 2
- 230000000767 anti-ulcer Effects 0.000 description 2
- 239000003529 anticholesteremic agent Substances 0.000 description 2
- 239000000935 antidepressant agent Substances 0.000 description 2
- 229940125708 antidiabetic agent Drugs 0.000 description 2
- 239000003472 antidiabetic agent Substances 0.000 description 2
- 229940030600 antihypertensive agent Drugs 0.000 description 2
- 239000002220 antihypertensive agent Substances 0.000 description 2
- 239000003524 antilipemic agent Substances 0.000 description 2
- 229940125710 antiobesity agent Drugs 0.000 description 2
- 239000002249 anxiolytic agent Substances 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 229960003515 bendroflumethiazide Drugs 0.000 description 2
- HDWIHXWEUNVBIY-UHFFFAOYSA-N bendroflumethiazidum Chemical compound C1=C(C(F)(F)F)C(S(=O)(=O)N)=CC(S(N2)(=O)=O)=C1NC2CC1=CC=CC=C1 HDWIHXWEUNVBIY-UHFFFAOYSA-N 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 108010031969 benzoyl-Ile-Glu-Gly-Arg-p-nitroanilide Proteins 0.000 description 2
- NDTSRXAMMQDVSW-UHFFFAOYSA-N benzthiazide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(S(N2)(=O)=O)=C1N=C2CSCC1=CC=CC=C1 NDTSRXAMMQDVSW-UHFFFAOYSA-N 0.000 description 2
- 229960001541 benzthiazide Drugs 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- MAEIEVLCKWDQJH-UHFFFAOYSA-N bumetanide Chemical compound CCCCNC1=CC(C(O)=O)=CC(S(N)(=O)=O)=C1OC1=CC=CC=C1 MAEIEVLCKWDQJH-UHFFFAOYSA-N 0.000 description 2
- 229960004064 bumetanide Drugs 0.000 description 2
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 239000011111 cardboard Substances 0.000 description 2
- 229940097217 cardiac glycoside Drugs 0.000 description 2
- 239000002368 cardiac glycoside Substances 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 235000010980 cellulose Nutrition 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 229960002155 chlorothiazide Drugs 0.000 description 2
- 229960001523 chlortalidone Drugs 0.000 description 2
- 230000015271 coagulation Effects 0.000 description 2
- 238000005345 coagulation Methods 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 239000007891 compressed tablet Substances 0.000 description 2
- 229920001577 copolymer Polymers 0.000 description 2
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000001162 cycloheptenyl group Chemical group C1(=CCCCCC1)* 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 238000006193 diazotization reaction Methods 0.000 description 2
- 229960001259 diclofenac Drugs 0.000 description 2
- DCOPUUMXTXDBNB-UHFFFAOYSA-N diclofenac Chemical compound OC(=O)CC1=CC=CC=C1NC1=C(Cl)C=CC=C1Cl DCOPUUMXTXDBNB-UHFFFAOYSA-N 0.000 description 2
- HSUGRBWQSSZJOP-RTWAWAEBSA-N diltiazem Chemical compound C1=CC(OC)=CC=C1[C@H]1[C@@H](OC(C)=O)C(=O)N(CCN(C)C)C2=CC=CC=C2S1 HSUGRBWQSSZJOP-RTWAWAEBSA-N 0.000 description 2
- 229960004166 diltiazem Drugs 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 229960001850 droxicam Drugs 0.000 description 2
- OEHFRZLKGRKFAS-UHFFFAOYSA-N droxicam Chemical compound C12=CC=CC=C2S(=O)(=O)N(C)C(C2=O)=C1OC(=O)N2C1=CC=CC=N1 OEHFRZLKGRKFAS-UHFFFAOYSA-N 0.000 description 2
- GLGOPUHVAZCPRB-LROMGURASA-N eptifibatide Chemical compound N1C(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CCCCNC(=N)N)NC(=O)CCSSC[C@@H](C(N)=O)NC(=O)[C@@H]2CCCN2C(=O)[C@@H]1CC1=CN=C2[C]1C=CC=C2 GLGOPUHVAZCPRB-LROMGURASA-N 0.000 description 2
- 229960004468 eptifibatide Drugs 0.000 description 2
- 229940011871 estrogen Drugs 0.000 description 2
- 239000000262 estrogen Substances 0.000 description 2
- 229960003199 etacrynic acid Drugs 0.000 description 2
- AVOLMBLBETYQHX-UHFFFAOYSA-N etacrynic acid Chemical compound CCC(=C)C(=O)C1=CC=C(OCC(O)=O)C(Cl)=C1Cl AVOLMBLBETYQHX-UHFFFAOYSA-N 0.000 description 2
- OHLRLMWUFVDREV-UHFFFAOYSA-N ethyl 4-chloro-3-oxobutanoate Chemical compound CCOC(=O)CC(=O)CCl OHLRLMWUFVDREV-UHFFFAOYSA-N 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 229960003028 flumethiazide Drugs 0.000 description 2
- RGUQWGXAYZNLMI-UHFFFAOYSA-N flumethiazide Chemical compound C1=C(C(F)(F)F)C(S(=O)(=O)N)=CC2=C1NC=NS2(=O)=O RGUQWGXAYZNLMI-UHFFFAOYSA-N 0.000 description 2
- 239000006260 foam Substances 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 2
- 229960003883 furosemide Drugs 0.000 description 2
- 208000021302 gastroesophageal reflux disease Diseases 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 229960004580 glibenclamide Drugs 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 239000003292 glue Substances 0.000 description 2
- ZNNLBTZKUZBEKO-UHFFFAOYSA-N glyburide Chemical compound COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZNNLBTZKUZBEKO-UHFFFAOYSA-N 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- 125000001188 haloalkyl group Chemical group 0.000 description 2
- 229910052736 halogen Inorganic materials 0.000 description 2
- 150000002367 halogens Chemical class 0.000 description 2
- 239000008240 homogeneous mixture Substances 0.000 description 2
- 238000002657 hormone replacement therapy Methods 0.000 description 2
- 150000002430 hydrocarbons Chemical group 0.000 description 2
- 229960002003 hydrochlorothiazide Drugs 0.000 description 2
- DMDGGSIALPNSEE-UHFFFAOYSA-N hydroflumethiazide Chemical compound C1=C(C(F)(F)F)C(S(=O)(=O)N)=CC2=C1NCNS2(=O)=O DMDGGSIALPNSEE-UHFFFAOYSA-N 0.000 description 2
- 229960003313 hydroflumethiazide Drugs 0.000 description 2
- 238000005984 hydrogenation reaction Methods 0.000 description 2
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 2
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 2
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 2
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 210000000936 intestine Anatomy 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 2
- 239000003055 low molecular weight heparin Substances 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 229940103185 mefenamate Drugs 0.000 description 2
- HYYBABOKPJLUIN-UHFFFAOYSA-N mefenamic acid Chemical compound CC1=CC=CC(NC=2C(=CC=CC=2)C(O)=O)=C1C HYYBABOKPJLUIN-UHFFFAOYSA-N 0.000 description 2
- 229960002137 melagatran Drugs 0.000 description 2
- DKWNMCUOEDMMIN-PKOBYXMFSA-N melagatran Chemical compound C1=CC(C(=N)N)=CC=C1CNC(=O)[C@H]1N(C(=O)[C@H](NCC(O)=O)C2CCCCC2)CC1 DKWNMCUOEDMMIN-PKOBYXMFSA-N 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- 239000002394 mineralocorticoid antagonist Substances 0.000 description 2
- HYIMSNHJOBLJNT-UHFFFAOYSA-N nifedipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1[N+]([O-])=O HYIMSNHJOBLJNT-UHFFFAOYSA-N 0.000 description 2
- 229960001597 nifedipine Drugs 0.000 description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- BHAAPTBBJKJZER-UHFFFAOYSA-N p-anisidine Chemical compound COC1=CC=C(N)C=C1 BHAAPTBBJKJZER-UHFFFAOYSA-N 0.000 description 2
- LXCFILQKKLGQFO-UHFFFAOYSA-N p-hydroxybenzoic acid methyl ester Natural products COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 2
- 239000003182 parenteral nutrition solution Substances 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 239000003208 petroleum Substances 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 239000002570 phosphodiesterase III inhibitor Substances 0.000 description 2
- 150000003904 phospholipids Chemical class 0.000 description 2
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical compound N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 2
- 229960002797 pitavastatin Drugs 0.000 description 2
- RHGYHLPFVJEAOC-FFNUKLMVSA-L pitavastatin calcium Chemical compound [Ca+2].[O-]C(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1.[O-]C(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1 RHGYHLPFVJEAOC-FFNUKLMVSA-L 0.000 description 2
- 230000010118 platelet activation Effects 0.000 description 2
- 229920000747 poly(lactic acid) Polymers 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 239000004633 polyglycolic acid Substances 0.000 description 2
- 239000004626 polylactic acid Substances 0.000 description 2
- 229960005483 polythiazide Drugs 0.000 description 2
- 229920000046 polythiazide Polymers 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 2
- BNRNXUUZRGQAQC-UHFFFAOYSA-N sildenafil Chemical compound CCCC1=NN(C)C(C(N2)=O)=C1N=C2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(C)CC1 BNRNXUUZRGQAQC-UHFFFAOYSA-N 0.000 description 2
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 2
- 239000004299 sodium benzoate Substances 0.000 description 2
- 235000010234 sodium benzoate Nutrition 0.000 description 2
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 2
- 235000010288 sodium nitrite Nutrition 0.000 description 2
- 239000012064 sodium phosphate buffer Substances 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 235000010356 sorbitol Nutrition 0.000 description 2
- LXMSZDCAJNLERA-ZHYRCANASA-N spironolactone Chemical compound C([C@@H]1[C@]2(C)CC[C@@H]3[C@@]4(C)CCC(=O)C=C4C[C@H]([C@@H]13)SC(=O)C)C[C@@]21CCC(=O)O1 LXMSZDCAJNLERA-ZHYRCANASA-N 0.000 description 2
- 229960002256 spironolactone Drugs 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 229930002534 steroid glycoside Natural products 0.000 description 2
- 150000008143 steroidal glycosides Chemical class 0.000 description 2
- 230000003637 steroidlike Effects 0.000 description 2
- 229960005202 streptokinase Drugs 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- MBGGBVCUIVRRBF-UHFFFAOYSA-N sulfinpyrazone Chemical compound O=C1N(C=2C=CC=CC=2)N(C=2C=CC=CC=2)C(=O)C1CCS(=O)C1=CC=CC=C1 MBGGBVCUIVRRBF-UHFFFAOYSA-N 0.000 description 2
- 229960003329 sulfinpyrazone Drugs 0.000 description 2
- MLKXDPUZXIRXEP-MFOYZWKCSA-N sulindac Chemical compound CC1=C(CC(O)=O)C2=CC(F)=CC=C2\C1=C/C1=CC=C(S(C)=O)C=C1 MLKXDPUZXIRXEP-MFOYZWKCSA-N 0.000 description 2
- 229960000894 sulindac Drugs 0.000 description 2
- 230000002195 synergetic effect Effects 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- JSNLQWAGJJGYOB-UHFFFAOYSA-N tert-butyl 5-amino-2,3-dihydroindole-1-carboxylate Chemical compound NC1=CC=C2N(C(=O)OC(C)(C)C)CCC2=C1 JSNLQWAGJJGYOB-UHFFFAOYSA-N 0.000 description 2
- 238000011287 therapeutic dose Methods 0.000 description 2
- 125000003396 thiol group Chemical group [H]S* 0.000 description 2
- 229960003766 thrombin (human) Drugs 0.000 description 2
- COKMIXFXJJXBQG-NRFANRHFSA-N tirofiban Chemical compound C1=CC(C[C@H](NS(=O)(=O)CCCC)C(O)=O)=CC=C1OCCCCC1CCNCC1 COKMIXFXJJXBQG-NRFANRHFSA-N 0.000 description 2
- 229960003425 tirofiban Drugs 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 238000000844 transformation Methods 0.000 description 2
- LMJSLTNSBFUCMU-UHFFFAOYSA-N trichlormethiazide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC2=C1NC(C(Cl)Cl)NS2(=O)=O LMJSLTNSBFUCMU-UHFFFAOYSA-N 0.000 description 2
- 229960004813 trichlormethiazide Drugs 0.000 description 2
- JLTRXTDYQLMHGR-UHFFFAOYSA-N trimethylaluminium Chemical compound C[Al](C)C JLTRXTDYQLMHGR-UHFFFAOYSA-N 0.000 description 2
- 239000002691 unilamellar liposome Substances 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 229960005080 warfarin Drugs 0.000 description 2
- PJVWKTKQMONHTI-UHFFFAOYSA-N warfarin Chemical compound OC=1C2=CC=CC=C2OC(=O)C=1C(CC(=O)C)C1=CC=CC=C1 PJVWKTKQMONHTI-UHFFFAOYSA-N 0.000 description 2
- 239000008096 xylene Substances 0.000 description 2
- 150000003738 xylenes Chemical class 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- XUFXOAAUWZOOIT-SXARVLRPSA-N (2R,3R,4R,5S,6R)-5-[[(2R,3R,4R,5S,6R)-5-[[(2R,3R,4S,5S,6R)-3,4-dihydroxy-6-methyl-5-[[(1S,4R,5S,6S)-4,5,6-trihydroxy-3-(hydroxymethyl)-1-cyclohex-2-enyl]amino]-2-oxanyl]oxy]-3,4-dihydroxy-6-(hydroxymethyl)-2-oxanyl]oxy]-6-(hydroxymethyl)oxane-2,3,4-triol Chemical compound O([C@H]1O[C@H](CO)[C@H]([C@@H]([C@H]1O)O)O[C@H]1O[C@@H]([C@H]([C@H](O)[C@H]1O)N[C@@H]1[C@@H]([C@@H](O)[C@H](O)C(CO)=C1)O)C)[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O XUFXOAAUWZOOIT-SXARVLRPSA-N 0.000 description 1
- XDIYNQZUNSSENW-UUBOPVPUSA-N (2R,3S,4R,5R)-2,3,4,5,6-pentahydroxyhexanal Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O XDIYNQZUNSSENW-UUBOPVPUSA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- YDMBNDUHUNWWRP-VJBWXMMDSA-N (2s)-1-[(2r)-2-amino-3-phenylpropanoyl]-n-[(2s)-5-(diaminomethylideneamino)-1-(4-nitroanilino)-1-oxopentan-2-yl]piperidine-2-carboxamide Chemical compound C([C@@H](N)C(=O)N1[C@@H](CCCC1)C(=O)N[C@@H](CCCN=C(N)N)C(=O)NC=1C=CC(=CC=1)[N+]([O-])=O)C1=CC=CC=C1 YDMBNDUHUNWWRP-VJBWXMMDSA-N 0.000 description 1
- NVXFXLSOGLFXKQ-JMSVASOKSA-N (2s)-1-[(2r,4r)-5-ethoxy-2,4-dimethyl-5-oxopentanoyl]-2,3-dihydroindole-2-carboxylic acid Chemical compound C1=CC=C2N(C(=O)[C@H](C)C[C@@H](C)C(=O)OCC)[C@H](C(O)=O)CC2=C1 NVXFXLSOGLFXKQ-JMSVASOKSA-N 0.000 description 1
- BIDNLKIUORFRQP-XYGFDPSESA-N (2s,4s)-4-cyclohexyl-1-[2-[[(1s)-2-methyl-1-propanoyloxypropoxy]-(4-phenylbutyl)phosphoryl]acetyl]pyrrolidine-2-carboxylic acid Chemical compound C([P@@](=O)(O[C@H](OC(=O)CC)C(C)C)CC(=O)N1[C@@H](C[C@H](C1)C1CCCCC1)C(O)=O)CCCC1=CC=CC=C1 BIDNLKIUORFRQP-XYGFDPSESA-N 0.000 description 1
- DUORBNTWCCJANU-UHFFFAOYSA-N (3-chloro-4-fluorophenyl)hydrazine Chemical compound NNC1=CC=C(F)C(Cl)=C1 DUORBNTWCCJANU-UHFFFAOYSA-N 0.000 description 1
- ZGGHKIMDNBDHJB-NRFPMOEYSA-M (3R,5S)-fluvastatin sodium Chemical compound [Na+].C12=CC=CC=C2N(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 ZGGHKIMDNBDHJB-NRFPMOEYSA-M 0.000 description 1
- DIWRORZWFLOCLC-HNNXBMFYSA-N (3s)-7-chloro-5-(2-chlorophenyl)-3-hydroxy-1,3-dihydro-1,4-benzodiazepin-2-one Chemical compound N([C@H](C(NC1=CC=C(Cl)C=C11)=O)O)=C1C1=CC=CC=C1Cl DIWRORZWFLOCLC-HNNXBMFYSA-N 0.000 description 1
- FQHCPFMTXFJZJS-UHFFFAOYSA-N (4-methoxyphenyl)hydrazine;hydrochloride Chemical compound Cl.COC1=CC=C(NN)C=C1 FQHCPFMTXFJZJS-UHFFFAOYSA-N 0.000 description 1
- HAKVNWBUOCSHTR-UHFFFAOYSA-N (9,9-dimethylxanthen-1-yl)-diphenylphosphane Chemical compound C=12C(C)(C)C3=CC=CC=C3OC2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 HAKVNWBUOCSHTR-UHFFFAOYSA-N 0.000 description 1
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- VOYADQIFGGIKAT-UHFFFAOYSA-N 1,3-dibutyl-4-hydroxy-2,6-dioxopyrimidine-5-carboximidamide Chemical compound CCCCn1c(O)c(C(N)=N)c(=O)n(CCCC)c1=O VOYADQIFGGIKAT-UHFFFAOYSA-N 0.000 description 1
- KNGYMKLFFAXRDV-UHFFFAOYSA-N 1-(3-chloro-4-fluorophenyl)-6-(4-iodophenyl)-3-(trifluoromethyl)-4,5-dihydropyrazolo[3,4-c]pyridin-7-one Chemical compound C1=C(Cl)C(F)=CC=C1N1C(C(=O)N(CC2)C=3C=CC(I)=CC=3)=C2C(C(F)(F)F)=N1 KNGYMKLFFAXRDV-UHFFFAOYSA-N 0.000 description 1
- ZMLCHORSMMSQJI-UHFFFAOYSA-N 1-(3-chloro-4-fluorophenyl)-6-[4-(2-oxopiperidin-1-yl)phenyl]-3-(trifluoromethyl)-4,5-dihydropyrazolo[3,4-c]pyridin-7-one Chemical compound C1=C(Cl)C(F)=CC=C1N1C(C(=O)N(CC2)C=3C=CC(=CC=3)N3C(CCCC3)=O)=C2C(C(F)(F)F)=N1 ZMLCHORSMMSQJI-UHFFFAOYSA-N 0.000 description 1
- XKWAQQLFBYYFFZ-UHFFFAOYSA-N 1-(3-chlorophenyl)-3-(2-hydroxypropan-2-yl)-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridin-7-one Chemical compound CC(C)(O)C1=NN(C=2C=C(Cl)C=CC=2)C(C2=O)=C1CCN2C(C=C1)=CC=C1N1CCCCC1=O XKWAQQLFBYYFFZ-UHFFFAOYSA-N 0.000 description 1
- QSOJYPHQMZUOOI-UHFFFAOYSA-N 1-(3-chlorophenyl)-3-methylsulfonyl-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridin-7-one Chemical compound CS(=O)(=O)C1=NN(C=2C=C(Cl)C=CC=2)C(C2=O)=C1CCN2C(C=C1)=CC=C1N1CCCCC1=O QSOJYPHQMZUOOI-UHFFFAOYSA-N 0.000 description 1
- OMABWPVFVPSLCB-UHFFFAOYSA-N 1-(3-chlorophenyl)-3-methylsulfonyl-6-[4-(2-oxopyridin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridin-7-one Chemical compound CS(=O)(=O)C1=NN(C=2C=C(Cl)C=CC=2)C(C2=O)=C1CCN2C(C=C1)=CC=C1N1C=CC=CC1=O OMABWPVFVPSLCB-UHFFFAOYSA-N 0.000 description 1
- APJJYTCHBBQKGD-UHFFFAOYSA-N 1-(3-cyano-4-fluorophenyl)-5-[5-(2-oxopiperidin-1-yl)-2,3-dihydroindole-1-carbonyl]pyrazole-3-carboxamide Chemical compound C=1C=C(F)C(C#N)=CC=1N1N=C(C(=O)N)C=C1C(=O)N(C1=CC=2)CCC1=CC=2N1CCCCC1=O APJJYTCHBBQKGD-UHFFFAOYSA-N 0.000 description 1
- HGHLWAZFIGRAOT-UHFFFAOYSA-N 1-(4-hydroxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide Chemical compound NC(=O)C1=NN(C=2C=CC(O)=CC=2)C(C2=O)=C1CCN2C(C=C1)=CC=C1N1CCCCC1=O HGHLWAZFIGRAOT-UHFFFAOYSA-N 0.000 description 1
- BTEBAPINMMQWQC-UHFFFAOYSA-N 1-(4-iodophenyl)-4-(2,2,2-trifluoroacetyl)piperidine-2,3-dione Chemical compound O=C1C(=O)C(C(=O)C(F)(F)F)CCN1C1=CC=C(I)C=C1 BTEBAPINMMQWQC-UHFFFAOYSA-N 0.000 description 1
- UWWMPOYYIGCWEH-UHFFFAOYSA-N 1-(4-iodophenyl)-5-morpholin-4-yl-2,3-dihydropyridin-6-one Chemical compound C1=CC(I)=CC=C1N1C(=O)C(N2CCOCC2)=CCC1 UWWMPOYYIGCWEH-UHFFFAOYSA-N 0.000 description 1
- LLFDSAJZAXQJFF-UHFFFAOYSA-N 1-(4-methoxyphenyl)-3-(4-methyl-1,3-oxazol-2-yl)-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridin-7-one Chemical compound C1=CC(OC)=CC=C1N1C(C(=O)N(CC2)C=3C=CC(=CC=3)N3C(CCCC3)=O)=C2C(C=2OC=C(C)N=2)=N1 LLFDSAJZAXQJFF-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- VOXZDWNPVJITMN-ZBRFXRBCSA-N 17β-estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 VOXZDWNPVJITMN-ZBRFXRBCSA-N 0.000 description 1
- QMQZIXCNLUPEIN-UHFFFAOYSA-N 1h-imidazole-2-carbonitrile Chemical class N#CC1=NC=CN1 QMQZIXCNLUPEIN-UHFFFAOYSA-N 0.000 description 1
- YMJLEPMVGQBLHL-UHFFFAOYSA-N 1h-pyrazole-5-carbonitrile Chemical class N#CC1=CC=NN1 YMJLEPMVGQBLHL-UHFFFAOYSA-N 0.000 description 1
- OIPQMHWGNURKQP-UHFFFAOYSA-N 2-(5-chloropyridin-2-yl)-7-methoxy-3-[4-(2-oxopyridin-1-yl)phenyl]isoquinolin-1-one Chemical compound C=1C=C(Cl)C=NC=1N1C(=O)C2=CC(OC)=CC=C2C=C1C(C=C1)=CC=C1N1C=CC=CC1=O OIPQMHWGNURKQP-UHFFFAOYSA-N 0.000 description 1
- DAXVNEIBNBFTAL-UHFFFAOYSA-N 2-[7-oxo-6-[4-(2-oxopyridin-1-yl)phenyl]-3-(trifluoromethyl)-4,5-dihydropyrazolo[3,4-c]pyridin-1-yl]benzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=CC=C1N1C(C(=O)N(CC2)C=3C=CC(=CC=3)N3C(C=CC=C3)=O)=C2C(C(F)(F)F)=N1 DAXVNEIBNBFTAL-UHFFFAOYSA-N 0.000 description 1
- 125000005273 2-acetoxybenzoic acid group Chemical group 0.000 description 1
- IEMMBWWQXVXBEU-UHFFFAOYSA-N 2-acetylfuran Chemical compound CC(=O)C1=CC=CO1 IEMMBWWQXVXBEU-UHFFFAOYSA-N 0.000 description 1
- YIJGCAUGPCNCCN-UHFFFAOYSA-N 2-fluoro-5-hydrazinylbenzonitrile;hydron;chloride Chemical compound Cl.NNC1=CC=C(F)C(C#N)=C1 YIJGCAUGPCNCCN-UHFFFAOYSA-N 0.000 description 1
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- UVQSQWZYJWNHSU-UHFFFAOYSA-N 2h-triazole-4-carbonitrile Chemical class N#CC1=CNN=N1 UVQSQWZYJWNHSU-UHFFFAOYSA-N 0.000 description 1
- ZPOODPZCZLCUAN-UHFFFAOYSA-N 3,4-dihydro-1h-pyridin-2-one Chemical compound O=C1CCC=CN1 ZPOODPZCZLCUAN-UHFFFAOYSA-N 0.000 description 1
- NJPUGNOZGNELFV-UHFFFAOYSA-N 3-[7-oxo-6-[4-(2-oxopyridin-1-yl)phenyl]-3-(trifluoromethyl)-4,5-dihydropyrazolo[3,4-c]pyridin-1-yl]benzamide Chemical compound NC(=O)C1=CC=CC(N2C=3C(=O)N(CCC=3C(=N2)C(F)(F)F)C=2C=CC(=CC=2)N2C(C=CC=C2)=O)=C1 NJPUGNOZGNELFV-UHFFFAOYSA-N 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- SWLAMJPTOQZTAE-UHFFFAOYSA-N 4-[2-[(5-chloro-2-methoxybenzoyl)amino]ethyl]benzoic acid Chemical class COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(C(O)=O)C=C1 SWLAMJPTOQZTAE-UHFFFAOYSA-N 0.000 description 1
- GAMYYCRTACQSBR-UHFFFAOYSA-N 4-azabenzimidazole Chemical compound C1=CC=C2NC=NC2=N1 GAMYYCRTACQSBR-UHFFFAOYSA-N 0.000 description 1
- YCQHTIDVLROXSL-UHFFFAOYSA-N 4-iodomorpholine Chemical compound IN1CCOCC1 YCQHTIDVLROXSL-UHFFFAOYSA-N 0.000 description 1
- 125000005986 4-piperidonyl group Chemical group 0.000 description 1
- ZMGMDXCADSRNCX-UHFFFAOYSA-N 5,6-dihydroxy-1,3-diazepan-2-one Chemical group OC1CNC(=O)NCC1O ZMGMDXCADSRNCX-UHFFFAOYSA-N 0.000 description 1
- HCEQQASHRRPQFE-UHFFFAOYSA-N 5-chloro-n-[2-[4-(cyclohexylcarbamoylsulfamoyl)phenyl]ethyl]-2-methoxybenzamide;3-(diaminomethylidene)-1,1-dimethylguanidine;hydrochloride Chemical compound Cl.CN(C)C(=N)N=C(N)N.COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 HCEQQASHRRPQFE-UHFFFAOYSA-N 0.000 description 1
- SUBDBMMJDZJVOS-UHFFFAOYSA-N 5-methoxy-2-{[(4-methoxy-3,5-dimethylpyridin-2-yl)methyl]sulfinyl}-1H-benzimidazole Chemical compound N=1C2=CC(OC)=CC=C2NC=1S(=O)CC1=NC=C(C)C(OC)=C1C SUBDBMMJDZJVOS-UHFFFAOYSA-N 0.000 description 1
- HBPVGJGBRWIVSX-UHFFFAOYSA-N 6-bromohexanoyl chloride Chemical compound ClC(=O)CCCCCBr HBPVGJGBRWIVSX-UHFFFAOYSA-N 0.000 description 1
- LPMXVESGRSUGHW-UHFFFAOYSA-N Acolongiflorosid K Natural products OC1C(O)C(O)C(C)OC1OC1CC2(O)CCC3C4(O)CCC(C=5COC(=O)C=5)C4(C)CC(O)C3C2(CO)C(O)C1 LPMXVESGRSUGHW-UHFFFAOYSA-N 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- OGSPWJRAVKPPFI-UHFFFAOYSA-N Alendronic Acid Chemical compound NCCCC(O)(P(O)(O)=O)P(O)(O)=O OGSPWJRAVKPPFI-UHFFFAOYSA-N 0.000 description 1
- 102100035991 Alpha-2-antiplasmin Human genes 0.000 description 1
- ITPDYQOUSLNIHG-UHFFFAOYSA-N Amiodarone hydrochloride Chemical compound [Cl-].CCCCC=1OC2=CC=CC=C2C=1C(=O)C1=CC(I)=C(OCC[NH+](CC)CC)C(I)=C1 ITPDYQOUSLNIHG-UHFFFAOYSA-N 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 241000030634 Anthene Species 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 description 1
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 description 1
- MREBEPTUUMTTIA-PCLIKHOPSA-N Azimilide Chemical compound C1CN(C)CCN1CCCCN1C(=O)N(\N=C\C=2OC(=CC=2)C=2C=CC(Cl)=CC=2)CC1=O MREBEPTUUMTTIA-PCLIKHOPSA-N 0.000 description 1
- GUBGYTABKSRVRQ-DCSYEGIMSA-N Beta-Lactose Chemical compound OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-DCSYEGIMSA-N 0.000 description 1
- XNCOSPRUTUOJCJ-UHFFFAOYSA-N Biguanide Chemical compound NC(N)=NC(N)=N XNCOSPRUTUOJCJ-UHFFFAOYSA-N 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- LRHPLDYGYMQRHN-UHFFFAOYSA-N Butanol Natural products CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 1
- 239000002083 C09CA01 - Losartan Substances 0.000 description 1
- 239000004072 C09CA03 - Valsartan Substances 0.000 description 1
- 239000002947 C09CA04 - Irbesartan Substances 0.000 description 1
- YDNKGFDKKRUKPY-JHOUSYSJSA-N C16 ceramide Natural products CCCCCCCCCCCCCCCC(=O)N[C@@H](CO)[C@H](O)C=CCCCCCCCCCCCCC YDNKGFDKKRUKPY-JHOUSYSJSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 108091006146 Channels Proteins 0.000 description 1
- 229920001268 Cholestyramine Polymers 0.000 description 1
- 206010070954 Congenital hypercoagulation Diseases 0.000 description 1
- 229930105110 Cyclosporin A Natural products 0.000 description 1
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 1
- 108010036949 Cyclosporine Proteins 0.000 description 1
- 102000002004 Cytochrome P-450 Enzyme System Human genes 0.000 description 1
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- 240000001879 Digitalis lutea Species 0.000 description 1
- 108010061435 Enalapril Proteins 0.000 description 1
- 108010008165 Etanercept Proteins 0.000 description 1
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 1
- 108010048049 Factor IXa Proteins 0.000 description 1
- 108010014172 Factor V Proteins 0.000 description 1
- 108010080805 Factor XIa Proteins 0.000 description 1
- 102000030914 Fatty Acid-Binding Human genes 0.000 description 1
- 108010088842 Fibrinolysin Proteins 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- FAEKWTJYAYMJKF-QHCPKHFHSA-N GlucoNorm Chemical compound C1=C(C(O)=O)C(OCC)=CC(CC(=O)N[C@@H](CC(C)C)C=2C(=CC=CC=2)N2CCCCC2)=C1 FAEKWTJYAYMJKF-QHCPKHFHSA-N 0.000 description 1
- 102000004366 Glucosidases Human genes 0.000 description 1
- 108010056771 Glucosidases Proteins 0.000 description 1
- 108010051696 Growth Hormone Proteins 0.000 description 1
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 1
- ALOBUEHUHMBRLE-UHFFFAOYSA-N Ibutilide Chemical compound CCCCCCCN(CC)CCCC(O)C1=CC=C(NS(C)(=O)=O)C=C1 ALOBUEHUHMBRLE-UHFFFAOYSA-N 0.000 description 1
- CZGUSIXMZVURDU-JZXHSEFVSA-N Ile(5)-angiotensin II Chemical class C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC=1C=CC=CC=1)C([O-])=O)NC(=O)[C@@H](NC(=O)[C@H](CCCNC(N)=[NH2+])NC(=O)[C@@H]([NH3+])CC([O-])=O)C(C)C)C1=CC=C(O)C=C1 CZGUSIXMZVURDU-JZXHSEFVSA-N 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 229940122199 Insulin secretagogue Drugs 0.000 description 1
- 229940122355 Insulin sensitizer Drugs 0.000 description 1
- YQEZLKZALYSWHR-UHFFFAOYSA-N Ketamine Chemical compound C=1C=CC=C(Cl)C=1C1(NC)CCCCC1=O YQEZLKZALYSWHR-UHFFFAOYSA-N 0.000 description 1
- QUOGESRFPZDMMT-UHFFFAOYSA-N L-Homoarginine Natural products OC(=O)C(N)CCCCNC(N)=N QUOGESRFPZDMMT-UHFFFAOYSA-N 0.000 description 1
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- QUOGESRFPZDMMT-YFKPBYRVSA-N L-homoarginine Chemical compound OC(=O)[C@@H](N)CCCCNC(N)=N QUOGESRFPZDMMT-YFKPBYRVSA-N 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 239000000867 Lipoxygenase Inhibitor Substances 0.000 description 1
- 108010007859 Lisinopril Proteins 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 1
- 125000003047 N-acetyl group Chemical class 0.000 description 1
- CRJGESKKUOMBCT-VQTJNVASSA-N N-acetylsphinganine Chemical compound CCCCCCCCCCCCCCC[C@@H](O)[C@H](CO)NC(C)=O CRJGESKKUOMBCT-VQTJNVASSA-N 0.000 description 1
- PHSPJQZRQAJPPF-UHFFFAOYSA-N N-alpha-Methylhistamine Chemical compound CNCCC1=CN=CN1 PHSPJQZRQAJPPF-UHFFFAOYSA-N 0.000 description 1
- 150000001204 N-oxides Chemical class 0.000 description 1
- 229910020889 NaBH3 Inorganic materials 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- MHABMANUFPZXEB-UHFFFAOYSA-N O-demethyl-aloesaponarin I Natural products O=C1C2=CC=CC(O)=C2C(=O)C2=C1C=C(O)C(C(O)=O)=C2C MHABMANUFPZXEB-UHFFFAOYSA-N 0.000 description 1
- REYJJPSVUYRZGE-UHFFFAOYSA-N Octadecylamine Chemical compound CCCCCCCCCCCCCCCCCCN REYJJPSVUYRZGE-UHFFFAOYSA-N 0.000 description 1
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 1
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 1
- LPMXVESGRSUGHW-GHYGWZAOSA-N Ouabain Natural products O([C@@H]1[C@@H](O)[C@@H](O)[C@@H](O)[C@H](C)O1)[C@H]1C[C@@H](O)[C@@]2(CO)[C@@](O)(C1)CC[C@H]1[C@]3(O)[C@@](C)([C@H](C4=CC(=O)OC4)CC3)C[C@@H](O)[C@H]21 LPMXVESGRSUGHW-GHYGWZAOSA-N 0.000 description 1
- 239000008118 PEG 6000 Substances 0.000 description 1
- 108010028924 PPAR alpha Proteins 0.000 description 1
- 102000023984 PPAR alpha Human genes 0.000 description 1
- 108010016731 PPAR gamma Proteins 0.000 description 1
- 102000000536 PPAR gamma Human genes 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 102100038831 Peroxisome proliferator-activated receptor alpha Human genes 0.000 description 1
- 102000004861 Phosphoric Diester Hydrolases Human genes 0.000 description 1
- 108090001050 Phosphoric Diester Hydrolases Proteins 0.000 description 1
- 108090000113 Plasma Kallikrein Proteins 0.000 description 1
- 102100034869 Plasma kallikrein Human genes 0.000 description 1
- 229920002584 Polyethylene Glycol 6000 Polymers 0.000 description 1
- 229920002594 Polyethylene Glycol 8000 Polymers 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 108010039918 Polylysine Proteins 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 description 1
- 102100038277 Prostaglandin G/H synthase 1 Human genes 0.000 description 1
- 108050003243 Prostaglandin G/H synthase 1 Proteins 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- 108010094028 Prothrombin Proteins 0.000 description 1
- 102100027378 Prothrombin Human genes 0.000 description 1
- 102000000033 Purinergic Receptors Human genes 0.000 description 1
- 108010080192 Purinergic Receptors Proteins 0.000 description 1
- 206010038563 Reocclusion Diseases 0.000 description 1
- 108010039286 S 2238 Proteins 0.000 description 1
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 1
- 206010059054 Shunt thrombosis Diseases 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- BCKXLBQYZLBQEK-KVVVOXFISA-M Sodium oleate Chemical compound [Na+].CCCCCCCC\C=C/CCCCCCCC([O-])=O BCKXLBQYZLBQEK-KVVVOXFISA-M 0.000 description 1
- 229940123518 Sodium/glucose cotransporter 2 inhibitor Drugs 0.000 description 1
- 102100038803 Somatotropin Human genes 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 244000166550 Strophanthus gratus Species 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 229940100389 Sulfonylurea Drugs 0.000 description 1
- 108010039185 Tenecteplase Proteins 0.000 description 1
- 102000003938 Thromboxane Receptors Human genes 0.000 description 1
- 108090000300 Thromboxane Receptors Proteins 0.000 description 1
- 108010069102 Thromboxane-A synthase Proteins 0.000 description 1
- 229940127428 Tissue Plasminogen Activator Inhibitors Drugs 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- GCTFWCDSFPMHHS-UHFFFAOYSA-M Tributyltin chloride Chemical compound CCCC[Sn](Cl)(CCCC)CCCC GCTFWCDSFPMHHS-UHFFFAOYSA-M 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- BBAWTPDTGRXPDG-UHFFFAOYSA-N [1,3]thiazolo[4,5-b]pyridine Chemical compound C1=CC=C2SC=NC2=N1 BBAWTPDTGRXPDG-UHFFFAOYSA-N 0.000 description 1
- CKUAXEQHGKSLHN-UHFFFAOYSA-N [C].[N] Chemical group [C].[N] CKUAXEQHGKSLHN-UHFFFAOYSA-N 0.000 description 1
- 229960002632 acarbose Drugs 0.000 description 1
- XUFXOAAUWZOOIT-UHFFFAOYSA-N acarviostatin I01 Natural products OC1C(O)C(NC2C(C(O)C(O)C(CO)=C2)O)C(C)OC1OC(C(C1O)O)C(CO)OC1OC1C(CO)OC(O)C(O)C1O XUFXOAAUWZOOIT-UHFFFAOYSA-N 0.000 description 1
- 229960000583 acetic acid Drugs 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 229940009456 adriamycin Drugs 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 150000001299 aldehydes Chemical group 0.000 description 1
- 229940062527 alendronate Drugs 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 239000002160 alpha blocker Substances 0.000 description 1
- 108090000183 alpha-2-Antiplasmin Proteins 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- IJQDXWNHUCEMBA-UHFFFAOYSA-N aminosulfanyl hydrogen sulfate Chemical group NSOS(O)(=O)=O IJQDXWNHUCEMBA-UHFFFAOYSA-N 0.000 description 1
- 229960005260 amiodarone Drugs 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 239000002333 angiotensin II receptor antagonist Substances 0.000 description 1
- 125000002490 anilino group Chemical group [H]N(*)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000002429 anti-coagulating effect Effects 0.000 description 1
- 230000001430 anti-depressive effect Effects 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000003064 anti-oxidating effect Effects 0.000 description 1
- 230000000702 anti-platelet effect Effects 0.000 description 1
- 229940005513 antidepressants Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 229960005475 antiinfective agent Drugs 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 239000004019 antithrombin Substances 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- KXNPVXPOPUZYGB-XYVMCAHJSA-N argatroban Chemical compound OC(=O)[C@H]1C[C@H](C)CCN1C(=O)[C@H](CCCN=C(N)N)NS(=O)(=O)C1=CC=CC2=C1NC[C@H](C)C2 KXNPVXPOPUZYGB-XYVMCAHJSA-N 0.000 description 1
- 229960003856 argatroban Drugs 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 150000004982 aromatic amines Chemical class 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 229960005370 atorvastatin Drugs 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- 229950001786 azimilide Drugs 0.000 description 1
- 125000004931 azocinyl group Chemical group N1=C(C=CC=CC=C1)* 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- 235000012216 bentonite Nutrition 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- 229940054066 benzamide antipsychotics Drugs 0.000 description 1
- 150000003936 benzamides Chemical class 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004935 benzoxazolinyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000005512 benztetrazolyl group Chemical group 0.000 description 1
- BAHFPJFBMJTOPU-UHFFFAOYSA-N benzyl 3-oxopiperazine-1-carboxylate Chemical compound C1CNC(=O)CN1C(=O)OCC1=CC=CC=C1 BAHFPJFBMJTOPU-UHFFFAOYSA-N 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- 239000002876 beta blocker Substances 0.000 description 1
- 150000004283 biguanides Chemical class 0.000 description 1
- 229920000080 bile acid sequestrant Polymers 0.000 description 1
- 229940096699 bile acid sequestrants Drugs 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- 229920001400 block copolymer Polymers 0.000 description 1
- 150000001642 boronic acid derivatives Chemical class 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 229940045348 brown mixture Drugs 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- QWCRAEMEVRGPNT-UHFFFAOYSA-N buspirone Chemical compound C1C(=O)N(CCCCN2CCN(CC2)C=2N=CC=CN=2)C(=O)CC21CCCC2 QWCRAEMEVRGPNT-UHFFFAOYSA-N 0.000 description 1
- 229960002495 buspirone Drugs 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 235000011132 calcium sulphate Nutrition 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 125000004623 carbolinyl group Chemical group 0.000 description 1
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 229940105329 carboxymethylcellulose Drugs 0.000 description 1
- 206010061592 cardiac fibrillation Diseases 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 229960000590 celecoxib Drugs 0.000 description 1
- RZEKVGVHFLEQIL-UHFFFAOYSA-N celecoxib Chemical compound C1=CC(C)=CC=C1C1=CC(C(F)(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 RZEKVGVHFLEQIL-UHFFFAOYSA-N 0.000 description 1
- 229940106189 ceramide Drugs 0.000 description 1
- ZVEQCJWYRWKARO-UHFFFAOYSA-N ceramide Natural products CCCCCCCCCCCCCCC(O)C(=O)NC(CO)C(O)C=CCCC=C(C)CCCCCCCCC ZVEQCJWYRWKARO-UHFFFAOYSA-N 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- HGAZMNJKRQFZKS-UHFFFAOYSA-N chloroethene;ethenyl acetate Chemical compound ClC=C.CC(=O)OC=C HGAZMNJKRQFZKS-UHFFFAOYSA-N 0.000 description 1
- 239000003354 cholesterol ester transfer protein inhibitor Substances 0.000 description 1
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 1
- 125000004230 chromenyl group Chemical group O1C(C=CC2=CC=CC=C12)* 0.000 description 1
- 229960001265 ciclosporin Drugs 0.000 description 1
- RRGUKTPIGVIEKM-UHFFFAOYSA-N cilostazol Chemical compound C=1C=C2NC(=O)CCC2=CC=1OCCCCC1=NN=NN1C1CCCCC1 RRGUKTPIGVIEKM-UHFFFAOYSA-N 0.000 description 1
- 229960004588 cilostazol Drugs 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 239000013066 combination product Substances 0.000 description 1
- 229940127555 combination product Drugs 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 238000007887 coronary angioplasty Methods 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 229940111134 coxibs Drugs 0.000 description 1
- 239000013058 crude material Substances 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 150000005245 cyanopyrroles Chemical class 0.000 description 1
- 150000003950 cyclic amides Chemical class 0.000 description 1
- 125000001047 cyclobutenyl group Chemical group C1(=CCC1)* 0.000 description 1
- XSYZCZPCBXYQTE-UHFFFAOYSA-N cyclodecylcyclodecane Chemical compound C1CCCCCCCCC1C1CCCCCCCCC1 XSYZCZPCBXYQTE-UHFFFAOYSA-N 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- FWFSEYBSWVRWGL-UHFFFAOYSA-N cyclohex-2-enone Chemical compound O=C1CCCC=C1 FWFSEYBSWVRWGL-UHFFFAOYSA-N 0.000 description 1
- SSJXIUAHEKJCMH-UHFFFAOYSA-N cyclohexane-1,2-diamine Chemical compound NC1CCCCC1N SSJXIUAHEKJCMH-UHFFFAOYSA-N 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000522 cyclooctenyl group Chemical group C1(=CCCCCCC1)* 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 239000003260 cyclooxygenase 1 inhibitor Substances 0.000 description 1
- 239000003255 cyclooxygenase 2 inhibitor Substances 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 229960004969 dalteparin Drugs 0.000 description 1
- 125000004856 decahydroquinolinyl group Chemical group N1(CCCC2CCCCC12)* 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 229960005227 delapril Drugs 0.000 description 1
- WOUOLAUOZXOLJQ-MBSDFSHPSA-N delapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N(CC(O)=O)C1CC2=CC=CC=C2C1)CC1=CC=CC=C1 WOUOLAUOZXOLJQ-MBSDFSHPSA-N 0.000 description 1
- 239000007933 dermal patch Substances 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 229940039227 diagnostic agent Drugs 0.000 description 1
- 239000000032 diagnostic agent Substances 0.000 description 1
- AAOVKJBEBIDNHE-UHFFFAOYSA-N diazepam Chemical compound N=1CC(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 AAOVKJBEBIDNHE-UHFFFAOYSA-N 0.000 description 1
- 229960003529 diazepam Drugs 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- ZHXTWWCDMUWMDI-UHFFFAOYSA-N dihydroxyboron Chemical compound O[B]O ZHXTWWCDMUWMDI-UHFFFAOYSA-N 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 229960002768 dipyridamole Drugs 0.000 description 1
- IZEKFCXSFNUWAM-UHFFFAOYSA-N dipyridamole Chemical compound C=12N=C(N(CCO)CCO)N=C(N3CCCCC3)C2=NC(N(CCO)CCO)=NC=1N1CCCCC1 IZEKFCXSFNUWAM-UHFFFAOYSA-N 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- CIYGMWIAXRMHQS-UHFFFAOYSA-N ditert-butyl oxalate Chemical compound CC(C)(C)OC(=O)C(=O)OC(C)(C)C CIYGMWIAXRMHQS-UHFFFAOYSA-N 0.000 description 1
- IXTMWRCNAAVVAI-UHFFFAOYSA-N dofetilide Chemical compound C=1C=C(NS(C)(=O)=O)C=CC=1CCN(C)CCOC1=CC=C(NS(C)(=O)=O)C=C1 IXTMWRCNAAVVAI-UHFFFAOYSA-N 0.000 description 1
- 229960002994 dofetilide Drugs 0.000 description 1
- 230000008406 drug-drug interaction Effects 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 229960000873 enalapril Drugs 0.000 description 1
- GBXSMTUPTTWBMN-XIRDDKMYSA-N enalapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)CC1=CC=CC=C1 GBXSMTUPTTWBMN-XIRDDKMYSA-N 0.000 description 1
- 229940073621 enbrel Drugs 0.000 description 1
- 229960000610 enoxaparin Drugs 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 229960005309 estradiol Drugs 0.000 description 1
- 229930182833 estradiol Natural products 0.000 description 1
- ZCRZCMUDOWDGOB-UHFFFAOYSA-N ethanesulfonimidic acid Chemical compound CCS(N)(=O)=O ZCRZCMUDOWDGOB-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 229960000301 factor viii Drugs 0.000 description 1
- XUFQPHANEAPEMJ-UHFFFAOYSA-N famotidine Chemical compound NC(N)=NC1=NC(CSCCC(N)=NS(N)(=O)=O)=CS1 XUFQPHANEAPEMJ-UHFFFAOYSA-N 0.000 description 1
- 229960001596 famotidine Drugs 0.000 description 1
- 108091022862 fatty acid binding Proteins 0.000 description 1
- 210000001105 femoral artery Anatomy 0.000 description 1
- 210000003191 femoral vein Anatomy 0.000 description 1
- 229940125753 fibrate Drugs 0.000 description 1
- 230000002600 fibrillogenic effect Effects 0.000 description 1
- 230000003480 fibrinolytic effect Effects 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 229960003765 fluvastatin Drugs 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 229960002490 fosinopril Drugs 0.000 description 1
- 125000003838 furazanyl group Chemical group 0.000 description 1
- YRSVDSQRGBYVIY-GJZGRUSLSA-N gemopatrilat Chemical compound O=C1N(CC(O)=O)C(C)(C)CCC[C@@H]1NC(=O)[C@@H](S)CC1=CC=CC=C1 YRSVDSQRGBYVIY-GJZGRUSLSA-N 0.000 description 1
- 229950006480 gemopatrilat Drugs 0.000 description 1
- WIGIZIANZCJQQY-RUCARUNLSA-N glimepiride Chemical compound O=C1C(CC)=C(C)CN1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)N[C@@H]2CC[C@@H](C)CC2)C=C1 WIGIZIANZCJQQY-RUCARUNLSA-N 0.000 description 1
- 229960004346 glimepiride Drugs 0.000 description 1
- ZJJXGWJIGJFDTL-UHFFFAOYSA-N glipizide Chemical compound C1=NC(C)=CN=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZJJXGWJIGJFDTL-UHFFFAOYSA-N 0.000 description 1
- 229960001381 glipizide Drugs 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- 229940112611 glucovance Drugs 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 239000000122 growth hormone Substances 0.000 description 1
- 239000003324 growth hormone secretagogue Substances 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 125000005343 heterocyclic alkyl group Chemical group 0.000 description 1
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 150000004680 hydrogen peroxides Chemical class 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- 150000002440 hydroxy compounds Chemical class 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- UWYVPFMHMJIBHE-OWOJBTEDSA-N hydroxymaleic acid group Chemical group O/C(/C(=O)O)=C/C(=O)O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 description 1
- 229960001560 hydroxyzine pamoate Drugs 0.000 description 1
- 229960004053 ibutilide Drugs 0.000 description 1
- BBPRUNPUJIUXSE-DXKRWKNPSA-N ifetroban Chemical compound CCCCCNC(=O)C1=COC([C@H]2[C@H]([C@@H]3CC[C@H]2O3)CC=2C(=CC=CC=2)CCC(O)=O)=N1 BBPRUNPUJIUXSE-DXKRWKNPSA-N 0.000 description 1
- 229950004274 ifetroban Drugs 0.000 description 1
- YAMHXTCMCPHKLN-UHFFFAOYSA-N imidazolidin-2-one Chemical compound O=C1NCCN1 YAMHXTCMCPHKLN-UHFFFAOYSA-N 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 230000000937 inactivator Effects 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000004926 indolenyl group Chemical group 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 239000004026 insulin derivative Substances 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- YCPOHTHPUREGFM-UHFFFAOYSA-N irbesartan Chemical compound O=C1N(CC=2C=CC(=CC=2)C=2C(=CC=CC=2)C=2[N]N=NN=2)C(CCCC)=NC21CCCC2 YCPOHTHPUREGFM-UHFFFAOYSA-N 0.000 description 1
- 229960002198 irbesartan Drugs 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 125000004936 isatinoyl group Chemical group N1(C(=O)C(=O)C2=CC=CC=C12)C(=O)* 0.000 description 1
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 1
- 125000003384 isochromanyl group Chemical group C1(OCCC2=CC=CC=C12)* 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 229960003299 ketamine Drugs 0.000 description 1
- 230000003907 kidney function Effects 0.000 description 1
- 108010051044 lanoteplase Proteins 0.000 description 1
- 229950010645 lanoteplase Drugs 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000008297 liquid dosage form Substances 0.000 description 1
- 229960002394 lisinopril Drugs 0.000 description 1
- RLAWWYSOJDYHDC-BZSNNMDCSA-N lisinopril Chemical compound C([C@H](N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(O)=O)C(O)=O)CC1=CC=CC=C1 RLAWWYSOJDYHDC-BZSNNMDCSA-N 0.000 description 1
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 1
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 1
- LRRRAKPXIKVNJE-UHFFFAOYSA-M lithium;4-(furan-2-yl)-1-[(2-methylpropan-2-yl)oxy]-1,4-dioxobut-2-en-2-olate Chemical compound [Li+].CC(C)(C)OC(=O)C([O-])=CC(=O)C1=CC=CO1 LRRRAKPXIKVNJE-UHFFFAOYSA-M 0.000 description 1
- 230000003908 liver function Effects 0.000 description 1
- 229960004391 lorazepam Drugs 0.000 description 1
- KJJZZJSZUJXYEA-UHFFFAOYSA-N losartan Chemical compound CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C=2[N]N=NN=2)C=C1 KJJZZJSZUJXYEA-UHFFFAOYSA-N 0.000 description 1
- 229960004773 losartan Drugs 0.000 description 1
- 229960004844 lovastatin Drugs 0.000 description 1
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 1
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- 229950004994 meglitinide Drugs 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 1
- 229960003105 metformin Drugs 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 1
- FTQWRYSLUYAIRQ-UHFFFAOYSA-N n-[(octadecanoylamino)methyl]octadecanamide Chemical class CCCCCCCCCCCCCCCCCC(=O)NCNC(=O)CCCCCCCCCCCCCCCCC FTQWRYSLUYAIRQ-UHFFFAOYSA-N 0.000 description 1
- ZKYPXKWRGLLYSO-UHFFFAOYSA-N n-[2-[7-oxo-6-[4-(2-oxopyridin-1-yl)phenyl]-3-(trifluoromethyl)-4,5-dihydropyrazolo[3,4-c]pyridin-1-yl]phenyl]sulfonylacetamide Chemical compound CC(=O)NS(=O)(=O)C1=CC=CC=C1N1C(C(=O)N(CC2)C=3C=CC(=CC=3)N3C(C=CC=C3)=O)=C2C(C(F)(F)F)=N1 ZKYPXKWRGLLYSO-UHFFFAOYSA-N 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- VRBKIVRKKCLPHA-UHFFFAOYSA-N nefazodone Chemical compound O=C1N(CCOC=2C=CC=CC=2)C(CC)=NN1CCCN(CC1)CCN1C1=CC=CC(Cl)=C1 VRBKIVRKKCLPHA-UHFFFAOYSA-N 0.000 description 1
- 229960001800 nefazodone Drugs 0.000 description 1
- VVGIYYKRAMHVLU-UHFFFAOYSA-N newbouldiamide Natural products CCCCCCCCCCCCCCCCCCCC(O)C(O)C(O)C(CO)NC(=O)CCCCCCCCCCCCCCCCC VVGIYYKRAMHVLU-UHFFFAOYSA-N 0.000 description 1
- JPWNXVRKIXSUEI-UHFFFAOYSA-N nitramidosulfanyl hydrogen sulfate Chemical group [N+](=O)([O-])NSOS(=O)(=O)O JPWNXVRKIXSUEI-UHFFFAOYSA-N 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 125000006574 non-aromatic ring group Chemical group 0.000 description 1
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 125000004930 octahydroisoquinolinyl group Chemical group C1(NCCC2CCCC=C12)* 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- LVRLSYPNFFBYCZ-VGWMRTNUSA-N omapatrilat Chemical compound C([C@H](S)C(=O)N[C@H]1CCS[C@H]2CCC[C@H](N2C1=O)C(=O)O)C1=CC=CC=C1 LVRLSYPNFFBYCZ-VGWMRTNUSA-N 0.000 description 1
- 229950000973 omapatrilat Drugs 0.000 description 1
- 229960000381 omeprazole Drugs 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 229940127234 oral contraceptive Drugs 0.000 description 1
- 239000003539 oral contraceptive agent Substances 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 229940126701 oral medication Drugs 0.000 description 1
- 239000007935 oral tablet Substances 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- AHLBNYSZXLDEJQ-FWEHEUNISA-N orlistat Chemical compound CCCCCCCCCCC[C@H](OC(=O)[C@H](CC(C)C)NC=O)C[C@@H]1OC(=O)[C@H]1CCCCCC AHLBNYSZXLDEJQ-FWEHEUNISA-N 0.000 description 1
- 229960001243 orlistat Drugs 0.000 description 1
- 229960003104 ornithine Drugs 0.000 description 1
- LPMXVESGRSUGHW-HBYQJFLCSA-N ouabain Chemical compound O[C@@H]1[C@H](O)[C@@H](O)[C@H](C)O[C@H]1O[C@@H]1C[C@@]2(O)CC[C@H]3[C@@]4(O)CC[C@H](C=5COC(=O)C=5)[C@@]4(C)C[C@@H](O)[C@@H]3[C@@]2(CO)[C@H](O)C1 LPMXVESGRSUGHW-HBYQJFLCSA-N 0.000 description 1
- 229960003343 ouabain Drugs 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000003431 oxalo group Chemical group 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- QNNHQVPFZIFNFK-UHFFFAOYSA-N oxazolo[4,5-b]pyridine Chemical compound C1=CC=C2OC=NC2=N1 QNNHQVPFZIFNFK-UHFFFAOYSA-N 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 125000004095 oxindolyl group Chemical group N1(C(CC2=CC=CC=C12)=O)* 0.000 description 1
- BSCHIACBONPEOB-UHFFFAOYSA-N oxolane;hydrate Chemical compound O.C1CCOC1 BSCHIACBONPEOB-UHFFFAOYSA-N 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 235000019629 palatability Nutrition 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 1
- 125000001312 palmitoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 229950008492 pentopril Drugs 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 108091008725 peroxisome proliferator-activated receptors alpha Proteins 0.000 description 1
- 239000008024 pharmaceutical diluent Substances 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 125000004934 phenanthridinyl group Chemical group C1(=CC=CC2=NC=C3C=CC=CC3=C12)* 0.000 description 1
- 125000004625 phenanthrolinyl group Chemical group N1=C(C=CC2=CC=C3C=CC=NC3=C12)* 0.000 description 1
- 125000001791 phenazinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3N=C12)* 0.000 description 1
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 1
- 125000004932 phenoxathinyl group Chemical group 0.000 description 1
- 125000001644 phenoxazinyl group Chemical group C1(=CC=CC=2OC3=CC=CC=C3NC12)* 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 150000008105 phosphatidylcholines Chemical class 0.000 description 1
- 239000002590 phosphodiesterase V inhibitor Substances 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 229960005095 pioglitazone Drugs 0.000 description 1
- 125000004928 piperidonyl group Chemical group 0.000 description 1
- 229960005235 piperonyl butoxide Drugs 0.000 description 1
- 125000004591 piperonyl group Chemical group C(C1=CC=2OCOC2C=C1)* 0.000 description 1
- VGYFMXBACGZSIL-MCBHFWOFSA-N pitavastatin Chemical compound OC(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1 VGYFMXBACGZSIL-MCBHFWOFSA-N 0.000 description 1
- 229940096701 plain lipid modifying drug hmg coa reductase inhibitors Drugs 0.000 description 1
- 229940012957 plasmin Drugs 0.000 description 1
- 229920001610 polycaprolactone Polymers 0.000 description 1
- 229920000656 polylysine Polymers 0.000 description 1
- 229920006324 polyoxymethylene Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 229960002965 pravastatin Drugs 0.000 description 1
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 1
- 229960004618 prednisone Drugs 0.000 description 1
- 230000035935 pregnancy Effects 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- GCYXWQUSHADNBF-AAEALURTSA-N preproglucagon 78-108 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 GCYXWQUSHADNBF-AAEALURTSA-N 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 108090000765 processed proteins & peptides Chemical class 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- JWHAUXFOSRPERK-UHFFFAOYSA-N propafenone Chemical compound CCCNCC(O)COC1=CC=CC=C1C(=O)CCC1=CC=CC=C1 JWHAUXFOSRPERK-UHFFFAOYSA-N 0.000 description 1
- 229960000203 propafenone Drugs 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 229960003712 propranolol Drugs 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 239000002599 prostaglandin synthase inhibitor Substances 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 230000017854 proteolysis Effects 0.000 description 1
- 229940039716 prothrombin Drugs 0.000 description 1
- 108010014806 prothrombinase complex Proteins 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- DNXIASIHZYFFRO-UHFFFAOYSA-N pyrazoline Chemical compound C1CN=NC1 DNXIASIHZYFFRO-UHFFFAOYSA-N 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- LBFMENANQYHPPH-UHFFFAOYSA-N pyrazolo[3,4-c]pyridin-7-one Chemical compound O=C1N=CC=C2C=NN=C12 LBFMENANQYHPPH-UHFFFAOYSA-N 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- 229940073095 questran Drugs 0.000 description 1
- 229960001455 quinapril Drugs 0.000 description 1
- JSDRRTOADPPCHY-HSQYWUDLSA-N quinapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CC2=CC=CC=C2C1)C(O)=O)CC1=CC=CC=C1 JSDRRTOADPPCHY-HSQYWUDLSA-N 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 1
- 229960004622 raloxifene Drugs 0.000 description 1
- 229960003401 ramipril Drugs 0.000 description 1
- HDACQVRGBOVJII-JBDAPHQKSA-N ramipril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](C[C@@H]2CCC[C@@H]21)C(O)=O)CC1=CC=CC=C1 HDACQVRGBOVJII-JBDAPHQKSA-N 0.000 description 1
- VMXUWOKSQNHOCA-LCYFTJDESA-N ranitidine Chemical compound [O-][N+](=O)/C=C(/NC)NCCSCC1=CC=C(CN(C)C)O1 VMXUWOKSQNHOCA-LCYFTJDESA-N 0.000 description 1
- 229960000620 ranitidine Drugs 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 230000011514 reflex Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 239000002461 renin inhibitor Substances 0.000 description 1
- 229940086526 renin-inhibitors Drugs 0.000 description 1
- 229960002354 repaglinide Drugs 0.000 description 1
- 125000006413 ring segment Chemical group 0.000 description 1
- 229960000371 rofecoxib Drugs 0.000 description 1
- RZJQGNCSTQAWON-UHFFFAOYSA-N rofecoxib Chemical compound C1=CC(S(=O)(=O)C)=CC=C1C1=C(C=2C=CC=CC=2)C(=O)OC1 RZJQGNCSTQAWON-UHFFFAOYSA-N 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- BPRHUIZQVSMCRT-VEUZHWNKSA-N rosuvastatin Chemical compound CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC(O)=O BPRHUIZQVSMCRT-VEUZHWNKSA-N 0.000 description 1
- 229960000672 rosuvastatin Drugs 0.000 description 1
- LALFOYNTGMUKGG-BGRFNVSISA-L rosuvastatin calcium Chemical compound [Ca+2].CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O.CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O LALFOYNTGMUKGG-BGRFNVSISA-L 0.000 description 1
- 108010073863 saruplase Proteins 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 229940076279 serotonin Drugs 0.000 description 1
- 229960002073 sertraline Drugs 0.000 description 1
- VGKDLMBJGBXTGI-SJCJKPOMSA-N sertraline Chemical compound C1([C@@H]2CC[C@@H](C3=CC=CC=C32)NC)=CC=C(Cl)C(Cl)=C1 VGKDLMBJGBXTGI-SJCJKPOMSA-N 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 229960003310 sildenafil Drugs 0.000 description 1
- 229960002855 simvastatin Drugs 0.000 description 1
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 1
- PHWXUGHIIBDVKD-UHFFFAOYSA-N sitaxentan Chemical compound CC1=NOC(NS(=O)(=O)C2=C(SC=C2)C(=O)CC=2C(=CC=3OCOC=3C=2)C)=C1Cl PHWXUGHIIBDVKD-UHFFFAOYSA-N 0.000 description 1
- 229960002578 sitaxentan Drugs 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- RYYKJJJTJZKILX-UHFFFAOYSA-M sodium octadecanoate Chemical compound [Na+].CCCCCCCCCCCCCCCCCC([O-])=O RYYKJJJTJZKILX-UHFFFAOYSA-M 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- ZBMZVLHSJCTVON-UHFFFAOYSA-N sotalol Chemical compound CC(C)NCC(O)C1=CC=C(NS(C)(=O)=O)C=C1 ZBMZVLHSJCTVON-UHFFFAOYSA-N 0.000 description 1
- 229960002370 sotalol Drugs 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 150000003413 spiro compounds Chemical class 0.000 description 1
- 208000010110 spontaneous platelet aggregation Diseases 0.000 description 1
- 239000004059 squalene synthase inhibitor Substances 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 125000003107 substituted aryl group Chemical group 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 1
- 229960000216 tenecteplase Drugs 0.000 description 1
- YUSMZDVTEOAHDL-NTMALXAHSA-N tert-butyl (3z)-3-(dimethylaminomethylidene)-4-oxopiperidine-1-carboxylate Chemical compound CN(C)\C=C1\CN(C(=O)OC(C)(C)C)CCC1=O YUSMZDVTEOAHDL-NTMALXAHSA-N 0.000 description 1
- RCESYRMKOMNBSI-UHFFFAOYSA-N tert-butyl 5-(2-oxopiperidin-1-yl)-2,3-dihydroindole-1-carboxylate Chemical compound C=1C=C2N(C(=O)OC(C)(C)C)CCC2=CC=1N1CCCCC1=O RCESYRMKOMNBSI-UHFFFAOYSA-N 0.000 description 1
- FWPYKWQELLYKJC-UHFFFAOYSA-N tert-butyl 6-(2-oxoazepan-1-yl)-2,3-dihydroindole-1-carboxylate Chemical compound C1=C2N(C(=O)OC(C)(C)C)CCC2=CC=C1N1CCCCCC1=O FWPYKWQELLYKJC-UHFFFAOYSA-N 0.000 description 1
- GCKFGIZRGKPDQJ-UHFFFAOYSA-N tert-butyl 6-(2-oxoazepan-1-yl)indole-1-carboxylate Chemical compound C1=C2N(C(=O)OC(C)(C)C)C=CC2=CC=C1N1CCCCCC1=O GCKFGIZRGKPDQJ-UHFFFAOYSA-N 0.000 description 1
- NWVDKZOUGBMLIH-UHFFFAOYSA-N tert-butyl 6-amino-2,3-dihydroindole-1-carboxylate Chemical compound C1=C(N)C=C2N(C(=O)OC(C)(C)C)CCC2=C1 NWVDKZOUGBMLIH-UHFFFAOYSA-N 0.000 description 1
- KYZMFSVVFOVRNK-UHFFFAOYSA-N tert-butyl 6-aminoindole-1-carboxylate Chemical compound C1=C(N)C=C2N(C(=O)OC(C)(C)C)C=CC2=C1 KYZMFSVVFOVRNK-UHFFFAOYSA-N 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 1
- UEUXEKPTXMALOB-UHFFFAOYSA-J tetrasodium;2-[2-[bis(carboxylatomethyl)amino]ethyl-(carboxylatomethyl)amino]acetate Chemical compound [Na+].[Na+].[Na+].[Na+].[O-]C(=O)CN(CC([O-])=O)CCN(CC([O-])=O)CC([O-])=O UEUXEKPTXMALOB-UHFFFAOYSA-J 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- VLLMWSRANPNYQX-UHFFFAOYSA-N thiadiazole Chemical compound C1=CSN=N1.C1=CSN=N1 VLLMWSRANPNYQX-UHFFFAOYSA-N 0.000 description 1
- 125000004627 thianthrenyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3SC12)* 0.000 description 1
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical class NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 108090000721 thyroid hormone receptors Proteins 0.000 description 1
- 102000004217 thyroid hormone receptors Human genes 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 230000037317 transdermal delivery Effects 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 150000003852 triazoles Chemical class 0.000 description 1
- PVFOMCVHYWHZJE-UHFFFAOYSA-N trichloroacetyl chloride Chemical compound ClC(=O)C(Cl)(Cl)Cl PVFOMCVHYWHZJE-UHFFFAOYSA-N 0.000 description 1
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 1
- JBWKIWSBJXDJDT-UHFFFAOYSA-N triphenylmethyl chloride Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(Cl)C1=CC=CC=C1 JBWKIWSBJXDJDT-UHFFFAOYSA-N 0.000 description 1
- NHDIQVFFNDKAQU-UHFFFAOYSA-N tripropan-2-yl borate Chemical compound CC(C)OB(OC(C)C)OC(C)C NHDIQVFFNDKAQU-UHFFFAOYSA-N 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- GXPHKUHSUJUWKP-UHFFFAOYSA-N troglitazone Chemical compound C1CC=2C(C)=C(O)C(C)=C(C)C=2OC1(C)COC(C=C1)=CC=C1CC1SC(=O)NC1=O GXPHKUHSUJUWKP-UHFFFAOYSA-N 0.000 description 1
- 229960001641 troglitazone Drugs 0.000 description 1
- GXPHKUHSUJUWKP-NTKDMRAZSA-N troglitazone Natural products C([C@@]1(OC=2C(C)=C(C(=C(C)C=2CC1)O)C)C)OC(C=C1)=CC=C1C[C@H]1SC(=O)NC1=O GXPHKUHSUJUWKP-NTKDMRAZSA-N 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 230000006433 tumor necrosis factor production Effects 0.000 description 1
- 150000003672 ureas Chemical class 0.000 description 1
- ACWBQPMHZXGDFX-QFIPXVFZSA-N valsartan Chemical compound C1=CC(CN(C(=O)CCCC)[C@@H](C(C)C)C(O)=O)=CC=C1C1=CC=CC=C1C1=NN=NN1 ACWBQPMHZXGDFX-QFIPXVFZSA-N 0.000 description 1
- 229960004699 valsartan Drugs 0.000 description 1
- MWOOGOJBHIARFG-UHFFFAOYSA-N vanillin Chemical compound COC1=CC(C=O)=CC=C1O MWOOGOJBHIARFG-UHFFFAOYSA-N 0.000 description 1
- 235000012141 vanillin Nutrition 0.000 description 1
- FGQOOHJZONJGDT-UHFFFAOYSA-N vanillin Natural products COC1=CC(O)=CC(C=O)=C1 FGQOOHJZONJGDT-UHFFFAOYSA-N 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 239000000230 xanthan gum Substances 0.000 description 1
- 235000010493 xanthan gum Nutrition 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
- 229940082509 xanthan gum Drugs 0.000 description 1
- 125000001834 xanthenyl group Chemical group C1=CC=CC=2OC3=CC=CC=C3C(C12)* 0.000 description 1
- BPICBUSOMSTKRF-UHFFFAOYSA-N xylazine Chemical compound CC1=CC=CC(C)=C1NC1=NCCCS1 BPICBUSOMSTKRF-UHFFFAOYSA-N 0.000 description 1
- 229960001600 xylazine Drugs 0.000 description 1
- 229960002769 zofenopril Drugs 0.000 description 1
- IAIDUHCBNLFXEF-MNEFBYGVSA-N zofenopril Chemical compound C([C@@H](C)C(=O)N1[C@@H](C[C@@H](C1)SC=1C=CC=CC=1)C(O)=O)SC(=O)C1=CC=CC=C1 IAIDUHCBNLFXEF-MNEFBYGVSA-N 0.000 description 1
- 229930195724 β-lactose Natural products 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61J—CONTAINERS SPECIALLY ADAPTED FOR MEDICAL OR PHARMACEUTICAL PURPOSES; DEVICES OR METHODS SPECIALLY ADAPTED FOR BRINGING PHARMACEUTICAL PRODUCTS INTO PARTICULAR PHYSICAL OR ADMINISTERING FORMS; DEVICES FOR ADMINISTERING FOOD OR MEDICINES ORALLY; BABY COMFORTERS; DEVICES FOR RECEIVING SPITTLE
- A61J1/00—Containers specially adapted for medical or pharmaceutical purposes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61J—CONTAINERS SPECIALLY ADAPTED FOR MEDICAL OR PHARMACEUTICAL PURPOSES; DEVICES OR METHODS SPECIALLY ADAPTED FOR BRINGING PHARMACEUTICAL PRODUCTS INTO PARTICULAR PHYSICAL OR ADMINISTERING FORMS; DEVICES FOR ADMINISTERING FOOD OR MEDICINES ORALLY; BABY COMFORTERS; DEVICES FOR RECEIVING SPITTLE
- A61J1/00—Containers specially adapted for medical or pharmaceutical purposes
- A61J1/03—Containers specially adapted for medical or pharmaceutical purposes for pills or tablets
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61J—CONTAINERS SPECIALLY ADAPTED FOR MEDICAL OR PHARMACEUTICAL PURPOSES; DEVICES OR METHODS SPECIALLY ADAPTED FOR BRINGING PHARMACEUTICAL PRODUCTS INTO PARTICULAR PHYSICAL OR ADMINISTERING FORMS; DEVICES FOR ADMINISTERING FOOD OR MEDICINES ORALLY; BABY COMFORTERS; DEVICES FOR RECEIVING SPITTLE
- A61J1/00—Containers specially adapted for medical or pharmaceutical purposes
- A61J1/05—Containers specially adapted for medical or pharmaceutical purposes for collecting, storing or administering blood, plasma or medical fluids ; Infusion or perfusion containers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4365—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system having sulfur as a ring hetero atom, e.g. ticlopidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/513—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim having oxo groups directly attached to the heterocyclic ring, e.g. cytosine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/08—Vasodilators for multiple indications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B65—CONVEYING; PACKING; STORING; HANDLING THIN OR FILAMENTARY MATERIAL
- B65D—CONTAINERS FOR STORAGE OR TRANSPORT OF ARTICLES OR MATERIALS, e.g. BAGS, BARRELS, BOTTLES, BOXES, CANS, CARTONS, CRATES, DRUMS, JARS, TANKS, HOPPERS, FORWARDING CONTAINERS; ACCESSORIES, CLOSURES, OR FITTINGS THEREFOR; PACKAGING ELEMENTS; PACKAGES
- B65D81/00—Containers, packaging elements, or packages, for contents presenting particular transport or storage problems, or adapted to be used for non-packaging purposes after removal of contents
- B65D81/32—Containers, packaging elements, or packages, for contents presenting particular transport or storage problems, or adapted to be used for non-packaging purposes after removal of contents for packaging two or more different materials which must be maintained separate prior to use in admixture
- B65D81/3216—Rigid containers disposed one within the other
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/75—Amino or imino radicals, acylated by carboxylic or carbonic acids, or by sulfur or nitrogen analogues thereof, e.g. carbamates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
- C07D213/82—Amides; Imides in position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- This invention relates generally to lactam-containing compounds and derivatives thereof which are inhibitors of trypsin-like serine protease enzymes, especially factor Xa, pharmaceutical compositions containing the same, and methods of using the same as anticoagulant agents for treatment of thromboembolic disorders .
- X can be a number of substituents and Het can be a nitrogen-containing heterobicycle.
- O94/20460 does not suggest Factor Xa inhibition or exemplify compounds like those of the present invention.
- WO96/12720 depicts phosphodiesterase type IV and TNF production inhibitors of the following formula:
- X can be oxygen and R 2 and R 3 can a number of substituents including heterocycle, heterocycloalkyl, and phenyl.
- the presently claimed compounds do not correspond to the compounds of WO96/12720.
- O96/12720 does not suggest Factor Xa inhibition.
- W098/52948 details inhibitors of ceramide-mediated signal transduction.
- One of the types of inhibitors described is of the following formula:
- Y 1 can be N-R 6
- R 6 can be unsubstituted aryl-alkyl or unsubstituted heterocyclic-alkyl
- R-j_ can be a substituted aryl group.
- 0 8/52948 does not mention factor Xa inhibition or show compounds like those of the present invention.
- U.S. Patent Nos. 3,365,459, 3,340,269, and 3,423,414 illustrate anti-inflammatory inhibitors of the following formula:
- WO00/39131 describes heterobicyclic Factor Xa inhibitors of which the following is an example formula: wherein Z is C or N, G is a mono- or bicyclic group, A is a cyclic moiety and B is a basic group or a cyclic moiety. Compounds specifically described in WO00/39131 are not considered to be part of the present invention.
- ring M is a heterocycle
- Z is a linker
- A is a ring
- B is a basic or cylic group
- D is a basic moiety
- E is a ring.
- ring M can be a variety of heterocycles and rings D-E represent a heterobicyclic group.
- Compounds specifically described in W098/57951 are not considered to be part of the present invention.
- ring M is a 6-membered heteroaryl
- Z is a linker
- A is a ring
- B is a basic or cylic group
- D is a basic moiety
- E is a ring.
- WO99/50255 and US 6,191,159 describe pyrazoline and triazoline Factor Xa inhibitors of the following formulas:
- WO00/59902 describes Factor Xa inhibitors of the following formula:
- ring M can be a variety of rings all of which are substituted with Z-A-B, Z is a linker, A is a ring, B is a sulfonyl-containing heterobicycle, and rings D-E represent a heterobicyclic group or a 6-membered ring.
- Z is a linker
- A is a ring
- B is a sulfonyl-containing heterobicycle
- rings D-E represent a heterobicyclic group or a 6-membered ring.
- WO01/32628 describes cyano-pyrroles, cyano-imidazoles, cyano-pyrazoles, and cyano-triazoles that are Factor Xa inhibitors. Compounds specifically described in WO01/32628 are not considered to be part of the present invention.
- WO01/05784 describes Factor Xa inhibitors of the following formulas:
- Z is C or N
- G is a mono- or bicyclic ring M
- A is a linker
- B is a basic or cyclic group.
- WO00/39108 describes Factor Xa inhibitors of the following formula:
- ring M can be a variety of heterocycles and rings
- D-E represent a heterobicyclic group.
- Compounds specifically described in WO00/39108 are not considered to be part of the present invention.
- WO01/19798 describes factor Xa inhibitors of the following formula:
- Activated factor Xa whose major practical role is the generation of thrombin by the limited proteolysis of prothrombin, holds a central position that links the intrinsic and extrinsic activation mechanisms in the final common pathway of blood coagulation.
- the generation of thrombin, the final serine protease in the pathway to generate a fibrin clot, from its precursor is amplified by formation of prothrombinase complex (factor Xa, factor V,
- factor Xa Ca2 + and phospholipid
- thrombin Since it is calculated that one molecule of factor Xa can generate 138 molecules of thrombin (Elodi, S., Varadi, K. : Optimization of conditions for the catalytic effect of the factor IXa-factor VIII Complex: Probable role of the complex in the amplification of blood coagulation. Thromb. Res . 1979, 15, 617-629), inhibition of factor Xa may be more efficient than inactivation of thrombin in interrupting the blood coagulation system.
- factor Xa factor Xa inhibitors
- new compounds with improved pharmacological characteristics compared with known factor Xa inhibitors. For example, it is preferred to find new compounds with improved factor Xa inhibitory activity and selectivity for factor Xa versus other serine proteases (i.e., trypsin) . It is also desirable and preferable to find compounds with advantageous and improved characteristics in one or more of the following categories, but are not limited to: (a) pharmaceutical properties (e.g., solubility, permeability, and amenability to sustained release formulations) ; (b) dosage requirements
- the present invention provides novel lactam-containing compounds and derivatives thereof that are useful as factor Xa inhibitors or pharmaceutically acceptable salts or prodrugs thereof.
- the present invention provides pharmaceutical compositions comprising a pharmaceutically acceptable carrier and a therapeutically effective amount of at least one of the compounds of the present invention or a pharmaceutically acceptable salt or prodrug form thereof.
- the present invention provides a method for treating thromboembolic disorders comprising administering to a host in need of such treatment a therapeutically effective amount of at least one of the compounds of the present invention or a pharmaceutically acceptable salt or prodrug form thereof .
- the present invention provides a novel method of treating a patient in need of thromboembolic disorder treatment, comprising: administering a compound of the present invention or a pharmaceutically acceptable salt form thereof in an amount effective to treat a thromboembolic disorder.
- the present invention provides a novel method, comprising: administering a compound of the present invention or a pharmaceutically acceptable salt form thereof in an amount effective to treat a thromboembolic disorder.
- the present invention provides novel lactam-containing compounds and derivatives thereof for use in therapy.
- the present invention provides the use of novel lactam-containing compounds for the manufacture of a medicament for the treatment of a thromboembolic disorder.
- P 4 , P, M, and M 4 are defined below, or pharmaceutically acceptable salt or prodrug forms thereof, are effective factor Xa inhibitors.
- the present invention provides a novel compound of Formula I:
- M is a 3-10 membered carbocycle or a 4-10 membered heterocycle, consisting of: carbon atoms and 1-3 heteroatoms selected from 0, S(0) p , N, and NZ 2 ;
- ring M is substituted with 0-3 R la and 0-2 carbonyl groups, and there are 0-3 ring double bonds;
- P is fused onto ring M and is a 5, 6, or 7 membered carbocycle or a 5, 6, or 7 membered heterocycle, consisting of: carbon atoms and 1-3 heteroatoms selected from 0, S(0) p , and N;
- ring P is substituted with 0-3 R la and 0-2 carbonyl groups, and there are 0-3 ring double bonds;
- ring P is absent and P is directly attached to ring M, provided that when ring P is absent, P and M 4 are attached to the 1,2, 1,3, or 1,4 positions of ring M; one of P and M is -Z-A-B and the other -G ⁇ G;
- G is a group of Formula Ila or lib:
- ring D including the two atoms of Ring E to which it is attached, is a 5-6 membered ring consisting of: carbon atoms and 0-2 heteroatoms selected from the group consisting of N, 0, and S(0) p ;
- ring D is substituted with 0-2 R and there are 0-3 ring double bonds;
- E is selected from phenyl, pyridyl, pyrimidyl, pyrazinyl, and pyridazinyl, and is substituted with 1-2 R;
- ring D is absent and ring E is selected from phenyl, pyridyl, pyrimidyl, pyrazinyl, pyridazinyl, pyrrolyl, pyrazolyl, imidazolyl, isoxazolyl, oxazolyl, triazolyl, thienyl, and thiazolyl, and ring E is substituted with 1-2 R;
- A is selected from:
- 5-12 membered heterocycle consisting of: carbon atoms and 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p and substituted with 0-2 R 4 ; provided that A is other than a dihydro-benzopyran;
- B is ; provided that Z and B are attached to different atoms on A and that the A-X-N moiety forms other than a N-N-N group;
- B is other than triazolone, quinolone, or isoquinolone, wherein the triazolone, quinolone, and isoquinolone groups are substituted or unsubstituted;
- ring Q is a 4-8 membered monocyclic or bicyclic ring consisting of, in addition to the N-Q ⁇ group shown, carbon atoms and 0-2 heteroatoms selected from NR 4c , 0, S, S(O), and S(0) 2 , wherein: 0-2 double bonds are present within the ring and the ring is substituted with 0-2 R 4a ;
- ring Q is a 4-8 membered monocyclic or bicyclic ring to which another ring is fused, wherein: the 4-7 membered ring consists of, in addition to the shown amide group, carbon atoms and 0-2 heteroatoms selected from NR 4c , 0, S, S(0), and S(0) 2 and 0-2 double bonds are present within the ring; the fusion ring is phenyl or a 5-6 membered heteroaromatic consisting of carbon atoms and 1-2 heteroatoms selected from NR c , 0, S, S(0), and S(0) ; ring Q, which includes the 4-7 membered ring and the fusion ring, is substituted with 0-3 R 4a ;
- two non-adjacent atoms of one of the rings of ring Q are bridged with 1-2 atoms selected from: carbon atoms, NR c , 0, S, S(O), and S(0) 2 , provided bonds other than 0-0, S(0) p -0, S(0) p -S(0) p , N-O, and N-S(0) p are present;
- X is absent or is selected from - (CR 2 R 2a ) 1 _ 4 -,
- Z is selected from a bond, - (CR 3 R 3e ) ⁇ _ 4 -,
- B-A-Z form other than a pyridone-phenyl-CH 2 , pyridone-pyridyl-CH 2 , or pyridone-pyrimidyl-CH 2 , wherein the pyridone, phenyl, pyridyl, and pyrimidyl groups are substituted or unsubstituted;
- Z 2 is selected from H, S(0) 2 NHR 3b , C(0)R 3b , C(0)NHR 3b ,
- R la at each occurrence, is selected from H, - (CR 3 R 3a ) r -R lb ,
- R l c i s selected from H, CH(CH 2 OR 2 ) 2 , C(0)R 2c , C(0)NR 2 R 2a , S(0)R 2 , S(0) 2 R 2 , and S0NR 2 R 2a ;
- R l is selected from C _g carbocycle substituted with 0-2
- R 4b and 5-10 membered heterocycle consisting of carbon atoms and from 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , and substituted with 0-2 R 4b , provided that R ld forms other than an N-S bond;
- R 2 at each occurrence, is selected from H, CF 3 , C ⁇ _ 6 alkyl, benzyl, - (CH 2 ) r -C 3 _ ⁇ o carbocycle substituted with 0-2 R 4b , and ⁇ (CH 2 ) r ⁇ 5-10 membered heterocycle consisting of: carbon atoms and 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , and substituted with 0-2 R b ;
- R 2a at each occurrence, is selected from H, CF 3 ,
- R 2 and R 2a together with the atom to which they are attached, combine to form a 5 or 6 membered saturated, partially saturated or unsaturated ring substituted with 0-2 R 4b and consisting of: 0-1 additional heteroatoms selected from- the group consisting of N, 0, and S(0) p ;
- R b at each occurrence, is selected from CF 3 , C ⁇ _ 4 alkoxy substituted with 0-2 R , C ⁇ _ 6 alkyl substituted with 0-2 R 4b , -(CH 2 ) r -C 3 _ ⁇ o carbocycle substituted with 0-2 R , and -(CH 2 ) r -5-10 membered heterocycle consisting of: carbon atoms and 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , and substituted with 0-2 R 4b ;
- R 2c at each occurrence, is selected from CF 3 , OH,
- R 3 at each occurrence, is selected from H, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH , CH(CH 3 ) 2 , CH 2 CH 2 CH 2 CH , CH 2 CH(CH 3 ) 2 , CH (CH 3 ) CH 2 CH 3 , C (CH 3 ) 3 , benzyl, and phenyl;
- R 3a at each occurrence, is selected from H, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , CH 2 CH 2 CH 2 CH 3 , CH 2 CH(CH 3 ) 2 , CH (CH 3 ) CH 2 CH 3 , C (CH 3 ) 3 , benzyl, and phenyl;
- R 3 and R 3a together with the nitrogen atom to which they are attached, combine to form a 5 or 6 membered saturated, partially unsaturated, or unsaturated ring consisting of: carbon atoms, the nitrogen atom to which R 3 and R 3a are attached, and 0-1 additional heteroatoms selected from the group consisting of N, 0, and S(0) p ;
- R 3b at each occurrence, is selected from H, C ⁇ _ 6 alkyl substituted with 0-2 R la , C 2 - 6 alkenyl substituted with 0-2 R la , C 2 - 6 alkynyl substituted with 0-2 R la , -(Co- 4 alkyl) -5-10 membered carbocycle substituted with 0-3 R la , and -(Co- 4 alkyl)- 5-10 membered heterocycle substituted with 0-3 R la and consisting of: carbon atoms and 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p ;
- R 3c at each occurrence, is selected from CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , CH 2 CH 2 CH 2 CH 3 , CH 2 CH(CH 3 ) 2 , CH(CH 3 )CH 2 CH 3 , C(CH 3 ) 3 , benzyl, and phenyl;
- R 3e at each occurrence, is selected from H, S0 2 NHR 3 ,
- R 3f is selected from: C ⁇ _ 6 alkyl substituted with 0-2 R la , C 2 _ 6 alkenyl substituted with 0-2 R la , C 2 _ 6 alkynyl substituted with 0-2 R la , -(Co- 4 alkyl) -5-10 membered carbocycle substituted with 0-3 R la , and -(Co- 4 alkyl) -5-10 membered heterocycle substituted with 0-3 R la and consisting of: carbon atoms and 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p ;
- R c at each occurrence, is selected from H, C ⁇ _ 4 alkyl • ((CCRR 33 RR 33aa ) r ⁇ OR 2 , (CR 3 R 3 ) r ⁇ F, (CR 3 R 3a ) r ⁇ Br, (CR 3 R 3a ) r ⁇ Cl,
- R 5a at each occurrence, is selected from C ⁇ _g alkyl
- R 7 at each occurrence, is selected from H, OH, C ⁇ _ 6 alkyl, C ⁇ - 6 alkyl-C(O)-, C ⁇ _ 6 alkyl-O-, (CH 2 ) n -pheny1 , C ⁇ _ 4 alkyl-OC(O) -, C ⁇ -io aryl-O-,
- R 8 at each occurrence, is selected from H, C ⁇ _ 6 alkyl, and (CH 2 ) n -phenyl;
- R 7 and R 8 when attached to the same nitrogen, combine to form a 5-10 membered heterocyclic ring consisting of carbon atoms and 0-2 additional heteroatoms selected from the group consisting of N, 0, and S(0) p ;
- R 9 at each occurrence, is selected from H, C ⁇ _ 6 alkyl, and (CH 2 ) n -phenyl;
- n at each occurrence, is selected from 0, 1, 2, and 3;
- p at each occurrence, is selected from 0, 1, and 2;
- r at each occurrence, is selected from 0, 1, 2, 3, 4, 5, and 6 ;
- rl at each occurrence, is selected from 1, 2, 3, 4, 5, and 6;
- t at each occurrence, is selected from 0, 1, 2, and 3;
- ring M is phenyl and is substituted 1,2 by M 4 and
- Z-A is other than NHC (0) -thienyl, NHCH 2 -thienyl, NHC (0) -benzothienyl, and NHCH 2 -benzothienyl; and, (b) B is 2-oxo-l-pyrrolidinyl and rings P-M are 1,7- dihydro-2-methyl-6H-purin-6-one, then G-G is other then unsubstituted phenyl.
- the present invention provides a novel compound of Formula II :
- ring M including Pi, P , Mi, and M 2 , is a 5, 6, or 7 membered carbocycle or a 5 , 6 , or 7 membered heterocycle, consisting of: carbon atoms and 1-3 heteroatoms selected from 0, S(0) p , N, and NZ 2 ;
- ring M is substituted with 0-2 R l and 0-2 carbonyl groups, and there are 0-3 ring double bonds;
- ring P including Pi, P 2 , and P 3 , is a 5 or 6 membered aromatic heterocycle, consisting of: carbon atoms and 1-3 heteroatoms selected from 0, S(0) p , and N;
- ring P including Pi, P , and P 3
- ring P is a 5 or 6 membered dihydro-aromatic heterocycle, consisting of: carbon atoms and 1-3 heteroatoms selected from 0, S(0) p , and N;
- ring P is substituted with 0-2 R la ;
- one of P 4 and M is -Z-A-B and the other -G ⁇ G;
- G is a group of Formula Ila or lib:
- Ha lib ring D including the two atoms of Ring E to which it is attached, is a 5-6 membered ring consisting of: carbon atoms and 0-2 heteroatoms selected from the group consisting of N, 0, and S(0) p ;
- ring D is substituted with 0-2 R and there are 0-3 ring double bonds;
- E is selected from phenyl, pyridyl, pyrimidyl, pyrazinyl, and pyridazinyl, and is substituted with 1-2 R;
- ring D is absent, and ring E is selected from phenyl, pyridyl, pyrimidyl, and thienyl, and ring E is substituted with 1-2 R;
- ring D is absent
- ring E is selected from phenyl, pyridyl, and thienyl
- ring E is substituted with 1 R and substituted with a 5 membered heterocycle consisting of: carbon atoms and 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , wherein the 5 membered heterocycle is substituted with 0-1 carbonyl and 1-2 R and there are 0-3 ring double bonds;
- A is selected from:
- C 5 _ ⁇ o carbocycle substituted with 0-2 R 4 and 5-10 membered heterocycle consisting of: carbon atoms and 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p and substituted with 0-2 R 4 ; provided that A is other than a dihydro-benzopyran;
- B is ; provided that Z and B are attached to different atoms on A and that the A-X-N moiety forms other than a N-N-N group;
- B is other than triazolone, quinolone, or isoquinolone, wherein the triazolone, quinolone, and isoquinolone groups are substituted or unsubstituted;
- ring Q is a 4-7 membered monocyclic or tricyclic ring consisting of, in addition to the N-Qi group shown, carbon atoms and 0-2 heteroatoms selected from NR 4c , 0,
- 0-2 double bonds are present within the ring and the ring is substituted with 0-2 R 4a ;
- ring Q is a 4-7 membered ring to which another ring is fused, wherein: the 4-7 membered ring consists of, in addition to the shown amide group, carbon atoms and 0-2 heteroatoms selected from NR 4c , 0, S, S(O), and S(0) 2 and 0-1 double bonds are present within the ring; the fusion ring is phenyl or a 5-6 membered heteroaromatic consisting of carbon atoms and 1-2 heteroatoms selected from NR 4c , 0, and S; ring Q, which includes the 4-7 membered ring and the fusion ring, is substituted with 0-3 R ;
- X is absent or is selected from - (CRR 2a ) ⁇ _ 4 -, -C(0)-, -C(0)CR 2 R 2 -, -CR 2 R 2a C(0), -S(0) 2 -, -S (0) 2 CR 2 R 2a -,
- Z is selected from a bond, CH , CH 2 CH 2 , CH 2 0, OCH 2 , C(0), NH, CH 2 NH, NHCH 2 , CH 2 C(0), C(0)CH 2 , C(0)NH, NHC(O), NHC(0)CH 2 C(0)NH, S(0) 2 , CH 2 S(0) 2 , S(0) 2 (CH 2 ), S0 2 NH, and NHSO 2 , provided that Z does not form a N-S, NCH 2 N, NCH 0, or NCH 2 S bond with either group to which it is . attached;
- Z 2 is selected from H, C ⁇ _ alkyl, phenyl, benzyl, C(0)R 3b , S(0)R 3f , and S(0) 2 R 3f ;
- R l a i s selected from H, -(CH 2 )r-R lb - (CH (CH 3 ) ) r -R lb , -(C(CH 3 ) 2 ) r -R lb , NHCH 2 R lc , 0CH 2 R lc , SCHR lc , NH(CH 2 ) 2 (CH 2 ) t R lb , and 0 (CH 2 ) 2 (CH 2 ) t R lb , provided that
- R la forms other than an N-halo, N-S, or N-CN bond
- R lb is selected from H, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , F,
- R lb forms other than an 0-0, N-halo, N-S, or N-CN bond;
- R lc is selected from H, CH(CH 2 OR 2 ) 2 , C(0)R 2c , C(0)NR 2 R 2a , S(0)R 2 , S(0) 2 R 2 , and S0 2 NR 2 R 2a ;
- R 2 at each occurrence, is selected from H, CF 3 , CH 3 ,
- R 2a at each occurrence, is selected from H, CF 3 , CH 3 ,
- R 2 and R 2a together with the atom to which they are attached, combine to form a 5 or 6 membered saturated, partially saturated or unsaturated ring substituted with 0-2 R 4b and consisting of: 0-1 additional heteroatoms selected from the group consisting of N, 0, and S(0) p ;
- R 2b at each occurrence, is selected from CF 3 , C ⁇ _ 4 alkoxy, CH 3 , CH 2 CH 3 , CHCH 2 CH 3 , CH(CH 3 ) , CH 2 CH 2 CH 2 CH , CH 2 CH(CH 3 ) 2 , CH(CH 3 )CH 2 CH 3 , C(CH 3 ) 3 , benzyl, C 5 - 6 carbocycle substituted with 0-2 R 4b , and 5-6 membered heterocycle consisting of: carbon atoms and 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , and substituted with 0-2 R 4b ;
- R 2c is selected from CF 3 , OH, C 1 - 4 alkoxy, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , CH 2 CH 2 CH 2 CH 3 , CH 2 CH(CH 3 ) 2 , CH (CH 3 ) CH 2 CH 3 , C(CH 3 ) 3 , benzyl, Cs_ 6 carbocycle substituted with 0-2 R b , and 5-6 membered heterocycle containing from 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , and substituted with 0-2 R 4b ;
- R 3 at each occurrence, is selected from H, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , benzyl, and phenyl;
- R 3a at each occurrence, is selected from H, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , benzyl, and phenyl;
- R 3 and R a together with the nitrogen atom to which they are attached, combine to form a 5 or 6 membered saturated, partially unsaturated, or unsaturated ring consisting of: carbon atoms and the nitrogen atom to which R 3 and R 3a are attached;
- R 3c at each occurrence, is selected from CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , benzyl, and phenyl;
- 5-6 membered carbocycle substituted with 0-1 R 5 and a 5-6 membered heterocycle consisting of: carbon atoms and 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , and substituted with 0-1 R 5 ;
- R 4c is selected from H, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , CH 2 CH 2 CH 2 CH 3 , CH 2 CH(CH 3 ) 2 , CH(CH 3 )CH 2 CH 3 , C(CH 3 ) 3 , CH 2 OR 2 , CH 2 F, CH 2 Br, CH 2 C1, CH 2 CN, CH 2 N0 2 , CH 2 NR 2 R 2a , C(0)R 2c , CH 2 C(0)R 2c , CH 2 NR 2 C(0)R 2b , C(0)NR 2 R 2a , CH 2 C (0) NR 2 R 2a , CH 2 NR 2 C(0)NR 2 R 2a , S0 2 NR 2 R 2a , CH 2 S0 2 NR 2 R 2a , CH 2 NR 2 S0NR 2 R 2a , CH 2 NR 2 S0NR 2 R 2a , CH 2 NR 2 S0NR 2 R 2 -C ⁇ _ 4 alkyl,
- CH 2 -5-6 membered carbocycle substituted with 0-1 R 5 5-6 membered heterocycle consisting of: carbon atoms and 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , and substituted with 0-1 R 5
- R 6 at each occurrence, is selected from H, OH, OR 2 , F, Cl, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH , CH(CH 3 ) 2 , CH 2 CH 2 CH 2 CH ,
- the present invention provides a novel compound, wherein:
- ring M is substituted with 0-2 R la and is selected from the group :
- ring P including P 1# P 2 , P 3 , and P is selected from group:
- one of P and M is -Z-A-B and the other -G ⁇ G;
- G is selected from the group:
- A is selected from one of the following carbocyclic and heterocyclic groups which are substituted with 0-2 R 4 ; phenyl, piperidinyl, piperazinyl, pyridyl, pyrimidyl, furanyl, morpholinyl, thienyl, pyrrolyl, pyrrolidinyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, pyrazolyl, imidazolyl, 1,2, 3-oxadiazolyl, 1,2, 4-oxadiazolyl, 1,2, 5-oxadiazolyl, 1,3, 4-oxadiazolyl , 1,2, 3-thiadiazolyl
- B is ; provided that Z and B are attached to different atoms on A;
- B is other than triazolone, quinolone, or isoquinolone, wherein the triazolone, quinolone, and isoquinolone groups are substituted or unsubstituted;
- ring Q is a 5-7 membered ring consisting of, in addition to the N-Qi group shown, carbon atoms and 0-2 heteroatoms selected from NR 4c , 0, S, S(O), and S(0) 2 , wherein:
- 0-2 double bonds are present within the ring and the ring is substituted with 0-2 R a ;
- ring Q is a 5-7 membered ring to which another ring is fused, wherein: the 5-7 membered ring consists of, in addition to the shown amide group, carbon atoms and 0-2 heteroatoms selected from NR 4c , 0, S, S(0), and S(0) 2 , and 0-1 double bonds are present within the ring; the fusion ring is phenyl or a 5-6 membered heteroaromatic consisting of carbon atoms and 1-2 heteroatoms selected from NR c , 0, and S; ring Q, which includes the 5-7 membered ring and the fusion ring, is substituted with 0-3 R 4a ;
- R l a is selected from H, R lb , CH(CH 3 )R lb , C(CH 3 ) 2 R lb , CH 2 R lb , and CH 2 CHR lb , provided that R la forms other than an N-halo, N-S, or N-CN bond;
- R la groups when two R la groups are attached to adjacent atoms, together with the atoms to which they are attached they form a 5-6 membered ring consisting of: carbon atoms and 0-2 heteroatoms selected from the group consisting of N, 0, and S(0) p , this ring being substituted with 0-2 R b and 0-3 ring double bonds;
- R lb is selected from H, CH 3 , CH 2 CH 3 , F, Cl, Br, -CN, -CHO, CF 3 , OR 2 , NR 2 R 2a , C(0)R 2b , C0 2 R 2 , 0C(0)R 2 , C0 2 R 2a , S(0) p R 2 , NR 2 (CH 2 ) r OR 2 , NR 2 C(0)R 2b , C(0)NR 2 R 2a , S0 2 NR 2 R 2a , NR 2 S0 2 R 2 , phenyl substituted with 0-2 R b , and 5-6 membered aromatic heterocycle consisting of carbon atoms and from 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , and substituted with
- R lb forms other than an 0-0, N- halo, N-S, or N-CN bond;
- R 2 at each occurrence, is selected from H, CF 3 , CH 3 ,
- R 2a at each occurrence, is selected from H, CF 3 , CH 3 ,
- R 2 and R 2a together with the atom to which they are attached, combine to form a 5 or 6 membered saturated, partially saturated or unsaturated ring substituted with 0-2 R b and consisting of: 0-1 additional heteroatoms selected from the group consisting of N, 0, and S(0) p ;
- R 2b at each occurrence, is selected from CF , C ⁇ _ 4 alkoxy, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , benzyl, phenyl' substituted with 0-2 R 4b , and 5-6 membered aromatic heterocycle consisting of: carbon atoms and 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , and substituted with 0-2 R 4b ;
- R 2c at each occurrence, is selected from CF 3 , OH, 0CH 3 ,
- (CH 2 ) 2 OR 2 OR 2 , F, Cl, Br, I, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , CH 2 CH 2 CH 2 CH 3 , CH 2 CH(CH 3 ) 2 , CH (CH 3 ) CH 2 CH 3 , C(CH 3 ) 3 , -CN, N0 2 , NR 2 R 2a , CH 2 NR 2 R 2a , (CH 2 ) 2 NR 2 R 2a , C(0)R 2c , NR 2 C(0)R 2b , C(0)NR 2 R 2a , S0 2 NR 2 R 2a , CF 3 , and
- NR 3 S0 2 -C ⁇ _ 4 alkyl CH 2 NR 3 S0 -C ⁇ _ alkyl, NR 3 S0 2 -phenyl, CH 2 NR 3 S0 2 -phenyl, S(0) p CF 3 , CH 2 S(0) p CF 3 ,
- R c at each occurrence, is selected from H, CH 3 , CH CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , CH 2 CH 2 CH 2 CH 3 , CH 2 CH(CH 3 ) 2 ,
- R 6 at each occurrence, is selected from H, OH, OR 2 , F, Cl, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , -CN, N0 2 , NR 2 R 2a ,
- the present invention provides a novel compound, wherein:
- ring M is substituted with 0-2 R la and is selected from the group :
- ring P including Pi, P 2 , P 3 , and P 4 is selected from group:
- one of P 4 and M is -A-B and the other -G;
- G is selected from the group :
- Gi is absent or is selected from CH 2 , CH 2 CH 2 , CH 2 0, OCH 2 , NH, CH 2 NH, NHCH , CH 2 C(0), C(0)CH 2 , C(0)NH, NHC(O), CH 2 S(0) 2 , S(0) 2 (CH 2 ), S0 2 NH, and NHS0 2 , provided that Gi does not form a N-S, NCH 2 N, NCH 2 0, or NCH 2 S bond with either group to which it is attached;
- A is selected from phenyl, pyridyl, and pyrimidyl, and is substituted with 0-2 R 4 ;
- B is ; provided that Z and B are attached to different atoms on A;
- B is other than triazolone, quinolone, or isoquinolone, wherein the triazolone, quinolone, and isoquinolone groups are substituted or unsubstituted;
- ring Q is a 6-7 membered ring consisting of, in addition to the N-Qi group shown, carbon atoms and 0-1 heteroatoms selected from NR 4c , 0, S, S(0), and S(0) 2 , wherein:
- ring Q is a 5-7 membered ring to which another ring is fused, wherein: the 5-7 membered ring consists of, in addition to the shown amide group, carbon atoms and 0-1 heteroatoms selected from NR 4c , 0, S, S(0), and S(0) 2 , and 0-1 double bonds are present within the ring; the fusion ring is phenyl; ring Q, which includes the 5-7 membered ring and the fusion ring, is substituted with 0-2R 4a ;
- R la is selected from H, R lb , C(CH 3 ) 2 R lb , and CH 2 R lb , provided that R la forms other than an N-halo, N-S, or N-CN bond;
- R lb is selected from CH 3 , CH 2 CH 3 , F, Cl, Br, -CN, CF 3 , OR 2 , NR 2 R 2a , C(0)R 2b , C0 2 R 2b , C0 2 R 2a , S(0) p R 2 , C(0)NR 2 R 2a , S ⁇ 2 NR 2 R 2a , NR 2 S0 2 R 2 , and 5-6 membered aromatic heterocycle consisting of carbon atoms and from 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , and substituted with 0-2 R b , provided that R lb forms other than an 0-0, N-halo, N-S, or N-CN bond;
- R 2 at each occurrence, is selected from H, CH 3 , CH 2 CH 3 ,
- R 2a at each occurrence, is selected from H, CH 3 , CH 2 CH 3 ,
- 0-1 R 4b and 5-6 membered aromatic heterocycle consisting of: carbon atoms and 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , and substituted with 0-1 R 4b ;
- R 2 and R 2a together with the atom to which they are attached, combine to form a 5 or 6 membered saturated, partially saturated or unsaturated ring substituted with 0-1 R b and consisting of: 0-1 additional heteroatoms selected from the group consisting of N, 0, and S(0) p ;
- R at each occurrence, is selected from 0CH 3 , OCH 2 CH 3 ,
- OCH 2 CH 2 CH 3 OCH(CH 3 ) 2 , CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , benzyl, phenyl substituted with 0-1 R 4b , and 5-6 membered aromatic heterocycle consisting of: carbon atoms and 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , and substituted with
- R 2c is selected from OH, 0CH 3 , OCH 2 CH 3 , OCH 2 CH 2 CH 3 , OCH(CH 3 ) 2 , CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , benzyl, phenyl substituted with 0-1 R 4b , and 5-6 membered aromatic heterocycle containing from 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , and substituted with 0-1 R 4b ;
- R 4 is selected from OH, OR 2 , CH 2 OR 2 , (CH 2 ) 2 ⁇ R 2 , F, Br, Cl, I, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , CH 2 CH 2 CH 2 CH 3 , CH 2 CH(CH 3 ) 2 , CH (CH 3 ) CH 2 CH 3 , C(CH 3 ) 3 , NR 2 R 2a , CH 2 NR 2 R 2a , (CH 2 ) 2 NR 2 R 2a , CF 3 , and CF 2 CF 3 ;
- R 4c is selected from H, CH 3 , CH 2 CH 3 , phenyl substituted with 0-1 R 5 , and benzyl substituted with 0-1 R 5 ;
- R 6 at each occurrence, is selected from H, OH, OR 2 , F, Cl, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , -CN, N0 2 , NR 2 R 2a , CH 2 NR 2 R 2a , C(0)R 2b , CHC(0)R 2b , NR 2 C(0)R 2b , and S0 2 NR 2 R 2a .
- the present invention provides a novel compound, wherein:
- ring M is substituted with 0-1 R la and is selected from the group :
- ring P including Pi, P 2 , P 3 , and P 4 is selected from group:
- one of P and M 4 is -A-B and the other -G;
- G is selected from:
- B is attached to a different atom on A than M and is selected from the group:
- Rla is selected from H, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH 2 F, CH 2 C1, Br, CH 2 Br, -CN, CH 2 CN, CF 3 , CH 2 CF 3 , OCH 3 , CH 2 OH,
- R 2 at each occurrence, is selected from H, CH 3 , CH 2 CH 3 ,
- R 2 and R a together with the atom to which they are attached, combine to form a 5 or 6 membered saturated, partially saturated or unsaturated ring substituted with 0-1 R 4b and consisting of: 0-1 additional heteroatoms selected from the group consisting of N, 0, and S(0) p ;
- R 2b at each occurrence, is selected from 0CH 3 , OCH 2 CH 3 , CH 3 , and CH 2 CH 3 ;
- R c at each occurrence, is selected from OH, 0CH 3 , OCH 2 CH 3 , CH 3 , and CH 2 CH 3 ;
- the present invention provides a novel compound, wherein the compound is selected from:
- M 4 is -A-B
- G is selected from:
- A-B is selected from:
- the present invention provides a novel compound, wherein the compound is selected from:
- P 4 is -G ;
- M 4 is -A-B ;
- A-B is selected from:
- the present invention provides a novel compound, wherein the compound is selected from the group :
- the present invention provides a novel compound, wherein the compound is of Formula Ilia, Illb, or IIIc:
- ring M including Mi, M 2 , and, if present, M 3 , is phenyl or a 3-10 membered carbocyclic or 4-10 membered heterocyclic ring consisting of: carbon atoms and 1-4 heteroatoms selected from 0, S(0) p , N, and NZ 2 ;
- ring M is substituted with 0-3 R l and 0-2 carbonyl groups, and there are 0-3 ring double bonds;
- one of P 4 and M 4 is -Z-A-B and the other -G ⁇ G;
- G is a group of Formula Ila or lib:
- ring D including the two atoms of Ring E to which it is attached, is a 5-6 membered ring consisting of: carbon atoms and 0-2 heteroatoms selected from the group consisting of N, 0, and S(0) p ;
- ring D is substituted with 0-2 R and there are 0-3 ring double bonds;
- E is selected from phenyl, pyridyl, pyrimidyl, pyrazinyl, and pyridazinyl, and is substituted with 1-2 R;
- ring D is absent, and ring E is selected from phenyl, pyridyl, pyrimidyl, and thienyl, and ring E is substituted with 1-2 R; alternatively, ring D is absent, ring E is selected from phenyl, pyridyl, and thienyl, and ring E is substituted with 1 R and substituted with a 5 membered heterocycle consisting of: carbon atoms and 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , wherein the 5 membered heterocycle is substituted with 0-1 carbonyl and 1-2 R and there are 0-3 ring double bonds;
- A is selected from:
- C 5 - 10 carbocycle substituted with 0-2 R 4 and 5-10 membered heterocycle consisting of: carbon atoms and 1-4 heteroatoms selected from. the group consisting of N, 0, and S(0) p and substituted with 0-2 R 4 ; provided that A is other than a dihydro-benzopyran;
- A-X-N moiety forms other than a N-N-N group
- B is other than triazolone, quinolone, or isoquinolone, wherein the triazolone, quinolone, and isoquinolone groups are substituted or unsubstituted
- ring Q is a 4-7 membered monocyclic or tricyclic ring consisting of, in addition to the N-Qi group shown, carbon atoms and 0-2 heteroatoms selected from NR c , 0, S, S(0), and S(0) 2 , wherein:
- 0-2 double bonds are present within the ring and the ring is substituted with 0-2 R a ;
- ring Q is a 4-7 membered ring to which another ring is fused, wherein: the 4-7 membered ring consists of, in addition to the shown amide group, carbon atoms and 0-2 heteroatoms selected from NR 4c , 0, S, S(0), and S(0) 2 and 0-1 double bonds are present within the ring; the fusion ring is phenyl or a 5-6 membered heteroaromatic consisting of carbon atoms and 1-2 heteroatoms selected from NR c , 0, and S; ring Q, which includes the 4-7 membered ring and the fusion ring, is substituted with 0-3 R 4a ;
- X is absent or is selected from - (CR 2 R 2a ) ⁇ _ 4 -, -C(0)-,
- Z is selected from a bond, CH 2 , CH 2 CH 2 , CH 2 0, OCH 2 , C(0), NH, CHNH, NHCH 2 , CH 2 C(0), C(0)CH 2 , C(0)NH, NHC(O), NHC(0)NH, NHC(0)CH 2 C(0)NH, C(0)NHS(0) 2 , S(0) 2 , CH S(0)2, S(0) 2 (CH 2 ), S0 2 NH, and NHS0 , provided that Z does not form a N-S, NCH 2 N, NCH 2 0, or NCH 2 S bond with either group to which it is attached;
- Z 2 is selected from H, C 1 - 4 alkyl, phenyl, benzyl, C(0)R 3b , S(0)R 3f , and S(0) 2 R 3f ;
- R la is selected from H, -(CH 2 ) r -R lb , - (CH(CH 3 ) ) r -R lb ,
- R lb is selected from H, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , F, Cl, Br, I, -CN, -CHO, CF 3 , OR 2 , NR 2 R 2a , C(0)R 2 , C0 2 R 2b , 0C(0)R 2 , C0 R 2a , S(0) p R 2 , NR 2 (CH 2 ) r OR 2 , NR 2 C(0)R 2b , NR 2 C(0)NHR 2 , NR 2 C(0) 2 R 2a , OC(0)NR 2 R 2a , C(0)NR 2 R 2a , C(0)NR 2 (CH 2 ) r OR 2 , S0 2 NR 2 R 2a , NR 2 S0 R 2 , C 5 _ 6 carbocycle substituted with 0-2 R b , and 5-6 membered heterocycle consisting of carbon atoms and from 1-4 heteroatoms selected from the group consisting of N
- R lb forms other than an 0-0, N-halo, N-S, or N-CN bond;
- R lc is selected from H, CH (CH 2 OR 2 ) 2 , C (0) R 2c , C (0) NR 2 R 2a , S (0) R 2 , S (0) 2 R 2 , and S0 2 NR 2 R 2a ; R 2 , at each occurrence, is selected from H, CF , CH 3 ,
- R 2a at each occurrence, is selected from H, CF 3 , CH 3 ,
- R 2 and R a together with the atom to which they are attached, combine to form a 5 or 6 membered saturated, partially saturated or unsaturated ring substituted with 0-2 R b and consisting of: 0-1 additional heteroatoms selected from the group consisting of N, 0, and S(0) p ;
- R 2b at each occurrence, is selected from CF 3 , C ⁇ _ 4 alkoxy, CH , CH 2 CH , CH 2 CH 2 CH 3 , CH (CH 3 ) 2 , CH 2 CH 2 CHCH 3 , CH 2 CH(CH 3 ) 2 , CH(CH 3 )CH 2 CH 3 , C(CH 3 ) 3 , benzyl, C 5 _ 6 carbocycle substituted with 0-2 R 4b , and 5-6 membered heterocycle consisting of: carbon atoms and 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , and substituted with 0-2 R 4b ;
- R 2 at each occurrence, is selected from CF , OH, C 1 - 4 alkoxy, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , CH 2 CH(CH 3 )CH 2 CH 3 ,
- R 3 at each occurrence, is selected from H, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , benzyl, and phenyl;
- R 3a at each occurrence, is selected from H, CH 3 , CHCH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , benzyl, and phenyl;
- R 3 and R 3a together with the nitrogen atom to which they are attached, combine to form a 5 or 6 membered saturated, partially unsaturated, or unsaturated ring consisting of: carbon atoms and the nitrogen atom to which R 3 and R a are attached;
- R 3c at each occurrence, is selected from CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , benzyl, and phenyl;
- R 4c at each occurrence, is selected from H, CH 3 , CHCH 3 ,
- CH 2 5-6 membered carbocycle substituted with 0-1 R 5 5-6 membered heterocycle consisting of: carbon atoms and 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , and substituted with 0-1 R 5
- the present invention provides a novel compound, wherein:
- ring M is substituted with 0-3 R la and 0-1 carbonyl group
- G is selected from the group:
- G is absent or is selected from (CR R 3a ) ⁇ _ 3 ,
- A is selected from one of the following carbocyclic and heterocyclic groups which are substituted with 0-2 R 4 ; phenyl, piperidinyl, piperazinyl, pyridyl, pyrimidyl, furanyl, morpholinyl, thienyl, pyrrolyl, pyrrolidinyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, pyrazolyl, imidazolyl, 1,2, 3-oxadiazolyl, 1,2, 4-oxadiazolyl, 1,2, 5-oxadiazolyl , 1,3, 4-oxadiazolyl, 1,2, 3-thiadiazolyl, 1,2, 4-thiadiazolyl, 1,2 , 5-thiadiazolyl, 1, 3 , 4-thiadiazolyl,
- 1,2, 3-triazolyl 1, 2 , 4-triazolyl, 1, 2 , 5-triazolyl, 1, 3 , 4-triazolyl, benzofuranyl, benzothiofuranyl, indolinyl, indolyl, benzimidazolyl, benzoxazolyl, benzthiazolyl, indazolyl, benzisoxazolyl, benzisothiazolyl, and isoindazolyl;
- B is ; provided that Z and B are attached to different atoms on A;
- B is other than triazolone, quinolone, or isoquinolone, wherein the triazolone, quinolone, and isoquinolone groups are substituted or unsubstituted;
- ring Q is a 5-7 membered ring consisting of, in addition to the N-Qi group shown, carbon atoms and 0-2 heteroatoms selected from NR 4c , 0, S, S(O), and S(0) 2 , wherein:
- 0-2 double bonds are present within the ring and the ring is substituted with 0-2 R 4a ;
- ring Q is a 5-7 membered ring to which another ring is fused, wherein: the 5-7 membered ring consists of, in addition to the shown amide group, carbon atoms and 0-2 heteroatoms selected from NR 4c , 0, S, S(O), and S(0) 2 and 0-1 double bonds are present within the ring; the fusion ring is phenyl or a 5-6 membered heteroaromatic consisting of carbon atoms and 1-2 heteroatoms selected from NR c , 0, and S; ring Q, which includes the 5-7 membered ring and the fusion ring, is substituted with 0-3 R a ;
- R l a is selected from H, R lb , CH(CH 3 )R l , C(CH 3 ) 2 R lb , CH 2 R lb , and CH 2 CH 2 R lb , provided that R la forms other than an N- halo, N-S, or N-CN bond; alternatively, when two R la groups are attached to adjacent atoms, together with the atoms to which they are attached they form a 5-6 membered ring consisting of: carbon atoms and 0-2 heteroatoms selected from the group consisting of N, 0, and S(0) p , this ring being substituted with 0-2 R 4b and 0-3 ring double bonds;
- R lb is selected from H, CH 3 , CH 2 CH 3 , F, Cl, Br, -CN, -CHO, CF 3 , OR 2 , NR 2 R 2a , C(0)R 2b , C0 2 R 2b , 0C(0)R 2 , C0 2 R 2a ,
- R l forms other than an 0-0, N- halo, N-S, or N-CN bond;
- R 2 at each occurrence, is selected from H, CF 3 , CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , phenyl substituted with
- 0-2 R b a benzyl substituted with 0-2 R 4b , and 5-6 membered aromatic heterocycle consisting of: carbon atoms and 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , and substituted with 0-2 R 4b ;
- R a at each occurrence, is selected from H, CF 3 , CH 3 ,
- R b at each occurrence, is selected from CF 3 , C 1 - 4 alkoxy, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , benzyl, phenyl substituted with 0-2 R 4b , and 5-6 membered aromatic heterocycle consisting of: carbon atoms and 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , and substituted with 0-2 R 4b ;
- R c at each occurrence, is selected from CF 3 , OH, 0CH 3 ,
- R 4 at each occurrence, is selected from H, CH 2 OR 2 ,
- NR 3 S0 2 -C ⁇ _ 4 alkyl CH 2 NR 3 S0 2 -C ⁇ -4 alkyl, NR 3 S0 -phenyl, CH 2 NR 3 S0 2 -phenyl, S(0) p CF 3 , CH 2 S(0) p CF 3 , S(0) p -Ci_4 alkyl, CH 2 S (0) p -C ⁇ _ alkyl, S (0) p -phenyl, CH 2 S(0) p -phenyl, and CF 3 ;
- R 4c is selected from H, CH 3 , CH CH 3 , CH 2 CH 2 CH 3 , CH ( CH 3 ) 2 , CH 2 CH 2 CH 2 CH 3 , CH CH(CH 3 ) 2 , CH(CH 3 )CH 2 CH 3 , C(CH 3 ) 3 , CH 2 OR 2 , CH 2 F, CH 2 Br, CH 2 Cl, CH 2 CN, CH 2 N0 2 , CH 2 NR 2 R 2 , C(0)R 2c , CH 2 C(0)R 2c ,
- R 6 at each occurrence, is selected from H, OH, OR 2 , F, Cl, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , -CN, N0 2 , NR 2 R 2a , CH 2 NR 2 R 2a , C(0)R 2b , CH 2 C(0)R 2b , NR 2 C(0)R 2b , S0 2 NR 2 R 2a , and NR 2 S0 2 C ⁇ _ 4 alkyl.
- the present invention provides a novel compound, wherein the compound is selected from:
- J is selected from 0, S, NH, and NR la ;
- G is selected from the group:
- Gi is absent or is selected from CH 2 , CH 2 CH 2 , CH 2 0, OCH , NH, CH 2 NH, NHCH 2 , CH 2 C(0), C(0)CH , C(0)NH, NHC(O), NHC(0)NH, C(0)NHS(0) 2 , CH 2 S(0) 2 , S(0) 2 (CH 2 ), S0 2 NH, and NHS0 2 , provided that Gi does not form a N-S, NCHN, NCH 2 0, or NCH 2 S bond with either group to which it is attached;
- A is selected from indolinyl, phenyl, pyridyl, and pyrimidyl, and is substituted with 0-2 R 4 ;
- B is ; provided that Z and B are attached to different atoms on A;
- B is other than triazolone, quinolone, or isoquinolone, wherein the triazolone, quinolone, and isoquinolone groups are substituted or unsubstituted;
- ring Q is a 6-7 membered ring consisting of, in addition to the N-Q group shown, carbon atoms and 0-1 heteroatoms selected from NR 4c , 0, S, S(0), and S(0) 2 , wherein:
- 0-2 double bonds are present within the ring and the ring is substituted with 0-2 R a ;
- ring Q is a 5-7 membered ring to which another ring is fused, wherein: the 5-7 membered ring consists of, in addition to the shown amide group, carbon atoms and 0-1 heteroatoms selected from NR 4c , 0, S, S(0), and S(0) 2 and 0-1 double bonds are present within the ring; the fusion ring is phenyl; ring Q, which includes the 5-7 membered ring and the fusion ring, is substituted with 0-2R 4a ;
- R la is selected from H, R lb , C(CH 3 ) 2 R lb , and CH 2 R lb , provided that R la forms other than an N-halo, N-S, or N-CN bond;
- R lb is selected from CH 3 , CH 2 CH 3 , F, Cl, Br, -CN, CF 3 , OR 2 , NR 2 R 2a , C(0)R 2 , C0 2 R 2b , C0 2 R 2 , S(0) p R 2 , C(0)NR 2 R 2a , S0 2 NR 2 R 2a , NR 2 S0 2 R 2 , and 5-6 membered aromatic heterocycle consisting of carbon atoms and from 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , and substituted with 0-2 R 4b , provided that R lb forms other than an 0-0, N-halo, N-S, or N-CN bond;
- R 2 at each occurrence, is selected from H, CH 3 , CH 2 CH 3 ,
- R 2a at each occurrence, is selected from H, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , benzyl, phenyl substituted with
- 0-1 R b and 5-6 membered aromatic heterocycle consisting of: carbon atoms and 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , and substituted with 0-1 R 4b ;
- R 2 and R a together with the atom to which they are attached, combine to form a 5 or 6 membered saturated, partially saturated or unsaturated ring substituted with 0-1 R 4b and consisting of: 0-1 additional heteroatoms selected from the group consisting of N, 0, and S(0) p ;
- R 2b at each occurrence, is selected from OH, 0CH 3 , OCH 2 CH 3 , 0CH 2 CH 2 CH 3 , OCH(CH 3 ) 2 , CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , benzyl, phenyl substituted with 0-1 R b , and 5-6 membered aromatic heterocycle consisting of: carbon atoms and 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) , and substituted with 0-1 R 4b ;
- R 2c at each occurrence, is selected from OH, OCH 3 , OCH 2 CH 3 , 0CH 2 CH 2 CH 3 , OCH(CH 3 ) 2 , CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , benzyl, phenyl substituted with 0-1 R 4b , and 5-6 membered aromatic heterocycle containing from 1-4 heteroatoms selected from the group consisting of N, 0, and S(0) p , and substituted with 0-1 R 4b ;
- R 4 at each occurrence, is selected from OH, OR 2 , CH OR 2 , (CH 2 ) 2 OR 2 , F, Br, Cl, I, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH(CH 3 ) 2 , CH 2 CH 2 CH 2 CH 3 , CH 2 CH(CH 3 ) 2 , CH (CH 3 ) CH 2 CH 3 , C(CH 3 ) 3 , NR 2
- R 4c is selected from H, CH 3 , CH CH 3 , phenyl substituted with 0-1 R 5 , and benzyl substituted with 0-1 R 5 ;
- R 6 at each occurrence , is selected from H, OH, OR 2 , F , Cl , CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH (CH 3 ) 2 , -CN, N0 2 , NR 2 R 2a , CH 2 NR 2 R 2a , C(0)R 2b , CH 2 C(0)R 2b , NR 2 C(0)R 2b , and S0 2 NR 2 R 2a .
- the present invention provides a novel compound, wherein the compound is selected from:
- J is selected from 0, S, NH, and NR la ;
- M is -Z-A-B
- G is selected from:
- Gi is absent or is selected from CH 2 NH, NHCH , CH 2 C(0), C(0)CH 2 , C(0)NH, NHC(O), NHC(0)NH, CH 2 S(0) 2 ,
- A is selected from the group: indolinyl, phenyl, 2-pyridyl, 3-pyridyl, 2-pyrimidyl, 2-Cl-phenyl, 3-Cl-phenyl, 2-F- phenyl, 3-F-phenyl, 2-methylphenyl, 2-aminophenyl, and 2-methoxyphenyl ;
- B is attached to a different atom on A than M and is selected from the group:
- R l a i s selected from H, CH 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , CH 2 F, CH 2 C1, Br, CH 2 Br, -CN, CH 2 CN, CF 3 , CH 2 CF 3 , OCH 3 , CH 2 OH, C(CH 3 ) 2 OH, CH 2 OCH 3 , NH 2 , CH 2 NH 2 , NHCH 3 , CH 2 NHCH 3 , N(CH 3 ) 2 , CH 2 N(CH 3 ) 2 , C0 2 H, COCH 3 , C0 CH 3 , CH 2 C ⁇ 2 CH 3 , SCH 3 , CH 2 SCH 3 , S(0)CH 3 , CH 2 S(0)CH 3 , S(0) 2 CH 3 , CH 2 S(0) 2 CH 3 , C(0)NH 2 , CH 2 C(0)NH 2 , S0 2 NH 2 , CH 2 S0 2 NH 2 , NHS0 2 CH 3 , CH 2 NHS0 2 CH 3 ,
- R 2 at each occurrence, is selected from H, CH 3 , CH 2 CH 3 ,
- R 2 and R 2a together with the atom to which they are attached, combine to form a 5 or 6 membered saturated, partially saturated or unsaturated ring substituted with 0-1 R b and consisting of: 0-1 additional heteroatoms selected from the group consisting of N, 0, and S(0) p ;
- R 2b at each occurrence, is selected from 0CH 3 , 0CH 2 CH 3 , CH 3 , and CH 2 CH 3 ;
- R c at each occurrence, is selected from OH, 0CH 3 , OCHCH 3 , CH 3 , and CH 2 CH 3 ;
- the present invention provides a novel compound, wherein the compound is selected from:
- G is selected from:
- A-B is selected from:
- the present invention provides a novel compound, wherein the compound is selected from:
- P A is -G
- A-B is selected from:
- the present invention provides a novel compound, wherein the compound is selected from the group :
- ring M is 1,2 substituted by M 4 and P 4 and Z is (CH 2 )NR 3 , NR 3 (CH 2 ),
- the present invention provides a novel method for treating a thromboembolic disorder, comprising: administering to a patient in need thereof a therapeutically effective amount of a compound of the present invention or a pharmaceutically acceptable salt form thereof .
- the present invention provides a novel method, wherein the thromboembolic disorder is selected from the group consisting of arterial cardiovascular thromboembolic disorders, venous cardiovascular thromboembolic disorders, and thromboembolic disorders in the chambers of the heart.
- the present invention provides a novel method, wherein the thromboembolic disorder is selected from unstable angina, an acute coronary syndrome, first myocardial infarction, recurrent myocardial infarction, ischemic sudden death, transient ischemic attack, stroke, atherosclerosis, peripheral occlusive arterial disease, venous thrombosis, deep vein thrombosis, thrombophlebitis, arterial embolism, coronary arterial thrombosis, cerebral arterial thrombosis, cerebral embolism, kidney embolism, pulmonary embolism, and thrombosis resulting from (a) prosthetic valves or other implants, (b) indwelling catheters, (c) stents, (d) cardiopulmonary bypass, (e) hemodialysis, or (f) other procedures in which blood is exposed to an artificial surface that promotes thrombosis.
- the thromboembolic disorder is selected from unstable angina, an acute coronary syndrome, first myocardial infarction,
- the present invention provides a novel method of treating a patient in need of thromboembolic disorder treatment, comprising: administering a compound of the present invention or a pharmaceutically acceptable salt form thereof in an amount effective to treat a thromboembolic disorder
- the present invention provides a novel method, comprising: administering a compound of the present invention or a pharmaceutically acceptable salt form thereof in an amount effective to treat a thromboembolic disorder.
- the present invention provides a novel method for treating a thromboembolic disorder, comprising: administering to a patient in need thereof a therapeutically effective amount of a first and second therapeutic agent, wherein the first therapeutic agent is compound of the present invention or a pharmaceutically acceptable salt thereof and the second therapeutic agent is at least one agent selected from a second factor Xa inhibitor, an anti-coagulant agent, an anti-platelet agent, a thrombin inhibiting agent, a thrombolytic agent, and a fibrinolytic agent.
- the present invention provides a novel method, wherein the second therapeutic agent is at least one agent selected from warfarin, unfractionated heparin, low molecular weight heparin, synthetic pentasaccharide, hirudin, argatrobanas , aspirin, ibuprofen, naproxen, sulindac, indomethacin, mefenamate, droxicam, diclofenac, sulfinpyrazone, piroxicam, ticlopidine, clopidogrel, tirofiban, eptifibatide, abciximab, melagatran, disulfatohirudin, tissue plasminogen activator, modified tissue plasminogen activator, anistreplase, urokinase, and streptokinase.
- the second therapeutic agent is at least one agent selected from warfarin, unfractionated heparin, low molecular weight heparin, synthetic pentasaccharide,
- the present invention provides a novel method, wherein the second therapeutic agent is at least one anti-platelet agent.
- the present invention provides a novel method, wherein the anti-platelet agent is aspirin and clopidogrel.
- the present invention provides a novel method, wherein the anti-platelet agent is clopidogrel .
- the present invention provides a novel article of manufacture, comprising:
- composition located within the first container, wherein the composition, comprises: a first therapeutic agent, comprising: a compound of the present invention or a pharmaceutically acceptable salt form thereof; and,
- the present invention provides a novel article of manufacture, further comprising: (d) a second container; wherein components (a) and (b) are located within the second container and component (c) is located within or outside of the second container.
- the present invention provides a novel article of manufacture, comprising:
- composition located within the first container, wherein the composition, comprises: a first therapeutic agent, comprising: a compound of the present invention or a pharmaceutically acceptable salt form thereof; and,
- the present invention provides a novel article of manufacture, further comprising:
- components (a) and (b) are located within the second container and component (c) is located within or outside of the second container.
- the present invention provides novel compounds as described above for use in therapy. In another embodiment, the present invention provides the use of novel compounds as described above for the manufacture of a medicament for the treatment of a thromboembolic disorder.
- the compounds herein described may have asymmetric centers .
- the molecular weight of compounds of the present invention is less than about 500, 550, 600, 650, 700, 750, or 800 grams per mole.
- the molecular weight is less than about 800 grams per mole. More preferably, the molecular weight is less than about 750 grams per mole. Even more preferably, the molecular weight is less than about 700 grams per mole.
- substituted, " as used herein, means that any one or more hydrogens on the designated atom is replaced with a selection from the indicated group, provided that the designated atom's normal valency is not exceeded, and that the substitution results in a stable compound.
- the present invention in general, does not cover groups such as N-halo, S(0)H, and S0 2 H.
- the present invention is intended to include all isotopes of atoms occurring in the present compounds .
- Isotopes include those atoms having the same atomic number but different mass numbers.
- isotopes of hydrogen include tritium and deuterium.
- isotopes of carbon include C-13 and C-14.
- any variable e.g., R 6
- its definition at each occurrence is independent of its definition at every other occurrence.
- R 6 at each occurrence is selected independently from the definition of R 6 .
- combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
- amines on the compounds of this invention can be converted to amine N-oxides by treatment with an oxidizing agent (e.g., MCPBA and/or ' hydrogen peroxides) to afford other compounds of this invention.
- an oxidizing agent e.g., MCPBA and/or ' hydrogen peroxides
- alkyl is intended to include both branched and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms.
- C ⁇ _ 6 alkyl is intended to include Ci, C 2 , C 3 , C 4 , C 5 , and CQ alkyl groups.
- alkyl include, but are not limited to, methyl, ethyl, n-propyl, i-propyl, n-butyl, s-butyl, t-butyl, n-pentyl, and s-pentyl.
- haloalkyl include, but are not limited to, trifluoromethyl, trichloromethyl, pentafluoroethyl, and pentachloroethyl .
- Alkoxy represents an alkyl group as defined above with the indicated number of carbon atoms attached through an oxygen bridge.
- C ⁇ _ 6 alkoxy is intended to include Ci, C 2 , C 3 , C 4 , C 5 , and Cg alkoxy groups.
- alkoxy include, but are not limited to, methoxy, ethoxy, n-propoxy, i-propoxy, n-butoxy, s-butoxy, t-butoxy, n-pentoxy, and s-pentoxy.
- Cycloalkyl is intended to include saturated ring groups, such as cyclopropyl, cyclobutyl, or cyclopentyl.
- C 3 _ 7 cycloalkyl is intended to include C 3 , C , C 5 , Cs, and C 7 cycloalkyl groups.
- Alkenyl is intended to include hydrocarbon chains of either straight or branched configuration and one or more unsaturated carbon-carbon bonds that may occur in any stable point along the chain, such as ethenyl and propenyl .
- C_ 6 alkenyl is intended to include C 2 , C 3 , C 4 , C 5 , and C 6 alkenyl groups.
- Alkynyl is intended to include hydrocarbon chains of either straight or branched configuration and one or more triple carbon-carbon bonds that may occur in any stable point along the chain, such as ethynyl and propynyl .
- C _ 6 Alkynyl is intended to include C 2 , C 3 , C 4 , C 5 , and C 6 alkynyl groups.
- Halo or "halogen” as used herein refers to fluoro, chloro, bromo, and iodo; and "counterion” is used to represent a small, negatively charged species such as chloride, bromide, hydroxide, acetate, and sulfate.
- carrier or “carbocyclic residue” is intended to mean any stable 3, 4, 5, 6, or 7-membered monocyclic or bicyclic or 7, 8, 9, 10, 11, 12, or 13-membered bicyclic or tricyclic ring, any of which may be saturated, partially unsaturated, or unsaturated
- carbocycles include, but are not limited to, cyclopropyl, cyclobutyl, cyclobutenyl, cyclopentyl, cyclopentenyl, cyclohexyl, cycloheptenyl, cycloheptyl, cycloheptenyl, adamantyl, cyclooctyl, cyclooctenyl, cyclooctadienyl, [3.3.0]bicyclooctane,
- bridged rings are also included in the definition of carbocycle (e.g., [2.2.2]bicyclooctane) .
- a bridged ring occurs when one or more carbon atoms link two non-adjacent carbon atoms .
- Preferred bridges are one or two carbon atoms . It is noted that a bridge always converts a monocyclic ring into a tricyclic ring. When a ring is bridged, the substituents recited for the ring may also be present on the bridge.
- heterocycle or “heterocyclic group” is intended to mean a stable 5, 6, or 7-membered monocyclic or bicyclic or 7, 8, 9, or 10- membered bicyclic heterocyclic ring which is saturated, partially unsaturated or unsaturated (aromatic) , and which consists of carbon atoms and 1, 2, 3, or 4 ring heteroatoms independently selected from the group consisting of N, 0 and S and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring.
- the nitrogen and sulfur heteroatoms may optionally be oxidized (i.e., N ⁇ O and S(0) p ) .
- the nitrogen atom may be substituted or unsubstituted (i.e., N or NR wherein R is H or another substituent, if defined) .
- the heterocyclic ring may be attached to its pendant group at any heteroatom or carbon atom that results in a stable structure.
- the heterocyclic rings described herein may be substituted on carbon or on a nitrogen atom if the resulting compound is stable.
- a nitrogen in the heterocycle may optionally be quaternized. It is preferred that when the total number of S and 0 atoms in the heterocycle exceeds 1, then these heteroatoms are not adjacent to one another. It is preferred that the total number of S and 0 atoms in the heterocycle is not more than 1.
- aromatic heterocyclic group or “heteroaryl” is intended to mean a stable 5, 6, or 7-membered monocyclic or bicyclic or 7, 8, 9, or 10-membered bicyclic heterocyclic aromatic ring which consists of carbon atoms and 1, 2, 3, or 4 heteroatoms independently selected from the group consisting of N, 0 and S.
- the nitrogen atom may be substituted or unsubstituted (i.e., N or NR wherein R is H or another substituent, if defined) .
- the nitrogen and sulfur heteroatoms may optionally be oxidized (i.e., N ⁇ O and S(0) p ) .
- bridged rings are also included in the definition of heterocycle.
- a bridged ring occurs when one or more atoms (i.e., C, 0, N, or S) link two non-adjacent carbon or nitrogen atoms.
- Preferred bridges include, but are not limited to, one carbon atom, two carbon atoms, one nitrogen atom, two nitrogen atoms, and a carbon-nitrogen group. It is noted that a bridge always converts a monocyclic ring into a tricyclic ring. When a ring is bridged, the substituents recited for the ring may also be present on the bridge .
- heterocycles include, but are not limited to, acridinyl, azocinyl, benzimidazolyl, benzofuranyl, benzothiofuranyl, benzothiophenyl, benzoxazolyl, benzoxazolinyl, benzthiazolyl, benztriazolyl, benztetrazolyl, benzisoxazolyl, benzisothiazolyl, benzimidazolinyl, carbazolyl, 4aff-carbazolyl, carbolinyl, chromanyl, chromenyl, cinnolinyl, decahydroquinolinyl,
- phrases "pharmaceutically acceptable” is employed herein to refer to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- pharmaceutically acceptable salts refer to derivatives of the disclosed compounds wherein the parent compound is modified by making acid or base salts thereof.
- pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like.
- the pharmaceutically acceptable salts include the conventional non-toxic salts or the quaternary ammonium salts of the parent compound formed, for example, from nontoxic inorganic or organic acids.
- such ⁇ consult «£1 O 03/026652 conventional non-toxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like; and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pa oic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicyclic, sulfanilic, 2- acetoxybenzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, isethionic, and the like.
- inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like
- organic acids such as acetic, propionic,
- the pharmaceutically acceptable salts of the present invention can be synthesized from the parent compound that contains a basic or acidic moiety by conventional chemical methods.
- such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, non-aqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are preferred.
- Lists of suitable salts are found in Remington 's Pharmaceutical Sciences, 17th ed. , Mack Publishing Company, Easton, PA, 1985, p. 1418, the disclosure of which is hereby incorporated by reference.
- prodrugs are known to enhance numerous desirable qualities of pharmaceuticals (e.g., solubility, bioavailability, manufacturing, etc.) the compounds of the present invention may be delivered in prodrug form.
- the present invention is intended to cover prodrugs of the presently claimed compounds, methods of delivering the same and compositions containing the same.
- Prodrugs are intended to include any covalently bonded carriers that release an active parent drug of the present invention in vivo when such prodrug is administered to a mammalian subject.
- Prodrugs the present invention are prepared by modifying functional groups present in the compound in such a way that the modifications are cleaved, either in routine manipulation or in vivo, to the parent compound.
- Prodrugs include compounds of the present invention wherein a hydroxy, amino, or sulfhydryl group is bonded to any group that, when the prodrug of the present invention is administered to a mammalian subject, it cleaves to form a free hydroxyl, free amino, or free sulfhydryl group, respectively.
- Examples of prodrugs include, but are not limited to, acetate, formate, and benzoate derivatives of alcohol and amine functional groups in the compounds of the present invention.
- “Stable compound” and “stable structure” are meant to indicate a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent. It is preferred that there presently recited compounds do not contain a N-halo, S(0) 2 H, or S(0)H group.
- Substituted is intended to indicate that one or more hydrogens on the atom indicated in the expression using “substituted” is replaced with a selection from the indicated group (s), provided that the indicated atom's normal valency is not exceeded, and that the substitution results in a stable compound.
- treating cover the treatment of a disease-state in a mammal, particularly in a human, and include: (a) preventing the disease-state from occurring in a mammal, in particular, when such mammal is predisposed to the disease-state but has not yet been diagnosed as having it; (b) inhibiting the disease-state, i.e., arresting it development; and/or (c) relieving the disease-state, i.e., causing regression of the disease state.
- Therapeutically effective amount is intended to include an amount of a compound of the present invention that is effective when administered alone or in combination to inhibit factor Xa.
- “Therapeutically effective amount” is also intended to include an amount of the combination of compounds claimed that is effective to inhibit factor Xa.
- the combination of compounds is preferably a synergistic combination. Synergy, as described, for example, by Chou and Talalay, Adv. Enzyme Regul . 1984, 22:27-55, occurs when the effect (in this case, inhibition of factor Xa) of the compounds when administered in combination is greater than the additive effect of the compounds when administered alone as a single agent. In general, a synergistic effect is most clearly demonstrated at sub-optimal concentrations of the compounds . Synergy can be in terms of lower cytotoxicity, increased antithrombotic effect, or some other beneficial effect of the combination compared with the individual components .
- the compounds of the present invention can be prepared in a number of ways known to one skilled in the art of organic synthesis.
- the compounds of the present invention can be synthesized using the methods described below, together with synthetic methods known in the art of synthetic organic chemistry, or by variations thereon as appreciated by those skilled in the art. Preferred methods include, but are not limited to, those described below.
- the reactions are performed in a solvent appropriate to the reagents and materials employed and suitable for the transformations being effected. It will be understood by those skilled in the art of organic synthesis that the functionality present on the molecule should be consistent with the transformations proposed. This will sometimes require a judgment to modify the order of the synthetic steps or to select one particular process scheme over another in order to obtain a desired compound of the invention.
- G-G- L —P—M (acid chloride, acid, sulfonylchloride, amino, alkylhalide, etc)
- Intermediates A-B wherein the B group contains an oxidizable group can be obtained by oxidation, e.g. S to SO and S0 2 .
- the pyridone analogs can also be prepared via the Ullman methodology.
- the Ullman coupling can also be applied to prepare urea analogs shown in scheme 3.
- Aminopyridyl and aminopyrimidyl A-B analogs can also be prepared using routes similar to those of scheme 2-4.
- Piperidone A-B intermediates shown above can also be can be further elaborated to afford other compounds of the present invention by numerous methods known to those skilled in the art (e.g., see scheme 5).
- Additional A-B intermediates can be synthesized by the chemical manipulation of the amino functionality of the compounds described above (see Scheme 6) .
- A-B intermediates can be synthesized by the methods shown in scheme 7.
- the iodo-ester intermediate can be subjected to the Ullman and/or the Buchwald coupling methodologies to afford A-B intermediates.
- Non-aromatic intermediates as shown in scheme 8 can be synthesized via procedures known to those skilled in the art . These intermediates can than be further manipulated to incorporate R a via procedures previously described.
- R OH, SH, NH 2 , Br, Ester, etc
- Schemes 2-9 describe methods of preparing A-B intermediates that can then be coupled with other appropriate intermediates to form compounds of the present invention.
- the halogenated intermediates illustrated in the schemes shown above when subjected to the Ullman or the Buchwald-Goldman coupling methodologies afford compounds of this invention.
- Schemes 2-13 describe how to make the A-B moieties of the present invention and how to couple them to prepare compounds of the present invention.
- the Z group may or may not be present depending on how the A-B group is coupled.
- the coupling portion of the A-B group could (a) be displaced by the incoming Z or M group, (b) become the Z group, or (c) be incorporated into ring M.
- the remaining portions of the compounds of the present invention, G-Gi-P-M-Z, G-G ⁇ M-P-Z, G-Gi-P-M, G-Gi-M-P, G-G ⁇ M-Z, and G-G ⁇ M can be prepared using methods known to those of ordinary skill in the art.
- G-Gi-P-M-Z 60/278,165 for starting materials and intermediates to form the present G-Gi-P-M-Z, G-Gi-M-P-Z, G-Gi-P-M-Z-A, and/or G-
- G ⁇ M-P-Z-A groups to which the present B and/or A-B groups can be coupled.
- G is a ring substituted with a non-basic group
- one of ordinary skill in the art can look to US 5,998,424, WO00/39131, WO00/059902, WO01/32628, USSN 09/892,319, USSN 60/313,552, USSN 60/246,108, USSN 60/246,125, USSN 60/292,665, USSN 60/278,173, and USSN 60/278,165 for starting materials and intermediates to form the present G-Gi-P-M-Z, G-Gi-M-P-Z, G- G-L-P-M-Z-A, and/or G-Gi-M-P-Z-A groups to which the present
- G is a bicyclic moiety, one of ordinary skill in the art can look to W098/57951 WO00/039108, WO00/39131, USSN 09/892,319, USSN 60/313,552, USSN 60/246,108, USSN
- Scheme 14 illustrates some of the numerous pyrrole intermediates that can be used to prepare compounds of the present invention (R z is the point of attachment for Z-A-B and can be H, a protecting group, a group modifiable to Z or Z-A, Z, Z-A, or A) .
- R z is the point of attachment for Z-A-B and can be H, a protecting group, a group modifiable to Z or Z-A, Z, Z-A, or A.
- Scheme 15 illustrates some of the numerous imidazole, triazole, and tetrazole intermediates that can be used to prepare compounds of the present invention. These intermediates are described in the above-noted patents and publications.
- V is nitro, amino, thio, hydroxy, sulfonic acid, sulfonic ester, sulfonyl chloride, ester, acid, or halide.
- U is aldehyde, ester, acid, amide, amino, thio, hydroxy, sulfonic acid, sulfonic ester, sulfonyl chloride, or methylene halide.
- Scheme 16 shows some of the numerous pyrazole intermediates that can be used to prepare compounds of the present invention. These intermediates are described in the above-noted patents and publications.
- Scheme 17 depicts some of the numerous oxazole, thiazole, isoxazole, oxadiazole, and thiadiazole intermediates that can be used to prepare compounds of the present invention. These intermediates are described in the above-noted patents and publications.
- V is nitro, amino, ester, or acid.
- Scheme 18 illustrates two intermediates useful for making a compound of the present invention wherein ring P is fused to ring M.
- Scheme 18 also illustrates a number of bicyclic compounds that can be made from these intermediates or derivatives thereof. These intermediates and their modification are described in the above-noted patents and publications .
- Scheme 19 depicts another intermediate useful for making a compound of the present invention wherein ring P is fused to ring M.
- Scheme 19 also illustrates a number of bicyclic compounds that can be made from this intermediate or derivatives thereof (e.g., the corresponding cyclohexenone) .
- U is OH or morpholine and V is H or C(0)R la .
- This intermediate, derivatives thereof, and their modification are described in the above-noted patents and publications.
- Scheme 20 shows another intermediate useful for making a compound of the present invention wherein ring P is fused to ring M.
- Scheme 20 also illustrates a number of bicyclic compounds that can be made from this intermediate or derivatives thereof. This intermediate, derivatives thereof, and their modification are described in the above- noted patents and publications .
- Scheme 21 illustrates a number of other bicyclic rings that are considered to be part of the present bicyclic group, rings P-M.
- Scheme 21 also describes a method of converting the shown rings to compounds of the present invention. As one of ordinary skill in the art would recognize, this method would be applicable to other heterobicyclics not shown.
- Scheme 23 depicts some of the numerous 6-membered aromatic ring intermediates that can be used to prepare compounds of the present invention. These intermediates are described in the above-noted patents and publications .
- V is nitro, protected sulfonamide, or ester group and is a precursor of group Z of the present invention.
- Benzo-fused dihydro-pyridone intermediates of the present invention can be prepared from readily available starting materials as shown in Scheme 24.
- anthranilates can be coupled with a suitable amine, aniline, or aminopyrimidyl to afford the corresponding benzamide.
- the benzamides can then be coupled with an appropriate B-A-V (wherein V is a acid chloride derivative, an alkyl halide, or a sulfonyl chloride) to afford additional compounds of the present invention (see scheme 29) .
- the compounds of this invention with general structure of Formula I can be synthesized by using similar methods as described previously and by those skilled in the art.
- One diastereomer of a compound of Formula I may display better activity compared with the others.
- the following stereochemistries are considered to be a part of the present invention.
- racemic material can be achieved by HPLC using a chiral column or by a resolution using a resolving agent such as camphonic chloride as in Wilen, S. H. Tables of Resolving Agents and Optical Resolutions 1972, 308 pp or using enantiomerically pure acids and bases.
- a chiral compound of Formula I may also be directly synthesized using a chiral catalyst or a chiral ligand, e.g., Jacobsen, E. Ace . Chem. Res . 2000, 33 , 421-431 or using other enantio- and diastereo-selective reactions and reagents known to one skilled in the art of asymmetric synthesis.
- thromboembolic disorders are a circulatory disease caused by blood clots (i.e., diseases involving fibrin formation, platelet activation, and/or platelet aggregation) .
- blood clots i.e., diseases involving fibrin formation, platelet activation, and/or platelet aggregation
- thromboembolic disorders as used herein includes arterial cardiovascular thromboembolic disorders, venous cardiovascular thromboembolic disorders, and thromboembolic disorders in the chambers of the heart.
- thromboembolic disorders also includes specific disorders selected from, but not limited to, unstable angina or other acute coronary syndromes, first or recurrent myocardial infarction, ischemic sudden death, transient ischemic attack, stroke, atherosclerosis, peripheral occlusive arterial disease, venous thrombosis, deep vein thrombosis, thrombophlebitis, arterial embolism, coronary arterial thrombosis, cerebral arterial thrombosis, cerebral embolism, kidney embolism, pulmonary embolism, and thrombosis resulting from (a) prosthetic valves or other implants, (b) indwelling catheters, (c) stents, (d) cardiopulmonary bypass, (e) hemodialysis, or (f) other procedures in which blood is exposed to an artificial surface that promotes thrombosis.
- thrombosis includes occlusion (e.g. after a bypass) and reocclusion (e.g., during or after percutaneous transluminal coronary angioplasty) .
- the thromboembolic disorders may result from conditions including but not limited to atherosclerosis, surgery or surgical complications, prolonged immobilization, arterial fibrillation, congenital thrombophilia, cancer, diabetes, effects of medications or hormones, and complications of pregnancy.
- the anticoagulant effect of compounds of the present invention is believed to be due to inhibition of factor Xa or thrombin.
- Ki values of Ki were determined by allowing 0.2-0.5 nM human factor Xa (Enzyme Research Laboratories, South Bend, IN) to react with the substrate (0.20 mM - 1 mM) in the presence of inhibitor. Reactions were allowed to go for 30 min and the velocities (rate of absorbance change vs. time) were measured in the time frame of 25-30 min. The following relationship was used to calculate Ki values:
- v 0 is the velocity of the control in the absence of inhibitor
- v s is the velocity in the presence of inhibitor
- I is the concentration of inhibitor
- Ki is the dissociation constant of the enzyme: inhibitor complex
- S is the concentration of substrate
- K is the Michaelis constant
- Preferred compounds of the present invention have K 's of ⁇ 1 uM. More preferred compounds of the present invention have Ki's of ⁇ 0.1 ⁇ M. Even more preferred compounds of the present invention have Ki's of ⁇ 0.01 ⁇ M. Still more preferred compounds of the present invention have Ki's of ⁇ 0.001 ⁇ M. Using the methodology described above, a number of compounds of the present invention were found to exhibit Ki's of ⁇ 10 ⁇ M, thereby confirming the utility of the compounds of the present invention as effective Xa inhibitors .
- the antithrombotic effect of compounds of the present invention can be demonstrated in a rabbit arterio-venous (AV) shunt thrombosis model.
- AV arterio-venous
- rabbits weighing 2-3 kg anesthetized with a mixture of xylazine (10 mg/kg i.m.) and ketamine (50 mg/kg i.m.) are used.
- a saline-filled AV shunt device is connected between the femoral arterial and the femoral venous cannulae.
- the AV shunt device consists of a piece of 6-cm tygon tubing that contains a piece of silk thread. Blood will flow from the femoral artery via the AV-shunt into the femoral vein.
- the exposure of flowing blood to a silk thread will induce the formation of a significant thrombus.
- the shunt is disconnected and the silk thread covered with thrombus is weighed.
- Test agents or vehicle will be given (i.v., i.p., s.c, or orally) prior to the opening of the AV shunt.
- the percentage inhibition of thrombus formation is determined for each treatment group.
- the compounds of the present invention may also be useful as inhibitors of serine proteases, notably human thrombin, Factor Vila, Factor IXa, Factor XIa, urokinase, plasma kallikrein, and plasmin. Because of their inhibitory action, these compounds are indicated for use in the prevention or treatment of physiological reactions, blood coagulation and inflammation, catalyzed by the aforesaid class of enzymes. Specifically, the compounds have utility as drugs for the treatment of diseases arising from elevated thrombin activity such as myocardial infarction, and as reagents used as anticoagulants in the processing of blood to plasma for diagnostic and other commercial purposes.
- serine proteases notably human thrombin, Factor Vila, Factor IXa, Factor XIa, urokinase, plasma kallikrein, and plasmin. Because of their inhibitory action, these compounds are indicated for use in the prevention or treatment of physiological reactions, blood coagulation and inflammation, cat
- Some compounds of the present invention were shown to be direct acting inhibitors of the serine protease thrombin by their ability to inhibit the cleavage of small molecule substrates by thrombin in a purified system.
- In vitro inhibition constants were determined by the method described by Kettner et al. in J " . Biol . Chem. 265, 18289- 18297 (1990) , herein incorporated by reference.
- thrombin-mediated hydrolysis of the chromogenic substrate S2238 Helena Laboratories, Beaumont, TX
- Addition of an inhibitor to the assay mixture results in decreased absorbance and is indicative of thrombin inhibition.
- Human thrombin (Enzyme Research Laboratories, Inc., South Bend, IN) at a concentration of 0.2 nM in 0.10 M sodium phosphate buffer, pH 7.5, 0.20 M NaCl, and 0.5% PEG 6000, was incubated with various substrate concentrations ranging from 0.20 to 0.02 mM. After 25 to 30 min of incubation, thrombin activity was assayed by monitoring the rate of increase in absorbance at 405 nm that arises owing to substrate hydrolysis. Inhibition constants were derived from reciprocal plots of the reaction velocity as a function of substrate concentration using the standard method of Lineweaver and Burk. Using the methodology described above, some compounds of this invention were evaluated and found to exhibit a K of less than 10 ⁇ M, thereby confirming the utility of the compounds of the present invention as effective thrombin inhibitors.
- the compounds are administered to a mammal in a therapeutically effective amount.
- therapeutically effective amount it is meant an amount of a compound of the present invention that, when administered alone or in combination with an additional therapeutic agent to a mammal, is effective to treat a thromboembolic condition or disease.
- the compounds of the present invention can be administered alone or in combination with one or more additional therapeutic agents.
- administered in combination or “combination therapy” it is meant that a compound of the present invention and one or more additional therapeutic agents are administered concurrently to the mammal being treated.
- each component may be administered at the same time or sequentially in any order at different points in time.
- each component may be administered separately but sufficiently closely in time so as to provide the desired therapeutic effect.
- Additional therapeutic agents include other anticoagulant or coagulation inhibitory agents, anti-platelet or platelet inhibitory agents, thrombin inhibitors, thrombolytic or fibrinolytic agents, anti-arrythmic agents, anti-hypertensive agents, calcium channel blockers (L-type and T-type) , cardiac glycosides, diruetics, mineralocorticoid receptor antagonists, phospodiesterase inhibitors, cholesterol/lipid lowering agents and lipid profile therapies, anti-diabetic agents, anti-depressants, anti-inflammatory agents (steroidal and non-steroidal) , anti-osteoporosis agents, hormone replacement therapies, oral contraceptives, anti-obesity agents, anti-anxiety agents, anti-proliferative agents, anti-tumor agents, anti- ulcer and gastroesophageal reflux disease agents, growth hormone and/or growth hormone secretagogues, thyroid mimetics (including thyroid receptor antagonist) , anti- infective agents, anti-viral agents, anti-bacterial agents
- anticoagulant agents or coagulation inhibitory agents
- warfarin and heparin either unfractionated heparin or any commercially available low molecular weight heparin
- synthetic pentasaccharide synthetic pentasaccharide
- direct acting thrombin inhibitors including hirudin and argatrobanas well as other factor Xa inhibitors such as those described in the publications identified above under Background of the Invention.
- anti-platelet agents denotes agents that inhibit platelet function, for example by inhibiting the aggregation, adhesion or granular secretion of platelets.
- Agents include, but are not limited to, the various known non-steroidal anti-inflammatory drugs (NSAIDS) such as aspirin, ibuprofen, naproxen, sulindac, indomethacin, mefenamate, droxicam, diclofenac, sulfinpyrazone, piroxicam, and pharmaceutically acceptable salts or prodrugs thereof.
- NSAIDS non-steroidal anti-inflammatory drugs
- aspirin acetylsalicyclic acid or ASA
- piroxicam are preferred.
- Suitable platelet inhibitory agents include lib/Ilia antagonists (e.g., tirofiban, eptifibatide, and abciximab), thromboxane-A2-receptor antagonists (e.g., ifetroban) , thromboxane-A2-synthetase inhibitors, PDE-III inhibitors
- anti-platelet agents or platelet inhibitory agents
- platelet inhibitory agents is also intended to include ADP (adenosine diphosphate) receptor antagonists, preferably antagonists of the purinergic receptors P 2 Y 1 and p 2 ⁇ i 2 ' with P 2 Yi 2 being even more preferred.
- Preferred P 2 Y 12 receptor antagonists include ticlopidine and clopidogrel, including pharmaceutically acceptable salts or prodrugs thereof. Clopidogrel is an even more preferred agent. Ticlopidine and clopidogrel are also preferred compounds since they are known to be gentle on the gastro-intestinal tract in use.
- thrombin inhibitors denotes inhibitors of the serine protease thrombin.
- various thrombin-mediated processes such as thrombin-mediated platelet activation (that is, for example, the aggregation of platelets, and/or the granular secretion of plasminogen activator inhibitor-1 and/or serotonin) and/or fibrin formation are disrupted.
- thrombin inhibitors are known to one of skill in the art and these inhibitors are contemplated to be used in combination with the present compounds.
- Such inhibitors include, but are not limited to, boroarginine derivatives, boropeptides , heparins, hirudin, argatroban, and melagatran, including pharmaceutically acceptable salts and prodrugs thereof .
- Boroarginine derivatives and boropeptides include N-acetyl and peptide derivatives of boronic acid, such as C-terminal ⁇ -aminoboronic acid derivatives of lysine, ornithine, arginine, homoarginine and corresponding isothiouronium analogs thereof.
- hirudin includes suitable derivatives or analogs of hirudin, referred to herein as hirulogs, such as disulfatohirudin.
- thrombolytics or fibrinolytic agents denote agents that lyse blood clots (thrombi) .
- agents include tissue plasminogen activator (natural or recombinant) and modified forms thereof, anistreplase, urokinase, streptokinase, tenecteplase (TNK) , lanoteplase (nPA) , factor Vila inhibitors, PAI-1 inhibitors (i.e., inactivators of tissue plasminogen activator inhibitors) , alpha2-antiplasmin inhibitors, and anisoylated plasminogen streptokinase activator complex, including pharmaceutically acceptable salts or prodrugs thereof.
- anistreplase refers to anisoylated plasminogen streptokinase activator complex, as described, for example, in EP 028,489, the disclosure of which is hereby incorporated herein by reference herein.
- urokinase as used herein, is intended to denote both dual and single chain urokinase, the latter also being referred to herein as prourokinase.
- Suitable anti-arrythmic agents for use in combination with the present compounds include: Class I agents (such as propafenone) ; Class II agents (such as carvadiol and propranolol) ; Class III agents (such as sotalol, dofetilide, amiodarone, azimilide and ibutilide) ; Class IV agents (such as ditiazem and verapamil) ; K + channel openers such as I ⁇ c inhibitors, and I u r inhibitors (e.g., compounds such as those disclosed in WO01/40231) .
- Class I agents such as propafenone
- Class II agents such as carvadiol and propranolol
- Class III agents such as sotalol, dofetilide, amiodarone, azimilide and ibutilide
- Class IV agents such as ditiazem and verapamil
- K + channel openers such as I ⁇ c inhibitors, and I u
- Suitable anti-hypertensive agents for use in combination with the compounds of the present invention include: alpha adrenergic blockers; beta adrenergic blockers; calcium channel blockers (e.g., diltiazem, verapamil, nifedipine, amlodipine and mybefradii) ; diruetics (e.g., chlorothiazide, hydrochlorothiazide, flumethiazide, hydroflumethiazide, bendroflumethiazide, methylchlorothiazide, trichloromethiazide, polythiazide, benzthiazide, ethacrynic acid tricrynafen, chlorthalidone, furosemide, musolimine, bumetanide, triamtrenene, amiloride, spironolactone) ; renin inhibitors; ACE inhibitors (e.g., captopril, zof
- NEP neutral endopeptidase
- vasopepsidase inhibitors dual NEP-ACE inhibitors
- omapatrilat, gemopatrilat and nitrates e.g., omapatrilat, gemopatrilat and nitrates
- Suitable calcium channel blockers for use in combination with the compounds of the present invention include diltiazem, verapamil, nifedipine, amlodipine and mybefradil.
- Suitable diruetics for use in combination with the compounds of the present invention include: chlorothiazide, hydrochlorothiazide, flumethiazide, hydroflumethiazide, bendroflumethiazide, methylchlorothiazide, trichloromethiazide, polythiazide, benzthiazide, ethacrynic acid tricrynafen, chlorthalidone, furosemide, musolimine, bumetanide, triamtrenene, amiloride, and spironolactone.
- Suitable phospodiesterase inhibitors for use in combination with the compounds of the present invention include: PDE III inhibitors (such as cilostazol) ; and PDE V inhibitors (such as sildenafil) .
- Suitable cholesterol/lipid lowering agents and lipid profile therapies for use in combination with the compounds of the present invention include: HMG-CoA reductase inhibitors (e.g., pravastatin, lovastatin, atorvastatin, simvastatin, fluvastatin, NK-104 (a.k.a. itavastatin, or nisvastatin or nisbastatin) and ZD-4522 (a.k.a.
- HMG-CoA reductase inhibitors e.g., pravastatin, lovastatin, atorvastatin, simvastatin, fluvastatin, NK-104 (a.k.a. itavastatin, or nisvastatin or nisbastatin)
- ZD-4522 a.k.a.
- rosuvastatin, or atavastatin or visastatin) ) ; squalene synthetase inhibitors; fibrates; bile acid sequestrants (such as questran) ; ACAT inhibitors; MTP inhibitors; lipooxygenase inhibitors; choesterol absorption inhibitors; and cholesterol ester transfer protein inhibitors (e.g., CP-529414) .
- Suitable anti-diabetic agents for use in combination with the compounds of the present invention include: biguanides (e.g., metformin) ; glucosidase inhibitors (e.g., acarbose) ; insulins (including insulin secretagogues or insulin sensitizers) ; meglitinides (e.g., repaglinide) ; sulfonylureas (e.g., glimepiride, glyburide and glipizide) ; biguanide/glyburide combinations (e.g., glucovance) , thiozolidinediones (e.g., troglitazone, rosiglitazone and pioglitazone), PPAR-alpha agonists, PPAR- gamma agonists, PPAR alpha/gamma dual agonists, SGLT2 inhibitors, inhibitors of fatty acid binding protein (aP2) such as those
- suitable anti-depressant agents for use in combination with the compounds of the present invention include nefazodone and sertraline.
- suitable anti-inflammatory agents for use in combination with the compounds of the present invention include: prednisone; dexamethasone; enbrel; protien tyrosine kinase (PTK) inhibitors; cyclooxygenase inhibitors (including NSAIDs , and COX-1 and/or COX-2 inhibitors); aspirin; indomethacin; ibuprofen; prioxicam; naproxen; celecoxib; and/or rofecoxib.
- Suitable anti-osteoporosis agents for use in combination with the compounds of the present invention include alendronate and raloxifene.
- suitable hormone replacement therapies for use in combination with the compounds of the present invention include estrogen (e.g., congugated estrogens) and estradiol .
- suitable anti-coagulants for use in combination with the compounds of the present invention include heparins (e.g., unfractioned and low molecular weight heparins such as enoxaparin and dalteparin) .
- Suitable anti-obesity agents for use in combination with the compounds of the present invention include orlistat and aP2 inhibitors (such as those disclosed in WO00/59506) .
- Suitable anti-anxiety agents for use in combination with the compounds of the present invention include diazepam, lorazepam, buspirone, and hydroxyzine pamoate .
- Suitable anti-proliferative agents for use in combination with the compounds of the present invention include cyclosporin A, paclitaxel, adriamycin; epithilones, cisplatin, and carboplatin.
- Suitable anti-ulcer and gastroesophageal reflux disease agents for use in combination with the compounds of the present invention include famotidine, ranitidine, and omeprazole.
- Administration of the compounds of the present invention i.e., a first therapeutic agent
- at least one additional therapeutic agent i.e., a second therapeutic agent
- a lower dosage minimizes the potential of side effects, thereby providing an increased margin of safety. It is preferred that at least one of the therapeutic agents is administered in a sub-therapeutic dose.
- Sub-therapeutic is intended to mean an amount of a therapeutic agent that by itself does not give the desired therapeutic effect for the condition or disease being treated.
- Synergistic combination is intended to mean that the observed effect of the combination is greater than the sum of the individual agents administered alone.
- the compounds of the present invention are also useful as standard- or reference compounds, for example as a quality standard or control, in tests or assays involving the inhibition of factor Xa.
- Such compounds may be provided in a commercial kit, for example, for use in pharmaceutical research involving factor Xa.
- a compound of the present invention could be used as a reference in an assay to compare its known activity to a compound with an unknown activity. This would ensure the experimenter that the assay was being performed properly and provide a basis for comparison, especially if the test compound was a derivative of the reference compound.
- compounds according to the present invention could be used to test their effectiveness .
- the compounds of the present invention may also be used in diagnostic assays involving factor Xa.
- the presence of factor Xa in an unknown sample could be determined by addition of chromogenic substrate S2222 to a series of solutions containing test sample and optionally one of the compounds of the present invention. If production of pNA is observed in the solutions containing test sample, but not in the presence of a compound of the present invention, then one would conclude factor Xa was present.
- Compounds of the present invention may further be useful as diagnostic agents and adjuncts.
- the present compounds may be useful in maintaining whole and fractionated blood in the fluid phase such as required for analytical and biological testing.
- the present invention also encompasses an article of manufacture.
- article of manufacture is intended to include, but not be limited to, kits and packages.
- the article of manufacture of the present invention comprises: (a) a first container; (b) a pharmaceutical composition located within the first container, wherein the composition, comprises: a first therapeutic agent, comprising: a compound of the present invention or a pharmaceutically acceptable salt form thereof; and, (c) a package insert stating that the pharmaceutical composition can be used for the treatment of a thromboembolic disorder (as defined previously) .
- the package insert states that the pharmaceutical composition can be used in combination (as defined previously) with a second therapeutic agent to treat a thromboembolic disorder.
- the article of manufacture can further comprise: (d) a second container, wherein components (a) and (b) are located within the second container and component (c) is located within or outside of the second container. Located within the first and second containers means that the respective container holds the item within its boundaries.
- the first container is a receptacle used to hold a pharmaceutical composition.
- This container can be for manufacturing, storing, shipping, and/or individual/bulk selling.
- First container is intended to cover a bottle, jar, vial, flask, syringe, tube (e.g., for a cream preparation) , or any other container used to manufacture, hold, store, or distribute a pharmaceutical product.
- the second container is one used to hold the first container and, optionally, the package insert.
- the second container include, but are not limited to, boxes (e.g., cardboard or plastic), crates, cartons, bags (e.g., paper or plastic bags), pouches, and sacks.
- the package insert can be physically attached to the outside of the first container via tape, glue, staple, or another method of attachment, or it can rest inside the second container without any physical means of attachment to the first container.
- the package insert is located on the outside of the second container. When located on the outside of the second container, it is preferable that the package insert is physically attached via tape, glue, staple, or another method of attachment. Alternatively, it can be adjacent to or touching the outside of the second container without being physically attached.
- the package insert is a label, tag, marker, etc. that recites information relating to the pharmaceutical composition located within the first container.
- the information recited will usually be determined by the regulatory agency governing the area in which the article of manufacture is to be sold (e.g., the United States Food and Drug Administration) .
- the package insert specifically recites the indications for which the pharmaceutical composition has been approved.
- the package insert may be made of any material on which a person can read information contained therein or thereon.
- the package insert. is a printable material (e.g., paper, plastic, cardboard, foil, adhesive-backed paper or plastic, etc.) on which the desired information has been formed (e.g., printed or applied).
- the compounds of this invention can be administered in such oral dosage forms as tablets, capsules (each of which includes sustained release or timed release formulations) , pills, powders, granules, elixirs, tinctures, suspensions, syrups, and emulsions. They may also be administered in intravenous (bolus or infusion) , intraperitoneal, subcutaneous, or intramuscular form, all using dosage forms well known to those of ordinary skill in the pharmaceutical arts. They can be administered alone, but generally will be administered with a pharmaceutical carrier selected on the basis of the chosen route of administration and standard pharmaceutical practice.
- the dosage regimen for the compounds of the present invention will, of course, vary depending upon known factors, such as the pharmacodynamic characteristics of the particular agent and its mode and route of administration; the species, age, sex, health, medical condition, and weight of the recipient; the nature and extent of the symptoms; the kind of concurrent treatment; the frequency of treatment; the route of administration, the renal and hepatic function of the patient, and the effect desired.
- a physician or veterinarian can determine and prescribe the effective amount of the drug required to prevent, counter, or arrest the progress of the thromboembolic disorder.
- the daily oral dosage of each active ingredient when used for the indicated effects, will range between about 0.001 to 1000 mg/kg of body weight, preferably between about 0.01 to 100 mg/kg of body weight per day, and most preferably between about 1.0 to 20 mg/kg/day. Intravenously, the most preferred doses will range from about 1 to about 10 mg/kg/min during a constant rate infusion.
- Compounds of this invention may be administered in a single daily dose, or the total daily dosage may be administered in divided doses of two, three, or four times daily.
- Compounds of this invention can be administered in intranasal form via topical use of suitable intranasal vehicles, or via transdermal routes, using transdermal skin patches. When administered in the form of a transdermal delivery system, the dosage administration will, of course, be continuous rather than intermittent throughout the dosage regimen.
- the compounds are typically administered in admixture with suitable pharmaceutical diluents, excipients, or carriers (collectively referred to herein as pharmaceutical carriers) suitably selected with respect to the intended form of administration, that is, oral tablets, capsules, elixirs, syrups and the like, and consistent with conventional pharmaceutical practices .
- suitable pharmaceutical diluents, excipients, or carriers suitably selected with respect to the intended form of administration, that is, oral tablets, capsules, elixirs, syrups and the like, and consistent with conventional pharmaceutical practices .
- the active drug component can be combined with an oral, non-toxic, pharmaceutically acceptable, inert carrier such as lactose, starch, sucrose, glucose, methyl cellulose, magnesium stearate, dicalcium phosphate, calcium sulfate, mannitol, sorbitol and the like; for oral administration in liquid form, the oral drug components can be combined with any oral, non-toxic, pharmaceutically acceptable inert carrier such as ethanol, glycerol, water, and the like.
- suitable binders, lubricants, disintegrating agents, and coloring agents can also be incorporated into the mixture.
- Suitable binders include starch, gelatin, natural sugars such as glucose or beta-lactose, corn sweeteners, natural and synthetic gums such as acacia, tragacanth, or sodium alginate, carboxymethylcellulose, polyethylene glycol, waxes, and the like.
- Lubricants used in these dosage forms include sodium oleate, sodium stearate, magnesium stearate, sodium benzoate, sodium acetate, sodium chloride, and the like.
- Disintegrators include, without limitation, starch, methyl cellulose, agar, bentonite, xanthan gum, and the like.
- the compounds of the present invention can also be administered in the form of liposome delivery systems, such as small unilamellar vesicles, large unilamellar vesicles, and multilamellar vesicles.
- Liposomes can be formed from a variety of phospholipids, such as cholesterol, stearylamine, or phosphatidylcholines .
- Compounds of the present invention may also be coupled with soluble polymers as targetable drug carriers.
- soluble polymers can include polyvinylpyrrolidone, pyran copolymer, polyhydroxypropylmethacrylamide-phenol , polyhydroxyethylaspartamidephenol, or polyethyleneoxide- polylysine substituted with palmitoyl residues.
- the compounds of the present invention may be coupled to a class of biodegradable polymers useful in achieving controlled release -of a drug, for example, polylactic acid, polyglycolic acid, copolymers of polylactic and polyglycolic acid, polyepsilon caprolactone, polyhydroxy butyric acid, polyorthoesters, polyacetals, polydihydropyrans, polycyanoacylates, and crosslinked or amphipathic block copolymers of hydrogels.
- a class of biodegradable polymers useful in achieving controlled release -of a drug
- a drug for example, polylactic acid, polyglycolic acid, copolymers of polylactic and polyglycolic acid, polyepsilon caprolactone, polyhydroxy butyric acid, polyorthoesters, polyacetals, polydihydropyrans, polycyanoacylates, and crosslinked or amphipathic block copolymers of hydrogels.
- Dosage forms suitable for administration may contain from about 1 mg to about 100 mg of active ingredient per dosage unit.
- the active ingredient will ordinarily be present in an amount of about 0.5-95% by weight based on the total weight of the composition.
- Gelatin capsules may contain the active ingredient and powdered carriers, such as lactose, starch, cellulose derivatives, magnesium stearate, stearic acid, and the like. Similar diluents can be used to make compressed tablets . Both tablets and capsules can be manufactured as sustained release products to provide for continuous release of medication over a period of hours. Compressed tablets can be sugar coated or film coated to mask any unpleasant taste and protect the tablet from the atmosphere, or enteric coated for selective disintegration in the gastrointestinal tract.
- powdered carriers such as lactose, starch, cellulose derivatives, magnesium stearate, stearic acid, and the like. Similar diluents can be used to make compressed tablets . Both tablets and capsules can be manufactured as sustained release products to provide for continuous release of medication over a period of hours. Compressed tablets can be sugar coated or film coated to mask any unpleasant taste and protect the tablet from the atmosphere, or enteric coated for selective disintegration in the gastrointestinal tract.
- Liquid dosage forms for oral administration can contain coloring and flavoring to increase patient acceptance .
- water a suitable oil, saline, aqueous dextrose (glucose) , and related sugar solutions and glycols such as propylene glycol or polyethylene glycols are suitable carriers for parenteral solutions.
- Solutions for parenteral administration preferably contain a water soluble salt of the active ingredient, suitable stabilizing agents, and if necessary, buffer substances.
- Antioxidizing agents such as sodium bisulfite, sodium sulfite, or ascorbic acid, either alone or combined, are suitable stabilizing agents.
- citric acid and its salts and sodium EDTA are also used.
- parenteral solutions can contain preservatives, such as benzalkonium chloride, methyl-or propyl-paraben, and chlorobutanol .
- Suitable pharmaceutical carriers are described in Remington's Pharmaceutical Sciences, Mack Publishing Company, a standard reference text in this field.
- Representative useful pharmaceutical dosage-forms for administration of the compounds of this invention can be illustrated as follows:
- a large number of unit capsules can be prepared by filling standard two-piece hard gelatin capsules each with 100 mg of powdered active ingredient, 150 mg of lactose, 50 mg of cellulose, and 6 mg magnesium stearate.
- a mixture of active ingredient in a digestible oil such as soybean oil, cottonseed oil or olive oil may be prepared and injected by means of a positive displacement pump into gelatin to form soft gelatin capsules containing 100 mg of the active ingredient.
- the capsules should be washed and dried.
- Tablets Tablets may be prepared by conventional procedures so that the dosage unit is 100 mg of active ingredient, 0.2 mg of colloidal silicon dioxide, 5 mg of magnesium stearate, 275 mg of microcrystalline cellulose, 11 mg of starch and 98.8 mg of lactose. Appropriate coatings may be applied to increase palatability or delay absorption.
- a parenteral composition suitable for administration by injection may be prepared by stirring 1.5% by weight of active ingredient in 10% by volume propylene glycol and water. The solution should be made isotonic with sodium chloride and sterilized.
- aqueous suspension can be prepared for oral administration so that each 5 mL contain 100 mg of finely divided active ingredient, 200 mg of sodium carboxymethyl cellulose, 5 mg of sodium benzoate, 1.0 g of sorbitol solution, U.S.P, and 0.025 mL of vanillin.
- a daily dosage may be about 0.1 to 100 mg of the compound of Formula I and about 1 to 7.5 mg of the second anticoagulant, per kilogram of patient body weight.
- the compounds of this invention generally may be present in an amount of about 5 to 10 mg per dosage unit, and the second anti-coagulant in an amount of about 1 to 5 mg per dosage unit.
- a daily dosage may be about 0.01 to 25 mg of the compound of Formula I and about 50 to 150 mg of the anti-platelet agent, preferably about 0.1 to 1 mg of the compound of Formula I and about 1 to 3 mg of antiplatelet agents, per kilogram of patient body 5 weight .
- a daily dosage may be about 0.1 to 1 mg of the compound of Formula I, per. kilogram of patient body weight and, in the case of
- the usual dosage of the thrombolyic agent when administered alone may be reduced by about 70-80% when administered with a compound of Formula I.
- agents are administered with the compound of Formula I, generally the amount of each component in a typical daily dosage and typical dosage form may be reduced relative to the usual dosage of the agent when administered alone, in view of the additive or synergistic effect of the
- one active ingredient may be enteric coated.
- One of the active ingredients may also be coated with a material that affects a sustained-release throughout the gastrointestinal tract and also serves to minimize physical contact between the combined active ingredients .
- the sustained-released component can be additionally enteric coated such that the release of this component occurs only in the intestine.
- Still another approach would involve the formulation of a combination product in which the one component is coated with a sustained and/or enteric release polymer, and the other component is also coated with a polymer such as a low- viscosity grade of hydroxypropyl -methylcellulose (HPMC) or other appropriate materials as known in the art, in order to further separate the active components .
- the polymer coating serves to form an additional barrier to interaction with the other component.
- Part B The product from part A (50 mg) was dissolved in 20 mL of MeOH in a hydrogenation bottle. To the solution was added 5% Pd/C (20 mg) and one drop of TFA. The reaction mixture was put on hydrogenation Parr shaker at rt ' under 50 psi for 5 h. The reaction mixture was filtered through Celite . The filtrate was concentrated and purified via HPLC (C18 RP., 0.5% TFA, H20/MeCN gradient) to give 40 mg of the title compound as its TFA salt (65%). LRMS (ESI+) : 502.4 (M+H) + .
- Acetohydroxamic acid 54 mg, 0.72 mmol
- K 2 C0 3 0.2 g, 1.45 mmol
- the mixture was stirred at rt for 15 min, followed by addition of a solution of 2-fluoro-5- [7-oxo-6- [4- (2-oxo-l- piperidinyl)phenyl] -3- (trifluoromethyl) -4,5,6,7,- tetrahydro-lH-pyrazolo [3 , 4-c]pyridin-l-yl]benzonitrile (0.12 g, 0.24 mmol) in DMF (2 mL) .
- Part B The product from part A (85.17 g, 282.8 mmol) and phosphorus pentachloride (205.91 g, 990.0 mmol) was dissolved into CHC1 3 (750 mL) and refluxed for 3 1 /. h. Reaction was poured over ice and then quenched further with water, extracted with CHC1 3 (3x400 mL) , washed with brine
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Hematology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Diabetes (AREA)
- Urology & Nephrology (AREA)
- Mechanical Engineering (AREA)
- Vascular Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Peptides Or Proteins (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Hydrogenated Pyridines (AREA)
- Pyrrole Compounds (AREA)
Abstract
Description

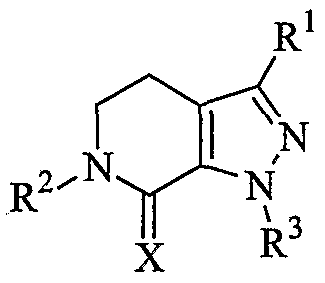
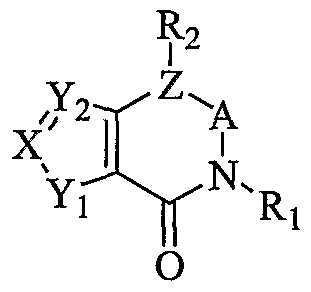



















































































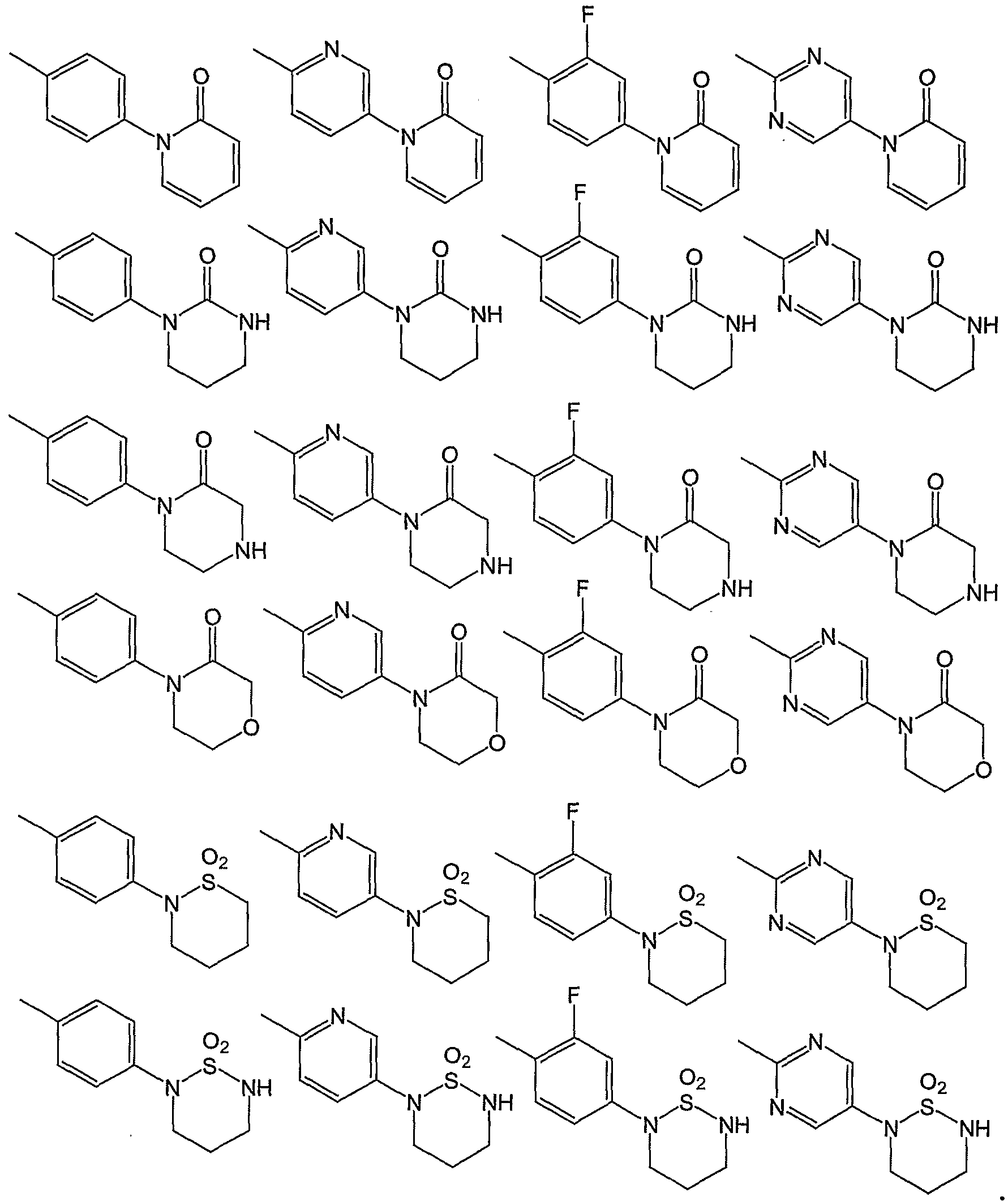

















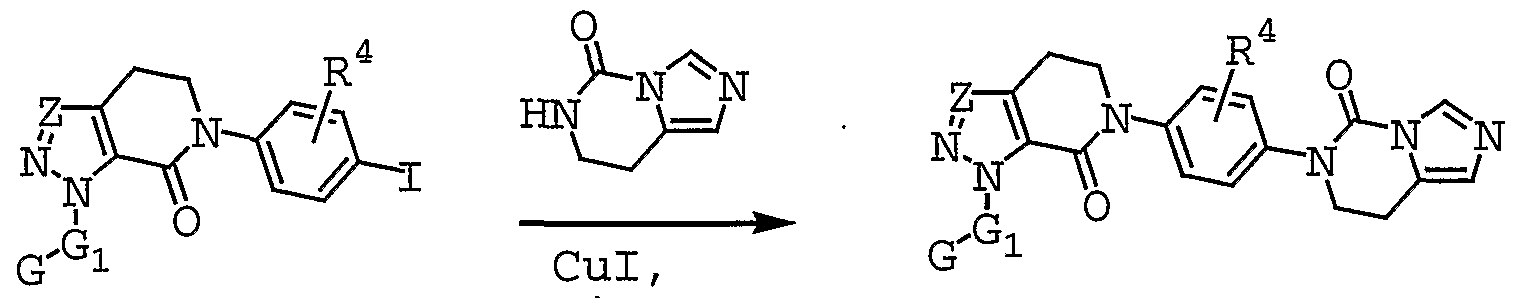
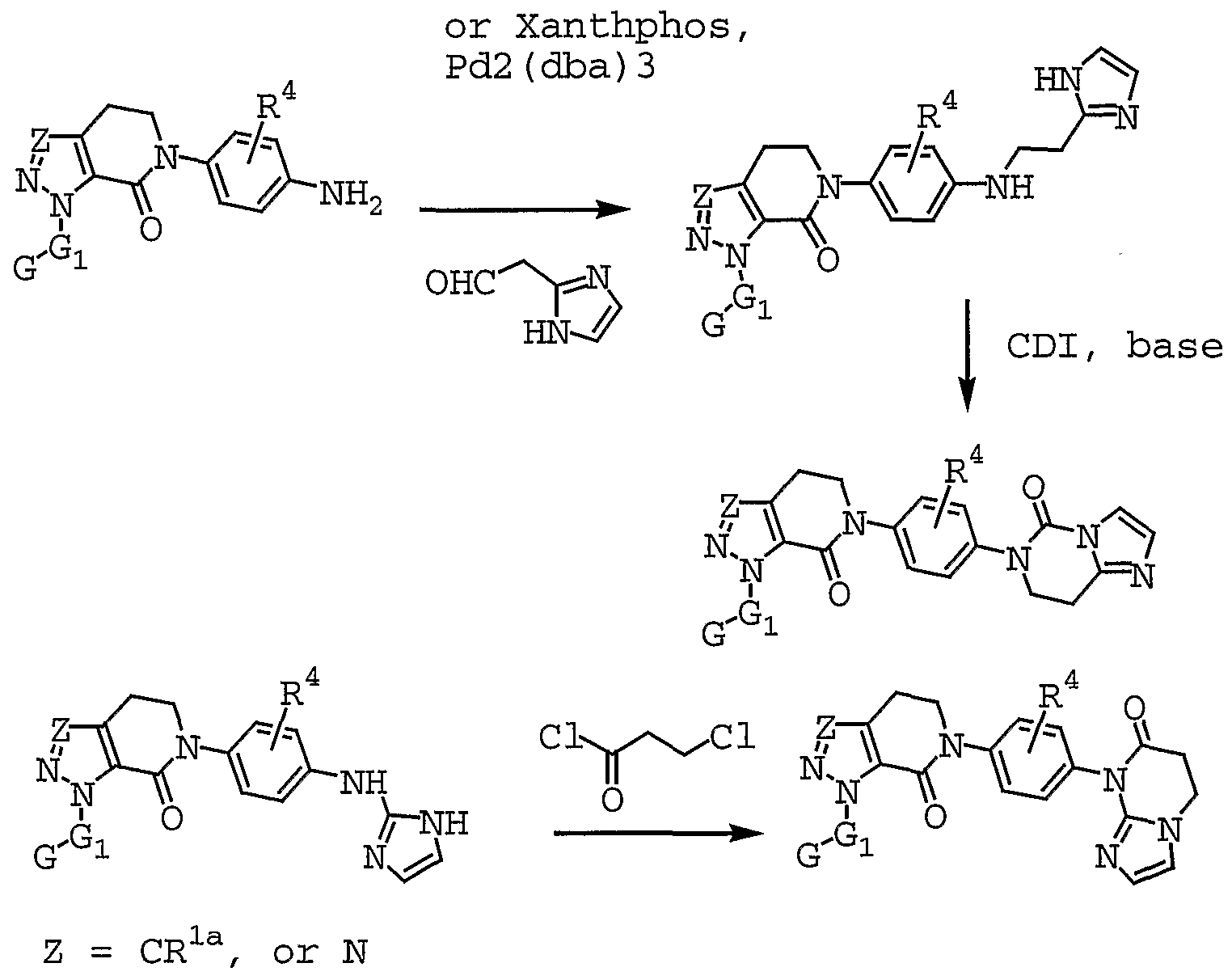













































Claims















































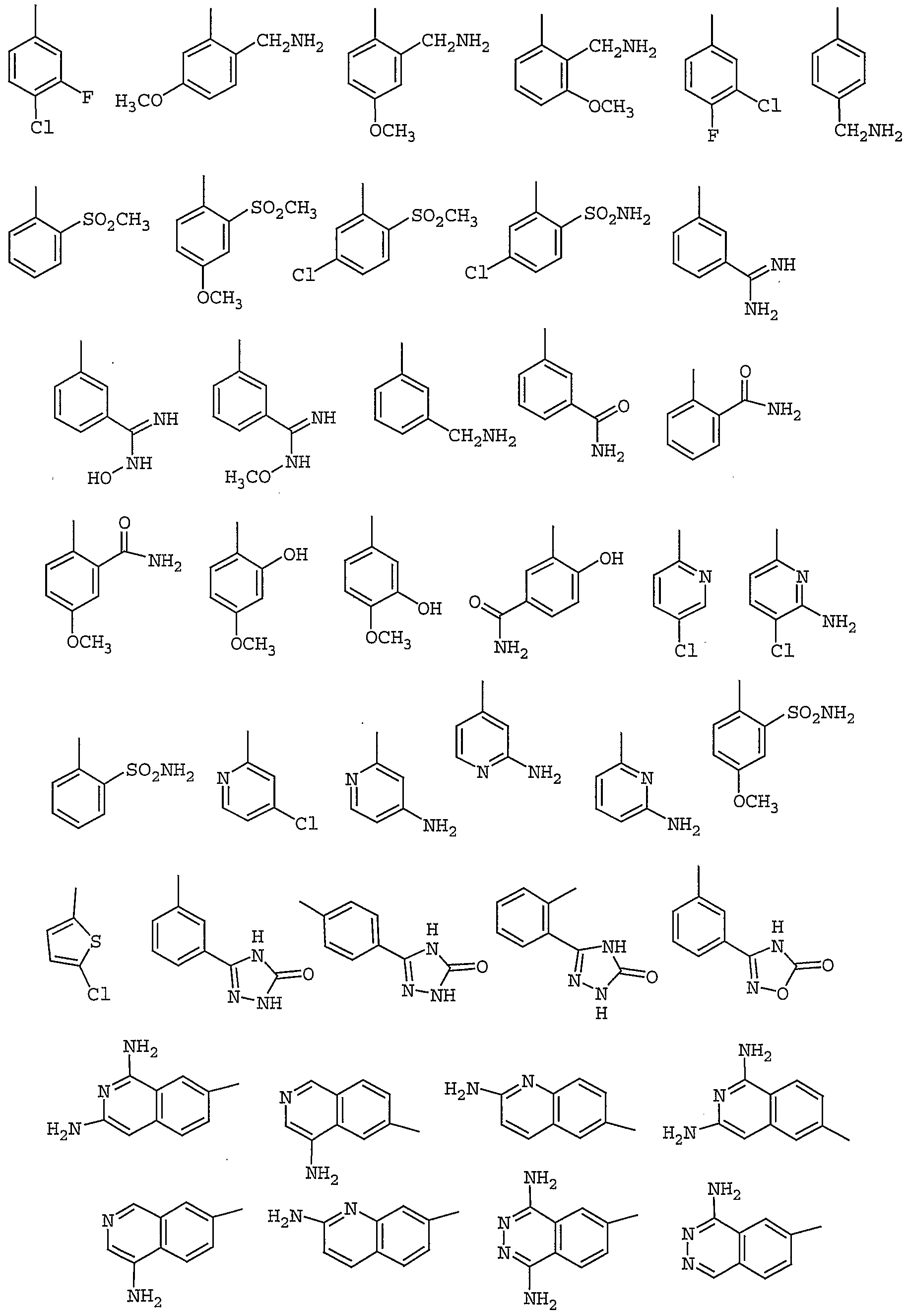

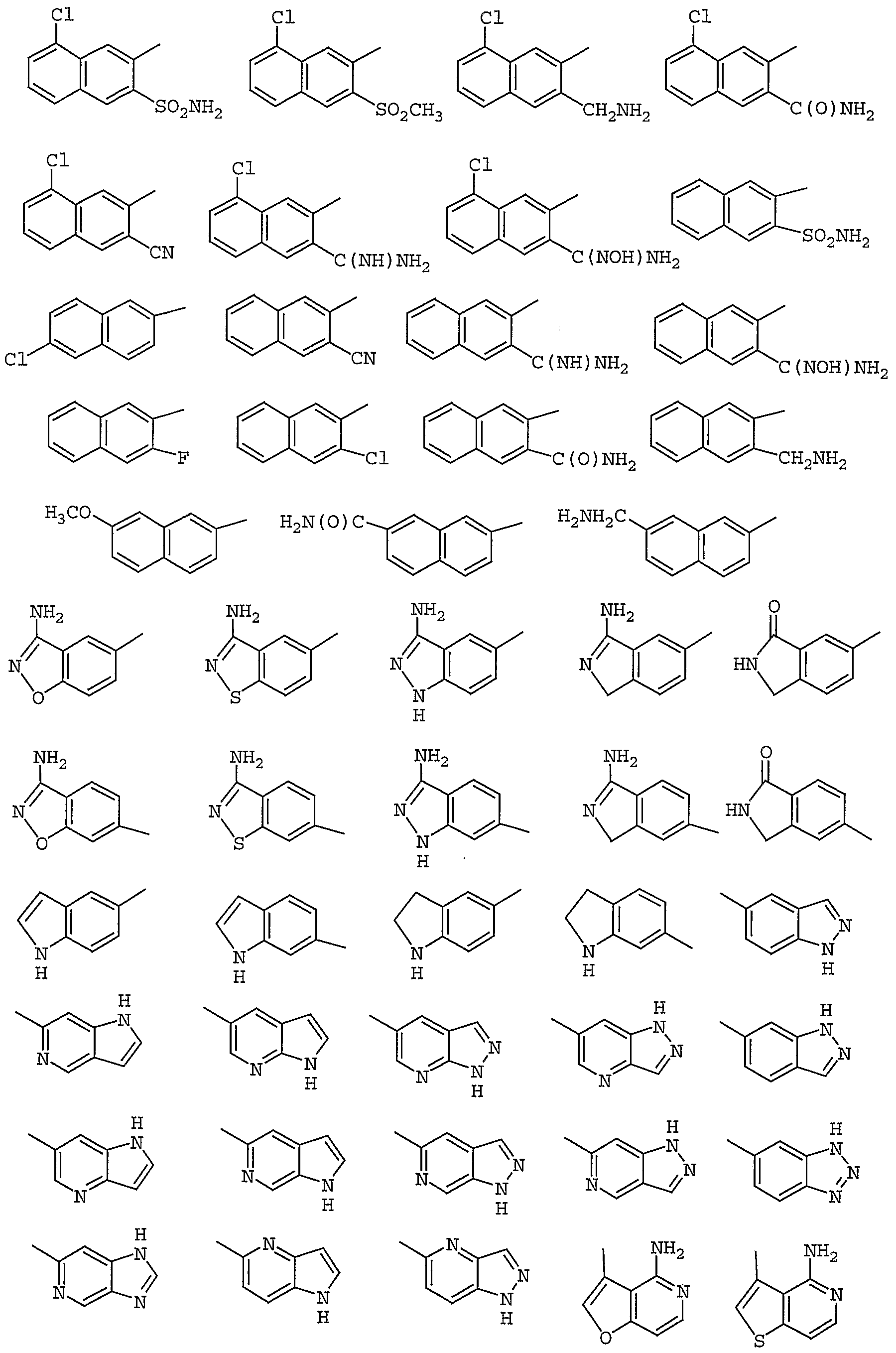

























Priority Applications (38)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE60233335T DE60233335D1 (en) | 2001-09-21 | 2002-09-17 | LACTOMATIC COMPOUNDS AND THEIR DERIVATIVES AS FACTOR XA HEMMER |
| AU2002341693A AU2002341693B2 (en) | 2001-09-21 | 2002-09-17 | Lactam-containing compounds and derivatives thereof as factor Xa inhibitors |
| YUP-227/04A RS51444B (en) | 2001-09-21 | 2002-09-17 | Lactam-containing compounds and derivatives thereof as factor xa inhibitors |
| SI200230849T SI1427415T1 (en) | 2001-09-21 | 2002-09-17 | Lactam-containing compounds and derivatives thereof as factor xa inhibitors |
| NZ531616A NZ531616A (en) | 2001-09-21 | 2002-09-17 | Lactam-containing compounds and derivatives thereof as factor Xa inhibitors |
| EP02775843A EP1427415B1 (en) | 2001-09-21 | 2002-09-17 | Lactam-containing compounds and derivatives thereof as factor xa inhibitors |
| IL16069302A IL160693A0 (en) | 2001-09-21 | 2002-09-17 | Lactam-containing compounds and derivatives thereof as factor xa inhibitors |
| KR1020047004025A KR100908176B1 (en) | 2001-09-21 | 2002-09-17 | Lactam-containing Compounds and Derivatives thereof as Factor Jaa Inhibitors |
| CH02775843T CH1427415H1 (en) | 2001-09-21 | 2002-09-17 | LACTAM-CONTAINING COMPOUNDS AND THEIR DERIVATIVES AS FACTOR XA INHIBITORS |
| BRPI0212726A BRPI0212726B8 (en) | 2001-09-21 | 2002-09-17 | compounds containing lactams, and derivatives thereof, pharmaceutical composition comprising them and their uses |
| CN028215370A CN1578660B (en) | 2001-09-21 | 2002-09-17 | Lactam-containing compounds and derivatives thereof as factor xa inhibitors |
| HU0402463A HU228195B1 (en) | 2001-09-21 | 2002-09-17 | Lactam derivatives having a pyrazolo-pyridine ring and pharmaceutical compositions thereof as factor xa inhibitors |
| MXPA04002526A MXPA04002526A (en) | 2001-09-21 | 2002-09-17 | Lactam-containing compounds and derivatives thereof as factor xa inhibitors. |
| DK02775843T DK1427415T3 (en) | 2001-09-21 | 2002-09-17 | Compounds containing lactam and derivatives thereof as factor Xa inhibitors |
| CA2461202A CA2461202C (en) | 2001-09-21 | 2002-09-17 | Lactam-containing compounds and derivatives thereof as factor xa inhibitors |
| KR1020087018676A KR100909141B1 (en) | 2001-09-21 | 2002-09-17 | Lactam-Containing Compounds and Derivatives thereof as Factor Xa Inhibitors |
| AT02775843T ATE439360T1 (en) | 2001-09-21 | 2002-09-17 | LACTAM-CONTAINING COMPOUNDS AND THEIR DERIVATIVES AS FACTOR XA INHIBITORS |
| MEP-2008-87A ME00090B (en) | 2001-09-21 | 2002-09-17 | Lactam-containing compounds and derivatives thereof as factor xa inhibitors |
| JP2003530289A JP4249621B2 (en) | 2001-09-21 | 2002-09-17 | Lactam-containing compounds and derivatives thereof as factor Xa inhibitors |
| UA20040402985A UA78232C2 (en) | 2001-09-21 | 2002-09-17 | Lactam-containing compounds and derivatives thereof as factor xa inhibitors |
| IL160693A IL160693A (en) | 2001-09-21 | 2004-03-02 | Lactam-containing compound or pharmaceutically acceptable salt thereof as factor xa inhibitor and pharmaceutical compositions containing it |
| IS7184A IS2824B (en) | 2001-09-21 | 2004-03-16 | Compounds containing lactam and their derivatives which inhibit Xa factor |
| ZA2004/02184A ZA200402184B (en) | 2001-09-21 | 2004-03-18 | Lactam-containing compounds and derivatives thereof as factor xa inhibitors |
| NO20041163A NO328558B1 (en) | 2001-09-21 | 2004-03-19 | Lactam-containing compounds, pharmaceutical compositions containing them, such compounds for use in therapy and the use thereof for the manufacture of medicaments for the treatment of disorders |
| HRP20040280AA HRP20040280B1 (en) | 2001-09-21 | 2004-03-19 | LACTAM-CONTAINING COMPOUNDS AND DERIVATIVES THEREOF AS FACTOR Xa INHIBITORS |
| HK04105041.4A HK1061973A1 (en) | 2001-09-21 | 2004-07-10 | Lactam-containing compounds and derivatives thereof as factor xa inhibitors |
| HR20080382A HRP20080382A2 (en) | 2001-09-21 | 2008-08-06 | LACTAM-CONTAINING COMPOUNDS AND DERIVATIVES THEREOF AS FACTOR Xa INHIBITORS |
| AU2008207537A AU2008207537B8 (en) | 2001-09-21 | 2008-08-26 | Lactam-containing compounds and derivatives thereof as factor Xa inhibitors |
| NO20083684A NO20083684L (en) | 2001-09-21 | 2008-08-27 | Lactam-containing compounds and derivatives thereof as factor XA inhibitors |
| IL203533A IL203533A (en) | 2001-09-21 | 2010-01-26 | Lactam-containing tetrahydro-pyrazolo [3,4-c] pyridine compounds useful as factor xa inhibitors and pharmaceutical compositions comprising the same for treating thromboembolic disorders |
| NO2011021C NO2011021I2 (en) | 2001-09-21 | 2011-09-26 | Apixaban and pharmaceutically acceptable salts thereof |
| FR11C0042C FR11C0042I2 (en) | 2001-09-21 | 2011-09-27 | COMPOUNDS CONTAINING LACTAME AND THEIR DERIVATIVES AS FACTOR XA INHIBITORS |
| BE2011C034C BE2011C034I2 (en) | 2001-09-21 | 2011-09-28 | |
| DE201112100050 DE122011100050I1 (en) | 2001-09-21 | 2011-09-28 | Lactam compounds and their derivatives as factor xa inhibitors. |
| CY2011016C CY2011016I2 (en) | 2001-09-21 | 2011-09-28 | LACTAMOUS COMPOUNDS AND THEIR DERIVATIVES AS INHIBITORS OF FACTOR HA |
| LTPA2011012C LTPA2011012I1 (en) | 2001-09-21 | 2011-09-29 | LACTAM-CONTAINING COMPOUNDS AND THEIR DERIVATIVES AS FACTOR XA INHIBITORS |
| LU91888C LU91888I2 (en) | 2001-09-21 | 2011-10-19 | Apixaban and its pharmaceutically acceptable salts in any of the forms benefiting from the protection of the original patent |
| HUS1300014C HUS1300014I1 (en) | 2001-09-21 | 2013-04-24 | Lactam derivatives having a pyrazolo-pyridine ring and pharmaceutical compositions thereof as factor xa inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US32416501P | 2001-09-21 | 2001-09-21 | |
| US60/324,165 | 2001-09-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2003026652A1 true WO2003026652A1 (en) | 2003-04-03 |
Family
ID=23262376
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2002/029491 WO2003026652A1 (en) | 2001-09-21 | 2002-09-17 | Lactam-containing compounds and derivatives thereof as factor xa inhibitors |
Country Status (40)
| Country | Link |
|---|---|
| US (11) | US7338963B2 (en) |
| EP (2) | EP2105436B1 (en) |
| JP (2) | JP4249621B2 (en) |
| KR (2) | KR100908176B1 (en) |
| CN (5) | CN104744461A (en) |
| AR (2) | AR037092A1 (en) |
| AT (2) | ATE544750T1 (en) |
| AU (2) | AU2002341693B2 (en) |
| BE (1) | BE2011C034I2 (en) |
| BR (1) | BRPI0212726B8 (en) |
| CA (2) | CA2461202C (en) |
| CH (1) | CH1427415H1 (en) |
| CL (1) | CL2008002717A1 (en) |
| CO (1) | CO5560567A2 (en) |
| CY (2) | CY1110509T1 (en) |
| DE (2) | DE60233335D1 (en) |
| DK (1) | DK1427415T3 (en) |
| ES (1) | ES2329881T3 (en) |
| FR (1) | FR11C0042I2 (en) |
| GE (1) | GEP20074098B (en) |
| HK (1) | HK1061973A1 (en) |
| HR (2) | HRP20040280B1 (en) |
| HU (2) | HU228195B1 (en) |
| IL (3) | IL160693A0 (en) |
| IS (2) | IS3006B (en) |
| LT (1) | LTPA2011012I1 (en) |
| LU (1) | LU91888I2 (en) |
| ME (2) | ME00090B (en) |
| MX (1) | MXPA04002526A (en) |
| MY (1) | MY137830A (en) |
| NO (4) | NO328558B1 (en) |
| NZ (1) | NZ531616A (en) |
| PL (2) | PL204263B1 (en) |
| PT (1) | PT1427415E (en) |
| RS (2) | RS20080517A (en) |
| RU (2) | RU2345993C2 (en) |
| SI (1) | SI1427415T1 (en) |
| UA (1) | UA78232C2 (en) |
| WO (1) | WO2003026652A1 (en) |
| ZA (1) | ZA200402184B (en) |
Cited By (113)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1415992A1 (en) * | 2001-08-09 | 2004-05-06 | Daiichi Pharmaceutical Co., Ltd. | Diamine derivatives |
| WO2004058715A1 (en) * | 2002-12-25 | 2004-07-15 | Daiichi Pharmaceutical Co., Ltd. | Diamine derivatives |
| EP1467984A2 (en) * | 2001-12-10 | 2004-10-20 | Bristol-Myers Squibb Company | Synthesis of 4,5-dihydro-pyrazolo (3,4-c) pyrid-2-ones |
| EP1602656A1 (en) * | 2004-05-24 | 2005-12-07 | NEUROSCIENZE PHARMANESS S.C. a R.L. | Pyrazole derivatives having affinity for cb1 and/or cb2 receptors |
| EP1603909A2 (en) * | 2003-03-18 | 2005-12-14 | Brystol-Myers Squibb Company | Linear chain substituted monocyclic and bicyclic derivatives as factor xa inhibitors |
| EP1603572A1 (en) * | 2003-03-18 | 2005-12-14 | Bristol-Myers Squibb Company | Lactam-containing cyclic diamines and derivatives as factor xa inhibitors |
| WO2006036926A2 (en) * | 2004-09-28 | 2006-04-06 | Bristol-Myers Squibb Company | Efficient synthesis of 4,5-dihydro-pyrazolo[3,4-c]pyrid-2-ones |
| WO2006047528A2 (en) * | 2004-10-26 | 2006-05-04 | Bristol-Myers Squibb Company | Pyrazolobenzamides and derivatives as factor xa inhibitors |
| WO2006061116A1 (en) * | 2004-12-09 | 2006-06-15 | Bayer Healthcare Ag | Pyrazine dicarboxamides and the use thereof |
| EP1670739A2 (en) * | 2003-10-08 | 2006-06-21 | Bristol-Myers Squibb Company | Cyclic diamines and derivatives as factor xa inhibitors |
| EP1684744A2 (en) * | 2003-10-01 | 2006-08-02 | Bristol-Myers Squibb Company | Pyrrolidine and piperidine derivatives as factor xa inhibitors |
| JPWO2005030706A1 (en) * | 2003-09-26 | 2006-12-07 | 田辺製薬株式会社 | Amide type carboxamide derivatives |
| WO2006135425A2 (en) * | 2004-09-28 | 2006-12-21 | Bristol-Myers Squibb Company | Preparation of 4,5-dihydro-pyrazolo[3,4-c]pyrid-2-ones |
| WO2007001385A2 (en) * | 2004-09-28 | 2007-01-04 | Bristol-Myers Squibb Company | Process for preparing 4,5-dihydro-pyrazolo [3,4-c] pyrid-2-ones |
| WO2007002635A2 (en) | 2005-06-27 | 2007-01-04 | Bristol-Myers Squibb Company | C-linked cyclic antagonists of p2y1 receptor useful in the treatment of thrombotic conditions |
| US7169926B1 (en) | 2003-08-13 | 2007-01-30 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| WO2007022165A2 (en) * | 2005-08-17 | 2007-02-22 | Bristol-Myers Squibb Company | Factor xa inhibitor inclusion complex with cyclodextrin |
| US7192968B2 (en) | 2000-04-05 | 2007-03-20 | Daiichi Pharmaceutical Co., Ltd. | Ethylenediamine derivatives |
| WO2007137801A1 (en) * | 2006-05-31 | 2007-12-06 | Bayer Healthcare Ag | Tetrahydropyrrolopyridine, tetrahydropyrazolopyridine, tetrahydro-imidazopyridine and tetrahydrotriazolopyridine derivatives and use thereof |
| US7342014B2 (en) | 2001-06-20 | 2008-03-11 | Daiichi Pharmaceutical Co., Ltd. | Diamine derivatives |
| US7371864B2 (en) * | 2004-12-15 | 2008-05-13 | Bristol-Myers Squibb Company | Crystalline forms of a factor Xa inhibitor |
| US7371743B2 (en) | 2004-02-28 | 2008-05-13 | Boehringer Ingelheim International Gmbh | Carboxylic acid amides, the preparation thereof and their use as medicaments |
| US7388096B2 (en) | 2004-09-28 | 2008-06-17 | Bristol-Myers Squibb Company | Crystalline forms of a factor Xa inhibitor |
| US7390908B2 (en) | 2001-08-17 | 2008-06-24 | Astrazeneca Ab | Compounds effecting glucokinase |
| WO2008076805A2 (en) | 2006-12-15 | 2008-06-26 | Bristol-Myers Squibb Company | Arylpropionamide, arylacrylamide, arylpropynamide, or arylmethylurea analogs as factor xia inhibitors |
| WO2008079836A2 (en) | 2006-12-20 | 2008-07-03 | Bristol-Myers Squibb Company | Macrocyclic factor viia inhibitors useful as anticoagulants |
| DE102007028406A1 (en) | 2007-06-20 | 2008-12-24 | Bayer Healthcare Ag | Substituted oxazolidinones and their use |
| DE102007028407A1 (en) | 2007-06-20 | 2008-12-24 | Bayer Healthcare Ag | Substituted oxazolidinones and their use |
| DE102007028319A1 (en) | 2007-06-20 | 2008-12-24 | Bayer Healthcare Ag | Substituted oxazolidinones and their use |
| DE102007032344A1 (en) | 2007-07-11 | 2009-01-15 | Bayer Healthcare Ag | Prodrug derivatives of 1- (4-methoxyphenyl) -7-oxo-6- [4- (2-oxopiperidin-1-yl) phenyl] -4,5,6,7-tetrahydro-1H-pyrazolo [3,4 -c] pyridine-3-carboxamide |
| US7550499B2 (en) | 2004-05-12 | 2009-06-23 | Bristol-Myers Squibb Company | Urea antagonists of P2Y1 receptor useful in the treatment of thrombotic conditions |
| US7645778B2 (en) | 2005-01-19 | 2010-01-12 | Bristol-Myers Squibb Company | Heteroaryl compounds as P2Y1 receptor inhibitors |
| US7678909B1 (en) | 2003-08-13 | 2010-03-16 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7687625B2 (en) | 2003-03-25 | 2010-03-30 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7687638B2 (en) | 2004-06-04 | 2010-03-30 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
| US7714002B2 (en) | 2005-06-27 | 2010-05-11 | Bristol-Myers Squibb Company | Carbocycle and heterocycle antagonists of P2Y1 receptor useful in the treatment of thrombotic conditions |
| US7723344B2 (en) | 2003-08-13 | 2010-05-25 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
| US7728008B2 (en) | 2005-06-27 | 2010-06-01 | Bristol-Myers Squibb Company | N-linked heterocyclic antagonists of P2Y1 receptor useful in the treatment of thrombotic conditions |
| US7732446B1 (en) | 2004-03-11 | 2010-06-08 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7745623B2 (en) | 2004-05-21 | 2010-06-29 | Takeda Pharmaceutical Company Limited | Cyclic amide derivative, and its production and use |
| US7781584B2 (en) | 2004-03-15 | 2010-08-24 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7790734B2 (en) | 2003-09-08 | 2010-09-07 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7816382B2 (en) | 2005-06-27 | 2010-10-19 | Bristol-Myers Squibb Company | Linear urea mimics antagonists of P2Y1 receptor useful in the treatment of thrombotic condition |
| US7825242B2 (en) | 2004-07-16 | 2010-11-02 | Takeda Pharmaceutical Company Limted | Dipeptidyl peptidase inhibitors |
| US7872124B2 (en) | 2004-12-21 | 2011-01-18 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7960569B2 (en) | 2006-10-17 | 2011-06-14 | Bristol-Myers Squibb Company | Indole antagonists of P2Y1 receptor useful in the treatment of thrombotic conditions |
| WO2011100401A1 (en) | 2010-02-11 | 2011-08-18 | Bristol-Myers Squibb Company | Macrocycles as factor xia inhibitors |
| US8022208B2 (en) | 2005-11-08 | 2011-09-20 | Astellas Pharma Inc. | Benzene derivative or salt thereof |
| US8084605B2 (en) | 2006-11-29 | 2011-12-27 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| US8093236B2 (en) | 2007-03-13 | 2012-01-10 | Takeda Pharmaceuticals Company Limited | Weekly administration of dipeptidyl peptidase inhibitors |
| US8222411B2 (en) | 2005-09-16 | 2012-07-17 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| WO2012098089A1 (en) | 2011-01-19 | 2012-07-26 | Bayer Pharma Aktiengesellschaft | Binding proteins to inhibitors of coagulation factors |
| EP2484209A1 (en) | 2004-02-24 | 2012-08-08 | Sumitomo Chemical Company, Limited | Insecticide compositions |
| US8263624B2 (en) | 2006-05-31 | 2012-09-11 | Bayer Intellectual Property Gmbh | Aryl-substituted heterocycles, and use thereof |
| US8324383B2 (en) | 2006-09-13 | 2012-12-04 | Takeda Pharmaceutical Company Limited | Methods of making polymorphs of benzoate salt of 2-[[6-[(3R)-3-amino-1-piperidinyl]-3,4-dihydro-3-methyl-2,4-dioxo-1(2H)-pyrimidinyl]methyl]-benzonitrile |
| ITMI20111047A1 (en) * | 2011-06-10 | 2012-12-11 | Dipharma Francis Srl | CRYSTAL FORM OF APIXABAN |
| EP2554159A1 (en) | 2011-08-04 | 2013-02-06 | ratiopharm GmbH | Dosage forms comprising apixaban and content uniformity enhancer |
| WO2013022818A1 (en) | 2011-08-05 | 2013-02-14 | Bristol-Myers Squibb Company | Novel macrocycles as factor xia inhibitors |
| WO2013022814A1 (en) | 2011-08-05 | 2013-02-14 | Bristol-Myers Squibb Company | Cyclic p1 linkers as factor xia inhibitors |
| WO2013055984A1 (en) | 2011-10-14 | 2013-04-18 | Bristol-Myers Squibb Company | Substituted tetrahydroisoquinoline compounds as factor xia inhibitors |
| WO2013056060A1 (en) | 2011-10-14 | 2013-04-18 | Bristol-Myers Squibb Company | Substituted tetrahydroisoquinoline compounds as factor xia inhibitors |
| WO2013056034A1 (en) | 2011-10-14 | 2013-04-18 | Bristol-Myers Squibb Company | Substituted tetrahydroisoquinoline compounds as factor xia inhibitors |
| US8426420B2 (en) | 2007-12-26 | 2013-04-23 | Sanofi | Heterocyclic pyrazole-carboxamidesas P2Y12 antagonists |
| WO2013174498A1 (en) | 2012-05-24 | 2013-11-28 | Ratiopharm Gmbh | Dosage forms comprising apixaban and matrix former |
| WO2014022766A1 (en) | 2012-08-03 | 2014-02-06 | Bristol-Myers Squibb Company | Dihydropyridone p1 as factor xia inhibitors |
| WO2014022767A1 (en) | 2012-08-03 | 2014-02-06 | Bristol-Myers Squibb Company | Dihydropyridone p1 as factor xia inhibitors |
| US8669266B2 (en) | 2007-04-23 | 2014-03-11 | Sanofi | Quinoline-carboxamide derivatives as P2Y12 antagonists |
| EP2752414A1 (en) | 2013-01-04 | 2014-07-09 | Sandoz AG | Crystalline form of apixaban |
| WO2014115020A1 (en) * | 2013-01-24 | 2014-07-31 | Universita' Degli Studi Di Bari | Heterocycles and their radiolabeled analogs useful as cox-1 selective inhibitors |
| WO2014160668A1 (en) | 2013-03-25 | 2014-10-02 | Bristol-Myers Squibb Company | Tetrahydroisoquinolines containing substituted azoles as factor xia inhibitors |
| WO2014173377A2 (en) | 2013-04-23 | 2014-10-30 | Zentiva, K.S. | New crystalline forms of apixaban and a method of their preparation |
| US8884016B2 (en) | 2011-06-10 | 2014-11-11 | Dipharma Francis S.R.L. | Apixaban preparation process |
| CZ304846B6 (en) * | 2012-11-13 | 2014-12-03 | Zentiva, K.S. | Process for preparing APIXABAN |
| US8906901B2 (en) | 2005-09-14 | 2014-12-09 | Takeda Pharmaceutical Company Limited | Administration of dipeptidyl peptidase inhibitors |
| WO2014203275A2 (en) | 2013-06-18 | 2014-12-24 | Cadila Healthcare Limited | An improved process for the preparation of apixaban and intermediates thereof |
| CN104277041A (en) * | 2014-09-19 | 2015-01-14 | 广东东阳光药业有限公司 | Methyl substituted aromatic ring substituent-containing pyrazolopiperidone compound and composition and use thereof |
| CN104277040A (en) * | 2014-09-19 | 2015-01-14 | 广东东阳光药业有限公司 | Acyl piperazinone substituent-containing pyrazolopiperidone compound and composition and use thereof |
| WO2015021902A1 (en) * | 2013-08-12 | 2015-02-19 | 药源药物化学(上海)有限公司 | Novel apixaban crystal form and preparation method thereof |
| US20150094296A1 (en) * | 2012-03-28 | 2015-04-02 | Takeda Pharmaceutical Company Limited | Heterocyclic compound |
| WO2015116885A1 (en) | 2014-01-31 | 2015-08-06 | Bristol-Myers Squibb Company | Macrocycles with aromatic p2' groups as factor xia inhibitors |
| WO2015116882A1 (en) | 2014-01-31 | 2015-08-06 | Bristol-Myers Squibb Company | Macrocyclic factor xia inhibitors condensed with heterocycles |
| US9193689B2 (en) | 2012-03-07 | 2015-11-24 | Institute Of Cancer Research: Royal Cancer Hospital (The) | 3-aryl-5-substituted-isoquinolin-1-one compounds and their therapeutic use |
| CN104327074B (en) * | 2014-09-19 | 2016-01-06 | 广东东阳光药业有限公司 | Containing lactan substituent pyrazolo piperidone compounds and composition thereof and purposes |
| WO2016020711A1 (en) | 2014-08-06 | 2016-02-11 | Egis Gyógyszergyár Zrt. | Process for the preparation of apixaban |
| WO2016053455A1 (en) | 2014-10-01 | 2016-04-07 | Bristol-Myers Squibb Company | Pyrimidinones as factor xia inhibitors |
| WO2016079549A1 (en) | 2014-11-19 | 2016-05-26 | Egis Gyógyszergyár Zrt. | Process and intermediate for the preparation of apixaban |
| EP3072892A4 (en) * | 2013-11-18 | 2016-09-28 | Chengdu Easton Biopharmaceuticals Co Ltd | Pyridine derivative and medical use thereof |
| WO2016205482A1 (en) | 2015-06-19 | 2016-12-22 | Bristol-Myers Squibb Company | Diamide macrocycles as factor xia inhibitors |
| WO2017023992A1 (en) | 2015-08-05 | 2017-02-09 | Bristol-Myers Squibb Company | Novel substituted glycine derived fxia inhibitors |
| US9611223B2 (en) | 2013-09-11 | 2017-04-04 | Institute Of Cancer Research: Royal Cancer Hospital (The) | 3-aryl-5-substituted-isoquinolin-1-one compounds and their therapeutic use |
| WO2017088841A1 (en) | 2015-11-26 | 2017-06-01 | Zentiva, K.S. | Preparation of a drug form containing amorphous apixaban |
| EP2099453B1 (en) | 2006-11-02 | 2017-09-06 | Bayer Intellectual Property GmbH | Combination therapy of substituted oxazolidinones |
| WO2017151746A1 (en) | 2016-03-02 | 2017-09-08 | Bristol-Myers Squibb Company | Diamide macrocycles having factor xia inhibiting activity |
| JP2017530104A (en) * | 2014-09-02 | 2017-10-12 | 石薬集団中奇制薬技術(石家庄)有限公司 | Pyrazolo [3,4-c] pyridine derivative |
| EP3147283A4 (en) * | 2014-05-22 | 2017-12-06 | North China Pharmaceutical Company., Ltd. | HYDRAZINE COMPOUND AS BLOOD COAGULATION FACTOR Xa INHIBITOR |
| WO2017221209A1 (en) | 2016-06-23 | 2017-12-28 | Lupin Limited | Pharmaceutical formulations of apixaban |
| US10144731B2 (en) | 2013-12-18 | 2018-12-04 | Glaxosmithkline Intellectual Property Development Limited | Nrf2 regulators |
| US10214519B2 (en) | 2016-09-23 | 2019-02-26 | Gilead Sciences, Inc. | Phosphatidylinositol 3-kinase inhibitors |
| US10227350B2 (en) | 2016-09-23 | 2019-03-12 | Gilead Sciences, Inc. | Phosphatidylinositol 3-kinase inhibitors |
| US10272095B2 (en) | 2015-06-15 | 2019-04-30 | GlaxoSmithKline Intellectual Propert Development Limited | NRF2 regulators |
| US10364256B2 (en) | 2015-10-06 | 2019-07-30 | Glaxosmithkline Intellectual Property Development Limited | Biaryl pyrazoles as NRF2 regulators |
| US10479770B2 (en) | 2016-09-23 | 2019-11-19 | Gilead Sciences, Inc. | Phosphatidylinositol 3-kinase inhibitors |
| US10604509B2 (en) | 2015-06-15 | 2020-03-31 | Glaxosmithkline Intellectual Property Development Limited | Nrf2 regulators |
| WO2020085616A1 (en) | 2018-10-24 | 2020-04-30 | 하나제약 주식회사 | Method for preparing apixaban |
| US11034669B2 (en) | 2018-11-30 | 2021-06-15 | Nuvation Bio Inc. | Pyrrole and pyrazole compounds and methods of use thereof |
| US11072610B2 (en) | 2018-09-12 | 2021-07-27 | Novartis Ag | Antiviral pyridopyrazinedione compounds |
| EP3868753A1 (en) | 2015-07-29 | 2021-08-25 | Bristol-Myers Squibb Company | Factor xia macrocyclic inhibitors bearing a non-aromatic p2' group |
| US11603523B2 (en) | 2019-01-18 | 2023-03-14 | Astrazeneca Ab | PCSK9 inhibitors and methods of use thereof |
| US11667613B2 (en) | 2019-09-26 | 2023-06-06 | Novartis Ag | Antiviral pyrazolopyridinone compounds |
| US11678659B2 (en) | 2014-06-11 | 2023-06-20 | Dietrich Gulba | Use as rodenticides of compounds that inhibit blood coagulation |
| US11827627B2 (en) | 2021-06-04 | 2023-11-28 | Vertex Pharmaceuticals Incorporated | N-(hydroxyalkyl (hetero)aryl) tetrahydrofuran carboxamides as modulators of sodium channels |
| US11834441B2 (en) | 2019-12-06 | 2023-12-05 | Vertex Pharmaceuticals Incorporated | Substituted tetrahydrofurans as modulators of sodium channels |
| WO2024084217A1 (en) * | 2022-10-19 | 2024-04-25 | Kalvista Pharmaceuticals Limited | 3a,4,5,6-tetrahydro-1 h-pyrazolo[3,4-c]pyridin-7(7ah)-one derivatives as factor xiia inhibitors |
Families Citing this family (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PL204263B1 (en) | 2001-09-21 | 2009-12-31 | Bristol Myers Squibb Co | Lactam-containing compounds and derivatives thereof as factor xa inhibitors |
| TWI331526B (en) * | 2001-09-21 | 2010-10-11 | Bristol Myers Squibb Pharma Co | Lactam-containing compounds and derivatives thereof as factor xa inhibitors |
| AU2003297441A1 (en) * | 2002-12-24 | 2004-07-22 | Arena Pharmaceuticals, Inc. | Diarylamine and arylheteroarylamine pyrazole derivatives as modulators of 5ht2a |
| JP4630267B2 (en) * | 2002-12-25 | 2011-02-09 | 第一三共株式会社 | Diamine derivatives |
| PT1558582E (en) | 2003-07-22 | 2006-05-31 | Arena Pharm Inc | DIARIL- AND ARIL-HETEROARIL-UREA DERIVATIVES AS SEROTONIN 5-HT2A RECEPTOR MODULES USEFUL FOR PROPHYLAXIS AND TREATMENT OF RELEVANT DISORDERS |
| ATE548353T1 (en) * | 2004-03-23 | 2012-03-15 | Arena Pharm Inc | METHOD FOR PRODUCING SUBSTITUTED N-ARYL-N'-Ä3-(1H-PYRAZOLE-5-YL)PHENYLÜ-UREAS AND INTERMEDIATE THEREOF. |
| CN101068812B (en) * | 2004-09-28 | 2010-09-29 | 布里斯托尔-迈尔斯斯奎布公司 | Efficient synthesis of 4,5-dihydro-pyrazolo[3,4-c]pyrid-2-ones |
| SA05260357B1 (en) * | 2004-11-19 | 2008-09-08 | ارينا فارماسيتو تيكالز ، أنك | 3-phenyle-pyrazole derivatives as modulators of the 5-ht 2a serotonin receptor useful for the treatment of disorders related thereto |
| US20060160841A1 (en) * | 2005-01-19 | 2006-07-20 | Chenkou Wei | Crystallization via high-shear transformation |
| JP2006298909A (en) * | 2005-03-25 | 2006-11-02 | Tanabe Seiyaku Co Ltd | Medicine composition |
| EP1978964A4 (en) * | 2006-01-24 | 2009-12-09 | Merck & Co Inc | Jak2 tyrosine kinase inhibition |
| WO2007087245A2 (en) * | 2006-01-24 | 2007-08-02 | Merck & Co., Inc. | Ret tyrosine kinase inhibition |
| USRE45337E1 (en) | 2006-05-18 | 2015-01-13 | Arena Pharmaceuticals, Inc. | Ethers, secondary amines and derivatives thereof as modulators of the 5-HT2A serotonin receptor useful for the treatment of disorders related thereto |
| CA2646081C (en) | 2006-05-18 | 2017-06-27 | Arena Pharmaceuticals, Inc. | Crystalline forms and processes for the preparation of phenyl-pyrazoles useful as modulators of the 5-ht2a serotonin receptor |
| USRE45336E1 (en) | 2006-05-18 | 2015-01-13 | Arena Pharmaceuticals, Inc. | Primary amines and derivatives thereof as modulators of the 5-HT2A serotonin receptor useful for the treatment of disorders related thereto |
| TWI415845B (en) * | 2006-10-03 | 2013-11-21 | Arena Pharm Inc | Pyrazole derivatives as modulators of the 5-ht2a serotonin receptor useful for the treatment of disorders related thereto |
| ES2421237T7 (en) | 2007-08-15 | 2013-09-30 | Arena Pharmaceuticals, Inc. | Imidazo [1,2-a] pyridine derivatives as modulators of the serotonergic 5ht2a receptor in the treatment of disorders related thereto |
| US20110021538A1 (en) * | 2008-04-02 | 2011-01-27 | Arena Pharmaceuticals, Inc. | Processes for the preparation of pyrazole derivatives useful as modulators of the 5-ht2a serotonin receptor |
| CN102066385A (en) * | 2008-04-30 | 2011-05-18 | 弗雷德里克·阿尔姆奎斯特 | New peptidomimetic compounds |
| US9126946B2 (en) | 2008-10-28 | 2015-09-08 | Arena Pharmaceuticals, Inc. | Processes useful for the preparation of 1-[3-(4-bromo-2-methyl-2H-pyrazol-3-yl)-4-methoxy-phenyl]-3-(2,4-difluoro-phenyl)urea and crystalline forms related thereto |
| PT2364142T (en) | 2008-10-28 | 2018-04-23 | Arena Pharm Inc | Compositions of a 5-ht2a serotonin receptor modulator useful for the treatment of disorders related thereto |
| WO2011075596A1 (en) | 2009-12-18 | 2011-06-23 | Arena Pharmaceuticals, Inc. | Crystalline forms of certain 3-phenyl-pyrazole derivatives as modulators of the 5-ht2a serotonin receptor useful for the treatment of disorders related thereto |
| SI3017811T1 (en) * | 2010-02-25 | 2019-04-30 | Bristol-Myers Squibb Holdings Ireland Unlimited Company | Apixaban formulations |
| CN202271910U (en) * | 2011-10-31 | 2012-06-13 | 潘磊 | Multifunctional rearview mirror of automobile |
| CN103242310A (en) * | 2012-02-10 | 2013-08-14 | 苏州迈泰生物技术有限公司 | Pyrazolo pyridone compound and its application in preparation of anticoagulant |
| WO2014044107A1 (en) * | 2012-09-18 | 2014-03-27 | 上海恒瑞医药有限公司 | Pyrazol[3,4-c] pyridine derivative, preparation method and use in medicine thereof |
| AU2013323435C1 (en) * | 2012-09-26 | 2018-04-12 | Bristol-Myers Squibb Holdings Ireland Unlimited Company | Apixaban liquid formulations |
| WO2014056434A1 (en) * | 2012-10-10 | 2014-04-17 | Sunshine Lake Pharma Co., Ltd. | Crystalline form and amorphous form of apixaban and preparation thereof |
| KR20150105302A (en) | 2012-11-05 | 2015-09-16 | 난트 홀딩스 아이피, 엘엘씨 | Cyclic sulfonamide containing derivatives as inhibitors of hedgehog signaling pathway |
| CN103539795A (en) * | 2013-03-18 | 2014-01-29 | 齐鲁制药有限公司 | Apixaban polymorph and preparation method thereof |
| CN104109165A (en) * | 2013-04-19 | 2014-10-22 | 四川海思科制药有限公司 | 4,5-dihydro-pyrazolo[3,4-c]pyrid-2-one derivatives, and preparation method and application thereof |
| CN103923080B (en) * | 2014-04-04 | 2016-06-22 | 苏州景泓生物技术有限公司 | A kind of method preparing antithrombotic reagent Eliquis |
| CN105085515B (en) * | 2014-05-22 | 2019-02-01 | 华北制药股份有限公司 | Hydrazide kind compound as coagulation factor xa inhibitors |
| WO2015177801A1 (en) * | 2014-05-23 | 2015-11-26 | Symed Labs Limited | Novel process for the preparation of a lactam-containing compound |
| CN104311557B (en) * | 2014-09-19 | 2016-01-06 | 广东东阳光药业有限公司 | Containing diketone substituent pyrazolo piperidone compounds and composition thereof and purposes |
| CN104311555B (en) * | 2014-09-19 | 2016-04-20 | 广东东阳光药业有限公司 | Pyrazolo piperidone compounds and composition thereof and purposes |
| CN104277039B (en) * | 2014-09-19 | 2016-06-01 | 广东东阳光药业有限公司 | Contain the pyrazoles piperidone compounds and composition thereof and purposes that replace butynyl |
| CN104311541B (en) * | 2014-09-19 | 2017-12-05 | 广东东阳光药业有限公司 | Containing substituted pyrazole compound of ketone and combinations thereof and purposes |
| US9603846B2 (en) | 2014-11-25 | 2017-03-28 | Cadila Healthcare Limited | Process for the preparation of apixaban |
| CN104513239B (en) * | 2014-12-10 | 2017-08-22 | 沈阳药科大学 | The ketone compounds of pyrazolo [3,4 c] pyridine 7 and its application |
| CN104530080B (en) * | 2014-12-10 | 2017-01-11 | 广东东阳光药业有限公司 | Oxazolidinone compounds and application thereof in drugs |
| EP3078378B1 (en) | 2015-04-08 | 2020-06-24 | Vaiomer | Use of factor xa inhibitors for regulating glycemia |
| CN104892601B (en) * | 2015-06-09 | 2017-09-19 | 江苏中邦制药有限公司 | A kind of preparation method of antithrombotic reagent Eliquis |
| RU2017145976A (en) | 2015-06-12 | 2019-07-15 | Аксовант Сайенсиз Гмбх | Diaryl- and arylheteroarylurea derivatives applicable for the prevention and treatment of behavioral disturbances during the REM phase of sleep |
| KR20180064373A (en) | 2015-07-15 | 2018-06-14 | 엑소반트 사이언시즈 게엠베하 | Diaryl and aryl heteroaryl urea derivatives as modulators of 5-HT2A serotonin receptors useful for the prevention and treatment of hallucinations associated with neurodegenerative diseases |
| SI3355890T1 (en) | 2015-10-01 | 2022-04-29 | Biocryst Pharmaceuticals, Inc. | Human plasma kallikrein inhibitors |
| WO2018067615A1 (en) * | 2016-10-03 | 2018-04-12 | Sigilon Therapeutics, Inc. | Compounds, devices, and uses thereof |
| CN107064367B (en) * | 2017-04-20 | 2019-09-13 | 青岛理工大学 | Method for analyzing and detecting four heterocyclic pesticides in environmental water sample |
| CN106940355B (en) * | 2017-04-24 | 2018-08-24 | 中国药科大学 | A kind of brufen, the detection method of its sodium salt and its preparation in relation to substance |
| WO2019123194A1 (en) * | 2017-12-20 | 2019-06-27 | Pi Industries Ltd. | Anthranilamides, their use as insecticide and processes for preparing the same. |
| EP3753924A1 (en) * | 2019-06-18 | 2020-12-23 | AnaMar AB | New tricyclic 5-ht2 antagonists |
| JP7477642B2 (en) * | 2020-04-09 | 2024-05-01 | ディスアーム セラピューティクス, インコーポレイテッド | SARM1 inhibitors |
| EP4251271A4 (en) | 2020-11-27 | 2024-07-31 | Santa Farma Ilac Sanayii A S | Direct compression method for non-micronised apixaban formulations |
| WO2022115052A1 (en) | 2020-11-27 | 2022-06-02 | Santa Farma Ilac Sanayii A.S. | Improved wet granulation processes for apixaban comprising formulations |
| NL2029536B1 (en) | 2021-10-27 | 2023-05-26 | Pharma Data S A | Apixaban suspension and preparation method |
| WO2023072967A1 (en) | 2021-10-27 | 2023-05-04 | Pharma-Data S.A. | Apixaban suspension and preparation method |
Family Cites Families (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3340269A (en) | 1964-09-08 | 1967-09-05 | Ciba Geigy Corp | 1-substituted 4-acyl-2, 3-dioxo-piperidine |
| US3365459A (en) | 1964-09-08 | 1968-01-23 | Ciba Geigy Corp | Certain tetrahydro pyrazolo-pyridine and pyrazolo-piperidine derivatives |
| US3423414A (en) | 1966-01-13 | 1969-01-21 | Ciba Geigy Corp | Pyrazolopyridines |
| EP0028489B1 (en) | 1979-11-05 | 1983-10-05 | Beecham Group Plc | Enzyme derivatives, and their preparation |
| WO1994020460A1 (en) | 1993-03-11 | 1994-09-15 | Smithkline Beecham Corporation | Chemical compounds |
| US5612359A (en) | 1994-08-26 | 1997-03-18 | Bristol-Myers Squibb Company | Substituted biphenyl isoxazole sulfonamides |
| CN1050129C (en) | 1994-10-20 | 2000-03-08 | 美国辉瑞有限公司 | Bicyclic tetrahydro pyrazolopyridines and their use as medicaments |
| US6323201B1 (en) | 1994-12-29 | 2001-11-27 | The Regents Of The University Of California | Compounds for inhibition of ceramide-mediated signal transduction |
| US5939418A (en) | 1995-12-21 | 1999-08-17 | The Dupont Merck Pharmaceutical Company | Isoxazoline, isothiazoline and pyrazoline factor Xa inhibitors |
| US5925635A (en) | 1996-04-17 | 1999-07-20 | Dupont Pharmaceuticals Company | N-(amidinophenyl) cyclourea analogs as factor XA inhibitors |
| US6057342A (en) | 1996-08-16 | 2000-05-02 | Dupont Pharmaceutical Co. | Amidinophenyl-pyrrolidines, -pyrrolines, and -isoxazolidines and derivatives thereof |
| US6548512B1 (en) | 1996-12-23 | 2003-04-15 | Bristol-Myers Squibb Pharma Company | Nitrogen containing heteroaromatics as factor Xa inhibitors |
| ES2196396T3 (en) | 1996-12-23 | 2003-12-16 | Bristol Myers Squibb Pharma Co | HETEROAROMATIC COMPOUNDS OF 5 MEMBERS CONTAINING OXYGEN OR SULFUR AS INHIBITORS OF THE XA FACTOR. |
| US6187797B1 (en) * | 1996-12-23 | 2001-02-13 | Dupont Pharmaceuticals Company | Phenyl-isoxazoles as factor XA Inhibitors |
| US6020357A (en) | 1996-12-23 | 2000-02-01 | Dupont Pharmaceuticals Company | Nitrogen containing heteroaromatics as factor Xa inhibitors |
| IL130286A0 (en) | 1996-12-23 | 2000-06-01 | Du Pont Pharm Co | Nitrogen containing heteroaromatics as factor Xa inhibitors |
| TW536540B (en) | 1997-01-30 | 2003-06-11 | Bristol Myers Squibb Co | Endothelin antagonists: N-[[2'-[[(4,5-dimethyl-3-isoxazolyl)amino]sulfonyl]-4-(2-oxazolyl)[1,1'-biphenyl]-2-yl]methyl]-N,3,3-trimethylbutanamide and N-(4,5-dimethyl-3-isoxazolyl)-2'-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl]-4'-(2-oxazolyl)[1,1'-biphe |
| US6060491A (en) | 1997-06-19 | 2000-05-09 | Dupont Pharmaceuticals | 6-membered aromatics as factor Xa inhibitors |
| ZA985247B (en) | 1997-06-19 | 1999-12-17 | Du Pont Merck Pharma | Guanidine mimics as factor Xa inhibitors. |
| WO1998057937A2 (en) | 1997-06-19 | 1998-12-23 | The Du Pont Merck Pharmaceutical Company | Inhibitors of factor xa with a neutral p1 specificity group |
| CA2293824A1 (en) | 1997-06-19 | 1998-12-23 | Mimi Lifen Quan | (amidino)6-membered aromatics as factor xa inhibitors |
| US5998424A (en) | 1997-06-19 | 1999-12-07 | Dupont Pharmaceuticals Company | Inhibitors of factor Xa with a neutral P1 specificity group |
| US6339099B1 (en) * | 1997-06-20 | 2002-01-15 | Dupont Pharmaceuticals Company | Guanidine mimics as factor Xa inhibitors |
| CA2315070A1 (en) | 1997-12-17 | 1999-07-01 | Schering Aktiengesellschaft | Ortho-anthranilamide derivatives as anti-coagulants |
| US6271237B1 (en) * | 1997-12-22 | 2001-08-07 | Dupont Pharmaceuticals Company | Nitrogen containing heteromatics with ortho-substituted P1s as factor Xa inhabitors |
| IL136637A0 (en) | 1997-12-22 | 2001-06-14 | Du Pont Pharm Co | Nitrogen containing heteroaromatics with ortho-substituted pi's as factor xa inhibitors |
| EP1064270B1 (en) | 1998-03-27 | 2004-10-06 | Bristol-Myers Squibb Pharma Company | Disubstituted pyrazolines and triazolines as factor xa inhibitors |
| IL140622A0 (en) | 1998-07-06 | 2002-02-10 | Bristol Myers Squibb Co | Biphenyl sufonamide derivatives, pharmaceutical compositions containing the same and methods for the preparation thereof |
| US6249205B1 (en) * | 1998-11-20 | 2001-06-19 | Steward, Inc. | Surface mount inductor with flux gap and related fabrication methods |
| US6858616B2 (en) | 1998-12-23 | 2005-02-22 | Bristol-Myers Squibb Pharma Company | Nitrogen containing heterobicycles as factor Xa inhibitors |
| AU3127900A (en) * | 1998-12-23 | 2000-07-31 | Du Pont Pharmaceuticals Company | Thrombin or factor xa inhibitors |
| SI1140941T1 (en) | 1998-12-23 | 2005-04-30 | Bristol-Myers Squibb Pharma Company | Nitrogen containing heterobicycles as factor xa inhibitors |
| FR2787708B1 (en) * | 1998-12-23 | 2002-09-13 | Oreal | DYEING PROCESS USING AN ACTIVE METHYLENE COMPOUND AND A COMPOUND CHOSEN FROM AN ALDEHYDE, A KETONE, A QUINONE AND A DERIVATIVE OF DI-IMINO-ISOINDOLINE OR 3-AMINO-ISOINDOLONE |
| ATE241621T1 (en) | 1999-04-02 | 2003-06-15 | Bristol Myers Squibb Pharma Co | ARYLSULFONYLS AS FACTOR XA INHIBITORS |
| DE19932813A1 (en) | 1999-07-14 | 2001-01-18 | Bayer Ag | Substituted phenyluracile |
| KR20030011268A (en) * | 1999-07-16 | 2003-02-07 | 브리스톨-마이어스 스퀴브 파마 컴퍼니 | Nitrogen Containing Heterobicycles As Factor Xa Inhibitors |
| NZ517828A (en) | 1999-09-17 | 2003-10-31 | Millennium Pharm Inc | Inhibitors having activity against mammalian factor Xa |
| WO2001032628A1 (en) | 1999-11-03 | 2001-05-10 | Bristol-Myers Squibb Pharma Company | Cyano compounds as factor xa inhibitors |
| US6407256B1 (en) * | 1999-11-03 | 2002-06-18 | Bristol Myers Squibb Co | Cyano-pyrrole, cyano-imidazole, cyano-pyrazole, and cyano-triazole compounds as factor Xa inhibitors |
| MY125533A (en) | 1999-12-06 | 2006-08-30 | Bristol Myers Squibb Co | Heterocyclic dihydropyrimidine compounds |
| EP1156569B1 (en) * | 2000-05-19 | 2003-07-09 | USM Holding AG | Wiring for modular furniture systems |
| DE10112768A1 (en) * | 2001-03-16 | 2002-09-19 | Merck Patent Gmbh | New heterocyclic-substituted phenyl compounds, are Factor Xa and Factor VIIa inhibitors useful e.g. for treating thrombosis, myocardial infarction, inflammation, restenosis or tumor diseases |
| US6998408B2 (en) * | 2001-03-23 | 2006-02-14 | Bristol-Myers Squibb Pharma Company | 6-5, 6-6, or 6-7 Heterobicycles as factor Xa inhibitors |
| US6960595B2 (en) * | 2001-03-23 | 2005-11-01 | Bristol-Myers Squibb Pharma Company | 5-6 to 5-7 Heterobicycles as factor Xa inhibitors |
| EP1379244A4 (en) * | 2001-04-18 | 2006-03-15 | Bristol Myers Squibb Co | 1,4,5,6-tetrahydropyrazolo- 3,4-c]-pyridin-7-ones as factor xa inhibitors |
| US6750225B2 (en) * | 2001-04-18 | 2004-06-15 | Bristol-Myers Squibb Pharms Company | 1,4,5,6-tetrahydropyrazolo-[3,4,-c]-pyridin-7-ones useful as factor Xa inhibitors |
| US7171416B2 (en) | 2001-07-20 | 2007-01-30 | Compulaw, Llc. | Method and apparatus for court date calculation engine |
| PL204263B1 (en) | 2001-09-21 | 2009-12-31 | Bristol Myers Squibb Co | Lactam-containing compounds and derivatives thereof as factor xa inhibitors |
| TWI331526B (en) | 2001-09-21 | 2010-10-11 | Bristol Myers Squibb Pharma Co | Lactam-containing compounds and derivatives thereof as factor xa inhibitors |
| WO2003048081A2 (en) | 2001-12-04 | 2003-06-12 | Bristol-Myers Squibb Company | Glycinamides as factor xa inhibitors |
| TW200302225A (en) * | 2001-12-04 | 2003-08-01 | Bristol Myers Squibb Co | Substituted amino methyl factor Xa inhibitors |
| TW200303201A (en) * | 2001-12-10 | 2003-09-01 | Bristol Myers Squibb Co | Synthesis of 4,5-dihydro-pyrazolo [3,4-c] pyrid-2-ones |
| EP1505966A4 (en) | 2002-05-10 | 2006-08-30 | Bristol Myers Squibb Co | 1,1-disubstituted cycloalkyl derivatives as factor xa inhibitors |
| US7135469B2 (en) | 2003-03-18 | 2006-11-14 | Bristol Myers Squibb, Co. | Linear chain substituted monocyclic and bicyclic derivatives as factor Xa inhibitors |
| US7122557B2 (en) | 2003-03-18 | 2006-10-17 | Bristol-Myers Squibb Company | Sulfonyl-amidino-containing and tetrahydropyrimidino-containing compounds as factor Xa inhibitors |
| US6927187B2 (en) | 2003-07-11 | 2005-08-09 | Exxonmobil Chemical Patents Inc. | Synthesis of silicoaluminophosphates |
-
2002
- 2002-09-17 PL PL373299A patent/PL204263B1/en unknown
- 2002-09-17 IS IS8803A patent/IS3006B/en unknown
- 2002-09-17 CN CN201510072782.4A patent/CN104744461A/en active Pending
- 2002-09-17 KR KR1020047004025A patent/KR100908176B1/en active IP Right Review Request
- 2002-09-17 KR KR1020087018676A patent/KR100909141B1/en not_active IP Right Cessation
- 2002-09-17 ME MEP-2008-87A patent/ME00090B/en unknown
- 2002-09-17 DE DE60233335T patent/DE60233335D1/en not_active Expired - Lifetime
- 2002-09-17 CN CNA200810210432XA patent/CN101357914A/en not_active Withdrawn
- 2002-09-17 AU AU2002341693A patent/AU2002341693B2/en active Active
- 2002-09-17 AT AT09075313T patent/ATE544750T1/en active
- 2002-09-17 GE GE5556A patent/GEP20074098B/en unknown
- 2002-09-17 RS RSP-2008/0517A patent/RS20080517A/en unknown
- 2002-09-17 CA CA2461202A patent/CA2461202C/en not_active Expired - Lifetime
- 2002-09-17 CN CN2012100339134A patent/CN102617567A/en active Pending
- 2002-09-17 PL PL386232A patent/PL204653B1/en not_active IP Right Cessation
- 2002-09-17 RU RU2004112191/04A patent/RU2345993C2/en active Protection Beyond IP Right Term
- 2002-09-17 BR BRPI0212726A patent/BRPI0212726B8/en active IP Right Grant
- 2002-09-17 JP JP2003530289A patent/JP4249621B2/en not_active Expired - Lifetime
- 2002-09-17 EP EP09075313A patent/EP2105436B1/en not_active Expired - Lifetime
- 2002-09-17 ME MEP-2008-581A patent/ME00384B/en unknown
- 2002-09-17 CA CA2726702A patent/CA2726702A1/en not_active Abandoned
- 2002-09-17 EP EP02775843A patent/EP1427415B1/en not_active Expired - Lifetime
- 2002-09-17 CN CN201910724975.1A patent/CN110894196A/en active Pending
- 2002-09-17 ES ES02775843T patent/ES2329881T3/en not_active Expired - Lifetime
- 2002-09-17 MX MXPA04002526A patent/MXPA04002526A/en active IP Right Grant
- 2002-09-17 AT AT02775843T patent/ATE439360T1/en active
- 2002-09-17 HU HU0402463A patent/HU228195B1/en active Protection Beyond IP Right Term
- 2002-09-17 IL IL16069302A patent/IL160693A0/en unknown
- 2002-09-17 RS YUP-227/04A patent/RS51444B/en unknown
- 2002-09-17 SI SI200230849T patent/SI1427415T1/en unknown
- 2002-09-17 DK DK02775843T patent/DK1427415T3/en active
- 2002-09-17 PT PT02775843T patent/PT1427415E/en unknown
- 2002-09-17 NZ NZ531616A patent/NZ531616A/en not_active IP Right Cessation
- 2002-09-17 WO PCT/US2002/029491 patent/WO2003026652A1/en active Search and Examination
- 2002-09-17 UA UA20040402985A patent/UA78232C2/en unknown
- 2002-09-17 CH CH02775843T patent/CH1427415H1/en unknown
- 2002-09-17 CN CN028215370A patent/CN1578660B/en not_active Expired - Lifetime
- 2002-09-20 AR ARP020103556A patent/AR037092A1/en active IP Right Grant
- 2002-09-20 MY MYPI20023503A patent/MY137830A/en unknown
-
2004
- 2004-03-02 IL IL160693A patent/IL160693A/en active Protection Beyond IP Right Term
- 2004-03-16 IS IS7184A patent/IS2824B/en unknown
- 2004-03-18 CO CO04025932A patent/CO5560567A2/en not_active Application Discontinuation
- 2004-03-18 ZA ZA2004/02184A patent/ZA200402184B/en unknown
- 2004-03-19 HR HRP20040280AA patent/HRP20040280B1/en active IP Right Review Request
- 2004-03-19 NO NO20041163A patent/NO328558B1/en active Protection Beyond IP Right Term
- 2004-07-10 HK HK04105041.4A patent/HK1061973A1/en not_active IP Right Cessation
-
2005
- 2005-08-05 US US11/198,801 patent/US7338963B2/en not_active Expired - Lifetime
-
2007
- 2007-12-13 US US11/955,678 patent/US7531535B2/en not_active Expired - Lifetime
-
2008
- 2008-08-06 HR HR20080382A patent/HRP20080382A2/en not_active Application Discontinuation
- 2008-08-15 AR ARP080103585A patent/AR067965A2/en active IP Right Grant
- 2008-08-22 RU RU2008134413/04A patent/RU2008134413A/en not_active Application Discontinuation
- 2008-08-26 AU AU2008207537A patent/AU2008207537B8/en not_active Ceased
- 2008-08-27 NO NO20083684A patent/NO20083684L/en not_active Application Discontinuation
- 2008-09-12 CL CL2008002717A patent/CL2008002717A1/en unknown
- 2008-10-14 JP JP2008265280A patent/JP4889705B2/en not_active Expired - Lifetime
-
2009
- 2009-03-10 US US12/401,401 patent/US7691846B2/en not_active Expired - Lifetime
- 2009-10-21 CY CY20091101090T patent/CY1110509T1/en unknown
-
2010
- 2010-01-22 US US12/691,895 patent/US7960411B2/en not_active Expired - Fee Related
- 2010-01-26 IL IL203533A patent/IL203533A/en not_active IP Right Cessation
-
2011
- 2011-05-05 US US13/101,536 patent/US8188120B2/en not_active Expired - Fee Related
- 2011-09-26 NO NO2011021C patent/NO2011021I2/en unknown
- 2011-09-27 FR FR11C0042C patent/FR11C0042I2/en active Active
- 2011-09-28 DE DE201112100050 patent/DE122011100050I1/en active Pending
- 2011-09-28 CY CY2011016C patent/CY2011016I2/en unknown
- 2011-09-28 BE BE2011C034C patent/BE2011C034I2/fr unknown
- 2011-09-29 LT LTPA2011012C patent/LTPA2011012I1/en unknown
- 2011-10-19 LU LU91888C patent/LU91888I2/en unknown
-
2012
- 2012-04-12 US US13/444,999 patent/US8470854B2/en not_active Expired - Lifetime
-
2013
- 2013-04-24 HU HUS1300014C patent/HUS1300014I1/en unknown
- 2013-05-10 US US13/891,548 patent/US20140113892A1/en not_active Abandoned
-
2015
- 2015-04-08 US US14/681,743 patent/US20150210691A1/en not_active Abandoned
-
2016
- 2016-11-07 US US15/345,045 patent/US9975891B2/en not_active Expired - Lifetime
-
2018
- 2018-04-30 US US15/967,149 patent/US20180244673A1/en not_active Abandoned
-
2020
- 2020-01-08 US US16/737,261 patent/US20200140436A1/en not_active Abandoned
-
2023
- 2023-08-17 NO NO2023030C patent/NO2023030I1/en unknown
Non-Patent Citations (2)
| Title |
|---|
| DATABASE CAPLUS [online] KUMAR ET AL.: "Ketene dithioacetals. Part II. Reaction of 3-cyano-4-methylthio-2(1H)-pyridones with hydrazine and guanidine: synthesis of novel substituted and fused pyrazolo[4,3-c]pyridone and pyrido(4,3-d)pyrimidine derivatives", XP002961179, accession no. STN Database accession no. 1979:54893 * |
| JOURNAL OF THE CHEMICAL SOCIETY, PERKIN TRANSACTIONS I: ORGANIC AND BIO-ORGANIC CHEMISTRY, no. 8, 1978, pages 857 - 862 * |
Cited By (169)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7192968B2 (en) | 2000-04-05 | 2007-03-20 | Daiichi Pharmaceutical Co., Ltd. | Ethylenediamine derivatives |
| US7365205B2 (en) | 2001-06-20 | 2008-04-29 | Daiichi Sankyo Company, Limited | Diamine derivatives |
| US7342014B2 (en) | 2001-06-20 | 2008-03-11 | Daiichi Pharmaceutical Co., Ltd. | Diamine derivatives |
| EP1415992A4 (en) * | 2001-08-09 | 2006-07-26 | Daiichi Seiyaku Co | Diamine derivatives |
| EP1415992A1 (en) * | 2001-08-09 | 2004-05-06 | Daiichi Pharmaceutical Co., Ltd. | Diamine derivatives |
| US7390908B2 (en) | 2001-08-17 | 2008-06-24 | Astrazeneca Ab | Compounds effecting glucokinase |
| EP1467984A2 (en) * | 2001-12-10 | 2004-10-20 | Bristol-Myers Squibb Company | Synthesis of 4,5-dihydro-pyrazolo (3,4-c) pyrid-2-ones |
| US6919451B2 (en) | 2001-12-10 | 2005-07-19 | Bristol-Myers Squibb Company | Synthesis of 4,5-dihydro-pyrazolo [3,4-c] pyrid-2-ones |
| US7153960B2 (en) | 2001-12-10 | 2006-12-26 | Bristol-Myers Squibb Company | Synthesis of 4,5-dihydro-pyrazolo[3,4-c]pyrid-2-ones |
| EP1467984A4 (en) * | 2001-12-10 | 2005-12-07 | Bristol Myers Squibb Co | Synthesis of 4,5-dihydro-pyrazolo (3,4-c) pyrid-2-ones |
| US7576135B2 (en) | 2002-12-25 | 2009-08-18 | Daiichi Pharmaceutical Co., Ltd. | Diamine derivatives |
| WO2004058715A1 (en) * | 2002-12-25 | 2004-07-15 | Daiichi Pharmaceutical Co., Ltd. | Diamine derivatives |
| EP1603572A4 (en) * | 2003-03-18 | 2008-06-25 | Bristol Myers Squibb Co | Lactam-containing cyclic diamines and derivatives as factor xa inhibitors |
| EP1603909A2 (en) * | 2003-03-18 | 2005-12-14 | Brystol-Myers Squibb Company | Linear chain substituted monocyclic and bicyclic derivatives as factor xa inhibitors |
| EP1603909A4 (en) * | 2003-03-18 | 2008-09-03 | Bristol Myers Squibb Co | Linear chain substituted monocyclic and bicyclic derivatives as factor xa inhibitors |
| EP1603572A1 (en) * | 2003-03-18 | 2005-12-14 | Bristol-Myers Squibb Company | Lactam-containing cyclic diamines and derivatives as factor xa inhibitors |
| US7687625B2 (en) | 2003-03-25 | 2010-03-30 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7790736B2 (en) | 2003-08-13 | 2010-09-07 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7723344B2 (en) | 2003-08-13 | 2010-05-25 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
| US7678909B1 (en) | 2003-08-13 | 2010-03-16 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7169926B1 (en) | 2003-08-13 | 2007-01-30 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7790734B2 (en) | 2003-09-08 | 2010-09-07 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| JP4571075B2 (en) * | 2003-09-26 | 2010-10-27 | 田辺三菱製薬株式会社 | Amide type carboxamide derivatives |
| JPWO2005030706A1 (en) * | 2003-09-26 | 2006-12-07 | 田辺製薬株式会社 | Amide type carboxamide derivatives |
| EP1684744A4 (en) * | 2003-10-01 | 2008-12-17 | Bristol Myers Squibb Co | Pyrrolidine and piperidine derivatives as factor xa inhibitors |
| EP1684744A2 (en) * | 2003-10-01 | 2006-08-02 | Bristol-Myers Squibb Company | Pyrrolidine and piperidine derivatives as factor xa inhibitors |
| EP1670739A2 (en) * | 2003-10-08 | 2006-06-21 | Bristol-Myers Squibb Company | Cyclic diamines and derivatives as factor xa inhibitors |
| EP1670739A4 (en) * | 2003-10-08 | 2007-08-08 | Bristol Myers Squibb Co | Cyclic diamines and derivatives as factor xa inhibitors |
| EP2484209A1 (en) | 2004-02-24 | 2012-08-08 | Sumitomo Chemical Company, Limited | Insecticide compositions |
| US7947700B2 (en) | 2004-02-28 | 2011-05-24 | Boehringer Ingelheim International Gmbh | Carboxylic acid amides, the preparation thereof and their use as medicaments |
| US8445525B2 (en) | 2004-02-28 | 2013-05-21 | Boehringer Ingelheim International Gmbh | Carboxylic acid amides, the preparation thereof and their use as medicaments |
| US8791103B2 (en) | 2004-02-28 | 2014-07-29 | Boehringer Ingelheim International Gmbh | Carboxylic acid amides, the preparation thereof and their use as medicaments |
| US7371743B2 (en) | 2004-02-28 | 2008-05-13 | Boehringer Ingelheim International Gmbh | Carboxylic acid amides, the preparation thereof and their use as medicaments |
| US7732446B1 (en) | 2004-03-11 | 2010-06-08 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8329900B2 (en) | 2004-03-15 | 2012-12-11 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8188275B2 (en) | 2004-03-15 | 2012-05-29 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7906523B2 (en) | 2004-03-15 | 2011-03-15 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8173663B2 (en) | 2004-03-15 | 2012-05-08 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7807689B2 (en) | 2004-03-15 | 2010-10-05 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7781584B2 (en) | 2004-03-15 | 2010-08-24 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8288539B2 (en) | 2004-03-15 | 2012-10-16 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7550499B2 (en) | 2004-05-12 | 2009-06-23 | Bristol-Myers Squibb Company | Urea antagonists of P2Y1 receptor useful in the treatment of thrombotic conditions |
| US7745623B2 (en) | 2004-05-21 | 2010-06-29 | Takeda Pharmaceutical Company Limited | Cyclic amide derivative, and its production and use |
| US8697865B2 (en) | 2004-05-21 | 2014-04-15 | Takeda Pharmaceutical Company Limited | Cyclic amide derivative, and its production and use |
| EP2208726A1 (en) | 2004-05-21 | 2010-07-21 | Takeda Pharmaceutical Company Limited | Cyclic amide derivatives, and their production and use as antithrombotic agents |
| US8227620B2 (en) | 2004-05-24 | 2012-07-24 | Neuroscienze Pharmaness S.C. A.R.L. | Pharmaceutical compounds |
| US7659407B2 (en) | 2004-05-24 | 2010-02-09 | Neuroscienze Pharmaness S.C.a.R.I. | Pharmaceutical compounds |
| EP1602656A1 (en) * | 2004-05-24 | 2005-12-07 | NEUROSCIENZE PHARMANESS S.C. a R.L. | Pyrazole derivatives having affinity for cb1 and/or cb2 receptors |
| US7687638B2 (en) | 2004-06-04 | 2010-03-30 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
| US7825242B2 (en) | 2004-07-16 | 2010-11-02 | Takeda Pharmaceutical Company Limted | Dipeptidyl peptidase inhibitors |
| US7304157B2 (en) | 2004-09-28 | 2007-12-04 | Bristol-Myers Squibb Company | Efficient synthesis of 4,5-dihydro-pyrazolo[3,4-c]pyrid-2-ones |
| US7435821B2 (en) | 2004-09-28 | 2008-10-14 | Bristol-Myers Squibb Company | Efficient synthesis of 4,5-dihydro-pyrazolo[3,4-c]pyrid-2-ones |
| WO2006036926A2 (en) * | 2004-09-28 | 2006-04-06 | Bristol-Myers Squibb Company | Efficient synthesis of 4,5-dihydro-pyrazolo[3,4-c]pyrid-2-ones |
| US7396932B2 (en) | 2004-09-28 | 2008-07-08 | Bristol-Myers Squibb Company | Process for preparing 4,5-dihydro-pyrazolo[3,4-c]pyrid-2-ones |
| WO2007001385A3 (en) * | 2004-09-28 | 2007-05-31 | Bristol Myers Squibb Co | Process for preparing 4,5-dihydro-pyrazolo [3,4-c] pyrid-2-ones |
| WO2006135425A3 (en) * | 2004-09-28 | 2007-02-22 | Bristol Myers Squibb Co | Preparation of 4,5-dihydro-pyrazolo[3,4-c]pyrid-2-ones |
| US7388096B2 (en) | 2004-09-28 | 2008-06-17 | Bristol-Myers Squibb Company | Crystalline forms of a factor Xa inhibitor |
| WO2007001385A2 (en) * | 2004-09-28 | 2007-01-04 | Bristol-Myers Squibb Company | Process for preparing 4,5-dihydro-pyrazolo [3,4-c] pyrid-2-ones |
| WO2006036926A3 (en) * | 2004-09-28 | 2007-01-18 | Bristol Myers Squibb Co | Efficient synthesis of 4,5-dihydro-pyrazolo[3,4-c]pyrid-2-ones |
| WO2006135425A2 (en) * | 2004-09-28 | 2006-12-21 | Bristol-Myers Squibb Company | Preparation of 4,5-dihydro-pyrazolo[3,4-c]pyrid-2-ones |
| US7579472B2 (en) | 2004-09-28 | 2009-08-25 | Bristol-Myers Squibb Company | Efficient synthesis of 4,5-dihydro-pyrazolo[3,4-c]pyrid-2-ones |
| WO2006047528A3 (en) * | 2004-10-26 | 2007-03-01 | Bristol Myers Squibb Co | Pyrazolobenzamides and derivatives as factor xa inhibitors |
| US7381732B2 (en) | 2004-10-26 | 2008-06-03 | Bristol-Myers Squibb Company | Pyrazolobenzamides and derivatives as factor Xa inhibitors |
| WO2006047528A2 (en) * | 2004-10-26 | 2006-05-04 | Bristol-Myers Squibb Company | Pyrazolobenzamides and derivatives as factor xa inhibitors |
| JP2008522992A (en) * | 2004-12-09 | 2008-07-03 | バイエル・ヘルスケア・アクチェンゲゼルシャフト | Pyrazinedicarboxamides and their use |
| WO2006061116A1 (en) * | 2004-12-09 | 2006-06-15 | Bayer Healthcare Ag | Pyrazine dicarboxamides and the use thereof |
| JP2008524228A (en) * | 2004-12-15 | 2008-07-10 | ブリストル−マイヤーズ スクイブ カンパニー | Crystal form of factor XA inhibitor |
| US7371864B2 (en) * | 2004-12-15 | 2008-05-13 | Bristol-Myers Squibb Company | Crystalline forms of a factor Xa inhibitor |
| US7872124B2 (en) | 2004-12-21 | 2011-01-18 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7645778B2 (en) | 2005-01-19 | 2010-01-12 | Bristol-Myers Squibb Company | Heteroaryl compounds as P2Y1 receptor inhibitors |
| US7816382B2 (en) | 2005-06-27 | 2010-10-19 | Bristol-Myers Squibb Company | Linear urea mimics antagonists of P2Y1 receptor useful in the treatment of thrombotic condition |
| US7728008B2 (en) | 2005-06-27 | 2010-06-01 | Bristol-Myers Squibb Company | N-linked heterocyclic antagonists of P2Y1 receptor useful in the treatment of thrombotic conditions |
| US8329718B2 (en) | 2005-06-27 | 2012-12-11 | Bristol-Myers Squibb Company | N-linked heterocyclic antagonists of P2Y1 receptor useful in the treatment of thrombotic conditions |
| WO2007002635A2 (en) | 2005-06-27 | 2007-01-04 | Bristol-Myers Squibb Company | C-linked cyclic antagonists of p2y1 receptor useful in the treatment of thrombotic conditions |
| US7714002B2 (en) | 2005-06-27 | 2010-05-11 | Bristol-Myers Squibb Company | Carbocycle and heterocycle antagonists of P2Y1 receptor useful in the treatment of thrombotic conditions |
| US7700620B2 (en) | 2005-06-27 | 2010-04-20 | Bristol-Myers Squibb Company | C-linked cyclic antagonists of P2Y1 receptor useful in the treatment of thrombotic conditions |
| WO2007022165A2 (en) * | 2005-08-17 | 2007-02-22 | Bristol-Myers Squibb Company | Factor xa inhibitor inclusion complex with cyclodextrin |
| WO2007022165A3 (en) * | 2005-08-17 | 2007-08-23 | Bristol Myers Squibb Co | Factor xa inhibitor inclusion complex with cyclodextrin |
| US8906901B2 (en) | 2005-09-14 | 2014-12-09 | Takeda Pharmaceutical Company Limited | Administration of dipeptidyl peptidase inhibitors |
| US8222411B2 (en) | 2005-09-16 | 2012-07-17 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8022208B2 (en) | 2005-11-08 | 2011-09-20 | Astellas Pharma Inc. | Benzene derivative or salt thereof |
| US8263624B2 (en) | 2006-05-31 | 2012-09-11 | Bayer Intellectual Property Gmbh | Aryl-substituted heterocycles, and use thereof |
| WO2007137801A1 (en) * | 2006-05-31 | 2007-12-06 | Bayer Healthcare Ag | Tetrahydropyrrolopyridine, tetrahydropyrazolopyridine, tetrahydro-imidazopyridine and tetrahydrotriazolopyridine derivatives and use thereof |
| US8324383B2 (en) | 2006-09-13 | 2012-12-04 | Takeda Pharmaceutical Company Limited | Methods of making polymorphs of benzoate salt of 2-[[6-[(3R)-3-amino-1-piperidinyl]-3,4-dihydro-3-methyl-2,4-dioxo-1(2H)-pyrimidinyl]methyl]-benzonitrile |
| US7960569B2 (en) | 2006-10-17 | 2011-06-14 | Bristol-Myers Squibb Company | Indole antagonists of P2Y1 receptor useful in the treatment of thrombotic conditions |
| EP2099453B1 (en) | 2006-11-02 | 2017-09-06 | Bayer Intellectual Property GmbH | Combination therapy of substituted oxazolidinones |
| US8084605B2 (en) | 2006-11-29 | 2011-12-27 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| WO2008076805A2 (en) | 2006-12-15 | 2008-06-26 | Bristol-Myers Squibb Company | Arylpropionamide, arylacrylamide, arylpropynamide, or arylmethylurea analogs as factor xia inhibitors |
| WO2008079836A2 (en) | 2006-12-20 | 2008-07-03 | Bristol-Myers Squibb Company | Macrocyclic factor viia inhibitors useful as anticoagulants |
| US8093236B2 (en) | 2007-03-13 | 2012-01-10 | Takeda Pharmaceuticals Company Limited | Weekly administration of dipeptidyl peptidase inhibitors |
| US8669266B2 (en) | 2007-04-23 | 2014-03-11 | Sanofi | Quinoline-carboxamide derivatives as P2Y12 antagonists |
| DE102007028406A1 (en) | 2007-06-20 | 2008-12-24 | Bayer Healthcare Ag | Substituted oxazolidinones and their use |
| DE102007028407A1 (en) | 2007-06-20 | 2008-12-24 | Bayer Healthcare Ag | Substituted oxazolidinones and their use |
| DE102007028319A1 (en) | 2007-06-20 | 2008-12-24 | Bayer Healthcare Ag | Substituted oxazolidinones and their use |
| DE102007032344A1 (en) | 2007-07-11 | 2009-01-15 | Bayer Healthcare Ag | Prodrug derivatives of 1- (4-methoxyphenyl) -7-oxo-6- [4- (2-oxopiperidin-1-yl) phenyl] -4,5,6,7-tetrahydro-1H-pyrazolo [3,4 -c] pyridine-3-carboxamide |
| US8426420B2 (en) | 2007-12-26 | 2013-04-23 | Sanofi | Heterocyclic pyrazole-carboxamidesas P2Y12 antagonists |
| WO2011100402A1 (en) | 2010-02-11 | 2011-08-18 | Bristol-Myers Squibb Company | Macrocycles as factor xia inhibitors |
| WO2011100401A1 (en) | 2010-02-11 | 2011-08-18 | Bristol-Myers Squibb Company | Macrocycles as factor xia inhibitors |
| EP3786165A1 (en) | 2010-02-11 | 2021-03-03 | Bristol-Myers Squibb Company | Synthetic intermediates for producing macrocycles as factor xia inhibitors |
| WO2012098089A1 (en) | 2011-01-19 | 2012-07-26 | Bayer Pharma Aktiengesellschaft | Binding proteins to inhibitors of coagulation factors |
| US8884016B2 (en) | 2011-06-10 | 2014-11-11 | Dipharma Francis S.R.L. | Apixaban preparation process |
| US8969561B2 (en) | 2011-06-10 | 2015-03-03 | Dipharma Francis S.R.L. | Apixaban preparation process |
| ITMI20111047A1 (en) * | 2011-06-10 | 2012-12-11 | Dipharma Francis Srl | CRYSTAL FORM OF APIXABAN |
| EP2554159A1 (en) | 2011-08-04 | 2013-02-06 | ratiopharm GmbH | Dosage forms comprising apixaban and content uniformity enhancer |
| WO2013022814A1 (en) | 2011-08-05 | 2013-02-14 | Bristol-Myers Squibb Company | Cyclic p1 linkers as factor xia inhibitors |
| EP3070087A1 (en) | 2011-08-05 | 2016-09-21 | Bristol-Myers Squibb Company | Intermediates for the synthesis of cyclic p1 linkers as factor xia inhibitors |
| WO2013022818A1 (en) | 2011-08-05 | 2013-02-14 | Bristol-Myers Squibb Company | Novel macrocycles as factor xia inhibitors |
| EP3309148A1 (en) | 2011-10-14 | 2018-04-18 | Bristol-Myers Squibb Company | Substituted tetrahydroisoquinoline compounds as factor xia inhibitors |
| WO2013056034A1 (en) | 2011-10-14 | 2013-04-18 | Bristol-Myers Squibb Company | Substituted tetrahydroisoquinoline compounds as factor xia inhibitors |
| WO2013056060A1 (en) | 2011-10-14 | 2013-04-18 | Bristol-Myers Squibb Company | Substituted tetrahydroisoquinoline compounds as factor xia inhibitors |
| WO2013055984A1 (en) | 2011-10-14 | 2013-04-18 | Bristol-Myers Squibb Company | Substituted tetrahydroisoquinoline compounds as factor xia inhibitors |
| EP2899183A1 (en) | 2011-10-14 | 2015-07-29 | Bristol-Myers Squibb Company | Substituted tetrahydroisoquinoline compounds as factor xia inhibitors |
| US9193689B2 (en) | 2012-03-07 | 2015-11-24 | Institute Of Cancer Research: Royal Cancer Hospital (The) | 3-aryl-5-substituted-isoquinolin-1-one compounds and their therapeutic use |
| US9187453B2 (en) * | 2012-03-28 | 2015-11-17 | Takeda Pharmaceutical Company Limited | Heterocyclic compound |
| US20150094296A1 (en) * | 2012-03-28 | 2015-04-02 | Takeda Pharmaceutical Company Limited | Heterocyclic compound |
| WO2013174498A1 (en) | 2012-05-24 | 2013-11-28 | Ratiopharm Gmbh | Dosage forms comprising apixaban and matrix former |
| WO2014022766A1 (en) | 2012-08-03 | 2014-02-06 | Bristol-Myers Squibb Company | Dihydropyridone p1 as factor xia inhibitors |
| WO2014022767A1 (en) | 2012-08-03 | 2014-02-06 | Bristol-Myers Squibb Company | Dihydropyridone p1 as factor xia inhibitors |
| CZ304846B6 (en) * | 2012-11-13 | 2014-12-03 | Zentiva, K.S. | Process for preparing APIXABAN |
| EP2752414A1 (en) | 2013-01-04 | 2014-07-09 | Sandoz AG | Crystalline form of apixaban |
| WO2014115020A1 (en) * | 2013-01-24 | 2014-07-31 | Universita' Degli Studi Di Bari | Heterocycles and their radiolabeled analogs useful as cox-1 selective inhibitors |
| WO2014160668A1 (en) | 2013-03-25 | 2014-10-02 | Bristol-Myers Squibb Company | Tetrahydroisoquinolines containing substituted azoles as factor xia inhibitors |
| WO2014173377A2 (en) | 2013-04-23 | 2014-10-30 | Zentiva, K.S. | New crystalline forms of apixaban and a method of their preparation |
| WO2014203275A2 (en) | 2013-06-18 | 2014-12-24 | Cadila Healthcare Limited | An improved process for the preparation of apixaban and intermediates thereof |
| WO2015021902A1 (en) * | 2013-08-12 | 2015-02-19 | 药源药物化学(上海)有限公司 | Novel apixaban crystal form and preparation method thereof |
| US9611223B2 (en) | 2013-09-11 | 2017-04-04 | Institute Of Cancer Research: Royal Cancer Hospital (The) | 3-aryl-5-substituted-isoquinolin-1-one compounds and their therapeutic use |
| EP3072892A4 (en) * | 2013-11-18 | 2016-09-28 | Chengdu Easton Biopharmaceuticals Co Ltd | Pyridine derivative and medical use thereof |
| US9815833B2 (en) | 2013-11-18 | 2017-11-14 | Chengdu Easton Biopharmaceuticals Co., Ltd. | Pyridine derivative and medical use thereof |
| US10144731B2 (en) | 2013-12-18 | 2018-12-04 | Glaxosmithkline Intellectual Property Development Limited | Nrf2 regulators |
| EP3392249A1 (en) | 2014-01-31 | 2018-10-24 | Bristol-Myers Squibb Company | Macrocycles with hetrocyclic p2' groups as factor xia inhibitors |
| WO2015116882A1 (en) | 2014-01-31 | 2015-08-06 | Bristol-Myers Squibb Company | Macrocyclic factor xia inhibitors condensed with heterocycles |
| WO2015116886A1 (en) | 2014-01-31 | 2015-08-06 | Bristol-Myers Squibb Company | Macrocycles with hetrocyclic p2' groups as factor xia inhibitors |
| EP3988549A1 (en) | 2014-01-31 | 2022-04-27 | Bristol-Myers Squibb Company | Macrocycles with heterocyclic p2' groups as factor xia inhibitors |
| WO2015116885A1 (en) | 2014-01-31 | 2015-08-06 | Bristol-Myers Squibb Company | Macrocycles with aromatic p2' groups as factor xia inhibitors |
| EP3147283A4 (en) * | 2014-05-22 | 2017-12-06 | North China Pharmaceutical Company., Ltd. | HYDRAZINE COMPOUND AS BLOOD COAGULATION FACTOR Xa INHIBITOR |
| US11678659B2 (en) | 2014-06-11 | 2023-06-20 | Dietrich Gulba | Use as rodenticides of compounds that inhibit blood coagulation |
| WO2016020711A1 (en) | 2014-08-06 | 2016-02-11 | Egis Gyógyszergyár Zrt. | Process for the preparation of apixaban |
| JP2017530104A (en) * | 2014-09-02 | 2017-10-12 | 石薬集団中奇制薬技術(石家庄)有限公司 | Pyrazolo [3,4-c] pyridine derivative |
| CN104277041A (en) * | 2014-09-19 | 2015-01-14 | 广东东阳光药业有限公司 | Methyl substituted aromatic ring substituent-containing pyrazolopiperidone compound and composition and use thereof |
| CN104277040A (en) * | 2014-09-19 | 2015-01-14 | 广东东阳光药业有限公司 | Acyl piperazinone substituent-containing pyrazolopiperidone compound and composition and use thereof |
| CN104277041B (en) * | 2014-09-19 | 2016-01-06 | 广东东阳光药业有限公司 | Aromatic nucleus containing substituent methyl substituent pyrazolo piperidone compounds and composition thereof and purposes |
| CN104327074B (en) * | 2014-09-19 | 2016-01-06 | 广东东阳光药业有限公司 | Containing lactan substituent pyrazolo piperidone compounds and composition thereof and purposes |
| WO2016053455A1 (en) | 2014-10-01 | 2016-04-07 | Bristol-Myers Squibb Company | Pyrimidinones as factor xia inhibitors |
| EP4286372A2 (en) | 2014-10-01 | 2023-12-06 | Bristol-Myers Squibb Company | Pyrimidinones as factor xia inhibitors |
| EP3828186A2 (en) | 2014-10-01 | 2021-06-02 | Bristol-Myers Squibb Company | Pyrimidinones as factor xia inhibitors |
| WO2016079549A1 (en) | 2014-11-19 | 2016-05-26 | Egis Gyógyszergyár Zrt. | Process and intermediate for the preparation of apixaban |
| US10604509B2 (en) | 2015-06-15 | 2020-03-31 | Glaxosmithkline Intellectual Property Development Limited | Nrf2 regulators |
| US10272095B2 (en) | 2015-06-15 | 2019-04-30 | GlaxoSmithKline Intellectual Propert Development Limited | NRF2 regulators |
| US10485806B2 (en) | 2015-06-15 | 2019-11-26 | Glaxosmithkline Intellectual Property Development Limited | Nrf2 regulators |
| WO2016205482A1 (en) | 2015-06-19 | 2016-12-22 | Bristol-Myers Squibb Company | Diamide macrocycles as factor xia inhibitors |
| EP3868753A1 (en) | 2015-07-29 | 2021-08-25 | Bristol-Myers Squibb Company | Factor xia macrocyclic inhibitors bearing a non-aromatic p2' group |
| WO2017023992A1 (en) | 2015-08-05 | 2017-02-09 | Bristol-Myers Squibb Company | Novel substituted glycine derived fxia inhibitors |
| US10364256B2 (en) | 2015-10-06 | 2019-07-30 | Glaxosmithkline Intellectual Property Development Limited | Biaryl pyrazoles as NRF2 regulators |
| WO2017088841A1 (en) | 2015-11-26 | 2017-06-01 | Zentiva, K.S. | Preparation of a drug form containing amorphous apixaban |
| WO2017151746A1 (en) | 2016-03-02 | 2017-09-08 | Bristol-Myers Squibb Company | Diamide macrocycles having factor xia inhibiting activity |
| WO2017221209A1 (en) | 2016-06-23 | 2017-12-28 | Lupin Limited | Pharmaceutical formulations of apixaban |
| US10479770B2 (en) | 2016-09-23 | 2019-11-19 | Gilead Sciences, Inc. | Phosphatidylinositol 3-kinase inhibitors |
| US10227350B2 (en) | 2016-09-23 | 2019-03-12 | Gilead Sciences, Inc. | Phosphatidylinositol 3-kinase inhibitors |
| US10214519B2 (en) | 2016-09-23 | 2019-02-26 | Gilead Sciences, Inc. | Phosphatidylinositol 3-kinase inhibitors |
| US11072610B2 (en) | 2018-09-12 | 2021-07-27 | Novartis Ag | Antiviral pyridopyrazinedione compounds |
| KR20200046290A (en) | 2018-10-24 | 2020-05-07 | 하나제약 주식회사 | Method for Preparation of Apixaban |
| WO2020085616A1 (en) | 2018-10-24 | 2020-04-30 | 하나제약 주식회사 | Method for preparing apixaban |
| US11034669B2 (en) | 2018-11-30 | 2021-06-15 | Nuvation Bio Inc. | Pyrrole and pyrazole compounds and methods of use thereof |
| US11603523B2 (en) | 2019-01-18 | 2023-03-14 | Astrazeneca Ab | PCSK9 inhibitors and methods of use thereof |
| US11667613B2 (en) | 2019-09-26 | 2023-06-06 | Novartis Ag | Antiviral pyrazolopyridinone compounds |
| US11834441B2 (en) | 2019-12-06 | 2023-12-05 | Vertex Pharmaceuticals Incorporated | Substituted tetrahydrofurans as modulators of sodium channels |
| US11919887B2 (en) | 2019-12-06 | 2024-03-05 | Vertex Pharmaceuticals Incorporated | Substituted tetrahydrofurans as modulators of sodium channels |
| US11827627B2 (en) | 2021-06-04 | 2023-11-28 | Vertex Pharmaceuticals Incorporated | N-(hydroxyalkyl (hetero)aryl) tetrahydrofuran carboxamides as modulators of sodium channels |
| WO2024084217A1 (en) * | 2022-10-19 | 2024-04-25 | Kalvista Pharmaceuticals Limited | 3a,4,5,6-tetrahydro-1 h-pyrazolo[3,4-c]pyridin-7(7ah)-one derivatives as factor xiia inhibitors |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2461202C (en) | Lactam-containing compounds and derivatives thereof as factor xa inhibitors | |
| AU2002341693A1 (en) | Lactam-containing compounds and derivatives thereof as factor Xa inhibitors | |
| US20040220174A1 (en) | Lactam-containing compounds and derivatives thereof as factor Xa inhibitors | |
| WO2003048081A2 (en) | Glycinamides as factor xa inhibitors | |
| WO2003047520A2 (en) | SUBSTITUTED AMINO METHYL FACTOR Xa INHIBITORS |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 1200400358 Country of ref document: VN Ref document number: P-227/04 Country of ref document: YU |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BY BZ CA CH CN CO CR CU CZ DE DM DZ EC EE ES FI GB GD GE GH HR HU ID IL IN IS JP KE KG KP KR LC LK LR LS LT LU LV MA MD MG MN MW MX MZ NO NZ OM PH PL PT RU SD SE SG SI SK SL TJ TM TN TR TZ UA UG US UZ VC VN YU ZA ZM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ UG ZM ZW AM AZ BY KG KZ RU TJ TM AT BE BG CH CY CZ DK EE ES FI FR GB GR IE IT LU MC PT SE SK TR BF BJ CF CG CI GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 160693 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1-2004-500320 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 531616 Country of ref document: NZ Ref document number: 00590/DELNP/2004 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/A/2004/002526 Country of ref document: MX Ref document number: PA/a/2004/002526 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 04025932A Country of ref document: CO Ref document number: 04025932 Country of ref document: CO Ref document number: 2002775843 Country of ref document: EP Ref document number: 2004/02184 Country of ref document: ZA Ref document number: 200402184 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P20040280A Country of ref document: HR Ref document number: 373299 Country of ref document: PL Ref document number: 2002341693 Country of ref document: AU Ref document number: 2461202 Country of ref document: CA Ref document number: 2003530289 Country of ref document: JP Ref document number: 1020047004025 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 8169 Country of ref document: GE Ref document number: 5556 Country of ref document: GE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20028215370 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2002775843 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020087018676 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P20080382A Country of ref document: HR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12008501844 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 203533 Country of ref document: IL |
|
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) |