WO2003018588A2 - Process for the preparation of a 14-hydroxynormorphinone derivative - Google Patents
Process for the preparation of a 14-hydroxynormorphinone derivative Download PDFInfo
- Publication number
- WO2003018588A2 WO2003018588A2 PCT/EP2002/009280 EP0209280W WO03018588A2 WO 2003018588 A2 WO2003018588 A2 WO 2003018588A2 EP 0209280 W EP0209280 W EP 0209280W WO 03018588 A2 WO03018588 A2 WO 03018588A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- derivative
- benzyl
- hydroxynormoφhinone
- moφhine
- Prior art date
Links
- 0 CC(C([C@@]1(C2)c3c4O[C@]11)C=CC1=O)C2(C*)Cc3ccc4O Chemical compound CC(C([C@@]1(C2)c3c4O[C@]11)C=CC1=O)C2(C*)Cc3ccc4O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D489/00—Heterocyclic compounds containing 4aH-8, 9 c- Iminoethano-phenanthro [4, 5-b, c, d] furan ring systems, e.g. derivatives of [4, 5-epoxy]-morphinan of the formula:
- C07D489/06—Heterocyclic compounds containing 4aH-8, 9 c- Iminoethano-phenanthro [4, 5-b, c, d] furan ring systems, e.g. derivatives of [4, 5-epoxy]-morphinan of the formula: with a hetero atom directly attached in position 14
- C07D489/08—Oxygen atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D489/00—Heterocyclic compounds containing 4aH-8, 9 c- Iminoethano-phenanthro [4, 5-b, c, d] furan ring systems, e.g. derivatives of [4, 5-epoxy]-morphinan of the formula:
- C07D489/02—Heterocyclic compounds containing 4aH-8, 9 c- Iminoethano-phenanthro [4, 5-b, c, d] furan ring systems, e.g. derivatives of [4, 5-epoxy]-morphinan of the formula: with oxygen atoms attached in positions 3 and 6, e.g. morphine, morphinone
Definitions
- the invention relates to a process for the production of 14-hydroxynormo ⁇ hinone derivatives, to a new synthetic route for producing noroxymorphone, as well as to new intermediates in said route.
- Noroxymorphone is a key intermediate for the production of important medicinal opioids, such as naltrexone and naloxone.
- the common starting material for the production of these opioids is thebaine from which they are readily synthesized.
- thebaine has only a low natural abundance in poppy heads and opium.
- many alternative approaches have been made for the preparation of 14- hydroxymo hine derivatives. See for example EP 0,158,476, US 5,922,876, and the references cited therein. Further, in an attempt to remove the requirement for (the preparation of) thebaine, Coop et al.
- Ri is (l-7C)alkyl optionally substituted with one or more chlorines (such as 1,1,1 - trichloroethyl), butenyl, vinyl, benzyl, phenyl or naphthyl; and R 2 is benzyl or benzyl substituted with one or more (l-6C)alkoxy group or benzyl substituted with one or more halogen.
- the oxidation process of the present invention is an efficient process with good yields, which are significantly improved when compared to the process described by Coop et al..
- the cobalt (II) oxidant according to the present invention may be selected from a range of cobalt (II) salts, such as CoF 2 , CoCl 2 , CoBr 2 , Co(II)sulfate, Co(II)nitrate, Co(II)acetate, Co(II)propionate, and the like, and mixtures thereof.
- the preferred oxidant in the process of this invention is Co(OAc) 2 and the preferred cooxidant is air.
- the reaction mixture of this oxidation process is a heterogeneous system; the oxidant dissolves only in minor amounts in the organic solvent that is used.
- the amount of cobalt (II) salts used is not very critical, as long as the system is heterogeneous, and a skilled person will know to choose sufficient amounts thereof.
- the cooxidant is introduced into the reaction mixture by bubbling it through the solution, while stirring.
- base A person skilled in the art is aware what type of base are meant with the term mild bases, however preferred bases are sodium acetate, potassium acetate, sodium phosphate and potassium phosphate. Most preferred is sodium acetate.
- R ⁇ is (l-7C)alkyl, and most preferred is ethyl.
- benzyl is most preferred.
- the oxidation process according to the present invention is performed in an organic solvent well-suited for dissolution of this type of compounds, preferably (l-4C)alcohols or mixtures thereof. Preferred is ethanol.
- the reaction temperature is usually higher than room temperature, and may be chosen dependent on the boiling point of the solvent used. However, the temperature may not be higher than about 100 °C in order to keep the oxygen sufficiently in solution.
- alkyl group is a branched or unbranched alkyl group having 1 to 7, 1 to 6 or 1 to 4 carbon atoms, respectively, such as methyl, ethyl, isopropyl, t-butyl, heptyl and the like.
- the compound of formula III may suitably prepared by methods well known in the art.
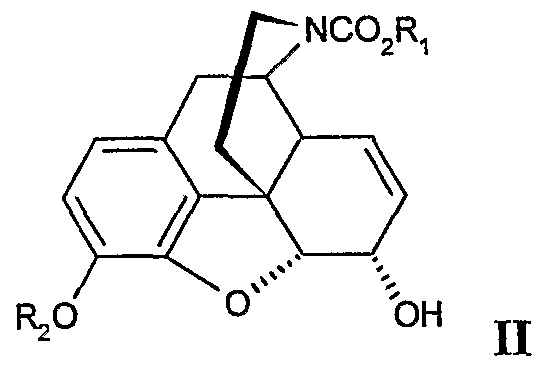
- the process for the preparation of a compound of formula III comprises reactively contacting a morphine derivative of formula II
- an oxidizing agent effective for oxidizing allylic hydroxy groups to form keto groups, where a morphinone compound of the formula III is prepared.
- the oxidizing agent is sodium dichromate.
- Ri is ethyl. For R 2 benzyl is most preferred.
- the new process of this invention may conveniently be used in the production of noroxymorphone. Therefore, another aspect of this invention is a process for the production of noroxymorphone, comprising a reaction step wherein a morphinone compound of formula LU is oxidized into the 14-hydroxynormorphinone derivative of formula IN. In particular preferred is the process further comprising the oxidation of a morphine derivative of formula II into the compound of formula III as described above.
- the novel intermediates of formula II, III and IV form each another aspect of the present invention.
- the intermediates of formula II, III and IV are in particular preferred wherein is ethyl.
- Most preferred are the intermediates of formula II, III and IV wherein Ri is ethyl and R 2 is benzyl.
- the invention is further illustrated by the following example.
- Mo ⁇ hine (1, 8 g) was dissolved in 80 ml of toluene and the solution was dried by azeotropic distillation of water.
- Sodium carbonate (15 g) and sodium hydrogen carbonate (6 g) were added and the solution was again dried by azeotropic distillation.
- Ethyl chloro formate (30 g) was slowly and in portions added over a period of approximately 4 h at 78°C. Completion of the reaction was checked with TLC. The excess of reagent and the salts were dissolved by addition of water. The layers were separated and the toluene layer was washed with water. The toluene solution was evaporated to dryness and the residue was dissolved in 70 ml of ethanol.
- the 3- carboxylic acid ethyl ester group was saponified by 6 g potassium hydroxide (dissolved in 18 ml of ethanol) and 5 g potassium carbonate at 55°C. The pH was checked (in a 1:1 dilution in water) and was >11. To this basic solution 5 g benzylchloride was added and the reaction was performed for 4 h at 75°C. The product was precipitated by the addition of water (70 ml), filtered, washed with water and dried. The yield of product (2) was 10 g.
Abstract
Description








Claims






Priority Applications (17)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| NZ531152A NZ531152A (en) | 2001-08-23 | 2002-08-15 | Process for the preparation of a 14 hydroxynormorphinone derivative |
| CA2457671A CA2457671C (en) | 2001-08-23 | 2002-08-15 | Process for the preparation of a 14-hydroxynormorphinone derivative |
| KR1020047002513A KR100901402B1 (en) | 2001-08-23 | 2002-08-15 | Process for the preparation of a 14-hydroxynormorphinone derivative |
| AU2002331163A AU2002331163B2 (en) | 2001-08-23 | 2002-08-15 | Process for the Preparation of a 14-hydroxynormorphinone Compound |
| JP2003523248A JP4559730B2 (en) | 2001-08-23 | 2002-08-15 | Preparation method of 14-hydroxynormorphinone derivative. |
| IL16019302A IL160193A0 (en) | 2001-08-23 | 2002-08-15 | Process for the preparation of a 14-hydroxynormorphinone derivative |
| US10/487,884 US7435817B2 (en) | 2001-08-23 | 2002-08-15 | C-14 oxidation of morphine derivatives |
| AT02767405T ATE466016T1 (en) | 2001-08-23 | 2002-08-15 | METHOD FOR PRODUCING A 14-HYDROXYNORMORPHINONE DERIVATIVE |
| HU0401600A HUP0401600A3 (en) | 2001-08-23 | 2002-08-15 | Process for the preparation of 14-hydroxymorphine derivatives |
| MXPA04001647A MXPA04001647A (en) | 2001-08-23 | 2002-08-15 | Process for the preparation of a 14-hydroxynormorphinone derivative. |
| DE60236171T DE60236171D1 (en) | 2001-08-23 | 2002-08-15 | PROCESS FOR FOR THE PREPARATION OF A 14-HYDROXYNORMORPHINONE DERIVATIVE |
| BRPI0211899A BRPI0211899B8 (en) | 2001-08-23 | 2002-08-15 | processes for the preparation of a 14-hydroxynormorphinone derivative and for the production of noroxymorphone |
| EP02767405A EP1421085B1 (en) | 2001-08-23 | 2002-08-15 | Process for the preparation of a 14-hydroxynormorphinone derivative |
| IS7153A IS7153A (en) | 2001-08-23 | 2004-02-13 | A process for the preparation of a 14-hydroxynormorphinone derivative |
| HR20040161A HRP20040161A2 (en) | 2001-08-23 | 2004-02-19 | Process for the preparation of a 14-hydroxynormorphinone derivative |
| NO20040724A NO328655B1 (en) | 2001-08-23 | 2004-02-19 | Morphine derivatives and processes for the preparation of such as well as intermediates |
| HK04106705.9A HK1063803A1 (en) | 2001-08-23 | 2004-09-06 | Process for the preparation of a 14-hydroxynormorphinone derivative |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP01203187 | 2001-08-23 | ||
| EP01203187.8 | 2001-08-23 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| WO2003018588A2 true WO2003018588A2 (en) | 2003-03-06 |
| WO2003018588A3 WO2003018588A3 (en) | 2003-11-27 |
| WO2003018588B1 WO2003018588B1 (en) | 2004-02-26 |
Family
ID=8180821
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2002/009280 WO2003018588A2 (en) | 2001-08-23 | 2002-08-15 | Process for the preparation of a 14-hydroxynormorphinone derivative |
Country Status (26)
| Country | Link |
|---|---|
| US (1) | US7435817B2 (en) |
| EP (1) | EP1421085B1 (en) |
| JP (1) | JP4559730B2 (en) |
| KR (1) | KR100901402B1 (en) |
| CN (1) | CN1271072C (en) |
| AR (1) | AR035289A1 (en) |
| AT (1) | ATE466016T1 (en) |
| AU (1) | AU2002331163B2 (en) |
| BR (1) | BRPI0211899B8 (en) |
| CA (1) | CA2457671C (en) |
| DE (1) | DE60236171D1 (en) |
| EC (1) | ECSP044984A (en) |
| ES (1) | ES2345327T3 (en) |
| HK (1) | HK1063803A1 (en) |
| HR (1) | HRP20040161A2 (en) |
| HU (1) | HUP0401600A3 (en) |
| IL (1) | IL160193A0 (en) |
| IS (1) | IS7153A (en) |
| MX (1) | MXPA04001647A (en) |
| NO (1) | NO328655B1 (en) |
| NZ (1) | NZ531152A (en) |
| PE (1) | PE20030460A1 (en) |
| PL (1) | PL368732A1 (en) |
| RU (1) | RU2297419C2 (en) |
| WO (1) | WO2003018588A2 (en) |
| ZA (1) | ZA200401016B (en) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006138020A2 (en) | 2005-06-16 | 2006-12-28 | Mallinckrodt Inc. | A synthetic route to 14-hydroxyl opiates through 1-halo-thebaine or analogs |
| WO2009078987A1 (en) * | 2007-12-17 | 2009-06-25 | Mallinckrodt Inc. | Process and compounds for the production of (+) opiates |
| WO2009078990A1 (en) | 2007-12-17 | 2009-06-25 | Mallinckrodt Inc. | N-demethylation of n-methyl morphinans |
| US9700508B2 (en) | 2010-05-10 | 2017-07-11 | Euro-Celtique S.A. | Pharmaceutical compositions comprising hydromorphone and naloxone |
| US9814710B2 (en) | 2013-11-13 | 2017-11-14 | Euro-Celtique S.A. | Hydromorphone and naloxone for treatment of pain and opioid bowel dysfunction syndrome |
| US9901540B2 (en) | 2010-05-10 | 2018-02-27 | Euro-Celtique S.A. | Combination of active loaded granules with additional actives |
| US9993433B2 (en) | 2010-05-10 | 2018-06-12 | Euro-Celtique S.A. | Manufacturing of active-free granules and tablets comprising the same |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8217175B2 (en) * | 2007-03-23 | 2012-07-10 | Mallinckrodt Llc | Preparation of oxymorphone from oripavine |
| CN101033228B (en) * | 2007-04-04 | 2010-05-19 | 复旦大学 | Method of preparing 14-hydroxy-7,8-dihydromorphone |
| WO2008130553A1 (en) | 2007-04-16 | 2008-10-30 | Mallinckrodt Inc. | Novel opiate reduction utilizing catalytic hydrogen transfer reaction |
| JP5579076B2 (en) * | 2007-12-17 | 2014-08-27 | マリンクロッド エルエルシー | Sinomenin derivatives and processes for their synthesis |
| EP2580218B1 (en) | 2010-06-11 | 2015-02-25 | Rhodes Technologies | Process for n-dealkylation of tertiary amines |
| AU2011263417B2 (en) | 2010-06-11 | 2014-03-27 | Rhodes Technologies | Transition metal-catalyzed processes for the preparation of N-allyl compounds and use thereof |
| RU2014112493A (en) | 2011-09-08 | 2015-10-20 | МАЛЛИНКРОДТ Эл-Эл-Си | PRODUCTION OF ALKALOIDS WITHOUT ISOLATION OF INTERMEDIATE PRODUCTS |
| GB2517000B (en) * | 2013-08-02 | 2018-05-09 | Johnson Matthey Plc | Process for the synthesis of oxymorphone alkaloid and oxymorphone salts |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000066588A1 (en) * | 1999-05-05 | 2000-11-09 | The Government Of The United States Of America, Represented By The Secretary, Departmentof Health A Nd Human Services | Direct c-14 oxidation of opioids |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2562072B1 (en) | 1984-03-27 | 1989-07-07 | Mallinckrodt Inc | PROCESS FOR THE PREPARATION OF NOROXYMORPHONE FROM MORPHINE, AND INTERMEDIATE COMPOUNDS USED IN THIS PROCESS |
| US5112975A (en) * | 1984-03-27 | 1992-05-12 | Mallinckrodt Specialty Chemicals Company | Preparation of noroxymorphone from morphine |
| US5869669A (en) * | 1996-07-26 | 1999-02-09 | Penick Corporation | Preparation of 14-hydroxynormorphinones from normorphinone dienol acylates |
-
2002
- 2002-08-15 HU HU0401600A patent/HUP0401600A3/en unknown
- 2002-08-15 JP JP2003523248A patent/JP4559730B2/en not_active Expired - Lifetime
- 2002-08-15 KR KR1020047002513A patent/KR100901402B1/en not_active IP Right Cessation
- 2002-08-15 EP EP02767405A patent/EP1421085B1/en not_active Expired - Lifetime
- 2002-08-15 RU RU2004108210/04A patent/RU2297419C2/en active
- 2002-08-15 CN CNB028163524A patent/CN1271072C/en not_active Expired - Lifetime
- 2002-08-15 US US10/487,884 patent/US7435817B2/en not_active Expired - Lifetime
- 2002-08-15 DE DE60236171T patent/DE60236171D1/en not_active Expired - Lifetime
- 2002-08-15 PL PL02368732A patent/PL368732A1/en not_active Application Discontinuation
- 2002-08-15 IL IL16019302A patent/IL160193A0/en unknown
- 2002-08-15 AU AU2002331163A patent/AU2002331163B2/en not_active Expired
- 2002-08-15 ES ES02767405T patent/ES2345327T3/en not_active Expired - Lifetime
- 2002-08-15 WO PCT/EP2002/009280 patent/WO2003018588A2/en active IP Right Grant
- 2002-08-15 AT AT02767405T patent/ATE466016T1/en active
- 2002-08-15 MX MXPA04001647A patent/MXPA04001647A/en active IP Right Grant
- 2002-08-15 NZ NZ531152A patent/NZ531152A/en not_active IP Right Cessation
- 2002-08-15 BR BRPI0211899A patent/BRPI0211899B8/en not_active IP Right Cessation
- 2002-08-15 CA CA2457671A patent/CA2457671C/en not_active Expired - Lifetime
- 2002-08-21 PE PE2002000800A patent/PE20030460A1/en not_active Application Discontinuation
- 2002-08-21 AR ARP020103130A patent/AR035289A1/en active IP Right Grant
-
2004
- 2004-02-06 ZA ZA200401016A patent/ZA200401016B/en unknown
- 2004-02-13 IS IS7153A patent/IS7153A/en unknown
- 2004-02-19 NO NO20040724A patent/NO328655B1/en not_active IP Right Cessation
- 2004-02-19 HR HR20040161A patent/HRP20040161A2/en not_active Application Discontinuation
- 2004-02-19 EC EC2004004984A patent/ECSP044984A/en unknown
- 2004-09-06 HK HK04106705.9A patent/HK1063803A1/en not_active IP Right Cessation
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000066588A1 (en) * | 1999-05-05 | 2000-11-09 | The Government Of The United States Of America, Represented By The Secretary, Departmentof Health A Nd Human Services | Direct c-14 oxidation of opioids |
Non-Patent Citations (1)
| Title |
|---|
| COOP A ET AL: "Studies into the Direct Oxidation of Codeinone to 14-Hydroxycodeinone" TETRAHEDRON, ELSEVIER SCIENCE PUBLISHERS, AMSTERDAM, NL, vol. 55, no. 38, 17 September 1999 (1999-09-17), pages 11429-11436, XP004181525 ISSN: 0040-4020 * |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006138020A2 (en) | 2005-06-16 | 2006-12-28 | Mallinckrodt Inc. | A synthetic route to 14-hydroxyl opiates through 1-halo-thebaine or analogs |
| WO2006138020A3 (en) * | 2005-06-16 | 2007-03-22 | Mallinckrodt Inc | A synthetic route to 14-hydroxyl opiates through 1-halo-thebaine or analogs |
| US8067597B2 (en) | 2005-06-16 | 2011-11-29 | Mallinckrodt Llc | Synthetic route to 14-hydroxyl opiates through 1-halo-thebaine or analogs |
| AU2006259780B2 (en) * | 2005-06-16 | 2012-04-12 | SpecGx LLC | A synthetic route to 14-hydroxyl opiates through 1-halo-thebaine or analogs |
| WO2009078987A1 (en) * | 2007-12-17 | 2009-06-25 | Mallinckrodt Inc. | Process and compounds for the production of (+) opiates |
| WO2009078990A1 (en) | 2007-12-17 | 2009-06-25 | Mallinckrodt Inc. | N-demethylation of n-methyl morphinans |
| US7671204B2 (en) | 2007-12-17 | 2010-03-02 | Mallinckrodt Inc. | N-demethylation of N-methyl morphinans |
| US9700508B2 (en) | 2010-05-10 | 2017-07-11 | Euro-Celtique S.A. | Pharmaceutical compositions comprising hydromorphone and naloxone |
| US9901540B2 (en) | 2010-05-10 | 2018-02-27 | Euro-Celtique S.A. | Combination of active loaded granules with additional actives |
| US9993433B2 (en) | 2010-05-10 | 2018-06-12 | Euro-Celtique S.A. | Manufacturing of active-free granules and tablets comprising the same |
| US9814710B2 (en) | 2013-11-13 | 2017-11-14 | Euro-Celtique S.A. | Hydromorphone and naloxone for treatment of pain and opioid bowel dysfunction syndrome |
| US10258616B2 (en) | 2013-11-13 | 2019-04-16 | Euro-Celtique S.A. | Hydromorphone and naloxone for treatment of pain and opioid bowel dysfunction syndrome |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1421085B1 (en) | Process for the preparation of a 14-hydroxynormorphinone derivative | |
| AU2002331163A1 (en) | Process for the Preparation of a 14-hydroxynormorphinone Compound | |
| EP2112153B1 (en) | Method of reducing alpha, beta- unsaturated ketones in opioid compositions | |
| US5922876A (en) | Preparation of oxymorphone from morphine | |
| EP1893616B1 (en) | A synthetic route to 14-hydroxyl opiates through 1-halo-thebaine or analogs | |
| US4795813A (en) | Synthesis of derivatives of codeine and other 3-O-alkylmorphines | |
| JP2001518444A5 (en) | ||
| CA2539659C (en) | Process for the synthesis of morphinane compounds and intermediates thereof | |
| Barber et al. | Conversion of thebaine to codeine | |
| US8106201B2 (en) | Process for the synthesis of morphinane compounds and intermediates thereof | |
| EP4267583A1 (en) | Novel process for the synthesis of noroxymorphone from morphine | |
| RU2236412C2 (en) | Method for preparing derivatives of morphinone, method for preparing derivatives of 14-hydroxymorphinone and method for preparing derivative of oxymorphone | |
| TWI292761B (en) | C-14 oxidation of morphine derivatives | |
| AU2004274038B2 (en) | Process for the synthesis of morphinane compounds and intermediates thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AU BA BB BR BZ CA CN CO CR CU DM DZ EC GD GE HR HU ID IL IN IS JP KE KP KR LC LK LR LT LV MA MG MK MN MX MZ NO NZ PH PL RO RU SG SI SK TT UA US UZ VN YU ZA Kind code of ref document: A2 Designated state(s): AE AG AL AU BA BB BR BZ CA CN CR CU DM DZ EC GD GE HR HU ID IL IS JP KE KP KR LC LK LR LT LV MA MK MN MX MZ NO NZ PH PL RO RU SI SK TT UA US UZ VN YU |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR IE IT LU MC NL PT SE SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ UG ZM ZW AM AZ BY KG KZ RU TJ TM AT BE BG CH CY CZ DK EE ES FI FR GB GR IE IT LU MC PT SE SK TR BF BJ CF CG CI GA GN GQ GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2002767405 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 160193 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004/01016 Country of ref document: ZA Ref document number: 200401016 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2457671 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 531152 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002331163 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P20040161A Country of ref document: HR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003523248 Country of ref document: JP Ref document number: 20028163524 Country of ref document: CN Ref document number: 364/CHENP/2004 Country of ref document: IN Ref document number: 1020047002513 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2004/001647 Country of ref document: MX |
|
| B | Later publication of amended claims |
Effective date: 20031212 |
|
| WWP | Wipo information: published in national office |
Ref document number: 2002767405 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10487884 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1-2004-500199 Country of ref document: PH |
|
| WWP | Wipo information: published in national office |
Ref document number: 531152 Country of ref document: NZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 531152 Country of ref document: NZ |