WO2003014085A1 - Solvates of lercanidipine hydrochloride and new crystalline forms of lercanidipine hydrochloride - Google Patents
Solvates of lercanidipine hydrochloride and new crystalline forms of lercanidipine hydrochloride Download PDFInfo
- Publication number
- WO2003014085A1 WO2003014085A1 PCT/EP2002/008700 EP0208700W WO03014085A1 WO 2003014085 A1 WO2003014085 A1 WO 2003014085A1 EP 0208700 W EP0208700 W EP 0208700W WO 03014085 A1 WO03014085 A1 WO 03014085A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- lercanidipine hydrochloride
- solvate
- lercanidipine
- ray diffraction
- anisole
- Prior art date
Links
- MQWDISMNBYOLAB-UHFFFAOYSA-N CC(C)(CN(C)CCC(c1ccccc1)c1ccccc1)O Chemical compound CC(C)(CN(C)CCC(c1ccccc1)c1ccccc1)O MQWDISMNBYOLAB-UHFFFAOYSA-N 0.000 description 1
- XUBNYSJZTPMZQV-JBASAIQMSA-N CC(C)(CN(C)CCC(c1ccccc1)c1ccccc1)OC1OC1/C(/C(C)=O)=C/c1cccc(C)c1 Chemical compound CC(C)(CN(C)CCC(c1ccccc1)c1ccccc1)OC1OC1/C(/C(C)=O)=C/c1cccc(C)c1 XUBNYSJZTPMZQV-JBASAIQMSA-N 0.000 description 1
- SCHZHHDQGRPRBN-UHFFFAOYSA-N CC(C)(CN(C)CCC(c1ccccc1)c1ccccc1)OCC(C)=O Chemical compound CC(C)(CN(C)CCC(c1ccccc1)c1ccccc1)OCC(C)=O SCHZHHDQGRPRBN-UHFFFAOYSA-N 0.000 description 1
- CSGZOFXLPPIGIJ-UHFFFAOYSA-N CC(C)(C[ClH]C)O Chemical compound CC(C)(C[ClH]C)O CSGZOFXLPPIGIJ-UHFFFAOYSA-N 0.000 description 1
- AKEGHAUFMKCWGX-UHFFFAOYSA-N CNCCC(c1ccccc1)c1ccccc1 Chemical compound CNCCC(c1ccccc1)c1ccccc1 AKEGHAUFMKCWGX-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/80—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members
- C07D211/84—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen directly attached to ring carbon atoms
- C07D211/90—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/12—Drugs for disorders of the metabolism for electrolyte homeostasis
- A61P3/14—Drugs for disorders of the metabolism for electrolyte homeostasis for calcium homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
Definitions
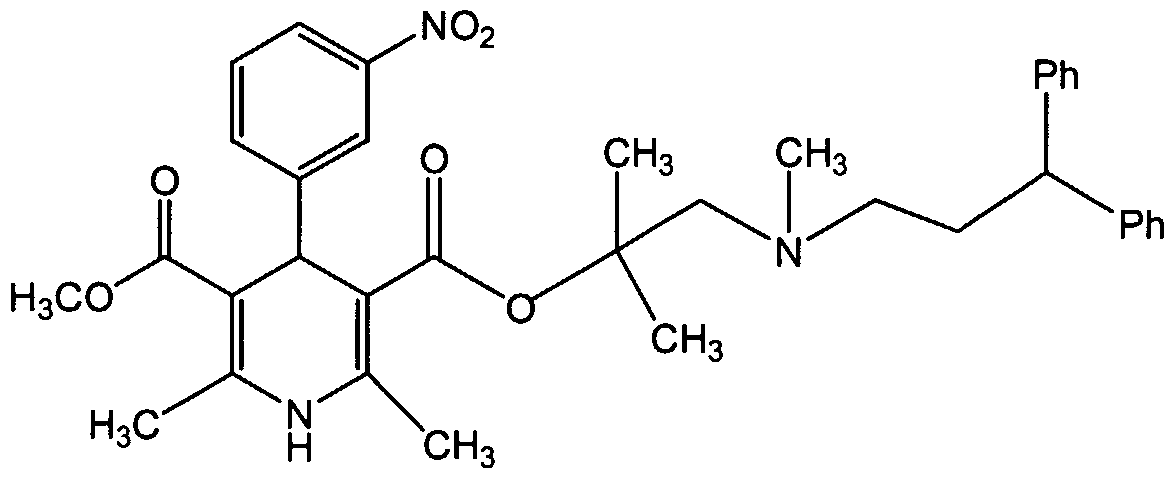
- the present invention relates to new solvates of lercanidipine hydrochloride with organic solvents, to new crystalline Forms (III) and (IV) of lercanidipine hydrochloride obtained from said solvates, to the process for preparing said solvates and Forms (III) and (IV) of lercanidipine hydrochloride by de-solvation, and to pharmaceutical compositions containing at least one of said crystalline Forms (III) and (IV).
- lercanidipine hydrochloride is a highly lipophylic dihydropyridine calcium antagonist with long action duration and high vascular selectivity. Its mechanism of antiihypertensive activity is attributed to a direct relaxant effect on vascular smooth muscle, which lowers total peripheral resistance .
- the recommended starting dose of lercanidipine hydrochloride as monotherapy is 10mg daily, by oral route, with a drug titration to 20 mg daily, lercanidipine is rapidly absorbed following oral administration with peak plasma levels occuring 2-3 hours following dosing, elimination is essentially via hepatic route.
- lercanidipine By virtue of its high lipophylicity and high membrane coefficient, lercanidipine combines a short plasma half life with a long duration of action. In fact, the preferential distribution of the drug into membranes of smooth muscle cells results in membrane controlled pharmacokinetics which is characterized by a prolonged pharmacological effect. In comparison to other calcium antagonists, lercandipine is characterized by gradual onset and long-lasting duration of action despite decreasing plasma levels. In vitro studies show that isolated rat aorta repsonse to high K + may be attenuated by lercanidipine, even after the drug has been removed from the environment of the aortic tissue for 6 hours, lercanidipine is commercially available from Recordati S.p.A. (Milan Italy) and has been described along with methods for making it and resolving it into individual enantiomers in US 4,705,797; 5,767,136; 4,968,832; 5,912,351 and 5,696,139.
- the preparation of said pharmaceutical agent can be obtained with different synthetic schemes.
- the mixture of the final reaction results in an oily residue which has to be purified by flash chromatography using as eluent chloroform with increasing amounts of acetone.
- the solvent is then evaporated from the eluate and the residue is dissolved in methanol adding a small amount of hydrochloric acid in ethanol.
- the hemi-hydrated hydrochloride salt is prepared by treatment with diluted hydrochloric acid and a saturated solution of sodium chloride.
- This esterification reaction is carried out after formation of 2,6-dimethyl-5- methoxycarbonyl-4-(3-nitrophenyl)-1 ,4-dihydropyridin-3-carboxylic acid chloride with thionyl chloride in dichloromethane and dimethylformamide at a temperature between -4 and +1°C and subsequent reaction with 2,N-dimethyl-N- (3,3-diphenylpropyl)-1-amino-2-propyl alcohol at a temperature between -10 and 0°C.
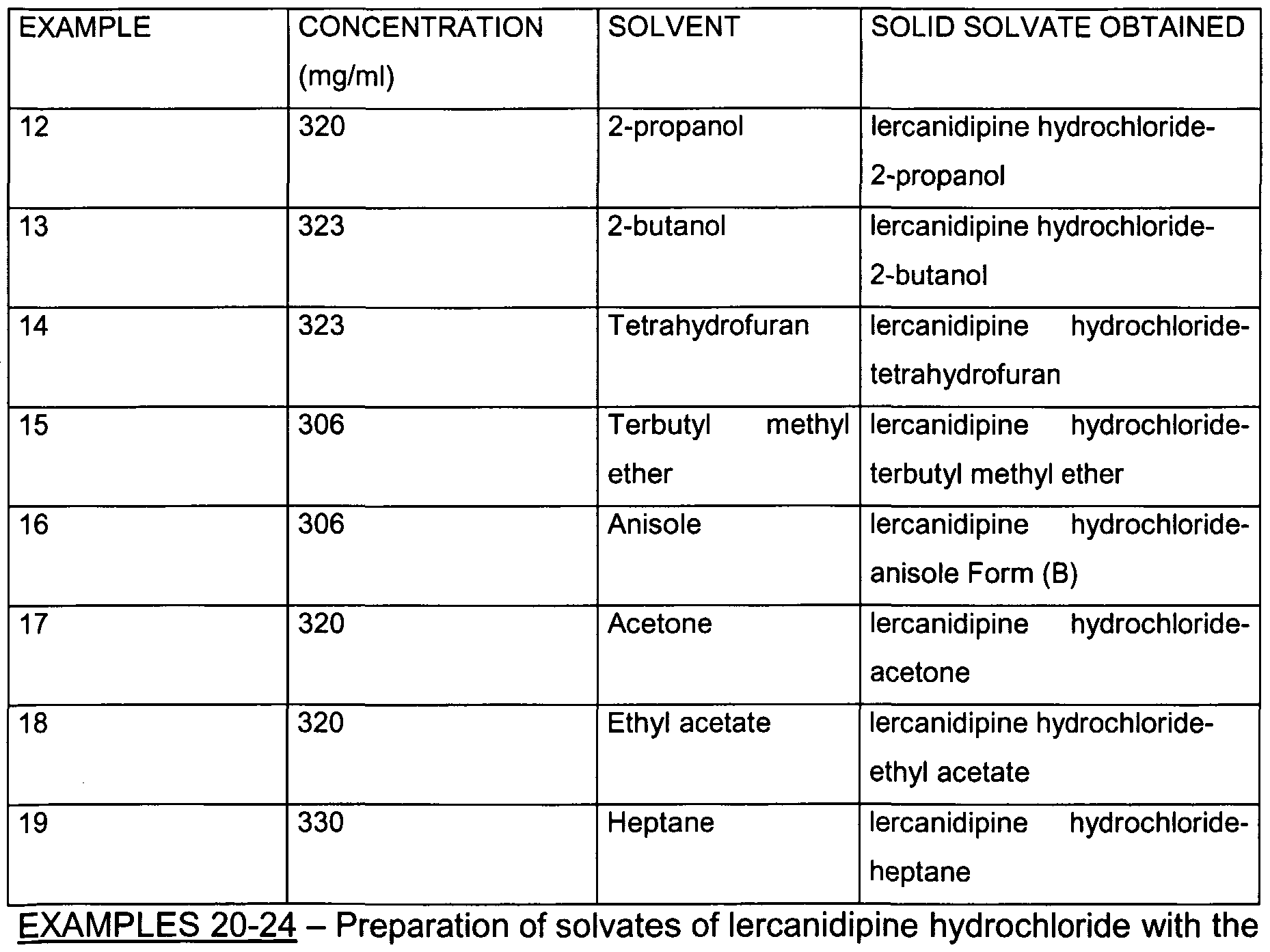
- lercanidipine hydrochloride can form solvates with solvents selected from the group consisting of: methylene chloride, acetone, anisole, tetrahydrofuran, terbutyl methyl ether, isopropanol, 2-butanol, heptane, methyl ethyl ketone, ethyl acetate.
- a further object of the present invention consists in pharmaceutical compositions containing as active agent at least one of the crystalline Forms (III) and (IV) of lercanidipine hydrochloride in combination with suitable excipients and/or diluents.
- Figure 1 shows the X-ray diffraction spectrum at wave length K ⁇ of the solvate of lercanidipine hydrochloride with methylene chloride having a lercanidipine hydrochloride-methylene chloride content of 1 :1 (mole/mole).
- the ordinate indicates the number of counts per second and the abscissa the values of 2 ⁇ angles.
- Figure 2 shows the X-ray diffraction spectrum at wave length K ⁇ of crystalline Form (III) of lercanidipine hydrochloride.
- Figures 3 and 4 show plots referring to the solvate of lercanidipine hydrochloride with methylene chloride having a lercanidipine hydrochloride-methylene chloride content of 1 :1 (mole/mole) and of lercanidipine hydrochloride crystalline Form (III) and concerning the thermogravimetric analysis carried out according to the operating modes described in Example 36B.
- the ordinate indicates % mass variation and the abscissa the temperature.
- Figures 5 and 6 show Raman spectrums referring to the solvate of lercanidipine hydrochloride with methylene chloride having a lercanidipine hydrochloride- methylene chloride content of 1 :1 (mole/mole) and of lercanidipine hydrochloride crystalline Form (III), respectively: the ordinate indicates Raman units and the abscissa wave number in cm "1 .
- Figure 7 shows the X-ray diffraction spectrum of lercanidipine hydrochloride crystalline Form (IV).
- Figure 8 shows the X-ray diffraction spectrum of the solvate lercanidipine hydrochloride-acetone having a lercanidipine hydrochloride-acetone content of
- Figure 9 shows the X-ray diffraction spectrum of the solvate lercanidipine hydrochloride-ethyl acetate having a lercanidipine hydrochloride-ethyl acetate content of 1 :1 (mole/mole).
- Figure 10 shows the X-ray diffraction spectrum of the solvate lercanidipine hydrochloride-tetrahydrofuran having a lercanidipine hydrochloride-tetrahydrofuran content of 1 :0.9(mole/mole).
- Figure 11 shows the X-ray diffraction spectrum of the solvate lercanidipine hydrochloride-terbutyl methyl ether having a lercanidipine hydrochloride-acetone content of (mole/mole).
- Figure 12 shows the X-ray diffraction spectrum of the solvate anisole-lercanidipine hydrochloride Form (A) having a lercanidipine hydrochloride-terbutyl methyl ether content of 1 :0.8 (mole/mole).
- Figure 13 shows the X-ray diffraction spectrum of the solvate anisole- lercanidipine hydrochloride Form (B) having a lercanidipine hydrochloride-anisole content of
- Figure 14 shows the X-ray diffraction spectrum of the solvate lercanidipine hydrochloride-isopropanol having a lercanidipine hydrochloride-isopropanol content of 1 :1 (mole/mole).
- Figure 15 shows the X-ray diffraction spectrum of the solvate lercanidipine hydrochloride-isobutanol having a lercanidipine hydrochloride-isobutanol content of 1 :0.8 (mole/mole).
- Figure 16 shows the X-ray diffraction spectrum of the solvate lercanidipine hydrochloride-heptane having a lercanidipine hydrochloride-heptane content of
- Figure 17 shows the Raman spectrum of lercanidipine hydrochloride crystalline
- Figure 18 shows the Raman spectrum of the solvate lercanidipine hydrochloride- acetone having a lercanidipine hydrochloride-acetone content of 1 :1.2(mole/mole).
- Figure 19 shows the Raman spectrum of the solvate lercanidipine hydrochloride- ethyl acetate having a lercanidipine hydrochloride-ethyl acetate content of 1 :1 (mole/mole).
- Figure 20 shows the Raman spectrum of the solvate lercanidipine hydrochloride- tetrahydrofuran having a lercanidipine hydrochloride-tetrahydrofuran content of 1 :0.9 (mole/mole).
- Figure 21 shows the Raman spectrum of the solvate lercanidipine hydrochloride- terbutyl methyl ether having a lercanidipine hydrochloride-terbutyl methyl ether content of 1 :0.8 (mole/mole).
- Figure 22 shows the Raman spectrum of the solvate anisole-lercanidipine hydrochloride Form (A) having a lercanidipine hydrochloride-anisole content of 1 :0.4(mole/mole).
- Figure 23 shows the Raman spectrum of the solvate anisole-lercanidipine hydrochloride Form (B) having a lercanidipine hydrochloride-anisole content of 1 :0.4(mole/mole).
- Figure 24 shows the Raman spectrum of the solvate lercanidipine hydrochloride- isopropanol having a lercanidipine hydrochloride-isopropanol content of 1 :1 (mole/mole).
- Figure 25 shows the Raman spectrum of the solvate lercanidipine hydrochloride- isobutanol having a lercanidipine hydrochloride-isobutanol content of 1 :0.8 (mole/mole).
- Figure 26 shows the Raman spectrum of the solvate lercanidipine hydrochloride- heptane having a lercanidipine hydrochloride-heptane content of 1 :0.9 (mole/mole).
- Figure 27 shows the results of the thermogravimetric analysis carried out on the solvate anisole-lercanidipine hydrochloride Form (B) having a lercanidipine hydrochloride-anisole content of 1 :0.4 (mole/mole).
- Figure 28 shows the results of the thermogravimetric analysis carried out on the solvate lercanidipine hydrochloride-ethyl acetate having a lercanidipine hydrochloride-ethyl acetate content of 1 :1 (mole/mole).
- Figure 29 shows the results of the thermogravimetric analysis carried out on the solvate lercanidipine hydrochloride-acetone having a lercanidipine hydrochloride- acetone content of 1 :1.2 (mole/mole).
- Figure 30 shows the results of the thermogravimetric analysis carried out on the solvate lercanidipine hydrochloride-tetrahydrofuran having a lercanidipine hydrochloride-tetrahydrofuran content of 1 :0.9 (mole/mole).
- Figure 31 shows the results of the thermogravimetric analysis carried out on the solvate anisole-lercanidipine hydrochloride Form (A) having a lercanidipine hydrochloride-anisole content of 1 :0.4 (mole/mole).
- Figure 32 shows the results of the thermogravimetric analysis carried out on the solvate lercanidipine hydrochloride-terbutyl methyl ether ether having a lercanidipine hydrochloride-terbutyl methyl ether content of 1 :0.8 (mole/mole).
- Figure 33 shows the results of the thermogravimetric analysis carried out on the solvate lercanidipine hydrochloride-isopropanol having a lercanidipine hydrochloride-isopropanol content of 1 :1 (mole/mole).
- Figure 34 shows the results of the thermogravimetric analysis carried out on the solvate lercanidipine hydrochloride-isobutanol having a lercanidipine hydrochloride-isobutanol content of 1 :0.8 (mole/mole).
- Figure 35 shows the results of the thermogravimetric analysis carried out on the solvate lercanidipine hydrochloride-heptane having a lercanidipine hydrochloride- heptane content of 1 :0.9 (mole/mole).
- Figure 36 shows the results of the thermogravimetric analysis carried out on lercanidipine hydrochloride crystalline Form (IV).
- Figure 37 shows the results of the thermogravimetric analysis carried out on the solvate lercanidipine hydrochloride-methyl ethyl ketone_having a lercanidipine hydrochloride-methyl ethyl ketone content of 1 : 0.7 (mole/mole).
- Figure 38 shows the X-ray spectrum of the solvate lercanidipine hydrochloride- methyl ethyl ketone having a lercanidipine hydrochloride-methyl ethyl ketone content of 1 : 0.7 (mole/mole).
- Figure 39 shows the Raman spectrum of the solvate lercanidipine hydrochloride- methyl ethyl ketone having a lercanidipine hydrochloride-methyl ethyl ketone content 1 : 0.7 (mole/mole).
- crude form refers to crystals of a compound that have not been washed and/or recrystallised to remove impurities that may be present .
- the crude forms refer to Form (A) and (B) of lercanidipine hydrocloride.
- crystalline form refers to crystals of a compound that have been washed and recrystallised to remove impurities.
- crystalline Forms (I) and (II) of lercanidipine hydrocloride were disclosed in the aforementioned copending Italian Patent Application MI2001A001726. Crystalline Forms (III) and (IV) are further described by their X-ray structure , which is discussed below.
- solvates refers both to real stable solvates, containing a specific number of solvent molecules pro molecule of lercanidipine hydrochloride, and inclusion complexes, which are less stable and contain a variable number of solvent molecules pro molecule of lercanidipine hydrochloride.
- solvates are written as "molecule-solvent". In other words a hyphen is used to separate the molecule and solvent that produces the whole solvate.
- the present invention contemplates novel solvates of lercanidipine hydrochloride with organic solvents, and specific methods of producing these solvates are disclosed herein. These solvates are advantageous as they can be easily obtained under defined conditions.
- the solvate of lercanidipine hydrochloride with methylene chloride is prepared with a method comprising suspending the crystalline Form (I) of lercanidipine hydrochloride (as identified in the aforeementioned copending Italian Patent application MI2001A001726) in methylene chloride, maintaining the mixture thus obtained in a closed vessel under mild stirring at a temperature between 20 and 50°C and filtering the precipitate thus obtained.
- solvate lercanidipine hydrochloride with methyl ethyl ketone is prepared with a process comprising: dissolving lercanidipine hydrochloride crystalline Form (I) at 80°C in said solvent containing up to 5% of water, and subsequently cooling under stirring to room temperature the solution thus obtained, which is then kept at said temperature for two days and filtering the precipitated solid and drying it in an oven at 60°C for 24 hours under vacuum.
- the other solvates constituting the object of the present invention can be obtained with a process comprising: suspending the solvate of lercanidipine hydrochloride with methylene chloride in a solvent selected from the group consisting of: acetone, anisole, ethyl acetate, tetrahydrofuran, terbutyl methyl ether, isopropanol, 2-butanol, heptane, mildly stirring at a temperature between 20 and 50°C and filtering the solid thus obtained.
- a solvent selected from the group consisting of: acetone, anisole, ethyl acetate, tetrahydrofuran, terbutyl methyl ether, isopropanol, 2-butanol, heptane
- anisole is used as solvent in said process, the solvate anisole-lercanidipine hydrochloride Form B is obtained.
- the solvates constituting the object of the present invention can be prepared with a process comprising suspending crystalline Form (III) of lercanidipine hydrochloride in a solvent selected from the group consisting of: anisole, ethyl acetate, tetrahydrofuran, terbutyl methyl ether, acetone, maintaining under mild stirring in a closed vessel at a temperature between 20 and 50°C and filtering the product obtained.
- a solvent selected from the group consisting of: anisole, ethyl acetate, tetrahydrofuran, terbutyl methyl ether, acetone
- the solvates constituting the object of the present invention can be prepared with a process comprising: suspending crude lercanidipine hydrochloride Form A or B in a solvent selected from the group consisting of anisole, ethyl acetate, tetrahydrofuran, terbutyl methyl ether, acetone, methylene chloride, maintaining under mild stirring in a closed vessel at a temperature between 20 and 50°C.
- the preparation of the aforesaid solvates is preferably carried out at room temperature.
- the method may include a series of thermal cycles performed after the solvent is added to lercanidipine hydrocloride.
- the length and number of the cooling and heating steps as well as the remperatures may be determined by one of ordinary skill in the art. In a preferred embodiment , the steps last about 3 hours each .
- the heating step is performed at 35°C and the cooling step is performed at 25°C.
- the thermal cycle is composed of a cooling step at 25°C, heating step at 35°C, and a cooling step at 25°C (25-35-25°C), where each thermal cycle lasts about 3 hours, the number of cycles can range from 10 to 20.
- the sample is stirred at a temperature of 25°C for a sufficient period of time, more preferably comprised between 24 to 240 hrs.
- the process for preparing the crystalline Form (III) is preferably carried out by evaporating the solvent from a solvate under a nitrogen stream or under a residual pressure comprised between 0,01 and 1 mbar at a temperature comprised between 50 and 90°C for a period of time comprised between 20 and 30 hours.
- the new compounds constituting the object of the present invention are hereinafter identified by means of their X-ray spectra.
- the new crystalline Form (III) of lercanidipine hydrochloride shows at the X-ray diffraction at wave length K ⁇ the image expressed in the following Table 1 and shown in Figure 2.
- the solvate of lercanidipine hydrochloride with methylene chloride shows at the X- ray diffraction at wavelength K ⁇ the image expressed in the following Table 3 and shown in Figure 1.
- the lercanidipine hydrochloride-methylene chloride content is 1 :1 (mole/mole).
- the solvate of anosole with lercanidipine hydrochloride Form (A) shows at the X- ray diffraction at wavelength K ⁇ the image expressed in the following Table 4 and shown in Figure 12.
- the lercanidipine hydrochloride-anisole content is 1 :0.4 (mole/mole).
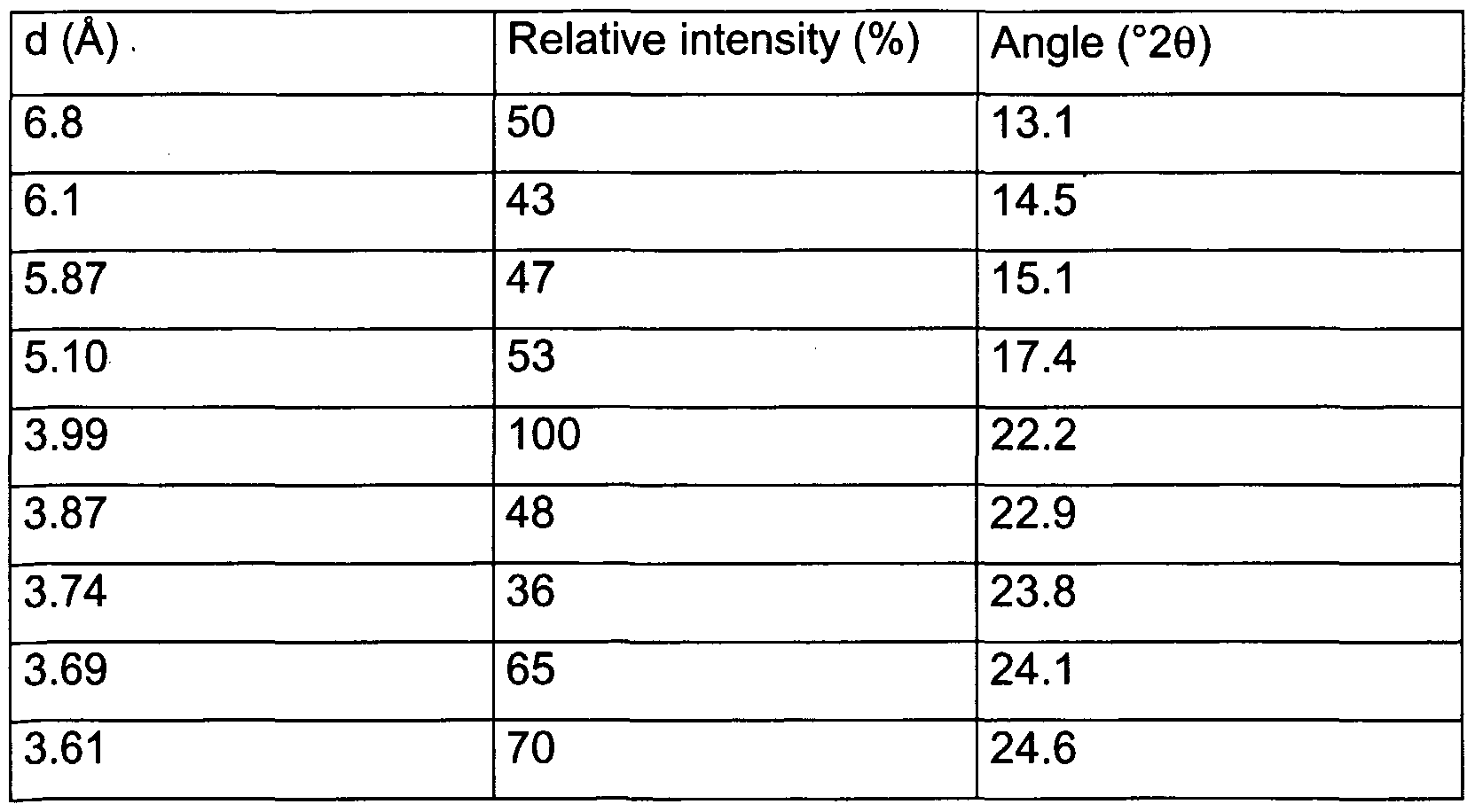
- the solvate of anisole with lercanidipine hydrochloride Form (B) shows at the X- ray diffraction at wavelength K ⁇ the image expressed in the following Table 5 and shown in Figure 13.
- the lercanidipine hydrochloride-anisole content is 1 :0.4 (mole/mole).
- the solvate of lercanidipine hydrochloride with acetone shows at the X-ray diffraction at wave length K ⁇ the image expressed in the following Table 6 and shown in Figure 8.
- the lercanidipine hydrochloride-acetone content is 1 :1.2 (mole/mole).
- the lercanidipine hydrochloride- terbutyl methyl ether content is 1 :0.8 (mole/mole).
- the lercanidipine hydrochloride-isopropanol content is 1 :1 (mole/mole).
- the solvate of lercanidipine hydrochloride with 2-butanol shows at the X-ray diffraction at wavelength K ⁇ the image expressed in the following Table 10 and shown in Figure 15.
- the lercanidipine hydrochloride-2-butanol content is 1 :0.8 (mole/mole).
- the solvate of lercanidipine hydrochloride with heptane shows at the X-ray diffraction at wavelength K ⁇ the image expressed in the following Table 11 and shown in Figure 16.
- the lercanidipine hydrochloride-heptane content is 1 :0.9 (mole/mole).
- the solvate of lercanidipine hydrochloride with tetrahydrofuran shows at the X-ray diffraction at wavelength K ⁇ the image expressed in the following Table 12 and shown in Figure 10.
- the lercanidipine hydrochloride-tetrahydrofuran content is 1 :0.9 (mole/mole).
- the solvate of lercanidipine hydrochloride with methyl ethyl ketone shows at the X- ray diffraction at wavelength K ⁇ the image expressed in the following Table 13 and shown in Figure 38.
- the lercanidipine hydrochloride-methyl ethyl ketone solvate content is 1 :0.7
- the crystalline Forms (III) and (IV) of the present invention may be formulated into a pharmaceutical composition.
- the pharmaceutical composition also may include optional additives, such as a pharmaceutically acceptable carrier or dilutant, a flavouring agent , a sweetener, a preservative, a dye, a binder, a suspending agent, a dispersing agent, a colorant, a disintegrant, an excipient, a film forming agent, a lubricant, a plasticizer, an edible oil or any combination of two or more of the foregoing.
- a pharmaceutically acceptable carrier or dilutant such as a pharmaceutically acceptable carrier or dilutant, a flavouring agent , a sweetener, a preservative, a dye, a binder, a suspending agent, a dispersing agent, a colorant, a disintegrant, an excipient, a film forming agent, a lubricant, a plasticizer, an edible oil or
- Both new crystalline forms of lercanidipine hydrochloride, i.e. (Ill) and (IV), constituting the object of the present invention preferably undergo micronization according to common techniques before being used in the preparation of the pharmaceutical compositions according to the present invention.
- the size of said particles is preferably of D(50%) 2-8 ⁇ m, D(90%) ⁇ 15 ⁇ m.
- Suitable pharmaceutically acceptable carriers or diluents include, but are not limited to ethanol, water, glycerol, propylene glycol, aloe vera gel, allantoin, glycerin, vitamin A and E oils, mineral oil, PPG2 myristyl propionate, magnesium carbonate, potassium phosphate, vegetable oil, animal oil, and solketal.
- Suitable binders include, but are not limited to, starch, gelatin, natural sugars, such as glucose, sucrose and lactose, corn sweeteners, natural and synthetic gums, such as acacia, tragacanth, vegetable gum, and sodium alginate, carboxymethylcellulose, hydroxypropylmethylcellulose, polyethylene glycol, povidone, waxes, and the like.
- Suitable disintegrators include, but are not limited to, starch, e.g., corn starch, methyl cellulose, agar, bentonite, xanthan gum, sodium starch glycolate, crosspovidone and the like.
- Suitable lubricants include, but are not limited to, sodium oleate, sodium stearate, sodium stearyl fumarate, magnesium stearate, sodium berizoate, sodium acetate, sodium chloride and the like.
- a suitable suspending agent is, but is not limited to, bentonite, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, agar-agar and tragacanth, or mixtures of two or more of these substances, and the like.
- Suitable dispersing and suspending agents include, but are not limited to, synthetic and natural gums, such as vegetable gum, tragacanth, acacia, alginate, dextran, sodium carboxymethylcellulose, methylcellulose, polyvinyl-pyrrolidone and gelatin.
- Suitable film forming agents include, but are not limited to, hydroxypropylmethylcellulose, ethylcellulose and polymethacrylates.
- Suitable plasticizers include, but are not limited to, polyethylene glycols of different molecular weights (e.g., 200-8000 Da) and propylene glycol.
- Suitable colorants include, but are not limited to, ferric oxide(s), titanium dioxide and natural and synthetic lakes.
- Suitable edible oils include, but are not limited to, cottonseed oil, sesame oil, coconut oil and peanut oil.
- additional additives include, but are not limited to, sorbitol, talc, stearic acid, dicalcium phosphate and polydextrose.
- the pharmaceutical composition may be formulated as unit dosage forms, such as tablets, pills, capsules, tablets, boluses, powders, granules, sterile parenteral solutions, sterile parenteral suspensions, sterile parenteral emulsions, elixirs, tinctures, metered aerosol or liquid sprays, drops, ampoules, autoinjector devices or suppositories.
- Unit dosage forms may be used for oral, parenteral, intranasal, sublingual or rectal administration, or for administration by inhalation or insufflation, transdermal patches, and a lyophilized composition. In general, any delivery of active ingredients that results in systemic availability of them can be used.
- the unit dosage form is an oral dosage form, most preferably a solid oral dosage form, therefore the preferred dosage forms are tablets, pills, caplets and capsules.
- parenteral preparations also are preferred.
- Solid unit dosage forms may be prepared by mixing the active agents of the present invention with a pharmaceutically acceptable carrier and any other desired additives as described above. The mixture is typically mixed until a homogeneous mixture of the active agents of the present invention and the carrier and any other desired additives is formed, i.e., until the active agents are dispersed evenly throughout the composition. In this case, the compositions can be formed as dry or moist granules.
- Tablets or pills can be coated or otherwise compounded to form a unit dosage form which has delayed and/or prolonged action, such as time release and sustained release unit dosage forms.
- the tablet or pill can comprise an inner dosage and an outer dosage component, the latter being in the form of a layer or envelope over the former.
- the two components can be separated by an enteric layer which serves to resist disintegration in the stomach and permits the inner component to pass intact into the duodenum or to be delayed in release.
- Biodegradable polymers for controlling the release of the active agents include, but are not limited to, polylactic acid, polyepsilon caprolactone, polyhydroxy butyric acid, polyorthoesters, polyacetals, polydihydro-pyrans, polycyanoacrylates and cross-linked or amphipathic block copolymers of hydrogels.
- the active substances or their physiologically acceptable salts are brought into solution, suspension or emulsion, optionally with the usually employed substances such as solubilizers, emulsifiers or other auxiliaries.
- Solvents for the active combinations and the corresponding physiologically acceptable salts can include water, physiological salt solutions or alcohols, e.g.
- Transdermal dosage form also is contemplated by the present invention.
- Transdermal forms may be a diffusion-driven transdermal system (transdermal patch) using either a fluid reservoir or a drug-in-adhesive matrix system.
- Other transdermal dosage forms include, but are not limited to, topical gels, lotions, ointments, transmucosal systems and devices, and iontohoretic (electrical diffusion) delivery system.
- Transdermal dosage forms may be used for timed release and sustained release of the active agents of the present invention.
- compositions and unit dosage forms of the present invention for administration parenterally, and in particular by injection typically include a pharmaceutically acceptable carrier, as described above.
- a preferred liquid carrier is vegetable oil.
- Injection may be, for example, intravenous, intrathecal, intramuscular, intraruminal, intratracheal, or subcutaneous.
- the active agent also can be administered in the form of liposome delivery systems, such as small unilamellar vesicles, large unilamellar vesicles and multilamellar vesicles.
- Liposomes can be formed from a variety of phospholipids, such as cholesterol, stearylamine or phosphatidylcholines.
- the crystalline forms of the present invention also may be coupled with soluble polymers as targetable drug carriers.
- Such polymers include, but are not limited to, polyvinyl-pyrrolidone, pyran copolymer, polyhydroxypropylmethacryl- amidephenol, polyhydroxy-ethyl-aspartamide-phenol, and polyethyl- eneoxideopolylysine substituted with palmitoyl residues.
- the pharmaceutical composition or unit dosage forms of the present invention may be administered by a variety of routes such as intravenous, intratracheal, subcutaneous, oral, mucosal parenteral, buccal, sublingual, opthalmic, pulmonary, transmucosal, transdermal, and intramuscular.
- Unit dosage forms also can be administered in intranasal form via topical use of suitable intranasal vehicles, or via transdermal routes, using those forms of transdermal skin patches known to those of ordinary skill in the art. Oral administration is preferred.
- the pharmaceutical composition or unit dosage forms of the present invention may be administered to an animal, preferably a human being, in need of antihypertensive treatment.
- the pharmaceutical composition or unit dosage form of the present invention may be administered according to a dosage and administration regimen defined by routine testing in light of the guidelines given above in order to obtain optimal antihypertensive activity and a decreased in blood pressure while minimizing toxicity or side-effects for a particular patient.
- a dosage and administration regimen defined by routine testing in light of the guidelines given above in order to obtain optimal antihypertensive activity and a decreased in blood pressure while minimizing toxicity or side-effects for a particular patient.
- fine turning of the therapeutic regimen is routine in light of the guidelines given herein.
- compositions containing polymorphs or mixtures of the present invention may vary according to a variety of factors such as underlying disease state, the individual's condition, weight, sex and age and the mode of administration.
- the pharmaceutical compositions can be provided in the form of scored or unscored solid unit dosage forms.
- a pharmaceutical composition comprising (1 ) lercanidipine hydrochloride, where the lercanidipine hydrochloride is selected from the group consisting of isolated lercanidipine hydrochloride crystalline Form (III), isolated lercanidipine hydrochloride crystalline Form (IV), or combinations thereof of predetermined polymorph composition; and (2) at least one component selected from the group consisting of a pharmaceutically acceptable carrier or diluent, a flavorant, a sweetener, a preservative, a dye, a binder, a suspending agent, a dispersing agent, a colorant, a disintegrant, an excipient, a diluent, a lubricant, a plasticizer, and an edible oil.
- a pharmaceutically acceptable carrier or diluent a flavorant, a sweetener, a preservative, a dye, a binder, a suspending agent, a dispersing agent, a colorant, a disintegrant, an
- the pharmaceutical composition or dosage form 0.1 to 400 mg lercanidipine hydrochloride.
- the composition or dosage form comprises 1 to 200 mg lercanidipine hydrochloride. More preferably, the composition or dosage form comprises 5 to 40 mg lercanidipine hydrochloride.
- the pharmaceutical composition or unit dosage form may be administered in a single daily dose, or the total daily dosage may be administered in divided doses. In addition, co-administration or sequential administration of other active agents may be desirable.
- the crystalline forms and mixtures thereof of the invention may be combined with any known drug therapy, preferably for treatment of hypertension.
- bimodal therapy involving in addition a diuretic, a ⁇ - receptor blocker, an ACE inhibitor or an angiotensin II receptor antagonist is contemplated by the present invention (see, e.g., U.S. Provisional Application No. 60/344,601 , filed October 23, 2001 ; Italian Application No. Ml 2001 A 002136 filed by the Applicant).
- the compounds may initially be provided as separate dosage forms until an optimum dosage combination and administration regimen is achieved. Therefore, the patient may be titrated to the appropriate dosages for his/her particular hypertensive condition. After the appropriate dosage of each of the compounds is determined to achieve a decrease of the blood pressure without untoward side effects, the patient then may be switched to a single dosage form containing the appropriate dosages of each of the active agents, or may continue with a dual dosage form.
- the exact dosage and administration regimen utilizing the combination therapy of the present invention is selected in accordance with a variety of factors including type, species, age, weight, sex and medical condition of the patient; the severity and etiology of the hypertension to be treated; the route of administration; the renal and hepatic function of the patient; the treatment history of the patient; and the responsiveness of the patient.
- Optimal precision in achieving concentrations of compounds within the range that yields efficacy without toxicity requires a regimen based on the kinetics of the drug's availability to target sites. This involves a consideration of the absorption, distribution, metabolism, excretion of a drug, and responsiveness of the patient to the dosage regimen.
- fine tuning of the therapeutic regimen is routine in light of the guidelines given herein.
- a pharmaceutical composition for parenteral administration contains not below 0.1 %, preferably from about 0.5% to about 30%, by weight of a polymorph or mixture of the present invention, based upon the total weight of the pharmaceutical composition. Individual isolated polymorphs are preferred for parenteral administration.
- transdermal dosage forms contain from about 0.01 % to about 100% by weight of the active agents, based upon 100% total weight of the dosage.
- the composition is administered daily to the patient.
- the pharmaceutical composition or dosage form 0.1 to 400 mg lercanidipine hydrochloride.
- the composition or dosage form comprises 1 to 200 mg lercanidipine hydrochloride. More preferably, the composition or dosage form comprises 5 to 40 mg lercanidipine hydrochloride.
- EXAMPLE 1 Preparation of the solvate of lercanidipine hydrochloride with methylene chloride 5.34 g of lercanidipine hydrochloride Form (I), prepared as described in the copending Italian Patent Application MI2001A001726, are introduced together with 20 ml of methylene chloride in a closed vessel and the suspension thus obtained is kept under mild stirring. After filtration with a glass filter G4 and washing with fresh methylene chloride 7.4 g of solvate of lercanidipine hydrochloride-methylene chloride are obtained (lercanidipine hydrochloride-methylene chloride content is 1 :1 (mole/mole)).
- EXAMPLE 2 Preparation of lercanidipine hydrochloride Form (III) 3.9 g of solvate of lercanidipine hydrochloride with methylene chloride are placed in a glove hood under constant nitrogen stream (25 l/h) at ambient temperature. The sample is then dried at 90°C and 1 millibar, then placed in a flask and isolated with parafilm.
- the solvate of lercanidipine hydrochloride with methylene chloride prepared as described in Example 1 , or the crystalline Form (III) of lercanidipine hydrochloride obtained as described in Example 2, or crude Form A or B are introduced in a closed vessel together with a solvent chosen among anisole, ethyl acetate, terbutyl methyl ether, under mild stirring, with several thermal cycles: 25°C-35°C- 25°C (3 hours of cooling, 3 hours of heating). After these thermal cycles the samples are kept at 25°C.
- the starting product and the solvent used, together with the concentrations of the starting product in the solvation solvent are shown in Table 14.
- EXAMPLE 10-11 Preparation of solvates of lercanidipine hydrochloride with acetone and tetrahydrofuran
- the solvate of lercanidipine hydrochloride with methylene chloride, prepared as described in Example 1 , and a solvent chosen between acetone and tetrahydrofuran are placed in a closed vessel, kept under mild stirring and subjected to 10-20 thermal cycles: 25°C-35°C-25°C (heating step 3 hours, cooling step 3 hours). After these thermal cycles the samples are kept at 25°C and then filtered.
- the solvent used and the concentrations of the starting product in the solvation solvent are shown in Table 14. All solvates are kept in closed vessels. This preparation can also be carried out starting from lercanidipine Form III or from crude Form A or B.
- the solvent is removed from the solvates by heating under vacuum.
- lercanidipine hydrochloride Form (I) 100 g of lercanidipine hydrochloride Form (I), are suspended in 250 ml of methyl ethyl ketone/water 95/5 and heated at 80°C, thus obtaining a complete dissolution. The solution is cooled spontaneously under magnetic stirring, kept at room temperature and then filtered. The whole is dried in an oven at 60°C under vacuum (about 200 mmHg), thus obtaining 93 g of the desired product (lercanidipine hydrochloride-methy ethyl ketone content is 1 :0.7 (mole/mole)).
- EXAMPLE 35 X-ray diffraction studies Philips PW 1710 and Philips X pert PW 3040 powder diffractometer (Copper K ⁇ radiation) were used, under the following typical conditions: about 5-70 mg sample (without any previous treatment) with application of a slight pressure to obtain a flat surface. Ambient air atmosphere. 0.02° 2 ⁇ step size, 2 sec step-1 , 2-50 2 ⁇ . The obtained spectra for the crystalline and solvate forms according to the present invention are given in figures 1 , 2, 7-16 and the most significant peaks are reported in tables:1-13.
- EXAMPLE 36 Description of the crystals and their thermal characterization EXAMPLE 36 A - Thermomicroscopic analysis Few mg of each sample are placed on a microscope slide provided with covering stripe and placed on a hot plate Mettler FP82 with a heating speed of 10°C/min, and analyzed with a Leitz microscope. The operating conditions in the thermomicroscope are similar to those of a common Buchi melting apparatus. The sample is not hermetically sealed, therefore the products of de-solvation or decomposition in gas phase can get out of the sample. Said analysis has given the following results.
- Example 2 the sample consists of small and very small birefringent crystals
- Example 3 the sample consists of small birefringent cylinders (examined with a crossed polarizer), having breaks and cracks. No transition phase can be observed up to the melting temperature of 144-146°C.
- ⁇ Solvate lercanidipine hvdrochloride-ethyl acetate obtained as described in Example 4: the sample consists of small birefringent cylinders (examined with a crossed polarizer), having breaks and cracks. Some small drops build up at 106°C. No transition phase can be observed up to the melting temperature of 135-145°C.
- ⁇ Solvate lercanidipine hvdrochloride-2-butanol (Ex. 8): the sample consists of birefringent cylinders (with a crossed polarizer) having several breaks and cracks. No transition phase can be observed when the crystals are heated up to their melting temperature of 125-145°C.
- p Solvate lercanidipine hydrochloride-heptane (Ex. 9): the sample consists of small irregular birefringent crystals (with a crossed polarizer). No transition phase can be observed when the crystals are heated up to their melting point at 125-150°C.
- ⁇ Solvate lercanidipine hydrochloride-acetone (Ex. 10): the sample consists of large irregular birefringent crystals (with a crossed polarizer). No transition phase can be observed when the crystals are heated up to their melting temperature at 125-135°C.
- ⁇ Lercanidipine hydrochloride crystalline Form IV (Ex. 29): the sample consists of large crystals having several breaks and cracks, which are practically non birefringent if examined with a crossed polarizer. No transition phase can be observed when the crystals are heated up to their melting temperature of 1 16- 135°C. Few crystals keep their solid form and melt only at 195°C.
- EXAMPLE 36B Thermogravimetric analysis (TG and TGFTIR) The thermogravimetric analysis is carried out according to the following operating modes. Each sample weighing 2 to 5 mg is placed in an aluminum crucible of an apparatus PERKIN ELMER TGS-2 Thermogravimetric System and heated in nitrogen stream at a rate of 10°C/min.
- thermogravimetric analysis together with an IR analysis in Fourier transform is carried out according to the following operating modes.
- Each sample weighing 2 to 5 mg is placed in an aluminum crucible of an apparatus Netzsch Thermomicrobalance TG209 coupled with a spectrometer in Fourier transform BRUKER FTIR Vector 22 and heated in nitrogen stream at a rate of 10°C/min.
- the aforesaid analyses give the following results: ⁇ Solvate of lercanidipine hydrochloride with methylene chloride prepared according to Example 1 : a weight loss of 10.1 % is observed in the temperature range between 25 and 150°C.
- Fig. 3 The volatile compound is identified by the corresponding IR spectrum and is found to be methylene chloride.
- the stechiometric compound monosolvate corresponds to a weight loss of 1 1.6%. Since methylene chloride has a high vapor pressure and since the sample already loses small amounts of dichloromethane at 25°C, it can be inferred that the product obtained in Example 1 corresponds to a solvate of lercanidipine hydrochloride with 1 molecule of methylene chloride.
- Example 4 a weight loss of 1 1.4% is observed in the temperature range between 25 and 160°C (Fig. 28).
- the volatile compound, as identified by the IR spectrum, is found to be ethyl acetate.
- Example 5 a weight loss of 5.9% is observed in the temperature range between 25 and 175°C (Fig. 31 ).
- the volatile compound is found to be anisole.
- ⁇ Solvate lercanidipine hydrochloride-terbutyl methyl ether obtained as described in Example 6 a weight loss of 10% is observed in the temperature range between 25 and 130°C (Fig. 32).
- the volatile compound, as identified by the IR spectrum, is found to be terbutyl methyl ether. Degradation can be observed at a temperature above 180°C (only CO 2 is present),
- the volatile component is found to be isopropanol.
- Example 8 a weight loss of 8.6% is observed in the temperature range 25- 155°C (Fig. 34). The volatile component is found to consist of 2-butanol. ⁇ Solvate lercanidipine hydrochloride-heptane obtained as described in Example
- mass loss does not correspond to stoichiometric values, which can be due to the presence of inclusion complexes.
- a Bruker FT-Raman RFS100 Spectrophotometer was utilized under the following typical conditions: about 10 mg sample (without any previous treatment), 64 scans 2 cm “1 resolution, 100 mW laser power, Ge-detector.
- the spectra of the crystalline (III) and solvate Form of lercanidipine hydrochloride- merthylene chloride are according to the present invention are reported in Figg. 5,6, 17-26 and 39.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Endocrinology (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Diabetes (AREA)
- Rheumatology (AREA)
- Hydrogenated Pyridines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description


















Claims












Priority Applications (14)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BR0211738-0A BR0211738A (en) | 2001-08-06 | 2002-08-05 | Lecandipine hydrochloride solvates and new crystalline forms of lecandipine hydrochloride obtained from said solvates |
| EP02767318A EP1423367B1 (en) | 2001-08-06 | 2002-08-05 | Solvates of lercanidipine hydrochloride and crystalline forms of lercanidipine hydrochloride |
| KR1020047001791A KR100912664B1 (en) | 2001-08-06 | 2002-08-05 | Solvates of lercanidipine hydrochloride and new crystalline forms of lercanidipine hydrochloride |
| DE60203919T DE60203919D1 (en) | 2001-08-06 | 2002-08-05 | SOLVATES OF LERCANIDIPINE HYDROCHLORIDE AND CRYSTALLINE FORMS OF LERCANIDIPINE HYDROCHLORIDE |
| MXPA04001073A MXPA04001073A (en) | 2001-08-06 | 2002-08-05 | Solvates of lercanidipine hydrochloride and new crystalline forms of lercanidipine hydrochloride. |
| EA200400279A EA200400279A1 (en) | 2001-08-06 | 2002-08-05 | SOLVATES OF LERCANIDIPINE HYDROCHLORIDE AND NEW CRYSTALLINE FORMS OF LERCANIDIPINE HYDROCHLORIDE OBTAINED FROM THE SPECIFIED SOLVATES |
| AT02767318T ATE294162T1 (en) | 2001-08-06 | 2002-08-05 | SOLVATES OF LERCANIDIPINE HYDROCHLORIDE AND CRYSTALLINE FORMS OF LERCANIDIPINE HYDROCHLORIDE |
| IL15391602A IL153916A0 (en) | 2001-08-06 | 2002-08-05 | Solvates of lercanidipine hydrochloride and new crystalline forms of lercanidipine hydrochloride obtained from said solvates |
| AU2002331390A AU2002331390B2 (en) | 2001-08-06 | 2002-08-05 | Solvates of lercanidipine hydrochloride and new crystalline forms of lercanidipine hydrochloride |
| HU0401161A HUP0401161A3 (en) | 2001-08-06 | 2002-08-05 | Solvates of lercanidipine hydrochloride and new crystalline forms of lercanidipine hydrochloride, process for their preparation and pharmaceutical compositions containing the crystalline forms |
| JP2003519035A JP2005502648A (en) | 2001-08-06 | 2002-08-05 | Solvate of lercanidipine hydrochloride and new crystalline form of lercanidipine hydrochloride |
| IL153916A IL153916A (en) | 2001-08-06 | 2003-01-13 | Solvates of lercanidipine hydrochloride and crystalline forms of lercanidipine hydrochloride |
| NO20040479A NO20040479L (en) | 2001-08-06 | 2004-02-03 | Solvates of lercanidipine hydrochloride and new crystalline forms of lercanidipine hydrochloride. |
| HR20040157A HRP20040157A2 (en) | 2001-08-06 | 2004-02-18 | Solvates of lercanidipine hydrochloride and new crystalline forms of lercanidipine hydrochloride |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ITMI2001A001727 | 2001-08-06 | ||
| IT2001MI001727A ITMI20011727A1 (en) | 2001-08-06 | 2001-08-06 | LERCANIDIPINE HYDROCHLORIDE SOLVATES AND NEW CRYSTALLINE FORMS OF LERCANIDIPINE HYDROCHLORIDE OBTAINED FROM THEM |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2003014085A1 true WO2003014085A1 (en) | 2003-02-20 |
Family
ID=11448246
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2002/008700 WO2003014085A1 (en) | 2001-08-06 | 2002-08-05 | Solvates of lercanidipine hydrochloride and new crystalline forms of lercanidipine hydrochloride |
Country Status (21)
| Country | Link |
|---|---|
| EP (1) | EP1423367B1 (en) |
| JP (1) | JP2005502648A (en) |
| KR (1) | KR100912664B1 (en) |
| CN (1) | CN100494175C (en) |
| AR (1) | AR037230A1 (en) |
| AT (1) | ATE294162T1 (en) |
| AU (1) | AU2002331390B2 (en) |
| BR (1) | BR0211738A (en) |
| DE (1) | DE60203919D1 (en) |
| EA (1) | EA200400279A1 (en) |
| ES (1) | ES2209684T1 (en) |
| HR (1) | HRP20040157A2 (en) |
| HU (1) | HUP0401161A3 (en) |
| IL (2) | IL153916A0 (en) |
| IT (1) | ITMI20011727A1 (en) |
| MX (1) | MXPA04001073A (en) |
| NO (1) | NO20040479L (en) |
| PE (1) | PE20030350A1 (en) |
| PL (1) | PL369449A1 (en) |
| UY (1) | UY27409A1 (en) |
| WO (1) | WO2003014085A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1856051A1 (en) | 2005-02-25 | 2007-11-21 | Recordati Ireland Limited | Amorphous lercanidipine hydrochloride |
| WO2008107797A2 (en) * | 2007-03-05 | 2008-09-12 | Actavis Group Ptc Ehf | Lercanidipine hydrochloride polymorphs and an improved process for preparation of 1,1,n-trimethyl-n-(3,3-diphenylpropyl)-2-aminoethyl acetoacetate |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR100651212B1 (en) | 2004-10-27 | 2006-12-01 | 제일약품주식회사 | Preparing Method for Amorphous Lercanidipine |
| US8097729B2 (en) | 2005-09-16 | 2012-01-17 | Glenmark Generics Ltd. | Polymorphic form of lercanidipine hydrochloride and process for the preparation thereof |
| WO2008136392A1 (en) * | 2007-04-27 | 2008-11-13 | Ajinomoto Co., Inc. | Preparation for oral administration |
| WO2011161223A2 (en) | 2010-06-23 | 2011-12-29 | Krka, Tovarna Zdravil, D.D., Novo Mesto | Pharmaceutical oral dosage forms comprising lercanidipine and enalapril and their pharmaceutically acceptable salts |
| SI2654729T1 (en) | 2010-12-24 | 2016-08-31 | Krka, D.D., Novo Mesto | Homogenous pharmaceutical oral dosage forms comprising lercanidipine and enalapril or their pharmaceutically acceptable salts together with an organic acid |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4705797A (en) * | 1984-02-14 | 1987-11-10 | Recordati S.A., Chemical And Pharmaceutical Company | N-(3,3-diphenylpropyl) aminoethyl esters of 1,4-dihydro-2,6-dimethyl-pyridine-3,5-dicarboxylic acid, compositions and use |
| US5767136A (en) * | 1995-05-12 | 1998-06-16 | Recordati, S.A. Chemical And Pharmaceutical Company | 1,4-Dihydropyridines useful for prevention or reduction of atherosclerotic lesions on arterial walls |
-
2001
- 2001-08-06 IT IT2001MI001727A patent/ITMI20011727A1/en unknown
-
2002
- 2002-08-02 AR ARP020102957A patent/AR037230A1/en not_active Application Discontinuation
- 2002-08-05 PL PL02369449A patent/PL369449A1/en unknown
- 2002-08-05 CN CNB028155114A patent/CN100494175C/en not_active Expired - Fee Related
- 2002-08-05 JP JP2003519035A patent/JP2005502648A/en active Pending
- 2002-08-05 HU HU0401161A patent/HUP0401161A3/en unknown
- 2002-08-05 KR KR1020047001791A patent/KR100912664B1/en not_active IP Right Cessation
- 2002-08-05 MX MXPA04001073A patent/MXPA04001073A/en not_active Application Discontinuation
- 2002-08-05 AU AU2002331390A patent/AU2002331390B2/en not_active Ceased
- 2002-08-05 IL IL15391602A patent/IL153916A0/en unknown
- 2002-08-05 AT AT02767318T patent/ATE294162T1/en not_active IP Right Cessation
- 2002-08-05 DE DE60203919T patent/DE60203919D1/en not_active Expired - Lifetime
- 2002-08-05 WO PCT/EP2002/008700 patent/WO2003014085A1/en active IP Right Grant
- 2002-08-05 ES ES02767318T patent/ES2209684T1/en active Pending
- 2002-08-05 BR BR0211738-0A patent/BR0211738A/en not_active IP Right Cessation
- 2002-08-05 EP EP02767318A patent/EP1423367B1/en not_active Expired - Lifetime
- 2002-08-05 EA EA200400279A patent/EA200400279A1/en unknown
- 2002-08-06 PE PE2002000716A patent/PE20030350A1/en not_active Application Discontinuation
- 2002-08-06 UY UY27409A patent/UY27409A1/en not_active Application Discontinuation
-
2003
- 2003-01-13 IL IL153916A patent/IL153916A/en unknown
-
2004
- 2004-02-03 NO NO20040479A patent/NO20040479L/en not_active Application Discontinuation
- 2004-02-18 HR HR20040157A patent/HRP20040157A2/en not_active Application Discontinuation
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4705797A (en) * | 1984-02-14 | 1987-11-10 | Recordati S.A., Chemical And Pharmaceutical Company | N-(3,3-diphenylpropyl) aminoethyl esters of 1,4-dihydro-2,6-dimethyl-pyridine-3,5-dicarboxylic acid, compositions and use |
| US5767136A (en) * | 1995-05-12 | 1998-06-16 | Recordati, S.A. Chemical And Pharmaceutical Company | 1,4-Dihydropyridines useful for prevention or reduction of atherosclerotic lesions on arterial walls |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1856051A1 (en) | 2005-02-25 | 2007-11-21 | Recordati Ireland Limited | Amorphous lercanidipine hydrochloride |
| EP1856051B1 (en) | 2005-02-25 | 2019-11-13 | Recordati Ireland Limited | Amorphous lercanidipine hydrochloride |
| WO2008107797A2 (en) * | 2007-03-05 | 2008-09-12 | Actavis Group Ptc Ehf | Lercanidipine hydrochloride polymorphs and an improved process for preparation of 1,1,n-trimethyl-n-(3,3-diphenylpropyl)-2-aminoethyl acetoacetate |
| WO2008107797A3 (en) * | 2007-03-05 | 2010-01-21 | Actavis Group Ptc Ehf | Lercanidipine hydrochloride polymorphs and an improved process for preparation of 1,1,n-trimethyl-n-(3,3-diphenylpropyl)-2-aminoethyl acetoacetate |
Also Published As
| Publication number | Publication date |
|---|---|
| KR100912664B1 (en) | 2009-08-17 |
| PL369449A1 (en) | 2005-04-18 |
| DE60203919D1 (en) | 2005-06-02 |
| AR037230A1 (en) | 2004-11-03 |
| NO20040479L (en) | 2004-02-03 |
| EP1423367B1 (en) | 2005-04-27 |
| ATE294162T1 (en) | 2005-05-15 |
| ITMI20011727A0 (en) | 2001-08-06 |
| MXPA04001073A (en) | 2005-02-17 |
| AU2002331390B2 (en) | 2008-06-05 |
| IL153916A0 (en) | 2003-07-31 |
| JP2005502648A (en) | 2005-01-27 |
| HUP0401161A2 (en) | 2004-09-28 |
| ITMI20011727A1 (en) | 2003-02-06 |
| BR0211738A (en) | 2004-09-28 |
| EA200400279A1 (en) | 2004-06-24 |
| ES2209684T1 (en) | 2004-07-01 |
| HUP0401161A3 (en) | 2009-04-28 |
| CN1538958A (en) | 2004-10-20 |
| PE20030350A1 (en) | 2003-06-06 |
| CN100494175C (en) | 2009-06-03 |
| KR20040030932A (en) | 2004-04-09 |
| EP1423367A1 (en) | 2004-06-02 |
| IL153916A (en) | 2008-07-08 |
| HRP20040157A2 (en) | 2004-08-31 |
| UY27409A1 (en) | 2003-03-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1799644B1 (en) | Lercanidipine salts | |
| US20050239847A1 (en) | Novel crude and crystalline forms of lercanidipine hydrochloride | |
| EP1432683B1 (en) | Novel crystalline polymorphic forms of lercanidipine hydrochloride and processes for their preparation | |
| AU2002327924A1 (en) | Novel crystalline polymorphic forms of lercanidipine hydrochloride and process for their preparation | |
| US20030069285A1 (en) | Novel solvate and crystalline forms of lercanidipine hydrochloride | |
| EP1423367B1 (en) | Solvates of lercanidipine hydrochloride and crystalline forms of lercanidipine hydrochloride | |
| AU2002331390A1 (en) | Solvates of lercanidipine hydrochloride and new crystalline forms of lercanidipine hydrochloride | |
| CA2399459C (en) | Novel crude and crystalline forms of lercanidipine hydrochloride | |
| CA2380202A1 (en) | Novel crude and crystalline forms of lercanidipine hydrochloride |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 153916 Country of ref document: IL |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BY BZ CH CN CO CR CU CZ DE DK DZ EC EE ES FI GB GD GE GH GM HR ID IL IN IS JP KE KG KP KR KZ LC LK LS LT LU LV MA MD MG MK MN MW MZ NO NZ OM PH PL PT RO RU SD SE SI SK SL TJ TM TN TR TT TZ UA UG VN YU ZA ZM Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SD SE SG SI SK SL TJ TM TN TR TT TZ UA UG UZ VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ UG ZM ZW AM AZ BY KG KZ RU TJ TM AT BE BG CH CY CZ DK EE ES FI FR GB GR IE IT LU MC PT SE SK TR BF BJ CF CG CI GA GN GQ GW ML MR NE SN TD TG Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR IE IT LU MC NL PT SE SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2004/001073 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P-114/04 Country of ref document: YU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020047001791 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003519035 Country of ref document: JP Ref document number: 20028155114 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P20040157A Country of ref document: HR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002767318 Country of ref document: EP Ref document number: 2002331390 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1200400193 Country of ref document: VN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 531559 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 491/CHENP/2004 Country of ref document: IN Ref document number: 200400279 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1-2004-500183 Country of ref document: PH |
|
| WWP | Wipo information: published in national office |
Ref document number: 2002767318 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2002767318 Country of ref document: EP |