WO2002085853A2 - Heterocyclylalkoxy-, -alkylthio- and -alkylaminobenzazole derivatives as 5-hydroxytryptamine-6 ligands - Google Patents
Heterocyclylalkoxy-, -alkylthio- and -alkylaminobenzazole derivatives as 5-hydroxytryptamine-6 ligands Download PDFInfo
- Publication number
- WO2002085853A2 WO2002085853A2 PCT/US2002/012512 US0212512W WO02085853A2 WO 2002085853 A2 WO2002085853 A2 WO 2002085853A2 US 0212512 W US0212512 W US 0212512W WO 02085853 A2 WO02085853 A2 WO 02085853A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- ylmethoxy
- sulfonyl
- formula
- optionally substituted
- compound
- Prior art date
Links
- 0 C*(C)CN(*)C(C)(*)C(C)(*)C(C)(*)* Chemical compound C*(C)CN(*)C(C)(*)C(C)(*)C(C)(*)* 0.000 description 7
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/06—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with radicals, containing only hydrogen and carbon atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- This invention relates to heterocyclylalkoxy-, -alkylthio- and -alkylaminobenzazole derivatives as 5- hydroxytryptamine-6 ligands, to processes for preparing them, to methods of using them and to pharmaceutical compositions containing them.
- 5-HT neurotransmitter 5- hydroxytryptamine
- serotonin is localized in the central and peripheral nervous systems and is known to affect many types of conditions including psychiatric disorders, motor activity, feeding behavior, sexual activity, and neuroendocrine regulation among others.
- the effects of serotonin are regulated by the various 5-HT receptor subtypes.
- 5-HT receptors include the 5-HTl family (e.g. 5-HTlA) , the 5-HT2 family (e.g. 5-HT2A) , 5-HT3, 5-HT4, 5-HT5, 5-HT6 and 5-HT7 subtypes .
- 5-HT6 receptor subtype The recently identified human 5-hydroxytryptamine-6 (5-HT6) receptor subtype has been cloned, and the extensive distribution of its mRNA has been reported. Highest levels of 5-HT6 receptor mRNA have been observed in the olfactory tubercle, the striatum, nucleus accumbens, dentate gyrus and CAl, CA2 and CA3 regions of the hippocampus. Lower levels of 5-HT6 receptor mRNA were seen in the granular layer of the cerebellum, several diencephalic nuclei, amygdala and in the cortex. Northern blots have revealed that 5-HT6 receptor mRNA appears to be exclusively present in the brain, with little evidence for its presence in peripheral tissues.
- 5-HT6 receptor ligands are believed to be of potential use in the treatment of certain CNS disorders such as anxiety, depression, epilepsy, obsessive compulsive disorders, attention deficit disorder, migraine, cognitive memory enhancement (e.g. for the treatment of Alzheimer's disease), sleep disorders, feeding disorders (e.g. anorexia and bulimia), panic attacks, withdrawal from drug abuse (e.g. cocaine, ethanol, nicotine and benzodiazepines) , schizophrenia, or the like; or in the treatment of certain gastrointestinal disorders such as irritable bowel syndrome.
- the present invention provides a compound of formula
- X is 0, SO y or NR 13 ; Y is CR 14 or N;
- Z is CR 15 or N with the proviso that when Y is N then Z must be CR 15 ; m and x are each independently 0 or an integer of 1,
- n and p are each independently an integer of 1, 2 or 3; n and p are each independently an integer of 1, 2 or
- R 1 is halogen, CN, 0R 16 , C0 2 R 17 , CONR 18 R 19 , CNR 20 NR 21 R 22 , S0 2 NR 23 R 21 , SO w R 25 , or a C.-C ⁇ lkyl, C 2 -C 6 alkenyl , C 2 -
- R 2 , R 3 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 and R 1X are each independently H or an optionally substituted C x - C 6 alkyl group;
- R is H, CNR 26 NR 27 R 28 or a C.,-C 6 alkyl, C 2 -C 6 alkenyl , C 2 - C 6 alkynyl, C 3 -C 6 cycloalkyl, cycloheteroalkyl, aryl or heteroaryl group each optionally substituted;
- R 12 is an optionally substituted C.-C 6 alkyl, aryl or heteroaryl grou ;
- y and w are each 0 or an integer of 1 or 2 ;
- R 13 is H or a C--C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl , C 3 - C 6 cycloalkyl, cycloheteroalkyl, aryl or heteroaryl group each optionally substituted;
- R 14 and R 15 are each independently H, halogen or a C ⁇ - C 6 alkyl, aryl, heteroaryl or C--C 6 alkoxy group each optionally substituted;
- R 16 is H, COR 29 or a C.-C 6 alkyl, C 2 -C 6 alkenyl , C 2 - C 6 alkynyl, aryl or heteroaryl group each optionally substituted;
- R 17 and R 29 are each independently H or a C..-C 6 alkyl,
- R 23 and R 24 are each independently H or a C--C 6 alkyl, aryl or heteroaryl group each optionally substituted; and R 25 is an optionally substituted C.,-C 6 alkyl, aryl, or heteroaryl group; or the stereoisomers thereof or the pharmaceutically acceptable salts thereof.
- the present invention also provides methods and compositions useful for the therapeutic treatment of central nervous system disorders related to or affected by the 5-HT6 receptor.
- the present invention further provides a method for the preparation of compounds of formula I and a compound useful therefor.
- the 5-hydroxytryptamine-6 (5-HT6) receptor is one of the most recent receptors to be identified by molecular cloning. Its ability to bind a wide range of therapeutic compounds used in psychiatry, coupled with its intriguing distribution in the brain has stimulated significant interest in new compounds which are capable of interacting with or affecting said receptor. At present, there are no known fully selective agonists. Significant efforts are being made to understand the possible role of the 5-HT6 receptor in psychiatry, cognitive dysfunction, motor function and control, memory, mood and the like. To that end, compounds which demonstrate a binding affinity for the 5-HT6 receptor are earnestly sought both as an aid in the study of the 5-HT6 receptor and as potential therapeutic agents in the treatment of central nervous system disorders.
- heterocyclylalkoxy-, -thioxy- or -aminobenzazole derivatives of formula I demonstrate 5-HT6 affinity.
- said benzazole derivatives may be used as effective therapeutic agents for the treatment of central nervous system (CNS) disorders associated with or affected by the 5-HT6 receptor.
- CNS central nervous system
- the present invention provides heterocyclylalkoxy-, -alkylthio- or -alkylaminobenzazole derivatives of formula I
- W is S0 2 , CO, CONH, CSNH or (CH 2 ) x ;
- X is 0, SO y or NR 13 ;
- Y is CR M or N;
- Z is CR 1S or N with the proviso that when Y is N then
- Z must be CR 1S ; m and x are each independently 0 or an integer of 1, 2 or 3; n and p are each independently an integer of 1, 2 or
- R. is halogen, CN, OR 16 , C0 2 R 17 , CONR 18 R 19 , CNR 20 NR 21 R 22 ,
- R 2 , R 3 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 and R u are each independently H or an optionally substituted C- C 6 alkyl group;
- R 4 is H, CNR 26 NR 27 R 28 or a C--C 6 alkyl, C 2 -C 6 alkenyl, C 2 -
- R 12 is an optionally substituted C--C 6 alkyl, aryl or heteroaryl group each optionally substituted;
- R 12 is an optionally substituted C--C 6 alkyl, aryl or heteroaryl group;
- y and w are each 0 or an integer of 1 or 2 ;
- R 13 is H or a C j -C j alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 3 - C 6 cycloalkyl, cycloheteroalkyl, aryl or heteroaryl group each optionally substituted;
- R 1(1 and R 15 are each independently H, halogen or a C x - C ⁇ alkyl, aryl, heteroaryl or C.,-C 6 alkoxy group each optionally substituted;
- R 16 is H, COR 29 or a C.-C 8 alkyl, C 2 -C 6 alkenyl, C 2
- R 17 and R 29 are each independently H or a C.-C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl , C 3 -C 6 cycloalkyl, cycloheteroalkyl, aryl or heteroaryl group each optionally substituted;
- R 1B , R 19 , R 20 , R 21 , R 22 , R 26 , R 27 and R 28 are each independently H or an optionally substituted C ⁇ - C 6 alkyl group;
- R 23 and R 2i are each independently H or a C--C 6 alkyl, aryl or heteroaryl group each optionally substituted; and
- R 25 is an optionally substituted C 1 -C 6 alkyl, aryl, or heteroaryl group; or the stereoisomers thereof or the pharmaceutically acceptable salts thereof.
- halogen designates Br, Cl, I or F
- cycloheteroalkyl designates a C 5 -C 7 cycloalkyl ring system containing 1 or 2 heteroatoms, which may be the same or different, selected from N, 0 or S and optionally containing one double bond.
- Exemplary of the cycloheteroalkyl ring systems included in the term as designated herein are the following rings wherein X x is NR, 0 or S and R is an optional substituent as described hereinbelow.
- heteroaryl designates a 5- to 10-membered aromatic ring system containing 1, 2 or 3 heteroatoms, which may be the same or different, selected from N, 0 or S.
- heteroaryl ring systems include pyrrolyl, azolyl, oxazolyl, thiazolyl, imidazolyl, furyl, thienyl, quinolinyl, isoguinolinyl, indolinyl, benzothienyl, benzofuranyl, benzisoxazolyl or the like.
- haloalkyl designates a C n H 2n+1 group having from one to 2n+l halogen atoms which may be the same or different and the term haloalkoxy as used herein designates an OC n H 2n+1 group having from one to 2n+l halogen atoms which may be the same or different.
- substituents include halogen atoms, nitro, cyano, thiocyanato, cyanato, hydroxyl, alkyl, haloalkyl, alkoxy, haloalkoxy, amino, alkylamino, dialkylamino , formyl, alkoxycarbonyl , carboxyl, alkanoyl, alkylthio, alkylsuphinyl, alkylsulphonyl, carbamoyl, alkylamido, phenyl, phenoxy, benzyl, benzyloxy, heterocyclyl (eg heteroaryl or cycloheteroalkyl) or cycloalkyl groups, preferably halogen atoms or lower alkyl groups.
- substituents include halogen atoms, nitro, cyano, thiocyanato, cyanato, hydroxyl, alkyl, haloalkyl, alkoxy, haloalkoxy, amino,
- substituents may be present .
- this may be linear or branched and may contain up to 12 , preferably up to 6, more preferably up to 4 carbon atoms.
- Pharmaceutically acceptable salts may be any acid addition salt formed by a compound of formula I and a pharmaceutically acceptable acid such as phosphoric, sulfuric, • hydrochloric, hydrobromic, citric, maleic, alonic, mandelic, succinic, fumaric, acetic, lactic, nitric, sulfonic, p-toluene sulfonic, methane sulfonic acid or the like.
- Compounds of the invention may exist as one or more stereoisomers .
- the various stereoisomers include enantiomers, diastereomers, atropisomers and geometric isomers.
- the present invention comprises compounds of Formula I, the stereoisomers thereof and the pharmaceutically acceptable salts thereof .
- the compounds of the invention may be present as a mixture of stereoisomers, individual stereoisomers, or as an opticaly active form.
- Preferred compounds of the invention are those compounds of formula I wherein W is S0 2 or CO. Also preferred are those compounds of formula I wherein X is 0.
- Another group of preferred compounds of the invention are those compounds of formula I wherein Y is CR 14 .
- Further preferred compounds of the invention are those compounds of formula I wherein R 12 is an aryl or heteroaryl group each optionally substituted; and n is 1.
- R 12 are aryl e.g., phenyl or naphthyl, or heteroaryl e.g., thienyl (such as thien-2-yl) or quinolyl (such as quinolin-8-yl) ; said aryl and heteroaryl groups being unsubstituted or optionally substituted by one or more (e.g., 1 to 3) substituents the same or different as described herein.
- Such substituents include halo, nitro, cyano, thiocyanato, cyanato, hydroxyl, alkyl of 1-6 carbon atoms, halo (C j -C 6 ) alkyl, (C.-C 6 ) alkoxy, halo(C ⁇ -
- C 6 )alkoxy amino, (C,-C 6 ) alkylamino, di- (C--C 6 alkyl) amino, formyl, (C ⁇ -C 6 alkoxy) carbonyl, carboxyl, (C.-C alkanoyl, (Ci-C 8 ) alkylthio, (C--C 6 ) alkylsulphinyl, (C,-C 5 ) alkyl- sulphonyl, carbamoyl, (C--C 5 ) alkylamido, phenyl, phenoxy, benzyl, benzyloxy, heteroaryl and cycloheteroalkyl or (C 3 - C 8 ) cycloalkyl groups.
- Such optionally substituted groups for R 12 are also examples of aryl or heteroaryl for each of R r R 4 , R 13 , R 14 , R 1S , R 16 , R 17 , R 23 , R 24 , R 25 and R 29 .
- More preferred compounds of the invention are those compounds of formula I having one or more, e.g. all, of the following values: W is S0 2 ; X is 0; and n is 1.
- Another group of more preferred compounds of the invention are those compounds of formula I having one or more, e.g. all, of the following values: W is S0 2 ; X is O; Y is CR 14 ; n is 1; and p is 1.
- R 2 and R 3 are hydrogen.
- An example of m is 0.
- R s -R ⁇ m Y a ll f° r example be hydrogen.
- This invention also provides a process for the preparation of a compound of formula I which comprises one of the following:
- R 12 is as defined above and W is S0 2 , CO, CONH, CSNH or (CH 2 ) x ; said reactants protected on reactive sites and/or on reactive substituent groups as required, and removing any protecting groups, to give a corresponding compound of formula (I) ; or b) removing a protecting group from a compound of formula (C)
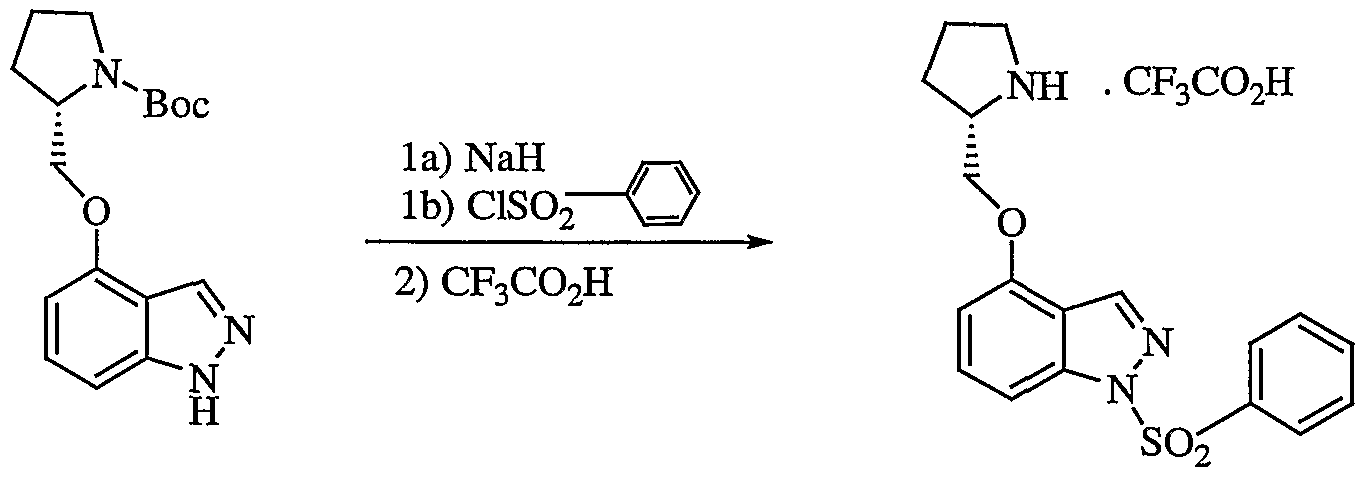
- compounds of formula I wherein W is S0 2 ; X is 0; Y is CR 13 ; Z is CR 14 ; and R 4 and R ⁇ are H (la) may be prepared by reacting an hydroxyindole of formula II with an N-protected-2-methoxyheterocycle of formula III in the presence of triphenylphosphine and diethyl azodicarboxylate to give the corresponding indol-4- yloxyalkylheterocycle of formula IV. Subsequent sulfonylation and deprotection of the formula IV compound gives the desired formula la product .
- the reaction sequence is illustrated in flow diagram I wherein P represents a protecting group.
- Commonly used protecting groups include t-butyl- carboxylate, benzyl, acetyl, benzyloxycarbonyl, or any conventional group known to protect a basic nitrogen in standard synthetic procedures.
- Compounds of formula I wherein W is S0 2 ; X is 0; Y is CH; Z is N and R 4 and R ⁇ 1 are H (lb) may be prepared by reacting a nitromethylphenol of formula V with an N- protected-2-alkoxyheterocyclic compound of formula III in the presence of triphenylphosphine and diethyl azodicarboxylate to give the corresponding heterocyclylalkoxybenzene of formula VI, reducing the nitro group of the formula VI compound, for example via catalytic hydrogenation, to give the amine of formula VII and reacting the formula VII amine with isoamylnitrite in the presence of potassium acetate and acetic anhydride to give the heterocyclyalkoxyindazole of formula VIII .
- Compounds of formula I wherein X is S and W is S0 2 may be prepared by employing the appropriate indolylthiol or thiophenol and utilizing the reactions shown in flow diagrams I and II, respectively.
- Compounds of formula I wherein W is CO may be prepared by reacting the benzazole precursor, for example a compound of formula IV, VIII, XIII or XV with the appropriate isocyanate, carbonyl halide or carbamoyl halide in the presence of a base.
- compounds of formula I wherein W is (CH 2 ) X and x is an integer of 1, 2 or 3 may be prepared by reacting the appropriately substituted alkylhalide with a compound of formula IV, VIII, XIII or XV in the presence of a base.
- Compounds of formula I wherein W is (CH 2 ) x and x is 0 may be prepared via a palladium-catalyzed N-arylation such as that descrited by D. W. Old et al , Organic Letters, 2000 (2) , pp 1403-1406. Using these and other conventional methods, compounds of formula I may be prepared from readily available starting materials.
- the present invention provides a compound of formula XVI
- X, Y, Z, m, n, p, R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 and R 11 are as defined for formula I .
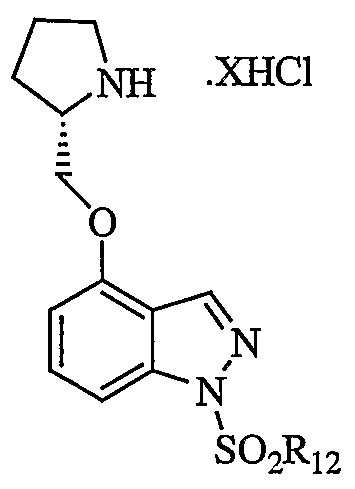
- Compounds of formula XVI are useful in the preparation of the therapeutic agents of formula I described hereinabove. Accordingly, the present invention also provides a method for the preparation of a compound of formula I wherein W is S0 2 (Ie) which comprises reacting a formula XVI compound with a sulfonyl chloride, R 12 S0 2 C1, wherein R 12 is as defined for formula I in the presence of a base optionally in the presence of a solvent. The reaction is shown in flow diagram V.
- Bases suitable for use in the method of invention are strong bases such as NaH, KOt-Bu, or any conventional base capable of removing a proton from a basic indole or benzazole nitrogen atom.
- the inventive compound of formula I may be utilized in the treatment of central nervous system disorders relating to or affected by the 5-HT6 receptor such as motor, mood, psychiatric, cognitive, neurodegenerative, or the like disorders, for example, Alzheimer's disease, Parkinson's disease, attention deficit disorder, anxiety, epilepsy, depression, obsessive compulsive disorder, migraine, sleep disorders, feeding disorders (such as anorexia or bulimia) , schizophrenia, memory loss, disorders associated with withdrawl from drug abuse, or the like or certain gastrointestinal disorders such as irritable bowel syndrome.
- the present invention provides a method for the treatment of a disorder of the central nervous system (CNS) related to or affected by the 5-HT6 receptor in a patient in need thereof which comprises providing said patient a therapeutically effective amount of a compound of formula I as described hereinabove.
- the compounds may be provided by oral or parenteral administration or in any common manner known to be an effective administration of a therapeutic agent to a patient in need thereof .
- the therapeutically effective amount provided in the treatment of a specific CNS disorder may vary according to the specific condition (s) being treated, the size, age and response pattern of the patient, the severity of the disorder, the judgment of the attending physician and the like.
- effective amounts for daily oral administration may be about 0.01 to 1,000 mg/kg, preferably about 0.5 to 500 mg/kg and effective amounts for parenteral administration may be about 0.1 to 100 mg/kg, preferably about 0.5 to 50 mg/kg.
- the compounds of the invention are provided by administering the compound or a precursor thereof in a solid or liquid form, either neat or in combination with one or more conventional pharmaceutical carriers or excipients. Accordingly, the present invention provides a pharmaceutical composition which comprises a pharmaceutically acceptable carrier and an effective amount of a compound of formula I as described hereinabove .
- Solid carriers suitable for use in the composition of the invention include one or more substances which may also act as flavoring agents, lubricants, solubilizers, suspending agents, fillers, glidants, compression aides, binders, tablet-disintegrating agents or encapsulating materials.
- the carrier may be a finely divided solid which is in admixture with a finely divided compound of formula I.
- the formula I compound may be mixed with a carrier having the necessary compression properties in suitable proportions and compacted in the shape and size desired. Said powders and tablets may contain up to 99% by weight of the formula I compound.
- Solid carriers suitable for use in the composition of the invention include calcium phosphate, magnesium stearate, talc, sugars, lactose, dextrin, starch, gelatin, cellulose, methyl cellulose, sodium carboxymethyl cellulose, polyvinylpyrrolidine, low melting waxes and ion exchange resins.
- Any pharmaceutically acceptable liquid carrier suitable for preparing solutions, suspensions, emulsions, syrups and elixirs may be employed in the composition of the invention.
- Compounds of formula I may be dissolved or suspended in a pharmaceutically acceptable liquid carrier such as water, an organic solvent, or a pharmaceutically acceptable oil or fat, or a mixture thereof.
- Said liquid composition may contain other suitable pharmaceutical additives such as solubilizers, emulsifiers, buffers, preservatives, sweeteners, flavoring agents, suspending agents, thickening agents, coloring agents, viscosity regulators, stabilizers, osmo- regulators, or the like.
- suitable pharmaceutical additives such as solubilizers, emulsifiers, buffers, preservatives, sweeteners, flavoring agents, suspending agents, thickening agents, coloring agents, viscosity regulators, stabilizers, osmo- regulators, or the like.
- suitable pharmaceutical additives such as solubilizers, emulsifiers, buffers, preservatives, sweeteners, flavoring agents, suspending agents, thickening agents, coloring agents, viscosity regulators, stabilizers, osmo- regulators, or the like.
- liquid carriers suitable for oral and parenteral administration include water (particularly containing additives as above, e.g.,
- compositions of the invention which are sterile solutions or suspensions are suitable for intramuscular, intraperitoneal or subcutaneous injection. Sterile solutions may also be administered intravenously.
- Inventive compositions suitable for oral administration may be in either liquid or solid composition form.
- N R designates nuclear magnetic resonance.
- THF and EtOAc designate tetrahydrofuran and ethyl acetate, respectively.
- the residue is purified by chromatography (silica gel, EtOAc :hexanes, 15:85).
- the purified oil (5.05 g) is dissolved in ethanol, treated with 40% aqueous NaOH, heated at reflux temperature for 45 min, cooled in an ice-water bath, neutralized to pH 8 with concentrated HCl and concentrated in vacuo to remove the ethanol .
- the resultant aqueous residue is extracted with EtOAc.
- the combined extracts are washed sequentially with water and brine, dried over MgS0 4 and concentrated in vacuo to give a yellow oil.
- the affinity of test compounds for the serotonin 5- HT6 receptor is evaluated in the following manner. Cultured Hela cells expressing human cloned 5-HT6 receptors are harvested and centrifuged at low speed (1,000 x g) for 10.0 min to remove the culture media. The harvested cells are suspended in half volume of fresh physiological phosphate buffered saline solution and recentrifuged at the same speed. This operation is repeated. The collected cells are then homogenized in ten volumes of 50 ⁇ i Tris.HCl (pH 7.4) and 0.5 mM EDTA. The homogenate is centrifuged at 40,000 x g for 30.0 min and the precipitate is collected.
- the obtained pellet is resuspended in 10 volumes of Tris.HCl buffer and recentrifuged at the same speed.
- the final pellet is suspended in a small volume of Tris.HCl buffer and the tissue protein content is determined in aliquots of 10-25 ⁇ l volumes.
- Bovine Serum Albumin is used as the standard in the protein determination according to the method described in Lowry et al . , J. Biol. Chem. , 193:265 (1951) .
- the volume of the suspended cell membranes is adjusted to give a tissue protein concentration of 1.0 mg/ml of suspension.
- the prepared membrane suspension (10 times concentrated) is aliquoted in 1.0 ml volumes and stored at -70° C until used in subsequent binding experiments .
- Binding experiments are performed in a 96 well microtiter plate format, in a total volume of 200 ⁇ l .
- To each well is added the following mixture: 80.0 ⁇ l of incubation buffer made in 50 mM Tris.HCl buffer (pH 7.4) containing 10.0 mM MgCl 2 and 0.5 mM EDTA and 20 ⁇ l of [ 3 H] -LSD (S.A., 86.0 Ci/mmol, available from Amersham Life Science), 3.0 nM.
- the dissociation constant, K-. of the [ 3 H]LSD at the human serotonin 5-HT6 receptor is 2.9 nM, as determined by saturation binding with increasing concentrations of [ 3 H]LSD.
- the reaction is initiated by the final addition of 100.0 ⁇ l of tissue suspension. Nonspecific binding is measured in the presence of 10.0 ⁇ M methiothepin.
- the test compounds are added in 20.0 ⁇ l volume .
- the reaction is allowed to proceed in the dark for 120 min at room temperature, at which time, the bound ligand-receptor complex is filtered off on a 96 well unifilter with a Packard Filtermate ® 196 Harvester.
- the bound complex caught on the filter disk is allowed to air dry and the radioactivity is measured in a Packard TopCount" equipped with six photomultiplier detectors, after the addition of 40.0 ⁇ l Microscint -20 scintillant to each shallow well.
- the unifilter plate is heat-sealed and counted in a PackardTopCount with a tritium efficiency of 31.0%.
- Specific binding to the 5-HT6 receptor is defined as the total radioactivity bound less the amount bound in the presence of lO.O ⁇ M unlabeled methiothepin. Binding in the presence of varying concentrations of test compound is expressed as a percentage of specific binding in the absence of test compound. The results are plotted as log % bound versus log concentration of test compound. Nonlinear regression analysis of data points with a computer assisted program Prism yielded both the IC 50 and the K £ values of test compounds with 95% confidence limits. A linear regression line of data points is plotted, from which the IC 50 value is determined and the K A value is determined based upon the following equation: where L is the concentration of the radioactive ligand used and K_.
- the compounds of the present invention have a high degree of affinity for the 5-HT6 receptor.
Abstract
Description





























Claims






Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| HU0303801A HUP0303801A2 (en) | 2001-04-20 | 2002-04-19 | Heterocyclylalkoxy-, alkylthio- and -alkylaminobenzazole derivatives as 5-hydroxytryptamine-6 ligands, process for their preparation and pharmaceutical compositions containing them |
| CA002444036A CA2444036A1 (en) | 2001-04-20 | 2002-04-19 | Heterocyclylalkoxy-, -alkylthio- and -alkylaminobenzazole derivatives as 5-hydroxytryptamine-6 ligands |
| BR0209047-3A BR0209047A (en) | 2001-04-20 | 2002-04-19 | Heterocyclylalkoxy, -alkylthio- and -alkylaminobenzazole derivatives as 5-hydroxytryptamine-6 binders |
| KR10-2003-7013584A KR20030088507A (en) | 2001-04-20 | 2002-04-19 | Heterocyclylalkoxy-, -alkylthio- and -alkylaminobenzazole derivatives as 5-hydroxytryptamine-6 ligands |
| EP02736592A EP1392682A2 (en) | 2001-04-20 | 2002-04-19 | Heterocyclylalkoxy-, -alkylthio- and -alkylaminobenzazole derivatives as 5-hydroxytryptamine-6 ligands |
| EA200301143A EA200301143A1 (en) | 2001-04-20 | 2002-04-19 | DERIVATIVES OF HETEROCYCLILALKOXY-, ALKYLTIO-AND-ALKYLAMINOBENZENOLE AS 5-HYDROXITRIPTAMINE-6 LIGANDS |
| IL15844802A IL158448A0 (en) | 2001-04-20 | 2002-04-19 | Heterocyclylalkoxy- alkylthio-and-alkylaminobenzazole derivatives as 5-hydroxytryptamine-6 ligands |
| JP2002583380A JP2004526781A (en) | 2001-04-20 | 2002-04-19 | Heterocyclylalkoxy-, -alkylthio- and -alkylaminobenzazole derivatives as 5-hydroxytryptoamine-6 ligands |
| MXPA03009490A MXPA03009490A (en) | 2001-04-20 | 2002-04-19 | Heterocyclylalkoxy-, -alkylthio- and -alkylaminobenzazole derivatives as 5-hydroxytryptamine-6 ligands. |
| AU2002309585A AU2002309585B2 (en) | 2001-04-20 | 2002-04-19 | Heterocyclylalkoxy-, -alkylthio- and -alkylaminobenzazole derivatives as 5-hydroxytryptamine-6 ligands |
| NO20034647A NO20034647L (en) | 2001-04-20 | 2003-10-17 | Hydroxyclylalkoxy, alkyloxy and alkylaminobenzazole derivatives as 5-hydroxytryptamine-6 ligands |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US28564401P | 2001-04-20 | 2001-04-20 | |
| US60/285,644 | 2001-04-20 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2002085853A2 true WO2002085853A2 (en) | 2002-10-31 |
| WO2002085853A3 WO2002085853A3 (en) | 2002-12-19 |
Family
ID=23095117
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2002/012512 WO2002085853A2 (en) | 2001-04-20 | 2002-04-19 | Heterocyclylalkoxy-, -alkylthio- and -alkylaminobenzazole derivatives as 5-hydroxytryptamine-6 ligands |
Country Status (17)
| Country | Link |
|---|---|
| US (2) | US6831094B2 (en) |
| EP (1) | EP1392682A2 (en) |
| JP (1) | JP2004526781A (en) |
| KR (1) | KR20030088507A (en) |
| CN (1) | CN1293073C (en) |
| AR (1) | AR034588A1 (en) |
| AU (1) | AU2002309585B2 (en) |
| BR (1) | BR0209047A (en) |
| CA (1) | CA2444036A1 (en) |
| EA (1) | EA200301143A1 (en) |
| HU (1) | HUP0303801A2 (en) |
| IL (1) | IL158448A0 (en) |
| MX (1) | MXPA03009490A (en) |
| NO (1) | NO20034647L (en) |
| PL (1) | PL366639A1 (en) |
| WO (1) | WO2002085853A2 (en) |
| ZA (1) | ZA200309009B (en) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003104193A1 (en) * | 2002-06-05 | 2003-12-18 | F. Hoffmann-La Roche Ag | 1-sulfonyl-4-aminoalkoxy indole derivatives as 5-ht6-receptor modulators for the treatment of cns-disorders |
| WO2004050085A1 (en) * | 2002-12-03 | 2004-06-17 | F. Hoffmann-La Roche Ag | Aminoalkoxyindoles as 5-ht6-receptor ligands for the treatment of cns-disorders |
| WO2005037834A1 (en) * | 2003-10-20 | 2005-04-28 | Biovitrum Ab | NOVEL TETRAYDROSPIRO{PIPERIDINE-2,7’ -PYRROLO[3,2-b]PYRIDINE DERIVATIVES AND NOVEL INDOLE DERIVATIVES USEFUL IN THE TREATMENT OF 5-HT6 RECEPTOR -RELATED DISORDERS |
| WO2007142905A2 (en) * | 2006-06-01 | 2007-12-13 | Wyeth | 1-sulfonylindazolylamine and -amide derivatives as 5-hydroxytryptamine-6 ligands |
| US8435988B2 (en) | 2010-10-06 | 2013-05-07 | Glaxosmithkline Llc | Benzimidazole derivatives as P13 kinase inhibitors |
| US9663498B2 (en) | 2013-12-20 | 2017-05-30 | Sunshine Lake Pharma Co., Ltd. | Aromatic heterocyclic compounds and their application in pharmaceuticals |
| US10392368B2 (en) | 2017-08-01 | 2019-08-27 | Theravance Biopharma R&D Ip, Llc | Pyrazolo and triazolo bicyclic compounds as JAK kinase inhibitors |
| US10851102B2 (en) | 2019-01-23 | 2020-12-01 | Theravance Biopharma R&D Ip, Llc | Imidazole and triazole containing bicyclic compounds as JAK inhibitors |
| US11697648B2 (en) | 2019-11-26 | 2023-07-11 | Theravance Biopharma R&D Ip, Llc | Fused pyrimidine pyridinone compounds as JAK inhibitors |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MXPA05008438A (en) * | 2003-02-14 | 2005-10-19 | Wyeth Corp | Heterocyclyl-3-sulfonylindazoles as 5-hydroxytryptamine-6 ligands. |
| CA2619010C (en) * | 2005-08-15 | 2013-10-01 | Wyeth | Substituted-3-sulfonylindazole derivatives as 5-hydroxytryptamine-6 ligands |
| JP2009504738A (en) * | 2005-08-15 | 2009-02-05 | ワイス | Azinyl-3-sulfonylindazole derivatives as 5-hydroxytryptamine-6 ligands |
| WO2007117413A1 (en) * | 2006-04-05 | 2007-10-18 | Wyeth | Sulfonyl-3-heterocyclylindazole derivatives as 5-hydroxytryptamine-6 ligands |
| WO2007120596A1 (en) * | 2006-04-12 | 2007-10-25 | Wyeth | DIHYDRO[1,4]DIOXINO[2,3-e]INDAZOLE DERIVATIVES AS 5-HYDROXYTRYPTAMINE-6 LIGANDS |
| US20090023925A1 (en) * | 2007-06-28 | 2009-01-22 | Wyeth | N'-(2-halobenzylidene)sulfonylhydrazides as intermediates in the manufacture of arylsulfonylindazoles |
| WO2011143430A1 (en) | 2010-05-12 | 2011-11-17 | Abbott Laboratories | Indazole inhibitors of kinase |
| AU2015286049B2 (en) | 2014-07-08 | 2018-03-01 | Sunshine Lake Pharma Co., Ltd. | Aromatic heterocyclic derivatives and pharmaceutical applications thereof |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995003298A1 (en) * | 1993-07-19 | 1995-02-02 | Fujisawa Pharmaceutical Co., Ltd. | BENZIMIDAZOLE DERIVATIVES USEFUL AS DOPAMINE RECEPTOR ANTAGONIST, 5-HT RECEPTOR AGONIST OR α1 RECEPTOR ANTAGONIST |
| US6054469A (en) * | 1995-11-16 | 2000-04-25 | Merck Sharp & Dohme Ltd. | Substituted tetrahydropyridine derivatives acting on 5-HT receptors |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3835291A1 (en) | 1988-04-19 | 1989-11-02 | Bayer Ag | 1,3-DISUBSTITUTED PYRROLIDINES |
| US5274097A (en) * | 1988-04-19 | 1993-12-28 | Bayer Aktiengesellschaft | 1,3-disubstituted pyrrolidines |
| GB9103862D0 (en) * | 1991-02-25 | 1991-04-10 | Glaxo Group Ltd | Chemical compounds |
| TW288010B (en) * | 1992-03-05 | 1996-10-11 | Pfizer | |
| US5578716A (en) * | 1993-12-01 | 1996-11-26 | Mcgill University | DNA methyltransferase antisense oligonucleotides |
| JPH0940646A (en) | 1995-07-27 | 1997-02-10 | Yamanouchi Pharmaceut Co Ltd | Condensed benzene ring derivative or its salt |
| WO1998050346A2 (en) * | 1997-04-18 | 1998-11-12 | Smithkline Beecham Plc | Acetamide and urea derivatives, process for their preparation and their use in the treatment of cns disorders |
| AU5960300A (en) | 1999-07-15 | 2001-02-05 | Nps Allelix Corp. | Heterocyclic compounds for the treatment of migraine |
| EP1355904B1 (en) * | 2000-12-22 | 2007-08-01 | Wyeth | Heterocyclindazole and azaindazole compounds as 5-hydroxytryptamine-6 ligands |
| TW593278B (en) * | 2001-01-23 | 2004-06-21 | Wyeth Corp | 1-aryl-or 1-alkylsulfonylbenzazole derivatives as 5-hydroxytryptamine-6 ligands |
| TW200401641A (en) * | 2002-07-18 | 2004-02-01 | Wyeth Corp | 1-Heterocyclylalkyl-3-sulfonylindole or-indazole derivatives as 5-hydroxytryptamine-6 ligands |
| MXPA05008438A (en) * | 2003-02-14 | 2005-10-19 | Wyeth Corp | Heterocyclyl-3-sulfonylindazoles as 5-hydroxytryptamine-6 ligands. |
-
2002
- 2002-04-19 EA EA200301143A patent/EA200301143A1/en unknown
- 2002-04-19 AU AU2002309585A patent/AU2002309585B2/en not_active Expired - Fee Related
- 2002-04-19 EP EP02736592A patent/EP1392682A2/en not_active Withdrawn
- 2002-04-19 CA CA002444036A patent/CA2444036A1/en not_active Abandoned
- 2002-04-19 WO PCT/US2002/012512 patent/WO2002085853A2/en active Application Filing
- 2002-04-19 IL IL15844802A patent/IL158448A0/en unknown
- 2002-04-19 KR KR10-2003-7013584A patent/KR20030088507A/en not_active Application Discontinuation
- 2002-04-19 US US10/126,805 patent/US6831094B2/en not_active Expired - Fee Related
- 2002-04-19 AR ARP020101443A patent/AR034588A1/en not_active Application Discontinuation
- 2002-04-19 JP JP2002583380A patent/JP2004526781A/en active Pending
- 2002-04-19 BR BR0209047-3A patent/BR0209047A/en not_active IP Right Cessation
- 2002-04-19 HU HU0303801A patent/HUP0303801A2/en unknown
- 2002-04-19 MX MXPA03009490A patent/MXPA03009490A/en active IP Right Grant
- 2002-04-19 CN CNB028123409A patent/CN1293073C/en not_active Expired - Fee Related
- 2002-04-19 PL PL02366639A patent/PL366639A1/en unknown
-
2003
- 2003-10-17 NO NO20034647A patent/NO20034647L/en not_active Application Discontinuation
- 2003-11-19 ZA ZA200309009A patent/ZA200309009B/en unknown
-
2004
- 2004-09-24 US US10/949,061 patent/US20050065185A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995003298A1 (en) * | 1993-07-19 | 1995-02-02 | Fujisawa Pharmaceutical Co., Ltd. | BENZIMIDAZOLE DERIVATIVES USEFUL AS DOPAMINE RECEPTOR ANTAGONIST, 5-HT RECEPTOR AGONIST OR α1 RECEPTOR ANTAGONIST |
| US6054469A (en) * | 1995-11-16 | 2000-04-25 | Merck Sharp & Dohme Ltd. | Substituted tetrahydropyridine derivatives acting on 5-HT receptors |
Cited By (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003104193A1 (en) * | 2002-06-05 | 2003-12-18 | F. Hoffmann-La Roche Ag | 1-sulfonyl-4-aminoalkoxy indole derivatives as 5-ht6-receptor modulators for the treatment of cns-disorders |
| US6774241B2 (en) | 2002-06-05 | 2004-08-10 | Roche Palo Alto Llc | 1-sulfonyl-4-aminoalkoxy indole derivatives and uses thereof |
| WO2004050085A1 (en) * | 2002-12-03 | 2004-06-17 | F. Hoffmann-La Roche Ag | Aminoalkoxyindoles as 5-ht6-receptor ligands for the treatment of cns-disorders |
| US7084169B2 (en) | 2002-12-03 | 2006-08-01 | Roche Palo Alto Llc | Aminoalkoxyindoles and methods of use |
| KR100755580B1 (en) * | 2002-12-03 | 2007-09-06 | 에프. 호프만-라 로슈 아게 | Aminoalkoxyindoles as 5-ht6-receptor ligands for the treatment of cns-disorders |
| AU2003289903B2 (en) * | 2002-12-03 | 2008-06-26 | F. Hoffmann-La Roche Ag | Aminoalkoxyindoles as 5-HT6-receptor ligands for the treatment of CNS-disorders |
| WO2005037834A1 (en) * | 2003-10-20 | 2005-04-28 | Biovitrum Ab | NOVEL TETRAYDROSPIRO{PIPERIDINE-2,7’ -PYRROLO[3,2-b]PYRIDINE DERIVATIVES AND NOVEL INDOLE DERIVATIVES USEFUL IN THE TREATMENT OF 5-HT6 RECEPTOR -RELATED DISORDERS |
| WO2007142905A2 (en) * | 2006-06-01 | 2007-12-13 | Wyeth | 1-sulfonylindazolylamine and -amide derivatives as 5-hydroxytryptamine-6 ligands |
| WO2007142905A3 (en) * | 2006-06-01 | 2008-01-24 | Wyeth Corp | 1-sulfonylindazolylamine and -amide derivatives as 5-hydroxytryptamine-6 ligands |
| US8865912B2 (en) | 2010-10-06 | 2014-10-21 | Glaxosmithkline Llc | Benzimidazole derivatives as PI3 kinase inhibitors |
| US9872860B2 (en) | 2010-10-06 | 2018-01-23 | Glaxosmithkline Llc | Benzimidazole derivatives as PI3 kinase inhibitors |
| US8674090B2 (en) | 2010-10-06 | 2014-03-18 | Glaxosmithkline Llc | Benzimidazole derivatives as PI3 kinase inhibitors |
| US8435988B2 (en) | 2010-10-06 | 2013-05-07 | Glaxosmithkline Llc | Benzimidazole derivatives as P13 kinase inhibitors |
| US9062003B2 (en) | 2010-10-06 | 2015-06-23 | Glaxosmithkline Llc | Benzimidazole derivatives as PI3 kinase inhibitors |
| US9156797B2 (en) | 2010-10-06 | 2015-10-13 | Glaxosmithkline Llc | Benzimidazole derivatives as PI3 kinase inhibitors |
| US10660898B2 (en) | 2010-10-06 | 2020-05-26 | Glaxosmithkline Llc | Benzimidazole derivatives as PI3 kinase inhibitors |
| US8541411B2 (en) | 2010-10-06 | 2013-09-24 | Glaxosmithkline Llc | Benzimidazole derivatives as PI3 kinase inhibitors |
| US10314845B2 (en) | 2010-10-06 | 2019-06-11 | Glaxosmithkline Llc | Benzimidazole derivatives as PI3 kinase inhibitors |
| US9663498B2 (en) | 2013-12-20 | 2017-05-30 | Sunshine Lake Pharma Co., Ltd. | Aromatic heterocyclic compounds and their application in pharmaceuticals |
| US10392368B2 (en) | 2017-08-01 | 2019-08-27 | Theravance Biopharma R&D Ip, Llc | Pyrazolo and triazolo bicyclic compounds as JAK kinase inhibitors |
| US10538513B2 (en) | 2017-08-01 | 2020-01-21 | Theravance Biopharma R&D Ip, Llc | Pyrazolo and triazolo bicyclic compounds as JAK kinase inhibitors |
| US10968205B2 (en) | 2017-08-01 | 2021-04-06 | Theravance Biopharma R&D Ip, Llc | Pyrazolo and triazolo bicyclic compounds as JAK kinase inhibitors |
| US10851102B2 (en) | 2019-01-23 | 2020-12-01 | Theravance Biopharma R&D Ip, Llc | Imidazole and triazole containing bicyclic compounds as JAK inhibitors |
| US11339160B2 (en) | 2019-01-23 | 2022-05-24 | Theravance Biopharma R&D Ip, Llc | Imidazole and triazole containing bicyclic compounds as JAK inhibitors |
| US11697648B2 (en) | 2019-11-26 | 2023-07-11 | Theravance Biopharma R&D Ip, Llc | Fused pyrimidine pyridinone compounds as JAK inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| EA200301143A1 (en) | 2004-02-26 |
| NO20034647D0 (en) | 2003-10-17 |
| IL158448A0 (en) | 2004-05-12 |
| PL366639A1 (en) | 2005-02-07 |
| CN1293073C (en) | 2007-01-03 |
| JP2004526781A (en) | 2004-09-02 |
| ZA200309009B (en) | 2005-02-21 |
| AR034588A1 (en) | 2004-03-03 |
| US6831094B2 (en) | 2004-12-14 |
| KR20030088507A (en) | 2003-11-19 |
| HUP0303801A2 (en) | 2004-03-01 |
| BR0209047A (en) | 2004-08-10 |
| US20050065185A1 (en) | 2005-03-24 |
| CA2444036A1 (en) | 2002-10-31 |
| AU2002309585B2 (en) | 2008-01-31 |
| US20030078286A1 (en) | 2003-04-24 |
| EP1392682A2 (en) | 2004-03-03 |
| NO20034647L (en) | 2003-11-20 |
| WO2002085853A3 (en) | 2002-12-19 |
| CN1518548A (en) | 2004-08-04 |
| MXPA03009490A (en) | 2004-02-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6815456B2 (en) | Heterocyclyloxy-, -thioxy- and -aminobenzazole derivatives as 5-hydroxytryptamine-6 ligands | |
| US6509357B1 (en) | 1-aryl or 1-alkylsulfonylbenzazole derivatives as 5-hydroxytryptamine-6 ligands | |
| US6831094B2 (en) | Heterocyclylalkoxy-,-alkylthio-and -alkylaminobenzazole derivatives as 5-hydroxytryptamine-6 ligands | |
| EP1355900B1 (en) | Heterocyclylalkylindole or -azaindole compounds as 5-hydroxytryptamine-6 ligands | |
| AU2002309585A1 (en) | Heterocyclylalkoxy-, -alkylthio- and -alkylaminobenzazole derivatives as 5-hydroxytryptamine-6 ligands | |
| KR20030040566A (en) | 1-Aryl- or 1-alkylsulfonyl-heterocyclylbenzazoles as 5-hydroxytryptamine-6 ligands | |
| AU2002251811A1 (en) | 1-aryl-or 1-alkylsulfonylbenzazole derivatives as 5-hydroxytryptamine-6 ligands | |
| JP2006518385A (en) | Heterocyclyl-3-sulfonylindazole as 5-hydroxytryptamine-6 ligand | |
| US7541358B2 (en) | 1-aryl-or 1-alkylsulfonylbenzazole derivatives as 5-hydroxytryptamine-6 ligands | |
| AU2002307424A1 (en) | Heterocyclyloxy-, -thioxy- and -aminobenzazole derivatives as 5-hydroxytrypltamine-6 ligands | |
| US20030225121A1 (en) | Heterocyclylalkylindole or-azaindole compounds as 5-hydroxytryptamine-6 ligands | |
| AU2008203389A1 (en) | Heterocyclyloxy-, -thioxy- and - aminobenzazole derivatives as 5- hydroxytryptamine-6 ligands | |
| AU2002245102A1 (en) | Heterocyclylalkylindole or -azaindole compounds as 5-hydroxytryptamine-6 ligands |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SD SE SG SI SK SL TJ TM TN TR TT TZ UA UG UZ VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SD SE SG SI SK SL TJ TM TN TR TT TZ UA UG UZ VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2002736592 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002309585 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1298/KOLNP/2003 Country of ref document: IN Ref document number: 01298/KOLNP/2003 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 528915 Country of ref document: NZ Ref document number: 2444036 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2003/009490 Country of ref document: MX Ref document number: 158448 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020037013584 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002583380 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003/09009 Country of ref document: ZA Ref document number: 200309009 Country of ref document: ZA Ref document number: 200301143 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 028123409 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2002736592 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |