WO2002006275A1 - Novel processes for the preparation of 4-phenylpiperidine derivatives - Google Patents
Novel processes for the preparation of 4-phenylpiperidine derivatives Download PDFInfo
- Publication number
- WO2002006275A1 WO2002006275A1 PCT/GB2001/003221 GB0103221W WO0206275A1 WO 2002006275 A1 WO2002006275 A1 WO 2002006275A1 GB 0103221 W GB0103221 W GB 0103221W WO 0206275 A1 WO0206275 A1 WO 0206275A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- toluene
- reaction
- minutes
- added
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
Definitions
- the present invention relates to new processes for preparing pharmaceutically active compounds and intermediates therefor.
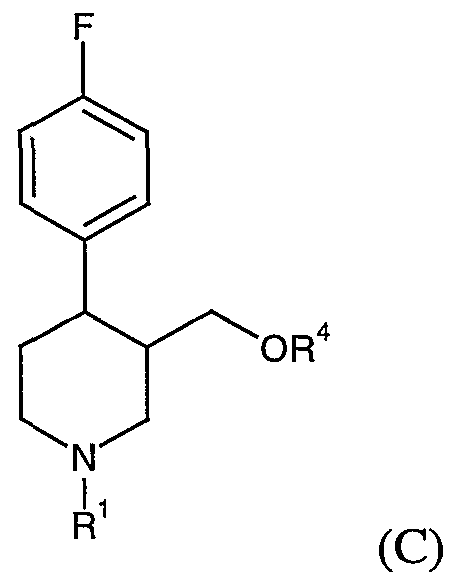
- the piperidine nitrogen is protected by a group R 1 , usually an alkyl (typically methyl) or aralkyl (such as benzyl) group.
- R 1 usually an alkyl (typically methyl) or aralkyl (such as benzyl) group.
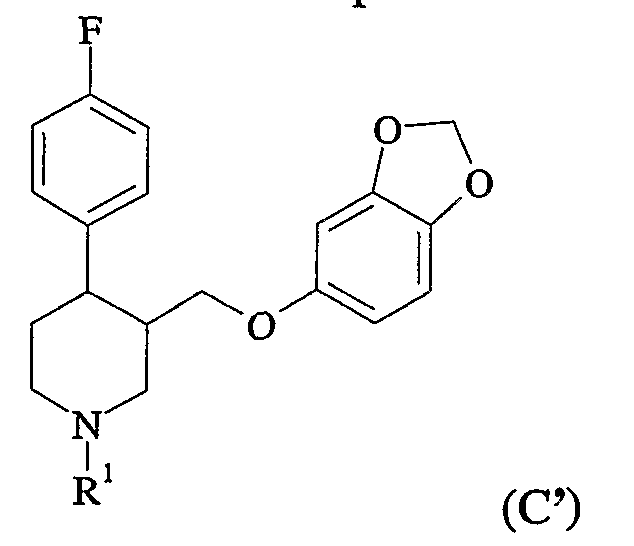
- the N-substituted piperidine must be coupled with sesamol to make an N-substituted paroxetine analogue (C)
- R ⁇ is typically a lower alkyl, aralkyl or aryl group, such as methyl, benzyl or phenyl.
- Example 1 a solution of 3-hydroxymethyl-l-methyl-4-phenyl piperidine in pyridine is reacted with methanesulphonyl chloride. The pyridine is removed and the crude resultant sulphonate ester is treated with sodium methoxide and 4- methoxyphenol in methanol under reflux.
- Example 5 of EP 0 152 273 4-(4- fluorophenyl)-3-hydroxymethyl-l -methyl pyridine is dissolved in toluene together with triethylamine and cooled. Benzenesulphonyl chloride is added to this mixture. The resultant solution of the benzenesulphonic ester is then mixed with sodium methoxide and 4-methoxyphenol in methyl isobutyl carbinol and heated.
- R 1 is typically lower alkyl (such as methyl) or aralkyl (such as benzyl) group,
- R2 is typically lower alkyl (such as methyl), aryl (such as phenyl), aralkyl (such as benzyl), or alkenyl (such as vinyl),
- R4 is a substituted phenyl group (especially 3,4-methylenedioxyphenyl)
- Example 2 a solution of 4-fluorophenyl-3-(3',4'- methylenedioxyphenoxymethyl)-l -methyl piperidine in dichloromethane was treated with phenyl chloroformate in dichloromethane at 0-5°C. After leaving overnight, the solution was washed with 1M NaOH and then 1M HCl, dried and evaporated. The solid residue was suspended in benzene, filtered and evaporated. The evaporation residue was heated at reflux with KOH and 2-methoxyethanol for 4 hours and then evaporated. Water was added and the mixture extracted with benzene, dried, and evaporated to give the N- deprotected compound.
- Example 9 of EP 0 152 273 4-(4'-fluorophenyl)-3-(4'-methoxy phenoxymethyl) -1- methyl piperidine was dissolved in toluene and treated at 0°C with a solution of 1.9 equivalents of phenyl chloroformate in toluene over 30 minutes. The mixture was allowed to stand at room temperature for 20 hours. A further 1.9 equivalents of phenyl chloroformate were added and the mixture left for 72 hours. The solution was washed with 2N NaOH, then water, then IN HCl and finally saturated aqueous NaCl. The organic phase was dried and concentrated to give an oil which was then crystallised as a white crystals from 96% ethanol.
- This intermediate was mixed with KOH and 2- methoxyethanol and stirred at 130-140°C for 4 hours and partitioned between water and toluene. The organic phase was dried and evaporated to give the N-deprotected compound as an oil which was then converted to the acetate salt.
- the present invention is based on the discovery of improvements in the above mentioned sulphonation, coupling and deprotection steps individually, and by combining steps, to provide reaction conditions which are more suitable for industrial scale production.
- the present invention provides a process for the preparation of a compound of structure (E):
- R ⁇ is a substituted phenyl group (especially 3,4-methylenedioxyphenyl), which comprises (a) providing a carbinol compound of structure (A) (b) reacting the carbinol with a sulphonyl chloride of structure R3s ⁇ 2 ⁇ to prepare a sulphonate derivative of stru
- R! is suitably an alkyl, arylalkyl, allyl, arylalkyloxycarbonyl, acyl or alkynyl group in which the alkyl groups have 1-6 carbon atoms, preferably an alkyl (typically methyl) or aralkyl (such as benzyl) group
- R2 is suitably an optionally substituted alkyl, aryl, allyl, alkenyl or arylalkyl group in which the alkyl groups have 1 to 6 carbon atoms, preferably a phenyl, methyl, ethyl, tertiary butyl, vinyl or benzyl group
- R3 is suitably an alkyl, aralkyl, alkaryl or aryl group in which the alkyl groups have 1 to 6 carbon atoms, preferably a Ph, CF3, CH3, CH 2 Ph, CH 2 COPh, CgH ⁇ -MeO, C6H 2 - 2,4,6-Me 3 , C6H4-4-Me, CH2PI1, or C ⁇ CgH ⁇ -Me group
- R4 is a substituted phenyl group, suitably substituted by C 1-4 alkyl, alkylthio, alkoxy, halogen, nitro, acylamino, methylsulfonyl, or preferably methylenedioxy,
- Compound (E) may be isolated as the free base, or more preferably is converted to a salt with a pharmaceutically acceptable acid before isolation.
- reaction sequence is carried out in toluene, starting with a solution of compound (A) in toluene and adding reagents neat or as solutions in toluene or with additional toluene as make-up, as appropriate.
- the above reaction sequence may be carried out by adding a solution of the sulphonyl chloride, such as benzenesulphonyl chloride, in the reaction solvent, suitably toluene, to the solution of compound (A) with a base, preferably an amine such as dimethylethylamine, and allowing the reaction to take place at reduced temperature, suitably less than 20 °C, for example between -10 and +5 °C.
- a base preferably an amine such as dimethylethylamine
- the reaction solution is subjected to an aqueous wash before proceeding, most suitably using aqueous sodium hydroxide.
- the substituted phenol such as sesamol is added to the reaction solution together with a suitable base, such as aqueous sodium or potassium hydroxide.
- a suitable base such as aqueous sodium or potassium hydroxide.
- a phase transfer catalyst is added.
- the temperature is suitably maintained at about 60-100 °C during this reaction.
- a further aqueous wash follows this reaction, and optionally drying of the reaction solution, for example by azeotropic distillation.
- the chloroformate such as phenyl chloroformate, is added.
- the temperature is maintained between 50-100 °C.
- the reaction solution is given a further aqueous wash, suitably with dilute sulphuric acid.
- a base such as sodium or potassium hydroxide is added to remove the nitrogen-protecting carbamate, suitably by heating the reaction solution under reflux.
- the product may be recovered from the solution by various means, such as evaporation of solvent, precipitation by addition of a non-solvent or adding an acid such as hydrochloric acid to form a salt and crystallising the salt.
- step (d) may suitably be carried out by adding chloroformate to the reaction solution containing compound (C).
- compound (C) in solid form or in solution with the reaction solvent may be added to chloroformate in the reaction solvent.
- the present invention also provides a process for preparing compound (E) from compound (A) by steps (a), (b), (c), (d) and (e) above, characterised by one or more of the following improvements:
- step (c) reacting the sulphonate compound (B) with the substituted phenol in the presence of a phase transfer catalyst and a base,
- step (d) reacting compound (C) with the haloformate and adding an HCl scavenging base, (3) in step (d) washing the reaction solution containing compound (D) with an aqueous acid selected from citric acid, phosphoric acid, acetic acid and formic acid,
- step (e) heating compound (D) with sodium hydroxide to remove the carbamate group.
- the invention also provides:
- a process for preparing compound (C) which comprises reacting a sulphonate compound (B) with a substituted phenol R ⁇ OH in the presence of a phase transfer catalyst and a base.
- a process for preparing compound (D) which comprises reacting a compound (C) and a haloformate R ⁇ OCOCl and adding an HCl scavenging base.
- a process for preparing compound (D) which comprises reacting a compound (C) with a haloformate R2OCOC1 and washing the reaction solution containing compound (D) with an aqueous acid selected from citric acid, phosphoric acid, acetic acid and formic acid.
- a process for preparing compound (E) which comprises heating a compound (D) with sodium hydroxide to remove the carbamate group.
- a solution of compound (B) is provided in a reaction vessel, and appropriate amounts of the substituted phenol e.g. sesamol, the phase transfer catalyst e.g. tetra- ⁇ -octylammonium bromide, and base e.g. aqueous sodium hydroxide, and optionally additional solvent, are added to the vessel. The mixture is heated and stirred to effect the reaction.
- the phase transfer catalyst e.g. tetra- ⁇ -octylammonium bromide
- base e.g. aqueous sodium hydroxide
- phase transfer catalyst for this process is tetra-n-octylammonium bromide, which may be used effectively when the reaction is carried out in toluene, a solvent especially suitable for use in commercial production.
- phase transfer catalyst for this process is tetra-n-butylammonium bromide, which may be used effectively when the reaction is carried out in toluene and may advantageously be removed from the reaction solution after step (c) without loss of yield.
- phase transfer catalysts that may be used for this reaction include tetra-n- dodecylammonium chloride, Aliquat/Adogen (R)464, tetra-n-butylammonium chloride, cetyltrimethylammonium bromide, and tetra- «-butylammonium fluoride hydrate.
- phase transfer catalysed reaction may also be carried in other solvents, such as benzene, xylene, mesitylene and other hydrocarbons.
- reaction preferably takes place in toluene.
- compound (B) is also formed by a reaction in toluene, for example by addition of a sulphonyl chloride in toluene to a solution of compound (A) in toluene in the presence of a base, such as an amine.
- a base such as an amine.
- the reaction solution containing compound (B) in toluene can be used directly for reaction with the phenol R ⁇ OH, without the need for intermediate isolation of compound (B).
- the base is preferably an amine, such as triethylamine, trimethylamine, diethylmethylamine or dimethylethylamine. More preferably, the amine is dimethylethylamine.
- the reaction mixture is then preferably washed with aqueous sodium hydroxide solution.
- a substituted phenol such as sesamol
- a phase transfer catalyst preferably tetra-n-octylammonium bromide
- aqueous typically 30-50 w/w% sodium hydroxide solution.
- the mixture is heated for reaction, typically at 70- 100 °C.
- the toluene solution is then separated, optionally washed with water, and concentrated.
- the product may be isolated for further reaction to form compound (D), or the reaction solution used directly in a non-isolation process as disclosed above.
- This aspect of the invention results from the finding that the reaction between the haloformate and the substituted phenol derivative (C) to prepare compound (D) does not always proceed to completion because any HCl present in the reaction mixture reacts with starting material to form its hydrochloride salt. This precipitates from solution and so does not contribute to the reaction product. As a result, we have found that a variable amount, typically from 5-12%, of the starting material may remain unreacted at the end of the reaction.
- This aspect of the invention is based on the surprising discovery that HCl scavenging bases, especially hindered amine bases such as Hunig's base (ethyldiwopropylamine) can be used successfully to allow the reaction to be driven to completion. This gives higher conversions and yields in the production of compound (D).
- HCl scavenging bases especially hindered amine bases such as Hunig's base (ethyldiwopropylamine) can be used successfully to allow the reaction to be driven to completion. This gives higher conversions and yields in the production of compound (D).
- the base is added after commencement of the reaction between the chloroformate and compound (C), most suitably after the initial reaction has been completed, for example about 30 minutes after addition of the chloroformate.
- the hindered amine may be selected by reference to possessing sufficient basicity to scavenge HCl but being insufficiently nucleophilic to react with the chloroformate. We believe that previous attempts to use HCl scavenging bases have failed due to their being present at the commencement of the reaction, and resultant reaction with the chloroformate.
- the base is added in amount of 0.1 to 1 equivalents.
- the reaction solution containing compound (D) resulting from the reaction of compound (C) with the haloformate is typically subjected to an aqueous wash as part of the work-up procedure. It has been proposed to wash with aqueous sulphuric acid which converts unreacted starting material to a sulphate or hydrogen sulphate, which separates as an oil. The oil must be removed, for example by Celite filtration, during work-up. Reducing the amount of unreacted starting material by the above-mentioned HCl-scavenging process will also have the effect of reducing the amount of oil to be removed during work-up. We have now found that it is more advantageous to wash the reaction solution with an aqueous acid selected from citric acid, phosphoric acid, acetic acid and formic acid, to remove traces of unreacted compound (C) before further treatment of compound (D).
- an aqueous acid selected from citric acid, phosphoric acid, acetic acid and formic acid
- This aspect of the invention is based on the surprising finding that use of citric acid, phosphoric acid, acetic acid and formic acid in the aqueous wash converts the unreacted starting material to salts that are sufficiently soluble in the aqueous phase to avoid the formation of oil, and so which can be extracted in the wash liquid.
- the selected acid is provided in the wash at 0.5 to 2M and washing is carried out with the wash solution at about 20-60 °C.
- Preferred acids are citric and phosphoric, especially citric acid.
- compound (D) is treated with a base to remove the carbamate group. Preferably this takes place by adding the base to the reaction solution containing compound (D). However, if desired, compound (D) may be isolated by evaporation of solvent, and the evaporation residue treated with the base, or the isolated compound (D) may be taken up in fresh solvent before reaction.
- potassium hydroxide may be added to a solution of compound (D), and the mixture is heated under reflux for several hours.
- the optimal amount of water depends on the reaction concentration and the amount of NaOH used.
- the final reaction solution is washed with water and optionally aqueous sodium hydroxide, before recovering compound (E) from the solution, for example by evaporation of solvent or crystallisation.
- the deprotection step is preferably carried out in toluene.
- a cosolvent such as propan-2-ol or industrial methylated spirits (IMS) may be added to the toluene solution, and hydrochloric acid added to allow crystallisation of paroxetine as the hydrochloride salt, optionally after seeding.
- IMS industrial methylated spirits
- the compound of structure (E) may isolated from the final solution as the free base for purification or for further reaction to create active compounds or salts as in US-A- 3912743 and US-A-4007196 and the references cited above.
- Preferably the above procedures are used to prepare paroxetine, in which case R 4 is a residue of sesamol.
- Paroxetine is the (-)-trans isomer of 4-(4'-fluorophenyl)-3-(3',4'-methylenedioxy- phenoxymethyl)-piperidine.
- optical resolution may be carried out prior to coupling with sesamol. Alternatively, resolution may be carried out at other stages, such as after deprotection of the piperidine nitrogen.
- the present invention includes within its scope the compound paroxetine and its pharmaceutically acceptable salts, particularly paroxetine hydrochloride, especially as an anhydrate or the hemihydrate, and paroxetine methanesulphonate, when obtained via any aspect of this invention, and any novel intermediates resulting from the described procedures.
- Paroxetine free base may be converted to paroxetine methanesulphonate by treatment with methanesulphonic acid or a labile derivative thereof, for example a soluble salt such as ammonium methanesulphonate.
- Paroxetine hydrochloride may be prepared by treatment of paroxetine free base with a source of hydrogen chloride, for example gaseous hydrogen chloride, or a solution thereof, or aqueous hydrochloric acid.
- Paroxetine and its salts obtained using this invention may be formulated for therapy in the dosage forms described in EP-A-0223403 or WO96/24595, either as solid formulations or as solutions for oral or parenteral use.
- paroxetine especially paroxetine hydrochloride or methanesulphonate, obtained using this invention
- therapeutic uses of paroxetine, especially paroxetine hydrochloride or methanesulphonate, obtained using this invention include treatment of: alcoholism, anxiety, depression, obsessive compulsive disorder, panic disorder, chronic pain, obesity, senile dementia, migraine, bulimia, anorexia, social phobia, pre-menstrual syndrome
- compositions using active compounds prepared in accordance with this invention are usually adapted for oral administration, but formulations for dissolution for parental administration are also within the scope of this invention.
- the composition is usually presented as a unit dose composition containing from 1 to 200mg of active ingredient calculated on a free base basis, more usually from 5 to 100 mg, for example 10 to 50 mg such as 10, 12.5, 15, 20, 25, 30 or 40 mg by a human patient. Most preferably unit doses contain 20 mg of active ingredient calculated on a free base basis. Such a composition is normally taken from 1 to 6 times daily, for example 2, 3 or 4 times daily so that the total amount of active agent administered is within the range 5 to 400 mg of active ingredient calculated on a free base basis. Most preferably the unit dose is taken once a day.
- Preferred unit dosage forms include tablets or capsules, including formulations adapted for controlled or delayed release.
- compositions of this invention may be formulated by conventional methods of admixture such as blending, filling and compressing.
- Suitable carriers for use in this invention include a diluent, a binder, a disintegrant, a colouring agent, a flavouring agent and/or preservative. These agents may be utilised in conventional manner, for example in a manner similar to that already used for marketed anti-depressant agents.
- the present invention also provides: a pharmaceutical composition for treatment or prophylaxis of one or more of the Disorders comprising paroxetine or a pharmaceutically acceptable salt such as the mesylate or hydrochloride obtained using the process of this invention and a pharmaceutically acceptable carrier; the use of paroxetine or a pharmaceutically acceptable salt such as the mesylate or hydrochloride obtained using the process of this invention to manufacture a medicament for the treatment or prophylaxis of one or more of the Disorders; and a method of treating the Disorders which comprises administering an effective or prophylactic amount of paroxetine or a pharmaceutically acceptable salt such as the mesylate or hydrochloride obtained using the process of this invention to a person suffering from one or more of the Disorders.
- a pharmaceutical composition for treatment or prophylaxis of one or more of the Disorders comprising paroxetine or a pharmaceutically acceptable salt such as the mesylate or hydrochloride obtained using the process of this invention and a pharmaceutically acceptable carrier; the
- Toluene (250 ml) and tran5-(-)-4-(4'-fluorophenyl)-3-hydroxymethyl-l-methylpiperidine (63 g) were charged to a 1 litre jacketed vessel at 20 C with an agitation rate of 300 r.p.m. The temperature was lowered to 5 C, then dimethylethylamine (42.8 ml) was charged to the vessel and the solution temperature was lowered to 0°C. A solution of benzenesulphonyl chloride (43.3 ml) in toluene (32 ml) was added to the vessel over 120 minutes, maintaining the solution temperature between +2 and -2°C.
- the reaction was cooled to 64°C and seeded with tra « ⁇ -(-)-4-(4'-fluorophenyl)-3-(3",4"- methylenedioxyphenoxymethyl)-l-methylpiperidine (20 mg). The reaction was then cooled slowly to 20°C and water (378 ml) was added over 1 hour. The reaction was cooled to 15 C and stirred for 1 hour. The suspension was filtered and the solid washed with water (150 ml) and dried in a vacuum oven at 40°C overnight. The yield of trans-(- )-4-(4'-fluorophenyl)-3-(3",4"-methylenedioxyphenoxymethyl)-l-methylpiperidine was 92.5 g, (95.5%) with a purity of 95.4%.
- Toluene (400 ml) and tr R5-(-)-4-(4'-fluorophenyl)-3-hydroxymethyl-l-methylpiperidine (100 g) were charged to a 1 litre jacketed vessel at 20 C with an agitation rate of 300 r.p.m. The temperature was lowered to 5°C, then dimethylethylamine (68.4 ml) was charged to the vessel and the solution temperature was lowered to 0 C. A solution of benzenesulphonyl chloride (69.1 ml) in toluene (50 ml) was added to the vessel over 65 minutes, maintaining the solution temperature between +2 and -2 C.
- the reaction was warmed to 75°C and stirred with an agitator speed set to 360 r.p.m. for 3 hours.
- the reaction was diluted with water (500 ml) stirred for 15 minutes then left to settle. After 15 minutes, the aqueous phase was removed.
- the reaction was cooled to 40 C and washed with water (100 ml). The toluene was then removed by distillation under reduced pressure.
- propan-2-ol 500 ml
- the resulting suspension was heated to 80°C to dissolve all the solid.
- the propan-2-ol was removed by distillation under reduced pressure to leave a solid that was left under reduced pressure at 50 C for 1 hour after the distillation was complete.
- reaction mixture was washed at 60°C with 2M citric acid (2 x 60 ml), followed by water (2 x 30 ml), then evaporated to dryness to give an oil. This was dissolved in hot propan-2-ol (110 ml) and again evaporated to dryness to give a white solid. The residue was redissolved in hot propan-2-ol (144 ml) and allowed to cool. The resulting solid was collected by filtration to give 19.9 g (76%) of tr ⁇ «s-(-)-4-(4'-fluorophenyl)-3-(3",4"- methylenedioxyphenoxymethyl)- 1 -phenoxycarbonylpiperidine.
- Example 4 rr n.y-(-)-4-(4'-fluorophenyl)-3-(3",4"-methylenedioxyphenoxymethyl)-l- methylpiperidine (22.5 g, 0.066 mol) and toluene (130 ml) were charged to a 500 ml 3- necked, round-bottomed flask. The mixture was dried by distillation of toluene (20 ml) and cooled to 60-65°C.
- Phenylchloroformate (9 ml, 0.072 mol, 1.1 eq.) was added over 45 minutes, then di-wo-propylethylamine (2.3 ml, 0.013 mol, 0.2 eq.) added and the mixture left to stir at 60-65°C for 1 hour. After cooling to 20°C the mixture was washed with 10% sulphuric acid (2 x 30 ml) and water (2 x 38 ml). The mixture was treated with celite (0.63g), filtered and the solvent evaporated. Propan-2-ol (125 ml) was added and the solvent again evaporated. Fresh propan-2-ol (160 ml) was added and the mixture heated to give a solution.
- Example 5 rranif-(-)-4-(4'-fluorophenyl)-3-(3",4"-methylenedioxyphenoxymethyl)-l- phenoxycarbonylpiperidine (75 g) and toluene (560 ml) were charged to a 1 L jacketed vessel and the agitator set to a speed of 400 rpm. Water (8.6 ml) and sodium hydroxide pearl (29.53 g) were added and the mixture was heated at reflux for 60 minutes then cooled to 75 °C. Water (170 ml) was added and the mixture stirred for 5 minutes at 70-75 °C then settled for 5 minutes.
- Toluene (375 ml) and tr ⁇ n5-(-)-4-(4'-fluorophenyl)-3-hydroxymethyl-l-methylpiperidine (63 g) were charged to a 1 litre jacketed vessel at 20°C with an agitation rate of 300 r.p.m. The temperature was lowered to 5 C, then dimethylethylamine (46 ml) was charged to the vessel and the solution temperature was lowered to 0°C. A solution of benzenesulphonyl chloride (43 ml) in toluene (51 ml) was added to the vessel over 1 hour 45 minutes, maintaining the solution temperature between 0 and +2°C.
- the reaction was cooled to room temperature, diluted with water (600 ml), stirred for 15 minutes then left to settle. After 15 minutes, the aqueous phase was removed. Toluene (330 ml) was then removed from the reaction by distillation. The reaction solution was then transferred to a clean 1 litre jacketed vessel using toluene (75 ml) as a wash. A further portion of toluene (80 ml) was removed by distillation. The mixture was cooled to 60 C and phenylchloroformate (42 ml) was added over 45 minutes. N,N-di-z5 ⁇ -propylethylamine (10 ml) was then added and the mixture left to stir at 60°C for 1 hour.
- Toluene (380 ml) and tr n5 , -(-)-4-(4'-fluorophenyI)-3-hydroxymethyl-l-methylpiperidine (63 g) were charged to a 1 litre jacketed vessel at 20°C with an agitation rate of 300 r.p.m. The temperature was lowered to 5°C, then dimethylethylamine (43.8 ml) was charged to the vessel and the solution temperature was lowered to 0 C. A solution of benzenesulphonyl chloride (42.6 ml) in toluene (51 ml) was added to the vessel over 2 hours 45 minutes, maintaining the solution temperature between +2 and -2 C.
- reaction was cooled to room temperature, diluted with water (283 ml), stirred for 15 minutes then left to settle. After 15 minutes, the aqueous phase was removed. To the reaction was added toluene (300 ml) and toluene (300 ml) was then removed from the reaction by distillation. The reaction solution was then transferred to a clean 1 litre jacketed vessel using toluene (100 ml) as a wash. A further 88 ml of toluene was removed by distillation. The mixture was cooled to 60 C and phenylchloroformate (39 ml) was added over 45 minutes.
- N,N-di-wo- propylethylamine (10 ml) was then added and the mixture left to stir at 60 C for 1 hour.
- the reaction was then cooled to 20°C and washed with 2M citric acid (2 x 380 ml) and water (2 x 166 ml).
- Toluene (468 ml) was then added, and removed (100 ml) by distillation.
- the reaction was cooled to 50°C and the agitator set to a speed of 400 rpm. Water (19.5 ml) and sodium hydroxide pellet (45.3 g) were added and the mixture was heated at reflux for 75 minutes then cooled to 80°C.
- the mixture was further cooled to 18°C, seeds of with tr ⁇ .s-(-)-4-(4'- fluorophenyl)-3-(3",4"-methylenedioxyphenoxymethyl)-piperidine hydrochloride hemihydrate (0.086 g) were added, followed immediately by concentrated hydrochloric acid (39 ml). The mixture was then cooled to 20-22°C and stirred for 1 hour. The toluene slurry was filtered and the resulting cake was washed with fresh toluene (80 ml). The product was dried in a vacuum oven at 55°C overnight.
- the lower aqueous layer was removed and a second portion of 2M citric acid (170 ml) added. The mixture was stirred for 5 minutes at 40-45 °C, settled for 5 minutes and the lower layer was removed. Water (70 ml) was then added and the mixture stirred at 40-45 °C for 5 minutes and settled for 10 minutes. The lower aqueous layer was removed and a second portion of water (70 ml) added. The mixture was stirred for 5 minutes at 40-45 °C, settled for 10 minutes and the lower layer was removed. The toluene solution was concentrated in vacuo keeping the internal temperature around 40-50 °C to give a thick oil.
- IPA 300 ml was added to the oil and heated to around 70 °C until a solution formed.
- the IPA solution was concentrated in vacuo keeping the internal temperature around 40-50 °C to give a solid.
- IPA 405 ml was added to the solid and heated to reflux and stirred until complete dissolution.
- the IPA solution was cooled to 60-65 °C, stirred for 30 minutes, then cooled to 20-22 °C and stirred for 1 hour.
- the IPA slurry was filtered and the resulting cake was washed with fresh IPA (70 ml).
- the product was dried in a vacuum oven (55 °C) overnight.
- the lower aqueous layer was removed and a second portion of 2M citric acid (170 ml) added. The mixture was stirred for 5 minutes at 40-45 °C, settled for 5 minutes and the lower layer was removed. Water (70 ml ) was then added and the mixture stirred at 40-45 °C for 5 minutes and settled for 10 minutes. The lower aqueous layer was removed and a second portion of water (70 ml) added. The mixture was stirred for 5 minutes at 40-45 °C, settled for 10 minutes and the lower layer was removed.
- the toluene solution was diluted with more toluene (440 ml), heated to reflux and dried by azeotropic distillation of toluene at atmospheric pressure collecting 135 ml of distillate.
- the toluene solution was cooled to 75 °C and the agitator was adjusted to a speed of 400 rpm.
- Water (9.6 ml) and sodium hydroxide pellet (33.12 g) were added and the mixture was heated at reflux for 60 minutes then cooled to 75 °C.
- Water (170 ml) was added and the mixture stirred for 5 minutes at 70-75 °C then settled for 5 minutes.
- Toluene (80 ml) and tra «5-(-)-4-(4'-fluorophenyl)-3-hydroxymethyl-l-methylpiperidine (20 g) were charged to a 250 ml jacketed vessel at 20 C. The temperature was lowered to 10 C, then dimethylethylamine (13.6 ml) was charged to the vessel and the solution temperature was lowered to 0 C. A solution of benzenesulphonyl chloride (13.7 ml) in toluene (10 ml) was added to the vessel over 60 minutes, maintaining the solution temperature between +2 and -2°C. On completion of the addition, the solution was stirred at +2 to -2°C for 1 hour.
- a solution of sodium hydroxide in water (10% w/w, 20 ml) was added to the reactor over 5 minutes. The mixture was warmed to 20 C, stirred for 15 minutes then left to settle. After 15 minutes, the aqueous phase was removed. To the toluene solution was added tetra-n-butylammonium bromide (1.45 g), sesamol (13.74 g), and aqueous sodium hydroxide (32 ml of 47% w/w solution). The reaction was warmed to 75°C and stirred for 2.5 hours. The reaction was diluted with water (100 ml) stirred for 15 minutes then left to settle. After 15 minutes, the aqueous phase was removed.
- the reaction was cooled to 55°C and washed with water (20 ml). The toluene was then removed by distillation under reduced pressure. To the resulting solid was added propan-2-ol (100 ml) and the resulting suspension was heated to 80 C to dissolve all the solid. The propan-2-ol was removed by distillation under reduced pressure to leave a solid. To the solid was added propan-2-ol (30 ml) and the suspension was heated to reflux and stirred until all the solid dissolved. The reaction was cooled to 60 C and seeded with tr n5-(-)-4-(4'-fluorophenyl)-3-(3",4"-methylenedioxyphenoxymethyl)-l- methylpiperidine (5 mg).
- Toluene (480 ml) was charged to a 1L jacketed vessel, dried by the azeotropic distillation of 80 ml, then cooled to 65°C. Phenyl chloroformate (35.5 ml) was charged to the vessel. The solution was maintained at 60 to 65 °C and (-) trans-4-(4'-fluoropheny ⁇ )-3-(3",4"- methylenedioxyphenoxymethyl)-N-methylpiperidine (80 g) was added in 10 equal portions over 45 minutes, then washed in with toluene (2 ml).
- the lower aqueous layer was removed and a second portion of water (80 ml) added. The mixture was stirred for 5 minutes at 40-45 °C, settled for 10 minutes and the lower layer was removed.
- the toluene solution was concentrated in vacuo keeping the internal temperature around 40-50 °C to give a thick oil.
- IPA 360 ml was added to the oil and heated to around 70 °C until a solution formed.
- the IPA solution was concentrated in vacuo keeping the internal temperature around 40-50 °C to give a solid.
- IPA (480 ml) was added to the solid and heated to reflux and stirred until complete dissolution.
- the IPA solution was cooled to about 65 °C, then stirred at 60-70 °C for 30 minutes.
- the resultant slurry was then cooled to 20-22 °C and stirred for 1 hour.
- the IPA slurry was filtered and the resulting cake was washed with fresh IPA (80 ml).
- the product was dried in a vacuum oven (55 °C) overnight.
- Toluene (350 ml) and (-) trans-4-(4'-fluorophenyl)-3-(3",4"- methylenedioxyphenoxymethyl)-N-methylpiperidine (100 g) were charged to a 500 ml jacketed vessel.
- the solution was dried by the azeotropic distillation of 50 ml of toluene, then cooled to about 65 °C.
- Toluene (150 ml) and phenyl chloroformate (44.4 ml) were charged to a 1 L jacketed vessel and heated to 60-65 °C.
- the toluene solution of (-) trans-4-(4'-fluorophenyl)-3-(3",4"-methylenedioxyphenoxymethyl)-N-methylpiperidine was then added to the toluene solution of phenyl chloroformate over 45 minutes.
- the transfer lines were washed through with toluene (50 ml).
- the mixture was then stirred for 30 minutes at 60-65 °C then di-wo-propylethylamine (4.9 ml) was added in one portion and the mixture stirred for another 30 minutes.
- the mixture was then cooled to 40-45 °C.
- the IPA solution was concentrated in vacuo keeping the internal temperature around 40-50 °C to give a solid.
- IPA 600 ml was added to the solid and heated to reflux and stirred until complete dissolution.
- the IPA solution was cooled to about 65 °C, then stirred at 60-70 °C for 30 minutes.
- the resultant slurry was then cooled to 20-22 °C and stirred for 1 hour.
- the IPA slurry was filtered and the resulting cake was washed with fresh IPA (100 ml). The product was dried in a vacuum oven (55 °C) overnight.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Psychology (AREA)
- Pain & Pain Management (AREA)
- Psychiatry (AREA)
- Anesthesiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Hydrogenated Pyridines (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description









Claims




Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP01949741A EP1301508A1 (en) | 2000-07-17 | 2001-07-17 | Novel processes for the preparation of 4-phenylpiperidine derivatives |
| JP2002512178A JP2004504319A (en) | 2000-07-17 | 2001-07-17 | New method for producing 4-phenylpiperidine derivative |
| US10/333,274 US20040087795A1 (en) | 2000-07-17 | 2001-07-17 | Novel processes for the preparation of 4-phenylpiperidine derivatives |
| AU2001270858A AU2001270858A1 (en) | 2000-07-17 | 2001-07-17 | Novel processes for the preparation of 4-phenylpiperidine derivatives |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0017540.6 | 2000-07-17 | ||
| GB0017540A GB0017540D0 (en) | 2000-07-17 | 2000-07-17 | Novel compounds |
| GB0018857.3 | 2000-08-01 | ||
| GB0018857A GB0018857D0 (en) | 2000-08-01 | 2000-08-01 | Novel process |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2002006275A1 true WO2002006275A1 (en) | 2002-01-24 |
Family
ID=26244675
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2001/003221 WO2002006275A1 (en) | 2000-07-17 | 2001-07-17 | Novel processes for the preparation of 4-phenylpiperidine derivatives |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20040087795A1 (en) |
| EP (1) | EP1301508A1 (en) |
| JP (1) | JP2004504319A (en) |
| AU (1) | AU2001270858A1 (en) |
| WO (1) | WO2002006275A1 (en) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002062790A1 (en) * | 2001-02-05 | 2002-08-15 | Teva Pharmaceutical Industries Ltd. | Preparation of n-methylparoxetine and related intermediate compounds |
| US7691882B2 (en) | 2005-10-31 | 2010-04-06 | Eisai R&D Management Co., Ltd. | Heterocycles substituted pyridine derivatives and antifungal agent containing thereof |
| US8513287B2 (en) | 2007-12-27 | 2013-08-20 | Eisai R&D Management Co., Ltd. | Heterocyclic ring and phosphonoxymethyl group substituted pyridine derivatives and antifungal agent containing same |
| US8609656B2 (en) | 2004-02-23 | 2013-12-17 | Chugai Seiyaku Kabushiki Kaisha | Heteroarylphenylurea derivative |
| US8946329B2 (en) | 2003-08-25 | 2015-02-03 | Dow Global Technologies Llc | Coating compositions |
| CN104447714A (en) * | 2014-11-18 | 2015-03-25 | 成都医路康医学技术服务有限公司 | Production process of paroxetine hydrochloride |
| US9169406B2 (en) | 2003-08-25 | 2015-10-27 | Dow Global Technologies Llc | Coating compositions |
| US9422444B2 (en) | 2012-12-28 | 2016-08-23 | Dow Global Technologies Llc | Coating compositions |
| US9938413B2 (en) | 2012-12-28 | 2018-04-10 | Dow Global Technologies Llc | Coating composition and articles made therefrom |
| CN112521377A (en) * | 2020-11-26 | 2021-03-19 | 北京福元医药股份有限公司 | Continuous preparation method of paroxetine hydrochloride |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI306858B (en) * | 2004-12-23 | 2009-03-01 | Teva Pharma | Process for preparing pharmaceutically acceptable salts of duloxetine and intermediates thereof |
| US20070142389A1 (en) * | 2005-12-20 | 2007-06-21 | Pfizer Inc. | Piperidine derivatives |
| ES2770591T3 (en) * | 2013-11-18 | 2020-07-02 | SpecGx LLC | Preparation of normorfinanos |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3912743A (en) * | 1973-01-30 | 1975-10-14 | Ferrosan As | 4-Phenylpiperidine compounds |
| EP0152273A2 (en) * | 1984-02-07 | 1985-08-21 | A/S Ferrosan | A phenylpiperidine derivative, and its salts, their preparation, compositions containing them, and their therapeutic use |
| WO1996036636A1 (en) * | 1995-05-17 | 1996-11-21 | Novo Nordisk A/S | Process for preparing 4-aryl-piperidine derivatives |
| EP0810225A1 (en) * | 1996-05-31 | 1997-12-03 | Asahi Glass Company Ltd. | Process for producing paroxetine |
| WO2001004113A2 (en) * | 1999-07-09 | 2001-01-18 | Smithkline Beecham Plc | Process for the preparation of paroxetine and synthetic intermeditates thereof |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6777554B2 (en) * | 2001-02-05 | 2004-08-17 | Teva Pharmaceutical Industries Ltd. | Preparation of N-methylparoxetine and related intermediate compounds |
-
2001
- 2001-07-17 US US10/333,274 patent/US20040087795A1/en not_active Abandoned
- 2001-07-17 WO PCT/GB2001/003221 patent/WO2002006275A1/en not_active Application Discontinuation
- 2001-07-17 JP JP2002512178A patent/JP2004504319A/en active Pending
- 2001-07-17 AU AU2001270858A patent/AU2001270858A1/en not_active Abandoned
- 2001-07-17 EP EP01949741A patent/EP1301508A1/en not_active Withdrawn
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3912743A (en) * | 1973-01-30 | 1975-10-14 | Ferrosan As | 4-Phenylpiperidine compounds |
| EP0152273A2 (en) * | 1984-02-07 | 1985-08-21 | A/S Ferrosan | A phenylpiperidine derivative, and its salts, their preparation, compositions containing them, and their therapeutic use |
| WO1996036636A1 (en) * | 1995-05-17 | 1996-11-21 | Novo Nordisk A/S | Process for preparing 4-aryl-piperidine derivatives |
| EP0810225A1 (en) * | 1996-05-31 | 1997-12-03 | Asahi Glass Company Ltd. | Process for producing paroxetine |
| WO2001004113A2 (en) * | 1999-07-09 | 2001-01-18 | Smithkline Beecham Plc | Process for the preparation of paroxetine and synthetic intermeditates thereof |
Non-Patent Citations (1)
| Title |
|---|
| ENGELSTOFT M ET AL: "SYNTHESIS AND RHT MODULATING ACTIVITY OF STEREOISOMERS OF 3-PHENOXYMETHYL-4-PHENYLPIPERIDINES", ACTA CHEMICA SCANDINAVICA, MUNKSGAARD, COPENHAGEN, DK, vol. 50, no. 2, 1 February 1996 (1996-02-01), pages 164 - 169, XP000645441, ISSN: 0904-213X * |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002062790A1 (en) * | 2001-02-05 | 2002-08-15 | Teva Pharmaceutical Industries Ltd. | Preparation of n-methylparoxetine and related intermediate compounds |
| US6777554B2 (en) | 2001-02-05 | 2004-08-17 | Teva Pharmaceutical Industries Ltd. | Preparation of N-methylparoxetine and related intermediate compounds |
| US9416291B2 (en) | 2003-08-25 | 2016-08-16 | Dow Global Technologies Llc | Coating compositions |
| US8946329B2 (en) | 2003-08-25 | 2015-02-03 | Dow Global Technologies Llc | Coating compositions |
| US9169406B2 (en) | 2003-08-25 | 2015-10-27 | Dow Global Technologies Llc | Coating compositions |
| US8609656B2 (en) | 2004-02-23 | 2013-12-17 | Chugai Seiyaku Kabushiki Kaisha | Heteroarylphenylurea derivative |
| US8841327B2 (en) | 2005-10-31 | 2014-09-23 | Eisai R&D Management Co., Ltd. | Heterocycles substituted pyridine derivatives and antifungal agent containing thereof |
| US7691882B2 (en) | 2005-10-31 | 2010-04-06 | Eisai R&D Management Co., Ltd. | Heterocycles substituted pyridine derivatives and antifungal agent containing thereof |
| US8513287B2 (en) | 2007-12-27 | 2013-08-20 | Eisai R&D Management Co., Ltd. | Heterocyclic ring and phosphonoxymethyl group substituted pyridine derivatives and antifungal agent containing same |
| US9422444B2 (en) | 2012-12-28 | 2016-08-23 | Dow Global Technologies Llc | Coating compositions |
| US9938413B2 (en) | 2012-12-28 | 2018-04-10 | Dow Global Technologies Llc | Coating composition and articles made therefrom |
| CN104447714A (en) * | 2014-11-18 | 2015-03-25 | 成都医路康医学技术服务有限公司 | Production process of paroxetine hydrochloride |
| CN112521377A (en) * | 2020-11-26 | 2021-03-19 | 北京福元医药股份有限公司 | Continuous preparation method of paroxetine hydrochloride |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2001270858A1 (en) | 2002-01-30 |
| US20040087795A1 (en) | 2004-05-06 |
| JP2004504319A (en) | 2004-02-12 |
| EP1301508A1 (en) | 2003-04-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU593295B2 (en) | Paroxetine hydrochloride hemihydrate | |
| JP2002503248A (en) | 4-phenylpiperidine compound | |
| US20040087795A1 (en) | Novel processes for the preparation of 4-phenylpiperidine derivatives | |
| JP3066083B2 (en) | Process for preparing 1-benzyl-4-((5,6-dimethoxy-1-indanone) -2-yl) methylpiperidine and intermediates therefor | |
| JP2010143925A (en) | METHOD FOR PRODUCING (R)-alpha-(2,3-DIMETHOXYPHENYL)-1-[2-(4-FLUOROPHENYL)ETHYL]-4-PIPERIDINEMETHANOL | |
| JP2002514222A (en) | New method | |
| JP2002532494A (en) | Derivatives of paroxetine | |
| WO2001029032A1 (en) | Process for the preparation of paroxetine | |
| KR19990008406A (en) | Alpha- (substituted alkylphenyl) -4- (hydroxydiphenylmethyl) -1-piperidine butanol derivatives, methods of their preparation and their use as antihistamines, anti-allergic and bronchial dilators | |
| US6686473B2 (en) | Process for the production of paroxetine | |
| JP2002531451A (en) | Preparation of paroxetine hydrochloride | |
| WO2000078753A1 (en) | Process for the preparation of paroxetine and structurally related compounds | |
| US20030187269A1 (en) | Novel process | |
| WO2001004113A2 (en) | Process for the preparation of paroxetine and synthetic intermeditates thereof | |
| US20020137938A1 (en) | Novel process | |
| KR100469030B1 (en) | Synthesis of cisapride | |
| JPH058196B2 (en) | ||
| WO2001014335A1 (en) | Process for preparation of paroxetin intermediate | |
| JP2003518097A (en) | New method | |
| WO1999047519A1 (en) | Crystalline form of paroxetine | |
| DK175352B1 (en) | New crystalline paroxetine hydrochloride hemi:hydrate - used in treating depression | |
| WO2001029002A1 (en) | Process for the preparation of 4-(fluorophenyl)piperidine esters | |
| WO2001025202A1 (en) | Process for the preparation of paroxetine intermediate | |
| EP1140912A1 (en) | Process for the preparation of an acetate salt of paroxetine or paroxetine analogues |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2001949741 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2001949741 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10333274 Country of ref document: US |
|
| ENP | Entry into the national phase |
Country of ref document: RU Kind code of ref document: A Format of ref document f/p: F |
|
| ENP | Entry into the national phase |
Country of ref document: RU Kind code of ref document: A Format of ref document f/p: F |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2001949741 Country of ref document: EP |