WO2001081340A2 - Heterocycles that are inhibitors of impdh enzyme - Google Patents
Heterocycles that are inhibitors of impdh enzyme Download PDFInfo
- Publication number
- WO2001081340A2 WO2001081340A2 PCT/US2001/012900 US0112900W WO0181340A2 WO 2001081340 A2 WO2001081340 A2 WO 2001081340A2 US 0112900 W US0112900 W US 0112900W WO 0181340 A2 WO0181340 A2 WO 0181340A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hydrogen
- substituted
- compound
- formula
- alkyl
- Prior art date
Links
- 0 CC1C(*CCC*)C(C2)C2C1 Chemical compound CC1C(*CCC*)C(C2)C2C1 0.000 description 18
- VUCJMKUSLAVWBY-UHFFFAOYSA-N CC(C)(Cc1ccc(C(C2)Nc(cc(c(-c3cnc[o]3)c3)OC)c3C2=O)cc11)C1OC Chemical compound CC(C)(Cc1ccc(C(C2)Nc(cc(c(-c3cnc[o]3)c3)OC)c3C2=O)cc11)C1OC VUCJMKUSLAVWBY-UHFFFAOYSA-N 0.000 description 2
- VZNLQFGFEZWYTH-CSMDKSQMSA-N CC(C)NC(O[C@H](Cc(c1c2)ccc2C(Nc2c3cc(-c4cnc[o]4)c(OC)c2)=CC3=O)C1N(C)C)=O Chemical compound CC(C)NC(O[C@H](Cc(c1c2)ccc2C(Nc2c3cc(-c4cnc[o]4)c(OC)c2)=CC3=O)C1N(C)C)=O VZNLQFGFEZWYTH-CSMDKSQMSA-N 0.000 description 2
- FUFKWNOESYQTTD-UHFFFAOYSA-N CN(C)C(CCc1c2)c1ccc2C(Nc1c2cc(-c3cnc[o]3)c(OC)c1)=CC2=O Chemical compound CN(C)C(CCc1c2)c1ccc2C(Nc1c2cc(-c3cnc[o]3)c(OC)c1)=CC2=O FUFKWNOESYQTTD-UHFFFAOYSA-N 0.000 description 2
- XZWXESOAUOUZRA-UHFFFAOYSA-N COC1c2cc(C(Nc(cc(c(-c3cnc[o]3)c3)OC)c3C3=O)=C3O)ccc2CC1 Chemical compound COC1c2cc(C(Nc(cc(c(-c3cnc[o]3)c3)OC)c3C3=O)=C3O)ccc2CC1 XZWXESOAUOUZRA-UHFFFAOYSA-N 0.000 description 2
- BFGZWDRZPVLPCS-UHFFFAOYSA-N COc(cc(c1c2)NC(c3cccc(OCCN4CCOCC4)c3)=CC1=O)c2-c1cnc[o]1 Chemical compound COc(cc(c1c2)NC(c3cccc(OCCN4CCOCC4)c3)=CC1=O)c2-c1cnc[o]1 BFGZWDRZPVLPCS-UHFFFAOYSA-N 0.000 description 2
- LZWRBIKNTNFEKD-UHFFFAOYSA-N CC(C)(C)OC(N(C)C(CC1)c2c1ccc(C(Nc1c3cc(-c4cnc[o]4)c(OC)c1)=CC3=O)c2)=O Chemical compound CC(C)(C)OC(N(C)C(CC1)c2c1ccc(C(Nc1c3cc(-c4cnc[o]4)c(OC)c1)=CC3=O)c2)=O LZWRBIKNTNFEKD-UHFFFAOYSA-N 0.000 description 1
- ZWDDHYUNYSIZRM-UHFFFAOYSA-N CC(C)(Oc1[o](C)[s]c(Nc2ccc(-c3cnc[o]3)c(OC)c2)c11)OC1=O Chemical compound CC(C)(Oc1[o](C)[s]c(Nc2ccc(-c3cnc[o]3)c(OC)c2)c11)OC1=O ZWDDHYUNYSIZRM-UHFFFAOYSA-N 0.000 description 1
- LOZLVXMFEDRDAT-UHFFFAOYSA-N CC(C)(c1cc(C(Nc2c3cc(-c4cnc[o]4)c(OC)c2)=CC3=O)ccc1)O Chemical compound CC(C)(c1cc(C(Nc2c3cc(-c4cnc[o]4)c(OC)c2)=CC3=O)ccc1)O LOZLVXMFEDRDAT-UHFFFAOYSA-N 0.000 description 1
- BXDFQNLVAIQCRS-UHFFFAOYSA-N CC(Nc(cc(c(-c1cnc[o]1)c1)OC)c1S(CC(c1ccccc1)=O)(=O)=O)=O Chemical compound CC(Nc(cc(c(-c1cnc[o]1)c1)OC)c1S(CC(c1ccccc1)=O)(=O)=O)=O BXDFQNLVAIQCRS-UHFFFAOYSA-N 0.000 description 1
- VMRNKUSSHZEASV-XFXZXTDPSA-N CCOC(/C=C(/c1cc(C(OC)=O)ccc1)\NC)=O Chemical compound CCOC(/C=C(/c1cc(C(OC)=O)ccc1)\NC)=O VMRNKUSSHZEASV-XFXZXTDPSA-N 0.000 description 1
- PWSNZXGOSSLDOM-UHFFFAOYSA-N CN(C)C(Nc(c1c2)cc(OC)c2-c2cnc[o]2)=CC1=O Chemical compound CN(C)C(Nc(c1c2)cc(OC)c2-c2cnc[o]2)=CC1=O PWSNZXGOSSLDOM-UHFFFAOYSA-N 0.000 description 1
- XDGXMQABDWTCLT-XQZUBTRRSA-N CN(C)C1c2cc(C(Nc(c3c4)cc(OC)c4-c4cnc[o]4)=CC3=O)ccc2C[C@H]1OC(NC(OC)=O)=O Chemical compound CN(C)C1c2cc(C(Nc(c3c4)cc(OC)c4-c4cnc[o]4)=CC3=O)ccc2C[C@H]1OC(NC(OC)=O)=O XDGXMQABDWTCLT-XQZUBTRRSA-N 0.000 description 1
- KVBWZUNOWSEEAA-AVJYQCBHSA-N CN(C)C1c2cc(C(Nc3c4cc(-c5cnc[o]5)c(OC)c3)=CC4=O)ccc2C[C@H]1[O](C(NCCCl)=O)=C Chemical compound CN(C)C1c2cc(C(Nc3c4cc(-c5cnc[o]5)c(OC)c3)=CC4=O)ccc2C[C@H]1[O](C(NCCCl)=O)=C KVBWZUNOWSEEAA-AVJYQCBHSA-N 0.000 description 1
- OBJIDFFFYDQSOA-UHFFFAOYSA-N CN(C1c2cc(C(Nc3c4cc(-c5cnc[o]5)c(OC)c3)=CC4=O)ccc2CC1)C(C[n]1cncc1)=O Chemical compound CN(C1c2cc(C(Nc3c4cc(-c5cnc[o]5)c(OC)c3)=CC4=O)ccc2CC1)C(C[n]1cncc1)=O OBJIDFFFYDQSOA-UHFFFAOYSA-N 0.000 description 1
- LNZWDIKBYYNRCE-UHFFFAOYSA-N CN(C1c2cc(C(Nc3c4cc(-c5cnc[o]5)c(OC)c3)=CC4=O)ccc2CC1)C(C[n]1nncc1)=O Chemical compound CN(C1c2cc(C(Nc3c4cc(-c5cnc[o]5)c(OC)c3)=CC4=O)ccc2CC1)C(C[n]1nncc1)=O LNZWDIKBYYNRCE-UHFFFAOYSA-N 0.000 description 1
- VIZWJAAZRHHVRU-UHFFFAOYSA-N COC(CC(C1=C2)NC(CCC3)=C3C1=O)=C2c1cnc[o]1 Chemical compound COC(CC(C1=C2)NC(CCC3)=C3C1=O)=C2c1cnc[o]1 VIZWJAAZRHHVRU-UHFFFAOYSA-N 0.000 description 1
- RTELUXAULALPQS-UHFFFAOYSA-N COC(c(c(N)c1)cc(-c2cnc[o]2)c1OC)=O Chemical compound COC(c(c(N)c1)cc(-c2cnc[o]2)c1OC)=O RTELUXAULALPQS-UHFFFAOYSA-N 0.000 description 1
- DZQJRMYNMVWVQB-UHFFFAOYSA-N COC1c2cc(C(Nc(c3c4)cc(OC)c4-c4cnc[o]4)=CC3=O)ccc2CC1 Chemical compound COC1c2cc(C(Nc(c3c4)cc(OC)c4-c4cnc[o]4)=CC3=O)ccc2CC1 DZQJRMYNMVWVQB-UHFFFAOYSA-N 0.000 description 1
- IEZACMFRGJVTBH-UHFFFAOYSA-N COc(c(-c1cnc[o]1)c1)cc(NC(c2ccc(C(CC3)N4CCCC4)c3c2)=C2)c1C2=O Chemical compound COc(c(-c1cnc[o]1)c1)cc(NC(c2ccc(C(CC3)N4CCCC4)c3c2)=C2)c1C2=O IEZACMFRGJVTBH-UHFFFAOYSA-N 0.000 description 1
- RELARHZKKKIXGE-UHFFFAOYSA-N COc(c(-c1cnc[o]1)c1)cc(NC(c2ccc(CCC3N4CCC4)c3c2)=C2)c1C2=O Chemical compound COc(c(-c1cnc[o]1)c1)cc(NC(c2ccc(CCC3N4CCC4)c3c2)=C2)c1C2=O RELARHZKKKIXGE-UHFFFAOYSA-N 0.000 description 1
- KDOVOYNQTDQUFK-UHFFFAOYSA-N COc(c(-c1cnc[o]1)c1)cc(NC(c2ccc(CCC3N4CCOCC4)c3c2)=C2)c1C2=O Chemical compound COc(c(-c1cnc[o]1)c1)cc(NC(c2ccc(CCC3N4CCOCC4)c3c2)=C2)c1C2=O KDOVOYNQTDQUFK-UHFFFAOYSA-N 0.000 description 1
- FLGZEQZDTOBPEG-UHFFFAOYSA-N COc(c(-c1cnc[o]1)c1)cc(NC(c2ccc(CCC3O)c3c2)=C2)c1C2=O Chemical compound COc(c(-c1cnc[o]1)c1)cc(NC(c2ccc(CCC3O)c3c2)=C2)c1C2=O FLGZEQZDTOBPEG-UHFFFAOYSA-N 0.000 description 1
- ZEZMSFIYAOVYGB-UHFFFAOYSA-N COc(c(-c1cnc[o]1)c1)cc(NC(c2cccc(N3CCCC3)c2)=C2)c1C2=O Chemical compound COc(c(-c1cnc[o]1)c1)cc(NC(c2cccc(N3CCCC3)c2)=C2)c1C2=O ZEZMSFIYAOVYGB-UHFFFAOYSA-N 0.000 description 1
- HAKAXHFVVQMIBK-UHFFFAOYSA-N COc(c(-c1cnc[o]1)c1)cc(NC(c2cccc(S(C)(=O)=O)c2)=C2O)c1C2=O Chemical compound COc(c(-c1cnc[o]1)c1)cc(NC(c2cccc(S(C)(=O)=O)c2)=C2O)c1C2=O HAKAXHFVVQMIBK-UHFFFAOYSA-N 0.000 description 1
- STNPZSCUDBKBJG-UHFFFAOYSA-N COc(cc(c1c2)NC(CCC3)=C3C1=O)c2-c1cnc[o]1 Chemical compound COc(cc(c1c2)NC(CCC3)=C3C1=O)c2-c1cnc[o]1 STNPZSCUDBKBJG-UHFFFAOYSA-N 0.000 description 1
- OGBXETHSOCNLCF-UHFFFAOYSA-N COc(cc(c1c2)NC(c3cccc(Br)c3)=CC1=O)c2-c1cnc[o]1 Chemical compound COc(cc(c1c2)NC(c3cccc(Br)c3)=CC1=O)c2-c1cnc[o]1 OGBXETHSOCNLCF-UHFFFAOYSA-N 0.000 description 1
- MOBIZEJQBCQKDC-UHFFFAOYSA-N COc(cc(c1c2)NC(c3ccccc3)=CS1(=O)=O)c2-c1cnc[o]1 Chemical compound COc(cc(c1c2)NC(c3ccccc3)=CS1(=O)=O)c2-c1cnc[o]1 MOBIZEJQBCQKDC-UHFFFAOYSA-N 0.000 description 1
- AUROZLIWYZDAGX-UHFFFAOYSA-N COc(cc1)ccc1C(Nc(cc(c(-c1cnc[o]1)c1)OC)c1C1=O)=C1O Chemical compound COc(cc1)ccc1C(Nc(cc(c(-c1cnc[o]1)c1)OC)c1C1=O)=C1O AUROZLIWYZDAGX-UHFFFAOYSA-N 0.000 description 1
- STJJOBFVJJPUDW-UHFFFAOYSA-N Cc(c(OCCN(C)C)c1)ccc1C(Nc1c2cc(-c3cnc[o]3)c(OC)c1)=CC2=O Chemical compound Cc(c(OCCN(C)C)c1)ccc1C(Nc1c2cc(-c3cnc[o]3)c(OC)c1)=CC2=O STJJOBFVJJPUDW-UHFFFAOYSA-N 0.000 description 1
- VQZUDCLEOKPOCJ-UHFFFAOYSA-N Cc(c(OCc1ccccc1)c1)ccc1-c1nc(cc(c(-c2cnc[o]2)c2)OC)c2c(OCOC)c1 Chemical compound Cc(c(OCc1ccccc1)c1)ccc1-c1nc(cc(c(-c2cnc[o]2)c2)OC)c2c(OCOC)c1 VQZUDCLEOKPOCJ-UHFFFAOYSA-N 0.000 description 1
- XTYAGVGNXRTYFF-UHFFFAOYSA-N Cc(c(OCc1ccccc1)c1)ccc1C(Nc1c2cc(-c3cnc[o]3)c(OC)c1)=CC2=O Chemical compound Cc(c(OCc1ccccc1)c1)ccc1C(Nc1c2cc(-c3cnc[o]3)c(OC)c1)=CC2=O XTYAGVGNXRTYFF-UHFFFAOYSA-N 0.000 description 1
- BTZCUOZJGZGRJW-UHFFFAOYSA-N Cc1cc(C(Nc(cc(c(-c2cnc[o]2)c2)OC)c2C2=O)=C2O)ccc1Cl Chemical compound Cc1cc(C(Nc(cc(c(-c2cnc[o]2)c2)OC)c2C2=O)=C2O)ccc1Cl BTZCUOZJGZGRJW-UHFFFAOYSA-N 0.000 description 1
- CWWVCKJBHLCVIR-UHFFFAOYSA-N Cc1cccc(C(Nc(cc(c(-c2cnc[o]2)c2)OC)c2C2=O)=C2O)c1 Chemical compound Cc1cccc(C(Nc(cc(c(-c2cnc[o]2)c2)OC)c2C2=O)=C2O)c1 CWWVCKJBHLCVIR-UHFFFAOYSA-N 0.000 description 1
- KPAFWCKSUFEEPT-UHFFFAOYSA-N Cc1cccc(CC(CC(c2c3)=O)Nc2cc(OC)c3-c2cnc[o]2)c1 Chemical compound Cc1cccc(CC(CC(c2c3)=O)Nc2cc(OC)c3-c2cnc[o]2)c1 KPAFWCKSUFEEPT-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/48—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- the present invention relates to novel compounds which inhibit IMPDH, and to methods of making such compounds.
- the invention also encompasses pharmaceutical compositions containing these compounds.
- the compounds and pharmaceutical compositions of the invention are particularly well suited for inhibiting IMPDH enzyme activity and, consequently, can be advantageously used as therapeutic agents for IMPDH-associated disorders.
- This invention also relates to methods for inhibiting the activity of IMPDH using the compounds of this invention alone or in combination with other pharmaceutically active agents .
- Inosine monophosphate dehydrogenase has been shown to be a key enzyme in the regulation of cell proliferation and differentiation. Nucleotides are required for cells to divide and replicate. In mammals, nucleotides may be synthesized through one of two pathways : the de novo synthesis pathway or the salvage pathway. The extent of utilization of each pathway is dependent on the cell type. This selectivity has ramifications with regard to therapeutic utility as described below.
- IMPDH is involved in the de novo synthesis of guanosine nucleotides.
- IMPDH catalyzes the irreversible NAD-dependent oxidation of inosine-5 ' -monophosphate ("IMP") to xanthosine-5' -monophosphate ("XMP"), Jackson et al., Nature 256:331-333 (1975).
- IMPDH is ubiquitous in eukaryotes, bacteria and protozoa. The prokaryotic forms share 30-40% sequence identity with the human enzyme.
- IMPDH type I and type II Two distinct cDNA's encoding IMPDH have been identified and isolated. These transcripts are labeled type I and type II and are of identical size (514 amino acids). Collart et al . , J. Biol. Chem. 263:15769-15772 (1988); Natsu eda et al . , J. Biol. Chem. 265:5292-5295 (1990); and U.S. Patent 5,665,583 to Collart et al . These isoforms share 84% sequence identity. IMPDH type I and type II form tetramers in solution, the enzymatically active unit.
- B and T-lymphocytes depend on the de novo, rather than salvage pathway, to generate sufficient levels of nucleotides necessary to initiate a proliferative response to mitogen or antigen. Due to the B and T cell's unique reliance on the de novo pathway, IMPDH is an attractive target for selectively inhibiting the immune system without also inhibiting the proliferation of other cells.
- Immunosuppression has been achieved by inhibiting a variety of enzymes .
- examples include phosphatase calcineurin (inhibited by cyclosporin and FK-506) ; dihydroorotate dehydrogenase (DHODase) , an enzyme involved in the biosynthesis of pyrimidines (inhibited by leflunomide and brequinar) ; the kinase FRAP (inhibited by rapamycin) ; and the heat shock protein hsp70 (inhibited by deoxyspergualin) .
- Inhibitors of IMPDH have also been described in the art.
- WO 97/40028 and U.S. Patent 5,807,876 describe a class of urea derivatives that possess a common urea backbone.
- WO 98/40381 describes a series of heterocyclic substituted anilines as inhibitors of IMPDH.
- MPA mycophenolic acid
- WO 94/01105 and WO 94/12184 describe mycophenolic acid (“MPA”) and some of its derivatives as potent, uncompetitive, reversible inhibitors of human IMPDH type I and type II. MPA has been demonstrated to block the response of B and T-cells to mitogen or antigen.
- Immunosuppressants such as MPA and derivatives of MPA, are useful drugs in the treatment of transplant rejection and autoimmune disorders, psoriasis, inflammatory diseases, including, rheumatoid arthritis, tumors and for the treatment of allograft rejection. These are described in U.S. Pat. Nos. 4,686,234,
- cyclic AMP agonists such as Rolipram (Schering AG) , a Type 4 Phosphodiesterase Inhibitor (PDE4) and immunomodulator, synergized with IMPDH inhibitor MPA by a cAMP- and IMPDH-dependent dependent mechanism.
- Rolipram Schering AG
- PDE4 Type 4 Phosphodiesterase Inhibitor
- immunomodulator synergized with IMPDH inhibitor MPA by a cAMP- and IMPDH-dependent dependent mechanism.
- Rolipram [4- [3- (cyclopentyloxy) -4-methoxy-phenyl] -2- pyrrolidinone] .
- cyclic AMP agonists such as the PDE4 inhibitor Rolipram (Rol)
- Rolipram PDE4 inhibitor
- the potential for a very low concentration of Rol (10 ⁇ 7 M, approximate ICio) to synergize with a variety of immunosuppressive agents used for the prevention and/or treatment of allograft rejection was defined.
- MPA is a selective, uncompetitive, and reversible inhibitor of IMPDH, a key enzyme in the purine salvage pathway, the potential for cAMP-mediated crosstalk at this locus was further investigated. It was found that gene expression for IMPDH types I and II (assessed by RT-PCR) remained unaffected by the administration of rolipram, MPA, or both at low and high concentrations. However, functional reversal of the synergistic effect was demonstrated with the use of deoxyguanosine, a specific antagonist of MPA on IMPDH (% inhibition of proliferation 81 ⁇ 16 vs. 35 ⁇ 12, p ⁇ 0.05).
- Mycophenolate mofetil sold under the trade name CELLCEPT, is a prodrug which liberates MPA in vivo . It is approved for use in preventing acute renal allograft rejection following kidney transplantation. The side effect profile limits the therapeutic potential of this drug.
- MPA is rapidly metabolized to the inactive glucuronide in vivo . In humans, the blood levels of glucuronide exceed that of MPA. The glucuronide undergoes enterohepatic recycling causing accumulation of MPA in the bile and subsequently in the gastrointestinal tract. This together with the production of the inactive glucuronide effectively lowers the drug's in vivo potency, while increasing its undesirable gastrointestinal side effects.
- type II mRNA is preferentially upregulated in human leukemic cell lines K562 and H -60. Weber, J. Biol. Chem. 266: 506-509 (1991).
- cells from human ovarian tumors and leukemic cells from patients with chronic granulocytic, lymphocytic and acute myeloid leukemias also display an up regulation type II mRNA. This disproportionate increase in IMPDH activity in malignant cells may be addressed through the use of an appropriate IMPDH inhibitor.
- IMPDH has also been shown to play a role in the proliferation of smooth muscle cells, indicating that inhibitors of IMPDH may be useful in preventing restenosis or other hyperproliferative vascular diseases.
- IMPDH has been shown to play a role in viral replication in some viral cell lines. Carr, J. Biol. Chem. 268:27286-27290 (1993).
- the IMPDH inhibitor VX- 497 is currently being evaluated for the treatment of hepatitis C virus in humans . Ribavirin has also been used in the treatment of hepatitis C and B viruses and when used in combination with interferon an enhancement in activity was observed.
- the IMPDH inhibitor ribavirin is limited by its lack of a sustained response in monotherapy and broad cellular toxicity.
- inhibitors of IMPDH with improved pharmacological properties, physical properties and fewer side effects.
- Such inhibitors would have therapeutic potential as immunosuppressants, anti-cancer agents, anti-vascular hyperproliferative agents, antiinflammatory agents, antifungal agents, antipsoriatic and anti-viral agents.
- the compounds of the present invention are effective inhibitors of IMPDH.
- X 2 is CR 3 or N;
- X 3 is-NH-, -0-, or -S-;
- X 4 is CR 4 or N
- X 5 is CR 5 or N
- X 6 is CR 6 or N;
- R 1 is alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, NR 8 R 9 , SR 20 , cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycloalkyl, or heteroaryl;
- R 2 is halogen, cyano, nitro, hydroxy, oxo (double bond is no longer present between CR 2 and X 6 ), SR 7 , S(0)R 7 , S0 2 R 7 , S0 2 NR 8 R 9 , C0 2 R 7 , C(0)NR 8 R 9 , or heteroaryl;
- R 3 is hydrogen, hydroxy, halogen, cyano, C0R 7 , NR 8 R 9 , alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycloalkyl or heteroaryl;
- R 4 , R 5 , and R 6 are independently selected from the group consisting of hydrogen, halogen, nitro, cyano, O-R 7 , NR 8 R 9 , SR 7 , S(0)R 7 , S0 2 R 7 , S0 3 R 7 , S0 2 NR 8 R 9 , C0 2 R 7 , C(0)NR 8 R 9 , C(O) lkyl, C (0) substituted alkyl, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl and substituted alkynyl;
- R 7 , R 10 , and R 11 are independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, alkynyl, cycloalkyl, substituted cycloalkyl, C(0) alkyl, C (0) substituted alkyl, C (O) cycloalkyl, C(0) substituted cycloalkyl, C(0)aryl, C (O) substituted aryl, C(0) Oalkyl, C (0) Osubstituted alkyl, C (O) heterocycloalkyl, C (0) heteroaryl, aryl, substituted aryl, heterocycloalkyl and heteroaryl ;
- R 8 and R 9 are independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, alkenyl, alkynyl, C(O) alkyl, C (0) substituted alkyl, C (0) cycloalkyl, C
- R 20 is alkyl, substituted alkyl, cycloalkyl, aryl, substituted aryl, heteroaryl or heterocycloalkyl;
- R 3 and R 1 may be taken together with the carbon atoms to which they are attached to form a monocyclic or substituted monocyclic ring system of 5 or 6 carbon atoms;
- R 4 and R 5 may be joined together by the chain -O-CH 2 -O- or -0-CH 2 -CH 2 -0- .
- the present invention provides a method of treating inosine monophosphate dehydrogenase associated disorders comprising: administering a therapeutically effective amount of a compound of formula (I)
- X 2 is CR 3 or N
- X 3 is-NH-, -0-, or -S-;
- X 4 is CR 4 or N
- X 5 is CR 5 or N
- X 6 is CR 6 or N
- R 1 is alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, NR 8 R 9 , SR 20 , cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycloalkyl, or heteroaryl;
- R 2 is halogen, cyano, nitro, hydroxy, oxo (double bond is no longer present between CR 2 and X 6 ), SR 7 , S(0)R 7 , S0 2 R 7 , S0 2 NR 8 R 9 , C0 2 R 7 , C(0)NR 8 R 9 , or heteroaryl;
- R 3 is hydrogen, hydroxy, halogen, cyano, C0 2 R 7 , NR 8 R 9 , alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycloalkyl or heteroaryl;
- R 4 , R 5 , and R 6 are independently selected from the group consisting of hydrogen, halogen, nitro, cyano, O-R 7 , NR 8 R 9 , SR 7 , S(0)R 7 , S0 2 R 7 , S0 3 R 7 , S0 2 NR 8 R 9 , C0 2 R 7 , C(0)NR 8 R 9 , C(0) lkyl, C (0) substituted alkyl, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl and substituted alkynyl;
- R 7 , R 10 , and R 11 are independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, alkynyl, cycloalkyl, substituted cycloalkyl, C(O) alkyl, C (0) substituted alkyl, C (O) cycloalkyl, C(0) substituted cycloalkyl, C(0)aryl, C (0) substituted aryl, C(0)0alkyl, C (0) Osubstituted alkyl, C (O) heterocycloalkyl , C (0) heteroaryl, aryl, substituted aryl, heterocycloalkyl and heteroaryl;
- R 8 and R 9 are independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, alkenyl, alkynyl, C(0) alkyl, C (0) substituted alkyl, C (0) cycloalkyl, C (0) substituted cycloalkyl, C(0)aryl, C (0) substituted aryl, C(0)0alkyl, C (0) Osubstituted alkyl, C (0) heterocycloalkyl, C (0) heteroaryl, aryl, substituted aryl, heterocycloalkyl, and heteroaryl or R 8 and R 9 taken together with the nitrogen atom to which they are attached complete a heterocycloalkyl or heteroaryl ring;
- R 20 is alkyl, substituted alkyl, cycloalkyl, aryl, substituted aryl, heteroaryl or heterocycloalkyl;
- R 3 and R 1 may be taken together with the carbon atoms to which they are attached to form a monocyclic or substituted monocyclic ring system of 5 or 6 carbon atoms;
- R 4 and R 5 may be joined together by the chain -0-CH 2 -0- or -O-CH2-CH 2 -O- .
- the present invention provides a method of treating inosine monophosphate dehydrogenase associated disorders comprising: administering a therapeutically effective amount of a compound of formula (II)
- R 2 is a monocyclic substituted or unsubstituted heteroaryl group .
- the present invention provides a method of treating inosine monophosphate dehydrogenase associated disorders comprising: administering a therapeutically effective amount of a compound of formula (III)
- R 2 is 4-oxazolyl, substituted 4-oxazolyl, 5-oxazolyl, or substituted 5-oxazolyl;
- R 3 is hydrogen, hydroxy, NR 8 R 9 , alkyl of 1 to 4 carbons, alkenyl of 2 to 4 carbons, alkynyl of 2 to 4 carbons, substituted alkyl of 1 to 4 carbons, phenyl, substituted phenyl, cycloalkyl of 5 to 7 carbons, substituted cycloalkyl of 5 to 7 carbons, monocyclic heterocycloalkyl and monocyclic heteroaryl;
- R 4 is hydrogen, halogen, nitro, hydroxy, alkyl of 1 to 4 carbons, cyano, CF 3 , OCF 3 , OCH 3 , SCH 3 , S(0)CH 3 , or S(0) 2 CH 3 ;
- R 6 is hydrogen, halogen, nitro, hydroxy, alkyl of 1 to 4 carbons, cyano, CF 3 , 0CH 3 , 0CF 3 , SCH 3 , S(0)CH 3 , and S(0) 2 CH 3 .
- the present invention provides a method of treating inosine monophosphate dehydrogenase associated disorders comprising: administering a therapeutically effective amount of a compound including isomers, enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, prodrugs and solvates wherein: R 2 is 4-oxazolyl, substituted 4-oxazolyl, 5-oxazolyl, substituted 5-oxazolyl or heteroaryl;
- R 3 is hydrogen, hydroxy, halogen, methyl or NR 8 R 9 ;
- R 4 is hydrogen;
- R 5 is halogen, methyl, ethyl, substituted alkenyl, alkyne, OMe or 0CF 3 ; and R 5 is hydrogen.
- the present invention provides a method of treating inosine monophosphate dehydrogenase associated disorders comprising: administering a therapeutically effective amount of a compound including isomers, enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, prodrugs and solvates wherein: R 2 is 4-oxazolyl, substituted 4-oxazolyl, 5-oxazolyl or substituted 5-oxazolyl;
- R 3 is hydrogen, hydroxy, halogen or methyl
- R 4 is hydrogen;
- R 5 is halogen, methyl or OMe;
- R 6 is hydrogen
- the present invention provides a method of treating inosine monophosphate dehydrogenase associated disorders comprising: administering a therapeutically effective amount of a phosphodiesterase Type 4 inhibitor and a compound of formula (X) :
- X 2 is CR 3 or N;
- X 3 is-NH-, -0-, or -S-;
- X 4 is CR 4 or N;
- X 5 is CR 5 or N;
- X 6 is CR 6 or N;
- R 1 is alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, NR 8 R 9 , SR 20 , cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycloalkyl, or heteroaryl;
- R 2 is halogen, cyano, nitro, hydroxy, oxo (double bond is no longer present between CR 2 and X 6 ), SR 7 , S(0)R 7 , S0 2 R 7 , S0 2 NR 8 R 9 , C0 2 R 7 , C(0)NR 8 R 9 , or heteroaryl;
- R 3 is hydrogen, hydroxy, halogen, cyano, C0 2 R 7 , NR 8 R 9 , alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted
- R 4 , R 5 , and R ⁇ are independently selected from the group consisting of hydrogen, halogen, nitro, cyano, O-R 7 , NR 8 R 9 , SR 7 , S(0)R 7 , S0 2 R 7 , S0 3 R 7 , S0 2 NR 8 R 9 , C0 2 R 7 , C(0)NR 8 R 9 , C(O) alkyl, C (0) substituted alkyl, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl and substituted alkynyl;
- R 7 , R 10 , and R 11 are independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, alkynyl, cycloalkyl, substituted cycloalkyl, C(O) alkyl, C (0) substituted alkyl, C (0) cycloalkyl, C(0) substituted cycloalkyl, C(0)aryl, C (0) substituted aryl, C(0)0alkyl, C (0) Osubstituted alkyl, C (0) heterocycloalkyl, C (0) heteroaryl, aryl, substituted aryl, heterocycloalkyl and heteroaryl;
- R 8 and R 9 are independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, alkenyl, alkynyl, C(O) alkyl, C (0) substituted alkyl, C (0) cycloalkyl, C (0)
- R 20 is alkyl, substituted alkyl, cycloalkyl, aryl, substituted aryl, heteroaryl or heterocycloalkyl;
- R 3 and R 1 may be taken together with the carbon atoms to which they are attached to form a monocyclic or substituted monocyclic ring system of 5 or 6 carbon atoms; and R 4 and R 5 may be joined together by the chain -O-CH2-O- or -O-CH2-CH2-O-.
- the present invention provides a method for the treatment or prevention of allograft rejection comprising: administering a therapeutically effective amount of a phosphodiesterase Type 4 inhibitor and a compound of formula (X) :
- X 2 is CR 3 or N
- X 3 is-NH-, -0-, or -S-;
- X 4 is CR 4 or N
- X 5 is CR 5 or N
- X 6 is CR 6 or N
- R 1 is alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, NR 8 R 9 , SR 20 , cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycloalkyl, or heteroaryl;
- R 2 is halogen, cyano, nitro, hydroxy, oxo (double bond is no longer present between CR 2 and X 6 ), SR 7 , S(0)R 7 , S0 2 R 7 , S0 2 NR 8 R 9 , C0 2 R 7 , C(0)NR 8 R 9 , or heteroaryl;
- R 3 is hydrogen, hydroxy, halogen, cyano, C0 2 R 7 , NR 8 R 9 , alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycloalkyl or heteroaryl;
- R 4 , R 5 , and R 6 are independently selected from the group consisting of hydrogen, halogen, nitro, cyano, 0-R 7 , NR 8 R 9 , SR 7 , S(0)R 7 , S0 2 R 7 , S0 3 R 7 , S0 2 NR 8 R 9 , C0 2 R 7 , C(0)NR 8 R 9 , C(O) alkyl, C (0) substituted alkyl, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl and substituted alkynyl;
- R 7 , R 10 , and R 11 are independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, alkynyl, cycloalkyl, substituted cycloalkyl, C(0)alkyl, C (0) ubstituted alkyl , C (0) cycloalkyl, C(O) substituted cycloalkyl, C(0)aryl, C (O) substituted aryl, C(0)0alkyl, C (0) Osubstituted alkyl, C (0) heterocycloalkyl , C (0) heteroaryl, aryl, substituted aryl, heterocycloalkyl and heteroaryl;
- R 8 and R 9 are independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, alkenyl, alkynyl, C(0)alkyl, C (0) substituted alkyl , C (0) cyclo
- R 20 is alkyl, substituted alkyl, cycloalkyl, aryl, substituted aryl, heteroaryl or heterocycloalkyl;
- R 3 and R 1 may be taken together with the carbon atoms to which they are attached to form a monocyclic or substituted monocyclic ring system of 5 or 6 carbon atoms;
- R 4 and R 5 may be joined together by the chain -0-CHa-O- or -O-CH2-CH2-O- .
- the phosphodiesterase Type 4 inhibitor is Rolipram.
- the phosphodiesterase Type 4 inhibitor is [4- [3- (cyclopentyloxy) -4-methoxy-phenyl] -2-pyrrolidinone] .
- the present invention provides a novel compound, comprising: a compound of formula (I)
- X 2 is CR 3 or N
- X 3 is-NH-, -0-, or -S-;
- X 4 is CR 4 or N
- X 5 is CR 5 or N
- X 6 is CR 6 or N
- R 1 is alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycloalkyl, or heteroaryl;
- R 2 is cyano, hydroxy, oxo (double bond is no longer present between CR 2 and X 6 ), SR 7 , S(0)R 7 , S0 2 R 7 , S0 2 NR 8 R 9 , C0 2 R 7 , C(0)NR 8 R 9 , or heteroaryl;
- R 3 is hydrogen, hydroxy, halogen, cyano, C0 2 R 7 , NR 8 R 9 , alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycloalkyl or heteroaryl;
- R 4 , R 5 , and R 6 are independently selected from the group consisting of hydrogen, halogen, nitro, cyano, 0-R 7 , NR 8 R 9 , SR 7 , S(0)R 7 , S0 2 R 7 , S0 3 R 7 , S0 2 NR 8 R 9 , C0 2 R 7 , C(0)NR 8 R 9 , C(O) alkyl, C (0) substituted alkyl, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl and substituted alkynyl;
- R 7 , R 10 , and R 11 are independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, alkynyl, cycloalkyl, substituted cycloalkyl, C(0) alkyl, C (0) substituted alkyl, C (0) cycloalkyl, C(0) substituted cycloalkyl, C(0)aryl, C (O) substituted aryl, C(0)0alkyl, C (0) Osubstituted alkyl, C (O) heterocycloalkyl, C (O) heteroaryl, aryl, substituted aryl, heterocycloalkyl and heteroaryl;
- R 8 and R 9 are independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, alkenyl, alkynyl, C(0)alkyl, C (0) substituted alkyl , C (O) cycloalkyl, C (
- R 3 and R 1 may be taken together with the carbon atoms to which they are attached to form a monocyclic or substituted monocyclic ring system of 5 or 6 carbon atoms ;
- R 4 and R 5 may be joined together by the chain -O-CH2-O- or -O-CH 2 -CH 2 -O-;
- Y is CH 2 , O or S, m and n are each greater than 1, and the sum of m and n is between 3 and 6 ;
- the present invention provides a compound of formula (II)
- R 2 is a monocyclic substituted or unsubstituted heteroaryl group.
- R 2 is 4-oxazolyl, substituted 4-oxazolyl, 5-oxazolyl, or substituted 5-oxazolyl;
- R 3 is hydrogen, hydroxy, NR 8 R 9 , alkyl of 1 to 4 carbons, alkenyl of 2 to 4 carbons, alkynyl of 2 to 4 carbons, substituted alkyl of 1 to 4 carbons, phenyl, substituted phenyl, cycloalkyl of 5 to 7 carbons, substituted cycloalkyl of 5 to 7 carbons, monocyclic heterocycloalkyl and monocyclic heteroaryl;
- R 4 is hydrogen, halogen, nitro, hydroxy, alkyl of 1 to 4 carbons, cyano, CF 3 , OCF 3 , OCH 3 , SCH 3 , S(0)CH 3 , or
- R 6 is hydrogen, halogen, nitro, hydroxy, alkyl of 1 to 4 carbons, cyano, CF 3 , 0CH 3 , OCF 3 , SCH 3 , S(0)CH 3 , and S(0) 2 CH 3 .
- the present invention provides a compound including isomers, enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, prodrugs and solvates wherein :
- R 2 is 4-oxazolyl, substituted 4-oxazolyl, 5-oxazolyl, substituted 5-oxazolyl or heteroaryl;
- R 3 is hydrogen, hydroxy, halogen, methyl or NR 8 R 9 ;
- R 4 is hydrogen
- R 5 is halogen, methyl, ethyl, substituted alkenyl, alkyne, OMe or OCF 3 ; and R 6 is hydrogen.
- the present invention provides a compound including isomers, enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, prodrugs and solvates wherein:
- R 2 is 4-oxazolyl, substituted 4-oxazolyl, 5-oxazolyl or substituted 5-oxazolyl;
- R 3 is hydrogen, hydroxy, halogen or methyl
- R 4 is hydrogen;
- R 5 is halogen, methyl or OMe;
- R 6 is hydrogen
- the present invention provides a compound of formula (V)
- R 3 is hydrogen
- R is hydrogen
- R is hydrogen
- R 1 is CH 3 and R 3 is hydrogen
- R is CH 3 ;
- R 3 is hydrogen
- R is hydrogen
- R 3 is hydrogen
- R is hydrogen
- R is hydrogen
- R- 1 - is
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R 3 is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is Br
- R is hydrogen
- R is hydrogen
- R 3 is hydrogen
- R 3 is hydrogen
- R 3 is hydrogen
- R ,3 is hydrogen
- R is hydrogen
- R is hydrogen
- R 3 is hydrogen
- R is hydrogen
- R 3 is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R 3 is hydrogen
- R is hydrogen
- R 3 is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R 3 is hydrogen
- R is hydrogen
- R ,3 is hydrogen
- R is hydrogen
- R is hydrogen
- R 3 is hydrogen
- R 3 is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R 3 is hydrogen
- R 3 is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R 3 is hydrogen
- R is hydrogen
- R is hydrogen
- R 3 is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R is hydrogen
- R 3 is hydrogen
- R is hydrogen
- R is hydrogen
- R 3 is hydrogen
- R is hydrogen
- the present invention provides a compound including isomers, enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, prodrugs and solvates thereof selected from:
- the present invention provides a pharmaceutical composition comprising: a compound of the invention and a pharmaceutically acceptable carrier.
- the present invention provides a method of treating inosine monophosphate dehydrogenase associated disorders comprising: administering an effective amount of the pharmaceutical composition of the invention.
- the present invention provides a method of treating inosine monophosphate dehydrogenase associated disorders comprising: administering an effective amount of the pharmaceutical composition of the invention and another agent known to be useful in treatment of such disorders .
- the present invention provides a method of treating inosine monophosphate dehydrogenase associated disorders comprising: administering a therapeutically effective amount of the pharmaceutical composition of the invention and a phosphodiesterase Type 4 inhibitor.
- the present invention provides a method for the treatment or prevention of allograft rejection comprising: administering a therapeutically effective amount of the pharmaceutical composition of the invention and a phosphodiesterase Type 4 inhibitor.
- alkyl refers to straight or branched chain hydrocarbon groups having 1 to 12 carbons atoms , preferably 1 to 8 carbon atoms, and most preferably 1 to 4 carbon atoms .
- substituted alkyl refers to an alkyl group as defined above having one, two, or three substituents selected from the group consisting of halo, cyano, O-R 7 , S-R 7 , NR 8 R 9 , nitro, cycloalkyl, substituted cycloalkyl, oxo, aryl, substituted aryl, heterocycloalkyl, heteroaryl, C0 2 R 7 , S(0)R 7 , S0 2 R 7 , S0 3 R 7 , S0NR 8 R 9 , C(0)NR 8 R 9 , C(O) alkyl, and C(0)H.
- alkenyl refers to straight or branched chain hydrocarbon groups having 2 to 12 carbon atoms and one, two or three double bonds, preferably 2 to 6 carbon atoms and one double bond.
- substituted alkenyl refers to an alkenyl group as defined above having one, two, or three substituents selected from the group consisting of halo, cyano, O-R 7 , S-R 7 , NR 8 R 9 , nitro, cycloalkyl, substituted cycloalkyl, oxo, aryl, substituted aryl, heterocycloalkyl, heteroaryl, C0 2 R 7 , S(0)R 7 , S0 2 R 7 , S0 3 R 7 , S0 2 NR 8 R 9 , C(0)NR 8 R 9 , C(O) alkyl, and C(0)H.
- alkynyl refers to straight or branched chain hydrocarbon group having 2 to 12 carbon atoms and one, two or three triple bonds, preferably 2 to 6 carbon atoms and one triple bond.
- substituted alkynyl refers to an alkynyl group as defined above having one, two or three substituents selected from the group consisting of halo, cyano, O-R 7 , S-R 7 , NR 8 R 9 , nitro, cycloalkyl, substituted cycloalkyl, oxo, aryl, substituted aryl, heterocycloalkyl, heteroaryl, C0 2 R 7 , S(0)R 7 , S0 2 R 7 , S0 3 R 7 , S0 2 NR 8 R 9 , C(0)NR 8 R 9 , C(O) alkyl, and C(0)H.
- halo refers to chloro, bromo, fluoro, and iodo.
- cycloalkyl refers to fully saturated and partially unsaturated monocyclic hydrocarbon rings of 3 to 9, preferably 3 to 7 carbon atoms. Also included in this definition are bicyclic rings where the cycloalkyl ring as defined above has a fused aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, heterocycloalkyl, or heteroaryl ring provided that the point of attachment is in the cycloalkyl ring, i.e.
- a cycloalkyl ring as defined above having a two or three carbon bridge or a spirocycloalkyl in which a carbon atom of the cycloallkyl ring has a carbon atom in common with a second cycloalkyl, substituted cycloalkyl, or heterocycloalkyl ring again provided that the point of attachment is in the
- substituted cycloalkyl refers to such cycloalkyl group as defined above having one, two or three substituents selected from the group consisting of halogen, nitro, alkyl, substituted alkyl, alkenyl, cyano, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycloalkyl, heteroaryl, oxo, OR 7 , CO 2 R 7 , C(0)NR 8 R 9 , OC(0)R 7 , OC(0)OR 7 , OC(0)NR 8 R 9 , OCH 2 C0 2 R 7 , C(0)R 7 , NR 8 R 9 , NR 10 C(O)R 7 , NR 10 C(O)OR 7 , NR 10 C (O) C (0) OR 7 , NR 10 C(O)C(O)NR 8 R 9 , NR 10 C (0) C (0) alkyl, NR 10 C (NCN) OR 7 ,
- aryl refers to the phenyl, 1-naphthyl, and 2-naphthyl, preferably phenyl, as well as an aryl ring having a fused cycloalkyl, substituted cycloalkyl, heterocycloalkyl, or heteroaryl ring provided that the point of attachment is in the aryl ring, i.e.
- substituted aryl refers to such aryl groups as defined above having one, two, or three substituents selected from the group consisting of halogen, nitro, alkyl, substituted alkyl, alkenyl, cyano, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycloalkyl, heteroaryl, OR 7 , C0 2 R 7 , C(0)NR 8 R 9 , 0C(0)R 7 , 0C(0)0R 7 , OC(0)NR 8 R 9 , OCH 2 C0 2 R 7 , C(0)R 7 , NR 8 R 9 , NR 10 C(O)R 7 , NR 10 C(O)OR 7 , NR 10 C (O) C (0) OR 7 , NR 10 C (0) C (0) NR 8 R 9 , NR 10 C(O)C(O) alkyl, NR 10 C (NCN) OR 7 , NR 10 C (O) C (0) OR
- NR 10 C( NC) (CR 12 R 13 )rR 7 , NR 10 CO (CR 12 R 13 ) rNR 8 R 9 , NR 10 (CR 12 R 13 )mOR 7 , NR 10 ( CR 12 R 13 ) rC0 R 7 , NR 10 ( CR 12 R 13 ) mNR 8 R 9 , NR 10 ( CR 12 R 13 ) nS0 2 ( CR 14 R 15 ) qR 7 , CONR 10 ( CR 1 R 13 ) nS0 2 ( CR 14 R 15 ) qR 7 , S0 2 NR 10 ( CR 12 R 13 ) nCO ( CR 14 R 15 ) qR 7 , and S0 2 NR 10 ( CR 12 R 13 ) mOR 7 as well as pentaf luorophenyl .
- substituted monocyclic ring system of 5 or 6 carbon atoms refers to one, two or three substituents selected from the group consisting of halogen, nitro, alkyl, substituted alkyl, alkenyl, cyano, oxo, OR 7 , cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycloalkyl, heteroaryl, C0 2 R 7 , C(0)NR 8 R 9 , OC(0)R 7 , OC(0)OR 7 , OC(0)NR 8 R 9 , OCH 2 C0 R 7 , C(0)R 7 , NR 8 R 9 ,
- heterocycloalkyl refers to substituted and unsubstituted saturated or partially saturated monocyclic rings of 3 to 7 members and bicyclic rings of 7 to 11 members having one or two 0 or S atoms and/or one to four N atoms provided that the total number of heteroatoms is four or less and that the heterocycloalkyl ring contains at least one carbon atom.
- the nitrogen and sulfur atoms may optionally be oxidized, and the nitrogen atoms may optionally be quaternized.
- the bicyclic heterocycloalkyl ring may also contain a two or three carbon bridge between available carbon or nitrogen atoms .
- the bicyclic heterocycloalkyl rings may also have a cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycloalkyl, or heteroaryl ring fused to the monocyclic ring provided that the point of attachment is through an available carbon or nitrogen atom of the heterocycloalkyl ring. Also included are spiroheterocycloalkyl rings wherein a carbon atom of the heterocycloalkyl ring is in common with a second heterocycloalkyl ring, a cycloalkyl ring, or a substituted cycloalkyl ring again provided that the point of attachment is through an available carbon or nitrogen atom of the heterocycloalkyl ring.
- the heterocycloalkyl ring can have one, two or three substituents on available carbon or nitrogen atoms selected from the group consisting of halogen, nitro, alkyl, substituted alkyl, alkenyl, cyano, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycloalkyl, heteroaryl, oxo, OR 7 , C0 2 R 7 , C(0)NR 8 R 9 , OC(0)R 7 , OC(0)OR 7 , OC(0)NR 8 R 9 , OCH 2 C0 2 R 7 , C(0)R 7 , NR 8 R 9 , NR 10 C(0)R 7 , NR 10 C(O)OR 7 , NR 10 C (0) C (0) C (0) OR 7 , NR 10 C(O)C(O)NR 8 R 9 , NR 10 C (0) C (0) alkyl , NR 10 C (NCN) OR 7 , NR 10 C(O)NR
- Exemplary monocyclic heterocycloalkyl groups include pyrrolidinyl, pyrrolinyl, pyrazolinyl, pyrazolidinyl , oxetanyl, imidazolinyl , imidazolidinyl, oxazolidinyl, isothiazolidinyl, isoxazolinyl, thiazolidinyl, tetrahydrofuryl, piperidinyl, piperazinyl, tetrahydrothiopyranyl, tetrahydropyranyl, morpholinyl, thiamorpholinyl, thiamorpholinyl sulfoxide, thiamorpholinyl sulfone, tetrahydrothiopyranylsulfone, 1, 3-dioxolanyl, tetrahydro-1, 1-dioxothienyl, dioxanyl, thietanyl, thiiranyl,
- Exemplary bicyclic heterocycloalkyl groups include indolinyl, quinuclidinyl, tetrahydroisoquinolinyl, benzimidazolinyl, chromanyl, dihydrobenzofuran, dihydrofuro [3 , 4-b] pyridinyl, dihydroisoindolyl, dihydroquinazolinyl (such as 3 , 4-dihydro-4-oxo- quinazolinyl) , benzofurazanyl, benzotriazolinyl, dihydrobenzofuryl , dihydrobenzothienyl , dihydrobenzothiopyranyl, dihydrobenzothiopyranyl sulfone, dihydrobenzopyranyl, isoindolinyl, isochromanyl, benzodioxolyl, tetrahydroquinolinyl, etc.
- Exemplary spirocyclic heterocycloalkyl groups include 1-aza [4.5] spirodecane, 2-aza [4.5] spirodecane, 1- aza [5.5] spiroundecane, 2-aza [5.5] spiroundecane, 3- aza [5.5] spiroundecane, etc.
- heteroaryl refers to substituted and unsubstituted aromatic 5 or 6 membered monocyclic groups and 9 or 10 membered bicyclic groups which have at least one heteroatom (0,S or N) in at least one of rings.
- Each ring of the heteroaryl groups containing a heteroatom can contain one or two 0 and S atoms and/or from one to four N atoms provided that the total number of heteroatoms in each ring is four or less.
- the bicyclic heteroaryl rings are formed by fusing a cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycloalkyl, or heteroaryl group to the monocyclic heteroaryl ring as defined above.
- the heteroaryl group is attached via an available carbon or nitrogen in the aromatic heteroaryl ring.
- the nitrogen and sulfur atoms may optionally be oxidized and the nitrogen atoms may optionally be quaternized.
- the heteroaryl ring system may be substituted at an available carbon or nitrogen by one, two, or three substituents selected from the group consisting of halogen, nitro, alkyl, substituted alkyl, alkenyl, cyano, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycloalkyl, heteroaryl, OR 7 , C0 2 R 7 , C(0)NR 8 R 9 , OC(0)R 7 , OC(0)OR 7 , OC(0)NR 8 R 9 , OCH 2 C0 2 R 7 , C(0)R 7 , NR 8 R 9 , NR 10 C(O)R 7 , NR 10 C (0) OR 7 , NR 10 C (0) C (0) OR 7 , NR 10 C (0) C (0) OR 7 , NR 10 C(O)C(O)NR 8 R 9 , NR 10 C (0) C (0) alkyl, NR 10 C (NCN) OR 7 , NR 10
- NR 10 NR 8 R 9 N(COR 7 )OR 10 , N (C0 2 R 7 ) OR 10 , C (0) NR 10 (CR 12 R 13 ) r R 7 , CO ( CR 12 R 13 ) pO ( CR 1 R 15 ) qC0 2 R 7 , CO ( CR 12 R 13 ) rOR 7 , CO ( CR 12 R 13 ) pO ( CR 14 R 15 ) qR 7 , CO ( CR 12 R 13 ).
- NR 10 C( NC) (CR 12 R 13 )rR 7 , NR 10 CO (CR 12 R 13 ) rNR 8 R 9 , NR 10 (CR 12 R 13 )mOR 7 , NR 10 (CR 12 R 13 ) rC0 2 R 7 , NR 10 (CR 12 R 13 ) mNR 8 R ,
- Exemplary monocyclic heteroaryl groups include pyrrolyl, pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, thiadiazolyl, isothiazolyl, pyridinyl, furyl, thienyl, oxadiazolyl, 2-oxazepinyl, azepinyl, pyrazinyl, pyrimidinyl, pyridazinyl, triazinyl, triazolyl, etc.
- Exemplary bicyclic heteroaryl groups include benzothiazolyl, benzoxazolyl, benzothienyl, benzofuryl, quinolinyl, quinolinyl-N-oxide, isoquinolinyl, benzimidazolyl, benzopyranyl, indolizinyl, cinnolinyl, quinoxalinyl , indazolyl, pyrrolopyridyl, furopyridinyl (such as furo [2 , 3-c] pyridinyl, furo [3 , 1-b] pyridinyl or furo [2 , 3-b] yridinyl) , benzisothiazolyl, benzisoxazolyl, benzodiazinyl, benzothiopyranyl, benzotriazolyl , benzpyrazolyl, naphthyridinyl , phthalazinyl, purinyl, pyridopyridy
- R 12 and R 14 are independently selected from hydrogen and alkyl of 1 to 4 carbons .
- R 13 and R 15 are independently selected from hydrogen, alkyl of 1 to 4 carbons, and substituted alkyl of 1 to 4 carbons .
- n is zero or an integer from 1 to 4.
- m is an integer from 2 to 6.
- p is an integer from 1 to 3.
- q is zero or an integer from 1 to 3.
- r is zero or an integer from 1 to 6.
- IMPDH-associated disorders refers to any disorder or disease state in which inhibition of the enzyme IMPDH (inosine monophosphate dehydrogenase, ECl.1.1.205, of which there are presently two known isozymes referred to as IMPDH type 1 and IMPDH type 2 ) would modulate the activity of cells (such as lymphocytes or other cells) and thereby ameliorate or reduce the symptoms or modify the underlying cause (s) of that disorder or disease. There may or may not be present in the disorder or disease an abnormality associated directly with the IMPDH enzyme.
- IMPDH inosine monophosphate dehydrogenase
- IMPDH-associated disorders include transplant rejection and autoimmune disorders, such as rheumatoid arthritis, multiple sclerosis, juvenile diabetes, asthma, and inflammatory bowel disease, as well as inflammatory disorders, cancer and tumor disorders, T- cell mediated hypersensitivity diseases, ischemic or reperfusion injury, viral replication diseases, proliferative disorders and vascular diseases.
- treating includes prophylactic and therapeutic uses, and refers to the alleviation of symptoms of a particular disorder in a patient, the improvement of an ascertainable measurement associated with a particular disorder, or the prevention of a particular immune response (such as transplant rejection) .
- patient refers to a mammal, preferably a human.
- the compounds of this invention may contain one or more asymmetric carbon atoms and thus may occur as racemates and racemic mixtures, .single enantiomers, diastereomeric mixtures and individual diastereomers. All such isomers of the compounds disclosed herein are expressly included within the scope of the present invention.
- Each stereogenic carbon may be of the R or S configuration.
- stable refers to compounds which possess stability sufficient to allow manufacture and which maintain their integrity for a sufficient period of time to be useful as a therapeutic or diagnostic agent.
- the compounds of this invention are defined to include pharmaceutically acceptable derivatives and prodrugs thereof.
- a "pharmaceutically acceptable derivative or prodrug” includes any pharmaceutically acceptable salt, ester, salt of an ester, or other derivative of a compound of the present invention which, upon administration to a subject, is capable of providing (directly or indirectly) a compound of the invention.
- Particularly favored derivatives and prodrugs are those that increase the bioavailability of the compounds of the present invention when such compound is administered to a subject (e.g., by allowing an orally administered compound to be more readily absorbed into the blood) or which enhance delivery of the parent compound to a biological compartment (e.g., the brain or lymphatic system) relative to the parent species.
- Preferred prodrugs include derivatives where a group that enhances aqueous solubility or active transport through the gut membrane is appended to a compound of the present invention.
- Pharmaceutically acceptable salts of the compounds disclosed herein include those derived from pharmaceutically acceptable inorganic and organic acids and bases known to those skilled in the art.
- suitable acid salts include, but are not limited to, the following: acetate, adipate, alginate, aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, citrate, camphorate, camphorsulfonate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptanoate, glycerophosphate, glycolate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethanesulfonate, lactate, maleate, malonate, methanesulfonate, 2- naphthalenesulfonate, nicotinate
- Salts derived from appropriate bases include, but are not limited to, the following: alkali metal (e.g., sodium), alkaline earth metal (e.g., magnesium), ammonium and N-(C ] __4 alkyl ) ⁇ + salts.
- alkali metal e.g., sodium
- alkaline earth metal e.g., magnesium
- ammonium e.g., ammonium
- the present invention also envisions the quaternization of any basic nitrogen- containing groups of the compounds disclosed herein. Water- or oil-soluble or dispersible products may be obtained by such quaternization.
- the compounds of the present invention may be synthesized using conventional techniques known in the art. Advantageously, these compounds are conveniently synthesized from readily available starting materials. Following are general synthetic schemes for manufacturing compounds of the present invention. These, schemes are illustrative and are not meant to limit the possible techniques one skilled in the art may use to manufacture compounds disclosed herein. Different methods will be evident to those skilled in the art. Additionally, the various steps in the synthesis may be performed in an alternate sequence or order to give the desired compound (s) . All documents cited herein are incorporated herein by reference in their entirety.
- a general method for the synthesis of the anilines such as (la.4) useful in this invention can be perfomed by metal catalyzed cross coupling methods known in the literature.
- the simplest case is a Suzuki type cross coupling (Miyaura, N. , Yanagi, T. Suzuki, A., Synth. Comm. 11(7) :513-519 (1981); A. Suzuki et. al . , J. Am. Chem. Soc. 111:513 (1989); and V. N. Kalinin, Russ. Chem. Rev.
- the product may be deprotected by treatment with aqueous potassium hydroxide at a concentration of 0.5N to 5 N at room temperature to 100 °C for a period between 0.5h and 24h.
- aqueous potassium hydroxide at a concentration of 0.5N to 5 N at room temperature to 100 °C for a period between 0.5h and 24h.
- aryl boronic acid (la.5) may react with the known 5-bromothiazole (la.6) in the presence of tetrakis (triphenylphosphine) palladium (0), to provide (la.7) which may be deprotected by an appropriate method.
- HET a 5 or 6 membered ring containing at least one O, N, S atom with an unsaturated bond directly attached to the bromine
- P 1 alkyl, O-benzyl, O-ferfbutyl, ect.
- aryl boronic acid (la.5) may react with oxazolone (la.8) in the presence of copper (II) acetate in the presence of an amine base such as pyridine to provide intermediate (la.9) which may be deprotected by an appropriate method
- aryl boronic acids and esters lb.3, where X is not Br or I, may be prepared as shown in Scheme lb, from the corresponding arylbromide (lb.l) by treatment with a palladium catalyst such as [1,1'- Bis (diphenylphosphino) -ferrocene] dichloropalladium (II) and bis (pinacolato) diboron, (lb.2) , as reported by Ishayama et al .
- a palladium catalyst such as [1,1'- Bis (diphenylphosphino) -ferrocene] dichloropalladium (II) and bis (pinacolato) diboron, (lb
- Aryl boronic esters may be converted to the corresponding boronic acid by several methods including treatment with aqueous HCI.
- the nitrogen may be masked as a nitro group and later reduced by several means including metal reductions, such as by treatment with tin chloride in HCI or by refluxing the nitro compound with zinc in the presence of CaCl 2 in a solvent such as ethanol, or in certain cases the nitro group may be reduced by catalytic hydrogenation in the presence of catalysts such as palladium on carbon.
- the conditions for the reduction of nitro groups are detailed in several references including Hudlicky, M.
- X H, OMe, Cl, etc.
- P1 alkyl Obenzyl, Otertbutyl, etc.
- a general method for the synthesis of 5-membered heterocycles includes the 1,3-dipolar cycloaddition reaction, which is well known to one skilled in the art of organic chemistry and is described by Padwa, Albert, Editor in ⁇ 1,3-Dipolar Cycloaddition Chemistry, Vol. 2" (1984) John Wiley and Sons, New York, N. Y. ; and Padwa, Albert; Editor. in "1,3-Dipolar Cycloaddition Chemistry, Vol. 1" (1984) John Wiley and Sons, New York, N. Y.
- oxazoles may be prepared by 1,3 dipolar cycloaddtion of the corrosponding aldehyde (lc.l) and (p- tolylsulfonyl) ethyl isocyanate (TOSMIC) (lc.2) as shown in scheme Ic .
- the aldehyde may be commercially available or prepared from the corresponding methyl group by oxidation with reagents such as Cr0 3 , Mn0 2 , and ammonium cerium (IV) nitrate by methods well known to one skilled in the art of organic chemistry and is described in Hudlicky, M. , "Oxidations in Organic Chemistry", ACS Monograph 186 (1990) , American Chemical Society, Washington, DC.
- the nitro group in intermediate (lc.3), is reduced to an amine (lc.4), as discussed above.
- Halonitrobenzenes are either commercially available or readily prepared by methods known to one skilled in the art of organic synthesis. Displacement with a variety of nucleophiles produce compounds of structure (Id.2). In one example heating (Id.3) with a nucleophilic heterocycle such as triazole with or without the addition of a base provides the intermediate nitro compound which may be reduced as previously described to provide amines (Id.4). Alternatively simple organic nucleophiles such as cyanide can be reacted with halonitrobenzene (Id.5) to provide an intermediate nitrocompound which can be reduced by many methods to an amine (Id.6) . Scheme Id
- bromination of anilines may be accomplished in many cases by simply dissolving the aniline, such as (la.4) and (lc.4), in a suitable solvent such as methylene chloride, chloroform, acetic acid or hydrochloric acid, and treating with one equivalent of bromine, at a temperature from -78 to 40°C to provide aniline (le.l and le.2) .
- the aniline may be protected with a group such as acetate.
- the bromination can often be accomplished by the addition of a Lewis acid catalyst such as iron, or FeBr .
- lodination can be effected in a manner analogous to that described for bromine, but may also benefit from the addition of silver salts such as silver benzoate, silver triflate, silver trifluoroacetate, or periodic acid, to provide aniline (le.3) and (le.4).
- silver salts such as silver benzoate, silver triflate, silver trifluoroacetate, or periodic acid, to provide aniline (le.3) and (le.4).
- X H, OMe, Cl, CH 3 , CF3,etc.
- aniline (2a.1) (of which anilines (la.4) and (lc.4) are examples) is heated in an inert solvent such as toluene, xylene, or diphenylether at a temperature up to the boiling point of the solvent with a ⁇ -ketoester (2a.2) to provide quinolone (2a.3).
- an inert solvent such as toluene, xylene, or diphenylether
- a ⁇ -ketoester (2a.2) to provide quinolone (2a.3).
- This method of quinolone synthesis has been described in the literature, for example see Kuo et al . in J. Med. Chem 1993, 36, 1146-1156.
- this cyclization can be aided by the use of an acid such as polyphosphoric acid or sulfuric acid to facilitate the intramolecular Friedel-Crafts acylation.
- isomereric cyclization products may be formed. In general these may be separated by flash column
- This can be cyclized to the quinolone by heating in an inert solvent such as xylene at a temperature between 150-300°C.
- an acid catalyst may be added to aid in the intramolecular Friedel-Crafts acylation.
- 2a.3 ⁇ -ketoesters such as (IIa.2) useful for this invention are either commercially available or readily prepared from the corresponding carboxylic acids or esters by several means including, that reported by Clay, R.J., et al., in Synthesis, 1993, 290-292, and those outlined in chapter 2 of . "Advanced Organic Chemistry” 3 rd edition (1990) by Carey, F. A., and Sundberg R. J. , Plenum Press, New York, N. Y.
- the aniline (le.3) is heated between 60-160°C in the presence of an acetylene (2d.l) and 0.1- 5 mol% of a palladium catalyst such as tetrakistriphenylphosphine palladium ( 0 ) , dichlorobis (triphenylphosphine) palladium (II), or dichloro [1, 1 ' -bis (diphenylphosphino) ferrocene palladium ( II ) , in an atmosphere of carbon monoxide (3-40 atmospheres) in a steel autoclave.
- a palladium catalyst such as tetrakistriphenylphosphine palladium ( 0 ) , dichlorobis (triphenylphosphine) palladium (II), or dichloro [1, 1 ' -bis (diphenylphosphino) ferrocene palladium ( II )
- Acetylenes 2d.l are either commercially available or may be prepared by several methods including, palladium catalyzed coupling with an aryl or vinyl bromide or iodide with trimethylsilylacetylene as initially described by Takahashi, S. et . al . in Synthesis 1980, 627.
- the trimethylsilyl protecting group may be removed by treatment with aqueous base or with a fluoride source such as tetrabutylammonium fluoride.
- An alternative sythesis of terminal acetylenes is commonly known as the Corey-Fuchs sythesis, for examples see Wang,Z. et al . J.Org.
- aldehydes useful for this invention are either commercially available or readily prepared by oxidation of an alcohol by many methods as described by Hudlicky, M. "Oxidations in Organic Chemistry", ACS Monograph 186, 1990, American Chemical Society, Washington, DC.
- Acids (2c.1) useful for this invention in their own right or for the preparation of beta ketoesters such as (2a.2) are either commercially available or readily prepared by a number of methods known to one skilled in the art of orgainic chemistry including, oxidation of an alcohol or hydrolysis of an ester. Transformations that produce acids from commercially available reagents are described by Larock, R.C. in "Comprehensive Organic Transformations: a Guide to Functional Group Preparations.” 1989, VCH Publishers, N.Y. , N.Y..
- the compounds of the present invention may be modified by appending appropriate functionalities to enhance selective biological properties . Such modifications are known in the art and include those which increase biological penetration into a given biological compartment (e.g., blood, lymphatic system, central nervous system) , increase oral availability, increase solubility to allow administration by injection, alter metabolism and alter rate of excretion.
- the compounds of the present invention inhibit IMPDH enzyme, and are thus useful in the treatment, including prevention and therapy of disorders which are mediated or effected by cells which are sensitive to IMPDH inhibition, as described previously.
- the present invention thus provides methods for the treatment of IMPDH-associated disorders, comprising the step of administering to a subject in need thereof at least one compound of the formula I, preferably at least one compound represented by formulas II and/or III, in an amount effective therefor.
- Other therapeutic agents such as those described below, may be employed with the inventive compounds in the present methods .
- such other therapeutic agent (s) may be administered prior to, simultaneously with or following the administration of the compound (s) of the present invention.
- the compounds of the present invention can be used in treating a range of disorders exemplified by, but not limited to, disorders such as: the treatment of transplant rejection (e.g., kidney, liver, heart, lung, pancreas (e.g., islet cells), bone marrow, cornea, small bowel, skin allografts, skin homografts (such as employed in burn treatment), heart valve xenografts, serum sickness, and graft vs.
- transplant rejection e.g., kidney, liver, heart, lung, pancreas (e.g., islet cells), bone marrow, cornea, small bowel, skin allografts, skin homografts (such as employed in burn treatment), heart valve xenografts, serum sickness, and graft vs.
- transplant rejection e.g., kidney, liver, heart, lung, pancreas (e.g., islet cells), bone marrow, cornea, small bowel, skin allografts, skin homograf
- autoimmune diseases such as rheumatoid arthritis, psoriatic arthritis, multiple sclerosis, juvenile diabetes, asthma, inflammatory bowel disease (such as Crohn's disease and ulcerative colitus) , pyoderma gangrenum, lupus (systemic lupus erythematosis) , myasthenia gravis, psoriasis, dermatitis, dermatomyositis; eczema, seborrhoea, pulmonary inflammation, eye uveitis, hepatitis, Grave's disease, Hashimoto's thyroiditis, autoimmune thyroiditis, Behcet's or Sjorgen's syndrome (dry eyes /mouth) , pernicious or immunohaemolytic anaemia, Addison's disease (autoimmune disease of the adrenal glands) , idiopathic adrenal insufficiency, autoimmune polyglandular disease (also known as autoimmune polyglandular disease (also known
- HSV-1 herpes simplex type 1
- HSV-2 herpes simplex type 2
- hepatitis including hepatitis B and hepatitis C
- cytomegalovirus Epstein-Barr
- HMV human immunodeficiency virus
- IMPDH is also known to be present in bacteria and thus may regulate bacterial growth.
- the IMPDH-inhibitor compounds of- the present invention may be useful in treatment or prevention of bacterial infection, alone or in combination with other antibiotic agents .
- the compounds of the present invention are useful for the treatment of the aforementioned exemplary disorders irrespective of their etiology, for example, for the treatment of transplant rejection, rheumatoid arthritis, inflammatory bowel disease, and viral infections.
- the present invention also provides pharmaceutical compositions comprising at least one of the compounds of formula I, preferably at least one of the compounds of formulas II and/or III, or a salt thereof, capable of treating an IMPDH-associated disorder in an amount effective therefor, alone or in combination with at least one additional therapeutic agent, and any pharmaceutically acceptable carrier, adjuvant or vehicle.
- Additional therapeutic agents encompasses, but is not limited to, an agent or agents selected from the group consisting of an immunosuppressant , an anti-cancer agent, an anti-viral agent, an anti-inflammatory agent, an anti- fungal agent, an antibiotic, or an anti-vascular hyperproliferation compound.
- pharmaceutically acceptable carrier, adjuvant or vehicle refers to a carrier, adjuvant or vehicle that may be administered to a subject, together with a compound of the present invention, and which does not destroy the pharmacological activity thereof.
- Pharmaceutically acceptable carriers, adjuvants and vehicles that may be used in the pharmaceutical compositions of the present invention include, but are not limited to, the following: ion exchangers, alumina, aluminum stearate, lecithin, self-emulsifying drug delivery systems ("SEDDS") such as d(-tocopherol polyethyleneglycol 1000 succinate) , surfactants used in pharmaceutical dosage forms such as Tweens or other similar polymeric delivery matrices, serum proteins such as human serum albumin, buffer substances such as phosphates, glycine, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen
- Cyclodextrins such as ⁇ - , ⁇ - and ⁇ -cyclodextrin, or chemically modified derivatives such as hydroxyalkylcyclodextrins, including 2- and 3-hydroxypropyl- ⁇ -cyclodextrins, or other solubilized derivatives may also be used to enhance delivery of the compounds of the present invention.
- the compositions of the present invention may contain other therapeutic agents as described below, and may be formulated, for example, by employing conventional solid or liquid vehicles or diluents, as well as pharmaceutical additives of a type appropriate to the mode of desired administration (for example, excipients, binders, preservatives, stabilizers, flavors, etc.) according to techniques such as those well known in the art of pharmaceutical formulation.
- the compounds of the formula I may be administered by any suitable means, for example, orally, such as in the form of tablets, capsules, granules or powders; sublingually; buccally; parenterally, such as by subcutaneous, intravenous, intramuscular, or intrasternal injection or infusion techniques (e.g., as sterile injectable aqueous or non-aqueous solutions or suspensions) ; nasally such as by inhalation spray; topically, such as in the form of a cream or ointment; or rectally such as in the form of suppositories; in dosage unit formulations containing non-toxic, pharmaceutically acceptable vehicles or diluents.
- suitable means for example, orally, such as in the form of tablets, capsules, granules or powders; sublingually; buccally; parenterally, such as by subcutaneous, intravenous, intramuscular, or intrasternal injection or infusion techniques (e.g., as sterile injectable aqueous
- the present compounds may, for example, be administered in a form suitable for immediate release or extended release. Immediate release or extended release may be achieved by the use of suitable pharmaceutical compositions comprising the present compounds, or, particularly in the case of extended release, by the use of devices such as subcutaneous implants or osmotic pumps.
- the present compounds may also be administered liposomally.
- compositions for oral administration include suspensions which may contain, for example, microcrystalline cellulose for imparting bulk, alginic acid or sodium alginate as a suspending agent, methylcellulose as a viscosity enhancer, and sweeteners or flavoring agents such as those known in the art; and immediate release tablets which may contain, for example, microcrystalline cellulose, dicalcium phosphate, starch, magnesium stearate and/or lactose and/or other excipients, binders, extenders, disintegrants , diluents and lubricants such as those known in the art.
- the present compounds may also be delivered through the oral cavity by sublingual and/or buccal administration.
- Molded tablets, compressed tablets or freeze-dried tablets are exemplary forms which may be used.
- Exemplary compositions include those formulating the present compound (s) with fast dissolving diluents such as mannitol, lactose, sucrose and/or cyclodextrins .
- Also included in such formulations may be high molecular weight excipients such as celluloses (avicel) or polyethylene glycols (PEG) .
- Such formulations may also include an excipient to aid mucosal adhesion such as hydroxy propyl cellulose (HPC) , hydroxy propyl methyl cellulose (HPMC) , sodium carboxy methyl cellulose (SCMC) , aleic anhydride copolymer (e.g., Gantrez) , and agents to control release such as polyacrylic copolymer (e.g., Carbopol 934).
- HPC hydroxy propyl cellulose
- HPMC hydroxy propyl methyl cellulose
- SCMC sodium carboxy methyl cellulose
- aleic anhydride copolymer e.g., Gantrez
- agents to control release such as polyacrylic copolymer (e.g., Carbopol 934).
- Lubricants, glidants, flavors, coloring agents and stabilizers may also be added for ease of fabrication and use.
- compositions for nasal aerosol or inhalation administration include solutions in saline which may contain, for example, benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, and/or other solubilizing or dispersing agents such as those known in the art.
- compositions for parenteral administration include injectable solutions or suspensions which may contain, for example, suitable non-toxic, parenterally acceptable diluents or solvents, such as mannitol, 1,3- butanediol, water, Ringer's solution, an isotonic sodium chloride solution, or other suitable dispersing or wetting and suspending agents, including synthetic mono- or diglycerides, and fatty acids, including oleic acid.
- suitable non-toxic, parenterally acceptable diluents or solvents such as mannitol, 1,3- butanediol, water, Ringer's solution, an isotonic sodium chloride solution, or other suitable dispersing or wetting and suspending agents, including synthetic mono- or diglycerides, and fatty acids, including oleic acid.
- parenteral as used herein includes subcutaneous, intracutaneous, intravenous, intramuscular, intraarticular, intraarterial, intrasynovial, intrasternal , intrathecal
- compositions for rectal administration include suppositories which may contain, for example, a suitable non-irritating excipient, such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ordinary temperatures, but liquify and/or dissolve in the rectal cavity to release the drug.
- a suitable non-irritating excipient such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ordinary temperatures, but liquify and/or dissolve in the rectal cavity to release the drug.
- compositions for topical administration include a topical carrier such as Plastibase (mineral oil gelled with polyethylene) .
- a topical carrier such as Plastibase (mineral oil gelled with polyethylene) .
- the effective amount of a compound of the present invention may be determined by one of ordinary skill in the art, and includes exemplary dosage amounts for an adult human of from about 0.1 to 500 mg/kg of body weight of active compound per day, which may be administered in a single dose or in the form of individual divided doses, such as from 1 to 5 times per day. It will be understood that the specific dose level and frequency of dosage for any particular subject may be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the species, age, body weight, general health, sex and diet of the subject, the mode and time of administration, rate of excretion, drug combination, and severity of the particular condition.
- Preferred subjects for treatment include animals, most preferably mammalian species such as humans, and domestic animals such as dogs, cats and the like, subject to IMPDH-associated disorders.
- the compounds of the present invention may be employed alone or in combination with each other and/or other suitable therapeutic agents useful in the treatment of IMPDH-associated disorders, such as IMPDH inhibitors other than those of the present invention, immunosuppressants, anti-cancer agents, anti-viral agents, anti-inflammatory agents, anti-fungal agents, antibiotics, or anti-vascular hyperproliferation agents.
- cyclosporins e.g., cyclosporin A
- CTLA4-Ig antibodies such as anti-ICAM-3, anti-IL-2 receptor (Anti- Tac) , anti-CD45RB, anti-CD2, anti-CD3 (OKT-3), anti-CD4, anti-CD80, anti-CD86, monoclonal antibody OKT3
- agents blocking the interaction between CD40 and CD154 a.k.a. "gp39"
- CDl54/gp39 e.g., CD40Ig and CD8gp39
- inhibitors such as nuclear translocation inhibitors, of NF-kappa B function, such as deoxyspergualin (DSG) , non-steroidal antiinflammatory drugs (NSAIDs) such as ibuprofen, celecoxib and rofecoxib, steroids such as prednisone or dexamethasone, gold compounds, antiviral agents such as abacavir, antiproliferative agents such as methotrexate, leflunomide, FK506 (tacrolimus, Prograf ) , cytotoxic drugs such as azathiprine and cyclophosphamide, TNF- ⁇ inhibitors such as tenidap, anti-TNF antibodies or soluble TNF receptor, and rapamycin (sirolimus or Rapamune) or derivatives thereof.
- DSG deoxyspergualin
- NSAIDs non-steroidal antiinflammatory drugs
- the compounds disclosed herein are capable of targeting and inhibiting IMPDH enzyme. Inhibition can be measured by various methods, including, for example, IMP dehydrogenase HPLC assays (measuring enzymatic production of XMP and NADH from IMP and NAD) and IMP dehydrogenase spectrophotometric assays (measuring enzymatic production of NADH from NAD) . See, e.g., Montero et al . , Clinica Chimica Acta 238:169-178 (1995). Additional assays known in the art can be used in ascertaining the degree of activity of a compound ("test compound”) as an IMPDH inhibitor.
- the inventors used the following assay to determine the degree of activity of the compounds disclosed herein as IMPDH inhibitors: Activity of IMPDH I and IMPDH II was measured following an adaptation of the method described in WO 97/40028.
- the reaction mixture was prepared containing 0.1M Tris pH 8.0, 0.1 M KCl, 3 mM EDTA, 2 mM DTT, 0.4 mM IMP and 40 nM enzyme (IMPDH I or IMPDH II) .
- the reaction was started by the addition of NAD to a final concentration of 0.4 mM.
- the enzymatic reaction was followed by measuring the increase in absorbance at 340 nM that results from the formation of NADH.
- the compounds disclosed herein are capable of inhibiting the enzyme IMPDH at a measurable level, under the above-described assay or an assay which can determine an effect of inhibition of the enzyme IMPDH.
- 9A was prepared from 3A by a route analogous to that used for the preparation of 3B, except in the purification step: the reaction mixture was concentrated in vacuo and carried to the next step without further purification.
- Example 11 Part D 2- (2 , 3-Dihydro-3-methoxy-lH-inden-5- yl) -7-methoxy-6- (5-oxazolyl) -4 (IH) -quinolinone 4A , 0.15 g (0.47 mmol), 0.163 g (0.94 mmol) of 11D, 0.02 g (0.028mmol) of bis ( triphenylphosphine ) palladium ( II ) dichloride and diethylamine (5 L) were reacted in a similar manner were reacted in a similar manner to Example 5, part D to yield the title compound as a solid (0.04 g, 21%) .
- Examples 12 to 103 are prepared by several routes .
- the method to prepare a specific example is noted in table 1.
- Examples prepared in a manner analogous to Example 1 starting with aniline ID and an appropriate j eta-ketoester are designated as method Al .
- Examples prepared in a manner analogous to Example 2 starting with aniline ID and a appropriate jbeta-ketoester, which may be obtained from commercially available acid chlorides, are designated as method A2.
- Example prepared from esters, which are readily converted to an acid by hydrolysis, or acids, followed by reaction of the acid with carbonyldiimidazole, and reacted with potassium ethyl malonate as described in Example 6, part A are designated as method A3.
- Compounds prepared in a manner analogous to Example 9 are designated as method B2
- Compounds prepared in a manner analogous to Example 7 are designated as method Dl .
- Compounds prepared in a manner analogous to Example 8 are designated as method D2.
- the compounds of these examples have structures outlined in Table 1 below.
- Example 104 Part A, 6-Bromo-2 , 3-dihydro-N-methyl-lH- inden-1-amine
- Example 104 Part C, [2 , 3-Dihydro-6- [ (trimethylsilyl) ethynyl] -lH-inden-l-yl]methylcarbamic acid phenylmethyl ester
- Example 104 Part D, (6-Ethynyl-2 , 3-dihydro-lH-inden-l- yl)methylcarbamic acid phenylmethyl ester
- Example 104 Part F, [6- [1 , 4-Dihydro-7-methoxy-6- (5- oxazolyl ) -4-oxo-2-quinolinyl] -2 , 3-dihydro-lH-inden-1- yl]methylcarbamic acid phenylmethyl ester
- Example 106 Part A, 1- (6-Bromo-2 , 3-dihydro-lH-inden-l- yl ) yrrolidine
- 6-bromoindanol (0.3 g, 1.39 mmol) was subjected to the same conditions as 104, part A by substituting methylamine with pyrrolidine (1.16 mL, 14 mmol) to yield the title compound (0.195 g, 52%).
- 1 H NMR (CDC1 3 ) 7.4 (s, IH) , 7.25 (d, IH) , 7.0 (d, IH) , 4.1 (t, IH) , 2.9 (m, IH) , 2.6 (m, IH) , 2.55 (m, 4H) , 2.1 (m, 2H) , 1.7 (brs, 4H) .
- 6-bromoindanol (0.3 g, 1.39 mmol) was subjected to the same conditions as 104, part A by substituting methylamine with morpholine (4.0 mL) in toluene (5 mL) to yield the title compound (0.35 g, 89%).
- 1 H NMR (CDC1 3 ) 7.5 (s, IH) , 7.3 (d, IH) , 7.0 (d, IH) , 4.2 (t, IH) , 3.7 (brs, 4H) , 2.8 (m, IH) , 2.7 (m, IH) , 2.4 (m, 4H) , 2.1 (m, 2H) .
- 6-bromoindanol (0.3 g, 1.39 mmol) was subjected to the same conditions as 104, part A by substituting methylamine with azetidine (0.9 mL 13.9 mmol) in toluene (5 mL) to yield the title compound (0.167 g, 47%).
- 1 H NMR (CDC1 3 ) 7.3 (s, IH) , 7.2 (d, IH) , 7.0 (d, IH) , 3.8 (m, IH), 3.2 (m, 4H) , 3.0 (m, IH) , 2.8 (m, IH) , 2.0 (m, 3H) , 1.8 (m, IH) .
- HOB Compound 110A (0.485g, 2.14 mmol) was subjected to the same conditions as 104, part B and C to yield the title compound (0.35 g, 95%).
- X H NMR (CDC1 3 ) D 7.5 (s, IH) , 7.3 (d, IH) , 7.0 (d, IH) , 4.7 (m, IH) , 3.3 (s, 3H) , 3.0 (m, IH) , 2.9 (s, IH) , 2.7 (m, IH) , 2.3 (m, IH) , 2.0 (m, IH) .
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Virology (AREA)
- Immunology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Pulmonology (AREA)
- Molecular Biology (AREA)
- Cardiology (AREA)
- Rheumatology (AREA)
- Heart & Thoracic Surgery (AREA)
- Biotechnology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Vascular Medicine (AREA)
- Urology & Nephrology (AREA)
- AIDS & HIV (AREA)
- Transplantation (AREA)
- Pain & Pain Management (AREA)
- Hematology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Quinoline Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description





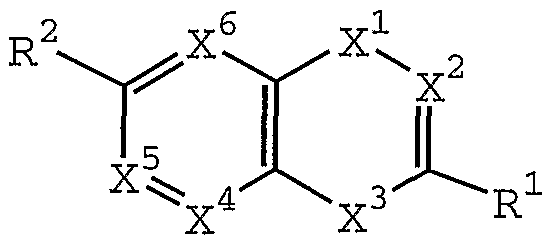

























































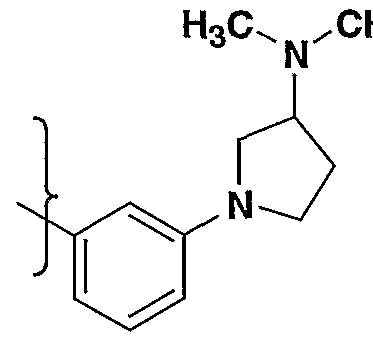















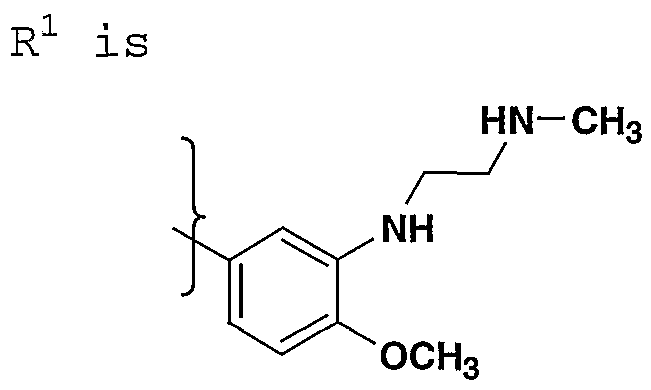































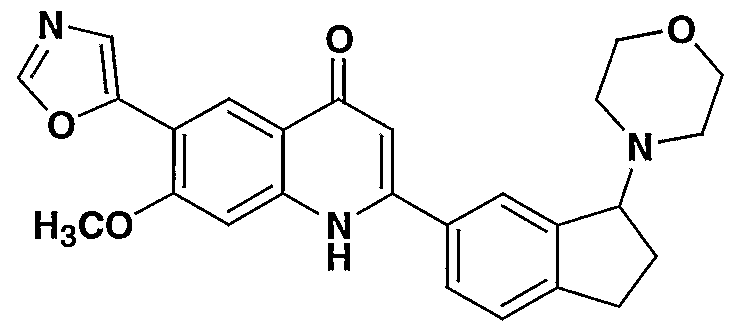
















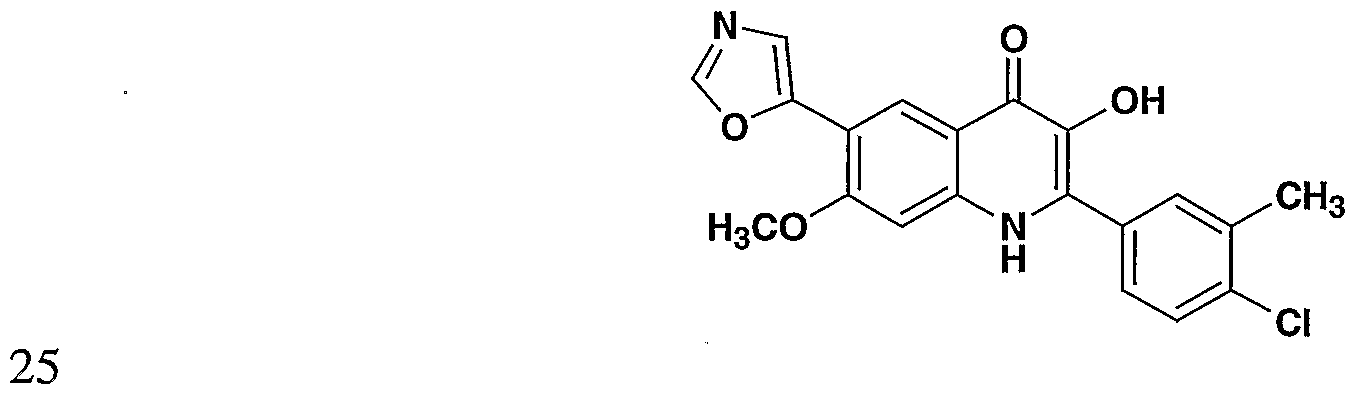








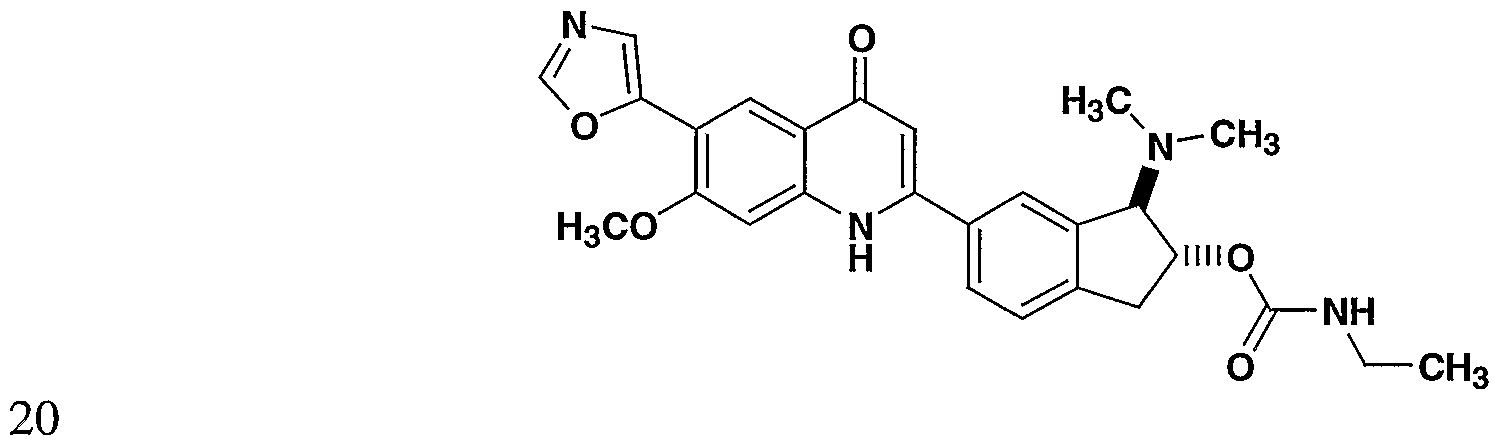





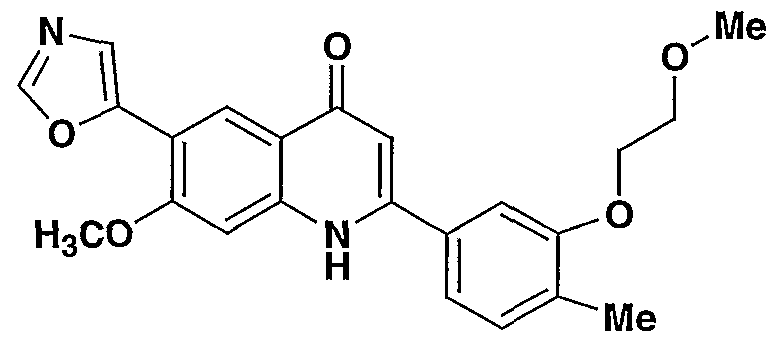




















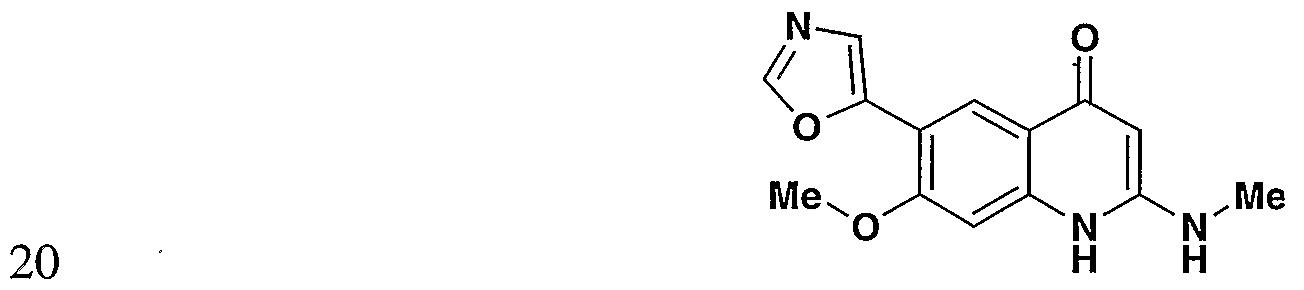

































































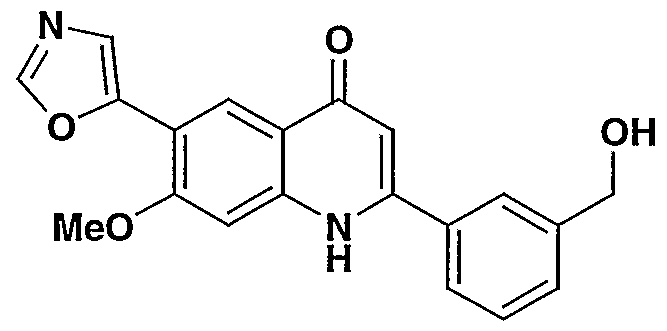


































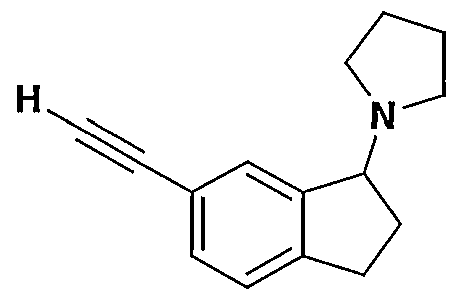

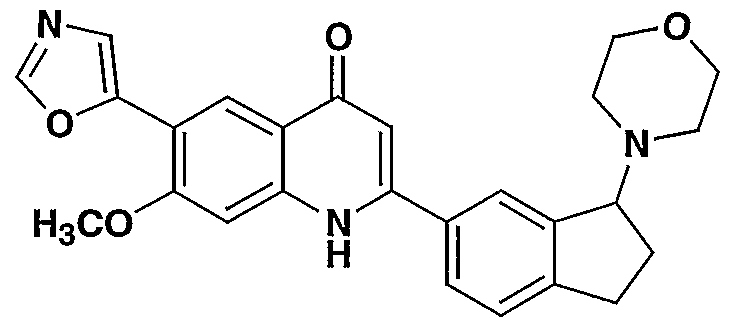









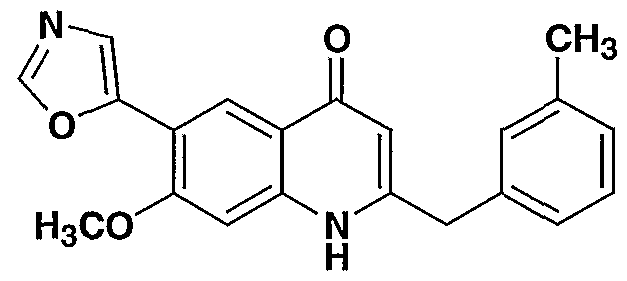
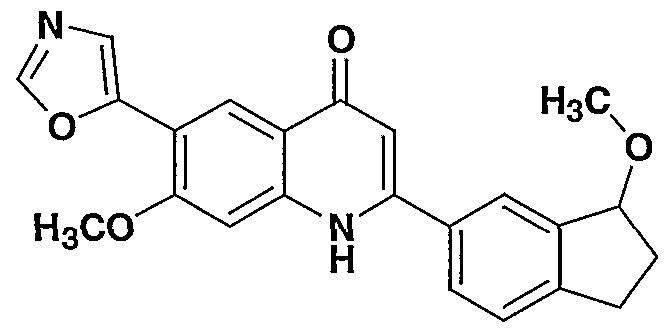
























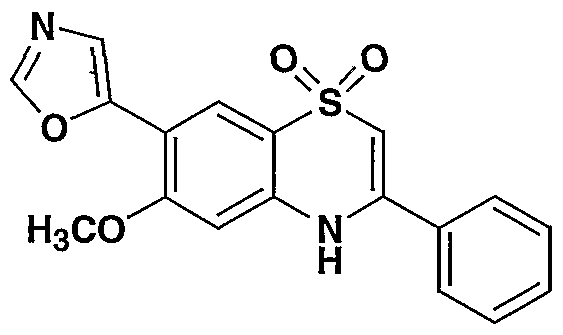






















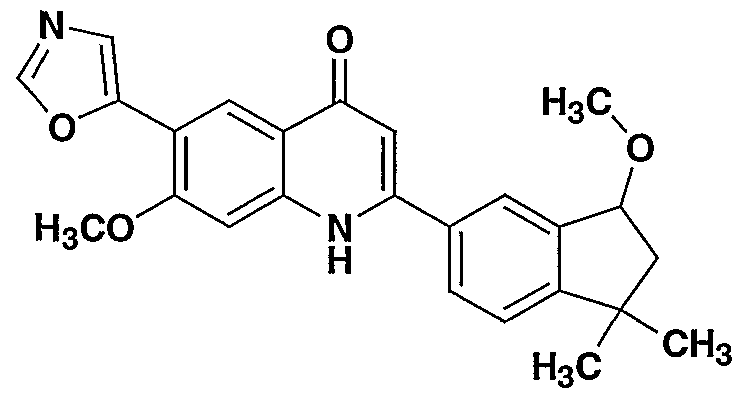















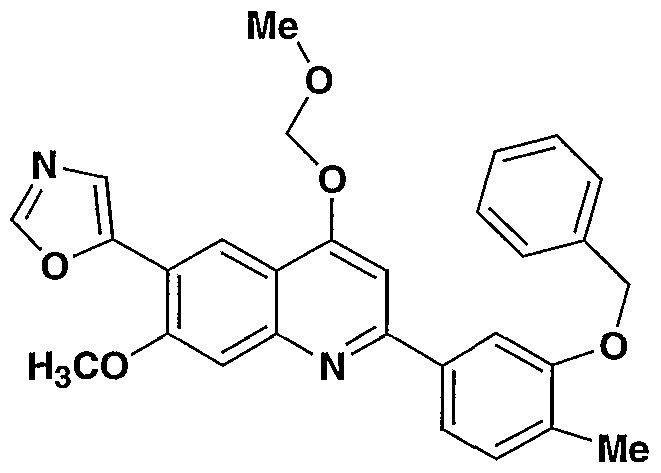






































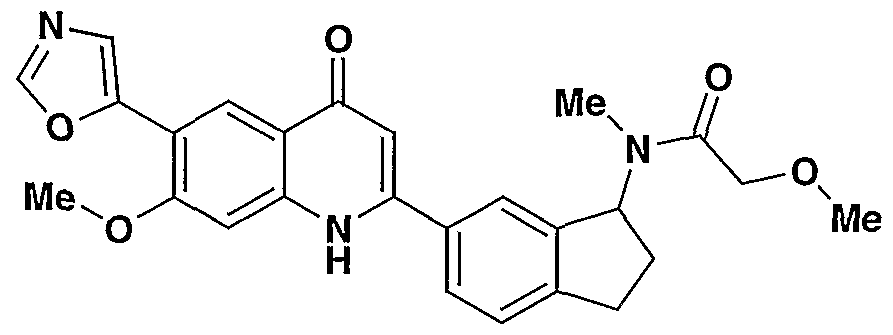










Claims
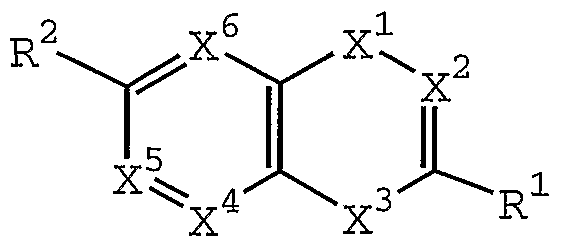














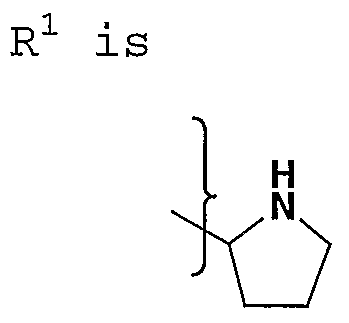















































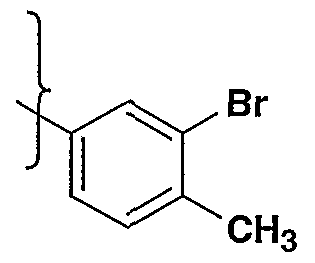





















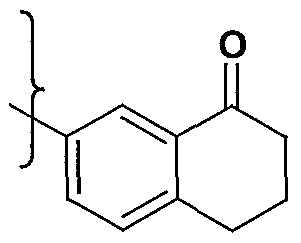












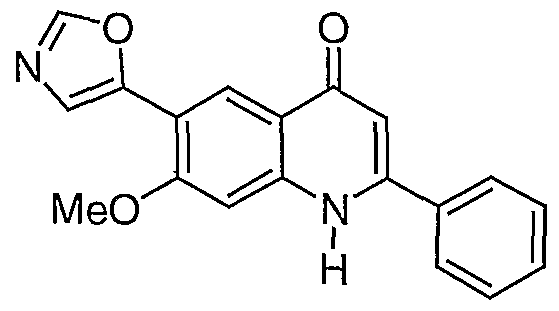
























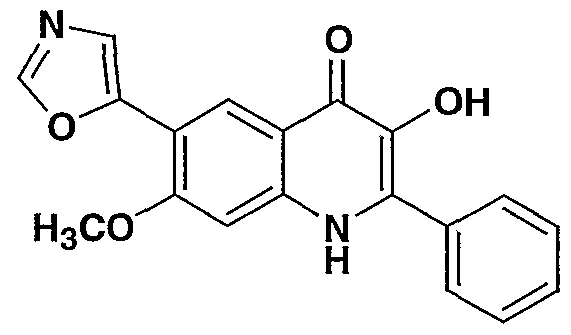





















































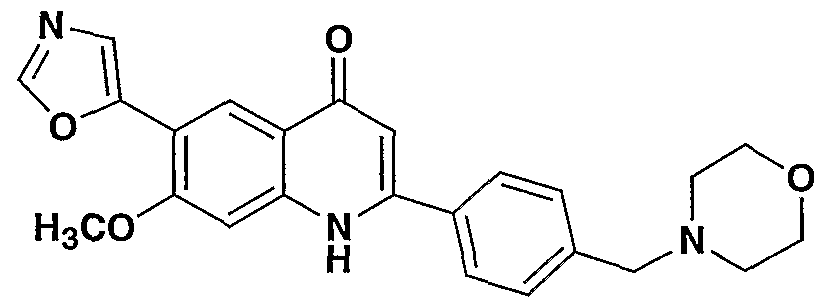
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU5553801A AU5553801A (en) | 2000-04-24 | 2001-04-19 | Heterocycles that are inhibitors of impdh enzyme |
| JP2001578430A JP2003531205A (en) | 2000-04-24 | 2001-04-19 | Heterocyclic compounds that are inhibitors of IMPDH enzyme |
| AU2001255538A AU2001255538B2 (en) | 2000-04-24 | 2001-04-19 | Heterocycles that are inhibitors of IMPDH enzyme |
| EP01928708A EP1276739A2 (en) | 2000-04-24 | 2001-04-19 | Heterocycles that are inhibitors of impdh enzyme |
| CA002407370A CA2407370A1 (en) | 2000-04-24 | 2001-04-19 | Heterocycles that are inhibitors of impdh enzyme |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US19942000P | 2000-04-24 | 2000-04-24 | |
| US60/199,420 | 2000-04-24 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2001081340A2 true WO2001081340A2 (en) | 2001-11-01 |
| WO2001081340A3 WO2001081340A3 (en) | 2002-05-23 |
Family
ID=22737412
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2001/012900 WO2001081340A2 (en) | 2000-04-24 | 2001-04-19 | Heterocycles that are inhibitors of impdh enzyme |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US6919335B2 (en) |
| EP (1) | EP1276739A2 (en) |
| JP (1) | JP2003531205A (en) |
| AU (2) | AU5553801A (en) |
| CA (1) | CA2407370A1 (en) |
| UY (1) | UY26677A1 (en) |
| WO (1) | WO2001081340A2 (en) |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003035066A1 (en) * | 2001-10-23 | 2003-05-01 | Celltech R & D Limited | 2-aminoquinolone derivatives for use as impdh inhibitors |
| WO2004018462A1 (en) * | 2002-08-23 | 2004-03-04 | Celltech R & D Limited | Quinazolinone derivatives |
| WO2004041165A2 (en) * | 2002-11-01 | 2004-05-21 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| US6858602B2 (en) | 2001-06-12 | 2005-02-22 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| US7109196B2 (en) | 2001-09-26 | 2006-09-19 | Bayer Pharmaceuticals Corporation | 1,8 Naphthyridine derivatives and their use to treat diabetes and related disorders |
| US7244738B2 (en) | 2003-07-02 | 2007-07-17 | Roche Palo Alto Llc | Arylamine-substituted quinazolinone compounds useful as alpha 1A/B adrenergic receptor antagonists |
| WO2009041521A1 (en) | 2007-09-26 | 2009-04-02 | Astellas Pharma Inc. | Quinolone derivative |
| US7592340B2 (en) | 2003-12-04 | 2009-09-22 | Vertex Pharmaceuticals Incorporated | Quinoxalines useful as inhibitors of protein kinases |
| EP2181704A2 (en) | 2002-12-30 | 2010-05-05 | Angiotech International Ag | Drug delivery from rapid gelling polymer composition |
| WO2011090095A1 (en) * | 2010-01-22 | 2011-07-28 | 富山化学工業株式会社 | Heterocyclic compound having azole group |
| CN102675200A (en) * | 2012-05-16 | 2012-09-19 | 中国药科大学 | 2-phenyl-4-carbostyril compounds with antineoplastic activity, and preparation method and usage thereof |
| WO2014014814A1 (en) * | 2012-07-16 | 2014-01-23 | Brown University | Compounds for the treatment and prevention of infections |
| EP2742033A4 (en) * | 2011-08-11 | 2015-07-29 | Neuprotect Pty Ltd | Flavonoid compounds, and methods of use thereof |
| WO2021184014A1 (en) * | 2020-03-13 | 2021-09-16 | Arizona Board Of Regetns On Behalf Of The University Of Arizona | Stable reactive compositions for bioconjugation, probes, and protein labeling |
| US11820747B2 (en) | 2021-11-02 | 2023-11-21 | Flare Therapeutics Inc. | PPARG inverse agonists and uses thereof |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0400895D0 (en) * | 2004-01-15 | 2004-02-18 | Smithkline Beecham Corp | Chemical compounds |
| AR050253A1 (en) * | 2004-06-24 | 2006-10-11 | Smithkline Beecham Corp | COMPOSITE DERIVED FROM INDAZOL CARBOXAMIDE, COMPOSITION THAT INCLUDES IT AND ITS USE FOR THE PREPARATION OF A MEDICINAL PRODUCT |
| JP5112621B2 (en) * | 2005-06-02 | 2013-01-09 | 日本曹達株式会社 | Purification method and production method of 5-substituted oxazole compounds |
| US8063071B2 (en) * | 2007-10-31 | 2011-11-22 | GlaxoSmithKline, LLC | Chemical compounds |
| US8440692B2 (en) * | 2006-12-07 | 2013-05-14 | China Medical University | Hydrophilic derivatives of 2-aryl-4-quinolones as anticancer agents |
| PE20081889A1 (en) * | 2007-03-23 | 2009-03-05 | Smithkline Beecham Corp | INDOL CARBOXAMIDES AS INHIBITORS OF IKK2 |
| JP2012520257A (en) | 2009-03-10 | 2012-09-06 | グラクソ グループ リミテッド | Indole derivatives as IKK2 inhibitors |
| BR112016013018A2 (en) * | 2013-12-09 | 2017-08-08 | Ucb Biopharma Sprl | FUSED BICYCLIC HETEROAROMATIC DERIVATIVES AS MODULATORS OF TNF ACTIVITY |
| EP3099673B1 (en) * | 2014-01-31 | 2018-09-26 | Bristol-Myers Squibb Company | Quinoline-based kinase inhibitors |
| CA3094324A1 (en) | 2018-03-26 | 2019-10-03 | Clear Creek Bio, Inc. | Compositions and methods for inhibiting dihydroorotate dehydrogenase |
| US20210300873A1 (en) | 2020-03-20 | 2021-09-30 | Clear Creek Bio, Inc. | Stable polymorphic compositions of brequinar sodium and methods of use and manufacture thereof |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3495009A (en) * | 1966-04-26 | 1970-02-10 | Ferlux Zone Ind De Cournon D A | Pharmaceutical compositions and methods for treating inflammatory conditions with halogenated flavones |
| WO1997040028A1 (en) * | 1996-04-23 | 1997-10-30 | Vertex Pharmaceuticals Incorporated | Urea derivatives as inhibitors of impdh enzyme |
| WO1998040381A1 (en) * | 1997-03-14 | 1998-09-17 | Vertex Pharmaceuticals Incorporated | Inhibitors of impdh enzyme |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4686234A (en) * | 1985-11-27 | 1987-08-11 | Syntex (U.S.A) Inc. | Mycophenolic acid derivatives in the treatment of inflammatory diseases, in particular rheumatoid arthritis |
| US4959387A (en) | 1986-01-23 | 1990-09-25 | Syntex (U.S.A.) Inc. | Mycophenolic acid derivatives in the treatment of rheumatoid arthritis |
| US4725622A (en) * | 1986-01-23 | 1988-02-16 | Syntex (U.S.A.) Inc. | Mycophenolic acid derivatives in the treatment of rheumatoid arthritis |
| US4753935A (en) * | 1987-01-30 | 1988-06-28 | Syntex (U.S.A.) Inc. | Morpholinoethylesters of mycophenolic acid and pharmaceutical compositions |
| US4861776A (en) * | 1987-01-30 | 1989-08-29 | Syntex (U.S.A) Inc. | Heterocyclic aminoalkyl esters of mycophenolic acid and derivatives thereof, compositions and use |
| US4727069A (en) * | 1987-01-30 | 1988-02-23 | Syntex (U.S.A.) Inc. | Heterocyclic aminoalkyl esters of mycophenolic acid, derivatives thereof and pharmaceutical compositions |
| US5665583A (en) * | 1988-08-12 | 1997-09-09 | Arch Dev Corp | Methods and materials relating to IMPDH and GMP production |
| US5283257A (en) | 1992-07-10 | 1994-02-01 | The Board Of Trustees Of The Leland Stanford Junior University | Method of treating hyperproliferative vascular disease |
| US5247083A (en) | 1992-07-10 | 1993-09-21 | Syntex (U.S.A.) Inc. | Direct esterification of mycophenolic acid |
| WO1994012184A1 (en) | 1992-11-24 | 1994-06-09 | Syntex (U.S.A.) Inc. | Use of mycophenolic acid, mycophenolate mofetil or derivate thereof to inhibit stenosis |
| US5380879A (en) * | 1994-02-18 | 1995-01-10 | Syntex (U.S.A.) Inc. | Derivatives of mycophenolic acid |
| US5444072A (en) * | 1994-02-18 | 1995-08-22 | Syntex (U.S.A.) Inc. | 6-substituted mycophenolic acid and derivatives |
| US5807876A (en) * | 1996-04-23 | 1998-09-15 | Vertex Pharmaceuticals Incorporated | Inhibitors of IMPDH enzyme |
| US20030105073A1 (en) | 2001-10-23 | 2003-06-05 | Haughan Alan Findlay | Quinolone derivatives |
-
2001
- 2001-04-19 CA CA002407370A patent/CA2407370A1/en not_active Abandoned
- 2001-04-19 EP EP01928708A patent/EP1276739A2/en not_active Withdrawn
- 2001-04-19 WO PCT/US2001/012900 patent/WO2001081340A2/en not_active Application Discontinuation
- 2001-04-19 AU AU5553801A patent/AU5553801A/en active Pending
- 2001-04-19 JP JP2001578430A patent/JP2003531205A/en active Pending
- 2001-04-19 AU AU2001255538A patent/AU2001255538B2/en not_active Ceased
- 2001-04-23 US US09/840,503 patent/US6919335B2/en not_active Expired - Lifetime
- 2001-04-24 UY UY26677A patent/UY26677A1/en not_active Application Discontinuation
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3495009A (en) * | 1966-04-26 | 1970-02-10 | Ferlux Zone Ind De Cournon D A | Pharmaceutical compositions and methods for treating inflammatory conditions with halogenated flavones |
| WO1997040028A1 (en) * | 1996-04-23 | 1997-10-30 | Vertex Pharmaceuticals Incorporated | Urea derivatives as inhibitors of impdh enzyme |
| WO1998040381A1 (en) * | 1997-03-14 | 1998-09-17 | Vertex Pharmaceuticals Incorporated | Inhibitors of impdh enzyme |
Non-Patent Citations (4)
| Title |
|---|
| CHEMICAL ABSTRACTS, vol. 116, no. 11, 16 March 1992 (1992-03-16) Columbus, Ohio, US; abstract no. 106220a, HABER, HANKA ET AL.: "Syntheis of 6-(4-pyridinyl)-substituted pyrido(2,3-d)pyrimidines and 1,8-naphthyridines" XP002187221 & J. PRAKT: CHEM., vol. 333, no. 4, - 1991 pages 637-642, -& DATABASE CHEMICAL ABSTRACTS [Online] CA 116: 106220a, XP002187224 * |
| CHEMICAL ABSTRACTS, vol. 122, no. 5, 30 January 1995 (1995-01-30) Columbus, Ohio, US; abstract no. 55978t, LEVIN, J.L. ET AL.: "6-Substituted quinazolinone angiotensin II receptor antagonists." XP002187220 -& DATABASE CHEMICAL ABSTRACTS [Online] CA 122: 55978, XP002187223 & BIOORG. MED. CHEM. LETT., vol. 4, no. 15, - 1994 pages 1819-1824, * |
| CHEMICAL ABSTRACTS, vol. 122, no. 5, 30 January 1995 (1995-01-30) Columbus, Ohio, US; abstract no. 55994v, SINGH, SARASWATI ET AL.: "Studies on imidazolinones: synthesis and antimicrobial activity of 6-(4'-substituted benzylidene-2'-methylphenyl-5'-imidazolino n-1'-yl)-2-methyl-4(3H)-quinazolinone." XP002187219 & J. INDIAN CHEM. SOC., vol. 71, no. 3, - 1994 pages 159-160, -& DATABASE CHEMICAL ABSTRACTS [Online] CA 122: 55994, XP002187222 * |
| V. HAGEN ET AL.: "Potentielle Kardiotonika 12. Mitteilung: 6-(pyrid-4-yl)-substituierte Pyrido(2,3-d)pyrimidine" PHARMAZIE., vol. 46, - 1991 pages 531-532, XP002187218 VEB VERLAG VOLK UND GESUNDHEIT. BERLIN., DD ISSN: 0031-7144 * |
Cited By (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7547802B2 (en) | 2001-06-12 | 2009-06-16 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| US7041659B2 (en) | 2001-06-12 | 2006-05-09 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| US6916848B2 (en) | 2001-06-12 | 2005-07-12 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| US8604083B2 (en) | 2001-06-12 | 2013-12-10 | Wellstat Therapeutics Corporation | Method for the treatment of metabolic disorders |
| US6858602B2 (en) | 2001-06-12 | 2005-02-22 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| US8552062B2 (en) | 2001-06-12 | 2013-10-08 | Wellstat Therapeutics Corporation | Methods for the treatment of metabolic disorders such as diabetes |
| US6924314B2 (en) | 2001-06-12 | 2005-08-02 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| US6946491B2 (en) | 2001-06-12 | 2005-09-20 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| US7012071B2 (en) | 2001-06-12 | 2006-03-14 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| US7863475B2 (en) | 2001-06-12 | 2011-01-04 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| US7045541B2 (en) | 2001-06-12 | 2006-05-16 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| US7101910B2 (en) | 2001-06-12 | 2006-09-05 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| US7329782B2 (en) | 2001-06-12 | 2008-02-12 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| US8487000B2 (en) | 2001-06-12 | 2013-07-16 | Wellstat Therapertics Corporation | Compound for the treatment of metabolic disorders |
| US7109196B2 (en) | 2001-09-26 | 2006-09-19 | Bayer Pharmaceuticals Corporation | 1,8 Naphthyridine derivatives and their use to treat diabetes and related disorders |
| WO2003035066A1 (en) * | 2001-10-23 | 2003-05-01 | Celltech R & D Limited | 2-aminoquinolone derivatives for use as impdh inhibitors |
| WO2004018462A1 (en) * | 2002-08-23 | 2004-03-04 | Celltech R & D Limited | Quinazolinone derivatives |
| WO2004041165A3 (en) * | 2002-11-01 | 2005-02-03 | Wellstat Therapeutics Corp | Compounds for the treatment of metabolic disorders |
| WO2004041165A2 (en) * | 2002-11-01 | 2004-05-21 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| US7645772B2 (en) | 2002-11-01 | 2010-01-12 | Wellstat Therapeutics Corporation | Treatment of metabolic disorders |
| EP2181704A2 (en) | 2002-12-30 | 2010-05-05 | Angiotech International Ag | Drug delivery from rapid gelling polymer composition |
| US7244738B2 (en) | 2003-07-02 | 2007-07-17 | Roche Palo Alto Llc | Arylamine-substituted quinazolinone compounds useful as alpha 1A/B adrenergic receptor antagonists |
| US7592340B2 (en) | 2003-12-04 | 2009-09-22 | Vertex Pharmaceuticals Incorporated | Quinoxalines useful as inhibitors of protein kinases |
| EP2194044A1 (en) * | 2007-09-26 | 2010-06-09 | Astellas Pharma Inc. | Quinolone derivative |
| WO2009041521A1 (en) | 2007-09-26 | 2009-04-02 | Astellas Pharma Inc. | Quinolone derivative |
| EP2194044A4 (en) * | 2007-09-26 | 2011-11-23 | Astellas Pharma Inc | Quinolone derivative |
| US8367702B2 (en) | 2007-09-26 | 2013-02-05 | Astellas Pharma Inc. | Quinolone derivative |
| JPWO2011090095A1 (en) * | 2010-01-22 | 2013-05-23 | 富山化学工業株式会社 | Heterocyclic compounds having an azole group |
| WO2011090095A1 (en) * | 2010-01-22 | 2011-07-28 | 富山化学工業株式会社 | Heterocyclic compound having azole group |
| EP2742033A4 (en) * | 2011-08-11 | 2015-07-29 | Neuprotect Pty Ltd | Flavonoid compounds, and methods of use thereof |
| CN102675200A (en) * | 2012-05-16 | 2012-09-19 | 中国药科大学 | 2-phenyl-4-carbostyril compounds with antineoplastic activity, and preparation method and usage thereof |
| WO2014014814A1 (en) * | 2012-07-16 | 2014-01-23 | Brown University | Compounds for the treatment and prevention of infections |
| US9695156B2 (en) | 2012-07-16 | 2017-07-04 | Brown University | Compounds for the treatment and prevention of infections |
| WO2021184014A1 (en) * | 2020-03-13 | 2021-09-16 | Arizona Board Of Regetns On Behalf Of The University Of Arizona | Stable reactive compositions for bioconjugation, probes, and protein labeling |
| US11820747B2 (en) | 2021-11-02 | 2023-11-21 | Flare Therapeutics Inc. | PPARG inverse agonists and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1276739A2 (en) | 2003-01-22 |
| US6919335B2 (en) | 2005-07-19 |
| AU2001255538B2 (en) | 2006-03-30 |
| AU5553801A (en) | 2001-11-07 |
| WO2001081340A3 (en) | 2002-05-23 |
| UY26677A1 (en) | 2001-11-30 |
| JP2003531205A (en) | 2003-10-21 |
| US20020040022A1 (en) | 2002-04-04 |
| CA2407370A1 (en) | 2001-11-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6919335B2 (en) | Heterocycles that are inhibitors of IMPDH enzyme | |
| AU2001255538A1 (en) | Heterocycles that are inhibitors of IMPDH enzyme | |
| US6624184B1 (en) | Amide and diamide inhibitors of IMPDH enzyme for use in treating IMPDH-associated disorders | |
| US6916809B2 (en) | Heterocyclic acridone inhibitors of IMPDH enzyme | |
| JP6258331B2 (en) | Amino-quinolines as kinase inhibitors | |
| CN111886233B (en) | Triazolone derivatives or salts thereof and pharmaceutical compositions containing the same | |
| CN111886227B (en) | Novel aryl or heteroaryl triazolone derivatives or salts thereof, or pharmaceutical compositions containing them | |
| US7008958B2 (en) | 2-substituted 5-oxazolyl indole compounds useful as IMPDH inhibitors and pharmaceutical compositions comprising same | |
| CA2983040C (en) | Heterocyclic-imidazole compounds, pharmaceutical compositions thereof, preparation method therefor and use thereof | |
| CN102816128B (en) | Containing the amino methanol derivant of heterocyclic radical and its esters and preparation method and use | |
| US6617323B2 (en) | Amino-substituted compounds useful as inhibitors of IMPDH enzyme | |
| US20200407361A1 (en) | Tricyclic compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001928708 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 2001 578430 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001255538 Country of ref document: AU Ref document number: 2407370 Country of ref document: CA |
|
| WWP | Wipo information: published in national office |
Ref document number: 2001928708 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2001255538 Country of ref document: AU |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2001928708 Country of ref document: EP |