WO2001044216A1 - Trans olefinic glucokinase activators - Google Patents
Trans olefinic glucokinase activators Download PDFInfo
- Publication number
- WO2001044216A1 WO2001044216A1 PCT/EP2000/012612 EP0012612W WO0144216A1 WO 2001044216 A1 WO2001044216 A1 WO 2001044216A1 EP 0012612 W EP0012612 W EP 0012612W WO 0144216 A1 WO0144216 A1 WO 0144216A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- methanesulfonyl
- compound
- mmol
- reaction mixture
- Prior art date
Links
- SQKAZYIFEWJVIG-HWQQAZHXSA-N CCC/C=C(/C=C\C(\Br)=C/C)\NC(/C(/c(cc1C(F)(F)F)ccc1S(C)(=O)=O)=C/C1CCCCC1)=O Chemical compound CCC/C=C(/C=C\C(\Br)=C/C)\NC(/C(/c(cc1C(F)(F)F)ccc1S(C)(=O)=O)=C/C1CCCCC1)=O SQKAZYIFEWJVIG-HWQQAZHXSA-N 0.000 description 1
- BCHXPOUTSCJBCH-FOWTUZBSSA-N CS(c(cc1)ccc1/C(/C(Nc1ncc[s]1)=O)=C\C1CCCC1)(=O)=O Chemical compound CS(c(cc1)ccc1/C(/C(Nc1ncc[s]1)=O)=C\C1CCCC1)(=O)=O BCHXPOUTSCJBCH-FOWTUZBSSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/75—Amino or imino radicals, acylated by carboxylic or carbonic acids, or by sulfur or nitrogen analogues thereof, e.g. carbamates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C275/00—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups
- C07C275/46—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups containing any of the groups, X being a hetero atom, Y being any atom, e.g. acylureas
- C07C275/48—Y being a hydrogen or a carbon atom
- C07C275/50—Y being a hydrogen or an acyclic carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/44—Sulfones; Sulfoxides having sulfone or sulfoxide groups and carboxyl groups bound to the same carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/38—Nitrogen atoms
- C07D277/44—Acylated amino or imino radicals
- C07D277/46—Acylated amino or imino radicals by carboxylic acids, or sulfur or nitrogen analogues thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/06—Systems containing only non-condensed rings with a five-membered ring
- C07C2601/08—Systems containing only non-condensed rings with a five-membered ring the ring being saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
Definitions
- Glucokinase is one of four hexokinases that are found in mammals [Colowick, S.P., in The Enzymes, Vol. 9 (P. Boyer, ed.) Academic Press, New York, NY, pages 1-48, 1973].
- the hexokinases catalyze the first step in the metabolism of glucose, i.e., the conversion of glucose to glucose-6-phosphate.
- Glucokinase has a limited cellular distribution, being found principally in pancreatic ⁇ -cells and liver parenchymal cells.
- GK is a rate-controlling enzyme for glucose metabolism in these two cell types that are known to play critical roles in whole-body glucose homeostasis [Chipkin, S.R., Kelly, K.L., and Ruderman, N.B. in Joslin's Diabetes (C.R. Khan and G.C. Wier, eds.), Lea and Febiger, Philadelphia, PA, pages 97-115, 1994].
- concentration of glucose at which GK demonstrates half-maximal activity is approximately 8 mM.
- the other three hexokinases are saturated with glucose at much lower concentrations ( ⁇ 1 mM).
- GK does indeed play a critical role in whole- body glucose homeostasis.
- Animals that do not express GK die within days of birth with severe diabetes while animals overexpressing GK have improved glucose tolerance (Grupe, A., Hultgren, B., Ryan, A. et al, Cell 83, 69-78, 1995; Ferrie, T., Riu, E., Bosch, F. et al, FASEB J., 10, 1213-1218, 1996).
- An increase in glucose exposure is coupled through GK in ⁇ -cells to increased insulin secretion and in hepatocytes to increased glycogen deposition and perhaps decreased glucose production.
- GK Gkinase activators
- Glucokinase activators will increase the flux of glucose metabolism in ⁇ -cells and hepatocytes, which will be coupled to increased insulin secretion. Such agents would be useful for treating type II diabetes.
- This invention provides a compound, comprising an amide of the formula:
- R 1 and R are independently hydrogen, halo, amino, nitro, perfluoro-lower alkyl, lower alkyl thio, perfluoro-lower alkyl thio, lower alkyl sulfonyl, perfluoro-lower alkyl sulfonyl, lower alkyl sulfonyl methyl or lower alkyl sulfinyl;
- R is -(CH 2 ) m -R 3 or lower alkyl containing from 2 to 4 carbon atoms;
- R 3 is cycloalkyl having from 3 to 8 carbon atoms
- R 4 is -C-NHR 7
- n 0 or 1
- n 0, 1, 2, 3 or 4;
- R 7 is hydrogen or lower alkyl
- ⁇ denotes a trans configuration across the double bond
- the compounds of formula I are glucokinase activators are useful for increasing insulin secretion in the treatment of type II diabetes.
- This invention provides a compound, comprising an amide of the formula:
- R and R are independently hydrogen, halo, amino, nitro, perfluoro-lower alkyl, lower alkyl thio, perfluoro-lower alkyl thio, lower alkyl sulfinyl, lower alkyl sulfonyl, lower alkyl sulfonyl methyl or perfluoro-lower alkyl sulfonyl;
- R is -(CH 2 ) m -R 3 or lower alkyl containing from 2 to 4 carbon atoms;
- R is cycloalkyl having from 3 to 8 carbon atoms
- n 0 or 1
- n O, 1, 2, 3 or 4;
- R 7 is hydrogen or lower alkyl
- ⁇ denotes a trans configuration across the double bond
- the present invention also relates to a pharmaceutical composition comprising a compound of formula I and a pharmaceutically acceptable carrier and/or adjuvant. Furthermore the present invention relates to the use of such compounds for the preparation of medicaments for the treatment of type II diabetes. The present invention also relates to processes for the preparation of the compounds of formula I. In addition, the present invention relates to a method for the therapeutic treatment of type II diabetes, which method comprises administering a compound of formula I to a human being or an animal.
- lower alkyl includes both straight chain and branched chain alkyl groups having from 1 to 7 carbon atoms, such as methyl, ethyl, propyl, isopropyl, preferably methyl and ethyl, most preferably methyl.
- halogen or halo designates all four halogens, i.e. fluorine, chlorine, bromine and iodine.
- perfluoro-lower alkyl means any lower alkyl group wherein all of the hydrogens of the lower alkyl group are substituted or replaced by fluoro.
- perfluoro-lower alkyl groups are trifluoromethyl, pentafluoroethyl, heptafluoropropyl, etc., most preferred is trifluoromethyl.
- aryl signifies mononuclear aromatic hydrocarbon groups such as phenyl, tolyl, etc. which can be unsubstituted or substituted in one or more positions with halogen, nitro, lower alkyl, or lower alkoxy substituents and polynuclear aryl groups, such as naphthyl, anthryl, and phenanthryl, which can be unsubstituted or substituted with one or more of the aforementioned groups.
- Preferred aryl groups are the substituted and unsubstituted mononuclear aryl groups, particularly phenyl.
- lower alkoxy includes both straight chain and branched chain alkoxy groups having from 1 to 7 carbon atoms, such as methoxy, ethoxy, propoxy, isopropoxy, preferably methoxy and ethoxy.
- arylalkyl denotes an alkyl group, preferably lower alkyl, in which one of the hydrogen atoms can be replaced by an aryl group. Examples of arylalkyl groups are benzyl, 2-phenylethyl, 3-phenylpropyl, 4-chlorobenzyl, 4- methoxybenzyl and the like.
- lower alkanoic acid denotes lower alkanoic acids containing from 2 to 7 carbon atoms such as propionic acid, acetic acid and the like.
- lower alkanoyl denotes monovalent alkanoyl groups having from 2 to 7 carbon atoms such as propionoyl, acetyl and the like.
- aromatic acids denotes aryl alkanoic acids where aryl is as defined above and alkanoic contains from 1 to 6 carbon atoms.
- aroyl denotes aroic acids wherein aryl is as defined hereinbefore, with the hydrogen group of the COOH moiety removed. Among the preferred aroyl groups is benzoyl.
- hydrolyzable ester or ether protecting groups designates any ester or ether conventionally used for protecting carboxylic acids or alcohols which can be hydrolyzed to yield the respective hydroxyl or carboxyl group.
- ester groups useful for those purposes are those in which the acyl moieties are derived from a lower alkanoic, aryl lower alkanoic, or lower alkane dicarboxcyclic acid.
- activated acids which can be utilized to form such groups are acid anhydrides, acid halides, preferably acid chlorides or acid bromides derived from aryl or lower alkanoic acids.
- Example of anhydrides are anhydrides derived from monocarboxylic acid such as acetic anhydride, benzoic acid anhydride, and lower alkane dicarboxcyclic acid anhydrides, e.g. succinic anhydride as well as chloro formates e.g. trichloro, ethylchloro formate being preferred.
- a suitable ether protecting group for alcohols are, for example, the tetrahydropyranyl ethers such as 4-methoxy-5,6-dihydroxy-2H-pyranyl ethers.
- aroylmethylethers such as benzyl, benzhydryl or trityl ethers or ⁇ -lower alkoxy lower alkyl ethers, for example, methoxymethyl or allylic ethers or alkyl silylethers such as trimethylsilylether.
- amino protecting group designates any conventional amino protecting group which can be cleaved to yield the free amino group.
- the preferred protecting groups are the conventional amino protecting groups utilized in peptide synthesis. Especially preferred are those amino protecting groups which are cleavable under mildly acidic conditions from about pH 2.0 to 3. Particularly preferred amino protecting groups such as t-butoxycarbonyl carbamate, benzyloxycarbonyl carbamate, 9-flurorenylmethyl carbamate.
- the heteroaromatic ring defined by R can be an unsubstituted or mono- substituted five- or six-membered heteroaromatic ring having from 1 to 2 heteroatoms selected from the group consisting of nitrogen, or sulfur and connected by a ring carbon to the amine of the amide group shown.
- the heteroaromatic ring contains a first nitrogen heteroatom adjacent to the connecting ring carbon atom and if present, the other heteroatoms can be sulfur, or nitrogen.
- the preferred heteroaromatic rings are pyridinyl, pyrimidinyl and thiazolyl, most preferred are pyridinyl and thiazolyl.
- heteroaromatic rings which constitute R 4 are connected via a ring carbon atom to the amide group to form the amides of formula I.
- the ring carbon atom of the heteroaromatic ring which is connected via the amide linkage to form the compound of formula I cannot contain any substituent.
- the preferred rings are those which contain a nitrogen heteroatom adjacent to the connecting ring carbon and a second heteroatom adjacent to the connecting ring carbon or adjacent to said first heteroatom.
- pharmaceutically acceptable salts include any salt with both inorganic or organic pharmaceutically acceptable acids such as hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid, citric acid, formic acid, maleic acid, acetic acid, succinic acid, tartaric acid, methanesulfonic acid, para-toluene sulfonic acid and the like.
- pharmaceutically acceptable salts also includes any pharmaceutically acceptable base salt such as amine salts, trialkyl amine salts and the like. Such salts can be formed quite readily by those skilled in the art using standard techniques.
- the compound of formula I of this invention constitutes two preferred species, i.e., the compound of formula
- R is an unsubstituted or a mono-substituted five- or six-membered heteroaromatic ring connected by a ring carbon atom to the amine group shown, which five- or six-membered heteroaromatic ring contains from 1 to 2 heteroatoms selected from the group consisting of sulfur or nitrogen, with one heteroatom being nitrogen which is adjacent to the connecting ring carbon atom; said mono- substituted heteroaromatic ring being monosubstituted at a position on a ring carbon atom other than adjacent to said connecting carbon atom with a substituent selected from the group consisting of halo or
- n 0, 1, 2, 3 or 4;
- R is hydrogen or lower alkyl.
- R can be lower alkyl containing from 2 to 4 carbon atoms.
- R can be -(CH 2 ) m -R 3 where R and m are as defined above.
- Preferred heterocyclic residues R " are cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl, more preferred are cyclopentyl, cyclohexyl and cycloheptyl.
- R 3 is cyclopentyl, in another preferred embodiment R is cyclohexyl.
- m is 1, in another preferable embodiment m is 0.
- Preferred heteroaromatic rings R 1 ' in accordance with the present invention are unsubstituted or mono-substituted pyridinyl or thiazolyl.
- heteroaromatic ring R is unsubstituted or mono-substituted pyridinyl
- heteroaromatic ring R 11 is unsubstituted or mono-substituted thiazolyl.
- the heteroaromatic ring R is either unsubstituted, mono-substituted with halogen or mono- substituted with -(CH 2 ) n -C(O)-OR 7 , wherein n and R 7 are as defined above.
- Preferred substituents R 1 and R 2 are independently selected from the group consisting of hydrogen, halo, nitro, perfluoro lower alkyl, lower alkyl sulfonyl and lower alkyl sulfonyl methyl.
- one of R 1 and R is halo, lower alkyl sulfonyl or lower alkyl sulfonyl methyl and the other is hydrogen, halo, nitro or perfluoro lower alkyl.
- one of R 1 and R is lower alkyl sulfonyl and the other is hydrogen, halo, nitro or perfluoro lower alkyl.
- Preferred residue R is lower alkyl.
- R can be a cycloalkyl group which contains from 3 to 8 carbon atoms, preferably cyclohexyl (compound I-Al).
- compound I-Al cyclohexyl
- the various embodiments of the cyclohexyl amides of compound I-Al are included, those compounds where one of R 1 and R 2 is hydrogen, halo, lower alkyl sulfonyl or perfluoro lower alkyl and the other of said R 1 and R 2 is halo, lower alkyl sulfonyl or perfluoro lower alkyl and particularly those compounds one of R 1 and R 2 is hydrogen or lower alkyl sulfonyl or perfluoro lower alkyl sulfonyl and the other is lower alkyl sulfonyl or perfluoro lower alkyl.
- Another embodiment of the compound of formula I-A are those compounds where R is a lower alkyl group containing from 2 to 4 carbon atoms (the compounds of formula I-A2).
- the compounds of formula I-A2 are those compounds where one of R and R is hydrogen, halo, lower alkyl sulfonyl or perfluoro lower alkyl and the other of said R 1 and R 2 is halo, lower alkyl sulfonyl or perfluoro lower alkyl.
- An embodiment of the compound of formula I-B are those compounds where R u is an unsubstituted or mono-substituted thiazole ring.
- R can be a lower alkyl group containing from 2 to 4 carbon atoms, (compound I-Bl)
- the compounds of the formula I-Bl are those compounds where one of R 1 or R 2 is hydrogen, lower alkyl sulfonyl, lower alkyl sulfonyl methyl, perfluoro lower alkyl, halo, nitro and the other of said R 1 or R 2 is lower alkyl sulfonyl, lower alkyl sulfonyl methyl, perfluoro lower alkyl, halo or nitro and preferably those compounds of formula IB-1 where one of R and R is hydrogen, lower alkyl sulfonyl and the other of said R 1 and R 2 is lower alkyl
- compounds of formula I-B2 are those compounds where the cycloalkyl group is cyclopentyl (IB-2a).
- the embodiment of compounds I-B2(a) are those compounds of formula IB-2(a) where R 11 is an unsubstituted thiazole ring (compounds IB-2a(l)).
- the compound IB-2a(l) are those compounds where one of said R 1 and R is hydrogen, lower alkyl sulfonyl, lower alkyl sulfonyl methyl, perfluoro lower alkyl, halo or nitro and the other of said R 1 and R 2 is lower alkyl sulfonyl, lower alkyl sulfonyl methyl, perfluoro lower alkyl, halo or nitro and particularly preferred embodiments of the compounds IB-2(a)( l) are those compounds wherein:
- R or R is lower alkyl sulfonyl and the other is hydrogen, nitro, lower alkyl sulfonyl, halo or perfluoro lower alkyl;
- R 1 and R are halo, hydrogen or perfluoro lower alkyl and the other is perfluoro lower alkyl or halogen;
- R 1 and R 2 are lower alkyl sulfonyl methyl and the other is hydrogen, lower alkyl sulfonyl methyl or halogen.
- compound of the formula IB-2a are those compounds where R is a mono-substituted thiazolyl ring which includes compounds where R is a halo substituted thiazole ring (compounds of the formula IB-2(a)(2)).
- compounds of the formula IB-2(a)(2) are those compounds where one of R 1 and R" is lower alkyl sulfonyl, hydrogen or halo and the other is lower alkyl sulfonyl halo or halo.
- compounds IB-2 are those compounds where R is cyclohexyl (compounds IB-2(b)).
- compounds IB-2(b) are those compounds where R is an unsubstituted thiazolyl ring (compound IB-2(b)(l).
- preferred compounds of IB-2(b) are those compounds where one of R or R is hydrogen, lower alkyl sulfonyl, lower alkyl sulfonyl methyl, perfluoro lower alkyl, halo, nitro and the other is lower alkyl sulfonyl, lower alkyl sulfonyl methyl, perfluoro lower alkyl, halo or nitro and particularly
- R l or R 2 is lower alkyl sulfonyl and the other is hydrogen, nitro, lower alkyl sulfonyl, halo or perfluoro lower alkyl;
- R 1 and R are halo, hydrogen or perfluoro lower alkyl and the other is perfluoro lower alkyl or halogen;
- R 1 and R 2 are lower alkyl sulfonyl methyl and the other is hydrogen, lower alkyl sulfonyl methyl or halogen.
- R 11 is a mono-substituted thiazolyl ring and particularly a halo substituted ring (compound IB-
- compounds IB-2(b)(2) are those compounds where one or R 1 and R is lower alkyl sulfonyl and the other is halogen, perfluoro lower alkyl or hydrogen.
- Another embodiment of the compound IB-2 are those compounds where R is cycloheptyl (compound IB-2(d)) or cyclooctyl (compound IB-2(e)).
- An embodiment of the compounds (compound IB-2(d) and compound IB-2(e)) are those compounds where R 11 is unsubstituted thiazolyl (compounds IB-2(d)(l) and IB-2(e)(l)) respectively.
- the compounds of IB-2(d)(l) and IB-2(e)(l) that are preferred are those compounds where one of R and R is lower alkyl, sulfonyl, hydrogen, halogen or perfluoro lower alkyl and the other is lower alkyl sulfonyl, halogen or perfluoro lower alkyl.
- R 11 is a mono-substituted thiazolyl ring and the substitution is a halo group.
- one of R 1 and R 2 can be hydrogen, lower alkyl sulfonyl, perfluoro lower alkyl or halogen and the other can be halogen, lower alkyl sulfonyl or perfluoro lower alkyl.
- R 11 is a monosubstituted thiazolyl, the substitution can be
- n and R are as above.
- these compounds are one of R 1 and R 2 in these compounds can be lower alkyl sulfonyl and the other of said R 1 and R 2 is lower alkyl sulfonyl or hydrogen.
- Another class of compounds of formula IB are those compounds where R is -CH 2 -R " and R 3 is as above.
- R is a -CH 2 -cyclohexyl group
- compounds IB-3 include compounds where R 1 1 is a substituted or unsubstituted thiazolyl ring and particularly those compounds where R 11 is an unsubstituted thiazolyl ring and where the substitution on the thiazolyl ring is: o
- n and R 7 are as above.
- R 1 and R 2 are lower alkyl sulfonyl and the other is lower alkyl sulfonyl or hydrogen are preferred.
- R can be cyclopentyl.
- An embodiment of this class includes compounds where R 11 is unsubstituted or mono-substituted pyridinyl ring.
- a preferred embodiment of this class is those compounds where one of R 1 and R 2 is hydrogen, lower alkyl sulfonyl or halogen and the other of said R 1 and R is lower alkyl sulfonyl or halogen.
- the compounds of formula IA and IB can be prepared from the following compounds of the formula:
- the compounds of formula IA and IB are prepared from the compounds of formula V via the following reaction scheme:
- R, R 1 , R 2 , R 7 and R u are as above;
- R 5 taken together with its attached oxygen atom forms a hydrolyzable acid protecting group and X is halogen.
- the compound of formula V or XIX wherein one of R 1 and R 2 is nitro, thio, amino, halo, and the other is hydrogen are known materials.
- the amino substituted compounds of formula V or XIX can be converted to other substituents either before or after conversion to the compounds of formula IA or IB.
- the amino groups can be diazotized to yield the corresponding diazonium compound, which in situ can be reacted with the desired lower alkyl thiol, perfluoro-lower alkyl thiol (see for example, Baleja, J.D. Synth. Comm.
- the corresponding halo substituted compounds of formula V or XIX can be used as starting materials.
- Any conventional method of converting an aromatic halo group to the corresponding perfluoro lower alkyl group see for example, Katayama, T.; Umeno, M., Chem. Lett. 1991, 2073; Reddy, G. S.; Tarn., Organometallics, 1984, 3, 630; Novak, J.; Salemink, C. A., Synthesis, 1983, 7, 597; Eapen, K. C.; Dua, S. S.; Tamboroski, C, /. Org. Chem.
- the compound of formula V or XIX where both R and R are amine groups can be used to prepare the corresponding compound of formula V or XIX where both R and R 2 are iodine or bromine via a diazotization reaction. Any conventional method of converting amino group to an iodo or bromo group (see for example, Lucas, H. J.;
- R 1 and R 2 are amino can be used as starting material. Any conventional method of converting aryl amino group to aryl thioalkyl group can be utilized to effect this conversion. If it is desired to produce compound of formula V or XIX where R 1 and R 2 are lower alkyl sulfonyl or lower perfluoro alkyl sulfonyl, the corresponding compounds of formula V or XIX where R 1 and R 2 are lower alkyl thio or perfluoro-lower alkyl thio can be used as starting material. Any conventional method of oxidizing alkyl thio substituents to sulfones can be utilized to effect this conversion.
- the compound of formula V or XIX where one of R 1 and R is nitro and the other is amino can be used as a starting material.
- the chloro substituent on the phenyl ring can be converted to an iodo substituent (see for example, Bunnett, J. F.; Conner, R. M.; Org. Synth. Coll Vol V, 1973, 478; Clark, J. H.; Jones, C. W. /. Chem. Soc. Chem. Commun.
- R and/or R are lower alkyl sulfonyl methyl in the compound of formula I
- the methyl groups in these compounds can be brominated by any conventional means for brominating the methyl groups on phenyl rings.
- This brominated compound is then treated with the sodium salt of a lower alkyl thiol (such as sodium thiomethoxide) to form the lower alkyl thio methyl compound.
- a lower alkyl thiol such as sodium thiomethoxide
- any conventional method of oxidizing lower alkyl thio substituents to sulfones such as described above, can be utilized to effect this conversion.
- R 1 and R 2 can be added to the ring after formation of the compounds of formulas IA and IB. Hence, all of the reactions described to produce various sustituents of R 1 and R in the compound of formula I can be carried out on the compounds of formulas IA and IB after their formation.
- the compounds of formula IA and IB are prepared from the compound of formula V or XIX as set forth in Schemes 1 or 2.
- the compound of formula V is reacted with oxalyl chloride wherein the free hydrolyzable organic acid group of the oxalyl chloride is protected by any conventional acid protecting groups.
- the preferred acid protecting groups are hydrolyzable esters of oxalyl chloride.
- the protecting group is formed by R 5 .
- the reaction of the protected oxalyl chloride with the compound of formula V to produce the compound of formula VI is carried out via a Friedel-Crafts reaction. In carrying out this reaction, any of the conditions conventional in carrying out a Friedel-Crafts reaction can be utilized.
- R 1 and R 2 cannot be a nitro group.
- R 1 and R 2 can be an amino group.
- this amino group must be protected with a conventional hydrolyzable amino protecting group prior to carrying out the reaction. At some later stage in the reaction, these amino groups can be removed and the amino groups converted to nitro groups as described hereinbefore.
- the compound of formula VI can be reacted with a triphenylphosphonium halide salt of formula IX via a Wittig reaction to produce the compound of formula VII.
- a Wittig reaction any of the conditions conventional in carrying out a Wittig reaction can be utilized to effect these synthesis of the compound of formula VI with the compound of formula IX to produce the compound of formula VII.
- the compound of formula VII is formed as a mixture of cis and trans isomers about the double bond formed through the Wittig reaction.
- the mixture of cis and trans isomers of the compound of formula VII is directly hydrolyzed to the compound of formula VIII. In this hydrolysis reaction, the compound of formula VIII is produced as predominantly the trans isomer in this mixture.
- the trans isomer produced by this hydrolysis reaction is formed as a solid whereas the cis isomer is formed as an oily material.
- This crystallization can take place at this stage or at later stages of the reaction in the formation of the compounds of formula IA or IB. Therefore, by this procedure, the compound of formula IA and IB can be produced in pure trans form substantially free of the corresponding cis isomer.
- R ' is as above
- This reaction can be carried out by converting the compound of formula VII to the corresponding free acid by removing the protecting group R 5 to form the carboxylic acid.
- the carboxylic acid of formula VIII can be converted to the corresponding amide by converting the acid to the acid chloride and thereafter reacting this acid chloride with ammonia. Conditions which are conventional for converting an acid to an acid chloride can be utilized in this procedure.
- This acid chloride is then reacted with an alkyl isocyanate of formula XV to form the urea adduct of formula IA. Any conventional method of reacting an alkyl isocyanate with an amide to form a urea linkage utilize the compound of formula IA.
- the compound of formula IA can be formed as a mixture of cis and trans provided the compound of formula VII has not been purified. If desired, purification can take place with respect to the compound of formula IA to produce the compound of formula IA as the all-trans isomer free of the cis isomer. In the same manner as the compound of formula IB or the compound of formula VIII can be purified, the compound of formula IA can be purified to produce this all trans isomer.
- the compound of formula VII can also be produced by the following reaction scheme 2.
- This reaction scheme is applicable for producing compounds of formula IA or IB where one or both R 1 and R 2 is nitro.
- the coupling reaction can be easily carried out with any of the designated R 1 and R" groups, particularly those where R 1 and R is nitro.
- R, R 1 , R 2 and ⁇ are as above.
- the compound of formula XI can be generated in situ from either the corresponding organomagnesium reagent or organozinc reagent and soluble copper reagent (CuCN and 2LiCl) (see for example, Knochel, P.; Singer, R.D, Chem. Rev. 1993, 93, 2117). Then, the compound of formula XI is added to the compound of formula XVII in a 1,4-conjugate addition in a highly regio- and stereoselective manner to obtain a vinylcopper intermediate, which upon iodolysis with iodine produced the compound of formula XVIII in which the R and iodide are in syn relationship to each other.
- organomagnesium reagent or organozinc reagent and soluble copper reagent CuCN and 2LiCl
- medicaments containing a compound of formula I are also an object of the present invention, as is a process for the manufacture of such medicaments, which process comprises bringing one or more compounds of formula I and, if desired, one or more other therapeutically valuable substances into a galenical administration form.
- compositions may be administered orally, for example in the form of tablets, coated tablets, dragees, hard or soft gelatine capsules, solutions, emulsions or suspensions.
- Administration can also be carried out rectally, for example using suppositories; locally or percutaneously, for example using ointments, creams, gels or solutions; or parenterally, e.g. intravenously, intramuscularly, subcutaneously, intrathecally or transdermally, using for example injectable solutions.
- administration can be carried out sublingually or as an aerosol, for example in the form of a spray.
- the compounds of the present invention may be admixed with pharmaceutically inert, inorganic or organic excipients.
- suitable excipients for tablets, dragees or hard gelatine capsules include lactose, maize starch or derivatives thereof, talc or stearic acid or salts thereof.
- suitable excipients for use with soft gelatine capsules include for example vegetable oils, waxes, fats, semi-solid or liquid polyols etc.; according to the nature of the active ingredients it may however be the case that no excipient is needed at all for soft gelatine capsules.
- excipients which may be used include for example water, polyols, saccharose, invert sugar and glucose.
- excipients which may be used include for example water, alcohols, polyols, glycerine, and vegetable oils.
- excipients which may be used include for example natural or hardened oils, waxes, fats and semi-solid or liquid polyols.
- the pharmaceutical compositions may also contain preserving agents, solubilising agents, stabilising agents, wetting agents, emulsifiers, sweeteners, colorants, odorants, salts for the variation of osmotic pressure, buffers, coating agents or antioxidants. As mentioned earlier, they may also contain other therapeutically valuable agents. It is a prerequisite that all adjuvants used in the manufacture of the preparations are non-toxic.
- Preferred forms of use are intravenous, intramuscular or oral administration, most preferred is oral administration.
- the dosages in which the compounds of formula (I) are administered in effective amounts depend on the nature of the specific active ingredient, the age and the requirements of the patient and the mode of application. In general, dosages of about 1-100 mg/kg body weight per day come into consideration.
- the assay was conducted at 25° C in a flat bottom 96-well tissue culture plate from Costar (Cambridge, MA) with a final incubation volume of 120 ⁇ l.
- the incubation mixture contained: 25 mM Hepes buffer (pH, 7.1), 25 mM KC1, 5 mM D-glucose, ImM ATP, 1.8 mM NAD, 2 mM MgCl 2) 1 ⁇ M sorbitol-6-phosphate, 1 mM dithiothreitol, test drug or 10% DMSO, 1.8 unit/ml G6PDH, and GK (see below).
- Glucokinase Activator in vivo Screen Protocol C57BL/6J mice are orally dosed via gavage with Glucokinase (GK) activator at 50 mg/kg body weight following a two hour fasting period. Blood glucose determinations are made five times during the six hour post-dose study period.
- GK Glucokinase
- GK activators are formulated at 6.76 mg/ml in Gelucire vehicle
- mice are dosed orally with 7.5 ⁇ l formulation per gram of body weight to equal a 50 mg/kg dose.
- a pre dose (time zero) blood glucose reading is acquired by snipping off a small portion of the animals tail ( ⁇ lmm) and collecting 15 ⁇ l blood into a heparinized capillary tube for analysis.
- additional blood glucose readings are taken at 1, 2, 4 and 6 hours post dose from the same tail wound. Results are interpreted by comparing the mean blood glucose values of six vehicle treated mice with six GK activator treated mice over the six hour study duration. Compounds are considered active when they exhibit a statistically significant (p ⁇ 0.05) decrease in blood glucose compared to vehicle for two consecutive assay time points.
- the resulting reaction mixture was again cooled back to -70°C and then slowly treated with methyl propiolate (1.52 g, 18 mmol).
- the reaction mixture was stirred for 4 h at -40°C to -30°C and then cooled to -70°C to -60°C, at which time, the reaction mixture was treated slowly with a solution of iodine (6.86 g, 27 mmol) in dry tetrahydrofuran (20 mL). After addition of the iodine solution, the cooling bath was removed, and the reaction mixture was allowed to warm to 25°C where it was stirred for 1 h.
- the reaction mixture was then poured into a solution consisting of a saturated aqueous ammonium chloride solution (90 mL) and ammonium hydroxide (10 mL), and the organic compound was extracted into diethyl ether (3 x 50 mL).
- the combined organic extracts were successively washed with a saturated aqueous sodium thiosulfate solution (1 x 100 mL) and a saturated aqueous sodium chloride solution (1 x 100 mL).
- the organic layer was dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo.
- reaction mixture was then treated dropwise with a solution of (E)-2-iodo-pentenoic acid methyl ester (2.9 g, 12 mmol) in dry tetrahydrofuran (3 mL) over 3 min.
- the reaction mixture was then stirred at 40-45°C for 1 h and then stirred overnight at 25°C.
- the reaction mixture was then diluted with dry tetrahydrofuran (10 mL), and the stirring was stopped to allow the excess zinc dust to settle down ( ⁇ 2 h).
- reaction mixture was then cooled to 25°C and then poured into a saturated aqueous ammonium chloride solution (100 mL), and the organic compound was extracted into ethyl acetate (3 x 50 mL).
- the combined organic extracts were washed with a saturated aqueous sodium chloride solution (1 x 100 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo.
- reaction mixture was then treated with 2-aminothiazole (829 mg, 8.28 mmol), and the resulting suspension was stirred for 15 h at 25°C.
- the reaction mixture was then concentrated in vacuo to remove methylene chloride, and the residue was diluted with ethyl acetate (100 mL) and a IN aqueous hydrochloric acid solution (100 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (1 x 50 mL).
- the resulting reaction mixture was again cooled back to -70°C and then slowly treated with methyl propiolate (1.52 g, 18 mmol).
- the reaction mixture was stirred for 4 h at -40°C to -30°C and then cooled to -70°C to -60°C, at which time, the reaction mixture was treated slowly with a solution of iodine (6.86 g, 27 mmol) in dry tetrahydrofuran (20 mL). After addition of the iodine solution, the cooling bath was removed, and the reaction mixture was allowed to warm to 25°C where it was stirred for 1 h.
- the reaction mixture was then poured into a solution consisting of a saturated aqueous ammonium chloride solution (90 mL) and ammonium hydroxide (10 mL), and the organic compound was extracted into diethyl ether (3 x 50 mL).
- the combined organic extracts were successively washed with a saturated aqueous sodium thiosulfate solution (1 x 100 mL) and a saturated aqueous sodium chloride solution ( 1 x 100 mL).
- the organic layer was dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo.
- reaction mixture was then treated dropwise with a solution of (E)-2-iodo-4-methyl-pentenoic acid methyl ester (2.22 g, 8.7 mmol) in dry tetrahydrofuran (3 mL) over 2 min.
- the reaction mixture was then stirred at 40-45°C for 1 h and then stirred overnight at 25°C.
- the reaction mixture was then diluted with dry tetrahydrofuran (8 mL), and the stirring was stopped to allow the excess zinc dust to settle down ( ⁇ 2 h).
- reaction mixture was then cooled to 25°C and then poured into a saturated aqueous ammonium chloride solution ( 100 mL), and the organic compound was extracted into ethyl acetate (3 x 50 mL). The combined organic extracts were washed with a saturated aqueous sodium chloride solution (1 x 100 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo.
- reaction mixture was then treated with 2- aminothiazole (636 mg, 6.36 mmol), and the resulting suspension was stirred for 15 h at 25°C.
- the reaction mixture was then concentrated in vacuo to remove methylene chloride, and the residue was diluted with ethyl acetate (100 mL) and a IN aqueous hydrochloric acid solution (100 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (1 x 50 mL).
- reaction mixture was then cooled back to 0°C and then slowly treated with ice/water (800 mL) over 1 h.
- the reaction mixture was then transferred to a separatory funnel in one-liter portions.
- the one-liter portions were continuously extracted with methylene chloride until the aqueous layer showed absence of product by thin layer chromatography.
- the resulting reaction mixture was stirred at 0°C for 30 min and then allowed to warm to 25°C where it was stirred for 6 h.
- the reaction mixture was concentrated in vacuo to remove tetrahydrofuran and then diluted with diethyl ether (1 L). A solid began to precipitate, and the reaction mixture was allowed to sit at 25°C for 1 h. The solid was filtered and washed well with diethyl ether.
- the resulting two-layer filtrate was transferred to a separatory funnel, and the layers ⁇ vere separated.
- the aqueous layer was further extracted with diethyl ether (1 x 500 mL).
- the combined organic layers were washed with a saturated aqueous sodium chloride solution (1 x 500 mL), dried over sodium sulfate, filtered, and concentrated in vacuo.
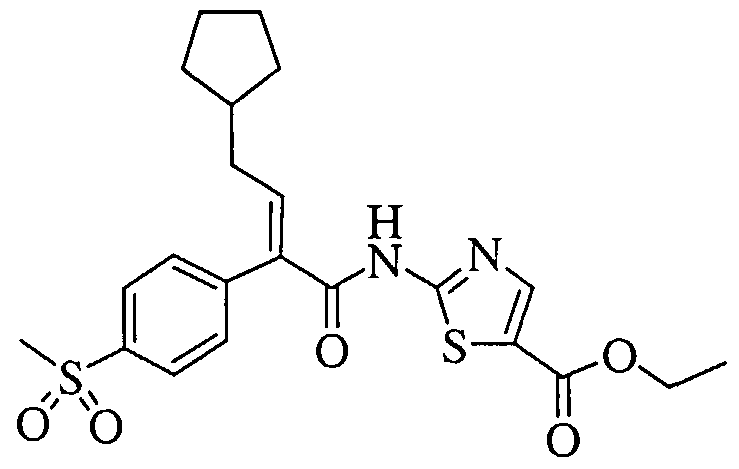
- reaction mixture was then cooled back to -25°C and then treated with a solution of the (E)-3-cyclopentyl-2-(4-methanesulfonyl-phenyl)-acrylic acid (41.18 g, 139.88 mmol) in dry tetrahydrofuran (300 mL) over a period of 10 min.
- the reaction mixture was allowed to warm to 0°C where it was stirred for 1 h. During the time at 0°C, the thick solids partially dissolved, leaving a fine suspension of white solids.
- reaction mixture After 1 h at 25°C, the reaction mixture was cooled to -45°C. The reaction mixture was then treated with a precooled (-45°C) solution of 2-aminothiazole (44.97 g, 449.02 mmol) and triethylamine (62.6 mL, 449.02 mmol) in dry tetrahyfdrofuran (280 mL) via cannulation over a period of 10 min. The reaction mixture changed from a white suspension to a light brown color after the complete addition of the 2- aminothiazole/triethylamine solution. The reaction mixture was then allowed to warm to 0°C over 15 min using an ice/water bath.
- reaction mixture was allowed to warm to 25°C over a period of 30 min and then stirred at 25°C for 1 h. After this time, the reaction mixture was cooled to -25°C and then treated with a 1M aqueous citric acid solution (250 mL), and the resulting reaction mixture was allowed to warm to 25°C.
- the reaction mixture was filtered through a plug of celite to remove the precipitated solids. The celite was washed well with ethyl acetate until the washings showed the absence of product by thin layer chromatography. The two-layer filtrate was transferred to a separatory funnel, and the layers were separated. The aqueous layer was extracted with ethyl acetate (1 x 500 mL).
- the organic layer was concentrated in vacuo to remove tetrahydrofuran, and the resulting residue was diluted with ethyl acetate (700 mL).
- the combined organic layers were washed successively with a 2M aqueous sodium hydrogen sulfate solution (3 x 200 mL), a saturated aqueous sodium chloride solution (1 x 200 mL), a 10% aqueous potassium carbonate solution (4 x 200 L), and a saturated aqueous sodium chloride solution (1 x 300 mL).
- the organic layer was then dried over magnesium sulfate, filtered, and concentrated in vacuo.
- reaction mixture was then treated dropwise with a solution of cyclohexyl iodide (21 g, 100 mmol) in dry tetrahydrofuran (30 mL) over 15 min. During the addition, the temperature rose to 60°C. The reaction mixture was then stirred for 3 h at 40-45°C. The reaction mixture was then cooled to 25°C and diluted with dry tetrahydrofuran (60 mL). The stirring was stopped to allow the excess zinc dust to settle down ( ⁇ 3 h).
- the resulting reaction mixture was stirred for 15 h at -70°C to -50°C and then slowly treated with a solution of iodine (34.26 g, 135 mmol) in dry tetrahydrofuran (30 mL), with the temperature kept at -70°C to -60°C. After addition of the iodine solution, the cooling bath was removed, and the reaction mixture was allowed to warm to 25°C where it was stirred for 2 h. The reaction mixture was then poured into a solution consisting of a saturated aqueous ammonium chloride solution (400 mL) and ammonium hydroxide (100 mL), and the organic compound was extracted into ethyl acetate (3 x 250 mL).
- reaction mixture was then treated dropwise with a solution of (E)-3-cyclohexyl-2-iodo-acrylic acid methyl ester (5.88 g, 20 mmol) in dry tetrahydrofuran (5 mL) over 5 min. During the addition, the temperature rose to 50°C. The reaction mixture was then stirred at 40-45°C for 1 h and then stirred overnight at 25°C. The reaction mixture was then diluted with dry tetrahydrofuran ( 10 mL), and the stirring was stopped to allow the excess zinc dust to settle down ( ⁇ 2 h).
- reaction mixture was cooled to 25°C and then poured into a saturated aqueous ammonium chloride solution (150 mL), and the organic compound was extracted into ethyl acetate (3 x 100 mL). The combined organic extracts were washed with a saturated aqueous sodium chloride solution (1 x 200 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo.
- reaction mixture was then treated with 2- aminothiazole (5.04 g, 50.3 mmol), and the resulting suspension was stirred for 2 d at 25°C.
- the reaction mixture was concentrated in vacuo to remove methylene chloride, and the residue was diluted with ethyl acetate (250 mL) and a IN aqueous hydrochloric acid solution (150 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (1 x 100 mL).
- reaction mixture was cooled to -70°C and then slowly treated with the freshly prepared cycloheptylmagnesium bromide. After the addition, the reaction mixture was allowed to warm to -10°C where it was stirred for 5 min. The resulting reaction mixture was again cooled back to -70°C and then treated with methyl propiolate (7.57 g, 90 mmol). The reaction mixture was stirred for 15 h at -70°C to -50°C and then slowly treated with a solution of iodine (34.3 g, 135 mmol) in dry tetrahydrofuran (30 mL), with the temperature kept at -70°C to -60°C.
- iodine 34.3 g, 135 mmol
- reaction mixture was then treated dropwise with a solution of (E)-3-cycloheptyl-2-iodo-acrylic acid methyl ester (6.16 g, 20 mmol) in dry tetrahydrofuran (5 mL) over 10 min.
- the reaction mixture was then stirred at 40-45°C for 1 h and then stirred overnight at 25°C.
- the reaction mixture was then diluted with dry tetrahydrofuran (10 mL), and the stirring was stopped to allow the excess zinc dust to settle down ( ⁇ 2 h).
- reaction mixture was cooled to 25°C and then poured into a saturated aqueous ammonium chloride solution (150 mL), and the organic compound was extracted into ethyl acetate (3 x 150 mL).
- the combined organic extracts were washed with a saturated aqueous sodium chloride solution (1 x 300 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo.
- reaction mixture was then treated with 2-aminothiazole (4.63 g, 46.23 mmol), and the resulting suspension was stirred for 2 d at 25°C.
- the reaction mixture was then concentrated in vacuo to remove methylene chloride, and the residue was diluted with ethyl acetate (250 mL) and a IN aqueous hydrochloric acid solution (150 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (1 x 150 mL).
- reaction mixture was cooled to -70°C and then slowly treated with the freshly prepared cyclooctylmagnesium bromide. After the addition, the reaction mixture was allowed to warm to -10°C where it was stirred for 5 min. The resulting reaction mixture was again cooled back to -70°C and then treated with methyl propiolate (3.02 g, 36 mmol). The reaction mixture was stirred for 15 h at -70°C to -50°C and then slowly treated with a solution of iodine (15.22 g, 60 mmol) in dry tetrahydrofuran (15 mL), with the temperature kept at -70°C to -60°C.
- reaction mixture was then treated dropwise with a solution of (E)-3-cyclooctyl-2-iodo-acrylic acid methyl ester (3.22 g, 10 mmol) in dry tetrahydrofuran (4 mL) over 10 min.
- the reaction mixture was then stirred at 40-45°C for 1 h and then stirred overnight at 25°C.
- the reaction mixture was then diluted with dry tetrahydrofuran (8 mL), and the stirring was stopped to allow the excess zinc dust to settle down ( ⁇ 2 h).
- reaction mixture was cooled to 25°C and then poured into a saturated aqueous ammonium chloride solution (100 mL), and the organic compound was extracted into ethyl acetate (3 x 75 mL). The combined organic extracts were washed with a saturated aqueous sodium chloride solution (1 x 200 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo.
- reaction mixture was then treated with 2-aminothiazole (1.2 g, 12 mmol), and the resulting suspension was stirred for 2 d at 25°C.
- the reaction mixture was then concentrated in vacuo to remove methylene chloride, and the residue was diluted with ethyl acetate (100 mL) and a IN aqueous hydrochloric acid solution (100 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate ( 1 x 50 mL).
- the layers were shaken and separated.
- the aqueous layer was further extracted with methylene chloride ( 1 x 200 mL).
- the combined organic layers were washed with a saturated aqueous sodium bicarbonate solution ( 1 x 200 mL) and water (1 x 100 mL), dried over magnesium sulfate, filtered, and concentrated in vacuo.
- the resulting reaction mixture was allowed to warm to 25°C where it was stirred for 64 h.
- the reaction mixture was then concentrated in vacuo to remove tetrahydrofuran.
- the residue was diluted with water (150 mL) and then extracted with diethyl ether (1 x 200 mL). The organic layer was dried over sodium sulfate, filtered, and concentrated in vacuo.
- reaction mixture was treated with 2-aminothiazole (46 mg, 0.46 mmol) and pyridine (0.044 mL, 0.55 mmol), and the resulting reaction mixture was stirred at 25°C for 16 h.
- the reaction was then diluted with water (10 mL) and extracted with methylene chloride (3 x 15 mL). The combined organic layers were dried over sodium sulfate, filtered and concentrated in vacuo.
- the resulting reaction mixture was again cooled back to -70°C and then slowly treated with methyl propiolate (7.99 g, 95 mmol).
- the reaction mixture was stirred for at -60°C to -50°C overnight and then cooled to -70°C to -60°C, at which time, the reaction mixture was treated slowly with a solution of iodine (34.3 g, 135 mmol) in dry tetrahydrofuran (30 mL). After addition of the iodine solution, the cooling bath was removed, and the reaction mixture was allowed to warm to 25°C where it was stirred for 2 h.
- the reaction mixture was then poured into a solution consisting of a saturated aqueous ammonium chloride solution (200 mL) and ammonium hydroxide (50 mL), and the organic compound was extracted into diethyl ether (3 x 100 mL).
- the combined organic extracts were successively washed with a saturated aqueous sodium thiosulfate solution (1 x 300 mL) and a saturated aqueous sodium chloride solution (1 x 300 mL).
- the organic layer was dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo.
- reaction mixture was then treated dropwise with a solution of (E)-3-cyclopentyl-2-iodo-acrylic acid methyl ester (660 mg, 2.25 mmol) in dry tetrahydrofuran (2 mL) over 3 min.
- the reaction mixture was then stirred at 40-45°C for 1 h and then stirred overnight at 25°C.
- the reaction mixture was then diluted with dry tetrahydrofuran (4 mL), and the stirring was stopped to allow the excess zinc dust to settle down ( ⁇ 2 h).
- reaction mixture was then cooled to 25°C and then poured into a saturated aqueous ammonium chloride solution (50 mL), and the organic compound was extracted into ethyl acetate (3 x 35 mL).
- the combined organic extracts were washed with a saturated aqueous sodium chloride solution (1 x 100 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo.
- reaction mixture was then treated with 2-aminothiazole (713 mg, 7.12 mmol), and the resulting suspension was stirred for 2 d at 25°C.
- the reaction mixture was then concentrated in vacuo to remove methylene chloride, and the residue was diluted with ethyl acetate (40 mL) and a IN aqueous hydrochloric acid solution (50 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate ( 1 x 25 mL).
- reaction mixture was then treated dropwise with a solution of (E)-3-cyclohexyl-2-iodo-acrylic acid methyl ester (prepared in Example 4, 1.47 g, 5 mmol) in dry tetrahydrofuran (1.5 mL) over 3 min. During the addition, the temperature rose to 45°C. The reaction mixture was then stirred at 40-45°C for 1 h and then stirred overnight at 25°C. The reaction mixture was then diluted with dry tetrahydrofuran (5 mL), and the stirring was stopped to allow the excess zinc dust to settle down ( ⁇ 2 h).
- reaction mixture was then poured into a saturated aqueous ammonium chloride solution (50 mL), and the organic compound was extracted into diethyl ether (2 x 50 mL). The combined organic extracts were washed with a saturated aqueous sodium chloride solution (1 x 50 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo.
- the reaction mixture was then treated with 2-aminothiazole (476 mg, 4.75 mmol), and the resulting suspension was stirred for 15 h at 25°C.
- the reaction mixture was concentrated in vacuo to remove methylene chloride, and the residue was diluted with ethyl acetate (75 mL).
- the organic layer was washed successively with a IN aqueous hydrochloric acid solution (2 x 30 mL), a saturated aqueous sodium bicarbonate solution (2 x 30 mL), and a saturated aqueous sodium chloride solution ( 1 x 50 mL).
- the organic layer was then dried over anhydrous magnesium sulfate, filtered, and, concentrated in vacuo.
- reaction mixture was then treated dropwise with a solution of (E)-3-cyclohexyl-2-iodo-acrylic acid methyl ester (prepared in Example 4, 2.5 g, 8.5 mmol) in dry tetrahydrofuran (3 mL) over 5 min. After the addition, the reaction mixture was stirred for 1 h at 40-45°C and then stirred overnight at 25°C. The reaction mixture was then diluted with dry tetrahydrofuran (4 mL), and the stirring was stopped to allow the excess zinc dust to settle down ( ⁇ 2 h).
- reaction mixture was cooled to 25°C and then poured into a saturated aqueous ammonium chloride solution (100 mL), and the organic compound was extracted into ethyl acetate (3 x 75 mL). The combined organic extracts were washed with a saturated aqueous sodium chloride solution (1 x 100 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo.
- reaction mixture was then treated with 2-aminothiazole (800 mg, 7.98 mmol), and the resulting suspension was stirred for 2 d at 25°C.
- the reaction mixture was concentrated in vacuo to remove methylene chloride, and the residue was diluted with ethyl acetate (100 mL) and a IN aqueous hydrochloric acid solution (100 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (1 x 50 mL).
- the zinc suspension was then heated with a heat gun to ebullition, allowed to cool, and heated again. This process was repeated three times to make sure the zinc dust was activated.
- the activated zinc dust suspension was then treated with trimethylsilyl chloride (110 mg, 1 mmol), and the suspension was stirred for 15 min at 25°C.
- the reaction mixture was then treated dropwise with a solution of (E)-3-cyclohexyl-2-iodo-acrylic acid methyl ester (prepared in Example 4, 1.2 g, 4.2 mmol) in dry tetrahydrofuran (2 mL) over 5 min. The reaction mixture was then stirred at 40-45°C for 1 h and then stirred overnight at 25°C.
- reaction mixture was then cooled to 25°C and then poured into a saturated aqueous ammonium chloride solution (70 mL), and the organic compound was extracted into ethyl acetate (3 x 50 mL).
- the combined organic extracts were washed with a saturated aqueous sodium chloride solution (1 x 100 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo.
- reaction mixture was then treated with 2-aminothiazole (412 mg, 4.12 mmol), and the resulting suspension was stirred for 2 d at 25°C.
- the reaction mixture was then concentrated in vacuo to remove methylene chloride, and the residue was diluted with ethyl acetate (70 mL) and a IN aqueous hydrochloric acid solution (50 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (1 x 50 mL).
- the zinc suspension was then heated with a heat gun to ebullition, allowed to cool, and heated again. This process was repeated three times to make sure the zinc dust was activated.
- the activated zinc dust suspension was then treated with trimethylsilyl chloride (110 mg, 1 mmol), and the suspension was stirred for 15 min at 25°C.
- the reaction mixture was then treated dropwise with a solution of (E)-3-cyclohexyl-2-iodo-acrylic acid methyl ester (prepared in Example 4, 1.17 g, 4 mmol) in dry tetrahydrofuran (2 mL) over 5 min. The reaction mixture was then stirred at 40-45°C for 1 h and then stirred overnight at 25°C.
- the resulting brick red solution was heated at 50°C for 2 d.
- the reaction mixture was then cooled to 25°C and then poured into a saturated aqueous ammonium chloride solution (50 mL), and the organic compound was extracted into ethyl acetate (3 x 30 mL).
- the combined organic extracts were washed with a saturated aqueous sodium chloride solution (1 x 100 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo.
- reaction mixture was then treated with 2-aminothiazole (702 mg, 7.02 mmol), and the resulting suspension was stirred for 2 d at 25°C.
- the reaction mixture was then concentrated in vacuo to remove methylene chloride, and the residue was diluted with ethyl acetate (70 mL) and a IN aqueous hydrochloric acid solution (50 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (1 x 50 mL).
- reaction mixture was then treated dropwise with a solution of (E)-3-cycloheptyl-2-iodo-acrylic acid methyl ester (prepared in Example 5, 616 mg, 2 mmol) in dry tetrahydrofuran (2 mL). After the addition, the reaction mixture was stirred for 1 h at 40-45°C and then stirred overnight at 25°C. The reaction mixture was then diluted with dry tetrahydrofuran (2 mL), and the stirring was stopped to allow the excess zinc dust to settle down ( ⁇ 2 h).
- reaction mixture was cooled to 25°C and then poured into a saturated aqueous ammonium chloride solution (30 mL), and the organic compound was extracted into ethyl acetate (3 x 25 mL).
- the combined organic extracts were washed with a saturated aqueous sodium chloride solution (1 x 100 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo.
- the aqueous layer was acidified with a IN aqueous hydrochloric acid solution.
- the resulting acid was extracted into ethyl acetate (2 x 35 mL).
- the combined organic layers were washed with a saturated aqueous sodium chloride solution (1 x 100 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo to afford the (E)-3-cycloheptyl-2-(4-(methanesulfonyl)-3-(trifluoromethyl)- phenyl) -acrylic acid (268 mg, 72%) as a brown solid: mp 151-156°C.
- reaction mixture was then treated with 2-aminothiazole (193 mg, 1.95 mmol), and the resulting suspension was stirred for 2 d at 25°C.
- the reaction mixture was concentrated in vacuo to remove methylene chloride, and the residue was diluted with ethyl acetate (50 mL) and a IN aqueous hydrochloric acid solution (50 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (1 x 30 mL).
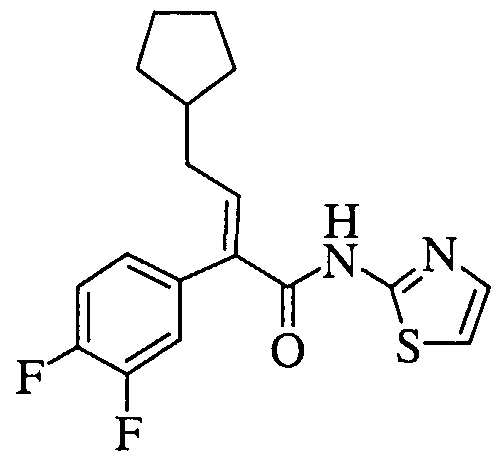
- reaction mixture was treated with 2-aminopyridine (95 mg, 1.01 mmol) and pyridine (0.098 mL, 1.22 mmol), and the resulting reaction mixture was stirred at 25°C for 16 h.
- the reaction was then diluted with water ( 10 mL) and extracted with methylene chloride (3 x 15 mL). The combined organic layers were dried over sodium sulfate, filtered and concentrated in vacuo.
- the reaction mixture was then treated with 2- amino-5-bromopyridine (519 mg, 3 mmol), and the resulting suspension was stirred for 3 d at 25°C.
- the reaction mixture was concentrated in vacuo to remove methylene chloride, and the residue was diluted with ethyl acetate (50 mL) and a IN aqueous hydrochloric acid solution (50 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate ( 1 x 30 mL). The combined organic extracts were successively washed with a saturated aqueous sodium bicarbonate solution ( 1 x 50 mL) and a saturated aqueous sodium chloride solution ( 1 x 50 mL).
- reaction mixture was then treated dropwise with a solution of cyclopentylmethyl iodide (4.2 g, 20 mmol) in dry tetrahydrofuran (7 mL) over 5 min. During the addition, the temperature rose to 50°C, and the reaction mixture was stirred overnight at 40-45°C. The reaction mixture was then cooled to 25°C and diluted with dry tetrahydrofuran (5 mL). The stirring was stopped to allow the excess zinc dust to settle down ( ⁇ 2 h).
- reaction mixture was stirred for 4 h at -40°C to -30°C and then slowly treated with a solution of iodine (6.85 g, 27 mmol) in dry tetrahydrofuran (10 mL), with the temperature kept at -70°C to -60°C. After the addition of the iodine solution, the cooling bath was removed, and the reaction mixture was allowed to warm to 25°C where it was stirred forl h. The reaction mixture was then poured into a solution consisting of a saturated aqueous ammonium chloride solution (90 mL) and ammonium hydroxide (10 mL), and the organic compound was extracted into diethyl ether (3 x 50 mL).
- reaction mixture was then treated dropwise with a solution of (E)-4-cyclopentyl-2-iodo-but-2-enoic acid methyl ester (1.47 g, 5 mmol) in dry tetrahydrofuran (1.5 mL) over 3 min. After the addition, the reaction mixture was stirred for 1 h at 40-45°C and then stirred overnight at 25°C. The reaction mixture was then diluted with dry tetrahydrofuran (5 mL), and the stirring was stopped to allow the excess zinc dust to settle down ( ⁇ 2 h).
- reaction mixture was cooled to 25°C and then poured into a saturated aqueous ammonium chloride solution (75 mL), and the organic compound was extracted into diethyl ether (3 x 50 mL). The combined ether extracts were washed with a saturated aqueous sodium chloride solution (1 x 100 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo.
- reaction mixture was then treated with 2-aminothiazole (378 mg, 3.76 mmol), and the resulting suspension was stirred at 25°C over the weekend.
- the reaction mixture was concentrated in vacuo to remove methylene chloride, and the residue was diluted with ethyl acetate (75 mL) and a IN aqueous hydrochloric acid solution (100 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (1 x 50 mL).
- reaction mixture was then treated with 2-aminothiazole-4- carboxylic acid methyl ester (400 mg, 2.52 mmol), and the resulting suspension was stirred at 25°C over the weekend.
- the reaction mixture was concentrated in vacuo to remove methylene chloride, and the residue was diluted with ethyl acetate (50 mL) and a IN aqueous hydrochloric acid solution (50 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (1 x 25 mL).
- reaction mixture was then treated with 2-aminothiazole-5- carboxylic acid ethyl ester (774 mg, 4.5 mmol), and the resulting suspension was stirred at 25°C over the weekend.
- the reaction mixture was concentrated in vacuo to remove methylene chloride, and the residue was diluted with ethyl acetate (70 mL) and a IN aqueous hydrochloric acid solution (70 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (1 x 50 mL).
- reaction mixture was then treated dropwise with a solution of (E)-4-cyclopentyl-2-iodo-but-2-enoic acid methyl ester (prepared in Example 21, 1.47 g, 5 mmol) in dry tetrahydrofuran ( 1.5 mL) over 3 min. After the addition, the reaction mixture was stirred for 1 h at 40-45°C and then stirred overnight at 25°C. The reaction mixture was then diluted with dry tetrahydrofuran (5 mL), and the stirring was stopped to allow the excess zinc dust to settle down ( ⁇ 2 h).
- reaction mixture was cooled to 25°C and then poured into a saturated aqueous ammonium chloride solution (50 mL), and the organic compound was extracted into diethyl ether (2 x 50 mL). The combined ether extracts were washed with a saturated aqueous sodium chloride solution (1 x 50 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo.
- reaction mixture was then treated with 2- aminothiazole (0.59 g, 5.9 mmol), and the resulting suspension was stirred at 25°C over the weekend.
- the reaction mixture was concentrated in vacuo to remove methylene chloride, and the residue was diluted with ethyl acetate (100 mL) and a IN aqueous hydrochloric acid solution (100 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (1 x 50 mL).
- reaction mixture was then treated dropwise with a solution of (E)-4-cyclopentyl-2-iodo-but-2-enoic acid methyl ester (prepared in Example 21, 1.03 g, 3.5 mmol) in dry tetrahydrofuran (1.5 mL) over 3 min. After the addition, the reaction mixture was stirred for 1 h at 40-45°C and then stirred overnight at 25°C. The reaction mixture was then diluted with dry tetrahydrofuran (3 mL), and the stirring was stopped to allow the excess zinc dust to settle down ( ⁇ 2 h).
- the solution was heated at 40°C for 15 h, at which time, thin layer chromatography analysis of the mixture indicated the absence of starting material.
- the reaction mixture was then concentrated in vacuo to remove ethanol, and the residue was diluted with water (30 mL) and extracted with diethyl ether (1 x 50 mL) to remove any neutral impurities.
- the aqueous layer was acidified with a IN aqueous hydrochloric acid solution. The resulting acid was extracted into ethyl acetate (2 x 50 mL).
- the reaction mixture was then treated with 2-aminothiazole (320 mg, 3.2 mmol), and the resulting suspension was stirred at 25°C over the weekend.
- the reaction mixture was concentrated in vacuo to remove methylene chloride, and the residue was diluted with ethyl acetate (50 mL) and a IN aqueous hydrochloric acid solution (50 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (1 x 30 mL).
- the combined organic extracts were successively washed with a saturated aqueous sodium bicarbonate solution (2 x 50 mL) and a saturated aqueous sodium chloride solution (1 x 100 mL).
- reaction mixture was quenched with water (10 mL) and then concentrated in vacuo to remove tetrahydrofuran. The residue was further diluted with water (50 mL) and then extracted with ethyl acetate (2 x 75 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated in vacuo.
- Flash chromatography (Merck Silica gel 60, 230-400 mesh, 9/1 hexanes/ethyl acetate) afforded impure l- [2-(3,4-dichloro-phenyl)-4-methyl-pent-2-enoyl]-3-methyl-urea (280.2 mg) as a white solid.
- a second flash chromatography (Merck Silica gel 60, 230-400 mesh, 3/2 hexanes/diethyl ether) again afforded impure l-[2-(3,4-dichloro-phenyl)-4-methyl-pent- 2-enoyl]-3-methyl-urea ( 114.6 mg) as a white solid.
- reaction mixture was then treated dropwise with a solution of cyclohexyl iodide (21 g, 100 mmol) in dry tetrahydrofuran (30 mL) over 15 min. During the addition, the temperature rose to 60°C. The reaction mixture was then stirred for 3 h at 40-45°C. The reaction mixture was then cooled to 25°C and diluted with dry tetrahydrofuran (60 mL). The stirring was stopped to allow the excess zinc dust to settle down ( ⁇ 3 h).
- the resulting reaction mixture was stirred for 15 h at -70°C to -50°C and then slowly treated with a solution of iodine (34.26 g, 135 mmol) in dry tetrahydrofuran (30 mL), with the temperature kept at -70°C to -60°C. After addition of the iodine solution, the cooling bath was removed, and the reaction mixture was allowed to warm to 25°C where it was stirred for 2 h. The reaction mixture was then poured into a solution consisting of a saturated aqueous ammonium chloride solution (400 mL) and ammonium hydroxide (100 mL), and the organic compound was extracted into ethyl acetate (3 x 250 mL).
- reaction mixture was then treated dropwise with a solution of (E)-3-cyclohexyl-2-iodo-acrylic acid methyl ester (2.5 g, 8.5 mmol) in dry tetrahydrofuran (3 mL) over 5 min. After the addition, the reaction mixture was stirred for 1 h at 40-45°C and then stirred overnight at 25°C. The reaction mixture was then diluted with dry tetrahydrofuran (4 mL), and the stirring was stopped to allow the excess zinc dust to settle down ( ⁇ 2 h).
- reaction mixture was cooled to 25°C and then poured into a saturated aqueous ammonium chloride solution (100 mL), and the organic compound was extracted into ethyl acetate (3 x 75 mL). The combined organic extracts were washed with a saturated aqueous sodium chloride solution (1 x 100 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo.
- the reaction mixture was then stirred at 70°C for 20 h.
- the reaction mixture was then cooled to 25°C and diluted with ethyl acetate (50 mL) and a 3N aqueous hydrochloric acid solution (40 mL).
- the two layers were separated, and the aqueous layer was extracted with ethyl acetate (1 x 20 mL).
- the combined organic extracts were successively washed with a saturated aqueous sodium bicarbonate solution (1 x 50 mL) and a saturated aqueous sodium chloride solution (1 x 50 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo.
- Example A Tablets containing the following ingredients can be produced in a conventional manner:
- Capsules containing the following ingredients can be produced in a conventional r:
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Diabetes (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Thiazole And Isothizaole Compounds (AREA)
- Pyridine Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Detergent Compositions (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Description
































Claims









Priority Applications (17)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP00987392A EP1242397B1 (en) | 1999-12-15 | 2000-12-12 | Trans olefinic glucokinase activators |
| DE60022903T DE60022903T2 (en) | 1999-12-15 | 2000-12-12 | OLEFINIC TRANS-GLUCOCINASE ACTIVATORS |
| AT00987392T ATE305461T1 (en) | 1999-12-15 | 2000-12-12 | OLEFINIC TRANS-GLUCOCINASE ACTIVATORS |
| SI200030752T SI1242397T1 (en) | 1999-12-15 | 2000-12-12 | Trans olefinic glucokinase activators |
| JP2001544706A JP3824936B2 (en) | 1999-12-15 | 2000-12-12 | trans olefin glucokinase activator |
| HU0203753A HUP0203753A3 (en) | 1999-12-15 | 2000-12-12 | Trans olefinic glucokinase activators, process for their preparation and their use |
| PL00355815A PL355815A1 (en) | 1999-12-15 | 2000-12-12 | Trans olefinic glucokinase activators |
| BR0016392-9A BR0016392A (en) | 1999-12-15 | 2000-12-12 | Compounds, use of these compounds, pharmaceutical composition comprising these compounds, process for the prophylactic or therapeutic treatment of type II diabetes and process for the preparation of these compounds |
| NZ518974A NZ518974A (en) | 1999-12-15 | 2000-12-12 | Trans olefinic glucokinase activators |
| MXPA02005874A MXPA02005874A (en) | 1999-12-15 | 2000-12-12 | Trans olefinic glucokinase activators. |
| AU23652/01A AU781029B2 (en) | 1999-12-15 | 2000-12-12 | Trans olefinic glucokinase activators |
| IL15008300A IL150083A0 (en) | 1999-12-15 | 2000-12-12 | Trans olefinic glucokinase activators |
| CA002392903A CA2392903C (en) | 1999-12-15 | 2000-12-12 | Trans olefinic glucokinase activators |
| IL150083A IL150083A (en) | 1999-12-15 | 2002-06-06 | Trans olefinic compounds and a process for their preparation |
| HR20020514A HRP20020514A2 (en) | 1999-12-15 | 2002-06-12 | Trans olefinic glucokinase activators |
| NO20022863A NO323142B1 (en) | 1999-12-15 | 2002-06-14 | Trans-olefinic glycokinase activators, their use as medicaments, pharmaceutical compositions containing them, their use in the manufacture of medicaments for the treatment or prevention of type II diabetes, and their method of preparation. |
| HK03106627.5A HK1054383B (en) | 1999-12-15 | 2003-09-16 | Trans olefinic glucokinase activators |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US17078399P | 1999-12-15 | 1999-12-15 | |
| US60/170,783 | 1999-12-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2001044216A1 true WO2001044216A1 (en) | 2001-06-21 |
Family
ID=22621235
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2000/012612 WO2001044216A1 (en) | 1999-12-15 | 2000-12-12 | Trans olefinic glucokinase activators |
Country Status (33)
| Country | Link |
|---|---|
| US (1) | US6353111B1 (en) |
| EP (1) | EP1242397B1 (en) |
| JP (1) | JP3824936B2 (en) |
| KR (1) | KR100502032B1 (en) |
| CN (1) | CN1185219C (en) |
| AR (1) | AR032752A1 (en) |
| AT (1) | ATE305461T1 (en) |
| AU (1) | AU781029B2 (en) |
| CA (1) | CA2392903C (en) |
| CO (1) | CO5050295A1 (en) |
| CZ (1) | CZ20022412A3 (en) |
| DE (1) | DE60022903T2 (en) |
| DK (1) | DK1242397T3 (en) |
| ES (1) | ES2249322T3 (en) |
| GC (1) | GC0000264A (en) |
| HK (1) | HK1054383B (en) |
| HR (1) | HRP20020514A2 (en) |
| HU (1) | HUP0203753A3 (en) |
| IL (2) | IL150083A0 (en) |
| JO (1) | JO2180B1 (en) |
| MA (1) | MA26855A1 (en) |
| MX (1) | MXPA02005874A (en) |
| MY (1) | MY125484A (en) |
| NO (1) | NO323142B1 (en) |
| NZ (1) | NZ518974A (en) |
| PE (1) | PE20011022A1 (en) |
| PL (1) | PL355815A1 (en) |
| RU (1) | RU2245332C2 (en) |
| TW (1) | TWI262916B (en) |
| UY (1) | UY26483A1 (en) |
| WO (1) | WO2001044216A1 (en) |
| YU (1) | YU41402A (en) |
| ZA (1) | ZA200203829B (en) |
Cited By (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003000267A1 (en) * | 2001-06-26 | 2003-01-03 | Astrazeneca Ab | Amino nicotinate derivatives as glucokinase (glk) modulators |
| WO2003000262A1 (en) * | 2001-06-26 | 2003-01-03 | Astrazeneca Ab | Vinyl phenyl derivatives as glk activators |
| WO2003015774A1 (en) * | 2001-08-17 | 2003-02-27 | Astrazeneca Ab | Compounds effecting glucokinase |
| WO2003055482A1 (en) | 2001-12-21 | 2003-07-10 | Novo Nordisk A/S | Amide derivatives as gk activators |
| WO2003080585A1 (en) * | 2002-03-26 | 2003-10-02 | Banyu Pharmaceutical Co., Ltd. | Novel aminobenzamide derivative |
| WO2004002481A1 (en) | 2002-06-27 | 2004-01-08 | Novo Nordisk A/S | Aryl carbonyl derivatives as therapeutic agents |
| WO2004046139A1 (en) * | 2002-11-19 | 2004-06-03 | Astrazeneca Ab | Benzofuran derivates, process for their preparation and intermediates thereof |
| WO2004063194A1 (en) * | 2003-01-06 | 2004-07-29 | Eli Lilly And Company | Heteroaryl compounds |
| WO2004072066A1 (en) * | 2003-02-11 | 2004-08-26 | Prosidion Limited | Tri(cyclo) substituted amide glucokinase activator compounds |
| WO2005103021A1 (en) * | 2004-04-21 | 2005-11-03 | Prosidion Limited | Tri(cyclo) substituted amide compounds |
| WO2005121110A1 (en) | 2004-06-05 | 2005-12-22 | Astrazeneca Ab | Hetroaryl benzamide derivatives for use as glk activators in the treatment of diabetes |
| JP2006509774A (en) * | 2002-10-03 | 2006-03-23 | ノバルティス アクチエンゲゼルシャフト | Substituted (thiazol-2-yl) -amides or sulfonamides as glucokinase activators useful in the treatment of type 2 diabetes |
| JP2006517590A (en) * | 2003-02-11 | 2006-07-27 | プロシディオン・リミテッド | Tri (cyclo) substituted amide compounds |
| WO2007007041A1 (en) | 2005-07-09 | 2007-01-18 | Astrazeneca Ab | Heteroaryl benzamide derivatives for use as glk activators in the treatment of diabetes |
| WO2007015805A1 (en) | 2005-07-20 | 2007-02-08 | Eli Lilly And Company | 1-amino linked compounds |
| WO2007039177A2 (en) | 2005-09-29 | 2007-04-12 | Sanofi-Aventis | Phenyl- and pyridinyl- 1, 2 , 4 - oxadiazolone derivatives, processes for their preparation and their use as pharmaceuticals |
| US7230108B2 (en) | 2002-11-19 | 2007-06-12 | Astrazeneca Ab | Quinoline derivatives as glucokinase ligands |
| WO2007123581A1 (en) | 2005-11-17 | 2007-11-01 | Eli Lilly And Company | Glucagon receptor antagonists, preparation and therapeutic uses |
| WO2007128761A2 (en) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Uses of dpp-iv inhibitors |
| WO2008078674A1 (en) | 2006-12-25 | 2008-07-03 | Kyorin Pharmaceutical Co., Ltd. | Glucokinase-activating substance |
| US7410956B2 (en) | 2002-02-11 | 2008-08-12 | Vertex Pharmaceuticals Incorporated | Caspase inhibitor prodrugs |
| WO2009039944A1 (en) * | 2007-09-21 | 2009-04-02 | Sanofi-Aventis | Phenothiazine derivative having a double bond, method for the production thereof, and use thereof as a pharmaceutical |
| EP2048137A1 (en) | 2004-02-18 | 2009-04-15 | AstraZeneca AB | Benzamide derivatives and their use as glucokinase activating agents. |
| US7541373B2 (en) | 2002-06-27 | 2009-06-02 | Novo Nordisk A/S | Aryl carbonyl derivatives as therapeutic agents |
| WO2009091014A1 (en) | 2008-01-18 | 2009-07-23 | Astellas Pharma Inc. | Phenyl acetamide derivative |
| US7582769B2 (en) | 2005-07-08 | 2009-09-01 | Novo Nordisk A/S | Dicycloalkyl urea glucokinase activators |
| US7598391B2 (en) | 2004-01-06 | 2009-10-06 | Novo Nordisk A/S | Heteroaryl-ureas and their use as glucokinase activators |
| WO2009133687A1 (en) | 2008-04-28 | 2009-11-05 | 杏林製薬株式会社 | Cyclopentylacrylic acid amide derivative |
| US7750020B2 (en) | 2004-04-02 | 2010-07-06 | Novartis Ag | Sulfonamide-thiazolpyridine derivatives as glucokinase activators useful the treatment of Type 2 diabetes |
| US7781451B2 (en) | 2004-04-02 | 2010-08-24 | Novartis Ag | Thiazolopyridine derivatives, pharmaceut ical conditions containing them and methods of treating glucokinase mediated conditions |
| US7795257B2 (en) | 2005-09-30 | 2010-09-14 | Novartis Ag | Organic compounds |
| WO2010150280A1 (en) | 2009-06-22 | 2010-12-29 | Cadila Healthcare Limited | Disubstituted benzamide derivatives as glucokinase (gk) activators |
| EP2272834A1 (en) | 2006-10-26 | 2011-01-12 | AstraZeneca AB | Benzoyl amino heterocyclyl compounds as glucokinase (GLK) activators |
| WO2011013141A2 (en) | 2009-07-31 | 2011-02-03 | Cadila Healthcare Limited | Substituted benzamide derivatives as glucokinase (gk) activators |
| US7902248B2 (en) | 2006-12-14 | 2011-03-08 | Hoffmann-La Roche Inc. | Oxime glucokinase activators |
| US8008332B2 (en) | 2006-05-31 | 2011-08-30 | Takeda San Diego, Inc. | Substituted indazoles as glucokinase activators |
| WO2011123572A1 (en) | 2010-03-31 | 2011-10-06 | The Scripps Research Institute | Reprogramming cells |
| US8034822B2 (en) | 2006-03-08 | 2011-10-11 | Takeda San Diego, Inc. | Glucokinase activators |
| US8124617B2 (en) | 2005-09-01 | 2012-02-28 | Takeda San Diego, Inc. | Imidazopyridine compounds |
| US8163779B2 (en) | 2006-12-20 | 2012-04-24 | Takeda San Diego, Inc. | Glucokinase activators |
| US8173645B2 (en) | 2007-03-21 | 2012-05-08 | Takeda San Diego, Inc. | Glucokinase activators |
| US8252931B2 (en) | 2005-09-30 | 2012-08-28 | Novartis Ag | Thiazolo[5,4-B]pyridine glucokinase activators |
| US8426465B2 (en) | 2006-08-24 | 2013-04-23 | University Of Tennesse Research Foundation | Substituted acylanilides and methods of use thereof |
| US8563730B2 (en) | 2008-05-16 | 2013-10-22 | Takeda San Diego, Inc. | Pyrazole and fused pyrazole glucokinase activators |
| WO2014077235A1 (en) | 2012-11-13 | 2014-05-22 | 大正製薬株式会社 | 2-pyridone compound |
| US8822503B2 (en) | 2009-12-04 | 2014-09-02 | Taisho Pharmaceutical Co., Ltd | 2-pyridone compounds |
| US8940900B2 (en) | 2007-02-28 | 2015-01-27 | Advinus Therapeutics Private Limited | 2,2,2-tri-substituted acetamide derivatives as glucokinase activators, their process and pharmaceutical application |
| US9604916B2 (en) | 2012-07-13 | 2017-03-28 | Gtx, Inc. | Method of treating androgen receptor (AR)-positive breast cancers with selective androgen receptor modulator (SARMs) |
| US9622992B2 (en) | 2012-07-13 | 2017-04-18 | Gtx, Inc. | Method of treating androgen receptor (AR)-positive breast cancers with selective androgen receptor modulator (SARMs) |
| US9744149B2 (en) | 2012-07-13 | 2017-08-29 | Gtx, Inc. | Method of treating androgen receptor (AR)-positive breast cancers with selective androgen receptor modulator (SARMs) |
| US9969683B2 (en) | 2012-07-13 | 2018-05-15 | Gtx, Inc. | Method of treating estrogen receptor (ER)-positive breast cancers with selective androgen receptor modulator (SARMS) |
| US10258596B2 (en) | 2012-07-13 | 2019-04-16 | Gtx, Inc. | Method of treating HER2-positive breast cancers with selective androgen receptor modulators (SARMS) |
| US10314807B2 (en) | 2012-07-13 | 2019-06-11 | Gtx, Inc. | Method of treating HER2-positive breast cancers with selective androgen receptor modulators (SARMS) |
| US10849873B2 (en) | 2012-07-13 | 2020-12-01 | Oncternal Therapeutics, Inc | Non-invasive method of evaluating breast cancers for selective androgen receptor modulator (SARM) therapy |
| US11690841B2 (en) | 2017-09-14 | 2023-07-04 | Queen Mary University Of London | Glycolysis-activating agents for treatment or prevention of disease |
| US11833136B2 (en) | 2018-06-12 | 2023-12-05 | Vtv Therapeutics Llc | Therapeutic uses of glucokinase activators in combination with insulin or insulin analogs |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PE20040801A1 (en) | 2002-12-12 | 2004-11-25 | Hoffmann La Roche | PYRAZINE AND PYRIDINE 5-SUBSTITUTED DERIVATIVES AS GLUCOKINASE ACTIVATORS |
| US20080026987A1 (en) * | 2004-06-17 | 2008-01-31 | Novo Nordisk A/S | Use of Liver-Selective Glucokinase Activators |
| KR20080105180A (en) * | 2004-08-12 | 2008-12-03 | 프로시디온 리미티드 | Substituted phenylacetamides and their use as glucokinase activators |
| US20090005391A1 (en) * | 2005-11-03 | 2009-01-01 | Matthew Colin Thor Fyfe | Tricyclo Substituted Amides |
| WO2007061923A2 (en) * | 2005-11-18 | 2007-05-31 | Takeda San Diego, Inc. | Glucokinase activators |
| MX2009009525A (en) * | 2007-03-07 | 2009-09-16 | Kyorin Seiyaku Kk | Glucokinase activator. |
| CL2009000004A1 (en) * | 2008-01-15 | 2010-02-19 | Lilly Co Eli | Crystal form of r-2- (4-cyclopropanesulfonyl-phenyl) -n-pyrazin-2-yl-3- (tetrahydropyran-4-yl) -propionamide; pharmaceutical composition comprising said crystalline form; and use for the treatment of diabetes or hyperglycemia. |
| KR100970871B1 (en) * | 2009-11-30 | 2010-07-20 | 케이제이알디 (주) | Flooring material assembly and method for installing the same |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000058293A2 (en) * | 1999-03-29 | 2000-10-05 | F. Hoffmann-La Roche Ag | Glucokinase activators |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB8909574D0 (en) | 1989-04-26 | 1989-06-14 | Ici Plc | Chemical process |
| US5169951A (en) | 1990-04-23 | 1992-12-08 | Ciba-Geigy Corporation | Process for preparing nematicidal compositions |
-
2000
- 2000-12-01 US US09/727,624 patent/US6353111B1/en not_active Expired - Fee Related
- 2000-12-11 JO JO2000197A patent/JO2180B1/en active
- 2000-12-12 RU RU2002118339/04A patent/RU2245332C2/en not_active IP Right Cessation
- 2000-12-12 KR KR10-2002-7007709A patent/KR100502032B1/en not_active IP Right Cessation
- 2000-12-12 PL PL00355815A patent/PL355815A1/en unknown
- 2000-12-12 DE DE60022903T patent/DE60022903T2/en not_active Expired - Lifetime
- 2000-12-12 DK DK00987392T patent/DK1242397T3/en active
- 2000-12-12 EP EP00987392A patent/EP1242397B1/en not_active Expired - Lifetime
- 2000-12-12 CZ CZ20022412A patent/CZ20022412A3/en unknown
- 2000-12-12 WO PCT/EP2000/012612 patent/WO2001044216A1/en active IP Right Grant
- 2000-12-12 YU YU41402A patent/YU41402A/en unknown
- 2000-12-12 IL IL15008300A patent/IL150083A0/en unknown
- 2000-12-12 CN CNB008172943A patent/CN1185219C/en not_active Expired - Fee Related
- 2000-12-12 JP JP2001544706A patent/JP3824936B2/en not_active Expired - Fee Related
- 2000-12-12 MX MXPA02005874A patent/MXPA02005874A/en active IP Right Grant
- 2000-12-12 PE PE2000001324A patent/PE20011022A1/en not_active Application Discontinuation
- 2000-12-12 AU AU23652/01A patent/AU781029B2/en not_active Ceased
- 2000-12-12 CA CA002392903A patent/CA2392903C/en not_active Expired - Fee Related
- 2000-12-12 AT AT00987392T patent/ATE305461T1/en not_active IP Right Cessation
- 2000-12-12 ES ES00987392T patent/ES2249322T3/en not_active Expired - Lifetime
- 2000-12-12 NZ NZ518974A patent/NZ518974A/en unknown
- 2000-12-12 HU HU0203753A patent/HUP0203753A3/en unknown
- 2000-12-13 MY MYPI20005851A patent/MY125484A/en unknown
- 2000-12-13 GC GCP20001095 patent/GC0000264A/en active
- 2000-12-13 AR ARP000106608A patent/AR032752A1/en active IP Right Grant
- 2000-12-13 CO CO00094817A patent/CO5050295A1/en unknown
- 2000-12-14 TW TW089126672A patent/TWI262916B/en active
- 2000-12-14 UY UY26483A patent/UY26483A1/en not_active Application Discontinuation
-
2002
- 2002-05-14 ZA ZA200203829A patent/ZA200203829B/en unknown
- 2002-06-06 IL IL150083A patent/IL150083A/en not_active IP Right Cessation
- 2002-06-12 HR HR20020514A patent/HRP20020514A2/en not_active Application Discontinuation
- 2002-06-14 MA MA26690A patent/MA26855A1/en unknown
- 2002-06-14 NO NO20022863A patent/NO323142B1/en not_active IP Right Cessation
-
2003
- 2003-09-16 HK HK03106627.5A patent/HK1054383B/en not_active IP Right Cessation
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000058293A2 (en) * | 1999-03-29 | 2000-10-05 | F. Hoffmann-La Roche Ag | Glucokinase activators |
Cited By (102)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7199140B2 (en) | 2001-06-26 | 2007-04-03 | Astrazeneca Ab | Vinyl phenyl derivatives as GLK activators |
| WO2003000262A1 (en) * | 2001-06-26 | 2003-01-03 | Astrazeneca Ab | Vinyl phenyl derivatives as glk activators |
| WO2003000267A1 (en) * | 2001-06-26 | 2003-01-03 | Astrazeneca Ab | Amino nicotinate derivatives as glucokinase (glk) modulators |
| EP1674097A1 (en) * | 2001-08-17 | 2006-06-28 | AstraZeneca AB | Compounds effecting glucokinase |
| EP1568367A1 (en) * | 2001-08-17 | 2005-08-31 | AstraZeneca AB | Compounds effecting glucokinase |
| US7390908B2 (en) | 2001-08-17 | 2008-06-24 | Astrazeneca Ab | Compounds effecting glucokinase |
| KR100920439B1 (en) * | 2001-08-17 | 2009-10-08 | 아스트라제네카 아베 | Compounds effecting glucokinase |
| EP1695705A1 (en) * | 2001-08-17 | 2006-08-30 | AstraZeneca AB | Compounds effecting glucokinase |
| EP1661567A1 (en) * | 2001-08-17 | 2006-05-31 | AstraZeneca AB | Compounds effecting Glucokinase |
| EP1529530A1 (en) * | 2001-08-17 | 2005-05-11 | AstraZeneca AB | Compounds affecting glucokinase |
| WO2003015774A1 (en) * | 2001-08-17 | 2003-02-27 | Astrazeneca Ab | Compounds effecting glucokinase |
| EP1987831A1 (en) | 2001-08-17 | 2008-11-05 | AstraZeneca AB | Compounds effecting glucokinase |
| EP1669069A1 (en) * | 2001-08-17 | 2006-06-14 | AstraZeneca AB | Compounds effecting glukokinase |
| EP1669068A1 (en) * | 2001-08-17 | 2006-06-14 | AstraZeneca AB | Compounds effecting Glucokinase |
| EP1661569A1 (en) * | 2001-08-17 | 2006-05-31 | AstraZeneca AB | Compounds effecting glucokinase |
| EP1661563A1 (en) * | 2001-08-17 | 2006-05-31 | AstraZeneca AB | Compound effecting Glucokinase |
| EP1661568A1 (en) * | 2001-08-17 | 2006-05-31 | AstraZeneca AB | Compounds effecting Glucokinase |
| EP2305648A1 (en) | 2001-12-21 | 2011-04-06 | Novo Nordisk A/S | Amide derivatives useful as glucokinase activators |
| WO2003055482A1 (en) | 2001-12-21 | 2003-07-10 | Novo Nordisk A/S | Amide derivatives as gk activators |
| US7410956B2 (en) | 2002-02-11 | 2008-08-12 | Vertex Pharmaceuticals Incorporated | Caspase inhibitor prodrugs |
| US7253188B2 (en) | 2002-03-26 | 2007-08-07 | Banyu Pharmaceutical Co., Ltd. | Aminobenzamide derivative |
| WO2003080585A1 (en) * | 2002-03-26 | 2003-10-02 | Banyu Pharmaceutical Co., Ltd. | Novel aminobenzamide derivative |
| US7897628B2 (en) | 2002-06-27 | 2011-03-01 | Novo Nordisk A/S | Aryl carbonyl derivatives as therapeutic agents |
| US7541373B2 (en) | 2002-06-27 | 2009-06-02 | Novo Nordisk A/S | Aryl carbonyl derivatives as therapeutic agents |
| US8063081B2 (en) | 2002-06-27 | 2011-11-22 | Novo Nordisk A/S | Aryl carbonyl derivatives as therapeutic agents |
| WO2004002481A1 (en) | 2002-06-27 | 2004-01-08 | Novo Nordisk A/S | Aryl carbonyl derivatives as therapeutic agents |
| USRE45670E1 (en) | 2002-06-27 | 2015-09-15 | Novo Nordisk A/S | Aryl carbonyl derivatives as therapeutic agents |
| US7384967B2 (en) | 2002-06-27 | 2008-06-10 | Novo Nordisk A/S | Aryl carbonyl derivatives as therapeutic agents |
| EP2471533A1 (en) | 2002-06-27 | 2012-07-04 | Novo Nordisk A/S | Aryl carbonyl derivatives as therapeutic agents |
| JP2006509774A (en) * | 2002-10-03 | 2006-03-23 | ノバルティス アクチエンゲゼルシャフト | Substituted (thiazol-2-yl) -amides or sulfonamides as glucokinase activators useful in the treatment of type 2 diabetes |
| US7812167B2 (en) | 2002-10-03 | 2010-10-12 | Novartis, Ag | Substituted (thiazol-2-yl)-amides or sulfonamides as glucokinase activators useful in the treatment of type 2 diabetes |
| US7230108B2 (en) | 2002-11-19 | 2007-06-12 | Astrazeneca Ab | Quinoline derivatives as glucokinase ligands |
| WO2004046139A1 (en) * | 2002-11-19 | 2004-06-03 | Astrazeneca Ab | Benzofuran derivates, process for their preparation and intermediates thereof |
| WO2004063194A1 (en) * | 2003-01-06 | 2004-07-29 | Eli Lilly And Company | Heteroaryl compounds |
| WO2004072066A1 (en) * | 2003-02-11 | 2004-08-26 | Prosidion Limited | Tri(cyclo) substituted amide glucokinase activator compounds |
| US7214681B2 (en) | 2003-02-11 | 2007-05-08 | Prosidion Limited | Tri(cyclo) substituted amide compounds |
| JP2006517590A (en) * | 2003-02-11 | 2006-07-27 | プロシディオン・リミテッド | Tri (cyclo) substituted amide compounds |
| US7582632B2 (en) | 2003-02-11 | 2009-09-01 | Prosidion Limited | Tri(cyclo) substituted amide compounds |
| JP4757188B2 (en) * | 2003-02-11 | 2011-08-24 | プロシディオン・リミテッド | Tri (cyclo) substituted amide compounds |
| US7598391B2 (en) | 2004-01-06 | 2009-10-06 | Novo Nordisk A/S | Heteroaryl-ureas and their use as glucokinase activators |
| USRE45183E1 (en) | 2004-01-06 | 2014-10-07 | Novo Nordisk A/S | Heteroaryl-ureas and their use as glucokinase activators |
| EP2048137A1 (en) | 2004-02-18 | 2009-04-15 | AstraZeneca AB | Benzamide derivatives and their use as glucokinase activating agents. |
| US7750020B2 (en) | 2004-04-02 | 2010-07-06 | Novartis Ag | Sulfonamide-thiazolpyridine derivatives as glucokinase activators useful the treatment of Type 2 diabetes |
| US7781451B2 (en) | 2004-04-02 | 2010-08-24 | Novartis Ag | Thiazolopyridine derivatives, pharmaceut ical conditions containing them and methods of treating glucokinase mediated conditions |
| EA012204B1 (en) * | 2004-04-21 | 2009-08-28 | Прозидион Лимитед | Tri(cyclo) substituted amide compounds |
| WO2005103021A1 (en) * | 2004-04-21 | 2005-11-03 | Prosidion Limited | Tri(cyclo) substituted amide compounds |
| EP1992623A1 (en) | 2004-06-05 | 2008-11-19 | Astra Zeneca AB | Heteroaryl benzamide derivative for use as GLK activator in the treatment of diabetes |
| WO2005121110A1 (en) | 2004-06-05 | 2005-12-22 | Astrazeneca Ab | Hetroaryl benzamide derivatives for use as glk activators in the treatment of diabetes |
| US7582769B2 (en) | 2005-07-08 | 2009-09-01 | Novo Nordisk A/S | Dicycloalkyl urea glucokinase activators |
| EP2305674A1 (en) | 2005-07-09 | 2011-04-06 | AstraZeneca AB | Heteroaryl benzamide derivatives for use as GLK activators in the treatment of diabetes |
| EP2301935A1 (en) | 2005-07-09 | 2011-03-30 | AstraZeneca AB (Publ) | Heteroaryl benzamide derivatives for use as GLK activators in the treatment of diabetes |
| WO2007007041A1 (en) | 2005-07-09 | 2007-01-18 | Astrazeneca Ab | Heteroaryl benzamide derivatives for use as glk activators in the treatment of diabetes |
| WO2007015805A1 (en) | 2005-07-20 | 2007-02-08 | Eli Lilly And Company | 1-amino linked compounds |
| US8124617B2 (en) | 2005-09-01 | 2012-02-28 | Takeda San Diego, Inc. | Imidazopyridine compounds |
| WO2007039177A2 (en) | 2005-09-29 | 2007-04-12 | Sanofi-Aventis | Phenyl- and pyridinyl- 1, 2 , 4 - oxadiazolone derivatives, processes for their preparation and their use as pharmaceuticals |
| US8252931B2 (en) | 2005-09-30 | 2012-08-28 | Novartis Ag | Thiazolo[5,4-B]pyridine glucokinase activators |
| US7795257B2 (en) | 2005-09-30 | 2010-09-14 | Novartis Ag | Organic compounds |
| WO2007123581A1 (en) | 2005-11-17 | 2007-11-01 | Eli Lilly And Company | Glucagon receptor antagonists, preparation and therapeutic uses |
| US8034822B2 (en) | 2006-03-08 | 2011-10-11 | Takeda San Diego, Inc. | Glucokinase activators |
| WO2007128761A2 (en) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Uses of dpp-iv inhibitors |
| EP2351568A2 (en) | 2006-05-04 | 2011-08-03 | Boehringer Ingelheim International GmbH | Uses of dpp-iv inhibitors |
| US8008332B2 (en) | 2006-05-31 | 2011-08-30 | Takeda San Diego, Inc. | Substituted indazoles as glucokinase activators |
| US8394843B2 (en) | 2006-05-31 | 2013-03-12 | Takeda California, Inc. | Substituted isoindoles as glucokinase activators |
| US8846756B2 (en) | 2006-08-24 | 2014-09-30 | University Of Tennessee Research Foundation | Substituted acylanilides and methods of use thereof |
| US8426465B2 (en) | 2006-08-24 | 2013-04-23 | University Of Tennesse Research Foundation | Substituted acylanilides and methods of use thereof |
| EP2272834A1 (en) | 2006-10-26 | 2011-01-12 | AstraZeneca AB | Benzoyl amino heterocyclyl compounds as glucokinase (GLK) activators |
| US7902248B2 (en) | 2006-12-14 | 2011-03-08 | Hoffmann-La Roche Inc. | Oxime glucokinase activators |
| US8163779B2 (en) | 2006-12-20 | 2012-04-24 | Takeda San Diego, Inc. | Glucokinase activators |
| WO2008078674A1 (en) | 2006-12-25 | 2008-07-03 | Kyorin Pharmaceutical Co., Ltd. | Glucokinase-activating substance |
| US8940900B2 (en) | 2007-02-28 | 2015-01-27 | Advinus Therapeutics Private Limited | 2,2,2-tri-substituted acetamide derivatives as glucokinase activators, their process and pharmaceutical application |
| US8173645B2 (en) | 2007-03-21 | 2012-05-08 | Takeda San Diego, Inc. | Glucokinase activators |
| US8207146B2 (en) | 2007-09-21 | 2012-06-26 | Sanofi-Aventis | Phenothiazine derivative having a double bond, method for the production thereof, and use thereof as a pharmaceutical |
| US20100261643A1 (en) * | 2007-09-21 | 2010-10-14 | Sanofi-Aventis | Phenothiazine derivative having a double bond, method for the production thereof, and use thereof as a pharmaceutical |
| WO2009039944A1 (en) * | 2007-09-21 | 2009-04-02 | Sanofi-Aventis | Phenothiazine derivative having a double bond, method for the production thereof, and use thereof as a pharmaceutical |
| US8329707B2 (en) | 2008-01-18 | 2012-12-11 | Astellas Pharma Inc. | Substituted pyrazine compounds |
| WO2009091014A1 (en) | 2008-01-18 | 2009-07-23 | Astellas Pharma Inc. | Phenyl acetamide derivative |
| WO2009133687A1 (en) | 2008-04-28 | 2009-11-05 | 杏林製薬株式会社 | Cyclopentylacrylic acid amide derivative |
| US8946440B2 (en) | 2008-04-28 | 2015-02-03 | Kyorin Pharmaceutical Co., Ltd. | Cyclopentylacrylamide derivative |
| US9452977B2 (en) | 2008-04-28 | 2016-09-27 | Kyorin Pharmaceutical Co., Ltd. | Cyclopentylacrylamide derivative |
| US8563730B2 (en) | 2008-05-16 | 2013-10-22 | Takeda San Diego, Inc. | Pyrazole and fused pyrazole glucokinase activators |
| WO2010150280A1 (en) | 2009-06-22 | 2010-12-29 | Cadila Healthcare Limited | Disubstituted benzamide derivatives as glucokinase (gk) activators |
| US8450494B2 (en) | 2009-06-22 | 2013-05-28 | Cadila Healthcare Limited | Disubstituted benzamide derivatives as glucokinase (GK) activators |
| WO2011013141A2 (en) | 2009-07-31 | 2011-02-03 | Cadila Healthcare Limited | Substituted benzamide derivatives as glucokinase (gk) activators |
| US8822503B2 (en) | 2009-12-04 | 2014-09-02 | Taisho Pharmaceutical Co., Ltd | 2-pyridone compounds |
| EP3199623A1 (en) | 2010-03-31 | 2017-08-02 | The Scripps Research Institute | Reprogramming cells |
| WO2011123572A1 (en) | 2010-03-31 | 2011-10-06 | The Scripps Research Institute | Reprogramming cells |
| EP3936608A1 (en) | 2010-03-31 | 2022-01-12 | The Scripps Research Institute | Reprogramming cells |
| US10258596B2 (en) | 2012-07-13 | 2019-04-16 | Gtx, Inc. | Method of treating HER2-positive breast cancers with selective androgen receptor modulators (SARMS) |
| US10314807B2 (en) | 2012-07-13 | 2019-06-11 | Gtx, Inc. | Method of treating HER2-positive breast cancers with selective androgen receptor modulators (SARMS) |
| US12115146B2 (en) | 2012-07-13 | 2024-10-15 | University Of Tennessee Research Foundation | Treatment of skeletal-related events for breast cancer patients |
| US10987334B2 (en) | 2012-07-13 | 2021-04-27 | University Of Tennessee Research Foundation | Method of treating ER mutant expressing breast cancers with selective androgen receptor modulators (SARMs) |
| US9744149B2 (en) | 2012-07-13 | 2017-08-29 | Gtx, Inc. | Method of treating androgen receptor (AR)-positive breast cancers with selective androgen receptor modulator (SARMs) |
| US9969683B2 (en) | 2012-07-13 | 2018-05-15 | Gtx, Inc. | Method of treating estrogen receptor (ER)-positive breast cancers with selective androgen receptor modulator (SARMS) |
| US9604916B2 (en) | 2012-07-13 | 2017-03-28 | Gtx, Inc. | Method of treating androgen receptor (AR)-positive breast cancers with selective androgen receptor modulator (SARMs) |
| US9622992B2 (en) | 2012-07-13 | 2017-04-18 | Gtx, Inc. | Method of treating androgen receptor (AR)-positive breast cancers with selective androgen receptor modulator (SARMs) |
| US10849873B2 (en) | 2012-07-13 | 2020-12-01 | Oncternal Therapeutics, Inc | Non-invasive method of evaluating breast cancers for selective androgen receptor modulator (SARM) therapy |
| WO2014077235A1 (en) | 2012-11-13 | 2014-05-22 | 大正製薬株式会社 | 2-pyridone compound |
| KR20150082301A (en) | 2012-11-13 | 2015-07-15 | 다이쇼 세이야꾸 가부시끼가이샤 | 2-pyridone compound |
| US9695148B2 (en) | 2012-11-13 | 2017-07-04 | Nissan Chemical Industries, Ltd. | 2-pyridone compound |
| US11690841B2 (en) | 2017-09-14 | 2023-07-04 | Queen Mary University Of London | Glycolysis-activating agents for treatment or prevention of disease |
| US11833136B2 (en) | 2018-06-12 | 2023-12-05 | Vtv Therapeutics Llc | Therapeutic uses of glucokinase activators in combination with insulin or insulin analogs |
| US11974989B2 (en) | 2018-06-12 | 2024-05-07 | Vtv Therapeutics Llc | Therapeutic uses of glucokinase activators in combination with insulin or insulin analogs |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1242397B1 (en) | Trans olefinic glucokinase activators | |
| EP1169312B1 (en) | Glucokinase activators | |
| EP1311504B1 (en) | Tetrazolyl-phenyl acetamide glucokinase activators | |
| US6320050B1 (en) | Heteroaromatic glucokinase activators | |
| US6610846B1 (en) | Heteroaromatic glucokinase activators | |
| EP1282611B1 (en) | Substituted phenylacetamides and their use as glucokinase activators | |
| US6528543B1 (en) | Urea derivatives | |
| AU2001283998A1 (en) | Tetrazolyl-phenyl acetamide glucokinase activators | |
| EP1501815A1 (en) | Substituted phenylacetamides and their use as glucokinase activators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: P-414/02 Country of ref document: YU |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2002/03829 Country of ref document: ZA Ref document number: 200203829 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 518974 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 23652/01 Country of ref document: AU Ref document number: 2392903 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 150083 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2000987392 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: IN/PCT/2002/879/CHE Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 2001 544706 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P20020514A Country of ref document: HR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2002/005874 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020027007709 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 008172943 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV2002-2412 Country of ref document: CZ |
|
| ENP | Entry into the national phase |
Ref document number: 2002 2002118339 Country of ref document: RU Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020027007709 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2000987392 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2002-2412 Country of ref document: CZ |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 518974 Country of ref document: NZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 518974 Country of ref document: NZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1020027007709 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 23652/01 Country of ref document: AU |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2000987392 Country of ref document: EP |