WO2001029004A1 - Oxidation process for the preparation of intermediates useful in the synthesis of diarylpyridines - Google Patents
Oxidation process for the preparation of intermediates useful in the synthesis of diarylpyridines Download PDFInfo
- Publication number
- WO2001029004A1 WO2001029004A1 PCT/EP2000/009995 EP0009995W WO0129004A1 WO 2001029004 A1 WO2001029004 A1 WO 2001029004A1 EP 0009995 W EP0009995 W EP 0009995W WO 0129004 A1 WO0129004 A1 WO 0129004A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- acid
- preparation
- formula
- synthesis
- oxidant
- Prior art date
Links
- 0 *Cc1ccc(*)cc1 Chemical compound *Cc1ccc(*)cc1 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/44—Radicals substituted by doubly-bound oxygen, sulfur, or nitrogen atoms, or by two such atoms singly-bound to the same carbon atom
- C07D213/46—Oxygen atoms
- C07D213/50—Ketonic radicals
Definitions
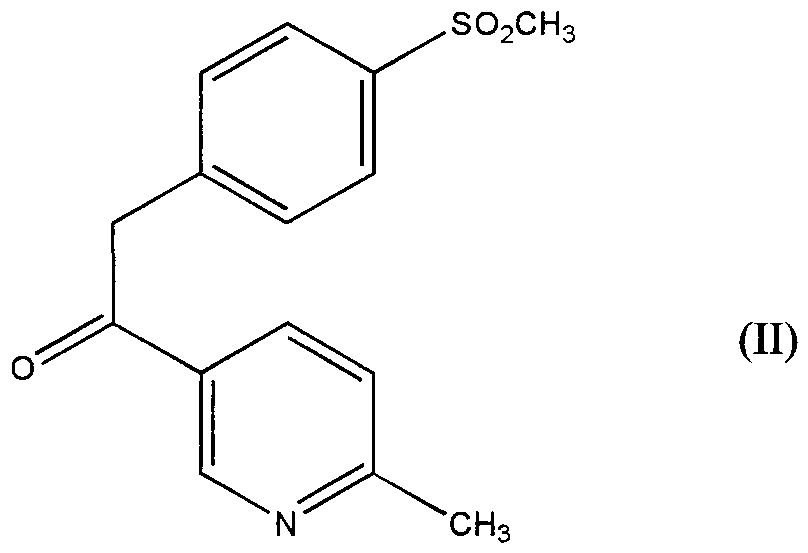
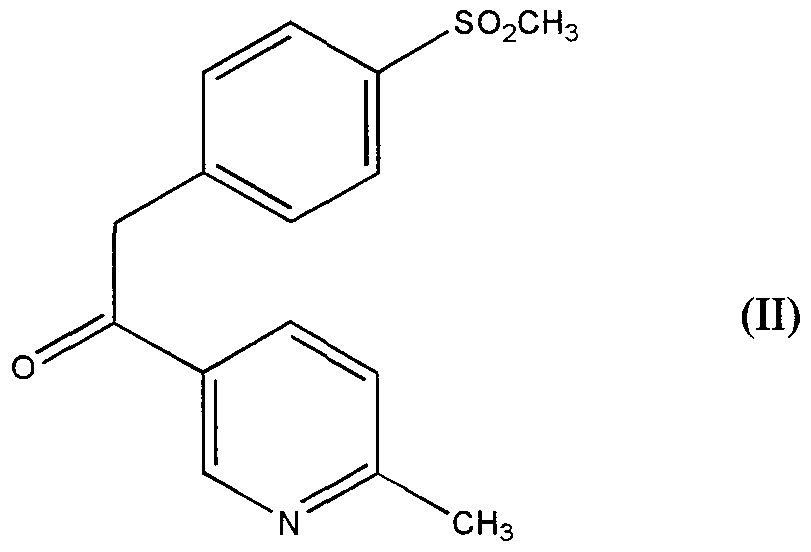
- the present invention relates to an oxidation process for the preparation of intermediates useful m the synthesis of diarylpy ⁇ dines and, more particularly, it relates to an oxidation process for the preparation of intermediates useful in the synthesis of compounds of formula
- R is chlo ⁇ ne, fluorine, bromine, iodine, CN or azide. useful as cyclooxygenase-2
- the oxidation reaction is earned out by using different oxidation systems such as hydrogen peroxide, oxone ® (2KHS0 5 * KHS0 4 -K 2 S0 4 ) or hydrogen peroxide/acetic acid, preferably by usmg oxone ® or hydrogen peroxide, m the presence of a catalyst, preferably Na W0 4 , under acid conditions
- a catalyst preferably Na W0 4
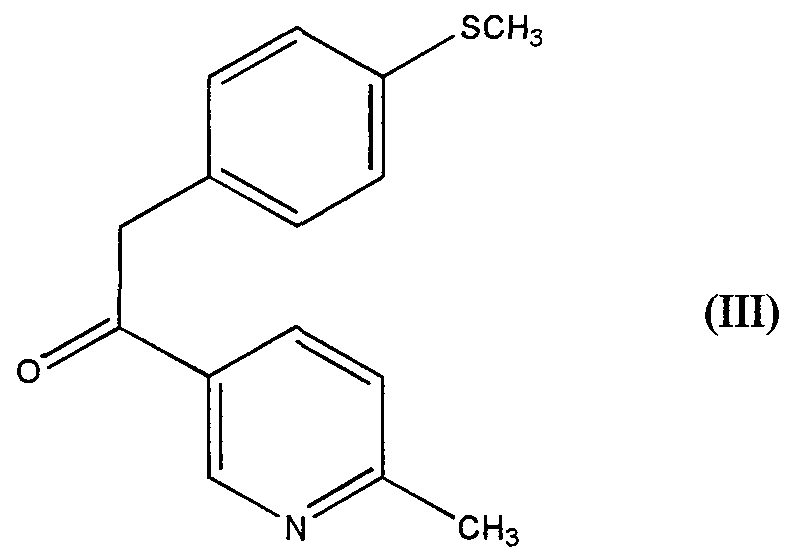
- a further oxidable function the nitrogen atom of py ⁇ dine
- object of the present mvention is a process for the preparation of the compound of formula
- the oxidant is used in excess and is a mixture of peracetic acid and hydrogen peroxide in a suitable solvent
- Methanesulfomc acid is used in slight excess with respect to the compound III, preferably in a molar ratio from 1 1 to 1 4
- Prefe ⁇ ed catalyst is Na 2 W0 4
- the oxidation process object of the present invention allows to selectively oxidize the methylthio group to methylsulfone group with good yields and without significant amounts of N-oxide as by-product This is a remarkable advantage m compa ⁇ son with the known processes wherein the obtained amount of N-oxide is about 1%, so that one or more punfication steps are required
- Example 1 the following examples are now given Example 1
Abstract
Description






Claims
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU79182/00A AU7918200A (en) | 1999-10-15 | 2000-10-11 | Oxidation process for the preparation of intermediates useful in the synthesis of diarylpyridines |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IT1999MI002156A IT1315243B1 (en) | 1999-10-15 | 1999-10-15 | OXIDATION PROCESS FOR THE PREPARATION OF USEFUL INTERMEDIATES IN THE SYNTHESIS OF DIARYLPYRIDIN |
| ITMI99A002156 | 1999-10-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2001029004A1 true WO2001029004A1 (en) | 2001-04-26 |
Family
ID=11383782
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2000/009995 WO2001029004A1 (en) | 1999-10-15 | 2000-10-11 | Oxidation process for the preparation of intermediates useful in the synthesis of diarylpyridines |
Country Status (3)
| Country | Link |
|---|---|
| AU (1) | AU7918200A (en) |
| IT (1) | IT1315243B1 (en) |
| WO (1) | WO2001029004A1 (en) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003051843A1 (en) * | 2001-12-19 | 2003-06-26 | Zambon Group S.P.A. | Oxidation process for the preparation of intermediates useful in the synthesis of diarylpyridines |
| WO2005016906A1 (en) * | 2003-08-14 | 2005-02-24 | Shasun Chemicals And Drugs Limited | Process for the manufacture of rofecoxib |
| WO2007039127A1 (en) * | 2005-09-26 | 2007-04-12 | Solmag S.P.A. | A process for the preparation and purification of bicalutamide |
| ITMI20110362A1 (en) * | 2011-03-09 | 2012-09-10 | F I S Fabbrica Italiana Sint P A | PROCEDURE FOR THE PREPARATION OF 1- (6-METHYLPYRIDIN-3-IL) -2- [4- (METHYLSOLFONYL) PHENYL] ETHANONE, AN INTERMEDIATE OF THE ETHORICOXIB. |
| EP2551265A1 (en) | 2011-07-29 | 2013-01-30 | F.I.S.- Fabbrica Italiana Sintetici S.p.A. | Novel process for the preparation of 1-(6-methylpyridin-3-yl)-2-[4-(methylsulfonyl)phenyl]ethanone, an intermediate of etoricoxib. |
| WO2014072143A1 (en) * | 2012-11-08 | 2014-05-15 | Evonik Industries Ag | Method for producing equilibrium peracetic acid and equilibrium peracetic acid obtainable by the method |
| ITMI20121947A1 (en) * | 2012-11-15 | 2014-05-16 | Erregierre Spa | PROCESS OF SYNTHESIS OF AN INTERMEDIATE IN THE PRODUCTION OF ETORICOXIB |
| CN108689917A (en) * | 2017-04-08 | 2018-10-23 | 深圳市华先医药科技有限公司 | A kind of Etoricoxib intermediate continuous flow production technology |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999015503A2 (en) * | 1997-09-25 | 1999-04-01 | Merck & Co., Inc. | Process for making diaryl pyridines useful as cox-2 inhibitors |
-
1999
- 1999-10-15 IT IT1999MI002156A patent/IT1315243B1/en active
-
2000
- 2000-10-11 WO PCT/EP2000/009995 patent/WO2001029004A1/en active Application Filing
- 2000-10-11 AU AU79182/00A patent/AU7918200A/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999015503A2 (en) * | 1997-09-25 | 1999-04-01 | Merck & Co., Inc. | Process for making diaryl pyridines useful as cox-2 inhibitors |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003051843A1 (en) * | 2001-12-19 | 2003-06-26 | Zambon Group S.P.A. | Oxidation process for the preparation of intermediates useful in the synthesis of diarylpyridines |
| WO2005016906A1 (en) * | 2003-08-14 | 2005-02-24 | Shasun Chemicals And Drugs Limited | Process for the manufacture of rofecoxib |
| WO2007039127A1 (en) * | 2005-09-26 | 2007-04-12 | Solmag S.P.A. | A process for the preparation and purification of bicalutamide |
| EP1777216A1 (en) * | 2005-09-26 | 2007-04-25 | SOLMAG S.p.A. | A process for the preparation and purification of bicalutamide |
| US8664402B2 (en) | 2011-03-09 | 2014-03-04 | F.I.S. Fabbrica Italiana Sintetici S.P.A. | Process for preparing 1-(6-methylpyridin-3-yl)-2-[4-(methylsulfonyl)phenyl]ethanone, an intermediate of etoricoxib |
| EP2497767A1 (en) | 2011-03-09 | 2012-09-12 | F.I.S. Fabbrica Italiana Sintetici S.p.A. | Improved process for preparing 1-(6-methylpyridin-3-yl)-2-[4-(methylsulfonyl)phenyl]ethanone, an intermediate of etoricoxib |
| CN102731374A (en) * | 2011-03-09 | 2012-10-17 | 意大利合成制造有限公司 | Improved process for preparing 1-(6-methylpyridin-3-yl)-2-[4-(methylsulfonyl)phenyl]ethanone, an intermediate of etoricoxib |
| ITMI20110362A1 (en) * | 2011-03-09 | 2012-09-10 | F I S Fabbrica Italiana Sint P A | PROCEDURE FOR THE PREPARATION OF 1- (6-METHYLPYRIDIN-3-IL) -2- [4- (METHYLSOLFONYL) PHENYL] ETHANONE, AN INTERMEDIATE OF THE ETHORICOXIB. |
| CN102731374B (en) * | 2011-03-09 | 2015-05-06 | 意大利合成制造有限公司 | Improved process for preparing 1-(6-methylpyridin-3-yl)-2-[4-(methylsulfonyl)phenyl]ethanone, an intermediate of etoricoxib |
| EP2551265A1 (en) | 2011-07-29 | 2013-01-30 | F.I.S.- Fabbrica Italiana Sintetici S.p.A. | Novel process for the preparation of 1-(6-methylpyridin-3-yl)-2-[4-(methylsulfonyl)phenyl]ethanone, an intermediate of etoricoxib. |
| WO2014072143A1 (en) * | 2012-11-08 | 2014-05-15 | Evonik Industries Ag | Method for producing equilibrium peracetic acid and equilibrium peracetic acid obtainable by the method |
| CN104684892A (en) * | 2012-11-08 | 2015-06-03 | 赢创工业集团股份有限公司 | Method for producing equilibrium peracetic acid and equilibrium peracetic acid obtainable by the method |
| US9573893B2 (en) | 2012-11-08 | 2017-02-21 | Evonik Degussa Gmbh | Method for producing equilibrium peracetic acid and equilibrium peracetic acid obtainable by the method |
| AU2013343772B2 (en) * | 2012-11-08 | 2017-06-15 | Evonik Operations Gmbh | Method for producing equilibrium peracetic acid and equilibrium peracetic acid obtainable by the method |
| CN104684892B (en) * | 2012-11-08 | 2017-08-29 | 赢创德固赛有限公司 | The preparation method of equilibrium peracetic acid and the equilibrium peracetic acid obtained by this method |
| ITMI20121947A1 (en) * | 2012-11-15 | 2014-05-16 | Erregierre Spa | PROCESS OF SYNTHESIS OF AN INTERMEDIATE IN THE PRODUCTION OF ETORICOXIB |
| CN108689917A (en) * | 2017-04-08 | 2018-10-23 | 深圳市华先医药科技有限公司 | A kind of Etoricoxib intermediate continuous flow production technology |
Also Published As
| Publication number | Publication date |
|---|---|
| ITMI992156A0 (en) | 1999-10-15 |
| ITMI992156A1 (en) | 2001-04-15 |
| AU7918200A (en) | 2001-04-30 |
| IT1315243B1 (en) | 2003-02-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2001237452B9 (en) | Method for oxidizing a thioether group into a sulfoxide group | |
| WO2001029004A1 (en) | Oxidation process for the preparation of intermediates useful in the synthesis of diarylpyridines | |
| EP1335913B1 (en) | A process for the preparation of pantoprazole and intermediates therefor | |
| WO2007034972A1 (en) | Method for recovering tungsten | |
| WO2007091391A1 (en) | Improved method for producing nitroguanidine derivative | |
| WO2001021617A1 (en) | Process for preparing sulfoxide compounds | |
| WO2007117027A1 (en) | Process for the production of organic oxides | |
| WO2007012183A1 (en) | Process for the production of bicalutamide | |
| JP5269031B2 (en) | Method for producing tribromomethylsulfonylpyridine | |
| US20050165238A1 (en) | Oxidation process for the preparation of intermediates useful in the synthesis of diarylpyridines | |
| JP6528772B2 (en) | Process for producing 3- (alkylsulfonyl) pyridine-2-carboxylic acid | |
| JPH07116153B2 (en) | Process for producing pyridine-2,3-dicarboxylic acid | |
| HU225921B1 (en) | Process and intermediates for preparing 3-(1-piperazinyl)-1,2-benzisothiazole | |
| WO2004039769A1 (en) | A process for the production of highly pure n-(4-cyano-3trifluoromethylphenyl)-3-(4-fluorophenylthio)-2-hydroxy-2methylpropanamide | |
| DK165832B (en) | METHOD OF PREPARING 2-CARBOXY-PYRAZINE-4-OXIDES | |
| JP2000344751A (en) | Production of 2,3-pyridinedicarboxylic acid compound | |
| WO2008032453A1 (en) | Method for producing hydrazone | |
| JP4000816B2 (en) | Method for producing 3-methyladipic acid | |
| EP1777216A1 (en) | A process for the preparation and purification of bicalutamide | |
| KR100537385B1 (en) | Process for preparing sodium thiofuroate | |
| JPH01238574A (en) | Production of guanidine derivative | |
| JPH02124872A (en) | Production of 4-alkylthiopyridine-n-oxide derivative |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| 122 | Ep: pct application non-entry in european phase | ||
| NENP | Non-entry into the national phase |
Ref country code: JP |