WO2001004100A1 - Process for preparing 2-amino-4-(4-fluorphenyl)-6-alkylpyrimidine-5-carboxylate - Google Patents
Process for preparing 2-amino-4-(4-fluorphenyl)-6-alkylpyrimidine-5-carboxylate Download PDFInfo
- Publication number
- WO2001004100A1 WO2001004100A1 PCT/EP2000/006099 EP0006099W WO0104100A1 WO 2001004100 A1 WO2001004100 A1 WO 2001004100A1 EP 0006099 W EP0006099 W EP 0006099W WO 0104100 A1 WO0104100 A1 WO 0104100A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- formula
- general formula
- fluorophenyl
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(C)c1nc(N)nc(-c(cc2)ccc2F)c1*(O*)=N Chemical compound CC(C)c1nc(N)nc(-c(cc2)ccc2F)c1*(O*)=N 0.000 description 3
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/42—One nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C229/00—Compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C229/02—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton
- C07C229/34—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/01—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms
- C07C311/02—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
- C07C311/03—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton having the nitrogen atoms of the sulfonamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C311/05—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton having the nitrogen atoms of the sulfonamide groups bound to hydrogen atoms or to acyclic carbon atoms to acyclic carbon atoms of hydrocarbon radicals substituted by nitrogen atoms, not being part of nitro or nitroso groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
Definitions
- the present invention relates to a novel process for the preparation of compounds of the general formula
- Compounds of the formula I are important intermediates for the preparation of pharmaceutically active compounds, for example of HMG-Co A reductase inhibitors.
- Japanese Patent publication JP-A 06 256318, and Watanabe M. et al., Bioorg. Med. Chem. 1997, Vol. 5, No. 2, 437-444 describe processes for the preparation of compounds of the formula I.
- JP-A 06 256318 has the disadvantage that three stages are needed in order to prepare 2-amino-4- (4-fluorophenyl) -6-isopropyl-pyri- midine-5-carboxylic acid.
- p-fluorobenzaldehyde is converted using ethyl iso- butyrylacetate into an unsaturated ketoester, which is then cyclocondensed in the second stage with S- methylisothiourea hydrogensulphate and subsequently dehydrated in the third stage to give the corresponding pyrimidine.
- this is then oxidized using m-chloroperbenzoic acid to give the corresponding sulphonylpyrimidine, which is then reacted in the fifth stage with methylamine and subsequent treatment with methanesulphonyl chloride to give ethyl 4- (4-fluorophenyl) -6-isopropyl-2- (N-methanesulphonyl-N-methyl- amino) pyrimidine-5-carboxylate.
- the object of the invention was to make available an economical, industrially feasible process for the preparation of compounds of the formula I.
- R is hydrogen or a group of the formula -S0 2 R x ;
- R 1 is Ci-e-alkyl;
- R 2 is hydrogen or C ⁇ _ 6 -alkyl
- R 3 is Ci-e-alkyl
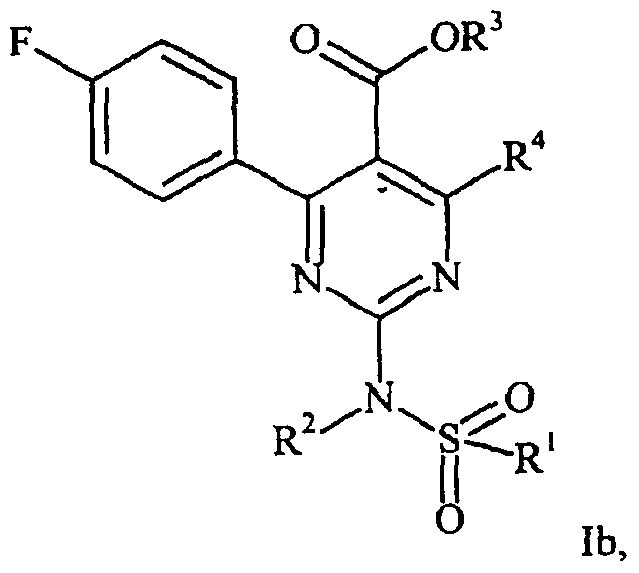
- R 4 is Ci-e-alkyl, are prepared in that, in a first stage, a compound of the general formula in which R 3 and R 4 have the abovementioned meaning, is reacted in the presence of a Lewis acid with 4-fluorobenzonitrile to give a compound of the general formula
- C ⁇ - 6 -alkyl is understood here and below as meaning linear and branched alkyl groups having 1-6 carbon atoms, such as, for example, methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, isobutyl, sec-butyl, pentyl and its iso ers and hexyl and its isomers.
- the compounds of the formula II can be prepared according to Chem . Berichte 1958, 91 , 759 or are commercially available organic synthetic chemicals. 4-Fluorobenzonitrile is a commercially available organic synthetic chemical.
- the Lewis acid employed in the first stage is expediently an aprotic Lewis acid such as, for example, tin tetrachloride, titanium tetrachloride or aluminium chloride. Tin tetrachloride is preferably employed.
- the first stage is expediently carried out in the presence of an organic solvent.
- the organic solvents employed can be, for example, aromatic hydrocarbons, chlorinated aromatic and aliphatic hydrocarbons.
- Aromatic hydrocarbons employed are preferably toluene, benzene or xylene.
- the chlorinated aromatic hydrocarbon employed is preferably chlorobenzene; the chlorinated aliphatic hydrocarbon employed is preferably 1, 2-dichloroethane .
- 1, 2-dichloroethane are particularly preferably employed.
- the reaction in the first stage is expediently carried out at a temperature from -5 to 140 °C, advantageously at 60 to 100 °C.
- the compounds of the formula III can be isolated by known methods such as, for example, by extraction or can be employed directly, without isolation, for the second stage.
- the intermediate (formula III) is preferably isolated.
- the invention comprises, on the one hand, compounds of the formula I in which R and R 2 are hydrogen. These compounds are prepared by reaction of compounds of the formula III with cyanamide .
- the reaction with cyanamide is expediently carried out in the presence of an organic solvent, a mixture of water with an organic solvent or in water.
- Water is particularly preferably employed.
- Organic solvents employed are advantageously toluene or ethyl acetate.
- Organic solvents employed as a mixture with water are advantageously alcohols such as, for example, methanol, ethers such as, for example, dioxane or aromatic hydrocarbons such as, for example, toluene or N,W-dimethylacetamide.
- the reaction with cyanamide is expediently carried out at a temperature of 10 to 120°C, advantageously at 40 to 100 °C.
- the pH is expediently in a range from 3 to 9, advantageously in a range from 4 to 7.
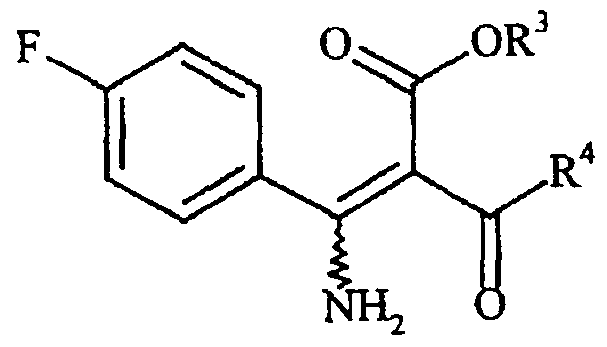
- R 3 has the meaning indicated in formula I are prepared in that, in a first stage, an alkyl iso- butyrylacetate of the general formula in which R has the meaning mentioned is reacted in the presence of a Lewis acid with 4-fluorobenzonitrile to give a 2- [1-amino-l- (4-fluorophenyl)methylene] -4- methyl-3-oxopentanoic acid ester of the general formula
- the radical R 3 is preferably methyl.
- the compounds of the formula III are novel and also a subject of the invention.
- the invention comprises, on the other hand, compounds of the formula I in which R is a group of the formula -S0 2 R 1 and R 1 and R 2 are C ⁇ _ 6 -alkyl.
- R is a group of the formula -S0 2 R 1 and R 1 and R 2 are C ⁇ _ 6 -alkyl.
- These 4- (4-fluorophenyl) -6-alkyl-2- (N-alkanesulphonyl-W- alkylamino) pyrimidine-5-carboxylic acid esters of the general formula in which R 1 , R 2 , R 3 and R 4 are identical or different and are a C ⁇ _ 6 -alkyl group can be prepared in that 2- [1-amino-l- (4-fluorophenyl)methylene] -4-alkyl-3-oxo- alkanoic acid esters of the general formula
- R 1 and R 2 are a C ⁇ - 6 -alkyl group.
- the reaction can be carried out either in the presence of a base or in the presence of a Lewis acid.
- Bases which can be employed are alkali metal compounds such as, for example, alkali metal hydrides, alkali metal carbonates, alkali metal alkoxides or alkali metal silazanes.
- Alkali metal carbonates which can be used are lithium, sodium or potassium carbonate.
- the alkali metal hydride employed can be potassium, lithium or sodium hydride; sodium hydride is preferably employed.
- the alkali metal alkoxide employed can be sodium or potassium tert-pentoxide or sodium or potassium tert-butoxide, preferably sodium tert- pentoxide or sodium tert-butoxide.
- the alkali metal silazane used can be sodium hexamethyldisilazane or potassium hexamethyldisilazane.
- the base preferably employed is an alkali metal hydride or an alkali metal alkoxide .
- the reaction is expediently carried out in the presence of a base in a polar organic solvent.
- the polar solvent used can be, for example, N,N-dimethyl- acetamide, isopropanol, tert-butanol, toluene, di- methylformamide, tetrahydrofuran, 1,4-dioxane or mixtures of these.
- N-Alkylalkanesulphonamides such as, for example, W-methylmethanesulphonamide are likewise suitable as solvents.
- the reaction is preferably carried out in W-alkylalkanesulphonamide and tert- butanol.
- the reaction can be carried out in the presence of a base at a temperature from -10 to 150 °C, preferably from 0 to 80°C.
- the reaction of compounds of the formula Illb with compounds of the formula IVb in the presence of a Lewis acid is expediently carried out in the solvent which is inert to the Lewis acid.
- Inert solvents which can be employed are, for example, aromatic hydrocarbons, and chlorinated aromatic and aliphatic hydrocarbons.
- Aromatic hydrocarbons employed are preferably toluene or xylene.
- the chlorinated aromatic hydrocarbon employed is preferably chlorobenzene; chlorinated aliphatic hydrocarbons employed are preferably dichloromethane or 1, 2-dichloroethane .
- the reaction in the presence of a Lewis acid can be carried out at a temperature from 20 to 150°C, preferably from 80 to 120°C.
- Suitable Lewis acids are for example TiCl 4 ,
- Titanium tetrachloride is preferred.
- the amount of Lewis acid is 0.1 to 2 molar equivalents based on the compound of the formula Illb.
- R 1 and R 2 are C ⁇ -6-alkyl
- Suitable bases are the bases described beforehand.
- the cyanogen halide employed can be cyanogen fluoride, cyanogen chloride, cyanogen bromide or cyanogen iodide. Cyanogen chloride or cyanogen bromide is preferably employed. This reaction can likewise be carried out in the polar organic solvents described beforehand. The reaction is preferably carried out in tetrahydrofuran.
- the reaction is expediently carried out at a temperature from -20 to 50°C, preferably at a temperature from -10 to +20°C.
- Examples of compounds of the formula IVb are: N-cyano-W-methylmethanesulphonamide, N-cyano-iV-ethyl- methanesulphonamide, W-cyano-N-propylmethanesulphon- amide, N-cyano-N-butylmethanesulphonamide, ⁇ J-cyano-N- pentylmethanesulphonamide and N-cyano-N-hexylmethane- sulphonamide.
- N-Cyano-N-methylmethanesulphonamide is preferred.
- the compounds of the formula I can also be prepared in that a compound of the general formula in which R 3 and R 4 have the meaning mentioned in Claim 1, is reacted with a compound of the formula IV.
- reaction is carried out analogously to the reaction of the compounds of the formula III with compounds of the formula IV, preferably in the presence of a base in a polar organic solvent at a temperature from -10 to 150°C.
- Suitable bases and solvents correspond to the bases and solvents which are listed under the reaction of compounds of the formula III with compounds of the formula IV.
- compounds of the general formula lb are prepared in that a compound of the formula VI is reacted with a compound of the formula IVb in a polar organic solvent at a temperature from 0 to 80°C in the presence of a base.
- the compounds of the formula VI can be prepared by reaction of C ⁇ - 6 -alkyl nitriles with C ⁇ _ 6 -alkyl 4-fluorobenzoylacetate in the presence of a Lewis acid.
- a compound of the formula VI in which R 3 is methyl and R 4 is isopropyl is preferred.
- the Lewis acid is preferably tin tetrachloride.
- the reaction is expediently carried out in a polar solvent. Suitable solvents correspond to the solvents which are listed under the reaction of compounds of the formula II with compounds of the formula III described above.
- the reaction in the first stage is expediently carried out at a temperature from -5 to 140°C, advantageously at 60 to 100°C.
- the compounds of the formula VI are novel and likewise a subject of the invention.
- R 3 methyl, toluene, SnCl 4
- R 3 methyl, toluene, SnCl 4 22.53 g of methyl isobutyrylacetate (0.15 mol, concentration 96%) and 18.54 g of 4-fluorobenzonitrile (0.15 mol, concentration 98%) were dissolved in 150 ml of toluene and treated with 43.32 g of tin tetrachloride (0.165 mol, concentration 99%) at room temperature over the course of 12 min. After half an hour at room temperature, the mixture was heated to 80°C. After 3 h, the suspension was cooled to room temperature and treated with 150 ml of water. It was diluted with 100 ml of ethyl acetate and the phases were separated.
- R 3 methyl, 1, 2-dichloroethane, A1C1 3 754 mg of methyl isobutyrylacetate (5.00 mmol, concentration 96%) and 618 mg of 4-fluorobenzonitrile (5.00 mmol, concentration 98%) were introduced into 5 ml of 1, 2-dichloroethane and treated with 673 mg of aluminium chloride (5.00 mol) at room temperature. After one hour at room temperature, the mixture was heated to 80°C. After 19 h - the mixture contained 13.9 area per cent of product according to HPLC analysis - the mixture was cooled to room temperature and treated with water (5 ml) .
- R 3 methyl, toluene, SnCl 4
- the solid contained an 84 : 16 product/starting material mixture, which corresponds to a yield of 27% of methyl 4- (4-fluorophenyl) -6-isopropyl-2- ( -methanesulphonyl-W- methylamino) pyrimidine-5-carboxylate .
- the reaction progressed exothermically.
- the red-orange- coloured suspension was heated to 110 °C and stirred for 4.5 h. It was then cooled to room temperature and treated with 30 ml of water.
- the organic phase was separated off and the aqueous phase was extracted with ethyl acetate (30 ml) .
- the combined organic phases were washed with 30 ml of water and dried over magnesium sulphate. After filtering and concentrating the solution in a water-jet vacuum and drying in vacuo, 11.54 g of crude product were obtained in the form of a tacky oil.
- the organic phase was separated off and the aqueous phase was extracted with methylene chloride (2 x 30 ml) .
- the combined organic phases were washed with 30 ml of water and dried over magnesium sulphate. After filtering and concentrating the solution in a water-jet vacuum and drying in vacuo, 12.02 g of crude product were obtained in the form of a tacky oil.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description













Claims















Priority Applications (14)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AT00949231T ATE307120T1 (en) | 1999-07-13 | 2000-06-30 | METHOD FOR PRODUCING 2-AMINO-4(4-FLUOROPHENYL)-6-ALKYLPYRIMIDINE-5-CARBONIC ACID RESIDUALS |
| HK02108215.0A HK1046682B (en) | 1999-07-13 | 2000-06-30 | Process for preparing 2-amino-4-(4-fluorphenyl)-6-alkylpyrimidine-5-carboxylate |
| SK22-2002A SK285993B6 (en) | 1999-07-13 | 2000-06-30 | Process for producing derivatives of pyrimidine substituted by amino group |
| HU0202006A HU228303B1 (en) | 1999-07-13 | 2000-06-30 | Process for preparing 2-amino-4-(4-fluorphenyl)-6-alkylpyrimidine-5-carboxylate |
| DE60023296T DE60023296T2 (en) | 1999-07-13 | 2000-06-30 | PROCESS FOR THE PREPARATION OF 2-AMINO-4 (4-FLUORPHENYL) -6-ALKYLPYRIMIDINE-5-CARBOXYLIC ACID ESTERS |
| ES00949231T ES2251392T3 (en) | 1999-07-13 | 2000-06-30 | PROCEDURE TO PREPARE 2-AMINO-4- (4-FLUOROPHENYL) -6-ALKYL PYRIMIDINE-5-CARBOXYLATES. |
| US10/030,077 US6579984B1 (en) | 1999-07-13 | 2000-06-30 | Process for preparing 2-amino-4-(4-fluorphenyl)-6-alkylpyrimidine-5-carboxylate |
| CA002378782A CA2378782C (en) | 1999-07-13 | 2000-06-30 | Process for preparing 2-amino-4-(4-fluorphenyl)-6-alkylpyrimidine-5-carboxylate |
| JP2001509711A JP4649813B2 (en) | 1999-07-13 | 2000-06-30 | Process for producing 2-amino-4- (4-fluorophenyl) -6-alkylpyrimidine-5-carboxylate |
| EP00949231A EP1194414B1 (en) | 1999-07-13 | 2000-06-30 | Process for preparing 2-amino-4-(4-fluorphenyl)-6-alkylpyrimidine-5-carboxylate |
| AU62666/00A AU6266600A (en) | 1999-07-13 | 2000-06-30 | Process for preparing 2-amino-4-(4-fluorphenyl)-6-alkylpyrimidine-5-carboxylate |
| NO20020163A NO328103B1 (en) | 1999-07-13 | 2002-01-11 | Process for the preparation of 2-amino-4- (4-fluorophenyl) -6-alkylpyrimidine-5-carboxylates and intermediates thereof |
| US10/806,315 US6984757B2 (en) | 1999-07-13 | 2004-03-23 | Process for preparing 2-amino-4-(4-fluorphenyl)-6-alkylpyrimidine-5-carboxylate |
| NO20092393A NO334937B1 (en) | 1999-07-13 | 2009-06-23 | Process for the preparation of N-cyano-N-alkylalkanesulfonamides and 4-phenylpyrimidines and such compounds |
Applications Claiming Priority (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP99113711.8 | 1999-07-13 | ||
| EP99113711 | 1999-07-13 | ||
| EP99120417.3 | 1999-10-14 | ||
| EP99120417 | 1999-10-14 | ||
| US18546500P | 2000-02-28 | 2000-02-28 | |
| US18537100P | 2000-02-28 | 2000-02-28 | |
| US60/185,465 | 2000-02-28 | ||
| US60/185,371 | 2000-02-28 | ||
| EP00106303.1 | 2000-03-23 | ||
| EP00106303 | 2000-03-23 |
Related Child Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10030077 A-371-Of-International | 2000-06-30 | ||
| US10/030,077 A-371-Of-International US6579984B1 (en) | 1999-07-13 | 2000-06-30 | Process for preparing 2-amino-4-(4-fluorphenyl)-6-alkylpyrimidine-5-carboxylate |
| US10/443,797 Division US6710178B2 (en) | 1999-07-13 | 2003-05-23 | Preparation of 4-(4-flourophenyl)-6-alkyl-2-N-alkansulfonyl-N-alkylamino)pyrimidine-5-carboxylic acid ester |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2001004100A1 true WO2001004100A1 (en) | 2001-01-18 |
Family
ID=27513029
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2000/006099 Ceased WO2001004100A1 (en) | 1999-07-13 | 2000-06-30 | Process for preparing 2-amino-4-(4-fluorphenyl)-6-alkylpyrimidine-5-carboxylate |
Country Status (17)
| Country | Link |
|---|---|
| US (3) | US6579984B1 (en) |
| EP (1) | EP1194414B1 (en) |
| JP (1) | JP4649813B2 (en) |
| KR (1) | KR100649927B1 (en) |
| CN (1) | CN1164577C (en) |
| AT (1) | ATE307120T1 (en) |
| AU (1) | AU6266600A (en) |
| CA (1) | CA2378782C (en) |
| CZ (1) | CZ304307B6 (en) |
| DE (1) | DE60023296T2 (en) |
| ES (1) | ES2251392T3 (en) |
| HK (1) | HK1046682B (en) |
| HU (1) | HU228303B1 (en) |
| NO (2) | NO328103B1 (en) |
| PL (1) | PL211797B1 (en) |
| SK (1) | SK285993B6 (en) |
| WO (1) | WO2001004100A1 (en) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001060804A1 (en) * | 2000-02-15 | 2001-08-23 | Astrazeneca Ab | Crystalline salts of 7-[4-(4-fluorophenyl)-6-isopropyl-2-[methyl(methylsulfonyl)amino]pyrimidin-5-yl]-(3r,5s)-3,5-dihydroxyhept-6-enoic acid |
| WO2004054986A2 (en) | 2002-12-16 | 2004-07-01 | Astrazeneca Uk Limited | Process for the preparation of pyrimidine compounds |
| CN1301977C (en) * | 2001-07-13 | 2007-02-28 | 阿斯特拉曾尼卡英国有限公司 | The preparation method of aminopyrimidine compound |
| RU2301801C2 (en) * | 2001-07-13 | 2007-06-27 | АстраЗенека Ю-Кей Лимитед | Method for preparing sulfonulaminopyrimidine compounds (variants), intermediate substances and methods for their preparing |
| CN100361979C (en) * | 2002-08-13 | 2008-01-16 | 阿斯利康(英国)有限公司 | Preparation method of rosuvastatin calcium salt |
| US8034932B2 (en) | 2004-12-24 | 2011-10-11 | Astrazeneca Uk Limited | Chemical process |
| WO2012013325A1 (en) | 2010-07-26 | 2012-02-02 | Lek Pharmaceuticals D.D. | Process for the preparation of key intermediates for the synthesis of statins or pharmaceutically acceptable salts thereof |
| US8609717B2 (en) | 2010-08-18 | 2013-12-17 | Samumed, Llc | β- and γ-diketones and γ-hydroxyketones as WNT/β-catenin signaling pathway activators |
| US9533976B2 (en) | 2013-02-22 | 2017-01-03 | Samumed, Llc | γ-diketones as WNT/β-catenin signaling pathway activators |
| US9795550B2 (en) | 2014-08-20 | 2017-10-24 | Samumed, Llc | Gamma-diketones for treatment and prevention of aging skin and wrinkles |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103910644B (en) * | 2014-04-21 | 2015-06-24 | 广西师范大学 | Beta-enaminone ester compounds, and synthesis method and application of beta-enaminone ester compounds |
| CN110483341A (en) * | 2019-07-23 | 2019-11-22 | 上海药明康德新药开发有限公司 | A kind of synthetic method of 1- cyano-N-methyl Methanesulfomide |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4013706A (en) * | 1973-11-14 | 1977-03-22 | Sucreries Du Soissonnais Et Compagnie Sucriere | Derivatives of substituted urea, acyl ureas, and sulphonyl ureas, and a process for producing the same |
| DE3717815A1 (en) * | 1987-05-27 | 1988-12-15 | Bayer Ag | Fungicidal compositions based on substituted cyanamides |
| EP0521471A1 (en) * | 1991-07-01 | 1993-01-07 | Shionogi Seiyaku Kabushiki Kaisha | Pyrimidine derivatives as HMG-CoA reductase inhibitors |
| JPH06256318A (en) * | 1993-03-01 | 1994-09-13 | Shionogi & Co Ltd | Method for synthesizing 5-carboalkoxypyrimidine derivative |
-
2000
- 2000-06-30 US US10/030,077 patent/US6579984B1/en not_active Expired - Lifetime
- 2000-06-30 SK SK22-2002A patent/SK285993B6/en not_active IP Right Cessation
- 2000-06-30 DE DE60023296T patent/DE60023296T2/en not_active Expired - Lifetime
- 2000-06-30 AU AU62666/00A patent/AU6266600A/en not_active Abandoned
- 2000-06-30 JP JP2001509711A patent/JP4649813B2/en not_active Expired - Lifetime
- 2000-06-30 CN CNB008116768A patent/CN1164577C/en not_active Expired - Lifetime
- 2000-06-30 AT AT00949231T patent/ATE307120T1/en active
- 2000-06-30 EP EP00949231A patent/EP1194414B1/en not_active Expired - Lifetime
- 2000-06-30 HU HU0202006A patent/HU228303B1/en not_active IP Right Cessation
- 2000-06-30 WO PCT/EP2000/006099 patent/WO2001004100A1/en not_active Ceased
- 2000-06-30 CA CA002378782A patent/CA2378782C/en not_active Expired - Lifetime
- 2000-06-30 HK HK02108215.0A patent/HK1046682B/en not_active IP Right Cessation
- 2000-06-30 ES ES00949231T patent/ES2251392T3/en not_active Expired - Lifetime
- 2000-06-30 KR KR1020027000488A patent/KR100649927B1/en not_active Expired - Fee Related
- 2000-06-30 CZ CZ2002-61A patent/CZ304307B6/en not_active IP Right Cessation
- 2000-06-30 PL PL353058A patent/PL211797B1/en unknown
-
2002
- 2002-01-11 NO NO20020163A patent/NO328103B1/en not_active IP Right Cessation
-
2003
- 2003-05-23 US US10/443,797 patent/US6710178B2/en not_active Expired - Lifetime
-
2004
- 2004-03-23 US US10/806,315 patent/US6984757B2/en not_active Expired - Lifetime
-
2009
- 2009-06-23 NO NO20092393A patent/NO334937B1/en not_active IP Right Cessation
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4013706A (en) * | 1973-11-14 | 1977-03-22 | Sucreries Du Soissonnais Et Compagnie Sucriere | Derivatives of substituted urea, acyl ureas, and sulphonyl ureas, and a process for producing the same |
| DE3717815A1 (en) * | 1987-05-27 | 1988-12-15 | Bayer Ag | Fungicidal compositions based on substituted cyanamides |
| EP0521471A1 (en) * | 1991-07-01 | 1993-01-07 | Shionogi Seiyaku Kabushiki Kaisha | Pyrimidine derivatives as HMG-CoA reductase inhibitors |
| JPH06256318A (en) * | 1993-03-01 | 1994-09-13 | Shionogi & Co Ltd | Method for synthesizing 5-carboalkoxypyrimidine derivative |
Non-Patent Citations (4)
| Title |
|---|
| BREAUX E J ET AL: "An improved general synthesis of 4-aryl-5-pyrimidinecarboxylates", JOURNAL OF HETEROCYCLIC CHEMISTRY,US,HETEROCORPORATION. PROVO, vol. 18, January 1981 (1981-01-01), pages 183 - 184, XP002117468, ISSN: 0022-152X * |
| KIN-YA AKIBA ET AL.: "Ring Transformation Equilibrium (Bond Switch) in the 5-(2-Aminovinyl)isothiazole System via Hypervalent Sulfurane. Synthesis, Structure Determination, and Kinetic Study", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 107, no. 9, 1985, pages 2721 - 30, XP002153123 * |
| PATENT ABSTRACTS OF JAPAN vol. 018, no. 656 (C - 1286) 13 December 1994 (1994-12-13) * |
| WATANABE M ET AL: "SYNTHESIS AND BIOLOGICAL ANTIVITY OF METHANESULFONAMIDE PYRIMIDINE-AND N-METHANESULFONYL PYRROLE-SUBSTITUTED 3,5-DIHYDROXY-6-HEPTENOATES, A NOVEL SERIES OF HMG-COA REDUCTASE INHIBITORS", BIOORGANIC & MEDICINAL CHEMISTRY,GB,ELSEVIER SCIENCE LTD, vol. 5, no. 2, 1997, pages 437 - 444, XP000882043, ISSN: 0968-0896 * |
Cited By (39)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1873148A1 (en) * | 2000-02-15 | 2008-01-02 | AstraZeneca AB | pharmaceutical compositions containing crystalline salts of 7-[4-(4-fluorophenyl)-6-isopropyl-2-[methyl(methylsulfonyl)amino]pyrimidin-5-yl]-(3R,5S)-3,5-dihydroxyhept-6-enoic acid |
| WO2001060804A1 (en) * | 2000-02-15 | 2001-08-23 | Astrazeneca Ab | Crystalline salts of 7-[4-(4-fluorophenyl)-6-isopropyl-2-[methyl(methylsulfonyl)amino]pyrimidin-5-yl]-(3r,5s)-3,5-dihydroxyhept-6-enoic acid |
| CZ302136B6 (en) * | 2000-02-15 | 2010-11-10 | Astrazeneca Ab | Crystalline salts of 7-{4-(4-fluorophenyl)-6-isopropyl-2-[methyl(methylsulfonyl)amino]pyrimidin-5-yl}-(3R,5S)-3,5-dihydroxyhept-6-enoic acid |
| US6841554B2 (en) | 2000-02-15 | 2005-01-11 | Astrazeneca Ab | Crystalline salts of 7-[4-(4-fluorophenyl)-6-isopropyl-2-[methyl(methylsulfonyl)amino]pyrimidin-5-yl]-(3r,5s)-3,5-dihydroxyhept-6-enoic acid |
| US7129352B2 (en) | 2000-02-15 | 2006-10-31 | Astrazeneca Ab | Crystalline salts of 7-′4-(4-fluorophenyl) -6-isopropyl-2-′methyl (methylsulfonyl) amino!pyrimidin-5-yl!- (3R, 5S) -3, 5-dihydroxyhept-6-enoic acid |
| CN1301977C (en) * | 2001-07-13 | 2007-02-28 | 阿斯特拉曾尼卡英国有限公司 | The preparation method of aminopyrimidine compound |
| US7816528B2 (en) | 2001-07-13 | 2010-10-19 | Astrazeneca Uk Limited | Preparation of aminopyrimidine compounds |
| CN100349877C (en) * | 2001-07-13 | 2007-11-21 | 阿斯特拉曾尼卡英国有限公司 | Preparation of aminopyrimidine compounds. |
| US7304156B2 (en) | 2001-07-13 | 2007-12-04 | Astrazeneca Uk Limited | Preparation of aminopyrimidine compounds |
| RU2301801C2 (en) * | 2001-07-13 | 2007-06-27 | АстраЗенека Ю-Кей Лимитед | Method for preparing sulfonulaminopyrimidine compounds (variants), intermediate substances and methods for their preparing |
| US8222412B2 (en) | 2001-07-13 | 2012-07-17 | Astrazeneca Uk Limited | Preparation of aminopyrimidine compounds |
| JP2009167167A (en) * | 2001-07-13 | 2009-07-30 | Astrazeneca Uk Ltd | Raw material compound for producing aminopyrimidine compound, and method for producing the raw material compound |
| US8614320B2 (en) | 2001-07-13 | 2013-12-24 | Astrazeneca Uk Limited | Preparation of aminopyrimidine compounds |
| CN100361979C (en) * | 2002-08-13 | 2008-01-16 | 阿斯利康(英国)有限公司 | Preparation method of rosuvastatin calcium salt |
| US8273878B2 (en) | 2002-12-16 | 2012-09-25 | Astrazeneca Uk Limited | Process for the preparation of pyrimidine compounds |
| WO2004054986A3 (en) * | 2002-12-16 | 2004-12-02 | Astrazeneca Uk Ltd | Process for the preparation of pyrimidine compounds |
| EP2088142A2 (en) | 2002-12-16 | 2009-08-12 | AstraZeneca UK Limited | Compounds and Processes |
| WO2004054986A2 (en) | 2002-12-16 | 2004-07-01 | Astrazeneca Uk Limited | Process for the preparation of pyrimidine compounds |
| US7524955B2 (en) | 2002-12-16 | 2009-04-28 | Astrazeneca Uk Limited | Process for the preparation of pyrimidine compounds |
| US8034932B2 (en) | 2004-12-24 | 2011-10-11 | Astrazeneca Uk Limited | Chemical process |
| EP2423195A1 (en) | 2010-07-26 | 2012-02-29 | LEK Pharmaceuticals d.d. | Process for the preparation of key intermediates for the synthesis of statins or pharmaceutically acceptable salts thereof |
| US9085538B2 (en) | 2010-07-26 | 2015-07-21 | Lek Pharmaceuticals D.D. | Process for the preparation of key intermediates for the synthesis of statins or pharmaceutically acceptable salts thereof |
| WO2012013325A1 (en) | 2010-07-26 | 2012-02-02 | Lek Pharmaceuticals D.D. | Process for the preparation of key intermediates for the synthesis of statins or pharmaceutically acceptable salts thereof |
| US9493437B2 (en) | 2010-08-18 | 2016-11-15 | Samumed, Llc | β- and γ-diketones and γ-hydroxyketones as Wnt/ β-catenin signaling pathway activators |
| US8921413B2 (en) | 2010-08-18 | 2014-12-30 | Samumed, Llc | β- and γ-diketones and γ-hydroxyketones as WNT/ β-catenin signaling pathway activators |
| US8629176B1 (en) | 2010-08-18 | 2014-01-14 | Samumed, Llc | β- and γ-diketones and γ-hydroxyketones as WNT/ β-catenin signaling pathway activators |
| US9303010B2 (en) | 2010-08-18 | 2016-04-05 | Samumed, Llc | β- and γ-diketones and γ-hydroxyketones as Wnt/β-catenin signaling pathway activators |
| US8609717B2 (en) | 2010-08-18 | 2013-12-17 | Samumed, Llc | β- and γ-diketones and γ-hydroxyketones as WNT/β-catenin signaling pathway activators |
| US9884053B2 (en) | 2010-08-18 | 2018-02-06 | Samumed, Llc | β- and γ-diketones and γ-hydroxyketones as WNT/β-catenin signaling pathway activators |
| US10314832B2 (en) | 2010-08-18 | 2019-06-11 | Samumed, Llc | β- and γ-diketones and γ-hydroxyketones as Wnt/β-catenin signaling pathway activators |
| US11034682B2 (en) | 2013-02-22 | 2021-06-15 | Samumed, Llc | Gamma-diketones as wnt/β-catenin signaling pathway activators |
| US9533976B2 (en) | 2013-02-22 | 2017-01-03 | Samumed, Llc | γ-diketones as WNT/β-catenin signaling pathway activators |
| US9951053B2 (en) | 2013-02-22 | 2018-04-24 | Samumed, Llc | γ-diketones as Wnt/β-catenin signaling pathway activators |
| US11673885B2 (en) | 2013-02-22 | 2023-06-13 | Biosplice Therapeutics, Inc. | γ-diketones as Wnt/β-catenin signaling pathway activators |
| US10457672B2 (en) | 2013-02-22 | 2019-10-29 | Samumed, Llc | γ-diketones as Wnt/β-catenin signaling pathway activators |
| US9795550B2 (en) | 2014-08-20 | 2017-10-24 | Samumed, Llc | Gamma-diketones for treatment and prevention of aging skin and wrinkles |
| US11077046B2 (en) | 2014-08-20 | 2021-08-03 | Biosplice Therapeutics, Inc. | Gamma-diketones for treatment and prevention of aging skin and wrinkles |
| US10434052B2 (en) | 2014-08-20 | 2019-10-08 | Samumed, Llc | Gamma-diketones for treatment and prevention of aging skin and wrinkles |
| US11839679B2 (en) | 2014-08-20 | 2023-12-12 | Biosplice Therapeutics, Inc. | Gamma-diketones for treatment and prevention of aging skin and wrinkles |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2002318041B2 (en) | Preparation of aminopyrimidine compounds | |
| NO334937B1 (en) | Process for the preparation of N-cyano-N-alkylalkanesulfonamides and 4-phenylpyrimidines and such compounds | |
| AU2002318041A1 (en) | Preparation of aminopyrimidine compounds | |
| AU2005317880B2 (en) | Process for preparing rosuvastatin | |
| CA2646795A1 (en) | Process for the preparation of bosentan | |
| KR100535450B1 (en) | Preparation of pyrimidine derivatives | |
| US6187926B1 (en) | Process for producing quinolone derivatives | |
| AU658965B2 (en) | N-5-protected 2,5-diamino-4,6-dichloro-pyrimidines and processes for their preparation | |
| WO2011021216A2 (en) | Improved process for the preparation of 4-(1,1-dimethylethyl)-n-[6-(2-hydroxyethoxy)-5-(2-methoxyphenoxy)[2,2'-bipyrimidin]-4-yl]benzenesulfonamide | |
| KR100586664B1 (en) | N-[5-diphenylphosphinoylmethyl-4-4-fluorphenyl-6-isopropylpyrimidin-2-yl]-n-methylmethanesulphonamide | |
| HK1160468A (en) | Process for preparing rosuvastatin |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 222002 Country of ref document: SK Ref document number: PV2002-61 Country of ref document: CZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2000949231 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2378782 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020027000488 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020027000488 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 008116768 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10030077 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 2000949231 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2002-61 Country of ref document: CZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2000949231 Country of ref document: EP |