WO2001003705A1 - Compositions and methods for raising hdl cholesterol levels - Google Patents
Compositions and methods for raising hdl cholesterol levels Download PDFInfo
- Publication number
- WO2001003705A1 WO2001003705A1 PCT/US2000/018533 US0018533W WO0103705A1 WO 2001003705 A1 WO2001003705 A1 WO 2001003705A1 US 0018533 W US0018533 W US 0018533W WO 0103705 A1 WO0103705 A1 WO 0103705A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- accordance
- alkyl
- group
- lxr
- lxr agonist
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/18—Sulfonamides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Definitions
- the present invention relates to LXR agonists and to methods of using such LXR agonists to raise high density lipoprotein (HDL) plasma levels in mammals and to prevent, halt or slow the progression of atherosclerotic cardiovascular diseases and related conditions.
- HDL high density lipoprotein
- Hyperlipidemia is a condition which is characterized by an abnormal increase in serum lipids, such as cholesterol, triglycerides and phospholipids. These lipids do not circulate freely in solution in plasma, but are bound to proteins and transported as macromolecular complexes called lipoproteins.
- lipoproteins There are five classifications of lipoproteins based on their degree of density: chylomicrons; very low density lipoproteins (VLDL); low density lipoproteins (LDL); intermediate density lipoproteins (LDL); and high density lipoproteins (HDL).
- VLDL very low density lipoproteins
- LDL low density lipoproteins
- LDL intermediate density lipoproteins
- HDL high density lipoproteins
- hyperlipidemia characterized by the existence of elevated LDL cholesterol levels.
- the initial treatment for hypercholesterolemia is often to modify the diet to one that is low in fat and cholesterol, coupled with appropriate physical exercise, followed by drug therapy when LDL-lowering goals are not met by diet and exercise alone.
- LDL is commonly known as the "bad” cholesterol
- HDL is the "good” cholesterol.
- it is desirable to lower elevated levels of LDL cholesterol it is also desirable to increase levels of HDL cholesterol.
- HDL coronary heart disease
- An example of an HDL raising agent is nicotinic acid, but the quantities needed to achieve HDL raising are associated with undesirable side effects, such as flushing.
- liver X receptors LXRs
- LXRs liver X receptors
- the LXRs were first identified as orphan members of the nuclear receptor superfamily whose ligands and functions were unknown.
- Two LXR proteins i.e., a and ⁇
- LXR ⁇ Two LXR proteins (i.e., a and ⁇ ) are known to exist in mammals.
- the expression of LXR ⁇ is restricted, with the highest levels being found in the liver and with lower levels being found in the kidneys, intestine, spleen, and adrenals (see, Willy, et al, Genes Dew, 9( j: 1033-45 (1995)).
- LXR ⁇ is rather ubiquitous, being found in nearly all tissues examined. Recent studies on the LXRs indicate that they are activated by certain naturally occurring, oxidized derivatives of cholesterol, including 22(R)-hydroxycholesterol, 24(S)-hydroxycholesterol, and 24,25(S)- epoxycholesterol (see, Lehmann, et al, J. Biol. Chem., 272 (6) :3137 -3140 (1997)).
- the expression pattern of LXRs and their oxysterol ligands provided the first hint that these receptors may play a role in cholesterol metabolism (see, Janowski, et al, Nature, 383:72%- 731 (1996)).
- LXRs Cholesterol metabolism in mammals occurs via conversion into steroid hormones or bile acids.
- the role of LXRs in cholesterol homeostasis was first postulated to involve the pathway of bile acid synthesis, wherein cholesterol 7 ⁇ -hydroxylase (CYP7 ⁇ ) operates in a rate-limiting manner. Support for this theory was provided when additional experiments found that the CYP7 ⁇ promoter contained a functional LXR response element that could be activated by RXR/LXR heterodimers in an oxysterol- and retinoid-dependent manner. Confirmation of LXR function as a transcriptional control point in cholesterol metabolism was made using knockout mice, particularly those lacking the oxysterol receptor, LXR ⁇ (see, Peet, et al, Cell, 93:693-704 (1998)).
- LXR ⁇ e.g. , knockout or (-/-) mice
- LXR ⁇ (-/-) mice did not induce transcription of the gene encoding CYP7 ⁇ when fed diets containing additional cholesterol. This resulted in an accumulation of large amounts of cholesterol in the livers of LXR ⁇ (- -) mice, and impaired hepatic function.
- LXR ⁇ could provide treatment for a variety of lipid disorders including obesity and diabetes.
- LXR ⁇ could provide treatment for a variety of lipid disorders including obesity and diabetes.
- compounds and methods that can be used to regulate LXRs and, in turn, to control the delicate balance of cholesterol metabolism and fatty acid biosynthesis.
- compounds and methods that can be used to increase HDL levels and, thus, to treat disorders associated with bile acid and cholesterol metabolism, including cholesterol gallstones, coronary heart disease, atherosclerosis, lipid storage diseases, obesity, diabetes, etc.
- the present invention fulfills these and other needs by providing such compounds and methods.
- the present invention provides methods for raising, i.e., increasing, HDL plasma levels in a mammal in need of such treatment, the methods comprising administering to the mammal, e.g.. a human, an HDL-raising amount of a LXR agonist and, in particular, a LXR ⁇ agonist.
- Any compound that activates and, therefore, is an agonist of LXRs can be used in the methods of the present invention.
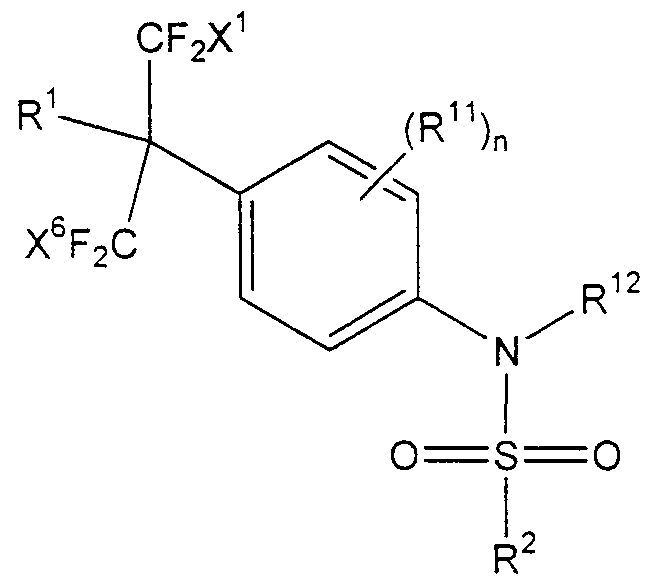
- LXR agonists have the following general formula:
- Ar is an aryl group
- R 1 is -OH, -0-(d-C 7 )alkyl, -OC(0)-(C r C 7 )alkyl, -C0 2 H, -NH 2 , -NH(d-C 7 )alkyl, -N((C C 7 )alkyl) 2 or -NH-S(0) 2 -(C 1 -C 5 )alkyl
- R 2 is (C r C 7 )alkyl, aryl and aryl(C ⁇ -C 7 )al yl
- X 1 , X 2 , X 3 , X 4 , X 5 and X 6 are each independently -H, (C ⁇ -C 5 )alkyl, -F and -Cl, with the proviso that no more than two of X 1 through X 6 are -H or (C ⁇ -C 5 )al yl
- Y is -N(R 12
- the present invention provides methods for preventing, halting or slowing the progression of atherosclerotic cardiovascular diseases and related conditions in a mammal in need of such treatment, the methods comprising administering to the mammal an HDL-raising amount of a LXR agonist.
- the present invention provides methods for preventing, halting or slowing the progression of atherosclerotic cardiovascular diseases and related conditions in a mammal in need of such treatment, the methods comprising administering to the mammal an HDL-raising amount of a LXR agonist in combination with one or more additional active agents, such as bile acid sequestrants, nicotinic acid, fabric acid derivatives. HMG CoA reductase inhibitors, etc.
- Figure 1 illustrates a schematic diagram of a strategy for the screening of LXR ⁇ agonists for use as cholesterol-lowering agents.
- HTS pharmacokinetic
- SAR structure-activity relationship
- FIG. 2 illustrates the results of a radioligand binding assay in which the radiolabeied LXR ⁇ agonist T314407 was demonstrated to directly bind to LXR ⁇ . Binding of the T314407 to a glutathione-S-transferase alone was negligible.
- Figure 3 illustrates the results of a ligand competition assay in which the ability of the LXR ⁇ agonist T314407 to compete with other molecules for binding to LXR ⁇ was tested.
- the amount of radiolabeied T314407 that bound to LXR ⁇ was competitively inhibited by T314407 itself, by the other agonists T900546 and T901433, and by the known LXR ⁇ ligand 24,25-epoxycholesterol.
- Figure 4 illustrates that the LXR ⁇ agonists T900546 and T314407 competitively inhibit the binding of the radiolabeied oxysterol 24,25-epoxycholesterol to the LXR ⁇ receptor.
- Figure 5 illustrates the results of a peptide sensor assay in which the LXR ⁇ agonists T900546, T314407 and T280404 were shown to induce a conformational change in the LXR ⁇ .
- the oxysterol 24,25-epoxycholesterol also induced a conformational change in the peptide sensor assay.
- Figure 6 illustrates the results of a mammalian two-hybrid assay in which the effect of LXR ⁇ agonists on transcription mediated by a fusion protein that includes a G AL4 DNA binding domain fused to a SRC-1 polypeptide and a second fusion protein that includes the LXR ⁇ ligand binding domain fused to a VP16 activation domain.
- the amount of expression of a luciferase gene under the control of a GAL 4 upstream activation sequence is shown.
- Figure 7 illustrates the results of an assay that tested the ability of LXR ⁇ agonists to activate LXR-mediated transcription in a cotransfection assay. The amount of expression of a luciferase gene is shown.
- Figure 8 presents data which demonstrate that LXR ⁇ agonists specifically activate LXR-mediated transcription.
- Figure 9 illustrates several analogs of the LXR ⁇ agonist T0314407, along with pharmacokinetic data for these analogs.
- Figure 10 illustrates the results of an experiment which demonstrates that oral administration of the LXR ⁇ agonist T0901317 increases total cholesterol, and also increases the fraction of HDL cholesterol, in mice.
- Figure 11 presents data which demonstrate that oral administration of the LXR ⁇ agonist T0901317 results in an increase in mouse plasma triglyceride levels.
- alkyl by itself or as part of another substituent, refers to, unless otherwise stated, a straight or branched chain, or cyclic hydrocarbon radical, or combination thereof, which may be fully saturated, mono- or polyunsaturated and can include di- and multi-radicals, having the number of carbon atoms designated (i.e., Cj-Cio means one to ten carbons).
- saturated hydrocarbon radicals include, but are not limited to, methyl, ethyl, n-propyl, isopropyl, n-butyl, t-butyl, isobutyl, sec-butyl, cyclohexyl, (cyclohexyl)methyl, cyclopropylmethyl, homologs and isomers of, for example, n-pentyl, n- hexyl, n-heptyl, n-octyl, and the like.
- An unsaturated alkyl group is one having one or more double bonds or triple bonds. Examples of unsaturated alkyl groups include, but are not limited to. vinyl, 2-propenyl.
- alkyl unless otherwise noted, is also meant to include those derivatives of alkyl defined in more detail below as “cycloalkyl” and “alkylene.”
- alkylene by itself or as part of another substituent, refers to a divalent radical derived from an alkane, as exemplified by -CH 2 CH CH 2 CH 2 -.
- an alkyl group will have from 1 to 24 carbon atoms, with those groups having 10 or fewer carbon atoms being preferred in the present invention.
- a “lower alkyl” or “lower alkylene” refers to a shorter chain alkyl or alkylene group, generally having eight or fewer carbon atoms, preferably four or fewer carbon atoms.
- alkoxy either alone or in combination with other terms, refers to. unless otherwise stated, an alkyl group, as defined above, connected to the remainder of the molecule via an oxygen atom, such as, for example, methoxy, ethoxy, 1 -propoxy. 2-propoxy and the higher homologs and isomers.
- heteroalkyl by itself or in combination with another term, refers to, unless otherwise stated, a stable straight or branched chain, or cyclic hydrocarbon radical, 7
- heteroatoms consisting of the stated number of carbon atoms and from one to three heteroatoms selected from the group consisting of 0, N, Si and S, and wherein the nitrogen and sulfur atoms may optionally be oxidized and the nitrogen heteroatom may optionally be quaternized.
- the heteroatom(s) 0, N and S may be placed at any interior position of the heteroalkyl group.
- the heteroatom Si may be piaced at any position of the heteroalkyl group, including the position at which the alkyl group is attached to the remainder of the molecule.
- heteroalkyl Up to two heteroatoms may be consecutive, such as, for example, -CH 2 -NH-OCH 3 and -CH 2 -0-Si(CH 3 ) 3 .
- heteroalkyl also included in the term “heteroalkyl” are those radicals described in more detail below as “heteroalkylene” and “heterocycloalkyl.”
- heteroatoms can also occupy either or both of the chain termini. Still further, for alkylene and heteroalkylene linking groups, as well as all other linking groups described herein, no specific orientation of the linking group is implied.
- cycloalkyl and heterocycloalkyl refer to, unless otherwise stated, cyclic versions of “alkyl” and “heteroalkyl,” respectively.
- cycloalkyl and heterocycloalkyl are intended to include bicyclic, tricyclic and polycyclic versions thereof. Additionally, for heterocycloalkyl, a heteroatom can occupy the position at which the heterocycle is attached to the remainder of the molecule.
- cycloalkyls include, but are not limited to, cyclopentyl, cyclohexyl, 1-cyclohexenyl, 3-cyclohexenyl, cycloheptyl, bicyclo[2.2.1]heptyl, bicyclo[2.2.2]octyl, adamantyl, and the like.
- heterocycloalkyls include, but are not limited to, l-(l,2,5,6-tetrahydropyridyl), 1-piperidinyl, 2-piperidinyl, 3-piperidinyl. 4- morpholinyl, 3-morpholinyl, l,4-diazabicyclo[2.2.2]oct-2-yl.
- halo or halogen
- fluorine chlorine, bromine, or iodine atom.
- fluoro alkyl are intended to include monofluoroalkyl and polyfluoroalkyl while, more generally, “haloalkyl” is intended to include monohaloalkvl and poly haloalkyl.
- aryl refers to, unless otherwise stated, an aromatic substituent that can be a single ring or multiple rings (up to three rings) that are fused together or linked covalently.
- the rings may each contain from zero to four heteroatoms selected from N, O, and S, wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaterized.
- the aryl groups that contain heteroatoms may be referred to as "heteroaryl” and can be attached to the remainder of the molecule through a carbon atom or a heteroatom.
- Non-limiting examples of aryl groups include phenyl, 1-naphthyl, 2- naphthyl, 4-biphenyl, 1-pyrrolyl, 2-pyrrolyl, 3-pyrrolyl, 3-pyrazolyl, 2-imidazolyl, 4- imidazolyl, pyrazinyl, 2-oxazolyl, 4-oxazolyl, 5-oxazolyl, 3-isoxazolyl, 4isoxazolyl, 5- isoxazolyi, 2-thiazolyl, 4-thiazolyl, 5-thiazolyl, 2-furyl, 3-furyl, 2-thienyl, 3-thienyl, 2- pyridyl, 3-pyridyl, 4-pyridyl, 2-pyrimidyl, 4-pyrimidyl, 5-benzothiazolyl, purinyl, 2- benzimidazolyl, 5-indolyl, 1-isoquinolyl, 5-isoquinolyl, 2-quinoxalinyl
- arylalkyl and arylhetero alkyl are intended to include those radicals in which an aryl group is attached to an alkyl group (e.g., benzyl, phenethyl, pyridyfmethyl and the like), or a heteroalkyl group (e.g., phenoxymethyl. 2- pyridyloxymethyl, l-naphthyloxy-3-propyl, and the like).
- arylalkyl and arylhetero alkyl groups will typically contain from 1 to 3 aryl moieties attached to the alkyl or heteroalkyl portion by a covalent bond or by fusing the ring to, for example, a cycloalkyl or heterocycloalkyl group.
- a heteroatom can occupy the position at which the group is attached to the remainder of the molecule.
- arylheteroalkyl is intended to include benzyloxy, 2-phenylethoxy. phenethylamine, and the like.
- substituted alkyl groups will have from one to six independently selected substituents, more preferably from one to four independently selected substituents, most preferably from one to three independently selected substituents.
- R', R" and R'" are each independently selected and are functional groups including, but not limited to, the following: hydrogen, unsubstituted(C ⁇ -Cg)alkyl and heteroalkyl, unsubstituted aryl, aryl substituted with 1-3 halogens, unsubstituted alkyl, alkoxy or thioalkoxy groups, or aryl(C
- R and R" When R and R" are attached to the same nitrogen atom, they can be combined with the nitrogen atom to form a 5-, 6-, or 7-membered ring.
- -NRR is meant to include 1 -pyrrolidinyl and 4-morpholinyl.
- substituents for the aryl groups are varied and include, but are not limited to the following: -halogen, -OR, -OC(0)R, -NR'R", -S -R, -CN, -N0 2 , -C0 2 R, -CONRR", -SiRR'R)", -C(0)R, -OC(0)NR'R".
- S(0) 2 NR'R S(0) 2 NR'R.
- substituted aryl groups will have from one to four independently selected substituents, more preferably from one to three independently selected substituents and. most preferably, from one to two independently selected substituents.
- Two of the substituents on adjacent atoms of the aryl ring may optionally be replaced with a substituent of the formula -T-C(0)-(CH?) q -U-, wherein T and U are independently -NH-, -0-, -CH?- or a single bond, and q is an integer of from 0 to 2.
- two of the substituents on adjacent atoms of the aryl ring may optionally be replaced with a substituent of the formula -A-(CH 2 ) r -B-. wherein A and B are independently 10
- r is an integer of from i to 3.
- One of the single bonds of the new ring so formed may optionally be replaced with a double bond.
- two of the substituents on adjacent atoms of the aryl ring may optionally be replaced with a substituent of the formula -(CH 2 ) s -X-(CH 2 ) t -, where s and t are independently integers of from 0 to 3, and X is -0-, NR-, -S-, -S(O)-, -S(0) 2 -, or - S(0) 2 NR'-.
- the substituent R in -NR- and -S(0) 2 NR- is selected from hydrogen or unsubstituted (C r C 6 )alkyl.
- heteroatom is intended to include oxygen (O), nitrogen (N), sulfur (S) and silicon (Si).
- pharmaceutically acceptable salt(s) is meant to include salts of the active compounds which are prepared with relatively nontoxic acids or bases, depending on the particular substituents found on the LXR agonists described herein.
- base addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired base, either neat or in a suitable inert solvent.
- pharmaceutically acceptable base addition salts include, but are not limited to, sodium, potassium, calcium, ammonium, organic amino or magnesium salts, or other similar salts.
- acid addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired acid, either neat or in a suitable inert solvent.
- pharmaceutically acceptable acid addition salts include those derived from inorganic acids, such as hydrochloric, hydrobromic, nitric, carbonic, monohydrogencarbonic, phosphoric, monohydrogenphosphoric, dihydrogenphosphoric, sulfuric, monohydrogensulfuric.
- hydriodic, or phosphorous acids and the like as well as those derived from relatively nontoxic organic acids, such as acetic, propionic, isobutyric, oxalic, maleic, malonic, benzoic, succinic, suberic, fumaric, mandelic, phthalic, benzenesulfonic, p-tolylsulfonic, citric, tartaiic, methanesulfonic, and the like.
- salts of amino acids such as arginate and the like
- salts of organic acids such as glucuronic or galactunoric acids and the like (see, for example, Berge, et al, "Pharmaceutical Salts", Journal of Pharmaceutical Science.
- Certain specific compounds of the present invention contain both basic and acidic functionalities that allow the compounds to be converted into either base or acid addition salts.
- the neutral forms of the compounds may be regenerated by contacting the salt with a base or acid and isolating the parent compound in the conventional manner.
- the parent form of the compound differs from the various salt forms in certain physical properties, such as solubility in polar solvents, but otherwise the salts are equivalent to the parent form of the compound for the purposes of the present invention.
- the present invention provides compounds that are in a prodrug form.
- Prodrugs of the compounds described herein are those compounds that readily undergo chemical changes under physiological conditions to provide a compound of Formula I.
- prodrugs can be converted to the compounds of the present invention by chemical or biochemical methods in an ex vivo environment. For example, prodrugs can be slowly converted to the compounds of the present invention when placed in a transdermal patch reservoir with, for example, a suitable enzyme.
- Certain compounds of the present invention can exist in unsolvated forms as well as solvated forms, including hydrated forms. In general, the solvated forms are equivalent to unsolvated forms and are intended to be encompassed within the scope of the present invention. Certain compounds of the present invention may exist in multiple crystalline or amorphous forms. In general, all physical forms are equivalent for the uses contemplated by the present invention and are intended to be within the scope of the present invention. Certain compounds of the present invention possess asymmetric carbon atoms
- the compounds of the present invention can also contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute such compounds.
- the compounds may be radiolabeied with radioactive isotopes, such as tritium ( ⁇ ), iodine- 125 ( 123 I) or carbon- 14 ( C). All isotopic variations of the compounds of the present invention, whether radioactive or not, are intended to be encompassed within the scope of the present invention.
- the present invention provides methods for preventing or reducing the risk of developing atherosclerosis, the methods comprising the administration of a prophylactically effective amount or, more particularly, an HDL-raising amount of a LXR ⁇ agonist, either alone or in combination with one or more additional pharmaceutically active agents, to a mammal, particularly a human who is at risk of developing atherosclerosis.
- LXR ⁇ agonists can also be used in methods for treating, halting or slowing the progression of atherosclerotic disease once it has become clinically evident, the methods comprising the administration of a therapeutically effective amount or, more particularly, an HDL-raising amount of a LXR ⁇ agonist, either alone or in combination with one or more additional pharmaceutically active agents, to a mammal, particularly a human, who already has atherosclerotic disease.
- Atherosclerosis encompasses vascular diseases and conditions that are recognized and understood by physicians practicing in the relevant fields of medicine.
- Atherosclerotic cardiovascular disease, coronary heart disease (also known as coronary artery disease or ischemic heart disease), cerebrovascular disease and peripheral vessel disease are all clinical manifestations of atherosclerosis and are, therefore, encompassed by the terms “atherosclerosis” and "atherosclerotic disease.”
- the present invention further provides methods for preventing or reducing the risk of a first or subsequent (where the potential exists for recurrence) atherosclerotic disease event, the methods comprising the administration of a prophylactically effective amount or, more particularly, an HDL-raising amount of a LXR ⁇ agonist, either alone or in combination with one or more additional pharmaceutically active agents, to a mammal, particularly a human, who is at risk for having an atherosclerotic disease event.
- the term "atherosclerotic disease event" as used herein, is intended to encompass coronary heart disease events, cerebrovascular events, and intermittent claudication. Coronary heart disease events are intended to include CHD death, myocardial infarction (i.e.
- Cerebrovascular events are intended to include ischemic or hemorrhagic stroke (also known as cerebrovascular accidents) and transient ischemic attacks. Intermittent claudication is a clinical manifestation of peripheral vessel disease. It is intended that persons who have previously experienced one or more non- fatal atherosclerotic disease event are those for whom the potential for recurrence of such an event exists.
- Persons to be treated with the instant therapy include those at risk of developing atherosclerotic disease and of having an atherosclerotic disease event.
- Standard atherosclerotic disease risk factors are known to the average physician practicing in the relevant fields of medicine. Such known risk factors include, but are not limited to, hypertension, smoking, diabetes, low levels of high density lipoprotein cholesterol, high levels of low density lipoprotein cholesterol, and a family history of atherosclerotic cardiovascular disease.
- People who are identified as having one or more of the above-noted risk factors are intended to be included in the group of people considered at risk for developing atherosclerotic disease. People identified as having one or more of the above-noted risk factors, as well as people who already have atherosclerosis, are intended to be included within the group of people considered to be at risk for having an atherosclerotic disease event.
- the present invention provides methods of raising, / ' . e. , increasing, the plasma level of high density lipoprotein (HDL) in a mammal, the methods comprising administering to the mammal an HDL-raising amount of an LXR agonist.
- HDL high density lipoprotein
- any compound that activates and, therefore, is an agonist of LXRs can be used in the methods of the present invention. More particularly, any compound that is found to be an agonist of LXR using in vitro or in vivo assay procedures, such as those described herein, can be used in the methods of the present invention.
- the present invention provides LXR agonists that are useful in the methods of the present invention, the LXR agonists having the following general formula:
- aryl group represents an aryl group.
- aryl groups are either monocyclic or fused-bicyclic aromatic rings.
- Particularly preferred aryl groups include, but are not limited to, benzene, naphthalene, pyridine, quinoline. isoquinoline, pyrrole, f ran and thiophene.
- the aryl group, i.e., Ar is either a benzene or pyridine ring.
- the aryl group is a benzene ring.
- the substituents will typically be selected from the following functional groups: -OH, -NH 2 , lower alkyl (e.g., methyl, butyl, trifluoromethyl, trifluoroethyl, and the like), lower alkoxy (e.g., methoxy, ethoxy, trifluoromethoxy, butoxy, and the like), -NR'R", -SR, -halogen, -SiRR'R"', -OC(0)R. - C0 2 R, -CONRR". -C(0)R.
- R, R" and R" are each independently selected from the group consisting of hydrogen, ( -Cg)alkyl, (C ⁇ -Cg)haloalkyl, (C ⁇ -Cg)heteroalkyl, unsubstituted aryl, (unsubstituted aryl)-(C ⁇ -C 4 )alkyl, and (unsubstituted aryl)oxy-(C ⁇ -C 4 )alkyl.
- the substitutents attached to aryl group can be in any spatial arrangement.
- Ar is a benzene ring
- the two groups illustrated in Formula I will preferably be attached to Ar in a 1,3-orientation (meta) or a 1 ,4-orientation (para). More preferably, the two groups illustrated in Formula I will be attached to a benzene or pyridine ring in a 1 ,4-orientation (para).
- R is a functional group including, but not limited to, the following: -OH, -O-(C r C 7 )alkyl. -OC(0)-(C r C 7 )alkyl. -C0 2 H, -NH 2 ,-NH(C r C 7 )alkyl. - N((C,-C 7 )alkyl) 2 or -NH-S(O) 2 -(C ⁇ -C 5 )alkyl.
- R 1 is -OH. -CO?H. -NH 2 , -NH(C r C 7 )alkyl.
- R is -OH.
- R ! is a diaikylamino group (-N((C ⁇ -C 7 )alkyl) 2 )
- the alkyl groups can either be the same or different.
- X , X X J , X , X 3 and X ,” in Formula I, are each independently selected and are functional groups including, but not limited to, the following: -H, (C ⁇ -C 5 )alkyl, -F and -Cl.
- X 1 through X 6 are selected such that no more than two of X 1 through X 6 are -H or (C ⁇ -C 5 )alkyl.
- X 1 through X 6 are selected such that no more than two of X 1 through X 6 are -H, with the remaining being -F.
- X 1 through X 6 are each -F.
- Y is a linking group and is selected from -N(R 12 )S(0) m -, - N(R 12 )S(0) m N(R i3 )-, -N(R 12 )C(0)-, -N(R I2 )C(0)N(R 13 )-, -N(R 12 )C(S)- or -N(R 12 )C(0)0-, wherein R 12 and R 13 are each independently hydrogen, (C ⁇ -C 7 )alkyl, aryl and aryl(C ⁇ - C 7 )alkyl, and optionally when Y is -N(R 12 )S(0) m - or -N(R 12 )S(0) m N(R 13 )-, R 12 forms a five- , six- or seven-membered ring fused to Ar through covalent attachment to Ar.
- the index "m" is an integer having a value ranging from 1 to 2.
- Y is -N(R I2 )S(0) m -, -N(R 12 )S(0)N(R 13 )- or -N(R 12 )C(0)0-.
- Y is -N(R )S(0) m -.
- R 12 and R lj are independently selected from the following functional groups: hydrogen, (C ⁇ -C 7 )alkyl, aryl or aryl(C ⁇ -C 7 )alkyi, which in the case of the latter two groups, can also be either substituted or unsubstituted.
- R 12 is hydrogen or (C ⁇ -C )alkyl, preferably fluoro(C ⁇ -C 4 )alkyl.
- a particularly preferred R 12 group is 2,2,2-trifluoroethyl.
- R 12 is attached to Ar to form a fused ring system, such as indoline, tetrahydroquinoline or tetrahydroisoquinoline .
- R which is attached to Y, is a functional group including, but not limited to, (C C 7 )alkyl, aryl or aryl(C ⁇ -C 7 )alkyl.
- R 2 groups can be either substituted or unsubstituted.
- R 2 is an aryl group. More preferably, R 2 is an aryl group, including, but not limited to, phenyl, thienyl, imidazolyi, oxazolyl and pyridyl.
- R " is phenyl or thienyl (including 2-thienyl and 3 -thienyl).
- Preferred substituted R 2 groups include 3-chloropf ⁇ enyi.
- the LXR agonists of the present invention will be selected from the group consisting of:
- index "n” represents an integer having a value ranging from 0 to 4; and each "R 11 " is independently selected and is a functional group including, but not limited to, -OH, -NH 2 , lower alkyl, lower alkoxy, -NR'R", -SR, -halogen, -SiRR'R'", -OC(0)R, -C(0)R.
- R, R" and R" are each independently selected from the group consisting of hydrogen, (C ⁇ -Cg)alkyl, (Ci- Cg)haloalkyl, (C ⁇ -Cg)heteroalkyl, unsubstituted aryl, (unsubstituted aryl)-(C ⁇ -C 4 )alkyl. and (unsubstituted aryl)oxy-(C ⁇ -C4)alkyl.
- the remaining groups in the above formulae are as defined above in connection with the LX
- the LXR agonists of the present invention will be selected from the group consisting of:
- the various groups are as defined above in connection with the LXR agonists of Formula I.
- R 1 is -OH or -NH .
- X 1 and X 6 are each independently hydrogen or fluorine.
- Still further preferred LXR agonists are those embodiments in which R 2 is a substituted aryl group and, more preferably, a substituted phenyl or substituted thienyl group.
- the LXR agonists of the present invention will be selected from the group consisting of:
- the LXR agonists of the present invention will have the following formula:
- R is -OH or -NH ; X and X are each independently hydrogen or fluorine; R 12 i • s fluoro(C ⁇ C 4 )alkyl; and R 2 is aryl (e.g., phenyl).
- R 1 is -OH or -NH 2 ; X 1 and X 6 are each independently hydrogen or fluorine; R 12 is hydrogen or (C C )alkyl; and R 2 is substituted or unsubstituted thienyl.
- R 1 is -OH or -NH 2 ;
- X 1 and X 6 are each independently hydrogen or fluorine,
- R " is (Cj-C 4 )alkyl; and
- R " is phenyl substituted with at least one member selected from the group consisting of -CN, -CF 3 , -O- (C ⁇ -C 4 )alkyl, -C(0)-(C,-C 4 )alkyl, -C(0)-0(C 1 -C 4 )alkyL -C(0)-NH(d-C 4 )alkyl and -
- LXR agonists are those in which the compound binds to the ligand binding domain of LXR, more preferably LXR ⁇ , with an affinity of at least 10 ⁇ M or less and, more preferably, 1 ⁇ M or less.
- the present invention provides compounds of Formula I, above, (wherein each of the recited substituents has the meaning provided above) with the proviso that when "-Y-R 2 " is -N(R 12 )S(0) m -R 2 or -N(R 12 )C(0)N(R 13 )-R : and is attached to a position para to the quaternary carbon attached to Ar, and when "R 2 " is substituted or unsubstituted phenyl, benzyl or benzoyl, then i) at least one of R 12 or R 13 is other than hydrogen or unsubstituted alkyl, or ii) R " is substituted with a moiety other than amino, acetamido, di(C ⁇ -C7)alkylamino, (C ⁇ -C 7 )alkylamino, halogen, hydroxy, nitro, or (Cp C 7 )alkyl, or iii) the benzene ring portion of R 2
- the compounds of Formula I may exist as stereoisomers, and the invention includes all active stereoisomeric forms of these compounds.
- optically active isomers such compounds may be obtained from corresponding optically active precursors using the procedures described above or by resolving racemic mixtures. The resolution may be carried out using various techniques, such as chromatography, repeated recrystallization of derived asymmetric salts, or derivatization, which techniques are well known to those of ordinary skill in the art.
- the LXR agonists of the present invention may be labeled in a variety of ways.
- the compounds may contain radioactive isotopes such as, for example, ⁇ (tritium) and 14 C (carbon-14).
- the compounds may be advantageously joined, covalently or noncovalently, directly or through a linker molecule, to a wide variety of other compounds, which may provide pro-drugs or function as carriers, labels, adjuvents, coactivators, stabilizers, etc. Such labeled and joined compounds are contemplated within the present invention.
- the LXR agonists of Formula I, supra, can be prepared using readily available starting materials or known intermediates.
- Scheme I provides a variety of synthesis avenues for the production of the LXR agonists of the present invention. One of skill in the art will understand that additional methods are also useful.
- Scheme 1
- aniline i. as representative of substituted anilines and other arvlamines
- Treatment of iii with an appropriate alkyiating group, acylating group or arylating group provides iv. which can be sulfonylated with, for example, an appropriate sulfonyl halide to form vi.
- the aniline derivative iii can be sulfonylated to form v.
- LXR agonists of the present invention can be formed by treating the substituted aniline iv (or, alternatively, iii), with reagents suitable for the formation of amides vii, carbamates viii, and ureas ix.
- any compound that activates and. therefore, is an agonist of LXR can be used in the methods of the present invention. More particularly, any compound that is found to be an agonist of LXRs and, in particular, LXR ⁇ using scientifically sound in vitro or in vivo assay procedures can be used in the methods of the present invention.
- Figure 1 illustrates a schematic diagram of an exemplar strategy that can be used for the identification and screening of LXR ⁇ agonists that are useful as cholesterol- lowering agents.
- a high throughput screen (HTS) is used to identify compounds that bind to LXR ⁇ . Compounds that exhibit binding are next tested for ability to enhance LXR ⁇ -mediated transactivation.
- the compounds and compositions can be evaluated for their ability to increase or decrease gene expression modulated by LXR, using western- blot analysis.
- Established animal models to evaluate hypocholesterolemic effects of the compounds are also known in the art.
- compounds disclosed herein can lower cholesterol levels in hamsters fed a high-cholesterol diet, using a protocol similar to that described in Spady, et al., J. Clin. Invest., 81:300 (1988); Evans, et al, J. Lipid Res., 55:1634 (1994), and Lin, et al, J. Med. Chem., 38:277 (1995).
- LXRa animal models e.g., LXR ⁇ (+/-) and (-/-) mice
- LXR ⁇ (+/-) and (-/-) mice can be used for evaluation of the present compounds and compositions (see, for example, Peet, et al, Cell, 95:693-704 (1998)).
- the assays are designed to screen large chemical libraries by automating the assay steps and providing compounds from any convenient source to assay, which are typically run in parallel (e.g., in micro titer formats on microtiter plates in robotic assays). It will be appreciated by those of skill in the art that there are many commercial suppliers of chemical compounds, including Sigma Chemical Co. (St. Louis, MO), Aldrich Chemical Co. (St.
- high throughput screening methods involve providing a combinatorial library containing a large number of potential therapeutic compounds (i.e., LXR agonists). Such "combinatorial chemical libraries" are then screened in one or more assays, as described herein, to identify those library members (particular chemical species or subclasses) that display a desired characteristic activity, i. e. , activate LXRs. The compounds thus identified can serve as conventional "lead compounds" or can themselves be used as potential or actual therapeutics.
- a combinatorial chemical library is a collection of diverse chemical compounds generated by either chemical synthesis or biological synthesis, by combining a number of chemical "building blocks,” such as reagents.
- a linear combinatorial chemical library such as a polypeptide library
- a linear combinatorial chemical library is formed by combining a set of chemical building blocks (amino acids) in every possible way for a given compound length (i.e.. the number of amino acids in a polypeptide compound). Millions of chemical compounds can be synthesized through such combinatorial mixing of chemical building blocks.
- combinatorial chemical libraries include, but are not limited to, peptide libraries (see, e.g., U.S. Patent No. 5,010,175, Furka, Int. J. Pept. Prot. Res., 57:487-493 (1991) and Houghton, et al, Nature, 554:84-88 (1991)).
- Other chemistries for generating chemical diversity libraries can also be used.
- Such chemistries include, but are not limited to, peptoids (PCT Publication No. WO 91/19735); encoded peptides (PCT Publication WO 93/20242); random bio-oligomers (PCT Publication No.
- WO 92/00091 benzodiazepines (U.S. Patent No. 5,288,514); diversomers, such as hydantoins, benzodiazepines and dipeptides (Hobbs, et al, Proc. Nat. Acad. Sci. USA, 90:6909-6913 (1993)); vinylogous polypeptides (Hagihara, et al, J. Amer. Chem. Soc. 114:656% (1992)); nonpeptidal peptidomimetics with ⁇ -D-glucose scaffolding (Hirschmann, et al. , J. Amer. Chem.
- Patent No. 5,593,853 small organic molecule libraries (see, e.g., benzodiazepines, Baum C&E News, Jan. 18, page 33 (1993); isoprenoids (U.S. Patent No. 5,569,588); thiazolidinones and metathiazanones (U.S. Patent No. 5,549,974); pyrrolidines (U.S. Patent Nos. 5,525,735 and 5,519,134); morpholino compounds (U.S. Patent No. 5,506,337); benzodiazepines (U.S. Patent No. 5,288,514); and the like.
- a number of well known robotic systems have also been developed for solution phase chemistries. These systems include automated workstations like the automated synthesis apparatus developed by Takeda Chemical Industries, LTD. (Osaka, Japan) and many robotic systems utilizing robotic arms (Zymate II, Zymark Corporation, Hopkinton, Mass.; Orca, Hewlett-Packard, Palo Alto, Calif), which mimic the manual synthetic operations performed by a chemist. Any of the above devices are suitable for use with the present invention. The nature and implementation of modifications to these devices (if any) so that they can operate as discussed herein will be apparent to persons skilled in the relevant art. In addition, numerous combinatorial libraries are themselves commercially available (see.
- each well of a microtiter plate can be used to run a separate assay against a selected potential LXR modulator, or if concentration or incubation time effects are to be observed, every 5-10 wells can test a single LXR modulator.
- a single standard microtiter plate can assay about 100 (96) modulators. If 1536 well plates are used, then a single plate can easily assay from about 100- about 1500 different compounds. It is possible to assay many different plates per day; assay screens for up to about 6,000-20,000, and even up to about 100,000-1,000,000 different compounds is possible using the integrated systems of the invention.
- High throughput screening systems are commercially available (see, e.g , Zymark Corp., Hopkinton, MA; Air Technical Industries, Mentor, OH; Beckman Instruments. Inc. Fullerton, CA; Precision Systems, Inc., Natick, MA, etc). These systems typically automate entire procedures, including all sample and reagent pipetting, liquid dispensing, timed incubations, and final readings of the microplate in detector(s) appropriate for the assa ⁇ . These configurable systems provide high throughput and rapid start up as well as a high degree of flexibility and customization. The manufacturers of such systems provide detailed protocols for various high throughput systems. Thus, for example, Zymark Corp. provides technical bulletins describing screening systems for detecting the modulation of gene transcription, ligand binding, and the like. LXR Agonist Compositions
- the compounds, i.e., the LXR agonists, of this invention can be incorporated into a variety of formulations for therapeutic administration. More particularly, the compounds of the present invention can be formulated into pharmaceutical compositions by combination with appropriate, pharmaceutically acceptable carriers or diluents. Suitable formulations for use in the present invention are found in Remington's Pharmaceutical Sciences (Mack Publishing Company, Philadelphia, PA, 17th ed. (1985)), which is incorporated herein by reference. In addition, for a brief review of methods for drug delivery (see, Langer, Science, 249:1527-1533 (1990), which is incorporated herein by reference). As used herein, the term "composition" is intended to encompass a product comprising the specified ingredients (e.g.
- LXR agonist in specified amounts where amounts are specified, as well as any product that results directly or indirectly from combination of the specified ingredients, in the specified amounts where amounts are specified.
- the active LXR agonist compounds of the present invention may be orally administered as a pharmaceutical composition, for example, with an inert diluent, or with an assimilable edible carrier, or they may be enclosed in hard or soft shell capsules, or they may be compressed into tablets, or they may be incorporated directly with the food of the diet.
- these active compounds may be incorporated with excipients and used in the form of tablets, pills, capsules, ampules, sachets, elixirs, suspensions, syrups, and the like.
- the active compounds can also be administered intranasally as, for example, liquid drops or spray. Oral administration is preferred.
- Such compositions and preparations should contain at least 0.1 percent of active compound, i.e.. the LXR agonist.
- the percentage of active compound in these compositions may, of course, be varied and may conveniently be between about 2% to about 60% of the weight of the unit.
- Therapeutically effective amounts, prophylactically effective amounts and/or high density lipoprotein-raising amounts of the LXR agonist are suitable for use in the compositions and methods of the present invention.
- terapéuticaally effective amount is intended to mean that amount of a drug or pharmaceutical agent that will elicit the biological or medical response of a tissue, a system, animal or human that is being sought by a clinician, such as a researcher, veterinarian, medical doctor or osteopathic doctor.
- prophylactically effective amount is intended to mean that amount of a drug or pharmaceutical agent that will prevent or reduce the risk of occurrence of a medical condition, such as atherosclerosis or an atherosclerotic disease event.
- high density lipoprotein-raising amount is intended to mean an amount of a drug or pharmaceutical agent that will elevate a subject's plasma HDL level above the level it was at prior to administration of the drug or pharmaceutical agent. Measurement of plasma HDL levels can be performed using any medically acceptable procedures known to those skilled in the medical arts, including assay kits designed for use directly by consumers.
- the dosage regimen utilizing a LXR agonist is selected in accordance with a variety of factors including type, species, age, weight, sex and medical condition of the patient; the severity of the condition to be treated; the route of administration; the renal and hepatic function of the patient; and the particular LXR agonist or derivative thereof employed. A consideration of these factors is well within the purview of the ordinarily skilled clinician for the purpose of determining an appropriate HDL-raising amount of the LXR agonist, as well as the therapeutically effective amounts of the LXR agonist needed to prevent, counter, or arrest the progress of the condition.
- the compounds of the present invention can be administered at a daily dosage of from about 0.1 milligram to about 100 milligram per kilogram of animal body weight, once a day or given in divided doses two to six times a day, or in sustained release form.
- the total daily dosage is from about 1.0 milligram to about 1000 milligrams, and preferably from about 1 milligram to about 50 milligrams.
- the total daily dose will generally be from about 7 milligrams to about 350 milligrams. This dosage regimen may be adjusted to provide the optimal therapeutic response.
- the tablets, pills, capsules, and the like may also contain a binder such as gum tragacanth, acacia, corn starch or gelatin; excipients such as dicalcium phosphate; a disintegrating agent such as corn starch, potato starch, alginic acid; a lubricant such as magnesium stearate; and a sweetening agent such as sucrose, lactose or saccharin.
- a dosage unit form is a capsule, it may contain, in addition to materials of the above type, a liquid carrier such as a fatty oil.
- tablets may be coated with shellac, sugar or both.
- a syrup or elixir may contain, in addition to the active ingredient, sucrose as a sweetening agent, methyl and propylparabens as preservatives, a dye and a flavoring such as cherry or orange flavor.
- active compounds i.e., the LXR agonists
- Solutions or suspensions of these active compounds can be prepared in water suitably mixed with a surfactant such as hydroxy-propylcellulose.
- Dispersions can also be prepared in glycerol, liquid polyethylene glycols and mixtures thereof in oils. Under ordinary conditions of storage and use, these preparations contain a preservative to prevent the growth of microorganisms.
- the pharmaceutical forms suitable for injectable use include sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions. In all cases, the form must be sterile and must be fluid to the extent that easy syringability exists.
- the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (e.g., glycerol, propylene glycol and liquid polyethylene glycol), suitable mixtures thereof, and vegetable oils.
- the LXR agonist may be administered either alone or in combination with one or more additional active agents.
- Combination therapy includes administration of a single pharmaceutical dosage formulation which contains a
- LXR asonist and one or more additional active agents as well as administration of the LXR agomst and each active agent in its own separate pharmaceutical dosage formulation
- a LXR agonist and an HMG-CoA reductase inhibitor can be administered to the patient together in a single oral dosage composition such as a tablet or capsule, or each agent administered in separate oral dosage formulations Where separate dosage formulations are used, the LXR agomst and one or more additional active agents can be administered at essentially the same time, i e , concurrently, or at separately staggered times, i e , sequentiall , combmation therapy is understood to include all these regimens
- the LXR agomst may be administered m combination with one or more of the following active agents an antihyperhpidemic agent, a plasma HDL-raismg agent, an antihypercholesterolemic agent, such as a cholesterol biosynthesis inhibitor, for example an HMG-CoA reductase inhibitor, an HMG-CoA synthase inhibitor, a squalene epoxidase inhibitor, or a squalene synthetase inhibitor (also known as squalene synthase inhibitor), an acyl-coenzyme A cholesterol acyltransferase (AC AT) inhibitor, such as melinamide, probucol.
- active agents an antihyperhpidemic agent, a plasma HDL-raismg agent, an antihypercholesterolemic agent, such as a cholesterol biosynthesis inhibitor, for example an HMG-CoA reductase inhibitor, an HMG-CoA synthase inhibitor, a squalene e
- nicotinic acid and the salts thereof and niacmamide a cholesterol absorption inhibitor such as ⁇ et ⁇ -sitosterol.
- a bile acid sequestrant anion exchange resm such as cholestyramme, colestipol or a dialkylammoalkyl derivatives of a cross-linked dextran.
- an LDL (low density lipoprotein) receptor mducer fibrates such as clofibrate, fenofibrate, and gemfibrizol, vitamin B 6 (also known as py ⁇ doxme) and the pharmaceutically acceptable salts thereof, such as the HC1 salt, vitamin B ⁇ 2 (also known as cyanocobalamin), anti-oxidant vitamins, such as vitamin C and E, and beta carotene a beta- blocker, an angiotensm II antagonist, an angiotensm converting enzyme inhibitor, and a platelet aggregation inhibitor, such as fibrinogen receptor antagonists (i e , glycoprote Ilb/IIIa fibrinogen receptor antagonists) and aspi ⁇ n
- the LXR agomst can be administered m combination with more than one additional active agent, for example, a combination of LXR agomst with an HMG-CoA reductase inhibitor and aspi ⁇ n, or LXR agomst w th an HMG
- the LXR agonist is preferably administered with a cholesterol biosynthesis inhibitor, particularly an HMG-CoA reductase inhibitor
- HMG-CoA reductase inhibitor is intended to include all pharmaceutically acceptable salt, ester, free acid and lactone forms of compounds which have HMG-CoA reductase inhibitory activity and. therefore, the use of such salts, esters, free acids and lactone forms is included within the scope of this invention.
- Compounds which have inhibitory activity for HMG-CoA reductase can be readily identified using assays well-known in the art. For instance, suitable assays are described or disclosed in U.S. Patent No. 4,231,938 and WO 84/02131, the teachings of which are incorporated herein by reference.
- HMG-CoA reductase inhibitors include, but are not limited to, lovastatin (MEVACOR ® ; see, U.S. Patent No. 4,231,938); simvastatin (ZOCOR ® ; see, U.S. Patent No. 4,444,784); pravastatin sodium (PRAVACHOL ® ; see, U.S. Patent No. 4,346,227); fluvastatin sodium (LESCOL ® ; see, U.S. Patent No. 5,354,772); atorvastatin calcium (LIPITOR ® ; see, U.S. Patent No. 5,273,995) and rivastatin (also known as cerivastatin; see, U.S. Patent No.
- HMG-CoA reductase inhibitors that can be used in the methods of the present invention are described at page 87 of M. Yalpani, "Cholesterol Lowering Drugs," Chemistry & Industry, pp. 85-89 (5 February 1996).
- the HMG-CoA reductase inhibitor is selected from lovastatin and simvastatin. Dosage information for HMG-CoA reductase inhibitors is well known in the art, since several HMG-CoA reductase inhibitors are marketed in the U.S.
- the daily dosage amounts of the HMG-CoA reductase inhibitor may be the same or similar to those amounts which are employed for anti-hypercholesterolemic treatment and which are described in the Physicians ' Desk Reference (PDR).
- PDR Physicians ' Desk Reference
- the oral dosage amount of HMG-CoA reductase inhibitor is from about 1 to 200 mg/day and, more preferably, from about 5 to 160 mg/day.
- dosage amounts will vary depending on the potency of the specific HMG-CoA reductase inhibitor used as well as other factors as noted above.
- An HMG-CoA reductase inhibitor which has sufficiently greater potency may be given in sub-milligram daily dosages.
- the daily dosage amount for simvastatin may be selected from 5 mg, 10 mg, 20 mg, 40 mg, 80 mg and 160 mg; for lovastatin, 10 mg, 20 mg, 40 mg and 80 mg; for fluvastatin sodium, 20 mg, 40 mg and 80 mg; and for pravastatin sodium. 10 mg, 20 mg. and 40 mg.
- the daily dosage amount for atorvastatin calcium may be in the range of from 1 mg to 160 mg and, more particularly, from 5 mg to 80 mg.
- Oral administration may be in single or divided doses of two, three, or four times daily, although a single daily dose of the HMG-CoA reductase inhibitor is preferred.
- an HDL-raising amount of a LXR agonist can be used for the preparation of a medicament useful for raising the plasma level of high density lipoprotein in mammals, particularly in humans.
- a prophylactically effective amount of a LXR agonist can be used for the preparation of a medicament useful for preventing or reducing the risk of developing atherosclerosis, and for preventing or reducing the risk of having a first or subsequent atherosclerotic disease event in mammals, particularly in humans.
- a therapeutically effective amount of a LXR agonist can be used for the preparation of a medicament useful for treating atherosclerosis in mammals, particularly in humans.
- the LXR agonist can be admixed together with a therapeutically effective amount of one or more additional active agents selected from the group consisting of: an LDL-lowering agent; an antihyperlipidemic agent; an HDL-raising agent; an HMG-CoA synthase inhibitor; a squalene epoxidase inhibitor; a squalene synthetase inhibitor; an acyl-coenzyme A: cholesterol acyltransferase inhibitor; probucol; nicotinic acid and the salts thereof; niacinamide; a cholesterol absorption inhibitor; a bile acid sequestrant anion exchange resin; a low density lipoprotein receptor inducer; clofibrate, fenofibrate, and gemfibrizol; vitamin B 6 and the pharmaceutically acceptable salts thereof; vitamin Bj ; an anti-oxidant vitamin; a ⁇ e/ -blocker; an angioten
- the LXR agonist and a therapeutically effective amount of an HMG-CoA reductase inhibitor can be admixed together for the preparation of a medicament useful for the above-described treatments. More particularly, the LXR agonist and a therapeutically effective amount of an HMG-CoA reductase inhibitor selected from the pharmaceutically acceptable lactone, free acid, ester and salt forms of lovastatin, simvastatin, pravastatin. fluvastatin, atorvastatin and rivastatin can be admixed together for the preparation of a medicament suitable for oral administration which is useful for the above- described treatments.
- the HMG-CoA reductase inhibitor used for the medicament preparation is lovastatin or simvastatin.
- LXR activation e.g., by LXR agonists
- HDL levels mediated by the activation of ABC family members by LXR.
- LXR agonists can induce the expression of ABC family members, in particular ABC family members involved in sterol transport.
- LXR activation leads to a dramatic increase in the transcription of ABC family members.
- This increased ABC activity causes an increase in the transport of sterols, e.g. , cholesterol, and other lipids across the membranes of cells, thereby leading to an overall increase in HDL levels.
- Example 1 is offered for illustrative purposes, and are not intended to limit the invention in any manner. Those of skill in the art will readily recognize a variety of noncritical parameters which can be changed or modified to yield essentially the same results.
- Example 1 is offered for illustrative purposes, and are not intended to limit the invention in any manner. Those of skill in the art will readily recognize a variety of noncritical parameters which can be changed or modified to yield essentially the same results.
- LXR ⁇ agonists are tested for their ability to act as LXR ⁇ agonists (see. Figure 1).
- the compounds were tested for ability to bind to LXR ⁇ .
- LXR ⁇ agonists were then identified in both a cell-based high throughput screen and a peptide sensor LXR assay.
- the compound T314407 was also tested for ability to compete with other ligands for binding to LXR ⁇ .
- the percentage of radiolabeied T314407 that bound to LXR ⁇ in the presence of varying concentrations of unlabeled T314407, T900546, T901.433, or 24,25-epoxycholesterol was determined. Results, which are shown in Figure 3, demonstrate that T314407 competitively binds to LXR ⁇ with a Ki of 0.2 ⁇ M.
- a peptide sensor assay was used to demonstrate the ability of a putative LXR ⁇ agonist to induce a conformational change in LXR ⁇ .
- a ligand- induced conformational change is believed to be a common property of nuclear hormone receptors such as LXR ⁇ .
- the peptide sensor assay which was carried out as described in PCT patent application PCT/US98/24969 (Publ. No. WO 99/27365), is an in vitro assay.
- LXR agonists to stimulate transcription of LXR ⁇ -mediated transcription was examined in the experiments described in this Example. First, the effect of the agonist T314407 on the recruitment of the coactivator SRC-1 to LXR ⁇ was examined in a mammalian two-hybrid assay. The ability of T314407 to activate LXR-mediated transcription was then tested in a reporter gene cotransfection assay was then tested. Finally, the effect of various LXR agonists on transcription was shown to be LXR-specific.
- a DNA binding domain of the nonreceptor transcription factor GAL4 was fused to the putative ligand binding domain of LXR ⁇ .
- the resultant construct was introduced into 293 cells, together with a luciferase reporter construct under the control of a GAL4 upstream activation sequence (UAS).
- UAS GAL4 upstream activation sequence
- the transfected cells were then treated with the compounds and luciferase activity was measured. Individual compounds were evaluated relative to a control (no additional compound) and the EC 50 was determined as the concentration necessary to produce 50% of the maximal luciferase activity.
- a fusion polypeptide in which the DNA binding domain of the GAL4 transcription factor was fused to the SRC-1 coactivator was fused to the SRC-1 coactivator.
- a plasmid expressing the GAL4 DNA-binding domain-SRC- 1 fusion protein and a plasmid expressing second fusion protein in which an LXR ⁇ ligand-binding domain was fused to VP-16 were used.
- HepG2 cells were transiently transfected with pG5 (a luciferase reporter coding sequence under the control of five GAL4 binding sites) luciferase reporter plasmid (0.25 ⁇ g/1-5 x 10 5 cells) and the plasmids expressing the two fusion proteins.
- Luciferase reporter activity was measured after treating cells with or without the LXR agonists (concentrations as indicated). Relative luciferase activity is a ratio of luciferase activity (normalized by ⁇ -galactosidase activity) between treated and untreated cells.
- the putative LXR agonists T314407 and T280404 markedly stimulated the recruitment of the SRC-1 coactivator to LXR ⁇ , as shown in Figure 6.
- the oxysterol 24,25- epoxycholesterol also stimulated recruitment, while the DMSO control indicated no enhancement of recruitment.
- the LXR ⁇ Agonists Specifically Activate LXR-mediated Transcription
- Plasmids that encode fusion proteins of the ligand binding domains of LXR ⁇ , LXR ⁇ , CPF, HNF4, FXR, and RXR were constructed by introducing the respective coding regions into the plasmid pM3 (Clontech, Inc.). which includes a GAL4 DNA binding domain. The resulting plasmids were individually co- transfected into the cells along with a reporter plasmid that contained a luciferase reporter gene under the control of a GAL4 upstream activation sequence.
- Relative luciferase activity was deierrnined in the presence or absence of the putative LXR ⁇ agonists Tl 70400, T280404, T314393, T314407, T513892, T210943, and T588142, as well as the known LXR ⁇ modulator 24,25-epoxycholesterol.
- each of the putative LXR ⁇ agonists strongly activated LXR-mediated transcription. A lesser amount of activation was observed for LXR ⁇ - mediated transcription.
- the LXR ⁇ agonists did not activate transcription mediated by CPF, HNF4, FXR, or RXR.
- the highest amount of activation was observed for the compound T314407, which activated LXR ⁇ -mediated transcription with an EC 5 o of 0.2 ⁇ M (EC 0 is defined as the amount of compound necessary to product 50% of the maximal luciferase activity).
- This compound directly binds to LXR ⁇ , as evidenced by the competition assay described above, with a Kj of 0.2 ⁇ m.
- T314407 is not cytotoxic, having an EC 50 of greater than 50 ⁇ m.
- T314407 was also demonstrated to transactivate expression of the human cholesterol 7 ⁇ -hydrolase (C YP7A) gene, which is a rate limiting enzyme in bile acid synthesis, which is a major pathway for cholesterol catabolism.
- C YP7A human cholesterol 7 ⁇ -hydrolase
- T314407 and other LXR ⁇ agonists are useful for lowering cholesterol levels and for treating other lipid disorders.
- This Example describes experiments to determine the effect of oral administration of the LXR agonist T0901317 on plasma triglyceride levels and HDL cholesterol levels.
- the study was conducted over two weeks, with twenty mice (C57BL/6) for each timepoint (10 males and 10 females).
- T090137 was administered to the mice once a day at doses of 5 or 50 mg/kg body weight.
- blood of the mice was analyzed for plasma lipid concentration and for hepatic gene expression.
- T090137 Oral administration of 5 mg/kg and 50 mg kg T090137 resulted in an increase in total plasma cholesterol in both male and female mice ( Figure 10, left panel). HDL cholesterol levels were also increased by administration of the LXR agonist ( Figure 10, right panel). The amount of the increase in total plasma cholesterol and in HDL cholesterol was dependent on the amount of T090137 administered in each dose, but 14 days of administration did not result in appreciable difference in effect compared to a 7 day administration regime. Oral administration of T0901317 also resulted in an increase in plasma triglyceride levels (Figure 11). These results were not changed significantly by feeding the mice a high cholesterol diet.
Landscapes
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Diabetes (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Child & Adolescent Psychology (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Epidemiology (AREA)
- Gastroenterology & Hepatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description







Claims








Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002377999A CA2377999A1 (en) | 1999-07-08 | 2000-07-07 | Compositions and methods for raising hdl cholesterol levels |
| AU60747/00A AU6074700A (en) | 1999-07-08 | 2000-07-07 | Compositions and methods for raising hdl cholesterol levels |
| EP00947080A EP1212065A4 (en) | 1999-07-08 | 2000-07-07 | Compositions and methods for raising hdl cholesterol levels |
| JP2001508985A JP2004500332A (en) | 1999-07-08 | 2000-07-07 | Compositions and methods for increasing HDL cholesterol levels |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14299499P | 1999-07-08 | 1999-07-08 | |
| US60/142,994 | 1999-07-08 | ||
| US61213500A | 2000-07-07 | 2000-07-07 | |
| US09/612,135 | 2000-07-07 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2001003705A1 true WO2001003705A1 (en) | 2001-01-18 |
Family
ID=26840588
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2000/018533 WO2001003705A1 (en) | 1999-07-08 | 2000-07-07 | Compositions and methods for raising hdl cholesterol levels |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP1212065A4 (en) |
| JP (1) | JP2004500332A (en) |
| AU (1) | AU6074700A (en) |
| CA (1) | CA2377999A1 (en) |
| WO (1) | WO2001003705A1 (en) |
Cited By (39)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001015676A2 (en) * | 1999-09-01 | 2001-03-08 | University Of British Columbia | Compositions and methods for modulating hdl cholesterol and triglyceride levels |
| WO2001082917A2 (en) * | 2000-05-03 | 2001-11-08 | Tularik Inc. | Treatment of hypertriglyceridemia and other conditions using lxr modulators |
| WO2002058690A2 (en) * | 2001-01-26 | 2002-08-01 | Chugai Seiyaku Kabushiki Kaisha | Methods for the treatment of diseases using malonyl-coa decarbox ylase inhibitors |
| WO2002058698A2 (en) | 2001-01-26 | 2002-08-01 | Chugai Seiyaku Kabushiki Kaisha | Malonyl-coa decarboxylase inhibitors useful as metabolic modulators |
| WO2002064136A2 (en) * | 2001-01-26 | 2002-08-22 | Chugai Seiyaku Kabushiki Kaisha | Malonyl-coa decarboxylase inhibitors useful as metabolic modulators |
| WO2002077229A2 (en) * | 2001-03-01 | 2002-10-03 | Lion Bioscience Ag | Cofactors of the liver x receptor alpha and methods of use |
| EP1285662A1 (en) * | 2001-08-20 | 2003-02-26 | ZLB Bioplasma AG | Reconstituted HDL for the treatment of stroke and ischemic conditions |
| WO2003018047A2 (en) * | 2001-08-20 | 2003-03-06 | Zlb Bioplasma Ag | Hdl for the treatment of stroke and other ischemic conditions |
| GB2381866A (en) * | 2001-11-12 | 2003-05-14 | Karobio Ab | Assays for liver X receptor (LXR) modulators |
| WO2003090732A1 (en) * | 2002-04-23 | 2003-11-06 | Chugai Seiyaku Kabushiki Kaisha | Lxr modulators for the treatment of cardiovascular diseases |
| WO2003090869A1 (en) * | 2002-04-23 | 2003-11-06 | Chugai Seiyaku Kabushiki Kaisha | Lxr modulators |
| WO2004001002A2 (en) * | 2002-06-19 | 2003-12-31 | Anagen Therapeutics, Inc. | Novel anticholesterol compositions and method for using same |
| WO2005005417A1 (en) * | 2003-07-11 | 2005-01-20 | Astrazeneca Ab | Pyrrole-2, 5-dione derivatives as liver x receptor modulators |
| WO2005005416A1 (en) * | 2003-07-11 | 2005-01-20 | Astrazeneca Ab | Pyrrole-2, 5-dithione derivatives as liver x receptor modulators |
| WO2005009383A2 (en) * | 2003-07-22 | 2005-02-03 | Glaxo Group Limited | Methods of treatment with lxr agonists |
| EP1511483A2 (en) * | 2002-03-27 | 2005-03-09 | Smithkline Beecham Corporation | Methods of treatment with lxr modulators |
| US6924311B2 (en) * | 2001-10-17 | 2005-08-02 | X-Ceptor Therapeutics, Inc. | Methods for affecting various diseases utilizing LXR compounds |
| WO2006073365A1 (en) * | 2005-01-10 | 2006-07-13 | Astrazeneca Ab | Non-anilinic derivatives of isothiazol-3(2h)-thione 1,1-dioxides as liver x receptor modulators |
| WO2006073363A1 (en) * | 2005-01-10 | 2006-07-13 | Astrazeneca Ab | Derivatives of isothiazol-3(2h)-one 1,1-dioxides as liver x receptor modulators |
| WO2006073364A1 (en) * | 2005-01-10 | 2006-07-13 | Astrazeneca Ab | Derivatives of isothiazol-3 (2h)-thione 1,1-dioxides as liver x receptor modulators |
| WO2006073366A1 (en) * | 2005-01-10 | 2006-07-13 | Astrazeneca Ab | Non-anilinic derivatives of isothiazol-3(2h)-one 1,1-dioxides as liver x receptor modulators |
| WO2007002559A1 (en) | 2005-06-27 | 2007-01-04 | Exelixis, Inc. | Pyrazole based lxr modulators |
| WO2007047991A1 (en) | 2005-10-21 | 2007-04-26 | Bristol-Myers Squibb Company | Tetrahydroisoquinoline as lxr modulators |
| WO2007050425A2 (en) | 2005-10-21 | 2007-05-03 | Bristol-Myers Squibb Company | Lxr modulators |
| US7261880B2 (en) | 2001-04-18 | 2007-08-28 | Genzyme Corporation | Methods of treating Syndrome X with aliphatic polyamines |
| US7285562B2 (en) | 2003-08-01 | 2007-10-23 | Chugai Seiyaku Kabushiki Kaisha | Cyanoamide compounds useful as malonyl-CoA decarboxylase inhibitors |
| US7449482B2 (en) | 2003-08-01 | 2008-11-11 | Chugai Seiyaku Kabushiki Kaisha | Piperidine compounds useful as malonyl-CoA decarboxylase inhibitors |
| US7696365B2 (en) | 2003-08-01 | 2010-04-13 | Chugai Seiyaku Kabushiki Kaisha | Heterocyclic compounds useful as malonyl-CoA decarboxylase inhibitors |
| US7709510B2 (en) | 2001-02-20 | 2010-05-04 | Chugai Seiyaku Kabushiki Kaisha | Azoles as malonyl-CoA decarboxylase inhibitors useful as metabolic modulators |
| US7723366B2 (en) | 2001-02-20 | 2010-05-25 | Chugai Seiyaku Kabushiki Kaisha | Azole compounds as malonyl-CoA decarboxylase inhibitors for treating metabolic diseases |
| US7786145B2 (en) | 2003-08-01 | 2010-08-31 | Chugai Seiyaku Kabushiki Kaisha | Cyanoguanidine-based azole compounds useful as malonyl-CoA decarboxylase inhibitors |
| US7998986B2 (en) | 2001-12-21 | 2011-08-16 | Exelixis Patent Company Llc | Modulators of LXR |
| US8013001B2 (en) | 2001-12-21 | 2011-09-06 | Exelixis, Inc. | Modulators of LXR |
| US8076376B2 (en) | 2005-07-22 | 2011-12-13 | Powers Jay P | Aniline sulfonamide derivatives and their uses |
| WO2012033353A2 (en) | 2010-09-07 | 2012-03-15 | 서울대학교 산학협력단 | Sesterterpene compounds and use thereof |
| WO2012135082A1 (en) | 2011-03-25 | 2012-10-04 | Bristol-Myers Squibb Company | Prodrugs of lxr modulating imidazole derivatives |
| CN105250253A (en) * | 2015-10-30 | 2016-01-20 | 黄恺 | Application of T0901317 serving as PARP1 inhibitor |
| US10011566B2 (en) | 2015-12-15 | 2018-07-03 | Astrazeneca Ab | Compounds |
| US11034654B2 (en) | 2017-06-14 | 2021-06-15 | Astrazeneca Ab | 2,3-dihydroisoindole-1-carboxamides useful as ROR-gamma modulators |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4159335A (en) * | 1974-12-02 | 1979-06-26 | Schering Corporation | Substituted anilino-2-thiazolines |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6835866B1 (en) * | 1998-12-10 | 2004-12-28 | Board Of Regents, The University Of Texas Systems | Compositions and methods of modulating cholesterol metabolism |
| US6316503B1 (en) * | 1999-03-15 | 2001-11-13 | Tularik Inc. | LXR modulators |
| KR20020012612A (en) * | 1999-06-18 | 2002-02-16 | 씨브이 쎄러퓨틱스, 인코포레이티드 | Compositions and methods for increasing cholesterol efflux and raising hdl using atp binding cassette transporter protein abc1 |
-
2000
- 2000-07-07 WO PCT/US2000/018533 patent/WO2001003705A1/en not_active Application Discontinuation
- 2000-07-07 JP JP2001508985A patent/JP2004500332A/en active Pending
- 2000-07-07 CA CA002377999A patent/CA2377999A1/en not_active Abandoned
- 2000-07-07 EP EP00947080A patent/EP1212065A4/en not_active Withdrawn
- 2000-07-07 AU AU60747/00A patent/AU6074700A/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4159335A (en) * | 1974-12-02 | 1979-06-26 | Schering Corporation | Substituted anilino-2-thiazolines |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP1212065A4 * |
Cited By (69)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001015676A2 (en) * | 1999-09-01 | 2001-03-08 | University Of British Columbia | Compositions and methods for modulating hdl cholesterol and triglyceride levels |
| WO2001015676A3 (en) * | 1999-09-01 | 2002-07-25 | Univ British Columbia | Compositions and methods for modulating hdl cholesterol and triglyceride levels |
| WO2001082917A2 (en) * | 2000-05-03 | 2001-11-08 | Tularik Inc. | Treatment of hypertriglyceridemia and other conditions using lxr modulators |
| WO2001082917A3 (en) * | 2000-05-03 | 2002-06-06 | Tularik Inc | Treatment of hypertriglyceridemia and other conditions using lxr modulators |
| WO2002058690A3 (en) * | 2001-01-26 | 2003-04-24 | Chugai Pharmaceutical Co Ltd | Methods for the treatment of diseases using malonyl-coa decarbox ylase inhibitors |
| US7524969B2 (en) | 2001-01-26 | 2009-04-28 | Chugai Seiyaku Kabushiki Kaisha | Malonyl-CoA decarboxylase inhibitors useful as metabolic modulators |
| WO2002064136A2 (en) * | 2001-01-26 | 2002-08-22 | Chugai Seiyaku Kabushiki Kaisha | Malonyl-coa decarboxylase inhibitors useful as metabolic modulators |
| US7279477B2 (en) | 2001-01-26 | 2007-10-09 | Chugai Seiyaku Kabushiki Kaisha | Malonyl-CoA decarboxylase inhibitors useful as metabolic modulators |
| US8119819B2 (en) | 2001-01-26 | 2012-02-21 | Chugai Seiyaku Kabushiki Kaisha | Malonyl-CoA decarboxylase inhibitors useful as metabolic modulators |
| WO2002058698A2 (en) | 2001-01-26 | 2002-08-01 | Chugai Seiyaku Kabushiki Kaisha | Malonyl-coa decarboxylase inhibitors useful as metabolic modulators |
| WO2002058690A2 (en) * | 2001-01-26 | 2002-08-01 | Chugai Seiyaku Kabushiki Kaisha | Methods for the treatment of diseases using malonyl-coa decarbox ylase inhibitors |
| JP2008001719A (en) * | 2001-01-26 | 2008-01-10 | Chugai Pharmaceut Co Ltd | Method for treatment of disease using malonyl-coa decarboxylase inhibitor |
| WO2002064136A3 (en) * | 2001-01-26 | 2003-10-30 | Chugai Pharmaceutical Co Ltd | Malonyl-coa decarboxylase inhibitors useful as metabolic modulators |
| WO2002058698A3 (en) * | 2001-01-26 | 2004-02-12 | Chugai Pharmaceutical Co Ltd | Malonyl-coa decarboxylase inhibitors useful as metabolic modulators |
| JP2010065054A (en) * | 2001-01-26 | 2010-03-25 | Chugai Pharmaceut Co Ltd | MALONYL-CoA DECARBOXYLASE INHIBITOR USEFUL AS METABOLIC MODULATOR |
| US7385063B2 (en) | 2001-01-26 | 2008-06-10 | Chugai Seiyaku Kabushiki Kaisha | Method for preparing imidazole derivatives |
| US8110686B2 (en) | 2001-02-20 | 2012-02-07 | Chugai Seiyaki Kabushiki Kaisha | Azoles as malonyl-CoA decarboxylase inhibitors useful as metabolic modulators |
| US7709510B2 (en) | 2001-02-20 | 2010-05-04 | Chugai Seiyaku Kabushiki Kaisha | Azoles as malonyl-CoA decarboxylase inhibitors useful as metabolic modulators |
| US7723366B2 (en) | 2001-02-20 | 2010-05-25 | Chugai Seiyaku Kabushiki Kaisha | Azole compounds as malonyl-CoA decarboxylase inhibitors for treating metabolic diseases |
| WO2002077229A3 (en) * | 2001-03-01 | 2003-12-04 | Lion Bioscience Ag | Cofactors of the liver x receptor alpha and methods of use |
| WO2002077229A2 (en) * | 2001-03-01 | 2002-10-03 | Lion Bioscience Ag | Cofactors of the liver x receptor alpha and methods of use |
| US7261880B2 (en) | 2001-04-18 | 2007-08-28 | Genzyme Corporation | Methods of treating Syndrome X with aliphatic polyamines |
| AU2002340825B2 (en) * | 2001-08-20 | 2007-07-05 | Csl Behring Ag | HDL for the treatment of stroke and other ischemic conditions |
| US7491693B2 (en) | 2001-08-20 | 2009-02-17 | Csl Behring Ag | HDL for the treatment of stroke and other ischemic conditions |
| EP1285662A1 (en) * | 2001-08-20 | 2003-02-26 | ZLB Bioplasma AG | Reconstituted HDL for the treatment of stroke and ischemic conditions |
| WO2003018047A2 (en) * | 2001-08-20 | 2003-03-06 | Zlb Bioplasma Ag | Hdl for the treatment of stroke and other ischemic conditions |
| WO2003018047A3 (en) * | 2001-08-20 | 2004-01-29 | Zlb Bioplasma Ag | Hdl for the treatment of stroke and other ischemic conditions |
| US6924311B2 (en) * | 2001-10-17 | 2005-08-02 | X-Ceptor Therapeutics, Inc. | Methods for affecting various diseases utilizing LXR compounds |
| GB2381866A (en) * | 2001-11-12 | 2003-05-14 | Karobio Ab | Assays for liver X receptor (LXR) modulators |
| US8013001B2 (en) | 2001-12-21 | 2011-09-06 | Exelixis, Inc. | Modulators of LXR |
| US7998986B2 (en) | 2001-12-21 | 2011-08-16 | Exelixis Patent Company Llc | Modulators of LXR |
| EP1511483A2 (en) * | 2002-03-27 | 2005-03-09 | Smithkline Beecham Corporation | Methods of treatment with lxr modulators |
| JP2005533007A (en) * | 2002-03-27 | 2005-11-04 | スミスクライン・ビーチャム・コーポレイション | Treatment method using LXR modulator |
| EP1511483A4 (en) * | 2002-03-27 | 2009-03-18 | Smithkline Beecham Corp | Methods of treatment with lxr modulators |
| WO2003090869A1 (en) * | 2002-04-23 | 2003-11-06 | Chugai Seiyaku Kabushiki Kaisha | Lxr modulators |
| WO2003090732A1 (en) * | 2002-04-23 | 2003-11-06 | Chugai Seiyaku Kabushiki Kaisha | Lxr modulators for the treatment of cardiovascular diseases |
| WO2004001002A3 (en) * | 2002-06-19 | 2004-05-06 | Anagen Therapeutics Inc | Novel anticholesterol compositions and method for using same |
| WO2004001002A2 (en) * | 2002-06-19 | 2003-12-31 | Anagen Therapeutics, Inc. | Novel anticholesterol compositions and method for using same |
| WO2005005416A1 (en) * | 2003-07-11 | 2005-01-20 | Astrazeneca Ab | Pyrrole-2, 5-dithione derivatives as liver x receptor modulators |
| WO2005005417A1 (en) * | 2003-07-11 | 2005-01-20 | Astrazeneca Ab | Pyrrole-2, 5-dione derivatives as liver x receptor modulators |
| US7432288B2 (en) | 2003-07-11 | 2008-10-07 | Astrazeneca Ab | Pyrrole-2,5-dione derivatives as Liver X receptor modulators |
| WO2005009383A2 (en) * | 2003-07-22 | 2005-02-03 | Glaxo Group Limited | Methods of treatment with lxr agonists |
| WO2005009383A3 (en) * | 2003-07-22 | 2005-12-08 | Glaxo Group Ltd | Methods of treatment with lxr agonists |
| US8080665B2 (en) | 2003-08-01 | 2011-12-20 | Chugai Seiyaku Kabushiki Kaisha | Piperidine compounds useful as malonyl-CoA decarboxylase inhibitors |
| US7449482B2 (en) | 2003-08-01 | 2008-11-11 | Chugai Seiyaku Kabushiki Kaisha | Piperidine compounds useful as malonyl-CoA decarboxylase inhibitors |
| US7285562B2 (en) | 2003-08-01 | 2007-10-23 | Chugai Seiyaku Kabushiki Kaisha | Cyanoamide compounds useful as malonyl-CoA decarboxylase inhibitors |
| US7786145B2 (en) | 2003-08-01 | 2010-08-31 | Chugai Seiyaku Kabushiki Kaisha | Cyanoguanidine-based azole compounds useful as malonyl-CoA decarboxylase inhibitors |
| US7696365B2 (en) | 2003-08-01 | 2010-04-13 | Chugai Seiyaku Kabushiki Kaisha | Heterocyclic compounds useful as malonyl-CoA decarboxylase inhibitors |
| US7897615B2 (en) | 2003-08-01 | 2011-03-01 | Chugai Sciyaku Kabushiki Kaisha | Cyanoamide compounds useful as malonyl-CoA decarboxylase inhibitors |
| US7582629B2 (en) | 2005-01-10 | 2009-09-01 | Astrazeneca Ab | Derivatives of isothiazol-3(2H)-one 1,1-dioxides as liver X receptor modulators |
| WO2006073364A1 (en) * | 2005-01-10 | 2006-07-13 | Astrazeneca Ab | Derivatives of isothiazol-3 (2h)-thione 1,1-dioxides as liver x receptor modulators |
| WO2006073365A1 (en) * | 2005-01-10 | 2006-07-13 | Astrazeneca Ab | Non-anilinic derivatives of isothiazol-3(2h)-thione 1,1-dioxides as liver x receptor modulators |
| WO2006073363A1 (en) * | 2005-01-10 | 2006-07-13 | Astrazeneca Ab | Derivatives of isothiazol-3(2h)-one 1,1-dioxides as liver x receptor modulators |
| US7960380B2 (en) | 2005-01-10 | 2011-06-14 | Astrazeneca Ab | Non-anilinic derivatives of isothiazol-3(2H)-one 1,1-dioxides as liver X receptor modulators |
| US7723333B2 (en) | 2005-01-10 | 2010-05-25 | Astrazeneca Ab | Non-anilinic derivatives of isothiazol-3(2H)-one 1,1-dioxides as liver X receptor modulators |
| WO2006073366A1 (en) * | 2005-01-10 | 2006-07-13 | Astrazeneca Ab | Non-anilinic derivatives of isothiazol-3(2h)-one 1,1-dioxides as liver x receptor modulators |
| WO2007002559A1 (en) | 2005-06-27 | 2007-01-04 | Exelixis, Inc. | Pyrazole based lxr modulators |
| US8076376B2 (en) | 2005-07-22 | 2011-12-13 | Powers Jay P | Aniline sulfonamide derivatives and their uses |
| EP2392567A1 (en) | 2005-10-21 | 2011-12-07 | Bristol-Myers Squibb Company | Benzothiazine derivatives and their use as lxr modulators |
| WO2007047991A1 (en) | 2005-10-21 | 2007-04-26 | Bristol-Myers Squibb Company | Tetrahydroisoquinoline as lxr modulators |
| WO2007050425A2 (en) | 2005-10-21 | 2007-05-03 | Bristol-Myers Squibb Company | Lxr modulators |
| WO2012033353A2 (en) | 2010-09-07 | 2012-03-15 | 서울대학교 산학협력단 | Sesterterpene compounds and use thereof |
| WO2012135082A1 (en) | 2011-03-25 | 2012-10-04 | Bristol-Myers Squibb Company | Prodrugs of lxr modulating imidazole derivatives |
| CN105250253A (en) * | 2015-10-30 | 2016-01-20 | 黄恺 | Application of T0901317 serving as PARP1 inhibitor |
| US10011566B2 (en) | 2015-12-15 | 2018-07-03 | Astrazeneca Ab | Compounds |
| US10526286B2 (en) | 2015-12-15 | 2020-01-07 | Astrazeneca Ab | Compounds |
| US10988445B2 (en) | 2015-12-15 | 2021-04-27 | Astrazeneca Ab | Compounds |
| US11453644B1 (en) | 2015-12-15 | 2022-09-27 | Astrazeneca, Ab | Compounds |
| US11034654B2 (en) | 2017-06-14 | 2021-06-15 | Astrazeneca Ab | 2,3-dihydroisoindole-1-carboxamides useful as ROR-gamma modulators |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2004500332A (en) | 2004-01-08 |
| EP1212065A1 (en) | 2002-06-12 |
| AU6074700A (en) | 2001-01-30 |
| CA2377999A1 (en) | 2001-01-18 |
| EP1212065A4 (en) | 2004-02-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2001003705A1 (en) | Compositions and methods for raising hdl cholesterol levels | |
| Cao et al. | Design, synthesis, and evaluation of in vitro and in vivo anticancer activity of 4-substituted coumarins: a novel class of potent tubulin polymerization inhibitors | |
| US6180660B1 (en) | Cholesterol-lowering therapy | |
| ES2544482T3 (en) | Rexinoid compound having an alkoxy group | |
| RU2765218C2 (en) | Fixed combinations and compositions containing etc1002 and one or more statins, and methods for treatment or reduction in risk of development of cardiovascular disease | |
| Abate et al. | Analogues of σ receptor ligand 1-cyclohexyl-4-[3-(5-methoxy-1, 2, 3, 4-tetrahydronaphthalen-1-yl) propyl] piperazine (PB28) with added polar functionality and reduced lipophilicity for potential use as positron emission tomography radiotracers | |
| EP2001897A2 (en) | Compounds and methods for treatment of disorders associated with er stress | |
| US20030073614A1 (en) | Methods for affecting various diseases utilizing LXR compounds | |
| AU2014340182A1 (en) | Cromolyn derivatives and related methods of imaging and treatment | |
| JP2007509842A (en) | Compounds containing nitric oxide moieties for inducing the expression of ApoA1 for the treatment of cardiovascular disorders | |
| PT1302461E (en) | Tnf-alpha production inhibitors | |
| Mantlo et al. | Update on the discovery and development of cholesteryl ester transfer protein inhibitors for reducing residual cardiovascular risk | |
| CN103096895A (en) | Niacin mimetics, and methods of use thereof | |
| AU639252B2 (en) | Partial agonists of the strychnine insensitive glycine modulatory site of the n-methyl-d-aspartate receptor complex as neuropsychopharmacological agents | |
| WO2004072041A1 (en) | Tetrahydroquinolines as agonists of liver- x receptors | |
| AU753657B2 (en) | Combination therapy for reducing the risks associated with cardio-and cerebrovascular disease | |
| WO2019001416A1 (en) | Thiazolinone heterocyclic compound and preparation method, pharmaceutical composition, and application thereof | |
| M Kamble et al. | In silico evidence for binding of pentacyclic triterpenoids to Keap1-Nrf2 protein-protein binding site | |
| Anand et al. | Prediction of activity spectra of substances assisted prediction of biological activity spectra of potential anti-Alzheimer’s phytoconstituents | |
| US5723465A (en) | Inhibitors for cell adhesion and cellular infiltration | |
| BRPI0606805A2 (en) | kit and dosage unit | |
| Ammazzalorso et al. | Synthesis, in vitro evaluation, and molecular modeling investigation of benzenesulfonimide peroxisome proliferator-activated receptors α antagonists | |
| WO2001022962A1 (en) | Anti-hypercholesterolemic drug combination | |
| Carty et al. | Modulation of arachidonic acid metabolism in the treatment of rheumatoid arthritis | |
| CN103087009A (en) | Carboxylic acid derivative compound and preparation method and application thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2377999 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2000947080 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 60747/00 Country of ref document: AU |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 2000947080 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2000947080 Country of ref document: EP |