WO2000075107A2 - Bradykinin receptor antagonists - Google Patents
Bradykinin receptor antagonists Download PDFInfo
- Publication number
- WO2000075107A2 WO2000075107A2 PCT/EP2000/005059 EP0005059W WO0075107A2 WO 2000075107 A2 WO2000075107 A2 WO 2000075107A2 EP 0005059 W EP0005059 W EP 0005059W WO 0075107 A2 WO0075107 A2 WO 0075107A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- compound
- formula
- phenyl
- bradykinin
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/18—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carboxylic acids, or sulfur or nitrogen analogues thereof
- C07D295/182—Radicals derived from carboxylic acids
- C07D295/192—Radicals derived from carboxylic acids from aromatic carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/30—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/37—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups having the sulfur atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring
- C07C311/38—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups having the sulfur atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring having sulfur atoms of sulfonamide groups and amino groups bound to carbon atoms of six-membered rings of the same carbon skeleton
- C07C311/44—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups having the sulfur atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring having sulfur atoms of sulfonamide groups and amino groups bound to carbon atoms of six-membered rings of the same carbon skeleton having the nitrogen atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/06—Antiabortive agents; Labour repressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/04—Drugs for skeletal disorders for non-specific disorders of the connective tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/02—Muscle relaxants, e.g. for tetanus or cramps
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P41/00—Drugs used in surgical methods, e.g. surgery adjuvants for preventing adhesion or for vitreum substitution
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/10—Antioedematous agents; Diuretics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/02—Non-specific cardiovascular stimulants, e.g. drugs for syncope, antihypotensives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/34—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/60—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D211/62—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/22—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with hetero atoms directly attached to ring nitrogen atoms
- C07D295/26—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
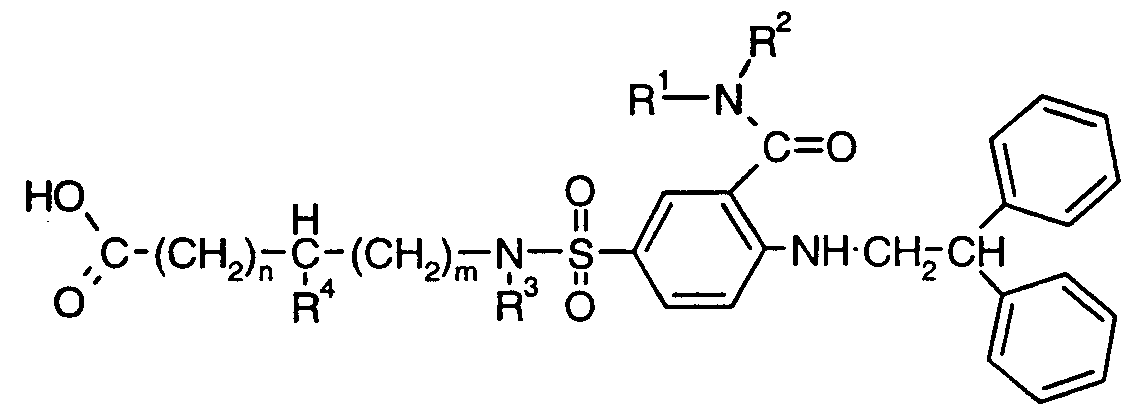
- the present invention relates to novel sulfonyl amine derivatives, to processes for their production, their use as pharmaceuticals and to pharmaceutical compositions comprising them.
- R 5A is -X A -R 6A or -N(R 7A )R 8A , wherein
- X A is piperidinylene or piperazinylene
- R 6A is H, d-C ⁇ lkyl, C 3 -C 4 alkenyl, C 3 -C 4 alkinyl, C C 4 (alkoxyalkyl), C C 4 (carboxyalkyl), a C 5 -C 7 heterocyclic group or phenyl-CrCalkyl;
- R 7A is amino-C 2 -C 4 alkyl or mono- or dKC CsalkylJamino-Cz-Csalkyl, and
- R 8A is H, C 1 -C 4 alkyl or has the meanings as given for R 7A ;
- R n is zero or 1 ;
- X 3 is CH or N;
- X 4 is a direct bond
- R 3A is H, C C 4 alkyl, C 3 -C 6 cycloalkyl, C 3 -C 6 alkenyl, C 3 -C 6 alkinyl, C 7 -C 10 aralkyl or C 6 -C 9 heteroaralkyl
- R 4A is H and m is 1 or 2 or 3; or
- X 4 is -CH(R 12 )-, R 3A is H and R 4A and R 12 together are propylene and m is 1 , or ethylene and m is 2;
- X ,2 is a divalent group of formula IA" wherein
- X 3 is CH or N
- R 11 is C ⁇ -C 4 alkyl, C 3 -C 6 cycloalkyl or -NR ⁇ R 2 , wherein
- R 1A and R ⁇ independently are d-C 4 alkyl or, together with the N-atom to which they are attached, represent a 5 to 7 membered heterocyclic ring; and R 9 and R 10 independently are a phenyl or pyridine ring; and salts thereof.
- substituents within their structure e.g. may bear appropriate phenyl ring or alkylene moiety substituents, e.g. phenyl and pyridine in the meaning of R 9 or R 10 may be unsubstituted or substituted by one or more halogen, and in the meaning of R 3A alkyl may be unsubstituted or substituted by halogen, C 3 -C 6 cycloalkyl or aryl; aralkyl may be unsubstituted or substituted by halogen, methoxy, nitro or C ⁇ -C alkyl which may be unsubstituted or substituted by halogen; heteroaralkyl may be unsubstituted or substituted by CrC 4 alkyl.
- the amino moiety of the defined aminocarb- onyl or amide groupings can be any appropriate amino grouping, e.g. cyclic or aliphatic or may bear further substituent groupings.
- the present invention provides a 2-(2,2-diphenylethylamino)- -5-(4-aminocarbonyl-piperidine-1-sulfonyl)-benzoic acid amide or -5-(aminocarbonyl- C 2 -C alkyleneaminosulfonyl)-benzoic acid amide, or salt thereof.
- R 1 and R 2 independently are d-C 4 alkyl or, together with the N-atom to which they are attached, represent a 5 to 7 membered heterocyclic ring;
- R 3 and R 4 together are ethylene and m is 2; or
- R 3 is H, C 1 -C 4 alkyl, C 5 -C 7 cycloalkyl or phenyl-C C 4 alkyl, R 4 is H and m is 1 or 2 or 3; n is zero or 1 ; and
- R 5 is -X-R 6 or -N(R 7 )R 8 , wherein X is -N N- ,
- R 6 is C C 4 alkyl, C 3 -C 4 alkenyl, C 3 -C 4 alkinyl, d-C ⁇ alkoxyalkyl), d-C 4 (carboxyalkyl), a
- R 7 is amino-C 2 -C 4 alkyl or mono- or di-(C ⁇ -C 5 alkyl)amino-C 2 -C 5 alkyl
- R 8 is H, C ⁇ -C 4 alkyl or has the meanings as given for R 7 ; and salts thereof.
- Alkyl groups and moieties in the compounds of formula IA or I may be branched or straight chain. Alkyl groups are suitably straight chain.
- Heterocyclic groups may be saturated or unsaturated and may contain one or more additional heterocyclic atoms, e.g. oxygen or sulfur.
- additional heterocyclic atoms e.g. oxygen or sulfur.
- Examples include piperidin-1-yl, morpholin- 1-yl, 3,6-dihydro-2.H.pyridin-1 -yl, thiomorpholin-1-yl, pyrrolin-1-yl, morpholin-4-yl, thiomor- pholin-4-yl, 3,6-dihydro-2H-pyridin-1 -yl, 2,5-dihydropyrrol-1-yl and 4-dif luoropiperidin-1 -yl, and may be unsubstituted or substituted by one or two halogen atoms.
- R 5A is unsubstituted piperazinyl or piperazinyl substituted by methyl, ethyl, benzyl, 2- pyridinyl, methoxyethyl, carboxymethyl or -CH 2 CHCH 2 ; or piperidine substituted by methyl;
- R 5A is N(R 7A )R 8A wherein R 7A is aminopropyl, aminobutyl, dimethylaminopropyl, diethylaminopropyl, dibutylaminoethyl, dimethylaminobutyl, or dimethylaminopentyl; and
- R 8A is H, methyl, aminopropyl, aminobutyl, dimethylaminopropyl or dimethylaminobutyl;
- X 1 is -(CH 2 ) n -CH(R 4A )-(CH 2 ) 2 -N(R 3A )- wherein n is zero or 1 and R 3A and R 4A together are ethylene;
- X 1 is -CH(R 4A )-(CH 2 ) m -CH(R 12 )-NH- wherein m is 1 or 2 and R 4A and R 12 together are propylene or ethylene;
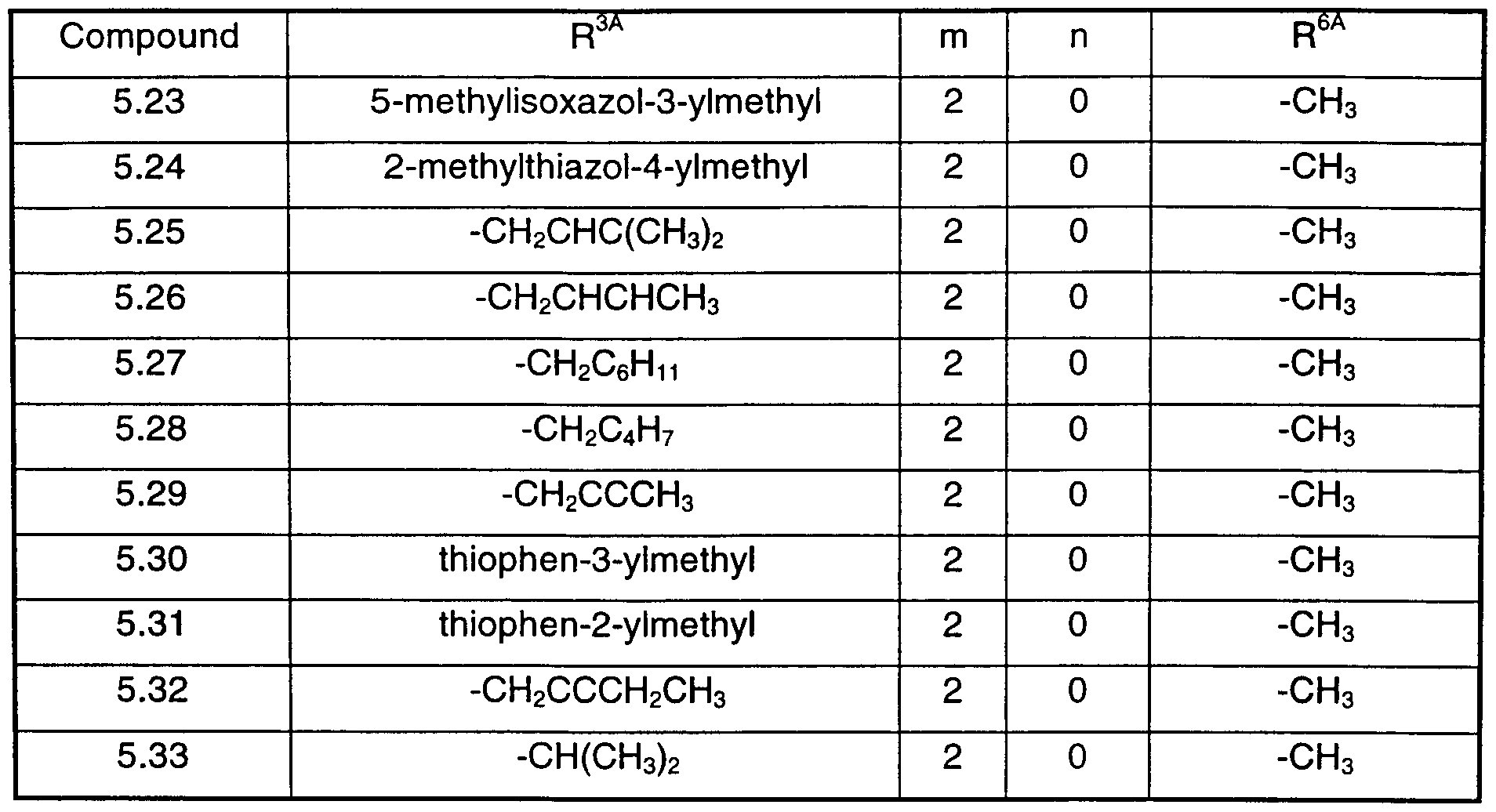
- X 1 is -(CH 2 ) n -CH 2 -(CH 2 ) m -N(R 3A )- wherein n is zero or 1 , m is 1 or 2 or 3 and R 3A is H, methyl, isopropyl, isobutyl, fluoroethyl, cyclopropylmethyl, cyclohexylmethyl, cyclobutylmethyl, -CH(CH 3 )C 6 H 5) cyclohexyl, propenyl, butenyl, pentenyl, propinyl, butinyl, pentinyl, benzyl, methylbenzyl, fluorobenzyl, trifluoromethylbenzyl, methoxybenzyl, nitrobenzyl, pyridinylmethyl, methylisoxazolylmethyl, methylthiazolylmethyl or thiophenmethyl;
- X 2 is a divalent group of formula IA" wherein X 3 is CH and R 11 is methyl, cyclopentyl, cyclohexyl, N(CH 3 )CH 2 CH 3 , piperidinyl, morpholinyl, thiomorpholinyl, dihydropyridinyl, dihydropyrrolyl or difluoropiperidinyl;
- X 2 is a divalent group of formula IA" wherein X 3 is N and R 11 is morpholinyl;
- R 9 and R 10 are phenyl which is unsubstituted or substituted by halogen
- R 9 is phenyl and R 10 is pyridine.
- R 1 and R 2 are independently methyl or ethyl.
- R 1 and R 2 together with the N-atom to which they are attached are piperidin-1-yl, morpholin-1-yl, 3,6-dihydro-2.H.pyridin-1 -yl, thiomorpholin-1-yl or pyrrolin-1 -yl.
- R 3 is H, methyl, cyclohexyl or benzyl; R 4 is H and m is 1 or 2 or 3, especially 1.
- R 5 is -X-R 6 , wherein R 6 is C ⁇ -C 4 alkyl (e.g. methyl, ethyl or isopropyl), 3-propenyl, methoxyethyl, carboxymethyl, 2-pyridyl, or benzyl, and X is as defined above.
- R 6 is C ⁇ -C 4 alkyl (e.g. methyl, ethyl or isopropyl), 3-propenyl, methoxyethyl, carboxymethyl, 2-pyridyl, or benzyl, and X is as defined above.
- R 5 is -N(R 7 )R 8 , wherein R 7 is aminopropyl, aminobutyl, dipropylaminoethyl, dimethylaminopropyl, dimethylaminobutyl, diethylaminopropyl or dimethylaminopentyl; and R 8 is H, methyl, aminopropyl, aminobutyl, dimethylaminopropyl or dimethylaminobutyl.
- the present invention also provides a process for the production of a compound of formula IA which process comprises reacting a compound of formula IIA (IIA) wherein X 1 , X 2 , R 9 and R 10 have the meanings given for formula IA, with an amine, e.g. of formula IIIA
- the present invention also provides a process for the production of a 2-(2,2-diphenylethyl- amino)- -5-(4-aminocarbonyl-piperidine-1-sulfonyl)-benzoic acid amide or -5-(aminocarbon- y ⁇ alkylene aminosulfonyl)-benzoic acid amide, for example a compound of formula I as defined above, or salt thereof, which process comprises reacting a 2-(2,2-diphenylethylamino)- -5-(4-carboxy-piperidine-1 -sulfonyl)-benzoic acid amide or -5-(carboxy-C 2 . 4 alkylene aminosulfonyl)-benzoic acid amide, for example a compound of formula II
- R 1 , R 2 , R 3 , R 4 , m and n have the meanings given for formula I, with an amine, e.g. of formula III
- the reaction may be earned out in accordance with standard procedures, for example by a first acid chloride formation step using e.g. thionyl chloride and catalytic DMF in an inert solvent, e.g. CH 2 CI 2 , at ambient temperature, followed by the coupling step involving addition of the acid chloride to a mixture of the amine and e.g. TEA, at a temperature of, e.g. -10°C.
- Aqueous workup followed by precipitation from, e.g. ethyl acetate gives the free base.
- the salt forms are made by standard procedures known to the skilled artisan.
- DMF dimethyl formamide
- DMSO dimethyl sulfoxide
- EDTA ethylenediamine-tetraacetic acid
- EtOAc ethylacetate
- IPA isopropanol
- RT room temperature
- TBME t-butyl methyl ether
- TBTU (O-(benzotriazol-1 - yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate
- TEA triethylamine
- TFA trifluoroacetic acid
- THF tetrahydrofuran.
- the solution of acid chloride is added to a mixture of isopropylpiperazine (47.60 g), TEA (98.09 g) and CH 2 CI 2 (1 I) at -10°C over 1 h.
- the reaction is worked up by removing volatiles via rotary evapor- tion.
- the residue is suspended in EtOAc (2.0 I) and washed with water (2 x 500 ml), brine (500 ml) and dried (Na 2 SO 4 ).
- the volume is reduced by rotary evaporation (35°C, house vacuum) to -500 ml and stirred at ambient temperature for 17 h.
- the suspension is filtered and dried (40°C, house vacuum) to give the title compound as free base.
- R 1A and R ⁇ independently are d-C alkyl or, together with the N-atom to which they are attached, represent a 5 to 7 membered heterocyclic ring, may be prepared applying known techniques, e.g. in accordance with the following reaction scheme:
- Hal is halogen, e.g. chlorine
- R 15 is d-C 4 alkyl, e.g. ethyl
- X 1 , X 2 , R A , R 2 *, R 9 and R 10 are as defined for formula IA.
- step (aA) the compound of formula XIIA may conveniently be reacted with e.g. chlorosulfonic acid or with chlorosulfonic acid followed by thionyl chloride.
- step (bA) the compound of formula XIA may be reacted with the compound of formula XA and e.g. triethyl- amine in the presence of a solvent like acetonitrile and acetone at 0°C.
- step (cA) the compound of formula IXA may be reacted with the compound of formula VINA at room temperature in the presence of a solvent like acetonitrile, acetone or ethyl acetate, ethyl acetate being preferred.
- a solvent like acetonitrile, acetone or ethyl acetate, ethyl acetate being preferred.
- an excess of the compound of formula VIIIA may be used, e.g. a 10 % excess.
- step (dA) the compound of formula VIIA may be reacted with an excess of the compound of formula VIA, e.g. a 10 % excess, in the presence of, e.g. trifluoroacetic acid and a desiccant, e.g. trimethyl orthoacetate.
- the reduction of step (eA) may, e.g.
- step (fA) may be accomplished via hydrogenation over 10 % palladium on carbon in the presence of a solvent, e.g. tetrahydrofuran.
- the hydrolysis of the ester of formula IVA [step (fA)] may be accomplished in the presence of a base like NaOH, in a solvent like ethanol, methanol, acetone or tetrahydrofuran, preferably tetrahydrofuran.
- the reac- tants may be warmed prior to reaction.
- R 11 is d-C 4 alkyl, C 3 -C 6 cycloalkyl, may be prepared applying known techniques, e.g. in accordance with the following reaction scheme:
- Hal is halogen, e.g. chlorine
- R 15 is d-C alkyl, e.g. ethyl
- y is 1 and M is a monovalent metal or y is 1/2 and M is a divalent metal
- X 1 , R 9 , R 10 and R 11 are as defined for formula IA.
- Examples of a metal include an alkali metal, e.g. lithium (Li), sodium (Na) and potassium (K), an alkaline earth metal, e.g. magnesium (Mg), or manganese (Mn), iron (Fe), zinc (Zn) or silver (Ag).
- an alkali metal e.g. lithium (Li), sodium (Na) and potassium (K)
- an alkaline earth metal e.g. magnesium (Mg), or manganese (Mn), iron (Fe), zinc (Zn) or silver (Ag).
- Hal is halogen, e.g. chlorine, R 15 ; i smartphoness r C. ⁇ - _C-. 4 a profession
- R ,-,10 are as defined for formula IA.
- R 11 is C ⁇ -C 4 alkyl, C 3 -C 6 cycloalkyl, may be prepared applying known techniques, e.g. in accordance with the following reaction scheme:
- Hal is halogen, e.g. chlorine
- R 15 is d-C 4 alkyl, e.g. ethyl
- R , R ,R , R , m and n are as defined for formula I.
- step (a) the compound of formula XII may conveniently be reacted with e.g. chlorosulfonic acid or with chlorosulfonic acid followed by thionyl chloride.
- step (b) the compound of formula XI is reacted with the compound of formula X and e.g. triethylamine in the presence of a solvent like acetonitrile and acetone at 0°C.
- step (c) the sulfonamide of formula IX is reacted with the compound of formula VIII at room temperature in the presence of a solvent like acetonitrile, acetone or ethyl acetate, ethyl acetate being preferred.
- a solvent like acetonitrile, acetone or ethyl acetate, ethyl acetate being preferred.
- an excess of the compound of formula VIII may be used, e.g. a 10 % excess.
- step (d) the compound of formula VII may be reacted with an excess of the compound of formula VI, e.g. a 10 % excess, in the presence of, e.g. trifluoroacetic acid and a desiccant, e.g. tri- methyl orthoacetate.
- step (e) may for example be accomplished via hydro- genation over 10 % palladium on carbon in the presence of a solvent, e.g. tetrahydrofuran.
- a solvent e.g. tetrahydrofuran.
- the hydrolysis of the ester of formula IV [step (f)] may be accomplished in the presence of a base like NaOH, in a solvent like ethanol, methanol, acetone or tetrahydrofuran, preferably tetrahydrofuran.
- the reactants may be warmed prior to reaction.
- Starting compounds of formula IIIA, VB, VIA, VIB, VIC, VID, VIIB, VINA, XA, XIB, XIC and XIIIB are known or may be prepared from corresponding known compounds.
- Starting compounds III, VI, VIII, X and XII are known or may be prepared from corresponding known compounds.
- compositions of the invention have pharmacological activity and are useful as pharmaceuticals.
- Pharmaceutical Compounds exhibit bradykinin antagonist activity.
- Pharmaceutical Compounds, e.g. compounds of Example 1 and 2 are active at the human Bi bradykinin receptor. Bradykinin receptor interaction of the Pharmaceutical Compounds is demonstrated by their ability to displace bradykinin at human bradykinin receptor sites, e.g. as demonstrated in accordance with the following test method.
- Test I Bradykinin receptor binding assay
- the human bradykinin B1 receptor is cloned from WI38 human foetal lung cell fibroblast cells by expression cloning in Xenopus laevis oocytes, which do not express bradykinin B1 receptors normally.
- a cDNA library is prepared in bacteriophage lambda ZAP express and grown in pools of approximately 10,000 clones per pool. Bacteriophage DNA is prepared from these pools and copy RNA is synthesised with T3 RNA polymerase and after phenol extraction and precipitation, the RNA is injected into Xenopus oocytes and allowed to be expressed for 3 days.
- the oocytes are then assayed electrophysiologically using two electrode voltage clamp, for a response in an endogenous chloride channel that can be activated by endogenous heterotrimeric GTP binding proteins of the Gq/G11 type that can couple to bradykinin receptors.
- a positive clone is isolated from a positive pool by several stages of splitting the pool into smaller pools and assaying, until a single clone is isolated.
- This cDNA is sequenced and subcloned into pcDNA3 (Clontech) and used to generate a cell-line which expresses the human bradykinin B1 receptor.
- the human bradykinin B1 receptor cDNA is subcloned into the Kpn1 and Not1 sites of pcDNA3 (HB1-pcDNA3), is grown up and transfected into human embryonic kidney fibroblast cell line, HEK 293 using the Calcium phosphate method.
- Cells are grown in Minimum Essential Medium with Earle's Salts (GIBCO) supplemented with 2 mM L-glutamine, 100 units/ml penicillin, 100 ⁇ g/ml streptomycin, 1 % nonessential amino acids and 10 % myoclone foetal calf serum (GIBCO) in a humidified atmosphere with 5 % C0 2 at 37°C.
- GEBCO Minimum Essential Medium with Earle's Salts
- the 293Hek cells are split 1 :2 the day prior to transfection.
- One 175 cm 2 flask with approximately 50 % confluent cells is transfected with approximately 30 ⁇ g/ml HB1-pcDNA3 DNA using the calcium phosphate precipitate method of transfection.
- the flask of transfected cells is split 1 :3 on day 2 post transfection to prevent overgrowth.
- the following day the cells are split 1 :5 and selection in 700 ⁇ g/ml G418 begins.
- the selective medium is changed every 3-4 days.
- the cells are cloned by limiting dilution and assayed for binding of [ 3 H]desArg 10 -kallidin. The clone with the highest binding is chosen for further use. Care is taken not to allow the cells to overgrow and to maintain G418 in the growth medium.
- HEK 293 cells expressing the human bradykinin B. receptor are used to prepare membranes.
- Cells are homogenised in 50 mM Tris-HCI, 1 mM EDTA pH 7.4 at 10,000 rpm for 30 sec in a Polytron homogeniser. All subsequent operations are carried out at 4°C.
- the resultant suspension is centrifuged for 30 min at 28,000 x g.
- the pellet is washed a further two times by resuspension in Tris-HCI (50 mM, pH 7.4) and recentrifugation.
- the final pellet is resuspended in Tris-HCI (50 mM, pH 7.4), containing 5 % glycerol and frozen rapidly on dry ice in 500 ⁇ l aliquots and stored at -80°C.
- membranes are thawed, homogenised, and diluted with physiological binding buffer (10 mM HEPES, HBSS ⁇ 137 mM NaCI, 5.4 mM KCI, 1.3 mM CaCI 2 , 0.4 mM KH 2 PO 4 , 0.3 mM NaHPO 4 , 0.5 mM MgCI 2 , 0.4 mM MgSO 4 , 5.6 mM glucose, pH 7.4 ⁇ containing 1 mM 1 -10 phenanthroline and 0J4 g/l bacitracin.
- physiological binding buffer 10 mM HEPES, HBSS ⁇ 137 mM NaCI, 5.4 mM KCI, 1.3 mM CaCI 2 , 0.4 mM KH 2 PO 4 , 0.3 mM NaHPO 4 , 0.5 mM MgCI 2 , 0.4 mM MgSO 4 , 5.6 mM glucose, pH 7.4 ⁇ containing 1 mM 1 -10 phenan
- Binding assays are performed in 1.2 ml polypropylene assay tubes (incorporated into a deep well block of 96 or individual Micronics tubes) containing a final volume of 0.5 ml.
- the assay composition is 425 ⁇ l membrane suspension (approximately 20 ⁇ g protein per tube) in physiological binding buffer, 50 ⁇ l [ 3 H]desArg 10 -kallidin (specific activity 95 Ci/mMol; 6.0 ⁇ 0.5 nM), 25 ⁇ l of either DMSO, or unlabelled desArg 10 kallidin (20 ⁇ M) or different concentrations of Pharmaceutical Compounds made up in DMSO. Specific binding to the bradykinin B.
- the receptor is defined as the difference between that found in total bound tubes and that found in non-specific binding tubes.
- the reaction is initiated with the addition of membranes and incubated at 4°C for 60 min.
- the reaction is terminated by rapid filtration of the assay mixture through Canberra Packard Unifilter-96 GF/B filterplates (which have been pre-soaked in 0.6% polyethylene- imine for 2 to 3 h at RT).
- the filters are washed 4 times with 1 ml aliquots of ice cold wash buffer.
- Microscintillant-40 liquid scintillant is added to the filters and radioactivity bound is determined in a Canberra Packard Topcount scintillation counter.
- Binding parameters are derived from non-linear iterative curve fitting of three or four data sets simultaneously, using a logistic model in MicrocalTM Origin.
- Activity specifically as analgesic agents may be demonstrated in accordance with standard test methods, e.g. as described in the following test.
- Test II Thermal antinociception in monkeys (warm water tail-withdrawal)
- Carrageenan at a dosage of 2 mg in 100 ⁇ l saline is injected subcutaneously into the terminal 1 to 4 cm of the tail of adult rhesus monkeys (Macaca mulatta) followed by administration of Pharmaceutical Compound in 100 ⁇ l vehicle (0.5 % methylcellulose in distilled water) or vehicle to the animal.
- the animals are seated in restraint chairs and the lower part of the shaved tail (approximately 15 cm) immersed into warm water maintained at temperatures of 42, 46, and 50°C.
- Tail-withdrawal latencies are recorded manually by a computerized timer.
- a maximum cutoff latency (20 sec) is recorded if the subjects fail to remove their tails by this time.
- a single dosing procedure is used in all test sessions. Each experimental session begins with control determinations at each temperature. Subsequent tail withdrawal latencies are determined based on each experimental condition.
- the subjects are tested 1 to 2 times at three temperatures in a varying order, with approximately 1 to 2 min interval between tests. Experimental sessions are conducted once per week.
- bradykinin Bi receptor antagonists are accordingly useful as bradykinin Bi receptor antagonists, e.g. in the treatment of diseases and conditions in which B. receptor activation plays a role or is implicated.
- diseases and conditions include in particular pain, e.g. bone and joint pain (osteoarthritis), cancer pain, myofascial pain (muscular injury, fibromyalgia) and perioperative pain (general surgery, gynecologic surgery).
- Pharmaceutical Compounds are particularly useful in the treatment or prevention of chronic pain, especially inflammatory, e.g. chronic inflammatory pain, inflammatory diseases for example inflammatory airways disease, e.g. COPD, or in asthma, rhinitis, inflammatory bowel disease, cystitis, e.g. interstitial cystistis, pancreatitis, uveitis, inflammatory skin disorders and rheumatoid arthritis.
- inflammatory for example inflammatory airways disease, e.g. COPD
- cystitis e.g. interstitial cystistis
- pancreatitis e.g. interstitial cystistis
- pancreatitis e.g. interstitial cystistis
- uveitis inflammatory skin disorders and rheumatoid arthritis.
- Pharmaceutical Compounds are thus useful as bradykinin B ⁇ receptor antagonists, e.g.
- bradykinin for the treatment of pain of various genesis or aetiology and as anti-inflammatory and/or anti-oedemic agents for the treatment of inflammatory reactions, diseases or conditions, as well as for the treatment of allergic responses mediated by bradykinin.
- analgesic/anti- inflammatory profile they are useful for the treatment of inflammatory pain, for the treatment of hyperalgesia and, in particular, for the treatment of severe chronic pain. They are, for example, useful for the treatment of pain, inflammation and or oedema consequential to trauma, e.g. associated with bums, sprains, fracture or the like, subsequent to surgical intervention, e.g.
- analgesics as post-operative analgesics, as well as for the treatment of inflammatory pain of diverse genesis, e.g. for the treatment of osteo and rheumatoid arthritis and rheumatic disease, teno-synovitis and gout. They are further suitable as analgesics for the treatment of pain associated with, e.g., angina, menstruation or cancer.
- anti-inflammatory/anti-oedema agents they are further useful, e.g., for the treatment of inflammatory skin disorders, for example psoriasis and eczema.
- bradykinin BK1 receptor antagonists Pharmaceutical Compounds are also useful as smooth muscle relaxants, e.g. for the treatment of spasm of the gastro-intestinal tract or uterus, e.g. in the therapy of Crohn's disease, ulcerative collitis or pancreatitis.
- Pharmaceutical Compounds are in particular useful as agents for the therapy of airways hyperreactivity and for the treatment of inflammatory events associated with airways disease, in particular asthma.
- Pharmaceutical Compounds may, for example, be used for the control, restriction or reversal of airways hyperreactivity in asthma.
- Inflammatory or obstructive airways diseases to which the present invention is applicable include asthma of whatever type or genesis including both intrinsic and, especially, extrinsic asthma.
- Pharmaceutical Compounds are useful for the treatment of allergic asthma, whether atopic (i.e. IgE-mediated) or non-atopic, as well as, for example, exercise induced asthma, occupational asthma, asthma induced following bacterial infection, other non-allergic asthmas and "whez-infant syndrome".
- Efficacy in the treatment of asthma will be evidenced by reduced frequency or severity of symptomatic attack, e.g. of acute asthmatic or bronchoconstrictor attack and by reduced requirement for other, symptomatic therapy, for example anti-inflammatory (e.g. corticosteroid) or bronchodilator (e.g. ⁇ 2 adrenergic) therapy.
- symptomatic attack e.g. of acute asthmatic or bronchoconstrictor attack
- other, symptomatic therapy for example anti-inflammatory (e.g. corticosteroid) or bronchodilator (e.g. ⁇ 2 adrenergic) therapy.
- anti-inflammatory e.g. corticosteroid
- bronchodilator e.g. ⁇ 2 adrenergic
- Inflammatory or obstructive airways diseases to which the present invention is applicable further include pneumoconiosis (an inflammatory, commonly occupational, disease of the lungs, frequently accompanied by repeated inhalation of dusts) of whatever type or genesis, including, for example, aluminosis, anthracosis, asbestosis, chalicosis, ptilosis, siderosis, silicosis, tabacosis and, in particular, byssinosis.
- pneumoconiosis an inflammatory, commonly occupational, disease of the lungs, frequently accompanied by repeated inhalation of dusts
- pneumoconiosis an inflammatory, commonly occupational, disease of the lungs, frequently accompanied by repeated inhalation of dusts
- aluminosis anthracosis
- asbestosis chalicosis
- ptilosis ptilosis
- siderosis silicosis
- tabacosis tabacosis
- ARDS adult respiratory distress syndrome
- COAD chronic obstructive pulmonary or airways disease
- bronchitis bronchitis
- Pharmaceutical Compounds may also be used for the treatment of allergic and vasomotor rhinitis.
- Compounds are also indicated for use in the therapy of septic shock, e.g. as anti-hypovolaemic and/or anti-hypotensive agents, in the treatment of inflammatory bowel disease cerebral oedema, headache, migraine and inflammatory skin disease such as eczema and psoriasis, and inflammatory disorders of the gut, e.g. irritable bowel syndrome, Crohn's disease, ulcerative colitis, cystitis, e.g. interstitial cystitis, nephritis, uveitis.
- septic shock e.g. as anti-hypovolaemic and/or anti-hypotensive agents
- inflammatory disorders of the gut e.g. irritable bowel syndrome, Crohn's disease, ulcerative colitis
- cystitis e.g. interstitial cystitis, nephritis, uveitis.
- the appropriate dosage of Pharmaceutical Compounds will, of course, vary depending upon, for example, the host, the mode of administration and the nature and severity of the condition being treated as well as the relative potency of the particular Pharmaceutical Compound employed.
- the amount of active agent required may be determined on the basis of known in vitro and in vivo techniques, determining how long a particular active agent concentration in the blood plasma remains at an acceptable level for a therapeutic effect.
- satisfactory results in animals are indicated to be obtained at daily dosages of from about 0.01 to about 20.0 mg/kg p.o.
- an indicated daily dosage is in the range of from about 0.7 to about 1400 mg/day p.o., e.g.
- Oral dosage forms accordingly suitably comprise from about 1.75 or 2.0 to about 700 or 1400 mg
- Pharmaceutical Compounds may alternatively be administered e.g. topically in the form of a cream, gel or the like for example for the treatment of conditions of the skin as hereinbefore described or by inhalation, e.g. in dry powder form, for example for the treatment of asthma.
- compositions comprising Pharmaceutical Compound include, e.g. a solid dispersion, an aqueous solution, e.g. containing a solubilising agent, e.g. cyclodextrin, a microemulsion and a suspension of, e.g. a micronized hydrochloride salt of a compound of formula IA in, e.g. aqueous methyl cellulose in the range of from OJ to 1 %, e.g. 0.5 %.
- the composition may be buffered to, e.g. a pH in the range of from 3.5 to 9.5, e.g. to pH 4.5, by a suitable buffer, e.g. malic acid.
- the present invention also provides:
- a pharmaceutical compound for use as a bradykinin BK, receptor antagonist for example for use in any of the particular indications hereinbefore set forth;
- composition comprising a pharmaceutical compound as under (1) as active ingredient together with a pharmaceutically acceptable diluent or carrier therefor;
- a pharmaceutical composition for the treatment or prevention of a disease or condition in which bradykinin Bi receptor activation plays a role or is implicated comprising a compound of formula IA and a carrier.
- a method for treating or preventing a disease or condition in which bradykinin Bi receptor activation plays a role or is implicated comprising administering to a mammal in need thereof a therapeutically effective amount of a compound of formula IA.
- a compound of formula IA for the manufacture of a medicament for the treatment or prevention of a disease or condition in which bradykinin Bi receptor activation plays a role or is implicated;
- the preferred Pharmaceutical Compounds for use in accordance with the invention are those of Examples 1 and 2.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Neurology (AREA)
- Pain & Pain Management (AREA)
- Biomedical Technology (AREA)
- Physical Education & Sports Medicine (AREA)
- Neurosurgery (AREA)
- Pulmonology (AREA)
- Dermatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Heart & Thoracic Surgery (AREA)
- Rheumatology (AREA)
- Cardiology (AREA)
- Immunology (AREA)
- Urology & Nephrology (AREA)
- Reproductive Health (AREA)
- Endocrinology (AREA)
- Pregnancy & Childbirth (AREA)
- Hematology (AREA)
- Communicable Diseases (AREA)
- Vascular Medicine (AREA)
- Ophthalmology & Optometry (AREA)
- Otolaryngology (AREA)
- Gynecology & Obstetrics (AREA)
- Surgery (AREA)
- Diabetes (AREA)
- Oncology (AREA)
Abstract
Description























Claims







Priority Applications (15)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE60022999T DE60022999T2 (en) | 1999-06-04 | 2000-06-02 | BRADYKININE RECEPTORANT AGONIST |
| PL00352056A PL352056A1 (en) | 1999-06-04 | 2000-06-02 | Antagonists of bradykinin receptor |
| NZ515304A NZ515304A (en) | 1999-06-04 | 2000-06-02 | Bradykinin receptor antagonists |
| US10/009,009 US6958331B1 (en) | 1999-06-04 | 2000-06-02 | Bradykinin receptor antagonists |
| CA2372575A CA2372575C (en) | 1999-06-04 | 2000-06-02 | Bradykinin receptor antagonists |
| MXPA01012473A MXPA01012473A (en) | 1999-06-04 | 2000-06-02 | Bradykinin receptor antagonists. |
| AU55282/00A AU765628B2 (en) | 1999-06-04 | 2000-06-02 | Bradykinin receptor antagonists |
| BR0011329-8A BR0011329A (en) | 1999-06-04 | 2000-06-02 | Bradykinin receptor antagonists |
| IL14611200A IL146112A0 (en) | 1999-06-04 | 2000-06-02 | Bradykinin receptor antagonists |
| SK1749-2001A SK17492001A3 (en) | 1999-06-04 | 2000-06-02 | Sulfonyl amino derivatives, process for the preparation thereof and pharmaceutical compositions comprising same |
| JP2001501588A JP3820148B2 (en) | 1999-06-04 | 2000-06-02 | Bradykinin receptor antagonist |
| AT00940304T ATE305920T1 (en) | 1999-06-04 | 2000-06-02 | BRADYKININ RECEPTOR ANTAGONIST |
| EP00940304A EP1183233B1 (en) | 1999-06-04 | 2000-06-02 | Bradykinin receptor antagonists |
| NO20015779A NO20015779D0 (en) | 1999-06-04 | 2001-11-27 | Bradykinin receptor antagonists |
| HK02105567.0A HK1045834A1 (en) | 1999-06-04 | 2002-07-29 | Bradykinin receptor antagonists |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB9913079.1 | 1999-06-04 | ||
| GBGB9913079.1A GB9913079D0 (en) | 1999-06-04 | 1999-06-04 | Organic compounds |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2000075107A2 true WO2000075107A2 (en) | 2000-12-14 |
| WO2000075107A3 WO2000075107A3 (en) | 2001-09-07 |
Family
ID=10854785
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2000/005059 WO2000075107A2 (en) | 1999-06-04 | 2000-06-02 | Bradykinin receptor antagonists |
Country Status (28)
| Country | Link |
|---|---|
| US (1) | US6958331B1 (en) |
| EP (1) | EP1183233B1 (en) |
| JP (1) | JP3820148B2 (en) |
| KR (1) | KR100479896B1 (en) |
| CN (1) | CN1165522C (en) |
| AR (1) | AR033945A1 (en) |
| AT (1) | ATE305920T1 (en) |
| AU (1) | AU765628B2 (en) |
| BR (1) | BR0011329A (en) |
| CA (1) | CA2372575C (en) |
| CZ (1) | CZ20014302A3 (en) |
| DE (1) | DE60022999T2 (en) |
| ES (1) | ES2250144T3 (en) |
| GB (1) | GB9913079D0 (en) |
| HK (1) | HK1045834A1 (en) |
| HU (1) | HUP0201524A3 (en) |
| IL (1) | IL146112A0 (en) |
| MX (1) | MXPA01012473A (en) |
| NO (1) | NO20015779D0 (en) |
| NZ (1) | NZ515304A (en) |
| PE (1) | PE20010215A1 (en) |
| PL (1) | PL352056A1 (en) |
| RU (1) | RU2001135802A (en) |
| SK (1) | SK17492001A3 (en) |
| TR (1) | TR200103108T2 (en) |
| TW (1) | TWI227224B (en) |
| WO (1) | WO2000075107A2 (en) |
| ZA (1) | ZA200109891B (en) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002092556A1 (en) * | 2001-05-14 | 2002-11-21 | Novartis Ag | Sulfonamide derivatives |
| FR2840897A1 (en) * | 2002-06-14 | 2003-12-19 | Fournier Lab Sa | New N-((N,N-disubstituted carbamoyl)-alkyl)-arylsulfonamides, are bradykinin B1 receptor antagonists useful e.g. for treating inflammatory diseases or especially pain |
| WO2004092164A1 (en) * | 2003-04-10 | 2004-10-28 | Amgen, Inc. | Cyclic amine derivatives and their use in the treatment of inflammation-related disorders mediated by bradykinin |
| US7211566B2 (en) | 2003-04-01 | 2007-05-01 | Universite De Sherbrooke | Selective bradykinin (BK) B1 peptidic receptor antagonists and uses thereof |
| WO2008050168A1 (en) | 2006-10-27 | 2008-05-02 | Richter Gedeon Nyrt. | New sulfonamide derivatives as bradykinin antagonists |
| EP2305668A1 (en) | 2007-03-23 | 2011-04-06 | Jerini AG | 8-oxy-quinoline derivatives as bradykinin B2 receptor modulators |
| EP2305652A2 (en) | 2005-12-08 | 2011-04-06 | Novartis AG | Trisubstituted quinazolinone derivatives as vanilloid antagonists |
| WO2011051375A1 (en) | 2009-10-28 | 2011-05-05 | Dompé S.p.A. | 2-aryl-propionamide derivatives useful as bradykinin receptor antagonists and pharmaceutical compositions containing them |
| US8034827B2 (en) | 2005-12-20 | 2011-10-11 | Richter Gegeon Nyrt. | Phenanthridine derivatives as bradykinin antagonists |
| WO2012164473A1 (en) | 2011-05-27 | 2012-12-06 | Novartis Ag | 3-spirocyclic piperidine derivatives as ghrelin receptor agonists |
| US8394858B2 (en) | 2009-12-03 | 2013-03-12 | Novartis Ag | Cyclohexane derivatives and uses thereof |
| US8481527B2 (en) | 2006-10-27 | 2013-07-09 | Richter Gedeon Nyrt. | Benzamide derivatives as bradykinin antagonists |
| WO2013164790A1 (en) | 2012-05-03 | 2013-11-07 | Novartis Ag | L-malate salt of 2, 7 - diaza - spiro [4.5 ] dec- 7 - yle derivatives and crystalline forms thereof as ghrelin receptor agonists |
| WO2019101906A1 (en) | 2017-11-24 | 2019-05-31 | Pharvaris B.V. | Novel bradykinin b2 receptor antagonists |
| WO2020234480A1 (en) | 2019-05-23 | 2020-11-26 | Pharvaris Gmbh | (r)-3-(chloro-5-fluoro-2-((4-(1h-pyrazol-1-yl)-2-methylquinolin-8-yloxy)methyl)phenyl)morpholine derivatives and related compounds as bradykinin (bk) b2 receptor antagonist for treating skin diseases |
| WO2020234479A1 (en) | 2019-05-23 | 2020-11-26 | Pharvaris Gmbh | 1-((s)-1-(3-chloro-5-fluoro-2-((4-(1h-pyrazol-1-yl)-2-methylquinolin-8-yloxy)methyl)phenyl)ethyl)-imidazolidine-2,4-dione derivatives and related compounds as bradykinin (bk) b2 receptor antagonist for treating skin diseases |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE69321845T2 (en) | 1992-08-25 | 1999-04-29 | Monsanto Co | HYDROXYETHYLAMINOSULPHONAMIDES USED AS INHIBITORS OF RETROVIRAL PROTEASES |
| US7141609B2 (en) | 1992-08-25 | 2006-11-28 | G.D. Searle & Co. | α- and β-amino acid hydroxyethylamino sulfonamides useful as retroviral protease inhibitors |
| WO2008153967A1 (en) * | 2007-06-08 | 2008-12-18 | Contec Therapeutics, Inc. | Bk1 antagonist conjugates |
| KR20230174415A (en) | 2022-06-21 | 2023-12-28 | 정인호 | Char-coal Meat Broiler Providing Evenly Doneness Status |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997025315A1 (en) * | 1996-01-11 | 1997-07-17 | Sanofi | Novel n-(arylsulphonyl)amino acid derivatives having bradykinin receptor affinity |
-
1999
- 1999-06-04 GB GBGB9913079.1A patent/GB9913079D0/en not_active Ceased
-
2000
- 2000-05-29 TW TW089110384A patent/TWI227224B/en not_active IP Right Cessation
- 2000-06-01 AR ARP000102710A patent/AR033945A1/en not_active Application Discontinuation
- 2000-06-02 IL IL14611200A patent/IL146112A0/en unknown
- 2000-06-02 EP EP00940304A patent/EP1183233B1/en not_active Expired - Lifetime
- 2000-06-02 KR KR10-2001-7015416A patent/KR100479896B1/en not_active IP Right Cessation
- 2000-06-02 NZ NZ515304A patent/NZ515304A/en unknown
- 2000-06-02 PL PL00352056A patent/PL352056A1/en not_active Application Discontinuation
- 2000-06-02 CN CNB008082405A patent/CN1165522C/en not_active Expired - Fee Related
- 2000-06-02 RU RU2001135802/04A patent/RU2001135802A/en not_active Application Discontinuation
- 2000-06-02 US US10/009,009 patent/US6958331B1/en not_active Expired - Fee Related
- 2000-06-02 AU AU55282/00A patent/AU765628B2/en not_active Ceased
- 2000-06-02 AT AT00940304T patent/ATE305920T1/en not_active IP Right Cessation
- 2000-06-02 BR BR0011329-8A patent/BR0011329A/en not_active IP Right Cessation
- 2000-06-02 WO PCT/EP2000/005059 patent/WO2000075107A2/en active IP Right Grant
- 2000-06-02 JP JP2001501588A patent/JP3820148B2/en not_active Expired - Fee Related
- 2000-06-02 SK SK1749-2001A patent/SK17492001A3/en unknown
- 2000-06-02 DE DE60022999T patent/DE60022999T2/en not_active Expired - Fee Related
- 2000-06-02 CA CA2372575A patent/CA2372575C/en not_active Expired - Fee Related
- 2000-06-02 HU HU0201524A patent/HUP0201524A3/en unknown
- 2000-06-02 ES ES00940304T patent/ES2250144T3/en not_active Expired - Lifetime
- 2000-06-02 MX MXPA01012473A patent/MXPA01012473A/en unknown
- 2000-06-02 TR TR2001/03108T patent/TR200103108T2/en unknown
- 2000-06-02 CZ CZ20014302A patent/CZ20014302A3/en unknown
- 2000-06-02 PE PE2000000549A patent/PE20010215A1/en not_active Application Discontinuation
-
2001
- 2001-11-27 NO NO20015779A patent/NO20015779D0/en not_active Application Discontinuation
- 2001-11-30 ZA ZA200109891A patent/ZA200109891B/en unknown
-
2002
- 2002-07-29 HK HK02105567.0A patent/HK1045834A1/en unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997025315A1 (en) * | 1996-01-11 | 1997-07-17 | Sanofi | Novel n-(arylsulphonyl)amino acid derivatives having bradykinin receptor affinity |
Cited By (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7109203B2 (en) | 2001-05-14 | 2006-09-19 | Novartis Ag | Sulfonamide derivatives |
| WO2002092556A1 (en) * | 2001-05-14 | 2002-11-21 | Novartis Ag | Sulfonamide derivatives |
| FR2840897A1 (en) * | 2002-06-14 | 2003-12-19 | Fournier Lab Sa | New N-((N,N-disubstituted carbamoyl)-alkyl)-arylsulfonamides, are bradykinin B1 receptor antagonists useful e.g. for treating inflammatory diseases or especially pain |
| WO2003106428A1 (en) * | 2002-06-14 | 2003-12-24 | Laboratoires Fournier Sa | Novel arylsulphonamide derivatives and use thereof as therapeutic agents |
| US7361687B2 (en) | 2002-06-14 | 2008-04-22 | Laboratoires Fournier Sa | Arylsulphonamide derivatives and methods of preparing |
| US7211566B2 (en) | 2003-04-01 | 2007-05-01 | Universite De Sherbrooke | Selective bradykinin (BK) B1 peptidic receptor antagonists and uses thereof |
| WO2004092164A1 (en) * | 2003-04-10 | 2004-10-28 | Amgen, Inc. | Cyclic amine derivatives and their use in the treatment of inflammation-related disorders mediated by bradykinin |
| US7199244B2 (en) | 2003-04-10 | 2007-04-03 | Amgen | Cyclic amine derivatives and methods of use |
| EP2305652A2 (en) | 2005-12-08 | 2011-04-06 | Novartis AG | Trisubstituted quinazolinone derivatives as vanilloid antagonists |
| US8034827B2 (en) | 2005-12-20 | 2011-10-11 | Richter Gegeon Nyrt. | Phenanthridine derivatives as bradykinin antagonists |
| WO2008050168A1 (en) | 2006-10-27 | 2008-05-02 | Richter Gedeon Nyrt. | New sulfonamide derivatives as bradykinin antagonists |
| US8481527B2 (en) | 2006-10-27 | 2013-07-09 | Richter Gedeon Nyrt. | Benzamide derivatives as bradykinin antagonists |
| EP2305668A1 (en) | 2007-03-23 | 2011-04-06 | Jerini AG | 8-oxy-quinoline derivatives as bradykinin B2 receptor modulators |
| WO2011051375A1 (en) | 2009-10-28 | 2011-05-05 | Dompé S.p.A. | 2-aryl-propionamide derivatives useful as bradykinin receptor antagonists and pharmaceutical compositions containing them |
| US8394858B2 (en) | 2009-12-03 | 2013-03-12 | Novartis Ag | Cyclohexane derivatives and uses thereof |
| WO2012164473A1 (en) | 2011-05-27 | 2012-12-06 | Novartis Ag | 3-spirocyclic piperidine derivatives as ghrelin receptor agonists |
| WO2013164790A1 (en) | 2012-05-03 | 2013-11-07 | Novartis Ag | L-malate salt of 2, 7 - diaza - spiro [4.5 ] dec- 7 - yle derivatives and crystalline forms thereof as ghrelin receptor agonists |
| WO2019101906A1 (en) | 2017-11-24 | 2019-05-31 | Pharvaris B.V. | Novel bradykinin b2 receptor antagonists |
| US10836748B2 (en) | 2017-11-24 | 2020-11-17 | Pharvaris Netherlands B.V. | Bradykinin B2 receptor antagonists |
| US11261173B2 (en) | 2017-11-24 | 2022-03-01 | Pharvaris Netherlands B.V. | Bradykinin B2 receptor antagonists |
| EP3998259A1 (en) | 2017-11-24 | 2022-05-18 | Pharvaris Netherlands B.V. | Novel bradykinin b2 receptor antagonists |
| US11820756B2 (en) | 2017-11-24 | 2023-11-21 | Pharvaris Netherlands B.V. | Bradykinin B2 receptor antagonists |
| WO2020234480A1 (en) | 2019-05-23 | 2020-11-26 | Pharvaris Gmbh | (r)-3-(chloro-5-fluoro-2-((4-(1h-pyrazol-1-yl)-2-methylquinolin-8-yloxy)methyl)phenyl)morpholine derivatives and related compounds as bradykinin (bk) b2 receptor antagonist for treating skin diseases |
| WO2020234479A1 (en) | 2019-05-23 | 2020-11-26 | Pharvaris Gmbh | 1-((s)-1-(3-chloro-5-fluoro-2-((4-(1h-pyrazol-1-yl)-2-methylquinolin-8-yloxy)methyl)phenyl)ethyl)-imidazolidine-2,4-dione derivatives and related compounds as bradykinin (bk) b2 receptor antagonist for treating skin diseases |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1183233B1 (en) | Bradykinin receptor antagonists | |
| US6294554B1 (en) | Muscarinic antagonists | |
| JP3390179B2 (en) | Ether muscarinic antagonist | |
| DE3854454T2 (en) | Pyrazolopyridine compounds and process for their preparation. | |
| JP4035163B2 (en) | Novel arylglycinamide derivative, process for producing the same, and pharmaceutical composition containing the compound | |
| PT1499589E (en) | De | |
| BRPI0508622B1 (en) | Biphenyl compounds useful as muscarinic receptor antagonists, pharmaceutical composition, process, medicament and use of the compound. | |
| CZ20012562A3 (en) | Amide compounds | |
| US20140309192A1 (en) | Modulators of the g protein-coupled mas receptor and the treatment of disorders related thereto | |
| BRPI0708788A2 (en) | compound, pharmaceutical composition, and methods for inhibiting a soluble epoxide hydrolase, the progression of nephropathy, and the obstruction of obstructive pulmonary disease, an interstitial lung disease or asthma in a patient, to treat soluble epoxide hydrolase-modulated diseases, to reduce renal deterioration, blood pressure, vascular inflammation, renal inflammation in a patient, and the formation of a biologically active diol, to regulate endothelial cell function in a patient, to decrease endothelial cell inflammation in a patient, and to monitor the activity of a soluble epoxide hydrolase | |
| PT99483A (en) | PROCESS FOR THE PREPARATION OF PIPERIDINILCANFRO-SULFONYL OXYTOCIN ANTAGONISTS | |
| TW201041868A (en) | Anthelmintic agents and their use | |
| ES2364727T3 (en) | CHLORHYDRATE SALT OF 5- [3- (3-HYDROXYPHENOXY) AZETIDIN-1-IL] -5-METHYL-2,2-DIFENYLHEXANAMIDE. | |
| JPWO2005009992A1 (en) | Cyclohexanecarboxylic acids | |
| DE69715891T2 (en) | PYRAZOLOPYRIDINE COMPOUND AND ITS PHARMACEUTICAL USE | |
| EP1214299B1 (en) | Muscarinic antagonists | |
| DE102007010815B3 (en) | New N-phenylsulfonyl-3-amidino-phenylalanine piperidide derivatives useful as antitumor agents | |
| PT99672A (en) | PROCESS FOR THE PREPARATION OF SUBSTITUTED N-BENZOYL-N '- (2-PHENYLETHYL) -PYERAZINES | |
| JPH0597845A (en) | Condensed diazepinone and medicine contain- ing same | |
| PT1330436E (en) | N-(4-aryloxypiperidin-1-ylalkyl) cinnamic amides as ccr3-3 receptor antagonists | |
| PT1401814E (en) | Derivatives of 3-phenyl-n-(2- (4-benzyl) piperidin-1-yl)-ethyl)-acrylamid with ccr-3 receptor antagonistic activity for use in the treatment of inflammations and allergic conditions | |
| WO2001010844A1 (en) | φ-AMINO-α-HYDROXYCARBOXYLIC ACID DERIVATIVES HAVING INTEGRIN αvβ3 ANTAGONISM | |
| AU1890400A (en) | Amide compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 1200101027 Country of ref document: VN Ref document number: 00808240.5 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2000940304 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2372575 Country of ref document: CA Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001/03108 Country of ref document: TR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 515304 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 55282/00 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: IN/PCT/2001/1674/CHE Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV2001-4302 Country of ref document: CZ Ref document number: 2001/09891 Country of ref document: ZA Ref document number: 17492001 Country of ref document: SK Ref document number: 1020017015416 Country of ref document: KR Ref document number: 200109891 Country of ref document: ZA |
|
| ENP | Entry into the national phase |
Ref document number: 2001 501588 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2001/012473 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10009009 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2001 2001135802 Country of ref document: RU Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020017015416 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2000940304 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2001-4302 Country of ref document: CZ |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWG | Wipo information: grant in national office |
Ref document number: 55282/00 Country of ref document: AU |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1020017015416 Country of ref document: KR |
|
| WWR | Wipo information: refused in national office |
Ref document number: PV2001-4302 Country of ref document: CZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2000940304 Country of ref document: EP |