WO2000056338A1 - Quinazoline formulations and therapeutic use thereof - Google Patents
Quinazoline formulations and therapeutic use thereof Download PDFInfo
- Publication number
- WO2000056338A1 WO2000056338A1 PCT/US2000/007066 US0007066W WO0056338A1 WO 2000056338 A1 WO2000056338 A1 WO 2000056338A1 US 0007066 W US0007066 W US 0007066W WO 0056338 A1 WO0056338 A1 WO 0056338A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- amino
- dimethoxyquinazoline
- pharmaceutical composition
- och
- whi
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/107—Emulsions ; Emulsion preconcentrates; Micelles
- A61K9/1075—Microemulsions or submicron emulsions; Preconcentrates or solids thereof; Micelles, e.g. made of phospholipids or block copolymers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/10—Alcohols; Phenols; Salts thereof, e.g. glycerol; Polyethylene glycols [PEG]; Poloxamers; PEG/POE alkyl ethers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/127—Liposomes
Definitions
- This application relates to new formulations for poorly water soluble quinazoline compounds and therapeutic methods for the treatment of cancers and treatment of allergic disorders by administering quinazoline formulations.
- Quinazoline compounds have been suggested as useful compounds in the treatment of cell growth and differentiation characterized by activity of the human epidermal growth factor receptor type2 (HER2). See, for example, Myers etal., U.S. Patent No. 5,721,237. Some quinazoline derivatives have been suggested as useful as anti-cancer agents for the treatment of specific receptor tyrosine kinase-expressing cancers, especially those expressing epithelial growth factor (EGF) receptor tyrosine kinase. See, for example, Barker et. al., U.S. Patent No. 5,457,105. It is generally taught that quinazolines exert their anti-tumor effects via tyrosine kinase inhibition.
- EGF epithelial growth factor
- a series of water soluble quinazoline formulations were prepared and analyzed for therapeutic activities, including anti-cancer activities, particularly against JAK3 receptor.
- the invention provides novel water soluble quinazoline formulations, as disclosed below, as well as therapeutic methods utilizing these formulations.
- One aspect of the invention is a pharmaceutical composition
- a pharmaceutical composition comprising a dialkoxyquinazoline compound, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable non-topic lipid based carrier, diluent or vehicle.
- Another aspect of the invention is a method of administering a dialkoxyquinazoline compound to a mammal, the method includes combining the dialkoxyquinazoline compound with a pharmaceutically acceptable lipid-based vehicle to form a pharmaceutical composition and administering the pharmaceutical composition to the mammal.
- Another aspect of the invention is a method of administering a dimethoxyquinazoline compound to a mammal.
- the method includes providing a pharmaceutical composition including dimethoxyquinazoline compound in the salt form, PEG phospholipids, a cosolvent system, and administering the pharmaceutical composition to the mammal.
- Figure 1 is a graph showing the solubility of WHI-P131 chloride as a function of PEG 300 and PEG 200 concentration.
- Figure 2 is a graph showing solubility of WHI-P131 chloride as a function of PEG2000-DPPE concentration.
- Figure 3 is a ternary phase diagram showing the location of a single phase microemulsion region.
- Figure 4 is a flow diagram of the cumulative solubilization enhancement of W ⁇ I-P131 with the formulations of the invention.
- Figure 5 is a graph showing the plasma concentration-time curves following i.v. bolus injection of WHI-P131 formulations of the invention in mice.
- Figure 6 is a graph showing mast cell inhibitory "anti-allergic" activity of the formulations of the invention in vitro.
- Halo is fluoro, chloro, bromo, or iodo.
- Alkyl, alkanoyl, etc. denote both straight and branched groups; but reference to an individual radical such as "propyl” embraces only the straight chain radical, a branched chain isomer such as “isopropyl” being specifically referred to.
- (C ⁇ -C 4 )alkyl includes methyl, ethyl, propyl, isopropyl, butyl, iso-butyl, and sec-butyl;
- (Q-C )alkoxy includes methoxy, ethoxy, propoxy, isopropoxy, butoxy, iso-butoxy, and sec-butoxy;
- (Q- C 4 )alkanoyl includes acetyl, propanoyl and butanoyl.
- pharmaceutically acceptable carrier means any material which, when combined with the compound of the invention, allows the compound to retain biological activity, such as the ability to potentiate antibacterial activity of mast cells and macrophages.
- conjugate means a compound formed as a composite between two or more molecules. More specifically, in the present invention, the quinazoline derivative is bonded, for example, covalently bonded, to cell-specific targeting moieties forming a conjugate compound for efficient and specific delivery of the agent to a cell of interest.
- targeting moiety means a molecule which serves to deliver the compound of the invention to a specific site for the desired activity. Targeting moieties include, for example, molecules that specifically bind molecules on a specific cell surface.
- targeting moieties useful in the invention include anti-cell surface antigen antibodies.
- Cytokines including interleukins and factors such as granulocyte/macrophage stimulating factor (GMCSF) are also specific targeting moieties, known to bind to specific cells expressing high levels of their receptors.
- GMCSF granulocyte/macrophage stimulating factor
- prodrug moiety is a substitution group which facilitates use of a compound of the invention, for example by facilitating entry of the drug into cells or administration of the compound.
- the prodrug moiety may be cleaved from the compound, for example by cleavage enzymes in vivo.
- prodrug moieties include phosphate groups, peptide linkers, and sugars, which moieties can be hydrolyzed in vivo.
- inhibitor means to reduce by a measurable amount, or to prevent entirely.
- to treat means to inhibit or block at least one symptom that characterizes a pathologic condition, in a mammal threatened by, or afflicted with, the condition.
- the invention is directed towards formulations for delivery of an effective amount of quinazoline to a treatment site.
- the formulations relate to pharmaceutical compositions that include a quinazoline compound or pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable, lipid— based vehicle or delivery system.
- the vehicle or delivery system of the quinazoline composition is a nontoxic delivery system or vehicle for parenteral administration.
- the formulations disclosed enhance the water solubility of quinazoline compounds without loss of biologic activity of the quinazoline compound at the treatment site.
- Quinazoline compounds include quinazolines having the formula:
- R a is hydrogen; halo; hydroxy; mercapto; ( -C )hydroxyalkyl, methylenedioxy, ethylenedioxy, benzyloxy,OCF 3> SCF 3 ⁇ SO 3 H, SO 2 F, SO 2 NR 2 R 3 in which R 2 is hydrogen or (C[-C )alkyl and R 3 is hydrogen, (C]-C 4 )alkyl, or phenyl, NR 2 R 4 in which R 2 is as defined above and R 4 is phenyl, or R a a group of the formula
- R 5 and R are each, independently, hydrogen, ( -C 4 )alkyl, or (C ⁇ -C 4 )perfluoroalkyl
- R 7 is hydrogen, halo, hydroxy, ( -C 4 )alkyl, (C ⁇ - C )alkoxy, (C ⁇ -C )hydroxyalkyl, or N(R ) 2 in which R is as defined above; n is an integer of 1-4;
- R b is each, independently, hydrogen; halo; hydroxy; mercapto; ( - C 4 )alkyl, (C[-C 4 )alkoxy, (C ⁇ -C 4 )thioalkyl, (C ⁇ -C )hydroxyalkyl, nitro, cyano, methylenedioxy, ethylenedioxy, COCH 3 , CF 3 ;, OCF 3 ; SCF 3 ; COOH; SO 3 H; SO 2 F; phenyl or phenyl substituted by a group selected from halo, hydroxy, mercapto, (Q- C 4 )alkyl, (C ⁇ -C 4 )alkoxy, (C ⁇ -C 4 )thioalkyl, (Cj-C 4 )hydroxyalkyl, amino, nitro, cyano, CF 3 , COOH, SO 3 H, SO 2 NR 2 R 3 in which R 2 and R 3 are as defined below, and SO 2 F;
- R a is also benzyloxy substituted on the phenyl portion by a group defined above, NR 2 R 3 in which R 2 is H or (C ⁇ -C 4 )alkyl and R 3 is H, (C ⁇ -C 4 )alkyl, phenyl or phenyl substituted by a group as defined above;
- R 1 is (C]-C 4 )alkyl, preferably methyl, or a pharmaceutically acceptable salt thereof, such as an acid addition salt.
- Preferred quinazoline compounds useful in the treatment of tumors are described more fully below and particularly in the Examples.
- the quinazoline compounds of the invention are useful as pharmaceutical compositions prepared with a therapeutically effective amount of a quinazoline compound and a pharmaceutically acceptable carrier.
- the quinazoline formulations of the invention can be administered to a mammalian host, such as a human patient in a variety of forms adapted to the chosen route of administration, i.e., orally or parenterally, by intravenous, intramuscular, transdermal or subcutaneous routes.
- the present invention is especially suitable for parenteral administration, particularly intravenous administration.
- the amount of quinazoline compounds in such therapeutically useful formulations is such that an effective dosage level will be obtained.
- the quinazoline formulations may be administered intravenously or intraperitoneally by infusion or injection.
- Solutions of the quinazoline compounds can be prepared in water, optionally mixed with a nontoxic surfactant.
- Dispersions can also be prepared in glycerol, liquid polyethylene glycols, triacetin, and mixtures thereof and in oils. Under ordinary conditions of storage and use, these preparations contain a preservative to prevent the growth of microorganisms.
- the pharmaceutical dosage forms suitable for injection or infusion can include sterile aqueous solutions or dispersions or sterile powders including the quinazoline compounds which are adapted for extemporaneous preparation of sterile injectable or infusible solutions or dispersions, or encapsulated in liposomes.
- the vehicle is a micellar solution, microemulsion or mixtures thereof.
- the ultimate dosage form must be sterile, fluid and stable under the conditions of manufacture and storage.
- the liquid carrier or vehicle can be a solvent or liquid dispersion medium comprising, for example, water, ethanol, a polyol (for example, glycerol, propylene glycol, liquid polyethylene glycols, and the like), vegetable oils, nontoxic glyceryl esters, and suitable mixtures thereof.
- a polyol for example, glycerol, propylene glycol, liquid polyethylene glycols, and the like
- vegetable oils nontoxic glyceryl esters, and suitable mixtures thereof.
- suitable mixtures thereof can be maintained, for example, by the formation of liposomes, by the maintenance of the required particle size in the case of dispersions, such as microemulsions, or by the use of surfactants, such as micellar solutions.
- Micelles are composed of aggregates consisting of generally 50 or more surfactant molecules. Micelles form in aqueous solutions at surfactant concentrations above the critical micellar concentration (CMC). Micelles have the ability to solubilize lipophilic or amphiphilic compounds. Thus, micellar systems can be used to enhance the solubility of poorly water soluble substances, such as some quinazoline compounds.
- CMC critical micellar concentration
- micellar solutions are good solubilizing vehicles for poorly water soluble quinazoline compounds.
- Micellar system formulations include a quinazoline compound, one or more surfactants, and a carrier.
- PEGylated phosphatidylethanolamines (1,2- Dipalmitoyl-sn-Glycero-3-Phosphoethanolamine-N-[Poly(ethylene glycol) 5000] and 1 ,2-Dipalmitoyl-sn-Glycero-3-Phosphoethanolamine-N-[Poly(ethylene glycol) 2000]) are effective in enhancing the solubilization of quinazoline compounds.
- the solubilization enhancement as represented by the amount of solubilized quinazoline compound (in milligram) per gram of surfactant varies with the type of surfactant used and depends on the hydrophobic chain length and polyoxyethylene number of the PEGylated phospholipid.
- Preferred PEGylated phospholipids include PEG2000-DPPE® and PEG5000-DPPE® and are commercially available from Avanti Polar-Lipids Inc., (Alabaster, AL.).
- the micellar solution may include a second surfactant such as, block copolymers of ethylene oxide and propylene oxide alone or in addition to the PEGylated phosphatidylethanolamine surfactant.
- a second surfactant such as, block copolymers of ethylene oxide and propylene oxide alone or in addition to the PEGylated phosphatidylethanolamine surfactant.
- Preferred block copolymers of ethylene oxide and propylene oxide include; Pluronic F-77, Pluronic F-87, and Pluronic F-88 and are commercially available form BASF Corp., (Mount Olive, NJ.)
- micellar solution may include a carrier.
- a preferred carrier is propylene glycol such as 1,2-propanediol.
- Microemulsions are thermodynamically stable, transparent, dispersions of water and oil, stabilized by an interfacial film of surfactant molecules. Microemulsions are characterized by their submicron particle size of 0.1 ⁇ m or below. Microemulsions and self-emulsifying drug delivery systems (SEDDS) can be used to enhance the solubility of poorly water soluble substances, such as some quinazoline compounds.
- SEDDS self-emulsifying drug delivery systems
- Microemulsion system formulations include a quinazoline compound, one or more surfactants, and a carrier.
- the microemulsion solution may include one or more surfactants. These include block copolymers of ethylene oxide and propylene oxide. Preferred block copolymers of ethylene oxide and propylene oxide include; Pluronic F-77, Pluronic F-87, and Pluronic F-88 and are commercially available from BASF Corp., (Mount Olive, NJ.)
- surfactants useful in microemulsion solutions include, ethoxylated castor oil such as Cremophor® EL castor oil commercially available from BASF Corp., (Mount Olive, NJ,) and purified soy bean phospholipid or lecithins such as phosphatidylcholine or Phospholipon® 90G commercially available from American Lecithin (Oxford, CT.)
- ethoxylated castor oil such as Cremophor® EL castor oil commercially available from BASF Corp., (Mount Olive, NJ,) and purified soy bean phospholipid or lecithins such as phosphatidylcholine or Phospholipon® 90G commercially available from American Lecithin (Oxford, CT.)
- the microemulsion solution may include one or more a carriers.
- Preferred carriers include, propylene glycol such as 1 ,2-propanediol, and medium chain triglycerides and monoglycerides such as, triglycerides of caprylic/capric acid such as, Captex® 355, Captex® 350 and Captex® 200 commercially available from Abitec Corp., (Columbus, OH.)
- the prevention of the action of microorganisms in the formulation can be brought about by various antibacterial and antifungal agents, for example, parabens, chlorobutanol, phenol, sorbic acid, thimerosal, and the like.
- isotonic agents for example, sugars, buffers or sodium chloride.
- Prolonged absorption of the injectable compositions can be brought about by the use in the compositions of agents delaying absorption, for example, aluminum monostearate and gelatin.
- Sterile injectable solutions are prepared by incorporating the quinazoline compounds in the required amount in the appropriate solvent with various of the other ingredients enumerated above, as required, followed by filter sterilization.
- the preferred methods of preparation are vacuum drying and the freeze drying techniques, which yield a powder of the active ingredient plus any additional desired ingredient present in the previously sterile- filtered solutions.
- the quinazoline formulations of the invention are useful for the treatment of animals, including humans.
- these quinazoline formulations have been found to be potent inhibitors of tumor cell proliferation and survival, and effective to induce apoptosis of malignant cells.
- Compounds of the invention have surprisingly been found to be effective for inducing apoptosis and/or cytotoxicity of leukemia cells.
- 4-(4'-hydroxyphenyl)amino-6,7-dimethoxyquinazoline compounds (WHI-P131) of the invention have been found to effectively induce apoptosis in multi-drug resistant leukemia.
- WHI-P131 is also a potent inhibitor of Janus kinase 3 (JAK 3) and shows considerable clinical potential for treatment of hematologic malignancies as well as allergic disorders.
- a preferred compound for the treatment of multi-drug resistant leukemia is 4-(3'-bromo-4'-hydroxyphenyl)amino-6,7- dimethoxyquinazoline.
- Compounds of the invention that are particularly useful for treating leukemia include:
- Compounds of the invention that are particularly useful for treating breast tumors include:
- Useful dosages of the quinazoline compounds can be determined by comparing their in vitro activity, and in vivo activity in animal models. Methods for the extrapolation of effective dosages in mice, and other animals, to humans are known to the art; for example, see U.S. Pat. No. 4,938,949.
- the amount of the quinazoline compounds required for use in treatment will vary not only with the particular salt selected but also with the route of administration, the nature of the condition being treated and the age and condition of the patient and will be ultimately at the discretion of the attendant physician or clinician.
- a suitable dose will be in the range of from about 0.5 to about 100 mg/kg, e.g., from about 10 to about 75 mg/kg of body weight per day, such as 3 to about 50 mg per kilogram body weight of the recipient per day, preferably in the range of 6 to 90 mg/kg/day, most preferably in the range of 15 to 60 mg/kg/day.
- the quinazoline compounds are conveniently administered in unit dosage form; for example, containing 5 to 1000 mg, conveniently 10 to 750 mg, most conveniently, 50 to 500 mg of active ingredient per unit dosage form.
- the quinazoline compounds should be administered to achieve peak plasma concentrations of from about 0.5 to about 75 ⁇ M, preferably, about 1 to 50 ⁇ M, most preferably, about 2 to about 30 ⁇ M. This may be achieved, for example, by the intravenous injection of a 0.05 to 5% solution of the quinazoline compounds. Desirable blood levels may be maintained by continuous infusion to provide about 0.01-5.0 mg/kg/hr or by intermittent infusions containing about 0.4- 15 mg/kg of the quinazoline compounds.
- the quinazoline compounds may conveniently be presented in a single dose or as divided doses administered at appropriate intervals, for example, as two, three, four or more sub-doses per day.
- the sub-dose itself may be further divided, e.g., into a number of discrete loosely spaced administrations.
- the quinazoline compound is targeted to cells where treatment is desired, for example, to leukemia cells, to breast cells, or to other tumor cells.
- the compound is targeted to the desired cell by conjugation to a targeting moiety that specifically binds the desired cell, thereby directing administration of a conjugated molecule.
- Useful targeting moieties are ligands which specifically bind cell antigens or cell surface ligands, for example, antibodies against the B cell antigen, CD 19 (such as B43) and the like.
- targeting moieties are covalently bonded to sites on the quinazoline compound.
- the targeting moiety which is often a polypeptide molecule, is bound to compounds of the invention at reactive sites, including NH , SH, CHO, COOH, and the like.
- Specific linking agents are used to join the compounds. Preferred linking agents are chosen according to the reactive site to which the targeting moiety is to be attached.
- quinazoline compounds may be administered prophylactically, i.e., prior to onset of the pathological condition, or the quinazoline compounds may be administered after onset of the reaction, or at both times.
- novel hydroxy-substituted quinazoline derivatives of the invention were created by reacting the appropriate substituted anilines with the key starting material, 4-chloro-6,7-dimethoxy quinazoline.
- Example 3 Chlorine Substituted Quinazoline Compounds Chlorine substituted quinazoline derivatives were synthesized and characterized as discussed above in Example 1. The structures and physical data are shown below:
- Iodine substituted quinazoline derivatives were synthesized as discussed above in Example 1, and analyzed. The structures and physical data are shown below: Iodine Substituted Quinazoline Compounds
- IR(KBR) ⁇ max 3391, 3139, 2938, 2850, 1633, 1607, 1567, 1509, 1447, 1359, 1220, 1189, 1055 cm “1 .
- GC/MS m/z: 314 (M 1, 13.00), 313 (m “ , 72.80), 312(m + -l, 10.20), 296 (5.24), 206(17.50).
- UV9MeOH 208.0, 215.0, 225.0, 240.0, 330.0 mn.
- IR(KBr) ⁇ max 3438, 321 1, 3061, 2932, 2834, 1633, 1576, 1509, 1437, 1380, 1276, 1215 cm "1 .
- GC/MS m z 281(51.00), 253(22.00), 207(88.00).
- C 2 oH 17 N 3 O 3 .HCl requires: C, 62.66; H, 4.70; N, 10.96%.
- UV(MeOH) 206.0, 210, 219.0, 225.0, 230.0, 340.0 nm.
- IR(KBr) ⁇ max 3391, 3165, 3051, 2938, 2840, 1628, 1576, 1504, 1437, 1281, 1215 cm “1 .
- GC/MS m/z 348(M ⁇ + 1, 7.00), 347(M " , 100.00), 346(M ⁇ 1.22.00), 331(15.00), 330(12.00), 281(23.00), 253(12.00), 207(49.00).
- IR(KBr) ⁇ max 3407, 3030, 2977, 2840, 1643, 1591 1514, 1463, 1370, 1282, 1230 cm “1 .
- GC/MS m z 325(M “ +1, 67.00), 324(M “ , 100.00), 323(M ⁇ 1.22.00), 308(17.00), 307(56.00), 306(21.00), 281(2.00), 280(8.00), 264(6.00).
- WHI-P131 free base was measured in water, propylene glycol, polyethylene glycols (PEGs), ethanol, and triglycerides. The results are summarized in Table 6.
- the solubility of WHI-P131 is very poor in water. It was about 35 times more soluble in C 8 -C ⁇ 0 medium chain triglyceride (Captex 300) than in water. It was much more soluble in ethanol and hydrophilic cosolvents such as propylene glycol and PEGs.
- WHI-P131 free base was most soluble in polyethylene glycols of greater than 10%, followed by propylene glycol (1.95%) and ethanol (1.86%). Parallel solubility measurements were also carried out using WHI-
- PEG300 the solubility continued to increase linearly with increasing PEG concentration, whereas for PEG200, a large increase in slopes occurred near 100%o PEG200. Since the solubility-PEG300 concentration curve is linear over the entire range of water- PEG300 mixtures, WHI-P131 solubilized in these mixtures at concentrations below its saturation point can be used as vehicles for this compound, since their dilution will not result in drug precipitation. In contrast, if one were to dilute by water a 2% WHI-P131 in PEG200, WHI-P131 concentration would fall above the solubility limit and precipitate out. Therefore, PEG300 is more appropriate for use as a cosolvent vehicle in the formulations of WHI-P 131.
- micellar solutions containing PEGylated phosphatidylethanolamines were exceptionally effective in enhancing the solubilization of WHI-P 131.
- Table 7 shows the compositions of several mixed micellar solutions containing various amounts of WHI-P131.
- Micellar solutions using purified soya lecithin (Phospholipon 90G) were feasible when an equal or higher amount of a nonionic surfactant (such as Cremophor EL for example) was also present. With PEGylated phospholipids, the presence of Cremophor EL was not necessary to form micellar solutions.
- solubility-surfactant concentration curves were plotted.
- Figure 2 depicts the amount of solubilized WHI-P131 chloride in a solution containing 20% of PEG300, and an increasing amount of PEG2000-DPPE. This figure indicates that, in the absence of surfactant, the solubility of WHI-P131 chloride salt in 20% PEG300 was 2.38 mg/ml. At low surfactant concentrations (below the CMC), the drug solubilization seems to remain unchanged, then increases linearily with surfactant concentration at higher PEG2000-DPPE concentration. The same solubilization characteristics were observed with other micellar solutions.
- Table 8 the slopes of the linear portions of the plot for a series of nonionic surfactants and cosolvents were used to calculate the solubilization enhancement per unit surfactant or cosolvent concentration.
- the solubilization enhancement as represented by the amount of solubilized WHI-P131 (in milligram) per gram of surfactant, are shown in Table 8 to vary with the type of surfactants used.
- the solubilization enhancement depended on the hydrophobic chain length and polyoxyethylene number of the PEGylated phospholipids.
- PEG2000-DPPE and PEG5000-DPPE seemed to be the most effective solubilizers for WHI-P131 of the three PEGylated phosphatidylethanolamines investigated.
- Table 8 for comparison purposes are the solubilization enhancements produced by the use of cosolvents. It can be seen that PEGylated surfactants were about 6 to 16 times more effective than cosolvents in producing solubilization enhancement of WHI-P131 chloride salt.
- a series of ternary phase diagrams were constructed at room temperature, and several microemulsions within the single phase microemulsion region were examined for their capacity to solubilize WHI-P 131.
- a representative ternary phase diagram depicted in Figure 3 shows the location of the single phase microemulsion region. In this phase diagram, it can be seen that microemulsions containing up to 30% of Captex 300 were possible. These microemulsions were transparent and tolerated dilution very well when mixed with aqueous phases.
- the drug was first solubilized in the microemulsions chosen from the one phase region of the phase diagram with mild heating, followed by dilution with water or buffer solution at room temperature.
- the microemulsion composition ME1 depicted in Table 9 was used in pharmacokinetic studies and biological activity assays. This microemulsion was prepared by first solubilizing WHI-P 131 in composition A in the ternary phase diagram, followed by a dilution with water (1 :9). Its volume-weighted average particle diameter as determined by dynamic light scattering was 24.8 nm prior to and 11.4 nm after the incorporation of WHI-P 131 chloride. Thus, the drug incorporation, in this case, resulted in the lowering of the particle size. The solubilization of WHI-P131 was at least 1.8 mg per ml of microemulsion. ME2 was a microemulsion composition obtained from a separate phase diagram not shown.
- This microemulsion can solubilize at least 2.8 mg of WHI-P131 per ml of microemulsion .
- ME2 had more than doubled the solubilization of WHI-P 131 in water.
- These microemulsions can readily be filtered through 0.2 ⁇ m filter, and stored at room temperature. The microemulsions and WHI-P 131 they contained were shown to be stable for an extended time at ambient temperature.
- WHI-P131 By converting WHI-P131 from its free base to its chloride salt form, a fifty fold increase in solubility was achieved raising the drug concentration from 0.025 mg/ml to 1.2 mg/ml. By adding 20% of PEG300 to the vehicle, the drug concentration further increased to 2.2 mg/ml. Furthermore, an incorporation of 3% of PEG2000-DPPE to the cosolvent vehicle brought the drug solubilization to 4.7mg/ml, which corresponds to a total solubilization enhancement of 190 fold. If a microemulsion formulation instead a cosolvent/micellar solution was used, a total solubilization enhancement of 110 fold. Lead micellar and microemulsion formulations of WHI-P 131 were as active as unformulated WHI-P 131 in DMSO. The miceller formulation inhibited allergic mast cell responses in vitro and prevented anaphy lactic shock in vivo.
- microemulsions can be used to enhance the solubilization of WHI-P131.
- the drug incorporation into the microemulsion seemed to be limited to the surfactant interfacial film only which resulted in a relatively small solubilization enhancement.
- the lipid cores of the microemulsion droplets in this case medium chain triglyceride, seemed to contribute very little to the solubilization enhancement.
- Table 9 Microemulsion compositions containing WHI-P131
- the cumulative solubilization enhancement obtained using a combination of solubilization methods is illustrated in Figure 4.
- the overall enhancement appears to be additive.
- WHI-P 131 from its free base to its chloride salt form
- a fifty fold increase in solubility was achieved raising the drug concentration from 0.025 mg/ml to 1.2 mg/ml.
- 20%o of PEG300 to the vehicle
- the drug concentration further increased to 2.2 mg/ml.
- an incorporation of 3%> of PEG200-DPPE to the cosolvent vehicle brought the drug solubilization to 4.7mg/ml, which corresponds to a total solubilization enhancement of 190 fold.
- a microemulsion formulation instead a cosolvent/micellar solution was used, one can reached a total solubilization enhancement of 110 fold.
- WHI-P131 drug containing solution 68 mg of WHI-P131 CI " was dissolved in 4 ml of the above propylene glycol solution and 0.6 ml DI water. This drug mixture was heated at 70°C for 10 min until all the WHI-P131 was dissolved and the solution was yellow and clear. This drug solution was mixed into 27.95 ml of DI water dropwise. The diluted solution was yellow and clear. This drug solution was filtered through 0.2 ⁇ m filter under a laminar flow hood for sterilization. The filtrate was collected in a liquid scintillation vial. The WHI-P 131 concentration in the solution was 1.97 mg/ml. The composition of the solution was:
- WHI-P131 220 mg was dissolved in 15.7 ml of the above microemulsion. The mixture was stirred and heated at 70°C for 30 min or until all solids were dissolved. WHI-P 131 concentration in this drug microemulsion was 14 mg/ml.
- Blood samples were obtained from the ocular venous plexus by retroorbital venupuncture prior to and at 3, 5, 10, 15, 30, 45 minutes, and 1, 2, 4, and 8 hours after administration of WHI-P 131. All collected blood samples were heparinized and centrifuged at 7,000 g for 10 min in a microcentrifuge to obtain plasma. The plasma samples were stored at -20°C until analysis. Aliquots of plasma were used for extraction and HPLC analysis.
- Pharmacokinetic modeling and parameter calculations were carried out using the software, WinNonlin Program, Version 2.0.
- An appropriate pharmacokinetic model was chosen on the basis of lowest weighted squared residuals, lowest Schwartz criterion, lowest Akaike's Information Criterion value, lowest standard errors of the fitted parameters, and dispersion of the residuals.
- the elimination half-life was estimated by linear regression analysis of the terminal phase of the plasma concentration profile.
- the area under the curve (AUC) was calculated by the trapezoidal rule between first (0 h) and last sampling time plus C/k, where C is the concentration of last sampling and k is the elimination rate constant.
- Systemic clearance (CLs) was determined by dividing the dose by the AUC.
- Statistical analysis was performed using the Instat program, 3.0. The significance of differences between pharmacokinetic parameters was analyzed using two-tailed t test, and P values ⁇ 0.05 were considered significant.
- HPLC system consisted of a Hewlett Packard series 1100 equipped with an automated electronic degasser, a quaternary pump, an autosampler, an automatic thermostatic column compartment, diode array detector and a computer with a Chemstation software program for data analysis.
- a 250 x 4 mm Lichrospher 100, RP-18 (5 ⁇ m) analytical and a 4 x 4 mm Lichrospher 100, RP-18 guard columns were obtained from Hewlett Packard Inc.
- Acetonitrile/water containing 0.1 % of trifluoroacetic acid and 0.1%o triethylamine (28:72, v/v) was used as the mobile phase.
- the wavelength of detection was set at 340 nm. Peak width, response time and slit were set at >0.03 min, 0.5 s and 8 nm, respectively.
- the cells were maintained as monolayer cultures in 75- or 150- cm flask in Eagle's essential medium supplemented with 20% fetal calf serum (Hamawy et. al., 1995, Cellular Signalling 7:535-544).
- RBL- 2H3 cells were sensitized with monoclonal anti-DNP IgE antibody (0.24 mg/ml) for lhour at 37 °C in a 48-well tissue culture plate.
- RBL-2H3 cells were allowed to adhere to the plate. Unbound IgE was removed by washing the cells with phosphate buffered saline.
- mice were sensitized with 2 mg BSA in 200 ⁇ l aluminum hydroxide gel (Reheis Inc., Berkeley, NJ), which induces the production of IgE response to the presented antigen.
- anaphylactic shock was induced by the i.v. injection of the animals with 200 ⁇ g BSA. Mice were continuously monitored for 3 hours for signs of anaphylaxis.
- mice Male Balb/c mice (6-8 weeks old) were purchased from Charles River Laboratories (Wilmington, MA). Breeder pairs of JAK3-null mice (Nosaka et. al., 1995) were obtained from Dr. J. Ihle (St. Jude Children's Research Hospital, Memphis, TN). Animals were caged in groups of five in a pathogen free environment in accordance with the rules and regulations of U. S. Animal Welfare Act, and National Institutes of Health (NIH). Animal care and the experimental procedures were carried out in agreement with institutional guidelines. Study We compared the pharmacokinetics of the lead micellar microemulsion formulations of WHI-P 131. The WHI-P 131 plasma concentration- time curves following i.v.
- Example 15 Mast cell inhibitory "anti-allergic" activity of formulated WHI- PI 31 in vitro.
- FIG. 6 Micellar solution and microemulsion formulations of WHI-P131 were active.
- Figure 6 shows the mast cell inhibitory "anti-allergic" activity of these formulations in vitro.
- Mast cell degranulation ⁇ -hexosaminidase release, % of total
- Unformulated WHI- P131 has been previously shown to prevent mast cell degranulation and release of preformed granule-associated ⁇ -hexosaminidase in a dose-dependent fashion with near to complete inhibition at >30 ⁇ M (Malaviya R et al., Targeting Janus kinase 3 in mast cells prevents immediate hypersensitivity reactions and anaphylaxis. J Biol Chem., 1999, 274, 27028-38). As shown in Figure 6, both formulations were as effective as unformulated WHIP131 in DMSO. Virtually complete inhibition of mast cell function was achieved at a WHI-P131 concentration of 30 ⁇ M.
- Example 16 In vivo anti-allergic activity formulated WHI-P131.
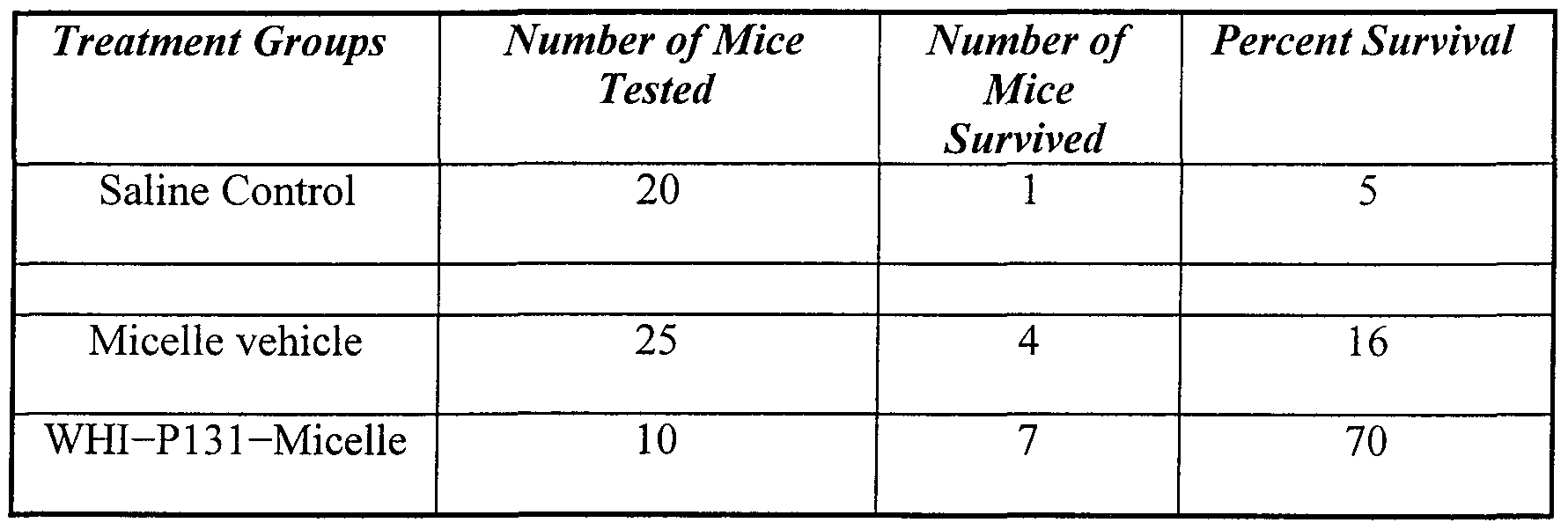
- mice were first injected with BSA in an aluminum hydroxide gel to trigger a BSA-specific IgE response. Ten days later, these BSA-sensitized mice were rechallenged with this antigen to induce anaphylaxis. Only one of 20 (5%) saline treated control mice and 4 of 25 (16%>) micelle vehicle (0% WHI-P131) treated control mice did not develop fatal anaphylaxis (Table 10). The remainder of these control mice (i.e., 40 of 45) developed anaphylaxis and died within 45 min after antigen challenge. In contrast, 7 of 10 (70 %) BSA-sensitized mice that were treated with WHI-P131 (micellar formulation) prior to antigen challenge survived without any signs of anaphylaxis, (PO.05 by log-rank test).
- Table 10 Protective activity of the WHI-P131 Micellar Formulation against Active Anaphylaxis in Mice.
- mice were sensitized with 100 mg/kg bovine serum albumin in 200 ⁇ l of the adjuvant aluminum hydroxide gel (Reheis Inc., Berkeley, NJ), which favors the production of IgE in response to the presented antigen.
- mice were treated with two doses of WHI-P131 formulations (50 mg/kg) or vehicle intraperitoneally 10 min before and 10 min after an intravenous injection of the 10 mg/kg BSA. Mice were continuously monitored for 3 hours for signs of anaphylaxis and the mice surviving the anaphylactic reaction were sacrificed.
- Figure 6 shows effects of WHI-P131 formulations on IgE receptor/Fc epsilon RI- mediated mast cell degranulation.
- RBL-2H3 cells were sensitized with monoclonal anti-DNP IgE, treated with WHI-P131 formulations or vehicle control compounds for lh, and then challenged with 20 ng/ml DNP-BSA for 30 min.
Abstract
Description
























Claims


Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP00914991A EP1162974A1 (en) | 1999-03-19 | 2000-03-17 | Quinazoline formulations and therapeutic use thereof |
| JP2000606242A JP2002539262A (en) | 1999-03-19 | 2000-03-17 | Quinazoline compound formulations and their use in therapy |
| AU36301/00A AU3630100A (en) | 1999-03-19 | 2000-03-17 | Quinazoline formulations and therapeutic use thereof |
| CA002366998A CA2366998A1 (en) | 1999-03-19 | 2000-03-17 | Quinazoline formulations and therapeutic use thereof |
| US09/960,464 US20020111360A1 (en) | 1999-03-19 | 2001-09-19 | Quinazoline formulations and therapeutic use thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12514799P | 1999-03-19 | 1999-03-19 | |
| US60/125,147 | 1999-03-19 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/960,464 Continuation US20020111360A1 (en) | 1999-03-19 | 2001-09-19 | Quinazoline formulations and therapeutic use thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2000056338A1 true WO2000056338A1 (en) | 2000-09-28 |
Family
ID=22418397
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2000/007066 WO2000056338A1 (en) | 1999-03-19 | 2000-03-17 | Quinazoline formulations and therapeutic use thereof |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20020111360A1 (en) |
| EP (1) | EP1162974A1 (en) |
| JP (1) | JP2002539262A (en) |
| AU (1) | AU3630100A (en) |
| CA (1) | CA2366998A1 (en) |
| WO (1) | WO2000056338A1 (en) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7148230B2 (en) | 2003-07-29 | 2006-12-12 | Astrazeneca Ab | Quinazoline derivatives |
| EP1890677A2 (en) * | 2005-06-16 | 2008-02-27 | Myriad Genetics, Inc. | Pharmaceutical compositions and use thereof |
| US7910731B2 (en) | 2002-03-30 | 2011-03-22 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Bicyclic heterocyclic compounds, pharmaceutical compositions containing these compounds, their use and process for preparing them |
| US7998949B2 (en) | 2007-02-06 | 2011-08-16 | Boehringer Ingelheim International Gmbh | Bicyclic heterocycles, drugs containing said compounds, use thereof, and method for production thereof |
| US8088782B2 (en) | 2008-05-13 | 2012-01-03 | Astrazeneca Ab | Crystalline 4-(3-chloro-2-fluoroanilino)-7 methoxy-6-{[1-(N-methylcarbamoylmethyl)piperidin-4-yl]oxy}quinazoline difumarate form A |
| US8399461B2 (en) | 2006-11-10 | 2013-03-19 | Boehringer Ingelheim International Gmbh | Bicyclic heterocycles, medicaments containing said compounds, use thereof, and method for production of same |
| US8497369B2 (en) | 2008-02-07 | 2013-07-30 | Boehringer Ingelheim International Gmbh | Spirocyclic heterocycles medicaments containing said compounds, use thereof and method for their production |
| US8648191B2 (en) | 2008-08-08 | 2014-02-11 | Boehringer Ingelheim International Gmbh | Cyclohexyloxy substituted heterocycles, pharmaceutical compositions containing these compounds and processes for preparing them |
| US8658654B2 (en) | 2002-07-15 | 2014-02-25 | Symphony Evolution, Inc. | Receptor-type kinase modulators and methods of use |
| US8877776B2 (en) | 2009-01-16 | 2014-11-04 | Exelixis, Inc. | (L)-malate salt of N-(4-{[6,7-bis(methyloxy) quinolin-4-yl]oxy}phenyl)-N'-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide |
| US10736886B2 (en) | 2009-08-07 | 2020-08-11 | Exelixis, Inc. | Methods of using c-Met modulators |
| US11124482B2 (en) | 2003-09-26 | 2021-09-21 | Exelixis, Inc. | C-met modulators and methods of use |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6126917A (en) * | 1999-06-01 | 2000-10-03 | Hadasit Medical Research Services And Development Ltd. | Epidermal growth factor receptor binding compounds for positron emission tomography |
| GB9922171D0 (en) * | 1999-09-21 | 1999-11-17 | Zeneca Ltd | Chemical compounds |
| CA2691914C (en) * | 2007-07-11 | 2012-06-26 | Pfizer Inc. | Pharmaceutical compositions and methods of treating dry eye disorders |
| BE1030538B1 (en) | 2022-05-18 | 2023-12-19 | Bogaert Gina Van | Liposomal preparation containing encapsulated hormones, method of production and use |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0082385A1 (en) * | 1981-12-18 | 1983-06-29 | Troponwerke GmbH & Co. KG | 1-(3-Nitrophenyl)pyrido(2,3-d)pyrimidine-2,4(1H,3H) diones and 1-(3-nitrophenyl)quinazoline-2-4(1H,3H)diones as cutaneous medicaments |
| US5411963A (en) * | 1988-01-29 | 1995-05-02 | Dowelanco | Quinazoline derivatives |
| US5449678A (en) * | 1994-01-11 | 1995-09-12 | Agricultural Research Organization, Ministry Of Agriculture | Anti-fibrotic quinazolinone-containing compositions and methods for the use thereof |
| WO1996006616A1 (en) * | 1994-08-31 | 1996-03-07 | Davidson, Clifford, M. | Quinazolinone pharmaceuticals and use thereof |
| US5792771A (en) * | 1992-11-13 | 1998-08-11 | Sugen, Inc. | Quinazoline compounds and compositions thereof for the treatment of disease |
| WO1998038984A2 (en) * | 1997-03-05 | 1998-09-11 | Sugen, Inc. | Formulations for hydrophobic pharmaceutical agents |
| WO1998051284A1 (en) * | 1997-05-13 | 1998-11-19 | Imarx Pharmaceutical Corp. | Novel acoustically active drug delivery systems |
-
2000
- 2000-03-17 AU AU36301/00A patent/AU3630100A/en not_active Abandoned
- 2000-03-17 EP EP00914991A patent/EP1162974A1/en not_active Withdrawn
- 2000-03-17 CA CA002366998A patent/CA2366998A1/en not_active Abandoned
- 2000-03-17 WO PCT/US2000/007066 patent/WO2000056338A1/en not_active Application Discontinuation
- 2000-03-17 JP JP2000606242A patent/JP2002539262A/en not_active Withdrawn
-
2001
- 2001-09-19 US US09/960,464 patent/US20020111360A1/en not_active Abandoned
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0082385A1 (en) * | 1981-12-18 | 1983-06-29 | Troponwerke GmbH & Co. KG | 1-(3-Nitrophenyl)pyrido(2,3-d)pyrimidine-2,4(1H,3H) diones and 1-(3-nitrophenyl)quinazoline-2-4(1H,3H)diones as cutaneous medicaments |
| US5411963A (en) * | 1988-01-29 | 1995-05-02 | Dowelanco | Quinazoline derivatives |
| US5792771A (en) * | 1992-11-13 | 1998-08-11 | Sugen, Inc. | Quinazoline compounds and compositions thereof for the treatment of disease |
| US5449678A (en) * | 1994-01-11 | 1995-09-12 | Agricultural Research Organization, Ministry Of Agriculture | Anti-fibrotic quinazolinone-containing compositions and methods for the use thereof |
| WO1996006616A1 (en) * | 1994-08-31 | 1996-03-07 | Davidson, Clifford, M. | Quinazolinone pharmaceuticals and use thereof |
| WO1998038984A2 (en) * | 1997-03-05 | 1998-09-11 | Sugen, Inc. | Formulations for hydrophobic pharmaceutical agents |
| WO1998051284A1 (en) * | 1997-05-13 | 1998-11-19 | Imarx Pharmaceutical Corp. | Novel acoustically active drug delivery systems |
Non-Patent Citations (4)
| Title |
|---|
| "VIDAL 1997", XP002143626 * |
| BRASSINNE C; ATASSI G; FRUHLING J; PENASSE W: "Anti-tumor activity of a water-insoluble compound entrapped in liposomes on L1210 Leukemia in mice", JOURNAL OF THE NATIONAL CANCER INSTITUTE, vol. 70, 1983, pages 1081 - 1086, XP002142785 * |
| SCWINN J; SPRINZ H; LEISTNER S: "The effects of a thio-containing quinazolinedione derivative (MECH) on the lipid oxidation in bilayer liposomes", JOURNAL OF RADIOANALYTICAL AND NUCLEAR CHEMISTRY, vol. 232, no. 1-2, 1998, pages 35 - 37, XP000929184 * |
| YIV SH; METZ M, LI M, LIU X-P: "Microemulsion, liposome and mixed micellar formulations for a poorly water soluble quinazoline derivative", ABSTRACTS OF PAPERS AMERICAN CHEMICAL SOCIETY, vol. 217, 21 March 1999 (1999-03-21), pages 148, XP000929164 * |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7910731B2 (en) | 2002-03-30 | 2011-03-22 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Bicyclic heterocyclic compounds, pharmaceutical compositions containing these compounds, their use and process for preparing them |
| US8343982B2 (en) | 2002-03-30 | 2013-01-01 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Bicyclic heterocyclic compounds pharmaceutical compositions containing these compounds, their use and process for preparing the same |
| US8658654B2 (en) | 2002-07-15 | 2014-02-25 | Symphony Evolution, Inc. | Receptor-type kinase modulators and methods of use |
| US9359332B2 (en) | 2002-07-15 | 2016-06-07 | Symphony Evolution, Inc. | Processes for the preparation of substituted quinazolines |
| US10266518B2 (en) | 2002-07-15 | 2019-04-23 | Symphony Evolution, Inc. | Solid dosage formulations of substituted quinazoline receptor-type kinase modulators and methods of use thereof |
| US9796704B2 (en) | 2002-07-15 | 2017-10-24 | Symphony Evolution, Inc. | Substituted quinazolines as receptor-type kinase inhibitors |
| US7148230B2 (en) | 2003-07-29 | 2006-12-12 | Astrazeneca Ab | Quinazoline derivatives |
| US11124482B2 (en) | 2003-09-26 | 2021-09-21 | Exelixis, Inc. | C-met modulators and methods of use |
| EP1890677A2 (en) * | 2005-06-16 | 2008-02-27 | Myriad Genetics, Inc. | Pharmaceutical compositions and use thereof |
| EP1890677A4 (en) * | 2005-06-16 | 2013-01-30 | Myriad Genetics Inc | Pharmaceutical compositions and use thereof |
| US8399461B2 (en) | 2006-11-10 | 2013-03-19 | Boehringer Ingelheim International Gmbh | Bicyclic heterocycles, medicaments containing said compounds, use thereof, and method for production of same |
| US7998949B2 (en) | 2007-02-06 | 2011-08-16 | Boehringer Ingelheim International Gmbh | Bicyclic heterocycles, drugs containing said compounds, use thereof, and method for production thereof |
| US8497369B2 (en) | 2008-02-07 | 2013-07-30 | Boehringer Ingelheim International Gmbh | Spirocyclic heterocycles medicaments containing said compounds, use thereof and method for their production |
| US8772298B2 (en) | 2008-02-07 | 2014-07-08 | Boehringer Ingelheim International Gmbh | Spirocyclic heterocycles medicaments containing said compounds, use thereof and method for their production |
| US8088782B2 (en) | 2008-05-13 | 2012-01-03 | Astrazeneca Ab | Crystalline 4-(3-chloro-2-fluoroanilino)-7 methoxy-6-{[1-(N-methylcarbamoylmethyl)piperidin-4-yl]oxy}quinazoline difumarate form A |
| US8648191B2 (en) | 2008-08-08 | 2014-02-11 | Boehringer Ingelheim International Gmbh | Cyclohexyloxy substituted heterocycles, pharmaceutical compositions containing these compounds and processes for preparing them |
| US8877776B2 (en) | 2009-01-16 | 2014-11-04 | Exelixis, Inc. | (L)-malate salt of N-(4-{[6,7-bis(methyloxy) quinolin-4-yl]oxy}phenyl)-N'-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide |
| US9809549B2 (en) | 2009-01-16 | 2017-11-07 | Exelixis, Inc. | Malate salt of N-(4-{[6,7-bis(methyloxy)quinolin-4-yl]oxy}phenyl)-N′(4-fluorophenyl)cyclopropane-1,1-dicarboxamide, and crystalline forms therof for the treatment of cancer |
| US11091439B2 (en) | 2009-01-16 | 2021-08-17 | Exelixis, Inc. | Malate salt of N-(4-{[6,7-bis(methyloxy) quinolin-4-yl]oxy}phenyl)-N′-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide, and crystalline forms therof for the treatment of cancer |
| US11091440B2 (en) | 2009-01-16 | 2021-08-17 | Exelixis, Inc. | Malate salt of N-(4-{[6,7-bis(methyloxy) quinolin-4-yl]oxy}phenyl)- N′-(4-fluorophenyl)cyclopropane-1,1 -dicarboxamide, and crystalline forms thereof for the treatment of cancer |
| US11098015B2 (en) | 2009-01-16 | 2021-08-24 | Exelixis, Inc. | Malate salt of N-(4-{[6,7-bis(methyloxy) quinolin-4-yl]oxy}phenyl)-N′-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide, and crystalline forms thereof for the treatment of cancer |
| US10736886B2 (en) | 2009-08-07 | 2020-08-11 | Exelixis, Inc. | Methods of using c-Met modulators |
| US11433064B2 (en) | 2009-08-07 | 2022-09-06 | Exelixis, Inc. | Methods of using c-Met modulators |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2002539262A (en) | 2002-11-19 |
| US20020111360A1 (en) | 2002-08-15 |
| CA2366998A1 (en) | 2000-09-28 |
| AU3630100A (en) | 2000-10-09 |
| EP1162974A1 (en) | 2001-12-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2000056338A1 (en) | Quinazoline formulations and therapeutic use thereof | |
| US6258820B1 (en) | Synthesis and anti-tumor activity of 6,7-dialkoxy-4-phenylamino-quinazolines | |
| AU690958B2 (en) | Treatment of platelet derived growth factor related disorders such as cancers using inhibitors of platelet derived growth factor receptor | |
| EP1682552B1 (en) | Tocopherol-modified therapeutic drug compounds | |
| US20080045559A1 (en) | Tocopherol-modified therapeutic drug compounds | |
| US20060276491A9 (en) | Therapeutic compounds | |
| US20060003976A1 (en) | Cholesterol/bile acid/bile acid derivative-modified therapeutic drug compounds | |
| US8258145B2 (en) | Method of treating brain cancer | |
| US10376591B2 (en) | Formulations and carrier systems including farnesylthiosalicylic moieties | |
| CN101287369A (en) | Method of treating brain cancer | |
| EP2697206B1 (en) | Heterocyclic compounds and uses thereof in the treatment of sexual disorders | |
| AU2007327048B2 (en) | Amino quinazolines derivatives with blood platelet reducing properties | |
| US8304420B2 (en) | Substituted quinazolines for reducing platelet count | |
| MXPA06004429A (en) | Tocopherol-modified therapeutic drug compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AT AU AZ BA BB BG BR BY CA CH CN CR CU CZ CZ DE DE DK DK DM DZ EE EE ES FI FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 36301/00 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2366998 Country of ref document: CA Ref country code: CA Ref document number: 2366998 Kind code of ref document: A Format of ref document f/p: F |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 2000 606242 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09960464 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2000914991 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2000914991 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2000914991 Country of ref document: EP |