WO2000049008A1 - Di- and tripeptide nitrile derivatives as inhibitors of cathepsin l and cathepsin s - Google Patents
Di- and tripeptide nitrile derivatives as inhibitors of cathepsin l and cathepsin s Download PDFInfo
- Publication number
- WO2000049008A1 WO2000049008A1 PCT/GB2000/000529 GB0000529W WO0049008A1 WO 2000049008 A1 WO2000049008 A1 WO 2000049008A1 GB 0000529 W GB0000529 W GB 0000529W WO 0049008 A1 WO0049008 A1 WO 0049008A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- optionally substituted
- carbamoyl
- formula
- sulphamoyl
- Prior art date
Links
- 0 CC*(**)C(*)C(C)(C1)C2[C@@]1CCC2 Chemical compound CC*(**)C(*)C(C)(C1)C2[C@@]1CCC2 0.000 description 4
- DQBUIZIKRCBBBF-UHFFFAOYSA-N CC(C(NC)SCc1ccccc1)=O Chemical compound CC(C(NC)SCc1ccccc1)=O DQBUIZIKRCBBBF-UHFFFAOYSA-N 0.000 description 2
- WIQCPQGRHPLSQV-UHFFFAOYSA-N CC(C(Cc1ccncc1)NC)=O Chemical compound CC(C(Cc1ccncc1)NC)=O WIQCPQGRHPLSQV-UHFFFAOYSA-N 0.000 description 1
- ZKUYJUWQZGWTAA-UHFFFAOYSA-N CC(C(NC)Sc1ccccc1)=O Chemical compound CC(C(NC)Sc1ccccc1)=O ZKUYJUWQZGWTAA-UHFFFAOYSA-N 0.000 description 1
- ACMODHAHLGAMJM-UHFFFAOYSA-N CC(C)SC(C(C)=O)NC Chemical compound CC(C)SC(C(C)=O)NC ACMODHAHLGAMJM-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/44—Sulfones; Sulfoxides having sulfone or sulfoxide groups and carboxyl groups bound to the same carbon skeleton
- C07C317/48—Sulfones; Sulfoxides having sulfone or sulfoxide groups and carboxyl groups bound to the same carbon skeleton the carbon skeleton being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups
- C07C317/50—Sulfones; Sulfoxides having sulfone or sulfoxide groups and carboxyl groups bound to the same carbon skeleton the carbon skeleton being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups at least one of the nitrogen atoms being part of any of the groups, X being a hetero atom, Y being any atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/01—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms
- C07C255/24—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms containing cyano groups and singly-bound nitrogen atoms, not being further bound to other hetero atoms, bound to the same saturated acyclic carbon skeleton
- C07C255/29—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms containing cyano groups and singly-bound nitrogen atoms, not being further bound to other hetero atoms, bound to the same saturated acyclic carbon skeleton containing cyano groups and acylated amino groups bound to the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/22—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of hydrocarbon radicals substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/50—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton
- C07C323/51—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton
- C07C323/60—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton with the carbon atom of at least one of the carboxyl groups bound to nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/34—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D307/38—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D307/54—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/06—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to the ring carbon atoms
- C07D333/24—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/06034—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms
- C07K5/06043—Leu-amino acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06078—Dipeptides with the first amino acid being neutral and aromatic or cycloaliphatic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06139—Dipeptides with the first amino acid being heterocyclic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- the present invention relates to compounds that are cysteine protease inhibitors and in particular compounds that are Cathepsin L inhibitors and or Cathepsin S inhibitors especially Cathepsin S inhibitors.
- the invention further relates to processes for their preparation, to intermediates useful in their preparation, to their use as therapeutic agents, to pharmaceutical compositions containing them and to a method of treating a Cathepsin L or Cathepsin S mediated disease state.
- Cysteine proteases are enzymes important in normal cell physiology, but they are also associated with several disease states including inflammation, metastasis, tissue damage following myocardial infarction, bone resorption and muscle wasting in dystrophic diseases.
- Cathepsins B, H, K, L, N and S are cysteinyl proteases involved in normal protein degradation and are normally located in the lysosomes of cells. However, when these enzymes are found outside the lysosomes they have been implicated as playing a causative role in a number of disease states including bone resorption disease such as osteoporosis.
- Living bone is continuously being remodelled and replenished by the process of resorption and deposition of the protein matrix and calcium minerals. These events are facilitated by the osteoclast, which has the ability to degrade and demineralise the bone, and the osteoblast which is responsible for new bone generation. In normal situations these processes are intimately linked resulting in little alteration of bone mass.
- pathological conditions exist in which there is an imbalance between their activities resulting in increased degradation and demineralisation of bone and the development of fragile and/or brittle bone structure, as seen during osteoporosis.
- Cathepsins B, H, K, L, N and S have been further implicated as playing a causative role in other diseases such as rheumatoid arthritis, osteoarthritis, tumour metastasis, pneumocystitis, Crithidia fusiculata, malaria, trypanosoma brucei brucei, schistosomiasis, periodontal disease, metachromatic leukodystrophy and muscular dystrophy.
- Cathepsins B, H, K, L, N and S either alone or together, have also been implicated as playing a causative role in chronic obstructive pulmonary disease (COPD).
- COPD chronic obstructive pulmonary disease
- the present invention discloses compounds with inhibitory activity of cysteine proteases and in particular of Cathepsin L and or Cathepsin S.
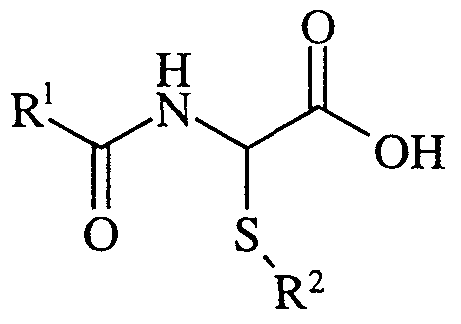
- the compounds of the invention are also useful in the treatment of chronic obstructive pulmonary disease (COPD). Accordingly the present invention provides a compound of formula (I):
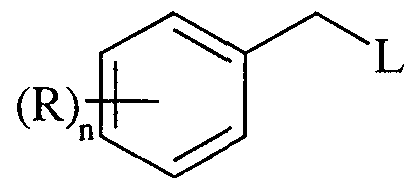
- R 1 is hydrogen, optionally substituted benzyl where said optional substituents are chosen from one or more of C 1- alkyl, halo, trifluoromethyl, hydroxy, trifluoromethoxy, cyano, C ⁇ , 6 alkoxy, C 1-6 alkanoyl, C 1-6 alkanoyloxy, amino, C 1-6 alkylamino, NN-(C ⁇ -6 alkyl) 2 amino, C 1-6 alkanoylamino, nitro, carboxy, carbamoyl, N-(C 1-6 alkyl)carbamoyl, NN-(C ⁇ -6 alkyl) 2 carbamoyl, C 1-6 alkoxycarbonyl, mercapto, C 1-6 alkylsulphanyl, C 1-6 alkylsulphinyl, C ⁇ -6 alkylsulphonyl, sulphamoyl, N-(C 1-6 alkyl)sulphamo
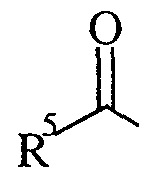
- R 5 is C ⁇ -6 alkyl (optionally substituted with an optionally substituted phenyl, an optionally substituted 5 or 6 membered heteroaryl ring, optionally substituted phenoxy, optionally substituted phenylsulphonyl, optionally substituted C 3 .
- R 2 is H, C ⁇ -6 alkyl [optionally substituted with one or more of hydroxy, C ⁇ -6 alkylsulphanyl, C ⁇ -6 alkylsulphinyl, C 1-6 alkylsulphonyl, R 4 , R 4 C ⁇ -6 alkylsulphanyl, R 4 C 1-6 alkylsulphinyl, R 4 C 1-6 alkylsulphonyl], or R 2 is C ⁇ -6 alkoxy [optionally substituted with one or more of C 2-6 alkenyl, C 2-6 alkynyl, R 4 , R 4 C 2-6 alkenyl, R 4 C 2-6 alkynyl, Het and trifluoromethyl], or R 2 is C 2-6 alkenyl, C 2-6 alkynyl, C 1-6 alkoxycarbonyl, carbamoyl, N-(C ⁇ -6 alkyl)carbamoyl, NN-(C 1-6 alkyl) 2 carbamoyl, R
- R 3 is H or Ci- ⁇ alkyl
- (AA 1 ) and (AA 2 ) are independently chosen from Ala, Arg, Cys, Gly, His, De, Leu, Lys, Met, Phe, Ser, Thr, Trp, Tyr, Val,
- Ring A is C 3-12 cycloalkyl
- Ring B is a 5 or 6 membered heteroaryl ring
- Ring C is Het
- V is Ci ⁇ alkyl excluding isopropyl
- the nitrogen of the amino acid may optionally be alkylated with C 1- alkyl
- the phenyl group of Phe(S) and Rings A and B are optionally substituted with one or more of C 1-6 alkyl, halo, trifluoromethyl, hydroxy, trifluoromethoxy, cyano, C 1-6 alkoxy, C ⁇ -6 alkanoyl, C 1-6 alkanoyloxy, amino, C 1-6 alkylamino, NN-(C 1- alkyl) 2 amino, C ⁇ -6 alkanoylamino, nitro, carboxy, carbamoyl, N-(C ⁇ -6 alkyl)carbamoyl, NN-(C ⁇ -6
- R 1 is optionally substituted benzyl where said optional substituents are chosen from one or more of C 1-6 alkyl, halo, trifluoromethyl, hydroxy, trifluoromethoxy, cyano, C 1-6 alkoxy, d- ⁇ alkanoyl, C 1-6 alkanoyloxy, amino, C 1-6 alkylamino, N,N-(C 1-6 alkyl) 2 amino, C 1-6 alkanoylamino, nitro, carboxy, carbamoyl, N-(C 1-6 alkyl)carbamoyl, NN-(C 1- alkyl) 2 carbamoyl, C 1-6 alkoxycarbonyl, mercapto, C 1-6 alkylsulphanyl, ⁇ alkylsulphinyl, C 1-6 alkylsulphonyl, sulphamoyl, N-(C 1-6 alkyl)sulphamoyl and
- R 5 is C 1-6 alkyl (optionally substituted with an optionally substituted phenyl, an optionally substituted 5 or 6 membered heteroaryl ring, optionally substituted phenoxy, optionally substituted phenylsulphonyl, optionally substituted C 3-12 cycloalkyl or Het), C 1-6 alkoxy, optionally substituted phenyl, optionally substituted naphthyl, optionally substituted 5 or 6 membered heteroaryl ring, optionally substituted C 3-12 cycloalkyl, Het, optionally substituted phenylC 1-6 alkoxy where said optional substituents are chosen from one or more of C ⁇ -6 alkyl, halo, trifluoromethyl, hydroxy, trifluoromethoxy, cyano, C 1-6 alkoxy, C ⁇ -6 alkanoyl, C ⁇ -6 alkanoyloxy, amino, Ci-ealkylamino, NN-(C] -6 alkyl) 2
- R 2 is an optionally substituted 5 or 6 membered heteroaryl ring containing a maximum of four heteroatoms said optional substituents being chosen from one or more of C 1-6 alkyl, halo, trifluoromethyl, hydroxy, trifluoromethoxy, cyano, C 1-6 alkoxy, C ⁇ -6 alkanoyl, C ⁇ -6 alkanoyloxy, amino, C 1-6 alkylamino, NN-(C ⁇ -6 alkyl) 2 amino, C 1-6 alkanoylamino, nitro, carboxy, carbamoyl, N-(C 1-6 alkyl)carbamoyl, NN-(C 1-6 alkyl) 2 carbamoyl, C 1-6 alkoxycarbonyl, mercapto, C ⁇ -6 alkylsulphanyl, C 1-6 alkylsulphonyl, sulphamoyl, N-(C 1-6 alkyl)sulphamoyl and NN-(C ⁇
- R 3 is H
- (AA 1 ) and (AA 2 ) are independently chosen from Ala, Arg, Cys, Gly, His, De, Leu, Lys, Met, Phe, Ser, Thr, Trp, Tyr, Nal,
- Ring A is C 3-12 cycloalkyl
- Ring B is a 5 or 6 membered heteroaryl ring
- C is Het
- V is C 1-6 alkyl excluding isopropyl
- the nitrogen of the amino acid may optionally be alkylated with C ⁇ -6 alkyl and the phenyl group of Phe(S) and Rings A and B may be optionally substituted with one or more of C ⁇ -6 alkyl, halo, trifluoromethyl, hydroxy, trifluoromethoxy, cyano, C 1-6 alkoxy, C 1-6 alkanoyl, C 1-6 alkanoyloxy, amino, C 1-6 alkylamino, NN-(C ⁇ . 6 alkyl) 2 amino, C 1-6 alkanoylamino, nitro, carboxy, carbamoyl,
- 'alkyl' includes straight chained and branched structures and ring systems.
- C ⁇ -6 alkyl includes propyl, isopropyl, t-butyl, cyclopropyl and cyclohexyl.
- references to individual alkyl groups such as 'propyl' are specific for the straight chained version only
- references to individual branched chain alkyl groups such as 'isopropyl' are specific for the branched chain version only
- references to individual cycloalkyl groups such as cyclohexyl are specific to the cyclic groups only.
- hydroxyC 1-6 alkyl includes 1-hydroxyethyl and 2-hydroxyethyl.
- Het means, unless otherwise further specified, a fully saturated monocyclic 5 - 8 membered heterocyclic ring, with up to 4 ring heteroatoms. Preferably these ring heteroatoms are selected from nitrogen, oxygen and sulphur. Examples of “Het” include pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidyl, piperazinyl and morpholinyl.
- 6- membered ring that contains some degree of unsaturation, with up to four ring heteroatoms selected from nitrogen, oxygen and sulphur.
- Examples of "5- or 6- membered heteroaryl ring” include thienyl, furyl, imidazolyl, thiazolyl, pyrimidinyl, pyridinyl, pyrrolyl and pyrazolyl.
- Examples of "6 membered heteroaryl ring” include pyrimidinyl, pyridinyl, pyrazinyl and pyridazinyl.
- Examples of “5 membered heteroaryl ring” include thienyl, furyl, imidazolyl, thiazolyl, pyrrolyl and oxadiazolyl.
- Examples of “C ⁇ -6 alkanoyloxy” are acetoxy and propionyloxy.
- Examples of “C ⁇ - alkoxycarbonyl” include methoxycarbonyl, ethoxycarbonyl, n- and t-butoxycarbonyl.
- Examples of “C 1-6 alkoxy” include methoxy, ethoxy and propoxy.
- Examples of “C ⁇ -6 alkanoylamino” include formamido, acetamido and propionylamino.
- Examples of “C 1-6 alkylsulphanyl” include methylthio and ethylthio.
- Examples of “C ⁇ -6 alkylsulphinyl” include methylsulphinyl and ethylsulphinyl.
- C 1- alkylsulphonyl examples include mesyl and ethylsulphonyl.
- C ⁇ -6 alkanoyl examples include acetyl and propionyl.
- C 1-6 alkylamino examples include methylamino and ethylamino.
- N,N-(C 1-6 alkyl) 2 amino examples include N,N-dimethylamino, NN-diethylamino and N-ethyl-N-methylamino.
- N-(C 1-6 alkyl)carbamoylC ⁇ examples include N,N-dimethylamino, NN-diethylamino and N-ethyl-N-methylamino.
- 6 alkyl are 2-(methylamino)carbonylethyl and 3-(ethylamino)carbonylpropyl.
- Examples of "N,N-(C 1-6 alkyl) 2 carbamoylC 1-6 alkyl” are 2-(dimethylamino)carbonylethyl and 3-(N-methyl-N-ethylamino)carbonylpropyl.
- Examples of "C 2-6 alkenyl” are vinyl, allyl and 1-propenyl.
- Examples of “C 2-6 alkynyl” are ethynyl, 1-propynyl and 2-propynyl.
- N-(C ⁇ , 6 alkyl)carbamoyl are N-methylaminocarbonyl and N-ethylaminocarbonyl.
- N,N-(C 1- alkyl) 2 carbamoyl are NN-dimethylaminocarbonyl and N-methyl-N- ethylaminocarbonyl.
- Examples of are N-methylsulphamoyl and N-ethylsulphamoyl.
- Examples of "NN-(C 1-6 alkyl) 2 sulphamoyl” are NN-dimethylsulphamoyl and NN-diethylsulphamoyl.
- R C ⁇ -6 alkylsulphanyl include R 4 methylthio and 2- R 4 ethylthio.
- R 4 C ⁇ -6 alkylsulphinyl include R 4 methylsulphinyl and 2- R 4 ethylsulphinyl.
- R 4 C ⁇ -6 alkylsulphonyl include R 4 mesyl and 2- R 4 ethylsulphonyl.
- R 4 C 2- alkenyl are 2-R 4 vinyl arid 3-R 4 allyl.
- R 4 C 2-6 alkynyl are 2-R 4 ethynyl and 3-R 4 propyn-l-yl.
- N-(R 4 C 1-6 alkyl)carbamoyl are R 4 methylaminocarbonyl and 2-R 4 ethylaminocarbonyl.
- N-(HetC ⁇ -6 alkyl)carbamoyl are morpholinomethylaminocarbonyl and 2- (piperidinoethyl)aminocarbonyl.
- C 3-12 cycloalkyl are cyclopropyl, cyclopentyl and cyclohexyl.
- substituents are chosen from "one or more” groups it is to be understood that this definition includes all substituents being chosen from one of the specified groups or the substituents being chosen from two or more of the specified groups. For example where optional substituents are chosen from one or more halo, C 1-6 alkoxy and
- C] -6 alkyl examples of possible combinations of substituents include 1) a bromo group, 2) two chloro groups, 3) a methoxy, ethoxy and propoxy substitutent, 4) a fluoro and a methoxy group, 5) a methoxy, a methyl and an ethyl group, and 6) a chloro, a methoxy and an ethyl group.
- substituents include 1) a bromo group, 2) two chloro groups, 3) a methoxy, ethoxy and propoxy substitutent, 4) a fluoro and a methoxy group, 5) a methoxy, a methyl and an ethyl group, and 6) a chloro, a methoxy and an ethyl group.
- R 1 is optionally substituted benzyl where said optional substituents are chosen from one or more of C ⁇ -6 alkyl, halo, trifluoromethyl, hydroxy, trifluoromethoxy, cyano, Cj -6 alkoxy, C 1-6 alkanoyl, C ⁇ -6 alkanoyloxy, amino, C 1-6 alkylamino, NN-(C ⁇ - alkyl) 2 amino, C ⁇ -6 alkanoylamino, nitro, carboxy, carbamoyl, N-(C 1-6 alkyl)carbamoyl, NN-(C 1-6 alkyl) 2 carbamoyl, C 1- alkoxycarbonyl, mercapto, Cr ⁇ alkylsulphanyl, C 1- alkylsulphinyl, C 1-6 alkylsulphonyl, sulphamoyl, N-(C 1- alkyl)sulphamoyl and N,N-(C 1-6 alkyl) 2 s
- R 2 is H, C 1-6 alkyl [optionally substituted with one or more of hydroxy, C 1-6 alkylsulphanyl, C 1-6 alkylsulphinyl, C 1- 6alkylsulphonyl, R 4 , R ⁇ alkylsulphanyl, R 4 C 1-6 alkylsulphinyl, R 4 C 1-6 alkylsulphonyl], or R 2 is C 1-6 alkoxy [optionally substituted with one or more of C 2- alkenyl, C 2-6 alkynyl, R 4 , R 4 C 2-6 alkenyl, R 4 C 2-6 alkynyl, Het and trifluoromethyl], or R 2 is C 2- alkenyl, C 2-6 alkynyl, C 1-6 alkoxycarbonyl, carbamoyl, N-(C ⁇ -6 alkyl)carbamoyl, NN-(C 1-6 alkyl) 2 carbamoyl, R 4 , R 4 S, R 4 C
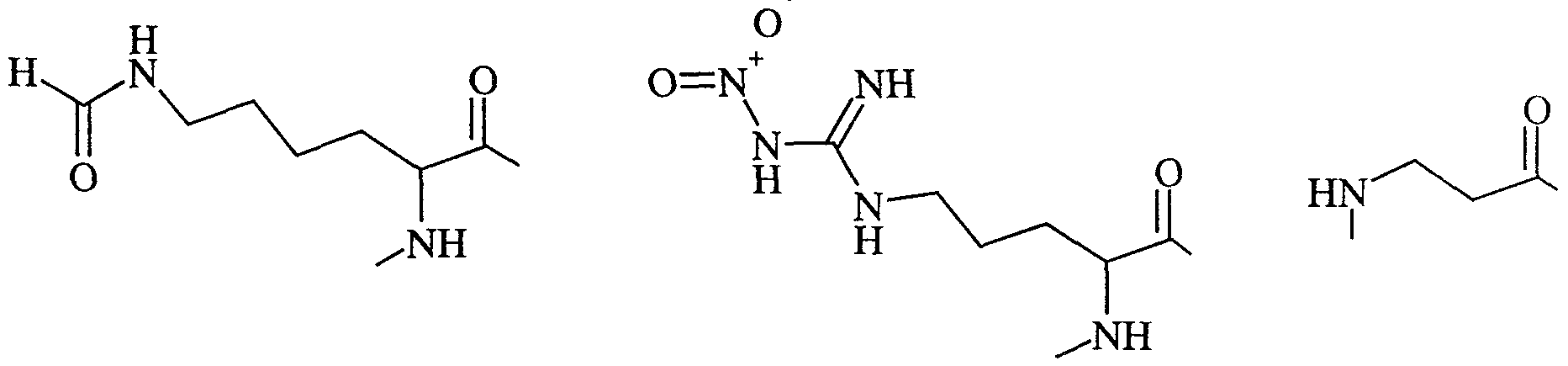
- R 3 is H or C ⁇ . 6 alkyl; and (AA 1 ) and (AA 2 ) are independently chosen from Ala, Arg, Cys, Gly, His, De, Leu, Lys, Met, Phe, Ser, Thr, Trp, Tyr, Nal, Lys(CHO), Arg( ⁇ O 2 ), ⁇ -Ala, Ser(Bzl), Ph-Gly, Nle,
- 6 alkyl and the phenyl group of Phe(S) may be optionally substituted with one or more of C ⁇ -6 alkyl, halo, trifluoromethyl, hydroxy, trifluoromethoxy, cyano, C 1- alkoxy, C 1-6 alkanoyl, amino, C 1-6 alkylamino, N,N-(C 1-6 alkyl) 2 amino, C 1-6 alkanoylamino, nitro, carboxy, carbamoyl, NN-(C 1-6 alkyl) carbamoyl, C ⁇ -6 alkoxycarbonyl, mercapto, C 1-6 alkylsulphanyl, C 1-6 alkylsulphinyl, C 1-6 alkylsulphonyl, sulphamoyl, and NN-(C 1-6 alkyl) 2 sulphamoyl or the phenyl group may be fused to another phenyl group to form a naphthyl group; or a pharmaceutically
- R 1 is optionally substituted benzyl where said optional substituents are chosen from one or more of C 1-6 alkyl, halo, trifluoromethyl, hydroxy, trifluoromethoxy, cyano, C 1-6 alkoxy, C 1-6 alkanoyl, C ]-6 alkanoyloxy, amino, C 1-6 alkylamino, NN-(C 1-6 alkyl) 2 amino, C 1- alkanoylamino, nitro, carboxy, carbamoyl, N-(C 1-6 alkyl)carbamoyl, NN-(C 1-6 alkyl) 2 carbamoyl, C 1- alkoxycarbonyl, mercapto, C ⁇ -6 alkylsulphanyl, C ⁇ - alkylsulphonyl, sulphamoyl, N-(C 1-6 alkyl
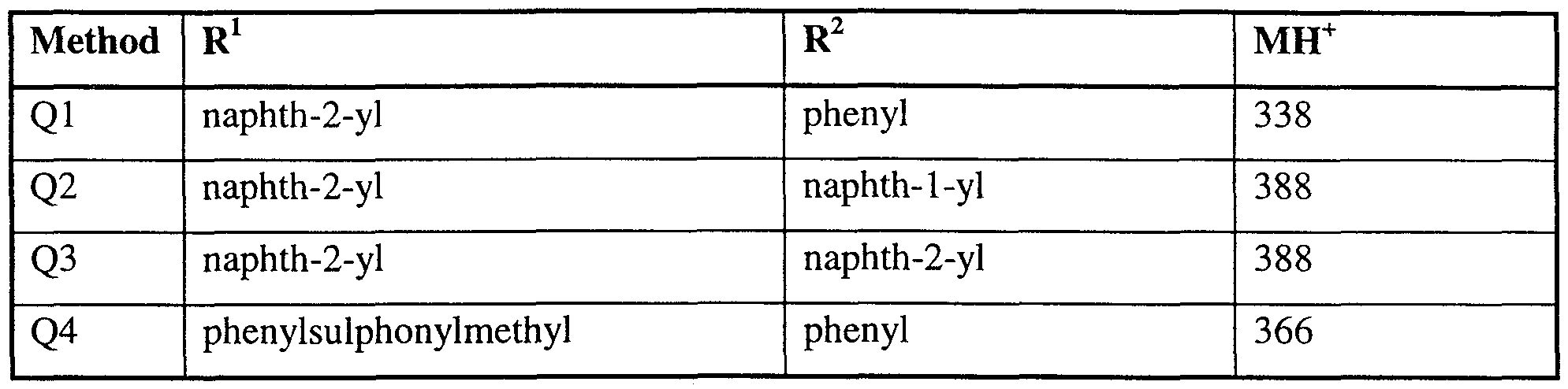
- R 1 is benzyl or a group of formula (II) wherein R 5 is C 1-6 alkyl (optionally substituted with a 6 membered heteroaryl ring, phenyl, phenylsulphonyl or phenoxy optionally substituted with one or more halo), C 1- alkoxy, phenyl (optionally substituted with one or more halo), naphthyl and phenylC 1-6 alkoxy.
- R 1 is benzyl or a group of formula (II) wherein R 5 is C 1-6 alkyl (optionally substituted with a 6 membered heteroaryl ring, phenyl, phenylsulphonyl, C 3-12 cycloalkyl or phenoxy optionally substituted with one or more halo), C 1-6 alkoxy, phenyl (optionally substituted with one or more halo), naphthyl, C 3- ⁇ 2 cycloalkyl, Het, and phenyl ⁇ alkoxy .
- R 1 is benzyl or a group of formula (II) wherein R 5 is methyl, methoxy, ethoxy, propoxy, 'butoxy, phenyl, 2,4-dichlorophenyl, naphthyl, benzyloxy, pyridylmethyl, benzyl, 2,4,6-trichlorophenoxymethyl and phenylsulphonylmethyl.
- R 1 is benzyl or a group of formula (II) wherein R 5 is methyl, methoxy, ethoxy, propoxy, 'butoxy, phenyl, 2,4-dichlorophenyl, naphthyl, benzyloxy, pyridylmethyl, benzyl, 2,4,6-trichlorophenoxymethyl, phenylsulphonylmethyl, morpholino, cyclohexyl, cyclopentyl, cyclohexylmethyl and piperidino.
- R 1 is a group of formula (II) wherein R 5 is methyl, oxy, benzyloxy and pyridylmethyl.
- R 1 is a group of formula (II) wherein R 5 is methyl, 'butoxy, benzyloxy, pyridylmethyl, morpholino, cyclohexyl, cyclopentyl, cyclohexylmethyl and piperidino.
- R 1 is a group of formula (II) wherein R 5 is methyl, 'butoxy, benzyloxy and 4-pyridylmethyl.
- R 1 is a group of formula (II) wherein R 5 is morpholino, cyclohexyl, cyclopentyl, cyclohexylmethyl and piperidino. In one aspect of the invention preferably r is 0.
- r is 1.
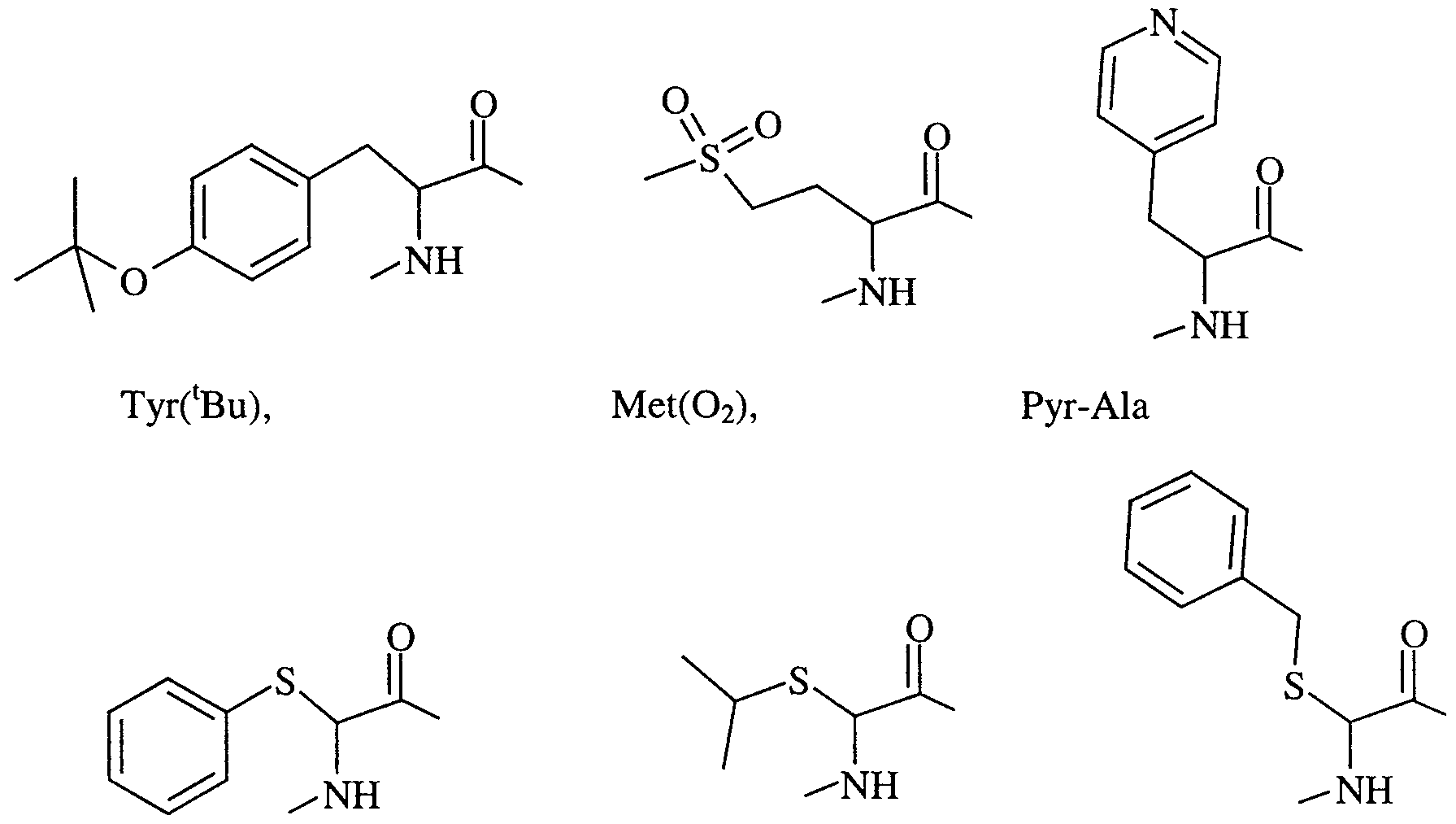
- AA 1 is Leu, Pyr-Ala and Phe wherein the nitrogen of the amino acid is optionally substituted with C 1-6 alkyl.
- AA 1 is Leu and the nitrogen of the amino acid is unsubstituted.
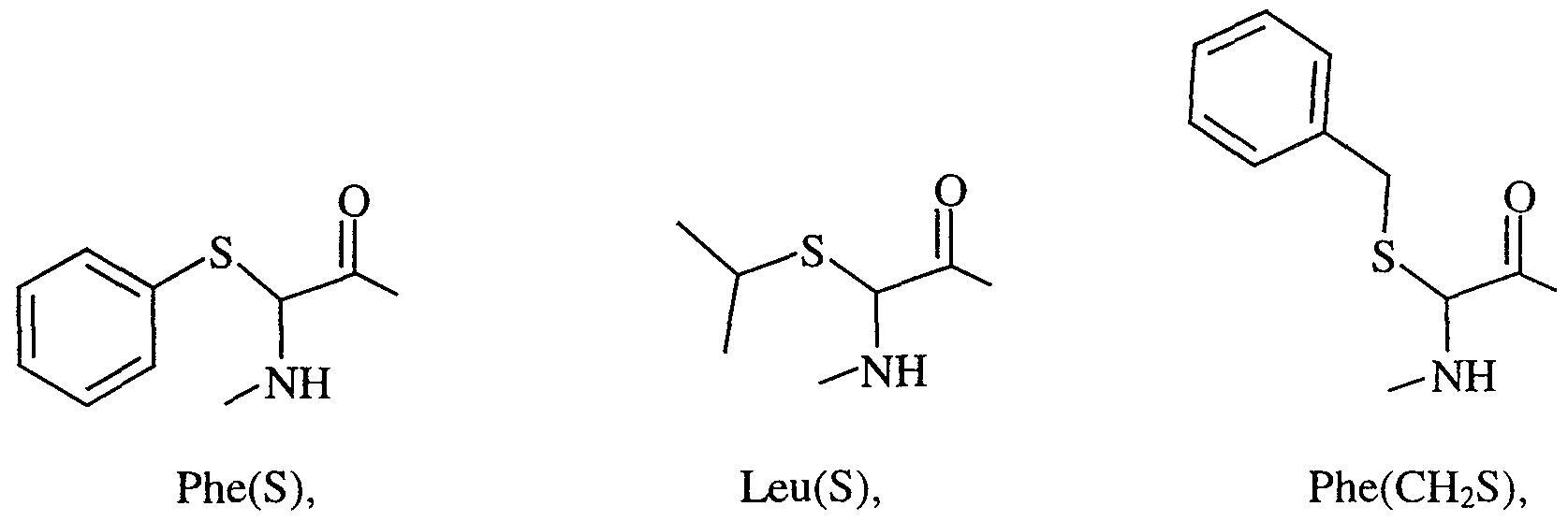
- AA 2 is Phe, Leu, De, Tyr, Tyr('Bu), Val, Cha, Leu(S), Phe(S) and
- Phe(CH 2 S) and the nitrogen of the amino acid is unsubstituted and the phenyl group of Phe(S) is optionally substituted with halo, C ⁇ -6 alkyl or is fused to another phenyl group to form a naphthyl group.
- AA 2 is Tyr, Leu and Phe and the nitrogen of the amino acid is unsubstituted.
- Preferred combinations of r, AA 1 and AA 2 are as follows.
- AA 2 is Phe, Leu, De, Val, Tyr, Tyr('Bu), Leu(S), Phe(S) and Phe(CH 2 S) and the nitrogen of the amino acid is unsubstituted and the phenyl group of Phe(S) is optionally substituted with halo, or is fused to another phenyl group to form a naphthyl group.
- NA 2 is Tyr.
- AA'-AA 2 is Leu-Leu, Pyr- Ala-Leu, Phe-Leu, Leu-Phe, Leu-De, Leu-Val, Leu-Cha and ( ⁇ -Me)Leu-Leu.
- AA*-NA 2 is Leu-Leu and Leu-Phe.
- (AA 1 ) and (AA 2 ) are both independently selected from Phe(S), Leu(S), Phe(CH 2 S), Cy(S)-Gly, Hetar(S)-Gly, alk(S)-Gly and Het(S)- Gly wherein Phe(S) and Rings A and B may be optionally substituted as hereinbefore defined and wherein the phenyl group of Phe(S) may be fused to another phenyl group to form a naphthyl group.
- r is 0 and (AA 2 ) is selected from Phe(S),
- Phe(S) and Rings A and B may be optionally substituted as hereinbefore defined and wherein the phenyl group of Phe(S) may be fused to another phenyl group to form a naphthyl group.
- r is 0 and (AA 2 ) is selected from Phe(S),
- R is C ⁇ _ 6 alkoxy [optionally substituted with one or more of C 2-6 alkenyl, C 2- alkynyl, R , R 4 C 2-6 alkenyl, R 4 C 2-6 alkynyl, Het and trifluoromethyl], or R 2 is C 2-6 alkenyl, C 2-6 alkynyl, carbamoyl, R 4 , R 4 S, R 4 C 1-6 alkylsulphanyl,
- R 4 is an optionally substituted phenyl, or an optionally substituted 5 or 6 membered heteroaryl ring containing a maximum of four heteroatoms said optional substituents being chosen from one or more of C 1-6 alkyl, halo, trifluoromethyl, hydroxy, trifluoromethoxy, cyano, C ⁇ -6 alkoxy,
- N N-(C ] -6 alkyl) 2 sulphamoyl N N-(C ] -6 alkyl) 2 sulphamoyl .
- R 2 is hydrogen, C ⁇ -6 alkyl [optionally substituted with C 1-6 alkylsulphanyl,
- R 2 is hydrogen, methyl, ethyl, propyl, isobutyl, furyl, thienyl, pyrazolyl (optionally substituted with one or more of methyl and bromo), imidazolyl, 1,2,4- triazolyl, phenyl, benzyl, 2-methylthioethyl, methylthio, ethylthio, isopropylthio, mesylethyl, methoxy, ethoxy, isopropoxy and 2-propynyloxy.
- R is furyl, pyrazolyl (optionally substituted with one or more of methyl and bromo), imidazolyl, 1,2,4-triazolyl, benzyl, 2-methylthioethyl, isopropylthio, methoxy, isopropoxy and 2-propynyloxy.
- R is fur-2-yl, pyrazol-1-yl, 3,5-dimethylpyrazol-l-yl, 4-bromo-3,5- dimethylpyrazol-1-yl, imidazol-1-yl, 1,2,4-triazol-l-yl, benzyl, methylthioethyl, isopropylthio, methoxy, isopropoxy and 2-propynyloxy.
- R 2 is an optionally substituted 5 membered heteroaryl ring containing a maximum of four heteroatoms said optional substituents being chosen from one or more of C 1-6 alkyl, halo, trifluoromethyl, hydroxy, trifluoromethoxy, cyano, C 1-6 alkoxy, C ⁇ -6 alkanoyl, C 1-6 alkanoyloxy, amino, C ⁇ -6 alkyl amino, NN-(Ci- alkyl) 2 amino, C ⁇ -6 alkanoylamino, nitro, carboxy, carbamoyl, N-(C )-6 alkyl)carbamoyl, NN-(C ⁇ -6 alkyl) 2 carbamoyl, C 1-6 alkoxycarbonyl, mercapto, C ⁇ -6 alkylsulphanyl, Cj -6 alkylsulphinyl, C 1-6 alkylsulphonyl, sulphamoyl, N-(C
- R 2 is thienyl
- R 3 is hydrogen
- R 1 is a group of formula (II) wherein R 5 is morpholino, cyclohexyl, cyclopentyl, cyclohexylmethyl and piperidino; r is O or 1;
- (AA 1 ) and (AA 2 ) are both independently selected from Phe(S), Leu(S), Phe(CH 2 S), Cy(S)-Gly, Hetar(S)-Gly, alk(S)-Gly and Het(S)-Gly wherein Phe(S) and Rings A and B may be optionally substituted as hereinbefore defined and wherein the phenyl group of Phe(S) may be fused to another phenyl group to form a naphthyl group;
- R 2 is an optionally substituted 5 membered heteroaryl ring containing a maximum of four heteroatoms said optional substituents being chosen from one or more of C ⁇ -6 alkyl, halo, trifluoromethyl, hydroxy, trifluoromethoxy, cyano, C ⁇ -6 alkoxy, C ⁇ -6 alkanoyl, C] -6 alkanoyloxy, amino, C 1-6 alkylamino, NN-(C ⁇ -6 alkyl) 2 amino, C ⁇ -6 alkanoylamino, nitro, carboxy, carbamoyl, N-(C 1- alkyl)carbamoyl, NN-(C ⁇ .
- R 3 is hydrogen; or a pharmaceutically acceptable salt thereof.
- R 1 is benzyl or a group of formula (II) wherein R 5 is C 1-6 alkyl (optionally substituted with a 6 membered heteroaryl ring, phenyl, phenylsulphonyl or phenoxy optionally substituted with one or more halo), C 1-6 alkoxy, phenyl (optionally substituted with one or more halo), naphthyl or phenylC i -6 alkoxy; r is 0 or 1 ;
- AA 1 is Leu, Pyr-Ala or Phe wherein the nitrogen of the amino acid is optionally substituted with C ⁇ -6 alkyl;
- AA 2 is Phe, Leu, De, Tyr, Tyr('Bu), Val, Cha, Leu(S), Phe(S) and Phe(CH 2 S) and the nitrogen of the amino acid is unsubstituted and the phenyl group of Phe(S) is optionally substituted with halo, C 1-6 alkyl or is fused to another phenyl group to form a naphthyl group;
- R 2 is hydrogen, C 1- alkyl [optionally substituted with C ⁇ -6 alkylsulphanyl, C ⁇ -6 alkylsulphonyl or R 4 ], C 1-6 alkoxy [optionally substituted with C 2-6 alkynyl,] or R 4 - wherein R 4 is an optionally substituted phenyl or an optionally substituted 5 membered heteroaryl ring containing a maximum of four heteroatoms said optional substituents being chosen from one or more of C 1-6 alkyl or halo; and
- R 3 is hydrogen; or a pharmaceutically acceptable salt thereof.
- a further preferred class of compounds is that of formula (I) wherein: R 1 is a group of formula (II) wherein R 5 is methyl, 'butoxy, benzyloxy or pyridylmethyl; r is O or 1;
- AA 1 is Leu wherein the nitrogen of the amino acid is unsubstituted
- AA 2 is Tyr, Leu or Phe wherein the nitrogen of the amino acid is unsubstituted;
- R 2 is furyl, pyrazolyl (optionally substituted with one or more methyl or bromo), imidazolyl, 1,2,4-triazolyl, benzyl, methylthioethyl, isopropylthio, methoxy, isopropoxy or propynyloxy; and
- R is hydrogen; or a pharmaceutically acceptable salt thereof.
- the present invention provides a compound of formula (la):
- R is optionally substituted benzyl, optionally substituted phenoxymethyl, optionally substituted phenylsulphonylmethyl, optionally substituted benzyloxy, optionally substituted naphthyl, optionally substituted phenyl or t-butoxy where said optional substituents are chosen from one or more of C ⁇ -6 alkyl, halo, trifluoromethyl, hydroxy, trifluoromethoxy, cyano, C ⁇ -6 alkoxy, C ⁇ -6 alkanoyl, C ⁇ -6 alkanoyloxy, amino, C ⁇ .
- R 6 is hydrogen, optionally substituted phenyl or optionally substituted 5 or 6 membered heteroaryl ring containing a maximum of four heteroatoms; said optional substituents being chosen from one or more of C 1-6 alkyl, halo, trifluoromethyl, hydroxy, trifluoromethoxy, cyano, C ⁇ -6 alkoxy, C ⁇ -6 alkanoyl, C ⁇ -6 alkanoyloxy, amino, C ⁇ -6 alkylamino, NN-(Cj -6 alkyl) 2 amino, C ⁇ - alkanoylamino, nitro, carboxy, carbamoyl, N-(C 1-6 alkyl)carbamoyl, NN-(C ⁇ -6 alkyl) 2 carbamoyl, C ⁇ -6 alkoxycarbonyl, mercapto, C 1-6 alkylsulphanyl, C ⁇ - alkylsulphinyl, C 1-6 alkylsulphonyl, sulphamo
- (AA 3 ) is selected from:
- Ring A is C 3-1 cycloalkyl
- Ring B is a 5 or 6 membered heteroaryl ring
- Ring C is Het
- V is C ⁇ -6 alkyl excluding isopropyl
- the nitrogen of the amino acid may optionally be alkylated with C ⁇ -6 alkyl
- the phenyl group of Phe(S) and Rings A and B may be optionally substituted with one or more of C ⁇ -6 alkyl, halo, trifluoromethyl, hydroxy, trifluoromethoxy, cyano, C ⁇ -6 alkoxy, C 1-6 alkanoyl, C ⁇ -6 alkanoyloxy, amino, C 1- alkylamino, NN-(C] -6 alkyl) amino, Ci- ⁇ alkanoylamino, nitro, carboxy, carbamoyl,
- Het is a fully saturated monocyclic 5 - 8 membered heterocyclic ring, with up to 4 ring heteroatoms; or a pharmaceutically acceptable salt thereof.
- the present invention provides a compound of formula (la):
- R 7 is optionally substituted benzyl, optionally substituted phenoxymethyl, optionally substituted phenylsulphonylmethyl, optionally substituted benzyloxy, optionally substituted naphthyl, optionally substituted phenyl or t-butoxy where said optional substituents are chosen from one or more of C ⁇ - alkyl, halo, trifluoromethyl, hydroxy, trifluoromethoxy, cyano, C ⁇ -6 alkoxy, C 1-6 alkanoyl, amino, C 1-6 alkylamino, NN-(C 1-6 alkyl) 2 amino, C 1-6 alkanoylamino, nitro, carboxy, carbamoyl, N-(C ⁇ -6 alkyl)carbamoyl, NN-(C ⁇ -6 alkyl) 2 carbamoyl, Ci- ⁇ alkoxycarbonyl, mercapto, C ⁇ .
- R 6 is hydrogen or an optionally substituted 5 or 6 membered heteroaryl ring containing a maximum of four heteroatoms said optional substituents being chosen from one or more of C 1-6 alkyl, halo, trifluoromethyl, hydroxy, trifluoromethoxy, cyano, C ⁇ _ 6 alkoxy, C 1-6 alkanoyl, C 1-6 alkanoyloxy, amino, C ⁇ -6 alkylamino, NN-(C 1-6 alkyl) 2 amino, C ⁇ -6 alkanoylamino, nitro, carboxy, carbamoyl, N-(C 1-6 alkyl)carbamoyl, N,N-(C].
- (AA 3 ) is selected from:
- Ring A is C 3-12 cycloalkyl
- Ring B is a 5 or 6 membered heteroaryl ring
- Ring C is Het
- V is C ⁇ -6 alkyl excluding isopropyl
- the nitrogen of the amino acid may optionally be alkylated with C ⁇ -6 alkyl and the phenyl group of Phe(S) and Rings A and B may be optionally substituted with one or more of C 1-6 alkyl, halo, trifluoromethyl, hydroxy, trifluoromethoxy, cyano, C ⁇ -6 alkoxy, C ⁇ -6 alkanoyl, C ⁇ -6 alkanoyloxy, amino, C 1-6 alkylamino, NN-(C ⁇ _ 6 alkyl) 2 amino, C ⁇ alkanoylamino, nitro, carboxy, carbamoyl, N-(C ⁇ _ 6 alkyl)carbamoyl, NN-(C 1-6 alkyl) 2
- the phenyl group of Phe(S) may be fused to another phenyl group to form a naphthyl group and the sulphur moiety in the ⁇ -position of the amino acid (AA) may be optionally oxidised to form an -S(O) 2 - or -S(O)- moiety; or a pharmaceutically acceptable salt thereof.
- the variable Het is a fully saturated monocyclic 5 - 8 membered heterocyclic ring, with up to 4 ring heteroatoms.
- R 7 , AA 3 and R 6 for the compound of formula (la) are as follows.
- the variable R 7 is, for example, benzyl (optionally substituted with halo (such as chloro)), ⁇ -(C ⁇ -4 alkyl)-benzyl (optionally substituted with halo (such as chloro)), ⁇ , ⁇ -di(C ⁇ _ alkyl)-benzyl (optionally substituted with halo (such as chloro)), optionally substituted phenoxy methyl, phenylsulphonylmethyl, benzyloxy, naphthyl or optionally substituted phenyl where said optional substituents are chosen from one or more halo.
- R 7 is benzyl, optionally substituted phenoxymethyl, phenylsulphonylmethyl, benzyloxy, naphthyl or optionally substituted phenyl where said optional substituents are chosen from one or more halo. More preferably R 7 is benzyl, phenoxymethyl optionally substituted with chloro, phenylsulphonylmethyl, benzyloxy, naphthyl or phenyl optionally substituted with chloro.
- R 7 is benzyl, 2,4,6-trichlorophenoxymethyl, phenylsulphonylmethyl, benzyloxy, naphthyl or 2,4-dichlorophenyl.
- R 7 is benzyl, 2,4,6-trichlorophenoxymethyl, phenylsulphonylmethyl, benzyloxy, naphth-2-yl or 2,4-dichlorophenyl.
- (NA 3 ) is Leu(S), Phe(S) optionally substituted with C 1-6 alkyl or halo and wherein the phenyl group of Phe(S) may be fused to another phenyl group to form a naphthyl group or the sulphur moiety in the ⁇ -position of the amino acid ( AA) may be optionally oxidised to form an -S(O) 2 - or Phe(CH 2 S).
- More preferably (AA 3 ) is Leu(S), Phe(S), 4-Cl-Phe(S), 3-Cl-Phe(S), 2-Cl-Phe(S),
- R 6 is hydrogen or a 5 membered heteroaryl ring containing a maximum of four heteroatoms.
- the heteroatoms are, for example, nitrogen, oxygen or sulphur.
- the 5 membered heteroaryl ring is, for example, thienyl, furyl, pyrazolyl, imidazolyl or 1 ,2,4- triazolyl.
- R 6 is hydrogen, optionally substituted phenyl or a 5 membered heteroaryl ring containing a maximum of four heteroatoms.
- the heteroatoms are, for example, nitrogen, oxygen or sulphur.
- the 5 membered heteroaryl ring is, for example, thienyl, furyl, pyrazolyl, imidazolyl or 1,2,4-triazolyl.
- Optional substituents include C 1-4 alkoxy (such as methoxy).
- variable R 6 is, for example, methoxyphenyl.
- R 6 is hydrogen or thienyl. Particularly R 6 is hydrogen or thien-2-yl.
- R 6 is thien-2-yl.
- R 6 is an optionally substituted 5 or 6 membered heteroaryl ring containing a maximum of four heteroatoms wherein said optional substituents are as defined hereinbefore. In another aspect of the invention more preferably R 6 is an optionally substituted 5 membered heteroaryl ring containing a maximum of four heteroatoms wherein said optional substituents are as defined hereinbefore.
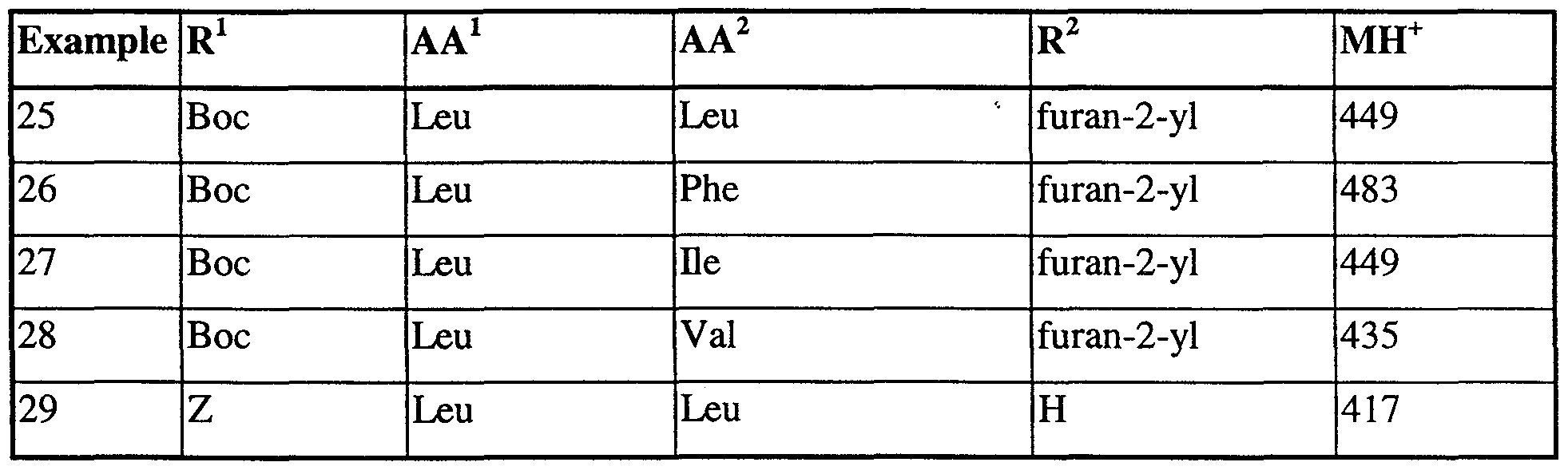
- Preferred compounds are those of Examples 1 - 58 or a pharmaceutically acceptable salt thereof. Especially preferred compounds are those of Examples 8, 13, 15, 17, 19, 25, 30, 31,
- Preferred compounds are those of Examples 59-66 or a pharmaceutically acceptable salt thereof.
- Especially preferred compounds are those of Examples 59-65.
- Preferred compounds are those of Examples 67-74 and 97-113 or a pharmaceutically acceptable salt thereof.
- Suitable pharmaceutically acceptable salts include acid addition salts such as the methanesulphonate, fumarate, hydrochloride, hydrobromide, citrate and maleate salts and salts formed with phosphoric and sulphuric acid.
- suitable salts are base salts such as an alkali metal salt for example a sodium salt, an alkaline earth metal salt for example a calcium or a magnesium salt, an organic amine salt for example a salt with triethylamine, morpholine, N-methylpiperidine, N-ethylpiperidine, procaine, dibenzylamine,
- NN-dibenzylethylamine or an amino acid for example a lysine salt There may be more than one cation or anion depending on the number of charged functions and the valency of the cations or anions.
- a preferred pharmaceutically acceptable salt is a sodium salt.
- Some compounds of formula (I) may possess chiral centres. It is to be understood that the invention encompasses all such optical isomers and diasteroisomers of compounds of formula (I) which possess cysteine protease inhibitory activity.
- the invention further relates to all tautomeric forms of the compounds of formula (I). It is also to be understood that certain compounds of the formula (I) can exist in solvated as well as unsolvated forms such as, for example, hydrated forms. It is to be understood that the invention encompasses all such solvated forms.
- Another aspect of the present invention provides a process for preparing a compound of formula (I) or a pharmaceutically acceptable salt thereof. According to this aspect of the invention there is provided a process (in which variable groups are as defined for formula (I) unless otherwise stated) which comprises:
- a suitable reactive derivative of an acid of the formula (III) is, for example, an acyl halide, for example an acyl chloride formed by the reaction of the acid and an inorganic acid chloride, for example thionyl chloride; a mixed anhydride, for example an anhydride formed by the reaction of the acid and a chloroformate such as isobutyl chloroformate; an active ester, for example an ester formed by the reaction of the acid and a phenol such as pentafluorophenol, an ester such as pentafluorophenyl trifluoroacetate, an alcohol such as 1 - hydroxybenzotriazole or a uronium salt such as 2-(l-benzotriazolyl)-l,l,3,3- tetramethyluronium hexafluorophosphate(V); an acyl azide, for example an azide formed by the reaction of the acid and an azide such as diphenylphosphoryl azide; an acyl azi
- the reaction is preferably carried out in the presence of a suitable base such as, for example, an alkali or alkaline earth metal carbonate, alkoxide or hydroxide, for example sodium carbonate or potassium carbonate, or, for example, an organic amine base such as, for example, pyridine, 2,6-lutidine, collidine, 4-dimethylaminopyridine, triethylamine, morpholine or diazabicyclo-[5.4.0]undec-7-ene.
- a suitable base such as, for example, an alkali or alkaline earth metal carbonate, alkoxide or hydroxide, for example sodium carbonate or potassium carbonate, or, for example, an organic amine base such as, for example, pyridine, 2,6-lutidine, collidine, 4-dimethylaminopyridine, triethylamine, morpholine or diazabicyclo-[5.4.0]undec-7-ene.
- the reaction is also preferably carried out in a suitable inert solvent or diluent, for example methylene chloride, acetonitrile, tetrahydrofuran, 1,2-dimethoxyethane, NN-dimethylformamide, NN-dimethylacetamide, N-methylpyrrolidin- 2-one or dimethylsulphoxide, and at a temperature in the range, for example, -78° to 150°C, conveniently at or near ambient temperature.
- a suitable inert solvent or diluent for example methylene chloride, acetonitrile, tetrahydrofuran, 1,2-dimethoxyethane, NN-dimethylformamide, NN-dimethylacetamide, N-methylpyrrolidin- 2-one or dimethylsulphoxide
- a dehydration reaction may conventionally be carried out by reaction with a reagent such as trifluoroacetic anhydride.
- the reaction can conveniently be conducted in the presence of a suitable base as defined hereinbefore such as, for example, triethylamine.
- the reaction is also preferably carried out in a suitable inert solvent or diluent, as defined hereinbefore such as dichloromethane and at a temperature in the range, for example, -10°C to reflux conveniently 10°C to reflux.
- reaction can conveniently be conducted under standard coupling conditions, such as those described in a) above.
- reaction can conveniently be conducted under standard coupling conditions, such as those described in a) above.
- a suitable displaceable group L is, for example, a halogeno or sulphonyloxy group, for example a chloro, bromo, methanesulphonyloxy or toluene -4-sulphonyloxy group.
- This reaction may be carried out under standard conditions such as, for example, those described in Synthesis 1993, 12, 1243-6; or ii) by reaction with an aldehyde of formula (XII):
- Another aspect of the present invention provides a process for preparing a compound of formula (la) or a pharmaceutically acceptable salt thereof. According to this aspect of the invention there is provided a process (in which variable groups are as defined for formula (la) unless otherwise stated) which comprises: a) coupling an acid of formula (Ilia):
- the necessary starting materials for the procedures described above may be made by procedures which are selected from standard organic chemical techniques, techniques which are analogous to the synthesis of known, structurally similar compounds, by techniques which are analogous to the above described procedures or by techniques which are analogous to the procedures described in the examples.
- certain of the optional substituents on a phenyl or naphthyl or a heteroaryl ring in the compounds of the present invention may be introduced by standard aromatic substitution reactions or generated by conventional functional group modifications either prior to or immediately following the processes mentioned above, and as such are included in the process aspect of the invention.
- Such reactions and modifications include, for example, introduction of a substituent by means of an aromatic substitution reaction, reduction of substituents, alkylation of substituents and oxidation of substituents.
- the reagents and reaction conditions for such procedures are well known in the chemical art.
- aromatic substitution reactions include the introduction of a nitro group using concentrated nitric acid, the introduction of an acyl group using, for example, an acyl halide and a Lewis acid (such as aluminium trichloride) under Friedel Crafts conditions; the introduction of an alkyl group using an alkyl halide and Lewis acid (such as aluminium trichloride) under Friedel Crafts conditions; and the introduction of a halogeno group.
- modifications include the reduction of a nitro group to an amino group by, for example, catalytic hydrogenation with a nickel catalyst or treatment with iron in the presence of hydrochloric acid with heating; oxidation of alkylthio to alkylsulphinyl or alkylsulphonyl.
- a suitable protecting group for an amino or alkylamino group is, for example, an acyl group, for example an alkanoyl group such as acetyl, an alkoxycarbonyl group, for example a methoxycarbonyl, ethoxycarbonyl or t-butoxycarbonyl group, an arylmethoxycarbonyl group, for example benzyloxycarbonyl, or an aroyl group, for example benzoyl.
- the deprotection conditions for the above protecting groups necessarily vary with the choice of protecting group.
- an acyl group such as an alkanoyl or alkoxycarbonyl group or an aroyl group may be removed for example, by hydrolysis with a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- an acyl group such as a t-butoxycarbonyl group may be removed, for example, by treatment with a suitable acid as hydrochloric, sulphuric or phosphoric acid or trifluoroacetic acid and an arylmethoxycarbonyl group such as a benzyloxycarbonyl group may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon, or by treatment with a Lewis acid for example boron tris(trifluoroacetate).
- a suitable alternative protecting group for a primary amino group is, for example, a phthaloyl group which may be removed by treatment with an alkylamine, for example dimethylaminopropylamine, or with hydrazine.
- a suitable protecting group for a hydroxy group is, for example, an acyl group, for example an alkanoyl group such as acetyl, an aroyl group, for example benzoyl, or an arylmethyl group, for example benzyl.
- the deprotection conditions for the above protecting groups will necessarily vary with the choice of protecting group.
- an acyl group such as an alkanoyl or an aroyl group may be removed, for example, by hydrolysis with a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- an arylmethyl group such as a benzyl group may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon.
- a suitable protecting group for a carboxy group is, for example, an esterifying group, for example a methyl or an ethyl group which may be removed, for example, by hydrolysis with a base such as sodium hydroxide, or for example a t-butyl group which may be removed, for example, by treatment with an acid, for example an organic acid such as trifluoroacetic acid, or for example a benzyl group which may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon.
- the protecting groups may be removed at any convenient stage in the synthesis using conventional techniques well known in the chemical art.
- a compound of the formula (I) or (la), or a pharmaceutically acceptable salt thereof for use in a method of treatment of the human or animal body by therapy.
- a compound of the formula (I) or (la) or a pharmaceutically acceptable salt thereof for the therapeutic treatment of mammals including humans, in particular in the inhibition of a cysteine protease, it is normally formulated in accordance with standard pharmaceutical practice as a pharmaceutical composition.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I) or (la), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable diluent or carrier.
- the present invention provides a compound of formula (I) or (la), or a pharmaceutically acceptable salt thereof, for use as a medicament.
- the present invention provides the use of a compound of formula (I) or (la), or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for use in the inhibition of a cysteine protease in a warm blooded animal, such as man.
- the present invention provides the use of a compound of formula (I) or (la), or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for use in the treatment of chronic obstructive pulmonary disease in a warm blooded animal, such as man.
- a method of treating a Cathepsin L or Cathepsin S mediated disease state in mammals which comprises administering to a mammal in need of such treatment an effective amount of a compound of formula (I) or (la), or a pharmaceutically acceptable salt thereof.
- a method for producing inhibition of a cysteine protease in a warm blooded animal, such as man, in need of such treatment which comprises administering to said animal an effective amount of a compound of the present invention, or a pharmaceutically acceptable salt thereof.
- the invention provides the use of a compound of the formula (I) or (la) of the present invention, or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for use in the inhibition of Cathepsin S in a warm blooded animal, such as man.
- compositions of this invention may be administered in standard manner for the disease condition that it is desired to treat, for example by oral, rectal or parenteral administration.
- the compounds of this invention may be formulated by means known in the art into the form of, for example, tablets, capsules, aqueous or oily solutions or suspensions, (lipid) emulsions, dispersible powders, suppositories, ointments, creams, drops and sterile injectable aqueous or oily solutions or suspensions.
- a suitable pharmaceutical composition of this invention is one suitable for oral administration in unit dosage form, for example a tablet or capsule which contains between 100 mg and 1 g of the compound of this invention.
- composition of the invention is one suitable for intravenous, subcutaneous or intramuscular injection.
- Each patient may receive, for example, an intravenous, subcutaneous or intramuscular dose of 1 mgkg "1 to 100 mgkg "1 of the compound, preferably in the range of 5 mgkg "1 to 20 mgkg “1 of this invention, the composition being administered 1 to 4 times per day.
- the intravenous, subcutaneous and intramuscular dose may be given by means of a bolus injection.
- the intravenous dose may be given by continuous infusion over a period of time.
- each patient will receive a daily oral dose which is approximately equivalent to the daily parenteral dose, the composition being administered 1 to 4 times per day.
- Buffers such as polyethylene glycol, polypropylene glycol, glycerol or ethanol or complexing agents such as hydroxy-propyl ⁇ cyclodextrin may be used to aid formulation.
- the above formulations may be obtained by conventional procedures well known in the pharmaceutical art.
- the tablets (a)-(c) may be enteric coated by conventional means, for example to provide a coating of cellulose acetate phthalate.
- the pharmaceutically-acceptable compounds of the present invention are useful in the inhibition of Cathepsin L and Cathepsin S, having a good activity in vitro against human Cathepsin L, human Cathepsin S and rabbit Cathepsin L.
- Recombinant human Cathepsin L was cloned and expressed in E Coli and purified using the method as described by Zeneca Limited, GB 2 306 961 A (published 14.05.1997).
- Rabbit Cathepsin L was purified from rabbit liver as described by Maciewicz R. A. and Etherington D. J. (Biochem. J. (1988) 256, 433-440) except the liver homogenate supernatant was concentrated by fractionation with (NH ) 2 SO 4 (20-80% saturation), and the pellet taken up and dialysed against 20mM NaAcetate pH 5.5, lmM ethylenediaminetetraacetic acid (EDTA).
- Cathepsin L activity was measured based on the method of Barrett and Kirschke (1981 Methods in Enzymology, 80, 535-561), using the fluorogenic substrates NCBz-Phe-Arg- NHMec. Inhibitors were identified by their ability to decrease the generation of the fluorescent leaving group (NHMec). Briefly the assay was as follows: rHuman Cathepsin L or rabbit Cathepsin L (0.025 pmoles) was pre-incubated with or without test compound in 0.1M sodium acetate buffer pH4.5, lOmM cysteine, 0.1% Brij 35 at 25°C for 15 minutes in a solid black 96 well plate.
- Synthetic substrate 20 ⁇ M NCBz-Phe-Arg- NHMec, was added and the mixture incubated at 37°C for 30 minutes. The reaction was stopped by the addition of 0.1 M sodium chloroacetate pH 4.3. Fluorescence was determined using a Fluoroskan II plate reader; excitation 355nm, emission " 460nm. Compound potency was determined from the raw fluorescence data by calculating the IC 50 against each enzyme using a PC graph drawing software package.
- Recombinant human Cathepsin S was cloned and expressed in Baculovirus, by the following method.
- the cDNA sequence for human Cathepsin S is available in the EMBL database Accession Number M90696. This database sequence was used to prepare, by PCR on mRNA from human tissues, a recombinant plasmid carrying an insert with a DNA sequence identical to that of Cathepsin S in the EMBL database (Ace No M90696).
- the techniques for mRNA isolation, PCR and cloning are standard techniques known by those skilled in the art. Sequence determination of the recombinant insert was carried out using established DNA sequencing techniques. The PCR was done so as to introduce an EcoRI cloning site 5' of the 'ATG' of
- Procathepsin S was found in the insect cell medium and acid activated.
- the medium was mixed with an equal volume of lOOmM Sodium Acetate buffer pH 4.5, 5mM dithiothreitol (DTT) and 5mM EDTA and incubated for one hour at 37°C method of Maubach et al (Eur. J. Biochem., 250, 745-750, 1997).
- Method 2
- the pH of insect cell medium (10ml) containing procathepsin S was adjusted to 4.5 with glacial acetic acid and DTT and EDTA added to 5mM. The sample was then incubated at 37°C for 150min to enable conversion to the active enzyme. Ammonium sulphate was then added to 80% saturation and a pellet obtained by centrifugation. This pellet was redissolved in 2ml buffer A (lOOmM Tris, 500mM NaCl, ImM EDTA, pH7.5) and mixed in a batchwise fashion with lOO ⁇ l thiopropyl-Sepharose for 15min at 4°C. The non bound fraction was removed by a brief centrifugation and the gel washed with 2x1 ml buffer A. Cathepsin S was then eluted by batch mixing with 0.4ml 20mM DTT in buffer A for 15min at 4°C.
- Cathepsin S activity was measured based on the method of Maubach et al (Eur. J. Biochem., 250, 745-750, 1997), using the fluorogenic substrate Z-Val-Val-Arg-NHMec. Inhibitors were identified by their ability to decrease the generation of the fluorescent leaving group (NHMec). Briefly the assay was as follows: rHuman Cathepsin S (1.5 nmoles) was pre-incubated with or without compounds in 50mM Potassium phosphate buffer pH 6.0-6.2, 20mM Na 2 EDTA, 0.1% Brij at 25°C for 5 minutes in a solid black 96 well plate.
- Synthetic substrate 20 ⁇ M Z-Val-Val-Arg-NHMec
- 20 ⁇ M Z-Val-Val-Arg-NHMec was added and the mixture incubated at 30°C for 20 minutes.
- the reaction was stopped by the addition of 0.1M sodium chloroacetate pH 4.3. Fluorescence was determined using a
- Fluoroskan H plate reader excitation 355nm, emission 460nm.
- Compound potency was determined from the raw fluorescence data by calculating the IC 50 against Cathepsin S using a PC graph drawing software package.
- NMR data is in the form of delta values for major diagnostic protons, given in parts per million (ppm) relative to tetramethylsilane (TMS) as an internal standard, determined at 250 MHz using perdeuterio dimethyl sulphoxide (DMSO- ⁇ 6 ) as the solvent unless otherwise stated;
- melting points are uncorrected and (dec) indicates decomposition; the melting points given are those obtained for the materials prepared as described; polymorphism may result in isolation of materials with different melting points in some preparations; and (xii) Z refers to benzyloxycarbonyl and Boc refers to tert-butoxycarbonyl.
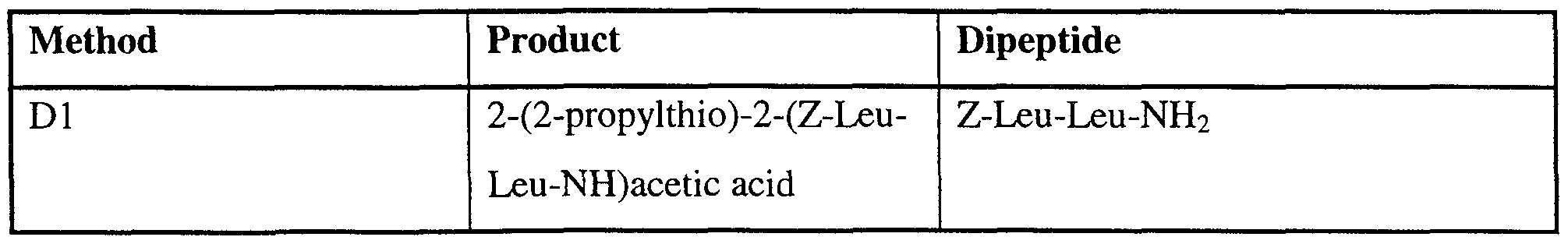
- Trifluoroacetic anhydride (0.28 ml) was added dropwise to a mixture of Z-Leu-Leu- Phe-NH 2 (0.9 g) and pyridine (10 ml) which was stirred under argon at -10°C. The mixture was allowed to warm to room temperature over 1 hour, diluted with water and extracted with ethyl acetate. The extract was washed successively with IM hydrochloric acid and brine, dried and evaporated to dryness and the residue was recrystallized from ethyl acetate/hexane to give (2S)-2-(Z-Leu-Leu-NH)-3-phenylpropionitrile (0.57 g).
- Phosphoryl chloride (0.07 ml) was added dropwise to stirred, ice-cooled NN- dimethylformamide (2 ml) and the resulting solution added to a stirred ice cooled solution of Z-Phe-Leu-Met- ⁇ H 2 (163 mg) in NN-dimethylformamide (2 ml) under an atmosphere of argon. The mixture was stirred for 0.5 hours then poured into ice-water and extracted with ethyl acetate. The extract was washed with water, dried and evaporated to dryness.
- Example 20 The process described in Example 20 was repeated using (2S)-2-[(N-phenylacetyl-N- methyl-Leu)-Leu-Met-NH 2 instead of Z-Phe-Leu-Met-NH 2 as starting material to give (2S)-2- [(N-phenylacetyl-N-methyl-Leu)-Leu-NH]-4-methylthiobutyronitrile.
- Example 20 The process described in Example 20 was repeated using (2S)-2-[(N-benzoyl-N- methyl-Leu)-Leu-Met-NH instead of Z-Phe-Leu-Met-NH 2 as starting material to give (2S)-2- [(N-benzoyl-N-methyl-Leu)-Leu-NH]-4-methylthiobutyronitrile.
- (2RS)-2-(Z-Leu-Leu-NH)-2-phenylacetonitrile A mixture of (2RS)-2-(BOC-LeuNH)-2-phenylacetonitrile (0.69 g), dichloromethane ( 10 ml) and NN-diisopropylethylamine (0.5 ml) was stirred under an argon atmosphere and iodotrimethylsilane (0.36 g) was added dropwise. The mixture was stirred for 1 hour and then additional iodotrimethylsilane (0.36 g) was added and the mixture was stirred for a further hour. N-mefhylmorpholine (0.5 ml) was added followed by methanol (0.5 ml) and then the solution was evaporated to dryness.
- N-bromosuccinimide (133 mg) was added to a stirred solution of (2RS)-2-(Z-Leu-Leu- NH)-2-(2-propylthio)acetonitrile (245 mg) in methanol (10 ml), the mixture was allowed to warm to room temperature and it was then stirred at room temperature for 1 hour. The mixture was evaporated to dryness and the residue was partitioned between ethyl acetate and water.
- Trimethylsilyl iodide (0.7 ml) was added to a solution of Boc-Leu-Leu-(2-furyl)- acetonitrile (1.68 g) in chloroform (50 ml) at 0°C. The mixture was stirred at 0°C for 15 minutes and the solvent was removed under reduced pressure. The residue was dissolved in pyridine (20 ml), the solution was cooled to 0°C and acetic anhydride (20 ml) was added and the reaction mixture was stirred at ambient temperature for 14 hours.
- Trifluoroacetic anhydride (0.84 g) was added dropwise to a mixture of BOC-Leu-Leu- NH 2 (1 g) and pyridine (10 ml) which was stirred under argon at -10°C. The mixture was allowed to warm to room temperature over lhour, diluted with water and extracted with diethyl ether. The extract was washed successively with IM hydrochloric acid and brine, dried and evaporated to dryness. The residue was recrystallized from ether/hexane to give (2S)-2- (BOC-Leu-NH)isovaleronitrile (0.66 g). Mp 117-118°C; m/z 326 (MH) + .
- Example 42 The process described in Example 42 was repeated using BOC-Leu-Met-NH 2 instead of BOC-Leu-Leu-NH 2 to give (2S)-2-(BOC-Leu-NH)-4-methylthiobutyronitrile. Mp 69-71°C; m/z 344 (MH) + .
- the ethyl acetate layer was separated and washed with 2M hydrochloric acid, brine, saturated aqueous sodium bicarbonate and dried and evaporated to dryness.
- the residue was purified by flash chromatography on silica (Merck, ART 9385) using increasingly polar mixtures of ethyl acetate and hexane as eluent followed by recrystallisation from a mixture of ethyl acetate and hexane to give 2-(2- benzyloxycarbonylamino-2-phenylthioacetamido)-2-(2-thienyl)-acetonitrile (4.53 g).
- reaction mixture was evaporated to dryness (high vac) and the residue was suspended in saturated aqueous sodium bicarbonate solution and extracted with ethyl acetate (2x50 ml). The combined ethyl acetate extracts were washed successively with 10% citric acid and brine and dried. The residue obtained on removal of the solvent was chromatographed on a Bond-elut column eluting with a mixture of ethyl acetate and dichloromethane (1/1 v/v) to give the title compound (0.159 g) as a 1/1 mixture of diastereoisomers.
- Carbonyl diimidazole (0.538g) was added to a solution of N-[(2-phenylacetylamino-2- phenylthio)acetic acid (lg) in THF (25 ml) and the mixture was stirred at 20 °C for 20 hours.
- 2-(2-Thienyl)-aminoacetonitrile (Method Al) (0.579 g) and triethylamine (0.664 g) were added and the mixture was stirred at 20 °C for 20 hours.
- Diastereoisomer 1 Faster running fraction: Mp 172 °C; m/z 422 (M+H) + ; NMR: 9.9 (d, IH), 8.95 (d, IH), 7.65 (m, IH), 7.2 (m, 1 IH), 6.45 (d, IH), 5.8 (d, IH), 3.5 (m, 2H).
- Carbonyl diimidazole (0.538g) was added to a solution of N-[(2-phenylacetylamino-2- phenylthio)acetic acid (Method O) (lg) in THF (25 ml) and the mixture was stirred at 20 °C for 20 hours.
- 2-(2-Thienyl)-aminoacetonitrile (Method Al) (0.579 g) and triethylamine (0.664 g) were added and the mixture was stirred at 20 °C for 20 hours.
- Diastereoisomer 1 Faster running fraction: Mp 172 °C; m/z 422 (M+H) + ; NMR: 9.9 (d, IH), 8.95 (d, IH), 7.65 (m, IH), 7.2 (m, 1 IH), 6.45 (d, IH), 5.8 (d, IH), 3.5 (m, 2H).
- Diastereoisomer 2 ; Slower running fraction: Mp 159 °C; m/z 422 (M+H) + ; NMR: 9.8 (d, IH), 8.95 (d, IH), 7.65 (m, IH), 7.2 (m, 1 IH), 6.45 (d, IH), 5.8 (d, IH), 3.5 (m, 2H).
- Diastereoisomer 1 Diastereoisomer 1 ; Faster running fraction.
- the ethyl acetate layer was separated and washed with 2M hydrochloric acid, brine, saturated aqueous sodium bicarbonate and dried and evaporated to dryness.
- the residue was purified by flash chromatography on silica (Merck, ART 9385) using increasingly polar mixtures of ethyl acetate and hexane as eluent followed by recrystallisation from a mixture of ethyl acetate and hexane to give 2-(2-benzyloxycarbonylamino-2-phenylthioacetamido)-2-(2-thienyl)- acetonitrile (4.53 g).
- N-[(2-Phenylacetylamino-2-(4-fluorophenylsulphonyl ⁇ )acetyl1 aminoacetonitrile m-Cloroperbenzoic acid (578 mg) was added to a suspension of N-[(2- Phenylacetylamino-2- ⁇ 4-fluorophenylthio ⁇ )acetyl] aminoacetonitrile (Example 69) (300 mg) in dichloromethane (30 ml) and the mixture was stirred at room temperature for 3 hours then washed successively with aqueous sodium bicarbonate (3 x 10 ml), aqueous sodium thiosulphate (1 x 10 ml) and the organic layer was collected and dried.
- Ammonium chloride (25 g) was added to a solution of 2-furfuraldehyde (25 g) in diethyl ether (250 ml). A solution of sodium cyanide (17 g) in water (80 ml) was added over 20 minutes. The reaction mixture was stirred at ambient temperature for 14 hours , the aqueous layer was removed and the organic layer was washed twice with saturated aqueous sodium hydrogen carbonate solution (100 ml each time), dried and evaporated to dryness. The residue was dissolved in diethyl ether (250 ml) and cooled to 0 °C. Hydrogen chloride gas was bubbled through the solution keeping the temperature below 10 °C.
- Lithium hydroxide solution (5.9 g in 40 ml water) was added to a solution of N-(N- mo ⁇ holinocarbonyl)-(L)-leucine methyl ester (Method N) (7.3 g) in THF (40 ml) and the mixture was stirred for 20 hours. The residue obtained on removal of the solvent was suspended in water (150 ml) and washed with ethyl acetate (50 ml). The aqueous layer was acidified to pH 1 with 2M HCl and extracted with ethyl acetate (3x70 ml).
- Triethylamine (8.4 ml) was added dropwise to L-leucine methyl ester hydrochloride (5 g) in dichloromethane (50 ml) at 0 °C followed by a solution of 4-mo ⁇ holine carbonyl chloride (5 g) in dichloromethane (10 ml) and the mixture was stirred at ambient temperature for 20 hours.
- the reaction mixture was diluted with dichloromethane (100 ml) and washed with water (100 ml). The organic layer was collected and washed with 2M HCl (50 ml), brine
Abstract
Description










































































Claims










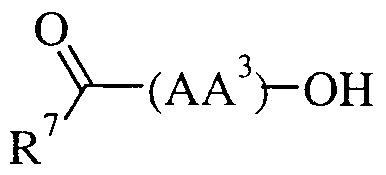














Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP00903845A EP1155011A1 (en) | 1999-02-20 | 2000-02-16 | Di- and tripeptide nitrile derivatives as inhibitors of cathepsin l and cathepsin s |
| AU25600/00A AU2560000A (en) | 1999-02-20 | 2000-02-16 | Di- and tripeptide nitrile derivatives as inhibitors of cathepsin l and cathepsin |
| JP2000599748A JP2002537294A (en) | 1999-02-20 | 2000-02-16 | Di- and tripeptide nitrile derivatives as inhibitors of cathepsin L and cathepsin S |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB9903853.1 | 1999-02-20 | ||
| GBGB9903853.1A GB9903853D0 (en) | 1999-02-20 | 1999-02-20 | Chemical compounds |
| GBGB9916099.6A GB9916099D0 (en) | 1999-07-10 | 1999-07-10 | Chemical compounds |
| GB9916099.6 | 1999-07-10 | ||
| GBGB9917171.2A GB9917171D0 (en) | 1999-07-23 | 1999-07-23 | Chemical compounds |
| GB9917171.2 | 1999-07-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2000049008A1 true WO2000049008A1 (en) | 2000-08-24 |
Family
ID=27269652
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2000/000529 WO2000049008A1 (en) | 1999-02-20 | 2000-02-16 | Di- and tripeptide nitrile derivatives as inhibitors of cathepsin l and cathepsin s |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP1155011A1 (en) |
| JP (1) | JP2002537294A (en) |
| AU (1) | AU2560000A (en) |
| WO (1) | WO2000049008A1 (en) |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002020002A2 (en) * | 2000-09-06 | 2002-03-14 | Ortho Mcneil Pharmaceutical, Inc. | A method for treating allergies |
| US6455502B1 (en) | 1999-03-15 | 2002-09-24 | Axys Pharmaceuticals, Inc. | Compounds and compositions as protease inhibitors |
| US6525036B2 (en) | 2000-01-06 | 2003-02-25 | Merck & Co., Inc. | Compounds and compositions as protease inhibitors |
| US6635633B2 (en) | 2000-08-14 | 2003-10-21 | Ortho-Pharmaceutical, Inc. | Substituted pyrazoles |
| US6642239B2 (en) | 2000-02-10 | 2003-11-04 | Novartis Ag | Dipeptide nitrile cathepsin K inhibitors |
| WO2004052921A1 (en) * | 2002-12-05 | 2004-06-24 | Axys Pharmaceuticals, Inc. | Cyanomethyl derivatives as cysteine protease inhibitors |
| EP1452522A2 (en) * | 1999-03-15 | 2004-09-01 | Axys Pharmaceuticals, Inc. | Novel compounds and compositions as protease inhibitors |
| US6953793B2 (en) | 2000-08-14 | 2005-10-11 | Ortho-Mcneil Pharmaceutical, Inc. | Substituted pyrazoles |
| US6977256B2 (en) | 2001-11-14 | 2005-12-20 | Aventis Pharmaceuticals Inc. | Compounds and compositions as cathepsin S inhibitors |
| US7012075B2 (en) | 2001-03-02 | 2006-03-14 | Merck & Co., Inc. | Cathepsin cysteine protease inhibitors |
| US7030116B2 (en) | 2000-12-22 | 2006-04-18 | Aventis Pharmaceuticals Inc. | Compounds and compositions as cathepsin inhibitors |
| US7064123B1 (en) | 2000-12-22 | 2006-06-20 | Aventis Pharmaceuticals Inc. | Compounds and compositions as cathepsin inhibitors |
| US7196099B2 (en) | 2001-09-14 | 2007-03-27 | Aventis Pharmaceuticals Inc. | Compounds and compositions as cathepsin inhibitors |
| US7279478B2 (en) | 2002-09-04 | 2007-10-09 | Merck Frosst Canada & Co. | Cathepsin cysteine protease inhibitors |
| US7309703B2 (en) | 2000-08-14 | 2007-12-18 | Ortho Mcneil Pharmaceutical, Inc. | Substituted pyrazoles |
| US7332494B2 (en) | 2000-08-14 | 2008-02-19 | Janssen Pharmaceutica, N.V. | Method for treating allergies using substituted pyrazoles |
| EP2198879A1 (en) | 2008-12-11 | 2010-06-23 | Institut Curie | CD74 modulator agent for regulating dendritic cell migration and device for studying the motility capacity of a cell |
| EP2251007A2 (en) | 2002-09-24 | 2010-11-17 | Novartis AG | Sphingosine-1-phosphate (S1P) receptor agonists for use in the treatment of demyelinating diseases |
| EP2719700A1 (en) | 2008-01-09 | 2014-04-16 | Amura Therapeutics Limited | Tetrahydrofuro(3,2-b)pyrrol-3-one derivatives as inhibitors of cysteine proteinases |
| WO2016007638A1 (en) | 2014-07-08 | 2016-01-14 | Dow Agrosciences Llc | Process for the preparation of 4-alkoxy-3-hydroxypicolinic acids |
| WO2017127794A1 (en) | 2016-01-22 | 2017-07-27 | Dow Agrosciences Llc | Process for the preparation of 4-alkoxy-3-hydroxypicolinic acids |
| EP3545953A1 (en) | 2008-06-20 | 2019-10-02 | Novartis AG | Paediatric compositions for treating1 multiple sclerosis |
| WO2023044171A1 (en) * | 2021-09-20 | 2023-03-23 | Pardes Biosciences, Inc. | Inhibitors of cysteine proteases and methods of use thereof |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1073288A (en) * | 1964-04-22 | 1967-06-21 | Kyorin Seiyaku Kk | Aminoacetonitriles, their preparation and compositions containing them |
| WO1999024460A2 (en) * | 1997-11-05 | 1999-05-20 | Novartis Ag | Dipeptide nitriles |
-
2000
- 2000-02-16 EP EP00903845A patent/EP1155011A1/en not_active Withdrawn
- 2000-02-16 WO PCT/GB2000/000529 patent/WO2000049008A1/en not_active Application Discontinuation
- 2000-02-16 JP JP2000599748A patent/JP2002537294A/en active Pending
- 2000-02-16 AU AU25600/00A patent/AU2560000A/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1073288A (en) * | 1964-04-22 | 1967-06-21 | Kyorin Seiyaku Kk | Aminoacetonitriles, their preparation and compositions containing them |
| WO1999024460A2 (en) * | 1997-11-05 | 1999-05-20 | Novartis Ag | Dipeptide nitriles |
Non-Patent Citations (8)
| Title |
|---|
| A.R. KATRITZKY, ET AL.: "Benzotriazole-assisted synthesis of alpha-aminonitriles and a conceptually novel method for peptide elongation", JOURNAL OF THE CHEMICAL SOCIETY, PERKIN TRANSACTIONS 1, no. 6, June 1990 (1990-06-01), Royal Society of Chemistry, Letchworth, GB, pages 1853 - 1857, XP002140042, ISSN: 0300-922X * |
| C.A. LEWIS, ET AL.: "Thiohemiacetal formation by inhibitory aldehydes at the active site", BIOCHEMISTRY, vol. 16, no. 22, 1 November 1977 (1977-11-01), American Chemical Society, Washinton, DC, US, pages 4890 - 4895, XP002140046, ISSN: 0006-2960 * |
| D.J. BUTTLE, ET AL.: "Affininty purification of the novel cysteine proteinase papaya proteinase IV, and papain from papaya latex", BIOCHEMICAL JOURNAL, vol. 261, no. 2, 15 July 1989 (1989-07-15), Portland Press, London, GB, pages 469 - 476, XP000913659, ISSN: 0264-6021 * |
| D.T. ELMORE, ET AL.: "Thioesters of amino acid deriviatives as thioacylating agents in thiopeptide synthesis", JOURNAL OF THE CHEMICAL SOCIETY, PERKIN TRANSACTIONS 1, no. 5, May 1988 (1988-05-01), Royal Society of Chemistry, Letchworth, GB, pages 1051 - 1055, XP002140044, ISSN: 0300-922X * |
| H. MOSER, ET AL.: "Poly(dipeptamidium)-Salze: Definition und Methoden zur präparativen Herstellung", HELVETICA CHIMICA ACTA, vol. 69, 1986, Verlag Helvetica Chimica Aata, Basle, CH, pages 1224 - 1262, XP002049723, ISSN: 0018-019X * |
| S. LIU, ET AL.: "Effects of ligand homologation and ligand reactivity on the apparent kinetic specificity of papain", BIOCHIMICA ET BIOPHYSICA ACTA, vol. 1250, 1995, Elsevier Science, Amsterdam, NL, pages 43 - 48, XP002140045, ISSN: 0006-3002 * |
| S. SUZUE, ET AL.: "Hepatic agents. I. Synthesis of aminoacyl (and hydroxyacyl) aminoacetonitriles", CHEMICAL AND PHARMACEUTICAL BULLETIN, vol. 16, no. 8, August 1968 (1968-08-01), Pharmaceutical Society of Japan, Tokyo, JP, pages 1417 - 1432, XP002108053, ISSN: 0009-2363 * |
| S.A. THOMPSON, ET AL.: "Carboxy-modified amino acids and peptides as protease inhibitors", JOURNAL OF MEDICINAL CHEMISTRY, vol. 29, no. 1, January 1986 (1986-01-01), American Chemical Society, Washington, DC, US, pages 104 - 111, XP002140043, ISSN: 0022-2623 * |
Cited By (48)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6593327B2 (en) | 1999-03-15 | 2003-07-15 | Axys Pharmaceuticals, Inc. | Compounds and compositions as protease inhibitors |
| US6455502B1 (en) | 1999-03-15 | 2002-09-24 | Axys Pharmaceuticals, Inc. | Compounds and compositions as protease inhibitors |
| US6476026B1 (en) | 1999-03-15 | 2002-11-05 | Axys Pharmaceuticals, Inc. | Compounds and compositions as protease inhibitors |
| EP1452522A3 (en) * | 1999-03-15 | 2005-02-09 | Axys Pharmaceuticals, Inc. | Novel compounds and compositions as protease inhibitors |
| EP1452522A2 (en) * | 1999-03-15 | 2004-09-01 | Axys Pharmaceuticals, Inc. | Novel compounds and compositions as protease inhibitors |
| US6525036B2 (en) | 2000-01-06 | 2003-02-25 | Merck & Co., Inc. | Compounds and compositions as protease inhibitors |
| US6642239B2 (en) | 2000-02-10 | 2003-11-04 | Novartis Ag | Dipeptide nitrile cathepsin K inhibitors |
| US7309703B2 (en) | 2000-08-14 | 2007-12-18 | Ortho Mcneil Pharmaceutical, Inc. | Substituted pyrazoles |
| US6635633B2 (en) | 2000-08-14 | 2003-10-21 | Ortho-Pharmaceutical, Inc. | Substituted pyrazoles |
| US7393850B2 (en) | 2000-08-14 | 2008-07-01 | Ortho Mcneil Pharmaceutical, Inc. | Substituted pyrazoles |
| US7772236B2 (en) | 2000-08-14 | 2010-08-10 | Ortho Mcneil Pharmaceutical, Inc. | Substituted pyrazoles |
| US7388011B2 (en) | 2000-08-14 | 2008-06-17 | Ortho Mcneil Pharmaceutical, Inc. | Substituted pyrazoles |
| US7332494B2 (en) | 2000-08-14 | 2008-02-19 | Janssen Pharmaceutica, N.V. | Method for treating allergies using substituted pyrazoles |
| US6936603B2 (en) | 2000-08-14 | 2005-08-30 | Ortho-Mcneil Pharmaceutical, Inc. | Substituted pyrazoles |
| US6949540B2 (en) | 2000-08-14 | 2005-09-27 | Ortho-Mcneil Pharmaceutical, Inc. | Substituted pyrazoles |
| US6951851B2 (en) | 2000-08-14 | 2005-10-04 | Hui Cai | Substituted pyrazoles |
| US6953793B2 (en) | 2000-08-14 | 2005-10-11 | Ortho-Mcneil Pharmaceutical, Inc. | Substituted pyrazoles |
| US7589202B2 (en) | 2000-08-14 | 2009-09-15 | Ortho Mcneil Pharmaceutical, Inc. | Substituted pyrazoles |
| US7417046B2 (en) | 2000-08-14 | 2008-08-26 | Ortho Mcneil Pharmaceutical, Inc. | Substituted pyrazoles |
| US7429591B2 (en) | 2000-08-14 | 2008-09-30 | Ortho Mcneil Pharmaceutical, Inc. | Substituted pyrazoles |
| US7265102B2 (en) | 2000-08-14 | 2007-09-04 | Ortho Mcneil Pharmaceutical, Inc. | Substituted pyrazoles |
| US7452890B2 (en) | 2000-08-14 | 2008-11-18 | Ortho Mcneil Pharmaceutical, Inc. | Substituted pyrazoles |
| WO2002020002A2 (en) * | 2000-09-06 | 2002-03-14 | Ortho Mcneil Pharmaceutical, Inc. | A method for treating allergies |
| WO2002020002A3 (en) * | 2000-09-06 | 2002-11-28 | Ortho Mcneil Pharm Inc | A method for treating allergies |
| US6579896B2 (en) | 2000-09-06 | 2003-06-17 | Ortho-Mcneil Pharmaceutical, Inc. | Method for treating allergies using substituted pyrazoles |
| US6583155B2 (en) | 2000-09-06 | 2003-06-24 | Ortho-Mcneil Pharmaceutical, Inc. | Method for treating allergies using substituted pyrazoles |
| US7064123B1 (en) | 2000-12-22 | 2006-06-20 | Aventis Pharmaceuticals Inc. | Compounds and compositions as cathepsin inhibitors |
| US7030116B2 (en) | 2000-12-22 | 2006-04-18 | Aventis Pharmaceuticals Inc. | Compounds and compositions as cathepsin inhibitors |
| US7012075B2 (en) | 2001-03-02 | 2006-03-14 | Merck & Co., Inc. | Cathepsin cysteine protease inhibitors |
| US7196099B2 (en) | 2001-09-14 | 2007-03-27 | Aventis Pharmaceuticals Inc. | Compounds and compositions as cathepsin inhibitors |
| US6977256B2 (en) | 2001-11-14 | 2005-12-20 | Aventis Pharmaceuticals Inc. | Compounds and compositions as cathepsin S inhibitors |
| US7279478B2 (en) | 2002-09-04 | 2007-10-09 | Merck Frosst Canada & Co. | Cathepsin cysteine protease inhibitors |
| EP2251007A2 (en) | 2002-09-24 | 2010-11-17 | Novartis AG | Sphingosine-1-phosphate (S1P) receptor agonists for use in the treatment of demyelinating diseases |
| EP2255798A2 (en) | 2002-09-24 | 2010-12-01 | Novartis AG | Sphingosine-1-phosphate receptor agonists for use in the treatment of optic neuritis |
| WO2004052921A1 (en) * | 2002-12-05 | 2004-06-24 | Axys Pharmaceuticals, Inc. | Cyanomethyl derivatives as cysteine protease inhibitors |
| EP2719700A1 (en) | 2008-01-09 | 2014-04-16 | Amura Therapeutics Limited | Tetrahydrofuro(3,2-b)pyrrol-3-one derivatives as inhibitors of cysteine proteinases |
| EP3545953A1 (en) | 2008-06-20 | 2019-10-02 | Novartis AG | Paediatric compositions for treating1 multiple sclerosis |
| EP2198879A1 (en) | 2008-12-11 | 2010-06-23 | Institut Curie | CD74 modulator agent for regulating dendritic cell migration and device for studying the motility capacity of a cell |
| WO2016007638A1 (en) | 2014-07-08 | 2016-01-14 | Dow Agrosciences Llc | Process for the preparation of 4-alkoxy-3-hydroxypicolinic acids |
| EP3166612A4 (en) * | 2014-07-08 | 2017-12-20 | Dow AgroSciences LLC | Process for the preparation of 4-alkoxy-3-hydroxypicolinic acids |
| EP3166611A4 (en) * | 2014-07-08 | 2018-02-21 | Dow AgroSciences LLC | Process for the preparation of 4-alkoxy-3-hydroxypicolinic acids |
| TWI682923B (en) | 2014-07-08 | 2020-01-21 | 美商陶氏農業科學公司 | Process for the preparation of 4-alkoxy-3-hydroxypicolinic acids |
| TWI684587B (en) | 2014-07-08 | 2020-02-11 | 美商陶氏農業科學公司 | Process for the preparation of 4-alkoxy-3-hydroxypicolinic acids |
| CN108495555A (en) * | 2016-01-22 | 2018-09-04 | 美国陶氏益农公司 | The method for preparing 4- alkoxy -3- hydroxy-picolinic acids |
| WO2017127794A1 (en) | 2016-01-22 | 2017-07-27 | Dow Agrosciences Llc | Process for the preparation of 4-alkoxy-3-hydroxypicolinic acids |
| EP3405034A4 (en) * | 2016-01-22 | 2019-10-23 | Dow Agrosciences LLC | Process for the preparation of 4-alkoxy-3-hydroxypicolinic acids |
| RU2744834C2 (en) * | 2016-01-22 | 2021-03-16 | ДАУ АГРОСАЙЕНСИЗ ЭлЭлСи | Method for producing 4-alkoxy-3-hydroxypicolinic acids |
| WO2023044171A1 (en) * | 2021-09-20 | 2023-03-23 | Pardes Biosciences, Inc. | Inhibitors of cysteine proteases and methods of use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2002537294A (en) | 2002-11-05 |
| AU2560000A (en) | 2000-09-04 |
| EP1155011A1 (en) | 2001-11-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2000049008A1 (en) | Di- and tripeptide nitrile derivatives as inhibitors of cathepsin l and cathepsin s | |
| EP1155010A1 (en) | Acetamido acetonitrile derivatives as inhibitors of cathepsin l and/or cathepsin s | |
| JP4201716B2 (en) | Cathepsin cysteine protease inhibitor | |
| US7012075B2 (en) | Cathepsin cysteine protease inhibitors | |
| US7217794B2 (en) | Compounds and methods for treatment of thrombosis | |
| US6255453B1 (en) | Peptoid and nonpeptoid containing alpha-keto oxadiazoles as serine protease inhibitors | |
| NO964514L (en) | angiogenesis inhibitor | |
| AU2004268707A1 (en) | Cathepsin inhibitors | |
| EP0950046B1 (en) | 3,4-disubstituted azetidin-2-one derivatives useful as cysteine proteinase regulators | |
| KR20010020391A (en) | Protease Inhibitors | |
| WO1997003060A1 (en) | Piperazine derivatives and use of the same | |
| US6518267B1 (en) | Protease inhibitors | |
| CA2092414A1 (en) | N-(2-alkyl-3-mercaptoglutaryl)-amino-diaza cycloalkanone derivatives and their use as collagenase inhibitors | |
| EP1157003A1 (en) | Acylated aminoacetonitriles as cysteine protease inhibitors | |
| US7001887B2 (en) | Peptide derivatives | |
| PL198827B1 (en) | N-ARYLSULFONYL AMINO ACID OMEGA AMIDES, method of making them, pharmaceutical agent and their application | |
| US8268789B2 (en) | PAR-2 antagonists | |
| EP1228054A1 (en) | Compounds and their use as cysteine protease inhibitors | |
| JP2006526586A (en) | Benzamidonitrile derivatives | |
| US6277871B1 (en) | Inhibitors of protein isoprenyl transferases | |
| WO2000009542A1 (en) | SERINE PROTEASE INHIBITORS COMPRISING α-KETO HETEROCYCLES | |
| EP1283825B1 (en) | N-substituted peptidyl nitriles as cysteine cathepsin inhibitors | |
| JPH0523259B2 (en) | ||
| JPH03148246A (en) | Peptidyldifluorodiol renin inhibiting substance | |
| JPH10511342A (en) | Malonic acid-based matrix metalloproteinase inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AL AM AT AU AZ BA BB BG BR BY CA CH CN CR CU CZ DE DK DM EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2000903845 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 2000 599748 Kind code of ref document: A Format of ref document f/p: F |
|
| WWP | Wipo information: published in national office |
Ref document number: 2000903845 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09913810 Country of ref document: US |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2000903845 Country of ref document: EP |