WO2000006722A1 - Eukaryotic cell-based gene interaction cloning - Google Patents
Eukaryotic cell-based gene interaction cloning Download PDFInfo
- Publication number
- WO2000006722A1 WO2000006722A1 PCT/EP1999/005491 EP9905491W WO0006722A1 WO 2000006722 A1 WO2000006722 A1 WO 2000006722A1 EP 9905491 W EP9905491 W EP 9905491W WO 0006722 A1 WO0006722 A1 WO 0006722A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- receptor
- cells
- ligand
- cell
- chimeric receptor
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6897—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids involving reporter genes operably linked to promoters
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/71—Receptors; Cell surface antigens; Cell surface determinants for growth factors; for growth regulators
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/715—Receptors; Cell surface antigens; Cell surface determinants for cytokines; for lymphokines; for interferons
- C07K14/7155—Receptors; Cell surface antigens; Cell surface determinants for cytokines; for lymphokines; for interferons for interleukins [IL]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/715—Receptors; Cell surface antigens; Cell surface determinants for cytokines; for lymphokines; for interferons
- C07K14/7156—Receptors; Cell surface antigens; Cell surface determinants for cytokines; for lymphokines; for interferons for interferons [IFN]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
Definitions
- the present invention relates to a method for screening compounds for their ability to bind a receptor and/or the screening of compounds that antagonise the binding of a ligand to a receptor.
- Receptors are defined as proteinaceous macromolecules that are often located on cell membranes and that perform a signal transducing function. Many receptors are located on the outer cell membrane. Several receptors possess three domains, the extracellular domain, the transmembrane domain and the cytoplasmic domain. The extracellular domain is capable of specifically binding to a compound, normally called "ligand". Signal transduction appears to occur in a variety of ways upon ligand binding, such as for example by a conformational change in the structure of the receptor, by clustering of two or more identical or related receptor-type molecules.
- receptors have been identified and the scientific literature has variously divided them into groups, superfamilies, families and/or classes of receptors based on common features such as tissue distribution of the receptors, nucleic acid or amino acid homology of the receptors, mechanisms of signalling by the receptors or the type of ligand that binds to the receptors.
- a uniform system of classifying or grouping receptors has not been used in the literature.
- polypeptide hormones elicit their biological effect by binding to receptors expressed on the surface of responsive cells.
- At least four families of polypeptide hormone receptors can be defined on the basis of similarity in primary sequence, predicted secondary and tertiary structure and biochemical function.
- haemopoietin/interferon receptor family the receptor kinase family, the tumour necrosis factor (TNF) / nerve growth factor (NGF) family and the family of G-protein coupled receptors.
- the haemopoietin/interferon family receptors have no intrinsic enzymatic activity;
- CONRR A ⁇ ON copy they can be recognised on the base of their "cytokine receptor homology" (CRH) region in their extracellular domains.
- This CRH region contains two conserved cystein bridges and a tryptophan - serine - X - tryptophan - serine motif.
- the defining features of members of the TNF-NGF receptor family are located in the extracellular domain and centre on a domain that contains 6 cysteine residues.
- the receptor kinase family is characterised by a conserved catalytic kinase domain in the cytoplasmic part of the receptor; the family is subdivided in tyrosine kinase and serine/threonine kinase receptors, on the base of their substrate specificity.
- G- protein coupled receptors traverse the membrane several times. With the exception of the G-protein coupled receptors, cytokine driven multimerization of the receptor subunits appears to be the initial event in signal transduction. While homo- or heterodimerization and trimerization are central to the function of haemopoietin / interferon receptors and TNF / NGF receptors, homodimerization appears a preferred way of receptor kinase action. A special case is that of the receptor-like protein tyrosine phosphatases.
- cytokine-d riven interaction between receptor subunits appears to be the initial event for haemopoietin / interferon receptors.
- the recognition of the ligand starts with one receptor subunit; this subunit is often called a-subunit in case of heteromeric receptors.
- there is an association of one or more additional receptor molecules which is essential for the initiation of the signal transduction and, as an additional effect can lead to an increase in affinity of the ligand binding.
- Receptor clustering leads to activation of the kinase function.
- the haemopoietin / interferon receptors which, contrary to the tyrosine kinase receptors, do not have an intrinsic kinase activity, are using the help of the associated "Janus kinases" (JAKs) to phosphorylate the tyrosine residues.
- JAKs Janus kinases
- Subsequent targets for the JAKs include the JAK molecules themselves, the cytoplasmic part of the receptor and the "Signal Transducers and Activators of Transcription" proteins (STAT). This pathway is called the "JAK / STAT pathway”. Additional pathways, such as the Ras - Raf - mitogen activated protein kinase pathway may also be activated.
- haemopoietin / interferon receptors are, amongst others, the interleukin-5 (IL-5) receptor, the erythropoietin receptor and the interferon receptor family.
- the IL-5 receptor is a heteromer consisting of two subunits.
- the IL-5 receptor ⁇ -chain is ligand specific and has a low to intermediate binding affinity. Association with the IL-5 receptor ⁇ -chain, that is common with other receptor complexes such as IL-3, results in a high affinity binding complex. Both receptor subunits are required for signalling. Furthermore, signalling requires the cytoplasmic tails of both receptor subunits. Interferons are classified into two classes.
- Type I interferons consist of the IFN ⁇ group, IFN ⁇ , IFN ⁇ and the bovine embryonic form, IFN ⁇ .
- IFN ⁇ belongs to the second group (type II interferon).
- the receptor complex of the type I interferons consists of an IFNaRI subunit and an IFNaR2 subunit. The latter receptor chain exists in three isoforms, resulting from alternative splicing: IFNaR2-1 and IFNaR2-2 are membrane associated but differ in length of the cytoplasmic domain, whereas IFNaR2-3 is a soluble form.
- the human 2fTGH cell line is hypoxanthine-guanine phosphoribosyl transferase (HGPRT) deficient, but is containing the xanthine guanine phosphoribosyl transferase (gpt) gene of E. coli, under the control of the type I IFN inducible 6-16 promoter.
- HGPRT hypoxanthine-guanine phosphoribosyl transferase
- gpt xanthine guanine phosphoribosyl transferase
- XGPRT xanthine guanine phosphoribosyl transferase
- US 5597693 describes a screening method in mammalian cells that is, however, limited to intracellular receptors of the steroid/thyroid superfamily and can not be used for cytokine receptors.
- WO 95/21930 describes a screening method for cytokine receptors. In this method, ligands are screened after random mutagenesis of a cell line. Only those ligands can be detected of which the expression can be activated by mutagenesis in the cell type used. Moreover, the isolation of the ligand encoding genes is rather complicated. This is a severe restriction for the usefulness of said screening method.
- a method is described to screen for ligands of the Denervated Muscle Kinase (DMK) receptor and chimeric variants thereof.
- DMK Denervated Muscle Kinase
- the applicability of this method is rather limited and there is no direct, rapid way provided to isolate the genetic material encoding the ligand.
- chimeric receptors are constructed, comprising an extracellular domain derived from one protein, preferentially the extracellular domain of a receptor, and a cytoplasmic part derived from another protein which should be a receptor; at least one chimeric receptor is expressed in a eukaryotic host cell which is not a yeast cell.
- the same eukaryotic host cell comprises a recombinant gene, encoding for a compound of which the expression creates an autocrinic loop, and a reporter system that is activated upon the creation of said autocrinic loop.
- the compound of which the expression creates an autocrinic loop is a ligand for the chimeric receptor.
- the reporter system is switched on, preferentially by the use of a promoter that can be activated as a result of binding said ligand to said chimeric receptor.
- All three elements can be either stably transformed into the eukaryotic cell, or transiently expressed.
- Transfection methods described in the art can be used to obtain this. Non-limiting examples are methods such as calcium-phosphate transfection (Graham and Van der Eb, 1973), lipofection (Loeffner and Behr, 1993) and retroviral gene transfer (Kitamura et al., 1995).
- the retroviral gene transfer is preferred since, depending on the virus/cell ratio, an average infection of one virus per cell can be obtained.
- the autocrinic loop can be more complex, and may consist of more than one loop.
- the recombinant gene may express the ligand of a first (chimeric or non-chimeric) receptor that activates a second gene, which upon activation expresses the ligand of a second receptor, of which the ligand binding results in the induction of the reporter system.
- the first and the second receptor are situated within the same cell: it is clear, for people skilled in the art, that one can work with two cell populations, the first one carrying a recombinant gene, expressing a ligand for a receptor for the second cell, which upon binding of the ligand starts to produce the ligand of the chimeric receptor, situated on the first cell. Binding of the latter ligand to the chimeric receptor then results in the expression of the reporter system.
- the gpt selection system can be applied to the screening and/or selection of orphan receptors.
- the extracellular domain of the receptor that is studied is fused to the intracellular domain(s) of IFNaR.
- the receptor studied may be an orphan receptor or a receptor from which not all the ligands are known.
- the use of the IFN receptor cytoplasmic tails is sufficient for signal transduction which is required for reporter activation, independent of the function (which may be unknown) of the receptor studied.
- the ligand is supplied by the creation of an autocrinic loop: cells are transfected by a DNA expression library, where genes, encoding for possible ligands for the orphan receptor, are placed preferentially after a strong, constitutive promoter. It is known, however, to people skilled in the art that other promoters can be used, such as inducible promoters and even an IFN inducible promoter.
- the production of the cognate ligand induces the transcription of the gpt gene, enabling a positive selection in HAT medium.
- candidate ligands can be added to the medium; survival of the cells in the HAT medium will only be detected when a ligand can activate the orphan receptor.
- secreted alkaline phosphatase SEAP may be used as reporter system.
- Cells expressing the reporter system can be identified by measuring the SEAP activity using CSPD (disodium 3-(4- methoxyspirol-1 ,2-dioxetane-3,2'-(5'-chloro)trichloro ⁇ 3.3.1.1 (3,7) ⁇ decan-4- yl)phenyl phosphate) as luminogenic substrate.
- CSPD disodium 3-(4- methoxyspirol-1 ,2-dioxetane-3,2'-(5'-chloro)trichloro ⁇ 3.3.1.1 (3,7) ⁇ decan-4- yl)phenyl phosphate
- the invention is not limited to the use of the cytoplasmic tails of the interferon receptor and the gpt selection system, but other receptor systems and/or other inducible promoters and/or other reporter systems and/or other cell lines, known to people skilled in the art may be used.
- PC12 cells Greene et al., 1976
- a chimeric receptor based on the leptin receptor Talaglia et al., 1995
- the inducible promoter from the Pancreatitis associated protein I gene may be used.
- the reporter system may be based upon the detection of the gene product of an inducible gene, as is the case for Green Fluorescent Protein (GFP) as a non limiting example, or may be based on modification of a protein already present in the cell (proteolytic cleavage, phosphorylation, complex formation%) such as the systems described by Mitra et al. (1995), Miyawaki et al. (1997) and Romoser et al. (1997). Moreover, optimal reporter activation may require a co-stimulus, as is the case for the leptin-forskolin system.
- GFP Green Fluorescent Protein
- a further aspect of the invention is the screening of compounds that are antagonists of the ligand-receptor binding. Due to the fact that can be screened for the toxicity of gpt expression in D-MEM + 6-TG medium, it is possible to set up an antagonistic screening system for compounds that inhibit and/or compete with the binding of the ligand to the chimeric receptor. This can be realized by using the autocrinic loop and adding possible inhibitors to the medium, but it is clear for people skilled in the art that, alternatively, the cell can be transformed with genes encoding candidate inhibitors. Expression of an inhibitor would create an anti-autocrinic loop.
- the ligand is produced either by an autocrinic loop, or added to the medium,or the receptor may be mutated and/or genetically modified to a form that constitutively initiates the signalling pathway.
- a screening may be useful in the identification of compounds with potential pharmaceutical applications.
- a further aspect of the invention is the screening of compounds in the signalling pathway: a host cell, carrying the chimeric receptor and the gene for its ligand, placed after a promoter, in principle inducible by the chimeric receptor, but where said host cell is missing one or more compounds of the signalling pathway, can be transfected by an expression library in order to complement the signalling pathway. Complemented cells will be detected by the activation of the reporter system.
- Still another aspect of the invention is the screening of compounds that are involved in the secretory pathway: as the ligand for the chimeric receptor needs to be secreted in order to activate the receptor, both compounds that block the secretion, or compounds that can complement a mutation in the secretory pathway can be screened.
- multimerizing receptor every receptor of which the interaction with or binding of the ligand results in the multime zation of receptor components, and/or every protein that can be identified by the people skilled in the art as such a receptor on the base of its amino acid sequence and/or protein structure. Interaction is often the binding to the receptor, but can for instance also be binding to one component of a receptor complex, which subsequently associates with other receptor components to form said receptor complex. Another example is the transient interaction of a ligand with a receptor component leading to a conformational change or allowing a specific enzymatic modification leading to signal transduction. Multime zation can be homo- or heterodimerization, homo- or heterothmerization, ..., up to complex formation of multiple proteins.
- Orphan receptor every receptor, preferentially a multimerizing receptor, or protein with known receptor components of which no ligand is known that is interacting or binding to this receptor and, as a consequence, initiating or inhibiting the signalling pathway.
- Ligand every compound that can interact with or bind to a receptor, preferentially a multimerizing receptor and that is initiating or inhibiting the signalling pathway by its interaction with or binding to said receptor.
- Unknown ligand every compound that can interact with or bind to a receptor, preferentially a multimerizing receptor and that is initiating or inhibiting the signalling pathway by its interaction with or binding to said receptor, but for which this interaction or binding has not yet been demonstrated.
- Compound means any chemical or biological compound, including simple or complex inorganic or organic molecules, peptides, peptido-mimetics, proteins, antibodies, carbohydrates, phospholipids, nucleic acids or derivatives thereof.
- Extracellular domain means the extracellular domain of a receptor and/or orphan receptor, or a functional fragment thereof characterised by the fact that it still can interact with or bind to a known and/or unknown ligand, or a fragment thereof fused to other amino acid sequences, characterised by the fact that it still can interact with or bind to a known and/or unknown ligand, or a fragment from a non-receptor protein that can interact with or bind to a known and/or unknown ligand.
- Bind(ing) means any interaction, be it direct ( direct interaction of the compound with the extracellular domain) or indirect (interaction of a compound with one or more identical and/or non-identical compounds resulting in a complex of which one or more compounds can interact with the extracellular domain), that result in initiating or inhibiting the signalling pathway of the chimeric receptor
- Cytoplasmic domain means the cytoplasmic part of a receptor, or a functional fragment thereof, or a fragment thereof fused to other amino acid sequences, capable of initiating the signalling pathway of said receptor and of inducing a reporter system.
- Chimeric receptor functional receptor comprising an extracellular domain of one receptor and the cytoplasmic domain of another receptor.
- Reporter system every compound of which the synthesis and/or modification and/or complex formation can be detected and/or be used in a screening and/or selection system.
- the reporter system can be, as a non limiting example, a gene product encoding an enzymatic activity, a coloured compound, a surface compound or a fluorescent compound.
- Autocrinic loop every succession of events by which a cell, carrying a receptor allows the synthesis of a known or unknown compound that, directly or indirectly, induces the activation of said receptor.
- Anti-autocrinic loop every succession of events by which a cell, carrying a receptor allows the synthesis of a known or unknown compound that, directly or indirectly, inhibits the binding of a ligand and/or unknown ligand to said receptor.
- Signalling pathway means every succession of events after the binding of a ligand and/or unknown ligand to an extracellular domain of a natural occurring or chimeric receptor whereby said binding can result in the induction and/or repression of a set of genes.
- Selection means isolation and/or identification of cells in which the reporter system is activated or isolation and/or identification of cells in which the reporter system is not activated.
- PCR polymerase chain reactions
- the sequence encoding the ⁇ c extracellular domain was PCR amplified using the forward primer MBU-O-39 which also contains a Kpnl site and the reverse primer MBU-O-40.
- a forward primer MBU-O-41 was used with a reverse primer MBU-O-42, which contains an Xhol site, to amplify the sequence that codes for the IFNaRI transmembrane (TM) and intracellular (IC) domain (amino acids 436-557, including the last residue of the extracellular domain, Lys436).
- the forward primer MBU-O-43 was used to amplify the sequence encoding the IFNaR2-1 transmembrane and intracellular domains (amino acids 243-331 , including the last residue of the extracellular domain, Lys243) and the IFNaR2-2 TM and IC domains (amino acids 243-515, including the last residue of the extracellular domain, Lys243), respectively in combination with the reverse primers MBU-O-44 and MBU-O- 45, containing an Xhol site.
- the resultant blunt PCR fragments, coding for the hybrid receptors, were isolated by agarose gel electrophoresis, digested with Kpnl - Xhol and ligated into the Kpnl-Xhol opened pcDNA3 vector (Invitrogen).
- the constructs were checked by DNA sequence analysis and named as follows: pcDNA3-IL-5R ⁇ /IFNaR1 , pcDNA3-IL-5R ⁇ /IFNaR2-1 , pcDNA3-IL-5R ⁇ /IFNaR2-2, pcDNA3- ⁇ c/IFNaR1 , pcDNA3- ⁇ c/IFNaR2-1 and pcDNA3- ⁇ c/IFNaR2-2.
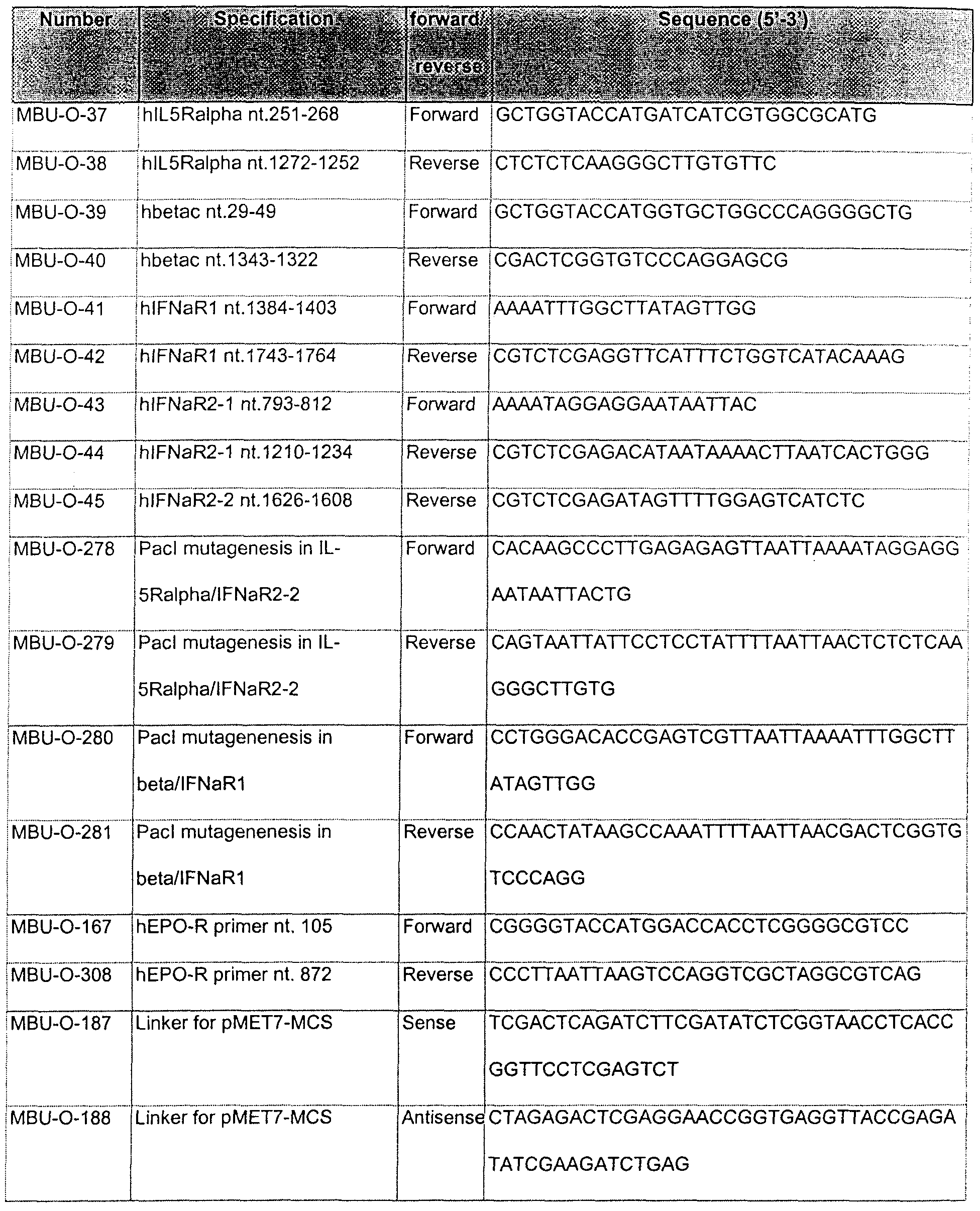
- Table 1 oligonucleotides used for construction of chimeric receptors and IL-5 expression vectors.
- pSV-SPORT expression vector contains an SV40 early promoter which is normally weaker as compared to the CMV promoter of the pcDNA3 plasmid.
- the genes for the chimeric receptors in pcDNA3-IL-5R ⁇ /IFNaR2-2 and pcDNA3- ⁇ c/IFNaR1 were isolated by Asp718 and Xhol digestion and agarose gelelectrophoresis, followed by insertion in the Asp718-Sall opened pSV-SPORT vector. The resulting constructs were verified by sequence analysis and named pSV-SPORT-IL-5R ⁇ /lFNaR2-2 and pSV-SPORT- ⁇ c/IFNaR1.
- Insertion mutagenesis was performed with the QuickChange site-directed mutagenesis kit (Stratagene), using the oligonucleotides MBU-O-278 (sense) and MBU-O-279 (antisense) for IL-5R ⁇ /IFNaR2-2 and MBU-O-280 (sense) and MBU-0-281 (antisense) for ⁇ c/IFNaR1 (tablel ).
- plasmids were named pSV-SPORT-IL5R ⁇ P/IFNaR2-2 and pSV- SPORT- ⁇ cP/IFNaR1
- RNA was prepared from 5x10 6 TF-1 cells according to the procedure of the RNeasy kit (Qiagen), and dissolved in 50 ⁇ l water from which 10 ⁇ l was used for RT-PCR. To these, 2 ⁇ l (2 ⁇ g) of oligodT (12-18 mer; Pharmacia) was added and incubated at 70°C for 10 min. After chilling on ice for 1 min., cDNA was prepared by adding 4 ⁇ l of RT buffer (10x; Life Sciences), 1 ⁇ l dNTP's (20 mM; Pharmacia), 2 ⁇ l DTT (0.1 M) and 1 ⁇ l of MMLV reverse transcriptase (200U; superscript; Life Technologies) so that the total volume was 20 ⁇ l.
- the PCR was started at 94°C for 2 min. during which 2 ⁇ l Pfu enzyme (5 U; Stratagene) was added (hot start) and followed by 40 cycles with denaturation at 92°C (1 min.), hybridization between 55 till 59°C (1 min.; with an increasing temperature gradient over 4°C during the 40 cycles) and polymerization at 72°C (3 min.; with an increasing time elongation of 0.05 min. during every cycle, but only in the last 25 cycles). To finalise, the reaction was hold on 72°C for 12 min. and chilled to 4°C.
- Pfu enzyme 5 U; Stratagene
- a band of correct size was isolated from an agarose gel and the DNA was digested with Pad and Kpnl and inserted into the Pacl-Kpnl opened pSV-SPORT-IL-5R ⁇ P/IFNaR2-2 or pSV-SPORT- ⁇ cP/IFNaR1 vectors.
- the resultant vectors were named pSV-SPORT-EPO-R/IFNaR2-2 and EPO-R/IFNaR1 , respectively.
- IL-5 can activate the 6-16 promoter via IL-5R/IFNaR chimeric receptors. 11.1.1. Activation of 6-16 gpt allows selection of stable colonies.
- Transfection was according to the calcium phosphate method (Graham and van der Eb (1973)). For each plasmid, 10 ⁇ g DNA was used (20 ⁇ g of pcDNA3 for mock transfection). The precipitate was made up in 1 ml and left on the cells overnight (5x10 5 cells/transfection/petridish). The dishes were then washed twice with Dulbecco's PBS (Life Technologies) and cells were left in DMEM (Life Technologies). 48 hours later, DMEM medium + G418 (Calbiochem; 400 ⁇ g/ml) was added.
- HAT medium (Life Technologies) alone + G418, 2) HAT medium + G418 + 500 U/ml IFN ⁇ 2b (PeproTech, Ine) or 3) HAT medium + G418 + 1 ng/ml IL-5 (produced in Sf9 cells using published methodologies) was added.
- 2ftGH cells that were stabile transfected with the pSV-SPORT IL-5R ⁇ /IFNaR2-2 + pSV-SPORT ⁇ c/IFNaR1 vectors were isolated essentially the same way with the exception that selection in G418 medium was omitted.
- 10 ⁇ g DNA was used (20 ⁇ g of pSV-SPORT for mock transfection).
- the precipitate was made up in 1 ml and left on the cells overnight (5x10 5 cells/transfection/petridish). The dishes were then washed twice with Dulbecco'sPBS and cells were left in DMEM.
- the survival or death was determined visually during a two-week period, using an inverted microscope A clone with the best response to IL-5 was called 2fTGH IL-5R ⁇ /R2-2 + ⁇ c/R1 CloneE.
- the cells developed at this stage could already serve as an assay system for the evaluation of exogeneously added ligands.
- the resultant plasmid was named p6-16SEAP.
- Stabile 6-16SEAP transfected 2fTGH cell lines were obtained by co- transfection of 20 ⁇ g p6-16SEAP with 2 ⁇ g pBSpac/deltap (obtained from the Belgian Coordinated Collections of Microorganisms, BCCM) in the 2fTGH cells.
- the latter plasmid contained a gene for puromycin resistence under control of the constitutive SV40 early promoter. Selection on puromycin was on the basis of methods described in the art. We choose 3 ⁇ g puromycin/ml as an optimal concentration for selection of puromycin-resistant 2ftGH cells.
- Single colonies were isolated by limited dilution in 96-well microtiterplates and investigated on SEAP production after treatment with IFN ⁇ or ⁇ versus no stimulus.
- the clones 2fTGH-6-16SEAPclone2 and 2ftGH-6-16SEAPclone5 were selected, based on an optimal stimulation window.
- Erythropoietin can activate the 6-16 promoter via Epo-R/IFNaR chimeric receptors.
- the precipitate was made up in 1 ml and left on the cells for six hours (5x10 5 cells/transfection/petridish). The dishes were then washed twice with Dulbecco's PBS and cells were further grown in DMEM. After 24 hours, cells from every transfection were trypsinized with 5 ml 0.05% trypsine / 0.02% EDTA solution (Life Technologies) and seeded in three wells of a 6-well microtiterplate. The next day, no stimulus, IFN ⁇ (500U/ml) or erythropoietin (EPO, 0.5 U/ml, R&D systems) was added and the cells were left for another 24 hours.
- IFN ⁇ 500U/ml
- EPO erythropoietin
- 2fTGH-6-16SEAP clone ⁇ cells were transfected with 20 ⁇ g of pSV-SPORT- EpoR/R2-2 and 2 ⁇ g pcDNA1/Neo.
- a calcium phosphate precipitate was made up in 1 ml according to the method of Graham and Van der Eb (1973), and left on the cells overnight (8x10 5 cells/transfection/petridish). The dishes were then washed twice with PBS and cells were left in DMEM. 48 hours later, DMEM medium + G418 (400 ⁇ g/ml) was added and refreshed every 3-4 days for a period up to 14 days. Individual cells were isolated by limited dilution in a 96-well microtiterplate.
- Degree of responsiveness of single colonies to Epo was determined by investigating growth in HAT medium supplemented with Epo, versus cell death in HAT medium alone. Alternatively, cell growth in medium containing 6-thioguanine (6-TG) versus cell death in 6-TG containing medium supplemented with Epo, was also determined. The survival or death was determined visually during a two-week period, using an inverted microscope. Furthermore, the 2fTGH 6-16SEAP clone 5 cells have the 6-16SEAP construct stabile transfected, allowing fast determination of Epo responsiveness by measurement of SEAP induction. On the basis of these assays, 2fTGH-6-16SEAP EpoR/2-2 clone 4 showed the highest responsiveness for Epo and was selected for further analysis.
- the gene for hlL-5syn was isolated from the pGEM1-hlL-5syn vector (Tavemier et al. 1989) by Sal I digestion and agarose gelelectrophoresis. The fragment was cloned into the Sal I opened pEFBOS vector (gift from Nagata.S., Osaha Bioscience Institute, Japan). As a result, the hlL-5syn gene was cloned downstream the promoter for human elongation factor 1 a (HEF1 a, Mizushima et al., 1990) and the resultant plasmid was named pEFBos-hlL-5syn.
- HEF1 a human elongation factor 1 a
- the Sal I fragment was also cloned into the pMET7MCS vector.
- This vector was constructed by replacing the DNA encoding the leptin receptor long form (Lrlo) in the plasmid pMET7-Lrlo (gift from L. Tartaglia, Millenium, Cambridge), with the DNA coding for a multicloning site (Sal l-Bgl ll-EcoR V-BstE ll-Age l-Xho l-Xba I), formed by hybridization of the oligonucleotides MBU-O-187 and MBU-O-188 (table 1 ).
- the hlL-5syn gene was cloned downstram the hybrid SR ⁇ promoter (Takebe et al. 1988) and the plasmid was named pMET7-hlL-5syn. 111.2. Construction of pMET7-moEpo for constitutive eukaryotic expression of monkey Epo.
- the plasmid pMFEpo2 (gift from Dr. C. Laker, Heinrich-Pette-institut), was used as input DNA for PCR amplification of monkey Epo cDNA, using a forward primer (GGAATTCGCCAGGCGCCACCATGGGGGTGCACGAATGTCCTG) that contains a kozak sequence and an EcoR1 site and a reverse primer (GCCTCGAGTCATCTGTCCCCTCTCCTGCAG), containing a Xhol site.
- the PCR was performed with Pfu polymerase (Stratagene) and the obtained product of + 600 bp was purified by gel extraction and digested with EcoRI- Xhol. This fragment was inserted into the pMET7m ⁇ c/SEAP vector.
- This plasmid encodes for a chimeric protein (alkaline phosphatase fused to the C- terminal end of the mouse IL-5 beta common (m ⁇ c) chain), downstream the SR ⁇ promoter.
- the m ⁇ c/SEAP gene was removed by an EcoRI-Xhol digest, allowing ligation of the moEpo fragment into the opened pMET7 vector.
- the resulting plasmid was named pMET7-moEpo.
- the plasmids pEFBOS-hlL-5syn or the pUC18 vector (mock) were used for transfection of 2ftGH cells that stabile expressed the IL-5R ⁇ /IFNaR2-2 + ⁇ c/IFNaR1 chimeras (2ftGH clone C cells). Transfection was performed overnight according to the Ca-phosphate method (Graham and Van der Eb,
- a 1 :10 dilution series of pEFBOS-hlL-5syn DNA in irrelevant DNA was set up : 1.5 (1/10), 0.15 (1/100), 0.015 (1/1000) and 0.0015 (1/10000) ⁇ g of pEFBOS- hlL-5syn DNA were added to 15 ⁇ g pcDNA3 DNA and transfected in the IL- 5R ⁇ /IFNaR2-2 + ⁇ c/IFNaR1 clone C cells.
- Positive and negative controls were 15 ⁇ g of pEFBOS-hlL-5syn and 15 ⁇ g of pcDNA3, respectively.
- Transfection was according to the Ca-phosphate procedure (Graham and Van der Eb, 1973). The precipitates were made up in 1 ml and left on the cells overnight (5 x 10 5 cells / transfection / petridish). Following washing (2 x with Dulbecco's PBS), DMEM medium was added for 24 hours after which it was changed to HAT medium. Cells were visually followed using an inverted microscope and 15 days after transfection, photographs of representative regions in every petri dish were taken.
- a dilution series of pMET7-hlL-5syn DNA in irrelevant DNA was set up : 4 ng (1/10 4 ), 400 pg (1/10 5 ), and 40 pg (1/10 6 ) of pMET7-hlL-5syn
- DNA were added to 40 ⁇ g pCDNA3 DNA and transfected in the 2fTGH IL-5R ⁇ /IFNaR2-2 + ⁇ c/IFNaR1 CloneE cells (stabile transfected with pSV-SPORT- IL-5R ⁇ /IFNaR2-2 + pSV-SPORT- ⁇ c/IFNaR1 ).
- 40 ⁇ g of pCDNA3 alone was used.
- 10 ⁇ g p6-16 SEAP was added to all samples.
- Every precipitate was prepared in 1 ml according to the Ca-phosphate procedure (Graham and Van der eb, 1973), from which 165 ⁇ l (6.8 ⁇ g of total DNA) was brought onto 10 5 cells in the well of a 6-well microtiterplate. The precipitate was left on the cells overnight after which cells were washed twice with Dulbecco's PBS. Cells were further grown in DMEM medium. After 24 hours, medium samples were taken from each well and SEAP activity was measured using the Phospha-Light assay (Tropix). Luminescence was measured in a Topcount luminometer.
- Every precipitate was prepared in 500 ⁇ l, according to the Ca-phosphate procedure (Graham and Van der eb, 1973), and 165 ⁇ l ( ⁇ 4 ⁇ g total DNA) was brought onto 10 5 2fTGH 6-16SEAP EpoR/IFNaR2-2 Clone 4 cells in the well of a 6-well microtiterplate. The precipitate was left on the cells for 6 hours after which cells were washed twice with Dulbecco's PBS. Cells were further grown in DMEM medium. After 18 hours, medium samples were taken from each well and SEAP activity was measured using the Phospha-Light assay (Tropix). Luminescence was measured in a Topcount luminometer.
- the precipitate was left on the cells for 6 hours after which cells were washed twice with Dulbecco's PBS. Cells were further grown in DMEM medium. After 18 hours, medium samples were taken from each well and SEAP activity was measured using the Phospha-Light assay (Tropix). Luminescence was measured in a Topcount luminometer. Transfection of the cells with 400 pg pMET7-hlL-5syn in 4 ⁇ g total DNA (1/10 4 dilution), still resulted in a clear SEAP production, as compared to the negative control, indicating that an autocrine loop was formed (figure 6).
- FIG. 1 Transient co-transfection of pSV-SPORT-IL-5R ⁇ /IFNaR2-2, pSV- SPORT- ⁇ c/IFNaR1 and p6-16SEAP in 2ftGH cells and analysis of induction of SEAP activity. 24 hours after transfection, cells were left unstimulated or were stimulated with IFN ⁇ (positive control) or IL-5 (1 and 2 ng/ml). Samples from the medium were taken 24 hours after stimulation and SEAP activity was measured using CSPD as a luminogenic substrate (phospha-light kit, Tropix). The amount of light produced was determined in a Topcount luminometer (Packard).
- FIG. 2 Transient transfection of pSV-SPORT-EpoR/IFNaR1 + pSV- SPORT-EpoR/IFNaR2-2, pSV-SPORT-EpoR/IFNaR1 or pSV-SPORT- EpoR/IFNaR2-2 in 2fTGH 6-16SEAP Clone 5 cells. 24 hours after transfection, cells were left unstimulated or were stimulated with IFN ⁇ (1 ng/ml; positive control) or Epo (5 ng/ml). Samples from the medium were taken 24 hours after stimulation and SEAP activity was measured using CSPD as luminogenic substrate (phospha-light kit, Tropix). The amount of light was determined in a Topcount luminometer (Packard).
- Figure 3 Survival of 2fTGH IL-5R ⁇ /IFNaR2-2 + ⁇ c/IFNaR1 clone C cells, transfected with dilutions of the vector pEFBOS-hlL-5syn in irrelevant DNA. Formation of an autocrinic loop results in survival of the cells in HAT medium. Fifteen days after transfection, photographs of representative regions in each petridish were taken.
- Figure 4 Induction of SEAP activity in IL-5R ⁇ /IFNaR2-2 + ⁇ c/IFNaR1 clone E, transfected with dilutions of the vector pMET7-hlL-5syn in irrelevant DNA and co-transfected with the p6-16 plasmid. Formation of an autocrinic loop results in activation of the 6-16 promoter followed by secretion of SEAP. Samples from the medium were taken 24 hours after transfection and SEAP activity was measured using CSPD as luminogenic substrate (phospha-light kit, Tropix). The amount of light produced was determined in a Topcount luminometer (Packard).
- Figure 5 A. Induction of SEAP activity in 2fTGH IL-5R ⁇ /IFNaR2-2 + ⁇ c/IFNaR1 clone E cells, transfected with dilutions of the vector pMET7-hlL- 5syn in an EL4 cDNA library that was expressed in the eukaryotic expression vector pACGGS. All dilutions were co-transfected with the p6-16 plasmid. Negative control was pACGGS-EL4cDNA + p6-16SEAP. Transfection was performed according to the Ca-phosphate method. Formation of an autocrinic loop results in activation of the 6-16 promoter followed by secretion of SEAP.
- Figure 6 Induction of SEAP activity in 2fTGH 6-16SEAP EpoR/IFNaR2-2 clone 4 cells, transfected with dilutions of the vector pMET7-moEpo in an EL4 cDNA library that was expressed in the eukaryotic expression vector pACGGS. All dilutions were co-transfected with the p6-16 plasmid. Negative control was pACGGS-EL4cDNA + p6-16SEAP. Formation of an autocrinic loop results in activation of the 6-16 promoter followed by secretion of SEAP. Samples from the medium were taken 24 hours after transfection and SEAP activity was measured using CSPD as luminogenic substrate (phospha-light kit, Tropix). The amount of light produced was determined in a Topcount luminometer (Packard). References
- SR alpha promoter an efficient and versatile mammalian cDNA expression system composed of the simian virus 40 early promoter and the R-U5 segment of human T-cell leukemia virus type I long terminal repeat. Mol. Cell. Biol., 8, 466-472.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Zoology (AREA)
- Genetics & Genomics (AREA)
- Immunology (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Toxicology (AREA)
- Cell Biology (AREA)
- Gastroenterology & Hepatology (AREA)
- Engineering & Computer Science (AREA)
- Wood Science & Technology (AREA)
- Biotechnology (AREA)
- Analytical Chemistry (AREA)
- Microbiology (AREA)
- Physics & Mathematics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
Description

Claims
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002337086A CA2337086A1 (en) | 1998-07-28 | 1999-07-27 | Eukaryotic cell-based gene interaction cloning |
| AU53726/99A AU765010B2 (en) | 1998-07-28 | 1999-07-27 | Eukaryotic cell-based gene interaction cloning |
| EP99939421A EP1100911A1 (en) | 1998-07-28 | 1999-07-27 | Eukaryotic cell-based gene interaction cloning |
| JP2000562504A JP2002522031A (en) | 1998-07-28 | 1999-07-27 | Eukaryotic cell-based gene interaction cloning |
| US09/771,425 US20010023062A1 (en) | 1998-07-28 | 2001-01-26 | Eukaryotic cell-based gene interaction cloning |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP98202528 | 1998-07-28 | ||
| EP98202528.0 | 1998-07-28 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/771,425 Continuation US20010023062A1 (en) | 1998-07-28 | 2001-01-26 | Eukaryotic cell-based gene interaction cloning |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2000006722A1 true WO2000006722A1 (en) | 2000-02-10 |
Family
ID=8233984
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP1999/005491 WO2000006722A1 (en) | 1998-07-28 | 1999-07-27 | Eukaryotic cell-based gene interaction cloning |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20010023062A1 (en) |
| EP (1) | EP1100911A1 (en) |
| JP (1) | JP2002522031A (en) |
| AU (1) | AU765010B2 (en) |
| CA (1) | CA2337086A1 (en) |
| WO (1) | WO2000006722A1 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001090188A2 (en) * | 2000-05-22 | 2001-11-29 | Vlaams Interuniversitair Instituut Voor Biotechnologie Vzw | Receptor-based interaction trap |
| WO2003002726A2 (en) * | 2001-06-28 | 2003-01-09 | Vlaams Interuniversitair Instituut Voor Biotechnologie Vzw | Cellular reporter gene assay with a ligand amplifying feedback loop |
| US7291458B2 (en) | 1998-07-28 | 2007-11-06 | Vlaams Interuniversitair Instituut Voor Biotechnologie Vzw | Leptin-mediated gene-induction |
| EP1975620A2 (en) | 2001-03-02 | 2008-10-01 | GPC Biotech AG | Three hybrid assay system |
| US7575878B2 (en) | 2004-11-18 | 2009-08-18 | Vib Vzw | Methods of inhibiting leptin-induced signaling with fibronectin III domain antibodies |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1912069A3 (en) * | 2002-06-05 | 2008-07-09 | Sopherion Therapeutics, Inc. | Method to screen ligands using eukaryotic cell display |
| EP1514114A4 (en) * | 2002-06-05 | 2007-03-21 | Sopherion Therapeutics Inc | Method to screen ligands using eukaryotic cell display |
| DE102004022484B4 (en) * | 2004-05-07 | 2007-12-20 | P.A.L.M. Microlaser Technologies Ag | microscope stage |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996023814A1 (en) * | 1995-02-03 | 1996-08-08 | Cell Genesys, Inc. | Chimeric receptor molecules for delivery of co-stimulatory signals |
| WO1998002542A1 (en) * | 1996-07-17 | 1998-01-22 | University Of Medicine And Dentistry Of New Jersey | Cytokine receptor signal transduction chain |
| WO1998013513A2 (en) * | 1996-09-24 | 1998-04-02 | Cadus Pharmaceutical Corporation | Methods and compositions for identifying receptor effectors |
| WO1998016557A1 (en) * | 1996-10-11 | 1998-04-23 | The General Hospital Corporation | Assays for g-protein-linked receptors |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5283173A (en) * | 1990-01-24 | 1994-02-01 | The Research Foundation Of State University Of New York | System to detect protein-protein interactions |
| CA2304367C (en) * | 1997-09-16 | 2009-06-09 | Fox Chase Cancer Center | An improved yeast interaction trap assay |
| US6332897B1 (en) * | 1998-03-27 | 2001-12-25 | Glaxo Wellcome Inc. | Assay methods |
| US6406863B1 (en) * | 2000-06-23 | 2002-06-18 | Genetastix Corporation | High throughput generation and screening of fully human antibody repertoire in yeast |
-
1999
- 1999-07-27 AU AU53726/99A patent/AU765010B2/en not_active Ceased
- 1999-07-27 CA CA002337086A patent/CA2337086A1/en not_active Abandoned
- 1999-07-27 JP JP2000562504A patent/JP2002522031A/en active Pending
- 1999-07-27 WO PCT/EP1999/005491 patent/WO2000006722A1/en not_active Application Discontinuation
- 1999-07-27 EP EP99939421A patent/EP1100911A1/en not_active Withdrawn
-
2001
- 2001-01-26 US US09/771,425 patent/US20010023062A1/en not_active Abandoned
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996023814A1 (en) * | 1995-02-03 | 1996-08-08 | Cell Genesys, Inc. | Chimeric receptor molecules for delivery of co-stimulatory signals |
| WO1998002542A1 (en) * | 1996-07-17 | 1998-01-22 | University Of Medicine And Dentistry Of New Jersey | Cytokine receptor signal transduction chain |
| WO1998013513A2 (en) * | 1996-09-24 | 1998-04-02 | Cadus Pharmaceutical Corporation | Methods and compositions for identifying receptor effectors |
| WO1998016557A1 (en) * | 1996-10-11 | 1998-04-23 | The General Hospital Corporation | Assays for g-protein-linked receptors |
Non-Patent Citations (2)
| Title |
|---|
| MUTHUKURMARAN G. ET AL.: "Chimeric Erythropoietin-Interferon gamma receptors reveal differences in functional architecture of intracellular domains for signal transduction", J. BIOL. CHEM., vol. 272, no. 8, 21 February 1997 (1997-02-21), pages 4993 - 4999, XP002122944 * |
| PELLEGRINI S ET AL: "USE OF A SELECTABLE MARKER REGULATED BY ALPHA INTERFERON TO OBTAIN MUTATIONS IN THE SIGNALLING PATHWAY", MOLECULAR AND CELLULAR BIOLOGY, vol. 9, no. 11, November 1989 (1989-11-01), pages 4605 - 4612, XP000673982 * |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7291458B2 (en) | 1998-07-28 | 2007-11-06 | Vlaams Interuniversitair Instituut Voor Biotechnologie Vzw | Leptin-mediated gene-induction |
| WO2001090188A2 (en) * | 2000-05-22 | 2001-11-29 | Vlaams Interuniversitair Instituut Voor Biotechnologie Vzw | Receptor-based interaction trap |
| WO2001090188A3 (en) * | 2000-05-22 | 2002-06-13 | Vlaams Interuniv Inst Biotech | Receptor-based interaction trap |
| EP1612221A3 (en) * | 2000-05-22 | 2008-07-23 | Vlaams Interuniversitair Instituut voor Biotechnologie vzw. | Receptor-based interaction trap |
| US7855270B2 (en) | 2000-05-22 | 2010-12-21 | Vlaams Interuniversitair Instituut Voor Biotechnologie Vzw | Receptor-based interaction trap |
| US8003757B2 (en) | 2000-05-22 | 2011-08-23 | Vlaams Interuniversitair Instituut Voor Biotechnologie Vzw | Receptor-based interaction trap |
| EP1975620A2 (en) | 2001-03-02 | 2008-10-01 | GPC Biotech AG | Three hybrid assay system |
| WO2003002726A2 (en) * | 2001-06-28 | 2003-01-09 | Vlaams Interuniversitair Instituut Voor Biotechnologie Vzw | Cellular reporter gene assay with a ligand amplifying feedback loop |
| WO2003002726A3 (en) * | 2001-06-28 | 2003-03-20 | Vlaams Interuniv Inst Biotech | Cellular reporter gene assay with a ligand amplifying feedback loop |
| US7575878B2 (en) | 2004-11-18 | 2009-08-18 | Vib Vzw | Methods of inhibiting leptin-induced signaling with fibronectin III domain antibodies |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1100911A1 (en) | 2001-05-23 |
| CA2337086A1 (en) | 2000-02-10 |
| US20010023062A1 (en) | 2001-09-20 |
| AU765010B2 (en) | 2003-09-04 |
| AU5372699A (en) | 2000-02-21 |
| JP2002522031A (en) | 2002-07-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Wisdon et al. | Transformation by Fos proteins requires a C-terminal transactivation domain | |
| US8003757B2 (en) | Receptor-based interaction trap | |
| US20030170656A1 (en) | Method of screening for factors that modulate gene expression | |
| Kim et al. | Mammalian type I interferon receptors consists of two subunits: IFNaR1 and IFNaR2 | |
| EP1003863A1 (en) | Mammalian cytokine receptor-11 | |
| Larner et al. | Protein tyrosine phosphorylation as a mechanism which regulates cytokine activation of early response genes | |
| CA2164623A1 (en) | Hybrid receptor molecules | |
| WO2007030803A9 (en) | Method for preparing trimeric proteins | |
| AU765010B2 (en) | Eukaryotic cell-based gene interaction cloning | |
| Oki et al. | Stat6 activation and Th2 cell proliferation driven by CD28 signals | |
| Müller et al. | Interferon response pathways—a paradigm for cytokine signalling? | |
| AU2001281784A1 (en) | Receptor-based interaction trap | |
| RU2139936C1 (en) | Method of determining modulation effect of substance on interleukin-5 receptor-linked signal-transmission route in human or animal cell | |
| AU704762B2 (en) | Human STAT4 | |
| US6605703B1 (en) | Deletion of the Hck binding region in the IL-6 receptor | |
| Jaster et al. | Analysis of cis-acting sequences and trans-acting factors regulating the interleukin-3 response element of the DUB-1 gene | |
| Piekorz et al. | Modulation of the activation status of Stat5a during LIF-induced differentiation of M1 myeloid leukemia cells | |
| HEMPSTEAD et al. | The nerve growth factor receptor: biochemical and structural analysis | |
| Hibi et al. | IL-6 receptor | |
| Starnes | Characterization of the interleukin-17 family of proteins | |
| WO2001025797A2 (en) | Methods of evaluating whether a test agent is an agent affecting a leptin receptor | |
| WO2004003509A2 (en) | A method to identify specific interaction between ligand and receptor | |
| US20070117182A1 (en) | Nucleic acids conferring transcriptional responsiveness on the RANKL gene promoter and uses thereof | |
| Cao et al. | The Coiled-Coil Domain of Stat3 Is | |
| US20020115168A1 (en) | Novel protein zlmda2. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SL SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1999939421 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 53726/99 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2337086 Country of ref document: CA Ref country code: JP Ref document number: 2000 562504 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09771425 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 1999939421 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| NENP | Non-entry into the national phase |
Ref country code: CA |
|
| WWG | Wipo information: grant in national office |
Ref document number: 53726/99 Country of ref document: AU |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1999939421 Country of ref document: EP |