WO1997025041A1 - Benzimidzolyl neuropeptide y receptor antagonists - Google Patents
Benzimidzolyl neuropeptide y receptor antagonists Download PDFInfo
- Publication number
- WO1997025041A1 WO1997025041A1 PCT/US1997/000511 US9700511W WO9725041A1 WO 1997025041 A1 WO1997025041 A1 WO 1997025041A1 US 9700511 W US9700511 W US 9700511W WO 9725041 A1 WO9725041 A1 WO 9725041A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkylenyl
- alkoxy
- heterocyclic
- preparation
- piperidin
- Prior art date
Links
- 239000002660 neuropeptide Y receptor antagonist Substances 0.000 title description 8
- 150000001875 compounds Chemical class 0.000 claims abstract description 98
- 101710151321 Melanostatin Proteins 0.000 claims abstract description 51
- URPYMXQQVHTUDU-OFGSCBOVSA-N nucleopeptide y Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 URPYMXQQVHTUDU-OFGSCBOVSA-N 0.000 claims abstract description 51
- 238000000034 method Methods 0.000 claims abstract description 38
- 239000008194 pharmaceutical composition Chemical class 0.000 claims abstract description 3
- 102400000064 Neuropeptide Y Human genes 0.000 claims abstract 2
- -1 phenoxy, naphthyl Chemical group 0.000 claims description 213
- 125000000623 heterocyclic group Chemical group 0.000 claims description 155
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 67
- 150000003839 salts Chemical class 0.000 claims description 55
- 125000003545 alkoxy group Chemical group 0.000 claims description 47
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims description 44
- 125000000217 alkyl group Chemical group 0.000 claims description 35
- 125000001624 naphthyl group Chemical group 0.000 claims description 34
- 125000000392 cycloalkenyl group Chemical group 0.000 claims description 33
- 125000003282 alkyl amino group Chemical group 0.000 claims description 32
- 125000003342 alkenyl group Chemical group 0.000 claims description 28
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 25
- 125000000304 alkynyl group Chemical group 0.000 claims description 24
- 125000001589 carboacyl group Chemical group 0.000 claims description 23
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 claims description 21
- 125000004414 alkyl thio group Chemical group 0.000 claims description 20
- 229910052739 hydrogen Inorganic materials 0.000 claims description 19
- 239000001257 hydrogen Substances 0.000 claims description 19
- 125000001188 haloalkyl group Chemical group 0.000 claims description 18
- 125000005186 naphthyloxy group Chemical group C1(=CC=CC2=CC=CC=C12)O* 0.000 claims description 18
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 17
- 125000004423 acyloxy group Chemical group 0.000 claims description 14
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 claims description 14
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 11
- 125000004400 (C1-C12) alkyl group Chemical group 0.000 claims description 10
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 10
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 9
- 239000012453 solvate Substances 0.000 claims description 9
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims description 7
- 125000003368 amide group Chemical group 0.000 claims description 4
- 241000124008 Mammalia Species 0.000 claims description 3
- 239000003085 diluting agent Substances 0.000 claims description 2
- 239000003937 drug carrier Substances 0.000 claims description 2
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 2
- 125000001475 halogen functional group Chemical group 0.000 claims 12
- 125000004356 hydroxy functional group Chemical group O* 0.000 claims 6
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims 1
- 125000004454 (C1-C6) alkoxycarbonyl group Chemical group 0.000 claims 1
- 150000001556 benzimidazoles Chemical class 0.000 abstract description 8
- 239000004480 active ingredient Substances 0.000 abstract 1
- 238000002360 preparation method Methods 0.000 description 522
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 412
- 238000005481 NMR spectroscopy Methods 0.000 description 307
- 239000000047 product Substances 0.000 description 263
- 238000000434 field desorption mass spectrometry Methods 0.000 description 233
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 210
- 239000000243 solution Substances 0.000 description 207
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 195
- 239000000203 mixture Substances 0.000 description 190
- 229910001868 water Inorganic materials 0.000 description 181
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 175
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 172
- 238000004458 analytical method Methods 0.000 description 163
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 163
- 239000002904 solvent Substances 0.000 description 150
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 140
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 126
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 114
- 239000011541 reaction mixture Substances 0.000 description 105
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 104
- 125000004483 piperidin-3-yl group Chemical group N1CC(CCC1)* 0.000 description 94
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 description 84
- 239000003921 oil Substances 0.000 description 78
- 235000019198 oils Nutrition 0.000 description 78
- 239000012299 nitrogen atmosphere Substances 0.000 description 74
- 238000006243 chemical reaction Methods 0.000 description 72
- 229910000027 potassium carbonate Inorganic materials 0.000 description 70
- 235000011181 potassium carbonates Nutrition 0.000 description 70
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 68
- 102000005962 receptors Human genes 0.000 description 68
- 108020003175 receptors Proteins 0.000 description 68
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 66
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 66
- 239000007787 solid Substances 0.000 description 63
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 52
- 239000012267 brine Substances 0.000 description 50
- 230000002829 reductive effect Effects 0.000 description 50
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 50
- 102100028427 Pro-neuropeptide Y Human genes 0.000 description 49
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 48
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 47
- 229910052938 sodium sulfate Inorganic materials 0.000 description 47
- 235000011152 sodium sulphate Nutrition 0.000 description 47
- 238000003818 flash chromatography Methods 0.000 description 45
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 42
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 42
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 39
- 239000012223 aqueous fraction Substances 0.000 description 37
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 29
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 29
- 239000012312 sodium hydride Substances 0.000 description 29
- 229910000104 sodium hydride Inorganic materials 0.000 description 29
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 28
- 239000006260 foam Substances 0.000 description 25
- 239000013058 crude material Substances 0.000 description 24
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 24
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 23
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 22
- 238000010898 silica gel chromatography Methods 0.000 description 22
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 21
- 238000003756 stirring Methods 0.000 description 21
- 238000004809 thin layer chromatography Methods 0.000 description 21
- 238000004587 chromatography analysis Methods 0.000 description 20
- 239000000706 filtrate Substances 0.000 description 20
- 239000000463 material Substances 0.000 description 20
- 238000010511 deprotection reaction Methods 0.000 description 19
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 17
- 125000004432 carbon atom Chemical group C* 0.000 description 17
- 238000005187 foaming Methods 0.000 description 17
- 239000002480 mineral oil Substances 0.000 description 17
- 235000010446 mineral oil Nutrition 0.000 description 17
- 239000012043 crude product Substances 0.000 description 16
- 108090000765 processed proteins & peptides Proteins 0.000 description 16
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 15
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 15
- YNXLOPYTAAFMTN-SBUIBGKBSA-N C([C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)C1=CC=C(O)C=C1 Chemical compound C([C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)C1=CC=C(O)C=C1 YNXLOPYTAAFMTN-SBUIBGKBSA-N 0.000 description 14
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 14
- 102100029909 Peptide YY Human genes 0.000 description 14
- 108010088847 Peptide YY Proteins 0.000 description 14
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 14
- 235000019341 magnesium sulphate Nutrition 0.000 description 14
- 125000004574 piperidin-2-yl group Chemical group N1C(CCCC1)* 0.000 description 14
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 13
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 13
- 229910052757 nitrogen Inorganic materials 0.000 description 13
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 12
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 12
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 11
- 125000005843 halogen group Chemical group 0.000 description 11
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 10
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 10
- 239000000908 ammonium hydroxide Substances 0.000 description 10
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 10
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 9
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 9
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 9
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 9
- 239000002585 base Substances 0.000 description 9
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 9
- 239000013078 crystal Substances 0.000 description 9
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 9
- 239000000543 intermediate Substances 0.000 description 9
- CYNYIHKIEHGYOZ-UHFFFAOYSA-N 1-bromopropane Chemical compound CCCBr CYNYIHKIEHGYOZ-UHFFFAOYSA-N 0.000 description 8
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 8
- 239000003054 catalyst Substances 0.000 description 8
- 210000004027 cell Anatomy 0.000 description 8
- 239000007788 liquid Substances 0.000 description 8
- 125000004115 pentoxy group Chemical group [*]OC([H])([H])C([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 8
- 239000002244 precipitate Substances 0.000 description 8
- 239000000651 prodrug Substances 0.000 description 8
- 229940002612 prodrug Drugs 0.000 description 8
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 description 7
- SNBSKIQOHSMQSX-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-phenylmethoxy-1h-benzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC1=NC2=CC=CC(OCC=3C=CC=CC=3)=C2N1 SNBSKIQOHSMQSX-UHFFFAOYSA-N 0.000 description 7
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 7
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 7
- DPBSXYRJZFUOIL-UHFFFAOYSA-N ethyl 3-piperidin-3-ylpropanoate Chemical compound CCOC(=O)CCC1CCCNC1 DPBSXYRJZFUOIL-UHFFFAOYSA-N 0.000 description 7
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 7
- 102000004196 processed proteins & peptides Human genes 0.000 description 7
- 229920006395 saturated elastomer Polymers 0.000 description 7
- NQRYJNQNLNOLGT-UHFFFAOYSA-N tetrahydropyridine hydrochloride Natural products C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 7
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 6
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 6
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 6
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 6
- 108050002826 Neuropeptide Y Receptor Proteins 0.000 description 6
- 102000012301 Neuropeptide Y receptor Human genes 0.000 description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 6
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 6
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 6
- 210000004556 brain Anatomy 0.000 description 6
- 229910052799 carbon Inorganic materials 0.000 description 6
- 238000004440 column chromatography Methods 0.000 description 6
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 6
- 208000035475 disorder Diseases 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 239000010410 layer Substances 0.000 description 6
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 6
- 229910052763 palladium Inorganic materials 0.000 description 6
- 238000006467 substitution reaction Methods 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- 210000001519 tissue Anatomy 0.000 description 6
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 5
- 206010020772 Hypertension Diseases 0.000 description 5
- 238000004566 IR spectroscopy Methods 0.000 description 5
- FZERHIULMFGESH-UHFFFAOYSA-N N-phenylacetamide Chemical compound CC(=O)NC1=CC=CC=C1 FZERHIULMFGESH-UHFFFAOYSA-N 0.000 description 5
- 102000030621 adenylate cyclase Human genes 0.000 description 5
- 108060000200 adenylate cyclase Proteins 0.000 description 5
- 125000001931 aliphatic group Chemical group 0.000 description 5
- 125000003277 amino group Chemical group 0.000 description 5
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 5
- 238000001914 filtration Methods 0.000 description 5
- 239000012634 fragment Substances 0.000 description 5
- 239000012458 free base Substances 0.000 description 5
- QMEZUZOCLYUADC-UHFFFAOYSA-N hydrate;dihydrochloride Chemical compound O.Cl.Cl QMEZUZOCLYUADC-UHFFFAOYSA-N 0.000 description 5
- 150000002431 hydrogen Chemical class 0.000 description 5
- IWYDHOAUDWTVEP-UHFFFAOYSA-N mandelic acid Chemical compound OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 5
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 5
- 238000007363 ring formation reaction Methods 0.000 description 5
- 125000001424 substituent group Chemical group 0.000 description 5
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 4
- IWYDHOAUDWTVEP-SSDOTTSWSA-N (R)-mandelic acid Chemical compound OC(=O)[C@H](O)C1=CC=CC=C1 IWYDHOAUDWTVEP-SSDOTTSWSA-N 0.000 description 4
- JOPZJEAGGYIFBP-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-methyl-1h-benzimidazole Chemical compound N1C=2C(C)=CC=CC=2N=C1COC1=CC=C(Cl)C=C1 JOPZJEAGGYIFBP-UHFFFAOYSA-N 0.000 description 4
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 4
- XMZQWZJMTBCUFT-UHFFFAOYSA-N 3-bromopropylbenzene Chemical compound BrCCCC1=CC=CC=C1 XMZQWZJMTBCUFT-UHFFFAOYSA-N 0.000 description 4
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 4
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 4
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 4
- 108090000189 Neuropeptides Proteins 0.000 description 4
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 4
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 4
- RJUHRQQXFYFPSL-UHFFFAOYSA-N [2-[(4-chlorophenoxy)methyl]-1-(trifluoromethylsulfonyl)benzimidazol-4-yl] trifluoromethanesulfonate Chemical compound N=1C=2C(OS(=O)(=O)C(F)(F)F)=CC=CC=2N(S(=O)(=O)C(F)(F)F)C=1COC1=CC=C(Cl)C=C1 RJUHRQQXFYFPSL-UHFFFAOYSA-N 0.000 description 4
- 230000009471 action Effects 0.000 description 4
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 4
- 210000000133 brain stem Anatomy 0.000 description 4
- 238000004090 dissolution Methods 0.000 description 4
- 239000000284 extract Substances 0.000 description 4
- 239000012530 fluid Substances 0.000 description 4
- 125000000524 functional group Chemical group 0.000 description 4
- 125000005842 heteroatom Chemical group 0.000 description 4
- 150000002430 hydrocarbons Chemical group 0.000 description 4
- 238000011534 incubation Methods 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 4
- 150000007524 organic acids Chemical class 0.000 description 4
- 230000003389 potentiating effect Effects 0.000 description 4
- 125000006308 propyl amino group Chemical group 0.000 description 4
- WGYKZJWCGVVSQN-UHFFFAOYSA-N propylamine Chemical compound CCCN WGYKZJWCGVVSQN-UHFFFAOYSA-N 0.000 description 4
- 230000004044 response Effects 0.000 description 4
- 238000012552 review Methods 0.000 description 4
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 4
- 235000017557 sodium bicarbonate Nutrition 0.000 description 4
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- BADQZTVDULCXNA-UHFFFAOYSA-N 2,3-diaminophenol;dihydrochloride Chemical compound Cl.Cl.NC1=CC=CC(O)=C1N BADQZTVDULCXNA-UHFFFAOYSA-N 0.000 description 3
- UQOQRFKGJJCZLY-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-1-(3-piperidin-4-ylpropyl)-4-prop-2-enylbenzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC1=NC2=C(CC=C)C=CC=C2N1CCCC1CCNCC1 UQOQRFKGJJCZLY-UHFFFAOYSA-N 0.000 description 3
- DKKBDCFSMBEBMH-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-1h-benzimidazol-4-ol Chemical compound N=1C=2C(O)=CC=CC=2NC=1COC1=CC=C(Cl)C=C1 DKKBDCFSMBEBMH-UHFFFAOYSA-N 0.000 description 3
- KHBMOUWRRWBYLU-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-cyclopentyloxy-1-(3-piperidin-4-ylpropyl)benzimidazole;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.C1=CC(Cl)=CC=C1OCC(N(C1=CC=C2)CCCC3CCNCC3)=NC1=C2OC1CCCC1 KHBMOUWRRWBYLU-UHFFFAOYSA-N 0.000 description 3
- KJIBYDDFYNLKEM-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-ethenyl-1-(trifluoromethylsulfonyl)benzimidazole Chemical compound N=1C2=C(C=C)C=CC=C2N(S(=O)(=O)C(F)(F)F)C=1COC1=CC=C(Cl)C=C1 KJIBYDDFYNLKEM-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 3
- 102000003797 Neuropeptides Human genes 0.000 description 3
- 102000018886 Pancreatic Polypeptide Human genes 0.000 description 3
- 108010081690 Pertussis Toxin Proteins 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- 241000700159 Rattus Species 0.000 description 3
- 229960001413 acetanilide Drugs 0.000 description 3
- 150000001350 alkyl halides Chemical class 0.000 description 3
- 230000029936 alkylation Effects 0.000 description 3
- 238000005804 alkylation reaction Methods 0.000 description 3
- 229940024606 amino acid Drugs 0.000 description 3
- 150000001413 amino acids Chemical group 0.000 description 3
- 239000012298 atmosphere Substances 0.000 description 3
- 230000027455 binding Effects 0.000 description 3
- 230000036772 blood pressure Effects 0.000 description 3
- 239000011575 calcium Substances 0.000 description 3
- 229910052791 calcium Inorganic materials 0.000 description 3
- 150000001721 carbon Chemical group 0.000 description 3
- IQFVPQOLBLOTPF-HKXUKFGYSA-L congo red Chemical compound [Na+].[Na+].C1=CC=CC2=C(N)C(/N=N/C3=CC=C(C=C3)C3=CC=C(C=C3)/N=N/C3=C(C4=CC=CC=C4C(=C3)S([O-])(=O)=O)N)=CC(S([O-])(=O)=O)=C21 IQFVPQOLBLOTPF-HKXUKFGYSA-L 0.000 description 3
- 238000002425 crystallisation Methods 0.000 description 3
- 230000008025 crystallization Effects 0.000 description 3
- 238000013461 design Methods 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 238000001704 evaporation Methods 0.000 description 3
- 230000008020 evaporation Effects 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 229940093915 gynecological organic acid Drugs 0.000 description 3
- 210000001320 hippocampus Anatomy 0.000 description 3
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 3
- 238000005984 hydrogenation reaction Methods 0.000 description 3
- 239000005457 ice water Substances 0.000 description 3
- 150000007529 inorganic bases Chemical class 0.000 description 3
- 238000002844 melting Methods 0.000 description 3
- 230000008018 melting Effects 0.000 description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 3
- HRDXJKGNWSUIBT-UHFFFAOYSA-N methoxybenzene Chemical group [CH2]OC1=CC=CC=C1 HRDXJKGNWSUIBT-UHFFFAOYSA-N 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- 210000002569 neuron Anatomy 0.000 description 3
- 239000002858 neurotransmitter agent Substances 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- 235000005985 organic acids Nutrition 0.000 description 3
- 210000001428 peripheral nervous system Anatomy 0.000 description 3
- SUSQOBVLVYHIEX-UHFFFAOYSA-N phenylacetonitrile Chemical compound N#CCC1=CC=CC=C1 SUSQOBVLVYHIEX-UHFFFAOYSA-N 0.000 description 3
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 3
- 125000006239 protecting group Chemical group 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 3
- 238000001953 recrystallisation Methods 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 239000012047 saturated solution Substances 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 3
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 3
- QIWRFOJWQSSRJZ-UHFFFAOYSA-N tributyl(ethenyl)stannane Chemical compound CCCC[Sn](CCCC)(CCCC)C=C QIWRFOJWQSSRJZ-UHFFFAOYSA-N 0.000 description 3
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 3
- SFLSHLFXELFNJZ-QMMMGPOBSA-N (-)-norepinephrine Chemical compound NC[C@H](O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-QMMMGPOBSA-N 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical class O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 2
- IWYDHOAUDWTVEP-ZETCQYMHSA-N (S)-mandelic acid Chemical class OC(=O)[C@@H](O)C1=CC=CC=C1 IWYDHOAUDWTVEP-ZETCQYMHSA-N 0.000 description 2
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 2
- UWYZHKAOTLEWKK-UHFFFAOYSA-N 1,2,3,4-tetrahydroisoquinoline Chemical compound C1=CC=C2CNCCC2=C1 UWYZHKAOTLEWKK-UHFFFAOYSA-N 0.000 description 2
- GEYOCULIXLDCMW-UHFFFAOYSA-N 1,2-phenylenediamine Chemical class NC1=CC=CC=C1N GEYOCULIXLDCMW-UHFFFAOYSA-N 0.000 description 2
- SKMHPZXWMNAWGC-UHFFFAOYSA-N 1-(2-piperidin-2-ylethyl)piperidine Chemical compound C1CCCNC1CCN1CCCCC1 SKMHPZXWMNAWGC-UHFFFAOYSA-N 0.000 description 2
- LQHMYBSWLQUCAD-UHFFFAOYSA-N 1-(3-bromopropyl)-2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazole Chemical compound N=1C=2C(C)=CC=CC=2N(CCCBr)C=1COC1=CC=C(Cl)C=C1 LQHMYBSWLQUCAD-UHFFFAOYSA-N 0.000 description 2
- YZWKKMVJZFACSU-UHFFFAOYSA-N 1-bromopentane Chemical compound CCCCCBr YZWKKMVJZFACSU-UHFFFAOYSA-N 0.000 description 2
- 125000006018 1-methyl-ethenyl group Chemical group 0.000 description 2
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 2
- PCAXITAPTVOLGL-UHFFFAOYSA-N 2,3-diaminophenol Chemical compound NC1=CC=CC(O)=C1N PCAXITAPTVOLGL-UHFFFAOYSA-N 0.000 description 2
- PTXUJRTVWRYYTE-UHFFFAOYSA-N 2-(4-chlorophenyl)-1h-benzimidazole Chemical compound C1=CC(Cl)=CC=C1C1=NC2=CC=CC=C2N1 PTXUJRTVWRYYTE-UHFFFAOYSA-N 0.000 description 2
- YUSPMXQHCNUCBA-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-1-(3-piperidin-1-ylpropyl)benzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC1=NC2=CC=CC=C2N1CCCN1CCCCC1 YUSPMXQHCNUCBA-UHFFFAOYSA-N 0.000 description 2
- WXJASILOPZMCHP-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-1-(3-piperidin-4-ylpropyl)-4-propan-2-yloxybenzimidazole;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.C=1C=C(Cl)C=CC=1OCC1=NC=2C(OC(C)C)=CC=CC=2N1CCCC1CCNCC1 WXJASILOPZMCHP-UHFFFAOYSA-N 0.000 description 2
- UPGSRCVWJKZSPX-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-(cyclohexylmethoxy)-1-(3-piperidin-4-ylpropyl)benzimidazole;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.C1=CC(Cl)=CC=C1OCC(N(C1=CC=C2)CCCC3CCNCC3)=NC1=C2OCC1CCCCC1 UPGSRCVWJKZSPX-UHFFFAOYSA-N 0.000 description 2
- LWJYERJAQCCDMR-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-ethenyl-1h-benzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC1=NC2=CC=CC(C=C)=C2N1 LWJYERJAQCCDMR-UHFFFAOYSA-N 0.000 description 2
- CHRRLNLDFQNCFP-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-methoxy-1-(3-piperidin-4-ylpropyl)benzimidazole;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.C=1C=C(Cl)C=CC=1OCC1=NC=2C(OC)=CC=CC=2N1CCCC1CCNCC1 CHRRLNLDFQNCFP-UHFFFAOYSA-N 0.000 description 2
- QIDBTGIHUCYGJK-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-methyl-1-(3-piperidin-1-ylpropyl)benzimidazole Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCN1CCCCC1 QIDBTGIHUCYGJK-UHFFFAOYSA-N 0.000 description 2
- VACJYSNLLCSLMB-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-prop-2-enyl-1-(trifluoromethylsulfonyl)benzimidazole Chemical compound N=1C2=C(CC=C)C=CC=C2N(S(=O)(=O)C(F)(F)F)C=1COC1=CC=C(Cl)C=C1 VACJYSNLLCSLMB-UHFFFAOYSA-N 0.000 description 2
- HACWSBYHROBTPH-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-prop-2-enyl-1h-benzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC1=NC2=CC=CC(CC=C)=C2N1 HACWSBYHROBTPH-UHFFFAOYSA-N 0.000 description 2
- COGUOPIIFAMLES-UHFFFAOYSA-N 2-[(4-chlorophenyl)methyl]-1h-benzimidazole Chemical compound C1=CC(Cl)=CC=C1CC1=NC2=CC=CC=C2N1 COGUOPIIFAMLES-UHFFFAOYSA-N 0.000 description 2
- UIPYWGXBBFFEEL-UHFFFAOYSA-N 2-[(4-methylphenoxy)methyl]-4-phenylmethoxy-1h-benzimidazole Chemical compound C1=CC(C)=CC=C1OCC1=NC2=CC=CC(OCC=3C=CC=CC=3)=C2N1 UIPYWGXBBFFEEL-UHFFFAOYSA-N 0.000 description 2
- INQULXLCLQROSC-UHFFFAOYSA-N 3-[1-[3-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]propyl]piperidin-4-yl]-n-propylpropanamide Chemical compound C1CC(CCC(=O)NCCC)CCN1CCCN1C2=CC=CC(C)=C2N=C1COC1=CC=C(Cl)C=C1 INQULXLCLQROSC-UHFFFAOYSA-N 0.000 description 2
- QVXRTXGWLZZBMZ-UHFFFAOYSA-N 3-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]propan-1-ol Chemical compound N=1C=2C(C)=CC=CC=2N(CCCO)C=1COC1=CC=C(Cl)C=C1 QVXRTXGWLZZBMZ-UHFFFAOYSA-N 0.000 description 2
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 2
- XZBXAYCCBFTQHH-UHFFFAOYSA-N 3-chloropropylbenzene Chemical compound ClCCCC1=CC=CC=C1 XZBXAYCCBFTQHH-UHFFFAOYSA-N 0.000 description 2
- HJKGBRPNSJADMB-UHFFFAOYSA-N 3-phenylpyridine Chemical compound C1=CC=CC=C1C1=CC=CN=C1 HJKGBRPNSJADMB-UHFFFAOYSA-N 0.000 description 2
- ZDYLEXHUSVXJPP-UHFFFAOYSA-N 4-(cyclohexylmethyl)piperidine Chemical compound C1CNCCC1CC1CCCCC1 ZDYLEXHUSVXJPP-UHFFFAOYSA-N 0.000 description 2
- YEJRWHAVMIAJKC-UHFFFAOYSA-N 4-Butyrolactone Chemical compound O=C1CCCO1 YEJRWHAVMIAJKC-UHFFFAOYSA-N 0.000 description 2
- IQXUIDYRTHQTET-UHFFFAOYSA-N 4-amino-3-nitrophenol Chemical compound NC1=CC=C(O)C=C1[N+]([O-])=O IQXUIDYRTHQTET-UHFFFAOYSA-N 0.000 description 2
- ABGXADJDTPFFSZ-UHFFFAOYSA-N 4-benzylpiperidine Chemical compound C=1C=CC=CC=1CC1CCNCC1 ABGXADJDTPFFSZ-UHFFFAOYSA-N 0.000 description 2
- UTBULQCHEUWJNV-UHFFFAOYSA-N 4-phenylpiperidine Chemical compound C1CNCCC1C1=CC=CC=C1 UTBULQCHEUWJNV-UHFFFAOYSA-N 0.000 description 2
- HFDKKNHCYWNNNQ-YOGANYHLSA-N 75976-10-2 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@@H](NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)N)C(C)C)[C@@H](C)O)C1=CC=C(O)C=C1 HFDKKNHCYWNNNQ-YOGANYHLSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- 125000006374 C2-C10 alkenyl group Chemical group 0.000 description 2
- 125000005865 C2-C10alkynyl group Chemical group 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- IVOMOUWHDPKRLL-KQYNXXCUSA-N Cyclic adenosine monophosphate Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-KQYNXXCUSA-N 0.000 description 2
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 108091006027 G proteins Proteins 0.000 description 2
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 2
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 2
- 102000030782 GTP binding Human genes 0.000 description 2
- 108091000058 GTP-Binding Proteins 0.000 description 2
- 206010019280 Heart failures Diseases 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- HOKKHZGPKSLGJE-GSVOUGTGSA-N N-Methyl-D-aspartic acid Chemical compound CN[C@@H](C(O)=O)CC(O)=O HOKKHZGPKSLGJE-GSVOUGTGSA-N 0.000 description 2
- 102100038991 Neuropeptide Y receptor type 2 Human genes 0.000 description 2
- 101710197945 Neuropeptide Y receptor type 2 Proteins 0.000 description 2
- BHHGXPLMPWCGHP-UHFFFAOYSA-N Phenethylamine Chemical compound NCCC1=CC=CC=C1 BHHGXPLMPWCGHP-UHFFFAOYSA-N 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- 101000983124 Sus scrofa Pancreatic prohormone precursor Proteins 0.000 description 2
- 206010072810 Vascular wall hypertrophy Diseases 0.000 description 2
- XAKBSHICSHRJCL-UHFFFAOYSA-N [CH2]C(=O)C1=CC=CC=C1 Chemical group [CH2]C(=O)C1=CC=CC=C1 XAKBSHICSHRJCL-UHFFFAOYSA-N 0.000 description 2
- 229960000583 acetic acid Drugs 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 210000001789 adipocyte Anatomy 0.000 description 2
- 230000001919 adrenal effect Effects 0.000 description 2
- 125000005466 alkylenyl group Chemical group 0.000 description 2
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- 239000011609 ammonium molybdate Substances 0.000 description 2
- 235000018660 ammonium molybdate Nutrition 0.000 description 2
- APUPEJJSWDHEBO-UHFFFAOYSA-P ammonium molybdate Chemical compound [NH4+].[NH4+].[O-][Mo]([O-])(=O)=O APUPEJJSWDHEBO-UHFFFAOYSA-P 0.000 description 2
- 229940010552 ammonium molybdate Drugs 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 210000001367 artery Anatomy 0.000 description 2
- 150000001502 aryl halides Chemical class 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- YTLQFZVCLXFFRK-UHFFFAOYSA-N bendazol Chemical compound N=1C2=CC=CC=C2NC=1CC1=CC=CC=C1 YTLQFZVCLXFFRK-UHFFFAOYSA-N 0.000 description 2
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 2
- 235000019445 benzyl alcohol Nutrition 0.000 description 2
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 2
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 2
- 125000002619 bicyclic group Chemical group 0.000 description 2
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- YNHIGQDRGKUECZ-UHFFFAOYSA-L bis(triphenylphosphine)palladium(ii) dichloride Chemical compound [Cl-].[Cl-].[Pd+2].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-L 0.000 description 2
- 230000036760 body temperature Effects 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 125000001246 bromo group Chemical group Br* 0.000 description 2
- RDHPKYGYEGBMSE-UHFFFAOYSA-N bromoethane Chemical compound CCBr RDHPKYGYEGBMSE-UHFFFAOYSA-N 0.000 description 2
- GZUXJHMPEANEGY-UHFFFAOYSA-N bromomethane Chemical compound BrC GZUXJHMPEANEGY-UHFFFAOYSA-N 0.000 description 2
- 210000004900 c-terminal fragment Anatomy 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 230000008878 coupling Effects 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 2
- 229940043279 diisopropylamine Drugs 0.000 description 2
- KOXOZQFRIOGJGW-UHFFFAOYSA-N ethyl 2-benzyl-4-bromobutanoate Chemical compound CCOC(=O)C(CCBr)CC1=CC=CC=C1 KOXOZQFRIOGJGW-UHFFFAOYSA-N 0.000 description 2
- RJFIWCWTENIBKC-UHFFFAOYSA-N ethyl 2-piperidin-3-ylacetate Chemical compound CCOC(=O)CC1CCCNC1 RJFIWCWTENIBKC-UHFFFAOYSA-N 0.000 description 2
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- RWTNPBWLLIMQHL-UHFFFAOYSA-N fexofenadine Chemical compound C1=CC(C(C)(C(O)=O)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 RWTNPBWLLIMQHL-UHFFFAOYSA-N 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 2
- 210000003016 hypothalamus Anatomy 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 229910052500 inorganic mineral Inorganic materials 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 230000037041 intracellular level Effects 0.000 description 2
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 229960002510 mandelic acid Drugs 0.000 description 2
- 238000004949 mass spectrometry Methods 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 210000004379 membrane Anatomy 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 229940098779 methanesulfonic acid Drugs 0.000 description 2
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical compound COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 2
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 2
- 235000010755 mineral Nutrition 0.000 description 2
- 239000011707 mineral Substances 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- XTEPTNIYBDNBFL-UHFFFAOYSA-N n-[3-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]propyl]-2-(2-phenylphenyl)-n-(3-piperidin-1-ylpropyl)acetamide Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCN(C(=O)CC=1C(=CC=CC=1)C=1C=CC=CC=1)CCCN1CCCCC1 XTEPTNIYBDNBFL-UHFFFAOYSA-N 0.000 description 2
- ZQXCNYHYNIXSAS-UHFFFAOYSA-N n-[3-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]propyl]-3-piperidin-1-ylpropan-1-amine Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCNCCCN1CCCCC1 ZQXCNYHYNIXSAS-UHFFFAOYSA-N 0.000 description 2
- 230000001537 neural effect Effects 0.000 description 2
- 230000001272 neurogenic effect Effects 0.000 description 2
- 229960002748 norepinephrine Drugs 0.000 description 2
- SFLSHLFXELFNJZ-UHFFFAOYSA-N norepinephrine Natural products NCC(O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-UHFFFAOYSA-N 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 239000012044 organic layer Substances 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 239000001301 oxygen Chemical group 0.000 description 2
- 125000004430 oxygen atom Chemical group O* 0.000 description 2
- 125000003395 phenylethylamino group Chemical group [H]N(*)C([H])([H])C([H])([H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 150000003141 primary amines Chemical group 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 239000001294 propane Substances 0.000 description 2
- 230000001681 protective effect Effects 0.000 description 2
- 239000002464 receptor antagonist Substances 0.000 description 2
- 229940044551 receptor antagonist Drugs 0.000 description 2
- 229910052703 rhodium Inorganic materials 0.000 description 2
- 239000010948 rhodium Substances 0.000 description 2
- 229930195734 saturated hydrocarbon Natural products 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- 239000012056 semi-solid material Substances 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 239000011877 solvent mixture Substances 0.000 description 2
- 125000000547 substituted alkyl group Chemical group 0.000 description 2
- 239000011593 sulfur Chemical group 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 230000002889 sympathetic effect Effects 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 2
- DLYUQMMRRRQYAE-UHFFFAOYSA-N tetraphosphorus decaoxide Chemical compound O1P(O2)(=O)OP3(=O)OP1(=O)OP2(=O)O3 DLYUQMMRRRQYAE-UHFFFAOYSA-N 0.000 description 2
- 210000001103 thalamus Anatomy 0.000 description 2
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 2
- 230000002792 vascular Effects 0.000 description 2
- 210000004509 vascular smooth muscle cell Anatomy 0.000 description 2
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 2
- 238000010792 warming Methods 0.000 description 2
- DOMQFIFVDIAOOT-ROUUACIJSA-N (2S,3R)-N-[4-(2,6-dimethoxyphenyl)-5-(5-methylpyridin-3-yl)-1,2,4-triazol-3-yl]-3-(5-methylpyrimidin-2-yl)butane-2-sulfonamide Chemical compound COC1=C(C(=CC=C1)OC)N1C(=NN=C1C=1C=NC=C(C=1)C)NS(=O)(=O)[C@@H](C)[C@H](C)C1=NC=C(C=N1)C DOMQFIFVDIAOOT-ROUUACIJSA-N 0.000 description 1
- MAYZWDRUFKUGGP-VIFPVBQESA-N (3s)-1-[5-tert-butyl-3-[(1-methyltetrazol-5-yl)methyl]triazolo[4,5-d]pyrimidin-7-yl]pyrrolidin-3-ol Chemical compound CN1N=NN=C1CN1C2=NC(C(C)(C)C)=NC(N3C[C@@H](O)CC3)=C2N=N1 MAYZWDRUFKUGGP-VIFPVBQESA-N 0.000 description 1
- KUMVUFICIDUBDZ-ZVYVYBDNSA-N (4S)-5-[[(2S)-1-[[(2S)-1-[(2S)-2-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S,3S)-1-[[(2S)-4-amino-1-[[(2S)-1-[[(2S,3S)-1-[[(2S,3R)-1-[[(2S)-1-[[(2S)-5-amino-1-[[(2S)-1-[[(1S)-1-carboxy-2-(4-hydroxyphenyl)ethyl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-1,5-dioxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-hydroxy-1-oxobutan-2-yl]amino]-3-methyl-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-1,4-dioxobutan-2-yl]amino]-3-methyl-1-oxopentan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amino]-3-(1H-imidazol-4-yl)-1-oxopropan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-1-oxopropan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-1-oxopropan-2-yl]amino]-4-methylsulfanyl-1-oxobutan-2-yl]amino]-3-carboxy-1-oxopropan-2-yl]amino]-4-carboxy-1-oxobutan-2-yl]amino]-1-oxopropan-2-yl]carbamoyl]pyrrolidin-1-yl]-1-oxopropan-2-yl]amino]-3-carboxy-1-oxopropan-2-yl]amino]-4-[[2-[[(2S)-1-[(2S)-4-amino-2-[[(2S)-2-[[(2S)-1-[(2S)-6-amino-2-[[(2S)-3-hydroxy-2-[[(2S)-pyrrolidine-2-carbonyl]amino]propanoyl]amino]hexanoyl]pyrrolidine-2-carbonyl]amino]-3-carboxypropanoyl]amino]-4-oxobutanoyl]pyrrolidine-2-carbonyl]amino]acetyl]amino]-5-oxopentanoic acid Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CC(=O)N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CCC(=O)N)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CC1=CC=C(C=C1)O)C(=O)O)NC(=O)[C@H](CC2=CC=C(C=C2)O)NC(=O)[C@H](CC3=CNC=N3)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CC4=CC=C(C=C4)O)NC(=O)[C@H](CC5=CC=C(C=C5)O)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](C)NC(=O)[C@H](CCSC)NC(=O)[C@H](CC(=O)O)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](C)NC(=O)[C@@H]6CCCN6C(=O)[C@H](C)NC(=O)[C@H](CC(=O)O)NC(=O)[C@H](CCC(=O)O)NC(=O)CNC(=O)[C@@H]7CCCN7C(=O)[C@H](CC(=O)N)NC(=O)[C@H](CC(=O)O)NC(=O)[C@@H]8CCCN8C(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@@H]9CCCN9 KUMVUFICIDUBDZ-ZVYVYBDNSA-N 0.000 description 1
- WNYKZOWMSHGMPJ-YGPTWXQHSA-N (4S)-5-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S,3S)-1-[[(2S)-4-amino-1-[[(2S)-1-[[(2S,3S)-1-[[(2S,3R)-1-[[(2S)-1-[[(2S)-5-amino-1-[[(2S)-1-[[(1S)-1-carboxy-2-(4-hydroxyphenyl)ethyl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-1,5-dioxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-hydroxy-1-oxobutan-2-yl]amino]-3-methyl-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-1,4-dioxobutan-2-yl]amino]-3-methyl-1-oxopentan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amino]-3-(1H-imidazol-4-yl)-1-oxopropan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-1-oxopropan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-1-oxopropan-2-yl]amino]-4-methylsulfanyl-1-oxobutan-2-yl]amino]-3-carboxy-1-oxopropan-2-yl]amino]-5-oxo-4-[[(2S)-2-[[(2S)-pyrrolidine-2-carbonyl]amino]propanoyl]amino]pentanoic acid Chemical compound CC[C@H](C)[C@H](NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@H](Cc1c[nH]cn1)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](C)NC(=O)[C@H](CCSC)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H]1CCCN1)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](Cc1ccc(O)cc1)C(O)=O WNYKZOWMSHGMPJ-YGPTWXQHSA-N 0.000 description 1
- 125000004455 (C1-C3) alkylthio group Chemical group 0.000 description 1
- 125000006274 (C1-C3)alkoxy group Chemical group 0.000 description 1
- 125000004890 (C1-C6) alkylamino group Chemical group 0.000 description 1
- GRZHHTYDZVRPIC-UHFFFAOYSA-N (benzyloxy)acetic acid Chemical compound OC(=O)COCC1=CC=CC=C1 GRZHHTYDZVRPIC-UHFFFAOYSA-N 0.000 description 1
- UKGJZDSUJSPAJL-YPUOHESYSA-N (e)-n-[(1r)-1-[3,5-difluoro-4-(methanesulfonamido)phenyl]ethyl]-3-[2-propyl-6-(trifluoromethyl)pyridin-3-yl]prop-2-enamide Chemical compound CCCC1=NC(C(F)(F)F)=CC=C1\C=C\C(=O)N[C@H](C)C1=CC(F)=C(NS(C)(=O)=O)C(F)=C1 UKGJZDSUJSPAJL-YPUOHESYSA-N 0.000 description 1
- ZGYIXVSQHOKQRZ-COIATFDQSA-N (e)-n-[4-[3-chloro-4-(pyridin-2-ylmethoxy)anilino]-3-cyano-7-[(3s)-oxolan-3-yl]oxyquinolin-6-yl]-4-(dimethylamino)but-2-enamide Chemical compound N#CC1=CN=C2C=C(O[C@@H]3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC(C=C1Cl)=CC=C1OCC1=CC=CC=N1 ZGYIXVSQHOKQRZ-COIATFDQSA-N 0.000 description 1
- MOWXJLUYGFNTAL-DEOSSOPVSA-N (s)-[2-chloro-4-fluoro-5-(7-morpholin-4-ylquinazolin-4-yl)phenyl]-(6-methoxypyridazin-3-yl)methanol Chemical compound N1=NC(OC)=CC=C1[C@@H](O)C1=CC(C=2C3=CC=C(C=C3N=CN=2)N2CCOCC2)=C(F)C=C1Cl MOWXJLUYGFNTAL-DEOSSOPVSA-N 0.000 description 1
- SCZNXLWKYFICFV-UHFFFAOYSA-N 1,2,3,4,5,7,8,9-octahydropyrido[1,2-b]diazepine Chemical compound C1CCCNN2CCCC=C21 SCZNXLWKYFICFV-UHFFFAOYSA-N 0.000 description 1
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 1
- JLIDRDJNLAWIKT-UHFFFAOYSA-N 1,2-dimethyl-3h-benzo[e]indole Chemical compound C1=CC=CC2=C(C(=C(C)N3)C)C3=CC=C21 JLIDRDJNLAWIKT-UHFFFAOYSA-N 0.000 description 1
- VEFLKXRACNJHOV-UHFFFAOYSA-N 1,3-dibromopropane Chemical compound BrCCCBr VEFLKXRACNJHOV-UHFFFAOYSA-N 0.000 description 1
- APWRZPQBPCAXFP-UHFFFAOYSA-N 1-(1-oxo-2H-isoquinolin-5-yl)-5-(trifluoromethyl)-N-[2-(trifluoromethyl)pyridin-4-yl]pyrazole-4-carboxamide Chemical compound O=C1NC=CC2=C(C=CC=C12)N1N=CC(=C1C(F)(F)F)C(=O)NC1=CC(=NC=C1)C(F)(F)F APWRZPQBPCAXFP-UHFFFAOYSA-N 0.000 description 1
- MJUVRTYWUMPBTR-MRXNPFEDSA-N 1-(2,2-difluoro-1,3-benzodioxol-5-yl)-n-[1-[(2r)-2,3-dihydroxypropyl]-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)indol-5-yl]cyclopropane-1-carboxamide Chemical compound FC=1C=C2N(C[C@@H](O)CO)C(C(C)(CO)C)=CC2=CC=1NC(=O)C1(C=2C=C3OC(F)(F)OC3=CC=2)CC1 MJUVRTYWUMPBTR-MRXNPFEDSA-N 0.000 description 1
- FTARUZICTYBUKL-UHFFFAOYSA-N 1-(3-bromopropyl)-2-[(4-chlorophenoxy)methyl]-5,6-dimethylbenzimidazole Chemical compound BrCCCN1C=2C=C(C)C(C)=CC=2N=C1COC1=CC=C(Cl)C=C1 FTARUZICTYBUKL-UHFFFAOYSA-N 0.000 description 1
- ABLXQHNRSLVWMX-UHFFFAOYSA-N 1-(3-bromopropyl)-2-[(4-chlorophenoxy)methyl]benzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC1=NC2=CC=CC=C2N1CCCBr ABLXQHNRSLVWMX-UHFFFAOYSA-N 0.000 description 1
- CRXKZZVSNNGYHR-UHFFFAOYSA-N 1-(3-bromopropyl)-5,6-dichloro-2-[(4-chlorophenoxy)methyl]benzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC1=NC2=CC(Cl)=C(Cl)C=C2N1CCCBr CRXKZZVSNNGYHR-UHFFFAOYSA-N 0.000 description 1
- ZGIDAOWYVMTRNU-UHFFFAOYSA-N 1-(3-bromopropyl)-5-chloro-2-[(4-chlorophenoxy)methyl]benzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC1=NC2=CC(Cl)=CC=C2N1CCCBr ZGIDAOWYVMTRNU-UHFFFAOYSA-N 0.000 description 1
- HDDNBUNZJIQDBQ-UHFFFAOYSA-N 1-(3-chloropropyl)piperidine Chemical compound ClCCCN1CCCCC1 HDDNBUNZJIQDBQ-UHFFFAOYSA-N 0.000 description 1
- WIYQVAALSCQYLV-UHFFFAOYSA-N 1-[2-[2-[(4-chlorophenoxy)methyl]benzimidazol-1-yl]ethyl]-n,n-dimethylpiperidin-4-amine Chemical compound C1CC(N(C)C)CCN1CCN1C2=CC=CC=C2N=C1COC1=CC=C(Cl)C=C1 WIYQVAALSCQYLV-UHFFFAOYSA-N 0.000 description 1
- WGXCEQYRKKEQPR-UHFFFAOYSA-N 1-[3-(1-benzylpiperidin-4-yl)propyl]-2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazole Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCC(CC1)CCN1CC1=CC=CC=C1 WGXCEQYRKKEQPR-UHFFFAOYSA-N 0.000 description 1
- OROHLGCHVFLYDX-UHFFFAOYSA-N 1-[3-(4-benzylpiperidin-1-yl)propyl]-2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazole Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCN(CC1)CCC1CC1=CC=CC=C1 OROHLGCHVFLYDX-UHFFFAOYSA-N 0.000 description 1
- DKRUGNIMZBDGTH-UHFFFAOYSA-N 1-[3-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]propyl]-1,2,3,4-tetrahydroisoquinoline Chemical compound N=1C=2C(C)=CC=CC=2N(CCCC2C3=CC=CC=C3CCN2)C=1COC1=CC=C(Cl)C=C1 DKRUGNIMZBDGTH-UHFFFAOYSA-N 0.000 description 1
- ZFBDIWYBEKXXIC-UHFFFAOYSA-N 1-[3-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]propyl]-n,n-dimethylpiperidin-4-amine Chemical compound C1CC(N(C)C)CCN1CCCN1C2=CC=CC(C)=C2N=C1COC1=CC=C(Cl)C=C1 ZFBDIWYBEKXXIC-UHFFFAOYSA-N 0.000 description 1
- FLYBPINQLJJCEG-UHFFFAOYSA-N 1-[3-[2-[(4-chlorophenoxy)methyl]benzimidazol-1-yl]propyl]-n,n-dimethylpiperidin-4-amine Chemical compound C1CC(N(C)C)CCN1CCCN1C2=CC=CC=C2N=C1COC1=CC=C(Cl)C=C1 FLYBPINQLJJCEG-UHFFFAOYSA-N 0.000 description 1
- LOHCPUCKPIQSAS-UHFFFAOYSA-N 1-[3-[5,6-dichloro-2-[(4-chlorophenoxy)methyl]benzimidazol-1-yl]propyl]-n,n-dimethylpiperidin-4-amine Chemical compound C1CC(N(C)C)CCN1CCCN1C2=CC(Cl)=C(Cl)C=C2N=C1COC1=CC=C(Cl)C=C1 LOHCPUCKPIQSAS-UHFFFAOYSA-N 0.000 description 1
- KIPGFAFVRBSEEX-UHFFFAOYSA-N 1-[3-[5-chloro-2-[(4-chlorophenoxy)methyl]benzimidazol-1-yl]propyl]-n,n-dimethylpiperidin-4-amine Chemical compound C1CC(N(C)C)CCN1CCCN1C2=CC=C(Cl)C=C2N=C1COC1=CC=C(Cl)C=C1 KIPGFAFVRBSEEX-UHFFFAOYSA-N 0.000 description 1
- YJMBDNAHIOHXPM-UHFFFAOYSA-N 1-[4-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]-1-phenylbutyl]-n-methyl-4-phenylpiperidine-4-carboxamide Chemical compound C1CC(C(=O)NC)(C=2C=CC=CC=2)CCN1C(C=1C=CC=CC=1)CCCN(C1=CC=CC(C)=C1N=1)C=1COC1=CC=C(Cl)C=C1 YJMBDNAHIOHXPM-UHFFFAOYSA-N 0.000 description 1
- BCQWSIPTFYPLER-UHFFFAOYSA-N 1-[4-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]butyl]-1,2,3,4-tetrahydroisoquinoline Chemical compound N=1C=2C(C)=CC=CC=2N(CCCCC2C3=CC=CC=C3CCN2)C=1COC1=CC=C(Cl)C=C1 BCQWSIPTFYPLER-UHFFFAOYSA-N 0.000 description 1
- JCERKCRUSDOWLT-UHFFFAOYSA-N 1-bromopropan-1-ol Chemical compound CCC(O)Br JCERKCRUSDOWLT-UHFFFAOYSA-N 0.000 description 1
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000004972 1-butynyl group Chemical group [H]C([H])([H])C([H])([H])C#C* 0.000 description 1
- 125000006039 1-hexenyl group Chemical group 0.000 description 1
- 125000006019 1-methyl-1-propenyl group Chemical group 0.000 description 1
- 125000006017 1-propenyl group Chemical group 0.000 description 1
- VTDIWMPYBAVEDY-UHFFFAOYSA-N 1-propylpiperidine Chemical compound CCCN1CCCCC1 VTDIWMPYBAVEDY-UHFFFAOYSA-N 0.000 description 1
- 125000000530 1-propynyl group Chemical group [H]C([H])([H])C#C* 0.000 description 1
- DODRSIDSXPMYQJ-UHFFFAOYSA-N 1h-benzimidazol-4-ol Chemical compound OC1=CC=CC2=C1N=CN2 DODRSIDSXPMYQJ-UHFFFAOYSA-N 0.000 description 1
- UTQNKKSJPHTPBS-UHFFFAOYSA-N 2,2,2-trichloroethanone Chemical group ClC(Cl)(Cl)[C]=O UTQNKKSJPHTPBS-UHFFFAOYSA-N 0.000 description 1
- 125000000453 2,2,2-trichloroethyl group Chemical group [H]C([H])(*)C(Cl)(Cl)Cl 0.000 description 1
- YUGDKEWUYZXXRU-UHFFFAOYSA-N 2-(4-chlorophenoxy)acetonitrile Chemical compound ClC1=CC=C(OCC#N)C=C1 YUGDKEWUYZXXRU-UHFFFAOYSA-N 0.000 description 1
- GOCOKJDTFHEYKP-UHFFFAOYSA-N 2-(4-methylphenoxy)acetonitrile Chemical compound CC1=CC=C(OCC#N)C=C1 GOCOKJDTFHEYKP-UHFFFAOYSA-N 0.000 description 1
- QSKOLRSBJZXQJO-UHFFFAOYSA-N 2-(phenoxymethyl)-1-(3-piperidin-3-ylpropyl)benzimidazole Chemical compound C=1C=CC=CC=1OCC1=NC2=CC=CC=C2N1CCCC1CCCNC1 QSKOLRSBJZXQJO-UHFFFAOYSA-N 0.000 description 1
- XATKRQREMKIRLA-UHFFFAOYSA-N 2-(phenoxymethyl)-1h-benzimidazole Chemical compound N=1C2=CC=CC=C2NC=1COC1=CC=CC=C1 XATKRQREMKIRLA-UHFFFAOYSA-N 0.000 description 1
- SBRDRCVRUYIRLG-UHFFFAOYSA-N 2-(phenylmethoxymethyl)-1h-benzimidazol-4-ol Chemical compound N=1C=2C(O)=CC=CC=2NC=1COCC1=CC=CC=C1 SBRDRCVRUYIRLG-UHFFFAOYSA-N 0.000 description 1
- 125000003821 2-(trimethylsilyl)ethoxymethyl group Chemical group [H]C([H])([H])[Si](C([H])([H])[H])(C([H])([H])[H])C([H])([H])C(OC([H])([H])[*])([H])[H] 0.000 description 1
- NNWUEBIEOFQMSS-UHFFFAOYSA-N 2-Methylpiperidine Chemical compound CC1CCCCN1 NNWUEBIEOFQMSS-UHFFFAOYSA-N 0.000 description 1
- XVTFHKPLRYPIMX-UHFFFAOYSA-N 2-[(2,4-dichlorophenoxy)methyl]-1-(3-piperidin-2-ylpropyl)benzimidazole;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.ClC1=CC(Cl)=CC=C1OCC1=NC2=CC=CC=C2N1CCCC1NCCCC1 XVTFHKPLRYPIMX-UHFFFAOYSA-N 0.000 description 1
- VSAWVRKGLBTQNT-UHFFFAOYSA-N 2-[(2,4-dichlorophenoxy)methyl]-1-(3-piperidin-3-ylpropyl)benzimidazole Chemical compound ClC1=CC(Cl)=CC=C1OCC1=NC2=CC=CC=C2N1CCCC1CNCCC1 VSAWVRKGLBTQNT-UHFFFAOYSA-N 0.000 description 1
- CXKZUNIQSJXPPO-UHFFFAOYSA-N 2-[(2,4-dichlorophenoxy)methyl]-1h-benzimidazole Chemical compound ClC1=CC(Cl)=CC=C1OCC1=NC2=CC=CC=C2N1 CXKZUNIQSJXPPO-UHFFFAOYSA-N 0.000 description 1
- DTFBGFNYJMSVMO-UHFFFAOYSA-N 2-[(2,6-dichlorophenoxy)methyl]-1-(3-piperidin-3-ylpropyl)benzimidazole Chemical compound ClC1=CC=CC(Cl)=C1OCC1=NC2=CC=CC=C2N1CCCC1CNCCC1 DTFBGFNYJMSVMO-UHFFFAOYSA-N 0.000 description 1
- QRFAHQFHXAQJLP-UHFFFAOYSA-N 2-[(2-chlorophenoxy)methyl]-1-(3-piperidin-3-ylpropyl)benzimidazole Chemical compound ClC1=CC=CC=C1OCC1=NC2=CC=CC=C2N1CCCC1CNCCC1 QRFAHQFHXAQJLP-UHFFFAOYSA-N 0.000 description 1
- UIPOKFUXNJLYAO-UHFFFAOYSA-N 2-[(2-chlorophenoxy)methyl]-1h-benzimidazole Chemical compound ClC1=CC=CC=C1OCC1=NC2=CC=CC=C2N1 UIPOKFUXNJLYAO-UHFFFAOYSA-N 0.000 description 1
- FVWIXADCKIOMHP-UHFFFAOYSA-N 2-[(3,5-dichlorophenoxy)methyl]-1h-benzimidazol-4-ol Chemical compound N=1C=2C(O)=CC=CC=2NC=1COC1=CC(Cl)=CC(Cl)=C1 FVWIXADCKIOMHP-UHFFFAOYSA-N 0.000 description 1
- GZVYRNVTBZRAON-UHFFFAOYSA-N 2-[(3,5-dichlorophenoxy)methyl]-1h-benzimidazole Chemical compound ClC1=CC(Cl)=CC(OCC=2NC3=CC=CC=C3N=2)=C1 GZVYRNVTBZRAON-UHFFFAOYSA-N 0.000 description 1
- GHFXFPQEQOXPOL-UHFFFAOYSA-N 2-[(3,5-dichlorophenoxy)methyl]-4-methyl-1h-benzimidazole Chemical compound N=1C=2C(C)=CC=CC=2NC=1COC1=CC(Cl)=CC(Cl)=C1 GHFXFPQEQOXPOL-UHFFFAOYSA-N 0.000 description 1
- OTEYEOMFXHFXPJ-UHFFFAOYSA-N 2-[(3-chlorophenoxy)methyl]-1-(3-piperidin-3-ylpropyl)benzimidazole Chemical compound ClC1=CC=CC(OCC=2N(C3=CC=CC=C3N=2)CCCC2CNCCC2)=C1 OTEYEOMFXHFXPJ-UHFFFAOYSA-N 0.000 description 1
- ILZKMNQNWFATLP-UHFFFAOYSA-N 2-[(3-chlorophenoxy)methyl]-1h-benzimidazol-4-ol Chemical compound N=1C=2C(O)=CC=CC=2NC=1COC1=CC=CC(Cl)=C1 ILZKMNQNWFATLP-UHFFFAOYSA-N 0.000 description 1
- KSNDOUJAQJUSRG-UHFFFAOYSA-N 2-[(3-chlorophenoxy)methyl]-1h-benzimidazole Chemical compound ClC1=CC=CC(OCC=2NC3=CC=CC=C3N=2)=C1 KSNDOUJAQJUSRG-UHFFFAOYSA-N 0.000 description 1
- NHIBYEVVQCYNAG-UHFFFAOYSA-N 2-[(3-chlorophenoxy)methyl]-4-methyl-1h-benzimidazole Chemical compound N=1C=2C(C)=CC=CC=2NC=1COC1=CC=CC(Cl)=C1 NHIBYEVVQCYNAG-UHFFFAOYSA-N 0.000 description 1
- YVMVPAVYSJBLEA-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-1-(3-piperazin-1-ylpropyl)benzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC1=NC2=CC=CC=C2N1CCCN1CCNCC1 YVMVPAVYSJBLEA-UHFFFAOYSA-N 0.000 description 1
- GDXPWEKZBFXGRD-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-1-(3-piperidin-3-ylpropyl)benzimidazole;2,2,2-trifluoroacetic acid Chemical compound [O-]C(=O)C(F)(F)F.C1=CC(Cl)=CC=C1OCC1=NC2=CC=CC=C2N1CCCC1C[NH2+]CCC1 GDXPWEKZBFXGRD-UHFFFAOYSA-N 0.000 description 1
- RUGWZHXHSGSHNH-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-1-(3-piperidin-4-ylpropyl)-4-prop-2-enylbenzimidazole;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.C1=CC(Cl)=CC=C1OCC1=NC2=C(CC=C)C=CC=C2N1CCCC1CCNCC1 RUGWZHXHSGSHNH-UHFFFAOYSA-N 0.000 description 1
- ITCRYYSSWFKZJM-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-1h-benzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC1=NC2=CC=CC=C2N1 ITCRYYSSWFKZJM-UHFFFAOYSA-N 0.000 description 1
- FVVVWNWCITXTGD-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4,5,6,7-tetramethyl-1-(3-piperidin-4-ylpropyl)benzimidazole Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC2=C(C)C(C)=C(C)C(C)=C2N1CCCC1CCNCC1 FVVVWNWCITXTGD-UHFFFAOYSA-N 0.000 description 1
- PJKFKCREACTZED-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4,5,6,7-tetramethyl-1h-benzimidazole Chemical compound N=1C2=C(C)C(C)=C(C)C(C)=C2NC=1COC1=CC=C(Cl)C=C1 PJKFKCREACTZED-UHFFFAOYSA-N 0.000 description 1
- PLOGOWKFAUTRCY-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4,5-dimethyl-1-(3-piperidin-3-ylpropyl)benzimidazole Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC2=C(C)C(C)=CC=C2N1CCCC1CCCNC1 PLOGOWKFAUTRCY-UHFFFAOYSA-N 0.000 description 1
- BWKLGIFIUCYSFA-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4,5-dimethyl-1-(3-piperidin-3-ylpropyl)benzimidazole;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.C=1C=C(Cl)C=CC=1OCC1=NC2=C(C)C(C)=CC=C2N1CCCC1CCCNC1 BWKLGIFIUCYSFA-UHFFFAOYSA-N 0.000 description 1
- MUIZNWFTOUQWAU-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4,5-dimethyl-1-(3-piperidin-4-ylpropyl)benzimidazole Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC2=C(C)C(C)=CC=C2N1CCCC1CCNCC1 MUIZNWFTOUQWAU-UHFFFAOYSA-N 0.000 description 1
- GMBYBBOHQPULBM-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4,5-dimethyl-1-(4-piperidin-4-ylbutyl)benzimidazole Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC2=C(C)C(C)=CC=C2N1CCCCC1CCNCC1 GMBYBBOHQPULBM-UHFFFAOYSA-N 0.000 description 1
- CFWVFYXAPTVFMF-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4,5-dimethyl-1h-benzimidazole Chemical compound N=1C2=C(C)C(C)=CC=C2NC=1COC1=CC=C(Cl)C=C1 CFWVFYXAPTVFMF-UHFFFAOYSA-N 0.000 description 1
- KWGDURXIKWPRNZ-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-(3-piperidin-3-ylpropoxy)-1-(3-piperidin-3-ylpropyl)benzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC(N(C1=CC=C2)CCCC3CNCCC3)=NC1=C2OCCCC1CNCCC1 KWGDURXIKWPRNZ-UHFFFAOYSA-N 0.000 description 1
- KWGDURXIKWPRNZ-MIHMCVIASA-N 2-[(4-chlorophenoxy)methyl]-4-(3-piperidin-3-ylpropoxy)-1-[3-[(3r)-piperidin-3-yl]propyl]benzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC(N(C1=CC=C2)CCC[C@@H]3CNCCC3)=NC1=C2OCCCC1CNCCC1 KWGDURXIKWPRNZ-MIHMCVIASA-N 0.000 description 1
- PINQLGGZAXPNMU-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-(5-piperidin-4-ylpentoxy)-1-(3-piperidin-3-ylpropyl)benzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC(N(C1=CC=C2)CCCC3CNCCC3)=NC1=C2OCCCCCC1CCNCC1 PINQLGGZAXPNMU-UHFFFAOYSA-N 0.000 description 1
- HRDDOHXNPUEETF-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-[3-(3-methylpiperidin-1-yl)propoxy]-1-(3-piperidin-4-ylpropyl)benzimidazole Chemical compound C1C(C)CCCN1CCCOC1=CC=CC2=C1N=C(COC=1C=CC(Cl)=CC=1)N2CCCC1CCNCC1 HRDDOHXNPUEETF-UHFFFAOYSA-N 0.000 description 1
- LOJPNRBJPTZWHR-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-[3-[2-(2-piperidin-1-ylethyl)piperidin-1-yl]propoxy]-1-(3-piperidin-4-ylpropyl)benzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC(N(C1=CC=C2)CCCC3CCNCC3)=NC1=C2OCCCN1C(CCN2CCCCC2)CCCC1 LOJPNRBJPTZWHR-UHFFFAOYSA-N 0.000 description 1
- VXHNHOIJRPBDQL-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-methyl-1-(3-methyl-4-piperidin-1-ylbutyl)benzimidazole Chemical compound C1CCCCN1CC(C)CCN(C1=CC=CC(C)=C1N=1)C=1COC1=CC=C(Cl)C=C1 VXHNHOIJRPBDQL-UHFFFAOYSA-N 0.000 description 1
- ZQQIEKHLZFQXKX-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-methyl-1-(3-piperidin-2-ylpropyl)benzimidazole;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCC1CCCCN1 ZQQIEKHLZFQXKX-UHFFFAOYSA-N 0.000 description 1
- WAIGOCMMTMZHTI-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-methyl-1-(3-piperidin-3-ylpropyl)benzimidazole Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCC1CCCNC1 WAIGOCMMTMZHTI-UHFFFAOYSA-N 0.000 description 1
- DEDJLZPXLMZMCJ-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-methyl-1-(3-piperidin-4-ylpropyl)benzimidazole Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCC1CCNCC1 DEDJLZPXLMZMCJ-UHFFFAOYSA-N 0.000 description 1
- DHUZWWFKGRRJNH-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-methyl-1-(4-piperidin-4-ylbutyl)benzimidazole Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCCC1CCNCC1 DHUZWWFKGRRJNH-UHFFFAOYSA-N 0.000 description 1
- IFWSPNMLIIJMPR-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-methyl-1-(5-piperidin-3-ylpentyl)benzimidazole Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCCCC1CCCNC1 IFWSPNMLIIJMPR-UHFFFAOYSA-N 0.000 description 1
- ZUMODXCRPYTCGU-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-methyl-1-(5-piperidin-4-ylpentyl)benzimidazole Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCCCC1CCNCC1 ZUMODXCRPYTCGU-UHFFFAOYSA-N 0.000 description 1
- WELVOHGDTPFQBL-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-methyl-1-[3-(4-phenylpiperidin-1-yl)propyl]benzimidazole Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCN(CC1)CCC1C1=CC=CC=C1 WELVOHGDTPFQBL-UHFFFAOYSA-N 0.000 description 1
- WYBPGXWPOTZEOX-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-methyl-1-[3-(4-pyridin-2-ylpiperazin-1-yl)propyl]benzimidazole Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCN(CC1)CCN1C1=CC=CC=N1 WYBPGXWPOTZEOX-UHFFFAOYSA-N 0.000 description 1
- KCOXJCXEIYTGHP-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-methyl-1-[3-(4-pyrimidin-2-ylpiperazin-1-yl)propyl]benzimidazole Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCN(CC1)CCN1C1=NC=CC=N1 KCOXJCXEIYTGHP-UHFFFAOYSA-N 0.000 description 1
- WUIAIAUKABFUBA-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-methyl-1-[3-[2-(2-piperidin-1-ylethyl)piperidin-1-yl]propyl]benzimidazole Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCN1CCCCC1CCN1CCCCC1 WUIAIAUKABFUBA-UHFFFAOYSA-N 0.000 description 1
- NQEDQEHQYHQJCD-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-methyl-1-[3-[4-(2-phenylethyl)piperazin-1-yl]propyl]benzimidazole Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCN(CC1)CCN1CCC1=CC=CC=C1 NQEDQEHQYHQJCD-UHFFFAOYSA-N 0.000 description 1
- FHSPOLLNVGMXGG-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-methyl-1-[3-methyl-4-[2-(2-piperidin-1-ylethyl)piperidin-1-yl]butyl]benzimidazole Chemical compound C1CCCC(CCN2CCCCC2)N1CC(C)CCN(C1=CC=CC(C)=C1N=1)C=1COC1=CC=C(Cl)C=C1 FHSPOLLNVGMXGG-UHFFFAOYSA-N 0.000 description 1
- PWIVREHOFICPOY-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-methyl-1-[3-methyl-4-[4-(2-phenylethyl)piperazin-1-yl]butyl]benzimidazole Chemical compound C1CN(CCC=2C=CC=CC=2)CCN1CC(C)CCN(C1=CC=CC(C)=C1N=1)C=1COC1=CC=C(Cl)C=C1 PWIVREHOFICPOY-UHFFFAOYSA-N 0.000 description 1
- INFSHYKYPPXSOG-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-methyl-1-[4-(4-phenyl-4-propoxypiperidin-1-yl)pentyl]benzimidazole Chemical compound C1CC(OCCC)(C=2C=CC=CC=2)CCN1C(C)CCCN(C1=CC=CC(C)=C1N=1)C=1COC1=CC=C(Cl)C=C1 INFSHYKYPPXSOG-UHFFFAOYSA-N 0.000 description 1
- IILBJDZLFICLTI-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-methyl-1-[4-(4-phenylpiperidin-1-yl)butyl]benzimidazole Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCCN(CC1)CCC1C1=CC=CC=C1 IILBJDZLFICLTI-UHFFFAOYSA-N 0.000 description 1
- GFUKFOJLEYBIGZ-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-nitro-1h-benzimidazole Chemical compound N=1C=2C([N+](=O)[O-])=CC=CC=2NC=1COC1=CC=C(Cl)C=C1 GFUKFOJLEYBIGZ-UHFFFAOYSA-N 0.000 description 1
- CMKHXUMREQPJJS-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-4-phenylmethoxy-1-(3-piperidin-4-ylpropyl)benzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC(N(C1=CC=C2)CCCC3CCNCC3)=NC1=C2OCC1=CC=CC=C1 CMKHXUMREQPJJS-UHFFFAOYSA-N 0.000 description 1
- QSGMPTKGVLCMSV-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-5,6-dimethyl-1-(3-piperidin-1-ylpropyl)benzimidazole Chemical compound C1CCCCN1CCCN1C=2C=C(C)C(C)=CC=2N=C1COC1=CC=C(Cl)C=C1 QSGMPTKGVLCMSV-UHFFFAOYSA-N 0.000 description 1
- VKOBCTYJDWGTHK-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-5,6-dimethyl-1-(3-piperidin-3-ylpropyl)benzimidazole Chemical compound C1CCNCC1CCCN1C=2C=C(C)C(C)=CC=2N=C1COC1=CC=C(Cl)C=C1 VKOBCTYJDWGTHK-UHFFFAOYSA-N 0.000 description 1
- LAJMDAUVVGOREV-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-5,6-dimethyl-1-(4-piperidin-4-ylbutyl)benzimidazole Chemical compound C1CNCCC1CCCCN1C=2C=C(C)C(C)=CC=2N=C1COC1=CC=C(Cl)C=C1 LAJMDAUVVGOREV-UHFFFAOYSA-N 0.000 description 1
- BGXIHVSHWAGGSI-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-5,6-dimethyl-1h-benzimidazole Chemical compound N1C=2C=C(C)C(C)=CC=2N=C1COC1=CC=C(Cl)C=C1 BGXIHVSHWAGGSI-UHFFFAOYSA-N 0.000 description 1
- GWQPLSDIPPMQQT-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-5-methoxy-1-(3-piperidin-3-ylpropyl)benzimidazole Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC2=CC(OC)=CC=C2N1CCCC1CCCNC1 GWQPLSDIPPMQQT-UHFFFAOYSA-N 0.000 description 1
- PFBWWVZUDGZUGR-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-5-methoxy-1-(3-piperidin-3-ylpropyl)benzimidazole;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.C=1C=C(Cl)C=CC=1OCC1=NC2=CC(OC)=CC=C2N1CCCC1CCCNC1 PFBWWVZUDGZUGR-UHFFFAOYSA-N 0.000 description 1
- YHGKNNJHEBAUJD-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-5-methyl-1-(3-piperidin-3-ylpropyl)benzimidazole;2,2,2-trifluoroacetic acid Chemical compound [O-]C(=O)C(F)(F)F.C=1C=C(Cl)C=CC=1OCC1=NC2=CC(C)=CC=C2N1CCCC1CCC[NH2+]C1 YHGKNNJHEBAUJD-UHFFFAOYSA-N 0.000 description 1
- ULRIMYYGMZDLNX-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-6,7-dimethyl-1-(4-piperidin-4-ylbutyl)benzimidazole Chemical compound C1CNCCC1CCCCN1C2=C(C)C(C)=CC=C2N=C1COC1=CC=C(Cl)C=C1 ULRIMYYGMZDLNX-UHFFFAOYSA-N 0.000 description 1
- KRWNCGDGRWTVDX-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-6-methoxy-1-(3-piperidin-3-ylpropyl)benzimidazole Chemical compound C1CCNCC1CCCN1C2=CC(OC)=CC=C2N=C1COC1=CC=C(Cl)C=C1 KRWNCGDGRWTVDX-UHFFFAOYSA-N 0.000 description 1
- ONFAPXZYPHQRKQ-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-6-methoxy-1h-benzimidazole Chemical compound N1C2=CC(OC)=CC=C2N=C1COC1=CC=C(Cl)C=C1 ONFAPXZYPHQRKQ-UHFFFAOYSA-N 0.000 description 1
- JOYDLNMJDVWJPY-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-6-methyl-1-(3-piperidin-3-ylpropyl)benzimidazole;2,2,2-trifluoroacetic acid Chemical compound [O-]C(=O)C(F)(F)F.C1CC[NH2+]CC1CCCN1C2=CC(C)=CC=C2N=C1COC1=CC=C(Cl)C=C1 JOYDLNMJDVWJPY-UHFFFAOYSA-N 0.000 description 1
- BZWDVMGDUAJJLM-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-6-methyl-1h-benzimidazole Chemical compound N1C2=CC(C)=CC=C2N=C1COC1=CC=C(Cl)C=C1 BZWDVMGDUAJJLM-UHFFFAOYSA-N 0.000 description 1
- JYHSAWORPZWUIB-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-7-methyl-1-(3-piperidin-3-ylpropyl)benzimidazole Chemical compound C1CCNCC1CCCN1C=2C(C)=CC=CC=2N=C1COC1=CC=C(Cl)C=C1 JYHSAWORPZWUIB-UHFFFAOYSA-N 0.000 description 1
- PNRDXHLYASKKKO-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-7-methyl-1-(3-pyridin-3-ylpropyl)benzimidazole Chemical compound C=1C=CN=CC=1CCCN1C=2C(C)=CC=CC=2N=C1COC1=CC=C(Cl)C=C1 PNRDXHLYASKKKO-UHFFFAOYSA-N 0.000 description 1
- FJRKBUPGBYGMLP-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-7-phenylmethoxy-1-(3-piperidin-4-ylpropyl)benzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC1=NC2=CC=CC(OCC=3C=CC=CC=3)=C2N1CCCC1CCNCC1 FJRKBUPGBYGMLP-UHFFFAOYSA-N 0.000 description 1
- WWXBUXBBPTURGC-UHFFFAOYSA-N 2-[(4-chlorophenoxy)methyl]-n-(2-piperidin-1-ylethyl)-1-(3-piperidin-4-ylpropyl)benzimidazole-5-carboxamide Chemical compound C1=CC(Cl)=CC=C1OCC1=NC2=CC(C(=O)NCCN3CCCCC3)=CC=C2N1CCCC1CCNCC1 WWXBUXBBPTURGC-UHFFFAOYSA-N 0.000 description 1
- QOZZNZRBDATTEJ-UHFFFAOYSA-N 2-[(4-chlorophenyl)methyl]-1-(3-piperidin-3-ylpropyl)benzimidazole Chemical compound C1=CC(Cl)=CC=C1CC1=NC2=CC=CC=C2N1CCCC1CNCCC1 QOZZNZRBDATTEJ-UHFFFAOYSA-N 0.000 description 1
- GILMZLZCSKKFKH-UHFFFAOYSA-N 2-[(4-methylphenoxy)methyl]-1h-benzimidazol-4-ol Chemical compound C1=CC(C)=CC=C1OCC1=NC2=CC=CC(O)=C2N1 GILMZLZCSKKFKH-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- KVLSJBYIXZBUTQ-UHFFFAOYSA-N 2-[2-(4-chlorophenoxy)ethyl]-4-methyl-1-(3-pyrrolidin-3-yloxypropyl)benzimidazole Chemical compound C=1C=C(Cl)C=CC=1OCCC1=NC=2C(C)=CC=CC=2N1CCCOC1CCNC1 KVLSJBYIXZBUTQ-UHFFFAOYSA-N 0.000 description 1
- LDKARUHMXBCYCM-UHFFFAOYSA-N 2-[4-(1h-benzimidazol-2-ylmethoxy)phenyl]-1,3-thiazole Chemical compound N=1C2=CC=CC=C2NC=1COC(C=C1)=CC=C1C1=NC=CS1 LDKARUHMXBCYCM-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- RCINRTQHLIDDIO-UHFFFAOYSA-N 2-[4-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]-2-methylbutyl]-3,4-dihydro-1h-isoquinoline Chemical compound C1CC2=CC=CC=C2CN1CC(C)CCN(C1=CC=CC(C)=C1N=1)C=1COC1=CC=C(Cl)C=C1 RCINRTQHLIDDIO-UHFFFAOYSA-N 0.000 description 1
- FDAXYSFHHIIBIX-UHFFFAOYSA-N 2-[[3-(trifluoromethyl)phenoxy]methyl]-1h-benzimidazole Chemical compound FC(F)(F)C1=CC=CC(OCC=2NC3=CC=CC=C3N=2)=C1 FDAXYSFHHIIBIX-UHFFFAOYSA-N 0.000 description 1
- NAMYKGVDVNBCFQ-UHFFFAOYSA-N 2-bromopropane Chemical compound CC(C)Br NAMYKGVDVNBCFQ-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- 125000000069 2-butynyl group Chemical group [H]C([H])([H])C#CC([H])([H])* 0.000 description 1
- IQWYYIQTIYLVMP-UHFFFAOYSA-N 2-ethylphosphanylacetic acid Chemical compound CCPCC(O)=O IQWYYIQTIYLVMP-UHFFFAOYSA-N 0.000 description 1
- 125000006022 2-methyl-2-propenyl group Chemical group 0.000 description 1
- NFCPRRWCTNLGSN-UHFFFAOYSA-N 2-n-phenylbenzene-1,2-diamine Chemical class NC1=CC=CC=C1NC1=CC=CC=C1 NFCPRRWCTNLGSN-UHFFFAOYSA-N 0.000 description 1
- APVQNDRXQVWGKX-UHFFFAOYSA-N 2-nitrosoaniline Chemical compound NC1=CC=CC=C1N=O APVQNDRXQVWGKX-UHFFFAOYSA-N 0.000 description 1
- 125000006024 2-pentenyl group Chemical group 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- 125000002774 3,4-dimethoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C(OC([H])([H])[H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 1
- HCDMJFOHIXMBOV-UHFFFAOYSA-N 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-4,7-dihydropyrrolo[4,5]pyrido[1,2-d]pyrimidin-2-one Chemical compound C=1C2=C3N(CC)C(=O)N(C=4C(=C(OC)C=C(OC)C=4F)F)CC3=CN=C2NC=1CN1CCOCC1 HCDMJFOHIXMBOV-UHFFFAOYSA-N 0.000 description 1
- YYAYTNPNFKPFNG-UHFFFAOYSA-N 3-(2-methylpiperidin-1-yl)propan-1-amine Chemical compound CC1CCCCN1CCCN YYAYTNPNFKPFNG-UHFFFAOYSA-N 0.000 description 1
- YGYGASJNJTYNOL-CQSZACIVSA-N 3-[(4r)-2,2-dimethyl-1,1-dioxothian-4-yl]-5-(4-fluorophenyl)-1h-indole-7-carboxamide Chemical compound C1CS(=O)(=O)C(C)(C)C[C@@H]1C1=CNC2=C(C(N)=O)C=C(C=3C=CC(F)=CC=3)C=C12 YGYGASJNJTYNOL-CQSZACIVSA-N 0.000 description 1
- DOECXGQQUPGHFC-UHFFFAOYSA-N 3-[1-[3-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]propyl]piperidin-4-yl]-1-piperidin-1-ylpropan-1-one Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCN(CC1)CCC1CCC(=O)N1CCCCC1 DOECXGQQUPGHFC-UHFFFAOYSA-N 0.000 description 1
- OBHDGSDTJJJSCZ-UHFFFAOYSA-N 3-[1-[3-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]propyl]piperidin-4-yl]-n-[(4-methoxyphenyl)methyl]propanamide Chemical compound C1=CC(OC)=CC=C1CNC(=O)CCC1CCN(CCCN2C3=CC=CC(C)=C3N=C2COC=2C=CC(Cl)=CC=2)CC1 OBHDGSDTJJJSCZ-UHFFFAOYSA-N 0.000 description 1
- ZYRZCEWYFJISGV-UHFFFAOYSA-N 3-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]-n-(2-phenylethyl)propan-1-amine Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCNCCC1=CC=CC=C1 ZYRZCEWYFJISGV-UHFFFAOYSA-N 0.000 description 1
- RNLWAGHHLYMVJA-UHFFFAOYSA-N 3-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]propanal Chemical compound N=1C=2C(C)=CC=CC=2N(CCC=O)C=1COC1=CC=C(Cl)C=C1 RNLWAGHHLYMVJA-UHFFFAOYSA-N 0.000 description 1
- VVPFUGAGUDODCX-UHFFFAOYSA-N 3-[2-[(4-chlorophenoxy)methyl]-5,6-dimethylbenzimidazol-1-yl]propan-1-ol Chemical compound OCCCN1C=2C=C(C)C(C)=CC=2N=C1COC1=CC=C(Cl)C=C1 VVPFUGAGUDODCX-UHFFFAOYSA-N 0.000 description 1
- GWNHLGBLPGESAD-UHFFFAOYSA-N 3-[2-[(4-chlorophenoxy)methyl]benzimidazol-1-yl]propan-1-ol Chemical compound N=1C2=CC=CC=C2N(CCCO)C=1COC1=CC=C(Cl)C=C1 GWNHLGBLPGESAD-UHFFFAOYSA-N 0.000 description 1
- WNEODWDFDXWOLU-QHCPKHFHSA-N 3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[(2s)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one Chemical compound C([C@@H](N(CC1)C=2C=NC(NC=3C(N(C)C=C(C=3)C=3C(=C(N4C(C5=CC=6CC(C)(C)CC=6N5CC4)=O)N=CC=3)CO)=O)=CC=2)C)N1C1COC1 WNEODWDFDXWOLU-QHCPKHFHSA-N 0.000 description 1
- SRVXSISGYBMIHR-UHFFFAOYSA-N 3-[3-[3-(2-amino-2-oxoethyl)phenyl]-5-chlorophenyl]-3-(5-methyl-1,3-thiazol-2-yl)propanoic acid Chemical compound S1C(C)=CN=C1C(CC(O)=O)C1=CC(Cl)=CC(C=2C=C(CC(N)=O)C=CC=2)=C1 SRVXSISGYBMIHR-UHFFFAOYSA-N 0.000 description 1
- KHFSYNCKFXEVRF-UHFFFAOYSA-N 3-[5,6-dichloro-2-[(4-chlorophenoxy)methyl]benzimidazol-1-yl]propan-1-ol Chemical compound N=1C2=CC(Cl)=C(Cl)C=C2N(CCCO)C=1COC1=CC=C(Cl)C=C1 KHFSYNCKFXEVRF-UHFFFAOYSA-N 0.000 description 1
- MBGPBMZNUMDNNT-UHFFFAOYSA-N 3-[5-chloro-2-[(4-chlorophenoxy)methyl]benzimidazol-1-yl]propan-1-ol Chemical compound N=1C2=CC(Cl)=CC=C2N(CCCO)C=1COC1=CC=C(Cl)C=C1 MBGPBMZNUMDNNT-UHFFFAOYSA-N 0.000 description 1
- WGEZJWMZNGUEHR-UHFFFAOYSA-N 3-amino-4-nitrophenol Chemical compound NC1=CC(O)=CC=C1[N+]([O-])=O WGEZJWMZNGUEHR-UHFFFAOYSA-N 0.000 description 1
- FWTJDTQXENOXPC-UHFFFAOYSA-N 3-benzyloxolan-2-one Chemical compound O=C1OCCC1CC1=CC=CC=C1 FWTJDTQXENOXPC-UHFFFAOYSA-N 0.000 description 1
- UUCLVSDUMKMBSM-UHFFFAOYSA-N 3-benzylpyridine Chemical compound C=1C=CN=CC=1CC1=CC=CC=C1 UUCLVSDUMKMBSM-UHFFFAOYSA-N 0.000 description 1
- LEQKPQQOCKLKHK-UHFFFAOYSA-N 3-cyclohexylpiperidine Chemical compound C1CCCCC1C1CNCCC1 LEQKPQQOCKLKHK-UHFFFAOYSA-N 0.000 description 1
- JEGMWWXJUXDNJN-UHFFFAOYSA-N 3-methylpiperidine Chemical compound CC1CCCNC1 JEGMWWXJUXDNJN-UHFFFAOYSA-N 0.000 description 1
- NZYBILDYPCVNMU-UHFFFAOYSA-N 3-phenylpiperidine Chemical compound C1CCNCC1C1=CC=CC=C1 NZYBILDYPCVNMU-UHFFFAOYSA-N 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000002103 4,4'-dimethoxytriphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)(C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H])C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H] 0.000 description 1
- KROFADHHHXIPDD-UHFFFAOYSA-N 4,5-diaminocyclohexa-2,4-dien-1-one Chemical compound NC1=C(N)C=CC(=O)C1 KROFADHHHXIPDD-UHFFFAOYSA-N 0.000 description 1
- ROLMZTIHUMKEAI-UHFFFAOYSA-N 4,5-difluoro-2-hydroxybenzonitrile Chemical compound OC1=CC(F)=C(F)C=C1C#N ROLMZTIHUMKEAI-UHFFFAOYSA-N 0.000 description 1
- VJPPLCNBDLZIFG-ZDUSSCGKSA-N 4-[(3S)-3-(but-2-ynoylamino)piperidin-1-yl]-5-fluoro-2,3-dimethyl-1H-indole-7-carboxamide Chemical compound C(C#CC)(=O)N[C@@H]1CN(CCC1)C1=C2C(=C(NC2=C(C=C1F)C(=O)N)C)C VJPPLCNBDLZIFG-ZDUSSCGKSA-N 0.000 description 1
- HCDPRLPVCDGFLJ-UHFFFAOYSA-N 4-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]-2-methyl-n-(3-piperidin-1-ylpropyl)butan-1-amine Chemical compound C1CCCCN1CCCNCC(C)CCN(C1=CC=CC(C)=C1N=1)C=1COC1=CC=C(Cl)C=C1 HCDPRLPVCDGFLJ-UHFFFAOYSA-N 0.000 description 1
- SUSXFZZWMXSFAR-UHFFFAOYSA-N 4-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]-2-methyl-n-[3-(2-methylpiperidin-1-yl)propyl]butan-1-amine Chemical compound C1CCCC(C)N1CCCNCC(C)CCN(C1=CC=CC(C)=C1N=1)C=1COC1=CC=C(Cl)C=C1 SUSXFZZWMXSFAR-UHFFFAOYSA-N 0.000 description 1
- UDLSYWIIEDWCPW-UHFFFAOYSA-N 4-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]-2-methylbutan-1-amine Chemical compound N=1C2=C(C)C=CC=C2N(CCC(CN)C)C=1COC1=CC=C(Cl)C=C1 UDLSYWIIEDWCPW-UHFFFAOYSA-N 0.000 description 1
- CDAZKPSZCQRXCK-UHFFFAOYSA-N 4-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]-n,n,2-trimethylbutan-1-amine Chemical compound N=1C2=C(C)C=CC=C2N(CCC(C)CN(C)C)C=1COC1=CC=C(Cl)C=C1 CDAZKPSZCQRXCK-UHFFFAOYSA-N 0.000 description 1
- UFJMFUAGMPWPFQ-UHFFFAOYSA-N 4-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]butan-1-ol Chemical compound N=1C=2C(C)=CC=CC=2N(CCCCO)C=1COC1=CC=C(Cl)C=C1 UFJMFUAGMPWPFQ-UHFFFAOYSA-N 0.000 description 1
- YFCIFWOJYYFDQP-PTWZRHHISA-N 4-[3-amino-6-[(1S,3S,4S)-3-fluoro-4-hydroxycyclohexyl]pyrazin-2-yl]-N-[(1S)-1-(3-bromo-5-fluorophenyl)-2-(methylamino)ethyl]-2-fluorobenzamide Chemical compound CNC[C@@H](NC(=O)c1ccc(cc1F)-c1nc(cnc1N)[C@H]1CC[C@H](O)[C@@H](F)C1)c1cc(F)cc(Br)c1 YFCIFWOJYYFDQP-PTWZRHHISA-N 0.000 description 1
- XYWIPYBIIRTJMM-IBGZPJMESA-N 4-[[(2S)-2-[4-[5-chloro-2-[4-(trifluoromethyl)triazol-1-yl]phenyl]-5-methoxy-2-oxopyridin-1-yl]butanoyl]amino]-2-fluorobenzamide Chemical compound CC[C@H](N1C=C(OC)C(=CC1=O)C1=C(C=CC(Cl)=C1)N1C=C(N=N1)C(F)(F)F)C(=O)NC1=CC(F)=C(C=C1)C(N)=O XYWIPYBIIRTJMM-IBGZPJMESA-N 0.000 description 1
- MLCYDVREEAHNTI-UHFFFAOYSA-N 4-amino-5-nitrocyclohexa-2,4-dien-1-one Chemical compound NC1=C([N+]([O-])=O)CC(=O)C=C1 MLCYDVREEAHNTI-UHFFFAOYSA-N 0.000 description 1
- AQHWLPCHOWKOOK-UHFFFAOYSA-N 4-benzyl-1-[3-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]propyl]piperidin-4-ol Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCN(CC1)CCC1(O)CC1=CC=CC=C1 AQHWLPCHOWKOOK-UHFFFAOYSA-N 0.000 description 1
- PXACTUVBBMDKRW-UHFFFAOYSA-N 4-bromobenzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=C(Br)C=C1 PXACTUVBBMDKRW-UHFFFAOYSA-N 0.000 description 1
- GJNGXPDXRVXSEH-UHFFFAOYSA-N 4-chlorobenzonitrile Chemical compound ClC1=CC=C(C#N)C=C1 GJNGXPDXRVXSEH-UHFFFAOYSA-N 0.000 description 1
- IVYMIRMKXZAHRV-UHFFFAOYSA-N 4-chlorophenylacetonitrile Chemical compound ClC1=CC=C(CC#N)C=C1 IVYMIRMKXZAHRV-UHFFFAOYSA-N 0.000 description 1
- HQSCLNJCVFZWCW-UHFFFAOYSA-N 4-cyclohexylpiperidine Chemical compound C1CCCCC1C1CCNCC1 HQSCLNJCVFZWCW-UHFFFAOYSA-N 0.000 description 1
- SJZRECIVHVDYJC-UHFFFAOYSA-N 4-hydroxybutyric acid Chemical compound OCCCC(O)=O SJZRECIVHVDYJC-UHFFFAOYSA-N 0.000 description 1
- RVUMXRHPFWOAST-UHFFFAOYSA-N 4-methyl-2-[[3-(trifluoromethyl)phenoxy]methyl]-1h-benzimidazole Chemical compound N=1C=2C(C)=CC=CC=2NC=1COC1=CC=CC(C(F)(F)F)=C1 RVUMXRHPFWOAST-UHFFFAOYSA-N 0.000 description 1
- UZOFELREXGAFOI-UHFFFAOYSA-N 4-methylpiperidine Chemical compound CC1CCNCC1 UZOFELREXGAFOI-UHFFFAOYSA-N 0.000 description 1
- JOOXCMJARBKPKM-UHFFFAOYSA-M 4-oxopentanoate Chemical compound CC(=O)CCC([O-])=O JOOXCMJARBKPKM-UHFFFAOYSA-M 0.000 description 1
- OBKXEAXTFZPCHS-UHFFFAOYSA-N 4-phenylbutyric acid Chemical compound OC(=O)CCCC1=CC=CC=C1 OBKXEAXTFZPCHS-UHFFFAOYSA-N 0.000 description 1
- 125000005986 4-piperidonyl group Chemical group 0.000 description 1
- AOTFAVHCWWVWCR-UHFFFAOYSA-N 5,6-dichloro-2-[(2,4-dichlorophenoxy)methyl]-1-(3-piperidin-2-ylpropyl)benzimidazole;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.ClC1=CC(Cl)=CC=C1OCC1=NC2=CC(Cl)=C(Cl)C=C2N1CCCC1NCCCC1 AOTFAVHCWWVWCR-UHFFFAOYSA-N 0.000 description 1
- XRNNRYIOLFUQES-UHFFFAOYSA-N 5,6-dichloro-2-[(2,4-dichlorophenoxy)methyl]-1-(3-piperidin-3-ylpropyl)benzimidazole Chemical compound ClC1=CC(Cl)=CC=C1OCC1=NC2=CC(Cl)=C(Cl)C=C2N1CCCC1CNCCC1 XRNNRYIOLFUQES-UHFFFAOYSA-N 0.000 description 1
- USVPUYBERMLTOE-UHFFFAOYSA-N 5,6-dichloro-2-[(2,4-dichlorophenoxy)methyl]-1h-benzimidazole Chemical compound ClC1=CC(Cl)=CC=C1OCC1=NC2=CC(Cl)=C(Cl)C=C2N1 USVPUYBERMLTOE-UHFFFAOYSA-N 0.000 description 1
- HUIWRTSOLSGRMA-UHFFFAOYSA-N 5,6-dichloro-2-[(4-chlorophenoxy)methyl]-1-(3-piperidin-1-ylpropyl)benzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC1=NC2=CC(Cl)=C(Cl)C=C2N1CCCN1CCCCC1 HUIWRTSOLSGRMA-UHFFFAOYSA-N 0.000 description 1
- YTECPMHZJWZELC-UHFFFAOYSA-N 5,6-dichloro-2-[(4-chlorophenoxy)methyl]-1-(3-piperidin-3-ylpropyl)benzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC1=NC2=CC(Cl)=C(Cl)C=C2N1CCCC1CNCCC1 YTECPMHZJWZELC-UHFFFAOYSA-N 0.000 description 1
- URHMOBIFMKDEBZ-UHFFFAOYSA-N 5,6-dichloro-2-[(4-chlorophenoxy)methyl]-1-(3-piperidin-3-ylpropyl)benzimidazole;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.C1=CC(Cl)=CC=C1OCC1=NC2=CC(Cl)=C(Cl)C=C2N1CCCC1CNCCC1 URHMOBIFMKDEBZ-UHFFFAOYSA-N 0.000 description 1
- YUOOQIVYHDUXAL-UHFFFAOYSA-N 5,6-dichloro-2-[(4-chlorophenoxy)methyl]-1h-benzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC1=NC2=CC(Cl)=C(Cl)C=C2N1 YUOOQIVYHDUXAL-UHFFFAOYSA-N 0.000 description 1
- IRPVABHDSJVBNZ-RTHVDDQRSA-N 5-[1-(cyclopropylmethyl)-5-[(1R,5S)-3-(oxetan-3-yl)-3-azabicyclo[3.1.0]hexan-6-yl]pyrazol-3-yl]-3-(trifluoromethyl)pyridin-2-amine Chemical compound C1=C(C(F)(F)F)C(N)=NC=C1C1=NN(CC2CC2)C(C2[C@@H]3CN(C[C@@H]32)C2COC2)=C1 IRPVABHDSJVBNZ-RTHVDDQRSA-N 0.000 description 1
- QJIPEANAVSFHHZ-UHFFFAOYSA-N 5-chloro-2-[(4-chlorophenoxy)methyl]-1-(3-piperidin-1-ylpropyl)benzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC1=NC2=CC(Cl)=CC=C2N1CCCN1CCCCC1 QJIPEANAVSFHHZ-UHFFFAOYSA-N 0.000 description 1
- XQVHOECPLIRACI-UHFFFAOYSA-N 5-chloro-2-[(4-chlorophenoxy)methyl]-1-(3-piperidin-3-ylpropyl)benzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC1=NC2=CC(Cl)=CC=C2N1CCCC1CNCCC1 XQVHOECPLIRACI-UHFFFAOYSA-N 0.000 description 1
- KCBWAFJCKVKYHO-UHFFFAOYSA-N 6-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-[[4-[1-propan-2-yl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methyl]pyrazolo[3,4-d]pyrimidine Chemical compound C1(CC1)C1=NC=NC(=C1C1=NC=C2C(=N1)N(N=C2)CC1=CC=C(C=C1)C=1N(C=C(N=1)C(F)(F)F)C(C)C)OC KCBWAFJCKVKYHO-UHFFFAOYSA-N 0.000 description 1
- AOVFOFHJYXPODM-UHFFFAOYSA-N 6-chloro-2-[(4-chlorophenoxy)methyl]-1h-benzimidazole Chemical compound C1=CC(Cl)=CC=C1OCC1=NC2=CC(Cl)=CC=C2N1 AOVFOFHJYXPODM-UHFFFAOYSA-N 0.000 description 1
- UNQYAAAWKOOBFQ-UHFFFAOYSA-N 7-[(4-chlorophenyl)methyl]-8-[4-chloro-3-(trifluoromethoxy)phenoxy]-1-(3-hydroxypropyl)-3-methylpurine-2,6-dione Chemical compound C=1C=C(Cl)C=CC=1CN1C=2C(=O)N(CCCO)C(=O)N(C)C=2N=C1OC1=CC=C(Cl)C(OC(F)(F)F)=C1 UNQYAAAWKOOBFQ-UHFFFAOYSA-N 0.000 description 1
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 1
- ZRPZPNYZFSJUPA-UHFFFAOYSA-N ARS-1620 Chemical compound Oc1cccc(F)c1-c1c(Cl)cc2c(ncnc2c1F)N1CCN(CC1)C(=O)C=C ZRPZPNYZFSJUPA-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 206010002383 Angina Pectoris Diseases 0.000 description 1
- 208000000103 Anorexia Nervosa Diseases 0.000 description 1
- 208000019901 Anxiety disease Diseases 0.000 description 1
- 208000027559 Appetite disease Diseases 0.000 description 1
- 108010001478 Bacitracin Proteins 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical class OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 206010006550 Bulimia nervosa Diseases 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 206010007559 Cardiac failure congestive Diseases 0.000 description 1
- 206010007572 Cardiac hypertrophy Diseases 0.000 description 1
- 208000006029 Cardiomegaly Diseases 0.000 description 1
- 208000009132 Catalepsy Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 206010010904 Convulsion Diseases 0.000 description 1
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 1
- QGLBZNZGBLRJGS-UHFFFAOYSA-N Dihydro-3-methyl-2(3H)-furanone Chemical compound CC1CCOC1=O QGLBZNZGBLRJGS-UHFFFAOYSA-N 0.000 description 1
- YFPJFKYCVYXDJK-UHFFFAOYSA-N Diphenylphosphine oxide Chemical compound C=1C=CC=CC=1[P+](=O)C1=CC=CC=C1 YFPJFKYCVYXDJK-UHFFFAOYSA-N 0.000 description 1
- 208000031637 Erythroblastic Acute Leukemia Diseases 0.000 description 1
- 208000036566 Erythroleukaemia Diseases 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- GISRWBROCYNDME-PELMWDNLSA-N F[C@H]1[C@H]([C@H](NC1=O)COC1=NC=CC2=CC(=C(C=C12)OC)C(=O)N)C Chemical compound F[C@H]1[C@H]([C@H](NC1=O)COC1=NC=CC2=CC(=C(C=C12)OC)C(=O)N)C GISRWBROCYNDME-PELMWDNLSA-N 0.000 description 1
- 208000019454 Feeding and Eating disease Diseases 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 238000005863 Friedel-Crafts acylation reaction Methods 0.000 description 1
- 238000003547 Friedel-Crafts alkylation reaction Methods 0.000 description 1
- 238000005727 Friedel-Crafts reaction Methods 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 description 1
- XKMLYUALXHKNFT-UUOKFMHZSA-N Guanosine-5'-triphosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O XKMLYUALXHKNFT-UUOKFMHZSA-N 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-L L-tartrate(2-) Chemical compound [O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O FEWJPZIEWOKRBE-JCYAYHJZSA-L 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- 239000004367 Lipase Substances 0.000 description 1
- 108090001060 Lipase Proteins 0.000 description 1
- 102000004882 Lipase Human genes 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- 102000004129 N-Type Calcium Channels Human genes 0.000 description 1
- 108090000699 N-Type Calcium Channels Proteins 0.000 description 1
- AYCPARAPKDAOEN-LJQANCHMSA-N N-[(1S)-2-(dimethylamino)-1-phenylethyl]-6,6-dimethyl-3-[(2-methyl-4-thieno[3,2-d]pyrimidinyl)amino]-1,4-dihydropyrrolo[3,4-c]pyrazole-5-carboxamide Chemical compound C1([C@H](NC(=O)N2C(C=3NN=C(NC=4C=5SC=CC=5N=C(C)N=4)C=3C2)(C)C)CN(C)C)=CC=CC=C1 AYCPARAPKDAOEN-LJQANCHMSA-N 0.000 description 1
- FEYNFHSRETUBEM-UHFFFAOYSA-N N-[3-(1,1-difluoroethyl)phenyl]-1-(4-methoxyphenyl)-3-methyl-5-oxo-4H-pyrazole-4-carboxamide Chemical compound COc1ccc(cc1)N1N=C(C)C(C(=O)Nc2cccc(c2)C(C)(F)F)C1=O FEYNFHSRETUBEM-UHFFFAOYSA-N 0.000 description 1
- 102100029549 Neuropeptide Y receptor type 5 Human genes 0.000 description 1
- 101710198055 Neuropeptide Y receptor type 5 Proteins 0.000 description 1
- QQEYATYNIKJIHD-UHFFFAOYSA-N O=C1OCCC1CC1=CC=CC=C1.O=C1OCCC1CC1=CC=CC=C1 Chemical compound O=C1OCCC1CC1=CC=CC=C1.O=C1OCCC1CC1=CC=CC=C1 QQEYATYNIKJIHD-UHFFFAOYSA-N 0.000 description 1
- IDRGFNPZDVBSSE-UHFFFAOYSA-N OCCN1CCN(CC1)c1ccc(Nc2ncc3cccc(-c4cccc(NC(=O)C=C)c4)c3n2)c(F)c1F Chemical compound OCCN1CCN(CC1)c1ccc(Nc2ncc3cccc(-c4cccc(NC(=O)C=C)c4)c3n2)c(F)c1F IDRGFNPZDVBSSE-UHFFFAOYSA-N 0.000 description 1
- 208000008589 Obesity Diseases 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 108700020479 Pancreatic hormone Proteins 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 208000018262 Peripheral vascular disease Diseases 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- 229920002873 Polyethylenimine Polymers 0.000 description 1
- 229920000037 Polyproline Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 208000032851 Subarachnoid Hemorrhage Diseases 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 206010049418 Sudden Cardiac Death Diseases 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical compound OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 206010047163 Vasospasm Diseases 0.000 description 1
- 206010047853 Waxy flexibility Diseases 0.000 description 1
- ZZXDRXVIRVJQBT-UHFFFAOYSA-M Xylenesulfonate Chemical compound CC1=CC=CC(S([O-])(=O)=O)=C1C ZZXDRXVIRVJQBT-UHFFFAOYSA-M 0.000 description 1
- LXRZVMYMQHNYJB-UNXOBOICSA-N [(1R,2S,4R)-4-[[5-[4-[(1R)-7-chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methylthiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxycyclopentyl]methyl sulfamate Chemical compound CC1=C(C=C(S1)C(=O)C1=C(N[C@H]2C[C@H](O)[C@@H](COS(N)(=O)=O)C2)N=CN=C1)[C@@H]1NCCC2=C1C=C(Cl)C=C2 LXRZVMYMQHNYJB-UNXOBOICSA-N 0.000 description 1
- JJSCUXAFAJEQGB-QMMMGPOBSA-N [(1s)-1-isocyanatoethyl]benzene Chemical compound O=C=N[C@@H](C)C1=CC=CC=C1 JJSCUXAFAJEQGB-QMMMGPOBSA-N 0.000 description 1
- NVVPXXCVMGSNPN-UHFFFAOYSA-N [2-[(4-chlorophenoxy)methyl]-1-(3-piperidin-2-ylpropyl)benzimidazol-5-yl]-phenylmethanone;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.C1=CC(Cl)=CC=C1OCC1=NC2=CC(C(=O)C=3C=CC=CC=3)=CC=C2N1CCCC1NCCCC1 NVVPXXCVMGSNPN-UHFFFAOYSA-N 0.000 description 1
- ZIDUBVIZGMMNEF-UHFFFAOYSA-N [2-[(4-chlorophenoxy)methyl]-1-(3-piperidin-3-ylpropyl)benzimidazol-5-yl]-phenylmethanone Chemical compound C1=CC(Cl)=CC=C1OCC1=NC2=CC(C(=O)C=3C=CC=CC=3)=CC=C2N1CCCC1CNCCC1 ZIDUBVIZGMMNEF-UHFFFAOYSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- IPBVNPXQWQGGJP-UHFFFAOYSA-N acetic acid phenyl ester Natural products CC(=O)OC1=CC=CC=C1 IPBVNPXQWQGGJP-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 208000021841 acute erythroid leukemia Diseases 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 150000001266 acyl halides Chemical class 0.000 description 1
- 125000005585 adamantoate group Chemical group 0.000 description 1
- 210000001943 adrenal medulla Anatomy 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910001854 alkali hydroxide Inorganic materials 0.000 description 1
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 125000005115 alkyl carbamoyl group Chemical group 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 238000005915 ammonolysis reaction Methods 0.000 description 1
- 210000004727 amygdala Anatomy 0.000 description 1
- DZHSAHHDTRWUTF-SIQRNXPUSA-N amyloid-beta polypeptide 42 Chemical compound C([C@@H](C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@H](C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)NCC(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(O)=O)[C@@H](C)CC)C(C)C)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC(O)=O)C(C)C)C(C)C)C1=CC=CC=C1 DZHSAHHDTRWUTF-SIQRNXPUSA-N 0.000 description 1
- 230000000202 analgesic effect Effects 0.000 description 1
- 238000005571 anion exchange chromatography Methods 0.000 description 1
- 239000003957 anion exchange resin Substances 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 230000001387 anti-histamine Effects 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000001262 anti-secretory effect Effects 0.000 description 1
- 230000036506 anxiety Effects 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 150000004982 aromatic amines Chemical class 0.000 description 1
- 238000006172 aromatic nitration reaction Methods 0.000 description 1
- 206010003119 arrhythmia Diseases 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 125000002785 azepinyl group Chemical group 0.000 description 1
- 229960003071 bacitracin Drugs 0.000 description 1
- 229930184125 bacitracin Natural products 0.000 description 1
- CLKOFPXJLQSYAH-ABRJDSQDSA-N bacitracin A Chemical compound C1SC([C@@H](N)[C@@H](C)CC)=N[C@@H]1C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]1C(=O)N[C@H](CCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2N=CNC=2)C(=O)N[C@H](CC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)NCCCC1 CLKOFPXJLQSYAH-ABRJDSQDSA-N 0.000 description 1
- 239000003637 basic solution Substances 0.000 description 1
- DULCUDSUACXJJC-UHFFFAOYSA-N benzeneacetic acid ethyl ester Natural products CCOC(=O)CC1=CC=CC=C1 DULCUDSUACXJJC-UHFFFAOYSA-N 0.000 description 1
- MYONAGGJKCJOBT-UHFFFAOYSA-N benzimidazol-2-one Chemical compound C1=CC=CC2=NC(=O)N=C21 MYONAGGJKCJOBT-UHFFFAOYSA-N 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000004619 benzopyranyl group Chemical group O1C(C=CC2=C1C=CC=C2)* 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 125000000440 benzylamino group Chemical group [H]N(*)C([H])([H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 230000000975 bioactive effect Effects 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- LKXYJYDRLBPHRS-UHFFFAOYSA-N bromocyclopropane Chemical compound BrC1CC1 LKXYJYDRLBPHRS-UHFFFAOYSA-N 0.000 description 1
- AEILLAXRDHDKDY-UHFFFAOYSA-N bromomethylcyclopropane Chemical compound BrCC1CC1 AEILLAXRDHDKDY-UHFFFAOYSA-N 0.000 description 1
- 230000003182 bronchodilatating effect Effects 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- PRUGMAWKMXYGFZ-UHFFFAOYSA-N butan-2-yl butan-2-yloxycarbonyl carbonate Chemical compound CCC(C)OC(=O)OC(=O)OC(C)CC PRUGMAWKMXYGFZ-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000006630 butoxycarbonylamino group Chemical group 0.000 description 1
- 125000005467 butylenyl group Chemical group 0.000 description 1
- 125000004744 butyloxycarbonyl group Chemical group 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 1
- 239000000920 calcium hydroxide Substances 0.000 description 1
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 125000005518 carboxamido group Chemical group 0.000 description 1
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 1
- 150000003943 catecholamines Chemical class 0.000 description 1
- 238000005277 cation exchange chromatography Methods 0.000 description 1
- 210000003710 cerebral cortex Anatomy 0.000 description 1
- 210000004720 cerebrum Anatomy 0.000 description 1
- VZDYWEUILIUIDF-UHFFFAOYSA-J cerium(4+);disulfate Chemical compound [Ce+4].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O VZDYWEUILIUIDF-UHFFFAOYSA-J 0.000 description 1
- 229910000355 cerium(IV) sulfate Inorganic materials 0.000 description 1
- DGLFSNZWRYADFC-UHFFFAOYSA-N chembl2334586 Chemical compound C1CCC2=CN=C(N)N=C2C2=C1NC1=CC=C(C#CC(C)(O)C)C=C12 DGLFSNZWRYADFC-UHFFFAOYSA-N 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- KVSASDOGYIBWTA-UHFFFAOYSA-N chloro benzoate Chemical compound ClOC(=O)C1=CC=CC=C1 KVSASDOGYIBWTA-UHFFFAOYSA-N 0.000 description 1
- 125000002668 chloroacetyl group Chemical group ClCC(=O)* 0.000 description 1
- 210000003737 chromaffin cell Anatomy 0.000 description 1
- 238000013375 chromatographic separation Methods 0.000 description 1
- 125000000490 cinnamyl group Chemical group C(C=CC1=CC=CC=C1)* 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 1
- 210000001100 crypt cell Anatomy 0.000 description 1
- 239000002178 crystalline material Substances 0.000 description 1
- 125000004210 cyclohexylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 description 1
- GHVNFZFCNZKVNT-UHFFFAOYSA-N decanoic acid Chemical compound CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 150000004985 diamines Chemical class 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- 229960001760 dimethyl sulfoxide Drugs 0.000 description 1
- ZZVUWRFHKOJYTH-UHFFFAOYSA-N diphenhydramine Chemical group C=1C=CC=CC=1C(OCCN(C)C)C1=CC=CC=C1 ZZVUWRFHKOJYTH-UHFFFAOYSA-N 0.000 description 1
- AASUFOVSZUIILF-UHFFFAOYSA-N diphenylmethanone;sodium Chemical compound [Na].C=1C=CC=CC=1C(=O)C1=CC=CC=C1 AASUFOVSZUIILF-UHFFFAOYSA-N 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- GUVUOGQBMYCBQP-UHFFFAOYSA-N dmpu Chemical compound CN1CCCN(C)C1=O GUVUOGQBMYCBQP-UHFFFAOYSA-N 0.000 description 1
- 230000003291 dopaminomimetic effect Effects 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 238000000921 elemental analysis Methods 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 206010015037 epilepsy Diseases 0.000 description 1
- 208000028329 epileptic seizure Diseases 0.000 description 1
- 210000000981 epithelium Anatomy 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- HHFAWKCIHAUFRX-UHFFFAOYSA-N ethoxide Chemical compound CC[O-] HHFAWKCIHAUFRX-UHFFFAOYSA-N 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- QJRNNBKGHRTALC-UHFFFAOYSA-N ethyl 1-[3-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]propyl]-4-phenylpiperidine-4-carboxylate Chemical compound C1CC(C(=O)OCC)(C=2C=CC=CC=2)CCN1CCCN(C1=CC=CC(C)=C1N=1)C=1COC1=CC=C(Cl)C=C1 QJRNNBKGHRTALC-UHFFFAOYSA-N 0.000 description 1
- BMROUTYGZLSSDT-UHFFFAOYSA-N ethyl 1-[3-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]propyl]piperidine-4-carboxylate Chemical compound C1CC(C(=O)OCC)CCN1CCCN1C2=CC=CC(C)=C2N=C1COC1=CC=C(Cl)C=C1 BMROUTYGZLSSDT-UHFFFAOYSA-N 0.000 description 1
- GFEVXHSNRVJDDL-UHFFFAOYSA-N ethyl 1-[4-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]-2-methylbutyl]-4-phenylpiperidine-4-carboxylate Chemical compound C1CC(C(=O)OCC)(C=2C=CC=CC=2)CCN1CC(C)CCN(C1=CC=CC(C)=C1N=1)C=1COC1=CC=C(Cl)C=C1 GFEVXHSNRVJDDL-UHFFFAOYSA-N 0.000 description 1
- FCZCIXQGZOUIDN-UHFFFAOYSA-N ethyl 2-diethoxyphosphinothioyloxyacetate Chemical compound CCOC(=O)COP(=S)(OCC)OCC FCZCIXQGZOUIDN-UHFFFAOYSA-N 0.000 description 1
- RPWXYCRIAGBAGY-UHFFFAOYSA-N ethyl 2-pyridin-3-ylacetate Chemical compound CCOC(=O)CC1=CC=CN=C1 RPWXYCRIAGBAGY-UHFFFAOYSA-N 0.000 description 1
- YRZASYKSRQTOJT-UHFFFAOYSA-N ethyl 3-[1-[3-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]propyl]piperidin-4-yl]propanoate Chemical compound C1CC(CCC(=O)OCC)CCN1CCCN1C2=CC=CC(C)=C2N=C1COC1=CC=C(Cl)C=C1 YRZASYKSRQTOJT-UHFFFAOYSA-N 0.000 description 1
- PIEQSBWGIODYPE-UHFFFAOYSA-N ethyl 3-pyridin-3-ylprop-2-enoate Chemical compound CCOC(=O)C=CC1=CC=CN=C1 PIEQSBWGIODYPE-UHFFFAOYSA-N 0.000 description 1
- OKNBNKCLBOFYGM-UHFFFAOYSA-N ethyl 4-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]butanoate Chemical compound N=1C2=C(C)C=CC=C2N(CCCC(=O)OCC)C=1COC1=CC=C(Cl)C=C1 OKNBNKCLBOFYGM-UHFFFAOYSA-N 0.000 description 1
- FYLONIWABFGHJB-UHFFFAOYSA-N ethyl 4-bromo-2-methylbutanoate Chemical compound CCOC(=O)C(C)CCBr FYLONIWABFGHJB-UHFFFAOYSA-N 0.000 description 1
- XBPOBCXHALHJFP-UHFFFAOYSA-N ethyl 4-bromobutanoate Chemical compound CCOC(=O)CCCBr XBPOBCXHALHJFP-UHFFFAOYSA-N 0.000 description 1
- HKTDXDMLWVJPRL-UHFFFAOYSA-N ethyl 4-chloro-2-phenylbutanoate Chemical compound CCOC(=O)C(CCCl)C1=CC=CC=C1 HKTDXDMLWVJPRL-UHFFFAOYSA-N 0.000 description 1
- 125000005469 ethylenyl group Chemical group 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 230000002964 excitative effect Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000001400 expression cloning Methods 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 239000012894 fetal calf serum Substances 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 235000012631 food intake Nutrition 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical compound [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 238000005227 gel permeation chromatography Methods 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 239000003365 glass fiber Substances 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 102000031617 guanyl nucleotide binding proteins Human genes 0.000 description 1
- 108091009875 guanyl nucleotide binding proteins Proteins 0.000 description 1
- 150000005171 halobenzenes Chemical class 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-M hexanoate Chemical compound CCCCCC([O-])=O FUZZWVXGSFPDMH-UHFFFAOYSA-M 0.000 description 1
- 125000003104 hexanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- JKXCZYCVHPKTPK-UHFFFAOYSA-N hydrate;trihydrochloride Chemical compound O.Cl.Cl.Cl JKXCZYCVHPKTPK-UHFFFAOYSA-N 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- QYRFJLLXPINATB-UHFFFAOYSA-N hydron;2,4,5,6-tetrafluorobenzene-1,3-diamine;dichloride Chemical class Cl.Cl.NC1=C(F)C(N)=C(F)C(F)=C1F QYRFJLLXPINATB-UHFFFAOYSA-N 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical group [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000000185 intracerebroventricular administration Methods 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000005468 isobutylenyl group Chemical group 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000005928 isopropyloxycarbonyl group Chemical group [H]C([H])([H])C([H])(OC(*)=O)C([H])([H])[H] 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000004628 isothiazolidinyl group Chemical group S1N(CCC1)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000003965 isoxazolidinyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 229940058352 levulinate Drugs 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 235000019421 lipase Nutrition 0.000 description 1
- 230000004130 lipolysis Effects 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- 230000006742 locomotor activity Effects 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 102000006240 membrane receptors Human genes 0.000 description 1
- 108020004084 membrane receptors Proteins 0.000 description 1
- 125000005341 metaphosphate group Chemical group 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- NBTOZLQBSIZIKS-UHFFFAOYSA-N methoxide Chemical compound [O-]C NBTOZLQBSIZIKS-UHFFFAOYSA-N 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- SBCZRDBNDPGONI-UHFFFAOYSA-N methyl 2-[(4-chlorophenoxy)methyl]-1-(3-piperidin-4-ylpropyl)benzimidazole-5-carboxylate Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC2=CC(C(=O)OC)=CC=C2N1CCCC1CCNCC1 SBCZRDBNDPGONI-UHFFFAOYSA-N 0.000 description 1
- KXXOSICJTOCLFF-UHFFFAOYSA-N methyl 2-[(4-chlorophenoxy)methyl]-3-(3-piperidin-4-ylpropyl)benzimidazole-5-carboxylate Chemical compound C1CNCCC1CCCN1C2=CC(C(=O)OC)=CC=C2N=C1COC1=CC=C(Cl)C=C1 KXXOSICJTOCLFF-UHFFFAOYSA-N 0.000 description 1
- LGJSGPHSWYDAPG-UHFFFAOYSA-N methyl 2-[(4-chlorophenoxy)methyl]-3h-benzimidazole-5-carboxylate Chemical compound N=1C2=CC(C(=O)OC)=CC=C2NC=1COC1=CC=C(Cl)C=C1 LGJSGPHSWYDAPG-UHFFFAOYSA-N 0.000 description 1
- IZYBEMGNIUSSAX-UHFFFAOYSA-N methyl benzenecarboperoxoate Chemical compound COOC(=O)C1=CC=CC=C1 IZYBEMGNIUSSAX-UHFFFAOYSA-N 0.000 description 1
- 229940095102 methyl benzoate Drugs 0.000 description 1
- 229940102396 methyl bromide Drugs 0.000 description 1
- MBABOKRGFJTBAE-UHFFFAOYSA-N methyl methanesulfonate Chemical compound COS(C)(=O)=O MBABOKRGFJTBAE-UHFFFAOYSA-N 0.000 description 1
- 125000004092 methylthiomethyl group Chemical group [H]C([H])([H])SC([H])([H])* 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 210000002464 muscle smooth vascular Anatomy 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- ZABAXZVBVUDJPW-UHFFFAOYSA-N n'-[3-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]propyl]-n,n,n'-trimethylheptane-1,7-diamine Chemical compound N=1C2=C(C)C=CC=C2N(CCCN(C)CCCCCCCN(C)C)C=1COC1=CC=C(Cl)C=C1 ZABAXZVBVUDJPW-UHFFFAOYSA-N 0.000 description 1
- GFIYUHUYJFZAIM-UHFFFAOYSA-N n-[3-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]propyl]-n-(2-phenylethyl)-3-piperidin-1-ylpropan-1-amine Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCN(CCC=1C=CC=CC=1)CCCN1CCCCC1 GFIYUHUYJFZAIM-UHFFFAOYSA-N 0.000 description 1
- XOKNAZJYJSMXPP-UHFFFAOYSA-N n-[4-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]-2-methylbutyl]-1,2,3,4-tetrahydronaphthalen-1-amine Chemical compound C1CCC2=CC=CC=C2C1NCC(C)CCN(C1=CC=CC(C)=C1N=1)C=1COC1=CC=C(Cl)C=C1 XOKNAZJYJSMXPP-UHFFFAOYSA-N 0.000 description 1
- DUUCQDABFOGBCC-UHFFFAOYSA-N n-[[1-[4-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]-2-methylbutyl]-4-phenylpiperidin-4-yl]methyl]acetamide Chemical compound C1CC(CNC(C)=O)(C=2C=CC=CC=2)CCN1CC(C)CCN(C1=CC=CC(C)=C1N=1)C=1COC1=CC=C(Cl)C=C1 DUUCQDABFOGBCC-UHFFFAOYSA-N 0.000 description 1
- VFBILHPIHUPBPZ-UHFFFAOYSA-N n-[[2-[4-(difluoromethoxy)-3-propan-2-yloxyphenyl]-1,3-oxazol-4-yl]methyl]-2-ethoxybenzamide Chemical compound CCOC1=CC=CC=C1C(=O)NCC1=COC(C=2C=C(OC(C)C)C(OC(F)F)=CC=2)=N1 VFBILHPIHUPBPZ-UHFFFAOYSA-N 0.000 description 1
- NOYHNDWYCFWMRV-UHFFFAOYSA-N n-benzyl-3-[1-[3-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]propyl]piperidin-4-yl]propan-1-amine Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCN(CC1)CCC1CCCNCC1=CC=CC=C1 NOYHNDWYCFWMRV-UHFFFAOYSA-N 0.000 description 1
- MBODBZZCCRMUNG-UHFFFAOYSA-N n-benzyl-3-[1-[3-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]propyl]piperidin-4-yl]propanamide Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCN(CC1)CCC1CCC(=O)NCC1=CC=CC=C1 MBODBZZCCRMUNG-UHFFFAOYSA-N 0.000 description 1
- NUIKVCZIOLPTHO-UHFFFAOYSA-N n-benzyl-3-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]propan-1-amine Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCNCC1=CC=CC=C1 NUIKVCZIOLPTHO-UHFFFAOYSA-N 0.000 description 1
- QLVBKRNLKAUJMJ-UHFFFAOYSA-N n-benzyl-n-[3-[2-[(4-chlorophenoxy)methyl]-4-methylbenzimidazol-1-yl]propyl]-3-piperidin-1-ylpropan-1-amine Chemical compound C=1C=C(Cl)C=CC=1OCC1=NC=2C(C)=CC=CC=2N1CCCN(CC=1C=CC=CC=1)CCCN1CCCCC1 QLVBKRNLKAUJMJ-UHFFFAOYSA-N 0.000 description 1
- DOWVMJFBDGWVML-UHFFFAOYSA-N n-cyclohexyl-n-methyl-4-(1-oxidopyridin-1-ium-3-yl)imidazole-1-carboxamide Chemical compound C1=NC(C=2C=[N+]([O-])C=CC=2)=CN1C(=O)N(C)C1CCCCC1 DOWVMJFBDGWVML-UHFFFAOYSA-N 0.000 description 1
- NNKPHNTWNILINE-UHFFFAOYSA-N n-cyclopropyl-3-fluoro-4-methyl-5-[3-[[1-[2-[2-(methylamino)ethoxy]phenyl]cyclopropyl]amino]-2-oxopyrazin-1-yl]benzamide Chemical compound CNCCOC1=CC=CC=C1C1(NC=2C(N(C=3C(=C(F)C=C(C=3)C(=O)NC3CC3)C)C=CN=2)=O)CC1 NNKPHNTWNILINE-UHFFFAOYSA-N 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- 125000001038 naphthoyl group Chemical group C1(=CC=CC2=CC=CC=C12)C(=O)* 0.000 description 1
- 230000007383 nerve stimulation Effects 0.000 description 1
- 108010058785 neuropeptide Y (13-36) Proteins 0.000 description 1
- 108010069562 neuropeptide Y (2-36) Proteins 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 230000009871 nonspecific binding Effects 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 150000004987 o-phenylenediamines Chemical class 0.000 description 1
- 235000020824 obesity Nutrition 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-M octanoate Chemical compound CCCCCCCC([O-])=O WWZKQHOCKIZLMA-UHFFFAOYSA-M 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 1
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 1
- 230000001734 parasympathetic effect Effects 0.000 description 1
- 210000005034 parasympathetic neuron Anatomy 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- DYUMLJSJISTVPV-UHFFFAOYSA-N phenyl propanoate Chemical compound CCC(=O)OC1=CC=CC=C1 DYUMLJSJISTVPV-UHFFFAOYSA-N 0.000 description 1
- 229940049953 phenylacetate Drugs 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N phenylbenzene Natural products C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 1
- 229950009215 phenylbutanoic acid Drugs 0.000 description 1
- 150000004986 phenylenediamines Chemical class 0.000 description 1
- DYFXGORUJGZJCA-UHFFFAOYSA-N phenylmethanediamine Chemical compound NC(N)C1=CC=CC=C1 DYFXGORUJGZJCA-UHFFFAOYSA-N 0.000 description 1
- 208000028591 pheochromocytoma Diseases 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- UHZYTMXLRWXGPK-UHFFFAOYSA-N phosphorus pentachloride Chemical compound ClP(Cl)(Cl)(Cl)Cl UHZYTMXLRWXGPK-UHFFFAOYSA-N 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 125000005544 phthalimido group Chemical group 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 125000005547 pivalate group Chemical group 0.000 description 1
- 239000003880 polar aprotic solvent Substances 0.000 description 1
- 229920001467 poly(styrenesulfonates) Polymers 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 229920000137 polyphosphoric acid Polymers 0.000 description 1
- LZMJNVRJMFMYQS-UHFFFAOYSA-N poseltinib Chemical compound C1CN(C)CCN1C(C=C1)=CC=C1NC1=NC(OC=2C=C(NC(=O)C=C)C=CC=2)=C(OC=C2)C2=N1 LZMJNVRJMFMYQS-UHFFFAOYSA-N 0.000 description 1
- 230000001242 postsynaptic effect Effects 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011736 potassium bicarbonate Substances 0.000 description 1
- 235000015497 potassium bicarbonate Nutrition 0.000 description 1
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- KCXFHTAICRTXLI-UHFFFAOYSA-N propane-1-sulfonic acid Chemical compound CCCS(O)(=O)=O KCXFHTAICRTXLI-UHFFFAOYSA-N 0.000 description 1
- 125000001325 propanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000005470 propylenyl group Chemical group 0.000 description 1
- 125000004742 propyloxycarbonyl group Chemical group 0.000 description 1
- UORVCLMRJXCDCP-UHFFFAOYSA-M propynoate Chemical compound [O-]C(=O)C#C UORVCLMRJXCDCP-UHFFFAOYSA-M 0.000 description 1
- 238000002731 protein assay Methods 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 210000005234 proximal tubule cell Anatomy 0.000 description 1
- 208000020016 psychiatric disease Diseases 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- QJZUKDFHGGYHMC-UHFFFAOYSA-N pyridine-3-carbaldehyde Chemical compound O=CC1=CC=CN=C1 QJZUKDFHGGYHMC-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 210000005227 renal system Anatomy 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000007790 scraping Methods 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 238000007423 screening assay Methods 0.000 description 1
- 229940116351 sebacate Drugs 0.000 description 1
- CXMXRPHRNRROMY-UHFFFAOYSA-L sebacate(2-) Chemical compound [O-]C(=O)CCCCCCCCC([O-])=O CXMXRPHRNRROMY-UHFFFAOYSA-L 0.000 description 1
- XIIOFHFUYBLOLW-UHFFFAOYSA-N selpercatinib Chemical compound OC(COC=1C=C(C=2N(C=1)N=CC=2C#N)C=1C=NC(=CC=1)N1CC2N(C(C1)C2)CC=1C=NC(=CC=1)OC)(C)C XIIOFHFUYBLOLW-UHFFFAOYSA-N 0.000 description 1
- 210000001044 sensory neuron Anatomy 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- XGVXKJKTISMIOW-ZDUSSCGKSA-N simurosertib Chemical compound N1N=CC(C=2SC=3C(=O)NC(=NC=3C=2)[C@H]2N3CCC(CC3)C2)=C1C XGVXKJKTISMIOW-ZDUSSCGKSA-N 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- 159000000000 sodium salts Chemical group 0.000 description 1
- KKVTYAVXTDIPAP-UHFFFAOYSA-M sodium;methanesulfonate Chemical compound [Na+].CS([O-])(=O)=O KKVTYAVXTDIPAP-UHFFFAOYSA-M 0.000 description 1
- 210000001679 solitary nucleus Anatomy 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- TYFQFVWCELRYAO-UHFFFAOYSA-L suberate(2-) Chemical compound [O-]C(=O)CCCCCCC([O-])=O TYFQFVWCELRYAO-UHFFFAOYSA-L 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 210000002820 sympathetic nervous system Anatomy 0.000 description 1
- 210000000225 synapse Anatomy 0.000 description 1
- 230000000946 synaptic effect Effects 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 239000002462 tachykinin receptor antagonist Substances 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- PUGUQINMNYINPK-UHFFFAOYSA-N tert-butyl 4-(2-chloroacetyl)piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCN(C(=O)CCl)CC1 PUGUQINMNYINPK-UHFFFAOYSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000006090 thiamorpholinyl sulfone group Chemical group 0.000 description 1
- 125000006089 thiamorpholinyl sulfoxide group Chemical group 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 231100000027 toxicology Toxicity 0.000 description 1
- 125000005270 trialkylamine group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- YLGRTLMDMVAFNI-UHFFFAOYSA-N tributyl(prop-2-enyl)stannane Chemical compound CCCC[Sn](CCCC)(CCCC)CC=C YLGRTLMDMVAFNI-UHFFFAOYSA-N 0.000 description 1
- DBODNPMIIRVQGW-UHFFFAOYSA-N trihydrate;dihydrochloride Chemical compound O.O.O.Cl.Cl DBODNPMIIRVQGW-UHFFFAOYSA-N 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 125000003774 valeryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000004218 vascular function Effects 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 238000002424 x-ray crystallography Methods 0.000 description 1
- 229940071104 xylenesulfonate Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/06—Benzimidazoles; Hydrogenated benzimidazoles with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 2
- C07D235/12—Radicals substituted by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- Neuropeptide Y is a peptide present in the central and peripheral nervous systems. The peptide co-exists with noradrenaline in many neurons and acts as a neurotransmitter per se or synergistically together with noradrenaline. Neuropeptide Y- containing fibers are numerous around arteries in the heart, but are also found around the arteries in the respiratory tract, the gastrointestinal tract, and the genitourinary tract. Neuropeptide Y is also present in the cerebrum with effects on blood pressure, feeding, and the release of different hormones. Alterations in central concentrations of neuropeptide Y have been implicated in the etiology of psychiatric disorders. Neuropeptide Y was discovered, isolated and sequenced in
- Neuropeptide Y is a member of the pancreatic family of peptides and shares significant sequence homology with pancreatic polypeptide and peptide YY.
- Neuropeptide Y and the other members of its family of peptides all feature a tertiary structure consisting of an N- terminal polyproline helix and an amphiphilic ⁇ -helix, connected with a ⁇ -turn, creating a hairpin-like loop, which is sometimes referred to as the pancreatic polypeptide (PP) fold.
- the helices are kept together by hydrophobic interactions.
- the amidated C-terminal end projects away from the hairpin loop.
- neuropeptide Y was identified as being the most abundant peptide in the central nervous system with widespread distribution including the cortex, brainstem, hippocampus, hypotahlamus, amygdala, and thalamus as well as being present in the peripheral nervous system in sympathetic neurons and adrenal chromaffin cells.
- Neuropeptide Y seems to fulfill the main criteria for a role as a neurotransmitter, as it is stored in synaptic granules, is released upon electrical nerve stimulation, and acts at specific receptors.
- neuropeptide Y is an important messenger in its own right, probably in the brain, where neuropeptide Y potently inhibits the activity of adenylate cyclase and induces an increase in the intracellular levels of calcium.
- Central injection of neuropeptide Y results in blood pressure changes, increased feeding, increased fat storage, elevated blood sugar and insulin, decreased locomotor activity, reduced body temperature, and catalepsy.
- Neuropeptide Y acts upon membrane receptors that are dependent on guanyl-nucleotide binding proteins, known as G protein-coupled receptors.
- G proteins are a family of membrane proteins that become activated only after binding guanosine triphosphate. Activated G proteins in turn activate an amplifier enzyme on the inner face of a membrane; the enzyme then converts precursor molecules into second messengers.
- Neuropeptide Y appears to interact with a family of closely related receptors. These receptors are generally classified into several subtypes based upon the ability of different tissues and receptors to bind different fragments of neuropeptide Y and other members of the PP family of peptides.
- the Yl receptor subtype appears to be the major vascular neuropeptide Y receptor.
- the Y2 receptor subtypes can also occur postjunctionally on vascular smooth muscle.
- the as-yet- unisolated Y3 receptor subtype appears to be neuropeptide Y-spe ⁇ fic, not binding peptide YY. This receptor is likely to be present in the adrenal tissues, medulla, heart, and brain stem, among other areas.
- neuropeptide Y receptor antagonists In view of the wide number of clinical maladies associated with an excess of neuropeptide Y, the development of neuropeptide Y receptor antagonists will serve to control these clinical conditions.
- the earliest such receptor antagonists such as Patent Cooperation Treaty Patent Publication WO 91/08223, published June 13, 1991, and Patent Cooperation Treaty Patent Publication WO 94/00486, published January 6, 1994, were peptide derivatives. These antagonists are of limited pharmaceutical utility because of their metabolic instability.
- This invention provides a class of potent non-peptide neuropeptide Y receptor antagonists.
- the compounds of the present invention do not suffer from the shortcomings, in terms of metabolic instability, of known peptide-based neuropeptide Y receptor antagonists.
- This invention encompasses methods for the treatment or prevention of a physiological disorder associated with an excess of neuropeptide Y, which method comprises administering to a mammal in need of said treatment an effective amount of a compound of Formula I
- R 1 is phenyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, phenyKCi-C ⁇ alkylenyl)-, phenyKCi-C ⁇ alkoxy), phenoxy(C ⁇ -C6 alkylenyl)-, phenyKCi-C ⁇ alkoxy )-(C ⁇ -C ⁇ alkylenyl)-, naphthyl, naphthyKCi-C ⁇ alkylenyl)-, naphthyKCi-C ⁇ alkoxy), naphthyloxy(C ⁇ -C6 alkylenyl)-, or naphthyl(C ⁇ -C ⁇ alkoxy )-(C ⁇ -C6 alkylenyl)-,
- any one of which phenyl, C 3 -C8 cycloalkyl, phenoxy, naphthyl, or naphthyloxy moieties may be substituted with one or groups selected from the group consisting of halo, trifluoromethyl, Ci-C ⁇ alkyl, C 2 -C7 alkenyl, C2-C7 alkynyl, Ci-C ⁇ alkoxy, Ci-C ⁇ alkylthio, Ci-C ⁇ alkylamino, heterocyclic, unsaturated heterocyclic, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, phenyl, phenoxy, phenyKCi-C ⁇ alkylenyl)-, phenyKCi-C ⁇ alkoxy)-, benzoyl, phenyl(C2-C7 alkanoyl)-, and phenyl(C2-C7 alkanoyloxy)-;
- R 2 is C 1 -C12 alkyl, C 2 -C 7 alkenyl, C 2 -C 7 alkynyl, C 2 -C 7 alkanoyl, Ci-C ⁇ alkoxy, heterocyclic(C ⁇ -C6 alkylenyl)-, C3-C8 cycloalkyl, C 3 -C 8 cycloalkenyl, unsaturated heterocyclic(C ⁇ -C6 alkylenyl)-, heterocyclic(C ⁇ -C 6 alkoxy)-, unsaturated heterocyclic(C ⁇ -C6 alkoxy)-, phenyl, phenyKCi-C ⁇ alkylenyl)-, naphthyl, naphthyKCi-C ⁇ alkylenyl)-, phenoxy(C ⁇ -C6 alkylenyl)-, naphthyloxy(C ⁇ -C 6 alkylenyl)-, benzoyKCi-C ⁇ alkylenyl)
- C 1 -C 12 alkyl, phenyl, naphthyl, phenoxy, naphthyloxy, benzoyl, C 3 -C8 cycloalkyl, C 3 -C 8 cycloalkenyl, heterocyclic(C ⁇ -C6 alkoxy)-, unsaturated heterocyclic(C ⁇ -C ⁇ alkoxy)-, heterocyclic, or unsaturated heterocyclic moieties may be substituted with one or more groups selected from the group consisting of Ci-C ⁇ alkyl, Ci-C ⁇ alkoxy, phenyl, naphthyl, phenyKCi-C ⁇ alkylenyl)-, naphthyKCi-C ⁇ alkylenyl)-, halo, trifluoromethyl, C2-C7 alkenyl, C2-C7 alkynyl, Ci-C ⁇ alkoxy, heterocyclic, unsaturated heterocyclic, heterocyclic(C ⁇ -C6 alkyleny
- R 2 may also be -(CH 2 ) n -NR 7 R 8 , where, n is 0 to 10, and
- R 7 and R 8 are independently hydrogen, Ci-C ⁇ alkyl, C 2 -C7 alkanoyl, Ci-C ⁇ alkoxy, heterocyclic(C ⁇ -C6 alkylenyl)-, unsaturated heterocyclic(C ⁇ -C6 alkylenyl)-, phenyl, phenyKCi-C ⁇ alkylenyl)-, naphthyl, naphthyl(C ⁇ -C 6 alkylenyl)-, phenoxy(C ⁇ -C ⁇ alkylenyl)-, naphthyloxy(C ⁇ -C6 alkylenyl)-, benzoyKCi-C ⁇ alkylenyl)-, heterocyclic(C ⁇ -C 6 alkoxy)-, unsaturated heterocyclic(C ⁇ -C6 alkoxy)-, Ci-C ⁇ haloalkyl, C2-C7 alkenyl, C2-C7 alkynyl, C 3 -C8 cycloalken
- R 3 , R 4 , R 5 , and R 6 are independently hydrogen, halo, C ⁇ -C 6 alkyl, Ci-C ⁇ alkoxy, C2-C7 alkenyl, C2-C7 alkynyl, C2-C7 alkanoyl,
- This invention also encompasses, in additional embodiments, the novel compounds of Formula I, and the salts and solvates thereof, as well as pharmaceutical formulations comprising a compound of Formula I, or a pharmaceutically acceptable salt or solvate thereof, in combination with one or more pharmaceutically acceptable carriers, excipients, or diluents therefor.
- the current invention concerns the discovery that a select group of substituted benzimidazoles, those of Formula I, are useful as neuropeptide Y receptor antagonists.
- C 1 -C12 alkyl refers to straight or branched, monovalent, saturated aliphatic chains of 1 to 12 carbon atoms and includes, but is not limited to, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t-butyl, pentyl, isopentyl, and hexyl.
- C1-C12 alkyl includes within its definition the terms "Ci-C ⁇ alkyl” and "C1-C4 alkyl”.
- C2-C7 alkanoyloxy represents a straight or branched alkyl chain having from one to six carbon atoms attached to a carbonyl moiety joined through an oxygen atom.
- Typical C 2 -C7 alkanoyloxy groups include acetoxy, propanoyloxy, isopropanoyloxy, butanoyloxy, J-butanoyloxy, pentanoyloxy, hexanoyloxy, 3-methylpentanoyloxy and the like.
- C 3 -C 8 cycloalkyl represents a saturated hydrocarbon ring structure containing from three to eight carbon atoms.
- Typical C 3 -C8 cycloalkyl groups include cyclopropyl, cyclopentyl, cyclohexyl, cycloheptyl, and the like.
- carbamoyl refers to a group of the structure -NH-C(O)-.
- C2-C7 carbamoyl refers to a group of the structure (Ci-C ⁇ alkyl )-NH-C(O)-
- Halo represents chloro, fluoro, bromo or iodo.
- Ci-C ⁇ haloalkyl refers to a straight or branched, monovalent, saturated aliphatic chains of 1 to 6 carbon atoms substituted with one or more halo groups.
- C1-C10 alkylthio represents a straight or branched alkyl chain having from one to ten carbon atoms attached to a sulfur atom.
- Typical Ci-Cin alkylthio groups include methylthio, ethylthio, propylthio, isopropylthio, butylthio and the like.
- the term “C 1 -C1 0 alkylthio” includes within its definition the term “Ci-C ⁇ alkylthio" and "C1-C3 alkylthio".
- C1-C12 alkylenyl refers to a straight or branched, divalent, saturated aliphatic chains of 1 to 12 carbon atoms and includes, but is not limited to, methylenyl, ethylenyl, propylenyl, isopropylenyl, butylenyl, isobutylenyl, J-butylenyl, pentylenyl, isopentylenyl, hexylenyl, octylenyl, 3-methyloctylenyl, decylenyl.
- the term "Ci-C ⁇ alkylenyl” is encompassed within the term "C1-C 1 2 alkylenyl”.
- C1-C 1 0 alkylamino represents a group of the formula
- C ⁇ -C ⁇ o alkyl wherein a chain having from one to ten carbon atoms is attached to an amino group.
- Typical C 1 -C4 alkylamino groups include methylamino, ethylamino, propylamino, isopropylamino, butylamino, sec-butylamino and the like.
- C2-C10 alkenyl as used herein represents a straight or branched, monovalent, unsaturated aliphatic chain having from two to ten carbon atoms.
- Typical C2-C10 alkenyl groups include ethenyl (also known as vinyl), 1-methylethenyl, 1-methyl- 1-propenyl, 1-butenyl, 1-hexenyl, 2-methyl-2-propenyl, 1-propenyl, 2-propenyl, 2-butenyl, 2-pentenyl, and the like.
- C2-C10 alkynyl represents a straight or branched, monovalent, unsaturated aliphatic chain having from two to ten carbon atoms with at least one triple bond.
- Typical C2-C 1 0 alkynyl groups include ethynyl, 1-methylethenyl, 1-propynyl,
- C 3 -C 8 cycloalkenyl represents a hydrocarbon ring structure containing from three to eight carbon atoms and having at least one double bond within that ring, which is unsubstituted or substituted with 1, 2 or 3 substituents independently selected from halo, halo(C ⁇ -C4 alkyl), C 1 -C4 alkyl, C 1 -C4 alkoxy, carboxy, C 1 -C 4 alkoxycarbonyl, carbamoyl, N-(C ⁇ -C4 alkyl )carbamoyl, amino, C 1 -C 4 alkylamino, di(C ⁇ -C 4 alkyDamino or -(CH 2 ) a -R y where a is 1, 2, 3 or 4 and Ry is hydroxy, C1-C 4 alkoxy, carboxy
- Ci-C ⁇ alkylamino represents a straight or branched alkylamino chain having from one to six carbon atoms attached to an amino group.
- Typical Ci-C ⁇ alkyl-amino groups include methylamino, ethylamino, propylamino, isopropylamino, butylamino, sec-butylamino and the like.
- Ci-C ⁇ alkylamino encompasses within this term “C1-C4 alkylamino”.
- Ci-C ⁇ alkoxy represents a straight or branched alkyl chain having from one to six carbon atoms attached to an oxygen atom.
- Typical Ci-C ⁇ alkoxy groups include methoxy, ethoxy, propoxy, isopropoxy, butoxy, J-butoxy, pentoxy and the like.
- the term "Ci-C ⁇ alkoxy” includes within its definition the term “C 1 -C3 alkoxy”.
- C2-C7 alkanoyl represents a straight or branched alkyl chain having from one to six carbon atoms attached to a carbonyl moiety.
- Typical C2-C7 alkanoyl groups include ethanoyl, propanoyl, isopropanoyl, butanoyl, £-butanoyl, pentanoyl, hexanoyl, 3-methylpentanoyl and the like.
- Ci-C ⁇ alkoxycarbonyl represents a straight or branched alkoxy chain having from one to six carbon atoms attached to a carbonyl moiety.
- Typical alkoxycarbonyl groups include methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, isopropoxycarbonyl, butoxycarbonyl, i-butoxycarbonyl and the like.
- C 3 -C 8 cycloalkyl represents a saturated hydrocarbon ring structure containing from three to eight carbon atoms.
- Typical C 3 -C8 cycloalkyl groups include cyclopropyl, cyclopentyl, cyclohexyl, cycloheptyl, and the like.
- heterocycle represents an unsubstituted or substituted stable 5- to 7-membered monocyclic or 7- to 10-membered bicyclic heterocyclic ring which is saturated and which consists of carbon atoms and from one to three heteroatoms selected from the group consisting of nitrogen, oxygen or sulfur, and wherein the nitrogen and sulfur heteroatoms may optionally be oxidized, and the nitrogen heteroatom may optionally be quaternized and including a bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring.
- the heterocyclic ring may be attached at any heteroatom or carbon atom which affords a stable structure.
- the hetero-cycle is unsubstituted or substituted with 1, 2 or 3 substituents independently selected from halo, halo(C ⁇ -C 4 )-alkyl, C 1 -C 4 alkyl, C1-C 4 alkoxy, carboxy, C 1 -C4 alkoxy ⁇ carbonyl, carbamoyl, N-(C ⁇ -C4)-alkylcarbamoyl, amino, C1-C4 alkylamino, di(C ⁇ -C 4 )alkylamino or -(CH 2 )a-R d where a is 1, 2, 3 or 4; and R d is hydroxy, C1-C4 alkoxy, carboxy, C 1 -C 4 alkoxycarbonyl, amino, carbamoyl, C1-C4 alkylamino or di(C ⁇ -C 4 )alkylamino.
- the term "unsaturated heterocycle” represents an unsubstituted or substituted stable 5- to 7-membered monocyclic or 7- to 10-membered bicyclic heterocyclic ring which has one or more double bonds and which consists of carbon atoms and from one to three heteroatoms selected from the group consisting of nitrogen, oxygen or sulfur, and wherein the nitrogen and sulfur heteroatoms may optionally be oxidized, and the nitrogen heteroatom may optionally be quartemized and including a bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring.
- the unsaturated heterocyclic ring may be attached at any heteroatom or carbon atom which affords a stable structure.
- the unsaturated heterocycle is unsubstituted or substituted with 1, 2 or 3 substituents independently selected from halo, Ci-C ⁇ haloalkyl, C1-C4 alkyl, C1-C4 alkoxy, carboxy, C1-C 4 alkoxycarbonyl, carbamoyl, N-(C ⁇ -C4)alkylcarbamoyl, amino, Ci- C4 alkylamino, di(C ⁇ -C 4 )alkylamino or -(CH2) a -R e where a is 1, 2, 3 or 4; and R e is hydroxy, C 1 -C 4 alkoxy, carboxy, C1-C4 alkoxycarbonyl, amino, carbamoyl, C 1 -C4 alkylamino or di(C ⁇ -C4)alkylamino.
- substituents independently selected from halo, Ci-C ⁇ haloalkyl, C1-C4 alkyl, C1-C4 alkoxy
- heterocycles and unsaturated heterocycles include piperidinyl, piperazinyl, azepinyl, pyrrolyl, 4- piperidonyl, pyrrolidinyl, pyrazolyl, pyrazolidinyl, imidazolyl, imidazolinyl, imidazolidinyl, pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, oxazolyl, oxazolidinyl, isoxazolyl, isoxazolidinyl, morpholinyl, thiazolyl, thiazolidinyl, isothiazolyl, quinuclidinyl, isothiazolidinyl, indolyl, quinolinyl, isoquinolinyl, benzimidazolyl, thiadiazolyl, benzopyranyl, benzothiazolyl, benzoazolyl, furyl, t
- amino-protecting group refers to substituents of the amino group commonly employed to block or protect the amino functionality while reacting other functional groups on the compound.
- amino-protecting groups include formyl, trityl, phthalimido, trichloroacetyl, chloroacetyl, bromoacetyl, iodoacetyl, and urethane-type blocking groups such as benzyloxycarbonyl, 4-phenylbenzyloxy carbonyl, 2-methylbenzyloxycarbonyl, 4-methoxybenzyloxycarbonyl, 4-fluorobenzyloxycarbonyl, 4-chlorobenzyloxycarbonyl, 3-chlorobenzyloxycarbonyl, 2-chlorobenzyloxycarbonyl, 2,4-dichlorobenzyloxycarbonyl, 4-bromobenzyloxycarbonyl, 3-bromobenzyloxycarbonyl, 4-nitrobenzyloxy carbonyl
- amino-protecting group employed is usually not critical so long as the derivatized amino group is stable to the condition of subsequent reactions on other positions of the intermediate molecule and can be selectively removed at the appropriate point without disrupting the remainder of the molecule including any other amino-protecting groups.
- Preferred amino-protecting groups are trityl, f-butoxycarbonyl (BoC), allyloxycarbonyl and benzyloxycarbonyl. Further examples of groups referred to by the above terms are described by E. Haslam,
- carboxy-protecting group refers to substituents of the carboxy group commonly employed to block or protect the carboxy functionality while reacting other functional groups on the compound.
- carboxy-protecting groups include methyl, jD-nitrobenzyl, p-methylbenzyl, p-methoxy-benzyl, 3,4-dimethoxybenzyl, 2,4-dimethoxybenzyl, 2,4,6-trimethoxybenzyl, 2,4,6-trimethylbenzyl, pentamethylbenzyl, 3,4-methylenedioxybenzyl, benzhydryl,
- hydroxy-protecting groups refers to substitents of the hydroxy group commonly employed to block or protect the hydroxy functionality while reacting other functional groups on the compound. Examples of such hydroxy-protecting groups include methoxymethyl, benzyloxymethyl, methoxyethoxymethyl,
- the compounds of the present invention may have one or more asymmetric centers. As a consequence of these chiral centers, those compounds of the present invention occur as racemates, mixtures of enantiomers and as individual enantiomers, as well as diastereomers and mixtures of diastereomers. All asymmetric forms, individual isomers and combinations thereof, are within the scope of the present invention.
- the terms "R” and “S” are used herein as commonly used in organic chemistry to denote specific configuration of a chiral center.
- the term “R” (rectus) refers to that configuration of a chiral center with a clockwise relationship of group priorities (highest to second lowest) when viewed along the bond toward the lowest priority group.
- S sinister
- S refers to that configuration of a chiral center with a counterclockwise relationship of group priorities (highest to second lowest) when viewed along the bond toward the lowest priority group.
- the priority of groups is based upon their atomic number (in order of decreasing atomic number).
- a partial list of priorities and a discussion of stereochemistry is contained in NOMENCLATURE OF ORGANIC COMPOUNDS: PRINCIPLES AND PRACTICE, (J.H. Fletcher, et al.. eds., 1974) at pages 103-120.
- the older D-L system may also be used in this document to denote absolute configuration, especially with reference to amino acids.
- a Fischer projection formula is oriented so that the number 1 carbon of the main chain is at the top.
- the prefix "D” is used to represent the absolute configuration of the isomer in which the functional (determimng) group is on the right side of the carbon atom at the chiral center and "L", that of the isomer in which it is on the left.
- the skilled practitioner can proceed by one of two routes.
- the practitioner may first prepare the mixture of enantiomers and then separate the two enantiomers.
- a commonly employed method for the resolution of the racemic mixture (or mixture of enantiomers) into the individual enantiomers is to first convert the enantiomers to diastereomers by way of forming a salt with an optically active salt or base. These diastereomers can then be separated using differential solubility, fractional crystallization, chromatography, or like methods. Further details regarding resolution of enantiomeric mixtures can be found in J. Jacques, et al.. ENANTIOMERS, RACEMATES, AND RESOLUTIONS, (1991).
- the practitioner of this invention may also choose an enantio specific protocol for the preparation of the compounds of Formula I.
- a protocol employs a synthetic reaction design which maintains the chiral center present in the starting material in a desired orientation.
- These reaction schemes usually produce compounds in which greater than 95 percent of the title product is the desired enantiomer.
- this invention includes methods employing the pharmaceutically acceptable salts of the compounds defined by Formula I as well as salts of the compounds of Formula II.
- a compound of this invention can possess a sufficiently acidic, a sufficiently basic, or both functional groups, and accordingly react with any of a number of organic and inorganic bases, and inorganic and organic acids, to form a pharmaceutically acceptable salt.
- pharmaceutically acceptable salt refers to salts of the compounds of the above formula which are substantially non-toxic to living organisms.
- Typical pharmaceutically acceptable salts include those salts prepared by reaction of the compounds of the present invention with a pharmaceutically acceptable mineral or organic acid or an organic or inorganic base. Such salts are known as acid addition and base addition salts.
- Acids commonly employed to form acid addition salts are inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, phosphoric acid, and the like, and organic acids such as p-toluenesulfonic, methanesulfonic acid, oxalic acid, p-bromophenylsulfonic acid, carbonic acid, succinic acid, citric acid, benzoic acid, acetic acid, and the like.
- inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, phosphoric acid, and the like
- organic acids such as p-toluenesulfonic, methanesulfonic acid, oxalic acid, p-bromophenylsulfonic acid, carbonic acid, succinic acid, citric acid, benzoic acid, acetic acid, and the like.
- salts examples include the sulfate, pyrosulfate, bisulfate, sulfite, bisulfite, phosphate, monohydrogenphosphate, dihydrogenphosphate, metaphosphate, pyrophosphate, chloride, bromide, iodide, acetate, propionate, decanoate, caprylate, acrylate, formate, hydrochloride, dihydrochloride, isobutyrate, caproate, heptanoate, propiolate, oxalate, malonate, succinate, suberate, sebacate, fumarate, maleate, butyne-l,4-dioate, hexyne-l,6-dioate, benzoate, chlorobenzoate, methylbenzoate, hydroxybenzoate, methoxybenzoate, phthalate, sulfonate, xylenesulfonate, phenylacetate, phenylprop
- Preferred pharmaceutically acceptable acid addition salts are those formed with mineral acids such as hydrochloric acid and hydrobromic acid, and those formed with organic acids such as maleic acid and methanesulfonic acid.
- Base addition salts include those derived from inorganic bases, such as ammonium or alkali or alkaline earth metal hydroxides, carbonates, bicarbonates, and the like.
- bases useful in preparing the salts of this invention thus include sodium hydroxide, potassium hydroxide, ammonium hydroxide, potassium carbonate, sodium carbonate, sodium bicarbonate, potassium bicarbonate, calcium hydroxide, calcium carbonate, and the like.
- the potassium and sodium salt forms are particularly preferred.
- any salt of this invention is usually not of a critical nature, so long as the salt as a whole is pharmacologically acceptable and as long as the counterion does not contribute undesired qualities to the salt as a whole.
- This invention further encompasses methods employing the pharmaceutically acceptable solvates of the compounds of Formula I.
- Many of the compounds of Formula I can combine with solvents such as water, methanol, ethanol and acetonitrile to form pharmaceutically acceptable solvates such as the corresponding hydrate, methanolate, ethanolate and acetonitrilate.
- a prodrug is a drug which has been chemically modified and may be biologically inactive at its site of action, but which may be degraded or modified by one or more enzymatic or other in vivo processes to the parent bioactive form.
- This prodrug should have a different pharmacokinetic profile than the parent, enabling easier absorption across the mucosal epithelium, better salt formation or solubility, or improved systemic stability (an increase in plasma half-life, for example).
- ester or amide derivatives which may be cleaved by esterases or lipases; 2) peptides which may be recognized by specific or nonspecific proteases; or
- the compounds of the present invention are derivatives of benzimidazole which are named and numbered according to the RING INDEX, The American Chemical Society, as follows.
- R 1 is phenyl, naphthyl, phenyKCi-C ⁇ alkylenyl)-, naphthyKCi-C ⁇ alkylenyl)-, phenyKCi-C ⁇ alkoxy)-, naphthyKCi-C ⁇ alkoxy)-, phenoxy(C ⁇ -C 6 alkylenyl)-, naphthyloxy(C ⁇ -C6 alkylenyl)-, or substituted derivatives thereof; b) R 2 is phenyl, heterocyclic, unsaturated heterocyclic, phenyKCi-C ⁇ alkylenyl)-, naphthyKCi-C ⁇ alkylenyl)-, phenyKCi-C ⁇ alkoxy)-, heterocyclic(Ci-Ce alkylenyl)-, unsaturated heterocyclic(C ⁇ -C6 alkylenyl)-, heterocyclic
- the preferred compounds of this invention are those compounds which are employed in the preferred methods of this invention.
- benzimidazole In the scientific literature derivatives of benzimidazole are already known to possess different biological activities, such as analgesic and antiinflammatory activity (Japan Kokai 75,126,682; United States Patent 4,925,853), gastric antisecretory activity (European Patent Publication 246,126), antihistaminic activity (United States Patents
- the compounds of Formula I can be prepared by processes known in the literature. See f e.g.. G.W.H. Cheeseman and R.F. Cookson, THE CHEMISTRY OF HETEROCYCLIC COMPOUNDS, (A. Weissberger, et al.. eds. 1979).
- Suitable solvents include ethanol, isopropanol, glacial acetic acid, benzene, toluene, chlorobenzene, glycol, ethylene glycol, dimethyl ether, diethyl ether, dimethylformamide, chloroform, ethyl acetate, and the like.
- a condensation agent such as phosphorous oxychloride, thionyl chloride, p-toluenesulfonic acid, hydrochloric acid, sulfuric acid, phosphoric acid, polyphosphoric acid, phosphorous pentoxide, methanesulfonyl hydroxide, methanesulfonyl chloride, and the like.
- the cyclization reaction may also optionally be performed in the presence of a base such as sodium hydroxide, sodium mesylate, or potassium tert-butylate.
- R 2 is phenyl a derivative of N- phenyl-o-phenylenediamine was used as the starting material for the cyclization reaction.
- the examples infra provide sufficient guidance in the preparation of those compounds of Formula I wherein all of R 3 , R 4 , R 5 , and R 6 are hydrogen.
- R 4 , R 5 , and R 6 is not hydrogen, can be prepared by methods taught in the literature.
- the compounds of this invention wherein phenyl portion ofthe benzimidazole is substituted with C 2 -C7 alkanoyl can be prepared from the appropriate keto o-phenylenediamine of the formula
- the keto benzimidazole reactants can be prepared from acetanilide by a Friedel-Crafts acylation with the appropriate derivative of C 2 -C7 alkanoic acid.
- the resulting 4-keto acetanilide is nitrated to give a 2-nitro-4-ketoacetanilide.
- the acetanilide is hydrolyzed to give a 2-nitro-4-ketoaniline, which can then be catalytically hydrogenated to yield a 4-keto-o-phenylenediamine which can then be ring closed to provide the 5 or 6-substituted benzimidazole.
- Those compounds of Formula III wherein phenyl portion of the benzimidazole is substituted with alkyl or alkylenyl may be prepared by means of a Friedel-Crafts alkylation with the appropriate derivative of the substituting moiety using standard procedures, usually employing an alkyl halide or an olefin in the presence of a catalyst such as aluminum chloride, aluminum bromide or another Lewis acid.
- a catalyst such as aluminum chloride, aluminum bromide or another Lewis acid.
- An alternative strategy for preparing those compounds of Formula I wherein R 5 is C ⁇ -C 6 alkoxy, R 7 R 8 N-(C ⁇ -C 6 alkoxy)-, or heterocyclic-(C ⁇ -C 6 alkoxy)-, or a substituted derivative thereof involves first reacting a 3-nitro-4-aminophenol with an acyl halide in the presence of a base
- the ester moiety serving as a hydroxy-protecting group for subsequent reactions.
- the nitro group is then reduced to an amino group, usually by catalytic hydrogenation.
- those compounds of Formula I in which R 2 is alkyl or substituted alkyl may be produced by alkylation of an aromatic amine with alkyl halide or tosylate, or the like, in the presence of a suitable base, such as trialkylamine, potassium carbonate, 1,8- diazabicyclo[5.4.0]undec-7-ene (DBU), and the like.
- a suitable base such as trialkylamine, potassium carbonate, 1,8- diazabicyclo[5.4.0]undec-7-ene (DBU), and the like.
- Suitable cyclization catalysts include phosphorous oxychloride, thionyl chloride, phosphorous pentoxide, phosphorous pentachloride, and other like strong dehydrating agents.
- a preferred method of cleaving this ester is by incubation of the intermediate in a basic solution, such as IN sodium hydroxide, or a weaker base such as potassium carbonate.
- a basic solution such as IN sodium hydroxide, or a weaker base such as potassium carbonate.
- the hydroxy group at the 6- position is then substituted using an alkyl or aryl halide, resulting in a compound of Formula I.
- Those compounds of Formula I wherein R 2 is alkyl or substituted alkyl may alternatively be prepared by the direct alkylation of a benzimidazole wherein the nitrogen at the 1-position is substituted with a hydrogen.
- This type of alkylation is usually performed by the reaction of the benzimidazole with an alkyl halide in the presence of a strong base, such as sodium hydride.
- This reaction is usually performed in a polar aprotic solvent, such as N,N-dimethylformamide, dimethyl sulf oxide, dimethylacetamide, hexamethylphosphoric triamide, and the like.
- Proton nuclear magnetic resonance (*H NMR) spectra were obtained on a GE E-300 spectrometer at 300.15 MHz, a Bruker AM-500 spectrometer at 500 MHz, or a Bruker AC-200P spectrometer at 200 MHz.
- NMR proton nuclear magnetic resonance
- Free atom bombardment mass spectroscopy (FAB) was performed on a VG ZAB-2SE instrument.
- Field desorption mass spectroscopy (FDMS) was performed using either a VG 70SE or a Varian MAT 731 instrument.
- Optical rotations were measured with a Perkin-Elmer 241 polarimeter. Chromatographic separation on a Waters Prep 500 LC was generally carried out using a linear gradient of the solvents indicated in the text unless otherwise specified. The reactions were generally monitored for completion using thin layer chromatography (TLC). Thin layer chromatography was performed using E. Merck Kieselgel 60 F254 plates, 5 cm x 10 cm,
- the title compound was prepared essentially as described in Preparation 1 except that an equimolar amount of 4- chlorobenzonitrile was employed instead of the benzyl cyanide employed therein.
- the title compound was prepared essentially as described in Preparation 1 except that an equimolar amount of 4-chlorobenzyl cyanide was employed instead of the benzyl cyanide employed therein.
- Ethyl-3-pyridylacetate (lOOg, 0.606 mol) was dissolved in ethanol (1.8 liters), treated with 5% rhodium on alumina (100 g) and hydrogenated at 60°C and 60 psi hydrogen gas overnight. The catalyst was removed by filtration and the solvent evaporated to give a brown liquid (101.4 g, 98%). The brown liquid was dissolved in ethyl acetate (600 ml) and treated with L-(+)-mandelic acid in warm ethyl acetate (600 ml). After cooling in the refrigerator for four hours, the solid was collected and the crystallization fluid reserved for processing to the other enantiomer, infra.
- the aqueous fraction was extracted with diethyl ether.
- the organic fractions were combined, washed with water, a saturated sodium bicarbonate solution, and then brine, and then dried over magnesium sulfate.
- the solvents were removed in vacuo to give the desired title product was a clear liquid. NMR was consistent with proposed title structure.
- triphenylphosphine (19.95 g, 76 mmol) in anhydrous methylene chloride (110 ml) was added bromine dropwise until the solution turned pale yellow. A few crystals of triphenylphosphine were added to the mixture to bring the color back to white.
- a suspension of (3'S) 3-[l-(t- butoxycarbonyl)piperidin-3-yl]propanol (13.2 g, 54.4 mmol) and pyridine (8.0 g, 76 mmol) in dry methylene chloride (110 ml). The resulting mixture was stirred for five hours while warming to room temperature. The reaction was stopped by adding water (200 ml).
- the reaction mixture was poured into water (20 ml).
- the organic fraction was extracted wtih diethyl ether (3 x 50 ml).
- the organic fractions were combined, washed with water (2 x 20 ml), and then brine (1 x 20 ml), and then dried over sodium sulfate.
- the solvents were removed in vacuo to yield a white solid as a crude product. No further purification was performed on this product.
- n 0, 1, or 2
- APG is an amino protecting group
- reaction mixture was then poured into water (10 ml).
- the organic fraction was extracted with diethyl ether (3 x 15 ml).
- the organic fractions were combined, washed with water (2 x 10 ml), brine (1 x 10 ml), and then dried over sodium sulfate.
- the solvents were removed in vacuo, leaving a light brown crude material which was further purified by flash chromatography to yield the desired title product as a white crystalline solid in 70-100% yield.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Pain & Pain Management (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description









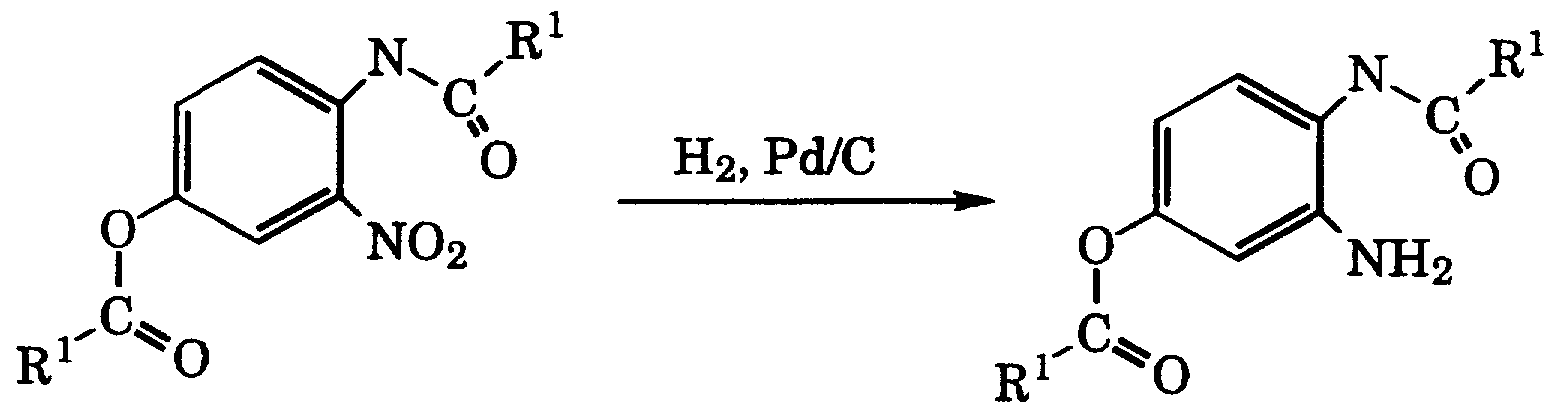







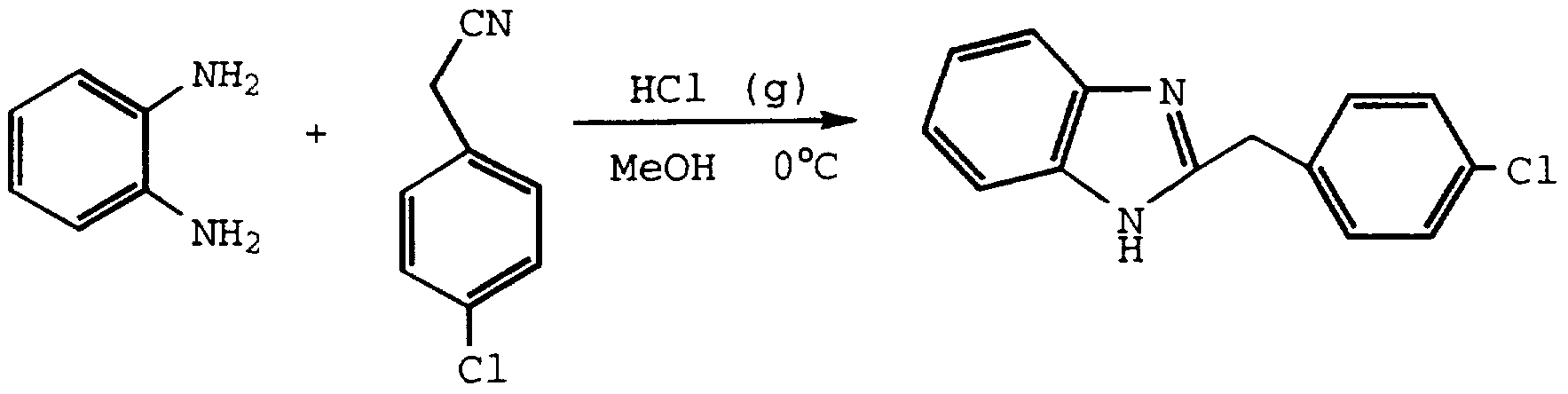











































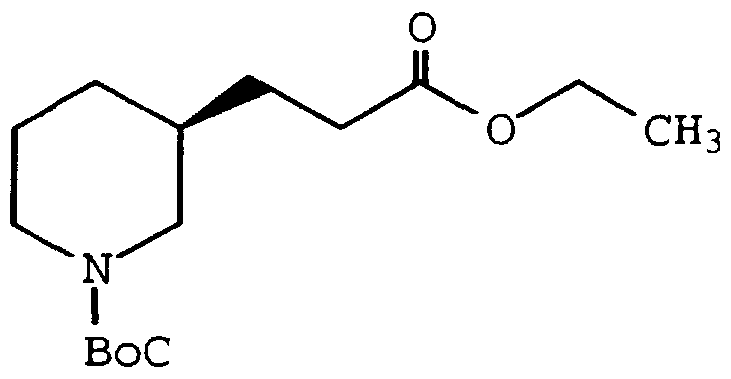
















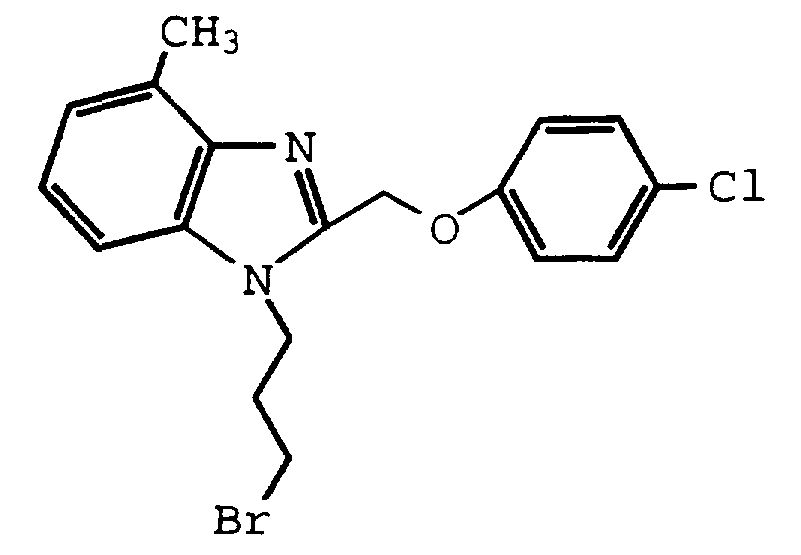































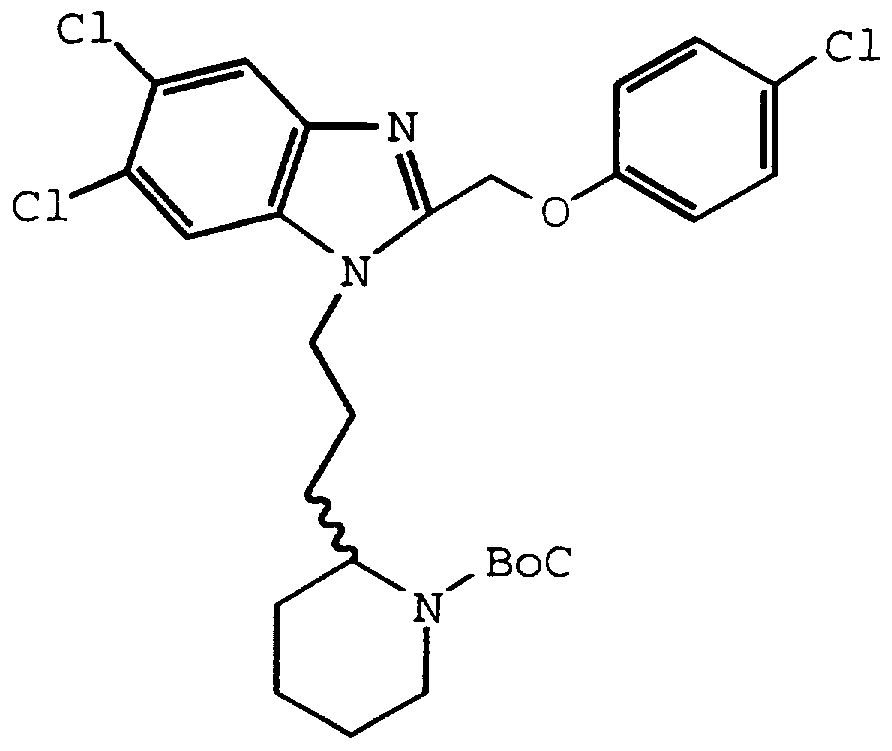








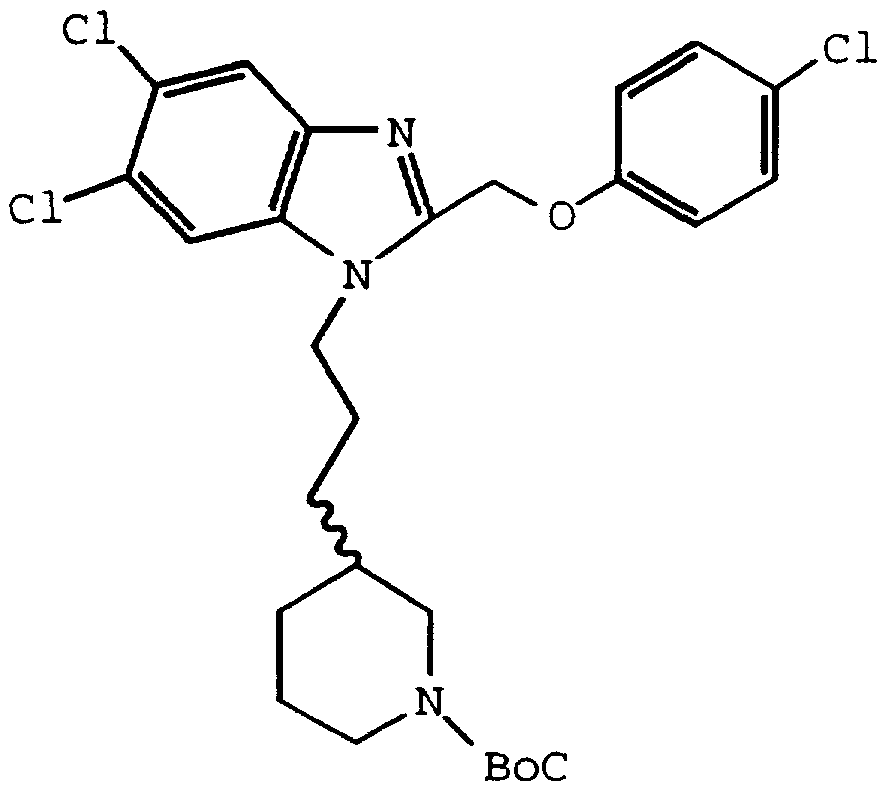














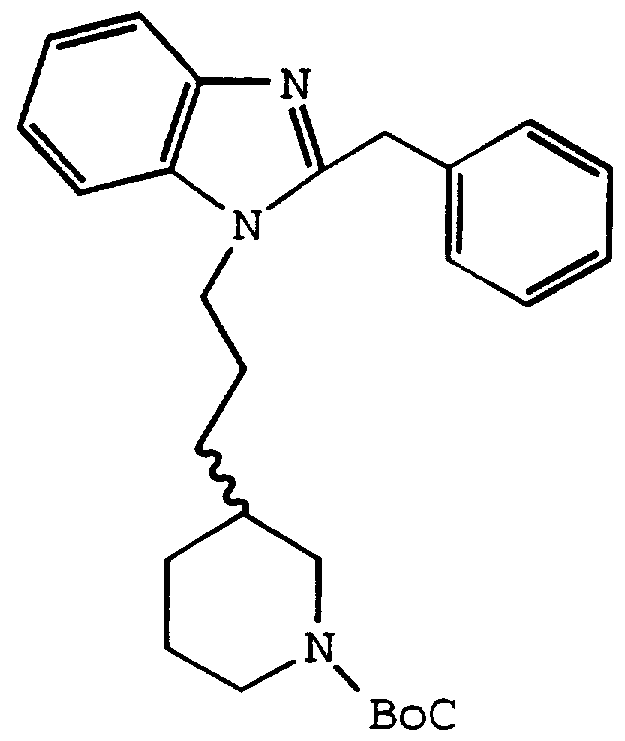





















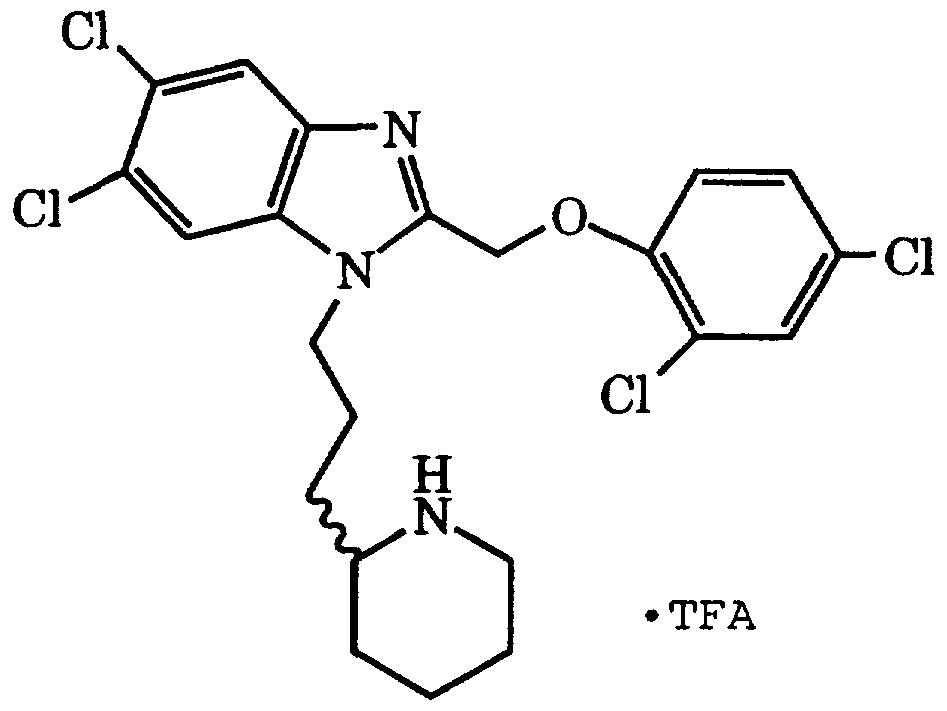















































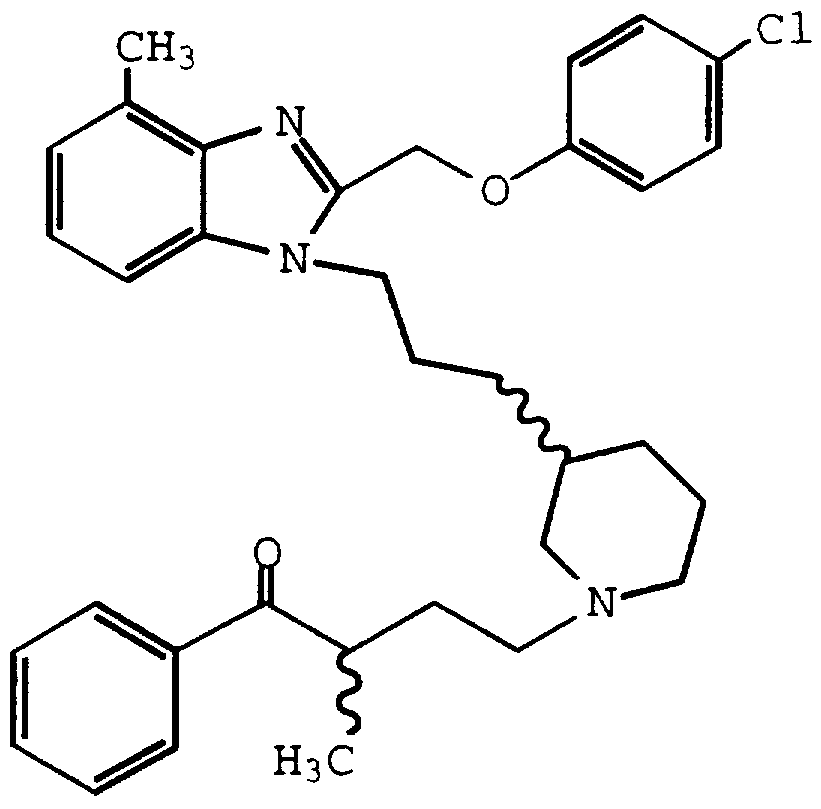
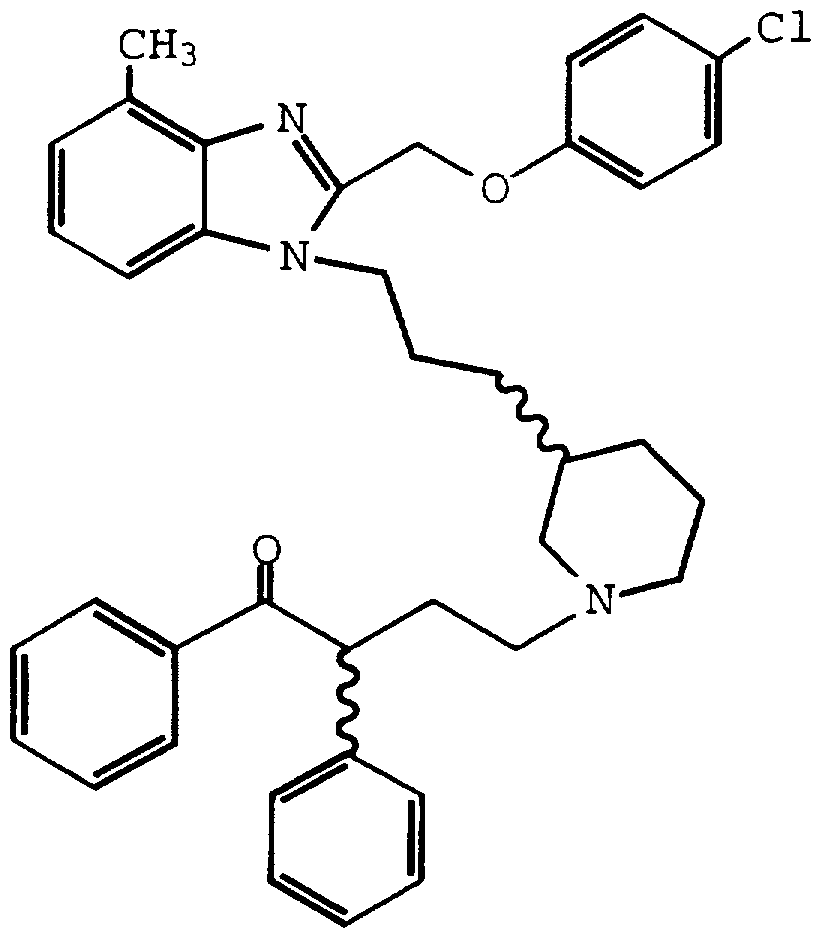


























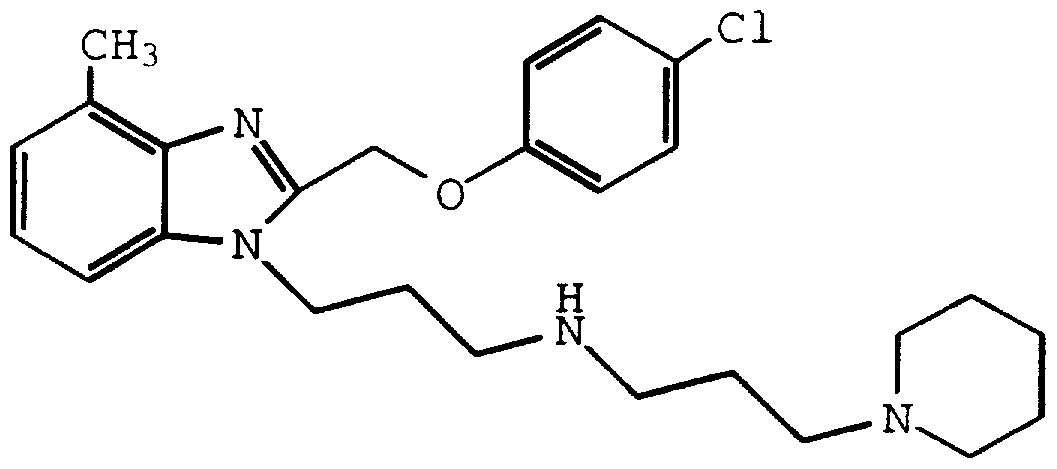












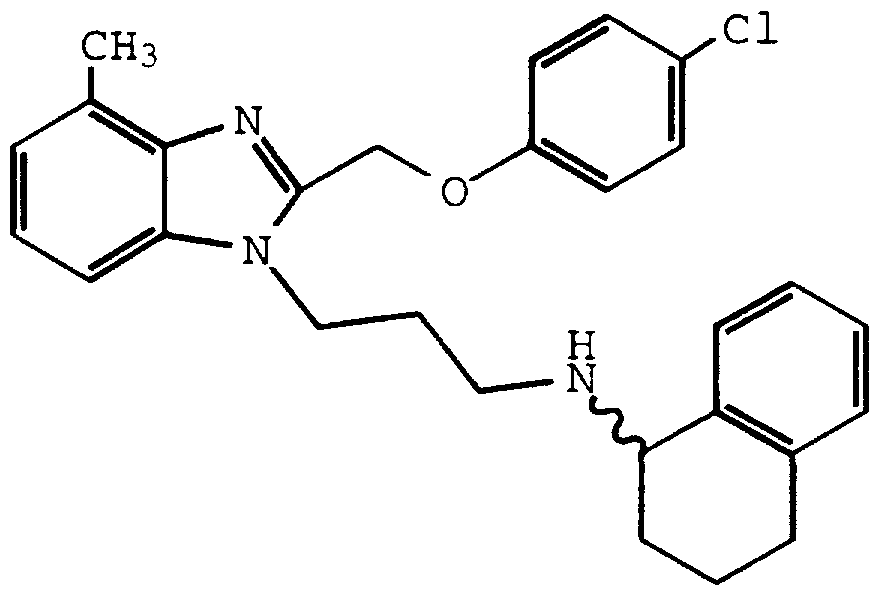
















































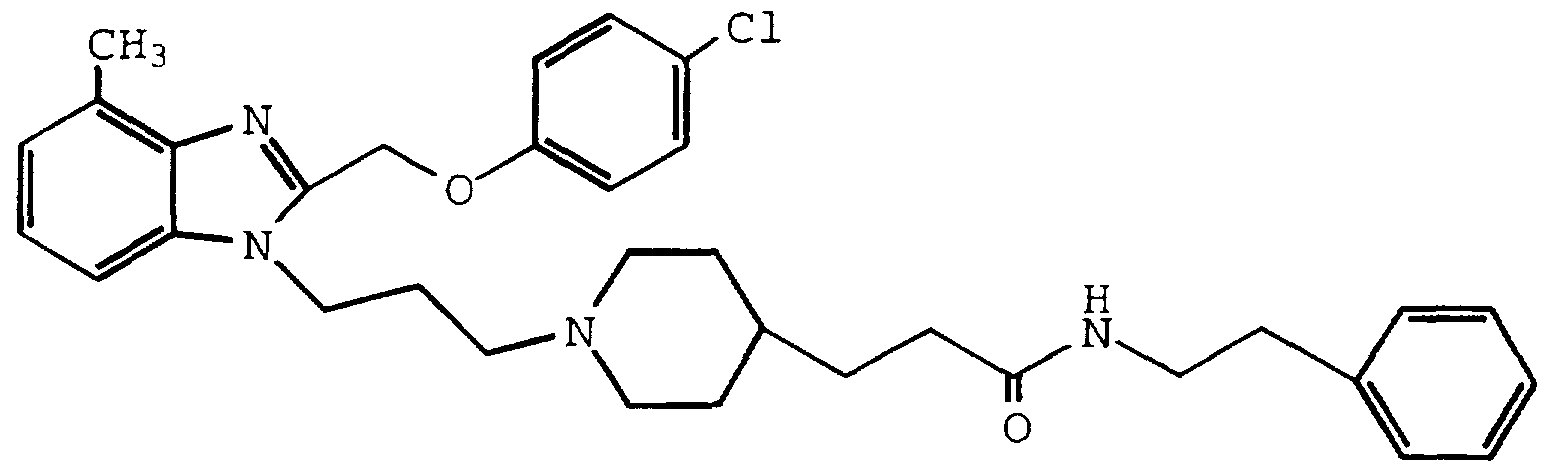

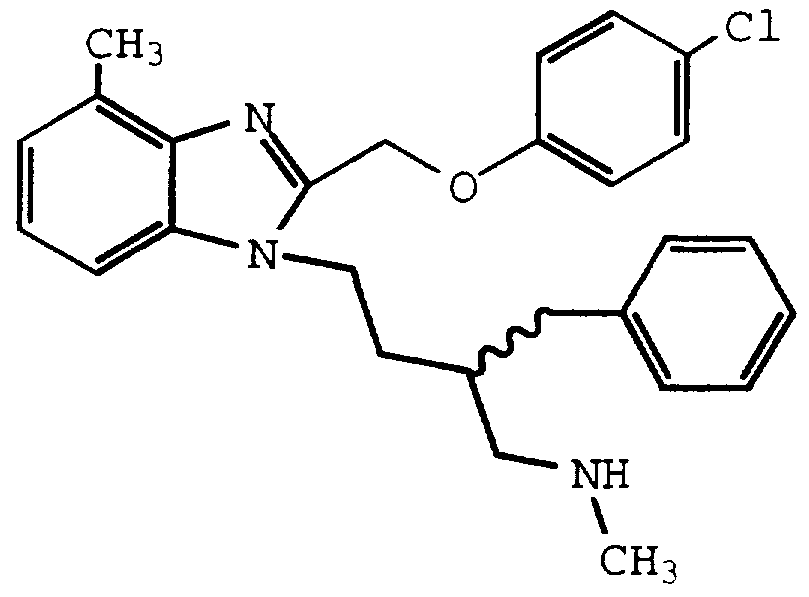











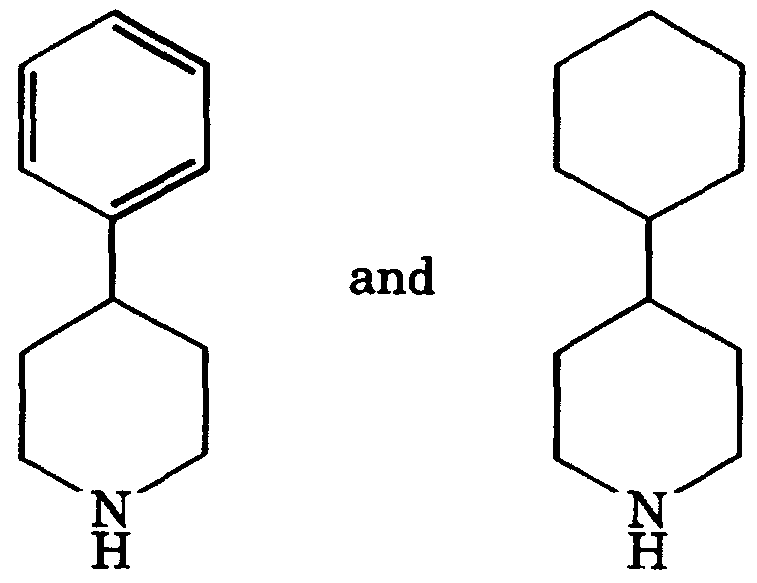







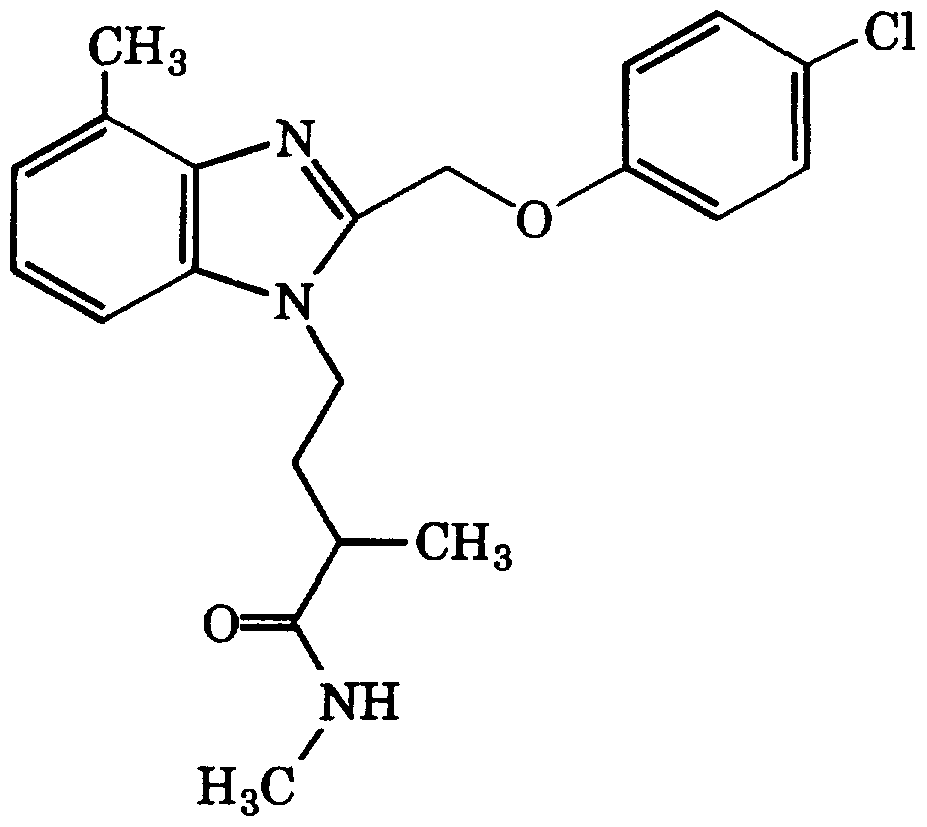
































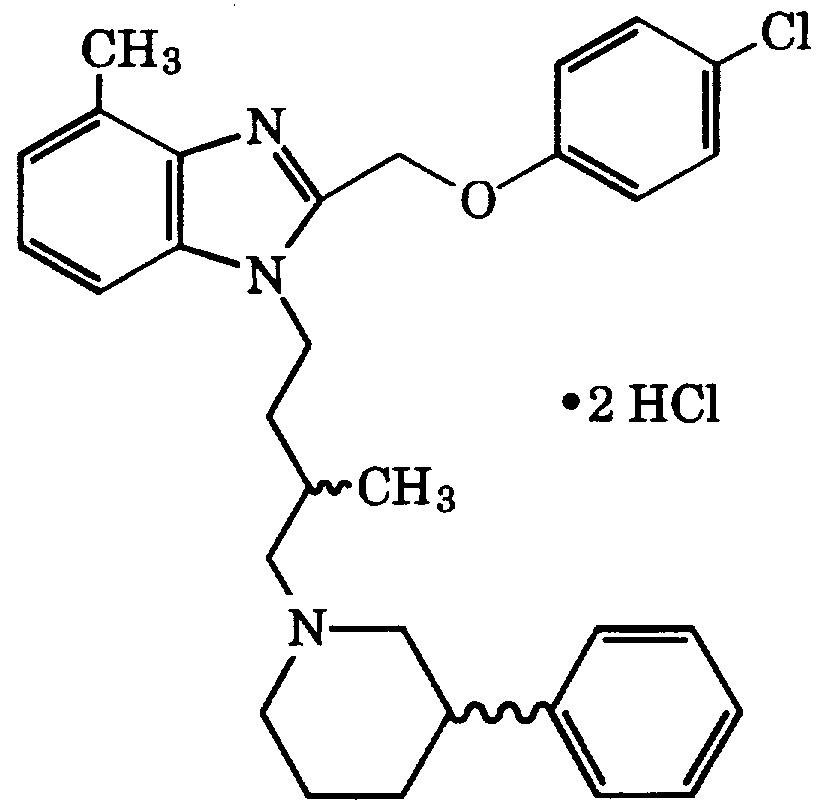

















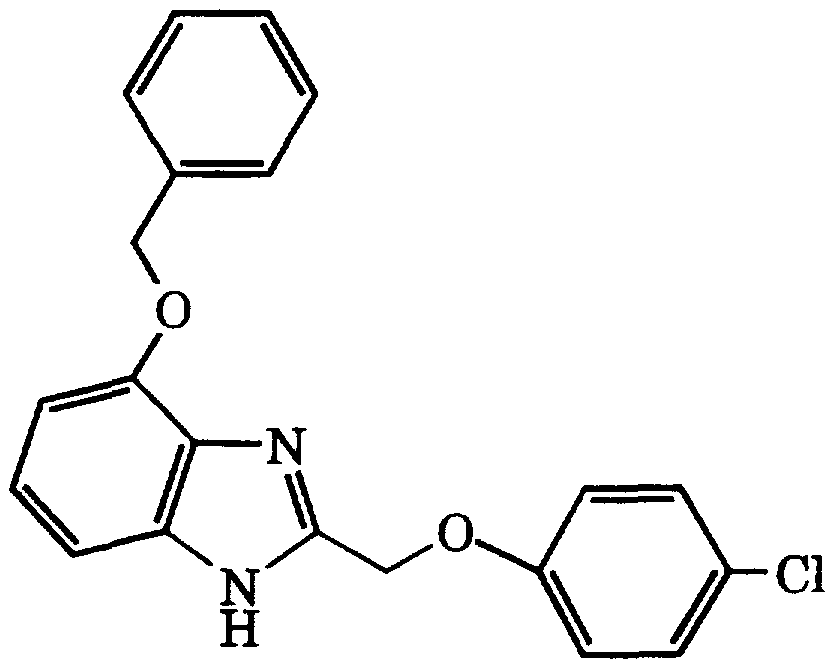


















































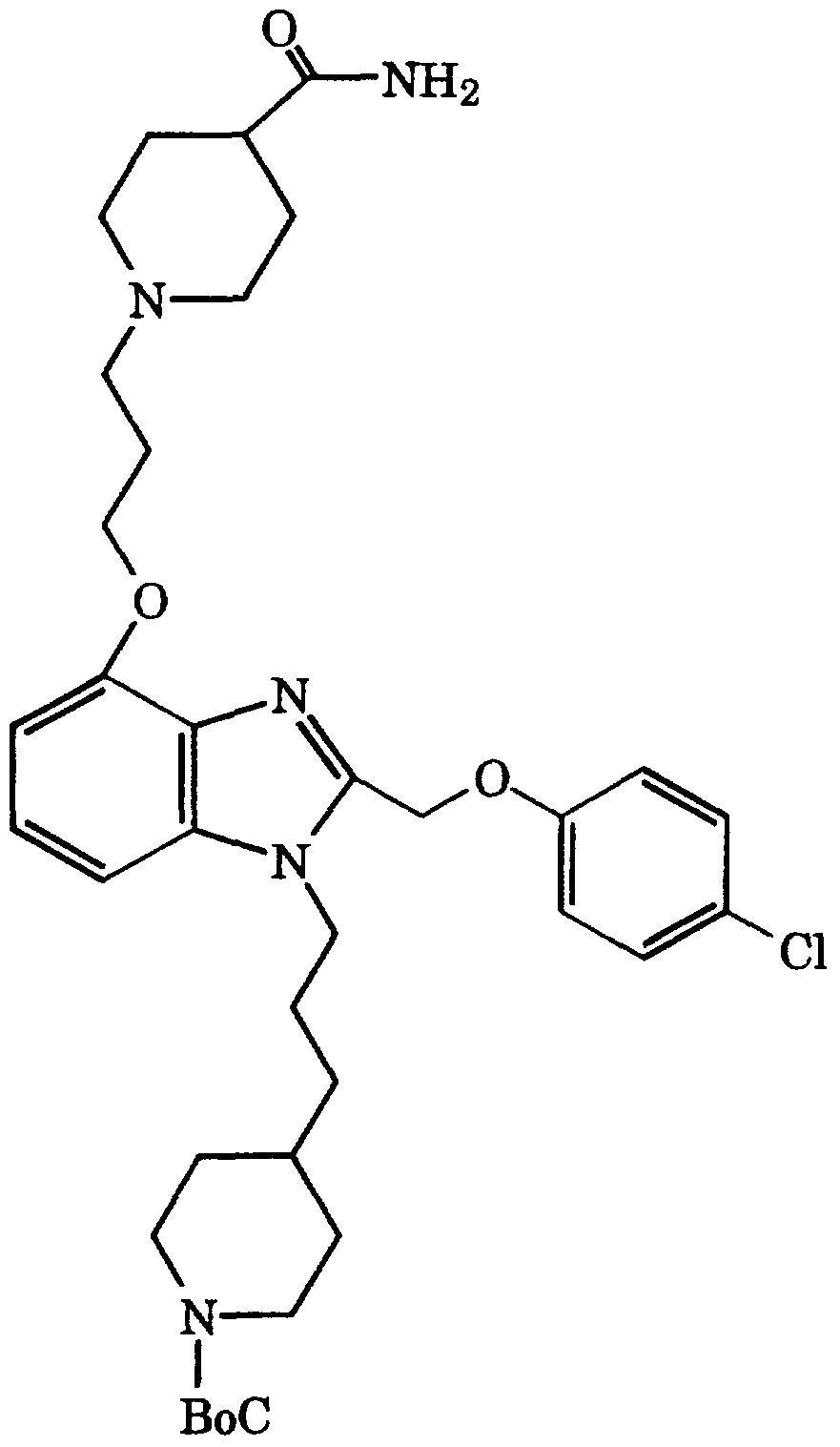


























Claims

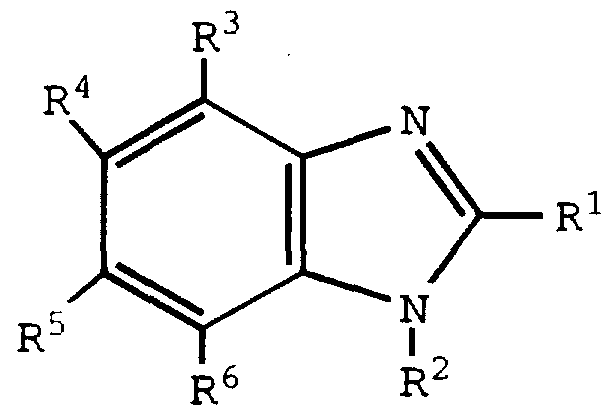

Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP97905573A EP0871442A1 (en) | 1996-01-09 | 1997-01-09 | Benzimidzolyl neuropeptide y receptor antagonists |
| JP9525457A JP2000501107A (en) | 1996-01-09 | 1997-01-09 | Benzimidazolyl neuropeptide Y receptor antagonist |
| AU22421/97A AU2242197A (en) | 1996-01-09 | 1997-01-09 | Benzimidzolyl neuropeptide y receptor antagonists |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB9600344.7 | 1996-01-09 | ||
| GBGB9600344.7A GB9600344D0 (en) | 1996-01-09 | 1996-01-09 | Benzimidzolyl neuropeptide y receptor antagonists |
| US2163696P | 1996-07-12 | 1996-07-12 | |
| US60/021,636 | 1996-07-12 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1997025041A1 true WO1997025041A1 (en) | 1997-07-17 |
Family
ID=26308442
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1997/000511 WO1997025041A1 (en) | 1996-01-09 | 1997-01-09 | Benzimidzolyl neuropeptide y receptor antagonists |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP0871442A1 (en) |
| JP (1) | JP2000501107A (en) |
| AU (1) | AU2242197A (en) |
| CA (1) | CA2242579A1 (en) |
| WO (1) | WO1997025041A1 (en) |
Cited By (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998040356A1 (en) * | 1997-03-12 | 1998-09-17 | Banyu Pharmaceutical Co., Ltd. | Drugs containing aminopyridine derivatives as the active ingredient |
| WO1999007703A1 (en) * | 1997-08-05 | 1999-02-18 | Pfizer Products Inc. | 4-AMINOPYRROLE(3,2-d) PYRIMIDINES AS NEUROPEPTIDE Y RECEPTOR ANTAGONISTS |
| WO1999036419A1 (en) * | 1998-01-17 | 1999-07-22 | Bayer Aktiengesellschaft | Substituted lactones as modulators of metabotropic glutamate receptors |
| WO1999040091A1 (en) * | 1998-02-06 | 1999-08-12 | Amgen Inc. | Bicyclic pyridine and pyrimidine derivatives as neuropeptide y receptor antagonists |
| US6127414A (en) * | 1997-09-23 | 2000-10-03 | Astra Aktiebolag | NPY antagonists |
| WO2002046168A1 (en) * | 2000-12-07 | 2002-06-13 | Astrazeneca Ab | Therapeutic benzimidazole compounds |
| WO2002059088A1 (en) * | 2001-01-23 | 2002-08-01 | Wyeth | 1-aryl-or 1-alkylsulfonylbenzazole derivatives as 5-hydroxytryptamine-6 ligands |
| US6774241B2 (en) | 2002-06-05 | 2004-08-10 | Roche Palo Alto Llc | 1-sulfonyl-4-aminoalkoxy indole derivatives and uses thereof |
| WO2004098591A2 (en) | 2003-05-05 | 2004-11-18 | Probiodrug Ag | Inhibitors of glutaminyl cyclase and their use in the treatment of neurological diseases |
| WO2005049027A2 (en) | 2003-11-03 | 2005-06-02 | Probiodrug Ag | Combinations useful for the treatment of neuronal disorders |
| WO2005075436A2 (en) | 2004-02-05 | 2005-08-18 | Probiodrug Ag | Novel inhibitors of glutaminyl cyclase |
| WO2005080348A1 (en) | 2004-02-19 | 2005-09-01 | Banyu Pharmaceutical Co., Ltd. | Novel sulfone amide derivative |
| WO2008055945A1 (en) | 2006-11-09 | 2008-05-15 | Probiodrug Ag | 3-hydr0xy-1,5-dihydr0-pyrr0l-2-one derivatives as inhibitors of glutaminyl cyclase for the treatment of ulcer, cancer and other diseases |
| WO2008065141A1 (en) | 2006-11-30 | 2008-06-05 | Probiodrug Ag | Novel inhibitors of glutaminyl cyclase |
| WO2008104580A1 (en) | 2007-03-01 | 2008-09-04 | Probiodrug Ag | New use of glutaminyl cyclase inhibitors |
| US7659263B2 (en) | 2004-11-12 | 2010-02-09 | Japan Tobacco Inc. | Thienopyrrole compound and use thereof as HCV polymerase inhibitor |
| WO2011029920A1 (en) | 2009-09-11 | 2011-03-17 | Probiodrug Ag | Heterocylcic derivatives as inhibitors of glutaminyl cyclase |
| US7977331B1 (en) | 2004-02-24 | 2011-07-12 | Japan Tobacco Inc. | Tetracyclic fused heterocyclic compound and use thereof as HCV polymerase inhibitor |
| WO2011107530A2 (en) | 2010-03-03 | 2011-09-09 | Probiodrug Ag | Novel inhibitors |
| WO2011110613A1 (en) | 2010-03-10 | 2011-09-15 | Probiodrug Ag | Heterocyclic inhibitors of glutaminyl cyclase (qc, ec 2.3.2.5) |
| WO2011131748A2 (en) | 2010-04-21 | 2011-10-27 | Probiodrug Ag | Novel inhibitors |
| WO2012123563A1 (en) | 2011-03-16 | 2012-09-20 | Probiodrug Ag | Benz imidazole derivatives as inhibitors of glutaminyl cyclase |
| WO2012130905A1 (en) | 2011-03-31 | 2012-10-04 | Bayer Pharma Aktiengesellschaft | Substituted benzimidazoles |
| US8435988B2 (en) | 2010-10-06 | 2013-05-07 | Glaxosmithkline Llc | Benzimidazole derivatives as P13 kinase inhibitors |
| US8653131B2 (en) | 2008-08-22 | 2014-02-18 | Baxter Healthcare S.A. | Polymeric benzyl carbonate-derivatives |
| CN103965113A (en) * | 2014-05-06 | 2014-08-06 | 四川大学 | 1-ethoxyl-2-substituted phenoxymethyl benzimidazole compound and preparation method thereof |
| EP2865670A1 (en) | 2007-04-18 | 2015-04-29 | Probiodrug AG | Thiourea derivatives as glutaminyl cyclase inhibitors |
| EP3461819A1 (en) | 2017-09-29 | 2019-04-03 | Probiodrug AG | Inhibitors of glutaminyl cyclase |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6400869B1 (en) * | 2018-02-23 | 2018-10-03 | 日本曹達株式会社 | Method for producing 4,5-dicyano-2- (fluoroalkyl) imidazole |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4772703A (en) * | 1986-01-27 | 1988-09-20 | American Home Products Corporation | 2-(phenoxymethyl)-quinazolines as antiallergic and antiinflammatory agents |
-
1997
- 1997-01-09 AU AU22421/97A patent/AU2242197A/en not_active Abandoned
- 1997-01-09 WO PCT/US1997/000511 patent/WO1997025041A1/en not_active Application Discontinuation
- 1997-01-09 EP EP97905573A patent/EP0871442A1/en not_active Withdrawn
- 1997-01-09 JP JP9525457A patent/JP2000501107A/en active Pending
- 1997-01-09 CA CA002242579A patent/CA2242579A1/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4772703A (en) * | 1986-01-27 | 1988-09-20 | American Home Products Corporation | 2-(phenoxymethyl)-quinazolines as antiallergic and antiinflammatory agents |
Non-Patent Citations (3)
| Title |
|---|
| DATABASE CAPLUS, (Columbus, Ohio, USA), Abstract No. 1983:46775, SHUKLA J.S. et al., "Synthesis of Some Newer 1-Heterocyclic Amino/iminomethyl-2-substituted Benzimidazoles as a Potent CNS, Anticonvulsant and Monoamine Oxidase Inhibitory Agents"; & CURR. SCI., 1982, 51(17), 820-822 (English), Registry Number * |
| DATABASE CAPLUS, (Columbus, Ohio, USA), Abstract No. 1984:591782, CHEN R. et al., "Synthesis of Some 1-Acylbenzimidazole and 1-Acylimidazole Derivatives"; & GAODENG XUEXIAO HUAXUE XUEBAO, 1984, 5(3), 361-365 (Chinese), Registry Numbers 92756-25-7, 92756-28-0, 92756-30-4, 92756-32-6, 92756-33-7, 92756-34-8 and 92772-55-9. * |
| DATABASE CAPLUS, (Columbus, Ohio, USA), Abstract No. 1988:198210, KULKARNI Y.D. et al., "Synthesis of 2-(1-Substituted Benzimidazol-2-yl)methylamino/methylenoxy-5 -chloro/nitrobenzo Phenones as Possible Anticonvulsants"; & BIOL. MEM. 1987, 13(2), 183-187 (English), Registry Number 114192-53-9. * |
Cited By (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998040356A1 (en) * | 1997-03-12 | 1998-09-17 | Banyu Pharmaceutical Co., Ltd. | Drugs containing aminopyridine derivatives as the active ingredient |
| WO1999007703A1 (en) * | 1997-08-05 | 1999-02-18 | Pfizer Products Inc. | 4-AMINOPYRROLE(3,2-d) PYRIMIDINES AS NEUROPEPTIDE Y RECEPTOR ANTAGONISTS |
| US6187778B1 (en) | 1997-08-05 | 2001-02-13 | Pfizer Inc. | 4-aminopyrrole (3, 2-D) pyrimidines as neuropeptide Y receptor antagonists |
| US6127414A (en) * | 1997-09-23 | 2000-10-03 | Astra Aktiebolag | NPY antagonists |
| WO1999036419A1 (en) * | 1998-01-17 | 1999-07-22 | Bayer Aktiengesellschaft | Substituted lactones as modulators of metabotropic glutamate receptors |
| WO1999040091A1 (en) * | 1998-02-06 | 1999-08-12 | Amgen Inc. | Bicyclic pyridine and pyrimidine derivatives as neuropeptide y receptor antagonists |
| US6187777B1 (en) | 1998-02-06 | 2001-02-13 | Amgen Inc. | Compounds and methods which modulate feeding behavior and related diseases |
| US6583154B1 (en) | 1998-02-06 | 2003-06-24 | Amgen Inc. | Compounds and methods which modulate feeding behavior and related diseases |
| WO2002046168A1 (en) * | 2000-12-07 | 2002-06-13 | Astrazeneca Ab | Therapeutic benzimidazole compounds |
| WO2002059088A1 (en) * | 2001-01-23 | 2002-08-01 | Wyeth | 1-aryl-or 1-alkylsulfonylbenzazole derivatives as 5-hydroxytryptamine-6 ligands |
| US6774241B2 (en) | 2002-06-05 | 2004-08-10 | Roche Palo Alto Llc | 1-sulfonyl-4-aminoalkoxy indole derivatives and uses thereof |
| WO2004098591A2 (en) | 2003-05-05 | 2004-11-18 | Probiodrug Ag | Inhibitors of glutaminyl cyclase and their use in the treatment of neurological diseases |
| EP2338490A2 (en) | 2003-11-03 | 2011-06-29 | Probiodrug AG | Combinations Useful for the Treatment of Neuronal Disorders |
| WO2005049027A2 (en) | 2003-11-03 | 2005-06-02 | Probiodrug Ag | Combinations useful for the treatment of neuronal disorders |
| WO2005075436A2 (en) | 2004-02-05 | 2005-08-18 | Probiodrug Ag | Novel inhibitors of glutaminyl cyclase |
| WO2005080348A1 (en) | 2004-02-19 | 2005-09-01 | Banyu Pharmaceutical Co., Ltd. | Novel sulfone amide derivative |
| US7977331B1 (en) | 2004-02-24 | 2011-07-12 | Japan Tobacco Inc. | Tetracyclic fused heterocyclic compound and use thereof as HCV polymerase inhibitor |
| US7659263B2 (en) | 2004-11-12 | 2010-02-09 | Japan Tobacco Inc. | Thienopyrrole compound and use thereof as HCV polymerase inhibitor |
| WO2008055945A1 (en) | 2006-11-09 | 2008-05-15 | Probiodrug Ag | 3-hydr0xy-1,5-dihydr0-pyrr0l-2-one derivatives as inhibitors of glutaminyl cyclase for the treatment of ulcer, cancer and other diseases |
| WO2008065141A1 (en) | 2006-11-30 | 2008-06-05 | Probiodrug Ag | Novel inhibitors of glutaminyl cyclase |
| EP2481408A2 (en) | 2007-03-01 | 2012-08-01 | Probiodrug AG | New use of glutaminyl cyclase inhibitors |
| WO2008104580A1 (en) | 2007-03-01 | 2008-09-04 | Probiodrug Ag | New use of glutaminyl cyclase inhibitors |
| EP2865670A1 (en) | 2007-04-18 | 2015-04-29 | Probiodrug AG | Thiourea derivatives as glutaminyl cyclase inhibitors |
| US8962549B2 (en) | 2008-08-22 | 2015-02-24 | Baxter International Inc. | Polymeric benzyl carbonate-derivatives |
| US8653131B2 (en) | 2008-08-22 | 2014-02-18 | Baxter Healthcare S.A. | Polymeric benzyl carbonate-derivatives |
| WO2011029920A1 (en) | 2009-09-11 | 2011-03-17 | Probiodrug Ag | Heterocylcic derivatives as inhibitors of glutaminyl cyclase |
| WO2011107530A2 (en) | 2010-03-03 | 2011-09-09 | Probiodrug Ag | Novel inhibitors |
| WO2011110613A1 (en) | 2010-03-10 | 2011-09-15 | Probiodrug Ag | Heterocyclic inhibitors of glutaminyl cyclase (qc, ec 2.3.2.5) |
| WO2011131748A2 (en) | 2010-04-21 | 2011-10-27 | Probiodrug Ag | Novel inhibitors |
| US9062003B2 (en) | 2010-10-06 | 2015-06-23 | Glaxosmithkline Llc | Benzimidazole derivatives as PI3 kinase inhibitors |
| US8541411B2 (en) | 2010-10-06 | 2013-09-24 | Glaxosmithkline Llc | Benzimidazole derivatives as PI3 kinase inhibitors |
| US8674090B2 (en) | 2010-10-06 | 2014-03-18 | Glaxosmithkline Llc | Benzimidazole derivatives as PI3 kinase inhibitors |
| US8865912B2 (en) | 2010-10-06 | 2014-10-21 | Glaxosmithkline Llc | Benzimidazole derivatives as PI3 kinase inhibitors |
| US8435988B2 (en) | 2010-10-06 | 2013-05-07 | Glaxosmithkline Llc | Benzimidazole derivatives as P13 kinase inhibitors |
| US9156797B2 (en) | 2010-10-06 | 2015-10-13 | Glaxosmithkline Llc | Benzimidazole derivatives as PI3 kinase inhibitors |
| US9872860B2 (en) | 2010-10-06 | 2018-01-23 | Glaxosmithkline Llc | Benzimidazole derivatives as PI3 kinase inhibitors |
| US10314845B2 (en) | 2010-10-06 | 2019-06-11 | Glaxosmithkline Llc | Benzimidazole derivatives as PI3 kinase inhibitors |
| US10660898B2 (en) | 2010-10-06 | 2020-05-26 | Glaxosmithkline Llc | Benzimidazole derivatives as PI3 kinase inhibitors |
| WO2012123563A1 (en) | 2011-03-16 | 2012-09-20 | Probiodrug Ag | Benz imidazole derivatives as inhibitors of glutaminyl cyclase |
| WO2012130905A1 (en) | 2011-03-31 | 2012-10-04 | Bayer Pharma Aktiengesellschaft | Substituted benzimidazoles |
| CN103965113A (en) * | 2014-05-06 | 2014-08-06 | 四川大学 | 1-ethoxyl-2-substituted phenoxymethyl benzimidazole compound and preparation method thereof |
| EP3461819A1 (en) | 2017-09-29 | 2019-04-03 | Probiodrug AG | Inhibitors of glutaminyl cyclase |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2000501107A (en) | 2000-02-02 |
| EP0871442A1 (en) | 1998-10-21 |
| CA2242579A1 (en) | 1997-07-17 |
| AU2242197A (en) | 1997-08-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO1997025041A1 (en) | Benzimidzolyl neuropeptide y receptor antagonists | |
| AU717422B2 (en) | Indolyl neuropeptide Y receptor antagonists | |
| US6329389B1 (en) | Amine compounds, their production and use | |
| US6245773B1 (en) | 5-(heterocyclic alkyl)-6-aryl-dihydropyrimidines | |
| US6727255B1 (en) | Piperidinyl and piperazinyl tachykinin receptor antagonists | |
| KR100748294B1 (en) | Propane-1,3-Dione Derivatives | |
| JP4870163B2 (en) | Indan derivatives as MCH receptor antagonists | |
| JP4527408B2 (en) | Muscarinic antagonist | |
| EP0714891A1 (en) | Heterocyclic tachykinin receptor antagonists | |
| DE60103375T2 (en) | 1,3-DIHYDRO-2H-INDOL-2-ON DERIVATIVES AND THEIR USE AS LIGANDS OF ARGININE VASOPRESSIN RECEPTORS V1B OR V1B AND V1A | |
| SK404491A3 (en) | Tricyclic lactam compounds, pharmaceutical compositions comprising same and their use | |
| KR19980703048A (en) | 5-substituted-3- (1,2,3,6-tetrahydropyridin-4-yl) - and 3- (piperidin-4-yl) -1H- indole: A novel 5-HT1F agonist | |
| TW201840550A (en) | Somatostatin modulators and uses thereof | |
| JPH08253474A (en) | Indolepiperidine derivative | |
| MX2007007894A (en) | 5-ht7 receptor antagonists. | |
| EP1373220B1 (en) | Benzimidazoles that are useful in treating sexual dysfunction | |
| US6255494B1 (en) | Benzimidzolyl neuropeptide Y receptor antagonists | |
| US6245761B1 (en) | Indolyl neuropeptide Y receptor antagonists | |
| US5776931A (en) | Naphthimidazolyl neuropeptide Y receptor antagonists | |
| US5563133A (en) | Hexamethyleneiminyl tachykinin receptor antagonists | |
| US6506756B2 (en) | Substituted imidazoles as dual histamine H1 and H3 agonists or antagonists | |
| EP0699665A1 (en) | Imidazoline derivatives, their preparation and their use as tachykinin receptor antagonists | |
| JPH10508321A (en) | Indolyl neuropeptide Y receptor antagonist | |
| US5607947A (en) | Pyrrolidinyl tachykinin receptor antagonists | |
| JP2005510475A6 (en) | Tetrahydroisoquinolines, methods for their production, and their use as anesthetics |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE HU IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK TJ TM TR TT UA UG UZ VN AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): KE LS MW SD SZ UG AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2242579 Country of ref document: CA Ref country code: CA Ref document number: 2242579 Kind code of ref document: A Format of ref document f/p: F |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 1997 525457 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1997905573 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1997905573 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1997905573 Country of ref document: EP |