WO1997018214A1 - Hemoregulatory compounds - Google Patents
Hemoregulatory compounds Download PDFInfo
- Publication number
- WO1997018214A1 WO1997018214A1 PCT/US1996/018247 US9618247W WO9718214A1 WO 1997018214 A1 WO1997018214 A1 WO 1997018214A1 US 9618247 W US9618247 W US 9618247W WO 9718214 A1 WO9718214 A1 WO 9718214A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- pyrazine
- alkyl
- xylenediyl
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 63
- 230000003612 virological effect Effects 0.000 claims abstract description 9
- 208000022362 bacterial infectious disease Diseases 0.000 claims abstract description 7
- 230000002538 fungal effect Effects 0.000 claims abstract description 6
- 239000001257 hydrogen Substances 0.000 claims description 15
- 229910052739 hydrogen Inorganic materials 0.000 claims description 15
- 238000000034 method Methods 0.000 claims description 14
- 125000000217 alkyl group Chemical group 0.000 claims description 10
- 241001465754 Metazoa Species 0.000 claims description 8
- 150000002431 hydrogen Chemical group 0.000 claims description 8
- 229910052757 nitrogen Inorganic materials 0.000 claims description 8
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 7
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 6
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 6
- 150000003839 salts Chemical class 0.000 claims description 6
- 125000005023 xylyl group Chemical group 0.000 claims description 6
- 229910052736 halogen Inorganic materials 0.000 claims description 5
- 230000003525 myelopoietic effect Effects 0.000 claims description 5
- 125000006239 protecting group Chemical group 0.000 claims description 5
- 239000002904 solvent Substances 0.000 claims description 5
- 230000004936 stimulating effect Effects 0.000 claims description 5
- 208000035143 Bacterial infection Diseases 0.000 claims description 4
- 208000031888 Mycoses Diseases 0.000 claims description 4
- -1 bicyclo[3.3.0]octanyl Chemical group 0.000 claims description 4
- 150000002367 halogens Chemical class 0.000 claims description 4
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 claims description 3
- CXHMDOZFANRPGH-UHFFFAOYSA-N 1,3,4,4a-tetrahydropyrrolo[2,3-b]pyrazin-2-one Chemical compound N1C(=O)CNC2N=CC=C21 CXHMDOZFANRPGH-UHFFFAOYSA-N 0.000 claims description 3
- 206010017533 Fungal infection Diseases 0.000 claims description 3
- 206010040047 Sepsis Diseases 0.000 claims description 3
- 208000036142 Viral infection Diseases 0.000 claims description 3
- 229910052740 iodine Inorganic materials 0.000 claims description 3
- BIPZVRUKFBUIMY-UHFFFAOYSA-N 4,4a,5,6-tetrahydro-1h-pyrrolo[2,3-b]pyrazine-2,3-dione Chemical compound N1C(=O)C(=O)NC2NCC=C21 BIPZVRUKFBUIMY-UHFFFAOYSA-N 0.000 claims description 2
- 230000002152 alkylating effect Effects 0.000 claims description 2
- 230000015572 biosynthetic process Effects 0.000 claims description 2
- 229910052794 bromium Inorganic materials 0.000 claims description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 2
- 230000008878 coupling Effects 0.000 claims description 2
- 238000010168 coupling process Methods 0.000 claims description 2
- 238000005859 coupling reaction Methods 0.000 claims description 2
- 150000004985 diamines Chemical class 0.000 claims description 2
- 125000001041 indolyl group Chemical group 0.000 claims description 2
- 238000004519 manufacturing process Methods 0.000 claims description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 2
- 125000001424 substituent group Chemical group 0.000 claims description 2
- 229910052801 chlorine Inorganic materials 0.000 claims 1
- 239000007822 coupling agent Substances 0.000 claims 1
- 229910052731 fluorine Inorganic materials 0.000 claims 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims 1
- 125000002801 octanoyl group Chemical group C(CCCCCCC)(=O)* 0.000 claims 1
- 238000011282 treatment Methods 0.000 abstract description 14
- 230000000694 effects Effects 0.000 abstract description 8
- 230000011132 hemopoiesis Effects 0.000 abstract description 5
- 208000024386 fungal infectious disease Diseases 0.000 abstract description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 18
- 239000000203 mixture Substances 0.000 description 17
- 210000004027 cell Anatomy 0.000 description 16
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 15
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 12
- 210000001185 bone marrow Anatomy 0.000 description 12
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 10
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 10
- 238000006243 chemical reaction Methods 0.000 description 9
- 208000015181 infectious disease Diseases 0.000 description 9
- 235000002639 sodium chloride Nutrition 0.000 description 9
- 239000007787 solid Substances 0.000 description 9
- 239000000243 solution Substances 0.000 description 9
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 8
- 239000003921 oil Substances 0.000 description 7
- 235000019198 oils Nutrition 0.000 description 7
- 239000008194 pharmaceutical composition Substances 0.000 description 7
- 238000000746 purification Methods 0.000 description 7
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- 230000003394 haemopoietic effect Effects 0.000 description 6
- 239000012071 phase Substances 0.000 description 6
- 206010065553 Bone marrow failure Diseases 0.000 description 5
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 5
- 235000019439 ethyl acetate Nutrition 0.000 description 5
- 238000003818 flash chromatography Methods 0.000 description 5
- 239000008187 granular material Substances 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 238000002156 mixing Methods 0.000 description 5
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 239000000741 silica gel Substances 0.000 description 5
- 229910002027 silica gel Inorganic materials 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 108010075031 Cytochromes c Proteins 0.000 description 4
- 108010010803 Gelatin Proteins 0.000 description 4
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 4
- 102000007651 Macrophage Colony-Stimulating Factor Human genes 0.000 description 4
- 108010046938 Macrophage Colony-Stimulating Factor Proteins 0.000 description 4
- 241001529936 Murinae Species 0.000 description 4
- 239000000969 carrier Substances 0.000 description 4
- 239000003937 drug carrier Substances 0.000 description 4
- 239000012636 effector Substances 0.000 description 4
- AEOCXXJPGCBFJA-UHFFFAOYSA-N ethionamide Chemical compound CCC1=CC(C(N)=S)=CC=N1 AEOCXXJPGCBFJA-UHFFFAOYSA-N 0.000 description 4
- 238000009472 formulation Methods 0.000 description 4
- 229920000159 gelatin Polymers 0.000 description 4
- 239000008273 gelatin Substances 0.000 description 4
- 235000019322 gelatine Nutrition 0.000 description 4
- 235000011852 gelatine desserts Nutrition 0.000 description 4
- 239000003102 growth factor Substances 0.000 description 4
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 4
- 208000004235 neutropenia Diseases 0.000 description 4
- 239000012044 organic layer Substances 0.000 description 4
- 239000000546 pharmaceutical excipient Substances 0.000 description 4
- BXRNXXXXHLBUKK-UHFFFAOYSA-N piperazine-2,5-dione Chemical compound O=C1CNC(=O)CN1 BXRNXXXXHLBUKK-UHFFFAOYSA-N 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 239000006228 supernatant Substances 0.000 description 4
- 229920001817 Agar Polymers 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- 102400000500 GRO-beta(5-73) Human genes 0.000 description 3
- 101500024488 Homo sapiens GRO-beta(5-73) Proteins 0.000 description 3
- 101500028514 Mus musculus KC(5-72) Proteins 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 3
- 239000012980 RPMI-1640 medium Substances 0.000 description 3
- 229910000564 Raney nickel Inorganic materials 0.000 description 3
- OUUQCZGPVNCOIJ-UHFFFAOYSA-M Superoxide Chemical compound [O-][O] OUUQCZGPVNCOIJ-UHFFFAOYSA-M 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 238000007792 addition Methods 0.000 description 3
- 239000008272 agar Substances 0.000 description 3
- 235000010419 agar Nutrition 0.000 description 3
- 210000002798 bone marrow cell Anatomy 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 239000006185 dispersion Substances 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 239000007903 gelatin capsule Substances 0.000 description 3
- 210000003714 granulocyte Anatomy 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 230000002265 prevention Effects 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 210000000130 stem cell Anatomy 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 239000006188 syrup Substances 0.000 description 3
- 235000020357 syrup Nutrition 0.000 description 3
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- IZHVBANLECCAGF-UHFFFAOYSA-N 2-hydroxy-3-(octadecanoyloxy)propyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)COC(=O)CCCCCCCCCCCCCCCCC IZHVBANLECCAGF-UHFFFAOYSA-N 0.000 description 2
- RCGIPGKJDFKYKW-UHFFFAOYSA-N 5,6-dibromo-5,6-dimethylcyclohexa-1,3-diene Chemical group CC1(Br)C=CC=CC1(C)Br RCGIPGKJDFKYKW-UHFFFAOYSA-N 0.000 description 2
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- 102000007644 Colony-Stimulating Factors Human genes 0.000 description 2
- 108010071942 Colony-Stimulating Factors Proteins 0.000 description 2
- 102100030497 Cytochrome c Human genes 0.000 description 2
- 102000004127 Cytokines Human genes 0.000 description 2
- 108090000695 Cytokines Proteins 0.000 description 2
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 2
- 108010016626 Dipeptides Proteins 0.000 description 2
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 description 2
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 240000007472 Leucaena leucocephala Species 0.000 description 2
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- ISKQADXMHQSTHK-UHFFFAOYSA-N [4-(aminomethyl)phenyl]methanamine Chemical compound NCC1=CC=C(CN)C=C1 ISKQADXMHQSTHK-UHFFFAOYSA-N 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 230000001580 bacterial effect Effects 0.000 description 2
- 210000000601 blood cell Anatomy 0.000 description 2
- 231100001018 bone marrow damage Toxicity 0.000 description 2
- 230000003915 cell function Effects 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 229940047120 colony stimulating factors Drugs 0.000 description 2
- 239000000824 cytostatic agent Substances 0.000 description 2
- 230000001085 cytostatic effect Effects 0.000 description 2
- 229940127089 cytotoxic agent Drugs 0.000 description 2
- 239000002254 cytotoxic agent Substances 0.000 description 2
- 231100000599 cytotoxic agent Toxicity 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 238000011049 filling Methods 0.000 description 2
- 239000012467 final product Substances 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 230000002458 infectious effect Effects 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- CFHGBZLNZZVTAY-UHFFFAOYSA-N lawesson's reagent Chemical compound C1=CC(OC)=CC=C1P1(=S)SP(=S)(C=2C=CC(OC)=CC=2)S1 CFHGBZLNZZVTAY-UHFFFAOYSA-N 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 210000000265 leukocyte Anatomy 0.000 description 2
- 239000012669 liquid formulation Substances 0.000 description 2
- 210000002540 macrophage Anatomy 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- 238000003801 milling Methods 0.000 description 2
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- IWDCLRJOBJJRNH-UHFFFAOYSA-N p-cresol Chemical compound CC1=CC=C(O)C=C1 IWDCLRJOBJJRNH-UHFFFAOYSA-N 0.000 description 2
- 238000010647 peptide synthesis reaction Methods 0.000 description 2
- PHEDXBVPIONUQT-RGYGYFBISA-N phorbol 13-acetate 12-myristate Chemical compound C([C@]1(O)C(=O)C(C)=C[C@H]1[C@@]1(O)[C@H](C)[C@H]2OC(=O)CCCCCCCCCCCCC)C(CO)=C[C@H]1[C@H]1[C@]2(OC(C)=O)C1(C)C PHEDXBVPIONUQT-RGYGYFBISA-N 0.000 description 2
- 239000002798 polar solvent Substances 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 238000010791 quenching Methods 0.000 description 2
- 230000008929 regeneration Effects 0.000 description 2
- 238000011069 regeneration method Methods 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 239000012086 standard solution Substances 0.000 description 2
- 230000000638 stimulation Effects 0.000 description 2
- 210000002536 stromal cell Anatomy 0.000 description 2
- 230000002195 synergetic effect Effects 0.000 description 2
- BNWCETAHAJSBFG-UHFFFAOYSA-N tert-butyl 2-bromoacetate Chemical compound CC(C)(C)OC(=O)CBr BNWCETAHAJSBFG-UHFFFAOYSA-N 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- OZFAFGSSMRRTDW-UHFFFAOYSA-N (2,4-dichlorophenyl) benzenesulfonate Chemical compound ClC1=CC(Cl)=CC=C1OS(=O)(=O)C1=CC=CC=C1 OZFAFGSSMRRTDW-UHFFFAOYSA-N 0.000 description 1
- KGKAYWMGPDWLQZ-UHFFFAOYSA-N 1,2-bis(bromomethyl)benzene Chemical group BrCC1=CC=CC=C1CBr KGKAYWMGPDWLQZ-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- 208000030507 AIDS Diseases 0.000 description 1
- 206010000830 Acute leukaemia Diseases 0.000 description 1
- 206010001052 Acute respiratory distress syndrome Diseases 0.000 description 1
- 201000010000 Agranulocytosis Diseases 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- 208000032467 Aplastic anaemia Diseases 0.000 description 1
- 208000018240 Bone Marrow Failure disease Diseases 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000222120 Candida <Saccharomycetales> Species 0.000 description 1
- 241000222122 Candida albicans Species 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 239000012591 Dulbecco’s Phosphate Buffered Saline Substances 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 208000009329 Graft vs Host Disease Diseases 0.000 description 1
- 239000012981 Hank's balanced salt solution Substances 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- XQFRJNBWHJMXHO-RRKCRQDMSA-N IDUR Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(I)=C1 XQFRJNBWHJMXHO-RRKCRQDMSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102000015696 Interleukins Human genes 0.000 description 1
- 108010063738 Interleukins Proteins 0.000 description 1
- RNKSNIBMTUYWSH-YFKPBYRVSA-N L-prolylglycine Chemical compound [O-]C(=O)CNC(=O)[C@@H]1CCC[NH2+]1 RNKSNIBMTUYWSH-YFKPBYRVSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 101001068640 Nicotiana tabacum Basic form of pathogenesis-related protein 1 Proteins 0.000 description 1
- XMGCCBQWHDYHEH-YFKPBYRVSA-N O=C([C@H]1N2CCC1)NCC2=S Chemical compound O=C([C@H]1N2CCC1)NCC2=S XMGCCBQWHDYHEH-YFKPBYRVSA-N 0.000 description 1
- SFXFLCYGXQYWRE-PMACEKPBSA-N O=C1N(Cc2ccccc2CN(CCN2[C@H]3CCC2)C3=O)CCN2[C@H]1CCC2 Chemical compound O=C1N(Cc2ccccc2CN(CCN2[C@H]3CCC2)C3=O)CCN2[C@H]1CCC2 SFXFLCYGXQYWRE-PMACEKPBSA-N 0.000 description 1
- JBIHMFNMKHDTEE-LURJTMIESA-N O=C1NCCN2[C@H]1CCC2 Chemical compound O=C1NCCN2[C@H]1CCC2 JBIHMFNMKHDTEE-LURJTMIESA-N 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- AFWBWPCXSWUCLB-WDSKDSINSA-N Pro-Ser Chemical compound OC[C@@H](C([O-])=O)NC(=O)[C@@H]1CCC[NH2+]1 AFWBWPCXSWUCLB-WDSKDSINSA-N 0.000 description 1
- 206010037660 Pyrexia Diseases 0.000 description 1
- 239000007868 Raney catalyst Substances 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 230000001464 adherent effect Effects 0.000 description 1
- 201000000028 adult respiratory distress syndrome Diseases 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 229940034982 antineoplastic agent Drugs 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 239000006286 aqueous extract Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 238000010936 aqueous wash Methods 0.000 description 1
- 230000003190 augmentative effect Effects 0.000 description 1
- 210000003651 basophil Anatomy 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 210000001772 blood platelet Anatomy 0.000 description 1
- 238000010322 bone marrow transplantation Methods 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 1
- PASHVRUKOFIRIK-UHFFFAOYSA-L calcium sulfate dihydrate Chemical compound O.O.[Ca+2].[O-]S([O-])(=O)=O PASHVRUKOFIRIK-UHFFFAOYSA-L 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000022131 cell cycle Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 230000027288 circadian rhythm Effects 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 238000011393 cytotoxic chemotherapy Methods 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 230000007123 defense Effects 0.000 description 1
- 230000000994 depressogenic effect Effects 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 231100000371 dose-limiting toxicity Toxicity 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 210000003979 eosinophil Anatomy 0.000 description 1
- 210000003743 erythrocyte Anatomy 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 210000000416 exudates and transudate Anatomy 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 238000005187 foaming Methods 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 229940074045 glyceryl distearate Drugs 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- 208000024908 graft versus host disease Diseases 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 210000000777 hematopoietic system Anatomy 0.000 description 1
- 230000002607 hemopoietic effect Effects 0.000 description 1
- 230000002008 hemorrhagic effect Effects 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical group O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 230000001506 immunosuppresive effect Effects 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 229940047122 interleukins Drugs 0.000 description 1
- 201000002364 leukopenia Diseases 0.000 description 1
- 231100001022 leukopenia Toxicity 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 239000008176 lyophilized powder Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 238000003760 magnetic stirring Methods 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 229940071648 metered dose inhaler Drugs 0.000 description 1
- KBYRFBWEIKLMLI-VIFPVBQESA-N methyl (2s)-1-[2-[(2-methylpropan-2-yl)oxycarbonylamino]acetyl]pyrrolidine-2-carboxylate Chemical compound COC(=O)[C@@H]1CCCN1C(=O)CNC(=O)OC(C)(C)C KBYRFBWEIKLMLI-VIFPVBQESA-N 0.000 description 1
- ZVWGEHPXUFGWAQ-VIFPVBQESA-N methyl (2s)-1-[2-[(2-methylpropan-2-yl)oxycarbonylamino]ethanethioyl]pyrrolidine-2-carboxylate Chemical compound COC(=O)[C@@H]1CCCN1C(=S)CNC(=O)OC(C)(C)C ZVWGEHPXUFGWAQ-VIFPVBQESA-N 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 230000003039 myelosuppressive effect Effects 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 210000002997 osteoclast Anatomy 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 238000009520 phase I clinical trial Methods 0.000 description 1
- 210000001778 pluripotent stem cell Anatomy 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 108010031719 prolyl-serine Proteins 0.000 description 1
- 108010029020 prolylglycine Proteins 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000001172 regenerating effect Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 238000006798 ring closing metathesis reaction Methods 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 239000012258 stirred mixture Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 230000002110 toxicologic effect Effects 0.000 description 1
- 231100000759 toxicological effect Toxicity 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 201000008827 tuberculosis Diseases 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D221/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00
- C07D221/02—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00 condensed with carbocyclic rings or ring systems
- C07D221/04—Ortho- or peri-condensed ring systems
Definitions
- the present invention relates to novel compounds which have hemoregulatory activities and can be used to stimulate hematopoiesis and for the treatment of viral, fungal and bacterial infectious diseases.
- the hematopoietic system is a life-long cell renewal process whereby a defined stem cell population gives rise to a larger population of mature, differentiated blood cells (Dexter TM. Stem cells in normal growth and disease Br Med J 1987; 195:1192-1194) of at least nine different cell lineages
- Stem cells are also ultimately responsible for regenerating bone marrow following treatment with cytotoxic agents or following bone marrow
- Myelosuppression is predictable and has been reported to be dose-limiting in greater than 50% of single-agent Phase I trials cytotoxic compounds (Merrouche Y, Catimel G, Clavel M. Hematopoietic growth factors and chemoprotectants; should we move toward a two-step process for phase I clinical trials in oncology? Ann Oncol 1993; 4:471 -474).
- the risk of infection is directly related to the degree of myelosuppression as measured by the severity and duration of neutropenia (Brody GP, Buckley M, Sathe YS, Freireich EJ. Quantitative relationship between circulating leukocytes and infections with acute leukemia. Ann In Med 1965; 64:328-334).
- the control of hematopoiesis involves the interplay of a variety of cytokines and growth factors during various stages of the hematopoietic cascade, including early pluripotent stem cells and mature circulating effector cells.
- These regulatory molecules include granulocyte colony stimulating factor (G-CSF), granulocyte-macrophage stimulating factor (GM-CSF), macrophage-colony stimulating factor (M-CSF), and a variety of interleukins which have overlapping, additive and synergistic actions which play major roles in host defense.
- myelotoxicity include the use of hematopoietic growth factors and/or other hematopoietic cytokines. Such treatments are becoming common practice, in that they offer the potential of increased doses of cytotoxic agents that may improve the therapeutic efficacy of antineoplastic agents, and reduce the morbidity associated with their use (Steward WP. Granulocyte and granulocyte-macrophage colony stimulating factors, Lancet 1993; 342:153- 157). Clinical studies have demonstrated the G-, GM- and/or M-CSF may reduce the duration of
- neutropenia accelerate myeloid recovery, and reduce neutropenia-associated infections and other infectious complications in patients with malignancies who are receiving cytotoxic chemotherapy or in high infectious-risk patients following bone marrow transplantation (Steward WP Granulocyte and granulocyte-macrophage colony stimulating factors, Lancet 1993; 342:153-157 and Munn DH, Cheung NKV. Prechnical and clinical studies of macrophage colony-stimulating factor Senun Oncol 1992; 19:395-407).
- Synthetic peptides have been reported to induce the synthesis and release of hematopoietic mediators, including m-CSF from bone marrow stromal elements (see U.S. Patent Application 08/001,905).
- novel non-peptide compounds which have a stimulative effect on myelopoietic cells. They are useful in stimulating myelopoiesis in patients suffering from reduced myelopoietic activity, including bone marrow damage, agranulocytosis and aplastic anemia including patients having depressed bone marrow function due to immunosuppressive treatment to suppress tissue reactions i.e. in bone marrow transplant surgery.
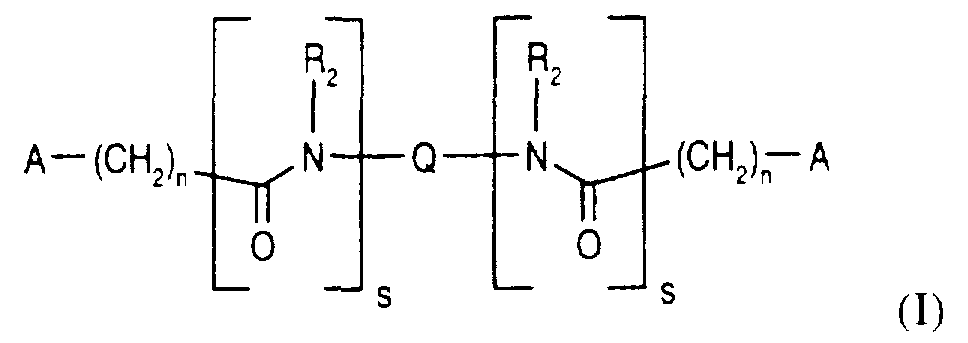
- This invention comprises compounds, hereinafter represented as Formula
- These compounds are useful in the restoration of leukocytes in patients with lowered cell counts resulting from a variety of clinical situations, such as surgical induced myelosuppression, AIDS, ARDS, congenital myelodysplacis, bone marrow and organ transplants; in the protection of patients with leukopenia from infection; in the treatment of severely burned patients and in the amelioration of the myelosuppression observed with some cell-cycle specific antiviral agents and in the treatment of infections in patients who have had bone marrow transplants, especially those with graft versus host disease, in the treatment of tuberculosis and in the treatment of fevers of unknown origin in humans and animals.
- the compounds are also useful in the treatment and prevention of viral, fungal and bacterial infectious diseases, particularly Candida and Herpes in both immunosuppressed and "normal" subjects. They are useful in the treatment of sepsis caused by gram negative and gram positive organisms. These compounds may also be used in combination with the
- cytostatic therapy can be given at periods of low bone marrow activity, thus reducing the risk of bone marrow damage, while regeneration will be promoted by the succeeding peak of activity.
- This invention is also a pharmaceutical composition, which comprises a compound of Formula (I) and a pharmaceutically acceptable carrier.
- This invention further constitutes a method for stimulating the
- myelopoietic system of an animal including humans, which comprises administering to an animal in need thereof, an effective amount of a compound of Formula (I).
- This invention also constitutes a method for preventing and treating viral, fungal and bacterial infections including sepsis in immunosuppressed and normal animals, including humans, which comprises administering to an animal in need thereof, an effective amount of a compound of Formula (I).
- A is or ;
- R 1 is independently NH 2 , OH, SH, CN, CO 2 H or hydrogen;
- R 2 is independently hydrogen, C 1-4 alkylC(O)R 5 , C 1 -4 alkyl or R 2 is benzyl which is optionally substituted by one or two C 1-4 alkyl, C 1 -4 alkoxy, F, C1, I, Br, OH, or N(R 4 ) 2 ;
- R 3 is independently hydrogen, C 1 -5 alkyl, C 1-5 alkylenehydroxy, C 1 - 5 alkyleneCO 2 H,
- R 4 is independently hydrogen, C 1-5 alkyl or benzyl
- Ar is independently phenyl or indolyl optionally substituted by one or two
- Q is bicyclo(3.3.0]octanyl, xylyl, benzophenonyl or 1,2,3,4-tetrahydronapthalyl; all of which are unsubstituted or substituted by one or two substituents chosen from:
- R 5 is -OR 6 , -N(R 6 ) 2 , or -SR 6 ;
- R 6 is hydrogen, C 1 -4 alkyl or benzyl;
- R 7 is halogen, R 5 or R 6 ;
- n is independently an integer from 0 to 3;
- n is independently an integer from 1 to 3;
- s is independently 0 or 1 ;
- y is independently an integer from 2 to 4.
- n is not 0 when s is 1 and further provided that the compound of Formula (I) is not:
- This invention is also a pharmaceutical composition, which comprises a compound of Formula (I) and a pharmaceutically acceptable carrier.
- Alkyl groups may be straight or branched.
- Halogen may be chloro, lodo, fluoro or bromo.
- the compounds of the present invention may contain one or more asymmetric carbon atoms and may exist in racemic and optically active forms. All the compounds and diastereomers are contemplated to be within the scope of the present invention.
- Preferred compounds of Formula (I) are those wherein R 1 is OH or hydrogen; R 3 is CH 2 OH or hydrogen and Q is xylyl or bicyclo[3.3.0]octanoyl. More preferred compounds are those wherein R 1 , R 2 and R 3 are hydrogen and Q is xylyl.
- the most preferred compounds of the invention are:
- diketopiperazine (such as 3 in Scheme 1 ).
- the diketopiperazines (such as 3 in
- Scheme 1 are alkylated with a suitably protected alkylating agent (such as t-butyl bromoacetate) in a suitable aprotic polar solvent (such as THF) to give the N-alkylated diketopiperazine (such as 4 in Scheme 1 ).
- a suitably protected alkylating agent such as t-butyl bromoacetate
- a suitable aprotic polar solvent such as THF
- the thioamide is reduced using conventional reagents (such as Raney-Nickel) in a suitable polar solvent (such as MeOH) to give the tetrahydropyrrolopyrazinone (such as 4 in Scheme 2).
- a suitable polar solvent such as MeOH
- compositions comprising as active ingredient one or more compounds of Formula (I) as herein before defined or physiologically compatible salts thereof, in association with a pharmaceutical carrier or excipient.
- the compositions according to the invention may be presented for example, in a form suitable for oral, nasal, parenteral or rectal administration.
- the term "pharmaceutical” includes veterinary applications of the invention These compounds may be encapsulated, tableted or prepared in an emulsion or syrup for oral administration.
- Pharmaceutically acceptable solid or liquid carriers may be added to enhance or stabilize the composition, or to facilitate preparation of the composition.
- Liquid carriers include syrup, peanut oil, olive oil, glycerin, saline and water.
- Solid carriers include starch, lactose, calcium sulfate dihydrate, terra alba, magnesium stearate or stearic acid, talc, pectin, acacia, agar or gelatin.
- the carrier may also include a sustained release material such a glyceryl monostearate or glyceryl distearate, alone or with a wax.
- a sustained release material such as a glyceryl monostearate or glyceryl distearate, alone or with a wax.
- the amount of solid carrier varies but, preferably will be between about 20 mg to about 1 g per dosage unit.
- the pharmaceutical preparations are made following the conventional techniques of pharmacy involving milling, mixing, granulating, and compressing, when necessary, for tablet forms; or milling, mixing and filling for hard gelatin capsule forms.
- Capsules containing one or several active ingredients may be produced, for example, by mixing the active ingredients with inert carriers, such as lactose or sorbitol, and filling the mixture into gelatin capsules.
- the preparation When a liquid carrier is used, the preparation will be in the form of a syrup, elixir, emulsion or an aqueous or non-aqueous suspension. Such a liquid formulation may be administered directly p.o. or filled into a soft gelatin capsule Organ specific carrier systems may also be used.
- compositions of the compounds of this invention, or derivatives thereof may be formulated as solutions of lyophilized powders for parenteral administration.
- Powders may be reconstituted by addition of a suitable diluent or other pharmaceutically acceptable carrier pnor to use.
- the liquid formulation is generally a buffered, isotonic, aqueous solution
- suitable diluents are normal isotonic saline solution, standard 5% dextrose in water or buffered sodium or ammonium acetate solution.
- Such formulation is especially suitable for parenteral administration, but may also be used for oral administration and contained in a metered dose inhaler or nebulizer for insufflation It may be desirable to add excipients such as
- polyvinylpyrrolidone gelatin, hydroxy cellulose, acacia, polyethylene glycol, mannitol, sodium chloride or sodium citrate.
- a pulverized powder of the compounds of this invention may be combined with excipients such as cocoa butter, glycerin, gelatin or polyethylene glycols and molded into a suppository.
- excipients such as cocoa butter, glycerin, gelatin or polyethylene glycols
- the pulverized powders may also be compounded with an oily preparation, gel, cream or emulsion, buffered or unbuffered, and administered through a transdermal patch.
- Nasal sprays may be formulated similarly in aqueous solution and packed into spray containers either with an aerosol propellant or provided with means for manual compression.
- Dosage units containing the compounds of this invention preferably contain 05-50 mg, for example .05-5 mg of the compound of formula (I) or salt thereof.
- the murine bone marrow derived stromal cell line, C6.4 is grown in 12 well plates in RPMI 1640 with 10% FBS Upon reaching confluence, the C6.4 cells are washed and the media exchanged with fresh RPMI 1640 without FBS Confluent cell layers of murine C6.4 cells are treated with compound. Cell-free supernatants are collected 18 hours later Supernatants are fractionated with a Centricon-30 molecular weight cut-off membrane. C6.4 cell hematopoietic synergistic factor (HSF) activity is measured in a murine CFU-C assay.
- HSF synergistic factor
- Bone marrow cells are obtained from C57B1/6 female mice and suspended in RPMI 1640 with 10% FBS Bone marrow cells (7.5E+4 cells/mL) are cultured with sub optimal levels of CFU plus dilutions of test C6.4 cell 30K-E supernatants from above in a standard murine soft agar CFU-C assay. Cell aggregates >50 cells are counted as colonies. The number of agar colonies counted is proportional to the amount of HSF present within the C6.4 bone marrow stromal line supernatant.
- mice Female C57B1 mice are administered test compound IP or PO daily for 8 days.
- Resident peritoneal exudate cells (PEC) utilized ex vivo from treated or untreated mice are harvested with cold calcium and magnesium-free DPBS supplemented with heparin and antibiotics within 2-4 hours following the last injection.
- Adherent PEM populations are prepared by incubating standardized PEC suspensions in microtiter dishes for 2 hours at 37 °C (5% CO2) and removing nonadherent cells by washing the wells with warm buffer.
- SOD superoxide dismutase-inhibitable
- PMA phorbol myristate acetate
- nmoles of cytochrome c reduced/well is calculated from spectrophotometric readings (550 nm) taken following a 1 hour incubation at 37 °C (5% CO 2 ).
- the amount of SOD-inhibitable cytochrome c reduced is determined by the inclusion of wells containing SOD (200 U/well). Baseline superoxide release is determined in the absence of stimuli. Experimental data are expressed as a percentage of the control group.
- 1,4-Xylylenediamine (0.14 g, 1.0 mmol), iPr 2 NEt (1.75 mL 10.0 mmol), HOBt (0.31 g, 2.3 mmol) and BOP reagent (1.02 g, 2.3 mmol) were added sequentially and the reaction was stirred at room temperature for 18 h.
- the reaction mixture was added to a rapidly-stirred mixture of EtOAc (100 mL), 1N HCl (100 mL), and sat NaCl ( 100 mL). After stirring for 1 h, the phases were separated and the aqueous layer was extracted with fresh EtOAc (2 ⁇ 50 mL).
- Example 3(a) To the compound of Example 3(a) (2.22 g, 7.35 mmol) in C ⁇ 2 Cl 2 (35 mL) at 0 °C was added TFA (35 mL). The ice bath was removed and the solution was allowed to stir for 1 h. The solvent was removed under vacuum and the residue was azeotroped from toluene (3 ⁇ 20 mL). The residue was suspended in toluene (500 mL) and iPr 2 NEt (3.84 mL, 22.0 mmol) was added. The reaction was heated to reflux for 18 h and then allowed to cool to RT.
- Example 3(b) To the compound of Example 3(b) (0.92 g, 5.41 mmol) in MeO ⁇ ( 180 mL) was added Ra-Ni (ca 9 g of a 50 % aqueous slurry). The mixture was heated to reflux for 2 h, cooled to room temperature and filtered through celite to remove the Ra-Ni. The filter cake was washed with EtO ⁇ and the combined filtrates were concentrated under vacuum to give 0.65 g (86 %) of the desired material as pale yellow solid. This material was homogeneous by 1 ⁇ NMR analysis and was used without further purification
- Example 3(c) To the compound of Example 3(c) (0.65 g, 4.64 mmol) and o-dibromoxylene (0.55 g, 2.08 mmol) in T ⁇ F (10 mL) was added Na ⁇ (0.19 g of a 60 % dispersion in oil, 4.75 mmol) portionwise. The reaction was allowed to stir at RT for 24 h and then carefully poured into 1N ⁇ Cl (75 mL). The aqueous phase was washed with hexane (20 mL) and Et 2 O (20 mL). The combined organic phases were back extracted with 1N ⁇ Cl (20 mL) and the combined aqueous phases were concentrated under vacuum to give a yellow residue.
- Formulations for pharmaceutical use incorporating compounds of the present invention can be prepared in various forms and with numerous excipients. Examples of such formulations are given below.
- Step 1 Blend ingredients No. 1, No. 2, No. 3 and No. 4
- Step 2 Add sufficient water portion-wise to the blend
- Step 3 The wet mass is converted to granules by passing
- Step 4 The wet granules are then dried in an oven at
- Step 5 The dry granules are lubricated with ingredient No. 5.
- Step 6 The lubricated granules are compressed on a suitable tablet press.
- a pharmaceutical composition for parenteral administration is prepared by dissolving an appropriate amount of a compound of formula I in polyethylene glycol with heating. This solution is then diluted with water for injections Ph Eur. (to 100 ml). The solution is then sterilized by filtration through a 0.22 micron membrane filter and sealed in sterile containers.
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Virology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description







Claims








Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US09/068,491 US6046197A (en) | 1995-11-03 | 1996-11-12 | Hemoregulatory compounds |
| JP9519062A JP2000500463A (en) | 1995-11-03 | 1996-11-12 | Blood regulatory compounds |
| EP96939701A EP0861255A4 (en) | 1995-11-03 | 1996-11-12 | Hemoregulatory compounds |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US664195P | 1995-11-03 | 1995-11-03 | |
| US60/006,641 | 1995-11-13 | ||
| US1553796P | 1996-04-17 | 1996-04-17 | |
| US60/015,537 | 1996-04-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1997018214A1 true WO1997018214A1 (en) | 1997-05-22 |
Family
ID=26675883
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1996/018247 WO1997018214A1 (en) | 1995-11-03 | 1996-11-12 | Hemoregulatory compounds |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US6046197A (en) |
| EP (1) | EP0861255A4 (en) |
| JP (1) | JP2000500463A (en) |
| WO (1) | WO1997018214A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002062797A2 (en) * | 2000-12-29 | 2002-08-15 | Celltech R & D, Inc. | Pharmaceutical uses and synthesis of diketopiperazines |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6197793B1 (en) * | 1995-11-13 | 2001-03-06 | Smithkline Beecham Corporation | Hemoregulatory compounds |
| EP1453500A1 (en) * | 2001-10-11 | 2004-09-08 | The Hospital For Sick Children | Styryl acrylonitrile compounds and their use to promote myelopoiesis |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3917626A (en) * | 1971-03-29 | 1975-11-04 | Ici Ltd | 3,6-Dioxa-1,8-octandiamido bis (pyridinium) compounds |
| FI77875C (en) * | 1982-11-26 | 1989-05-10 | Nyegaard & Co As | FOERFARANDE FOER FRAMSTAELLNING AV NYA TERAPEUTISKT ANVAENDBARA PEPTIDER. |
| TW222280B (en) * | 1991-11-26 | 1994-04-11 | Smithkline Beecham Corp | |
| US5830867A (en) * | 1993-05-24 | 1998-11-03 | Smithkline Beecham Corporation | Hemoregulatory peptides for stimulating the myelopoietic system |
-
1996
- 1996-11-12 WO PCT/US1996/018247 patent/WO1997018214A1/en not_active Application Discontinuation
- 1996-11-12 US US09/068,491 patent/US6046197A/en not_active Expired - Fee Related
- 1996-11-12 JP JP9519062A patent/JP2000500463A/en active Pending
- 1996-11-12 EP EP96939701A patent/EP0861255A4/en not_active Ceased
Non-Patent Citations (1)
| Title |
|---|
| CHEMICAL ABSTRACTS, Vol. 97, No. 17, 25 October 1982 (Columbus, OH., USA), page 731, Abstract No. 145258C, TOMIYASU H. et al., "Syntheses, Conformation and Interactions With Small Molecules of Bis(cyclic dipeptides)"; & HELV. CHIM. ACTA, 1982, 65(3), 775-784 (Eng). * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002062797A2 (en) * | 2000-12-29 | 2002-08-15 | Celltech R & D, Inc. | Pharmaceutical uses and synthesis of diketopiperazines |
| WO2002062797A3 (en) * | 2000-12-29 | 2002-12-19 | Celltech R & D Inc | Pharmaceutical uses and synthesis of diketopiperazines |
| US6815214B2 (en) | 2000-12-29 | 2004-11-09 | Celltech R & D, Inc. | Pharmaceutical uses and synthesis of diketopiperazines |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2000500463A (en) | 2000-01-18 |
| US6046197A (en) | 2000-04-04 |
| EP0861255A1 (en) | 1998-09-02 |
| EP0861255A4 (en) | 2000-06-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6077857A (en) | Hemoregulatory compounds | |
| US6191146B1 (en) | Hemoregulatory compounds | |
| US6194413B1 (en) | Hemoregulatory compounds | |
| US6200986B1 (en) | Hemoregulatory compounds | |
| US6046197A (en) | Hemoregulatory compounds | |
| US6046198A (en) | Hemoregulatory compounds | |
| US6077855A (en) | Hemoregulatory compounds | |
| WO1997017985A1 (en) | Hemoregulatory compounds | |
| EP0861077B1 (en) | Hemoregulatory compounds | |
| US6107309A (en) | Hemoregulatory compounds | |
| US6051584A (en) | Hemoregulatory compounds | |
| US6054465A (en) | Hemoregulatory compounds | |
| WO1997017963A1 (en) | Hemoregulatory compounds | |
| US6114357A (en) | Hemoregulatory compounds | |
| US6077856A (en) | Hemoregulatory compounds | |
| MXPA98003773A (en) | Hemoregulated compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): JP US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 1997 519062 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1996939701 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1996939701 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09068491 Country of ref document: US |
|
| WWR | Wipo information: refused in national office |
Ref document number: 1996939701 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1996939701 Country of ref document: EP |