WO1996036636A1 - Process for preparing 4-aryl-piperidine derivatives - Google Patents
Process for preparing 4-aryl-piperidine derivatives Download PDFInfo
- Publication number
- WO1996036636A1 WO1996036636A1 PCT/DK1996/000185 DK9600185W WO9636636A1 WO 1996036636 A1 WO1996036636 A1 WO 1996036636A1 DK 9600185 W DK9600185 W DK 9600185W WO 9636636 A1 WO9636636 A1 WO 9636636A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- treatment
- alkyl
- defined above
- Prior art date
Links
- 0 *c1ccc([C@](CC2)[C@](CO)CN2*=I)cc1 Chemical compound *c1ccc([C@](CC2)[C@](CO)CN2*=I)cc1 0.000 description 4
- WVVMCKRQHBAUPW-UHFFFAOYSA-N CN(CC1)CC(COc(cc2)ccc2OC)C1c(cc1)ccc1F Chemical compound CN(CC1)CC(COc(cc2)ccc2OC)C1c(cc1)ccc1F WVVMCKRQHBAUPW-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
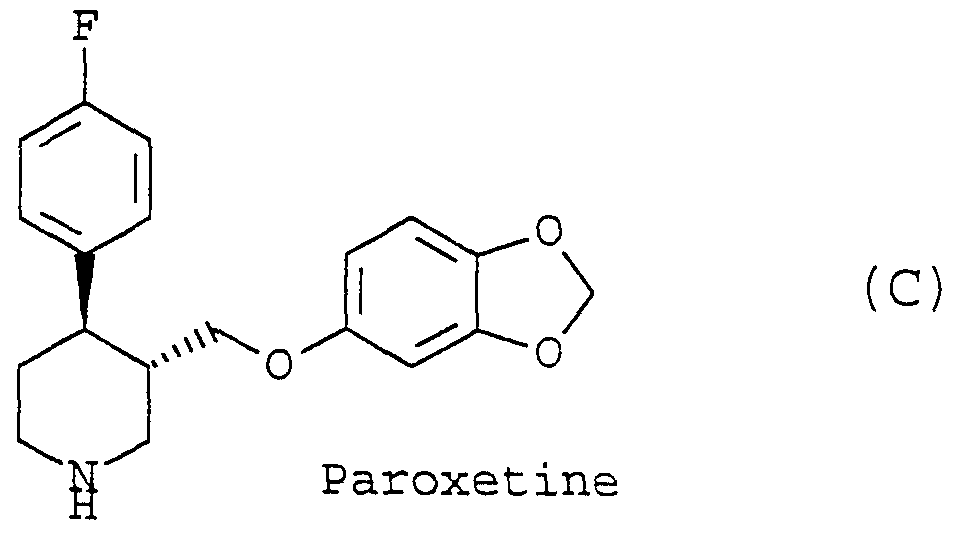
- the present invention relates to a novel process for preparing 4-aryl- piperidine derivatives.
- R 1 represents hydrogen, alkyl having 1 -4 carbon atoms and F may be in any of the available positions.
- US Patent No. 4,585,777 and US Patent No. 4, 593,036 describes a compound of the following formula B:
- the compounds of formula A and B are described as inhibitors of reup- take of 5-hydroxytryptamine (5-HT) which induces a potentiation of 5- HT induced neurotransmission.
- 5-HT 5-hydroxytryptamine
- Paroxetine which is the pure enantiomer (3S,4R)-4-(4-fluorophenyl)-3- (3,4-methylenedioxyphenoxymethyl)piperidine has been found to be a potent inhibitor of serotonin reuptake and to be an effective antide- pressant in man [ S. M. Holliday and G. L. Plosker, Drugs and Aging 3: 278-299 (1993)].
- the pharmacological activity resides in this isomer and the corresponding stereoisomer is considerably less potent with respect to inhibition of 5-HT uptake in vitro [P. Plenge, E. T. Mellerup, T. Honore, and P. L. Honore, J. Pharm. Pharmacol. 39: 877-882 (1987)].
- the Grignard reaction involves the use of ether solvents and is further ⁇ more complicated by the use of the toxic starting material arecoline.
- the intermediary D1 is prepared by reduction of the imide (F2), prepared from benzaldehyde and methyl N-methylamidomalonate.
- the reduction involves the use of lithium aluminium hydride, aluminium hydride or diborane using ether solvents like diethyl ether, tetrahydrofurane and dimethoxyethane, scheme F:
- the intermediate D1 is prepared by reacting methylamine, formaldehyde and ⁇ -methylstyrene (G 1 ). Intermediates in this synthesis is the oxazine derivative (G2) and the potent neurotoxic compound 1 -methyl-4-phenyl-1 ,2,3,6-tetrahydropyridine (MPTP) [USP 2,748, 140, C. J. Schmidle and R. C. Mansfield, J. Am. Chem. Soc. 11 5698-5700 (1955); C. J. Schmidle and R. C. Mansfield, J. Am. Chem. Soc.
- MPTP has in primates and in humans been found to cause anatomical and behavioral changes analogous to those of Parkinson's disease [M. Gerlach, P. Riederer, H. Przuntek, and M. B. H. Youdin, EUR. J. Pharma ⁇ col. Mol. Pharm, 208, 273-286, (1991 ); S. P. Markey and N. R. Schnuff, Medicinal Res. /?ev.6.386, ( 1 986)]. It is known that the 1 -methyl group causes MPTP to be toxic and that substitution of the methyl group with longer alkyl groups will abolish the toxicity [S. K. Youngster, P. K. Sonsalla, and R. E. Heikkila, J. Neurochem. 48, 929- 934, (1987)], scheme G:
- Paroxetine is one of four possible isomers, the use of the practi ⁇ cally and economically best procedure for the isolation of this isomer is of high importance.
- the procedure will involve the use of the appropriate isomer of D1 in combination with the use of the right conditions for reaction as well as separations by recrystallizations using optically active acids, e.g. mandelic acid, tartaric acid, and dibenzoyltartaric acid.
- optically active acids e.g. mandelic acid, tartaric acid, and dibenzoyltartaric acid.
- R 1 can be C 2 . 5 -alkyl, phenyl-C ⁇ -alkyl, or substituted phenyl-C ⁇ -alkyl, preferentially ethyl.
- R 1 can be C 2 . 5 -alkyl, phenyl-C ⁇ -alkyl, or substituted phenyl-C ⁇ -alkyl, preferentially ethyl.
- the intermediary 1 -alkyl-1 ,2,3,6-tetrahydro-4-phenylpyridine will in comparison with MPTP be non-toxic as described in:S. K. Youngster, P.
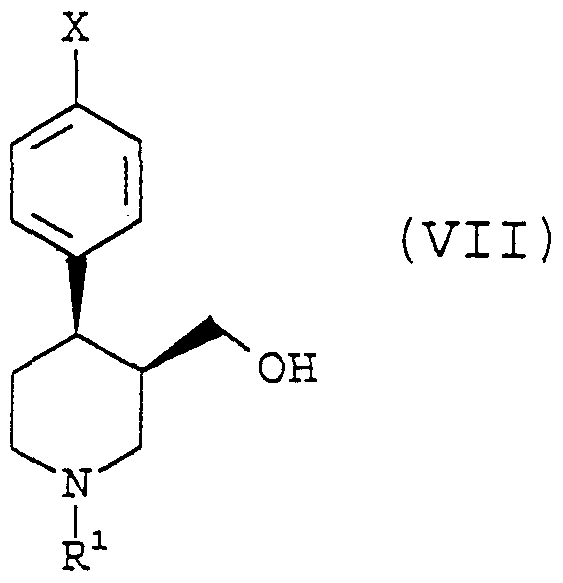
- the present invention provides a process for the preparation of a compound of formula VIII,
- R is C 2 . 5 -alkyl, phenyl-C ⁇ -alkyl, or substituted phenyl-C ⁇ -alkyl, by
- R 1 is C 2 . 5 -alkyl, phenyl-C ⁇ -alkyl, or substituted phenyl-C ⁇ -alkyl, with a compound of formula (II)
- X is halogen, preferably F, to form a compound of formula
- R 1 and X are as defined above, c) by treatment of a compound of formula IV, wherein R 1 and X are as defined above with metal hydrides, preferably LiAIH 4 or NaAIH 4 , to form a compound of formula VI,
- R 1 and X are as defined above, with benzene sulfonylchloride, or another suitable reagent, which reacts with the hydroxy group to trans ⁇ form it into a leaving group, which subsequently can be removed by treatment with 3,4-methylene dioxyphenolate, prepared by treatment of 3,4-methylenedioxyphenol with a base, preferably sodium methanolate to give a compound of formula VIII,
- R 1 and X are as defined above with chlorethylchloroformate or another similar reagent, followed by decomposition of the intermediary carbamate by methanol to form a compound of formula IX,
- Ethylamine hydrochloride 132.2 g was dissolved in formaldehyde (500 ml, 37 %) and the mixture heated to 70°C. 1 -methyl-4'-fluorostyrene (200 ml) was added over 1 hour keeping the temperature about 70°C.
- the phases were separated and the toluene phase extracted with hydrochloric acid (1 6 times 100 ml, 0.5 M).
- hydrochloric acid 1 6 times 100 ml, 0.5 M.
- the toluene phases were pooled and evaporated to an oil (1 64 g). The oil was dissolved in 2-propanol (300 ml) and the hydrochloride of the title compound precipitated with con ⁇ centrated hydrochloric acid.
- the aqueous phase was extracted with another portion of toluene (50 ml).
- the combined toluene extract was washed with water (50 ml), dried over potassium carbonate and evaporated.
- the aqueous phase was separated and extracted with another portion of toluene (100 ml) .
- the combined toluene phase was dried over potassium carbonate and evaporated to an oil (47 g).
- the oil was dissolved in acetone (900 ml) with ( + )-O,O'-ditoluoyltartaric acid (59 g).
- Formic acid ( 2.2 g) was added and the mixture stirred until next day.
- the precipitate was filtered off, washed with acetone and dried.
- the aqueous phase was extracted with another portion of toluene (50 ml), washed with water (50 ml) and evaporated.
- Lithium aluminium hydride (3 g) and sodium hydride 60 % (3 g) was dispersed in dry tetrahydrofuran (80 ml). The mixture was heated at 60°C for 1 hour and then cooled to 20°C. To this mixture was added a solution of ( + )-1 -ethyl-3-hydroxymethyl-4-(4-fluorophenyl)-1 , 2,3,6- tetrahydropyridine (20 g) in tetrahydrofuran (40 ml) over 1 hour. The mixture was stirred at 50°C for 1 hour.
- Benzene sulfonyl ⁇ chloride (16.6 g) was added over 1 hour keeping the temperature between 20 and 30°C with external cooling with ice and water. After the addition the reaction mixture was stirred at ambient temperature for 3 hours. Water was added (50 ml) and the toluene phase was separated. A solution of 3,4-methylenedioxyphenol (17 g) in methylisobutylcarbinol (4-methyl-2-pentanol) (90 ml) was added to the toluene phase together with sodium hydroxide (17.2 g, 32.5 %). The mixture was refluxed for 4 hours and stirred overnight at ambient temperature.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Hydrogenated Pyridines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description



















Claims









Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU56845/96A AU721257B2 (en) | 1995-05-17 | 1996-04-25 | Process for preparing 4-aryl-piperidine derivatives |
| EP96914861A EP0828735A1 (en) | 1995-05-17 | 1996-04-25 | Process for preparing 4-aryl-piperidine derivatives |
| NZ307479A NZ307479A (en) | 1995-05-17 | 1996-04-25 | A process for preparing 4-(4-halo substituted phenyl)-3-(3,4-methylenedioxy phenoxy methyl)piperidine derivatives |
| JP8534464A JPH11505229A (en) | 1995-05-17 | 1996-04-25 | Method for producing 4-aryl-piperidine derivatives |
| BR9608471A BR9608471A (en) | 1995-05-17 | 1996-04-25 | Process for the preparation of a compound |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DK56395 | 1995-05-17 | ||
| DK0563/95 | 1995-05-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1996036636A1 true WO1996036636A1 (en) | 1996-11-21 |
Family
ID=8094959
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/DK1996/000185 WO1996036636A1 (en) | 1995-05-17 | 1996-04-25 | Process for preparing 4-aryl-piperidine derivatives |
Country Status (11)
| Country | Link |
|---|---|
| EP (1) | EP0828735A1 (en) |
| JP (1) | JPH11505229A (en) |
| CN (1) | CN1068597C (en) |
| AU (1) | AU721257B2 (en) |
| BR (1) | BR9608471A (en) |
| CA (1) | CA2220963A1 (en) |
| HU (1) | HUP9900318A3 (en) |
| IL (1) | IL118294A0 (en) |
| NZ (1) | NZ307479A (en) |
| WO (1) | WO1996036636A1 (en) |
| ZA (1) | ZA963951B (en) |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997024323A1 (en) * | 1995-12-28 | 1997-07-10 | Chirotech Technology | Process for the preparation of optically enriched 4-aryl-3-hydromethyl substituted piperidines to be used as intermediates in the synthesis of paroxetine |
| WO1997031915A1 (en) * | 1996-02-29 | 1997-09-04 | Ferrer Internacional, S.A. | NEW PROCESS FOR PREPARING (-)-TRANS-N-p-FLUOROBENZOYLMETHYL-4-(p-FLUOROPHENYL)-3-[[3,4-(METHYLENEDIOXY)PHENOXY]METHYL]-PIPERIDINE |
| EP0812827A1 (en) * | 1996-06-13 | 1997-12-17 | SUMIKA FINE CHEMICALS Co., Ltd. | Piperidine derivative as intermediates for the preparation of paroxetine and process for their preparation |
| WO1998052920A1 (en) * | 1997-05-17 | 1998-11-26 | Knoll Aktiengesellschaft | Chemical process for the reduction of 1-substituted -3-hydroxymethyl-4- (4-fluorophenyl)tetrahydropyridines |
| US5874447A (en) * | 1997-06-10 | 1999-02-23 | Synthon B. V. | 4-Phenylpiperidine compounds for treating depression |
| US6063927A (en) * | 1998-07-02 | 2000-05-16 | Smithkline Beecham Plc | Paroxetine derivatives |
| WO2000032591A1 (en) * | 1998-11-28 | 2000-06-08 | Smithkline Beecham Plc | Process for the preparation of paroxetine hydrochloride |
| WO2000039090A1 (en) * | 1998-12-29 | 2000-07-06 | Smithkline Beecham Plc | Process for the preparation of paroxetine acetate and analogues thereof |
| WO2000050422A1 (en) * | 1999-02-23 | 2000-08-31 | Recordati S.A. Chemical And Pharmaceutical Company | Process for the production of paroxetine |
| US6153755A (en) * | 1996-11-09 | 2000-11-28 | Smithkline Beecham Plc | Process for preparing pharmaceutically active compounds and intermediates thereof |
| US6172233B1 (en) | 1997-01-15 | 2001-01-09 | Smithkline Beecham Plc | Process for making paroxetine |
| EP1074550A1 (en) * | 1999-08-02 | 2001-02-07 | CHEMI S.p.A. | Process for the preparation of 3-substituted 4-phenyl-piperidine derivatives |
| WO2001032178A1 (en) * | 1999-10-29 | 2001-05-10 | Novo Nordisk A/S | Use of 3,4-substituted piperidines |
| WO2002006275A1 (en) * | 2000-07-17 | 2002-01-24 | Smithkline Beecham P.L.C. | Novel processes for the preparation of 4-phenylpiperidine derivatives |
| WO2002018338A1 (en) * | 2000-08-30 | 2002-03-07 | Basf Aktiengesellschaft | Process for the racemisation of 1-benzyl-4-(4-fluorophenyl)-3-hydroxymethyl-1,2,3,6-tetrahydropyridine to be used as intermediate in the synthesis of paroxetine |
| WO2002018337A1 (en) * | 2000-08-30 | 2002-03-07 | Basf Aktiengesellschaft | Process for the racemisation of an intermediate useful in the preparation of paroxetine |
| WO2002028834A1 (en) * | 2000-10-06 | 2002-04-11 | Smithkline Beecham P.L.C. | Process for the preparation of aryl-piperidine carbinols and intermediates thereof |
| WO2002053537A1 (en) * | 2001-01-04 | 2002-07-11 | Ferrer Internacional, S.A. | Process for preparing (±) trans-4-p-fluorophenyl-3-hydroxymethyl-1-methylpiperidine |
| EP1242378A1 (en) * | 1999-12-23 | 2002-09-25 | SmithKline Beecham Corporation | Novel processes |
| US6489347B1 (en) | 1997-05-29 | 2002-12-03 | Smithkline Beecham Plc | Process |
| US6521758B2 (en) | 2000-05-12 | 2003-02-18 | Synthon Bv | Tosylate salts of 4-(p-fluorophenyl)-piperidine-3-carbinols |
| US6657062B1 (en) * | 1996-07-08 | 2003-12-02 | Richter Gedeon Vegyesseti Gyar Rt. | N-benzylpiperidine and tetrahydropyridine derivatives |
| WO2015071831A1 (en) * | 2013-11-18 | 2015-05-21 | Piramal Enterprises Limited | An improved process for minimising the formation of dehalogenated byproducts |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4007196A (en) * | 1973-01-30 | 1977-02-08 | A/S Ferrosan | 4-Phenylpiperidine compounds |
| US4593036A (en) * | 1984-02-07 | 1986-06-03 | A/S Ferrosan | (-)-Trans-4-(4-fluorophenyl)-3-[(4-methoxyphenoxy)methyl]-1-methylpiperidine useful as 5-HT potentiator |
| EP0266574A2 (en) * | 1986-11-03 | 1988-05-11 | Novo Nordisk A/S | Piperidine compounds and their preparation and use |
| EP0374674A2 (en) * | 1988-12-22 | 1990-06-27 | A/S Ferrosan | Etherification and dealkylation of piperidine derivatives and intermediates |
-
1996
- 1996-04-25 JP JP8534464A patent/JPH11505229A/en active Pending
- 1996-04-25 BR BR9608471A patent/BR9608471A/en not_active Application Discontinuation
- 1996-04-25 HU HU9900318A patent/HUP9900318A3/en unknown
- 1996-04-25 CN CN96193942A patent/CN1068597C/en not_active Expired - Fee Related
- 1996-04-25 CA CA002220963A patent/CA2220963A1/en not_active Abandoned
- 1996-04-25 NZ NZ307479A patent/NZ307479A/en unknown
- 1996-04-25 AU AU56845/96A patent/AU721257B2/en not_active Ceased
- 1996-04-25 WO PCT/DK1996/000185 patent/WO1996036636A1/en not_active Application Discontinuation
- 1996-04-25 EP EP96914861A patent/EP0828735A1/en not_active Ceased
- 1996-05-16 IL IL11829496A patent/IL118294A0/en unknown
- 1996-05-17 ZA ZA963951A patent/ZA963951B/en unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4007196A (en) * | 1973-01-30 | 1977-02-08 | A/S Ferrosan | 4-Phenylpiperidine compounds |
| US4593036A (en) * | 1984-02-07 | 1986-06-03 | A/S Ferrosan | (-)-Trans-4-(4-fluorophenyl)-3-[(4-methoxyphenoxy)methyl]-1-methylpiperidine useful as 5-HT potentiator |
| EP0266574A2 (en) * | 1986-11-03 | 1988-05-11 | Novo Nordisk A/S | Piperidine compounds and their preparation and use |
| EP0374674A2 (en) * | 1988-12-22 | 1990-06-27 | A/S Ferrosan | Etherification and dealkylation of piperidine derivatives and intermediates |
Cited By (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997024323A1 (en) * | 1995-12-28 | 1997-07-10 | Chirotech Technology | Process for the preparation of optically enriched 4-aryl-3-hydromethyl substituted piperidines to be used as intermediates in the synthesis of paroxetine |
| WO1997031915A1 (en) * | 1996-02-29 | 1997-09-04 | Ferrer Internacional, S.A. | NEW PROCESS FOR PREPARING (-)-TRANS-N-p-FLUOROBENZOYLMETHYL-4-(p-FLUOROPHENYL)-3-[[3,4-(METHYLENEDIOXY)PHENOXY]METHYL]-PIPERIDINE |
| US6476227B1 (en) | 1996-06-13 | 2002-11-05 | Sumika Fine Chemicals Co., Ltd. | Piperidine derivative and process for preparing the same |
| EP0812827A1 (en) * | 1996-06-13 | 1997-12-17 | SUMIKA FINE CHEMICALS Co., Ltd. | Piperidine derivative as intermediates for the preparation of paroxetine and process for their preparation |
| US6610851B1 (en) | 1996-06-13 | 2003-08-26 | Sumika Fine Chemicals Co., Ltd. | Process for preparing a piperidine derivative |
| US5948914A (en) * | 1996-06-13 | 1999-09-07 | Sumika Fine Chemicals Co., Ltd. | Piperidine derivative and process for preparing the same |
| US6815548B2 (en) | 1996-06-13 | 2004-11-09 | Sumika Fine Chemicals Co., Ltd. | Process for preparing a piperidine derivative |
| US6657062B1 (en) * | 1996-07-08 | 2003-12-02 | Richter Gedeon Vegyesseti Gyar Rt. | N-benzylpiperidine and tetrahydropyridine derivatives |
| US6153755A (en) * | 1996-11-09 | 2000-11-28 | Smithkline Beecham Plc | Process for preparing pharmaceutically active compounds and intermediates thereof |
| US6172233B1 (en) | 1997-01-15 | 2001-01-09 | Smithkline Beecham Plc | Process for making paroxetine |
| US6326496B1 (en) | 1997-05-17 | 2001-12-04 | Knoll Aktiengesellschaft | Process for preparing an intermediate in the production of paroxetine |
| CN1121386C (en) * | 1997-05-17 | 2003-09-17 | 克诺尔有限公司 | Chemical process for reduction of 1-substituted-3--hydroxymethyl-4-(4-fluorophenyl) tetrahydropyridines |
| WO1998052920A1 (en) * | 1997-05-17 | 1998-11-26 | Knoll Aktiengesellschaft | Chemical process for the reduction of 1-substituted -3-hydroxymethyl-4- (4-fluorophenyl)tetrahydropyridines |
| US6716985B2 (en) | 1997-05-29 | 2004-04-06 | Smithkline Beecham P.L.C. | Process for making paroxetine |
| US6489347B1 (en) | 1997-05-29 | 2002-12-03 | Smithkline Beecham Plc | Process |
| US7598271B1 (en) | 1997-06-10 | 2009-10-06 | Noven Therapeutics, Llc | Crystalline paroxetine methane sulfonate |
| US6900327B2 (en) | 1997-06-10 | 2005-05-31 | Synthon Bct Technologies, Llc | 4-phenylpiperidine compounds |
| US5874447A (en) * | 1997-06-10 | 1999-02-23 | Synthon B. V. | 4-Phenylpiperidine compounds for treating depression |
| US6063927A (en) * | 1998-07-02 | 2000-05-16 | Smithkline Beecham Plc | Paroxetine derivatives |
| WO2000032591A1 (en) * | 1998-11-28 | 2000-06-08 | Smithkline Beecham Plc | Process for the preparation of paroxetine hydrochloride |
| WO2000039090A1 (en) * | 1998-12-29 | 2000-07-06 | Smithkline Beecham Plc | Process for the preparation of paroxetine acetate and analogues thereof |
| US6583287B1 (en) | 1999-02-23 | 2003-06-24 | Recordati S.A. Chemical And Pharmaceutical Company | Process for the production of paroxetine |
| JP2002537394A (en) * | 1999-02-23 | 2002-11-05 | レコーダチ エス.エイ.ケミカル アンド ファルマチェウティカル カンパニー | Method for producing paroxetine |
| WO2000050422A1 (en) * | 1999-02-23 | 2000-08-31 | Recordati S.A. Chemical And Pharmaceutical Company | Process for the production of paroxetine |
| US6444822B1 (en) | 1999-08-02 | 2002-09-03 | Chemi S.P.A. | Process for the preparation of 3-substituted 4-phenyl-piperidine derivative |
| EP1074550A1 (en) * | 1999-08-02 | 2001-02-07 | CHEMI S.p.A. | Process for the preparation of 3-substituted 4-phenyl-piperidine derivatives |
| WO2001032178A1 (en) * | 1999-10-29 | 2001-05-10 | Novo Nordisk A/S | Use of 3,4-substituted piperidines |
| EP1242378A1 (en) * | 1999-12-23 | 2002-09-25 | SmithKline Beecham Corporation | Novel processes |
| EP1242378A4 (en) * | 1999-12-23 | 2003-01-15 | Smithkline Beecham Corp | Novel processes |
| US6521758B2 (en) | 2000-05-12 | 2003-02-18 | Synthon Bv | Tosylate salts of 4-(p-fluorophenyl)-piperidine-3-carbinols |
| WO2002006275A1 (en) * | 2000-07-17 | 2002-01-24 | Smithkline Beecham P.L.C. | Novel processes for the preparation of 4-phenylpiperidine derivatives |
| WO2002018338A1 (en) * | 2000-08-30 | 2002-03-07 | Basf Aktiengesellschaft | Process for the racemisation of 1-benzyl-4-(4-fluorophenyl)-3-hydroxymethyl-1,2,3,6-tetrahydropyridine to be used as intermediate in the synthesis of paroxetine |
| WO2002018337A1 (en) * | 2000-08-30 | 2002-03-07 | Basf Aktiengesellschaft | Process for the racemisation of an intermediate useful in the preparation of paroxetine |
| US6949650B2 (en) | 2000-08-30 | 2005-09-27 | Aesica Pharmaceuticals Ltd. | Process for the racemization of 1-benzyl-4-(4-fluorophenyl)-3-hydroxymethyl-1,2-3,6-tetrahydropyridine to be used as intermediate in the synthesis of paroxetine |
| WO2002028834A1 (en) * | 2000-10-06 | 2002-04-11 | Smithkline Beecham P.L.C. | Process for the preparation of aryl-piperidine carbinols and intermediates thereof |
| US6881845B2 (en) | 2001-01-04 | 2005-04-19 | Ferrer Internacional, S.A. | Process for preparing (±)trans-4-p-fluorophenyl-3-hydroxymethyl-1-methylpiperidine |
| WO2002053537A1 (en) * | 2001-01-04 | 2002-07-11 | Ferrer Internacional, S.A. | Process for preparing (±) trans-4-p-fluorophenyl-3-hydroxymethyl-1-methylpiperidine |
| WO2015071831A1 (en) * | 2013-11-18 | 2015-05-21 | Piramal Enterprises Limited | An improved process for minimising the formation of dehalogenated byproducts |
Also Published As
| Publication number | Publication date |
|---|---|
| AU5684596A (en) | 1996-11-29 |
| BR9608471A (en) | 1998-12-29 |
| ZA963951B (en) | 1997-01-21 |
| EP0828735A1 (en) | 1998-03-18 |
| CA2220963A1 (en) | 1996-11-21 |
| NZ307479A (en) | 1999-08-30 |
| AU721257B2 (en) | 2000-06-29 |
| HUP9900318A3 (en) | 2001-09-28 |
| CN1068597C (en) | 2001-07-18 |
| CN1184476A (en) | 1998-06-10 |
| JPH11505229A (en) | 1999-05-18 |
| HUP9900318A2 (en) | 1999-09-28 |
| IL118294A0 (en) | 1996-09-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU721257B2 (en) | Process for preparing 4-aryl-piperidine derivatives | |
| IE59901B1 (en) | Piperidine derivative, its preparation, and its use as medicament | |
| JP2007231024A (en) | Manufacturing process of 3,3-diarylpropylamine | |
| US7939660B2 (en) | Process and intermediates for preparing emtricitabine | |
| EP0759921B1 (en) | Process for the preparation of azabicyclic derivatives | |
| US4914208A (en) | Optically active salts of a substituted thiazolidine-4-carboxylate and 3-chloro-2-hydroxypropyltrimethyl ammonium, their preparation and use | |
| US5965734A (en) | Processes and intermediates for preparing 2-substituted piperidine stereoisomers | |
| AU696875B2 (en) | New process for the preparation of ropivacaine hydrochloride monohydrate | |
| JP2546624B2 (en) | Process for producing optically pure amino alcohol | |
| FI89042C (en) | Process for the preparation of novel therapeutically useful 2-acyloxy propylamine derivatives | |
| JP3037592B2 (en) | (-)-N-methyl-N- [4- (4-phenyl-4-acetylaminopiperidin-1-yl) -2- (3,4-dichlorophenyl) butyl] benzamide and pharmaceutically acceptable salts thereof Manufacturing method | |
| AU6086599A (en) | Method for producing (-)-alpha-(difluoromethyl)ornithine-monohydrochloride monohydrate | |
| EP1341762A1 (en) | Process for resolving racemic mixtures of piperidine derivatives | |
| EP2040697A2 (en) | Polymorphic form of duloxetine hydrochloride | |
| WO2009037718A2 (en) | Process for preparing 3-(2-(dimethylamino)ethyl)-n- methyl-1h-indole-5-methanesulfonamide and product thereof | |
| WO2006131773A1 (en) | Process for the preparation of s-(-)-amlodipine | |
| EP1140912A1 (en) | Process for the preparation of an acetate salt of paroxetine or paroxetine analogues |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 96193942.7 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BB BG BR BY CA CH CN CZ DE DK EE ES FI GB GE HU IS JP KE KG KP KR KZ LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK TJ TM TR TT UA UG US UZ VN AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): KE LS MW SD SZ UG AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 1997 945704 Country of ref document: US Date of ref document: 19971029 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1996914861 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 307479 Country of ref document: NZ |
|
| ENP | Entry into the national phase |
Ref document number: 2220963 Country of ref document: CA Ref document number: 2220963 Country of ref document: CA Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 1996 534464 Country of ref document: JP Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1996914861 Country of ref document: EP |
|
| WWR | Wipo information: refused in national office |
Ref document number: 1996914861 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1996914861 Country of ref document: EP |