WO1995015299A1 - Process for preparing a chiral tetralone - Google Patents
Process for preparing a chiral tetralone Download PDFInfo
- Publication number
- WO1995015299A1 WO1995015299A1 PCT/IB1994/000263 IB9400263W WO9515299A1 WO 1995015299 A1 WO1995015299 A1 WO 1995015299A1 IB 9400263 W IB9400263 W IB 9400263W WO 9515299 A1 WO9515299 A1 WO 9515299A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- process according
- tetralone
- formula
- chiral
- phenyl
- Prior art date
Links
- 0 CCC=CCC(*C(*)(*)[O+]B1*)N1CNC Chemical compound CCC=CCC(*C(*)(*)[O+]B1*)N1CNC 0.000 description 1
- GZQAUCBRPSENQB-BDJLRTHQSA-N O[C@H](CC1)c2ccccc2[C@H]1c(cc1Cl)ccc1Cl Chemical compound O[C@H](CC1)c2ccccc2[C@H]1c(cc1Cl)ccc1Cl GZQAUCBRPSENQB-BDJLRTHQSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/687—Unsaturated compounds containing a keto groups being part of a ring containing halogen
- C07C49/697—Unsaturated compounds containing a keto groups being part of a ring containing halogen containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B53/00—Asymmetric syntheses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/132—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of an oxygen containing functional group
- C07C29/136—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of an oxygen containing functional group of >C=O containing groups, e.g. —COOH
- C07C29/143—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of an oxygen containing functional group of >C=O containing groups, e.g. —COOH of ketones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C35/00—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a ring other than a six-membered aromatic ring
- C07C35/22—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a ring other than a six-membered aromatic ring polycyclic, at least one hydroxy group bound to a condensed ring system
- C07C35/23—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a ring other than a six-membered aromatic ring polycyclic, at least one hydroxy group bound to a condensed ring system with hydroxy on a condensed ring system having two rings
- C07C35/36—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a ring other than a six-membered aromatic ring polycyclic, at least one hydroxy group bound to a condensed ring system with hydroxy on a condensed ring system having two rings the condensed ring system being a (4.4.0) system, e.g. naphols
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/27—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by oxidation
- C07C45/30—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by oxidation with halogen containing compounds, e.g. hypohalogenation
- C07C45/305—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by oxidation with halogen containing compounds, e.g. hypohalogenation with halogenochromate reagents, e.g. pyridinium chlorochromate
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/67—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2602/00—Systems containing two condensed rings
- C07C2602/02—Systems containing two condensed rings the rings having only two atoms in common
- C07C2602/04—One of the condensed rings being a six-membered aromatic ring
- C07C2602/10—One of the condensed rings being a six-membered aromatic ring the other ring being six-membered, e.g. tetraline
Definitions
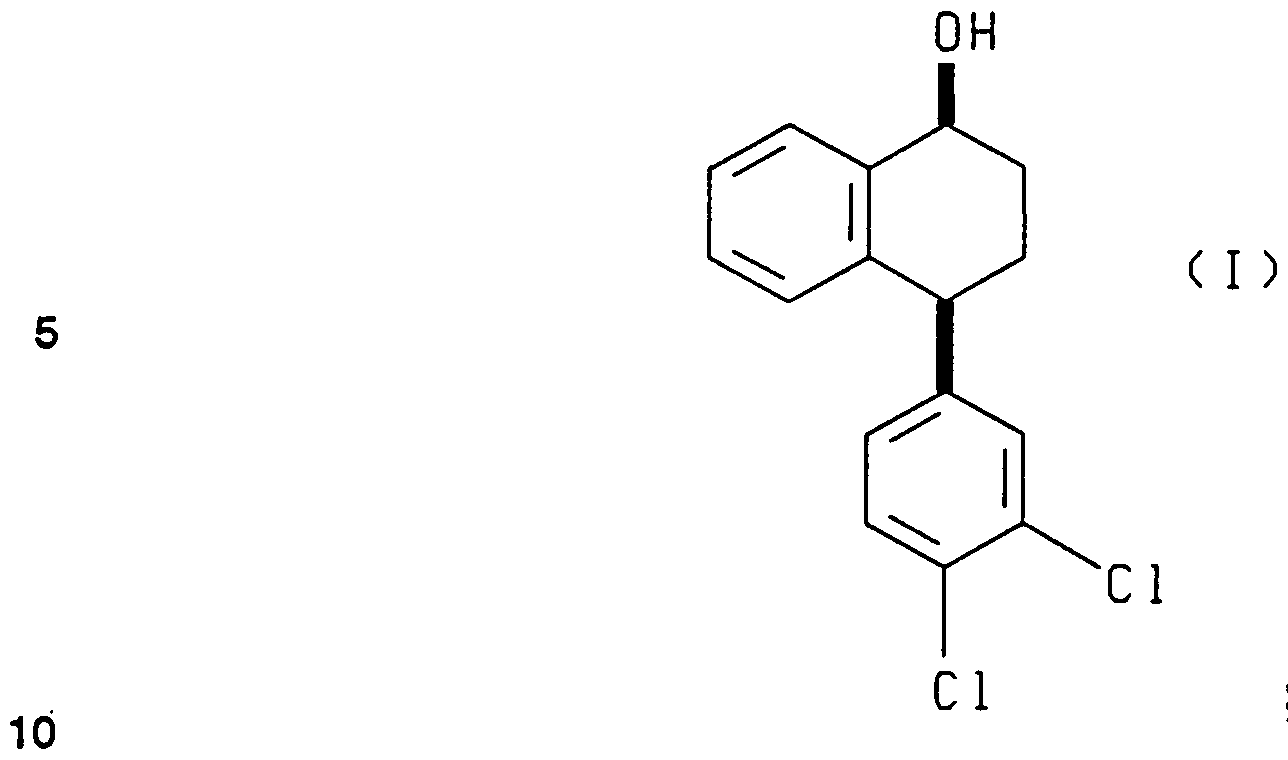
- the present invention relates to a novel process for asymmetrically reducing racemic 4-(3,4-dichlorophenyl)-3,4-dihydro-1 (2H)-naphthalenone (hereinafter also referred to as “the tetralone” or “the racemic tetralone”) and for preparing chiral (4S)- (3,4-dichlorophenyl)-3,4-dihydro-1 (2H)-naphthalenone (hereinafter also referred to as "the chiral tetralone”), which has utility as an intermediate in the production of pure s- (1 S)(4S)-N-methyl-4-(3,4-dichlorophenyl)-1 ,2,3,4-tetrahydro-l -naphthaleneamine (sertraline).
- Sertraline is a known antidepressant agent.
- This invention also relates to novel intermediates in the synthesis of chiral tetralone.
- Several documents relate to the synthesis of pure racemic N-methyl-4-(3,4- dichlorophenyl)-1 ,2, 3, 4-tetrahydro-1 -naphthaleneamine starting with 3,4- dichlorobenzophenone and proceeding via racemic ( ⁇ )-4-(3,4-dichlorophenyl)-4- butanoic acid and then to (+)-4-(3,4-dichlorophenyl)-3,4-dihydro-1 (2H)-naphthalenone. See, e.g., U.S.
- Patent Nos.4,536,518 (August 20, 1985); 4,556,676 (December 3, 1985); 4,777,288 (October 11 , 1988); and 4,839,104 (June 13, 1989); and Journal of Medicinal Chemistry, Vol. 27, No. 11 , p. 1508 (1984).
- Tetrahedron. Vol. 48, No. 47, pp. 10239-10248 (1992) relates to a process for preparing the (4S)-enantiomer of 4-(3,4-dichIorophenyl)-3,4-dihydro-1 (2H)- naphthalenone comprising reducing the 4-ketobutanoic acid ester with a carbonyl reducing agent, as outlined in E. J. Corey et al., Journal of Organic Chemistry. Vol. 53, p. 2861 (1988), to ultimately afford chiral tetralone.
- the present invention relates to a process for asymmetrically reducing racemic 4-(3,4-dichlorophenyl)-3,4-dihydro-1 (2H)-naphthalenone comprising reacting the racemic 4-(3,4-dichlorophenyl)-3,4-dihydro-1 (2H)-naphthalenone with an asym ⁇ metric ketone reducing agent.
- the asymmetric ketone reducing agent is preferably a catalytic chiral oxazaborolidine compound.
- Preferred chiral oxazaborolidine compounds have the formula:
- R 1 is hydrogen, (C,-C 8 ) alkyl, benzyl, phenyl or phenyl substituted with up to three substituents independently selected from (C,-C 8 )alkyl, (C C 8 )alkoxy and halo; and
- R 2 and R 3 are syn and the same and are each phenyl or phenyl substituted with up to three substituents independently selected from (C,-C 8 )alkyl, (C,-C 8 )alkoxy and halo.
- R 4 is hydrogen, lower alkyl or aralkyl; n is 2, 3, or 4, such that the group (CH 2 ) n forms, together with the oxazaborolidine nitrogen and adjacent carbon, a 4-, 5- or 6-membered ring; and
- R 5 and R ⁇ are phenyl.
- Another preferred asymmetric ketone reducing agent comprises either enantiomer of the compound having the formula: lpc 2 BX wherein Ipc is isopinocampheyl, B is boron and X is halo.
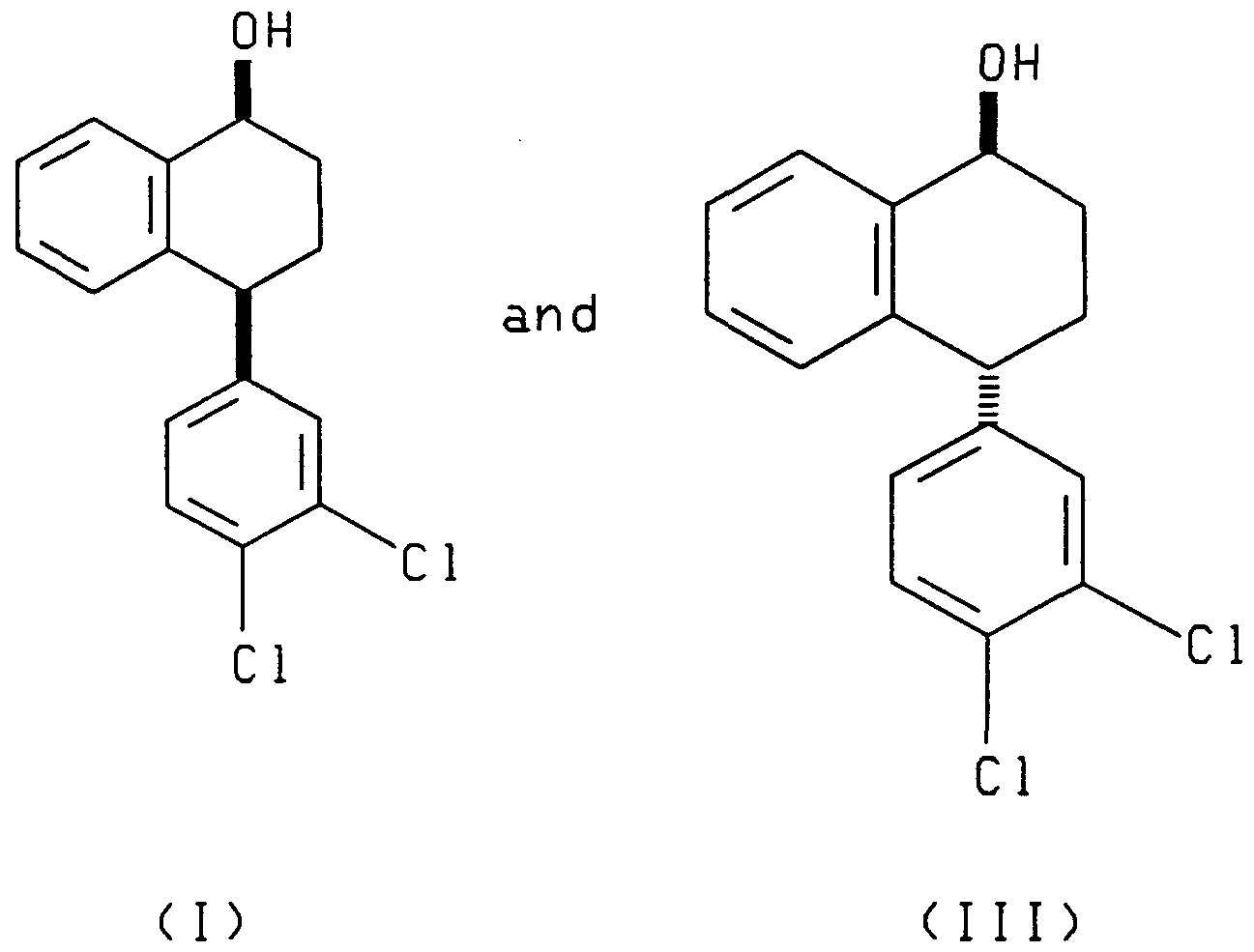
- the enantiomer of the asymmetric reducing agent determines whether (I) and (III) or (II) and (IV) is produced.
- the present invention also relates to each of the two reduction processes described above (i.e., that which produces compounds (I) and (III) and that which produces compounds (II) and (IV)), further comprising separating, respectively, the cis alcohol (I) from the trans alcohol (III) or the cis alcohol (IV) from the trans alcohol (II) and oxidizing, respectively, the resulting cis alcohol (I) or trans alcohol (II) to produce chiral tetralone.
- the present invention also relates to a process comprising reacting racemic tetralone with an asymmetric ketone reducing agent to produce compounds having the formulae:
- said process further comprising the steps of oxidizing the compounds having, respectively, formula (III) or (IV) to produce the 4(R) enantiomer of the tetralone ((4R)- (3,4-dichlorophenyl)-3,4-dihydro-1 (2H)-naphthalenone) I and contacting the resulting 4(R) tetralone with a base to produce racemic tetralone.
- the present invention also relates to compounds having the following formulae:
- halo as used herein, unless otherwise indicated, includes chloro, fluoro, bromo and iodo.
- alkyl as used herein, unless otherwise indicated, includes saturated monovalent hydrocarbon radicals having straight, branched or cyclic moieties or combinations thereof.
- alkoxy includes O-alkyl groups wherein “alkyl” is defined as above.
- aralkyl includes aryl groups, wherein “aryl” is defined as below, terminating in an alkyl group, as defined above, which is the point of attachment.
- aryl means mononuclear aromatic hydrocarbon groups such as phenyl, which can be unsubstituted or substituted in one or more positions, and polynuclear aryl groups such as naphthyl, anthryl, phenanthryl, and so forth, which can be unsubstituted or substituted with one or more groups.
- one or more substituents includes from one to the maximum number of substituents possible based on the number of available bonding sites.
- racemic 4-(3,4-dichlorophenyl)-3,4-dihydro-1(2H)- naphthalenone (tetralone) is asymmetrically reduced by reacting the racemic tetralone with an asymmetric reagent (A) or (B), wherein (A) and (B) are enantiomers.
- Reduction of racemic tetralone with enantiomer A yields compounds of formulae I and III.
- Reduction of racemic tetralone with enantiomer B yields compounds of formulae II and IV.
- the reduction is performed in a suitable solvent such as tetrahydrofuran, toluene, or an alternative etherial solvent.
- the reduction is performed at a temperature of from about -20°C to about 50°C, preferably from 20°C to 25°C.
- the ratio of racemic tetralone to asymmetric reagent is from about 1.0:0.025 to about 1.0:1.5.
- the asymmetric reagent is a compound of formula (V), (Va) or (VI)
- the ratio of racemic tetralone to asymmetric reagent is preferably from about 1.0:0.025 to about 1.0:0.1.
- the ratio of racemic tetralone to asymmetric reagent is preferably from about 1.0:1.0 to about 1.0:1.5.
- the asymmetric reduction of racemic tetralone produces a mixture of cis and trans alcohols of the formulae (I) and (III) or of the formulae II and IV depending upon the chirality of the asymmetric reagent employed.
- the cis alcohol (I) can be separated from the trans alcohol (III) by methods known in the art, such as chromatography.
- the trans alcohol II can be separated from the cis alcohol IV by methods known in the art.
- the desired product possesses the chirality desired for sertraline.
- the (4S) tetralone can be prepared by Jones oxidation, Swem oxidation, Manganese dioxide, pyridium chlorochromate, and pyridium dichromate of the resulting cis alcohol (I) and trans alcohol (II).
- Suitable asymmetric reducing reagents include chiral oxazaborolidine compounds of the formula:
- R 1 is hydrogen, (C,-C 8 )alkyl, benzyl, phenyl or phenyl substituted with up to three substituents independently selected from (C,-C 8 )alkyl, (C C 8 )aIkoxy or halo; and R 2 and R 3 are syn and the same and are each phenyl or phenyl substituted with up to three substituents independently selected from (C,-C 8 )alkyl, (C,-C 8 )alkoxy or halo groups such as chloro or fluoro.
- a preferred number of substituents is zero.
- a preferred group of such compounds is the group of compounds wherein R ⁇ R 2 and R 3 are all unsubstituted phenyl.
- Suitable asymmetric reagents also include a chiral 1 ,3,2-oxazaborolidine of the formula:
- R 4 is hydrogen, lower alkyl or aralkyl; n is 2, 3, or 4, such that the group (CH 2 ) n forms, together with the oxazaborolidine nitrogen and adjacent carbon, a 4-, 5- or 6-membered ring; and R 5 and R ⁇ are phenyl.
- Aralkyl is as defined above.
- Preferred alkyl groups of the aralkyl are CH 2 .
- Preferred aralkyl groups are phenylalkyl groups.
- Suitable asymmetric reagents also include a haloborane represented by the formula: lpc 2 BX, wherein Ipc is isopinocampheyl, B is boron and X is halo. Additional suitable asymmetric reagents are disclosed in U.S. Patent No.
- the process for making (4S)-(3,4-dichlorophenyI)-3,4- dihydro-1 (2H)-naphthalenone may optionally contain one or more additional steps wherein alcohols (III) and/or (IV) are recycled.
- the alcohols (III) and/or (IV) are oxidized to produce 4(R) enantiomer of the tetralone, which is then reacted with a base to produce the racemic tetralone.
- the oxidation can be done by methods known to those skilled in the art.
- the racemization reaction is performed at a temperature of from about 0°C to about 100°C, preferably 25 °C to 65 °C.
- the 4(R) enantiomer of the tetralone is reacted with a base at a temperature of from about 25 °C to about 65°C, preferably 50°C to 80°C.
- Suitable bases for this reaction include potassium t-butoxide, sodium hydroxide, sodium methoxide, and potassium hydroxide.
- a preferred base is potassium t-butoxide.
- the (4S)-4-(3,4-dichlorophenyl)-3,4-dihydro-1 (2H)-naphthalenone final product afforded by the process of this invention is a valuable intermediate that can be used to synthesize the antidepressant agent known as sertraline or cis-(1 S)(4S)-N-methyl-4-(3,4- dichlorophenyl)-1 ,2,3,4-tetrahydro-1 -naphthaleneamine by methods disclosed in the previously discussed prior art.
- (4S)-4-(3,4-dichIorophenyl)-3,4- dihydro-1 (2H)-naphthalenone is first converted to (4S)-N-[4-(3,4-dichlorophenyl)-3,4- dihydro-1(2H)-naphthalenylidine]methanamine and then finally to the desired cjs- (1 S)(4S)-N-methyl-4-(3,4-dichlorophenyl)-1 ,2,3,4-tetrahydro-l -naphthaleneamine by the known methods of the prior art process, as earlier described in U.S. Patent No. 4,536,518 (August 20, 1985).
- the optically-active ketone viz., (4S)-4-(3,4-dichlorophenyl)-1 (2H)-naphthalenone
- pressure is not critical unless otherwise indicated. Pressures from about 0.9 atmospheres to about 2 atmospheres are generally acceptable and ambient pressure, i.e., about 1 atmosphere, is preferred as a matter of convenience.
- Racemic tetralone (5.0 g, 17 mmol) as a solution in THF was added over 1 hour, the reaction stirred 15 minutes after the addition was completed, cooled to 0°C and quenched with methanol. After stirring the quenched reaction for 18 hours the solvents were removed under vacuum, the contents dissolved in methylene chloride (100 mL), and washed sequentially with pH4 phosphate buffer (100 mL), water (100 mL), treated with magnesium sulfate, and solvent removed to afford a mixture of the cis and trans alcohols (5.01 g). Chromatography with ethyl acetate/hexanes provided the less polar cis alcohol.
- EXAMPLE 4 4-(3.4-dichlorophenvQ-3,4-dihvdro-1 (2H)-naphthalenone Pyridium chlorochromate oxidation of the trans alcohols 3 and 4 with the same procedure employed on alcohols 1 and 2 provided the chiral tetralone. Racemization of the chiral tetralone into racemic tetralone was achieved as follows. Potassium t- butoxide (90 mg, 0.80 mmol) was added to a solution of chiral tetralone (1.12 g, 3.84 mmol) in THF (4 mL).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description












Claims












Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/652,485 US5750794A (en) | 1993-11-30 | 1994-09-02 | Process for preparing chiral tetralone |
| CA002176500A CA2176500C (en) | 1993-11-30 | 1994-09-02 | Process for preparing a chiral tetralone |
| DE69406553T DE69406553T2 (en) | 1993-11-30 | 1994-09-02 | METHOD FOR PRODUCING CHIRAL TETRALONES |
| EP94924378A EP0724552B1 (en) | 1993-11-30 | 1994-09-02 | Process for preparing a chiral tetralone |
| FI962250A FI115397B (en) | 1993-11-30 | 1996-05-29 | Process for making chiral tetralone |
| GR970402783T GR3025223T3 (en) | 1993-11-30 | 1997-10-30 | Process for preparing a chiral tetralone |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US15915693A | 1993-11-30 | 1993-11-30 | |
| US08/159,156 | 1993-11-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1995015299A1 true WO1995015299A1 (en) | 1995-06-08 |
Family
ID=22571320
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB1994/000263 WO1995015299A1 (en) | 1993-11-30 | 1994-09-02 | Process for preparing a chiral tetralone |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US5750794A (en) |
| EP (1) | EP0724552B1 (en) |
| JP (1) | JP2686366B2 (en) |
| AT (1) | ATE159706T1 (en) |
| CA (1) | CA2176500C (en) |
| DE (1) | DE69406553T2 (en) |
| DK (1) | DK0724552T3 (en) |
| ES (1) | ES2108484T3 (en) |
| FI (1) | FI115397B (en) |
| GR (1) | GR3025223T3 (en) |
| WO (1) | WO1995015299A1 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6103938A (en) * | 1998-04-23 | 2000-08-15 | Ciba Specialty Chemicals Corporation | Process for preparing 4-(substituted phenyl)-3,4-dihydro-2H-naphthalen-1-ones |
| WO2001049638A2 (en) * | 2000-01-04 | 2001-07-12 | Sun Pharmaceutical Industries Ltd. | A process for converting stereoisomers of sertraline into sertraline |
| US7648991B2 (en) | 2005-02-16 | 2010-01-19 | H. Lundbeck A/S | Crystalline base of trans-1-((1R,3S)-6-chloro-3-phenylindan-1-yl)-3,3-dimethylpiperazine |
| US7767683B2 (en) | 2003-08-18 | 2010-08-03 | H. Lundbeck A/S | Hydrogen succinate salts of trans-4-((1R, 3S)-6-chloro-3-phenylindan-1-YL)-1,2,2-trimethylpiperazine and the use as a medicament |
| US8569499B2 (en) | 2005-02-16 | 2013-10-29 | H. Lundbeck A/S | Process for making trans-1-((1R,3S)-6-chloro-3-phenylindan-1-yl)-3,3-dimethylpiperazine |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06329679A (en) * | 1993-01-20 | 1994-11-29 | Nissan Chem Ind Ltd | Optically active beta-aminoalkoxyborane complex |
| US5831132A (en) * | 1994-10-28 | 1998-11-03 | Sumika Fine Chemicals Company, Ltd. | Process for producing optically active carbinols |
| US6410794B1 (en) | 1994-12-16 | 2002-06-25 | Uop Llc | Process for preparation of pharmaceutically desired chiral tetralone from tetralones |
| IL132500A0 (en) * | 1998-10-29 | 2001-03-19 | Pfizer Prod Inc | Stereoselective microbial reduction of a racemic tetralone |
| US6720003B2 (en) * | 2001-02-16 | 2004-04-13 | Andrx Corporation | Serotonin reuptake inhibitor formulations |
| US6683220B2 (en) * | 2001-10-31 | 2004-01-27 | Pfizer, Inc. | Process for the preparation of optically pure or enriched racemic tetralone |
| EP1545485A2 (en) * | 2002-09-16 | 2005-06-29 | Sepracor Inc. | Treatment of cns disorders with trans-4- (3,4-dichlorophenyl)-1,2,3,4-tetrahydro-n-methyl-1-napthalenamine |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4772752A (en) * | 1986-08-29 | 1988-09-20 | Aldrich-Boranes, Inc. | Mono- and diisopinocampheylhaloboranes as new chiral reducing agents |
| EP0305180A2 (en) * | 1987-08-27 | 1989-03-01 | The President And Fellows Of Harvard College | Enantioselective reduction of ketones |
| WO1993023408A1 (en) * | 1992-05-14 | 1993-11-25 | Pfizer Inc. | Enantioselective oxazaborolidine catalysts |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4536518A (en) * | 1979-11-01 | 1985-08-20 | Pfizer Inc. | Antidepressant derivatives of cis-4-phenyl-1,2,3,4-tetrahydro-1-naphthalenamine |
| US4556676A (en) * | 1979-11-01 | 1985-12-03 | Pfizer Inc. | Antidepressant derivatives of trans-4-phenyl-1,2,3,4-tetrahydro-1-naphthalenamine |
| JPH0621109B2 (en) * | 1984-07-09 | 1994-03-23 | ダイキン工業株式会社 | Fluorine-containing acrylic acid ester |
| US4839104A (en) * | 1987-06-11 | 1989-06-13 | Pfizer, Inc. | Process for preparing sertraline intermediates |
| US4777288A (en) * | 1987-06-11 | 1988-10-11 | Pfizer Inc. | Process for preparing a 4,4-diphenylbutanoic acid derivative |
| FI95572C (en) * | 1987-06-22 | 1996-02-26 | Eisai Co Ltd | Process for the preparation of a medicament useful as a piperidine derivative or its pharmaceutical salt |
| FR2632633B1 (en) * | 1988-06-08 | 1991-04-05 | Delalande Sa | PROCESS FOR THE PREPARATION OF 4-ARYL-1-TETRALONES |
| US5189177A (en) * | 1990-04-18 | 1993-02-23 | Merck & Co., Inc. | Chiral boron catalysts for reduction of ketones and process for their preparation |
| US5082970A (en) * | 1991-03-06 | 1992-01-21 | Pfizer Inc. | Process for recycling amine isomer |
| US5196607A (en) * | 1992-02-14 | 1993-03-23 | Pfizer Inc. | Process for preparing ketone enantiomer |
| SE470242B (en) * | 1992-05-12 | 1993-12-13 | Ericsson Telefon Ab L M | Device for generating random numbers |
-
1994
- 1994-09-02 US US08/652,485 patent/US5750794A/en not_active Expired - Fee Related
- 1994-09-02 EP EP94924378A patent/EP0724552B1/en not_active Expired - Lifetime
- 1994-09-02 AT AT94924378T patent/ATE159706T1/en not_active IP Right Cessation
- 1994-09-02 DK DK94924378.6T patent/DK0724552T3/en active
- 1994-09-02 DE DE69406553T patent/DE69406553T2/en not_active Expired - Fee Related
- 1994-09-02 ES ES94924378T patent/ES2108484T3/en not_active Expired - Lifetime
- 1994-09-02 JP JP7512276A patent/JP2686366B2/en not_active Expired - Fee Related
- 1994-09-02 CA CA002176500A patent/CA2176500C/en not_active Expired - Fee Related
- 1994-09-02 WO PCT/IB1994/000263 patent/WO1995015299A1/en active IP Right Grant
-
1996
- 1996-05-29 FI FI962250A patent/FI115397B/en active IP Right Grant
-
1997
- 1997-10-30 GR GR970402783T patent/GR3025223T3/en unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4772752A (en) * | 1986-08-29 | 1988-09-20 | Aldrich-Boranes, Inc. | Mono- and diisopinocampheylhaloboranes as new chiral reducing agents |
| EP0305180A2 (en) * | 1987-08-27 | 1989-03-01 | The President And Fellows Of Harvard College | Enantioselective reduction of ketones |
| WO1993023408A1 (en) * | 1992-05-14 | 1993-11-25 | Pfizer Inc. | Enantioselective oxazaborolidine catalysts |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6103938A (en) * | 1998-04-23 | 2000-08-15 | Ciba Specialty Chemicals Corporation | Process for preparing 4-(substituted phenyl)-3,4-dihydro-2H-naphthalen-1-ones |
| WO2001049638A2 (en) * | 2000-01-04 | 2001-07-12 | Sun Pharmaceutical Industries Ltd. | A process for converting stereoisomers of sertraline into sertraline |
| WO2001049638A3 (en) * | 2000-01-04 | 2001-12-06 | Sun Pharmaceutical Ind Ltd | A process for converting stereoisomers of sertraline into sertraline |
| US6506940B1 (en) | 2000-01-04 | 2003-01-14 | Sun Pharmaceuticals Industries Ltd. | Process for converting stereoisomers of sertraline into sertraline |
| US7767683B2 (en) | 2003-08-18 | 2010-08-03 | H. Lundbeck A/S | Hydrogen succinate salts of trans-4-((1R, 3S)-6-chloro-3-phenylindan-1-YL)-1,2,2-trimethylpiperazine and the use as a medicament |
| US7772240B2 (en) | 2003-08-18 | 2010-08-10 | H. Lundbeck A/S | Trans-1(6-chloro-3-phenylindan-1-yl)-3,3-dimethylpiperazine |
| US8076342B2 (en) | 2003-08-18 | 2011-12-13 | Lopez De Diego Heidi | Malonate salt of 4-((1R,3S)-6-chloro-3-phenylindan-1-yl)-1,2,2-trimethylpiperazine and uses of same |
| US8227607B2 (en) | 2003-08-18 | 2012-07-24 | H. Lundbeck A/S | Processes for 4-((1R,35)-6-Chloro-3-phenylindan-1-yl)-1,2,2-trimethylpiperazine or a salt thereof |
| US7648991B2 (en) | 2005-02-16 | 2010-01-19 | H. Lundbeck A/S | Crystalline base of trans-1-((1R,3S)-6-chloro-3-phenylindan-1-yl)-3,3-dimethylpiperazine |
| US8450324B2 (en) | 2005-02-16 | 2013-05-28 | H. Lunbeck A/S | Crystalline base of trans-1-((1R,3S)-6-chloro-3-phenylindan-1-yl)-3,3-dimethylpiperazine |
| US8569499B2 (en) | 2005-02-16 | 2013-10-29 | H. Lundbeck A/S | Process for making trans-1-((1R,3S)-6-chloro-3-phenylindan-1-yl)-3,3-dimethylpiperazine |
Also Published As
| Publication number | Publication date |
|---|---|
| FI962250A (en) | 1996-05-29 |
| GR3025223T3 (en) | 1998-02-27 |
| DE69406553D1 (en) | 1997-12-04 |
| DE69406553T2 (en) | 1998-02-26 |
| FI962250A0 (en) | 1996-05-29 |
| EP0724552A1 (en) | 1996-08-07 |
| CA2176500A1 (en) | 1995-06-08 |
| DK0724552T3 (en) | 1997-12-22 |
| JPH09500390A (en) | 1997-01-14 |
| CA2176500C (en) | 1999-09-28 |
| ATE159706T1 (en) | 1997-11-15 |
| ES2108484T3 (en) | 1997-12-16 |
| FI115397B (en) | 2005-04-29 |
| US5750794A (en) | 1998-05-12 |
| EP0724552B1 (en) | 1997-10-29 |
| JP2686366B2 (en) | 1997-12-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Corey et al. | A new system for catalytic enantioselective reduction of achiral ketones to chiral alcohols. Synthesis of chiral α-hydroxy acids | |
| Corey et al. | Catalytic diastereoselective synthesis of cis-1, 2-disubstituted cyclopropanols from esters using a vicinal dicarbanion equivalent | |
| Botteghi et al. | Optically active nitrogen ligans: III. Enantioface-discriminating transfer hydrogenation of acetophenone catalyzed by rhodium (I) complexes with chiral 2-(2′-pyridyl) pyridines | |
| Chandrasekharan et al. | Diisopinocampheylchloroborane, a remarkably efficient chiral reducing agent for aromatic prochiral ketones | |
| Wirth | Enantioselective alkylation of aldehydes catalyzed by new chiral diselenides | |
| EP0724552B1 (en) | Process for preparing a chiral tetralone | |
| Schmalz et al. | On the enantioselective deprotonation/silylation of prochiral mono-and 1, 2-dimethoxybenzene-Cr (CO) 3 derivatives | |
| Kim et al. | Stereocontrolled catalytic asymmetric reduction of ketones with oxazaborolidines derived from new chiral amino alcohols | |
| EP1029852B1 (en) | Process for obtaining enantiomers of cis-olirtine | |
| CA2134096C (en) | Enantioselective oxazaborolidine catalysts | |
| US6005133A (en) | Enantioselective oxazaborolidine catalysts | |
| Davies et al. | Application of the combined C–H activation/Cope rearrangement as a key step in the total syntheses of the assigned structure of (+)-elisabethadione and a (+)-p-benzoquinone natural product | |
| US6593496B1 (en) | Process for preparing sertraline from chiral tetralone | |
| US5367073A (en) | Asymmetric synthesis of β-amino alcohols from chiral or achiral enamines | |
| CN108373398B (en) | Preparation method of organic ketone compound | |
| Suda et al. | Asymmetric reduction of aromatic ketones with borane-amine complexes modified with optically pure 2, 2′-dihydroxy-6, 6′-dimethylbiphenyl | |
| Chemla et al. | Synthesis of diastereoenriched acetylenic α-N-tert-butanesulfinamidoalkyl methoxymethyl ethers | |
| US4918242A (en) | Novel optically 1,3-phenoxypropylhalides | |
| JP4519255B2 (en) | Process for producing optically active 3,7-dimethyl-6-octenol | |
| US4918246A (en) | Process for preparing optically active haloalcohols | |
| US4918207A (en) | Novel optically active halo-substituted tetrahydrofurans | |
| US7816533B2 (en) | Asymmetric imine hydrogenation processes | |
| Takahashi et al. | Preparation of enantiomerically pure trans-and cis-2-(1-naphthyl) cyclohexan-1-ols | |
| CN113527078B (en) | Polysubstituted chiral cyclohexane derivative, preparation method and application thereof | |
| CN115650824B (en) | Chiral diol and preparation method thereof, prepared catalyst and preparation method and application thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): CA FI JP US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1994924378 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2176500 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 962250 Country of ref document: FI Ref document number: 08652485 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 1994924378 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1994924378 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 962250 Country of ref document: FI |