WO1995008986A1 - Camptothecin formulations - Google Patents
Camptothecin formulations Download PDFInfo
- Publication number
- WO1995008986A1 WO1995008986A1 PCT/US1994/010898 US9410898W WO9508986A1 WO 1995008986 A1 WO1995008986 A1 WO 1995008986A1 US 9410898 W US9410898 W US 9410898W WO 9508986 A1 WO9508986 A1 WO 9508986A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- upid
- pharmaceutical composition
- camptothecin
- lipid
- sterol
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/127—Liposomes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Definitions
- This invention relates to liposomal formulations of Camptothecin and related analogs exhibiting reduced toxicity and improved efficacy.
- liposomal encapsulation alters the pharmacokinetic parameters of the entrapped antineoplastics (see review by Daoud, S.S. et al; Liposomes in Cancer Therapy. Adv. Drug Deliv. Rev. 3: 405-418, 1989).
- Drug delivery advantages offered by liposomes include protection of liposome contents against the host, prolonged circulation/sustained release, and the possibility of targetting (passive, compartmental, ligand-mediated and physical).
- liposomes include: alkylating agents, nitrosoureas, cisplatin, antimetabolites (cytosine arabinoside, methotrexate, 5-fluoro- uracil) and anthracyclines.
- alkylating agents include nitrosoureas, cisplatin, antimetabolites (cytosine arabinoside, methotrexate, 5-fluoro- uracil) and anthracyclines.
- liposomes containing anthracycline antibiotics have clearly shown reduction of cardiotoxicity and dermal toxicity and prolonged survival of tumor-bearing animals compared to controls receiving free drug (Sells, R.A. et al. Cancer Treat. Rep. 14: 383-387 (1987)).
- Doxorubicin a widely used anthracycline antibiotic is now in Phase HI clinical trials.
- the anticancer drug camptothecin and a related analogue, topotecan, a water-soluble analogue are among a promising group of antineoplastics exhibiting a broad spectrum of activity against several human tumors.
- This class of therapeutic agents are specific inhibitors of topoisomerase I, an enzyme that intimately involves in DNA replication as it relieves the torsional strain introduced ahead of the moving replication fork. Inhibition of this enzyme results in lethal DNA damage during the course of replication.
- Inhibitors of topoisomerases are unique in that they do not cause cytotoxicity by depleting the product of their target enzymes, but by producing DNA damage by interfering with topoisomerase function (Hertzberg, R.P et al; Biochemistry 28: 4629-4638, (1989)).
- Camptothecin the parent compound, is both insoluble and unstable in water, undergoing a rapid pH dependent but reversible hydrolysis to the inactive carboxylate ion that is rapidly cleared in vivo.
- the lactone to open ring conversion is particularly favored at alkaline pH and the open form predominates at physiological pH (7.4).
- Topotecan a water-soluble analog of camptothecin, now in Phase II clinical studies, is also inactivated through reversible lactone hydrolysis and conversion to the carboxylate form (Underberg, W. J. M. fit al; I. Pharmac. Biomed. Analysis 8: 681- 683, (1990)).
- camptothecin and topotecan are limited by a dose-dependent toxicity.
- the determined toxic dose for camptothecin in a suspension is 12 mg/kg and for a saline solution of topotecan (pH 3.0) is 15 mg/kg, respectively.
- the pharmacokinetics and pharmacodynamics of camptothecin/topotecan would change in a liposome formulation.
- improving both the stability and antitumor activity of camptothecin and related analogues formulated in liposomes would certainly increase the commercial viability of these anticancer agents.
- the present invention provides for the novel liposomal formulations of camptothecin and its structurally related analogues. These novel formulations provide improved pharmacokinetics and pharmacodynamics for the compounds herein and thereby lowering the dose-dependent toxicity for use in anticancer treatments.
- the formulations of camptothecin and its structurally related analogues are in the form of multilamellar vesicles dispersed in an aqueous phase with at least 80% of the total drag present incorporated in the lipid bilayer of the liposome at a high drag to lipid ratio (molar) of at least 1/100, preferably of about 1/10, and the lipid is of natural sources.
- a liposome is defined as a structure consisting of one or more concentric spheres of lipid bilayers separated by water or aqueous buffer compartments. The number of bilayers is dependent on both the lipid composition and method of preparation. With additional energy, multilamellar vesicles (MLVs) can form unilamellar vesicles (ULVs).
- MLVs multilamellar vesicles
- UUVs unilamellar vesicles
- the present invention provides for novel liposomal formulations of camptothecin, and its structurally related analogues, to be administered parenterally, in the form of multilamellar vesicles or unilamellar vesicles dispersed in an aqueous phase, preferably 0
- the drag incorporated in the lipid bi-layer if it is a lipid-soluble drag, at a drag to lipid ratio (molar) of at least 1/100, preferably from 1/50 to 1/5 and more preferably from 1/20 to 1/10.
- the total lipid concentration is from 10-1000 mg/ml, preferably from 100-500 mg/ml, more preferably from 200-400 mg/ml, and still more preferably from 100-200 mg/ml.
- the vesicle is multilamellar.
- the particular size can be altered and readily determined -by one skilled in the ait
- the particle size for such administration is generally from 0.5 microns to 10 and preferably from 0.2 - 2.0 microns.
- the particle size is preferably less than 0.2 microns.
- the multilamellar vesicles of the present invention can be prepared by standard methods (Cullis, P. R. g£ al; Generating and loading of liposomal systems for drug- delivery applications. Adv. Drug Deliv. Rev. 3: 267-282, 1989) whose disclosure is incorporated by reference in its entirety herein.
- One particular method that is suitable for lipophilic drags involves: a) dissolving the lipids and drug in organic solvent(s) in a suitable lipid flask, b) slowly removing the organic solvents under vacuum to deposit on the inside walls of the lipid flask a dried lipid-drag film, and c) hydrating the dried lipid- drag film with an aqueous buffer or saline followed by mechanical agitation, such as vortexing, to disperse the drug-lipid suspension and produce the liposomes.
- These multilamellar liposomes can be freezed-thawed and then sized down, if necessary, by extrusion through polycarbonate membranes (Cullis P. R. et al: Adv. Drag Deliv.
- the lipophilic drag can be mixed with lipids using an ionizable hydrating agent to form a "preliposome gel".
- liposomes can be formed incorporating the lipophilic drug (EPO application No. 86306014.1, published 25 Feb. 1987, now US Patent No. 5,230,899) the disclosure of which are incorporated herein by reference in their entirety.
- the lipid may be obtained as a crude extract from natural sources.
- the crude extract comprises many different components, such as those indicated in the two tables below.
- Paltauf et al. "Phospholipids- Natural, Semisynthetic, Synthetic” in Phospholipids:Biochemical, Pharmaceutical and Analytical Considerations, pp 1-12, Plenum Publishers, NY (1990)).
- the crade extract is often highly purified to a single component if desired, i.e. such as lecithin.
- EPG egg phosphatidyl glycerol
- the lipid contains phosphorus, i.e. is a phospholipid, then it is preferably fluid at ambient tempartures, more preferably at physiological temperatures.
- the mixture may contain a small blend of synthetic lipids.
- Synthetic for use herein, means a homogeneous lipid which is not found in natural sources, such as the di-saturated phospholipids dimyristoyl-, dipalmitoyl-, and distearoyl-.
- the lipids for use herein may contain unsaturation. This unsaturation may be on a single fatty acyl chain or in both chains (where applicable), the chain may include more than one double bond (i.e., di-unsaturated) or the chains may be partially hydrogenated, or contain mixtures thereof.
- the phospholipids are unsaturated, then they are di-unsaturated products, such as di-oleoyl (C18:l) or di-linoleoyl (C18:2) phosphotidylcholine or glycerol.
- the lipid bilayer contains lipids which may have at least 30% (molar) unsaturated fatty acyl chains.
- fatty acyl chain as used herein is the ester of a fatty acid. Alternatively, the term “fatty acid chain” may be used.
- the lipids used herein may be of negative, neutral, or positive charge.
- Such products include, but are not limited to, lipids such, phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine, phosphatidylglycerol, disphosphatidylglycerol (cardiolipin), phosphatidic acid, phosphatidylinositol, sphingolipids, glycolipids, sulfatides, lysolipids and fatty acids, sterols such as cholesterol, polymerizable lipids, and combinations thereof.
- Commercial products such as, soy and egg sources are preferred, preferably soy and egg lecithin.
- Phospholipids such as phosphatidylglycerol which can be obtained from egg lecithin are also commercially available.
- Suitable hydrogenated lecithins are products under the trade name Phospholipon H and G, (American Lecithin, Danbury CT); as well as Asahi's Type 5, 20, 40 and 65 egg phosphatides (Asahi Chemical Industry Co., Tokyo, Japan).
- Other lecithin products for use herein are Centrophase 31 and Centrolex P (Central Soya, Fort Wayne, IN).
- lecithins and phosphatidylglycerols are bilayer-forming lipids, whereas certain types of phospholipids, such as phosphatidylethanolamine and cardiolipin, sphingolipids, sulfatides, lysolipids, glycolipids and glycerides, and fatty acids do not from stable bilayers. Therefore, the ratio of the bilayer-forming lipids to the non- bilayer-forming lipids can vary from 9:1 to 1:1, preferably from 9:1 to 7:1.
- phosphatidylcholine and phosphatidylglycerol derivatives include, but are not limited to natural soybean or egg-yolk; or hydrogenated products obtained by hydrogenation of the natural soybean or egg-yolk phosphatidylcholine and phosphatidyl- glycerol.
- Suitable phosphatidylglycerol derivatives are egg phosphatidylglycerol or salts forms thereof.
- Semi-synthetic products such as dimyristoyl-, dipalmitoyl-, distearoyl-, dioleoyl-, dilinoleoyl, and mixtures thereof, such as l-palmitoyl-2-oleoyl-, for instance, may be added.
- a semi-synthetic product it is l-palmitoyl-2-oleoyl-, dioleoyl- or di-linoleoyl; or mixtures thereof.
- the polar head group for the lipids is choline or glycerol.
- a single component or an admixture of these two may be in amounts similar to that of their natural analogs.
- Preferred components are 1-palmitoyl- 2-oleoyl- phosphatidylcholine or phosphatidylglycerol, dioleoyl phosphatidylcholine or phosphatidylglycerol (DOPC or DOPG).
- DOPC or DOPG dioleoyl phosphatidylcholine or phosphatidylglycerol
- the other di-saturated semi-synthetic components may be added in small amounts. Small amounts for use herein is less than 10% (molar), preferably less than 5% (molar).
- lipids such as phosphatidylcholine and phosphatidylglycerol derivatives with C-14, C-16 and C-18 fatty acyl chains; or lipids with ester-linked fatty acids such as the mono-, di-, and tri-glycerides, or combinations thereof, may also be added.
- Preferred lipids are the phosphatidylcholines (PC's) and phosphatidylglycerols (PG's) having a) mono-unsaturated (C18: 1) or (C18:2 ) fatty acyl chains; b) di-unsaturated with (C18:l) or (C18:2) fatty acyl chains; c) one particular class of mono-unsaturated PC or PG's are those having long (C16 or greater ) saturated fatty acyl chain, on the 1-position of the glycerol backbone, with oleic or linoleic acid being esterified on the 2-position.
- One such product is l-palmitoyl-2-oleoyl. This class is particularly preferred, since the fatty acid composition in these phospholipids is similar to the fatty acid distribution found in phospholipids from natural sources.
- Preferred disaturated phospholipids are dimyristoylphosphatidylglycerol (DMPG), and dimyristoylphosphatidylcholine (DMPC).
- DMPG dimyristoylphosphatidylglycerol
- DMPC dimyristoylphosphatidylcholine
- the lipid bilayer may preferably include lipids selected from phosphatidylcholine, phosphatidylserine, phosphatidylinositol, phosphatidylglycerol, cardiolipin, phosphatidic acid, and sterols such as cholesterol or combinations thereof. More preferably the lipid bilayer includes phosphatidylcholine, phosphatidylglycerol, and sterols such as cholesterol or combinations thereof.
- Suitable neutral lipids include, but are not limited to, phosphatidylcholine and sterols, such as cholesterol and cholesterol derivatives thereof.
- Suitable negatively charged phospholipids include, but are not limited to such as phosphatidylglycerol, phosphatidylserine, phosphatidylinositol, phosphatidic acid and disphosphatidylglycerol (cardiolipin) and cholesterol analogs, such as cholesterol sulfate and hemisuccinate.
- Suitable positive lipids include, but are not limited to stearylamine, or sphingosine.
- the lipids preferably are not solely a sterol derivative but will include a sterol derivative, such as but not limited to, cholesterol and cholesterol analogs.
- the lipid formulation may additionally comprise a negatively charged cholesterol salt such as cholesterol sulfate or hemisuccinate which may be in combination with a neutral sterol as well.
- the formulation may also contain additional ingredients such as antioxidants, i.e. vitamin A ( ⁇ -tocopherol) or tocopherol-hemisuccinate, or other conventional antioxidants may be used.
- antioxidants i.e. vitamin A ( ⁇ -tocopherol) or tocopherol-hemisuccinate, or other conventional antioxidants may be used.
- polymerizable lipids is well known to those of skill in the art and suitably these lipids are used to help keep the aggregation and fusion down to a minimum for the sonicated vesicles by covalently linking the individual lipid molecules which form the membrane of the liposome.
- Suitable groups for such polymerization include, but are not limited to, vinyl, acryloylic, methacryloylic, butadienic, diacetylenic and H2NCC(O)OCH3.
- the reactivity of the polymerizable group is influences by the state of the bilayer.
- the linking group is a diacetylenic moiety.
- Phospholipids containing diacetylene groups may do so in one or both acyl chains, for instance.
- Phosphatidylcholines containing a diacetylene group polymerize when dispersed as large multi- or unilamellar vesicles, but not as small ones.
- These polymerizable groups may be placed in all parts of the surfactant molecule, for the polar head to the extremity of the hydrocarbon chain.
- Cross-linking of the constituent molecules of a vesicle has been shown to increase the stability of the vesicle.
- a general review of polymerizable lipids may be found in Liposome Technology, Vol.
- Liposome Chapter 9, Gregoriadis, Gregory, (1984) CRC Press whose disclosure is herein incorporated by reference.
- the surface of the liposomes may also be modified with a polymer, such as, for example with polyethylene glycol (PEG), using procedures readily apparent to those skilled in the art. (Woodle, M.C. and Lasic, D.O., Biochim. Biophvs. Acta 1113:171-193, 1992; or Blume et al., Biochim. Biophvsica Acta. 1029: 91-97, 1990).
- PEG polyethylene glycol
- Modifications of the liposome may be made by using any type of lipid available which does not have deleterious effects to the mammal, provided the lipid or combination of lipids along with other materials incorporated within the lipid matrix, should form a bilayer phase under physiologically relevant conditions.
- the composition of the liposomes may be modified to modulate the biodistribution and pharmacokinetics of the resulting liposomes.
- Negatively charged liposomes may comprise from about 0% to about 50% (molar) negatively charged lipid; and more preferably from about 0 to about 30% (molar) negatively charged lipid in the composition.
- the ratio of the neutral to negatively charged lipid can very from 9:1 to 1:1 preferably from 3:1 to 1:1, more preferably from 9:1 to 3:1.
- cholesterol is also incorporated into the liposome its levels can vary 0 to about 50%, preferably from about 10% to about 30%, and more preferably from about 15 to 25%.
- the ratio of neutral lipid to cholesterol can vary from about 8:1 to about 1:1, more preferably from 4:1 to 1:1.
- Preferred molar ratios of neutral:negative:sterol containing lipids include 4:2:4,
- the neutral lipid is phosphatidylcholine
- the negative lipid is phosphatidylglycerol
- the sterol is cholesterol or a derivative thereof.
- a preferred embodiment of the present invention is a pharmaceutical composition
- a pharmaceutical composition comprising Camptothecin or structurally related analogues thereof, in the form of multilamellar or unilamellar vesicles dispersed in an aqueous phase with a drag to lipid ratio (molar) of at least 1/100 and wherein the lipid is selected from phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine, phosphatidylglycerol, disphosphatidylglycerol (cardiolipin), phosphatidic acid, phosphatidylinositol, sphingolipids, glycolipids, sulfatides, lysolipids and fatty acids, sterols, polymerizable lipids, or combinations thereof;and wherein the lipid bilayer must contain either at least one charged lipid component present in an amount of at least 5% (molar), or the salt form of a water-soluble analog in an amount
- the charged component is preferably negatively charged. Further the amount present of the negatively charged component is at least 10%, preferably at least 20% and more preferably at least 30%.
- the preferred negative component is a phosphatidylglycerol, charged salt of cholesterol, such as cholesterol sulfate, or hemi-succinate, or phosphatidylserine, more preferably egg phosphatidylglycerol (EPG).
- EPG egg phosphatidylglycerol
- This formulation while usable for lipid soluble analogs is preferred for use herein for water-soluble analogs, such as topotecan.
- the water-soluble analog is not present in high amounts in the aqueous phase but in the lipid bilayer, preferably at least 30%, more preferably 50%, most preferably 80%.
- the vesicle may consist of nonionic surfactants.
- This class of vesicles is known as "niosomes".
- ⁇ isomes have been prepared from several classes of non-ionic surfactants, e.g. poly glycerol alkylethers, glucosyl dialkylethers, crown ethers, and polyoxyethylene alkyl ethers.
- non-ionic surfactants e.g. poly glycerol alkylethers, glucosyl dialkylethers, crown ethers, and polyoxyethylene alkyl ethers.
- One possible lipid combination is the use of cholesterol and a non-ionic surfactant.
- a charged surfactant is intercalated in the bilayers in order to introduce electrostatic repulsion between the vesicles, thus increasing their stability.
- camptothecin and related analogs or congeners means camptothecin and compounds which have the same core ring system with various substitutions but preferably have such modifications or substitutions in rings A & B.
- camptothecin The basic structure for camptothecin is
- the analogue is a lipid soluble analogue. Since the initial isolation of Camptotheca ac minata of the novel alkaloid camptothecin in 1966 many related compounds have been made and are well known to those skilled in the ait It would be difficult to list all of the papers and patents covering such compounds but a representative grouping of reference showing additional congeners useful herein include but are not limited to, Wall et al., J. Med. Chem., Vol. 29, pp 1553- 1555 (1986) disclosing 11-hydroxycamptothecin; US Patent No.'s 4,981, 968, 4,894,456 and 5, 122,526 to Wall et al.; Hertzberg et al., J. Med. Chem., Vol. 32, pp 715-720
- lipid composition and/or charge of the liposome, or lipid derivatization of the analog may be necessary to improve incorporation into the liposomal lipid bilayer.
- Modification of the lipid composition of the liposome means herein, increasing the amount of negatively charged lipid or lipids present in the liposome; modifying the degree of fatty acid chain unsaturation and altering the levels of cholesterol in the bilayer.
- Modification of the charge of the liposome means herein, increasing the levels of the negatively charged lipids such that the overall liposome charge is increased, i.e. on the surface.
- a water-soluble analog of camptothecin, herein means an analog or drag which is charged when in the physiological pH range.
- negatively charged lipids such as cholesterol sulfate or hemisuccinate may be employed. Formation of an ion-pair with a water-soluble analog may be prepared in-situ during liposome formation or prior to incorporation into the liposome.
- parenterally as used herein is meant by intravenous, intramuscular, subcutaneous, or intraperitoneal administration. It will be recognized by one of skill in the art that the optimal quantity and spacing of individual dosages of the formulations herein will be determined by the nature and extent of the condition being treated, the form, route and site of administration, and the particular patient being treated, and that such optimums can be deteimined by conventional techniques. It will also be appreciated by one of skill in the art that the optimal course of treatment, i.e., the number of doses given per day for a defined number of days, can be ascertained by those skilled in the art using conventional course of treatment determination tests.
- camptothecin be administered in a dosage range per course of therapy (one month) from 20 mg/M-2 to 800 rng M* ⁇ .
- Prior treatment has generally accorded that topotecan, IV, be administered in a dosage range per course of therapy , daily X 5 days at 1.5 rng/M ⁇ for small cell lung carcinoma (SCLC) and Ovarian Cancer.
- SCLC small cell lung carcinoma
- the drag concentration in the liposome may vary from 0.1 mg/ml to 10 mg/ml, preferably from about 4 mg/ml to 6 mg/ml. It is recognized in any instance, that the total volume of the liposome may be adjusted to meet the desired dose requirements. For example, for camptothecin administration of 60 mg/kg a liposome volume of 10 ml/kg was employed.
- EXAMPLE 1 Preparation of camptothecin-containing DMPGDMPG multilamellar liposomes 1.440 g or 0.540 g of dimyristoylphosphatidylcholine (DMPC) were mixed with 0.160 g or 0.060 g dimyristoylphospatidylglycerol (DMPG) sodium salt and without or with 0.018 g camptothecin, respectively, in two separate lipid flasks. Six ml of methylene chloride:methanol (9/1, v/v) were added to solubilize camptothecin plus sufficient volume of chloroform (5-10 ml) to completely solubilize the lipids and produce a yellowish solution.
- DMPC dimyristoylphosphatidylcholine
- DMPG dimyristoylphospatidylglycerol
- the drug-free lipids were treated in a similar manner.
- the organic solvents were subsequently removed under vacuum in a rotary evaporator to leave behind a thin film of the drug-lipid mixture. Residual solvents were removed in dessicator under vacuum overnight.
- the composition of the resulting liposomes was DMPQDMPG (90:10) and DMPC:DMPG:Camptothecin (84.6:9.4:6.0) for the drag-free and ctrag-cont ⁇ ining liposomes, respectively.
- the newly formed liposomes were further equilibrated at 37°C for about an hour and then stored at ambient temperature.
- the total lipid and drag concentration were 200 and 6 milligrams/milliliters (hereinafter referred to as mg/ml), respectively.
- these multilamellar liposomes can be sized down by extrusion through 0.4 ⁇ m and 0.2 ⁇ m polycarbonate membranes.
- EPC egg phosphatidylcholine
- EPG egg phosphatidylglycerol
- EPC egg phosphatidylcholine
- EPG egg phosphatidylglycerol sodium salt
- CHOL cholesterol
- the resulting liposomes were: a) EPC:EPG:CHOL (70:10:20); and b) EPC:EPG:CHOL: camptothecin (65.8:9.4:18.8:6.0) at total lipid and drug concentration of 200 and 6 mg/ml, respectively.
- the drag-free and drag-lipid mixtures were then centrifuged at 50,000 rpm for 2 hrs at 25°C using a Ti 80 fixed angle rotor. Under these conditions, the lipids separated from the aqueous medium and the concentration of camptothecin in the supernatant was determined upon proper dilution by measuring the absorbence at 253 nm using a Beckman DU70 spectrophotometer. The amount of the drag in liposomes was calculated as follows:
- EPC:EPG:CHOL:camptothecin 97 (65.8:9.4:18.8:6.0)
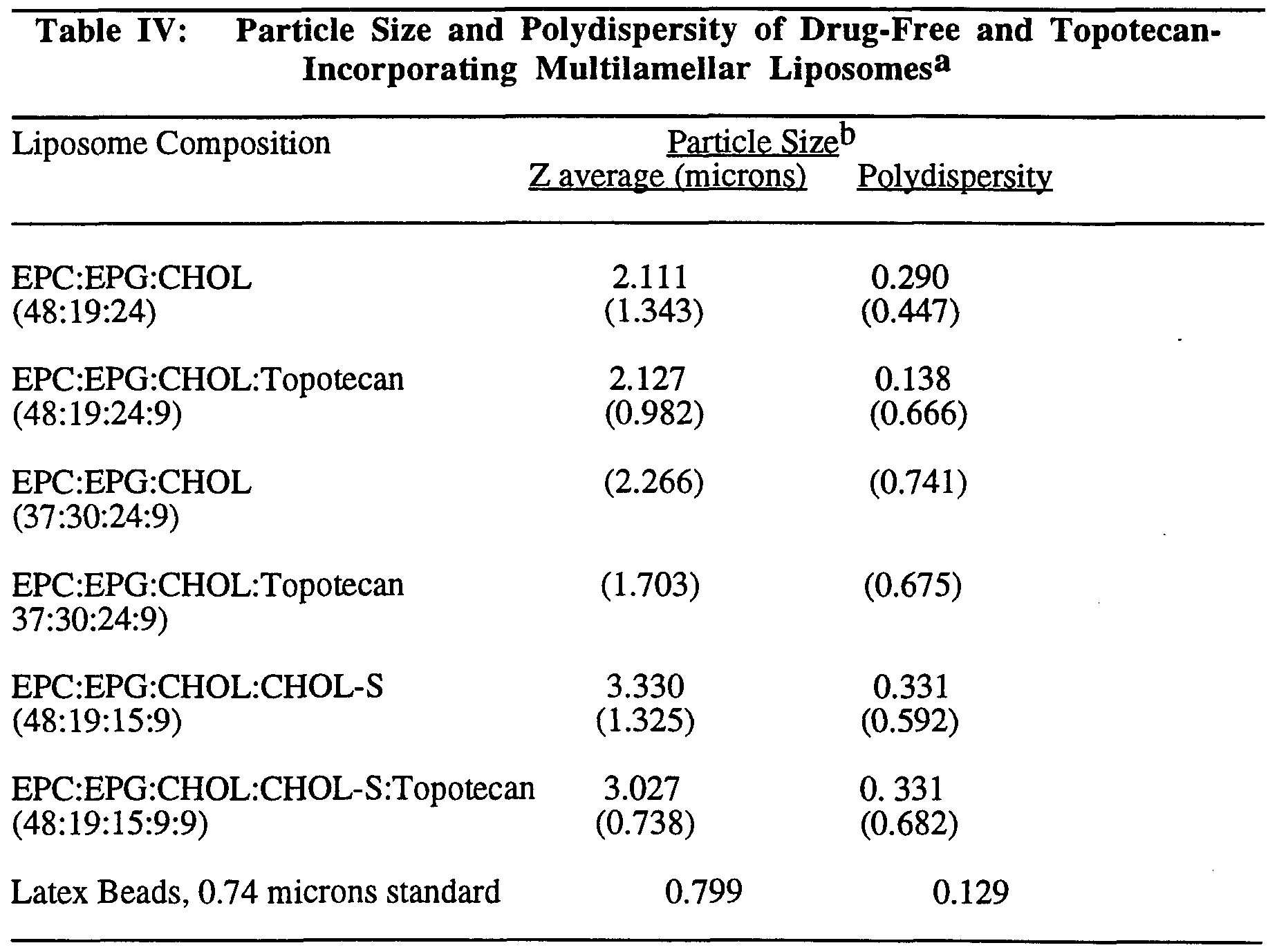
- Example 5 Particle Size Determination of Camptothecin Liposomes
- the particle size of camptothecin liposomes along with the corresponding drag-free liposomes prepared as described in Examples 1 to 3 was determined upon a 400-fold dilution with physiological saline using a laser light scattering.
- a Malvern Photon Correlation Spectrometer model 4700 equipped with an argon laser model 2000 from Spectra Physics was employed to monitor particle size of the various liposomal formulations. Light scattering was monitored at 90° angle and temperature of 25°C. Polystyrene (latex) beads were used as particle size standard. The light scattering data is summarized in Table ⁇ .
- the average particle size of these liposomes was about 1 micron ( ⁇ m) (Table II) with the size distribution varied between 0.5 to 5.0 ⁇ m.
- the size heterogeneity of these liposomes is also evident from the high polydispersity value as compared to that obtained with the latex beads standard.
- the polydispersity index is a measure of particle homogeneity. It varies from 0 to 1; the closer to zero the value the more homogeneous (monodispersed) the particles.
- EPC egg phosphatidylcholine
- EPG egg phosphatidylglycerol
- CPG egg phosphatidylglycerol
- CHOL cholesterol
- CHOL-S cholesterol
- Topotecan cholesterol sulfate
- CHOL 25.8 mUUmolar
- cholesterol sulfate (13.7 mUlimolar
- Topotecan (10.9 miUimolar) and cholesterol-sulfate-Topotecan salt (2.3 miUimolar) .
- EPC:EPG:CHOL:Topotecan 48:19:24:9
- EPC:EPG:CHOL:Topotecan 37:30:24:9
- EPC:EPG:CHOL:CHOL-S:Topotecan 48:19:15:9:9
- EPC:CHOL:CHOL-S: Topotecan 67:24:4.5:4.5
- EPC:CHOL:CHOL-S-Topotecan 67:24:9).
- mult ameUar Uposomes incorporating Topotecan were also prepared at high Upid concentration (200 mg ml or 270 miUimolar). A drag to Upid ratio (molar) of 1 to 11 was maintained at both low and high total Upid concentration.
- Liposome Composition Drag Loading (mol %) (% of total)
- a based on monomodal analysis.
- b numbers in parentheses are obtained at low lipid concentration (2.0 mg/ml). Other values were obtained at high Upid concentration (200 mg/ml).
- Free-topotecan was removed from aU liposome samples before particle size determination. At low Upid concentration, Topotecan reduced the particle size of the corresponding liposomes whereas, at high Upid concentration, smaU differences in particle size were observed between drag-free and drag-containing Uposomes. Once again, the size heterogeneity of these Uposomes is also evident from the high polydispersity value as compared to that obtained with the latex beads standard. Topotecan appears to have a smaU but variable effect on the polydispersity.
- camptothecin Liposomally formulated camptothecin (Examples 1-3) was evaluated for toxicity in B6D2F1 mice upon intraperitoneal administration as a single bolus injection on days 1 and 5 and comparison to a suspension formulation of camptothecin (N,N-dimethyl- acetamide:Cremophor EL: Water, 10:10:80, v/v). Dosing volume and total lipid dose were maintained at 10 ml/kg and 2.0 g/kg, respectively. No apparent toxicity was observed when drag-free liposomes were dosed at 2 g/kg. In fact, animals gained some weight in response to Upid administration.
- Camptothecin in Uposomes was dosed at 60, 30, 15, 7.5 and 3.75 mg/kg and in suspension at 24, 12, 6, 3 and 1.5 mg/kg. Toxicity evaluation was based on clinical signs i.e number of survivors and body weight lost during a three-week study. The results from these studies are summarized in Table V:
- Table V Comparison of the Toxicity of Camptothecin Formulated in Liposomes or Suspension upon i.p. Administration to Mice a
- L1210 was harvested, pooled, dUuted and counted using a ZBI Coulter Counter. The ceU suspension was adjusted to 5x10 ⁇ ceUs/ml. An inoculum of 0.2 ml of L1210 was given intravenously (i.v.) via lateral tail vein into 20-22 g B6D2F1 female mice using a 25-gauge needle. Tumor inoculum was tested for bacterial contamination by 24-hr incubation in thioglycoUate broth. Animals were randomized into groups of 5 and housed in shoe box cages. Food and water were provided ad libitum. All experiments included 3 groups of animals as untreated controls.
- a titration of tumor ceUs (10 ⁇ -10 ⁇ ) in untreated animals was included so that drug-induced cell k l (NCK) could be calculated.
- Administration of Uposomal and suspended camptothecin (see Example 6) commenced at Day 2 after tumor implantation. Camptothecin was administered i.p. as described in Example 6, that is at 4-5 dose levels, each being 50% of the preceding higher dose.
- MTD maximum tolerated dose
- the EPC:EPG Uposomal formulation reduced the toxicity of camptothecin without affecting antitumor activity. This resulted in greater efficacy at the higher dose levels achieved with the Uposomal formulation.
- Plasma samples were collected at 5, 15, 30, 60, 90, 120, 240, 360, 480, 600, 720, 840, 960, 1080, 1260 and 1440 min using a group of three animals per time point (with camptothecin in suspension at a dose of 60 mg/kg plasma samples were coUected up to 240 min). Plasma samples were assayed for total camptothecin (lactone and hydroxy acid) and lactone form by HPLC using a modification of the method by Grochow ej al; Drag Metabolism Disposition 20; 706-713, 1992. Briefly, 100 ml of plasma was added to cold methanol, the samples were vortexed and then centrifuged at 14,000 ⁇ ra for 30 seconds.
- the supernatants were removed and assayed immediately or stored at -70°C.
- 100 ml of the methanoUc supernatant was added to 50 ml of 0.050 M phosphoric acid and incubated at room temperature for 2 hrs prior to chromatographic analysis.
- 100 ml of the methanoUc supernatant was added to 0.010 M sodium phosphate buffer (pH 6.0) and immediately injected into the HPLC.
- Fig. 2 The results from the pharmacokinetic study are shown in Fig. 2. As can be seen high and sustained plasma levels of camptothecin were observed over a 4-hr period after administration of the liposomal formulation as compared to those obtained with the drag suspension. Significantly larger area under the plasma concentration-time curve (AUC) and longer eUmination half-time (t ⁇ /2) were obtained from Uposomal versus suspended camptothecin. Despite this higher exposure there was reduced toxicity for the Uposomal formulation.
- AUC plasma concentration-time curve
- t ⁇ /2 eUmination half-time
- mice Female BDFi mice weighed 22-26 gm at the start of the experiment
- mice Female BDFi mice weighed 18-21 gm at the start of the experiment
Abstract
Description






Claims
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP94929892A EP0721328A4 (en) | 1993-09-27 | 1994-09-27 | Camptothecin formulations |
| JP7510405A JPH09504517A (en) | 1993-09-27 | 1994-09-27 | Camptothecin formulation |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12720893A | 1993-09-27 | 1993-09-27 | |
| US08/127,208 | 1993-09-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1995008986A1 true WO1995008986A1 (en) | 1995-04-06 |
Family
ID=22428860
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1994/010898 WO1995008986A1 (en) | 1993-09-27 | 1994-09-27 | Camptothecin formulations |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP0721328A4 (en) |
| JP (1) | JPH09504517A (en) |
| WO (1) | WO1995008986A1 (en) |
Cited By (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0785772A1 (en) * | 1994-10-14 | 1997-07-30 | Pharmacia, Inc. | Lyophilizate of lipid complex of water insoluble camptothecins |
| WO1999065466A1 (en) * | 1998-06-18 | 1999-12-23 | Duke University | Temperature-sensitive liposomal formulation |
| WO2000023052A1 (en) * | 1998-09-16 | 2000-04-27 | Alza Corporation | Liposome-entrapped topoisomerase inhibitors |
| EP1037610A1 (en) * | 1997-09-16 | 2000-09-27 | NeXstar Pharmaceuticals, Inc. | Liposomal camptothecin formulations |
| US6143321A (en) * | 1997-02-06 | 2000-11-07 | Duke University | Liposomes containing active agents |
| WO2002002078A2 (en) * | 2000-06-30 | 2002-01-10 | Inex Pharmaceuticals Corporation | Improved liposomal camptothecins and uses thereof |
| WO2002009678A2 (en) * | 2000-07-31 | 2002-02-07 | Ottawa Heart Institute Research Corporation | Charged phospholipid compositions and methods for their use |
| WO2002094269A1 (en) * | 2001-05-25 | 2002-11-28 | G.O.T. Therapeutics Gmbh | Liposomally encapsulated hydrophobic active ingredients with a high active ingredient content ⊃ 50 % and method for the production of pharmaceutical preparations containing liposomally encapsulated hydrophobic active ingredients |
| WO2003030864A1 (en) * | 2001-05-29 | 2003-04-17 | Neopharm, Inc. | Liposomal formulation of irinotecan |
| EP1355634A2 (en) * | 2000-11-09 | 2003-10-29 | Neopharm, Inc. | Sn-38 lipid complexes and methods of use |
| WO2004002454A1 (en) * | 2002-06-26 | 2004-01-08 | Medigene Oncology Gmbh | Camptothecin-carboxylate formulations |
| US6726925B1 (en) | 1998-06-18 | 2004-04-27 | Duke University | Temperature-sensitive liposomal formulation |
| EP1676568A1 (en) * | 2004-12-30 | 2006-07-05 | Merckle Gmbh | Particles comprising phospholipids, process of preparation and use as medicament |
| WO2006052767A3 (en) * | 2004-11-05 | 2007-08-02 | Inex Pharmaceuticals Corp | Compositions and methods for stabilizing liposomal camptothecin formulations |
| US7311924B2 (en) | 1999-04-01 | 2007-12-25 | Hana Biosciences, Inc. | Compositions and methods for treating cancer |
| CN100356919C (en) * | 2004-05-31 | 2007-12-26 | 上海医药工业研究院 | Hydroxycamptothecin liposome and its preparation |
| US7452550B2 (en) | 2000-06-30 | 2008-11-18 | Hana Biosciences, Inc. | Liposomal antineoplastic drugs and uses thereof |
| US7691872B2 (en) * | 2001-03-20 | 2010-04-06 | University Of Kentucky Research Foundation | Methods and compositions for optimizing blood and tissue stability of camptothecin and other albumin-binding therapeutic compounds |
| US7807350B2 (en) | 2003-05-30 | 2010-10-05 | The University Of Chicago | Methods for predicting irinotecan toxicity |
| US7811602B2 (en) | 2004-05-17 | 2010-10-12 | Tekmira Pharmaceuticals Corporation | Liposomal formulations comprising dihydrosphingomyelin and methods of use thereof |
| US7846473B2 (en) | 2004-06-01 | 2010-12-07 | Terumo Kabushiki Kaisha | Irinotecan preparation |
| US7906139B2 (en) * | 2002-04-03 | 2011-03-15 | Lamellar Therapeutics Limited | Compositions and methods of using lamellar bodies for therapeutic purposes |
| WO2011076807A2 (en) | 2009-12-23 | 2011-06-30 | Novartis Ag | Lipids, lipid compositions, and methods of using them |
| US8206750B2 (en) | 2005-03-24 | 2012-06-26 | Cerenis Therapeutics Holding S.A. | Charged lipoprotein complexes and their uses |
| WO2014136086A1 (en) | 2013-03-08 | 2014-09-12 | Novartis Ag | Lipids and lipid compositions for the delivery of active agents |
| WO2015095340A1 (en) | 2013-12-19 | 2015-06-25 | Novartis Ag | Lipids and lipid compositions for the delivery of active agents |
| WO2015095346A1 (en) | 2013-12-19 | 2015-06-25 | Novartis Ag | Lipids and lipid compositions for the delivery of active agents |
| US9173901B2 (en) | 2003-09-25 | 2015-11-03 | Lamellar Therapeutics Limited | Compositions and methods of using lamellar bodies for modifying linear biological macromolecules |
| WO2016037053A1 (en) | 2014-09-05 | 2016-03-10 | Novartis Ag | Lipids and lipid compositions for the delivery of active agents |
| US9801874B2 (en) | 2012-11-20 | 2017-10-31 | Spectrum Pharmaceuticals | Method for the preparation of liposome encapsulated vincristine for therapeutic use |
| US10342761B2 (en) | 2014-07-16 | 2019-07-09 | Novartis Ag | Method of encapsulating a nucleic acid in a lipid nanoparticle host |
| US11559486B2 (en) | 2015-07-22 | 2023-01-24 | Acrotech Biopharma, LLC | Ready-to-use formulation for Vincristine Sulfate Liposome Injection |
| WO2023096511A1 (en) * | 2021-11-23 | 2023-06-01 | Uniwersytet Warszawski | Drug lipid carrier, pharmaceutical formulation, uses thereof and method for preparation of drug lipid carrier, carrier obtained by this method and uses thereof |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ATE286741T1 (en) * | 1999-01-27 | 2005-01-15 | Telik Inc | THERAPEUTIC COMPOSITIONS CONTAINING GLUTATHIONE ANALOGUES |
| ATE490763T1 (en) * | 2002-06-26 | 2010-12-15 | Medigene Ag | NEW METHOD FOR STABILIZING DIAGNOSTIC AND THERAPEUTIC COMPOUNDS IN A CATIONIC CARRIER SYSTEM |
| WO2005002546A1 (en) * | 2003-06-27 | 2005-01-13 | Smithkline Beecham Corporation | Stabilized topotecan liposomal composition and methods |
| AU2005253852B2 (en) * | 2004-06-18 | 2008-05-29 | Kabushiki Kaisha Yakult Honsha | Liposome preparation containing slightly water-soluble camptothecin |
| CN102516258B (en) * | 2011-11-11 | 2014-06-25 | 正大天晴药业集团股份有限公司 | Water-soluble vitamin E derivative modified fat-soluble anti-cancer drug compound and preparation, preparation method and application of compound |
| JP2015524410A (en) * | 2012-07-18 | 2015-08-24 | オニキス セラピューティクス, インク.Onyx Therapeutics, Inc. | Liposome composition of epoxy ketone-based proteasome inhibitor |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4721612A (en) * | 1984-04-12 | 1988-01-26 | The Liposome Company, Inc. | Steroidal liposomes |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU6832794A (en) * | 1993-05-19 | 1994-12-12 | Liposome Company, Inc., The | Liposome having a multicomponent bilayer which contains a bioactive agent as an integral component of the bilayer |
-
1994
- 1994-09-27 JP JP7510405A patent/JPH09504517A/en active Pending
- 1994-09-27 WO PCT/US1994/010898 patent/WO1995008986A1/en not_active Application Discontinuation
- 1994-09-27 EP EP94929892A patent/EP0721328A4/en not_active Withdrawn
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4721612A (en) * | 1984-04-12 | 1988-01-26 | The Liposome Company, Inc. | Steroidal liposomes |
Non-Patent Citations (5)
| Title |
|---|
| BIOCHIMICA ET BIOPHYSICA ACTA, Volume 1029, 1990, (BLUME et al.), "Liposomes for the sustained drug release in vivo". * |
| JOURNAL OF AMERICAN CHEMICAL SOCIETY, Volume 114, 1992, (BURKE et al.), "Liposomal Stabilization of Camptothecin's Lactone Ring", see pages 114-115. * |
| LIPOSOMES: FROM BIOPHYSICS TO THERAPEUTICS, (WEINSTEIN), "Liposomes in the Diagnosis and Treatment of Cancer", 1987, see pages 279, 280, 289 and 296. * |
| PROCEEDINGS OF NATIONAL ACADEMY OF SCIENCES USA, Volume 85, September 1988, (GABIZON et al.), "Liposome formulations with prolonged circulation time in blood and enhanced uptake by tumors", see page 6949, column 2, Table 2 on page 6950. * |
| See also references of EP0721328A4 * |
Cited By (70)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0785772A1 (en) * | 1994-10-14 | 1997-07-30 | Pharmacia, Inc. | Lyophilizate of lipid complex of water insoluble camptothecins |
| EP0785772A4 (en) * | 1994-10-14 | 2005-12-21 | Pharmacia & Upjohn Co Llc | Lyophilizate of lipid complex of water insoluble camptothecins |
| US6143321A (en) * | 1997-02-06 | 2000-11-07 | Duke University | Liposomes containing active agents |
| US6296870B1 (en) | 1997-02-06 | 2001-10-02 | Duke University | Liposomes containing active agents |
| KR100711315B1 (en) * | 1997-09-16 | 2007-04-27 | 오에스아이 파마슈티컬스, 인코포레이티드 | Liposomal camptothecin formulations |
| EP1037610A1 (en) * | 1997-09-16 | 2000-09-27 | NeXstar Pharmaceuticals, Inc. | Liposomal camptothecin formulations |
| EP1037610A4 (en) * | 1997-09-16 | 2004-07-07 | Osi Pharm Inc | Liposomal camptothecin formulations |
| US7901709B2 (en) | 1998-06-18 | 2011-03-08 | Duke University | Temperature-sensitive liposomal formulation |
| US6200598B1 (en) | 1998-06-18 | 2001-03-13 | Duke University | Temperature-sensitive liposomal formulation |
| WO1999065466A1 (en) * | 1998-06-18 | 1999-12-23 | Duke University | Temperature-sensitive liposomal formulation |
| US9492385B2 (en) | 1998-06-18 | 2016-11-15 | Duke University | Temperature-sensitive liposomal formulation |
| US6726925B1 (en) | 1998-06-18 | 2004-04-27 | Duke University | Temperature-sensitive liposomal formulation |
| US6355268B1 (en) | 1998-09-16 | 2002-03-12 | Alza Corporation | Liposome-entrapped topoisomerase inhibitors |
| US6465008B1 (en) | 1998-09-16 | 2002-10-15 | Alza Corporation | Liposome-entrapped topoisomerase inhibitors |
| WO2000023052A1 (en) * | 1998-09-16 | 2000-04-27 | Alza Corporation | Liposome-entrapped topoisomerase inhibitors |
| US7279179B2 (en) | 1998-09-16 | 2007-10-09 | Alza Corporation | Liposome-entrapped topoisomerase inhibitors |
| US7244449B2 (en) | 1998-09-16 | 2007-07-17 | Alza Corporation | Liposome-entrapped topoisomerase inhibitors |
| AU774715B2 (en) * | 1998-09-16 | 2004-07-08 | Alza Corporation | Liposome-entrapped topoisomerase inhibitors |
| US7311924B2 (en) | 1999-04-01 | 2007-12-25 | Hana Biosciences, Inc. | Compositions and methods for treating cancer |
| WO2002002078A3 (en) * | 2000-06-30 | 2002-12-27 | Inex Pharmaceuticals Corp | Improved liposomal camptothecins and uses thereof |
| WO2002002078A2 (en) * | 2000-06-30 | 2002-01-10 | Inex Pharmaceuticals Corporation | Improved liposomal camptothecins and uses thereof |
| US7244448B2 (en) | 2000-06-30 | 2007-07-17 | Tekmira Pharmaceuticals Corporation | Liposomal antineoplastic drugs and uses thereof |
| US7452550B2 (en) | 2000-06-30 | 2008-11-18 | Hana Biosciences, Inc. | Liposomal antineoplastic drugs and uses thereof |
| US7060828B2 (en) | 2000-06-30 | 2006-06-13 | Inex Pharmaceuticals Corporation | Liposomal camptothecins and uses thereof |
| US6828306B2 (en) | 2000-07-31 | 2004-12-07 | Ottawa Heart Institute Research Corporation | Charged lipid compositions and methods for their use |
| WO2002009678A2 (en) * | 2000-07-31 | 2002-02-07 | Ottawa Heart Institute Research Corporation | Charged phospholipid compositions and methods for their use |
| US7390783B2 (en) | 2000-07-31 | 2008-06-24 | Ottawa Heart Institute Research Corporation | Charged phospholipid compositions and methods for their use |
| WO2002009678A3 (en) * | 2000-07-31 | 2003-04-10 | Ottawa Heart Inst Res Corp | Charged phospholipid compositions and methods for their use |
| KR100869824B1 (en) * | 2000-11-09 | 2008-11-21 | 네오팜 인코포레이티드 | 38 lipid complexes and methods of use |
| US7390502B2 (en) * | 2000-11-09 | 2008-06-24 | Neopharm, Inc. | SN-38 lipid complexes and their methods of use |
| JP2004529086A (en) * | 2000-11-09 | 2004-09-24 | ネオファーム、インコーポレイティッド | SN-38 lipid complex and method of use |
| EP1355634A2 (en) * | 2000-11-09 | 2003-10-29 | Neopharm, Inc. | Sn-38 lipid complexes and methods of use |
| EP1355634A4 (en) * | 2000-11-09 | 2005-07-06 | Neopharm Inc | Sn-38 lipid complexes and methods of use |
| AU2002246510B2 (en) * | 2000-11-09 | 2007-09-20 | Neopharm, Inc. | SN-38 lipid complexes and methods of use |
| US7691872B2 (en) * | 2001-03-20 | 2010-04-06 | University Of Kentucky Research Foundation | Methods and compositions for optimizing blood and tissue stability of camptothecin and other albumin-binding therapeutic compounds |
| WO2002094269A1 (en) * | 2001-05-25 | 2002-11-28 | G.O.T. Therapeutics Gmbh | Liposomally encapsulated hydrophobic active ingredients with a high active ingredient content ⊃ 50 % and method for the production of pharmaceutical preparations containing liposomally encapsulated hydrophobic active ingredients |
| WO2003030864A1 (en) * | 2001-05-29 | 2003-04-17 | Neopharm, Inc. | Liposomal formulation of irinotecan |
| US7906139B2 (en) * | 2002-04-03 | 2011-03-15 | Lamellar Therapeutics Limited | Compositions and methods of using lamellar bodies for therapeutic purposes |
| WO2004002454A1 (en) * | 2002-06-26 | 2004-01-08 | Medigene Oncology Gmbh | Camptothecin-carboxylate formulations |
| EP1393719A1 (en) * | 2002-08-23 | 2004-03-03 | Munich Biotech AG | Camptothecin-carboxylate formulations |
| US7807350B2 (en) | 2003-05-30 | 2010-10-05 | The University Of Chicago | Methods for predicting irinotecan toxicity |
| US9750766B2 (en) | 2003-09-25 | 2017-09-05 | Lamellar Biomedical Limited | Compositions and methods of using lamellar bodies for modifying linear biological macromolecules |
| US9173901B2 (en) | 2003-09-25 | 2015-11-03 | Lamellar Therapeutics Limited | Compositions and methods of using lamellar bodies for modifying linear biological macromolecules |
| US7811602B2 (en) | 2004-05-17 | 2010-10-12 | Tekmira Pharmaceuticals Corporation | Liposomal formulations comprising dihydrosphingomyelin and methods of use thereof |
| CN100356919C (en) * | 2004-05-31 | 2007-12-26 | 上海医药工业研究院 | Hydroxycamptothecin liposome and its preparation |
| US7846473B2 (en) | 2004-06-01 | 2010-12-07 | Terumo Kabushiki Kaisha | Irinotecan preparation |
| WO2006052767A3 (en) * | 2004-11-05 | 2007-08-02 | Inex Pharmaceuticals Corp | Compositions and methods for stabilizing liposomal camptothecin formulations |
| AU2005304914B2 (en) * | 2004-11-05 | 2012-02-16 | Tekmira Pharmaceuticals Corporation | Compositions and methods for stabilizing liposomal camptothecin formulations |
| EP1676568A1 (en) * | 2004-12-30 | 2006-07-05 | Merckle Gmbh | Particles comprising phospholipids, process of preparation and use as medicament |
| WO2006072457A1 (en) * | 2004-12-30 | 2006-07-13 | Merckle Gmbh | Particles containing phospholipids, production and use thereof as medicaments |
| US8617615B2 (en) | 2005-03-24 | 2013-12-31 | Jean-Louis Dasseux | Charged lipoprotein complexes and their uses |
| US11801282B2 (en) | 2005-03-24 | 2023-10-31 | Abionyx Pharma Sa | Charged lipoprotein complexes and their uses |
| US8206750B2 (en) | 2005-03-24 | 2012-06-26 | Cerenis Therapeutics Holding S.A. | Charged lipoprotein complexes and their uses |
| US9567388B2 (en) | 2005-03-24 | 2017-02-14 | Cerenis Therapeutics Holding S.A. | Charged lipoprotein complexes and their uses |
| WO2011076807A2 (en) | 2009-12-23 | 2011-06-30 | Novartis Ag | Lipids, lipid compositions, and methods of using them |
| EP3721943A1 (en) | 2009-12-23 | 2020-10-14 | Novartis AG | Lipids, lipid compositions and methods of using them |
| US9801874B2 (en) | 2012-11-20 | 2017-10-31 | Spectrum Pharmaceuticals | Method for the preparation of liposome encapsulated vincristine for therapeutic use |
| EP3608308A1 (en) | 2013-03-08 | 2020-02-12 | Novartis AG | Lipids and lipid compositions for the delivery of active agents |
| WO2014136086A1 (en) | 2013-03-08 | 2014-09-12 | Novartis Ag | Lipids and lipid compositions for the delivery of active agents |
| WO2015095340A1 (en) | 2013-12-19 | 2015-06-25 | Novartis Ag | Lipids and lipid compositions for the delivery of active agents |
| US10059655B2 (en) | 2013-12-19 | 2018-08-28 | Novartis Ag | Lipids and lipid compositions for the delivery of active agents |
| WO2015095346A1 (en) | 2013-12-19 | 2015-06-25 | Novartis Ag | Lipids and lipid compositions for the delivery of active agents |
| US10906867B2 (en) | 2013-12-19 | 2021-02-02 | Novartis Ag | Lipids and lipid compositions for the delivery of active agents |
| EP3872066A1 (en) | 2013-12-19 | 2021-09-01 | Novartis AG | Lipids and lipid compositions for the delivery of active agents |
| EP4019506A1 (en) | 2013-12-19 | 2022-06-29 | Novartis AG | Lipids and lipid compositions for the delivery of active agents |
| US11420933B2 (en) | 2013-12-19 | 2022-08-23 | Novartis Ag | Lipids and lipid compositions for the delivery of active agents |
| US10342761B2 (en) | 2014-07-16 | 2019-07-09 | Novartis Ag | Method of encapsulating a nucleic acid in a lipid nanoparticle host |
| WO2016037053A1 (en) | 2014-09-05 | 2016-03-10 | Novartis Ag | Lipids and lipid compositions for the delivery of active agents |
| US11559486B2 (en) | 2015-07-22 | 2023-01-24 | Acrotech Biopharma, LLC | Ready-to-use formulation for Vincristine Sulfate Liposome Injection |
| WO2023096511A1 (en) * | 2021-11-23 | 2023-06-01 | Uniwersytet Warszawski | Drug lipid carrier, pharmaceutical formulation, uses thereof and method for preparation of drug lipid carrier, carrier obtained by this method and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| JPH09504517A (en) | 1997-05-06 |
| EP0721328A4 (en) | 1997-09-17 |
| EP0721328A1 (en) | 1996-07-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0721328A1 (en) | Camptothecin formulations | |
| KR100869824B1 (en) | 38 lipid complexes and methods of use | |
| Zhang et al. | Development and characterization of a novel liposome-based formulation of SN-38 | |
| Johnston et al. | Characterization of the drug retention and pharmacokinetic properties of liposomal nanoparticles containing dihydrosphingomyelin | |
| KR890004689B1 (en) | Compositions consisting of phospholipid encapsulated anthracyline anti-neoplastic agents | |
| JP4885715B2 (en) | Irinotecan formulation | |
| US20050238706A1 (en) | Pharmaceutically active lipid based formulation of SN-38 | |
| WO2010009186A1 (en) | Liposome formulation having hydrophilic and hydrophobic pharmaceutical compounds co-encapsulated therein | |
| WO2011066684A1 (en) | Liposome of irinotecan or its hydrochloride and preparation method thereof | |
| Elbialy et al. | Ehrlich tumor inhibition using doxorubicin containing liposomes | |
| CA2303366A1 (en) | Liposomal camptothecin formulations | |
| Liu et al. | Liposomes in solubilization | |
| CA2570329C (en) | Liposome preparation containing slightly water-soluble camptothecin | |
| Sugarman et al. | Lipid-complexed camptothecin: formulation and initial biodistribution and antitumor activity studies | |
| CN106109415A (en) | A kind of load camptothecin antineoplastic agents liposome, preparation method and applications | |
| Hao et al. | In-vitro cytotoxicity, in-vivo biodistribution and anti-tumour effect of PEGylated liposomal topotecan | |
| Lidgate et al. | In vitro and in vivo studies evaluating a liposome system for drug solubilization | |
| Hao et al. | In vitro and in vivo studies of different liposomes containing topotecan | |
| CA2052164A1 (en) | Lipid membrance structures for oral administration | |
| CN102670509A (en) | Lipid preparation containing insoluble camptothecin drug and preparation method thereof | |
| CN103520159B (en) | Quinine drug-vincristine drug co-carried liposome and preparation method thereof | |
| JPH06183954A (en) | Liposome pharmaceutical preparation | |
| WO2010118200A2 (en) | Liposomal formulations of tocopheryl amides | |
| Law et al. | Antitumor effect of mitoxantrone-containing liposomes | |
| JPH11171772A (en) | Liposome preparation of antitumor medicine |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): JP US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref country code: US Ref document number: 1996 619472 Date of ref document: 19960322 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1994929892 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1994929892 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1994929892 Country of ref document: EP |