WO1994003445A1 - 3-benzylamino-2-phenyl-piperidine derivatives as substance p receptor antagonists - Google Patents
3-benzylamino-2-phenyl-piperidine derivatives as substance p receptor antagonists Download PDFInfo
- Publication number
- WO1994003445A1 WO1994003445A1 PCT/US1993/005077 US9305077W WO9403445A1 WO 1994003445 A1 WO1994003445 A1 WO 1994003445A1 US 9305077 W US9305077 W US 9305077W WO 9403445 A1 WO9403445 A1 WO 9403445A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- carbon
- phenyl
- mammal
- nitrogen
- Prior art date
Links
- 0 CCCCC(C)*=CC(*C#N)=CC(C)(C)* Chemical compound CCCCC(C)*=CC(*C#N)=CC(C)(C)* 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/06—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention relates to novel substituted derivatives of nitrogen containing heterocycles, pharmaceutical compositions comprising such compounds and the use of such compounds in the treatment and prevention of inflammatory and central nervous system disorders, as well as several other disorders.
- the pharmaceutically active compounds of this invention are substance P receptor antagonists.
- Substance P is a naturally occurring undecapeptide belonging to the tachykinin family of peptides, the latter being named because of their prompt stimulatory action on smooth muscle tissue. More specifically, substance P is a pharmacologically active neuropeptide that is produced in mammals (having originally been isolated from gut) and possesses a characteristic amino acid sequence that is illustrated by D. F. Veber et al. in U.S. Patent No. 4,680,283. The wide involvement of substance P and other tachykinins in the pathophysiology of numerous diseases has been amply demonstrated in the art. For instance, substance P has recently been shown to be involved in the transmission of pain or migraine (see B.E.B. Sandberg et al., Journal of Medicinal Chemistry.
- the present invention relates to compounds of the formula
- m is an integer from 1 to 8
- any one of the carbon-carbon single bonds of (CH 2 ) m may optionally be replaced by a carbon-carbon double bond or a carbon-carbon triple bond, and any one of the carbon atoms of said (CH 2 ) m may optionally be substituted with R 11 ;
- w is an integer from zero to four;
- x is an integer from zero to four;
- y is an integer from zero to four;
- z is an integer from zero to six and wherein the ring containing (CH 2 ) z may contain from zero to three double bonds, and one of the carbons of (CH 2 ) z may optionally be replaced by oxygen, sulfur or nitrogen;
- R 1 is hydrogen or (C 1 -C 8 ) alkyl optionally substituted with hydroxy, alkoxy or fluoro;
- R 3 is aryl selected from phenyl, indanyl, and naphthyl; heteroaryl selected from benzothienyl, benzofuryl, thienyl, furyl, pyridyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, triazolyl, tetrazolyl, and quinolyl; or cycloalkyl having from three to seven carbon atoms, wherein one of said carbon atoms may optionally be replaced by nitrogen, oxygen or sulfur; wherein each of said aryl and heteroaryl groups may optionally be substituted with one or more substituents, and said (C 3 -C 7 ) cycloalkyl may optionally be substituted with one or two substituents, said substituents being independently selected from halo, nitro,
- (C 3 -C 10 ) alkyl optionally substituted with from one to three fluorine atoms
- (C 3 -C 10 ) alkoxy optionally substituted with from one to three fluorine atoms, trifluoromethyl, amino
- R 6 is a functionality selected from hydrogen, (C 1 -C 6 )straight or branched alkyl, (C 3 -C 7 )cycloalkyl wherein one of the carbon atoms may optionally be replaced by nitrogen, oxygen or sulfur; aryl selected from biphenyl, phenyl, indanyl and naphthyl; heteroaryl selected from benzothienyl, thienyl, furyl, benzofuryl, pyridyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, triazolyl, tetrazolyl and quinolyl; phenyl (C 2 -C 6 )alkyl, benzhydryl and benzyl, wherein each of said aryl and heteroaryl groups and the phenyl moieties of said benzyl, phenyl (C 2 -C 6 )alkyl and benzhydryl may optionally be substituted
- R 7 is hydrogen, phenyl or (C 1 -C 6 ) alkyl
- R 6 and R 7 together with the carbon to which they are attached, form a saturated carbocyclic ring having from 3 to 7 carbon atoms wherein one of said carbon atoms may optionally be replaced by oxygen, nitrogen or sulfur;
- R 8 may be attached to any atom of the nitrogen containing ring having an available bonding site and R 9 may be attached to any atom of the (CH 2 ) z containing ring having an available bonding site or to any carbon atom of the nitrogen containing ring having an available bonding site;
- A is selected from the group consisting of CH 2 , nitrogen, oxygen, sulfur and carbonyl; G is nitrogen, oxygen or sulfur;
- R 10 is a monocyclic or bicyclic heterocycle selected from the group consisting of pyrimidinyl, benzoxazolyl, 2,3-dihydro-3-oxobenzisosulfonazol-2-yl, morpholin-1-yl, thiomorpholin-1-yl, benzofuranyl, benzothienyl, indolyl, isoindolyl, isoquinolinyl, furyl, pyridyl, isothiazolyl, oxazolyl, triazolyl, tetrazolyl, quinolyl, thiazolyl, thienyl, and groups of the formulae and
- B and D are selected from carbon, oxygen and nitrogen, and at least one of B and D is other than carbon; E is carbon or nitrogen; n is an integer from 1 to 5; any one of the carbon atoms of said (CH 2 ) n and (CH 2 ) n+1 may be optionally substituted with (C 1 -C 6 ) alkyl or (C 2 -C 6 ) spiroalkyl; and either any one pair of the carbon atoms of said (CH 2 ) n and (CH 2 ) n+1 may be bridged by a one or two carbon atom linkage, or any one pair of adjacent carbon atoms of said (CH 2 ) n and (CH 2 ) n+1 may form, together with from one to three carbon atoms that are not members of the carbonyl containing ring, a (C 3 -C 5 ) fused carbocyclic ring;
- R 8 and R 9 is independently selected from hydrogen, fluoro, (C 1 -C 6 )alkyl, hydroxy-(C 1 -C 6 )alkyl and (C 1 -C C )alkoxy- (C 1 -C 6 ) alkyl, or R 8 and R 9 , together with the carbon to which they are attached, form a (C 3 -C 6 ) saturated carbocyclic ring that forms a spiro compound with the nitrogen containing ring to which they are attached, (d) when A is nitrogen,
- Preferred compounds of the formula I are those wherein z is zero, G is nitrogen, and R 9 is attached to the ring to which R 6 and R 7 are attached.
- Preferred compounds of the formula I are those wherein m is an integer from 4 to 6; G is nitrogen; R 3 is phenyl optionally substituted with one or two substituents, said substituents being independently selected from halo, nitro, (C 1 -C 10 ) alkyl optionally substituted with from one to three fluorine atoms, (C 1 -C 10 ) alkoxy optionally substituted with from one to three fluorine atoms, trifluoromethyl, amino,
- More preferred compounds of formula I are the foregoing compounds wherein x is zero to two, w, y and z are zero and R 8 , R 9 and R 11 are hydrogen.
- Specific preferred compounds of the formula I are: (2S,3S)-3-(2-methoxybenzyl)amino-2-phenyl-1-[4- (thiazol-2-yl)aminobutyl]piperidine; I (2S,3S)-3-(2-methoxybenzyl)amino-2-phenyl-1-[4- (pyrimidin-2-yl)aminobutyl]piperidine;
- the present invention also relates to the pharmaceutically acceptable acid addition salts of compounds of the formula I.
- the acids which are used to prepare the pharmaceutically acceptable acid addition salts of the basic compounds of this invention are those which form non-toxic acid addition salts, i.e., salts containing pharmacologically acceptable anions, such as the hydrochloride, hydrobromide, hydroiodide, nitrate, sulfate, bisulfate, phosphate, acid phosphate, acetate, lactate, citrate, acid citrate, tartrate, bitartrate, succinate, maleate, fumarate, gluconate, saccharate, benzoate, methanesulfonate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate and pamoate [i.e. , 1,1'-methylene-bis-(2-hydroxy-3- naphthoate)]saltsaltsalts
- halo as used herein, unless otherwise indicated, includes chloro, fluoro, bromo and iodo.
- alkyl as used herein, unless otherwise indicated, includes saturated monovalent hydrocarbon radicals having straight, branched or cyclic moieties or combinations thereof.
- one or more substituents includes from one to the maximum number of substituents possible based on the number of available bonding sites.
- the present invention also relates to a pharmaceutical composition for treating or preventing a condition selected from the group consisting of urinary incontinence, inflammatory diseases (e.g., arthritis, psoriasis, asthma and inflammatory bowel disease), reflux gastroesophogal disease, hypertension, anxiety, depression or dysthymic disorders, cluster headache, colitis, psychosis, pain, allergies such as eczema and rhinitis, chronic obstructive airways disease, hypersensitivity disorders such as poison ivy, vasospastic diseases such as angina, migraine and Reynaud's disease, fibrosing and collagen diseases such as scleroderma and eosinophilic fascioliasis, reflex sympathetic dystrophy such as shoulder/hand syndrome, addiction disorders such as alcoholism, stress related somatic disorders, peripheral neuropathy, neuralgia, neuropathological disorders such as Alzheimer's disease, AIDS related dementia, diabetic neuropathy and multiple sclerosis, disorders related to immune enhancement or suppression such as system
- the present invention also relates to a method of treating or preventing a condition selected from the group consisting of urinary incontinence, inflammatory diseases (e.g., arthritis, psoriasis, asthma and inflammatory bowel disease), reflux gastroesophogal disease, hypertension, anxiety, depression or dysthymic disorders, cluster headache, colitis, psychosis, pain, allergies such as eczema and rhinitis, chronic obstructive airways disease, hypersensitivity disorders such as poison ivy, vasospastic diseases such as angina, migraine and Reynaud's disease, fibrosing and collagen diseases such as scleroderma and eosinophilic fascioliasis, reflex sympathetic dystrophy such as shoulder/hand syndrome, addiction disorders such as alcoholism, stress related somatic disorders, peripheral neuropathy, neuralgia, neuropathological disorders such as Alzheimer's disease, AIDS related dementia, diabetic neuropathy and multiple sclerosis, disorders related to immune enhancement or suppression such as systemic
- the present invention also relates to a pharmaceutical composition for antagonizing the effects of substance P in a mammal, including a human, comprising a substance P antagonizing amount of a compound of the formula I, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- the present invention also relates to a method of antagonizing the effects of substance P in a mammal, including a human, comprising administering to said mammal a substance P antagonizing amount of a compound of the formula I, or a pharmaceutically acceptable salt thereof.
- the present invention also relates to a pharmaceutical composition for treating or preventing a condition selected from the group consisting of urinary incontinence, inflammatory diseases (e.g., arthritis, psoriasis, asthma and inflammatory bowel disease), anxiety, depression or dysthymic disorders, cluster headache, colitis, psychosis, pain, allergies such as eczema and rhinitis, chronic obstructive airways disease, hypersensitivity disorders such as poison ivy, vasospastic diseases such as angina, migraine and Reynaud's disease, fibrosing and collagen diseases such as scleroderma and eosinophilic fascioliasis, reflex sympathetic dystrophy such as shoulder/hand syndrome, addiction disorders such as alcoholism, stress related somatic disorders, peripheral neuropathy, neuralgia, neuropathological disorders such as Alzheimer's disease, AIDS related dementia, diabetic neuropathy and multiple sclerosis, disorders related to immune enhancement or suppression such as systemic lupus erythematosus
- the present invention also relates to a method of treating or preventing a condition selected from the group consisting of urinary incontinence, inflammatory diseases (e.g., arthritis, psoriasis, asthma and inflammatory bowel disease), anxiety, depression or dysthymic disorders, cluster headache, colitis, psychosis, pain, allergies such as eczema and rhinitis, chronic obstructive airways disease, hypersensitivity disorders such as poison ivy, vasospastic diseases such as angina, migraine and Reynaud's disease, fibrosing and collagen diseases such as scleroderma and eosinophilic fascioliasis, reflex sympathetic dystrophy such as shoulder/hand syndrome, addiction disorders such as alcoholism, stress related somatic disorders, peripheral neuropathy, neuralgia, neuropathological disorders such as Alzheimer's disease, AIDS related dementia, diabetic neuropathy and multiple sclerosis, disorders related to immune enhancement or suppression such as systemic lupus erythematosus,
- the present invention also relates to a pharmaceutical composition for treating or preventing a disorder in a mammal, including a human, the treatment or prevention of which is effected or facilitated by a decrease in substance P mediated neurotransmission, comprising an amount of a compound of the formula I, or a pharmaceutically acceptable salt thereof, effective in antagonizing the effect of substance P at its receptor site, and a pharmaceutically acceptable carrier.
- the present invention also relates to a method of treating or preventing a disorder in mammal, including a human, the treatment or prevention of which is effected or facilitated by a decrease in substance P mediated neurotransmission, comprising administering to said mammal an amount of a compound of the formula I, or a pharmaceutically acceptable salt thereof, effective in antagonizing the effect of substance P at its receptor site.
- the present invention also relates to a pharmaceutical composition for treating or preventing a disorder in a mammal, including a human, the treatment or prevention of which is effected or facilitated by a decrease in substance P mediated neurotransmission, comprising an amount of a compound of the formula I, or a pharmaceutically acceptable salt thereof, effective in treating or preventing such disorder, and a pharmaceutically acceptable carrier.
- the present invention also relates to a method of treating or preventing a disorder in mammal, including a human, the treatment or prevention of which is effected or facilitated by a decrease in substance P mediated neurotransmission, comprising administering to said mammal an amount of a compound of the formula I, or a pharmaceutically acceptable salt thereof, effective in treating or preventing such disorder.
- the compounds of the formula I have chiral centers and therefore exist in different enantiomeric forms.
- This invention relates to all optical isomers and all stereoisomers of compounds of the formula I, and mixtures thereof.
- the compounds of the formula I may be prepared as described in the following reaction schemes and discussion.
- R 1 , R 3 , R 6 , R 7 , R 8 , R 9 , R 10 , R 11 , m, w, x, y, and z in the reaction schemes and discussion that follow are defined as above.
- the compounds of formula III may be converted to compounds of the formula I having the same stereochemistry by reacting them with the appropriate compound of the formula , wherein L is halo,
- This reaction is typically carried out in the presence of a base such as triethylamine, lithium diisopropylamine, sodium methoxide, potassium hydroxide or potassium t-butoxide, in a polar solvent such as t-butanol, dimethyl formamide (DMF), methylene chloride or dichloroethane, at a temperature from about room temperature to about 150°C.
- a base such as triethylamine, lithium diisopropylamine, sodium methoxide, potassium hydroxide or potassium t-butoxide
- a polar solvent such as t-butanol, dimethyl formamide (DMF), methylene chloride or dichloroethane
- the reaction is carried out at the reflux temperature in methylene chloride in the presence of triethylamine.
- Scheme 2 illustrates an alternative method of converting compounds of formula III into compounds of the formula I having the same stereochemistry, and in which R 10 is a heteroaromatic group and A is selected from oxygen, nitrogen and sulfur, by first converting compounds of formula III into intermediates of formula II. These intermediates of formula II can then be converted into compounds of formula I.
- Preferred protecting groups for the hydroxyl, amino and thiol groups are t-butyldimethylsilyl, t-butoxycarbonyl and acetyl, respectively.
- This reaction is typically carried out in the presence of a base such as triethylamine or potassium t-butoxide, in a polar solvent such as methylene chloride, dichloroethane, tetrahydrofuran or chloroform, at a temperature from about room temperature to about 150°C.
- a base such as triethylamine or potassium t-butoxide
- a polar solvent such as methylene chloride, dichloroethane, tetrahydrofuran or chloroform
- the reaction is carried out at the reflux temperature in methylene chloride in the presence of triethylamine.
- the reaction is generally carried out for about 0.5 to about 72 hours.
- a protecting group When a protecting group is present, it is then removed from the compound of formula II.
- deprotection is accomplished by reacting the protected compound of formula II with an acid such as hydrochloric acid, trifluoroacetic acid or perchloric acid, to yield a compound of the formula II having the same stereochemistry in which the protecting group has been replaced with hydrogen.
- acid such as hydrochloric acid, trifluoroacetic acid or perchloric acid
- solvents for this reaction include polar solvents such as methylene chloride, dioxane, ether or THF, preferably dioxane.
- a t-butyldimethylsilyl ether is cleaved by similar conditions or by using tetrabutylammonium fluoride, in tetrahydrofuran (THF).
- An acetyl-protected thiol is cleaved using methanolic sodium methoxide or aqueous ammonia.
- the deprotection reaction is typically run at a temperature from about -10°C to about 50°C, preferably about 25°C, for about 0.5 to about 24 hours.
- Intermediate compounds of formula II so formed can be converted into compounds of formula I by reacting them with the appropriate monocyclic or bicyclic heterocycle of the formula R 10 -X wherein X is halo, mesylate, or tosylate and R 10 is defined as above.
- This reaction is typically carried out in the presence of a base such as triethylamine, lithium diisopropylamine, sodium methoxide, potassium hydroxide or potassium t-butoxide, in a polar solvent such as methylene chloride, t-butanol, dimethyl formamide (DMF) or dichloroethane, at a temperature from about room temperature to about 150°C.
- a base such as triethylamine, lithium diisopropylamine, sodium methoxide, potassium hydroxide or potassium t-butoxide
- a polar solvent such as methylene chloride, t-butanol, dimethyl formamide (DMF) or dichloroethane
- the reaction is
- compounds of formula II in which R 13 is amino may be converted into compounds of formula I in which R 10 is a cyclic imido group such as succinimido by treating the compound of formula II with an appropriate dicarboxylic acid, an activated derivative of a dicarboxylic acid (e.g., dihalo, mesylate or tosylate), or an anhydride.
- This reaction is typically carried out in a non-polar solvent such as xylene, hexanes, cyclohexane, ether, tetrahydrofuran or toluene at a temperature from 60°C to about the reflux temperature of the solvent.
- Scheme 3 illustrates an alternative method of converting compounds of formula III into compounds of formula I, in which A is oxygen or nitrogen, by first treating compounds of formula III with a compound of formula , wherein L' is halo, mesylate
- reaction is typically carried out in the presence of a base such as triethylamine, lithium diisopropylamine, sodium methoxide, potassium hydroxide or potassium t-butoxide, in a polar solvent such as t-butanol, dimethyl formamide (DMF), methylene chloride or dichloroethane, at a temperature from about room temperature to about 150°C.
- a base such as triethylamine, lithium diisopropylamine, sodium methoxide, potassium hydroxide or potassium t-butoxide
- a polar solvent such as t-butanol, dimethyl formamide (DMF), methylene chloride or dichloroethane
- the reaction is carried out at the reflux temperature in methylene chloride in the presence of triethylamine.
- the hydroxyl group may be protected as appropriate, preferably with the t-butyl dimethylsilyl group.
- This reaction is typically carried out in the presence of a base such as triethylamine, lithium diisopropylamine, sodium methoxide, potassium hydroxide or potassium t-butoxide, in a polar solvent such as t-butanol, dimethyl formamide (DMF), methylene chloride or dichloroethane, at a temperature from about room temperature to about 150°C.
- a base such as triethylamine, lithium diisopropylamine, sodium methoxide, potassium hydroxide or potassium t-butoxide
- a polar solvent such as t-butanol, dimethyl formamide (DMF), methylene chloride or dichloroethane
- the reaction is carried out at the reflux temperature in methylene chloride in the presence of triethylamine.
- the protecting group is t-butyldimethylsilyl
- deprotection is carried out with tetrabutylammonium fluoride in tetrahydrofuran or with an acid such as hydrochloric acid (HCl) or acetic acid in a polar solvent such as water or tetrahydrofuran, at a temperature from about 0°C to about 60°C, preferably at about room temperature.
- HCl hydrochloric acid
- acetic acid in a polar solvent such as water or tetrahydrofuran
- the free hydroxyl can then be converted into a leaving group by any of the conventional means.
- Treatment of the hydroxyl group with an agent such as methanesulfonyl chloride is preferred.
- Compounds of formula IV are converted into compounds of formula I by reacting them with the appropriate compound of the formula R 10 -A-H.
- This reaction is typically carried out in the presence of a base such as triethylamine or potassium t-butoxide, in a polar solvent such as methylene chloride, dichloroethane, tetrahydrofuran or chloroform, at a temperature from about room temperature to about 150°C.
- a base such as triethylamine or potassium t-butoxide
- a polar solvent such as methylene chloride, dichloroethane, tetrahydrofuran or chloroform
- the reaction is carried out at the reflux temperature in methylene chloride in the presence of triethylamine.
- the reaction is generally carried out for about 0.5 to about 72 hours.
- compounds of formula IV are converted into compounds of formula I by reacting them with the corresponding anion derived from treatment of R 10 -A-H with a base.
- the anion can be formed with a reagent such as sodium hydride or butyl lithium in a solvent such as tetrahydrofuran or ether.
- This reaction is typically carried out in the presence of a base such as triethylamine, lithium diisopropylamine, sodium methoxide, potassium hydroxide or potassium t-butoxide, in a polar solvent such as methylene chloride, t-butanol, dimethyl formamide (DMF) or dichloroethane, at a temperature from about room temperature to about 150°C.
- the reaction is carried out at the reflux temperature in methylene chloride in the presence of triethylamine.
- Compounds of formula III may also be converted into the corresponding compounds of the formula I by first reacting them with the appropriate compound of the formula wherein L is defined as above or is imidazole, and then reducing the resulting amide.
- This reaction is typically carried out in an inert solvent such as THF or dichloromethane at a temperature from about -20°C to about 60°C. It is preferably carried out in dichloromethane at about 0°C.
- Reduction of the resulting amide is accomplished by treatment with a reducing agent such as borane dimethylsulfide complex, lithium aluminum hydride or diisobutylaluminum hydride in an inert solvent such as ethyl ether or THF.
- the reaction temperature may range from about 0°C to about 60°C.
- the reduction is accomplished using borane dimethylsulfide complex in THF at about 60°C.
- Scheme 4 illustrates a method of preparing compounds of formula III wherein G is sulfur or oxygen, and R 1 is absent.
- Esters of formula VI are hydrolyzed to form acids of formula VI, wherein R 12 is hydrogen, by methods well known to those skilled in the art, for example, by treatment of the ester of formula VI with an acid or a base in a solvent such as water.
- the acids of formula VI, wherein R 12 is hydrogen, are oxidized to form a compound of formula V wherein G is oxygen by reacting the compound of formula VI with lead tetraacetate in an inert solvent such as cyclohexane, hexane, methylene chloride, or benzene at a temperature of 0°C to a temperature of 90°C.
- an inert solvent such as cyclohexane, hexane, methylene chloride, or benzene
- the oxidation of the compounds of formula is facilitated by the addition of copper (II) salts such as copper (II) acetate (Cu(OCOCH 3 ) 2 ) and pyridine.
- the compound of formula V wherein G is oxygen is converted to a compound of formula III wherein R 1 is absent by alkylating the compound of formula V with a compound of formula R 3 CH 2 X and a base, wherein X is a leaving group selected from halo and -SO 3 R 12 , wherein R 12 is (C 1 -C 4 )alkyl or phenyl, and R 3 is defined as above.
- the reaction of the compound of formula III with the compound of formula R 3 CH 2 X is typically carried out in a solvent such as dichloromethane, chloroform, carbon tetrachloride, ether, hexane, cyclohexane or tetrahydrofuran, preferably tetrahydrofuran, at a temperature from about 0°C to about 60°C, preferably at about 25°C.
- Suitable bases include sodium hydride, organolithium bases such as butyl lithium, alkali metal alkoxides such as potassium or sodium t-butoxide and organic bases such as triethylamine, diisopropylethylamine and hexamethyldisilazide.
- Non-nucleophilic bases such as triethylamine, diisopropylethylamine and hexamethyldisilazide are preferred because they will not react with the compound of formula II and this will not form the unwanted byproducts that result from such reaction.
- the conversion of the compound of formula V to the compound of formula III is facilitated by preforming the anion of formula V by the addition of a strong base such as sodium hydride.
- the amine of formula III can be converted to compounds of formula I by the procedures described in schemes 1 through 3 above.
- compounds of formula V can be prepared by reducing a ketone of formula VII.
- Ketones of formula VII can be reduced with lithium aluminium hydride, borane dimethylsulfide in tetrahydrofuran (THF), borane in THF and sodium borohydride titanium tetrachloride. Best results are obtained using sodium borohydride in THF.
- the reaction may be carried out at temperatures from about -78 °C to about 80°C, and are preferably carried out at about 0 °C temperature of the solvent.
- Compounds of formula V so formed may be converted to compounds of formula III as described above.
- Compounds of formula III wherein G is sulfur and R 1 is absent can be formed from compounds of formula V wherein G is sulfur.
- Compounds of formula V wherein G is sulfur may be prepared from compounds of formula VII wherein G is oxygen by reaction with phosphorus pentasulfide (P 4 S 10 ) in pyridine, followed by reduction with sodium borohydride (NaBH 4 ).
- P 4 S 10 phosphorus pentasulfide
- NaBH 4 sodium borohydride
- the temperature during the reaction with P 4 S 10 is preferably about 90°C, but can range between about 0°C to about 110°C.
- compounds of formula V wherein G is sulfur can be prepared from compounds of formula VII wherein the ketone of formula VII is reacted with Lawesson's reagent in the presence of a base followed by reduction with sodium borohydride.
- the compounds of formula V wherein G is sulfur can be converted to compounds of formula III wherein G is sulfur by reaction of the compound of formula V with a compound of the formula R 3 CH 2 X wherein X is a leaving group selected from halo and -SO 3 R 12 , R 3 is defined as above and R 12 is (C 1 -C 6 )alkyl or phenyl.
- the reaction of the compound of formula V with a compound of formula R 3 CH 2 X is typically carried out in a solvent such as dichloromethane, chloroform, carbon tetrachloride, hexane, cyclohexane or tetrahydrofuran, preferably dichloromethane at a temperature from about 0°C to about 60°C, preferably at about 25°C.
- a solvent such as dichloromethane, chloroform, carbon tetrachloride, hexane, cyclohexane or tetrahydrofuran, preferably dichloromethane at a temperature from about 0°C to about 60°C, preferably at about 25°C.
- the compound of formula III so formed is deprotected by the methods described above.
- compounds of formula V wherein G is oxygen may be converted to compounds of formula III by reaction of the compound of formula V with mesylchloride followed by reaction with a thiol of formula R 3 CH 2 SH, wherein R 3 is defined as above.
- the reaction of the compound of formula V with the compound of formula R 3 CH 2 SH is typically carried out in solvents such as dichloromethane, chloroform, carbon tetrachloride, hexane, cyclohexane or tetrahydrofuran, preferably dichloromethane at a temperature from about 0°C to about 60°C, preferably at about 25°C.
- the compounds of formula III so formed can be deprotected to form compounds of formula III by the methods described above.
- the compounds of formula III so formed may be converted to the final products of formula I by schemes 1 through 3, described above.
- pressure is not critical unless otherwise indicated. Pressures from about 0.5 atmospheres to about 5 atmospheres are generally acceptable, and ambient pressure, i.e. about 1 atmosphere, is preferred as a matter of convenience.
- novel compounds of the formula I and the pharmaceutically acceptable salts thereof are useful as substance P antagonists, i.e., they possess the ability to antagonize the effects of substance P at its receptor site in mammals, and therefore they are able to function as therapeutic agents in the treatment of the aforementioned disorders and diseases in an afflicted mammal.
- the compounds of the formula I which are basic in nature are capable of forming a wide variety of different salts with various inorganic and organic acids. Although such salts must be pharmaceutically acceptable for administration to animals, it is often desirable in practice to initially isolate a compound of the Formula I from the reaction mixture as a pharmaceutically unacceptable salt and then simply convert the latter back to the free base compound by treatment with an alkaline reagent and subsequently convert the latter free base to a pharmaceutically acceptable acid addition salt.
- the acid addition salts of the base compounds of this invention are readily prepared by treating the base compound with a substantially equivalent amount of the chosen mineral or organic acid in an aqueous solvent medium or in a suitable organic solvent, such as methanol or ethanol. Upon careful evaporation of the solvent, the desired solid salt is readily obtained.
- the compounds of formula I and their pharmaceutically acceptable salts exhibit substance P receptor-binding activity and therefore are of value in the treatment and prevention of a wide variety of clinical conditions the treatment or prevention of which are effected or facilitated by a decrease in substance P mediated neurotransmission.
- Such conditions include urinary incontinence, inflammatory diseases (e.g., arthritis, psoriasis, asthma and inflammatory bowel disease), reflux gastroesophogal disease, hypertension, anxiety, depression or dysthymic disorders, colitis, psychosis, pain, allergies such as eczema and rhinitis, chronic obstructive airways disease, hypersensitivity disorders such as poison ivy, vasospastic diseases such as angina, migraine and Reynaud's disease, fibrosing and collagen diseases such as scleroderma and eosinophilic fascioliasis, reflex sympathetic dystrophy such as shoulder/hand syndrome, addiction disorders such as alcoholism, stress related somatic disorders, peripheral neuropathy, neuralgia, neuropathological disorders such as Alzheimer's disease, AIDS related dementia, diabetic neuropathy and multiple sclerosis, disorders related to immune enhancement or suppression such as systemic lupus erythematosus, and rheumatic diseases such as fibrosit
- the compounds of the formula I and the pharmaceutically acceptable salts thereof can be administered via either the oral, parenteral or topical routes.
- these compounds are most desirably administered in dosages ranging from about 5.0 mg up to about 1500 mg per day, although variations will necessarily occur depending upon the weight and condition of the subject being treated and the particular route of administration chosen.
- a dosage level that is in the range of about 0.07 mg to about 21 mg per kg of body weight per day is most desirably employed. Variations may nevertheless occur depending upon the species of animal being treated and its individual response to said medicament, as well as on the type of pharmaceutical formulation chosen and the time period and interval at which such administration is carried out.
- dosage levels below the lower limit of the aforesaid range may be more than adequate, while in other cases still larger doses may be employed without causing any harmful side effect, provided that such larger doses are first divided into several small doses for administration throughout the day.
- the compounds of the invention may be administered alone or in combination with pharmaceutically acceptable carriers or diluents by any one of the three routes previously indicated, and such administration may be carried out in single or multiple doses.
- the novel therapeutic agents of this invention can be administered in a wide variety of different dosage forms, i.e., they may be combined with various pharmaceutically acceptable inert carriers in the form of tablets, capsules, lozenges, troches, hard candies, powders, sprays, creams, salves, suppositories, jellies, gels, pastes, lotions, ointments, aqueous suspensions, injectable solutions, elixirs, syrups, and the like.
- Such carriers include solid diluents or fillers, sterile aqueous media and various non-toxic organic solvents, etc.
- oral pharmaceutical compositions can be suitably sweetened and/or flavored.
- the therapeutically-effective compounds of this invention are present in such dosage forms at concentration levels ranging from about 5.0% to about 70% by weight.
- tablets containing various excipients such as microcrystalline cellulose, sodium citrate, calcium carbonate, dicalcium phosphate and glycine may be employed along with various disintegrants such as starch (and preferably corn, potato or tapioca starch), alginic acid and certain complex silicates, together with granulation binders like polyvinylpyrrolidone, sucrose, gelatin and acacia.
- disintegrants such as starch (and preferably corn, potato or tapioca starch), alginic acid and certain complex silicates, together with granulation binders like polyvinylpyrrolidone, sucrose, gelatin and acacia.
- lubricating agents such as magnesium stearate, sodium lauryl sulfate and talc are often very useful for tabletting purposes.
- compositions of a similar type may also be employed as fillers in gelatin capsules; preferred materials in this connection also include lactose or milk sugar as well as high molecular weight polyethylene glycols.
- preferred materials in this connection also include lactose or milk sugar as well as high molecular weight polyethylene glycols.
- the active ingredient may be combined with various sweetening or flavoring agents, coloring matter or dyes, and, if so desired, emulsifying and/or suspending agents as well, together with such diluents as water, ethanol, propylene glycol, glycerin and various like combinations thereof.
- solutions of a therapeutic compound of the present invention in either sesame or peanut oil or in aqueous propylene glycol may be employed.
- the aqueous solutions should be suitably buffered if necessary and the liquid diluent first rendered isotonic.
- These aqueous solutions are suitable for intravenous injection purposes.
- the oily solutions are suitable for intraarticular, intramuscular and subcutaneous injection purposes. The preparation of all these solutions under sterile conditions is readily accomplished by standard pharmaceutical techniques well known to those skilled in the art.
- the activity of the compounds of the present invention as substance P antagonists may be determined by their ability to inhibit the binding of substance P at its receptor sites in bovine caudate tissue, employing radioactive ligands to visualize the tachykinin receptors by means of autoradiography.
- the substance P antagonizing activity of the herein described compounds may be evaluated by using the standard assay procedure described by M. A. Cascieri et al., as reported in the Journal of Biological Chemistry. Vol. 258, p. 5158 (1983). This method essentially involves determining the concentration of the individual compound required to reduce by 50% the amount of radiolabelled substance P ligands at their receptor sites in said isolated cow tissues, thereby affording characteristic IC 50 values for each compound tested.
- bovine caudate tissue is removed from a -70°C freezer and homogenized in 50 volumes (w./v.) of an ice-cold 50 mM Tris (i.e., trimethamine which is 2-amino-2-hydroxymethyl-1,3-propanediol) hydrochloride buffer having a pH of 7.7.
- Tris i.e., trimethamine which is 2-amino-2-hydroxymethyl-1,3-propanediol
- the homogenate is centrifuged at 30,000 ⁇ G for a period of 20 minutes.
- the pellet is resuspended in 50 volumes of Tris buffer, rehomogenized and then recentrifuged at 30,000 ⁇ G for another twenty-minute period.
- the pellet is then resuspended in 40 volumes of ice-cold 50 mM Tris buffer (pH 7.7) containing 2 mM of calcium chloride, 2 mM of magnesium chloride, 40 g/ml of bacitracin, 4 ⁇ g/ml of leupeptin, 2 ⁇ g of chymostatin and 200 g/ml of bovine serum albumin. This step completes the production of the tissue preparation.
- the radioligand binding procedure is then carried out in the following manner, viz., by initiating the reaction via the addition of 100 ⁇ l of the test compound made up to a concentration of 1 ⁇ M, followed by the addition of
- the anti-psychotic activity of the compounds of the present invention as neuroleptic agents for the control of various psychotic disorders may be determined by a study of their ability to suppress substance P-induced or substance P agonist induced hypermotility in guinea pigs. This study is carried out by first dosing the guinea pigs with a control compound or with an appropriate test compound of the present invention, then injecting the guinea pigs with substance P or a substance P agonist by intracerebral administration via canula and thereafter measuring their individual locomotor response to said stimulus.
- the mixture was partitioned between chloroform and saturated aqueous sodium bicarbonate and extracted with two portions of chloroform.
- the combined chloroform extracts were dried (Na 2 SO 4 ) and concentrated.
- the crude brown oil was purified by flash column chromatography (35 g of silica gel) using 1:3 methanol/chloroform as the eluant to obtain 38 mg of product.
- This material was dissolved in ethyl acetate, and ether saturated with hydrogen chloride (HCl) was added to the solution. The solvent was removed with a pipet and the residue was subjected to high vacuum to obtain 21 mg of the title compound, mp 90-95°C.
- the crude product was purified by flash column chromotography (20 g of silica gel) using 1:19 methanol/chloroform as the eluant to obtain pure title compound as its free base. This material was dissolved in ethyl acetate, and the ether saturated with HCl was added to the solution. Filtration of the resulting suspension afforded the title compound as a hygroscopic solid, mp 69-74°C.
- the title compound was prepared in a similar manner to the compound of Example 4 by replacing cis-3-(2-methoxybenzylamino)-2-phenylpiperidine with the corresponding (2S, 3S)-enantiomer and the substituted chlorobutane with 1-bromo-4- (2 , 3-dihydro-3-oxobenzisosulfonazol-2-yl)butane: mp 120-122°C.
- the title compound was prepared in a similar manner to the compound of Example 4 by replacing the substituted chlorobutane with 4-(succinimido-1-yl)-1-methylsufonyloxybutane [prepared from 4-amino-1-butanol by sequential treatment with succinic anhydride (xylenes, acetic anhydride, reflux, 2 hours), sodium methoxide (methanol, 3 hours) and methanesulfonyl chloride (triethylamine, THF, 3h)].
- succinic anhydride xylenes, acetic anhydride, reflux, 2 hours
- sodium methoxide methanol, 3 hours
- methanesulfonyl chloride triethylamine, THF, 3h
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pain & Pain Management (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Rheumatology (AREA)
- Psychiatry (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description




































Claims










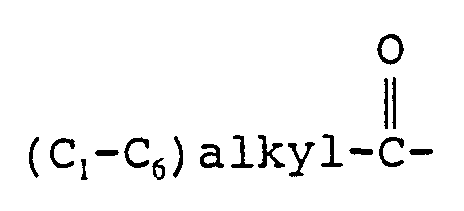















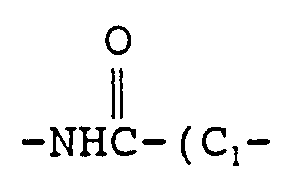
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP6505270A JPH07506379A (en) | 1992-08-04 | 1993-06-03 | 3-benzylamino-2-phenyl-piperidine as substance P receptor antagonist |
| US08/379,625 US5688804A (en) | 1992-08-04 | 1993-06-03 | 3-Benzylamino-2-phenyl-piperidine derivatives as substance P receptor antagonists |
| AU43961/93A AU4396193A (en) | 1992-08-04 | 1993-06-03 | 3-benzylamino-2-phenyl-piperidine derivatives as substance p receptor antagonists |
| EP93914220A EP0654029A1 (en) | 1992-08-04 | 1993-06-03 | 3-benzylamino-2-phenyl-piperidine derivatives as substance p receptor antagonists |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US92477392A | 1992-08-04 | 1992-08-04 | |
| US07/924,773 | 1992-08-04 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1994003445A1 true WO1994003445A1 (en) | 1994-02-17 |
Family
ID=25450702
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1993/005077 WO1994003445A1 (en) | 1992-08-04 | 1993-06-03 | 3-benzylamino-2-phenyl-piperidine derivatives as substance p receptor antagonists |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US5688804A (en) |
| EP (1) | EP0654029A1 (en) |
| JP (1) | JPH07506379A (en) |
| AU (1) | AU4396193A (en) |
| CA (1) | CA2141051A1 (en) |
| FI (1) | FI933455A (en) |
| HU (1) | HU9302246D0 (en) |
| IL (1) | IL106532A0 (en) |
| MX (1) | MX9304698A (en) |
| WO (1) | WO1994003445A1 (en) |
Cited By (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995020575A1 (en) * | 1994-01-28 | 1995-08-03 | Merck Sharp & Dohme Limited | Aralkylamino substituted azacyclic therapeutic agents |
| US5610165A (en) * | 1994-02-17 | 1997-03-11 | Merck & Co., Inc. | N-acylpiperidine tachykinin antagonists |
| WO2000047562A1 (en) * | 1999-02-09 | 2000-08-17 | Merck Sharp & Dohme Limited | Spirocyclic ketones and their use as tachykinin antagonists |
| WO2006123182A2 (en) | 2005-05-17 | 2006-11-23 | Merck Sharp & Dohme Limited | Cyclohexyl sulphones for treatment of cancer |
| WO2007011820A2 (en) | 2005-07-15 | 2007-01-25 | Amr Technology, Inc. | Aryl-and heteroaryl-substituted tetrahydrobenzazepines and use thereof to block reuptake of norepinephrine, dopamine, and serotonin |
| WO2007041052A2 (en) | 2005-09-29 | 2007-04-12 | Merck & Co., Inc. | Acylated spiropiperidine derivatives as melanocortin-4 receptor modulators |
| WO2007093827A1 (en) | 2006-02-15 | 2007-08-23 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Thiophene and thiazole substituted trifluoroethanone derivatives as histone deacetylase (hdac) inhibitors |
| WO2008120653A1 (en) | 2007-04-02 | 2008-10-09 | Banyu Pharmaceutical Co., Ltd. | Indoledione derivative |
| WO2009002495A1 (en) | 2007-06-27 | 2008-12-31 | Merck & Co., Inc. | 4-carboxybenzylamino derivatives as histone deacetylase inhibitors |
| WO2009111354A2 (en) | 2008-03-03 | 2009-09-11 | Tiger Pharmatech | Tyrosine kinase inhibitors |
| WO2010114780A1 (en) | 2009-04-01 | 2010-10-07 | Merck Sharp & Dohme Corp. | Inhibitors of akt activity |
| WO2010132487A1 (en) | 2009-05-12 | 2010-11-18 | Bristol-Myers Squibb Company | CRYSTALLINE FORMS OF (S)-7-([1,2,4]TRIAZOLO[1,5-a]PYRIDIN-6-YL)-4-(3,4-DICHLOROHPHENYL)-1,2,3,4-TETRAHYDROISOQUINOLINE AND USE THEREOF |
| WO2010132442A1 (en) | 2009-05-12 | 2010-11-18 | Albany Molecular Reserch, Inc. | 7-([1,2,4,]triazolo[1,5,-a]pyridin-6-yl)-4-(3,4-dichlorophenyl)-1,2,3,4- tetrahydroisoquinoline and use thereof |
| WO2011046771A1 (en) | 2009-10-14 | 2011-04-21 | Schering Corporation | SUBSTITUTED PIPERIDINES THAT INCREASE p53 ACTIVITY AND THE USES THEREOF |
| EP2336120A1 (en) | 2007-01-10 | 2011-06-22 | Istituto di ricerche di Biologia Molecolare P. Angeletti S.R.L. | Combinations containing amide substituted indazoles as poly(ADP-ribose)polymerase (PARP) inhibitors |
| WO2011163330A1 (en) | 2010-06-24 | 2011-12-29 | Merck Sharp & Dohme Corp. | Novel heterocyclic compounds as erk inhibitors |
| WO2012018754A2 (en) | 2010-08-02 | 2012-02-09 | Merck Sharp & Dohme Corp. | RNA INTERFERENCE MEDIATED INHIBITION OF CATENIN (CADHERIN-ASSOCIATED PROTEIN), BETA 1 (CTNNB1) GENE EXPRESSION USING SHORT INTERFERING NUCLEIC ACID (siNA) |
| WO2012027236A1 (en) | 2010-08-23 | 2012-03-01 | Schering Corporation | NOVEL PYRAZOLO[1,5-a]PYRIMIDINE DERIVATIVES AS mTOR INHIBITORS |
| WO2012030685A2 (en) | 2010-09-01 | 2012-03-08 | Schering Corporation | Indazole derivatives useful as erk inhibitors |
| WO2012036997A1 (en) | 2010-09-16 | 2012-03-22 | Schering Corporation | Fused pyrazole derivatives as novel erk inhibitors |
| WO2012087772A1 (en) | 2010-12-21 | 2012-06-28 | Schering Corporation | Indazole derivatives useful as erk inhibitors |
| WO2012145471A1 (en) | 2011-04-21 | 2012-10-26 | Merck Sharp & Dohme Corp. | Insulin-like growth factor-1 receptor inhibitors |
| WO2013063214A1 (en) | 2011-10-27 | 2013-05-02 | Merck Sharp & Dohme Corp. | Novel compounds that are erk inhibitors |
| WO2013165816A2 (en) | 2012-05-02 | 2013-11-07 | Merck Sharp & Dohme Corp. | SHORT INTERFERING NUCLEIC ACID (siNA) COMPOSITIONS |
| EP2698157A1 (en) | 2006-09-22 | 2014-02-19 | Merck Sharp & Dohme Corp. | Method of treatment using fatty acid synthesis inhibitors |
| WO2014052563A2 (en) | 2012-09-28 | 2014-04-03 | Merck Sharp & Dohme Corp. | Novel compounds that are erk inhibitors |
| WO2014085216A1 (en) | 2012-11-28 | 2014-06-05 | Merck Sharp & Dohme Corp. | Compositions and methods for treating cancer |
| WO2014100065A1 (en) | 2012-12-20 | 2014-06-26 | Merck Sharp & Dohme Corp. | Substituted imidazopyridines as hdm2 inhibitors |
| WO2014120748A1 (en) | 2013-01-30 | 2014-08-07 | Merck Sharp & Dohme Corp. | 2,6,7,8 substituted purines as hdm2 inhibitors |
| WO2015034925A1 (en) | 2013-09-03 | 2015-03-12 | Moderna Therapeutics, Inc. | Circular polynucleotides |
| US9034899B2 (en) | 2009-05-12 | 2015-05-19 | Albany Molecular Research, Inc. | Aryl, heteroaryl, and heterocycle substituted tetrahydroisoquinolines and use thereof |
| US9085531B2 (en) | 2004-07-15 | 2015-07-21 | Albany Molecular Research, Inc. | Aryl- and heteroaryl-substituted tetrahydroisoquinolines and use thereof to block reuptake of norepinephrine, dopamine, and serotonin |
| US9156812B2 (en) | 2008-06-04 | 2015-10-13 | Bristol-Myers Squibb Company | Crystalline form of 6-[(4S)-2-methyl-4-(2-naphthyl)-1,2,3,4-tetrahydroisoquinolin-7-yl]pyridazin-3-amine |
| WO2018071283A1 (en) | 2016-10-12 | 2018-04-19 | Merck Sharp & Dohme Corp. | Kdm5 inhibitors |
| EP3327125A1 (en) | 2010-10-29 | 2018-05-30 | Sirna Therapeutics, Inc. | Rna interference mediated inhibition of gene expression using short interfering nucleic acids (sina) |
| US10154988B2 (en) | 2012-11-14 | 2018-12-18 | The Johns Hopkins University | Methods and compositions for treating schizophrenia |
| WO2019094311A1 (en) | 2017-11-08 | 2019-05-16 | Merck Sharp & Dohme Corp. | Prmt5 inhibitors |
| WO2020033284A1 (en) | 2018-08-07 | 2020-02-13 | Merck Sharp & Dohme Corp. | Prmt5 inhibitors |
| WO2020033282A1 (en) | 2018-08-07 | 2020-02-13 | Merck Sharp & Dohme Corp. | Prmt5 inhibitors |
| US11096950B2 (en) | 2006-11-01 | 2021-08-24 | Barbara Brooke Jennings | Compounds, methods, and treatments for abnormal signaling pathways for prenatal and postnatal development |
| EP4079856A1 (en) | 2010-08-17 | 2022-10-26 | Sirna Therapeutics, Inc. | Rna interference mediated inhibition of hepatitis b virus (hbv) gene expression using short interfering nucleic acid (sina) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| UA39168C2 (en) * | 1991-06-20 | 2001-06-15 | Пфайзер, Інк. | Fluoroalkoxyphenyl derivatives of pyperidine or quinuclidine AS antagonists of P substance and pharmaceutical composition based thereon |
| US5988870A (en) * | 1998-03-02 | 1999-11-23 | Partsky; Howard | Apparatus and method for diluting nasal sprays containing addictive compounds |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0436334A2 (en) * | 1990-01-04 | 1991-07-10 | Pfizer Inc. | 3-Aminopiperidine derivatives and related nitrogen containing heterocycles |
| WO1992006079A1 (en) * | 1990-09-28 | 1992-04-16 | Pfizer Inc. | Fused ring analogs of nitrogen containing nonaromatic heterocycles |
| WO1993000331A1 (en) * | 1991-06-20 | 1993-01-07 | Pfizer Inc. | Fluoroalkoxybenzylamino derivatives of nitrogen containing heterocycles |
| WO1993000330A2 (en) * | 1991-06-21 | 1993-01-07 | Pfizer Inc. | Azanorbornane derivatives |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3560510A (en) * | 1969-03-05 | 1971-02-02 | Aldrich Chem Co Inc | 2-benzhydrylquinuclidines |
| CA1160229A (en) * | 1979-03-13 | 1984-01-10 | Pieter T. Haken | Pyridyliminomethylbenzene derivatives |
| CA1231710A (en) * | 1979-07-19 | 1988-01-19 | Haken Pieter Ten | Heterocyclic compounds having fungicidal, herbicidal and plant-growth regulating properties |
| EP0100158A3 (en) * | 1982-07-28 | 1985-03-27 | The Upjohn Company | (3-pyridinyl)heteroalkarylalkanols, alkanoic acids and esters |
| US4552960A (en) * | 1983-06-20 | 1985-11-12 | Eli Lilly And Company | Fungicidal amines |
| US4680283A (en) * | 1984-09-26 | 1987-07-14 | Merck & Co., Inc. | Analogs of substance P and eledoisin |
| US5232929A (en) * | 1990-11-28 | 1993-08-03 | Pfizer Inc. | 3-aminopiperidine derivatives and related nitrogen containing heterocycles and pharmaceutical compositions and use |
| US5364943A (en) * | 1991-11-27 | 1994-11-15 | Pfizer Inc. | Preparation of substituted piperidines |
| CA2086434C (en) * | 1990-07-23 | 1998-09-22 | John A. Lowe, Iii | Quinuclidine derivatives |
| US5138060A (en) * | 1991-01-03 | 1992-08-11 | Pfizer Inc. | Process and intermediates for preparing azabicyclo(2.2.2)octan-3-imines |
| AU652407B2 (en) * | 1991-01-10 | 1994-08-25 | Pfizer Inc. | N-alkyl quinuclidinium salts as substance P antagonists |
| DE69200921T2 (en) * | 1991-03-01 | 1995-05-04 | Pfizer | 1-AZABICYCLO [3.2.2] NONAN-3-AMINE DERIVATIVES. |
| BR9205807A (en) * | 1991-03-26 | 1994-06-28 | Pfizer | Stereo-selective preparation of substituted piperidines |
-
1993
- 1993-06-03 AU AU43961/93A patent/AU4396193A/en not_active Abandoned
- 1993-06-03 CA CA002141051A patent/CA2141051A1/en not_active Abandoned
- 1993-06-03 WO PCT/US1993/005077 patent/WO1994003445A1/en not_active Application Discontinuation
- 1993-06-03 US US08/379,625 patent/US5688804A/en not_active Expired - Fee Related
- 1993-06-03 JP JP6505270A patent/JPH07506379A/en active Pending
- 1993-06-03 EP EP93914220A patent/EP0654029A1/en not_active Withdrawn
- 1993-07-29 IL IL106532A patent/IL106532A0/en unknown
- 1993-08-03 HU HU939302246A patent/HU9302246D0/en unknown
- 1993-08-03 MX MX9304698A patent/MX9304698A/en unknown
- 1993-08-03 FI FI933455A patent/FI933455A/en unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0436334A2 (en) * | 1990-01-04 | 1991-07-10 | Pfizer Inc. | 3-Aminopiperidine derivatives and related nitrogen containing heterocycles |
| WO1992006079A1 (en) * | 1990-09-28 | 1992-04-16 | Pfizer Inc. | Fused ring analogs of nitrogen containing nonaromatic heterocycles |
| WO1993000331A1 (en) * | 1991-06-20 | 1993-01-07 | Pfizer Inc. | Fluoroalkoxybenzylamino derivatives of nitrogen containing heterocycles |
| WO1993000330A2 (en) * | 1991-06-21 | 1993-01-07 | Pfizer Inc. | Azanorbornane derivatives |
Cited By (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995020575A1 (en) * | 1994-01-28 | 1995-08-03 | Merck Sharp & Dohme Limited | Aralkylamino substituted azacyclic therapeutic agents |
| US5728716A (en) * | 1994-01-28 | 1998-03-17 | Merck Sharp & Dohme Limited | Aralkylamino substituted azacyclic therapeutic agents |
| US5610165A (en) * | 1994-02-17 | 1997-03-11 | Merck & Co., Inc. | N-acylpiperidine tachykinin antagonists |
| WO2000047562A1 (en) * | 1999-02-09 | 2000-08-17 | Merck Sharp & Dohme Limited | Spirocyclic ketones and their use as tachykinin antagonists |
| US6372754B1 (en) | 1999-02-09 | 2002-04-16 | Merck Sharp & Dohme Ltd. | Spirocyclic ketones and their use as tachykinin antagonists |
| US9085531B2 (en) | 2004-07-15 | 2015-07-21 | Albany Molecular Research, Inc. | Aryl- and heteroaryl-substituted tetrahydroisoquinolines and use thereof to block reuptake of norepinephrine, dopamine, and serotonin |
| US9499531B2 (en) | 2004-07-15 | 2016-11-22 | Albany Molecular Research, Inc. | Aryl- and heteroaryl-substituted tetrahydroisoquinolines and use thereof to block reuptake of norepinephrine, dopamine, and serotonin |
| WO2006123182A2 (en) | 2005-05-17 | 2006-11-23 | Merck Sharp & Dohme Limited | Cyclohexyl sulphones for treatment of cancer |
| WO2007011820A2 (en) | 2005-07-15 | 2007-01-25 | Amr Technology, Inc. | Aryl-and heteroaryl-substituted tetrahydrobenzazepines and use thereof to block reuptake of norepinephrine, dopamine, and serotonin |
| US9403776B2 (en) | 2005-07-15 | 2016-08-02 | Albany Molecular Research, Inc. | Aryl- and heteroaryl-substituted tetrahydrobenzazepines and use thereof to block reuptake of norepinephrine, dopamine, and serotonin |
| WO2007041052A2 (en) | 2005-09-29 | 2007-04-12 | Merck & Co., Inc. | Acylated spiropiperidine derivatives as melanocortin-4 receptor modulators |
| WO2007093827A1 (en) | 2006-02-15 | 2007-08-23 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Thiophene and thiazole substituted trifluoroethanone derivatives as histone deacetylase (hdac) inhibitors |
| EP2698157A1 (en) | 2006-09-22 | 2014-02-19 | Merck Sharp & Dohme Corp. | Method of treatment using fatty acid synthesis inhibitors |
| EP2946778A1 (en) | 2006-09-22 | 2015-11-25 | Merck Sharp & Dohme Corp. | Method of treatment using fatty acid synthesis inhibitors |
| US11096950B2 (en) | 2006-11-01 | 2021-08-24 | Barbara Brooke Jennings | Compounds, methods, and treatments for abnormal signaling pathways for prenatal and postnatal development |
| EP2805945A1 (en) | 2007-01-10 | 2014-11-26 | MSD Italia S.r.l. | Amide substituted indazoles as poly(ADP-ribose)polymerase (PARP) inhibitors |
| EP2336120A1 (en) | 2007-01-10 | 2011-06-22 | Istituto di ricerche di Biologia Molecolare P. Angeletti S.R.L. | Combinations containing amide substituted indazoles as poly(ADP-ribose)polymerase (PARP) inhibitors |
| WO2008120653A1 (en) | 2007-04-02 | 2008-10-09 | Banyu Pharmaceutical Co., Ltd. | Indoledione derivative |
| EP3103791A1 (en) | 2007-06-27 | 2016-12-14 | Merck Sharp & Dohme Corp. | 4-carboxybenzylamino derivatives as histone deacetylase inhibitors |
| WO2009002495A1 (en) | 2007-06-27 | 2008-12-31 | Merck & Co., Inc. | 4-carboxybenzylamino derivatives as histone deacetylase inhibitors |
| WO2009111354A2 (en) | 2008-03-03 | 2009-09-11 | Tiger Pharmatech | Tyrosine kinase inhibitors |
| US9498476B2 (en) | 2008-06-04 | 2016-11-22 | Albany Molecular Research, Inc. | Crystalline form of 6-[(4S)-2-methyl-4-(2-naphthyl)-1,2,3,4-tetrahydroisoquinolin-7-yl]pyridazin-3-amine |
| US9156812B2 (en) | 2008-06-04 | 2015-10-13 | Bristol-Myers Squibb Company | Crystalline form of 6-[(4S)-2-methyl-4-(2-naphthyl)-1,2,3,4-tetrahydroisoquinolin-7-yl]pyridazin-3-amine |
| WO2010114780A1 (en) | 2009-04-01 | 2010-10-07 | Merck Sharp & Dohme Corp. | Inhibitors of akt activity |
| US9604960B2 (en) | 2009-05-12 | 2017-03-28 | Albany Molecular Research, Inc. | Aryl, heteroaryl, and heterocycle substituted tetrahydroisoquinolines and use thereof |
| WO2010132487A1 (en) | 2009-05-12 | 2010-11-18 | Bristol-Myers Squibb Company | CRYSTALLINE FORMS OF (S)-7-([1,2,4]TRIAZOLO[1,5-a]PYRIDIN-6-YL)-4-(3,4-DICHLOROHPHENYL)-1,2,3,4-TETRAHYDROISOQUINOLINE AND USE THEREOF |
| WO2010132442A1 (en) | 2009-05-12 | 2010-11-18 | Albany Molecular Reserch, Inc. | 7-([1,2,4,]triazolo[1,5,-a]pyridin-6-yl)-4-(3,4-dichlorophenyl)-1,2,3,4- tetrahydroisoquinoline and use thereof |
| US9173879B2 (en) | 2009-05-12 | 2015-11-03 | Bristol-Myers Squibb Company | Crystalline forms of (S)-7-([1,2,4]triazolo[1,5-a ]pyridin-6-yl)-4-(3,4-dichlorophenyl)-1,2,3,4-tetrahydroisoquinoline and use thereof |
| US9034899B2 (en) | 2009-05-12 | 2015-05-19 | Albany Molecular Research, Inc. | Aryl, heteroaryl, and heterocycle substituted tetrahydroisoquinolines and use thereof |
| WO2011046771A1 (en) | 2009-10-14 | 2011-04-21 | Schering Corporation | SUBSTITUTED PIPERIDINES THAT INCREASE p53 ACTIVITY AND THE USES THEREOF |
| WO2011163330A1 (en) | 2010-06-24 | 2011-12-29 | Merck Sharp & Dohme Corp. | Novel heterocyclic compounds as erk inhibitors |
| EP3330377A1 (en) | 2010-08-02 | 2018-06-06 | Sirna Therapeutics, Inc. | Rna interference mediated inhibition of catenin (cadherin-associated protein), beta 1 (ctnnb1) gene expression using short interfering nucleic acid (sina) |
| WO2012018754A2 (en) | 2010-08-02 | 2012-02-09 | Merck Sharp & Dohme Corp. | RNA INTERFERENCE MEDIATED INHIBITION OF CATENIN (CADHERIN-ASSOCIATED PROTEIN), BETA 1 (CTNNB1) GENE EXPRESSION USING SHORT INTERFERING NUCLEIC ACID (siNA) |
| EP4079856A1 (en) | 2010-08-17 | 2022-10-26 | Sirna Therapeutics, Inc. | Rna interference mediated inhibition of hepatitis b virus (hbv) gene expression using short interfering nucleic acid (sina) |
| WO2012027236A1 (en) | 2010-08-23 | 2012-03-01 | Schering Corporation | NOVEL PYRAZOLO[1,5-a]PYRIMIDINE DERIVATIVES AS mTOR INHIBITORS |
| WO2012030685A2 (en) | 2010-09-01 | 2012-03-08 | Schering Corporation | Indazole derivatives useful as erk inhibitors |
| WO2012036997A1 (en) | 2010-09-16 | 2012-03-22 | Schering Corporation | Fused pyrazole derivatives as novel erk inhibitors |
| EP3766975A1 (en) | 2010-10-29 | 2021-01-20 | Sirna Therapeutics, Inc. | Rna interference mediated inhibition of gene expression using short interfering nucleic acid (sina) |
| EP3327125A1 (en) | 2010-10-29 | 2018-05-30 | Sirna Therapeutics, Inc. | Rna interference mediated inhibition of gene expression using short interfering nucleic acids (sina) |
| WO2012087772A1 (en) | 2010-12-21 | 2012-06-28 | Schering Corporation | Indazole derivatives useful as erk inhibitors |
| WO2012145471A1 (en) | 2011-04-21 | 2012-10-26 | Merck Sharp & Dohme Corp. | Insulin-like growth factor-1 receptor inhibitors |
| WO2013063214A1 (en) | 2011-10-27 | 2013-05-02 | Merck Sharp & Dohme Corp. | Novel compounds that are erk inhibitors |
| EP3919620A1 (en) | 2012-05-02 | 2021-12-08 | Sirna Therapeutics, Inc. | Short interfering nucleic acid (sina) compositions |
| WO2013165816A2 (en) | 2012-05-02 | 2013-11-07 | Merck Sharp & Dohme Corp. | SHORT INTERFERING NUCLEIC ACID (siNA) COMPOSITIONS |
| WO2014052563A2 (en) | 2012-09-28 | 2014-04-03 | Merck Sharp & Dohme Corp. | Novel compounds that are erk inhibitors |
| US10154988B2 (en) | 2012-11-14 | 2018-12-18 | The Johns Hopkins University | Methods and compositions for treating schizophrenia |
| EP3610890A1 (en) | 2012-11-14 | 2020-02-19 | The Johns Hopkins University | Methods and compositions for treating schizophrenia |
| US10624875B2 (en) | 2012-11-14 | 2020-04-21 | The Johns Hopkins University | Methods and compositions for treating schizophrenia |
| WO2014085216A1 (en) | 2012-11-28 | 2014-06-05 | Merck Sharp & Dohme Corp. | Compositions and methods for treating cancer |
| WO2014100065A1 (en) | 2012-12-20 | 2014-06-26 | Merck Sharp & Dohme Corp. | Substituted imidazopyridines as hdm2 inhibitors |
| WO2014120748A1 (en) | 2013-01-30 | 2014-08-07 | Merck Sharp & Dohme Corp. | 2,6,7,8 substituted purines as hdm2 inhibitors |
| WO2015034925A1 (en) | 2013-09-03 | 2015-03-12 | Moderna Therapeutics, Inc. | Circular polynucleotides |
| WO2018071283A1 (en) | 2016-10-12 | 2018-04-19 | Merck Sharp & Dohme Corp. | Kdm5 inhibitors |
| WO2019094311A1 (en) | 2017-11-08 | 2019-05-16 | Merck Sharp & Dohme Corp. | Prmt5 inhibitors |
| WO2020033284A1 (en) | 2018-08-07 | 2020-02-13 | Merck Sharp & Dohme Corp. | Prmt5 inhibitors |
| WO2020033282A1 (en) | 2018-08-07 | 2020-02-13 | Merck Sharp & Dohme Corp. | Prmt5 inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| MX9304698A (en) | 1994-02-28 |
| US5688804A (en) | 1997-11-18 |
| CA2141051A1 (en) | 1994-02-17 |
| FI933455A (en) | 1994-02-05 |
| EP0654029A1 (en) | 1995-05-24 |
| IL106532A0 (en) | 1993-11-15 |
| JPH07506379A (en) | 1995-07-13 |
| HU9302246D0 (en) | 1993-10-28 |
| FI933455A0 (en) | 1993-08-03 |
| AU4396193A (en) | 1994-03-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US5688804A (en) | 3-Benzylamino-2-phenyl-piperidine derivatives as substance P receptor antagonists | |
| EP0594636B1 (en) | 3-aminopiperidine derivatives and related nitrogen containing heterocycles | |
| US6222038B1 (en) | Quinuclidine derivatives | |
| US5688806A (en) | Spiroazacyclic derivatives as substance P antagonists | |
| JP2535134B2 (en) | Fused tricyclic nitrogen-containing heterocycle | |
| CA2149242C (en) | Quinuclidine derivative for treatment of inflammatory and gastrointestinal disorders | |
| EP0436334B1 (en) | 3-Aminopiperidine derivatives and related nitrogen containing heterocycles | |
| EP0589924B1 (en) | Fluoroalkoxybenzylamino derivatives of nitrogen containing heterocycles | |
| US5703065A (en) | Heteroarylamino and heteroarylsulfonamido substituted 3-benyzlaminomethyl piperidines and related compounds | |
| CZ247993A3 (en) | Substituted 3-aminoquinuclidine derivatives, process of their preparation and use thereof | |
| US5716965A (en) | Substituted 3-aminoquinuclidines |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU CA JP KR NO NZ US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1993914220 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2141051 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 08379625 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 1993914220 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1993914220 Country of ref document: EP |