WO1983003606A1 - Improved melaminylthioarsenites - Google Patents
Improved melaminylthioarsenites Download PDFInfo
- Publication number
- WO1983003606A1 WO1983003606A1 PCT/US1983/000505 US8300505W WO8303606A1 WO 1983003606 A1 WO1983003606 A1 WO 1983003606A1 US 8300505 W US8300505 W US 8300505W WO 8303606 A1 WO8303606 A1 WO 8303606A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- pharmaceutically acceptable
- treatment
- trypanocidally
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 claims abstract description 40
- 239000002253 acid Substances 0.000 claims abstract description 17
- 208000015181 infectious disease Diseases 0.000 claims abstract description 12
- 150000003839 salts Chemical class 0.000 claims abstract description 12
- 238000000034 method Methods 0.000 claims description 10
- YDKGKLBKFOIXPU-UHFFFAOYSA-N 2-n-(4-arsorosophenyl)-1,3,5-triazine-2,4,6-triamine Chemical compound NC1=NC(N)=NC(NC=2C=CC(=CC=2)[As]=O)=N1 YDKGKLBKFOIXPU-UHFFFAOYSA-N 0.000 claims description 5
- 239000000203 mixture Substances 0.000 claims description 5
- 239000007788 liquid Substances 0.000 claims description 3
- 241000124008 Mammalia Species 0.000 claims 12
- 241000239183 Filaria Species 0.000 claims 6
- 239000004480 active ingredient Substances 0.000 claims 6
- 239000003937 drug carrier Substances 0.000 claims 6
- 239000008194 pharmaceutical composition Substances 0.000 claims 5
- 238000007792 addition Methods 0.000 abstract description 7
- 230000000654 trypanocidal effect Effects 0.000 abstract description 5
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 35
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 25
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 24
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 18
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 18
- 239000000243 solution Substances 0.000 description 17
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 14
- 238000009835 boiling Methods 0.000 description 9
- 0 C*c(cc1)ccc1N(*)C(N)NC(N)N[C@](C)N1C#C1 Chemical compound C*c(cc1)ccc1N(*)C(N)NC(N)N[C@](C)N1C#C1 0.000 description 8
- 238000006243 chemical reaction Methods 0.000 description 8
- 238000001816 cooling Methods 0.000 description 8
- 239000000843 powder Substances 0.000 description 8
- 150000003573 thiols Chemical class 0.000 description 8
- 239000000047 product Substances 0.000 description 7
- 238000012360 testing method Methods 0.000 description 7
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 6
- 241000699670 Mus sp. Species 0.000 description 6
- 238000010908 decantation Methods 0.000 description 5
- 230000004083 survival effect Effects 0.000 description 5
- 208000000230 African Trypanosomiasis Diseases 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 239000002244 precipitate Substances 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- XDTMQSROBMDMFD-UHFFFAOYSA-N C1CCCCC1 Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 3
- 241000243985 Onchocerca volvulus Species 0.000 description 3
- 239000007795 chemical reaction product Substances 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 230000034994 death Effects 0.000 description 3
- 231100000517 death Toxicity 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 239000006228 supernatant Substances 0.000 description 3
- 231100000331 toxic Toxicity 0.000 description 3
- 230000002588 toxic effect Effects 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 2
- QXNVGIXVLWOKEQ-UHFFFAOYSA-N Disodium Chemical class [Na][Na] QXNVGIXVLWOKEQ-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Chemical compound OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 241001442397 Trypanosoma brucei rhodesiense Species 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 125000002091 cationic group Chemical group 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 239000003814 drug Substances 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 229940093499 ethyl acetate Drugs 0.000 description 2
- 235000019439 ethyl acetate Nutrition 0.000 description 2
- 239000012458 free base Substances 0.000 description 2
- 229910052500 inorganic mineral Inorganic materials 0.000 description 2
- 230000001638 macrofilaricidal effect Effects 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 239000011707 mineral Substances 0.000 description 2
- 208000002042 onchocerciasis Diseases 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 231100001274 therapeutic index Toxicity 0.000 description 2
- 238000001665 trituration Methods 0.000 description 2
- 201000002311 trypanosomiasis Diseases 0.000 description 2
- FMCAFXHLMUOIGG-JTJHWIPRSA-N (2s)-2-[[(2r)-2-[[(2s)-2-[[(2r)-2-formamido-3-sulfanylpropanoyl]amino]-3-methylbutanoyl]amino]-3-(4-hydroxy-2,5-dimethylphenyl)propanoyl]amino]-4-methylsulfanylbutanoic acid Chemical compound O=CN[C@@H](CS)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(=O)N[C@@H](CCSC)C(O)=O)CC1=CC(C)=C(O)C=C1C FMCAFXHLMUOIGG-JTJHWIPRSA-N 0.000 description 1
- FMCAFXHLMUOIGG-IWFBPKFRSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2r)-2-formamido-3-sulfanylpropanoyl]amino]-3-methylbutanoyl]amino]-3-(4-hydroxy-2,5-dimethylphenyl)propanoyl]amino]-4-methylsulfanylbutanoic acid Chemical compound O=CN[C@@H](CS)C(=O)N[C@@H](C(C)C)C(=O)N[C@H](C(=O)N[C@@H](CCSC)C(O)=O)CC1=CC(C)=C(O)C=C1C FMCAFXHLMUOIGG-IWFBPKFRSA-N 0.000 description 1
- DXBSRDIXJYTKNV-SEPHDYHBSA-N 4-[(e)-2-(4-carbamimidoylphenyl)ethenyl]benzenecarboximidamide;2-hydroxyethanesulfonic acid Chemical compound OCCS(O)(=O)=O.OCCS(O)(=O)=O.C1=CC(C(=N)N)=CC=C1\C=C\C1=CC=C(C(N)=N)C=C1 DXBSRDIXJYTKNV-SEPHDYHBSA-N 0.000 description 1
- UTTWZSKBJCYOBM-UHFFFAOYSA-N Cc(cc1)ccc1[AsH2] Chemical compound Cc(cc1)ccc1[AsH2] UTTWZSKBJCYOBM-UHFFFAOYSA-N 0.000 description 1
- 201000006353 Filariasis Diseases 0.000 description 1
- 241000699694 Gerbillinae Species 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 241000243994 Litomosoides carinii Species 0.000 description 1
- HPZAAXYRUZVFPA-UHFFFAOYSA-N NC1NC(Nc(cc2)ccc2[AsH2])NC(N)N1 Chemical compound NC1NC(Nc(cc2)ccc2[AsH2])NC(N)N1 HPZAAXYRUZVFPA-UHFFFAOYSA-N 0.000 description 1
- 208000030852 Parasitic disease Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 241000224017 Plasmodium berghei Species 0.000 description 1
- 206010039509 Scab Diseases 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 241001442399 Trypanosoma brucei gambiense Species 0.000 description 1
- 229960000583 acetic acid Drugs 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 229960005475 antiinfective agent Drugs 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000007810 chemical reaction solvent Substances 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 239000000356 contaminant Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 102000038379 digestive enzymes Human genes 0.000 description 1
- 108091007734 digestive enzymes Proteins 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 230000000641 filaricidal effect Effects 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 208000029080 human African trypanosomiasis Diseases 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 238000011081 inoculation Methods 0.000 description 1
- 239000002054 inoculum Substances 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 244000045947 parasite Species 0.000 description 1
- 230000003071 parasitic effect Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 238000001226 reprecipitation Methods 0.000 description 1
- 201000002612 sleeping sickness Diseases 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 239000008174 sterile solution Substances 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 125000004014 thioethyl group Chemical group [H]SC([H])([H])C([H])([H])* 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/66—Arsenic compounds
- C07F9/70—Organo-arsenic compounds
- C07F9/80—Heterocyclic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/10—Anthelmintics
Definitions
- novel melaminylthioar senites which are trypanocidal and macrofilaricidal agents can be prepared. They can be provided as the principal active agent in therapeutically useful compositions in association with pharmaceutically acceptable excipients.
- the compounds of this invention may be represented by the formula:
- Y is selected from the group concisting of and n and m are both integers from 2 to
- the invention also includes pharmaceutically acceptable acid addition salts of the free bases within its scope.
- the compounds of the invention are obtained by reaction of melarsenoxide with an appropriate thiol.
- the reaction can be represented by the equation:
- n and m have the same meaning as above.
- the following compounds are the preferred thiols for use in this invention. They are preferred because they are readily available, known to be non-toxic and because they produce compounds with a high order of activity, a high therapeutic index and low toxicity.
- Pharmaceutically acceptable acid addition salts are those containing non-toxic anions and include, for example, hydrochloric, hydrobromic, sulfuric, phosphoric, acetic, lactic, citric, tartaric, succinic, maleic, gluconic and saccharic acids. These acid addition salts are prepared by standard reactions either directly from the initial reaction or from the free base by reaction with the selected acid.
- the general procedure for the preparation of the compounds of this invention is to react melarsenoxide with the selected thiol in an aqueous or lower alkanol medium at a temperature of from 40o to 100°C until all of the reactants have dissolved.
- the reaction products separate as a viscous bottom layer.
- the reaction solvent which may be a mixture of water and a lower alkanol, preferably methanol or ethanol is separated, suitably by decantation.
- the product may be purified by repeated recrystallization together with trituration with a dehydrating solvent, preferably ethanol.
- the preferred recrystallization solvent is water which may contain a small amount of ethanol, e.g. up to 20% by weight.
- thiol i.e. more than two moles of thiol per mole of melarsenoxide. This insures as complete a reaction as possible of the toxic melarsenoxide and limits the amount which may be present as a contaminant in the final product. It is preferred to use at least 10% molar excess of thiol, and for most purposes an excess of from 10 to 20% is acceptable.
- the thiols may be reacted either as free thiols, phosphothionites, or as acid salts such as the hydrochloride or hydrobromide. Accordingly, a product of the invention may be obtained as a free base or an acid addition salt.
- the free cationic melaminylthioarsenites can be prepared according to conventional methods.
- the cationic thioarsenites of the present invention are white powders, soluble in water, dilute acids and alkali, insoluble in acetone, chloroform, and ether. They have no sharp melting point and decompose above
- the products of this invention may be administered alone but will generally be administered with pharmaceutically acceptable, non-toxic carriers, the proportions of which are determined by the suitability and chemical nature of the particular carrier, the chosen route of administration, and standard pharmaceutical practice.
- pharmaceutically acceptable, non-toxic carriers the proportions of which are determined by the suitability and chemical nature of the particular carrier, the chosen route of administration, and standard pharmaceutical practice.
- in combatting various infections or in maintaining therapeutically effective levels in the blood or tissue thay may be administered orally in the form of tablets or capsules containing such excipients as starch, milk sugar, certain types of clay, etc. They may be enteric coated so as to be more resistant to the acid and digestive enzymes of the stomach.
- intravenous and intramuscular administration they may be used in the form of a sterile solution containing other solutes, for example, enough saline or glucose to make the solution inotonic.
- the compounds of this invention are orders of magnitude more stable than related prior art compounds which have been similarly employed.
- the disodium salt of Compound I the first formula shown above
- the solution becomes slightly cloudy after only 12 hours.
- a precipitate forms in 5 days.
- Compounds X and XIII of this invention in 1% aqueous solution have been held at 37°C for 5 days while still remaining clear. In fact, the solutions remain clear for as long as 3 months at 100°C.
- the compounds of this invention have been screened for useful activity.
- This test system is patterned after the one developed and employed in testing of compounds for activity against Plasmodium berghei maleria in mice.
- the trypanosomiasis system is based on comparisons of responses to test compounds by ICR/HA Swiss mice infected with the Wellcome CT strain of T. rhodesiense as expressed in mean survival times compared with mean survival times of untreated controls.
- Using a standard inoculum it is possible to produce a uniform disease fatal to 100% of untreated animals within 4 to 6 days with a mean survival time of 4.45 ⁇ .25 days.
- Test mice six weeks of age, weighing 30-32 grams receive an intraperitoneal injection of 0.5 ml of a 1:50,000 dilution of heparinized heart blood drawn from donor mice infected 3 days earlier. Compounds are given as a single dose in peanut oil about two hours after parasite inoculation. Five mice per drug level, 20 infected untreated (negative) controls, and 10 infected positive controls are routinely used per test. Positive controls are mice infected and treated at 40 mg/kg with Stilbamidine Isethionate. Routinely compounds are first tested by the subcutaneous route of administration. retested for confirmation at selected drug levels and if the activity is confirmed it is tested by the oral route of administration.
- Example 2 Compound XII 10g III are disolved in a solution of 20.2g VI in 200 ml boiling water. On cooling to 0° of the filtered reaction, mixture XII precipitates, is purified by 3 reprecipitations by cooling of the hot aqueous solution, filtered off, washed with acetone and dried in vacuo. Yield: 7.1g.
- XII is a white hygroscopic powder, soluble in water, EtOH and MeOH, insoluble in acetone, ethylacetate, ether, chloroform. A 1% solution of XII in water remains unchanged for 4 weeks.
- Example 3 Compound XII
- XII 5g III are dissolved, with stirring, in a solution of 5.6g VI in 100 ml boiling MeOH. After cooling to 0°, the reaction mixture is filtered and added to 200 ml ether. The reaction product precipitates as a viscous colorless syrup, which is separated by decantation, washed with 3 portions of 50 ml ether and dried in vacuo. The resulting crusts are soluble in water, EtOH and MeOH, insoluble in ether, chloroform and ethylacetate.
- Example 4 Compound XII 13g III are dissolved with stirring in a solution of 15g VII in 150 ml water warmed to 80°. On cooling of the filtered reaction mixture to 0° , XII forms a viscous transparent bottom layer. The supernatant is decanted, the bottom layer washed with 3 portions of 50 ml ice water. After the last decantation the product is triturated with 200 ml hot EtOH, whereupon it becomes granular. It is filtered off, washed with EtOH and dried over P 2 O 5 in vacuo. Yield 18.6g. XII is a white powder, soluble in water and dilute mineral acids. It is insoluble in EtOH, MeOH, acetone, ether and chloroform. A 1% solution of XII in water remains unchanged at 100 for 4 weeks.
- Example 5 Compound XIII
- Example 6 - Compound XIV 10g III are dissolved with stirring in a solution of 10.1g IX in 100 ml boiling water.
- the filtered solution on standing at 0°, deposits a clear viscous bottom layer which is separated from the supernatant by decantation. On trituration with 100 ml EtOH, the bottom layer becomes solid, granular. It is filtered off and dried in vacuo. The yield is 18g.
- the reaction product is purified by dissolving in the minimal amount of boiling water, followed by cooling and treatment with EtOH as above.
- XIV is a white powder, soluble in water, insoluble in EtOH, MeOH, acetone, chloroform and ether. A 1% solution of XIV in water remains unchanged at 100° for 4 weeks.
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Tropical Medicine & Parasitology (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Compounds of the formula I wherein Y is selected from the group consisting of NH2$(6,)$and n and m are both integers from 2 to 12 with the proviso that the value of n plus m is no more than 12, and the pharmaceutically acceptable acid additions salts thereof, are useful in the treatment of trypanocide and macrofilaricide infections.
Description
IMPROVED MELAMINYLTHIOARSENITES
BACKGROUND OF THE INVENTION The compound represented by the formula

It has now been discovered that novel melaminylthioar senites which are trypanocidal and macrofilaricidal agents can be prepared. They can be provided as the principal active agent in therapeutically useful compositions in association with pharmaceutically acceptable excipients. The compounds of this invention may be represented by the formula:

wherein Y is selected from the group concisting of
 and n and m are both integers from 2 to
and n and m are both integers from 2 to

12 with the proviso that the value of n plus m is no more than 12. The invention also includes pharmaceutically acceptable acid addition salts of the free bases within its scope.
The compounds of the invention are obtained by reaction of melarsenoxide with an appropriate thiol. The reaction can be represented by the equation:

In the equation Y, n and m have the same meaning as above. The following compounds are the preferred thiols for use in this invention. They are preferred because they are readily available, known to be non-toxic and because they produce compounds with a high order of activity, a high therapeutic index and low toxicity. NH2CH2CH2SH · HCl V
NH2C(=NH)CH2CH2SH · 2HBr VI
NH2C( NH)NH(CH2)3S H2PO3 VII
NH2(CH2)3NH(CH2)2 · S H2PO3 VIII
NH2(CH2)3NH(CH2)3 ·S H2PO3 IX It will be noted that the first two compounds are acid addition salts. The last three compounds are esters. In the course of the reaction, the esters rearrange so that the final products are
acid addition salts.
Pharmaceutically acceptable acid addition salts are those containing non-toxic anions and include, for example, hydrochloric, hydrobromic, sulfuric, phosphoric, acetic, lactic, citric, tartaric, succinic, maleic, gluconic and saccharic acids. These acid addition salts are prepared by standard reactions either directly from the initial reaction or from the free base by reaction with the selected acid.
The general procedure for the preparation of the compounds of this invention is to react melarsenoxide with the selected thiol in an aqueous or lower alkanol medium at a temperature of from 40º to 100°C until all of the reactants have dissolved. The reaction products separate as a viscous bottom layer. The reaction solvent, which may be a mixture of water and a lower alkanol, preferably methanol or ethanol is separated, suitably by decantation. The product may be purified by repeated recrystallization together with trituration with a dehydrating solvent, preferably ethanol. The preferred recrystallization solvent is water which may contain a small amount of ethanol, e.g. up to 20% by weight. It is preferred to utilize a molar excess of thiol, i.e. more than two moles of thiol per mole of melarsenoxide. This insures as complete a reaction as possible of the toxic melarsenoxide and limits the amount which may be present as a contaminant in the final product. It is preferred to use at least 10% molar excess of thiol, and for most purposes an excess of from 10 to 20% is acceptable.
The thiols may be reacted either as free thiols, phosphothionites, or as acid salts such as the hydrochloride or hydrobromide. Accordingly, a product of the invention may be obtained as a free base or an acid addition salt.
The preferred products of the invention are those represented by the formulas:





200°C. They are useful for the treatment of parasitic diseases such as trypanosomiasis and filariasis, and onchocerciasis.
The products of this invention may be administered alone but will generally be administered with pharmaceutically acceptable, non-toxic carriers, the proportions of which are determined by the suitability and chemical nature of the particular carrier, the chosen route of administration, and standard pharmaceutical practice. For example, in combatting various infections or in maintaining therapeutically effective levels in the blood or tissue thay may be administered orally in the form of tablets or capsules containing such excipients as starch, milk sugar, certain types of clay, etc. They may be enteric coated so as to be more resistant to the acid and digestive enzymes of the stomach. For intravenous and intramuscular administration they may be used in the form of a sterile solution containing other solutes, for example, enough saline or glucose to make the solution inotonic.
The compounds of this invention are orders of magnitude more stable than related prior art compounds which have been similarly employed. For example, if the disodium salt of Compound I, the first formula shown above, is dissolved in 1% water solution and held at 37°C, the solution becomes slightly cloudy after only 12 hours. A precipitate forms in 5 days. In contrast, Compounds
X and XIII of this invention in 1% aqueous solution have been held at 37°C for 5 days while still remaining clear. In fact, the solutions remain clear for as long as 3 months at 100°C.
The compounds of this invention have been screened for useful activity.
The test for trypanocidal activity is as described by Rane et al., Amer. J. Trop. Med. Hug. 25:394(1976).
This test system is patterned after the one developed and employed in testing of compounds for activity against Plasmodium berghei maleria in mice. The trypanosomiasis system is based on comparisons of responses to test compounds by ICR/HA Swiss mice infected with the Wellcome CT strain of T. rhodesiense as expressed in mean survival times compared with mean survival times of untreated controls. Using a standard inoculum, it is possible to produce a uniform disease fatal to 100% of untreated animals within 4 to 6 days with a mean survival time of 4.45 ± .25 days.
Test mice, six weeks of age, weighing 30-32 grams receive an intraperitoneal injection of 0.5 ml of a 1:50,000 dilution of heparinized heart blood drawn from donor mice infected 3 days earlier. Compounds are given as a single dose in peanut oil about two hours after parasite inoculation. Five mice per drug level, 20 infected untreated (negative) controls, and 10 infected positive controls are routinely used per test. Positive controls are mice infected and treated at 40 mg/kg with Stilbamidine Isethionate. Routinely compounds are first tested by the subcutaneous route of administration.
retested for confirmation at selected drug levels and if the activity is confirmed it is tested by the oral route of administration.
Deaths prior to the fourth day, when untreated controls begin to die, are regarded as non-parasitic and are scored as "toxic deaths". Treated animals are kept under observation for 30 days. Survivors at the end of this period of time are considered as "cured". An increase of 100% in mean survival time is considered the minimum effective ("active") response for a candidate compound. In calculating mean survival times, toxic deaths and 30-day survivors are not included.
Using this test it was found that with Compound XII the LD50 was of the order of 400 mg/kg of body weight and the DCur50 (dose curative - 50%) was of the order of 0.16 mg/kg of body weight. This corresponds to a therapeutic index of more than 2,000. The filaricidal activity of Compound XIII was tested in gerbils infected with L. carinii according to the standard method employed at the WHO screening center at the Justes Liebig University in Giessen, West Germany. The results, as a function of dose, are shown in the following table.

The following non-limiting examples are given by way of illustration only. All temperatures are in degrees Centigrade. Example 1 - Compound X
A solution of 3.6g III in 72 ml boiling EtOH is stirred into a solution of 3.0g of V in 30 ml boiling EtOH. A white precipitate is formed which, after cooling to 10°, is filtered off, washed with EtOH and dried in vacuo over -P2O5. X is reprecipitated by cooling out of boiling water. Yield 5.0g. White powder, soluble in water, insoluble in MeOH, EtOH, chloroform, ether, unchanged dissolved in boiling water for 1 month.
Example 2 - Compound XII 10g III are disolved in a solution of 20.2g VI in 200 ml boiling water. On cooling to 0° of the filtered reaction, mixture XII precipitates, is purified by 3 reprecipitations by cooling of the hot aqueous solution, filtered off, washed with
acetone and dried in vacuo. Yield: 7.1g. XII is a white hygroscopic powder, soluble in water, EtOH and MeOH, insoluble in acetone, ethylacetate, ether, chloroform. A 1% solution of XII in water remains unchanged for 4 weeks. Example 3 - Compound XII
XII 5g III are dissolved, with stirring, in a solution of 5.6g VI in 100 ml boiling MeOH. After cooling to 0°, the reaction mixture is filtered and added to 200 ml ether. The reaction product precipitates as a viscous colorless syrup, which is separated by decantation, washed with 3 portions of 50 ml ether and dried in vacuo. The resulting crusts are soluble in water, EtOH and MeOH, insoluble in ether, chloroform and ethylacetate.
Example 4 - Compound XII 13g III are dissolved with stirring in a solution of 15g VII in 150 ml water warmed to 80°. On cooling of the filtered reaction mixture to 0° , XII forms a viscous transparent bottom layer. The supernatant is decanted, the bottom layer washed with 3 portions of 50 ml ice water. After the last decantation the product is triturated with 200 ml hot EtOH, whereupon it becomes granular. It is filtered off, washed with EtOH and dried over P2O5 in vacuo. Yield 18.6g. XII is a white powder, soluble in water and dilute mineral acids. It is insoluble in EtOH, MeOH, acetone, ether and chloroform. A 1% solution of XII in water remains unchanged at 100 for 4 weeks. Example 5 - Compound XIII
12g VII are dissolved in 100 ml of water. 8g III are added with stirring, while the mixture is brought to a boil. The resulting
solution is filtered hot. On standing at 2º a colorless viscous bottom layer is formed. The supernatant is decanted. The bottom layer is dissolved in 50 ml boiling water and precipitated again by cooling. This procedure is repeated twice. After the last decantation the bottom layer has turned into a white powder, which is filtered off and dried in vacuo over P2O5. Yield 14.3g. XIII is a white powder, sparingly soluble in cold water, soluble in hot water, dilute mineral acids, glacial acetic acid, and insoluble in EtOH, MeOH, acetone, chloroform and ether. A 1% solution of XIII in water remains unchanged at 100° for 1 month.
Example 6 - Compound XIV 10g III are dissolved with stirring in a solution of 10.1g IX in 100 ml boiling water. The filtered solution, on standing at 0°, deposits a clear viscous bottom layer which is separated from the supernatant by decantation. On trituration with 100 ml EtOH, the bottom layer becomes solid, granular. It is filtered off and dried in vacuo. The yield is 18g. The reaction product is purified by dissolving in the minimal amount of boiling water, followed by cooling and treatment with EtOH as above. XIV is a white powder, soluble in water, insoluble in EtOH, MeOH, acetone, chloroform and ether. A 1% solution of XIV in water remains unchanged at 100° for 4 weeks.
Claims
1. A compound represented by the formula



12 with the proviso that the value of n plus m is no more than 12, and the pharmaceutically acceptable acid addition salts thereof. 2. A compound as in Claim 1 of the formula

3. A compound as in Claim 1 of the formula
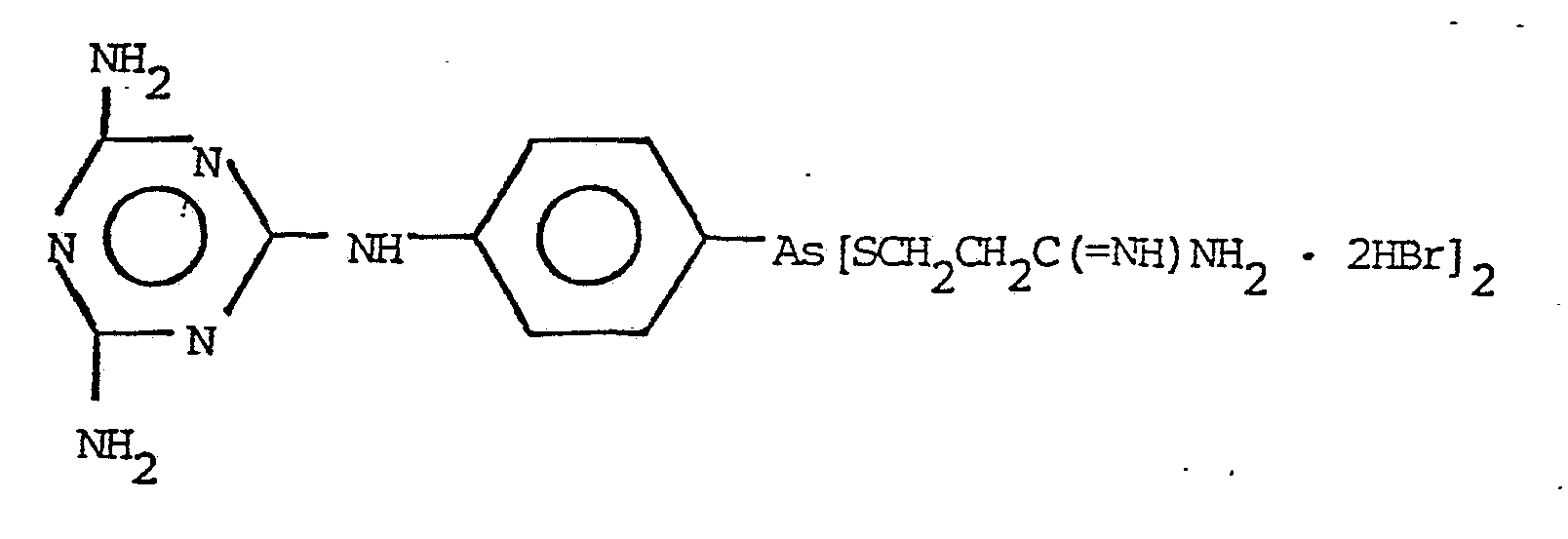
4. A compound as in Claim 1 of the formula

5. A compound as in Claim 1 of the formula

6. A compound as in Claim 1 of the formula 
7. A pharmaceutical composition comprising a pharmaceutically acceptable carrier and, as the principal active ingredient, a compound of the formula:



8. A pharmaceutical composition of Claim 7 comprising a pharmaceutically acceptable carrier and, as the principal active ingredient a compound of the formula:

9. A pharmaceutical composition of Claim 7 comprising a pharmaceutically acceptable carrier and, as the principal active ingredient, a compound of the formula:

10. A pharmaceutical composition of Claim 7 comprising a pharmaceutically acceptable carrier and, as the principal active ingredient, a compound of the formula: 
11. A pharmaceutical, composition of Claim 7 comprising a pharmaceutically acceptable carrier and, as the principal active ingredient, a compound of the formula:

12. A pharmaceutical composition of Claim 7 comprising a pharmaceutically acceptable carrier and, as the principal active ingredient, a compound of the formula:

13. A method of treating infections caused by trypanosomes or filaria of mammals which comprises treating a mammal in need of such treatment with a trypanocidally or macrofilaricidally effective amount of a compound represented by the formula: 
wherein Y is selected from the group consisting of  and n and m are both integers from 2
and n and m are both integers from 2  to 12 with the proviso that the value of n plus m is no more than 12, and the pharmaceutically acceptable acid addition salts thereof.
to 12 with the proviso that the value of n plus m is no more than 12, and the pharmaceutically acceptable acid addition salts thereof.
14. A method of treating infections caused by trypanosomes or filaria as in Claim 13 of mammals which comprises treating a mammal in need of such treatment with a trypanocidally or macrofilaricidally effective amount of a compound represented by the formula:
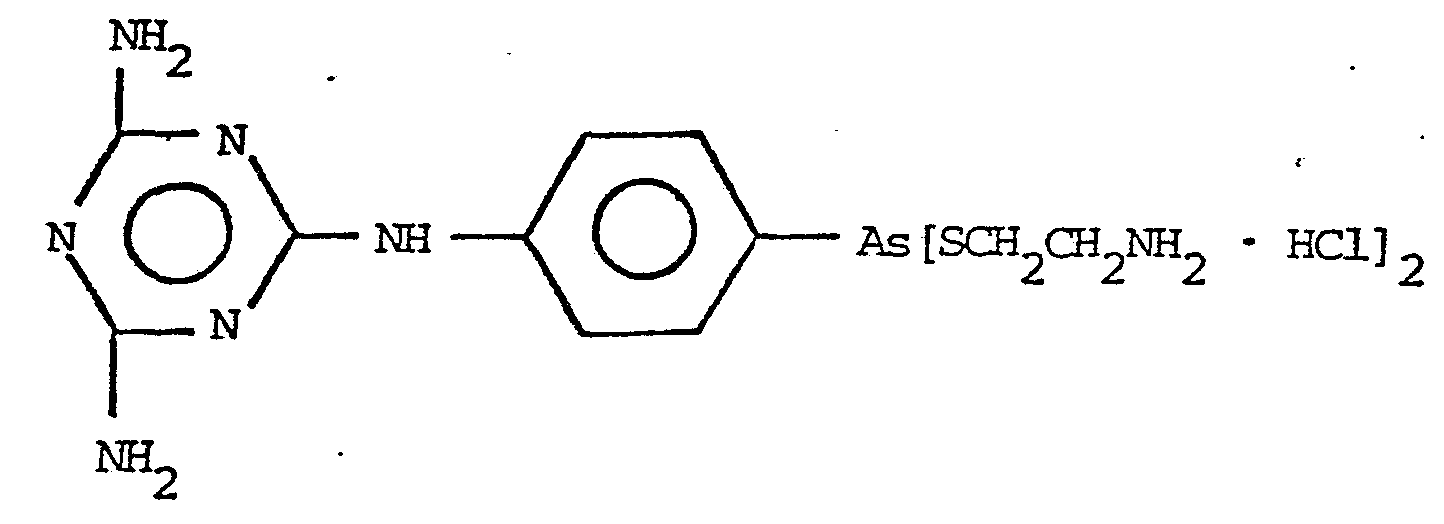
15. A method of treating infections caused by trypanosomes or filaria as in Claim 13 of mammals which comprises treating a mammal in need of such treatment with a trypanocidally or macrofilaricidally effective amount of a compound represented by the formula: 
16. A method of treating infections caused by trypanosomes or filaria as in Claim 13 of mammals which comprises treating a mammal in need of such treatment with a trypanocidally or macrofilaricidally effective amount of a compound represented by the formula:
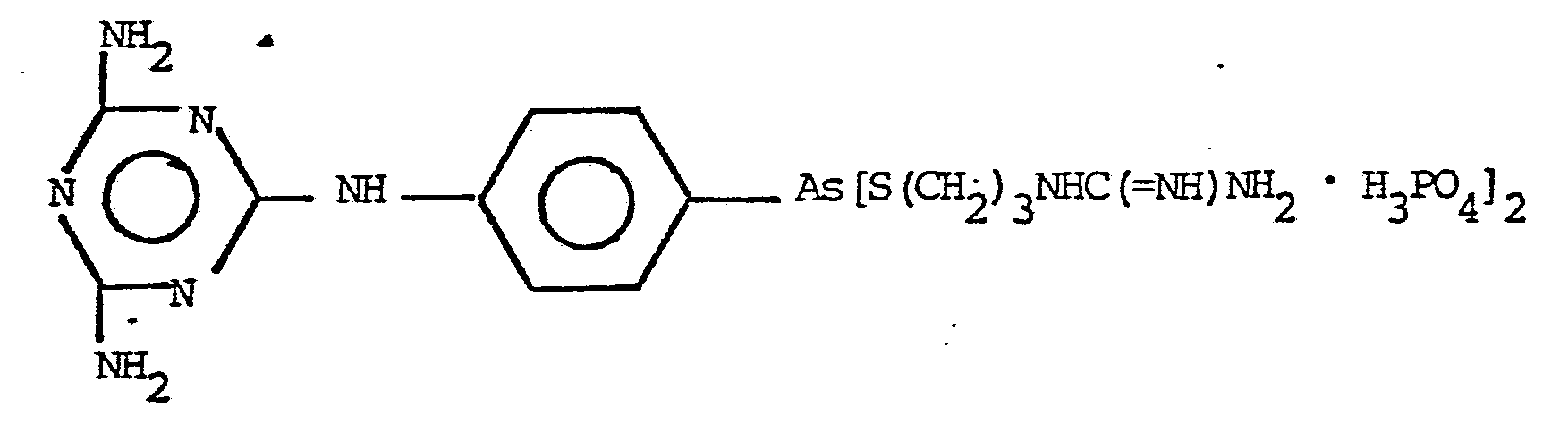
17. A method of treating infections caused by trypanosomes or filaria as in Claim 13 of mammals which comprises treating a mammal in need of such treatment with a trypanocidally or macrofilaricidally effective amount of a compound represented by the formula:

18. A method of treating infections caused by trypanosomes or filaria as in Claim 13 of mammals which comprises treating a mammal in need of such treatment with a trypanocidally or macrofilaricidally effective amount of a compound represented by the formula:

19. A process for the production of a compound of Claim 1 which comprises reacting melarsenoxide with a compound of the formula: HS(CH2)nY in a liquid medium at a temperature of from 40°C to 100°C.
20. A process as in Claim 19 wherein the liquid medium is an aqueous or lower alkanol medium.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AT83901552T ATE20348T1 (en) | 1982-04-12 | 1983-04-11 | IMPROVED MELAMINYLTHIOARSENITES. |
| DE8383901552T DE3364051D1 (en) | 1982-04-12 | 1983-04-11 | Improved melaminylthioarsenites |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US367,562820412 | 1982-04-12 | ||
| US06/367,562 US4514390A (en) | 1982-04-12 | 1982-04-12 | Melaminylthioarsenites |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1983003606A1 true WO1983003606A1 (en) | 1983-10-27 |
Family
ID=23447686
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1983/000505 WO1983003606A1 (en) | 1982-04-12 | 1983-04-11 | Improved melaminylthioarsenites |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US4514390A (en) |
| EP (1) | EP0105912B1 (en) |
| JP (1) | JPS59500519A (en) |
| DE (1) | DE3364051D1 (en) |
| OA (1) | OA07604A (en) |
| WO (1) | WO1983003606A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2684382A1 (en) * | 1991-12-02 | 1993-06-04 | Rhone Merieux | Pure preparations and medicaments of melarsomine dihydrochloride, process for their preparation and intermediates obtained |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE602004026759D1 (en) * | 2003-10-24 | 2010-06-02 | Chemo Sero Therapeut Res Inst | NEW RECOMBINANT PET CELLS HAVING HIGH PROTEIN PRODUCTION, METHOD OF CONSTRUCTING THEM AND PROCESSING THE MASS PRODUCTION OF PROTEIN USING THEREOF |
| US20140051840A1 (en) * | 2012-08-17 | 2014-02-20 | The Curators Of The University Of Missouri | Arsenic complexes for potential diagnostic applications |
| US9593052B2 (en) | 2012-08-17 | 2017-03-14 | The Curators Of The University Of Missouri | Arsenic complexes for potential diagnostic applications |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2400547A (en) * | 1944-01-20 | 1946-05-21 | Ernst A H Friedheim | Substituted 1, 3, 5-triazinyl-(6)-aminophenyl-arsenic compounds |
| US2422724A (en) * | 1944-01-20 | 1947-06-24 | Ernst A H Friedheim | Substituted 1,3,5-triazinyl-(6)-aminophenyl-arsenic compounds |
| US3974148A (en) * | 1971-07-20 | 1976-08-10 | Friedheim Ernst A H | Filaricidal and trypanocidal phenylarsenodithio compounds |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NL54891C (en) * | 1938-12-23 | 1900-01-01 | ||
| US2390091A (en) * | 1944-06-03 | 1945-11-27 | Ernst A H Friedheim | Substituted 1,3,5-triazinyl-(6)-aminophenyl-arsenic compounds |
| US2659723A (en) * | 1947-01-28 | 1953-11-17 | Ernst A H Friedheim | Triazine organometallic compounds and process for preparing same |
| US2593434A (en) * | 1947-02-19 | 1952-04-22 | Ernst A H Friedheim | Propylene glycol solution of arsenic medicaments |
-
1982
- 1982-04-12 US US06/367,562 patent/US4514390A/en not_active Expired - Lifetime
-
1983
- 1983-04-11 EP EP83901552A patent/EP0105912B1/en not_active Expired
- 1983-04-11 JP JP58501605A patent/JPS59500519A/en active Granted
- 1983-04-11 DE DE8383901552T patent/DE3364051D1/en not_active Expired
- 1983-04-11 WO PCT/US1983/000505 patent/WO1983003606A1/en active IP Right Grant
- 1983-12-08 OA OA58176A patent/OA07604A/en unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2400547A (en) * | 1944-01-20 | 1946-05-21 | Ernst A H Friedheim | Substituted 1, 3, 5-triazinyl-(6)-aminophenyl-arsenic compounds |
| US2422724A (en) * | 1944-01-20 | 1947-06-24 | Ernst A H Friedheim | Substituted 1,3,5-triazinyl-(6)-aminophenyl-arsenic compounds |
| US3974148A (en) * | 1971-07-20 | 1976-08-10 | Friedheim Ernst A H | Filaricidal and trypanocidal phenylarsenodithio compounds |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP0105912A4 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2684382A1 (en) * | 1991-12-02 | 1993-06-04 | Rhone Merieux | Pure preparations and medicaments of melarsomine dihydrochloride, process for their preparation and intermediates obtained |
| GR920100526A (en) * | 1991-12-02 | 1993-08-31 | Rhone Merieux | Medicaments and pure dichlorhydrate melarsomine compounds, method for obtaining them and intermediate receptive products. |
| ES2039311A1 (en) * | 1991-12-02 | 1993-09-16 | Rhone Merieux | Medicinal products and pure preparations of melarsomine dihydrochloride, process for obtaining them and intermediate products obtained |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0105912A4 (en) | 1984-08-23 |
| JPS59500519A (en) | 1984-03-29 |
| JPS6365073B2 (en) | 1988-12-14 |
| EP0105912A1 (en) | 1984-04-25 |
| OA07604A (en) | 1985-03-31 |
| EP0105912B1 (en) | 1986-06-11 |
| US4514390A (en) | 1985-04-30 |
| DE3364051D1 (en) | 1986-07-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR910002837B1 (en) | Process for the preparation of pyrrolo (1,2-a) imidazo (1,2-a) pyridine derivatives | |
| CS235320B2 (en) | Method of sultamiciline's addition salt preparation | |
| US3758684A (en) | Treating dna virus infections with amino purine derivatives | |
| JP3207200B2 (en) | N, N'-substituted imidocarboximide diamides derived from hydroxylamine | |
| EP0105912B1 (en) | Improved melaminylthioarsenites | |
| EP0038161A1 (en) | 2,6-Diaminonebularines, their production and use | |
| US5281588A (en) | Organometallic compounds, methods for their preparation and pharmaceutical compositions containing them | |
| GB2189239A (en) | Furosemide salts | |
| KR930005261B1 (en) | Process for producing new platinium complexes | |
| JPS63297364A (en) | Antitumoral compound | |
| EP0224121A2 (en) | 7-[4-amino-piperazinyl]- or 7-[4-chloro-piperazinyl]quinolinone derivatives, a process for the preparation thereof and pharmaceutical compositions containing them | |
| EP0138374B1 (en) | Salts of antimalarial phenanthrenemethanol compounds | |
| US5475028A (en) | 2-aminoethanesulfonic acid zinc complex | |
| EP0040420A1 (en) | 4-Bis((phenylmethyl)amino)benzenesulfonic acids and virus inhibiting compositions containing them | |
| US4507288A (en) | β-Glycerophosphate salts of antimalarial phenanthrenemethanol compounds | |
| KR850000210B1 (en) | Process for preparing oxazolines | |
| US5489609A (en) | 2-aminoethanesulfonic acid zinc complex compound | |
| US20050143400A1 (en) | Process for preparing famciclovir | |
| KR810000473B1 (en) | Process for preparing-2,6-dichlorophenyl-2-amino pyrimidine | |
| SU589747A1 (en) | Potassium salt of s-[benzymydazolyl-(2)-methyl]-thiosulfuric acid manifesting antispasmodic activity | |
| EA005929B1 (en) | Antitumoral, antiviral, antibacterial, antiparasitic, anti-inflammatory, immunomodulatory and antimicotic medical preparation, method for production and dosage forms thereof | |
| KR840000323B1 (en) | Process for preparing 2-alkoxy phenyl-imidazo(4,5-b)pyridines | |
| NZ199917A (en) | 1,3,4-thiadiazoles | |
| US3433789A (en) | S-benzoyloxymethyl-thiamines | |
| SU1547252A1 (en) | 2-oxy-3,5-diiodo-n-@-/1-bromonaphxhoxy-2)-3-chlorophenyl/-benzamide displaying antimalaria activity |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Designated state(s): JP |
|
| AL | Designated countries for regional patents |
Designated state(s): AT BE CF CG CH CM DE FR GA GB LU NL SE SN TD TG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1983901552 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1983901552 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1983901552 Country of ref document: EP |