US8669260B2 - Ketoconazole-derivative antagonist of human pregnane X receptor and uses thereof - Google Patents
Ketoconazole-derivative antagonist of human pregnane X receptor and uses thereof Download PDFInfo
- Publication number
- US8669260B2 US8669260B2 US12/735,368 US73536809A US8669260B2 US 8669260 B2 US8669260 B2 US 8669260B2 US 73536809 A US73536809 A US 73536809A US 8669260 B2 US8669260 B2 US 8669260B2
- Authority
- US
- United States
- Prior art keywords
- pxr
- cells
- compound
- rifampicin
- ketoconazole
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active, expires
Links
- 0 *C1=CC=C(OC[C@H]2COC(B)(C)O2)C=C1 Chemical compound *C1=CC=C(OC[C@H]2COC(B)(C)O2)C=C1 0.000 description 17
- YSDBJKNOEWSFGA-UHFFFAOYSA-N CC(=O)N1CCN(C)CC1 Chemical compound CC(=O)N1CCN(C)CC1 YSDBJKNOEWSFGA-UHFFFAOYSA-N 0.000 description 2
- JPBCNLIDWJVQEU-XUZZJYLKSA-N CC(=O)N1CCN(C2=CC=C(OC[C@H]3CO[C@@](C)(C4=C(F)C=C(C)C=C4)O3)C=C2)CC1 Chemical compound CC(=O)N1CCN(C2=CC=C(OC[C@H]3CO[C@@](C)(C4=C(F)C=C(C)C=C4)O3)C=C2)CC1 JPBCNLIDWJVQEU-XUZZJYLKSA-N 0.000 description 2
- SGHSSXBYBNNMPF-UHFFFAOYSA-N CCCCCC1(C2=CC=CC2)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1 Chemical compound CCCCCC1(C2=CC=CC2)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1 SGHSSXBYBNNMPF-UHFFFAOYSA-N 0.000 description 2
- PAMIQIKDUOTOBW-UHFFFAOYSA-N CN1CCCCC1 Chemical compound CN1CCCCC1 PAMIQIKDUOTOBW-UHFFFAOYSA-N 0.000 description 2
- LJTYGHLQHHWKHQ-UHFFFAOYSA-N C.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Br)C=C(OC)C=C4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Cl)C=C(OC)C=C4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(F)C=C(OC)C=C4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(OC)C=C(OC)C=C4)O3)C=C2)CC1 Chemical compound C.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Br)C=C(OC)C=C4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Cl)C=C(OC)C=C4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(F)C=C(OC)C=C4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(OC)C=C(OC)C=C4)O3)C=C2)CC1 LJTYGHLQHHWKHQ-UHFFFAOYSA-N 0.000 description 1
- LAAURYUVGQVNSK-UHFFFAOYSA-N C.C=C(C)N1CCN(C2=CC=C(OCC3COC(CCCCC)(C4=CC=CC4)O3)C=C2)CC1.CCCC1(C2=CC=CC2)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1 Chemical compound C.C=C(C)N1CCN(C2=CC=C(OCC3COC(CCCCC)(C4=CC=CC4)O3)C=C2)CC1.CCCC1(C2=CC=CC2)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1 LAAURYUVGQVNSK-UHFFFAOYSA-N 0.000 description 1
- SKGCSYVZPZHTQK-UHFFFAOYSA-N C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Br)C=C(Br)C=C4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Cl)C=C(I)C=C4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(F)C=C(F)C=C4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(I)C=C(I)C=C4)O3)C=C2)CC1 Chemical compound C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Br)C=C(Br)C=C4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Cl)C=C(I)C=C4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(F)C=C(F)C=C4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(I)C=C(I)C=C4)O3)C=C2)CC1 SKGCSYVZPZHTQK-UHFFFAOYSA-N 0.000 description 1
- OVGDYZIBDOUTAZ-UHFFFAOYSA-N C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Br)C=C(F)C=C4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Br)C=C(I)C=C4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Cl)C=C(F)C=C4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(F)C=C(I)C=C4)O3)C=C2)CC1 Chemical compound C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Br)C=C(F)C=C4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Br)C=C(I)C=C4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Cl)C=C(F)C=C4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(F)C=C(I)C=C4)O3)C=C2)CC1 OVGDYZIBDOUTAZ-UHFFFAOYSA-N 0.000 description 1
- NQPVWFZUHGXCTF-UHFFFAOYSA-N C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(F)C=C(F)C=C4)O3)C=C2)CC1 Chemical compound C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(F)C=C(F)C=C4)O3)C=C2)CC1 NQPVWFZUHGXCTF-UHFFFAOYSA-N 0.000 description 1
- XHZOTSKFGSJTQQ-UHFFFAOYSA-N C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=CC=CC4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(CC)(C4CCCCC4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(CCC)(C4CCCC4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(CCC)(C4CCCCC4)O3)C=C2)CC1 Chemical compound C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4=CC=CC4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(CC)(C4CCCCC4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(CCC)(C4CCCC4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(CCC)(C4CCCCC4)O3)C=C2)CC1 XHZOTSKFGSJTQQ-UHFFFAOYSA-N 0.000 description 1
- DUNSJWOTXJQMBD-UHFFFAOYSA-N C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4CCCC4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C4=CC=CC4)(C4CCCC4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C4=CC=CC4)(C4CCCCC4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(CCCC)(C4CCCC4)O3)C=C2)CC1 Chemical compound C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4CCCC4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C4=CC=CC4)(C4CCCC4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C4=CC=CC4)(C4CCCCC4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(CCCC)(C4CCCC4)O3)C=C2)CC1 DUNSJWOTXJQMBD-UHFFFAOYSA-N 0.000 description 1
- JROMTPCXSGZABT-UHFFFAOYSA-N C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4CCCCC4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(CCC)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(CCCC)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(CCCCCCCC)O3)C=C2)CC1 Chemical compound C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(C4CCCCC4)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(CCC)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(CCCC)O3)C=C2)CC1.C=C(C)N1CCN(C2=CC=C(OCC3COC(C)(CCCCCCCC)O3)C=C2)CC1 JROMTPCXSGZABT-UHFFFAOYSA-N 0.000 description 1
- GWPXOKDCCVVJRS-KWTMUBMQSA-N CC(=O)C1=C(F)C=C(F)C=C1.CC(=O)N1CCN(C2=CC=C(OC[C@H]3CO[C@@](C)(C4=C(F)C=C(F)C=C4)O3)C=C2)CC1.CC(=O)N1CCNCC1.COC1=CC(F)=C([C@]2(C)OC[C@H](COC3=CC=C(N4CCN(C(C)=O)CC4)C=C3)O2)C=C1.COC1=CC=C([C@]2(C)OC[C@H](COC3=CC=C(N4CCN(C(C)=O)CC4)C=C3)O2)C=C1Cl.C[C@@]1(C2=C(F)C=C(F)C=C2)OC[C@H](COC2=CC=C(Br)C=C2)O1.OC1=CC=C(Br)C=C1.OC[C@@H](O)COC1=CC=C(Br)C=C1.OC[C@@H]1CO1 Chemical compound CC(=O)C1=C(F)C=C(F)C=C1.CC(=O)N1CCN(C2=CC=C(OC[C@H]3CO[C@@](C)(C4=C(F)C=C(F)C=C4)O3)C=C2)CC1.CC(=O)N1CCNCC1.COC1=CC(F)=C([C@]2(C)OC[C@H](COC3=CC=C(N4CCN(C(C)=O)CC4)C=C3)O2)C=C1.COC1=CC=C([C@]2(C)OC[C@H](COC3=CC=C(N4CCN(C(C)=O)CC4)C=C3)O2)C=C1Cl.C[C@@]1(C2=C(F)C=C(F)C=C2)OC[C@H](COC2=CC=C(Br)C=C2)O1.OC1=CC=C(Br)C=C1.OC[C@@H](O)COC1=CC=C(Br)C=C1.OC[C@@H]1CO1 GWPXOKDCCVVJRS-KWTMUBMQSA-N 0.000 description 1
- WTCCGXXLYWIDPP-UHFFFAOYSA-N CC(=O)N1CCN(C)CC1.CCC(=O)NC.CCOC(=O)NC.CN.CN(C)C.CN1CCCC1.CN1CCN(C=O)CC1.CN1CCNCC1.CN1CCOCC1.CN=C=S.CNC(=O)C(Cl)Cl.CNC(=O)C1=CC=C(Br)C=C1.CNC(=O)C1=CC=C(Cl)C=C1.CNC(=O)C1=CC=C(F)C=C1.CNC(=O)C1=CC=C(OC)C=C1.CNC(=O)C1=CC=CC=C1.CNC(=O)NC.CNC(=O)NC.CNC(=O)OC.CNC(=O)OC1=CC=CC=C1.CNC(=S)NC.CNC(=S)NC.CNC(C)=O.CNC(N)=S.CNC=O.COC(=O)N1CCN(C)CC1.[HH].[HH].[HH].[HH] Chemical compound CC(=O)N1CCN(C)CC1.CCC(=O)NC.CCOC(=O)NC.CN.CN(C)C.CN1CCCC1.CN1CCN(C=O)CC1.CN1CCNCC1.CN1CCOCC1.CN=C=S.CNC(=O)C(Cl)Cl.CNC(=O)C1=CC=C(Br)C=C1.CNC(=O)C1=CC=C(Cl)C=C1.CNC(=O)C1=CC=C(F)C=C1.CNC(=O)C1=CC=C(OC)C=C1.CNC(=O)C1=CC=CC=C1.CNC(=O)NC.CNC(=O)NC.CNC(=O)OC.CNC(=O)OC1=CC=CC=C1.CNC(=S)NC.CNC(=S)NC.CNC(C)=O.CNC(N)=S.CNC=O.COC(=O)N1CCN(C)CC1.[HH].[HH].[HH].[HH] WTCCGXXLYWIDPP-UHFFFAOYSA-N 0.000 description 1
- LBHOJELCKWPPIQ-UHFFFAOYSA-N CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C)O3)C=C2)CC1.CCCC1(C)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1.CCCCC1(C)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1.CCCCCCCCC1(C)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1 Chemical compound CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C)O3)C=C2)CC1.CCCC1(C)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1.CCCCC1(C)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1.CCCCCCCCC1(C)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1 LBHOJELCKWPPIQ-UHFFFAOYSA-N 0.000 description 1
- JXEWTEAPJMWYKJ-UHFFFAOYSA-N CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Br)C=C(Br)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Cl)C=C(I)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(F)C=C(F)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(I)C=C(I)C=C4)O3)C=C2)CC1 Chemical compound CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Br)C=C(Br)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Cl)C=C(I)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(F)C=C(F)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(I)C=C(I)C=C4)O3)C=C2)CC1 JXEWTEAPJMWYKJ-UHFFFAOYSA-N 0.000 description 1
- SYHGWAXMLNZXHM-UHFFFAOYSA-N CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Br)C=C(F)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Br)C=C(I)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Cl)C=C(F)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(F)C=C(I)C=C4)O3)C=C2)CC1 Chemical compound CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Br)C=C(F)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Br)C=C(I)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Cl)C=C(F)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(F)C=C(I)C=C4)O3)C=C2)CC1 SYHGWAXMLNZXHM-UHFFFAOYSA-N 0.000 description 1
- MZNFCEJNZJPUMT-UHFFFAOYSA-N CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Br)C=C(O)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Cl)C=C(O)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(F)C=C(O)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(O)C=C(O)C=C4)O3)C=C2)CC1 Chemical compound CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Br)C=C(O)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(Cl)C=C(O)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(F)C=C(O)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(O)C=C(O)C=C4)O3)C=C2)CC1 MZNFCEJNZJPUMT-UHFFFAOYSA-N 0.000 description 1
- ZRVROXFXYAFZIF-UHFFFAOYSA-N CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(CO)C=C(Br)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(CO)C=C(F)C=C4)O3)C=C2)CC1 Chemical compound CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(CO)C=C(Br)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(CO)C=C(F)C=C4)O3)C=C2)CC1 ZRVROXFXYAFZIF-UHFFFAOYSA-N 0.000 description 1
- PZJMHKWUKLMIKJ-UHFFFAOYSA-N CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(CO)C=C(Cl)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(CO)C=C(I)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(CO)C=C(O)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(OCO)C=C(O)C=C4)O3)C=C2)CC1 Chemical compound CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(CO)C=C(Cl)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(CO)C=C(I)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(CO)C=C(O)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(OCO)C=C(O)C=C4)O3)C=C2)CC1 PZJMHKWUKLMIKJ-UHFFFAOYSA-N 0.000 description 1
- IQSYYGLESCQEGF-UHFFFAOYSA-N CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(I)C=C(O)C=C4)O3)C=C2)CC1.COC1=CC(Cl)=C(C2(C)OCC(COC3=CC=C(N4CCN(C(C)=O)CC4)C=C3)O2)C=C1.COC1=CC(O)=C(C2(C)OCC(COC3=CC=C(N4CCN(C(C)=O)CC4)C=C3)O2)C=C1.COC1=CC(OC)=C(C2(C)OCC(COC3=CC=C(N4CCN(C(C)=O)CC4)C=C3)O2)C=C1 Chemical compound CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(I)C=C(O)C=C4)O3)C=C2)CC1.COC1=CC(Cl)=C(C2(C)OCC(COC3=CC=C(N4CCN(C(C)=O)CC4)C=C3)O2)C=C1.COC1=CC(O)=C(C2(C)OCC(COC3=CC=C(N4CCN(C(C)=O)CC4)C=C3)O2)C=C1.COC1=CC(OC)=C(C2(C)OCC(COC3=CC=C(N4CCN(C(C)=O)CC4)C=C3)O2)C=C1 IQSYYGLESCQEGF-UHFFFAOYSA-N 0.000 description 1
- VMNKROWUKUFDJJ-UHFFFAOYSA-N CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(S)C=C(O)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C([N+](=O)[O-])C=C(O)C=C4)O3)C=C2)CC1.COC1=CC(Br)=C(C2(C)OCC(COC3=CC=C(N4CCN(C(C)=O)CC4)C=C3)O2)C=C1.COC1=CC(F)=C(C2(C)OCC(COC3=CC=C(N4CCN(C(C)=O)CC4)C=C3)O2)C=C1 Chemical compound CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C(S)C=C(O)C=C4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=C([N+](=O)[O-])C=C(O)C=C4)O3)C=C2)CC1.COC1=CC(Br)=C(C2(C)OCC(COC3=CC=C(N4CCN(C(C)=O)CC4)C=C3)O2)C=C1.COC1=CC(F)=C(C2(C)OCC(COC3=CC=C(N4CCN(C(C)=O)CC4)C=C3)O2)C=C1 VMNKROWUKUFDJJ-UHFFFAOYSA-N 0.000 description 1
- VMIWXYGYKPOXJM-UHFFFAOYSA-N CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=CC=CC4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4CCCCC4)O3)C=C2)CC1.CCC1(C2CCCCC2)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1.CCCCC1(C2CCCC2)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1 Chemical compound CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4=CC=CC4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4CCCCC4)O3)C=C2)CC1.CCC1(C2CCCCC2)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1.CCCCC1(C2CCCC2)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1 VMIWXYGYKPOXJM-UHFFFAOYSA-N 0.000 description 1
- MAMJXZALNMYIFI-UHFFFAOYSA-N CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4CCCC4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4CCCC4)O3)C=C2)CC1.CCCC1(C2CCCC2)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1.CCCC1(C2CCCC2)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1 Chemical compound CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4CCCC4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4CCCC4)O3)C=C2)CC1.CCCC1(C2CCCC2)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1.CCCC1(C2CCCC2)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1 MAMJXZALNMYIFI-UHFFFAOYSA-N 0.000 description 1
- DQBFZRYKVJSLLO-UHFFFAOYSA-N CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4CCCCC4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C4=CC=CC4)(C4CCCC4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C4=CC=CC4)(C4CCCCC4)O3)C=C2)CC1.CCCC1(C2=CC=CC2)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1 Chemical compound CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4CCCCC4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C4=CC=CC4)(C4CCCC4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC(C4=CC=CC4)(C4CCCCC4)O3)C=C2)CC1.CCCC1(C2=CC=CC2)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1 DQBFZRYKVJSLLO-UHFFFAOYSA-N 0.000 description 1
- QALBRIIYVDYRKE-UHFFFAOYSA-N CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4CCCCC4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC4(CCCC4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC4(CCCC4)O3)C=C2)CC1.CCCC1(C2CCCCC2)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1 Chemical compound CC(=O)N1CCN(C2=CC=C(OCC3COC(C)(C4CCCCC4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC4(CCCC4)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC4(CCCC4)O3)C=C2)CC1.CCCC1(C2CCCCC2)OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1 QALBRIIYVDYRKE-UHFFFAOYSA-N 0.000 description 1
- LDNZTROUWNTCOZ-UHFFFAOYSA-N CC(=O)N1CCN(C2=CC=C(OCC3COC(C)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC4(CCCCC4)O3)C=C2)CC1.CCC1OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1.CCCC1OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1 Chemical compound CC(=O)N1CCN(C2=CC=C(OCC3COC(C)O3)C=C2)CC1.CC(=O)N1CCN(C2=CC=C(OCC3COC4(CCCCC4)O3)C=C2)CC1.CCC1OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1.CCCC1OCC(COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1 LDNZTROUWNTCOZ-UHFFFAOYSA-N 0.000 description 1
- IHYBIHABUSEHNT-UHFFFAOYSA-N CC(C)CN1CCN(C)CC1.CC(C)N1CCN(C)CC1.CCCCN1CCN(C)CC1.CCCN1CCN(C)CC1.CCN1CCN(C)CC1.CCNC.CCOC(=O)N1CCN(C)CC1.CCOC(=O)N1CCN(C)CC1.CN1CCN(C(N)=O)CC1.CN1CCN(C)CC1.CN1CCN(SO(C)O)CC1.CNC.CNC(=O)N1CCN(C)CC1.CNC(=O)N1CCN(C)CC1.CNC(=S)N1CCN(C)CC1.COC(=O)N1CCN(C)CC1 Chemical compound CC(C)CN1CCN(C)CC1.CC(C)N1CCN(C)CC1.CCCCN1CCN(C)CC1.CCCN1CCN(C)CC1.CCN1CCN(C)CC1.CCNC.CCOC(=O)N1CCN(C)CC1.CCOC(=O)N1CCN(C)CC1.CN1CCN(C(N)=O)CC1.CN1CCN(C)CC1.CN1CCN(SO(C)O)CC1.CNC.CNC(=O)N1CCN(C)CC1.CNC(=O)N1CCN(C)CC1.CNC(=S)N1CCN(C)CC1.COC(=O)N1CCN(C)CC1 IHYBIHABUSEHNT-UHFFFAOYSA-N 0.000 description 1
- LPGKTOALOLBBFK-UHFFFAOYSA-N CC.CC1=CC=C(C)C=C1.CC1=CC=CC=C1.CN1CCCC1.CN1CCOCC1.COC1=CC=CC=C1 Chemical compound CC.CC1=CC=C(C)C=C1.CC1=CC=CC=C1.CN1CCCC1.CN1CCOCC1.COC1=CC=CC=C1 LPGKTOALOLBBFK-UHFFFAOYSA-N 0.000 description 1
- BCXILLAVLLWCEJ-QTMGEFODSA-N CCCCCCCC[C@]1(C)OC[C@H](COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1.COC1=CC=C([C@]2(C)OC[C@H](COC3=CC=C(N4CCN(C(C)=O)CC4)C=C3)O2)C=C1I Chemical compound CCCCCCCC[C@]1(C)OC[C@H](COC2=CC=C(N3CCN(C(C)=O)CC3)C=C2)O1.COC1=CC=C([C@]2(C)OC[C@H](COC3=CC=C(N4CCN(C(C)=O)CC4)C=C3)O2)C=C1I BCXILLAVLLWCEJ-QTMGEFODSA-N 0.000 description 1
- UDZYAUJQSOQBMZ-UHFFFAOYSA-N CCCNC(=O)N1CCN(C)CC1.CN1CCN(CC2=CC=CC=C2)CC1.CN1CCN(SO(O)CC2=CC=CC=C2)CC1 Chemical compound CCCNC(=O)N1CCN(C)CC1.CN1CCN(CC2=CC=CC=C2)CC1.CN1CCN(SO(O)CC2=CC=CC=C2)CC1 UDZYAUJQSOQBMZ-UHFFFAOYSA-N 0.000 description 1
- MCUSAHRAWAKHHR-SECBINFHSA-N CC[C@@H](O)COC1=CC=C(Br)C=C1 Chemical compound CC[C@@H](O)COC1=CC=C(Br)C=C1 MCUSAHRAWAKHHR-SECBINFHSA-N 0.000 description 1
- ITQHNWFKRPAHQM-UHFFFAOYSA-N CNC(=O)C1=CC=C(C)C=C1.CNC(=O)C1=CC=C(Cl)C=C1.CNC(=O)C1=CC=C(F)C=C1.CNC(=O)C1=CC=CC=C1 Chemical compound CNC(=O)C1=CC=C(C)C=C1.CNC(=O)C1=CC=C(Cl)C=C1.CNC(=O)C1=CC=C(F)C=C1.CNC(=O)C1=CC=CC=C1 ITQHNWFKRPAHQM-UHFFFAOYSA-N 0.000 description 1
- FZGZDDRZAZBANV-MRVPVSSYSA-N OC[C@@H](O)COC1=CC=C(Br)C=C1 Chemical compound OC[C@@H](O)COC1=CC=C(Br)C=C1 FZGZDDRZAZBANV-MRVPVSSYSA-N 0.000 description 1
Images
















Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Definitions
- the present invention relates to ketoconazole derivatives that are antagonists of the human pregnane X receptor (PXR), methods of preparing the derivatives, uses of the derivatives with drug therapy, and methods of inhibiting tumor cell proliferation and multidrug resistance using inhibitors of PXR.
- PXR human pregnane X receptor
- PXR Pregnane X Receptor
- NR1I2 also termed SXR, PAR
- SXR Pregnane X Receptor
- NR1I2 also termed SXR, PAR
- PXR Pregnane X Receptor
- NR1I2 also termed SXR, PAR
- SXR Pregnane X Receptor
- a large number of commonly encountered environmental toxins e.g., phthalates
- chemotherapy e.g., taxanes
- drug vehicles e.g., DEHP
- co-medications e.g., dexamethasone
- herbals e.g., curcumin, hyperforin
- ketoconazole and related azoles; 11
- suphoraphane (12)
- ecteinascidin-743 (ET-743)
- Ketoconazole was first described as a PXR antagonist by Takeshita et al. (14), and was subsequently shown to disrupt the binding of coregulators (including both coactivators and corepressors) to the surface of PXR in an agonist-dependent fashion (15).
- coregulators including both coactivators and corepressors
- ketoconazole and related azoles were shown to prevent the activation of the receptor both in cell-based assays as well as in a humanized PXR mouse model (16).
- Ketoconazole binds to at least a region outside the ligand-binding pocket.
- the revertant activation function 2 (AF-2) region double mutant of PXR (T248E/K277Q) activates with rifampicin but is not inhibited by ketoconazole (11, 17).
- the present invention is directed to ketoconazole derivatives having the formula:
- the invention also provides a method of preparing a compound of formula (3)
- the invention further provides methods of preventing or reducing proliferation of tumor cells and methods of preventing or reducing multidrug resistance in tumor cells, where the methods comprise contacting the tumor cells with an inhibitor of the human pregnane X receptor (PXR).
- PXR human pregnane X receptor
- the invention also provides pharmaceutical compositions for antagonizing the human pregnane X receptor (PXR) comprising a compound of the present invention and a pharmaceutically acceptable carrier or diluent.
- PXR human pregnane X receptor
- the invention also provides a method of inhibiting the human pregnane X receptor (PXR) in a subject comprising administering to the subject a compound of the present invention in an amount and manner effective to inhibit the human pregnane X receptor (PXR).
- PXR human pregnane X receptor
- FIG. 1A-1H Concentration-response curves for PXR activation with (a) rifampicin, (c) ketoconazole, (e) FL-B-12 and (g) UCL2158H. The concentration-response curves for activated PXR antagonism are shown with (b) rifampicin, (d) ketoconazole, (f) FL-B-12 and (h) UCL2158H.
- FIG. 2A-2C Chemical 2D structures of (A) ketoconazole, (B) UCL2158H and (C) FL-B-12.
- FIG. 4A-4C SKOV-3 cell proliferation in the presence of PXR ligands.
- SKOV-3 cells were treated with (A) rifampicin (0.49-120 ⁇ M) or (B) hyperforin (0.05-1 ⁇ M) or (C) T0901317 (T1317) or GSK1385 (both ranging between 0.05-1 ⁇ M) over a duration ranging between 24-96 h.
- A rifampicin (0.49-120 ⁇ M) or
- B hyperforin
- T0901317 T1317)
- GSK1385 both ranging between 0.05-1 ⁇ M
- the Y-axis represents a Fractional Survival value defined by the Fold survival (S) observed with compound T1317 (denoted, S T1317 ) divided by the Fold Survival observed with compound GSK1385 (denoted, S GSK ).
- the X-axis shows the concentrations for both drugs (e.g., 0.05 means 0.05 ⁇ M for T1317 and for GSK).
- the fractional survival when no drug(s) are present ⁇ S T1317 /S GSK ⁇ by definition is 1. Any positive value thereafter implies a growth advantage for T1317 over GSK.
- cells were subject to MTT assay (see Materials and Methods).
- % survival was calculated from OD 490 ratios of treated divided by control (vehicle treated) wells (minus the blank) ⁇ 100 (expressed and plotted as a percentage growth over control cells). Experiments were repeated four separate times each in triplicate. Individual points represent mean ( ⁇ SD).
- FIG. 5 HepG2 cell proliferation in the presence of Rifampicin and/or Ketoconazole.
- HepG2 cells were treated with rifampicin (0-50 ⁇ M) or ketoconazole (0-50 ⁇ M) or both drugs each at the same concentration over a 48 hour duration.
- rifampicin (0-50 ⁇ M) or ketoconazole (0-50 ⁇ M) or both drugs each at the same concentration over a 48 hour duration.
- MTT assay see Materials and Methods.
- the fractional survival was calculated from OD 490 ratios of treated divided by control (vehicle treated) wells (minus the blank). Experiments were repeated four separate times each in triplicate. Individual points represent mean ( ⁇ SD).
- FIG. 6A-6C SKOV-3 mouse xenografts treated with rifampicin or vehicle.
- Sixteen NOD.SCID mice carrying SKOV-3 xenografts in both flanks were treated with rifampicin three times per week (as indicated by arrows in B) continuously from days 35-60. Tumor volumes were assessed as described in Materials and Methods.
- FIG. 7A-7C Cell cytotoxicity from anti-cancer xenobiotics in SKOV-3 cells pretreated with rifampicin or vehicle.
- SKOV-3 cells were pretreated (for 24 hours) with 15 ⁇ M rifampicin or vehicle and then exposed to (A) ixabepilone (0.1-100 ⁇ M) or (B) paclitaxel (0.05-50 ⁇ M) or (C) SN-38 (0.05-20 ⁇ M) for another 24 hours.
- B(i) and B(ii) show the effects of 3 ⁇ -OH and 6 ⁇ -OH paclitaxel treatment of SKOV-3 cells for 24 hours, respectively.
- Each experiment was performed three times in triplicate. Individual points and Bar, mean ( ⁇ SD). The statistical significance is shown as a *p-value on the figure across the annotated concentration range.
- FIG. 8A-8D PXR activation in LS174T cells induces cell proliferation.
- A Doubling time of LS174T cells in the presence and absence of PXR agonist, rifampicin. * p ⁇ 0.001, student t test.
- B Survival of LS174T cells (as in A), as determined using the MTT assay, in the presence and absence of rifampicin. Data shown as fold survival (values normalized to vehicle control).
- the invention provides a compound having the formula:
- X is H, CHO, COCH 3 , CO—OCH 3 , CO—OC 2 H 5 , CONH 2 , CONHCH 3 , CONHC 2 H 5 , CSNHCH 3 , CH 2 CH 3 , COO—CH 3 , COO—C 2 H 5 , CH 3 , CH 2 —CH(CH 3 ) 2 , CO—NH—(CH 2 ) 2 CH 3 , CH(CH 3 ) 2 , (CH 2 ) 3 CH 3 , (CH 2 ) 2 CH 3 , SO 2 CH 3 , CH 2 C 6 H 5 , or SO 2 CH 2 C 6 H 5 ; and
- each R 1 , R 2 , R 3 , R 4 and R 5 is independently H, F, Cl, Br, I, OCH 3 , OC 2 H 5 , O-alkyl, SH, S-alkyl, NH 2 , NH-alkyl, alkyl, phenyl or heterocyclic;
- the compound is not ketoconazole or 1-(4-(4-((2R,4S)-2-(2,4-dichlorophenyl)-2-methyl-1,3-dioxolan-4-yl)methoxy)phenyl)piperazin-1-yl)ethanone.
- X is H, CHO or COCH 3 .
- A is
- the invention also provides a compound having the formula:
- A is —N ⁇ C ⁇ S; —N(CH 3 ) 2 ; or —NHD, where D is H, CHO, CS—NH 2 , CS—NH—CH 3 , CS—NH—C 2 H 5 , CH 2 CH 3 , or CH 3 ; or —NHCOE, where E is C 2 H 5 , OCH 3 , OC 2 H 5 , NH—CH 3 , NH—C 2 H 5 , CH(G) 2 where G is H, F, Cl, Br or I; or
- each R 1 , R 2 , R 3 , R 4 and R 5 is independently H, F, Cl, Br, I, OCH 3 , OC 2 H 5 , O-alkyl, SH, S-alkyl, NH 2 , NH-alkyl, alkyl, phenyl or heterocyclic; or a pharmaceutically acceptable salt thereof.
- variable A Preferred structures of variable A include:
- B is CH 3 .
- C is
- each R 1 , R 2 , R 3 , R 4 and R 5 is independently H, F, Cl, Br or I, or wherein each R 1 , R 2 , R 3 , R 4 and R 5 is independently H, F, Br or I.
- at least two of R 1 , R 2 , R 3 , R 4 and R 5 are F, Br or I, and the remainder are H.
- at least R 3 is F.
- Examples of the structure of A include:
- the compound can have a structure selected from the group consisting of:
- the compound can also have a structure selected from the group consisting of:
- a preferred compound has the structure:
- the compounds of the present invention exclude compounds UCL2112H, UCL2134D, UCL2135, UCL2202D, UCL2245 and UCL2238.
- the invention also provides a method of preparing a compound of formula (3)
- each R 1 , R 2 , R 3 , R 4 and R 5 is independently H, F, Cl, Br, I, OCH 3 , OC 2 H 5 , O-alkyl, SH, S-alkyl, NH 2 , NH-alkyl, alkyl, phenyl or heterocyclic.
- the method comprises three steps, steps (a), (b) and (c).
- Step (a) comprises reacting (S)-( ⁇ )-glycidol and 4-bromophenol in the presence of a mild base and dimethyl formamide (DMF) to produce a compound of formula (1):
- mild bases examples include, but are not limited to, sodium carbonate, potassium carbonate, lithium carbonate, cesium carbonate, sodium hydride, potassium hydride, and lithium hydride.
- a preferred base is K 2 CO 3 .
- the reaction is carried out between 100° C.-120° C. for 5-10 hours.
- Step (b) comprises reacting the compound of formula (1) with an acetophenone derivative in the presence of p-toluenesulfonic acid monohydrate and either benzene or toluene to produce a compound of formula (2)
- each R 1 , R 2 , R 3 , R 4 and R 5 is independently H, F, Cl, Br, I, OCH 3 , OC 2 H 5 , O-alkyl, SH, S-alkyl, NH 2 , NH-alkyl, alkyl, phenyl or heterocyclic.
- the reaction is carried out at 100° C.-120° C. for 10-30 hours.
- Step (c) comprises reacting the compound of formula (2) with 1-acetylpiperazine in the presence of a palladium catalyst or a copper catalyst to produce the compound of formula (3).
- a palladium catalyst or a copper catalyst examples include, but are not limited to, Pd(dba) 2 , palladium carbonate, palladium acetate, copper acetate and copper carbonate.
- the reaction is carried out between 80° C.-100° C. for 10-24 hours.
- the invention further provides a compound prepared by the method of the present invention.
- the compounds of the present invention have reduced cytotoxicity compared to ketoconazole and/or compared to 1-(4-(4-((2R,4S)-2-(2,4-dichlorophenyl)-2-methyl-1,3-dioxolan-4-yl)methoxy)phenyl)piperazin-1-yl)ethanone.
- Cytotoxicity can be assayed in an in vitro assay using an epithelial cell line, such as for example those described herein in Experimental Details.
- the invention also provides a pharmaceutical composition for antagonizing the human pregnane X receptor (PXR) comprising a compound of the present invention and a pharmaceutically acceptable carrier or diluent.
- Pharmaceutically acceptable carriers and diluents encompasses any of the standard pharmaceutical carriers or diluents, such as, for example, a sterile isotonic saline, phosphate buffered saline solution, water, and emulsions, such as an oil/water or water/oil emulsions.
- the invention further provides a method of preventing or reducing proliferation of tumor cells comprising contacting the tumor cells with an inhibitor of the human pregnane X receptor (PXR).
- the invention also provides a method of preventing or reducing multidrug resistance in tumor cells comprising contacting the tumor cells with an inhibitor of the human pregnane X receptor (PXR).
- the inhibitor of the human pregnane X receptor (PXR) can be administered to a subject in an amount and manner effective to inhibit the human pregnane X receptor (PXR) on the tumor cells.
- the tumor cells can be epithelial or hematologic tumor cells.
- the tumor cells can be ovarian, colon, cervical or hepatic tumor cells.
- Preferred inhibitors of the human pregnane X receptor (PXR) include compounds of the present invention.
- the effectiveness of different compounds on tumor cells can be assayed using tumor cells grown in in vitro cell culture or in in vivo xenograft models.
- the invention also provides a method of inhibiting the human pregnane X receptor (PXR) in a subject comprising administering to the subject a compound of the present invention in an amount and manner effective to inhibit the human pregnane X receptor (PXR).
- administration of the PXR inhibitor increases the transport of a therapeutic drug across the blood-brain barrier and/or increases bioavailability of a therapeutic drug administered through the gastrointestinal tract and/or decreases PXR-mediated cell proliferation and/or decreases PXR-mediated neoplastic transformation.
- the compounds of the present invention can be administered to subjects using routes of administration known in the art.
- the administration can be systemic or localized to a tumor site.
- Routes of administration include, but are not limited to, intravenous, intramuscular, intrathecal or subcutaneous injection, oral or rectal administration, and injection into a tumor site.
- Dulbecco's modified eagle medium DMEM
- LipofectamineTM 2000 heat-inactivated fetal bovine serum (FBS), trypsin-EDTA (0.25%), and penicillin-streptomycin were purchased from GIBCO/Invitrogen (Carlsbad, Calif.).
- Charcoal/dextran treated fetal bovine serum (FBS) was purchased from Hyclone (Logan, Utah).
- HepG2 cells were obtained from ATCC (Manassas, Va.).
- White TC-surface 384-well plates were purchased from Perkin Elmer (Boston, Mass.). Luciferase substrate (STEADY-GLO®) was purchased from Promega (Madison, Wis.). Alamar-Blue reagent was purchased from Trek Diagnostics (Clevland, Ohio). Rifampicin, ketoconazole and midazolam were purchased from Sigma (St. Louis, Mo.). Human liver microsome and 1-hydroxymidazolam were obtained from BD Biosciences (San Jose, Calif.).
- HepG2 cells were cultured in DMEM containing 10% FBS.
- a transfection mixture containing 1 ⁇ g of PXR-pcDNA3 plasmid DNA, 20 ⁇ g of Cyp3A-Luc plasmid DNA and 90 ⁇ l of LipofectamineTM 2000 was prepared in 1-ml of serum-free DMEM. The transfection mixture was then applied to the cells in fresh medium (20 ml per flask) and incubated at 37° C. (5% CO 2 ) overnight. The transfected cells were trypsinized and cryopreserved for long-term storage.
- % ⁇ ⁇ Act Compound ⁇ ⁇ signal - Blank ⁇ ⁇ signal Total ⁇ ⁇ signal - Blank ⁇ ⁇ signal ⁇ 100 ⁇ %
- UCL2112H (21) did not transactivate PXR nor inhibit the rifampicin-mediated activation of the receptor
- UCL2238 (24) was a potent activator of PXR but was not an inhibitor.
- other compounds behaved like ketoconazole, being a weak activator of PXR and a moderate antagonist of activated PXR.
- UCL2158H (21) and UCL2202D (21) appeared to be the most potent inhibitors of PXR.
- UCL2158H appeared to be a better PXR antagonist than UCL2202D since UCL2202D moderately activated PXR (43%) while UCL2158H did not elicit a significant PXR activation (10%).
- the FL compounds in Table 1 are fluorinated derivatives of ketoconazole lacking the imidazole group, synthesized at Albert Einstein College of Medicine. Five FL compounds were tested and all showed a varying degree of PXR antagonism. However, except for FL-B-12, all showed a moderate potent activation of PXR in addition to their activities against rifampicin-mediated PXR activation.
- Ketoconazole a prototypical reversible inhibitor of CYP3A4 (and thus midazolam 1-hydroxylase activity), completely inhibits conversion of midazolam to its metabolite (103% inhbition with an IC 50 of 0.020 ⁇ M) with no indication of time dependency (no IC 50 change).
- UCL2158H was shown to be a weak inhibitor (IC 50 of 10.8 or 19.6 ⁇ M) of CYP3A4 which demonstrated no time-dependency.
- FL-B-12 appears to be a very weak activator of CYP3A4 (IC 50 >40M), again without a time-dependency of its CYP3A4 inhibition.
- the elimination of midazolam group in these analogues markedly improved CYP3A4 inhibition compared to ketoconazole without introducing new liability (time-dependent inhibition).
- FL-B-12 was the least cytotoxic ( ⁇ 98% viability across all cell lines tested) in the effective concentration range for PXR inhibition (e.g., two times the IC 50 ⁇ 30 ⁇ M). However, in the same concentration range, ketoconazole is cytotoxic (viability is ⁇ 65-75% across all cell lines tested).
- CREMOPHOR®EL and rifampicin were obtained from Sigma Chemical Co. (St. Louis, Mo.). Hyperforin was purchased from Cayman Chemical Co. (Ann Arbor, Mich.). GlaxoSmithKline (Dr. J. Collins, Research Triangle, Durham, N.C.) and SN-38 by Enzon Pharmaceuticals (Dr. O, Sachdev, Piscataway, N.J.) supplied T0901317 (T1317) and GSK1385, respectively. Bristol-Myers Squibb (Dr. F. Lee, Princeton, N.J.) supplied BMS-247550 (ixabepilone). Clinical grade paclitaxel was obtained from the Albert Einstein College of Medicine (AECOM) Pharmacy (Bronx, N.Y.).
- AECOM Albert Einstein College of Medicine
- Paclitaxel was formulated in CREMOPHOR® EL and hyperforin in methanol. All the other compounds were dissolved in 100% dimethyl sulfoxide (DMSO) and stored at ⁇ 20° C. The final concentration of DMSO was ⁇ 0.2% in all experiments.
- DMSO dimethyl sulfoxide
- the polyclonal PXR antibody used for Western blot analysis was a kind gift from Dr. R. K Tyagi (Jawaharlal University, New Delhi, India).
- the polyclonal PXR antibody used for immunohistochemistry was purchased from BioLegend, Inc (San Diego, Calif.).
- HepG2 (ATCC, Mansassas, Va.) and OVCAR-8 cells (kind gift from I. David Goldman, Bronx, N.Y.) were maintained in RPMI 1640 supplemented with 10% FBS.
- SKOV-3 cells were a gift from Dr. Gloria Huang (Bronx, N.Y.) and were maintained in MEM alpha supplemented with 13% FBS. When indicated, cells were propagated in charcoal-adsorbed sera and phenol-free media.
- the presence of PXR in SKOV-3 and HepG2 nuclear protein fraction was determined by Western blot analysis. Protein concentration was determined via the modified Bradford assay using the NanoDrop ND-100 Spectrophotometer (Wilmington, Del.). Nuclear fraction was isolated using the BioSource Nuclear Extraction Kit (Invitrogen, Carlsbad, Calif.) per manufacturer's instructions. 40-200 ⁇ g of nuclear protein was resolved by 12% SDS-PAGE and transferred to nitrocellulose. The blot was probed with a 1:10,000 dilution of a polyclonal PXR antibody as described previously (25) and developed using the LI-COR Odyssey Infrared Imager (Lincoln, Nebr.). The Western blot analysis was performed in duplicate.
- HepG2 cells were processed into slides using histogel (Richard-Allan Scientific, Kalamazoo, Mich.). The slides were placed in a 59° C. oven in the morning of the experiments. For cells and tissue specimens, after de-waxing and re-hydrating the slides, slides were placed in a sodium citrate solution, pH 6.0 (Vector labs, Burlingame, Calif.) for 20 minutes. The cells were then sequentially blocked with a 3% peroxidase blocking solution and 2% BSA and 5% donkey serum in TBS for 1-2 hours.
- the forward primer for PXR was 5′-GAGCTGATGGACGCTCAG-3′ (SEQ ID NO:1) and reverse 5′-TGGCAAAGCTGATGATGC-3′ (SEQ ID NO:2).
- GAPDH was used as an internal control with the following forward and reverse primers: 5′-TGCATCCTGCACCACCAAC-3′ (SEQ ID NO:3) and 5′-CGCCTGCTTCACCACCTTC-3′ (SEQ ID NO:4), respectively.
- Real time PCR for cDNA quantification was performed using TaqMan universal PCR master mix and TaqMan probes using, VIC as the 5′ reporter fluorochrome and tetramethylrhodamine (TAMRA) as the 3′ quencher fluorochrome.
- the reference gene, PPIA (Cyclophilin A), was a VIC/MGB probe (Applied Biosystems, Foster City, Calif.).
- the standard curves for PXR, CYP3A4, CYP2B6, UGT1A1, MDR1, and MRP-2 cDNA were constructed to ensure linearity in the concentration range studied.
- the PXR assay (ID #Hs01114267_m1) and the UGT1A1 assay (ID # Hs02511055_s1) were ordered through Applied Biosystems.
- the CYP3A4 forward primer sequence was 5′-TGGTGAATGAAACGCTCAGATT-3′ (SEQ ID NO:5)
- the CYP3A4 reverse primer sequence was 5′-CATCTTTTTTGCAGACCCTCTCA-3′ (SEQ ID NO:6)
- the CYP3A4 probe sequence was 5′-VIC-TTCCCAATTGCTATGAGAC (SEQ ID NO:7)-TAMRA-3′, all spanning exon junctions, thus preventing amplification of genomic DNA.
- the CYP2B6 forward primer sequence was 5′-GACCGAGCCAAAATGCCATA-3′ (SEQ ID NO:8)
- the reverse primer sequence was 5′-GGTCGGAAAATCTCTGAATCTCA-3′ (SEQ ID NO:9)
- the probe sequence was 5′-VIC-ACAGAGGCAGTCATC (SEQ ID NO:10)-TAMRA-3′.
- the MDR-1 Forward primer sequence was 5′GGAAGCCAATGCCTATGACTTTA-3′ (SEQ ID NO:11), the reverse primer sequence was 5′-ACTCAACTGGGCCCCTCTCT-3′ (SEQ ID NO:12), and the probe sequence was 5′-VIC-CATGAAACTGCCTCATAAATTTGACACCCTG (SEQ ID NO:13)-TAMRA-3′.
- the MRP-2 forward primer sequence was 5′-GGCTGTTGAGCGAATAACTGAGT-3′ (SEQ ID NO:14), the reverse primer sequence was 5′-GCCTTTGCTGGGCCAAT-3′ (SEQ ID NO:15), and the probe sequence was 5′-VIC-AAAATGAGGCACCCTGGGTGACTGATAAGA (SEQ ID NO:16)-TAMRA-3′.
- Amplification was detected and analyzed using the ABI PRISM 7700 sequence detector with SDS 2.1 analysis software (Applied Biosystems, Foster City, Calif.). The relative fold increase in mRNA in samples compared with controls was calculated using the comparative CT method.
- SKOV-3 cells were incubated in complete media with and without 20 ⁇ M rifampicin for 48 hours. At 48 hours, cells were trypsinized, and aliquots of 5 ⁇ 10 3 cells were plated in 96-well plates in triplicates with or without 20 ⁇ M of rifampicin. After 24 hours of incubation at 37° C., cells were treated with serial dilutions of the chemotherapeutic agent. After a further 48 hours of incubation, the MTT assay was performed as described previously (19).
- SKOV-3 cells were implanted by subcutaneous route into both flanks of NOD.SCID mice.
- Sixteen mice were selected from this pool (mean tumor volume [ ⁇ SD] 6.1 [ ⁇ 3.4]) for rifampicin injections and another sixteen (mean tumor volume [ ⁇ SD] 10.1 [ ⁇ 4.8]) for control injections.
- Rifampicin was formulated in 30% polyethylene glycol and dosed by direct venous (tail vein) injection at 40 mg/kg/day over three consecutive days repeated every 7 days. Control intravenous injections consisted of 30% polyethylene glycol.
- the tumor volume was calculated twice a week using the following formula: Length (mm) ⁇ [Width (mm)] 2 ⁇ /6 (25).
- PXR is expressed in two ovarian cancer cell lines, SKOV-3 and OVCAR-8.
- MB-468 and SKOV3 cells express PXR mRNA.
- SKOV-3 cells express PXR mRNA approximately 3.8-fold over that observed in OVCAR-8 cells.
- PXR is expressed at the protein level in both SKOV-3 and OVCAR-8 cells; however, no such expression is observed for MB-468 (MDA-MB-468) cells.
- the latter cell line has been shown to have very low or undetectable levels of PXR mRNA (26).
- PXR is clearly detected by immunochemistry as speckled bodies within the nucleus of human ovarian carcinoma tissue.
- PXR Activation Induces PXR Target Genes in SKOV-3 Cells.
- PXR protein kinase inhibitor
- the latter is an investigational anti-cholesterol drug that is a dual PXR/LXR agonist (27, 28); however, at a concentration range between 0-1.0 ⁇ M serves as a more potent PXR agonist.
- Rifampicin (0.49-60 ⁇ M), a known PXR agonist, induces a significant increase in cell survival and the longer the duration of exposure to rifampicin, the greater is the effect on cell survival (compare 48, 72 and 96 hr exposures, FIG. 4A ) (29). Similar data using rifampicin has been shown for OVCAR-8 cells. Hyperforin (0.05-1 ⁇ M), a known potent PXR agonist, also induces a significant increase in cell survival and the longer the duration of exposure to hyperforin, the greater is the effect on cell survival (compare 48, 72 and 96 hr exposures, FIG. 4B ) (30).
- SKOV-3 cells were treated with T1317 or GSK1385 (0-1.0 ⁇ M) for 48 hours and then MTT assay was performed to determine cell survival fraction. The data were then expressed as fold survival when treated with the PXR agonist, T1317 normalized to survival at the same concentration of GSK1385. There is a significant increase in cell survival with T1317 over the concentration range 0.05-1.0 ⁇ M, suggesting that PXR activation directly contributes to cell survival ( FIG. 4C ).
- HepG2 cells were treated with rifampicin, ketoconazole or a concentration range of ketoconazole in the presence of rifampicin for 48 hours.
- the MTT survival assay was performed to determine viability of cells.
- rifampicin induces a concentration dependent proliferation of cells.
- Ketoconazole has minimal effects until a concentration of 12.5 ⁇ M, when ⁇ 15% of cells are non-viable and by 50 ⁇ M, >35% of cells are non-viable.
- cell proliferation is clearly decreased ( FIG. 5 ).
- NOD.SCID mice carrying SKOV-3 xenografts were treated with or without rifampicin.
- Rifampicin treated mice consistently had significantly larger tumors on both visual inspection at necropsy (day 60) as well as by tumor weight ( FIG. 6A ).
- the tumor volumes were assessed twice weekly and these values were significantly higher for the rifampicin treated group as compared with controls (days 45-60) ( FIG. 6B ).
- Assessment for proliferation in tumor cells was performed using Ki67 antibody.
- the resistance to ixabepilone (0.1-50 ⁇ M) in the presence of rifampicin is best represented as % increased survival relative to ixabepilone without rifampicin. This averages to ⁇ 20% ( FIG. 7A ).
- the resistance index induced by rifampicin is ⁇ 10-fold (IC 50 with rifampicin (R)/IC 50 without rifampicin (no R) ⁇ 40 ⁇ M/4 ⁇ M) ( FIG. 7B ).
- the resistance index induced by rifampicin is ⁇ 17.5 (IC 50 (R)/IC 50 (no R) ⁇ 3.5/0.2).
- FIG. 8A shows the doubling time of LS174T cells in the presence and absence of the PXR agonist, rifampicin.
- Vector Li-CMV-GFP
- PXR knockdown Li-CMV-PXRshRNA-GFP
- LS174T cells were seeded into 24-well plates at a density of 17,500 viable cells/well. After seeding, cells were treated with rifampicin or vehicle control as indicated. At 24, 48 and 72 hours, cells were trypsinized and counted using trypan blue staining. Each assay was performed as six replicates and experiments were repeated eight times.
- FIG. 8B shows survival of LS174T cells as determined using the MTT assay, in the presence and absence of rifampicin. The data are shown as fold survival (values normalized to vehicle control). Each assay was performed four separate times each in triplicate.
- clinically palpable ( ⁇ 15-20 mm 3 ) tumors on each flank were treated with rifampicin (i.p., 3 days/week till day 50). Tumors were measured every other day and plotted in 5-day intervals.
- all animals were sacrificed, and tumors excised and weighed (FIG. 8 C(II)).
- Transwell migration assay was determined at three time points, 5, 12 and 24 hrs in the presence or absence of rifampicin (25 ⁇ M) or EGF (5 nM) or both ( FIG. 8D ). Each assay was performed four separate times each in triplicate.
- the vehicle control for all experiments was 0.2% DMSO.
- PXR activation has the potential to interfere with drug therapy in a number of ways.
- PXR is a key regulator of the permissibility of drugs across the blood-brain barrier (32, 35).
- PXR activation upregulates p-glycoprotein, as well as other transporters such as MRP2. Since many commonly used anticancer drugs are transported by these efflux pumps, drugs are immediately pumped back into the circulation. Hence, activation of PXR tightens the blood-brain barrier and limits influx of drugs into brain tumors. For patients with primary brain tumors or metastases to the brain, this represents a hurdle for effective therapeutics.
- PXR is also a know orphan receptor engaged in drug-drug interaction effects.
- St. John's Wort hyperfoin, activates PXR and lowers the therapeutic drug concentrations of CPT-11, a FDA approved drug for the treatment of colon cancer (36).
- Controlled inhibition of PXR activation would thus preserve inadvertent lowering of beneficial drug concentrations. This would serve to lower initial drug doses for patients and reduce the cost of chemotherapy.
- PXR activation in the gut can lower drug bioavailability. Inhibition of PXR in the context of oral drug therapy can improve overall drug availability, and reduce waste and costs associated with therapy.
- PXR activation induces cell proliferation and multi-drug resistance in ovarian cancer cells.
- the data regarding PXR mediated induction of drug resistance demonstrates that PXR activation can increase the cytotoxic threshold of cells to chemotherapy.
- Down-regulation of PXR has been reported to inhibit endometrial cancer cell growth and induce apoptosis (33, 34).
- PXR activation was also shown to induce cell proliferation in colon carcinoma cells.
- ketoconazole analogs have been developed that antagonize activated PXR yet have reduced cytotoxicity compared with ketoconazole. Accordingly, PXR-inhibiting compounds of the present invention are expected to be useful for improving delivery of chemotherapy to the brain, preserving concentrations of active drugs in chronic therapy, preventing drug-drug interactions during chemotherapy, and inhibiting potential growth and drug resistance of tumors.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description

the method comprising:
-
- (a) reacting (S)-(−)-glycidol and 4-bromophenol in the presence of a mild base and dimethylformamide (DMF) to produce a compound of formula (1)

-
- (b) reacting the compound of formula (1) with an acetophenone derivative in the presence of p-toluenesulfonic acid monohydrate and either benzene or toluene to produce a compound of formula (2)

-
-
- wherein each R1, R2, R3, R4 and R5 is independently H, F, Cl, Br, I, OCH3, OC2H5, O-alkyl, SH, S-alkyl, NH2, NH-alkyl, alkyl, phenyl or heterocyclic; and
- (c) reacting the compound of formula (2) with 1-acetylpiperazine in the presence of a palladium catalyst or a copper catalyst to produce the compound of formula (3).
-

wherein each R1, R2, R3, R4 and R5 is independently H, F, Cl, Br, I, OCH3, OC2H5, O-alkyl, SH, S-alkyl, NH2, NH-alkyl, alkyl, phenyl or heterocyclic;


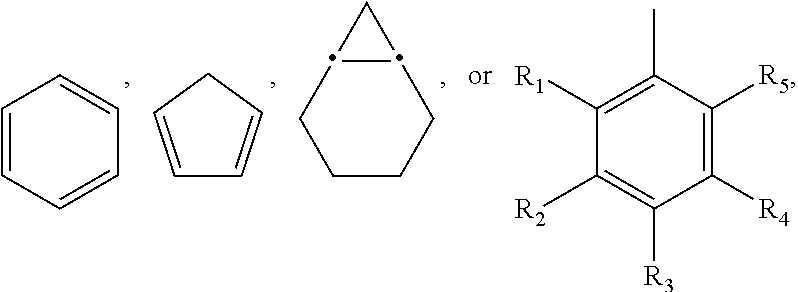
wherein each R1, R2, R3, R4 and R5 is independently H, F, Cl, Br, I, OCH3, OC2H5, O-alkyl, SH, S-alkyl, NH2, NH-alkyl, alkyl, phenyl or heterocyclic; or a pharmaceutically acceptable salt thereof.


wherein each R1, R2, R3, R4 and R5 is independently H, F, Cl, Br or I, or wherein each R1, R2, R3, R4 and R5 is independently H, F, Br or I. Preferably, at least two of R1, R2, R3, R4 and R5 are F, Br or I, and the remainder are H. Preferably, at least R3 is F.



















wherein each R1, R2, R3, R4 and R5 is independently H, F, Cl, Br, I, OCH3, OC2H5, O-alkyl, SH, S-alkyl, NH2, NH-alkyl, alkyl, phenyl or heterocyclic. The method comprises three steps, steps (a), (b) and (c).

Examples of mild bases that can be used include, but are not limited to, sodium carbonate, potassium carbonate, lithium carbonate, cesium carbonate, sodium hydride, potassium hydride, and lithium hydride. A preferred base is K2CO3. Preferably, the reaction is carried out between 100° C.-120° C. for 5-10 hours.

wherein each R1, R2, R3, R4 and R5 is independently H, F, Cl, Br, I, OCH3, OC2H5, O-alkyl, SH, S-alkyl, NH2, NH-alkyl, alkyl, phenyl or heterocyclic. Preferably, the reaction is carried out at 100° C.-120° C. for 10-30 hours.
CDMSO and Ccompound are concentrations of 1-hydroxymidazolam in the vehicle control and a compound tested, respectively. A plot of concentration-% Inh was then created and from the plot, IC50 and Maximum % Inhibition were reported.
Synthesis of Ketoconazole Analogues

Synthesis of FL-B-12 and Derivatives


| TABLE 1 |
| Maximum activation and inhibition of PXR transactivation by |
| ketoconazole and its derivatives. |
| Maximum % Activation | Maximum % Inhibition | |
| Substance (Ref) | Observed | Observed |
| |
20 | 93 |
| UCL2112H (21) | 0 | 0 |
| UCL2134D (23) | 2 | 82 |
| UCL2135 (23) | 10 | 32 |
| UCL2158H (21) | 10 | 109 |
| UCL2202D (21) | 43 | 108 |
| UCL2245 (24) | 2 | 58 |
| UCL2238 (24) | 91 | 1 |
| FL-B-12 | 10 | 103 |
| FL-B-15 | 36 | 90 |
| FL-B-24 | 50 | 75 |
| FL-B-31 | 33 | 59 |
| FL-B-45 | 72 | 48 |
| Rifampicin | 107 | 18 |
| EC50, concentration required for activation of PXR to 50% of maximum as seen with 10 μM rifampicin; | ||
| Emax, maximal activation observed compared to that seen with 10 μM rifampicin; | ||
| IC50, concentration required to antagonize 10 μM rifampicin mediated PXR activation by 50%; | ||
| Imax, maximal inhibition of PXR activation with 10 μM. | ||
| TABLE 2 |
| Non-linear regression analysis of PXR activation and PXR inhibition |
| by rifampicin and ketoconazole, FL-B-12 and UCL2158H. |
| PXR Activation | PXR Inhibition |
| Substance | EC50 | Emax | IC50 | Imax | ||
| Ketoconazole | >50 μM | — | 18.73 μM | 93% | ||
| UCL2158H | >50 μM | — | 14.61 |
128% | ||
| FL-B-12 | >50 μM | — | 13.68 μM | 104% | ||
| Rifampicin | 0.78 μM | 94% | >50 μM | — | ||
| IC50: Concentration required to inhibit 50% of effect (activation by rifampicin); | ||||||
| Imax: maximal inhibition of PXR activation with 10 μM rifampicin. | ||||||
| TABLE 3 |
| Inhibition of midazolam-1-hyxroxylase activity by ketoconazole, |
| FL-B-12 and UCL2158H in human liver microsome with and without |
| preincubation with the test articles. |
| −preincubation | +preincubation |
| Maximum | Maximum | |||
| Substance | IC50 | % Inhibition | IC50 | % Inhibition |
| Ketoconazole | 0.020 μM | 103% | 0.026 μM | 102% |
| FL-B-12 | >40 μM | 20% | >40 μM | 37% |
| UCL2158H | 10.8 μM | 80% | 19.6 μM | 67% |
- (1) Chawla, A., Repa, J. J., Evans, R. M., and Mangelsdorf, D. J. (2001) Nuclear receptors and lipid physiology: opening the X-files. Science 294, 1866-70.
- (2) Gronemeyer, H., Gustafsson, J. A., and Laudet, V. (2004) Principles for modulation of the nuclear receptor superfamily. Nat
Rev Drug Discov 3, 950-64. - (3) Mangelsdorf, D. J., and Evans, R. M. (1995) The RXR heterodimers and orphan receptors. Cell 83, 841-50.
- (4) Mangelsdorf, D. J., Thummel, C., Beato, M., Herrlich, P., Schutz, G., Umesono, K., Blumberg, B., Kastner, P., Mark, M., Chambon, P., and et al. (1995) The nuclear receptor superfamily: the second decade. Cell 83, 835-9.
- (5) Ingraham, H. A., and Redinbo, M. R. (2005) Orphan nuclear receptors adopted by crystallography. Curr
Opin Struct Biol 15, 708-15. - (6) McDonnell, D. P., Connor, C. E., Wijayaratne, A., Chang, C. Y., and Norris, J. D. (2002) Definition of the molecular and cellular mechanisms underlying the tissue-selective agonist/antagonist activities of selective estrogen receptor modulators. Recent Prog Horm Res 57, 295-316.
- (7) Shiau, A. K., Barstad, D., Loria, P. M., Cheng, L., Kushner, P. J., Agard, D. A., and Greene, G. L. (1998) The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 95, 927-937.
- (8) Kliewer, S. A., Moore, J. T., Wade, L., Staudinger, J. L., Watson, M. A., Jones, S. A., McKee, D. D., Oliver, B. B., Willson, T. M., Zetterstrom, R. H., Perlmann, T., and Lehmann, J. M. (1998) An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell 92, 73-82.
- (9) Fayard, E., Auwerx, J., and Schoonjans, K. (2004) LRH-1: an orphan nuclear receptor involved in development, metabolism and steroidogenesis. Trends Cell Biol 14, 250-60.
- (10) Bertilsson, G., Heidrich, J., Svensson, K., Asman, M., Jendeberg, L., Sydow-Backman, M., Ohlsson, R., Postlind, H., Blomquist, P., and Berkenstam, A. (1998) Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proceedings of the National Academy of Sciences of the United States of America 95, 12208-13.
- (11) Wang H, Huang H, Li H, Teotico D G, Sinz M, Baker S D, Staudinger J, Kalpana G, Redinbo M R, Mani S (2007) Activated Pregnenolone X-Receptor Is a Target for Ketoconazole and Its Analogs. Clin Cancer Res 13:2488-2495.
- (12) Zhou C, Poulton E-J, Grun F, Bammler T K, Blumberg B, Thummel K E, Eaton D L (2007) The dietary isothiocyanate sulforaphane is an antagonist of the human steroid and xenobiotic nuclear receptor. Mol Pharmacol 71:220-9.
- (13) Synold T W, Dussault I, Forman B M (2001) The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat Med 7:584-90.
- (14) Takeshita A, Taguchi M, Koibuchi N, Ozawa Y (2002) Putative role of the orphan nuclear receptor SXR (steroid and xenobiotic receptor) in the mechanism of CYP3A4 inhibition by xenobiotics. J Biol Chem 277:32453-8.
- (15) Huang H, Wang H, Sinz M, Zoeckler M, Staudinger J, Redinbo M R, Teotico D G, Locker J, Kalpana G V, Mani S (2007) Inhibition of drug metabolism by blocking the activation of nuclear receptors by ketoconazole. Oncogene 26: 258-68.
- (16) Xue Y C E, Zuercher W J, Willson T M, Collins J L, Redinbo M R. (2007) Crystal structure of the PXR-T1317 complex provides a scaffold to examine the potential for receptor antagonism. Bioorg Med. Chem. 15:2156-66.
- (17) Ekins S, Chang C, Mani S, Krasowski M D, Reschly E J, Iyer M, Kholodovch V, Ai Ni, William J W, Sinz M, Swaan P W, Patel R, Bachman K. (2007) Human pregnane x receptor antagonists and agonists define molecular requirements for different binding sites. Mol. Pharmacol. September; 72(3):592-603.
- (18) Estébanez-Perpiñá E, Arnold A A, Nguyen P, Rodrigues E D, Mar E, Bateman R, Pallai P, Shokat K M, Baxter J D, Guy R K, Webb P, Fletterick R J (2007) A surface on the androgen receptor that allosterically regulates coactivator binding. Proc Natl Acad Sci USA. October 9; 104(41):16074-9.
- (19) Wu K M, Wang C G, D'Amico M, Lee R J, Albanese C, Pestell, R, Mani S. (2002) Flavopiridol and Trastuzumab Synergistically Inhibit Proliferation of Breast Cancer Cells: Association with Selective Cooperative Inhibition of Cyclin D1 Dependent Kinase and Akt Signaling Pathways. Mol Cancer Ther. 1: 695-706.
- (20) Wu K M, Wang C G, D'Amico M, Albanese C, Pestell, R, Mani S. (2005) Flavopiridol synergizes with UCN-01: Role of surviving in reversal of resistance to UCN-01. Invest New Drugs 23: 299-309.
- (21) Power E C, Ganellin C R, Benton C H (2006) Partial structures of ketoconazole as modulators of the large conductance calcium-activated potassium channel (BKCa) Bioorganic & Medicinal Chemistry Letters 16: 887-890.
- (22) Verras, A.; Alian, A.; de Montellano P R. (2006) Cytochrome P450 active site plasticity: attenuation of imidazole binding in cytochrome P450(cam) by an L244A mutation. Protein Eng Des Sel. 19(11):491-6. Epub 2006 Aug. 30.
- (23) Heeres J, Backx, L. J.; Mostmans, J. H.; Van Cutsem, J. (1979) Antimycotic imidazoles. part 4. Synthesis and antifungal activity of ketoconazole, a new potent orally active broad-spectrum antifungal agent. J Med. Chem. 22: 1003-5.
- (24) Pelayo Camps, Xavier FarrésMa Luisa García, Joan Ginesta, Jaume Pascual, David Mauleón and Germano Carganico. (1995) Stereoselective synthesis of both enantiomers of ketoconazole from (R)- and (S)-epichlorohydrin Tetrahedron: Asymmetry 6: 283.
- (25) Lu D, Zhang H, Koo H, et al. A Fully Human Recombinant IgG-like Bispecific Antibody to Both the Epidermal Growth Factor Receptor and the Insulin-like Growth Factor Receptor for Enhanced Antitumor Activity. JBC 2005; 280:19665-72.
- (26) Miki Y, Suzuki T, Kitada K, et al. Expression of the Steroid and Xenobiotic Receptor and Its Possible Target Gene, Organic Anion Transporting Polypeptide-A, in Human Breast Carcinoma. Cancer Res 2006; 66:535-42.
- (27) Mitro N, Vargas L, Romeo R, et al. T0901317 is a potent PXR ligand: implications for the biology ascribed to LXR. FEBS Lett 2007; 581:1721-6.
- (28) Xue Y, Chao E, Zuercher W J, et al. Crystal structure of the PXR-T1317 complex provides a scaffold to examine the potential for receptor antagonism. Bioorg Med Chem 2007; 15:2156-66.
- (29) Watkins R E, Noble S M, Redinbo M R. Structural insights into the promiscuity and function of the human pregnane X receptor. Curr Opin Drug Discov Devel 2002; 5:150-8.
- (30) Watkins R E, Maglich J M, Moore L B, et al. 2.1 A crystal structure of human PXR in complex with the St. John's wort compound hyperforin. Biochemistry 2003; 42:1430-8.
- (31) Mani S, Huang H, Sundarababu S, et al. Activation of the steroid and xenobiotic receptor (human pregnane X receptor) by nontaxane microtubule-stabilizing agents. Clin Cancer Res. 2005 Sep. 1; 11(17):6359-69.
- (32) Bauer B, Hartz A M, Fricker G, et al. Pregnane X receptor up-regulation of P-glycoprotein expression and transport function at the blood-brain barrier. Mol Pharm 2004; 66:413-9.
- (33) Masuyama H, Nakatsukasa H, Takamoto N, et al. Down-regulation of Pregnane X Receptor Contributes to Cell Growth Inhibition and Apoptosis by Anti-cancer Agents in Endometrial Cancer Cells. Mol. Pharmacol. 2007 October; 72(4):1045-53. Epub 2007 Jul. 17.
- (34) Masuyama H, Hiramatsu Y, Kodama, et al. Expression and Potential Roles of Pregnane X Receptor in Endometrial Cancer. J Clin Endo & Metab 2003; 88:4446-54.
- (35) Bauer B, Yang X, Hartz A M, Olson E R, Zhao R, Kalvass J C, Pollack G M, Miller D S. In vivo activation of human pregnane X receptor tightens the blood-brain barrier to methadone through P-glycoprotein up-regulation. Mol. Pharmacol. 2006 October; 70(4):1212-9. Epub 2006 Jul. 12.
- (36) Mannel M. Drug interactions with St John's wort: mechanisms and clinical implications. Drug Saf. 2004; 27(11):773-97. Review.
Claims (14)















Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/735,368 US8669260B2 (en) | 2008-02-29 | 2009-01-27 | Ketoconazole-derivative antagonist of human pregnane X receptor and uses thereof |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US6768808P | 2008-02-29 | 2008-02-29 | |
| PCT/US2009/000524 WO2009110955A2 (en) | 2008-02-29 | 2009-01-27 | Ketoconazole-derivative antagonists of human pregnane x receptor and uses thereof |
| US12/735,368 US8669260B2 (en) | 2008-02-29 | 2009-01-27 | Ketoconazole-derivative antagonist of human pregnane X receptor and uses thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| US20110105522A1 US20110105522A1 (en) | 2011-05-05 |
| US8669260B2 true US8669260B2 (en) | 2014-03-11 |
Family
ID=41056506
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/735,368 Active 2029-08-31 US8669260B2 (en) | 2008-02-29 | 2009-01-27 | Ketoconazole-derivative antagonist of human pregnane X receptor and uses thereof |
Country Status (2)
| Country | Link |
|---|---|
| US (1) | US8669260B2 (en) |
| WO (1) | WO2009110955A2 (en) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3049084A4 (en) * | 2013-09-25 | 2017-03-15 | Cortendo AB (publ) | Novel functionalized 5-(phenoxymethyl)-1,3-dioxane analogs exhibitng cytochrome p450 inhibition |
| CA3073228A1 (en) | 2017-08-20 | 2019-02-28 | University Of Connecticut | Azole analogues and methods of use thereof |
| US12435067B2 (en) | 2019-08-09 | 2025-10-07 | University Of Connecticut | Truncated itraconazole analogues and methods of use thereof |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4590282A (en) | 1984-09-24 | 1986-05-20 | Sandoz Ltd. | Pest control agents |
| US5495052A (en) | 1994-09-29 | 1996-02-27 | Arco Chemical Technology, L.P. | Process for producing enantiomerically enriched guaifenesin |
| US6376514B1 (en) | 2000-10-17 | 2002-04-23 | The Procter & Gamble Co. | Substituted six-membered heterocyclic compounds useful for treating multidrug resistance and compositions and methods thereof |
| US6630475B2 (en) | 2000-05-26 | 2003-10-07 | Schering Corporation | Adenosine A2a receptor antagonists |
| WO2007078615A2 (en) | 2005-12-15 | 2007-07-12 | Cavit Sciences, Inc | Methods and compositions for treatment of cancer |
-
2009
- 2009-01-27 WO PCT/US2009/000524 patent/WO2009110955A2/en not_active Ceased
- 2009-01-27 US US12/735,368 patent/US8669260B2/en active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4590282A (en) | 1984-09-24 | 1986-05-20 | Sandoz Ltd. | Pest control agents |
| US5495052A (en) | 1994-09-29 | 1996-02-27 | Arco Chemical Technology, L.P. | Process for producing enantiomerically enriched guaifenesin |
| US6630475B2 (en) | 2000-05-26 | 2003-10-07 | Schering Corporation | Adenosine A2a receptor antagonists |
| US6376514B1 (en) | 2000-10-17 | 2002-04-23 | The Procter & Gamble Co. | Substituted six-membered heterocyclic compounds useful for treating multidrug resistance and compositions and methods thereof |
| WO2007078615A2 (en) | 2005-12-15 | 2007-07-12 | Cavit Sciences, Inc | Methods and compositions for treatment of cancer |
Non-Patent Citations (10)
| Title |
|---|
| Biswas A et al., entitled "Acetylation of Pregnane X Receptor protein determines selective function independent of ligand activation," Biochem Biophys Res Commun., Mar. 18, 2011;406(3):371-376. |
| Das B C, et al., entitled "Synthesis of novel ketoconazole derivatives as inhibitors of the human Pregnane X Receptor (PXR;NR112; also termed SXR, PAR)," Bioorganic & Medicinal Chemistry Letters, 18 (2008) 3974-3977. |
| Ekins S et al., entitled "Human Pregnane X Receptor Antagonists and Agonists Define Molecular Requirements for Different Binding Sites," Mol Pharmacol 72:592-603, 2007. |
| Fotakis et al. Toxicology letters, vol. 160, pp. 171-177 (2006). * |
| Marques-Gallego et al. BMC Biotechnology, vol. 10, pp. 1-7 (2010). * |
| PCT Notification Concerning Transmittal of International Preliminary Report on Patentability and Written Opinion of the International Searching Authority in connection with PCT International Patent Application No. PCT/US2009/000524, 8 pages, (Mailed Sep. 10, 2010). |
| Pondugula S R et al., entitled "Pregnane xenobiotic receptor in cancer pathogenesis and therapeutic response," Cancer Letters, 328 (2013), 1-9. |
| Power et al. Bioorganic & Medicinal Chemistry letters vol. 16, p. 887-890 (2006). * |
| Venkatesh M, et al., entitled "In Vivo and In Vitro Characterization of First-in-Class Novel Azole Analog That Targets Pregnane X Receptor Activation," Mol Pharmacol, 80:124-135, 2011. |
| Wang H, et al., entitled "Pregnane X receptor activation induces FGF19-dependent tumor aggressiveness in humans and mice," J Clin Invest doi:10.1172/JCI41514. Epub Jul. 11, 2011. |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2009110955A3 (en) | 2009-12-30 |
| US20110105522A1 (en) | 2011-05-05 |
| WO2009110955A2 (en) | 2009-09-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Purushottamachar et al. | Systematic structure modifications of multitarget prostate cancer drug candidate galeterone to produce novel androgen receptor down-regulating agents as an approach to treatment of advanced prostate cancer | |
| CN101861151B (en) | Cancer combination therapy with a selective inhibitor of histone deacetylase HDAC1, HDAC2 and/or HDAC3 and a microtubule stabilizer | |
| Soares et al. | Reactivation of wild-type and mutant p53 by tryptophanolderived oxazoloisoindolinone SLMP53-1, a novel anticancer small-molecule | |
| Demirbaş et al. | Synthesis of 3-alkyl (Aryl)-4-alkylidenamino-4, 5-dihydro-1H-1, 2, 4-triazol-5-ones and 3-alkyl-4-alkylamino-4, 5-dihydro-1H-1, 2, 4-triazol-5-ones as antitumor agents | |
| Hou et al. | Synthesis and antitumor activity of 1, 2, 4-triazoles having 1, 4-benzodioxan fragment as a novel class of potent methionine aminopeptidase type II inhibitors | |
| Raffa et al. | Synthesis, cytotoxicity, and inhibitory effects on tubulin polymerization of a new 3-heterocyclo substituted 2-styrylquinazolinones | |
| Bariwal et al. | Design of Hedgehog pathway inhibitors for cancer treatment | |
| US20110105436A1 (en) | Heteroaryl compounds, compositions, and methods of use in cancer treatment | |
| US20120258975A1 (en) | Potent Small Molecule Inhibitors of Autophagy, and Methods of Use Thereof | |
| US10220033B2 (en) | Substituted benzoxazepine for cancer therapy | |
| JP2013500255A5 (en) | ||
| US8669260B2 (en) | Ketoconazole-derivative antagonist of human pregnane X receptor and uses thereof | |
| Wu et al. | Design, synthesis, and biological evaluation of 5-((4-(pyridin-3-yl) pyrimidin-2-yl) amino)-1 H-Indole-2-Carbohydrazide derivatives: the methuosis inducer 12A as a Novel and selective anticancer agent | |
| Wang et al. | Design, synthesis and evaluation of antiproliferative and antitubulin activities of 5-methyl-4-aryl-3-(4-arylpiperazine-1-carbonyl)-4H-1, 2, 4-triazoles | |
| US20090062222A1 (en) | Methods for Sensitizing Cancer Cells to Inhibitors | |
| US8242160B2 (en) | Inhibitors of ubiquitin E1 | |
| Angelone et al. | Indenopyrazole oxime ethers: Synthesis and β1-adrenergic blocking activity | |
| JP5599385B2 (en) | Small molecule inhibitors of androgen receptor | |
| Singh et al. | A review of the recent developments in synthetic anti-breast cancer agents | |
| Coleman et al. | Kinesin spindle protein (KSP) inhibitors. Part 6: Design and synthesis of 3, 5-diaryl-4, 5-dihydropyrazole amides as potent inhibitors of the mitotic kinesin KSP | |
| Das et al. | Synthesis of novel ketoconazole derivatives as inhibitors of the human pregnane X receptor (PXR; NR1I2; also termed SXR, PAR) | |
| Madoux et al. | Modulators of STAT transcription factors for the targeted therapy of cancer (STAT3 activators) | |
| US10093642B2 (en) | Multitarget hedgehog pathway inhibitors and uses thereof | |
| US20220202785A1 (en) | Small molecule inhibitors of gpcr gpr68 and related receptors for treating cancer, glioblastoma, and other indications | |
| Liao et al. | Discovery of Thiadiazoleamide Derivatives as Potent, Selective, and Orally Available Antagonists Disrupting Androgen Receptor Homodimer |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| STCF | Information on status: patent grant |
Free format text: PATENTED CASE |
|
| AS | Assignment |
Owner name: NATIONAL INSTITUTES OF HEALTH (NIH), U.S. DEPT. OF Free format text: CONFIRMATORY LICENSE;ASSIGNOR:ALBERT EINSTEIN COLLEGE OF MEDICINE;REEL/FRAME:036041/0946 Effective date: 20150625 |
|
| AS | Assignment |
Owner name: COM AFFILIATION, INC., NEW YORK Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:ALBERT EINSTEIN COLLEGE OF MEDICINE OF YESHIVA UNIVERSITY;REEL/FRAME:036875/0147 Effective date: 20150909 |
|
| AS | Assignment |
Owner name: ALBERT EINSTEIN COLLEGE OF MEDICINE, INC., NEW YOR Free format text: CHANGE OF NAME;ASSIGNOR:COM AFFILIATION, INC.;REEL/FRAME:036888/0939 Effective date: 20150909 |
|
| CC | Certificate of correction | ||
| MAFP | Maintenance fee payment |
Free format text: PAYMENT OF MAINTENANCE FEE, 4TH YR, SMALL ENTITY (ORIGINAL EVENT CODE: M2551) Year of fee payment: 4 |
|
| AS | Assignment |
Owner name: ALBERT EINSTEIN COLLEGE OF MEDICINE, NEW YORK Free format text: MERGER AND CHANGE OF NAME;ASSIGNORS:ALBERT EINSTEIN COLLEGE OF MEDICINE, INC.;ALBERT EINSTEIN COLLEGE OF MEDICINE;REEL/FRAME:048438/0275 Effective date: 20190101 |
|
| MAFP | Maintenance fee payment |
Free format text: PAYMENT OF MAINTENANCE FEE, 8TH YR, SMALL ENTITY (ORIGINAL EVENT CODE: M2552); ENTITY STATUS OF PATENT OWNER: SMALL ENTITY Year of fee payment: 8 |
|
| MAFP | Maintenance fee payment |
Free format text: PAYMENT OF MAINTENANCE FEE, 12TH YR, SMALL ENTITY (ORIGINAL EVENT CODE: M2553); ENTITY STATUS OF PATENT OWNER: SMALL ENTITY Year of fee payment: 12 |