US8063240B2 - Prostaglandin E1 and E2 analogs for the treatment of various medical conditions - Google Patents
Prostaglandin E1 and E2 analogs for the treatment of various medical conditions Download PDFInfo
- Publication number
- US8063240B2 US8063240B2 US12/271,764 US27176408A US8063240B2 US 8063240 B2 US8063240 B2 US 8063240B2 US 27176408 A US27176408 A US 27176408A US 8063240 B2 US8063240 B2 US 8063240B2
- Authority
- US
- United States
- Prior art keywords
- compound
- enyl
- benzo
- thiophen
- hept
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related, expires
Links
- GMVPRGQOIOIIMI-DWKJAMRDSA-N prostaglandin E1 Chemical compound CCCCC[C@H](O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1CCCCCCC(O)=O GMVPRGQOIOIIMI-DWKJAMRDSA-N 0.000 title description 3
- XEYBRNLFEZDVAW-UHFFFAOYSA-N prostaglandin E2 Natural products CCCCCC(O)C=CC1C(O)CC(=O)C1CC=CCCCC(O)=O XEYBRNLFEZDVAW-UHFFFAOYSA-N 0.000 title description 3
- GMVPRGQOIOIIMI-UHFFFAOYSA-N (8R,11R,12R,13E,15S)-11,15-Dihydroxy-9-oxo-13-prostenoic acid Natural products CCCCCC(O)C=CC1C(O)CC(=O)C1CCCCCCC(O)=O GMVPRGQOIOIIMI-UHFFFAOYSA-N 0.000 title 1
- 229960000711 alprostadil Drugs 0.000 title 1
- 150000003166 prostaglandin E2 derivatives Chemical class 0.000 title 1
- 102000008866 Prostaglandin E receptors Human genes 0.000 claims abstract description 29
- 108010088540 Prostaglandin E receptors Proteins 0.000 claims abstract description 25
- 230000001404 mediated effect Effects 0.000 claims abstract description 12
- 210000003958 hematopoietic stem cell Anatomy 0.000 claims abstract description 6
- 150000001875 compounds Chemical class 0.000 claims description 172
- 150000003839 salts Chemical class 0.000 claims description 38
- 238000000034 method Methods 0.000 claims description 31
- 229910052739 hydrogen Inorganic materials 0.000 claims description 30
- 239000001257 hydrogen Substances 0.000 claims description 29
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 20
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 15
- MBIFONNOELWXGT-GRZQHZHKSA-N (z)-7-[(1r,2r,3r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-3-hydroxy-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O MBIFONNOELWXGT-GRZQHZHKSA-N 0.000 claims description 14
- 239000008194 pharmaceutical composition Substances 0.000 claims description 14
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 13
- 239000011737 fluorine Substances 0.000 claims description 13
- 229910052731 fluorine Inorganic materials 0.000 claims description 13
- 229910052799 carbon Inorganic materials 0.000 claims description 12
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 12
- 150000002431 hydrogen Chemical class 0.000 claims description 9
- 239000011203 carbon fibre reinforced carbon Substances 0.000 claims description 8
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 7
- 229910052760 oxygen Inorganic materials 0.000 claims description 7
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 6
- 125000001072 heteroaryl group Chemical group 0.000 claims description 6
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 6
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 5
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims description 5
- 150000001721 carbon Chemical group 0.000 claims description 5
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 5
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 5
- 125000000217 alkyl group Chemical group 0.000 claims description 4
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 4
- 125000001153 fluoro group Chemical group F* 0.000 claims description 4
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 4
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 3
- 125000004070 6 membered heterocyclic group Chemical group 0.000 claims description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 2
- 125000003342 alkenyl group Chemical group 0.000 claims description 2
- 125000000304 alkynyl group Chemical group 0.000 claims description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 2
- 229910052794 bromium Inorganic materials 0.000 claims description 2
- CREMABGTGYGIQB-UHFFFAOYSA-N carbon carbon Chemical compound C.C CREMABGTGYGIQB-UHFFFAOYSA-N 0.000 claims description 2
- 239000000460 chlorine Substances 0.000 claims description 2
- 229910052801 chlorine Inorganic materials 0.000 claims description 2
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims description 2
- 125000004356 hydroxy functional group Chemical group O* 0.000 claims 3
- 208000006673 asthma Diseases 0.000 abstract description 28
- 239000000556 agonist Substances 0.000 abstract description 25
- 230000000694 effects Effects 0.000 abstract description 18
- 150000003180 prostaglandins Chemical class 0.000 abstract description 8
- 206010020751 Hypersensitivity Diseases 0.000 abstract description 7
- 208000026935 allergic disease Diseases 0.000 abstract description 7
- 230000007815 allergy Effects 0.000 abstract description 7
- 208000010392 Bone Fractures Diseases 0.000 abstract description 5
- 208000020084 Bone disease Diseases 0.000 abstract description 5
- 206010013935 Dysmenorrhoea Diseases 0.000 abstract description 5
- 206010057671 Female sexual dysfunction Diseases 0.000 abstract description 5
- 208000010412 Glaucoma Diseases 0.000 abstract description 5
- 206010057672 Male sexual dysfunction Diseases 0.000 abstract description 5
- 206010030043 Ocular hypertension Diseases 0.000 abstract description 5
- 208000001132 Osteoporosis Diseases 0.000 abstract description 5
- 208000006399 Premature Obstetric Labor Diseases 0.000 abstract description 5
- 208000007107 Stomach Ulcer Diseases 0.000 abstract description 5
- 201000005917 gastric ulcer Diseases 0.000 abstract description 5
- 208000026278 immune system disease Diseases 0.000 abstract description 5
- 206010022000 influenza Diseases 0.000 abstract description 5
- 208000017169 kidney disease Diseases 0.000 abstract description 5
- 208000028169 periodontal disease Diseases 0.000 abstract description 5
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 89
- 238000002360 preparation method Methods 0.000 description 56
- 239000000543 intermediate Substances 0.000 description 52
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 48
- 239000011541 reaction mixture Substances 0.000 description 46
- 210000004027 cell Anatomy 0.000 description 42
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 38
- 102000005962 receptors Human genes 0.000 description 34
- 108020003175 receptors Proteins 0.000 description 34
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 31
- -1 —(C1-C6)-alkyl amide Chemical class 0.000 description 28
- 239000012453 solvate Substances 0.000 description 27
- XEYBRNLFEZDVAW-ARSRFYASSA-N dinoprostone Chemical compound CCCCC[C@H](O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O XEYBRNLFEZDVAW-ARSRFYASSA-N 0.000 description 26
- 239000000243 solution Substances 0.000 description 26
- 238000003556 assay Methods 0.000 description 25
- 238000003756 stirring Methods 0.000 description 25
- 0 [1*]CccCC1CCCC1c(C)c(C)C(C)(C)C([2*])(C)C Chemical compound [1*]CccCC1CCCC1c(C)c(C)C(C)(C)C([2*])(C)C 0.000 description 24
- 239000000203 mixture Substances 0.000 description 23
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 22
- 239000000741 silica gel Substances 0.000 description 22
- 229910002027 silica gel Inorganic materials 0.000 description 22
- 239000012043 crude product Substances 0.000 description 21
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 18
- 239000012267 brine Substances 0.000 description 18
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 18
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 18
- 238000006243 chemical reaction Methods 0.000 description 17
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 16
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 16
- 229910052938 sodium sulfate Inorganic materials 0.000 description 16
- 235000011152 sodium sulphate Nutrition 0.000 description 16
- 238000003818 flash chromatography Methods 0.000 description 15
- 238000002825 functional assay Methods 0.000 description 15
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 14
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 14
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 14
- 239000000872 buffer Substances 0.000 description 14
- 239000000047 product Substances 0.000 description 14
- 239000002904 solvent Substances 0.000 description 14
- 230000008484 agonism Effects 0.000 description 13
- 201000010099 disease Diseases 0.000 description 13
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 12
- 239000012044 organic layer Substances 0.000 description 12
- 239000012981 Hank's balanced salt solution Substances 0.000 description 11
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 11
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical class CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 11
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 11
- 239000012299 nitrogen atmosphere Substances 0.000 description 11
- 230000004044 response Effects 0.000 description 11
- 125000000623 heterocyclic group Chemical group 0.000 description 10
- 238000012360 testing method Methods 0.000 description 10
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 9
- 239000005557 antagonist Substances 0.000 description 9
- 125000004432 carbon atom Chemical group C* 0.000 description 9
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 9
- 239000010410 layer Substances 0.000 description 9
- 238000005259 measurement Methods 0.000 description 9
- HNQIVZYLYMDVSB-UHFFFAOYSA-N methanesulfonimidic acid Chemical compound CS(N)(=O)=O HNQIVZYLYMDVSB-UHFFFAOYSA-N 0.000 description 9
- 229940127293 prostanoid Drugs 0.000 description 9
- 150000003814 prostanoids Chemical class 0.000 description 9
- 239000007787 solid Substances 0.000 description 9
- 238000002474 experimental method Methods 0.000 description 8
- 238000001914 filtration Methods 0.000 description 8
- CGIGDMFJXJATDK-UHFFFAOYSA-N indomethacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 CGIGDMFJXJATDK-UHFFFAOYSA-N 0.000 description 8
- 229910052757 nitrogen Inorganic materials 0.000 description 8
- BVIORTDFNGRUCP-HYJHBKHRSA-N (z)-7-[(1r,2r,3r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-3-hydroxy-5-methylidenecyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@H]1[C@H](O)CC(=C)[C@@H]1C\C=C/CCCC(O)=O BVIORTDFNGRUCP-HYJHBKHRSA-N 0.000 description 7
- VIKSLCBVJMJXPH-GCTAJMIWSA-N (z)-7-[(1r,2r,5s)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-5-hydroxy-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=O VIKSLCBVJMJXPH-GCTAJMIWSA-N 0.000 description 7
- ZVJMKFXJQIELFG-VOJWTERLSA-N (z)-7-[(1r,5r)-5-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-2-methyl-4-oxocyclopent-2-en-1-yl]hept-5-enoic acid Chemical compound OC(=O)CCC\C=C/C[C@H]1C(C)=CC(=O)[C@@H]1\C=C\[C@@H](O)C1=CC2=CC=CC=C2S1 ZVJMKFXJQIELFG-VOJWTERLSA-N 0.000 description 7
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 7
- UXODJZIERWRNPZ-LKXVCVDBSA-N [(3ar,4r,5r,6as)-4-[(e)-3-(1-benzothiophen-2-yl)-3-oxoprop-1-enyl]-2-oxo-3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-5-yl] benzoate Chemical compound O([C@H]1[C@@H]([C@H]2CC(=O)O[C@H]2C1)\C=C\C(=O)C=1SC2=CC=CC=C2C=1)C(=O)C1=CC=CC=C1 UXODJZIERWRNPZ-LKXVCVDBSA-N 0.000 description 7
- 230000005764 inhibitory process Effects 0.000 description 7
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 7
- 235000019341 magnesium sulphate Nutrition 0.000 description 7
- UPJIHIKZXUPUHJ-UVAJNVRCSA-N methyl (z)-7-[(1r,2r,3r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-[tert-butyl(diphenyl)silyl]oxyprop-1-enyl]-3-(oxan-2-yloxy)-5-oxocyclopentyl]hept-5-enoate Chemical compound O([C@H](/C=C/[C@@H]1[C@H](C(C[C@H]1OC1OCCCC1)=O)C\C=C/CCCC(=O)OC)C=1SC2=CC=CC=C2C=1)[Si](C(C)(C)C)(C=1C=CC=CC=1)C1=CC=CC=C1 UPJIHIKZXUPUHJ-UVAJNVRCSA-N 0.000 description 7
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 7
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 7
- MPJJIBCCKNOJRA-VOJWTERLSA-N (z)-7-[(1r,2r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-3-methyl-5-oxocyclopent-3-en-1-yl]hept-5-enoic acid Chemical compound CC1=CC(=O)[C@H](C\C=C/CCCC(O)=O)[C@H]1\C=C\[C@@H](O)C1=CC2=CC=CC=C2S1 MPJJIBCCKNOJRA-VOJWTERLSA-N 0.000 description 6
- MSSHMDUQFDMVKG-BLVLESEMSA-N (z)-7-[(1r,2r,5s)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-[tert-butyl(diphenyl)silyl]oxyprop-1-enyl]-5-hydroxy-3-methylidenecyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O[Si](C(C)(C)C)(C=1C=CC=CC=1)C=1C=CC=CC=1)C=1SC2=CC=CC=C2C=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=C MSSHMDUQFDMVKG-BLVLESEMSA-N 0.000 description 6
- OHCQJHSOBUTRHG-KGGHGJDLSA-N FORSKOLIN Chemical compound O=C([C@@]12O)C[C@](C)(C=C)O[C@]1(C)[C@@H](OC(=O)C)[C@@H](O)[C@@H]1[C@]2(C)[C@@H](O)CCC1(C)C OHCQJHSOBUTRHG-KGGHGJDLSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 6
- ITCPUEWUYNWVPF-WNDXBJOVSA-N [(3ar,4r,5r,6as)-4-[(e)-3-(1-benzothiophen-2-yl)-3-oxoprop-1-enyl]-2-oxo-3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-5-yl] 4-phenylbenzoate Chemical compound O([C@H]1[C@@H]([C@H]2CC(=O)O[C@H]2C1)\C=C\C(=O)C=1SC2=CC=CC=C2C=1)C(=O)C(C=C1)=CC=C1C1=CC=CC=C1 ITCPUEWUYNWVPF-WNDXBJOVSA-N 0.000 description 6
- 238000001514 detection method Methods 0.000 description 6
- 239000003480 eluent Substances 0.000 description 6
- 238000011156 evaluation Methods 0.000 description 6
- 238000011534 incubation Methods 0.000 description 6
- SMICUBOVSVDINS-OTEQTRORSA-N methyl (z)-7-[(1r,2r,3r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-[tert-butyl(diphenyl)silyl]oxyprop-1-enyl]-5-methylidene-3-(oxan-2-yloxy)cyclopentyl]hept-5-enoate Chemical compound O([C@H](/C=C/[C@@H]1[C@H](C(C[C@H]1OC1OCCCC1)=C)C\C=C/CCCC(=O)OC)C=1SC2=CC=CC=C2C=1)[Si](C(C)(C)C)(C=1C=CC=CC=1)C1=CC=CC=C1 SMICUBOVSVDINS-OTEQTRORSA-N 0.000 description 6
- 125000004433 nitrogen atom Chemical group N* 0.000 description 6
- 239000000546 pharmaceutical excipient Substances 0.000 description 6
- CHKVPAROMQMJNQ-UHFFFAOYSA-M potassium bisulfate Chemical compound [K+].OS([O-])(=O)=O CHKVPAROMQMJNQ-UHFFFAOYSA-M 0.000 description 6
- 229910000343 potassium bisulfate Inorganic materials 0.000 description 6
- 229920006395 saturated elastomer Polymers 0.000 description 6
- OSBRXMZCWPMCQV-HLMHMNLASA-N (z)-7-[(1r,2r,3r)-2-(3,3-difluoro-4-phenoxybutyl)-3-hydroxy-5-oxocyclopentyl]hept-5-enoic acid Chemical compound O[C@@H]1CC(=O)[C@H](C\C=C/CCCC(O)=O)[C@H]1CCC(F)(F)COC1=CC=CC=C1 OSBRXMZCWPMCQV-HLMHMNLASA-N 0.000 description 5
- 238000005160 1H NMR spectroscopy Methods 0.000 description 5
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 5
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 5
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 5
- 239000007995 HEPES buffer Substances 0.000 description 5
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 5
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Natural products CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 5
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 5
- RJZBEYWEIHMXKP-PQTHUQGOSA-N [(3ar,4r,5r,6as)-4-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-2-oxo-3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-5-yl] 4-phenylbenzoate Chemical compound O([C@H]1[C@@H]([C@H]2CC(=O)O[C@H]2C1)/C=C/[C@@H](O)C=1SC2=CC=CC=C2C=1)C(=O)C(C=C1)=CC=C1C1=CC=CC=C1 RJZBEYWEIHMXKP-PQTHUQGOSA-N 0.000 description 5
- 239000011575 calcium Substances 0.000 description 5
- 229910052791 calcium Inorganic materials 0.000 description 5
- 231100000673 dose–response relationship Toxicity 0.000 description 5
- 239000012035 limiting reagent Substances 0.000 description 5
- HXITXNWTGFUOAU-UHFFFAOYSA-N phenylboronic acid Chemical compound OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 5
- 239000000651 prodrug Substances 0.000 description 5
- 229940002612 prodrug Drugs 0.000 description 5
- 239000000018 receptor agonist Substances 0.000 description 5
- 229940044601 receptor agonist Drugs 0.000 description 5
- IJPVAAORJYOJPD-USSDDLBYSA-N (z)-7-[(1r,2r,3r)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-3-hydroxy-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O IJPVAAORJYOJPD-USSDDLBYSA-N 0.000 description 4
- CUOMTEAOMVCIFF-NJDSCPHLSA-N (z)-7-[(1r,2r,3r)-2-[(e)-3-(1-benzothiophen-2-yl)-3,3-difluoroprop-1-enyl]-3-hydroxy-5-oxocyclopentyl]hept-5-enoic acid Chemical compound O[C@@H]1CC(=O)[C@H](C\C=C/CCCC(O)=O)[C@H]1\C=C\C(F)(F)C1=CC2=CC=CC=C2S1 CUOMTEAOMVCIFF-NJDSCPHLSA-N 0.000 description 4
- OCMRUIYXTPMEQD-CNFLNUCUSA-N (z)-7-[(1r,2r,3r)-2-[(e,3r)-3-(1,3-benzothiazol-2-yl)-3-hydroxyprop-1-enyl]-3-hydroxy-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2N=1)=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O OCMRUIYXTPMEQD-CNFLNUCUSA-N 0.000 description 4
- BGQUYQWHESXNBS-DHRDONBFSA-N (z)-7-[(1r,2r,3r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-[tert-butyl(diphenyl)silyl]oxyprop-1-enyl]-3-hydroxy-5-methylidenecyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O[Si](C(C)(C)C)(C=1C=CC=CC=1)C=1C=CC=CC=1)C=1SC2=CC=CC=C2C=1)=C\[C@H]1[C@H](O)CC(=C)[C@@H]1C\C=C/CCCC(O)=O BGQUYQWHESXNBS-DHRDONBFSA-N 0.000 description 4
- LEPZOYPFXATQAU-FIXVKWTQSA-N (z)-7-[(1r,2r,3r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-[tert-butyl(diphenyl)silyl]oxyprop-1-enyl]-5-methylidene-3-(oxan-2-yloxy)cyclopentyl]hept-5-enoic acid Chemical compound O([C@H]1[C@@H]([C@H](C(=C)C1)C\C=C/CCCC(O)=O)/C=C/[C@@H](O[Si](C(C)(C)C)(C=1C=CC=CC=1)C=1C=CC=CC=1)C=1SC2=CC=CC=C2C=1)C1CCCCO1 LEPZOYPFXATQAU-FIXVKWTQSA-N 0.000 description 4
- OKPBSCRDOFNOFX-NBFJYYDFSA-N (z)-7-[(1r,2r,3s)-2-[(e,3s)-3-(2,3-dihydro-1h-inden-2-yl)-3-hydroxyprop-1-enyl]-3-fluoro-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C1CC2=CC=CC=C2C1)=C\[C@H]1[C@@H](F)CC(=O)[C@@H]1C\C=C/CCCC(O)=O OKPBSCRDOFNOFX-NBFJYYDFSA-N 0.000 description 4
- ZCFSOKSBUYOGGK-OWELLUKLSA-N (z)-7-[(1r,2r,5s)-2-[(e,3r)-3-(1,3-benzothiazol-2-yl)-3-hydroxyprop-1-enyl]-5-hydroxy-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2N=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=O ZCFSOKSBUYOGGK-OWELLUKLSA-N 0.000 description 4
- WNNSBUDKWLEWTK-OXPYUZCASA-N (z)-7-[(1r,2r,5s)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-5-hydroxy-3-methylidenecyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=C WNNSBUDKWLEWTK-OXPYUZCASA-N 0.000 description 4
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 4
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 4
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 4
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 4
- 101000929799 Homo sapiens Acyl-CoA-binding protein Proteins 0.000 description 4
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 4
- ZTAKTAUQEHDDRZ-TUPUNYPXSA-N [(3ar,4r,5r,6as)-4-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-2-oxo-3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-5-yl] benzoate Chemical compound O([C@H]1[C@@H]([C@H]2CC(=O)O[C@H]2C1)/C=C/[C@@H](O)C=1SC2=CC=CC=C2C=1)C(=O)C1=CC=CC=C1 ZTAKTAUQEHDDRZ-TUPUNYPXSA-N 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- YZXBAPSDXZZRGB-DOFZRALJSA-N arachidonic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O YZXBAPSDXZZRGB-DOFZRALJSA-N 0.000 description 4
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 4
- 230000027455 binding Effects 0.000 description 4
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 4
- 230000003491 cAMP production Effects 0.000 description 4
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 4
- 238000010367 cloning Methods 0.000 description 4
- 238000001816 cooling Methods 0.000 description 4
- 239000013058 crude material Substances 0.000 description 4
- 230000001086 cytosolic effect Effects 0.000 description 4
- GWQVMPWSEVRGPY-UHFFFAOYSA-N europium cryptate Chemical compound [Eu+3].N=1C2=CC=CC=1CN(CC=1N=C(C=CC=1)C=1N=C(C3)C=CC=1)CC(N=1)=CC(C(=O)NCCN)=CC=1C(N=1)=CC(C(=O)NCCN)=CC=1CN3CC1=CC=CC2=N1 GWQVMPWSEVRGPY-UHFFFAOYSA-N 0.000 description 4
- 239000001963 growth medium Substances 0.000 description 4
- 238000002868 homogeneous time resolved fluorescence Methods 0.000 description 4
- 229960000905 indomethacin Drugs 0.000 description 4
- 230000006698 induction Effects 0.000 description 4
- 238000001819 mass spectrum Methods 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- 239000012528 membrane Substances 0.000 description 4
- 239000012074 organic phase Substances 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 150000003254 radicals Chemical class 0.000 description 4
- 239000002287 radioligand Substances 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 229910052717 sulfur Inorganic materials 0.000 description 4
- 238000012546 transfer Methods 0.000 description 4
- AKLHKYGBGHMGMN-LFPNZXLMSA-N (3ar,4r,5r,6as)-4-[(3r)-3-(1-benzothiophen-2-yl)-3-[tert-butyl(diphenyl)silyl]oxypropyl]-3,3a,4,5,6,6a-hexahydro-2h-cyclopenta[b]furan-2,5-diol Chemical compound O([C@H](CC[C@@H]1[C@H]2CC(O)O[C@H]2C[C@H]1O)C=1SC2=CC=CC=C2C=1)[Si](C(C)(C)C)(C=1C=CC=CC=1)C1=CC=CC=C1 AKLHKYGBGHMGMN-LFPNZXLMSA-N 0.000 description 3
- AOANZOUJCZSACF-TYUZFQBTSA-N (3ar,4r,5r,6as)-4-[(e)-3-(1-benzothiophen-2-yl)-3,3-difluoroprop-1-enyl]-5-hydroxy-3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-2-one Chemical compound C1=CC=C2SC(C(F)(F)/C=C/[C@@H]3[C@H]4CC(=O)O[C@H]4C[C@H]3O)=CC2=C1 AOANZOUJCZSACF-TYUZFQBTSA-N 0.000 description 3
- HHIFBQWQRXEUHZ-UFKQFPNGSA-N (3ar,4r,5r,6as)-4-[(e)-3-(1-benzothiophen-2-yl)-3-hydroxybut-1-enyl]-5-hydroxy-3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-2-one Chemical compound C1=CC=C2SC(C(O)(\C=C\[C@@H]3[C@H]4CC(=O)O[C@H]4C[C@H]3O)C)=CC2=C1 HHIFBQWQRXEUHZ-UFKQFPNGSA-N 0.000 description 3
- AKQCSGBQIJEURS-RQDKSWMHSA-N (3ar,4r,5r,6as)-4-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-5-hydroxy-3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-2-one Chemical compound C1=CC=C2SC([C@@H](\C=C\[C@@H]3[C@H]4CC(=O)O[C@H]4C[C@H]3O)O)=CC2=C1 AKQCSGBQIJEURS-RQDKSWMHSA-N 0.000 description 3
- CWHMNVDLGITRKJ-VBRJGFDYSA-N (3ar,4s,5s,6as)-4-[[tert-butyl(diphenyl)silyl]oxymethyl]-5-fluoro-3,3a,4,5,6,6a-hexahydro-2h-cyclopenta[b]furan-2-ol Chemical compound C([C@@H]1[C@H]2CC(O)O[C@H]2C[C@@H]1F)O[Si](C(C)(C)C)(C=1C=CC=CC=1)C1=CC=CC=C1 CWHMNVDLGITRKJ-VBRJGFDYSA-N 0.000 description 3
- ICMDJQNVBUZZGZ-CZYKHXBRSA-N (3ar,4s,5s,6as)-4-[[tert-butyl(diphenyl)silyl]oxymethyl]-5-fluoro-3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-2-one Chemical compound C([C@@H]1[C@H]2CC(=O)O[C@H]2C[C@@H]1F)O[Si](C(C)(C)C)(C=1C=CC=CC=1)C1=CC=CC=C1 ICMDJQNVBUZZGZ-CZYKHXBRSA-N 0.000 description 3
- VIPOETDMBDGARX-QUXANAIZSA-N (z)-7-[(1r,2r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-[tert-butyl(diphenyl)silyl]oxyprop-1-enyl]-3-methyl-5-oxocyclopent-3-en-1-yl]hept-5-enoic acid Chemical compound CC1=CC(=O)[C@H](C\C=C/CCCC(O)=O)[C@H]1\C=C\[C@H](C=1SC2=CC=CC=C2C=1)O[Si](C(C)(C)C)(C=1C=CC=CC=1)C1=CC=CC=C1 VIPOETDMBDGARX-QUXANAIZSA-N 0.000 description 3
- XOTKSJPQOJOTQZ-BNPXPVJVSA-N (z)-7-[(1r,2r,3r)-2-[(e)-3-(1-benzothiophen-2-yl)-3-hydroxybut-1-enyl]-3-hydroxy-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C=1C2=CC=CC=C2SC=1C(O)(C)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O XOTKSJPQOJOTQZ-BNPXPVJVSA-N 0.000 description 3
- FTUCGIXXGGZHHW-PSRWLAKFSA-N (z)-7-[(1r,2r,3r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-[tert-butyl(diphenyl)silyl]oxyprop-1-enyl]-3-hydroxy-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O[Si](C(C)(C)C)(C=1C=CC=CC=1)C=1C=CC=CC=1)C=1SC2=CC=CC=C2C=1)=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O FTUCGIXXGGZHHW-PSRWLAKFSA-N 0.000 description 3
- LJMOQDHYDMYKBT-NJIIROJJSA-N (z)-7-[(1r,2r,3r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-3-hydroxy-5-oxocyclopentyl]-n-methylsulfonylhept-5-enamide Chemical compound O[C@@H]1CC(=O)[C@H](C\C=C/CCCC(=O)NS(=O)(=O)C)[C@H]1\C=C\[C@@H](O)C1=CC2=CC=CC=C2S1 LJMOQDHYDMYKBT-NJIIROJJSA-N 0.000 description 3
- AVVLVAANBKUOGB-ZBACHXLQSA-N (z)-7-[(1r,2r,5s)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-[tert-butyl(diphenyl)silyl]oxyprop-1-enyl]-5-hydroxy-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O[Si](C(C)(C)C)(C=1C=CC=CC=1)C=1C=CC=CC=1)C=1SC2=CC=CC=C2C=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=O AVVLVAANBKUOGB-ZBACHXLQSA-N 0.000 description 3
- MZEZLMNTIJYOCD-QUXANAIZSA-N (z)-7-[(1r,5r)-5-[(e,3r)-3-(1-benzothiophen-2-yl)-3-[tert-butyl(diphenyl)silyl]oxyprop-1-enyl]-2-methyl-4-oxocyclopent-2-en-1-yl]hept-5-enoic acid Chemical compound OC(=O)CCC\C=C/C[C@H]1C(C)=CC(=O)[C@@H]1\C=C\[C@H](C=1SC2=CC=CC=C2C=1)O[Si](C(C)(C)C)(C=1C=CC=CC=1)C1=CC=CC=C1 MZEZLMNTIJYOCD-QUXANAIZSA-N 0.000 description 3
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 3
- LLHBJNWMKOAJDD-UHFFFAOYSA-N 5-bromo-n-methylsulfonylpentanamide Chemical compound CS(=O)(=O)NC(=O)CCCCBr LLHBJNWMKOAJDD-UHFFFAOYSA-N 0.000 description 3
- NKASQOYWICTEQV-SOAMZJECSA-N 7-[(1r,2r,3r)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-3-hydroxy-5-oxocyclopentyl]heptanoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@H]1[C@H](O)CC(=O)[C@@H]1CCCCCCC(O)=O NKASQOYWICTEQV-SOAMZJECSA-N 0.000 description 3
- CNBRMDAOJWMDKI-FRPYLKKPSA-N 7-[(1r,2r,3r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-3-hydroxy-5-oxocyclopentyl]heptanoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@H]1[C@H](O)CC(=O)[C@@H]1CCCCCCC(O)=O CNBRMDAOJWMDKI-FRPYLKKPSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- QCMHGCDOZLWPOT-FMNCTDSISA-N COC1=C(CC[C@@H]2CCC3=C(C2)C=CC(=C3)[C@H]2CC[C@](N)(CO)C2)C=CC=C1 Chemical compound COC1=C(CC[C@@H]2CCC3=C(C2)C=CC(=C3)[C@H]2CC[C@](N)(CO)C2)C=CC=C1 QCMHGCDOZLWPOT-FMNCTDSISA-N 0.000 description 3
- 229910004664 Cerium(III) chloride Inorganic materials 0.000 description 3
- SUZLHDUTVMZSEV-UHFFFAOYSA-N Deoxycoleonol Natural products C12C(=O)CC(C)(C=C)OC2(C)C(OC(=O)C)C(O)C2C1(C)C(O)CCC2(C)C SUZLHDUTVMZSEV-UHFFFAOYSA-N 0.000 description 3
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 3
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 3
- OUVXYXNWSVIOSJ-UHFFFAOYSA-N Fluo-4 Chemical compound CC1=CC=C(N(CC(O)=O)CC(O)=O)C(OCCOC=2C(=CC=C(C=2)C2=C3C=C(F)C(=O)C=C3OC3=CC(O)=C(F)C=C32)N(CC(O)=O)CC(O)=O)=C1 OUVXYXNWSVIOSJ-UHFFFAOYSA-N 0.000 description 3
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 3
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium on carbon Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 3
- NFHFRUOZVGFOOS-UHFFFAOYSA-N Pd(PPh3)4 Substances [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 3
- KCSRFTZHGLZZNZ-YSFYHYPLSA-N [(3ar,4r,5r,6as)-2-oxo-4-(3-oxo-4-phenoxybutyl)-3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-5-yl] benzoate Chemical compound O([C@H]1[C@@H]([C@H]2CC(=O)O[C@H]2C1)CCC(=O)COC=1C=CC=CC=1)C(=O)C1=CC=CC=C1 KCSRFTZHGLZZNZ-YSFYHYPLSA-N 0.000 description 3
- MSLKSGJPVJYHJO-JWVPBWIXSA-N [(3ar,4r,5r,6as)-2-oxo-4-[(e)-3-oxo-4-phenoxybut-1-enyl]-3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-5-yl] benzoate Chemical compound C(\[C@@H]1[C@H]2CC(=O)O[C@H]2C[C@H]1OC(=O)C=1C=CC=CC=1)=C/C(=O)COC1=CC=CC=C1 MSLKSGJPVJYHJO-JWVPBWIXSA-N 0.000 description 3
- TYPZPOPSHRRIFM-BDHOTLMUSA-N [(3ar,4r,5r,6as)-4-[(e)-3-(1-benzothiophen-2-yl)-3,3-difluoroprop-1-enyl]-2-oxo-3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-5-yl] benzoate Chemical compound O([C@H]1[C@@H]([C@H]2CC(=O)O[C@H]2C1)/C=C/C(F)(F)C=1SC2=CC=CC=C2C=1)C(=O)C1=CC=CC=C1 TYPZPOPSHRRIFM-BDHOTLMUSA-N 0.000 description 3
- UOIFTOBIGNZZSO-UHFFFAOYSA-N acetic acid;ethyl acetate;hexane Chemical compound CC(O)=O.CCCCCC.CCOC(C)=O UOIFTOBIGNZZSO-UHFFFAOYSA-N 0.000 description 3
- 230000008485 antagonism Effects 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 3
- OHCQJHSOBUTRHG-UHFFFAOYSA-N colforsin Natural products OC12C(=O)CC(C)(C=C)OC1(C)C(OC(=O)C)C(O)C1C2(C)C(O)CCC1(C)C OHCQJHSOBUTRHG-UHFFFAOYSA-N 0.000 description 3
- 238000009109 curative therapy Methods 0.000 description 3
- 208000035475 disorder Diseases 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 125000005843 halogen group Chemical group 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 230000003834 intracellular effect Effects 0.000 description 3
- 201000010659 intrinsic asthma Diseases 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 239000004973 liquid crystal related substance Substances 0.000 description 3
- GRWIABMEEKERFV-UHFFFAOYSA-N methanol;oxolane Chemical compound OC.C1CCOC1 GRWIABMEEKERFV-UHFFFAOYSA-N 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 239000008188 pellet Substances 0.000 description 3
- 238000003359 percent control normalization Methods 0.000 description 3
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 230000002265 prevention Effects 0.000 description 3
- DBABZHXKTCFAPX-UHFFFAOYSA-N probenecid Chemical compound CCCN(CCC)S(=O)(=O)C1=CC=C(C(O)=O)C=C1 DBABZHXKTCFAPX-UHFFFAOYSA-N 0.000 description 3
- 230000027425 release of sequestered calcium ion into cytosol Effects 0.000 description 3
- 230000029058 respiratory gaseous exchange Effects 0.000 description 3
- 239000000377 silicon dioxide Substances 0.000 description 3
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 3
- 229910000033 sodium borohydride Inorganic materials 0.000 description 3
- 239000012279 sodium borohydride Substances 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 230000003319 supportive effect Effects 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 3
- 125000003396 thiol group Chemical group [H]S* 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 3
- AZGAENRQOWMEQT-PAVKGVHNSA-N (3ar,4r,5r,6as)-4-(3,3-difluoro-4-phenoxybutyl)-5-(oxan-2-yloxy)-3,3a,4,5,6,6a-hexahydro-2h-cyclopenta[b]furan-2-ol Chemical compound O([C@@H]1C[C@H]2[C@@H]([C@H]1CCC(F)(F)COC=1C=CC=CC=1)CC(O2)O)C1CCCCO1 AZGAENRQOWMEQT-PAVKGVHNSA-N 0.000 description 2
- RPGNZRSECYSMTC-KEMYJPCSSA-N (3ar,4r,5r,6as)-4-[(e)-3-(1-benzothiophen-2-yl)-3,3-difluoroprop-1-enyl]-5-(oxan-2-yloxy)-3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-2-one Chemical compound O([C@H]1[C@@H]([C@H]2CC(=O)O[C@H]2C1)/C=C/C(F)(F)C=1SC2=CC=CC=C2C=1)C1CCCCO1 RPGNZRSECYSMTC-KEMYJPCSSA-N 0.000 description 2
- KRBZYMCSTPUCRL-ZXJYTPBHSA-N (3ar,4r,5r,6as)-4-[(e)-3-(1-benzothiophen-2-yl)-3-(oxan-2-yloxy)but-1-enyl]-5-(oxan-2-yloxy)-3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-2-one Chemical compound C(\[C@@H]1[C@H]2CC(=O)O[C@H]2C[C@H]1OC1OCCCC1)=C/C(C=1SC2=CC=CC=C2C=1)(C)OC1CCCCO1 KRBZYMCSTPUCRL-ZXJYTPBHSA-N 0.000 description 2
- HZRFYNFWBCFEHG-UXCUCUQXSA-N (3ar,4r,5r,6as)-4-[(e,3r)-3-(1-benzothiophen-2-yl)-3-(oxan-2-yloxy)prop-1-enyl]-5-(oxan-2-yloxy)-3,3a,4,5,6,6a-hexahydro-2h-cyclopenta[b]furan-2-ol Chemical compound O([C@@H]1C[C@H]2[C@@H]([C@H]1\C=C\[C@@H](OC1OCCCC1)C=1SC3=CC=CC=C3C=1)CC(O2)O)C1CCCCO1 HZRFYNFWBCFEHG-UXCUCUQXSA-N 0.000 description 2
- HADQGLMDSWTTCW-OMZKKNHASA-N (3ar,4r,5r,6as)-4-[(e,3r)-3-(1-benzothiophen-2-yl)-3-[tert-butyl(diphenyl)silyl]oxyprop-1-enyl]-3,3a,4,5,6,6a-hexahydro-2h-cyclopenta[b]furan-2,5-diol Chemical compound O([C@H](\C=C\[C@@H]1[C@H]2CC(O)O[C@H]2C[C@H]1O)C=1SC2=CC=CC=C2C=1)[Si](C(C)(C)C)(C=1C=CC=CC=1)C1=CC=CC=C1 HADQGLMDSWTTCW-OMZKKNHASA-N 0.000 description 2
- KSDMISMEMOGBFU-UHFFFAOYSA-N (all-Z)-7,10,13-Eicosatrienoic acid Natural products CCCCCCC=CCC=CCC=CCCCCCC(O)=O KSDMISMEMOGBFU-UHFFFAOYSA-N 0.000 description 2
- WCVRPFDTUZWPRO-CMMHURNUSA-N (z)-7-[(1r,2r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-3,3-difluoro-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)C(=O)CC1(F)F WCVRPFDTUZWPRO-CMMHURNUSA-N 0.000 description 2
- QJLJSUFNWOSHOO-VOJWTERLSA-N (z)-7-[(1r,2r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-3-methylidene-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)C(=O)CC1=C QJLJSUFNWOSHOO-VOJWTERLSA-N 0.000 description 2
- YTPVAKBJFINMCZ-HJZGFFBBSA-N (z)-7-[(1r,2r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-5-fluoro-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)C(F)CC1=O YTPVAKBJFINMCZ-HJZGFFBBSA-N 0.000 description 2
- UNSJIUPNGWVQGE-LWOOZIKRSA-N (z)-7-[(1r,2r,3r,5s)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-[tert-butyl(diphenyl)silyl]oxypropyl]-3,5-dihydroxycyclopentyl]hept-5-enoic acid Chemical compound C([C@@H](O[Si](C(C)(C)C)(C=1C=CC=CC=1)C=1C=CC=CC=1)C=1SC2=CC=CC=C2C=1)C[C@H]1[C@H](O)C[C@H](O)[C@@H]1C\C=C/CCCC(O)=O UNSJIUPNGWVQGE-LWOOZIKRSA-N 0.000 description 2
- ZEYUMNDUEKQFEG-LVKGEYFXSA-N (z)-7-[(1r,2r,3r,5s)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-(oxan-2-yloxy)prop-1-enyl]-5-hydroxy-3-(oxan-2-yloxy)cyclopentyl]-n-methylsulfonylhept-5-enamide Chemical compound O([C@@H]1C[C@H](O)[C@@H]([C@H]1\C=C\[C@@H](OC1OCCCC1)C=1SC2=CC=CC=C2C=1)C\C=C/CCCC(=O)NS(=O)(=O)C)C1CCCCO1 ZEYUMNDUEKQFEG-LVKGEYFXSA-N 0.000 description 2
- KLZNPBCTILCCEC-FVILAGQQSA-N (z)-7-[(1r,2r,5s)-2-[(e,3r)-3-(1,3-benzothiazol-2-yl)-3-[tert-butyl(diphenyl)silyl]oxyprop-1-enyl]-5-hydroxy-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O[Si](C(C)(C)C)(C=1C=CC=CC=1)C=1C=CC=CC=1)C=1SC2=CC=CC=C2N=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=O KLZNPBCTILCCEC-FVILAGQQSA-N 0.000 description 2
- JHDNIQZEKBOLEY-NLUXKXRRSA-N (z)-7-[(1r,2r,5s)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-[tert-butyl(diphenyl)silyl]oxyprop-1-enyl]-3-methylidene-5-(oxan-2-yloxy)cyclopentyl]hept-5-enoic acid Chemical compound O([C@@H]1[C@H](C\C=C/CCCC(O)=O)[C@H](C(C1)=C)/C=C/[C@@H](O[Si](C(C)(C)C)(C=1C=CC=CC=1)C=1C=CC=CC=1)C=1SC2=CC=CC=C2C=1)C1CCCCO1 JHDNIQZEKBOLEY-NLUXKXRRSA-N 0.000 description 2
- QSTFLAZWBOIYBI-WAGHVWTHSA-N (z)-7-[(1r,2s,3s,5s)-2-[[tert-butyl(diphenyl)silyl]oxymethyl]-3-fluoro-5-hydroxycyclopentyl]hept-5-enoic acid Chemical compound C=1C=CC=CC=1[Si](C=1C=CC=CC=1)(C(C)(C)C)OC[C@H]1[C@@H](F)C[C@H](O)[C@@H]1C\C=C/CCCC(O)=O QSTFLAZWBOIYBI-WAGHVWTHSA-N 0.000 description 2
- IMQSRZOFKBJBNL-CMMHURNUSA-N (z)-7-[(1r,5r)-5-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-2,2-difluoro-4-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)C(F)(F)CC1=O IMQSRZOFKBJBNL-CMMHURNUSA-N 0.000 description 2
- MLOSJPZSZWUDSK-UHFFFAOYSA-N 4-carboxybutyl(triphenyl)phosphanium;bromide Chemical compound [Br-].C=1C=CC=CC=1[P+](C=1C=CC=CC=1)(CCCCC(=O)O)C1=CC=CC=C1 MLOSJPZSZWUDSK-UHFFFAOYSA-N 0.000 description 2
- CHDJXXUUPCANRW-MMVKXINWSA-N CC(C)OC(=O)CCC/C=C\C[C@H]1[C@@H](OC2CCCCO2)C[C@H](F)[C@@H]1/C=C/C(=O)C1CC2=C(C=CC=C2)C1 Chemical compound CC(C)OC(=O)CCC/C=C\C[C@H]1[C@@H](OC2CCCCO2)C[C@H](F)[C@@H]1/C=C/C(=O)C1CC2=C(C=CC=C2)C1 CHDJXXUUPCANRW-MMVKXINWSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 2
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 2
- 238000011891 EIA kit Methods 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 239000003810 Jones reagent Substances 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- COTFRVCHGYSFNO-TUVASFSCSA-N O=C1C[C@H]2[C@H](C[C@@H](O)[C@@H]2CCC(F)(F)COC2=CC=CC=C2)O1 Chemical compound O=C1C[C@H]2[C@H](C[C@@H](O)[C@@H]2CCC(F)(F)COC2=CC=CC=C2)O1 COTFRVCHGYSFNO-TUVASFSCSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 206010047924 Wheezing Diseases 0.000 description 2
- JRUCAYGIUANHRO-NCYKPQTJSA-N [(3ar,4r,5r,6as)-4-(3,3-difluoro-4-phenoxybutyl)-2-oxo-3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-5-yl] benzoate Chemical compound O([C@H]1[C@@H]([C@H]2CC(=O)O[C@H]2C1)CCC(F)(F)COC=1C=CC=CC=1)C(=O)C1=CC=CC=C1 JRUCAYGIUANHRO-NCYKPQTJSA-N 0.000 description 2
- YIAKSKVMQHTYHH-RSQODDFBSA-N [(3ar,4r,5r,6as)-4-[(e)-3-(1-benzothiophen-2-yl)-3-hydroxybut-1-enyl]-2-oxo-3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-5-yl] benzoate Chemical compound O([C@H]1[C@@H]([C@H]2CC(=O)O[C@H]2C1)/C=C/C(O)(C)C=1SC2=CC=CC=C2C=1)C(=O)C1=CC=CC=C1 YIAKSKVMQHTYHH-RSQODDFBSA-N 0.000 description 2
- JYJFITFFGKGGCD-ZLLMDIIPSA-N [(3ar,4r,5r,6as)-4-[(e,3r)-3-(1-benzothiophen-2-yl)-3-[tert-butyl(diphenyl)silyl]oxyprop-1-enyl]-2-oxo-3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-5-yl] benzoate Chemical compound O([C@H]1[C@@H]([C@H]2CC(=O)O[C@H]2C1)/C=C/[C@@H](O[Si](C(C)(C)C)(C=1C=CC=CC=1)C=1C=CC=CC=1)C=1SC2=CC=CC=C2C=1)C(=O)C1=CC=CC=C1 JYJFITFFGKGGCD-ZLLMDIIPSA-N 0.000 description 2
- NDHMOBCVFGMXRK-FVCCEPFGSA-N [(3ar,4r,5r,6as)-4-formyl-2-oxo-3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-5-yl] benzoate Chemical compound O([C@H]1[C@@H]([C@H]2CC(=O)O[C@H]2C1)C=O)C(=O)C1=CC=CC=C1 NDHMOBCVFGMXRK-FVCCEPFGSA-N 0.000 description 2
- BOHICIHVVCHUIS-UHFFFAOYSA-N [5-(methanesulfonamido)-5-oxopentyl]-triphenylphosphanium;bromide Chemical compound [Br-].C=1C=CC=CC=1[P+](C=1C=CC=CC=1)(CCCCC(=O)NS(=O)(=O)C)C1=CC=CC=C1 BOHICIHVVCHUIS-UHFFFAOYSA-N 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 229940114079 arachidonic acid Drugs 0.000 description 2
- 235000021342 arachidonic acid Nutrition 0.000 description 2
- 125000003118 aryl group Chemical group 0.000 description 2
- 238000000065 atmospheric pressure chemical ionisation Methods 0.000 description 2
- TWJVNKMWXNTSAP-UHFFFAOYSA-N azanium;hydroxide;hydrochloride Chemical class [NH4+].O.[Cl-] TWJVNKMWXNTSAP-UHFFFAOYSA-N 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 229930189065 blasticidin Natural products 0.000 description 2
- 235000011089 carbon dioxide Nutrition 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 239000013592 cell lysate Substances 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- WGLUMOCWFMKWIL-UHFFFAOYSA-N dichloromethane;methanol Chemical compound OC.ClCCl WGLUMOCWFMKWIL-UHFFFAOYSA-N 0.000 description 2
- HOBAELRKJCKHQD-QNEBEIHSSA-N dihomo-γ-linolenic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/CCCCCCC(O)=O HOBAELRKJCKHQD-QNEBEIHSSA-N 0.000 description 2
- 229960002986 dinoprostone Drugs 0.000 description 2
- 238000006073 displacement reaction Methods 0.000 description 2
- 239000003814 drug Substances 0.000 description 2
- 238000000132 electrospray ionisation Methods 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 208000024711 extrinsic asthma Diseases 0.000 description 2
- 230000035558 fertility Effects 0.000 description 2
- 239000007850 fluorescent dye Substances 0.000 description 2
- 239000012634 fragment Substances 0.000 description 2
- 230000035876 healing Effects 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 229940125425 inverse agonist Drugs 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- WCRWHLBFTKOTGO-KVKHEUIFSA-N methyl (z)-7-[(1r,2r,3r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-[tert-butyl(diphenyl)silyl]oxyprop-1-enyl]-3-hydroxy-5-oxocyclopentyl]hept-5-enoate Chemical compound O[C@@H]1CC(=O)[C@H](C\C=C/CCCC(=O)OC)[C@H]1\C=C\[C@H](C=1SC2=CC=CC=C2C=1)O[Si](C(C)(C)C)(C=1C=CC=CC=1)C1=CC=CC=C1 WCRWHLBFTKOTGO-KVKHEUIFSA-N 0.000 description 2
- KNFIYFGKIOWFGM-OUHWBAKHSA-N methyl (z)-7-[(1r,2r,5s)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-[tert-butyl(diphenyl)silyl]oxyprop-1-enyl]-5-hydroxy-3-oxocyclopentyl]hept-5-enoate Chemical compound COC(=O)CCC\C=C/C[C@H]1[C@@H](O)CC(=O)[C@@H]1\C=C\[C@H](C=1SC2=CC=CC=C2C=1)O[Si](C(C)(C)C)(C=1C=CC=CC=1)C1=CC=CC=C1 KNFIYFGKIOWFGM-OUHWBAKHSA-N 0.000 description 2
- XTEGVFVZDVNBPF-UHFFFAOYSA-N naphthalene-1,5-disulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1S(O)(=O)=O XTEGVFVZDVNBPF-UHFFFAOYSA-N 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 229940094443 oxytocics prostaglandins Drugs 0.000 description 2
- 238000002638 palliative care Methods 0.000 description 2
- YBYRMVIVWMBXKQ-UHFFFAOYSA-N phenylmethanesulfonyl fluoride Chemical compound FS(=O)(=O)CC1=CC=CC=C1 YBYRMVIVWMBXKQ-UHFFFAOYSA-N 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 2
- SDGTXFKKLSIRSI-RXSTTXKKSA-N propan-2-yl (z)-7-[(1r,2r,3s,5s)-2-[(e,3s)-3-(2,3-dihydro-1h-inden-2-yl)-3-hydroxyprop-1-enyl]-3-fluoro-5-(oxan-2-yloxy)cyclopentyl]hept-5-enoate Chemical compound O([C@@H]1[C@@H]([C@H]([C@@H](F)C1)\C=C\[C@@H](O)C1CC2=CC=CC=C2C1)C\C=C/CCCC(=O)OC(C)C)C1CCCCO1 SDGTXFKKLSIRSI-RXSTTXKKSA-N 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 239000012679 serum free medium Substances 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 238000000935 solvent evaporation Methods 0.000 description 2
- 125000004434 sulfur atom Chemical group 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 125000000037 tert-butyldiphenylsilyl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1[Si]([H])([*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- RZWIIPASKMUIAC-VQTJNVASSA-N thromboxane Chemical compound CCCCCCCC[C@H]1OCCC[C@@H]1CCCCCCC RZWIIPASKMUIAC-VQTJNVASSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- DZUXGQBLFALXCR-UHFFFAOYSA-N (+)-(9alpha,11alpha,13E,15S)-9,11,15-trihydroxyprost-13-en-1-oic acid Natural products CCCCCC(O)C=CC1C(O)CC(O)C1CCCCCCC(O)=O DZUXGQBLFALXCR-UHFFFAOYSA-N 0.000 description 1
- JFKQASGNSCLFLO-YSFYHYPLSA-N (3ar,4s,5r,6as)-4-[[tert-butyl(diphenyl)silyl]oxymethyl]-5-hydroxy-3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-2-one Chemical compound C([C@@H]1[C@H]2CC(=O)O[C@H]2C[C@H]1O)O[Si](C(C)(C)C)(C=1C=CC=CC=1)C1=CC=CC=C1 JFKQASGNSCLFLO-YSFYHYPLSA-N 0.000 description 1
- VMKAFJQFKBASMU-KRWDZBQOSA-N (3as)-1-methyl-3,3-diphenyl-3a,4,5,6-tetrahydropyrrolo[1,2-c][1,3,2]oxazaborole Chemical compound C([C@H]12)CCN1B(C)OC2(C=1C=CC=CC=1)C1=CC=CC=C1 VMKAFJQFKBASMU-KRWDZBQOSA-N 0.000 description 1
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 description 1
- 125000002733 (C1-C6) fluoroalkyl group Chemical group 0.000 description 1
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 description 1
- PXGPLTODNUVGFL-BRIYLRKRSA-N (E,Z)-(1R,2R,3R,5S)-7-(3,5-Dihydroxy-2-((3S)-(3-hydroxy-1-octenyl))cyclopentyl)-5-heptenoic acid Chemical compound CCCCC[C@H](O)C=C[C@H]1[C@H](O)C[C@H](O)[C@@H]1CC=CCCCC(O)=O PXGPLTODNUVGFL-BRIYLRKRSA-N 0.000 description 1
- OVFKTEWPWKWGEZ-RSIXOJRVSA-N (z)-7-[(1r,2r)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-3,3-difluoro-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@@H]1[C@@H](C\C=C/CCCC(O)=O)C(=O)CC1(F)F OVFKTEWPWKWGEZ-RSIXOJRVSA-N 0.000 description 1
- LJGZVMXJEKSYLK-LUMNEJPKSA-N (z)-7-[(1r,2r)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-3-methyl-5-oxocyclopent-3-en-1-yl]hept-5-enoic acid Chemical compound CC1=CC(=O)[C@H](C\C=C/CCCC(O)=O)[C@H]1CC[C@@H](O)C1=CC2=CC=CC=C2S1 LJGZVMXJEKSYLK-LUMNEJPKSA-N 0.000 description 1
- IJSFIYCPPSLCKA-LUMNEJPKSA-N (z)-7-[(1r,2r)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-3-methylidene-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@@H]1[C@@H](C\C=C/CCCC(O)=O)C(=O)CC1=C IJSFIYCPPSLCKA-LUMNEJPKSA-N 0.000 description 1
- KEKBQFVJZWOCMA-LUMNEJPKSA-N (z)-7-[(1r,2r)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-5-methylidene-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@@H]1[C@@H](C\C=C/CCCC(O)=O)C(=C)CC1=O KEKBQFVJZWOCMA-LUMNEJPKSA-N 0.000 description 1
- ITWFRKOEGVGTCW-VDCQVKDQSA-N (z)-7-[(1r,2r)-2-[(e)-3-(1-benzothiophen-2-yl)-3,3-difluoroprop-1-enyl]-3-methyl-5-oxocyclopent-3-en-1-yl]hept-5-enoic acid Chemical compound CC1=CC(=O)[C@H](C\C=C/CCCC(O)=O)[C@H]1\C=C\C(F)(F)C1=CC2=CC=CC=C2S1 ITWFRKOEGVGTCW-VDCQVKDQSA-N 0.000 description 1
- QXQIJEWJLWWSAX-VDCQVKDQSA-N (z)-7-[(1r,2r)-2-[(e)-3-(1-benzothiophen-2-yl)-3,3-difluoroprop-1-enyl]-3-methylidene-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C=C1CC(=O)[C@H](C\C=C/CCCC(=O)O)[C@H]1\C=C\C(F)(F)C1=CC2=CC=CC=C2S1 QXQIJEWJLWWSAX-VDCQVKDQSA-N 0.000 description 1
- OEPYECJWXTWAQZ-MXQFKEPZSA-N (z)-7-[(1r,2r)-2-[(e)-3-(1-benzothiophen-2-yl)-3-methylbut-1-enyl]-3-methyl-5-oxocyclopent-3-en-1-yl]hept-5-enoic acid Chemical compound CC1=CC(=O)[C@H](C\C=C/CCCC(O)=O)[C@H]1\C=C\C(C)(C)C1=CC2=CC=CC=C2S1 OEPYECJWXTWAQZ-MXQFKEPZSA-N 0.000 description 1
- YOEFSYYJGMEPFY-MXQFKEPZSA-N (z)-7-[(1r,2r)-2-[(e)-3-(1-benzothiophen-2-yl)-3-methylbut-1-enyl]-3-methylidene-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C=1C2=CC=CC=C2SC=1C(C)(C)\C=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)C(=O)CC1=C YOEFSYYJGMEPFY-MXQFKEPZSA-N 0.000 description 1
- SHCUXKSLCZRVLY-VDCQVKDQSA-N (z)-7-[(1r,2r)-2-[(e)-3-(1-benzothiophen-2-yl)-3-oxoprop-1-enyl]-3-methyl-5-oxocyclopent-3-en-1-yl]hept-5-enoic acid Chemical compound CC1=CC(=O)[C@H](C\C=C/CCCC(O)=O)[C@H]1\C=C\C(=O)C1=CC2=CC=CC=C2S1 SHCUXKSLCZRVLY-VDCQVKDQSA-N 0.000 description 1
- AKVJORRNLLMMNB-VDCQVKDQSA-N (z)-7-[(1r,2r)-2-[(e)-3-(1-benzothiophen-2-yl)-3-oxoprop-1-enyl]-3-methylidene-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C=C1CC(=O)[C@H](C\C=C/CCCC(=O)O)[C@H]1\C=C\C(=O)C1=CC2=CC=CC=C2S1 AKVJORRNLLMMNB-VDCQVKDQSA-N 0.000 description 1
- CGUVNQYIHVNGRU-AQWQHNGQSA-N (z)-7-[(1r,2r)-2-[(e,3r)-3-(1,3-benzothiazol-2-yl)-3-hydroxyprop-1-enyl]-3-methyl-5-oxocyclopent-3-en-1-yl]hept-5-enoic acid Chemical compound CC1=CC(=O)[C@H](C\C=C/CCCC(O)=O)[C@H]1\C=C\[C@@H](O)C1=NC2=CC=CC=C2S1 CGUVNQYIHVNGRU-AQWQHNGQSA-N 0.000 description 1
- PHOYXQOITMNKEB-AQWQHNGQSA-N (z)-7-[(1r,2r)-2-[(e,3r)-3-(1,3-benzothiazol-2-yl)-3-hydroxyprop-1-enyl]-3-methylidene-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2N=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)C(=O)CC1=C PHOYXQOITMNKEB-AQWQHNGQSA-N 0.000 description 1
- IUCNEZVKVUFQDC-AQWQHNGQSA-N (z)-7-[(1r,2r)-2-[(e,3r)-3-(1,3-benzoxazol-2-yl)-3-hydroxyprop-1-enyl]-3-methyl-5-oxocyclopent-3-en-1-yl]hept-5-enoic acid Chemical compound CC1=CC(=O)[C@H](C\C=C/CCCC(O)=O)[C@H]1\C=C\[C@@H](O)C1=NC2=CC=CC=C2O1 IUCNEZVKVUFQDC-AQWQHNGQSA-N 0.000 description 1
- ONGWYEXUYGAULR-AQWQHNGQSA-N (z)-7-[(1r,2r)-2-[(e,3r)-3-(1,3-benzoxazol-2-yl)-3-hydroxyprop-1-enyl]-3-methylidene-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1OC2=CC=CC=C2N=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)C(=O)CC1=C ONGWYEXUYGAULR-AQWQHNGQSA-N 0.000 description 1
- HBCKDDDBRSWWIU-XGCRWXAESA-N (z)-7-[(1r,2r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxybut-1-enyl]-3-methyl-5-oxocyclopent-3-en-1-yl]hept-5-enoic acid Chemical compound CC1=CC(=O)[C@H](C\C=C/CCCC(O)=O)[C@H]1\C=C\[C@@](C)(O)C1=CC2=CC=CC=C2S1 HBCKDDDBRSWWIU-XGCRWXAESA-N 0.000 description 1
- NUFMMXPJHRAERQ-XGCRWXAESA-N (z)-7-[(1r,2r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxybut-1-enyl]-3-methylidene-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@](O)(C)C=1SC2=CC=CC=C2C=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)C(=O)CC1=C NUFMMXPJHRAERQ-XGCRWXAESA-N 0.000 description 1
- OODZJPFRQSNESB-VOJWTERLSA-N (z)-7-[(1r,2r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-5-methylidene-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)C(=C)CC1=O OODZJPFRQSNESB-VOJWTERLSA-N 0.000 description 1
- RQWPGPPVCAFPMJ-QXYWQFJLSA-N (z)-7-[(1r,2r)-2-[(e,3r)-3-(1h-benzimidazol-2-yl)-3-hydroxyprop-1-enyl]-3-methyl-5-oxocyclopent-3-en-1-yl]hept-5-enoic acid Chemical compound CC1=CC(=O)[C@H](C\C=C/CCCC(O)=O)[C@H]1\C=C\[C@@H](O)C1=NC2=CC=CC=C2N1 RQWPGPPVCAFPMJ-QXYWQFJLSA-N 0.000 description 1
- DTQCUHNLGWQEDM-QXYWQFJLSA-N (z)-7-[(1r,2r)-2-[(e,3r)-3-(1h-benzimidazol-2-yl)-3-hydroxyprop-1-enyl]-3-methylidene-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1NC2=CC=CC=C2N=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)C(=O)CC1=C DTQCUHNLGWQEDM-QXYWQFJLSA-N 0.000 description 1
- VHNBCHCILOADRL-JYZOCMRUSA-N (z)-7-[(1r,2r)-2-[(e,3r)-3-hydroxy-3-(1-methylbenzimidazol-2-yl)prop-1-enyl]-3-methyl-5-oxocyclopent-3-en-1-yl]hept-5-enoic acid Chemical compound CC1=CC(=O)[C@H](C\C=C/CCCC(O)=O)[C@H]1\C=C\[C@@H](O)C1=NC2=CC=CC=C2N1C VHNBCHCILOADRL-JYZOCMRUSA-N 0.000 description 1
- BMLOQTQQSRSXJC-JYZOCMRUSA-N (z)-7-[(1r,2r)-2-[(e,3r)-3-hydroxy-3-(1-methylbenzimidazol-2-yl)prop-1-enyl]-3-methylidene-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1N(C2=CC=CC=C2N=1)C)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)C(=O)CC1=C BMLOQTQQSRSXJC-JYZOCMRUSA-N 0.000 description 1
- XYMREDDUNOUGKI-OMMVWFPWSA-N (z)-7-[(1r,2r)-2-[(e,3r)-3-hydroxy-4-[3-(trifluoromethyl)phenoxy]but-1-enyl]-3-methyl-5-oxocyclopent-3-en-1-yl]hept-5-enoic acid Chemical compound CC1=CC(=O)[C@H](C\C=C/CCCC(O)=O)[C@H]1\C=C\[C@@H](O)COC1=CC=CC(C(F)(F)F)=C1 XYMREDDUNOUGKI-OMMVWFPWSA-N 0.000 description 1
- LZYFEOIPGXXJRG-OMMVWFPWSA-N (z)-7-[(1r,2r)-2-[(e,3r)-3-hydroxy-4-[3-(trifluoromethyl)phenoxy]but-1-enyl]-3-methylidene-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C([C@H](O)\C=C\[C@H]1C(CC(=O)[C@@H]1C\C=C/CCCC(O)=O)=C)OC1=CC=CC(C(F)(F)F)=C1 LZYFEOIPGXXJRG-OMMVWFPWSA-N 0.000 description 1
- VSSDAOWIINQZKN-FOGSTIQSSA-N (z)-7-[(1r,2r)-2-[(e,3s)-5-(2,3-dihydro-1h-inden-2-yl)-3-hydroxypent-1-enyl]-3-methyl-5-oxocyclopent-3-en-1-yl]hept-5-enoic acid Chemical compound CC1=CC(=O)[C@H](C\C=C/CCCC(O)=O)[C@H]1\C=C\[C@@H](O)CCC1CC2=CC=CC=C2C1 VSSDAOWIINQZKN-FOGSTIQSSA-N 0.000 description 1
- IDABRTIJUAGPIB-FOGSTIQSSA-N (z)-7-[(1r,2r)-2-[(e,3s)-5-(2,3-dihydro-1h-inden-2-yl)-3-hydroxypent-1-enyl]-3-methylidene-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@H](CCC1CC2=CC=CC=C2C1)O)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)C(=O)CC1=C IDABRTIJUAGPIB-FOGSTIQSSA-N 0.000 description 1
- NOKBOVOALOLKGN-AENWUBLQSA-N (z)-7-[(1r,2r)-2-[(e,3s)-6-(1-benzothiophen-2-yl)-3-hydroxyhex-1-enyl]-3-methyl-5-oxocyclopent-3-en-1-yl]hept-5-enoic acid Chemical compound CC1=CC(=O)[C@H](C\C=C/CCCC(O)=O)[C@H]1\C=C\[C@@H](O)CCCC1=CC2=CC=CC=C2S1 NOKBOVOALOLKGN-AENWUBLQSA-N 0.000 description 1
- KQZIVFHNMLNHON-AENWUBLQSA-N (z)-7-[(1r,2r)-2-[(e,3s)-6-(1-benzothiophen-2-yl)-3-hydroxyhex-1-enyl]-3-methylidene-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@H](CCCC=1SC2=CC=CC=C2C=1)O)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)C(=O)CC1=C KQZIVFHNMLNHON-AENWUBLQSA-N 0.000 description 1
- MKLBCUXQDPSPGY-QMRSPPQLSA-N (z)-7-[(1r,2r)-2-[(e,3s)-6-(2,3-dihydro-1h-inden-2-yl)-3-hydroxyhex-1-enyl]-3-methyl-5-oxocyclopent-3-en-1-yl]hept-5-enoic acid Chemical compound CC1=CC(=O)[C@H](C\C=C/CCCC(O)=O)[C@H]1\C=C\[C@@H](O)CCCC1CC2=CC=CC=C2C1 MKLBCUXQDPSPGY-QMRSPPQLSA-N 0.000 description 1
- HELLBYZMIOWJGA-QMRSPPQLSA-N (z)-7-[(1r,2r)-2-[(e,3s)-6-(2,3-dihydro-1h-inden-2-yl)-3-hydroxyhex-1-enyl]-3-methylidene-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@H](CCCC1CC2=CC=CC=C2C1)O)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)C(=O)CC1=C HELLBYZMIOWJGA-QMRSPPQLSA-N 0.000 description 1
- XEGQTVCLLDDBLK-KMVIMSPISA-N (z)-7-[(1r,2r,3r)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-[tert-butyl(diphenyl)silyl]oxypropyl]-3-hydroxy-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C([C@@H](O[Si](C(C)(C)C)(C=1C=CC=CC=1)C=1C=CC=CC=1)C=1SC2=CC=CC=C2C=1)C[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O XEGQTVCLLDDBLK-KMVIMSPISA-N 0.000 description 1
- VKWFXKPHWXJZNB-SDPYNCHPSA-N (z)-7-[(1r,2r,3r)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-3-fluoro-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@H]1[C@H](F)CC(=O)[C@@H]1C\C=C/CCCC(O)=O VKWFXKPHWXJZNB-SDPYNCHPSA-N 0.000 description 1
- OMPOCGBYFCMXDJ-VCRPCSLDSA-N (z)-7-[(1r,2r,3r)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-3-hydroxy-5-methylidenecyclopentyl]hept-5-enoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@H]1[C@H](O)CC(=C)[C@@H]1C\C=C/CCCC(O)=O OMPOCGBYFCMXDJ-VCRPCSLDSA-N 0.000 description 1
- WEURLISWANBBHH-QYXMGJOPSA-N (z)-7-[(1r,2r,3r)-2-[(e)-3-(1-benzothiophen-2-yl)-3-methylbut-1-enyl]-3-hydroxy-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C=1C2=CC=CC=C2SC=1C(C)(C)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O WEURLISWANBBHH-QYXMGJOPSA-N 0.000 description 1
- CAIGMNRJQLUCQE-LZMVQSPFSA-N (z)-7-[(1r,2r,3r)-2-[(e)-3-(1-benzothiophen-2-yl)-3-oxoprop-1-enyl]-3-hydroxy-5-oxocyclopentyl]hept-5-enoic acid Chemical compound O[C@@H]1CC(=O)[C@H](C\C=C/CCCC(O)=O)[C@H]1\C=C\C(=O)C1=CC2=CC=CC=C2S1 CAIGMNRJQLUCQE-LZMVQSPFSA-N 0.000 description 1
- IEZPEFCRPJHORM-CNFLNUCUSA-N (z)-7-[(1r,2r,3r)-2-[(e,3r)-3-(1,3-benzoxazol-2-yl)-3-hydroxyprop-1-enyl]-3-hydroxy-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1OC2=CC=CC=C2N=1)=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O IEZPEFCRPJHORM-CNFLNUCUSA-N 0.000 description 1
- XOTKSJPQOJOTQZ-DAADMISMSA-N (z)-7-[(1r,2r,3r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxybut-1-enyl]-3-hydroxy-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@](O)(C)C=1SC2=CC=CC=C2C=1)=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O XOTKSJPQOJOTQZ-DAADMISMSA-N 0.000 description 1
- LHVJOKSIYAWVKX-JKFLSUOJSA-N (z)-7-[(1r,2r,3r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-3-fluoro-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@H]1[C@H](F)CC(=O)[C@@H]1C\C=C/CCCC(O)=O LHVJOKSIYAWVKX-JKFLSUOJSA-N 0.000 description 1
- SVUBUTGSAAGRBH-KAOSKRLXSA-N (z)-7-[(1r,2r,3r)-2-[(e,3r)-3-(1h-benzimidazol-2-yl)-3-hydroxyprop-1-enyl]-3-hydroxy-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1NC2=CC=CC=C2N=1)=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O SVUBUTGSAAGRBH-KAOSKRLXSA-N 0.000 description 1
- DRFGNEHBUTVWEP-RYSUORISSA-N (z)-7-[(1r,2r,3r)-2-[(e,3s)-5-(2,3-dihydro-1h-inden-2-yl)-3-hydroxypent-1-enyl]-3-hydroxy-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@H](CCC1CC2=CC=CC=C2C1)O)=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O DRFGNEHBUTVWEP-RYSUORISSA-N 0.000 description 1
- KRIRZUCAQJNVAF-QLHOKORLSA-N (z)-7-[(1r,2r,3r)-2-[(e,3s)-6-(1-benzothiophen-2-yl)-3-hydroxyhex-1-enyl]-3-hydroxy-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@H](CCCC=1SC2=CC=CC=C2C=1)O)=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O KRIRZUCAQJNVAF-QLHOKORLSA-N 0.000 description 1
- CKJNBDFTYBGQRY-MQPRVQJTSA-N (z)-7-[(1r,2r,3r)-2-[(e,3s)-6-(2,3-dihydro-1h-inden-2-yl)-3-hydroxyhex-1-enyl]-3-hydroxy-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@H](CCCC1CC2=CC=CC=C2C1)O)=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O CKJNBDFTYBGQRY-MQPRVQJTSA-N 0.000 description 1
- WXXQLBUOFYIWSI-HNUYCGMQSA-N (z)-7-[(1r,2r,3r)-3-hydroxy-2-[(e,3r)-3-hydroxy-3-(1-methylbenzimidazol-2-yl)prop-1-enyl]-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1N(C2=CC=CC=C2N=1)C)=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O WXXQLBUOFYIWSI-HNUYCGMQSA-N 0.000 description 1
- YXMXFAFWASXMKY-QJJPCVRTSA-N (z)-7-[(1r,2r,3r)-3-hydroxy-2-[(e,3r)-3-hydroxy-3-pyridin-2-ylprop-1-enyl]-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1N=CC=CC=1)=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O YXMXFAFWASXMKY-QJJPCVRTSA-N 0.000 description 1
- VKWFXKPHWXJZNB-UMQUAKBUSA-N (z)-7-[(1r,2r,3s)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-3-fluoro-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@H]1[C@@H](F)CC(=O)[C@@H]1C\C=C/CCCC(O)=O VKWFXKPHWXJZNB-UMQUAKBUSA-N 0.000 description 1
- LHVJOKSIYAWVKX-VCXOPIFBSA-N (z)-7-[(1r,2r,3s)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-3-fluoro-5-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@H]1[C@@H](F)CC(=O)[C@@H]1C\C=C/CCCC(O)=O LHVJOKSIYAWVKX-VCXOPIFBSA-N 0.000 description 1
- CGDHMUFMUAROGC-SDPYNCHPSA-N (z)-7-[(1r,2r,5r)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-5-fluoro-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@H](F)CC1=O CGDHMUFMUAROGC-SDPYNCHPSA-N 0.000 description 1
- YTPVAKBJFINMCZ-JKFLSUOJSA-N (z)-7-[(1r,2r,5r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-5-fluoro-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@H](F)CC1=O YTPVAKBJFINMCZ-JKFLSUOJSA-N 0.000 description 1
- JSHQQUAEUPPSJS-VMXSXOFESA-N (z)-7-[(1r,2r,5s)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-[tert-butyl(diphenyl)silyl]oxypropyl]-5-hydroxy-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C([C@@H](O[Si](C(C)(C)C)(C=1C=CC=CC=1)C=1C=CC=CC=1)C=1SC2=CC=CC=C2C=1)C[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=O JSHQQUAEUPPSJS-VMXSXOFESA-N 0.000 description 1
- HAKCLEVBSBNODO-VTPTUHDASA-N (z)-7-[(1r,2r,5s)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-3,3-difluoro-5-hydroxycyclopentyl]hept-5-enoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1(F)F HAKCLEVBSBNODO-VTPTUHDASA-N 0.000 description 1
- CGDHMUFMUAROGC-UMQUAKBUSA-N (z)-7-[(1r,2r,5s)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-5-fluoro-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](F)CC1=O CGDHMUFMUAROGC-UMQUAKBUSA-N 0.000 description 1
- IRAGVSGWJQXVDJ-HYBUZTIPSA-N (z)-7-[(1r,2r,5s)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-5-hydroxy-3-methylidenecyclopentyl]hept-5-enoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=C IRAGVSGWJQXVDJ-HYBUZTIPSA-N 0.000 description 1
- LYGBHMHJDRMLFA-VTPTUHDASA-N (z)-7-[(1r,2r,5s)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-5-hydroxy-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=O LYGBHMHJDRMLFA-VTPTUHDASA-N 0.000 description 1
- DFGINWUFJGHFIT-AGEXTVMASA-N (z)-7-[(1r,2r,5s)-2-[(e)-3-(1-benzothiophen-2-yl)-3,3-difluoroprop-1-enyl]-5-hydroxy-3-oxocyclopentyl]hept-5-enoic acid Chemical compound OC(=O)CCC\C=C/C[C@H]1[C@@H](O)CC(=O)[C@@H]1\C=C\C(F)(F)C1=CC2=CC=CC=C2S1 DFGINWUFJGHFIT-AGEXTVMASA-N 0.000 description 1
- RKASWMNPHJYVKQ-JKRVCFJGSA-N (z)-7-[(1r,2r,5s)-2-[(e)-3-(1-benzothiophen-2-yl)-3-methylbut-1-enyl]-5-hydroxy-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C=1C2=CC=CC=C2SC=1C(C)(C)\C=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=O RKASWMNPHJYVKQ-JKRVCFJGSA-N 0.000 description 1
- CIQXKBCADIANLL-VQEDUQJOSA-N (z)-7-[(1r,2r,5s)-2-[(e)-3-(1-benzothiophen-2-yl)-3-oxoprop-1-enyl]-5-hydroxy-3-oxocyclopentyl]hept-5-enoic acid Chemical compound OC(=O)CCC\C=C/C[C@H]1[C@@H](O)CC(=O)[C@@H]1\C=C\C(=O)C1=CC2=CC=CC=C2S1 CIQXKBCADIANLL-VQEDUQJOSA-N 0.000 description 1
- RFMVYPNLUCDVKU-OWELLUKLSA-N (z)-7-[(1r,2r,5s)-2-[(e,3r)-3-(1,3-benzoxazol-2-yl)-3-hydroxyprop-1-enyl]-5-hydroxy-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1OC2=CC=CC=C2N=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=O RFMVYPNLUCDVKU-OWELLUKLSA-N 0.000 description 1
- MAQCHSUUJYGQDM-YQECFWACSA-N (z)-7-[(1r,2r,5s)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxybut-1-enyl]-5-hydroxy-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@](O)(C)C=1SC2=CC=CC=C2C=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=O MAQCHSUUJYGQDM-YQECFWACSA-N 0.000 description 1
- SGLCDNMEQCIROL-GCTAJMIWSA-N (z)-7-[(1r,2r,5s)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-3,3-difluoro-5-hydroxycyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1(F)F SGLCDNMEQCIROL-GCTAJMIWSA-N 0.000 description 1
- YTPVAKBJFINMCZ-VCXOPIFBSA-N (z)-7-[(1r,2r,5s)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-5-fluoro-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](F)CC1=O YTPVAKBJFINMCZ-VCXOPIFBSA-N 0.000 description 1
- BMCCGYAHMQYJOP-KILLSRIUSA-N (z)-7-[(1r,2r,5s)-2-[(e,3r)-3-(1h-benzimidazol-2-yl)-3-hydroxyprop-1-enyl]-5-hydroxy-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1NC2=CC=CC=C2N=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=O BMCCGYAHMQYJOP-KILLSRIUSA-N 0.000 description 1
- UWKAOJPSDGXUMD-HGOLIGCMSA-N (z)-7-[(1r,2r,5s)-2-[(e,3s)-5-(2,3-dihydro-1h-inden-2-yl)-3-hydroxypent-1-enyl]-5-hydroxy-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@H](CCC1CC2=CC=CC=C2C1)O)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=O UWKAOJPSDGXUMD-HGOLIGCMSA-N 0.000 description 1
- MWBKMWCHTPKOOF-RUOHXLKZSA-N (z)-7-[(1r,2r,5s)-2-[(e,3s)-6-(1-benzothiophen-2-yl)-3-hydroxyhex-1-enyl]-5-hydroxy-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@H](CCCC=1SC2=CC=CC=C2C=1)O)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=O MWBKMWCHTPKOOF-RUOHXLKZSA-N 0.000 description 1
- QAZMWJRCTJEJFV-HULZQETDSA-N (z)-7-[(1r,2r,5s)-2-[(e,3s)-6-(2,3-dihydro-1h-inden-2-yl)-3-hydroxyhex-1-enyl]-5-hydroxy-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@H](CCCC1CC2=CC=CC=C2C1)O)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=O QAZMWJRCTJEJFV-HULZQETDSA-N 0.000 description 1
- ZHYBDPWZIBOTGX-LTLAXFGGSA-N (z)-7-[(1r,2r,5s)-5-hydroxy-2-[(e,3r)-3-hydroxy-3-(1-methylbenzimidazol-2-yl)prop-1-enyl]-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1N(C2=CC=CC=C2N=1)C)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=O ZHYBDPWZIBOTGX-LTLAXFGGSA-N 0.000 description 1
- BZLAYCLDCHAUEZ-HWCQALDPSA-N (z)-7-[(1r,2r,5s)-5-hydroxy-2-[(e,3r)-3-hydroxy-3-pyridin-2-ylprop-1-enyl]-3-oxocyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1N=CC=CC=1)=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=O BZLAYCLDCHAUEZ-HWCQALDPSA-N 0.000 description 1
- UFMPMBHQIPZCOC-SDPYNCHPSA-N (z)-7-[(1r,4r,5r)-5-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-2,2-difluoro-4-hydroxycyclopentyl]hept-5-enoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@H]1[C@H](O)CC(F)(F)[C@@H]1C\C=C/CCCC(O)=O UFMPMBHQIPZCOC-SDPYNCHPSA-N 0.000 description 1
- AEZLQVAUCQLMQB-JKFLSUOJSA-N (z)-7-[(1r,4r,5r)-5-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-2,2-difluoro-4-hydroxycyclopentyl]hept-5-enoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@H]1[C@H](O)CC(F)(F)[C@@H]1C\C=C/CCCC(O)=O AEZLQVAUCQLMQB-JKFLSUOJSA-N 0.000 description 1
- DWPIWJPZFATELE-RSIXOJRVSA-N (z)-7-[(1r,5r)-5-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-2,2-difluoro-4-oxocyclopentyl]hept-5-enoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@@H]1[C@@H](C\C=C/CCCC(O)=O)C(F)(F)CC1=O DWPIWJPZFATELE-RSIXOJRVSA-N 0.000 description 1
- NPKAGEUCVXKJKV-LUMNEJPKSA-N (z)-7-[(1r,5r)-5-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-2-methyl-4-oxocyclopent-2-en-1-yl]hept-5-enoic acid Chemical compound OC(=O)CCC\C=C/C[C@H]1C(C)=CC(=O)[C@@H]1CC[C@@H](O)C1=CC2=CC=CC=C2S1 NPKAGEUCVXKJKV-LUMNEJPKSA-N 0.000 description 1
- LOWBJKKAGZXFFS-UHFFFAOYSA-N 1-(1-benzothiophen-2-yl)-2-diethoxyphosphorylethanone Chemical compound C1=CC=C2SC(C(=O)CP(=O)(OCC)OCC)=CC2=C1 LOWBJKKAGZXFFS-UHFFFAOYSA-N 0.000 description 1
- UAWXZBOUZHGQHD-UHFFFAOYSA-N 1-(2,7a-dihydro-1-benzothiophen-2-yl)-2-diethoxyphosphorylethanone Chemical compound C1=CC=CC2=CC(C(=O)CP(=O)(OCC)OCC)SC21 UAWXZBOUZHGQHD-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical group C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- NQTSTBMCCAVWOS-UHFFFAOYSA-N 1-dimethoxyphosphoryl-3-phenoxypropan-2-one Chemical compound COP(=O)(OC)CC(=O)COC1=CC=CC=C1 NQTSTBMCCAVWOS-UHFFFAOYSA-N 0.000 description 1
- YGTUPRIZNBMOFV-UHFFFAOYSA-N 2-(4-hydroxybenzoyl)benzoic acid Chemical compound OC(=O)C1=CC=CC=C1C(=O)C1=CC=C(O)C=C1 YGTUPRIZNBMOFV-UHFFFAOYSA-N 0.000 description 1
- SXGZJKUKBWWHRA-UHFFFAOYSA-N 2-(N-morpholiniumyl)ethanesulfonate Chemical compound [O-]S(=O)(=O)CC[NH+]1CCOCC1 SXGZJKUKBWWHRA-UHFFFAOYSA-N 0.000 description 1
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 1
- APIXJSLKIYYUKG-UHFFFAOYSA-N 3 Isobutyl 1 methylxanthine Chemical group O=C1N(C)C(=O)N(CC(C)C)C2=C1N=CN2 APIXJSLKIYYUKG-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- HCLQARMRCPEALF-DNQXCXABSA-N 3-[[(2r)-2-[(1r)-2-[[1-(1-benzothiophen-2-yl)-2-methylpropan-2-yl]amino]-1-hydroxyethyl]pyrrolidin-1-yl]methyl]benzonitrile Chemical compound C([C@@H]1[C@H](O)CNC(C)(CC=2SC3=CC=CC=C3C=2)C)CCN1CC1=CC=CC(C#N)=C1 HCLQARMRCPEALF-DNQXCXABSA-N 0.000 description 1
- CQBVTZDISUKDSX-UHFFFAOYSA-N 3-chloro-n'-[3-(furan-2-ylmethylsulfanyl)propanoyl]-6h-benzo[b][1,4]benzoxazepine-5-carbohydrazide Chemical group C12=CC(Cl)=CC=C2OC2=CC=CC=C2CN1C(=O)NNC(=O)CCSCC1=CC=CO1 CQBVTZDISUKDSX-UHFFFAOYSA-N 0.000 description 1
- RGRNSTGIHROKJB-UHFFFAOYSA-N 3-hydroxy-3-methylhexanoic acid Chemical compound CCCC(C)(O)CC(O)=O RGRNSTGIHROKJB-UHFFFAOYSA-N 0.000 description 1
- WCDLCPLAAKUJNY-UHFFFAOYSA-N 4-[4-[3-(1h-pyrazol-4-yl)pyrazolo[1,5-a]pyrimidin-6-yl]phenyl]morpholine Chemical compound C1COCCN1C1=CC=C(C2=CN3N=CC(=C3N=C2)C2=CNN=C2)C=C1 WCDLCPLAAKUJNY-UHFFFAOYSA-N 0.000 description 1
- 229940124125 5 Lipoxygenase inhibitor Drugs 0.000 description 1
- OKRUMSWHDWKGHA-UHFFFAOYSA-N 5-bromopentanoyl chloride Chemical compound ClC(=O)CCCCBr OKRUMSWHDWKGHA-UHFFFAOYSA-N 0.000 description 1
- ODHCTXKNWHHXJC-VKHMYHEASA-N 5-oxo-L-proline Chemical compound OC(=O)[C@@H]1CCC(=O)N1 ODHCTXKNWHHXJC-VKHMYHEASA-N 0.000 description 1
- TWMFZTGWLKKREH-KZNAEPCWSA-N 7-[(1r,2r)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-3,3-difluoro-5-oxocyclopentyl]heptanoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@@H]1[C@@H](CCCCCCC(O)=O)C(=O)CC1(F)F TWMFZTGWLKKREH-KZNAEPCWSA-N 0.000 description 1
- UJUDCFYGOQNXAK-XUVXKRRUSA-N 7-[(1r,2r)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-3-methyl-5-oxocyclopent-3-en-1-yl]heptanoic acid Chemical compound CC1=CC(=O)[C@H](CCCCCCC(O)=O)[C@H]1CC[C@@H](O)C1=CC2=CC=CC=C2S1 UJUDCFYGOQNXAK-XUVXKRRUSA-N 0.000 description 1
- IJULVZCLQJHSKK-XUVXKRRUSA-N 7-[(1r,2r)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-3-methylidene-5-oxocyclopentyl]heptanoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@@H]1[C@@H](CCCCCCC(O)=O)C(=O)CC1=C IJULVZCLQJHSKK-XUVXKRRUSA-N 0.000 description 1
- ZPWUKBWJZZCWIN-XUVXKRRUSA-N 7-[(1r,2r)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-5-methylidene-3-oxocyclopentyl]heptanoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@@H]1[C@@H](CCCCCCC(O)=O)C(=C)CC1=O ZPWUKBWJZZCWIN-XUVXKRRUSA-N 0.000 description 1
- IKVRTCBXZOGUGD-BECVJJOHSA-N 7-[(1r,2r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-3,3-difluoro-5-oxocyclopentyl]heptanoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@@H]1[C@@H](CCCCCCC(O)=O)C(=O)CC1(F)F IKVRTCBXZOGUGD-BECVJJOHSA-N 0.000 description 1
- MQVRMPYWDYBFNQ-ANRYODRRSA-N 7-[(1r,2r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-5-methylidene-3-oxocyclopentyl]heptanoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@@H]1[C@@H](CCCCCCC(O)=O)C(=C)CC1=O MQVRMPYWDYBFNQ-ANRYODRRSA-N 0.000 description 1
- XUTURJQWAADEBG-PPRJIQIQSA-N 7-[(1r,2r)-2-[(e,3s)-3-(2,3-dihydro-1h-inden-2-yl)-3-hydroxyprop-1-enyl]-3-methyl-5-oxocyclopent-3-en-1-yl]heptanoic acid Chemical compound CC1=CC(=O)[C@H](CCCCCCC(O)=O)[C@H]1\C=C\[C@@H](O)C1CC2=CC=CC=C2C1 XUTURJQWAADEBG-PPRJIQIQSA-N 0.000 description 1
- UVMUENDKJQVSFA-PPRJIQIQSA-N 7-[(1r,2r)-2-[(e,3s)-3-(2,3-dihydro-1h-inden-2-yl)-3-hydroxyprop-1-enyl]-3-methylidene-5-oxocyclopentyl]heptanoic acid Chemical compound C(/[C@@H](O)C1CC2=CC=CC=C2C1)=C\[C@@H]1[C@@H](CCCCCCC(O)=O)C(=O)CC1=C UVMUENDKJQVSFA-PPRJIQIQSA-N 0.000 description 1
- VYGNXFHQKAKLPF-NCXUSEDFSA-N 7-[(1r,2r,3r)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-3-fluoro-5-oxocyclopentyl]heptanoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@H]1[C@H](F)CC(=O)[C@@H]1CCCCCCC(O)=O VYGNXFHQKAKLPF-NCXUSEDFSA-N 0.000 description 1
- YNAPPWJDVZHDOH-DOIPELPJSA-N 7-[(1r,2r,3r)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-3-hydroxy-5-methylidenecyclopentyl]heptanoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@H]1[C@H](O)CC(=C)[C@@H]1CCCCCCC(O)=O YNAPPWJDVZHDOH-DOIPELPJSA-N 0.000 description 1
- DVGLEQCDJIUNIR-BWKHYTDRSA-N 7-[(1r,2r,3r)-2-[(3r)-5-(1-benzothiophen-2-yl)-3-hydroxypentyl]-3-hydroxy-5-oxocyclopentyl]heptanoic acid Chemical compound C([C@@H](O)CCC=1SC2=CC=CC=C2C=1)C[C@H]1[C@H](O)CC(=O)[C@@H]1CCCCCCC(O)=O DVGLEQCDJIUNIR-BWKHYTDRSA-N 0.000 description 1
- JAHMPXXLBBRLGV-GDNMGRSTSA-N 7-[(1r,2r,3r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-3-fluoro-5-oxocyclopentyl]heptanoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@H]1[C@H](F)CC(=O)[C@@H]1CCCCCCC(O)=O JAHMPXXLBBRLGV-GDNMGRSTSA-N 0.000 description 1
- YAKPFSCELNSRQI-HUKCMAGJSA-N 7-[(1r,2r,3r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-3-hydroxy-5-methylidenecyclopentyl]heptanoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@H]1[C@H](O)CC(=C)[C@@H]1CCCCCCC(O)=O YAKPFSCELNSRQI-HUKCMAGJSA-N 0.000 description 1
- HOOGEKTYNWNHEP-WVXAJDQPSA-N 7-[(1r,2r,3r)-2-[(e,3s)-3-(2,3-dihydro-1h-inden-2-yl)-3-hydroxyprop-1-enyl]-3-hydroxy-5-oxocyclopentyl]heptanoic acid Chemical compound C(/[C@@H](O)C1CC2=CC=CC=C2C1)=C\[C@H]1[C@H](O)CC(=O)[C@@H]1CCCCCCC(O)=O HOOGEKTYNWNHEP-WVXAJDQPSA-N 0.000 description 1
- YPOFITWUBDEMER-CKQPALCZSA-N 7-[(1r,2r,3r,5s)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-[tert-butyl(diphenyl)silyl]oxypropyl]-3,5-dihydroxycyclopentyl]heptanoic acid Chemical compound C([C@@H](O[Si](C(C)(C)C)(C=1C=CC=CC=1)C=1C=CC=CC=1)C=1SC2=CC=CC=C2C=1)C[C@H]1[C@H](O)C[C@H](O)[C@@H]1CCCCCCC(O)=O YPOFITWUBDEMER-CKQPALCZSA-N 0.000 description 1
- VYGNXFHQKAKLPF-AKHDSKFASA-N 7-[(1r,2r,3s)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-3-fluoro-5-oxocyclopentyl]heptanoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@H]1[C@@H](F)CC(=O)[C@@H]1CCCCCCC(O)=O VYGNXFHQKAKLPF-AKHDSKFASA-N 0.000 description 1
- JAHMPXXLBBRLGV-KXCTWPOZSA-N 7-[(1r,2r,3s)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-3-fluoro-5-oxocyclopentyl]heptanoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@H]1[C@@H](F)CC(=O)[C@@H]1CCCCCCC(O)=O JAHMPXXLBBRLGV-KXCTWPOZSA-N 0.000 description 1
- FFGXWCNITYQYCE-NCXUSEDFSA-N 7-[(1r,2r,5r)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-5-fluoro-3-oxocyclopentyl]heptanoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@@H]1[C@@H](CCCCCCC(O)=O)[C@H](F)CC1=O FFGXWCNITYQYCE-NCXUSEDFSA-N 0.000 description 1
- UKYZZPVQPAXPKM-GDNMGRSTSA-N 7-[(1r,2r,5r)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-5-fluoro-3-oxocyclopentyl]heptanoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@@H]1[C@@H](CCCCCCC(O)=O)[C@H](F)CC1=O UKYZZPVQPAXPKM-GDNMGRSTSA-N 0.000 description 1
- BXPYODXDFOULSL-MKXGPGLRSA-N 7-[(1r,2r,5s)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-3,3-difluoro-5-hydroxycyclopentyl]heptanoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@@H]1[C@@H](CCCCCCC(O)=O)[C@@H](O)CC1(F)F BXPYODXDFOULSL-MKXGPGLRSA-N 0.000 description 1
- FFGXWCNITYQYCE-AKHDSKFASA-N 7-[(1r,2r,5s)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-5-fluoro-3-oxocyclopentyl]heptanoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@@H]1[C@@H](CCCCCCC(O)=O)[C@@H](F)CC1=O FFGXWCNITYQYCE-AKHDSKFASA-N 0.000 description 1
- RIIDVNXZRZBOJM-BQJUDKOJSA-N 7-[(1r,2r,5s)-2-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-5-hydroxy-3-methylidenecyclopentyl]heptanoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@@H]1[C@@H](CCCCCCC(O)=O)[C@@H](O)CC1=C RIIDVNXZRZBOJM-BQJUDKOJSA-N 0.000 description 1
- GZUHECZBUZYUOA-PDGJWGCVSA-N 7-[(1r,2r,5s)-2-[(3r)-5-(1-benzothiophen-2-yl)-3-hydroxypentyl]-5-hydroxy-3-oxocyclopentyl]heptanoic acid Chemical compound C([C@@H](O)CCC=1SC2=CC=CC=C2C=1)C[C@@H]1[C@@H](CCCCCCC(O)=O)[C@@H](O)CC1=O GZUHECZBUZYUOA-PDGJWGCVSA-N 0.000 description 1
- QZCYVBCBBHDYIN-REOHDZAKSA-N 7-[(1r,2r,5s)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-3,3-difluoro-5-hydroxycyclopentyl]heptanoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@@H]1[C@@H](CCCCCCC(O)=O)[C@@H](O)CC1(F)F QZCYVBCBBHDYIN-REOHDZAKSA-N 0.000 description 1
- UKYZZPVQPAXPKM-KXCTWPOZSA-N 7-[(1r,2r,5s)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-5-fluoro-3-oxocyclopentyl]heptanoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@@H]1[C@@H](CCCCCCC(O)=O)[C@@H](F)CC1=O UKYZZPVQPAXPKM-KXCTWPOZSA-N 0.000 description 1
- PRUBGTNFIVOOCW-VONQFSBDSA-N 7-[(1r,2r,5s)-2-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-5-hydroxy-3-methylidenecyclopentyl]heptanoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@@H]1[C@@H](CCCCCCC(O)=O)[C@@H](O)CC1=C PRUBGTNFIVOOCW-VONQFSBDSA-N 0.000 description 1
- GHFTYHBQXIUKJQ-JWVPBWIXSA-N 7-[(1r,2r,5s)-2-[(e,3s)-3-(2,3-dihydro-1h-inden-2-yl)-3-hydroxyprop-1-enyl]-5-hydroxy-3-oxocyclopentyl]heptanoic acid Chemical compound C(/[C@@H](O)C1CC2=CC=CC=C2C1)=C\[C@@H]1[C@@H](CCCCCCC(O)=O)[C@@H](O)CC1=O GHFTYHBQXIUKJQ-JWVPBWIXSA-N 0.000 description 1
- MRRJQPXMCORNAB-NCXUSEDFSA-N 7-[(1r,4r,5r)-5-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-2,2-difluoro-4-hydroxycyclopentyl]heptanoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@H]1[C@H](O)CC(F)(F)[C@@H]1CCCCCCC(O)=O MRRJQPXMCORNAB-NCXUSEDFSA-N 0.000 description 1
- IYVMZJSHJWNXFR-GDNMGRSTSA-N 7-[(1r,4r,5r)-5-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-2,2-difluoro-4-hydroxycyclopentyl]heptanoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@H]1[C@H](O)CC(F)(F)[C@@H]1CCCCCCC(O)=O IYVMZJSHJWNXFR-GDNMGRSTSA-N 0.000 description 1
- UJYQZVASIOFEOF-KZNAEPCWSA-N 7-[(1r,5r)-5-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-2,2-difluoro-4-oxocyclopentyl]heptanoic acid Chemical compound C([C@@H](O)C=1SC2=CC=CC=C2C=1)C[C@@H]1[C@@H](CCCCCCC(O)=O)C(F)(F)CC1=O UJYQZVASIOFEOF-KZNAEPCWSA-N 0.000 description 1
- AOZGRHQSHHPIAP-XUVXKRRUSA-N 7-[(1r,5r)-5-[(3r)-3-(1-benzothiophen-2-yl)-3-hydroxypropyl]-2-methyl-4-oxocyclopent-2-en-1-yl]heptanoic acid Chemical compound OC(=O)CCCCCC[C@H]1C(C)=CC(=O)[C@@H]1CC[C@@H](O)C1=CC2=CC=CC=C2S1 AOZGRHQSHHPIAP-XUVXKRRUSA-N 0.000 description 1
- QNVDAVLIQYITSM-BECVJJOHSA-N 7-[(1r,5r)-5-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-2,2-difluoro-4-oxocyclopentyl]heptanoic acid Chemical compound C(/[C@@H](O)C=1SC2=CC=CC=C2C=1)=C\[C@@H]1[C@@H](CCCCCCC(O)=O)C(F)(F)CC1=O QNVDAVLIQYITSM-BECVJJOHSA-N 0.000 description 1
- WMEXXEFTIXOMNO-ANRYODRRSA-N 7-[(1r,5r)-5-[(e,3r)-3-(1-benzothiophen-2-yl)-3-hydroxyprop-1-enyl]-2-methyl-4-oxocyclopent-2-en-1-yl]heptanoic acid Chemical compound OC(=O)CCCCCC[C@H]1C(C)=CC(=O)[C@@H]1\C=C\[C@@H](O)C1=CC2=CC=CC=C2S1 WMEXXEFTIXOMNO-ANRYODRRSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 206010001052 Acute respiratory distress syndrome Diseases 0.000 description 1
- 108010000239 Aequorin Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 206010003557 Asthma exercise induced Diseases 0.000 description 1
- 206010003645 Atopy Diseases 0.000 description 1
- GMFDJWMJDIXPTE-LVQVYYBASA-N B=[O][C@H](C1)[C@H](CCC(COc2ccccc2)=O)[C@@H](C2)[C@H]1OC2=O Chemical compound B=[O][C@H](C1)[C@H](CCC(COc2ccccc2)=O)[C@@H](C2)[C@H]1OC2=O GMFDJWMJDIXPTE-LVQVYYBASA-N 0.000 description 1
- 229910015845 BBr3 Inorganic materials 0.000 description 1
- 208000035143 Bacterial infection Diseases 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 206010006458 Bronchitis chronic Diseases 0.000 description 1
- 206010006482 Bronchospasm Diseases 0.000 description 1
- PFTNMGUMMYQJPF-UHFFFAOYSA-N C.CC1=C(C)C(C)=C([V]C(C)(C)C)C([Y])=C1C.CC1=C(C)C(C)=C([Y])C2=[U][U]=C(C(C)(C)C)[U]=C21.CC1=C(C)C2=C(CC(C(C)(C)C)C2)C([Y])=C1C.CC1=C(C)C2=C([V]C(C(C)(C)C)=[U]2)C([Y])=C1C.CC1=C(C)C2=C([V][U]=C2C(C)(C)C)C([Y])=C1C.CC1=C([W])C(C)=C([Y])C([V]C(C)(C)C)=N1 Chemical compound C.CC1=C(C)C(C)=C([V]C(C)(C)C)C([Y])=C1C.CC1=C(C)C(C)=C([Y])C2=[U][U]=C(C(C)(C)C)[U]=C21.CC1=C(C)C2=C(CC(C(C)(C)C)C2)C([Y])=C1C.CC1=C(C)C2=C([V]C(C(C)(C)C)=[U]2)C([Y])=C1C.CC1=C(C)C2=C([V][U]=C2C(C)(C)C)C([Y])=C1C.CC1=C([W])C(C)=C([Y])C([V]C(C)(C)C)=N1 PFTNMGUMMYQJPF-UHFFFAOYSA-N 0.000 description 1
- JCUUWFDVTUGOSV-PTQHSSTGSA-N C1=COCCC1.C1CCOC1.ClCCl.O=C(O)CCC/C=C\C[C@H]1C(=O)C[C@@H](O)[C@@H]1/C=C/[C@@H](O)C1=CC2=C(C=CC=C2)S1.O=C(O)CCC/C=C\C[C@H]1C(=O)C[C@@H](OC2CCCCO2)[C@@H]1/C=C/[C@@H](OC1CCCCO1)C1=CC2=C(C=CC=C2)S1.O=C(O)CCC/C=C\C[C@H]1[C@@H](O)C[C@@H](OC2CCCCO2)[C@@H]1/C=C/[C@@H](OC1CCCCO1)C1=CC2=C(C=CC=C2)S1.O=C(O)CCCC[P+](C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1.O=C1C[C@H]2[C@H](C[C@@H](O)[C@@H]2/C=C/[C@@H](O)C2=CC3=C(C=CC=C3)S2)O1.O=C1C[C@H]2[C@H](C[C@@H](OC(=O)C3=CC=C(C4=CC=CC=C4)C=C3)[C@@H]2/C=C/C(=O)C2=CC3=C(C=CC=C3)S2)O1.O=C1C[C@H]2[C@H](C[C@@H](OC(=O)C3=CC=C(C4=CC=CC=C4)C=C3)[C@@H]2/C=C/[C@@H](O)C2=CC3=C(C=CC=C3)S2)O1.O=C1C[C@H]2[C@H](C[C@@H](OC3CCCCO3)[C@@H]2/C=C/[C@@H](OC2CCCCO2)C2=CC3=C(C=CC=C3)S2)O1.O=COO[K].OC1C[C@H]2[C@H](C[C@@H](OC3CCCCO3)[C@@H]2/C=C/[C@@H](OC2CCCCO2)C2=CC3=C(C=CC=C3)S2)O1.[Br-].[KH] Chemical compound C1=COCCC1.C1CCOC1.ClCCl.O=C(O)CCC/C=C\C[C@H]1C(=O)C[C@@H](O)[C@@H]1/C=C/[C@@H](O)C1=CC2=C(C=CC=C2)S1.O=C(O)CCC/C=C\C[C@H]1C(=O)C[C@@H](OC2CCCCO2)[C@@H]1/C=C/[C@@H](OC1CCCCO1)C1=CC2=C(C=CC=C2)S1.O=C(O)CCC/C=C\C[C@H]1[C@@H](O)C[C@@H](OC2CCCCO2)[C@@H]1/C=C/[C@@H](OC1CCCCO1)C1=CC2=C(C=CC=C2)S1.O=C(O)CCCC[P+](C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1.O=C1C[C@H]2[C@H](C[C@@H](O)[C@@H]2/C=C/[C@@H](O)C2=CC3=C(C=CC=C3)S2)O1.O=C1C[C@H]2[C@H](C[C@@H](OC(=O)C3=CC=C(C4=CC=CC=C4)C=C3)[C@@H]2/C=C/C(=O)C2=CC3=C(C=CC=C3)S2)O1.O=C1C[C@H]2[C@H](C[C@@H](OC(=O)C3=CC=C(C4=CC=CC=C4)C=C3)[C@@H]2/C=C/[C@@H](O)C2=CC3=C(C=CC=C3)S2)O1.O=C1C[C@H]2[C@H](C[C@@H](OC3CCCCO3)[C@@H]2/C=C/[C@@H](OC2CCCCO2)C2=CC3=C(C=CC=C3)S2)O1.O=COO[K].OC1C[C@H]2[C@H](C[C@@H](OC3CCCCO3)[C@@H]2/C=C/[C@@H](OC2CCCCO2)C2=CC3=C(C=CC=C3)S2)O1.[Br-].[KH] JCUUWFDVTUGOSV-PTQHSSTGSA-N 0.000 description 1
- LDKGRXSHVHDIIN-ZJWPJHLXSA-N C=C1C[C@@H](OC2CCCCO2)[C@H](/C=C/[C@@H](O)C2=CC3=C(C=CC=C3)S2)[C@H]1C/C=C\CCCC(=O)O Chemical compound C=C1C[C@@H](OC2CCCCO2)[C@H](/C=C/[C@@H](O)C2=CC3=C(C=CC=C3)S2)[C@H]1C/C=C\CCCC(=O)O LDKGRXSHVHDIIN-ZJWPJHLXSA-N 0.000 description 1
- GTNQFXPMUPHNJD-DWCUOBTKSA-N C=C1C[C@H](OC2CCCCO2)[C@H](C/C=C\CCCC(=O)O)[C@H]1/C=C/[C@@H](O)C1=CC2=C(C=CC=C2)S1 Chemical compound C=C1C[C@H](OC2CCCCO2)[C@H](C/C=C\CCCC(=O)O)[C@H]1/C=C/[C@@H](O)C1=CC2=C(C=CC=C2)S1 GTNQFXPMUPHNJD-DWCUOBTKSA-N 0.000 description 1
- IOAUEYBAXFACEG-JUYDJMGZSA-N C=C1C[C@H](OC2CCCCO2)[C@H](C/C=C\CCCC(=O)OC)[C@H]1/C=C/[C@@H](O[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C)C1=CC2=C(C=CC=C2)S1 Chemical compound C=C1C[C@H](OC2CCCCO2)[C@H](C/C=C\CCCC(=O)OC)[C@H]1/C=C/[C@@H](O[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C)C1=CC2=C(C=CC=C2)S1 IOAUEYBAXFACEG-JUYDJMGZSA-N 0.000 description 1
- KFVAYJOXYARNHH-HYKWTKIMSA-N CC(/C=C/[C@H]1[C@H](OC2CCCCO2)CC(=O)[C@@H]1C/C=C\CCCC(=O)O)(OC1CCCCO1)C1=CC2=C(C=CC=C2)S1 Chemical compound CC(/C=C/[C@H]1[C@H](OC2CCCCO2)CC(=O)[C@@H]1C/C=C\CCCC(=O)O)(OC1CCCCO1)C1=CC2=C(C=CC=C2)S1 KFVAYJOXYARNHH-HYKWTKIMSA-N 0.000 description 1
- WUBKCRCHYPMRPH-ZSVYZSLBSA-N CC(/C=C/[C@H]1[C@H](OC2CCCCO2)C[C@@H]2OC(O)C[C@@H]21)(OC1CCCCO1)C1=CC2=C(C=CC=C2)S1 Chemical compound CC(/C=C/[C@H]1[C@H](OC2CCCCO2)C[C@@H]2OC(O)C[C@@H]21)(OC1CCCCO1)C1=CC2=C(C=CC=C2)S1 WUBKCRCHYPMRPH-ZSVYZSLBSA-N 0.000 description 1
- HTOQAKTZAURNPI-BFBPRCINSA-N CC(/C=C/[C@H]1[C@H](OC2CCCCO2)C[C@H](O)[C@@H]1C/C=C\CCCC(=O)O)(OC1CCCCO1)C1=CC2=C(C=CC=C2)S1 Chemical compound CC(/C=C/[C@H]1[C@H](OC2CCCCO2)C[C@H](O)[C@@H]1C/C=C\CCCC(=O)O)(OC1CCCCO1)C1=CC2=C(C=CC=C2)S1 HTOQAKTZAURNPI-BFBPRCINSA-N 0.000 description 1
- URAAFNPXYSWIGV-UHFFFAOYSA-N CC(C)(C)C1=CC2=C(C=CC=C2)S1 Chemical compound CC(C)(C)C1=CC2=C(C=CC=C2)S1 URAAFNPXYSWIGV-UHFFFAOYSA-N 0.000 description 1
- ZQWHWRQRGKZTSE-UHFFFAOYSA-N CC(C)(C)C1=NC2=C(C=CC=C2)N1 Chemical compound CC(C)(C)C1=NC2=C(C=CC=C2)N1 ZQWHWRQRGKZTSE-UHFFFAOYSA-N 0.000 description 1
- IORNPTDHRCBFER-UHFFFAOYSA-N CC(C)(C)C1=NC2=C(C=CC=C2)O1 Chemical compound CC(C)(C)C1=NC2=C(C=CC=C2)O1 IORNPTDHRCBFER-UHFFFAOYSA-N 0.000 description 1
- YCAIAEVWVDGLAV-UHFFFAOYSA-N CC(C)(C)C1=NC2=C(C=CC=C2)S1 Chemical compound CC(C)(C)C1=NC2=C(C=CC=C2)S1 YCAIAEVWVDGLAV-UHFFFAOYSA-N 0.000 description 1
- OKQVUMMFRMUJMX-UHFFFAOYSA-N CC(C)(C)C1=NN=CN1 Chemical compound CC(C)(C)C1=NN=CN1 OKQVUMMFRMUJMX-UHFFFAOYSA-N 0.000 description 1
- YCGIUKPDDDXMDE-UHFFFAOYSA-N CC(C)(C)C1CC2=C(C=CC=C2)C1 Chemical compound CC(C)(C)C1CC2=C(C=CC=C2)C1 YCGIUKPDDDXMDE-UHFFFAOYSA-N 0.000 description 1
- NJIACBGKUBWJIF-UHFFFAOYSA-N CC(C)(C)Oc1cccc(C(F)(F)F)c1 Chemical compound CC(C)(C)Oc1cccc(C(F)(F)F)c1 NJIACBGKUBWJIF-UHFFFAOYSA-N 0.000 description 1
- BEVFKGWVXOFYAM-XOTBXEHSSA-N CC(C)(C)[Si](C)(C)O[C@H](/C=C/[C@H]1[C@@H](F)CC(=O)[C@@H]1C/C=C\CCCC(=O)O)C1CC2=C(C=CC=C2)C1 Chemical compound CC(C)(C)[Si](C)(C)O[C@H](/C=C/[C@H]1[C@@H](F)CC(=O)[C@@H]1C/C=C\CCCC(=O)O)C1CC2=C(C=CC=C2)C1 BEVFKGWVXOFYAM-XOTBXEHSSA-N 0.000 description 1
- PLCWCADCSALGSP-GORRPPPJSA-N CC(C)(C)[Si](O[C@H](/C=C/[C@H]1[C@H](O)CC(=O)[C@@H]1C/C=C\CCCC(=O)O)C1=NC2=C(C=CC=C2)S1)(C1=CC=CC=C1)C1=CC=CC=C1 Chemical compound CC(C)(C)[Si](O[C@H](/C=C/[C@H]1[C@H](O)CC(=O)[C@@H]1C/C=C\CCCC(=O)O)C1=NC2=C(C=CC=C2)S1)(C1=CC=CC=C1)C1=CC=CC=C1 PLCWCADCSALGSP-GORRPPPJSA-N 0.000 description 1
- QKXSQLMLSFDKQD-VFWNRNBTSA-N CC(C)(C)[Si](O[C@H](/C=C/[C@H]1[C@H](O)C[C@@H]2OC(=O)C[C@@H]21)C1=CC2=C(C=CC=C2)S1)(C1=CC=CC=C1)C1=CC=CC=C1 Chemical compound CC(C)(C)[Si](O[C@H](/C=C/[C@H]1[C@H](O)C[C@@H]2OC(=O)C[C@@H]21)C1=CC2=C(C=CC=C2)S1)(C1=CC=CC=C1)C1=CC=CC=C1 QKXSQLMLSFDKQD-VFWNRNBTSA-N 0.000 description 1
- NFERLMFKRQEHAS-NWWVMPTLSA-N CC(C)(C)[Si](O[C@H](/C=C/[C@H]1[C@H](O)C[C@H](O)[C@@H]1C/C=C\CCCC(=O)O)C1=CC2=C(C=CC=C2)S1)(C1=CC=CC=C1)C1=CC=CC=C1 Chemical compound CC(C)(C)[Si](O[C@H](/C=C/[C@H]1[C@H](O)C[C@H](O)[C@@H]1C/C=C\CCCC(=O)O)C1=CC2=C(C=CC=C2)S1)(C1=CC=CC=C1)C1=CC=CC=C1 NFERLMFKRQEHAS-NWWVMPTLSA-N 0.000 description 1
- JVUNMIAIEFJELD-QYWSSVFWSA-N CC(C)(C)[Si](O[C@H](CC[C@H]1C(=O)C[C@H](O)[C@@H]1C/C=C\CCCC(=O)O)C1=CC2=C(C=CC=C2)S1)(C1=CC=CC=C1)C1=CC=CC=C1.CC(C)(C)[Si](O[C@H](CC[C@H]1[C@H](O)CC(=O)[C@@H]1C/C=C\CCCC(=O)O)C1=CC2=C(C=CC=C2)S1)(C1=CC=CC=C1)C1=CC=CC=C1 Chemical compound CC(C)(C)[Si](O[C@H](CC[C@H]1C(=O)C[C@H](O)[C@@H]1C/C=C\CCCC(=O)O)C1=CC2=C(C=CC=C2)S1)(C1=CC=CC=C1)C1=CC=CC=C1.CC(C)(C)[Si](O[C@H](CC[C@H]1[C@H](O)CC(=O)[C@@H]1C/C=C\CCCC(=O)O)C1=CC2=C(C=CC=C2)S1)(C1=CC=CC=C1)C1=CC=CC=C1 JVUNMIAIEFJELD-QYWSSVFWSA-N 0.000 description 1
- BSXWLVPTZWHWGC-BQNLJHGISA-N CC(C)OC(=O)CCC/C=C\C[C@H]1C(=O)C[C@H](F)[C@@H]1/C=C/[C@@H](O[Si](C)(C)C(C)(C)C)C1CC2=C(C=CC=C2)C1 Chemical compound CC(C)OC(=O)CCC/C=C\C[C@H]1C(=O)C[C@H](F)[C@@H]1/C=C/[C@@H](O[Si](C)(C)C(C)(C)C)C1CC2=C(C=CC=C2)C1 BSXWLVPTZWHWGC-BQNLJHGISA-N 0.000 description 1
- NHXJEEIAJJWMNC-FUERGKQZSA-N CC(C)OC(=O)CCC/C=C\C[C@H]1[C@@H](O)C[C@H](F)[C@@H]1/C=C/[C@@H](O)C1CC2=C(C=CC=C2)C1 Chemical compound CC(C)OC(=O)CCC/C=C\C[C@H]1[C@@H](O)C[C@H](F)[C@@H]1/C=C/[C@@H](O)C1CC2=C(C=CC=C2)C1 NHXJEEIAJJWMNC-FUERGKQZSA-N 0.000 description 1
- VUSWVLCVWOVEGD-KLTCJQAYSA-N CC(C)OC(=O)CCC/C=C\C[C@H]1[C@@H](O)C[C@H](F)[C@@H]1/C=C/[C@@H](O[Si](C)(C)C(C)(C)C)C1CC2=C(C=CC=C2)C1 Chemical compound CC(C)OC(=O)CCC/C=C\C[C@H]1[C@@H](O)C[C@H](F)[C@@H]1/C=C/[C@@H](O[Si](C)(C)C(C)(C)C)C1CC2=C(C=CC=C2)C1 VUSWVLCVWOVEGD-KLTCJQAYSA-N 0.000 description 1
- FNRZBNJXOUIZBU-DJQNUGANSA-N CC(C)OC(=O)CCC/C=C\C[C@H]1[C@@H](O)C[C@H](F)[C@@H]1CO[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C Chemical compound CC(C)OC(=O)CCC/C=C\C[C@H]1[C@@H](O)C[C@H](F)[C@@H]1CO[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C FNRZBNJXOUIZBU-DJQNUGANSA-N 0.000 description 1
- APRVACVBLIDAHO-GJHGIRJZSA-N CC(C)OC(=O)CCC/C=C\C[C@H]1[C@@H](OC2CCCCO2)C[C@H](F)[C@@H]1C=O Chemical compound CC(C)OC(=O)CCC/C=C\C[C@H]1[C@@H](OC2CCCCO2)C[C@H](F)[C@@H]1C=O APRVACVBLIDAHO-GJHGIRJZSA-N 0.000 description 1
- MLRRBKGINFORAI-GJHGIRJZSA-N CC(C)OC(=O)CCC/C=C\C[C@H]1[C@@H](OC2CCCCO2)C[C@H](F)[C@@H]1CO Chemical compound CC(C)OC(=O)CCC/C=C\C[C@H]1[C@@H](OC2CCCCO2)C[C@H](F)[C@@H]1CO MLRRBKGINFORAI-GJHGIRJZSA-N 0.000 description 1
- AGFHWNJJXIHUDN-VUYIIYOJSA-N CC(C)OC(=O)CCC/C=C\C[C@H]1[C@@H](OC2CCCCO2)C[C@H](F)[C@@H]1CO[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C Chemical compound CC(C)OC(=O)CCC/C=C\C[C@H]1[C@@H](OC2CCCCO2)C[C@H](F)[C@@H]1CO[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C AGFHWNJJXIHUDN-VUYIIYOJSA-N 0.000 description 1
- XPILIICSUJKOEA-UHFFFAOYSA-N CC1=CC(OC(C)(C)C)=CC=C1 Chemical compound CC1=CC(OC(C)(C)C)=CC=C1 XPILIICSUJKOEA-UHFFFAOYSA-N 0.000 description 1
- KBGRDMQIZNTBMC-NYPDDCMTSA-N CCCC(=O)CCC/C=C\C[C@H]1C(=O)C[C@@H](O)[C@@H]1/C=C/[C@@H](O)C1=CC2=C(C=CC=C2)S1 Chemical compound CCCC(=O)CCC/C=C\C[C@H]1C(=O)C[C@@H](O)[C@@H]1/C=C/[C@@H](O)C1=CC2=C(C=CC=C2)S1 KBGRDMQIZNTBMC-NYPDDCMTSA-N 0.000 description 1
- HFHHJBNFOGRDMA-UHFFFAOYSA-N CN1C(C(C)(C)C)=NC2=C1C=CC=C2 Chemical compound CN1C(C(C)(C)C)=NC2=C1C=CC=C2 HFHHJBNFOGRDMA-UHFFFAOYSA-N 0.000 description 1
- ZWNAPYWBCSZOCY-KTMAPIEGSA-N COC(=O)CCC/C=C\C[C@H]1C(=O)C[C@@H](O)[C@@H]1/C=C/[C@@H](O[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C)C1=CC2=C(C=CC=C2)S1.COC(=O)CCC/C=C\C[C@H]1[C@@H](O)CC(=O)[C@@H]1/C=C/[C@@H](O[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C)C1=CC2=C(C=CC=C2)S1 Chemical compound COC(=O)CCC/C=C\C[C@H]1C(=O)C[C@@H](O)[C@@H]1/C=C/[C@@H](O[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C)C1=CC2=C(C=CC=C2)S1.COC(=O)CCC/C=C\C[C@H]1[C@@H](O)CC(=O)[C@@H]1/C=C/[C@@H](O[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C)C1=CC2=C(C=CC=C2)S1 ZWNAPYWBCSZOCY-KTMAPIEGSA-N 0.000 description 1
- DLRIKXGCOTWIRC-NRXOXWLLSA-N COC(=O)CCC/C=C\C[C@H]1C(=O)C[C@@H](O)[C@@H]1/C=C/[C@@H](O[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C)C1=NC2=C(C=CC=C2)S1.COC(=O)CCC/C=C\C[C@H]1[C@@H](O)CC(=O)[C@@H]1/C=C/[C@@H](O[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C)C1=NC2=C(C=CC=C2)S1 Chemical compound COC(=O)CCC/C=C\C[C@H]1C(=O)C[C@@H](O)[C@@H]1/C=C/[C@@H](O[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C)C1=NC2=C(C=CC=C2)S1.COC(=O)CCC/C=C\C[C@H]1[C@@H](O)CC(=O)[C@@H]1/C=C/[C@@H](O[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C)C1=NC2=C(C=CC=C2)S1 DLRIKXGCOTWIRC-NRXOXWLLSA-N 0.000 description 1
- WZZAGMJBXNXNEC-HDDSOSNCSA-N COC(=O)CCC/C=C\C[C@H]1[C@@H](O)C[C@@H](O)[C@@H]1/C=C/[C@@H](O[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C)C1=CC2=C(C=CC=C2)S1 Chemical compound COC(=O)CCC/C=C\C[C@H]1[C@@H](O)C[C@@H](O)[C@@H]1/C=C/[C@@H](O[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C)C1=CC2=C(C=CC=C2)S1 WZZAGMJBXNXNEC-HDDSOSNCSA-N 0.000 description 1
- JPLCDZBFSACWJU-RREWPFIPSA-N COC(=O)CCC/C=C\C[C@H]1[C@@H](OC2CCCCO2)CC(=O)[C@@H]1/C=C/[C@@H](O[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C)C1=CC2=C(C=CC=C2)S1 Chemical compound COC(=O)CCC/C=C\C[C@H]1[C@@H](OC2CCCCO2)CC(=O)[C@@H]1/C=C/[C@@H](O[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C)C1=CC2=C(C=CC=C2)S1 JPLCDZBFSACWJU-RREWPFIPSA-N 0.000 description 1
- MQYXVAFMKDQOBR-ZCBCPHFHSA-N CS(=O)(=O)NC(=O)CCC/C=C\C[C@H]1C(=O)C[C@@H](OC2CCCCO2)[C@@H]1/C=C/[C@@H](OC1CCCCO1)C1=CC2=C(C=CC=C2)S1 Chemical compound CS(=O)(=O)NC(=O)CCC/C=C\C[C@H]1C(=O)C[C@@H](OC2CCCCO2)[C@@H]1/C=C/[C@@H](OC1CCCCO1)C1=CC2=C(C=CC=C2)S1 MQYXVAFMKDQOBR-ZCBCPHFHSA-N 0.000 description 1
- SGBMDPNLMADWOL-UHFFFAOYSA-O CS(=O)(=O)NC(=O)CCCC[P+](C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1.[Br-] Chemical compound CS(=O)(=O)NC(=O)CCCC[P+](C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1.[Br-] SGBMDPNLMADWOL-UHFFFAOYSA-O 0.000 description 1
- MEYALKZDMJNLKK-SMYMCJIOSA-N C[C@@H]1C[C@@H]2OC(=O)C[C@@H]2[C@H]1/C=C/[C@@H](O[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C)C1=CC2=C(C=CC=C2)S1 Chemical compound C[C@@H]1C[C@@H]2OC(=O)C[C@@H]2[C@H]1/C=C/[C@@H](O[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C)C1=CC2=C(C=CC=C2)S1 MEYALKZDMJNLKK-SMYMCJIOSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 108020004635 Complementary DNA Proteins 0.000 description 1
- 206010010744 Conjunctivitis allergic Diseases 0.000 description 1
- 206010011224 Cough Diseases 0.000 description 1
- 206010011703 Cyanosis Diseases 0.000 description 1
- IVOMOUWHDPKRLL-KQYNXXCUSA-N Cyclic adenosine monophosphate Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-KQYNXXCUSA-N 0.000 description 1
- DSLZVSRJTYRBFB-LLEIAEIESA-N D-glucaric acid Chemical compound OC(=O)[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O DSLZVSRJTYRBFB-LLEIAEIESA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- 206010012438 Dermatitis atopic Diseases 0.000 description 1
- BUDQDWGNQVEFAC-UHFFFAOYSA-N Dihydropyran Chemical compound C1COC=CC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 description 1
- 239000012983 Dulbecco’s minimal essential medium Substances 0.000 description 1
- 208000000059 Dyspnea Diseases 0.000 description 1
- 206010013975 Dyspnoeas Diseases 0.000 description 1
- 206010014561 Emphysema Diseases 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 208000004657 Exercise-Induced Asthma Diseases 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 206010017533 Fungal infection Diseases 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 206010019233 Headaches Diseases 0.000 description 1
- 229940122236 Histamine receptor antagonist Drugs 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 208000016300 Idiopathic chronic eosinophilic pneumonia Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-L L-tartrate(2-) Chemical compound [O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O FEWJPZIEWOKRBE-JCYAYHJZSA-L 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 239000000867 Lipoxygenase Inhibitor Substances 0.000 description 1
- 208000019693 Lung disease Diseases 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 229930045534 Me ester-Cyclohexaneundecanoic acid Natural products 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 1
- 229940122694 Muscarinic M3 receptor antagonist Drugs 0.000 description 1
- 229940121948 Muscarinic receptor antagonist Drugs 0.000 description 1
- 208000031888 Mycoses Diseases 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 206010028735 Nasal congestion Diseases 0.000 description 1
- 206010052319 Nasal flaring Diseases 0.000 description 1
- 206010028741 Nasal inflammation Diseases 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- JFHZPHRQEDRWDL-WMJZDSFQSA-N O=C(O)CCC/C=C\C[C@H]1C(=O)C[C@@H](OC2CCCCO2)[C@@H]1/C=C/C(F)(F)C1=CC2=C(C=CC=C2)S1 Chemical compound O=C(O)CCC/C=C\C[C@H]1C(=O)C[C@@H](OC2CCCCO2)[C@@H]1/C=C/C(F)(F)C1=CC2=C(C=CC=C2)S1 JFHZPHRQEDRWDL-WMJZDSFQSA-N 0.000 description 1
- LFHMKSWCIGAYAT-HGWAAWLGSA-N O=C(O)CCC/C=C\C[C@H]1C(=O)C[C@@H](OC2CCCCO2)[C@@H]1/C=C/[C@@H](OC1CCCCO1)C1=CC2=C(C=CC=C2)S1 Chemical compound O=C(O)CCC/C=C\C[C@H]1C(=O)C[C@@H](OC2CCCCO2)[C@@H]1/C=C/[C@@H](OC1CCCCO1)C1=CC2=C(C=CC=C2)S1 LFHMKSWCIGAYAT-HGWAAWLGSA-N 0.000 description 1
- DUCLMDQBPUQJQW-UDSDMIASSA-N O=C(O)CCC/C=C\C[C@H]1C(=O)C[C@@H](OC2CCCCO2)[C@@H]1CCC(F)(F)COC1=CC=CC=C1 Chemical compound O=C(O)CCC/C=C\C[C@H]1C(=O)C[C@@H](OC2CCCCO2)[C@@H]1CCC(F)(F)COC1=CC=CC=C1 DUCLMDQBPUQJQW-UDSDMIASSA-N 0.000 description 1
- RFCGJYLBRNGHFJ-IUNDKUODSA-N O=C(O)CCC/C=C\C[C@H]1[C@@H](O)C[C@@H](OC2CCCCO2)[C@@H]1/C=C/C(F)(F)C1=CC2=C(C=CC=C2)S1 Chemical compound O=C(O)CCC/C=C\C[C@H]1[C@@H](O)C[C@@H](OC2CCCCO2)[C@@H]1/C=C/C(F)(F)C1=CC2=C(C=CC=C2)S1 RFCGJYLBRNGHFJ-IUNDKUODSA-N 0.000 description 1
- PZMYFZMHOJFDDQ-LBHAUNBFSA-N O=C(O)CCC/C=C\C[C@H]1[C@@H](O)C[C@@H](OC2CCCCO2)[C@@H]1/C=C/[C@@H](OC1CCCCO1)C1=CC2=C(C=CC=C2)S1 Chemical compound O=C(O)CCC/C=C\C[C@H]1[C@@H](O)C[C@@H](OC2CCCCO2)[C@@H]1/C=C/[C@@H](OC1CCCCO1)C1=CC2=C(C=CC=C2)S1 PZMYFZMHOJFDDQ-LBHAUNBFSA-N 0.000 description 1
- BRTGCTGOPSGWLJ-WBBGFGDFSA-N O=C(O)CCC/C=C\C[C@H]1[C@@H](O)C[C@@H](OC2CCCCO2)[C@@H]1CCC(F)(F)COC1=CC=CC=C1 Chemical compound O=C(O)CCC/C=C\C[C@H]1[C@@H](O)C[C@@H](OC2CCCCO2)[C@@H]1CCC(F)(F)COC1=CC=CC=C1 BRTGCTGOPSGWLJ-WBBGFGDFSA-N 0.000 description 1
- CNGBBCZWLBKHQN-YGFGSHOLSA-N O=C1C[C@H]2[C@H](C[C@@H](OC3CCCCO3)[C@@H]2/C=C/[C@@H](OC2CCCCO2)C2=CC3=C(C=CC=C3)S2)O1 Chemical compound O=C1C[C@H]2[C@H](C[C@@H](OC3CCCCO3)[C@@H]2/C=C/[C@@H](OC2CCCCO2)C2=CC3=C(C=CC=C3)S2)O1 CNGBBCZWLBKHQN-YGFGSHOLSA-N 0.000 description 1
- TUIYYKHIPJJPAG-YNAZQSIESA-N O=C1C[C@H]2[C@H](C[C@@H](OC3CCCCO3)[C@@H]2CCC(F)(F)COC2=CC=CC=C2)O1 Chemical compound O=C1C[C@H]2[C@H](C[C@@H](OC3CCCCO3)[C@@H]2CCC(F)(F)COC2=CC=CC=C2)O1 TUIYYKHIPJJPAG-YNAZQSIESA-N 0.000 description 1
- ZZDYFLARJVOLCF-QAUBHBCPSA-N OC1C[C@H]2[C@H](C[C@@H](OC3CCCCO3)[C@@H]2/C=C/C(F)(F)C2=CC3=C(C=CC=C3)S2)O1 Chemical compound OC1C[C@H]2[C@H](C[C@@H](OC3CCCCO3)[C@@H]2/C=C/C(F)(F)C2=CC3=C(C=CC=C3)S2)O1 ZZDYFLARJVOLCF-QAUBHBCPSA-N 0.000 description 1
- 208000027771 Obstructive airways disease Diseases 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 208000002193 Pain Diseases 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 208000013616 Respiratory Distress Syndrome Diseases 0.000 description 1
- 208000037656 Respiratory Sounds Diseases 0.000 description 1
- 206010039085 Rhinitis allergic Diseases 0.000 description 1
- 206010039094 Rhinitis perennial Diseases 0.000 description 1
- 208000036071 Rhinorrhea Diseases 0.000 description 1
- 206010039101 Rhinorrhoea Diseases 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- IVOMOUWHDPKRLL-UHFFFAOYSA-N UNPD107823 Natural products O1C2COP(O)(=O)OC2C(O)C1N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-UHFFFAOYSA-N 0.000 description 1
- 206010047139 Vasoconstriction Diseases 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- KBOZUCMAAWBGQV-AKHDSKFASA-N [(3ar,4r,5r,6as)-4-formyl-2-oxo-3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-5-yl] 4-phenylbenzoate Chemical compound O([C@H]1[C@@H]([C@H]2CC(=O)O[C@H]2C1)C=O)C(=O)C(C=C1)=CC=C1C1=CC=CC=C1 KBOZUCMAAWBGQV-AKHDSKFASA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 206010069351 acute lung injury Diseases 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000003470 adrenal cortex hormone Substances 0.000 description 1
- 208000011341 adult acute respiratory distress syndrome Diseases 0.000 description 1
- 201000000028 adult respiratory distress syndrome Diseases 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 239000013566 allergen Substances 0.000 description 1
- 201000009961 allergic asthma Diseases 0.000 description 1
- 208000002205 allergic conjunctivitis Diseases 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 230000009285 allergic inflammation Effects 0.000 description 1
- 201000010105 allergic rhinitis Diseases 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 239000000739 antihistaminic agent Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 208000024998 atopic conjunctivitis Diseases 0.000 description 1
- 201000008937 atopic dermatitis Diseases 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 208000022362 bacterial infectious disease Diseases 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 239000000090 biomarker Substances 0.000 description 1
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 201000009267 bronchiectasis Diseases 0.000 description 1
- 206010006451 bronchitis Diseases 0.000 description 1
- 230000007885 bronchoconstriction Effects 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 230000001593 cAMP accumulation Effects 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 239000000812 cholinergic antagonist Substances 0.000 description 1
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 1
- 208000007451 chronic bronchitis Diseases 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 201000009323 chronic eosinophilic pneumonia Diseases 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 230000008602 contraction Effects 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 229940109275 cyclamate Drugs 0.000 description 1
- 229940095074 cyclic amp Drugs 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- HCAJEUSONLESMK-UHFFFAOYSA-N cyclohexylsulfamic acid Chemical compound OS(=O)(=O)NC1CCCCC1 HCAJEUSONLESMK-UHFFFAOYSA-N 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 238000005828 desilylation reaction Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 229950010286 diolamine Drugs 0.000 description 1
- 125000000532 dioxanyl group Chemical group 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 238000002651 drug therapy Methods 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 150000002066 eicosanoids Chemical class 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 238000011067 equilibration Methods 0.000 description 1
- 235000004626 essential fatty acids Nutrition 0.000 description 1
- 150000002148 esters Chemical group 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- NBEMQPLNBYYUAZ-UHFFFAOYSA-N ethyl acetate;propan-2-one Chemical compound CC(C)=O.CCOC(C)=O NBEMQPLNBYYUAZ-UHFFFAOYSA-N 0.000 description 1
- 230000005713 exacerbation Effects 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 208000024695 exercise-induced bronchoconstriction Diseases 0.000 description 1
- 239000013604 expression vector Substances 0.000 description 1
- 229940044170 formate Drugs 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical compound [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 229960001731 gluceptate Drugs 0.000 description 1
- 239000003862 glucocorticoid Substances 0.000 description 1
- KWMLJOLKUYYJFJ-VFUOTHLCSA-N glucoheptonic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)C(O)=O KWMLJOLKUYYJFJ-VFUOTHLCSA-N 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 229940097042 glucuronate Drugs 0.000 description 1
- 231100000869 headache Toxicity 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229950000177 hibenzate Drugs 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 239000003395 histamine H3 receptor antagonist Substances 0.000 description 1
- 239000011539 homogenization buffer Substances 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical group O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 description 1
- 230000035874 hyperreactivity Effects 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 230000001524 infective effect Effects 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 208000030603 inherited susceptibility to asthma Diseases 0.000 description 1
- UEXQBEVWFZKHNB-UHFFFAOYSA-N intermediate 29 Natural products C1=CC(N)=CC=C1NC1=NC=CC=N1 UEXQBEVWFZKHNB-UHFFFAOYSA-N 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 125000000468 ketone group Chemical group 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- YEESKJGWJFYOOK-IJHYULJSSA-N leukotriene D4 Chemical compound CCCCC\C=C/C\C=C/C=C/C=C/[C@H]([C@@H](O)CCCC(O)=O)SC[C@H](N)C(=O)NCC(O)=O YEESKJGWJFYOOK-IJHYULJSSA-N 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 238000004020 luminiscence type Methods 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-L malate(2-) Chemical compound [O-]C(=O)C(O)CC([O-])=O BJEPYKJPYRNKOW-UHFFFAOYSA-L 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- JBVVQOOOWFKMBL-JGYVQLPLSA-N methyl (z)-7-[(7s,8r,9r)-9-[(e,3r)-3-(1-benzothiophen-2-yl)-3-[tert-butyl(diphenyl)silyl]oxyprop-1-enyl]-7-hydroxy-1,4-dioxaspiro[4.4]nonan-8-yl]hept-5-enoate Chemical compound C([C@H](O)[C@@H]([C@H]1\C=C\[C@@H](O[Si](C=2C=CC=CC=2)(C=2C=CC=CC=2)C(C)(C)C)C=2SC3=CC=CC=C3C=2)C\C=C/CCCC(=O)OC)C21OCCO2 JBVVQOOOWFKMBL-JGYVQLPLSA-N 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 1
- GRVDJDISBSALJP-UHFFFAOYSA-N methyloxidanyl Chemical compound [O]C GRVDJDISBSALJP-UHFFFAOYSA-N 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 239000003681 muscarinic M3 receptor antagonist Substances 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005487 naphthalate group Chemical group 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 231100000706 no observed effect level Toxicity 0.000 description 1
- 230000009871 nonspecific binding Effects 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 208000007892 occupational asthma Diseases 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229950004864 olamine Drugs 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- PXQPEWDEAKTCGB-UHFFFAOYSA-M orotate Chemical compound [O-]C(=O)C1=CC(=O)NC(=O)N1 PXQPEWDEAKTCGB-UHFFFAOYSA-M 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000000466 oxiranyl group Chemical group 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- BSCHIACBONPEOB-UHFFFAOYSA-N oxolane;hydrate Chemical compound O.C1CCOC1 BSCHIACBONPEOB-UHFFFAOYSA-N 0.000 description 1
- 230000036407 pain Effects 0.000 description 1
- 230000001991 pathophysiological effect Effects 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 238000003752 polymerase chain reaction Methods 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 229960003081 probenecid Drugs 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- LBDZRNCMTKAOEQ-NVVPFNIESA-N propan-2-yl (z)-7-[(1r,2r,3s,5s)-2-[(e)-3-(1-benzothiophen-2-yl)-3-oxoprop-1-enyl]-3-fluoro-5-(oxan-2-yloxy)cyclopentyl]hept-5-enoate Chemical compound O([C@@H]1[C@@H]([C@H]([C@@H](F)C1)\C=C\C(=O)C=1SC2=CC=CC=C2C=1)C\C=C/CCCC(=O)OC(C)C)C1CCCCO1 LBDZRNCMTKAOEQ-NVVPFNIESA-N 0.000 description 1
- 150000003815 prostacyclins Chemical class 0.000 description 1
- BHMBVRSPMRCCGG-OUTUXVNYSA-N prostaglandin D2 Chemical compound CCCCC[C@H](O)\C=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=O BHMBVRSPMRCCGG-OUTUXVNYSA-N 0.000 description 1
- KAQKFAOMNZTLHT-OZUDYXHBSA-N prostaglandin I2 Chemical compound O1\C(=C/CCCC(O)=O)C[C@@H]2[C@@H](/C=C/[C@@H](O)CCCCC)[C@H](O)C[C@@H]21 KAQKFAOMNZTLHT-OZUDYXHBSA-N 0.000 description 1
- 102000017953 prostanoid receptors Human genes 0.000 description 1
- 108050007059 prostanoid receptors Proteins 0.000 description 1
- 208000028172 protozoa infectious disease Diseases 0.000 description 1
- 208000002815 pulmonary hypertension Diseases 0.000 description 1
- 239000012264 purified product Substances 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 229940043131 pyroglutamate Drugs 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 239000002469 receptor inverse agonist Substances 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000029865 regulation of blood pressure Effects 0.000 description 1
- 230000004648 relaxation of smooth muscle Effects 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 238000003345 scintillation counting Methods 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 208000013220 shortness of breath Diseases 0.000 description 1
- 238000006884 silylation reaction Methods 0.000 description 1
- 201000009890 sinusitis Diseases 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 description 1
- 238000009120 supportive therapy Methods 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- FRACPXUHUTXLCX-BELIEFIBSA-N tert-butyl N-{1-[(1S)-1-{[(1R,2S)-1-(benzylcarbamoyl)-1-hydroxy-3-[(3S)-2-oxopyrrolidin-3-yl]propan-2-yl]carbamoyl}-2-cyclopropylethyl]-2-oxopyridin-3-yl}carbamate Chemical compound CC(C)(C)OC(=O)NC1=CC=CN(C1=O)[C@@H](CC2CC2)C(=O)N[C@@H](C[C@@H]3CCNC3=O)[C@H](C(=O)NCC4=CC=CC=C4)O FRACPXUHUTXLCX-BELIEFIBSA-N 0.000 description 1
- MHYGQXWCZAYSLJ-UHFFFAOYSA-N tert-butyl-chloro-diphenylsilane Chemical compound C=1C=CC=CC=1[Si](Cl)(C(C)(C)C)C1=CC=CC=C1 MHYGQXWCZAYSLJ-UHFFFAOYSA-N 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 150000003595 thromboxanes Chemical class 0.000 description 1
- 125000003944 tolyl group Chemical group 0.000 description 1
- 239000012096 transfection reagent Substances 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 229960005486 vaccine Drugs 0.000 description 1
- 230000025033 vasoconstriction Effects 0.000 description 1
- 230000024883 vasodilation Effects 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 210000001835 viscera Anatomy 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
- 239000011995 wilkinson's catalyst Substances 0.000 description 1
- UTODFRQBVUVYOB-UHFFFAOYSA-P wilkinson's catalyst Chemical compound [Cl-].C1=CC=CC=C1P(C=1C=CC=CC=1)(C=1C=CC=CC=1)[Rh+](P(C=1C=CC=CC=1)(C=1C=CC=CC=1)C=1C=CC=CC=1)P(C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 UTODFRQBVUVYOB-UHFFFAOYSA-P 0.000 description 1
- 239000002023 wood Substances 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C405/00—Compounds containing a five-membered ring having two side-chains in ortho position to each other, and having oxygen atoms directly attached to the ring in ortho position to one of the side-chains, one side-chain containing, not directly attached to the ring, a carbon atom having three bonds to hetero atoms with at the most one bond to halogen, and the other side-chain having oxygen atoms attached in gamma-position to the ring, e.g. prostaglandins ; Analogues or derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
Definitions
- the present invention relates to pharmaceutically active compounds and more particularly to prostaglandin analogs with selectivity for prostaglandin E (EP) receptors and demonstrating EP agonist activity, and the use of such compounds and compositions thereof for the treatment of various medical conditions.
- EP prostaglandin E
- Prostanoids are ubiquitous lipid mediator biomolecules involved in numerous physiological processes, such as the contraction and relaxation of smooth muscle, vasodilation, vasoconstriction, pain, regulation of blood pressure, and modulation of inflammation.
- Prostanoids are a family of eicosanoids that comprise prostaglandins (PGs), prostacyclins (PGIs), and thromboxanes (Txs).
- PGs prostaglandins
- PKIs prostacyclins
- Txs thromboxanes
- GPCR G-protein coupled receptor
- DP prostaglandin D
- EP prostaglandin E
- FP prostaglandin F
- IP prostaglandin I
- TP Thromboxane A
- Prostanoids are synthesized from essential fatty acids comprising twenty carbon atoms, such as arachidonic acid and 8,11,14-eicosatrienoic acid. Prostanoids are synthesized in response to both extracellular and intracellular stimuli and are then rapidly released from the cells. In general, the short half-lives of most prostanoids ensure they act near the sites of their biosynthesis.
- Prostaglandin E 2 is a potent endogenous EP receptor agonist derived from arachidonic acid and possesses two carbon-carbon double bonds, one in each the ⁇ -chain and ⁇ -chain, and is thus called a “Series 2” prostaglandin.
- Prostaglandin E 1 (PGE 1 ) is derived from 8,11,14-eicosatrienoic acid and possesses only one carbon-carbon double bond, located in the co-chain, and is thus called a “Series 1” prostaglandin.
- EP receptor agonists may have a number of utilities. These include, but are not limited to treatment of influenza (WO 2008/058766), bone fracture healing (Li, M., et al., J. Bone Miner. Res., 18(11), 2003, 2033-2042; Paralkar, V.
- EP receptor agonists may also be useful for expansion of hematopoietic stem cell populations (WO 2008/073748; North, T. E., et al., Nature, 447, 2007, 1007-1011).
- the exemplary embodiments may be directed to compounds of structural formula (I) that may be used to expand hematopoietic stem cell populations or to treat or prevent influenza, bone fracture, bone disease, glaucoma, ocular hypertension, dysmenorrhoea, pre-term labor, immune disorders, osteoporosis, asthma, allergy, male sexual dysfunction, female sexual dysfunction, periodontal disease, gastric ulcer, renal disease, or other EP receptor-mediated conditions wherein C9, C 11 , R 1 , R 2 , Z 1 , Z 2 , Z 3 , Z 4 , Z 5 , Z 6 , m and n are defined herein:
- Another aspect of the embodiment may be a pharmaceutical composition
- a pharmaceutical composition comprising a pharmaceutically effective amount of a compound according to formula (I), any stereoisomer or geometric isomer thereof, or a prodrug thereof, or a hydrate or solvate thereof, or a pharmaceutically acceptable salt thereof, in admixture with a pharmaceutically acceptable carrier.
- Another aspect of the embodiment may be directed to a method of expanding hematopoietic stem cell populations in a culture or patient in need thereof by administering to the culture or patient a compound according to formula (I), any stereoisomer or geometric isomer thereof, or a prodrug thereof, or a hydrate or solvate thereof, or a pharmaceutically acceptable salt thereof.
- Another aspect of the embodiment may be directed to a method of treating or preventing influenza, bone fracture, bone disease, glaucoma, ocular hypertension, dysmenorrhoea, pre-term labor, immune disorders, osteoporosis, asthma, allergy, male sexual dysfunction, female sexual dysfunction, periodontal disease, gastric ulcer, renal disease, or other EP receptor-mediated conditions in a patient in need thereof by administering to the patient a compound according to formula (I), any stereoisomer or geometric isomer thereof, or a prodrug thereof, or a hydrate or solvate thereof, or a pharmaceutically acceptable salt thereof.
- the exemplary embodiments may be directed to a compound of formula (I), their preparation, pharmaceutical compositions comprising these compounds, and their pharmaceutical use in the prevention and treatment of EP receptor-mediated diseases or conditions.
- the compounds of formula (I) are shown below:
- dashed bonds may each independently represent a second carbon-carbon bond in order to give a carbon-carbon double bond with either (E) or (Z) geometry or may be ignored in order to give a carbon-carbon single bond;
- C 9 and C 11 each is independently C ⁇ CH 2 , C ⁇ O, CF 2 , CHF (any stereoisomer), or C(H)OH (any stereoisomer) with the proviso that C 9 does not equal C 11 and also with the proviso that when one of either C9 or C 11 is CHF, the other is not C(H)OH;
- R 1 is CO 2 R 3 , CH 2 OR 3 , CONR 4 R 5 , COCH 2 OH, CONR 4 SO 2 R 5 , P(O)(OR 4 ) 2 , or
- the exemplary embodiment above may also include any stereoisomer or geometric isomer thereof, or an equivalent thereof, or a prodrug thereof, or a hydrate or solvate thereof, or a pharmaceutically acceptable salt thereof.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein C 9 is C ⁇ O and C 11 is C(H)OH.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein C 9 is C(H)OH and C 11 is C ⁇ O.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein C 9 is C ⁇ O and C 11 is C ⁇ CH 2 .
- Another exemplary embodiment may be directed to a compound of formula (I) wherein C 9 is C(H)OH and C 11 is C ⁇ CH 2 .
- Another exemplary embodiment may be directed to a compound of formula (I) wherein C 9 is C ⁇ CH 2 and C 11 is C ⁇ O.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein C 9 is C ⁇ CH 2 and C 11 is C(H)OH.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein C 9 is C ⁇ O and C 11 is CF 2 .
- Another exemplary embodiment may be directed to a compound of formula (I) wherein C 9 is C(H)OH and C 11 is CF 2 .
- Another exemplary embodiment may be directed to a compound of formula (I) wherein C 9 is CF 2 and C 11 is C ⁇ O.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein C 9 is CF 2 and C 11 is C(H)OH.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein C 9 is C ⁇ O and C 11 is CHF.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein C 9 is CHF and C 11 is C ⁇ O.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein R 1 is CO 2 H.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein R 1 is CO 2 i Pr.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein R 1 is CON(H)Et.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein R 1 is CON(H)SO 2 Me.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein R 1 is CH 2 OH.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein R 1 is
- Another exemplary embodiment may be directed to a compound of formula (I) wherein m is 3.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein Z 1 is hydroxy and Z 2 is hydrogen.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein Z 1 is hydroxy and Z 2 is methyl.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein Z 1 is fluorine and Z 2 is hydrogen.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein each Z 1 and Z 2 is hydrogen.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein each Z 1 and Z 2 is fluorine.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein each Z 1 and Z 2 is methyl.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein Z 1 and Z 2 together is an oxygen atom that form a carbonyl with the adjoining carbon atom.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein each Z 3 and Z 4 is hydrogen.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein each Z 3 and Z 4 is fluorine.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein each Z 3 and Z 4 is methyl.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein Z 3 is hydroxy and Z 4 is hydrogen.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein Z 3 is hydroxy and Z 4 is methyl.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein Z 3 is methyl and Z 4 is hydrogen.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein Z 3 and Z 4 together is an oxygen atom that form a carbonyl with the adjoining carbon atom.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein n is 0.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein n is 1.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein Z 5 and Z 6 are hydrogen.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein Z 5 is hydrogen and Z 6 is fluorine.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein Z 5 is fluorine and Z 6 is hydrogen.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein R 2 is phenyl.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein R 2 is —OPh.
- Another exemplary embodiment may be directed to a compound of formula (I) wherein R 2 is
- Another exemplary embodiment may be directed to a compound of formula (I) wherein R 2 is
- Another exemplary embodiment may be directed to a compound of formula (I) wherein R 2 is
- Another exemplary embodiment may be directed to a compound of formula (I) wherein R 2 is
- Another exemplary embodiment may be directed to a compound of formula (I) wherein R 2 is
- Another exemplary embodiment may be directed to a compound of formula (I) wherein R 2 is
- Another exemplary embodiment may be directed to a compound of formula (I) wherein R 2 is
- Another exemplary embodiment may be directed to a more specific version of the compound of formula (I), namely to a compound of formula (II):
- Another exemplary embodiment may be directed to a more specific version of the compound of formula (I), namely to a compound of formula (III):
- Another exemplary embodiment may be directed to a more specific version of the compound of formula (I), namely to a compound of formula (IV):
- Another exemplary embodiment may be directed to a more specific version of the compound of formula (I), namely to a compound of formula (V):
- Another exemplary embodiment may be a more specific compound of formula (I) selected from the group consisting of: (Z)-7-((1R,2R,3R)-2-((E)-3-(benzo[b]thiophen-2-yl)-3,3-difluoroprop-1-enyl)-3-hydroxy-5-oxocyclopentyl)hept-5-enoic acid; (Z)-7-((1R,2R,3R)-2-((E)-3-(benzo[b]thiophen-2-yl)-3-methylbut-1-enyl)-3-hydroxy-5-oxocyclopentyl)hept-5-enoic acid; (Z)-7-((1R,2R,3R)-2-((E)-3-(benzo[b]thiophen-2-yl)-3-oxoprop-1-enyl)-3-hydroxy-5-oxocyclopentyl)hept-5-enoic acid; (Z)-7-((1R
- Another exemplary embodiment may be a more specific compound of formula (I) selected from the group consisting of: (Z)-7-((1R,2R,5S)-2-((E)-3-(benzo[b]thiophen-2-yl)-3,3-difluoroprop-1-enyl)-5-hydroxy-3-oxocyclopentyl)hept-5-enoic acid; (Z)-7-((1R,2R,5S)-2-((E)-3-(benzo[b]thiophen-2-yl)-3-methylbut-1-enyl)-5-hydroxy-3-oxocyclopentyl)hept-5-enoic acid; (Z)-7-((1R,2R,5S)-2-((E)-3-(benzo[b]thiophen-2-yl)-3-oxoprop-1-enyl)-5-hydroxy-3-oxocyclopentyl)hept-5-enoic acid; (Z)-7-((1R
- Another exemplary embodiment may be a more specific compound of formula (I) selected from the group consisting of: (Z)-7-((1R,2R)-2-((E)-3-(benzo[b]thiophen-2-yl)-3,3-difluoroprop-1-enyl)-3-methylene-5-oxocyclopentyl)hept-5-enoic acid and its tautomer (Z)-7-((1R,2R)-2-((E)-3-(benzo[b]thiophen-2-yl)-3,3-difluoroprop-1-enyl)-3-methyl-5-oxocyclopent-3-enyl)hept-5-enoic acid; (Z)-7-((1R,2R)-2-((E)-3-(benzo[b]thiophen-2-yl)-3-methylbut-1-enyl)-3-methylene-5-oxocyclopentyl)hept-5-enoic acid and its tauto
- Another exemplary embodiment may be a more specific compound of formula (I) selected from the group consisting of: (Z)-7-((1R,2R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-3,3-difluoro-5-oxocyclopentyl)hept-5-enoic acid and its tautomer (Z)-7-((1R,5R)-5-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-2-methyl-4-oxocyclopent-2-enyl)hept-5-enoic acid; (Z)-7-((1R,2R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-5-methylene-3-oxocyclopentyl)hept-5-enoic acid; (Z)-7-
- Another exemplary embodiment may be a more specific compound of formula (I) selected from the group consisting of: 7-((1R,2R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-3,3-difluoro-5-oxocyclopentyl)heptanoic acid and its tautomer 7-((1R,5R)-5-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-2-methyl-4-oxocyclopent-2-enyl)heptanoic acid; 7-((1R,2R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-5-methylene-3-oxocyclopentyl)heptanoic acid; 7-((1R,2R,3R)-2-((R,E
- Another exemplary embodiment may be a more specific compound of formula (I) selected from the group consisting of: (Z)-7-((1R,2R)-2-((R)-3-(benzo[b]thiophen-2-yl)-3-hydroxypropyl)-3,3-difluoro-5-oxocyclopentyl)hept-5-enoic acid; (Z)-7-((1R,2R)-2-((R)-3-(benzo[b]thiophen-2-yl)-3-hydroxypropyl)-5-methylene-3-oxocyclopentyl)hept-5-enoic acid and its tautomer (Z)-7-((1R,5R)-5-((R)-3-(benzo[b]thiophen-2-yl)-3-hydroxypropyl)-2-methyl-4-oxocyclopent-2-enyl)hept-5-enoic acid; (Z)-7-((1R,2R,3R)-2-((
- Another exemplary embodiment may be a more specific compound of formula (I) selected from the group consisting of: 7-((1R,2R)-2-((R)-3-(benzo[b]thiophen-2-yl)-3-hydroxypropyl)-3,3-difluoro-5-oxocyclopentyl)heptanoic acid; 7-((1R,2R)-2-((R)-3-(benzo[b]thiophen-2-yl)-3-hydroxypropyl)-5-methylene-3-oxocyclopentyl)heptanoic acid and its tautomer 7-((1R,5R)-5-((R)-3-(benzo[b]thiophen-2-yl)-3-hydroxypropyl)-2-methyl-4-oxocyclopent-2-enyl)heptanoic acid; 7-((1R,2R,3R)-2-((R)-3-(benzo[b]thiophen-2-yl)
- the exemplary embodiments may also be directed to a method of preventing or treating a disease or condition mediated at least in part by agonism of an EP receptor, in a subject in need of such treatment, comprising administering to the subject a therapeutically effective amount of a compound of any of the exemplary embodiments formula (I), or a pharmaceutically acceptable salt, hydrate, or solvate thereof; the use of a compound of any of the exemplary embodiments of formula (I), or a pharmaceutically acceptable salt, hydrate, or solvate thereof, for the manufacture of a medicament for preventing or treating a disease or condition mediated at least in part by agonism of an EP receptor; a compound of any of the exemplary embodiments of formula (I), or a pharmaceutically acceptable salt, hydrate, or solvate thereof, for use as a medicament; a compound of any of the exemplary embodiments of formula (I), or a pharmaceutically acceptable salt, hydrate, or solvate thereof, for use in the prevention or treatment of a disease or condition mediated at least in part by ago
- the diseases and conditions mediated at least in part by agonism of an EP receptor may include allergy and allergic inflammation.
- Diseases and conditions of this kind may be allergic respiratory conditions such as allergic rhinitis, nasal congestion, rhinorrhea, perennial rhinitis, nasal inflammation, asthma of all types, chronic obstructive pulmonary disease (COPD), chronic or acute bronchoconstriction, chronic bronchitis, small airways obstruction, emphysema, chronic eosinophilic pneumonia, adult respiratory distress syndrome, exacerbation of airways hyper-reactivity consequent to other drug therapy, airways disease that may be associated with pulmonary hypertension, acute lung injury, bronchiectasis, sinusitis, allergic conjunctivitis, or atopic dermatitis, particularly asthma or chronic obstructive pulmonary disease.
- allergic respiratory conditions such as allergic rhinitis, nasal congestion, rhinorrhea, perennial rhinitis, nasal inflammation, asthma of all types, chronic obstructive pulmonary
- Types of asthma may include atopic asthma, non-atopic asthma, allergic asthma, atopic bronchial IgE-mediated asthma, bronchial asthma, essential asthma, true asthma, intrinsic asthma caused by pathophysiologic disturbances, extrinsic asthma caused by environmental factors, essential asthma of unknown or inapparent cause, bronchitic asthma, emphysematous asthma, exercise-induced asthma, exertion asthma, allergen-induced asthma, cold air induced asthma, occupational asthma, infective asthma caused by bacterial, fungal, protozoal, or viral infection, non-allergic asthma, incipient asthma, whez infant syndrome, and bronchiolytis.
- the compounds of any of the exemplary embodiments of formula (I) for the treatment of asthma may be palliative treatment for the symptoms and conditions of asthma such as wheezing, coughing, shortness of breath, tightness in the chest, shallow or fast breathing, nasal flaring (nostril size increases with breathing), retractions (neck area and between or below the ribs moves inward with breathing), cyanosis (gray or bluish tint to skin, beginning around the mouth), runny or stuffy nose, and headache.
- the exemplary embodiments may also be directed to any of the uses, methods, or compositions as defined above wherein the compound of any of the exemplary embodiments of formula (I), or a pharmaceutically acceptable salt, hydrate, or solvate thereof, may be used in combination with another pharmacologically active compound.
- Specific combinations useful for the treatment of allergy or asthma may include combinations comprising a compound of any of the exemplary embodiments of formula (I), or a pharmaceutically acceptable salt, hydrate, or solvate thereof, and (i) a glucocorticosteroid or DAGR (dissociated agonist of the corticoid receptor); (ii) a ⁇ 2 agonist, an example of which is a long-acting ⁇ 2 agonist; (iii) a muscarinic M3 receptor antagonist or anticholinergic agent; (iv) a histamine receptor antagonist or inverse agonist, which may be an H1 or an H3 antagonist or inverse agonist; (v) a 5-lipoxygenase inhibitor; (vi) a thromboxane inhibitor; (vii) an LTD 4 inhibitor; (viii) a kinase inhibitor; or (ix) a vaccine.
- the compounds of the combination may be administered together as a formulation in association with one or more pharmaceutically acceptable excipients.
- EP receptor agonists may also be useful for expansion of hematopoietic stem cell populations.
- compounds of formula (I) may also be useful for veterinary treatment of companion animals, exotic animals, and farm animals.
- terapéuticaally effective is intended to qualify the amount of compound or pharmaceutical composition, or the combined amount of active ingredients in the case of combination therapy. This amount or combined amount may achieve the goal of treating the relevant condition.
- treatment means administration of the compound, pharmaceutical composition, or combination to effect preventative, palliative, supportive, restorative, or curative treatment.
- treatment encompasses any objective or subjective improvement in a subject with respect to a relevant condition or disease.
- prevention treatment means that the compound, pharmaceutical composition, or combination may be administered to a subject to inhibit or stop the relevant condition from occurring in a subject, particularly in a subject or member of a population that may be significantly predisposed to the relevant condition.
- support treatment means that the compound, pharmaceutical composition, or combination may be administered to a subject as part of a regimen of therapy, but that such therapy is not limited to administration of the compound, pharmaceutical composition, or combination.
- supportive treatment may embrace preventative, palliative, restorative, or curative treatment, particularly when the compounds or pharmaceutical compositions are combined with another component of supportive therapy.
- restorative treatment means that the compound, pharmaceutical composition, or combination may be administered to a subject to modify the underlying progression or etiology of a condition.
- Non-limiting examples include an increase in forced expiratory volume in one second (FEV 1) for lung disorders, inhibition of progressive nerve destruction, reduction of biomarkers associated and correlated with diseases or disorders, a reduction in relapses, improvement in quality of life, and the like.
- curative treatment means that the compound, pharmaceutical composition, or combination may be administered to a subject for the purpose of bringing the disease or disorder into complete remission, or that the disease or disorder in undetectable after such treatment.
- alkyl alone or in combination, means an acyclic radical, linear or branched, preferably containing from 1 to about 6 carbon atoms.
- examples of such radicals include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, pentyl, iso-amyl, hexyl, heptyl, octyl, and the like.
- alkyl radicals may be optionally substituted with groups consisting of hydroxy, sulfhydryl, methoxy, ethoxy, amino, cyano, chloro, and fluoro.
- the carbon atom content of various hydrocarbon-containing moieties is indicated by a prefix designating a lower and upper number of carbon atoms in the moiety, that is, the prefix C i -C j indicates a moiety of the integer “i” to the integer “j” carbon atoms, inclusive.
- the prefix C i -C j indicates a moiety of the integer “i” to the integer “j” carbon atoms, inclusive.
- ‘(C 1 -C 8 )-alkyl’ refers to alkyl of one to eight carbon atoms, inclusive.
- hydroxy and “hydroxyl,” as used herein, mean an OH radical.
- sulfhydryl means an SH radical
- oxo means a doubly bonded oxygen
- alkoxy means a radical comprising an alkyl radical that is bonded to an oxygen atom, such as a methoxy radical.
- aryl means a fully unsaturated mono- or multi-ring cycloalkyl having a cyclic array of p-orbitals containing 4n+2 electrons, including, but not limited to, substituted or unsubstituted phenyl, naphthyl, or anthracenyl optionally fused to a carbocyclic radical wherein aryl may be optionally substituted with one or more substituents from the group consisting of halo, methoxy, ethoxy, (C 1 -C 6 )-alkyl, phenyl, O-phenyl, cyano, nitro, hydroxyl, sulfhydryl, or trifluoromethyl.
- halo means one of the following group consisting of fluoro, chloro, bromo, or iodo.
- heterocycle refers to a saturated or unsaturated mono- or multi-ring cycloalkyl wherein one or more carbon atoms is replaced by N, S, or O.
- heterocycle refers to a saturated or unsaturated mono- or multi-ring cycloalkyl wherein one or more carbon atoms is replaced by N, S, or O.
- heterocycle refers to a saturated or unsaturated mono- or multi-ring cycloalkyl wherein one or more carbon atoms is replaced by N, S, or O.
- heterocycle include fully saturated ring structures such as piperazinyl, dioxanyl, tetrahydrofuranyl, oxiranyl, aziridinyl, morpholinyl, pyrrolidinyl, piperidinyl, thiazolidinyl, and others.
- heterocycle also include partially unsaturated ring structures such as dihydrofuranyl, pyrazolinyl, imidazolinyl, pyrrolinyl, chromanyl, dihydrothiphenyl, and others.
- heteroaryl refers to an aromatic heterocyclic group.
- Heteroaryl is preferably: (a) a five-membered aromatic heterocyclic group containing either (i) 1-4 nitrogen atoms or (ii) 0-3 nitrogen atoms and 1 oxygen or 1 sulfur atom; (b) a six-membered aromatic heterocyclic group containing 1-3 nitrogen atoms; (c) a nine-membered bicyclic heterocyclic group containing either (i) 1-5 nitrogen atoms or (ii) 0-4 nitrogen atoms and 1 oxygen or 1 sulfur atom; or (d) a ten-membered bicyclic aromatic heterocyclic group containing 1-6 nitrogen atoms; each of said groups (a)-(d) being optionally substituted by one or more of (C 1 -C 6 )-alkyl, (C 1 -C 6 )-fluoroalkyl, (C 3 -C 6 )-cycloalkyl, hydroxy(C 3 -C
- heteroaryl examples include pyridyl, pyrimidinyl, pyridazinyl, pyrazinyl, thionyl, furanyl, pyrrolyl, pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, triazolyl, oxadiazolyl, thiadiazolyl, and tetrazolyl, optionally substituted as specified above.
- heterocycle or “heteroaryl,” the point of attachment to the molecule of interest may be at a heteroatom or elsewhere within the ring.
- cycloalkyl means a mono- or multi-ringed cycloalkyl wherein each ring contains three to ten carbon atoms, preferably three to six carbon atoms. “Cycloalkyl” is preferably a monocyclic cycloalkyl containing from three to six carbon atoms. Examples include cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl.
- excipient is used herein to describe any ingredient other than a compound of any of the exemplary embodiments of formula (I). The choice of excipient will to a large extent depend on factors such as the particular mode of administration, the effect of the excipient on solubility and stability, and the nature of the dosage form.
- excipient encompasses diluents, carrier, or adjuvant.
- compositions of any of the exemplary embodiments of formula (I) include the acid addition and base salts thereof.
- Suitable acid addition salts are formed by acids which form non-toxic salts. Examples include the acetate, adipate, aspartate, benzoate, besylate, bicarbonate/carbonate, bisulfate/sulfate, borate, camsylate, citrate, cyclamate, edisylate, esylate, formate, fumarate, gluceptate, gluconate, glucuronate, hexafluorophosphate, hibenzate, hydrochloride/chloride, hydrobromide/bromide, hydroiodide/iodide, isethionate, lactate, malate, maleate, malonate, mesylate, methylsulfate, naphthylate, 2-napsylate, nicotinate, nitrate, orotate, oxalate, palmitate, pamoate, phosphate/hydrogen phosphate/dihydrogen phosphate, propionate, pyroglutamate
- Suitable base salts are formed from bases which form non-toxic salts. Examples include the aluminum, arginine, benzathine, calcium, choline, diethylamine, diolamine, glycine, lysine, magnesium, meglumine, olamine, potassium, sodium, tromethamine, and zinc salts.
- Hemisalts of acids and bases may also be formed, for example, hemisulfate and hemicalcium salts.
- suitable salts see Handbook of Pharmaceutical Salts: Properties, Selection, and Use, by Stahl and Wermuth (Wiley-VCH, 2002).
- the compounds of any of the exemplary embodiments of formula (I) may also exist in unsolvated and solvated forms.
- solvate is used herein to describe a molecular complex comprising the compound of any of the exemplary embodiments of formula (I), or a pharmaceutically acceptable salt thereof, and one or more pharmaceutically acceptable solvent molecules, for example, ethanol.
- solvent molecules for example, ethanol.
- hydrate is employed when said solvent is water.
- multi-component complexes other than salts and solvates wherein the compound of any of the exemplary embodiments of formula (I) and at least one other component are present in stoichiometric or non-stoichiometric amounts.
- the compounds of any exemplary embodiment of formula (I) may exist in a continuum of solid states ranging from fully amorphous to fully crystalline.
- the compounds of any exemplary embodiment of formula (I) may also exist in a mesomorphic state (mesophase or liquid crystal) when subjected to suitable conditions.
- the mesomorphic state is intermediate between the true crystalline state and the true liquid state (either melt or solution).
- Compounds of any of the exemplary embodiments of formula (I) may be administered orally, topically, transdermally, intranasally, by inhalation, directly into the bloodstream, into muscle, into an internal organ, into the eye, into the ear, into the rectum, or by other means.
- Mass spectra (MS) methods include positive electrospray ionization (ESI + ), negative electrospray ionization (ESI ⁇ ), positive atmospheric pressure chemical ionization (APCI + ), or negative atmospheric pressure chemical ionization (APCI ⁇ ).
- Step A Preparation of (3aR,4R,5R,6aS)-2-oxo-4-((E)-3-oxo-4-phenoxybut-1-enyl)hexahydro-2H-cyclopent[b]furan-5-yl benzoate
- Step B Preparation of (3aR,4R,5R,6aS)-2-oxo-4-(3-oxo-4-phenoxybutyl)hexahydro-2H-cyclopenta[b]furan-5-yl benzoate
- Step C Preparation of (3aR,4R,5R,6aS)-4-(3,3-difluoro-4-phenoxybutyl)-2-oxohexahydro-2H-cyclopenta[b]furan-5-yl benzoate
- the product is extracted with ethyl acetate and the aqueous layer is back-extracted with ethyl acetate.
- the combined organic layers are dried over sodium sulfate, filtered, and evaporated.
- the product is purified by flash chromatography on regular silica gel, eluted with ethyl acetate-hexanes (35:75). The pure fractions are combined and concentrated under reduced pressure to afford the title intermediate.
- Step D Preparation of (3aR,4R,5R,6aS)-4-(3,3-difluoro-4-phenoxybutyl)-5-hydroxyhexahydro-2H-cyclopenta[b]furan-2-one
- Step E Preparation of (3aR,4R,5R,6aS)-4-(3,3-difluoro-4-phenoxybutyl)-5-(tetrahydro-2H-pyran-2-yloxy)hexahydro-2H-cyclopenta[b]furan-2-one
- Step F Preparation of (3aR,4R,5R,6aS)-4-(3,3-difluoro-4-phenoxybutyl)-5-(tetrahydro-2H-pyran-2-yloxy)hexahydro-2H-cyclopenta[b]furan-2-ol
- the lactol ((3aR,4R,5R,6aS)-4-(3,3-difluoro-4-phenoxybutyl)-5-(tetrahydro-2H-pyran-2-yloxy)hexahydro-2H-cyclopenta[b]furan-2-ol) (limiting reagent, prepared in Step F) is dissolved in THF and added dropwise to the ylide. The reaction mixture becomes lighter orange in color. The mixture continues stirring for 2 hours at ⁇ 15° C. and is subsequently allowed to warm to room temperature and stir overnight as the reaction mixture becomes dark red.
- the reaction mixture is acidified with 5% KHSO 4 , diluted with brine (250 mL) and extracted with ethyl acetate (200 mL).
- the aqueous layer is extracted with another portion of ethyl acetate (50 mL) and the combined organic extracts are washed twice with brine (250 mL), dried over sodium sulfate and evaporated to yield crude product.
- the crude product is purified by flash chromatography on regular silica gel using ethyl acetate-hexane-acetic acid as eluent to afford the title intermediate.
- Step H Preparation of (Z)-7-((1R,2R,3R)-2-(3,3-difluoro-4-phenoxybutyl)-5-oxo-3-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoic acid
- Step I Preparation of (Z)-7-((1R,2R,3R)-2-(3,3-difluoro-4-phenoxybutyl)-3-hydroxy-5-oxocyclopentyl)hept-5-enoic acid
- Scheme 1 illustrates a synthetic route that may be utilized to prepare compounds of general structure 13-14, 21-22, and 25-26.
- Examples 2, 3, 4, and 5 describe the preparation of exemplary embodiment compounds 21a, 25a, 22a, and 26a, respectively, as illustrated in Scheme 1.
- Examples 6 and 9 describe the preparation of exemplary embodiment compounds 13a and 13b, respectively, as illustrated in Scheme 1.
- Examples 7 and 8 describe the preparation of exemplary embodiment compounds 14a and 14 b, respectively, as illustrated in Scheme 1.
- Step A Preparation of (3aR,4R,5R,6aS)-4-((E)-3-(benzo[b]thiophen-2-yl)-3-oxoprop-1-enyl)-2-oxohexahydro-2H-cyclopenta[b]furan-5-yl benzoate (4a)
- a reactor equipped with a mechanical stirrer, under nitrogen, is charged with (3aR,4R,5R,6aS)-4-formyl-2-oxohexahydro-2H-cyclopenta[b]furan-5-yl benzoate (99.2 g, 0.362 mol) in DCM and lithium chloride (1 molar equivalent) dissolved in THF. Diethyl 2-(benzo[b]thiophen-2-yl)-2-oxoethylphosphonate (1 molar equivalent) is added neat and rinsed with DCM. Some lithium chloride precipitates from solution when the THF and DCM solutions are mixed. The mixture is stirred under nitrogen and cooled to ⁇ 20° C.
- Step B Preparation of (3aR,4R,5R,6aS)-4-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-2-oxohexahydro-2H-cyclopenta[b]furan-5-yl benzoate (5a)
- reaction mixture slowly warms to room temperature.
- the reaction mixture is diluted with brine and acidified with a small amount of 5% aqueous KHSO 4 .
- the product is extracted with ethyl acetate twice and the combined organic layers are dried over sodium sulfate, filtered, and evaporated under reduced pressure.
- the crude product is purified by flash chromatography on regular silica gel eluted with hexanes-ethyl acetate to afford the title intermediate 5a.
- Step C Preparation of (3aR,4R,5R,6aS)-4-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-2-oxohexahydro-2H-cyclopenta[b]furan-5-yl benzoate (6a)
- a stirring mixture consisting of (3aR,4R,5R,6aS)-4-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-2-oxohexahydro-2H-cyclopenta[b]furan-5-yl benzoate (5a, limiting reagent) and imidazole (1.1 molar equivalents) in DMF (5 M in 5a) is cooled to 0° C. under a nitrogen atmosphere. A solution consisting of TBDPSCl (1.1 molar equivalents) in DMF is added slowly.
- reaction mixture Upon completion of the reaction, as judged by TLC, the reaction mixture is diluted with ethyl acetate and is washed with water and brine. The organic layer is dried over sodium sulfate, filtered, and evaporated under reduced pressure. The crude product is purified by flash chromatography on regular silica gel eluted with hexanes-ethyl acetate to afford the title intermediate 6a.
- Step D Preparation of (3aR,4R,5R,6aS)-4-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-5-hydroxyhexahydro-2H-cyclopenta[b]furan-2-one (7a)
- Step E Preparation of (3aR,4R,5R,6aS)-4-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)hexahydro-2H-cyclopenta[b]furan-2,5-diol (8a)
- Step F Preparation of (Z)-7-((1R,2R,3R,5S)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-3,5-dihydroxycyclopentyl)hept-5-enoic Acid (9a)
- lactol (3aR,4R,5R,6aS)-4-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)hexahydro-2H-cyclopenta[b]furan-2,5-diol (8a, limiting reagent) is dissolved in THF and added dropwise to the ylide.
- the reaction mixture becomes lighter orange in color and is additionally stirred for 2 hours at ⁇ 15° C. and is then allowed to warm to room temperature and stir overnight.
- the reaction mixture becomes dark red.
- the reaction mixture is acidified with 5% aqueous KHSO 4 , diluted with brine (250 mL) and extracted with ethyl acetate (200 mL).
- the aqueous layer is extracted with another portion of ethyl acetate (50 mL) and the combined organic extracts are washed twice with brine (250 mL), dried over sodium sulfate, and evaporated to yield crude product.
- the crude product is purified by flash chromatography on regular silica gel using hexanes-ethyl acetate with 0.4% acetic acid as eluent to afford the title intermediate 9a.
- Step G Preparation of (Z)-methyl 7-((1R,2R,3R,5S)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-3,5-dihydroxycyclopentyl)hept-5-enoate (10a)
- Step H Preparation, separation, and isolation of (Z)-methyl 7-((1R,2R,5S)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-5-hydroxy-3-oxocyclopentyl)hept-5-enoate (11a) and (Z)-methyl 7-((1R,2R,3R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-3-hydroxy-5-oxocyclopentyl)hept-5-enoate (12a)
- Step I Preparation of (Z)-methyl 7-((1R,2R,5S)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-3-oxo-5-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoate (15a)
- Step J Preparation of (Z)-methyl 7-((1R,2R,5S)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-3-methylene-5-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoate (17a)
- a solution consisting of zinc methylene dibromide titanium tetrachloride is prepared by combining zinc dust (2.3 g) in THF (40 mL) with methylene dibromide (0.81 mL) at ⁇ 40° C. under a nitrogen atmosphere. To the suspension is slowly added TiCl 4 (0.92 mL). Portions (2 mL) of the zinc methylene dibromide titanium tetrachloride solution are added until the reaction is complete as judged by TLC. Upon completion, the reaction mixture is diluted with ethyl acetate and filtered twice through a bed of Celite.
- Step K Preparation of (Z)-7-((1R,2R,5S)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-3-methylene-5-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoic Acid (19a)
- Step L Preparation of (Z)-7-((1R,2R,5S)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-3-methylene-5-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoic acid
- Step M Preparation of (Z)-7-((1R,2R,5S)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-5-hydroxy-3-methylenecyclopentyl)hept-5-enoic Acid (21a)
- Step A Preparation of (Z)-7-((1R,2R,5S)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-5-hydroxy-3-methylenecyclopentyl)hept-5-enoic acid (23a)
- the title intermediate 23a is prepared from (Z)-7-((1R,2R,5S)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-3-methylene-5-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoic acid (prepared in Example 2, Step K) in a THP-deprotection procedure similar to that which is described in Example 2, Step M.
- Step B Preparation of (Z)-7-((1R,2R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-3-methyl-5-oxocyclopent-3-enyl)hept-5-enoic acid
- Step C Preparation of (Z)-7-((1R,2R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-3-methyl-5-oxocyclopent-3-enyl)hept-5-enoic Acid (25a)
- the title compound 25a is prepared from (Z)-7-((1R,2R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-3-methyl-5-oxocyclopent-3-enyl)hept-5-enoic acid (prepared in Step B) with a procedure similar to that which is described in Example 2, Step L.
- Step A Preparation of (Z)-methyl 7-((1R,2R,3R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-5-oxo-3-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoate (16a)
- the title intermediate 16a is prepared from (Z)-methyl 7-((1R,2R,3R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-3-hydroxy-5-oxocyclopentyl)hept-5-enoate (intermediate 12a prepared in Example 2, Step H) with a procedure similar to that which is described in Example 2, Step I.
- Step B Preparation of (Z)-methyl 7-((1R,2R,3R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-5-methylene-3-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoate (18a)
- the title intermediate 18a is prepared from (Z)-methyl 7-((1R,2R,3R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-5-oxo-3-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoate (16a) with a procedure similar to that which is described in Example 2, Step J.
- Step C Preparation of (Z)-7-((1R,2R,3R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-5-methylene-3-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoic Acid (20a)
- the title intermediate 20a is prepared from (Z)-methyl 7-((1R,2R,3R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-5-methylene-3-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoate (18a) with a procedure similar to that which is described in Example 2, Step K.
- Step D Preparation of (Z)-7-((1R,2R,3R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-5-methylene-3-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoic acid
- Step E Preparation of (Z)-7-((1R,2R,3R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-3-hydroxy-5-methylenecyclopentyl)hept-5-enoic Acid (22a)
- the title compound 22a is prepared from (Z)-7-((1R,2R,3R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-5-methylene-3-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoic acid, prepared in Step D, with a procedure to that which is described in Example 2, Step M.
- Step A Preparation of (Z)-7-((1R,2R,3R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-3-hydroxy-5-methylenecyclopentyl)hept-5-enoic Acid (24a)
- the title intermediate 24a is prepared from (Z)-7-((1R,2R,3R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-3-hydroxy-5-methylenecyclopentyl)hept-5-enoic acid (22a) with a procedure similar to that which is described in Example 2, Step M.
- Step B Preparation of (Z)-7-((1R,5R)-5-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-2-methyl-4-oxocyclopent-2-enyl)hept-5-enoic Acid
- Step C Preparation of (Z)-7-((1R,5R)-5-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-2-methyl-4-oxocyclopent-2-enyl)hept-5-enoic Acid (26a)
- the title compound 26a is prepared from (Z)-7-((1R,5R)-5-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-2-methyl-4-oxocyclopent-2-enyl)hept-5-enoic acid, prepared in Step B, with a procedure similar to that which is described in Example 2, Step L.
- Step A Preparation of (Z)-7-((1R,2R,5S)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-5-hydroxy-3-oxocyclopentyl)hept-5-enoic Acid
- the title intermediate is prepared from (Z)-methyl 7-((1R,2R,5S)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-5-hydroxy-3-oxocyclopentyl)hept-5-enoate (11a, prepared in Example 2, Step H) using a hydrolysis procedure described in Example 2, Step K.
- Step B Preparation of (Z)-7-((1R,2R,5S)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-5-hydroxy-3-oxocyclopentyl)hept-5-enoic Acid (13a)
- the title compound 13a is prepared from (Z)-7-((1R,2R,5S)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-5-hydroxy-3-oxocyclopentyl)hept-5-enoic acid, prepared in Step A, with a procedure similar to that which is described in Example 2, Step L.
- Step A Preparation of (Z)-7-((1R,2R,3R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-3-hydroxy-5-oxocyclopentyl)hept-5-enoic Acid
- Step B Preparation of (Z)-7-((1R,2R,3R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-3-hydroxy-5-oxocyclopentyl)hept-5-enoic Acid (14a)
- the title compound 14a is prepared from (Z)-7-((1R,2R,3R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-3-hydroxy-5-oxocyclopentyl)hept-5-enoic acid, prepared in Step A, with a procedure similar to that which is described in Example 2, Step L.
- Step A Preparation of (3aR,4R,5R,6aS)-4-((E)-3-(benzo[b]thiophen-2-yl)-3-oxoprop-1-enyl)-2-oxohexahydro-2H-cyclopenta[b]furan-5-yl biphenyl-4-carboxylate (27)
- Step B Preparation of (3aR,4R,5R,6aS)-4-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-2-oxohexahydro-2H-cyclopenta[b]furan-5-yl biphenyl-4-carboxylate (28)
- the reaction mixture was diluted with ethyl acetate (300 mL) and subsequently washed with 1:1 saturated ammonium chloride-water (3 ⁇ 50 mL), followed by brine (100 mL).
- the organic layer was separated, dried over sodium sulfate, filtered, and the filtrate was subsequently evaporated to give a foamy solid (3.55 g).
- the crude material was dissolved in DCM and then purified on a 120-g Analogix flash silica cartridge eluted with DCM (100%, 1 L) to DCM-methanol ((300:1), 1 L) to DCM-methanol ((200:1), 2 L) to afford product (1.43 g).
- Step C Preparation of (3aR,4R,5R,6aS)-4-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-5-hydroxyhexahydro-2H-cyclopenta[b]furan-2-one (29)
- reaction mixture was diluted with ethyl acetate (300 mL), washed with 1:1 saturated ammonium chloride-water (2 ⁇ 50 mL) and brine (50 mL). The combined organic layers were dried over sodium sulfate, filtered and evaporated to give a yellow solid. The material was triturated in 10% ethyl acetate in hexanes (200 mL) for three days.
- Step D Preparation of (3aR,4R,5R,6aS)-4-((3R,E)-3-(benzo[b]thiophen-2-yl)-3-(tetrahydro-2H-pyran-2-yloxy)prop-1-enyl)-5-(tetrahydro-2H-pyran-2-yloxy)hexahydro-2H-cyclopenta[b]furan-2-one (30)
- the reaction mixture was stirred at room temperature for 18 hours.
- the reaction mixture was diluted with DCM (200 mL) and was subsequently washed with 50% brine in water (2 ⁇ 50 mL).
- the organic layer was separated, dried over sodium sulfate, filtered, and the filtrate was evaporated to give a crude product (626 mg) as a yellow oil.
- the crude product was purified on a 40-g Analogix flash silica cartridge.
- Step E Preparation of (3aR,4R,5R,6aS)-4-((3R,E)-3-(benzo[b]thiophen-2-yl)-3-(tetrahydro-2H-pyran-2-yloxy)prop-1-enyl)-5-(tetrahydro-2H-pyran-2-yloxy)hexahydro-2H-cyclopenta[b]furan-2-ol (31)
- reaction mixture was stirred for two hours and was complete as judged by TLC.
- the reaction mixture was warmed to 10° C. and was subsequently quenched with a 7:1 mixture of THF-water (8 mL).
- the reaction mixture was allowed to stir overnight (18 hours). After stirring overnight a white solid had formed.
- the reaction mixture was decanted away and the solid was washed with 50% brine in water (3 ⁇ 30 mL).
- Step F Preparation of (Z)-7-((1R,2R,3R,5S)-2-((3R,E)-3-(benzo[b]thiophen-2-yl)-3-(tetrahydro-2H-pyran-2-yloxy)prop-1-enyl)-5-hydroxy-3-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoic Acid (32)
- Step G Preparation of (Z)-7-((1R,2R,3R)-2-((3R,E)-3-(benzo[b]thiophen-2-yl)-3-(tetrahydro-2H-pyran-2-yloxy)prop-1-enyl)-5-oxo-3-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoic Acid (33)
- Step H Preparation of (Z)-7-((1R,2R,3R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-3-hydroxy-5-oxocyclopentyl)hept-5-enoic Acid (14a)
- Steps A-H Preparation of a mixture of (Z)-methyl-7-((1R,2R,5S)-2-((R,E)-3-(benzo[d]thiazol-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-5-hydroxy-3-oxocyclopentyl)hept-5-enoate (11b) and (Z)-methyl-7-((1R,2R,3R)-2-((R,E)-3-(benzo[d]thiazol-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-3-hydroxy-5-oxocyclopentyl)hept-5-enoate (12b)
- a mixture of the intermediates 11b and 12b are prepared from Corey lactone aldehyde benzoate, the mixture separated and each intermediate isolated using the series of procedures described for Example 2, Steps A-H, except that diethyl 2-(benzo[d]thiazol-2-yl)-2-oxoethylphosphonate (prepared from ethyl benzo[d]thiazole-2-carboxylate and diethyl methylphosphate according to a method described in the Journal of Organic Chemistry, 73(12), 2008, 4568-4574) is used instead of diethyl 2-(benzo[b]thiophen-2-yl)-2-oxoethylphosphonate in Step A.
- Step I Preparation of (Z)-7-((1R,2R,3R)-2-((R,E)-3-(benzo[d]thiazol-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-3-hydroxy-5-oxocyclopentyl)hept-5-enoic acid
- Step J Preparation of (Z)-7-((1R,2R,3R)-2-((R,E)-3-(benzo[d]thiazol-2-yl)-3-hydroxyprop-1-enyl)-3-hydroxy-5-oxocyclopentyl)hept-5-enoic Acid (14b)
- the title compound 14b is prepared from (Z)-7-((1R,2R,3R)-2-((R,E)-3-(benzo[d]thiazol-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-3-hydroxy-5-oxocyclopentyl)hept-5-enoic acid with a procedure similar to that which is described in Example 2, Step L.
- Step A Preparation of (Z)-7-((1R,2R,5S)-2-((R,E)-3-(benzo[d]thiazol-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-5-hydroxy-3-oxocyclopentyl)hept-5-enoic Acid
- Step B Preparation of (Z)-7-((1R,2R,5S)-2-((R,E)-3-(benzo[d]thiazol-2-yl)-3-hydroxyprop-1-enyl)-5-hydroxy-3-oxocyclopentyl)hept-5-enoic Acid (13b)
- the title compound 13b is prepared from (Z)-7-((1R,2R,5S)-2-((R,E)-3-(benzo[d]thiazol-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-5-hydroxy-3-oxocyclopentyl)hept-5-enoic acid, prepared in Step A, with a procedure similar to that which is described in Example 2, Step L.
- the crude product is purified using a filtration column on silica gel, eluted with 0.4% acetic acid in ethyl acetate.
- a second flash silica column eluted with an isopropanol-hexanes-acetic acid solvent system affords the title compound.
- Step A Preparation of (3aR,4R,5R,6aS)-4-((R)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)propyl)hexahydro-2H-cyclopenta[b]furan-2,5-diol
- Step B Preparation of (Z)-7-((1R,2R,3R,5S)-2-((R)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)propyl)-3,5-dihydroxycyclopentyl)hept-5-enoic Acid
- lactol (3aR,4R,5R,6aS)-4-((R)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)propyl)hexahydro-2H-cyclopenta[b]furan-2,5-diol (1 molar equivalent) is dissolved in THF and added dropwise to the ylide.
- the reaction mixture becomes lighter orange in color and is further stirred for 2 hours at ⁇ 15° C. and is then allowed to warm to room temperature and stir overnight.
- the reaction mixture becomes dark red.
- the reaction mixture is acidified with 5% aqueous KHSO 4 , diluted with brine and extracted with ethyl acetate.
- the aqueous layer is extracted with another portion of ethyl acetate and the combined organic extracts are washed twice with brine, dried over sodium sulfate, and evaporated to yield crude product.
- the crude product is purified by flash chromatography on regular silica gel using ethyl acetate-hexane-acetic acid as eluent to afford the title intermediate.
- Step C Preparation, separation, and isolation of (Z)-7-((1R,2R,5S)-2-((R)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)propyl)-5-hydroxy-3-oxocyclopentyl)hept-5-enoic acid and (Z)-7-((1R,2R,3R)-2-((R)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)propyl)-3-hydroxy-5-oxocyclopentyl)hept-5-enoic acid
- Step D Preparation of (Z)-7-((1R,2R,3R)-2-((R)-3-(Benzo[b]thiophen-2-yl)-3-hydroxypropyl)-3-hydroxy-5-oxocyclopentyl)hept-5-enoic Acid
- Step A Preparation of (3aR,4R,5R,6aS)-4-((E)-3-(benzo[b]thiophen-2-yl)-3,3-difluoroprop-1-enyl)-2-oxohexahydro-2H-cyclopenta[b]furan-5-yl Benzoate
- the reaction Upon completion the reaction is cooled to 0° C. and quenched by the slow addition of a saturated aqueous solution of sodium bicarbonate. The layers are separated and the aqueous phase is extracted with ethyl acetate. The organic layers are combined and dried over magnesium sulfate. The solvents are evaporated and the crude material is purified on regular silica gel eluted with hexanes-ethyl acetate solvent system to afford the title intermediate.
- Step B Preparation of (3aR,4R,5R,6aS)-4-((E)-3-(benzo[b]thiophen-2-yl)-3,3-difluoroprop-1-enyl)-5-hydroxyhexahydro-2H-cyclopenta[b]furan-2-one
- Step C Preparation of (3aR,4R,5R,6aS)-4-((E)-3-(benzo[b]thiophen-2-yl)-3,3-difluoroprop-1-enyl)-5-(tetrahydro-2H-pyran-2-yloxy)hexahydro-2H-cyclopenta[b]furan-2-one
- Step D Preparation of (3aR,4R,5R,6aS)-4-((E)-3-(benzo[b]thiophen-2-yl)-3,3-difluoroprop-1-enyl)-5-(tetrahydro-2H-pyran-2-yloxy)hexahydro-2H-cyclopenta[b]furan-2-ol
- Step E Preparation of (Z)-7-((1R,2R,3R,5S)-2-((E)-3-(benzo[b]thiophen-2-yl)-3,3-difluoroprop-1-enyl)-5-hydroxy-3-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoic acid
- Step F Preparation of (Z)-7-((1R,2R,3R)-2-((E)-3-(benzo[b]thiophen-2-yl)-3,3-difluoroprop-1-enyl)-5-oxo-3-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoic acid
- Step G Preparation of (Z)-7-((1R,2R,3R)-2-((E)-3-(benzo[b]thiophen-2-yl)-3,3-difluoroprop-1-enyl)-3-hydroxy-5-oxocyclopentyl)hept-5-enoic Acid
- Step A Preparation of (3aR,4R,5R,6aS)-4-((E)-3-(benzo[b]thiophen-2-yl)-3-hydroxybut-1-enyl)-2-oxohexahydro-2H-cyclopenta[b]furan-5-yl benzoate
- Step B Preparation of (3aR,4R,5R,6aS)-4-((E)-3-(benzo[b]thiophen-2-yl)-3-hydroxybut-1-enyl)-5-hydroxyhexahydro-2H-cyclopenta[b]furan-2-one
- Step C Preparation of (3aR,4R,5R,6aS)-4-((E)-3-(benzo[b]thiophen-2-yl)-3-(tetrahydro-2H-pyran-2-yloxy)but-1-enyl)-5-(tetrahydro-2H-pyran-2-yloxy)hexahydro-2H-cyclopenta[b]furan-2-one
- Step D Preparation of (3aR,4R,5R,6aS)-4-((E)-3-(benzo[b]thiophen-2-yl)-3-(tetrahydro-2H-pyran-2-yloxy)but-1-enyl)-5-(tetrahydro-2H-pyran-2-yloxy)hexahydro-2H-cyclopenta[b]furan-2-ol
- Step E Preparation of (Z)-7-((1R,2R,3R,5S)-2-((E)-3-(benzo[b]thiophen-2-yl)-3-(tetrahydro-2H-pyran-2-yloxy)but-1-enyl)-5-hydroxy-3-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoic acid
- Step F Preparation of (Z)-7-((1R,2R,3R)-2-((E)-3-(benzo[b]thiophen-2-yl)-3-(tetrahydro-2H-pyran-2-yloxy)but-1-enyl)-5-oxo-3-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoic acid
- Step G Preparation of (Z)-7-((1R,2R,3R)-2-((E)-3-(benzo[b]thiophen-2-yl)-3-hydroxybut-1-enyl)-3-hydroxy-5-oxocyclopentyl)hept-5-enoic acid
- the title compound is prepared from (Z)-7-((1R,2R,3R)-2-((E)-3-(benzo[b]thiophen-2-yl)-3-(tetrahydro-2H-pyran-2-yloxy)but-1-enyl)-5-oxo-3-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoic acid, prepared in Step F, in a manner similar to that as is described in Example 1, Step I.
- the title compound may be prepared from intermediate 15a with a series of five procedures comprising the following sequence: Example 2, Steps I, M, H, K, and L.
- the title compound may be prepared from intermediate 16a with a series of five procedures comprising the following sequence: Example 2, Steps I, M, H, K, and L.
- Step A Preparation of (3aR,4S,5S,6aS)-4-((tert-butyldiphenylsilyloxy)methyl)-5-fluorohexahydro-2H-cyclopenta[b]furan-2-one
- the DAST (1 molar equivalent) was placed in a 2-liter round bottom flask flushed with nitrogen and was dissolved in anhydrous CH 2 Cl 2 (200 mL). This mixture was then cooled to ⁇ 78° C. using an acetone/dry ice bath. After 20 minutes of cooling, a solution consisting of (3aR,4S,5R,6aS)-4-((tert-butyldiphenylsilyloxy)methyl)-5-hydroxyhexahydro-2H-cyclopenta[b]furan-2-one (prepared by appropriate silylation of the ( ⁇ )-Corey lactone diol, 1 molar equivalent) in anhydrous CH 2 Cl 2 (800 mL) was added slowly over a 30 minute period using an addition funnel.
- Step B Preparation of (3aR,4S,5S,6aS)-4-((tert-butyldiphenylsilyloxy)methyl)-5-fluorohexahydro-2H-cyclopenta[b]furan-2-ol
- Step C Preparation of (Z)-7-((1R,2S,3S,5S)-2-((tert-butyldiphenylsilyloxy)methyl)-3-fluoro-5-hydroxycyclopentyl)hept-5-enoic acid
- the reaction mixture was allowed to slowly warm to room temperature overnight with stirring.
- the reaction was acidified with 5% KHSO 4 , extracted with ethyl acetate, and washed twice with brine.
- the crude material was dried over sodium sulfate, filtered and evaporated to an oil.
- the crude product was purified on silica gel, eluted with hexanes-ethyl acetate (4:1 with 0.4% acetic acid) to afford the title intermediate (30.8 g, 97%).
- Step D Preparation of (Z)-isopropyl 7-((1R,2S,3S,5S)-2-((tert-butyldiphenylsilyloxy)methyl)-3-fluoro-5-hydroxycyclopentyl)hept-5-enoate
- the reaction mixture was acidified with 5% KHSO 4 , extracted with ethyl acetate, washed with brine, dried over sodium sulfate, filtered and evaporated.
- the crude product was purified on silica gel, eluted with hexanes-ethyl acetate (4:1) to afford the title intermediate (28.6 g, 86%).
- Step E Preparation of (Z)-isopropyl 7-((1R,2S,3S,5S)-2-((tert-butyldiphenylsilyloxy)methyl)-3-fluoro-5-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoate
- the reaction mixture was then allowed to warm to room temperature and stirring was continued for another 2 hours.
- the crude reaction mixture was transferred to a separatory funnel, washed with saturated aqueous sodium bicarbonate solution and then with brine.
- the organic phase was dried over sodium sulfate, filtered, and the solvents were removed by evaporation to afford the crude title intermediate (32.7 g).
- the crude intermediate was carried on with no further purification.
- Step F Preparation of (Z)-isopropyl 7-((1R,2S,3S,5S)-3-fluoro-2-(hydroxymethyl)-5-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoate
- the reaction mixture was acidified with 5% KHSO 4 , extracted into ethyl acetate, washed with brine, and dried over sodium sulfate, filtered, and concentrated under reduced pressure.
- the crude product was purified on silica gel, eluted with hexanes-ethyl acetate (3:1) to afford the title intermediate 12.94 g, 64%).
- Step G Preparation of (Z)-isopropyl 7-((1R,2S,3S,5S)-3-fluoro-2-formyl-5-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoate
- Oxalyl chloride 25 mL, 50 mmol was dissolved in DCM (400 mL) and cooled to ⁇ 78° C. under a nitrogen atmosphere, to which a solution consisting of DMSO (4.5 mL) in DCM (150 mL) was added. The reaction mixture was stirred for 15 minutes at ⁇ 78° C. and a solution consisting of (Z)-isopropyl 7-((1R,2S,3S,5S)-3-fluoro-2-(hydroxymethyl)-5-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoate (prepared in Step F, 12.94 g, 33.36 mmol) in DCM was subsequently added slowly.
- reaction mixture was stirred for 40 minutes and triethylamine (28 mL) was subsequently added. Stirring was continued as the reaction mixture was allowed to warm to room temperature for 2 hours, monitoring by TLC.
- the reaction was acidified with 5% KHSO 4 , extracted with DCM, and the organic layers were washed twice with brine. After drying over sodium sulfate, filtration, and solvent removal under reduced pressure, the crude product was purified on silica gel, eluted with hexanes-ethyl acetate (85:15) to afford the title intermediate (10.31 g, 80%).
- Step H Preparation of (Z)-isopropyl 7-((1R,2R,3S,5S)-2-((E)-3-(2,3-dihydro-1H-inden-2-yl)-3-oxoprop-1-enyl)-3-fluoro-5-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoate
- Step I Preparation of (Z)-isopropyl 7-((1R,2R,3S,5S)-2-((S,E)-3-(2,3-dihydro-1H-inden-2-yl)-3-hydroxyprop-1-enyl)-3-fluoro-5-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)hept-5-enoate
- Step J Preparation of (Z)-isopropyl 7-((1R,2R,3S,5S)-2-((S,E)-3-(2,3-dihydro-1H-inden-2-yl)-3-hydroxyprop-1-enyl)-3-fluoro-5-hydroxycyclopentyl)hept-5-enoate
- reaction mixture was evaporated to dryness and redissolved in ethyl acetate.
- organic solution was washed twice with brine, dried over sodium sulfate, filtered, and concentrated under reduced pressure.
- the crude product was purified on silica gel. Elution with hexanes-ethyl acetate (9:1) afforded the title intermediate (2.2 g).
- Step K Preparation of (Z)-isopropyl 7-((1R,2R,3S,5S)-2-((S,E)-3-(tert-butyldimethylsilyloxy)-3-(2,3-dihydro-1H-inden-2-yl)prop-1-enyl)-3-fluoro-5-hydroxycyclopentyl)hept-5-enoate
- Step L Preparation of (Z)-isopropyl 7-((1R,2R,3S)-2-((S,E)-3-(tert-butyldimethylsilyloxy)-3-(2,3-dihydro-1H-inden-2-yl)prop-1-enyl)-3-fluoro-5-oxocyclopentyl)hept-5-enoate
- reaction mixture was acidified with saturated ammonium chloride, extracted into ethyl acetate, washed with brine, and dried over sodium sulfate, filtered, and the solvents were evaporated under reduced pressure.
- the crude product was purified on silica gel. Elution with hexanes-ethyl acetate (4:1 with 0.4% acetic acid) afforded the title intermediate (0.58 g, 96%).
- Step M Preparation of (Z)-7-((1R,2R,3S)-2-((S,E)-3-(tert-butyldimethylsilyloxy)-3-(2,3-dihydro-1H-inden-2-yl)prop-1-enyl)-3-fluoro-5-oxocyclopentyl)hept-5-enoic acid
- Step N Preparation of (Z)-7-((1R,2R,3S)-2-((S,E)-3-(2,3-dihydro-1H-inden-2-yl)-3-hydroxyprop-1-enyl)-3-fluoro-5-oxocyclopentyl)hept-5-enoic acid
- the title compound was prepared from (Z)-7-((1R,2R,3S)-2-((S,E)-3-(tert-butyldimethylsilyloxy)-3-(2,3-dihydro-1H-inden-2-yl)prop-1-enyl)-3-fluoro-5-oxocyclopentyl)hept-5-enoic acid (prepared in Step M) utilizing the conditions described in Example 2, Step L.
- the product was purified by semipreparative HPLC with conditions for purification as follows:
- Peak concentration sample was dissolved at 100 mg/mL in 70:30 MeOH/H 2 O Peaks were collected manually at 0° C. and purified batches were kept at ⁇ 20° C. at the end of each day. Peak 2 (title compound, 14.6 minute retention time) degrades about 6% when left at 4° C. overnight in mobile phase and degrades minimally ( ⁇ 1%) when left at ⁇ 20° C. overnight. Peak 1 (HF-eliminated by-product) possesses a retention time of 13 minutes. LCMS data of both products confirm masses consistent with the designations presented herein.
- Instrument used was a Gilson HPLC equipped with 321 binary pump, 215 liquid handler and 155 dual wavelength detector.
- the title compound may be prepared from intermediate 11a (prepared in Example 2, Step H) by: (A) Protecting the keto group by preparing the intermediate (Z)-methyl 7-((6R,7R,8S)-6-((R,E)-3-(benzo[b]thiophen-2-yl)-3-(tert-butyldiphenylsilyloxy)prop-1-enyl)-8-hydroxy-1,4-dioxaspiro[4.4]nonan-7-yl)hept-5-enoate from 11a and ethane-1,2-diol using conditions known to those skilled in the art, (B) Treating the intermediate from (A) with DAST under conditions similar to those presented for Example 17, Step A, (C) Hydrolyzing the ester moiety of the intermediate prepared in Step (B) using conditions similar to those presented for Example 2, Step K, (D) Deprotecting the ketone on the intermediate prepared in Step (C) using methods known to those skilled in the art, and (E) Desilylation of the
- Example 19 exemplifies a procedure that may generally be used to prepare compounds for which the terminal group of the ⁇ chain is the N-methanesulfonylamide functional group.
- Step B Preparation of (5-(methylsulfonamido)-5-oxopentyl)triphenylphosphonium bromide
- Step C Preparation of (Z)-7-((1R,2R,3R,5S)-2-((3R,E)-3-(benzo[b]thiophen-2-yl)-3-(tetrahydro-2H-pyran-2-yloxy)prop-1-enyl)-5-hydroxy-3-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)-N-(methylsulfonyl)hept-5-enamide
- the title intermediate is prepared from (5-(methylsulfonamido)-5-oxopentyl)triphenylphosphonium bromide (prepared in Step B) and (3aR,4R,5R,6aS)-4-((3R,E)-3-(benzo[b]thiophen-2-yl)-3-(tetrahydro-2H-pyran-2-yloxy)prop-1-enyl)-5-(tetrahydro-2H-pyran-2-yloxy)hexahydro-2H-cyclopenta[b]furan-2-ol (intermediate 31 prepared in Example 13, Step E) in a manner similar to that as described in Example 1, Step G.
- Step D Preparation of (Z)-7-((1R,2R,3R)-2-((3R,E)-3-(benzo[b]thiophen-2-yl)-3-(tetrahydro-2H-pyran-2-yloxy)prop-1-enyl)-5-oxo-3-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)-N-(methylsulfonyl)hept-5-enamide
- Step E Preparation of (Z)-7-((1R,2R,3R)-2-((R,E)-3-(benzo[b]thiophen-2-yl)-3-hydroxyprop-1-enyl)-3-hydroxy-5-oxocyclopentyl)-N-(methylsulfonyl)hept-5-enamide
- the title compound is prepared from (Z)-7-((1R,2R,3R)-2-((3R,E)-3-(benzo[b]thiophen-2-yl)-3-(tetrahydro-2H-pyran-2-yloxy)prop-1-enyl)-5-oxo-3-(tetrahydro-2H-pyran-2-yloxy)cyclopentyl)-N-(methylsulfonyl)hept-5-enamide (prepared in Step D) in a manner similar to that described in Example 1, Step I.
- Example 20 exemplifies a procedure that may generally be used to convert carboxylic acid compounds to their respective carboxamides.
- the filtrate is evaporated to give the NHS-ester intermediate.
- the crude NHS-ester is dissolved in anhydrous DMF (0.2 M) under a nitrogen atmosphere.
- Ethylamine 1.5 molar equivalents
- the reaction mixture is stirred at room temperature overnight.
- the reaction mixture is diluted with brine and the product is extracted with ethyl acetate.
- the organic extracts are washed with brine, dried over sodium sulfate and evaporated.
- the product is purified by flash chromatography on regular silica gel. Elution with an ethyl acetate-hexanes solvent system affords the title compound.
- Human prostaglandin E 2 receptor subtypes EP 1 , EP 2 , EP 3 and EP 4 are PCR amplified from commercially available full length human cDNAs (Open Biosystems, Origene).
- the MCS region of a mammalian expression vector pcDNA6/V5-HisA (Invitrogen) has been modified in-house to include a sequence cassette compatible with the USER Friendly PCR cloning system (New England Biolabs), and renamed pcDNA6/V5-HisA-USER.
- the PCR fragments of human EP 1 , EP 2 , EP 3 and EP 4 receptor subtypes with fragment ends compatible to the USER cassette are cloned into pcDNA6/V5-HisA-USER. All four EP receptor clones are sequence-confirmed prior to mammalian expression studies.
- Mammalian expression is performed in HEK-293 cells using FuGENE® 6 Transfection Reagent (Roche) following the manufacturer's standard protocol.
- Cells expressing each of the EP receptor subtypes are selected using blasticidin. Foci of cells surviving blasticidin treatment are picked and expanded to allow the development of clonal EP-overexpressing stable cell lines. Highly expressing cell lines is identified by western blot analysis using antibodies selective for each of the four isotypes of the EP receptor (available in-house) and/or by assessing binding of [ 3 H]-PGE 2 , as described below.
- the ability of compounds to bind the EP receptors and their selectivity for each receptor can be demonstrated in radioligand competition displacement binding experiments using the cell lines described above which stably overexpress the human EP receptors.
- the ability of compounds to activate the receptors can be demonstrated in second messenger functional assays, measuring changes in intracellular calcium for EP 1 and changes in cAMP formation for EP 2 , EP 3 and EP 4 .
- Membranes are prepared from cells stably transfected with human EP receptor DNA. In brief, cells are cultured to confluence, scraped from culture flasks and centrifuged to pellet (800 ⁇ g, 5 minutes, 4° C.). Cells are washed twice with ice-cold homogenization buffer containing 10 mM Tris-HCl, 1 mM EDTA, 250 mM sucrose, 1 mM PMSF, 300 ⁇ M indomethacin, pH 7.4, homogenized by sonication and centrifuged as before. The supernatant is stored on ice; the pellets are rehomogenized and respun. Supernatants are pooled and centrifuged at 100,000 ⁇ g for 10 minutes at 4° C. The resultant membrane pellet is stored at ⁇ 80° C. until use.
- membranes from cells expressing human EP 1 , EP 2 , EP 3 or EP 4 receptors are added to assay buffer (10 mM MES, pH 6.0, 10 mM MgCl 2 , 1 mM EDTA, 3 ⁇ M indomethacin) containing 5 nM [ 3 H]-PGE 2 (GE Healthcare) and 0.1 to 10,000 nM concentrations of compounds to be tested. Incubations are performed at suitable temperatures and times to allow equilibration to be reached. Non-specific binding is determined in the presence of 10 ⁇ M PGE 2 . Reactions are terminated by the addition of ice-cold buffer followed by rapid filtration through Whatman GF/B filters. The filters are dried after washing, and membrane-bound radioactivity is quantified by scintillation counting.
- EP 1 Receptor Agonism Assay Intracellular Calcium Assay
- Calcium fluorescence is measured using an Analyst AD (Molecular Devices) with an excitation wavelength of 485 nm, emission wavelength of 560 nm, and emission cutoff of 505 nm. Responses are quantified as peak fluorescence intensity minus basal fluorescence intensity.
- Evaluation of the agonist activity of compounds at the human EP 1 receptor in transfected HEK-293 cells determined by measuring their effect on cytosolic Ca 2+ ion mobilization using a fluorimetric detection method.
- the cells are suspended in DMEM buffer (Invitrogen), then distributed in microplates at a density of 3.10 4 cells/well.
- the fluorescent probe (Fluo4 NR, Invitrogen) mixed with probenicid in HBSS buffer (Invitrogen) complemented with 20 mM Hepes (Invitrogen) (pH 7.4) is then added into each well and equilibrated with the cells for 30 minutes at 37° C. then 30 minutes at 22° C.
- the assay plates are positioned in a microplate reader (CellLux, PerkinElmer) which is used for the addition of the test compound, reference agonist or HBSS buffer (basal control), and the measurements of changes in fluorescence intensity which varies proportionally to the free cytosolic Ca 2+ ion concentration.
- a microplate reader CellLux, PerkinElmer
- PGE 2 at 100 nM is added in separate assay wells.
- the results are expressed as a percent of the control response to 100 nM PGE2.
- the standard reference agonist is PGE2, which is tested in each experiment at several concentrations to generate a concentration-response curve from which its EC50 value is calculated.
- the cells are suspended in DMEM buffer (Invitrogen), then distributed in microplates at a density of 3.10 4 cells/well.
- the fluorescent probe (Fluo4 NR, Invitrogen) mixed with probenicid in HBSS buffer (Invitrogen) complemented with 20 mM Hepes (Invitrogen) (pH 7.4) is then added into each well and equilibrated with the cells for 30 minutes at 37° C. then 30 minutes at 22° C.
- the assay plates are positioned in a microplate reader (CellLux, PerkinElmer) which is used for the addition of the test compound, reference antagonist or HBSS buffer (basal control), then 5 minutes later 3 nM PGE 2 , and the measurements of changes in fluorescence intensity which varies proportionally to the free cytosolic Ca 2+ ion concentration. The results are expressed as a percent inhibition of the control response to 3 nM PGE 2 .
- the standard reference antagonist is SC 51322, which is tested in each experiment at several concentrations to generate a concentration-response curve from which its IC 50 value is calculated.
- the cells are suspended in HBSS buffer (Invitrogen) complemented with HEPES 20 mM (pH 7.4) and 500 ⁇ M IBMX, then distributed in microplates at a density of 10 4 cells/well and incubated for 30 minutes at 37° C. in the absence (control) or presence of the test compound or the reference agonist. For stimulated control measurements, separate assay wells contain 10 ⁇ M PGE 2 . Following incubation, the cells are lysed and the fluorescence acceptor (D2-labeled cAMP) and fluorescence donor (anti-cAMP antibody labeled with europium cryptate) are added.
- D2-labeled cAMP fluorescence acceptor
- fluorescence donor anti-cAMP antibody labeled with europium cryptate
- the cAMP concentration is determined by dividing the signal measured at 665 nm by that measured at 620 nm (ratio). The results are expressed as a percent of the control response to 10 ⁇ M PGE 2 .
- the standard reference agonist is PGE 2 , which is tested in each experiment at several concentrations to generate a concentration-response curve from which its EC 50 value is calculated.
- the cells are suspended in HBSS buffer (Invitrogen) complemented with HEPES 20 mM (pH 7.4) and 500 ⁇ M IBMX, then distributed in microplates at a density of 10 4 cells/well and preincubated for 5 minutes at room temperature in the absence (control) or presence of the test compound or the reference antagonist. Thereafter, the reference agonist PGE 2 is added at a final concentration of 300 nM. For basal control measurements, separate assay wells do not contain PGE 2 . Following 30 minutes incubation at 37° C., the cells are lysed and the fluorescence acceptor (D2-labeled cAMP) and fluorescence donor (anti-cAMP antibody labeled with europium cryptate) are added.
- HBSS buffer Invitrogen
- HEPES 20 mM pH 7.4
- IBMX 500 ⁇ M IBMX
- the cAMP concentration is determined by dividing the signal measured at 665 nm by that measured at 620 nm (ratio). The results are expressed as a percent inhibition of the control response to 300 nM PGE 2 .
- the standard reference antagonist is AH 6809, which is tested in each experiment at several concentrations to generate a concentration-response curve from which its IC 50 value is calculated.
- EP 3 Receptor Agonism Assay Inhibition of Forskolin-Induced cAMP Generation Assay
- cAMP accumulation induced by forskolin following treatment with compounds is measured.
- Cells expressing the EP 3 receptor are plated in 24-well plates in normal growth medium and allowed to come to confluence. When the cells have come to confluence, the medium is replaced with 450 ⁇ l of serum-free medium containing 0.25 mM IBMX and 20 ⁇ M indomethacin. Cells are incubated in this medium for one hour. Fifty microliters of this same buffer containing 3 ⁇ M forskolin and various concentrations of PGE 2 or compounds to be tested are subsequently added to the cells. After incubation at 37° C. for 10 minutes, reactions are terminated by the addition of 500 ⁇ l of 10% TCA.
- cAMP measurements of the cell lysates are performed using Cayman Chemical's cAMP EIA Kit following the instructions provided in the kit booklet.
- the cells are suspended in HBSS buffer (Invitrogen) complemented with HEPES 20 mM (pH 7.4) and 500 ⁇ M IBMX, then distributed in microplates at a density of 2.10 4 cells/well and incubated for 10 minutes at room temperature in the absence (control) or presence of the test compound or the reference agonist. For stimulated control measurements, separate assay wells contain 1 ⁇ M PGE 2 . Following incubation, the cells are lysed and the fluorescence acceptor (D2-labeled cAMP) and fluorescence donor (anti-cAMP antibody labeled with europium cryptate) are added.
- D2-labeled cAMP fluorescence acceptor
- fluorescence donor anti-cAMP antibody labeled with europium cryptate
- the cAMP concentration is determined by dividing the signal measured at 665 nm by that measured at 620 nm (ratio). The results are expressed as a percent of the control response to 1 ⁇ M PGE 2 .
- the standard reference agonist is PGE 2 , which is tested in each experiment at several concentrations to generate a concentration-response curve from which its EC 50 value is calculated.
- the cells are suspended in HBSS buffer (Invitrogen) complemented with HEPES 20 mM (pH 7.4) and 500 ⁇ M IBMX, then distributed in microplates at a density of 2.10 4 cells/well and preincubated for 5 minutes at room temperature in the absence (control) or presence of the test compound or the reference antagonist. Thereafter, the reference agonist PGE 2 is added at a final concentration of 10 nM. For basal control measurements, separate assay wells do not contain PGE 2 . Following 10 minutes incubation at room temperature, the cells are lysed and the fluorescence acceptor (D2-labeled cAMP) and fluorescence donor (anti-cAMP antibody labeled with europium cryptate) are added.
- HBSS buffer Invitrogen
- HEPES 20 mM pH 7.4
- IBMX 500 ⁇ M IBMX
- the cAMP concentration is determined by dividing the signal measured at 665 nm by that measured at 620 nm (ratio). The results are expressed as a percent inhibition of the control response to 10 nM PGE 2 . There is no standard reference antagonist for this assay.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Ophthalmology & Optometry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description


wherein:
dashed bonds may each independently represent a second carbon-carbon bond in order to give a carbon-carbon double bond with either (E) or (Z) geometry or may be ignored in order to give a carbon-carbon single bond;
C9 and C11 each is independently C═CH2, C═O, CF2, CHF (any stereoisomer), or C(H)OH (any stereoisomer) with the proviso that C9 does not equal C11 and also with the proviso that when one of either C9 or C11 is CHF, the other is not C(H)OH;
R1 is CO2R3, CH2OR3, CONR4R5, COCH2OH, CONR4SO2R5, P(O)(OR4)2, or

-
- wherein R3 is hydrogen or (C1-C6)-alkyl, and
- wherein R4 and R5 each is independently hydrogen or (C1-C6)-alkyl;
m is 0, 1, 2, or 3;
Z1 and Z2 each is independently hydrogen, fluorine, hydroxy, or methyl, or together are an oxygen atom that form a carbonyl group with the adjoining carbon atom of the ω chain; Z3 and Z4 each is independently hydrogen, fluorine, hydroxy, or methyl;
n is 0 or 1;
Z5 and Z6 each is independently hydrogen or fluorine;
R2 is

-
- wherein V, if present, is O, S, or NR6,
- wherein any U is CH or N.
- wherein M, Q, W, X, and Y are independently hydrogen, fluorine, chlorine, bromine, iodine, hydroxy, methoxy, trifluoromethoxy, cyano, trifluoromethyl, (C1-C6)-alkyl, (C3-C6)-cycloalkyl, (C2-C6)-alkenyl, or (C2-C6)-alkynyl wherein any alkyl, cycloalkyl, alkenyl, or alkynyl is optionally substituted with one or more fluorine atoms, and
- wherein R6 is hydrogen, (C1-C6)-alkyl, (C3-C6)-cycloalkyl, (C2-C6)-alkenyl, (C2-C6)-alkynyl, phenyl, benzyl, three- to six-membered heterocycle, or five- to six-membered heteroaryl.




or an equivalent thereof, or a hydrate, solvate, or a pharmaceutically acceptable salt thereof.

or an equivalent thereof, or a hydrate, solvate, or a pharmaceutically acceptable salt thereof.

or an equivalent thereof, or a hydrate, solvate, or a pharmaceutically acceptable salt thereof.

or an equivalent thereof, or a hydrate, solvate, or a pharmaceutically acceptable salt thereof.

























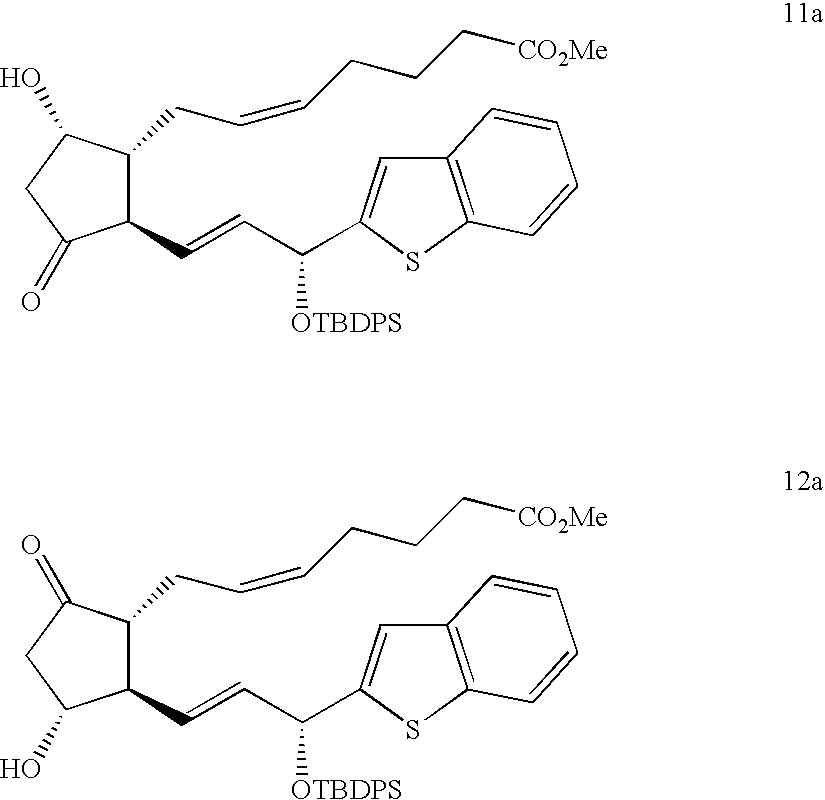























































































K i =IC 50/[1+(radioligand concentration/radioligand K d)]
Functional Assays Effect of Compounds on Second Messenger Generation
Claims (14)







Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/271,764 US8063240B2 (en) | 2007-11-14 | 2008-11-14 | Prostaglandin E1 and E2 analogs for the treatment of various medical conditions |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US98785907P | 2007-11-14 | 2007-11-14 | |
| US3749308P | 2008-03-18 | 2008-03-18 | |
| US12/271,764 US8063240B2 (en) | 2007-11-14 | 2008-11-14 | Prostaglandin E1 and E2 analogs for the treatment of various medical conditions |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| US20090221654A1 US20090221654A1 (en) | 2009-09-03 |
| US8063240B2 true US8063240B2 (en) | 2011-11-22 |
Family
ID=40624365
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/271,764 Expired - Fee Related US8063240B2 (en) | 2007-11-14 | 2008-11-14 | Prostaglandin E1 and E2 analogs for the treatment of various medical conditions |
| US12/271,798 Abandoned US20090124695A1 (en) | 2007-11-14 | 2008-11-14 | Prostaglandin e1 and e2 analogs for the treatment of various medical conditions |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/271,798 Abandoned US20090124695A1 (en) | 2007-11-14 | 2008-11-14 | Prostaglandin e1 and e2 analogs for the treatment of various medical conditions |
Country Status (3)
| Country | Link |
|---|---|
| US (2) | US8063240B2 (en) |
| EP (1) | EP2220057A4 (en) |
| WO (2) | WO2009065081A2 (en) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112645861A (en) * | 2020-12-21 | 2021-04-13 | 上海彩迩文生化科技有限公司 | Method for separating carboprost 15-position isomer |
| CN112979599A (en) * | 2021-03-01 | 2021-06-18 | 河北赛谱睿思医药科技有限公司 | Preparation method of carboprost tromethamine intermediate |
Citations (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3886206A (en) | 1972-09-08 | 1975-05-27 | Syntex Inc | 10,11-Methylene-substituted phostaglandin derivatives |
| US4138590A (en) | 1977-01-13 | 1979-02-06 | American Home Products Corporation | Prostaglandin derivatives |
| US5576347A (en) | 1994-11-14 | 1996-11-19 | Sredni; Benjamin | Method of treating gastric ulcers |
| WO1998034916A1 (en) | 1997-02-10 | 1998-08-13 | Ono Pharmaceutical Co., Ltd. | 11,15-o-dialkylprostaglandin e derivatives, process for producing the same, and drugs containing the same as the active ingredient |
| WO2000031084A1 (en) | 1998-11-20 | 2000-06-02 | Merck Frosst Canada & Co. | Prostaglandin conjugates for treating or preventing bone disease |
| WO2000040248A1 (en) | 1999-01-08 | 2000-07-13 | Synphora Ab | Method and composition for treatment of female sexual dysfunction |
| WO2001046140A1 (en) | 1999-12-22 | 2001-06-28 | Pfizer Products Inc. | Ep4 receptor selective agonists in the treatment of osteoporosis |
| US6426359B1 (en) | 1996-12-20 | 2002-07-30 | Pfizer Inc. | Prevention and treatment of skeletal disorder with EP2 receptor subtype selective prostaglandin E2 agonists |
| US6476064B1 (en) * | 2001-06-13 | 2002-11-05 | Allergan, Inc. | Cyclopentane heptan(ene) acyl sulfonamide, 2-alkyl or 2-arylalkyl, or 2-heteroarylalkenyl derivatives as therapeutic agents |
| US20020177620A1 (en) | 2001-05-17 | 2002-11-28 | Allergan Sales, Inc. | Prostanoic acid derivatives as agents for lowering intraocular pressure |
| WO2003037433A1 (en) | 2001-10-26 | 2003-05-08 | Allergan, Inc. | Co-cycloalkyl 17-heteroaryl prostaglandin e2 analogs as ep2-receptor agonists |
| WO2003040126A1 (en) | 2001-11-05 | 2003-05-15 | Allergan, Inc. | φ-CYCLOALKYL 17-HETEROARYL PROSTAGLANDIN E2 ANALOGS AS EP2-RECEPTOR AGONISTS |
| US6894175B1 (en) | 1999-08-04 | 2005-05-17 | The Procter & Gamble Company | 2-Decarboxy-2-phosphinico prostaglandin derivatives and methods for their preparation and use |
| WO2007027468A1 (en) | 2005-08-29 | 2007-03-08 | Allergan, Inc. | Ep2 receptor agonists for treating glaucoma |
| US20070112067A1 (en) * | 1999-03-01 | 2007-05-17 | Nitromed, Inc. | Nitrosated and nitrosylated prostaglandins, compositions and methods of use |
| US7326732B2 (en) | 2004-02-12 | 2008-02-05 | Pharmagene Laboratories Limited | EP2 receptor agonists |
| WO2008073748A1 (en) | 2006-12-08 | 2008-06-19 | University Of Rochester | Expansion of hematopoietic stem cells |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4011262A (en) * | 1972-07-13 | 1977-03-08 | Pfizer Inc. | 13,14-Dihydro-15-substituted-ω-pentanorprostaglandins of the two series |
| US3982016A (en) * | 1975-08-06 | 1976-09-21 | Pfizer Inc. | Bone deposition by 16-aryl-13,14-dihydro-PGE2 p-biphenyl esters |
-
2008
- 2008-11-14 EP EP08850884A patent/EP2220057A4/en not_active Withdrawn
- 2008-11-14 WO PCT/US2008/083694 patent/WO2009065081A2/en not_active Ceased
- 2008-11-14 US US12/271,764 patent/US8063240B2/en not_active Expired - Fee Related
- 2008-11-14 US US12/271,798 patent/US20090124695A1/en not_active Abandoned
- 2008-11-14 WO PCT/US2008/083679 patent/WO2009065070A2/en not_active Ceased
Patent Citations (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3886206A (en) | 1972-09-08 | 1975-05-27 | Syntex Inc | 10,11-Methylene-substituted phostaglandin derivatives |
| US4138590A (en) | 1977-01-13 | 1979-02-06 | American Home Products Corporation | Prostaglandin derivatives |
| US5576347A (en) | 1994-11-14 | 1996-11-19 | Sredni; Benjamin | Method of treating gastric ulcers |
| US6426359B1 (en) | 1996-12-20 | 2002-07-30 | Pfizer Inc. | Prevention and treatment of skeletal disorder with EP2 receptor subtype selective prostaglandin E2 agonists |
| WO1998034916A1 (en) | 1997-02-10 | 1998-08-13 | Ono Pharmaceutical Co., Ltd. | 11,15-o-dialkylprostaglandin e derivatives, process for producing the same, and drugs containing the same as the active ingredient |
| WO2000031084A1 (en) | 1998-11-20 | 2000-06-02 | Merck Frosst Canada & Co. | Prostaglandin conjugates for treating or preventing bone disease |
| WO2000040248A1 (en) | 1999-01-08 | 2000-07-13 | Synphora Ab | Method and composition for treatment of female sexual dysfunction |
| US6562868B1 (en) | 1999-01-08 | 2003-05-13 | Synphora Ab | Method for treatment of female sexual dysfunction |
| US20070112067A1 (en) * | 1999-03-01 | 2007-05-17 | Nitromed, Inc. | Nitrosated and nitrosylated prostaglandins, compositions and methods of use |
| US6894175B1 (en) | 1999-08-04 | 2005-05-17 | The Procter & Gamble Company | 2-Decarboxy-2-phosphinico prostaglandin derivatives and methods for their preparation and use |
| WO2001046140A1 (en) | 1999-12-22 | 2001-06-28 | Pfizer Products Inc. | Ep4 receptor selective agonists in the treatment of osteoporosis |
| US20020177620A1 (en) | 2001-05-17 | 2002-11-28 | Allergan Sales, Inc. | Prostanoic acid derivatives as agents for lowering intraocular pressure |
| US6476064B1 (en) * | 2001-06-13 | 2002-11-05 | Allergan, Inc. | Cyclopentane heptan(ene) acyl sulfonamide, 2-alkyl or 2-arylalkyl, or 2-heteroarylalkenyl derivatives as therapeutic agents |
| WO2003037433A1 (en) | 2001-10-26 | 2003-05-08 | Allergan, Inc. | Co-cycloalkyl 17-heteroaryl prostaglandin e2 analogs as ep2-receptor agonists |
| WO2003040126A1 (en) | 2001-11-05 | 2003-05-15 | Allergan, Inc. | φ-CYCLOALKYL 17-HETEROARYL PROSTAGLANDIN E2 ANALOGS AS EP2-RECEPTOR AGONISTS |
| US7326732B2 (en) | 2004-02-12 | 2008-02-05 | Pharmagene Laboratories Limited | EP2 receptor agonists |
| WO2007027468A1 (en) | 2005-08-29 | 2007-03-08 | Allergan, Inc. | Ep2 receptor agonists for treating glaucoma |
| WO2008073748A1 (en) | 2006-12-08 | 2008-06-19 | University Of Rochester | Expansion of hematopoietic stem cells |
Non-Patent Citations (14)
| Title |
|---|
| "Crystallization" in Kirk-Othmer Encyclopedia of Chemical Technology Copyright © 2002 by John Wiley & Sons, Inc., pp. 95-147, Article Online Posting Date: Aug. 16, 2002. * |
| Asai et al., "Synthesis of New Stable Fluoroprostacyclin Analogs with Potent Anti-Anginal Activity", Tetrahedron Letters, vol. 36, No. 2, (1995), pp. 273-276. |
| Breyer et al., "Structure-Function Analyses of Eicosanoid Receptors", Annals of New York Academy of Sciences, 905, (2000), pp. 221-231. |
| Grieco et al., "Fluoroprostaglandins: Total Synthesis of (+)-13-Fluoroprostaglandin F2alpha Methyl Ester", Journal of Organic Chemistry, 50, (1985), pp. 3111-3115. |
| Klimko et al., "15-Fluoro Prostaglandin FP Agonists: A New Class of Topical Ocular Hypotensives", Bioorganic & Medicinal Chemistry 12 (2004), pp. 3451-3469. |
| Li et al., "A Novel, Non-Prostanoid EP2 Receptor-Selective Prostaglandin E2 Agonist Stimulates Local Bone Formation and Enhances Fracture Healing", Journal of Bone and Mineral Research, vol. 18, No. 11, 2003, pp. 2033-2042. |
| Matsumara et al., "Novel Fluoroprostacyclin Analogs with Modified Cycloalkylenyl Chains. Highly Potent and Orally Active Anti-Aging Agents", Chem. Pharm. Bull 43(2), (1995), pp. 353-355U. |
| Matsumura et al., "Synthesis of 7-Fluoro-2, 4-methylene-17, 20-dimethylprostacyclins. Novel Stable Prostacyclin Analogs as Potent Anti-anginal Agents", Tetrahedron, vol. 51, No. 32, (1995), pp. 8771-8782. |
| Matsumura et al., "Synthesis of the Highly Potent Prostanoid FP Receptor Agonist, AFP-168: a novel 15-deoxy-15, 15-difluoroprostaglandin F2alpha Derivative", Tetrahedron Letters 45 (2004), pp. 1527-1529. |
| North et al., "Prostaglandin E2 Regulates Vertebrate Haematopoietic Stem Cell Homeostasis," Nature, vol. 447, Jun. 21, 2007, pp. 1007-1011. |
| Paralkar et al., "An EP2 Receptor-Selective Prostaglandin E2 Agonist Induces Bone Healing," Proceedings of the National Academy of Sciences, vol. 100, No. 11, May 27, 2003, pp. 6736-6740. |
| Rouhi, "The Right Stuff, from research and development to the clinic, getting drug crystals right is full of pitfalls", Chemical & Engineering News, Feb. 24, 2003, pp. 32-35. * |
| Ullmann's Encyclopedia of Industrial Chemistry, Copyright © 2002 by Wiley-VCH Verlag GmbH & Co. KGaA , pp. 1-51. * |
| Waterbury et al., "EP3 But Not EP2, FP, or TP Prostanoid-Receptor Stimulation May Reduce Intraocular Pressure", Investigative Opthalmology & Visual Science, vol. 31, No. 12, Dec. 1990, pp. 2560-2567. |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2009065081A3 (en) | 2009-07-30 |
| WO2009065070A4 (en) | 2009-10-01 |
| EP2220057A2 (en) | 2010-08-25 |
| WO2009065070A3 (en) | 2009-07-30 |
| US20090124695A1 (en) | 2009-05-14 |
| WO2009065081A2 (en) | 2009-05-22 |
| EP2220057A4 (en) | 2011-10-12 |
| WO2009065070A2 (en) | 2009-05-22 |
| US20090221654A1 (en) | 2009-09-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2019825B1 (en) | Inhibitors of human immunodeficiency virus replication | |
| US20100179137A1 (en) | Pyridone compound | |
| KR20090005383A (en) | Inhibitors of C-MFS Kinase | |
| JP2021533101A (en) | Deuterated derivative of lanifibranol | |
| US20230348441A1 (en) | Bicyclic compound that acts as crbn protein regulator | |
| KR20140061475A (en) | EP1 receptor ligand | |
| WO2023084158A1 (en) | Cyp11a1 inhibitors | |
| US11357755B2 (en) | Compounds for controlling arthropods | |
| KR20080072688A (en) | Oxadiazole derivatives | |
| US8063240B2 (en) | Prostaglandin E1 and E2 analogs for the treatment of various medical conditions | |
| EP2414367A1 (en) | Prostaglandin e receptor antagonists | |
| JPH044312B2 (en) | ||
| US20080200490A1 (en) | Alpha-(Aryl-or Heteroaryl-Methyl)-Beta-Piperidino Propanamide Compounds as Orl-1-Receptor Antagonists | |
| EP0284202B1 (en) | Substituted hydantoin compounds | |
| CN115160259A (en) | Roxatidine or its acetate related substances, preparation method and detection method | |
| KR910000443B1 (en) | Process for preparing haloalkylthiazol and its products | |
| US20080207665A1 (en) | Alpha-(Aryl-or Heteroaryl-Methyl)-Beta-Piperidinopropanoic Acid Compounds as Orl-1-Receptor Antagonists | |
| JP2007522181A (en) | Substituted azetidine compounds as cyclooxygenase-1 and cyclooxygenase-2 inhibitors, and their preparation and use as pharmaceuticals | |
| HK40110761B (en) | Therapeutic uses of deuterated derivatives of lanifibranor | |
| JP2008512384A (en) | Fibrate compounds having PPAR agonist activity | |
| JPH10511115A (en) | Novel imidazole lipoxygenase inhibitors | |
| US20170362235A1 (en) | Adenylyl cyclase inhibitors, pharmaceutical compositions and method of use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: CAYMAN CHEMICAL COMAPNY, MICHIGAN Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:BARTA, NANCY S.;BARRETT, STEPHEN D.;ENDRES, GERGORY W.;AND OTHERS;REEL/FRAME:022156/0180 Effective date: 20081113 |
|
| AS | Assignment |
Owner name: BROADOAK CAPITAL PARTNERS, LLC, AS AGENT, MARYLAND Free format text: SECURITY AGREEMENT;ASSIGNOR:CAYMAN CHEMICAL COMPANY, INCORPORATED;REEL/FRAME:024710/0623 Effective date: 20100714 |
|
| AS | Assignment |
Owner name: CAYMAN CHEMICAL COMPANY, INCORPORATED, MICHIGAN Free format text: CORRECTIVE ASSIGNMENT TO CORRECT THE ASSIGNEE NAME TO CAYMAN CHEMICAL COMPANY, INCORPORATED PREVIOUSLY RECORDED ON REEL 022156 FRAME 180. ASSIGNOR(S) HEREBY CONFIRMS THE ASSIGNMENT;ASSIGNORS:BARTA, NANCY S.;BARRETT, STEPHEN D.;ENDRES, GREGORY W.;AND OTHERS;REEL/FRAME:026436/0720 Effective date: 20081113 |
|
| AS | Assignment |
Owner name: CAYMAN CHEMICAL COMPANY, MICHIGAN Free format text: RELEASE BY SECURED PARTY;ASSIGNOR:BROADOAK CAPITAL PARTNERS, LLC;REEL/FRAME:026574/0808 Effective date: 20110705 |
|
| STCF | Information on status: patent grant |
Free format text: PATENTED CASE |
|
| CC | Certificate of correction | ||
| FPAY | Fee payment |
Year of fee payment: 4 |
|
| FEPP | Fee payment procedure |
Free format text: MAINTENANCE FEE REMINDER MAILED (ORIGINAL EVENT CODE: REM.); ENTITY STATUS OF PATENT OWNER: SMALL ENTITY |
|
| LAPS | Lapse for failure to pay maintenance fees |
Free format text: PATENT EXPIRED FOR FAILURE TO PAY MAINTENANCE FEES (ORIGINAL EVENT CODE: EXP.); ENTITY STATUS OF PATENT OWNER: SMALL ENTITY |
|
| STCH | Information on status: patent discontinuation |
Free format text: PATENT EXPIRED DUE TO NONPAYMENT OF MAINTENANCE FEES UNDER 37 CFR 1.362 |
|
| FP | Lapsed due to failure to pay maintenance fee |
Effective date: 20191122 |