US7727981B2 - Benzamides and related inhibitors of factor Xa - Google Patents
Benzamides and related inhibitors of factor Xa Download PDFInfo
- Publication number
- US7727981B2 US7727981B2 US11/924,480 US92448007A US7727981B2 US 7727981 B2 US7727981 B2 US 7727981B2 US 92448007 A US92448007 A US 92448007A US 7727981 B2 US7727981 B2 US 7727981B2
- Authority
- US
- United States
- Prior art keywords
- group
- alkyl
- member selected
- och
- phenyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 [*+]C(c(cc1)ccc1C(CCCc(c(*)cc(Cl)c1)c1C(C(C1)N1C(C=CC(*)=C1)=I1=C)=Cl)=O)=N Chemical compound [*+]C(c(cc1)ccc1C(CCCc(c(*)cc(Cl)c1)c1C(C(C1)N1C(C=CC(*)=C1)=I1=C)=Cl)=O)=N 0.000 description 111
- YGQPFQDPVUTEHL-WVXUZJBCSA-N C/N=C(\C)N1CC1.C/N=C(\C)N1CCC1.CC(=N)N.CC(=N)N(C)C.CC(=N)N(C)C.CC(=N)N1CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCCC1.CC1=NCCN1C.CCN(C)C(C)=N Chemical compound C/N=C(\C)N1CC1.C/N=C(\C)N1CCC1.CC(=N)N.CC(=N)N(C)C.CC(=N)N(C)C.CC(=N)N1CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCCC1.CC1=NCCN1C.CCN(C)C(C)=N YGQPFQDPVUTEHL-WVXUZJBCSA-N 0.000 description 8
- ONWKVBBKBHITDI-UHFFFAOYSA-N CC(=N)N.CC(=N)N(C)C.CC(=N)N1CCCC1.CC(=N)N1CCCCC1.CC(=O)N1CCCCC1.CC(=O)N1CCN(C)CC1.CC(=O)N1CCN(C)CC1.CC(=O)N1CCNCC1.CC(=O)N1CCOCC1.CC(=O)N1CCS(=O)(=O)CC1.CC(=O)N1CCS(=O)CC1.CC(=O)N1CCSCC1.CC1=NCCC1.CC1=NCCN1C.CC1=NCCN1C.CC1=NCCO1.CC1=NCCS1.CCNC1=NCCN1.CCNC1=NCCN1.CCOC(=O)C1CCN(C)CC1.CN1CCC(C(=O)O)CC1.CN1CCC(C(N)=O)CC1.CN1CCC(N)CC1.CN1CCC(O)CC1.CN1CCCC1.CN1CCCCC1.CN1CCCCCC1.CN1CCCN(C)CC1.CN1CCN(C(=O)OC(C)(C)C)CC1.CN1CCN(C)CC1.CN1CCN(S(C)(=O)=O)CC1.CN1CCNCC1.CN1CCOCC1.CN1CCSCC1.CNC(=N)N.CNC(=N)N(C)C.CNC(=N)NC.CNC(C)=N.CNC1=NCCN1.CNC1=NCCN1C Chemical compound CC(=N)N.CC(=N)N(C)C.CC(=N)N1CCCC1.CC(=N)N1CCCCC1.CC(=O)N1CCCCC1.CC(=O)N1CCN(C)CC1.CC(=O)N1CCN(C)CC1.CC(=O)N1CCNCC1.CC(=O)N1CCOCC1.CC(=O)N1CCS(=O)(=O)CC1.CC(=O)N1CCS(=O)CC1.CC(=O)N1CCSCC1.CC1=NCCC1.CC1=NCCN1C.CC1=NCCN1C.CC1=NCCO1.CC1=NCCS1.CCNC1=NCCN1.CCNC1=NCCN1.CCOC(=O)C1CCN(C)CC1.CN1CCC(C(=O)O)CC1.CN1CCC(C(N)=O)CC1.CN1CCC(N)CC1.CN1CCC(O)CC1.CN1CCCC1.CN1CCCCC1.CN1CCCCCC1.CN1CCCN(C)CC1.CN1CCN(C(=O)OC(C)(C)C)CC1.CN1CCN(C)CC1.CN1CCN(S(C)(=O)=O)CC1.CN1CCNCC1.CN1CCOCC1.CN1CCSCC1.CNC(=N)N.CNC(=N)N(C)C.CNC(=N)NC.CNC(C)=N.CNC1=NCCN1.CNC1=NCCN1C ONWKVBBKBHITDI-UHFFFAOYSA-N 0.000 description 7
- OHTQMBOMIVODOY-UHFFFAOYSA-N CC1=C(CN(C)C)C=C(F)C=C1.CC1=C(CN(C)C)C=N(O)C=C1.CC1=CC=NC(CN(C)C)=N1.CC1=CC=NC(N)=N1.CC1=CC=NC=N1.COC1CCN(C(C)=N)C1.COC1CCN(C(C)=N)CC1.COCCNC1=NC(C)=CC=N1 Chemical compound CC1=C(CN(C)C)C=C(F)C=C1.CC1=C(CN(C)C)C=N(O)C=C1.CC1=CC=NC(CN(C)C)=N1.CC1=CC=NC(N)=N1.CC1=CC=NC=N1.COC1CCN(C(C)=N)C1.COC1CCN(C(C)=N)CC1.COCCNC1=NC(C)=CC=N1 OHTQMBOMIVODOY-UHFFFAOYSA-N 0.000 description 6
- ZJDLURIRFXRZFH-GDEFNOCQSA-N C/C(=N/C#N)N(C)C.CC1=CC=CC=C1.CC1=CC=CS1.CN(C)CCN(C)/C(=N/C#N)N(C)C.CN(C)CCN1CCCC1.CN(C)CCN1CCCCC1.CN(C)CCN1CCOCC1.CN(C)CCN1CCS(=O)(=O)CC1.CNCCN(C)C1=NCCN1.CNCCN(C)C1=NCCN1C.CNCCNC1=NCCN1.COCCN.COCCN(C)C(=N)N.COCCN/C(N)=N/C#N.COCCN/C(N)=N/[N+](=O)[O-].COCCNC(=N)N.COCCNC(=N)N(C)C Chemical compound C/C(=N/C#N)N(C)C.CC1=CC=CC=C1.CC1=CC=CS1.CN(C)CCN(C)/C(=N/C#N)N(C)C.CN(C)CCN1CCCC1.CN(C)CCN1CCCCC1.CN(C)CCN1CCOCC1.CN(C)CCN1CCS(=O)(=O)CC1.CNCCN(C)C1=NCCN1.CNCCN(C)C1=NCCN1C.CNCCNC1=NCCN1.COCCN.COCCN(C)C(=N)N.COCCN/C(N)=N/C#N.COCCN/C(N)=N/[N+](=O)[O-].COCCNC(=N)N.COCCNC(=N)N(C)C ZJDLURIRFXRZFH-GDEFNOCQSA-N 0.000 description 5
- URALKZJOFBLYKY-UHFFFAOYSA-N CC(=O)N(C)CCN(C)C.CC(=O)N(C)CCS(C)(=O)=O.CC(=O)NCCN(C)C.CC(=O)NCCS(C)(=O)=O.CNC(=N)NCCOC.CNCCN(C)C(C)=O.CNCCNC(C)=O.COCCC(=N)N(C)C.COCCC1=NCCN1C.COCCCN1C=CN=C1.COCCCN1C=NN=N1.COCCCN1CCCC1.COCCCN1CCCCC1.COCCCN1CCN(C)CC1.COCCCN1CCNCC1.COCCCN1CCOCC1.COCCN(C)C.COCCN(C)C(C)=O.COCCN1C=CN=C1.COCCN1C=NN=N1.COCCN1CCCC1.COCCN1CCCCC1.COCCN1CCN(C)CC1.COCCN1CCOCC1.COCCNC(=N)C1=CC=CC=C1.COCCNC(=N)C1CC1.COCCNC(C)=O.COCCNC1=NCCN1C Chemical compound CC(=O)N(C)CCN(C)C.CC(=O)N(C)CCS(C)(=O)=O.CC(=O)NCCN(C)C.CC(=O)NCCS(C)(=O)=O.CNC(=N)NCCOC.CNCCN(C)C(C)=O.CNCCNC(C)=O.COCCC(=N)N(C)C.COCCC1=NCCN1C.COCCCN1C=CN=C1.COCCCN1C=NN=N1.COCCCN1CCCC1.COCCCN1CCCCC1.COCCCN1CCN(C)CC1.COCCCN1CCNCC1.COCCCN1CCOCC1.COCCN(C)C.COCCN(C)C(C)=O.COCCN1C=CN=C1.COCCN1C=NN=N1.COCCN1CCCC1.COCCN1CCCCC1.COCCN1CCN(C)CC1.COCCN1CCOCC1.COCCNC(=N)C1=CC=CC=C1.COCCNC(=N)C1CC1.COCCNC(C)=O.COCCNC1=NCCN1C URALKZJOFBLYKY-UHFFFAOYSA-N 0.000 description 5
- RMYVTIBCGUEFQD-UHFFFAOYSA-N CC.CC(=O)N(C)CCN(C)C.CC(=O)N1CCCCC1.CC1=NCCC1.CC1=NCCN1.CCNC(=N)N.CNC1=NCCC1.COCCN(C)C(C)=O.NCCN1CCOCC1 Chemical compound CC.CC(=O)N(C)CCN(C)C.CC(=O)N1CCCCC1.CC1=NCCC1.CC1=NCCN1.CCNC(=N)N.CNC1=NCCC1.COCCN(C)C(C)=O.NCCN1CCOCC1 RMYVTIBCGUEFQD-UHFFFAOYSA-N 0.000 description 4
- ALLSQHSRMVUDKR-LXKVIBFUSA-N C/N=C(\C)N(C)C.C/N=C(\C)NC.CC(=N)N.CC(=N)N(C)C.CC(=N)N1CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCCC1.CC(=N)N1CCCN(C)CC1.CC(=N)N1CCN(C)CC1.CC(=N)N1CCNCC1.CC(=N)N1CCOCC1.CC(=N)N1CCSCC1.CC(=N)NN.CC(=N)NO.CC1=CC=NC=C1.CC1=CC=[N+]([O-])C=C1.CC1=NCCCN1C.CC1=NCCN1C.CCCCN(C)CCN.CCN1CCN=C1C.CNNC(C)=N.CONC(C)=N Chemical compound C/N=C(\C)N(C)C.C/N=C(\C)NC.CC(=N)N.CC(=N)N(C)C.CC(=N)N1CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCCC1.CC(=N)N1CCCN(C)CC1.CC(=N)N1CCN(C)CC1.CC(=N)N1CCNCC1.CC(=N)N1CCOCC1.CC(=N)N1CCSCC1.CC(=N)NN.CC(=N)NO.CC1=CC=NC=C1.CC1=CC=[N+]([O-])C=C1.CC1=NCCCN1C.CC1=NCCN1C.CCCCN(C)CCN.CCN1CCN=C1C.CNNC(C)=N.CONC(C)=N ALLSQHSRMVUDKR-LXKVIBFUSA-N 0.000 description 3
- AYXLOLFFWZGKPP-UHFFFAOYSA-N CC1=C(CN(C)C)C=C(F)C=C1.CC1=C(CN(C)C)C=[N+](O)C=C1.CC1=CC=NC(CN(C)C)=N1.CC1=CC=NC(N)=N1.CC1=CC=NC=N1.COC1CCC(C(C)=N)C1.COC1CCN(C(C)=N)CC1.COCCNC1=NC(C)=CC=N1 Chemical compound CC1=C(CN(C)C)C=C(F)C=C1.CC1=C(CN(C)C)C=[N+](O)C=C1.CC1=CC=NC(CN(C)C)=N1.CC1=CC=NC(N)=N1.CC1=CC=NC=N1.COC1CCC(C(C)=N)C1.COC1CCN(C(C)=N)CC1.COCCNC1=NC(C)=CC=N1 AYXLOLFFWZGKPP-UHFFFAOYSA-N 0.000 description 3
- UHRBYYHNHCLWCP-UHFFFAOYSA-N CC1=C(CN(C)C)C=C(F)C=C1.CC1=C(CN(C)C)C=[N+](O)C=C1.CC1=CC=NC(CN(C)C)=N1.CC1=CC=NC(N)=N1.CC1=CC=NC=N1.COC1CCN(C(C)=N)C1.COC1CCN(C(C)=N)CC1.COCCNC1=NC(C)=CC=N1 Chemical compound CC1=C(CN(C)C)C=C(F)C=C1.CC1=C(CN(C)C)C=[N+](O)C=C1.CC1=CC=NC(CN(C)C)=N1.CC1=CC=NC(N)=N1.CC1=CC=NC=N1.COC1CCN(C(C)=N)C1.COC1CCN(C(C)=N)CC1.COCCNC1=NC(C)=CC=N1 UHRBYYHNHCLWCP-UHFFFAOYSA-N 0.000 description 3
- HOMJMSHONGIVIN-UHFFFAOYSA-N NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=NC=C(Cl)C=C4)SC=C3)C=C2)C=CC=C1 Chemical compound NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=NC=C(Cl)C=C4)SC=C3)C=C2)C=CC=C1 HOMJMSHONGIVIN-UHFFFAOYSA-N 0.000 description 3
- IOJGGHPOTAINMB-PFONDFGASA-N [H]/C(C(=O)NC1=CC=C(OC)C=C1)=C(\[H])C(=O)NC1=CC=C(C2=CC=CC=C2S(N)(=O)=O)C=C1 Chemical compound [H]/C(C(=O)NC1=CC=C(OC)C=C1)=C(\[H])C(=O)NC1=CC=C(C2=CC=CC=C2S(N)(=O)=O)C=C1 IOJGGHPOTAINMB-PFONDFGASA-N 0.000 description 3
- MCRFADJTQBNURO-KNMRYIDXSA-N C/N=C(\C)N(C)C.CC(=N)N.CC(=N)N(C)C.CC(=N)N1CC1.CC(=N)N1CCC(C)CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCCC1.CC(=N)NO.CC1=CC=C(F)C=C1S(C)(=O)=O.CC1=CC=C(F)C=C1S(N)(=O)=O.CC1=CC=CC=C1C(=N)N.CC1=CC=CC=C1C(=N)N(C)C.CC1=CC=CC=C1CN(C)C.CC1=CC=CC=C1S(C)(=O)=O.CC1=CC=CC=C1S(N)(=O)=O.CC1=CC=NC(N(C)C)=C1.CC1=CC=NC(N)=C1.CC1=CC=NC(N)=C1.CC1=CC=NC=C1.CC1=CC=NC=C1S(C)(=O)=O.CC1=CC=NC=C1S(N)(=O)=O.CC1=CC=[N+]([O-])C=C1.CC1=NCCN1C.CC1CCN(C(=N)N)CC1.CC1CCN(C)CC1.CCN(C)C.CCN1CCCC1.CC[N+](C)(C)C.CN1CCCN(C)CC1.CN1CCN(C)CC1.CNC(C)=N Chemical compound C/N=C(\C)N(C)C.CC(=N)N.CC(=N)N(C)C.CC(=N)N1CC1.CC(=N)N1CCC(C)CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCCC1.CC(=N)NO.CC1=CC=C(F)C=C1S(C)(=O)=O.CC1=CC=C(F)C=C1S(N)(=O)=O.CC1=CC=CC=C1C(=N)N.CC1=CC=CC=C1C(=N)N(C)C.CC1=CC=CC=C1CN(C)C.CC1=CC=CC=C1S(C)(=O)=O.CC1=CC=CC=C1S(N)(=O)=O.CC1=CC=NC(N(C)C)=C1.CC1=CC=NC(N)=C1.CC1=CC=NC(N)=C1.CC1=CC=NC=C1.CC1=CC=NC=C1S(C)(=O)=O.CC1=CC=NC=C1S(N)(=O)=O.CC1=CC=[N+]([O-])C=C1.CC1=NCCN1C.CC1CCN(C(=N)N)CC1.CC1CCN(C)CC1.CCN(C)C.CCN1CCCC1.CC[N+](C)(C)C.CN1CCCN(C)CC1.CN1CCN(C)CC1.CNC(C)=N MCRFADJTQBNURO-KNMRYIDXSA-N 0.000 description 2
- NMXKBXCHEVAUOV-DIEZXOIBSA-N C/N=C(\C)N(C)C.CC(=N)N.CC(=N)N(C)C.CC(=N)N1CC1.CC(=N)N1CCC(C)CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCCC1.CC(=N)NO.CC1=CC=C(F)C=C1S(C)(=O)=O.CC1=CC=C(F)C=C1S(N)(=O)=O.CC1=CC=CC=C1C(=N)N.CC1=CC=CC=C1C(=N)N(C)C.CC1=CC=CC=C1S(=O)(=O)N(C)C.CC1=CC=CC=C1S(C)(=O)=O.CC1=CC=CC=C1S(N)(=O)=O.CC1=CC=NC(N)=C1.CC1=CC=NC=C1.CC1=CC=NC=C1S(C)(=O)=O.CC1=CC=NC=C1S(N)(=O)=O.CC1=CC=[N+](O)C=C1.CC1=NCCN1C.CC1CCN(C(=N)N)CC1.CC1CCN(C)CC1.CCN(C)C.CCN1CCCC1.CC[N-](C)(C)C.CN1CCCN(C)CC1.CN1CCN(C)CC1.CNC(C)=N Chemical compound C/N=C(\C)N(C)C.CC(=N)N.CC(=N)N(C)C.CC(=N)N1CC1.CC(=N)N1CCC(C)CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCCC1.CC(=N)NO.CC1=CC=C(F)C=C1S(C)(=O)=O.CC1=CC=C(F)C=C1S(N)(=O)=O.CC1=CC=CC=C1C(=N)N.CC1=CC=CC=C1C(=N)N(C)C.CC1=CC=CC=C1S(=O)(=O)N(C)C.CC1=CC=CC=C1S(C)(=O)=O.CC1=CC=CC=C1S(N)(=O)=O.CC1=CC=NC(N)=C1.CC1=CC=NC=C1.CC1=CC=NC=C1S(C)(=O)=O.CC1=CC=NC=C1S(N)(=O)=O.CC1=CC=[N+](O)C=C1.CC1=NCCN1C.CC1CCN(C(=N)N)CC1.CC1CCN(C)CC1.CCN(C)C.CCN1CCCC1.CC[N-](C)(C)C.CN1CCCN(C)CC1.CN1CCN(C)CC1.CNC(C)=N NMXKBXCHEVAUOV-DIEZXOIBSA-N 0.000 description 2
- YEXVWUPFTYJVLM-UHFFFAOYSA-N C1=CC=C(CN2CCOCC2)C=C1.CC1=C(C#N)C=CC=C1.CC1=C(C(=N)N(C)C)C=CC=C1.CC1=C(C(=N)N)C=CC=C1.CC1=C(C(=O)N(C)C)C=CC=C1.CC1=C(C(N)=O)C=CC=C1.CC1=C(CN(C)C)C=CC=C1.CC1=C(CN)C=CC=C1.CC1=C(CN2CCCC2)C=CC=C1.CC1=C(CN2CCCCC2)C=CC=C1.CC1=C(S(C)(=O)=O)C=C(F)C=C1.CC1=C(S(C)(=O)=O)C=CC=C1.CC1=C(S(C)(=O)=O)C=NC=C1.CC1=C(S(N)(=O)=O)C=CC=C1.CC1=CC(C#N)=NC=C1.CC1=CC(C(N)=O)=CC=C1.CC1=CC(C(N)=O)=NC=C1.CC1=CC(CN(C)C)=CC=C1.CC1=CC(CN)=CC=C1.CC1=CC(CN)=NC=C1.CC1=CC(CN)=NC=N1.CC1=CC(N)=NC=C1.CC1=CC(N)=NC=N1.CC1=CC(NN(C)C)=NC=C1.CC1=CC(O)=NC=C1.CC1=CC=NC(CN)=N1.CC1=CC=NC(N)=N1.CC1=CC=NC=C1 Chemical compound C1=CC=C(CN2CCOCC2)C=C1.CC1=C(C#N)C=CC=C1.CC1=C(C(=N)N(C)C)C=CC=C1.CC1=C(C(=N)N)C=CC=C1.CC1=C(C(=O)N(C)C)C=CC=C1.CC1=C(C(N)=O)C=CC=C1.CC1=C(CN(C)C)C=CC=C1.CC1=C(CN)C=CC=C1.CC1=C(CN2CCCC2)C=CC=C1.CC1=C(CN2CCCCC2)C=CC=C1.CC1=C(S(C)(=O)=O)C=C(F)C=C1.CC1=C(S(C)(=O)=O)C=CC=C1.CC1=C(S(C)(=O)=O)C=NC=C1.CC1=C(S(N)(=O)=O)C=CC=C1.CC1=CC(C#N)=NC=C1.CC1=CC(C(N)=O)=CC=C1.CC1=CC(C(N)=O)=NC=C1.CC1=CC(CN(C)C)=CC=C1.CC1=CC(CN)=CC=C1.CC1=CC(CN)=NC=C1.CC1=CC(CN)=NC=N1.CC1=CC(N)=NC=C1.CC1=CC(N)=NC=N1.CC1=CC(NN(C)C)=NC=C1.CC1=CC(O)=NC=C1.CC1=CC=NC(CN)=N1.CC1=CC=NC(N)=N1.CC1=CC=NC=C1 YEXVWUPFTYJVLM-UHFFFAOYSA-N 0.000 description 2
- LGIMPUJXZYFQIQ-UHFFFAOYSA-N CC1=C(CN(C)C)C=C(F)C=C1.CC1=C(CN(C)C)C=[N+]([O-])C=C1.CC1=CC=N(=O)C(CN(C)C)=C1.CC1=CC=NC(CN(C)C)=C1.CC1=CC=NC(CN(C)C)=N1.CC1=CC=NC(CN)=C1.CC1=CC=NC(N)=N1.CC1=CC=NC=N1.CCOC1=NC(C)=CC=N1.COC1CCN(C(C)=N)C1.COC1CCN(C(C)=N)CC1.COCCNC1=NC(C)=CC=N1 Chemical compound CC1=C(CN(C)C)C=C(F)C=C1.CC1=C(CN(C)C)C=[N+]([O-])C=C1.CC1=CC=N(=O)C(CN(C)C)=C1.CC1=CC=NC(CN(C)C)=C1.CC1=CC=NC(CN(C)C)=N1.CC1=CC=NC(CN)=C1.CC1=CC=NC(N)=N1.CC1=CC=NC=N1.CCOC1=NC(C)=CC=N1.COC1CCN(C(C)=N)C1.COC1CCN(C(C)=N)CC1.COCCNC1=NC(C)=CC=N1 LGIMPUJXZYFQIQ-UHFFFAOYSA-N 0.000 description 2
- OFIUSIMCKQIRNO-UHFFFAOYSA-N CC1=CC(Br)=C(C)C=C1.CC1=CC(C)=C(C)C=C1.CC1=CC(Cl)=C(C)C=C1.CC1=CC(F)=C(C)C(Cl)=C1.CC1=CC(F)=C(C)C(F)=C1.CC1=CC(F)=C(C)C=C1.CC1=CC(F)=C(C)C=C1F.CC1=CC=C(C)C(F)=C1.CC1=CC=C(C)C=C1.CC1=CC=C(C)N=C1.CC1=CN(C)C(C)=C1.CC1=CN=C(C)N=C1.CC1=CNC(C)=C1.CC1=COC(C)=C1.CC1=CSC(C)=C1.CC1=CSC(C)=C1.CC1=CSC(C)=C1Cl.CC1=CSC(C)=N1.CC1CCC(C)CC1.CC1CCN(C)CC1.CC1CCN(C)CC1.CN1CCN(C)CC1 Chemical compound CC1=CC(Br)=C(C)C=C1.CC1=CC(C)=C(C)C=C1.CC1=CC(Cl)=C(C)C=C1.CC1=CC(F)=C(C)C(Cl)=C1.CC1=CC(F)=C(C)C(F)=C1.CC1=CC(F)=C(C)C=C1.CC1=CC(F)=C(C)C=C1F.CC1=CC=C(C)C(F)=C1.CC1=CC=C(C)C=C1.CC1=CC=C(C)N=C1.CC1=CN(C)C(C)=C1.CC1=CN=C(C)N=C1.CC1=CNC(C)=C1.CC1=COC(C)=C1.CC1=CSC(C)=C1.CC1=CSC(C)=C1.CC1=CSC(C)=C1Cl.CC1=CSC(C)=N1.CC1CCC(C)CC1.CC1CCN(C)CC1.CC1CCN(C)CC1.CN1CCN(C)CC1 OFIUSIMCKQIRNO-UHFFFAOYSA-N 0.000 description 2
- GGOZXFGJUJTKJP-UHFFFAOYSA-N CC1=CC=C(F)C=C1CN(C)C.CC1=CC=NC(CN(C)C)=C1.CC1=CC=NC(CN(C)C)=N1.CC1=CC=NC(CN)=C1.CC1=CC=NC(N(C)C)=C1.CC1=CC=NC(N)=N1.CC1=CC=NC=N1.CC1=CC=[N+](O)C(CN(C)C)=C1.CC1=CC=[N+](O)C=C1CN(C)C.CCOC1=NC(C)=CC=N1.COC1CCN(C(C)=N)C1.COC1CCN(C(C)=N)CC1.COCCNC1=NC(C)=CC=N1 Chemical compound CC1=CC=C(F)C=C1CN(C)C.CC1=CC=NC(CN(C)C)=C1.CC1=CC=NC(CN(C)C)=N1.CC1=CC=NC(CN)=C1.CC1=CC=NC(N(C)C)=C1.CC1=CC=NC(N)=N1.CC1=CC=NC=N1.CC1=CC=[N+](O)C(CN(C)C)=C1.CC1=CC=[N+](O)C=C1CN(C)C.CCOC1=NC(C)=CC=N1.COC1CCN(C(C)=N)C1.COC1CCN(C(C)=N)CC1.COCCNC1=NC(C)=CC=N1 GGOZXFGJUJTKJP-UHFFFAOYSA-N 0.000 description 2
- HBBKWRGGGRGEFP-UHFFFAOYSA-N CC1=CC=CC=C1C(=O)NC1=CC=C(C2=C(S(N)(=O)=O)C=CC=C2)C=C1.CC1=CC=CC=C1NC(=O)C1=CC=C(C2=C(S(N)(=O)=O)C=CC=C2)C=C1 Chemical compound CC1=CC=CC=C1C(=O)NC1=CC=C(C2=C(S(N)(=O)=O)C=CC=C2)C=C1.CC1=CC=CC=C1NC(=O)C1=CC=C(C2=C(S(N)(=O)=O)C=CC=C2)C=C1 HBBKWRGGGRGEFP-UHFFFAOYSA-N 0.000 description 2
- DRBLXJUCFQXZMY-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=NN2C)C=C1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=NN2C)C=C1 DRBLXJUCFQXZMY-UHFFFAOYSA-N 0.000 description 2
- XAMPEEXUUCSGJH-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(F)C=C3)C=CC=C2)C=C1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(F)C=C3)C=CC=C2)C=C1 XAMPEEXUUCSGJH-UHFFFAOYSA-N 0.000 description 2
- NTATYANFBIDWNW-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1 NTATYANFBIDWNW-UHFFFAOYSA-N 0.000 description 2
- LWIFOPXSAAMUJO-UHFFFAOYSA-N COC(=O)C1=C(NC(=O)C2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(C)(=O)=O)C=C3)C=CC=C2)C=CC(Cl)=C1 Chemical compound COC(=O)C1=C(NC(=O)C2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(C)(=O)=O)C=C3)C=CC=C2)C=CC(Cl)=C1 LWIFOPXSAAMUJO-UHFFFAOYSA-N 0.000 description 2
- OGQUMKSYOWTUNA-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(/C(N)=N/O)C=C2)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(/C(N)=N/O)C=C2)C=C1 OGQUMKSYOWTUNA-UHFFFAOYSA-N 0.000 description 2
- LMFPOQCLAWEMKA-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N(C)C)C=C2F)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N(C)C)C=C2F)C=C1 LMFPOQCLAWEMKA-UHFFFAOYSA-N 0.000 description 2
- NKRWSXUHADSJPZ-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCC3)C=C2F)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCC3)C=C2F)C=C1 NKRWSXUHADSJPZ-UHFFFAOYSA-N 0.000 description 2
- ZNZYTQLDGWBOHG-UHFFFAOYSA-N COC1=CC(NC(=O)C2=CC=C(C3=NCCN3C)C=C2)=C(C(=O)NC2=CC=C(Br)C=N2)C=C1OC Chemical compound COC1=CC(NC(=O)C2=CC=C(C3=NCCN3C)C=C2)=C(C(=O)NC2=CC=C(Br)C=N2)C=C1OC ZNZYTQLDGWBOHG-UHFFFAOYSA-N 0.000 description 2
- WADKRYXREGDOAB-UHFFFAOYSA-N CS(=O)(=O)NC1=CC(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)=C(C(=O)NC2=CC=C(Cl)C=N2)C=C1 Chemical compound CS(=O)(=O)NC1=CC(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)=C(C(=O)NC2=CC=C(Cl)C=N2)C=C1 WADKRYXREGDOAB-UHFFFAOYSA-N 0.000 description 2
- MACWTBHYFBJRMY-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1)N1CCCCC1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1)N1CCCCC1 MACWTBHYFBJRMY-UHFFFAOYSA-N 0.000 description 2
- IJMHKZXAQYOMHV-UHFFFAOYSA-N N=C(N)C1=CC=CC(OC2=C(C(=O)NC3=C(F)C=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=C([N+](=O)[O-])C=C2)=C1 Chemical compound N=C(N)C1=CC=CC(OC2=C(C(=O)NC3=C(F)C=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=C([N+](=O)[O-])C=C2)=C1 IJMHKZXAQYOMHV-UHFFFAOYSA-N 0.000 description 2
- OGQRZBLGQNDTOI-UHFFFAOYSA-N N=C(N)C1=CC=CC(OC2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=C(F)C=C2)=C1 Chemical compound N=C(N)C1=CC=CC(OC2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=C(F)C=C2)=C1 OGQRZBLGQNDTOI-UHFFFAOYSA-N 0.000 description 2
- BDJJUYHQEZYGIQ-UHFFFAOYSA-N NC(=O)C1=CC2=CC=CC=C2C=C1OC1=C(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)C=CC=C1 Chemical compound NC(=O)C1=CC2=CC=CC=C2C=C1OC1=C(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)C=CC=C1 BDJJUYHQEZYGIQ-UHFFFAOYSA-N 0.000 description 2
- ODRSBTUNOBGADL-UHFFFAOYSA-N NC1=C(C(=O)NC2=NC=C(Cl)C=C2)C=C(Cl)C=C1 Chemical compound NC1=C(C(=O)NC2=NC=C(Cl)C=C2)C=C(Cl)C=C1 ODRSBTUNOBGADL-UHFFFAOYSA-N 0.000 description 2
- RKZSYQBVECWLRX-UHFFFAOYSA-N NCC1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CC=C2)C=C1 Chemical compound NCC1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CC=C2)C=C1 RKZSYQBVECWLRX-UHFFFAOYSA-N 0.000 description 2
- LJFXEGHUWQRJHE-UHFFFAOYSA-N NCC1=CC=CC(OC2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=CC=C2)=C1 Chemical compound NCC1=CC=CC(OC2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=CC=C2)=C1 LJFXEGHUWQRJHE-UHFFFAOYSA-N 0.000 description 2
- MDOVXAOHKAENNT-UHFFFAOYSA-N NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=N4)C=C(F)C=C3)C=C2)C=CC=C1 Chemical compound NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=N4)C=C(F)C=C3)C=C2)C=CC=C1 MDOVXAOHKAENNT-UHFFFAOYSA-N 0.000 description 2
- FCCVBKGVSLKKQE-UHFFFAOYSA-N NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=N4)C=C(N4C=CN=C4)C=C3)C=C2)C=CC=C1 Chemical compound NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=N4)C=C(N4C=CN=C4)C=C3)C=C2)C=CC=C1 FCCVBKGVSLKKQE-UHFFFAOYSA-N 0.000 description 2
- WPFVTXUNMVSMKY-UHFFFAOYSA-N NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=NC=C(Br)C=C4)SC=C3)C=C2)C=CC=C1 Chemical compound NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=NC=C(Br)C=C4)SC=C3)C=C2)C=CC=C1 WPFVTXUNMVSMKY-UHFFFAOYSA-N 0.000 description 2
- YJNRLVXURAMOIW-UHFFFAOYSA-N NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC([N+](=O)[O-])=C2)C=C1 Chemical compound NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC([N+](=O)[O-])=C2)C=C1 YJNRLVXURAMOIW-UHFFFAOYSA-N 0.000 description 2
- AKTHLSKCYBESTM-UHFFFAOYSA-N NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(OC3=CC4=CC=CC=C4C=C3C(=O)O)C=CC=C2)C=C1 Chemical compound NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(OC3=CC4=CC=CC=C4C=C3C(=O)O)C=CC=C2)C=C1 AKTHLSKCYBESTM-UHFFFAOYSA-N 0.000 description 2
- HORYUBJWSRXLGX-UHFFFAOYSA-N O=C(NC1=C(C(=O)NC2=CC=C(Br)C=N2)C=CC=C1)C1=CC=C(C2=NN=NN2)C=C1 Chemical compound O=C(NC1=C(C(=O)NC2=CC=C(Br)C=N2)C=CC=C1)C1=CC=C(C2=NN=NN2)C=C1 HORYUBJWSRXLGX-UHFFFAOYSA-N 0.000 description 2
- MAUIXWVGNSGLLB-YPKPFQOOSA-N [H]/C(C(=O)NC1=CC=C(Br)C=C1)=C(\[H])C(=O)NC1=CC=C(C2=CC=CC=C2S(N)(=O)=O)C=C1 Chemical compound [H]/C(C(=O)NC1=CC=C(Br)C=C1)=C(\[H])C(=O)NC1=CC=C(C2=CC=CC=C2S(N)(=O)=O)C=C1 MAUIXWVGNSGLLB-YPKPFQOOSA-N 0.000 description 2
- URAMRDIOBUCBLZ-APPBQSGCSA-N [H]/C(C(=O)NC1=CC=C(Br)C=N1)=C(\C)C(=O)NC1=CC=C(C2=CC=CC=C2S(N)(=O)=O)C=C1.[H]/C(C(=O)NC1=CC=C(C2=CC=CC=C2S(N)(=O)=O)C=C1)=C(\C)C(=O)NC1=CC=C(Br)C=N1 Chemical compound [H]/C(C(=O)NC1=CC=C(Br)C=N1)=C(\C)C(=O)NC1=CC=C(C2=CC=CC=C2S(N)(=O)=O)C=C1.[H]/C(C(=O)NC1=CC=C(C2=CC=CC=C2S(N)(=O)=O)C=C1)=C(\C)C(=O)NC1=CC=C(Br)C=N1 URAMRDIOBUCBLZ-APPBQSGCSA-N 0.000 description 2
- BPYUHUHCSMPTNM-UHFFFAOYSA-N C#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#C.C#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(Cl)C=C2)C=C1)N1CCCCC1.NN=Br(Cl)(N=O)=NN=O.NN=Br(Cl)(N=O)=NN=O.[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH] Chemical compound C#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#C.C#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(Cl)C=C2)C=C1)N1CCCCC1.NN=Br(Cl)(N=O)=NN=O.NN=Br(Cl)(N=O)=NN=O.[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH] BPYUHUHCSMPTNM-UHFFFAOYSA-N 0.000 description 1
- DIJVPMVFTTWOBA-UHFFFAOYSA-N C#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCCC1.NN=Cl(F)(Cl)(N=O)=NN=O.NN=Cl(F)(Cl)(N=O)=NN=O.[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH] Chemical compound C#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCCC1.NN=Cl(F)(Cl)(N=O)=NN=O.NN=Cl(F)(Cl)(N=O)=NN=O.[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH] DIJVPMVFTTWOBA-UHFFFAOYSA-N 0.000 description 1
- QIXVHEVMNFWFLI-UHFFFAOYSA-N C#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.C#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Br)C=C2)C=C1)N1CCC1.NN=Br(Cl)(N=O)=NN=O.NN=Cl(F)(Cl)(N=O)=NN=O.[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH] Chemical compound C#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.C#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Br)C=C2)C=C1)N1CCC1.NN=Br(Cl)(N=O)=NN=O.NN=Cl(F)(Cl)(N=O)=NN=O.[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH] QIXVHEVMNFWFLI-UHFFFAOYSA-N 0.000 description 1
- HLAUXBGZESAZQP-UHFFFAOYSA-N C#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(Cl)C=C2)C=C1)N1CCCC1.NN=Br(Cl)(N=O)=NN=O.NN=Br(Cl)(N=O)=NN=O.[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH] Chemical compound C#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(Cl)C=C2)C=C1)N1CCCC1.NN=Br(Cl)(N=O)=NN=O.NN=Br(Cl)(N=O)=NN=O.[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH] HLAUXBGZESAZQP-UHFFFAOYSA-N 0.000 description 1
- HMMLTERUYPFOPE-UHFFFAOYSA-N C#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCC1.N=C(NO)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.NN=Cl(F)(Cl)(N=O)=NN=O.O=NN=Cl(F)(Cl)(N=O)=NN=O.[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH] Chemical compound C#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCC1.N=C(NO)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.NN=Cl(F)(Cl)(N=O)=NN=O.O=NN=Cl(F)(Cl)(N=O)=NN=O.[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH] HMMLTERUYPFOPE-UHFFFAOYSA-N 0.000 description 1
- DYGAHJPZVGEIRO-UHFFFAOYSA-N C#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.C#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#C.CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.CNC(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.NN=Cl(F)(Cl)(N=O)=NN=O.NN=Cl(F)(Cl)(N=O)=NN=O.[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH] Chemical compound C#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.C#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#C.CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.CNC(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.NN=Cl(F)(Cl)(N=O)=NN=O.NN=Cl(F)(Cl)(N=O)=NN=O.[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH] DYGAHJPZVGEIRO-UHFFFAOYSA-N 0.000 description 1
- WBHPIAVFPSIUIJ-UHFFFAOYSA-N C#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(Cl)C=C2)C=C1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(Cl)C=C2)C=C1.NN=Br(Cl)(N=O)=NN=O.NN=Br(Cl)(N=O)=NN=O.[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH] Chemical compound C#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(Cl)C=C2)C=C1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(Cl)C=C2)C=C1.NN=Br(Cl)(N=O)=NN=O.NN=Br(Cl)(N=O)=NN=O.[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH] WBHPIAVFPSIUIJ-UHFFFAOYSA-N 0.000 description 1
- CNSONICXZBFYSZ-UHFFFAOYSA-N C.C.C.C.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=CC=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=CC=C2)C=C1)N1CCCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CC=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CC=C2)C=C1)N1CCCCC1 Chemical compound C.C.C.C.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=CC=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=CC=C2)C=C1)N1CCCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CC=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CC=C2)C=C1)N1CCCCC1 CNSONICXZBFYSZ-UHFFFAOYSA-N 0.000 description 1
- BYQCXSKWWAAAPJ-UHFFFAOYSA-N C.C.C.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2Cl)C=C1)N1CCOCC1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2Cl)C=C1.N=C(NO)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2Cl)C=C1 Chemical compound C.C.C.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2Cl)C=C1)N1CCOCC1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2Cl)C=C1.N=C(NO)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2Cl)C=C1 BYQCXSKWWAAAPJ-UHFFFAOYSA-N 0.000 description 1
- RSYXAKUCDUISJB-UHFFFAOYSA-N C.C.CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Br)C=C2)C=C1.CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Br)C=C2)C=C1 Chemical compound C.C.CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Br)C=C2)C=C1.CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Br)C=C2)C=C1 RSYXAKUCDUISJB-UHFFFAOYSA-N 0.000 description 1
- CMMTVLQTEIVECZ-UHFFFAOYSA-N C.C.CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCCC1 Chemical compound C.C.CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCCC1 CMMTVLQTEIVECZ-UHFFFAOYSA-N 0.000 description 1
- PGSOIOPLWBSEOK-UHFFFAOYSA-N C.C.CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2Cl)C=C1.CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2Cl)C=C1 Chemical compound C.C.CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2Cl)C=C1.CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2Cl)C=C1 PGSOIOPLWBSEOK-UHFFFAOYSA-N 0.000 description 1
- JVZFVEPBKPMYAV-UHFFFAOYSA-N C.C.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Br)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCSCC1 Chemical compound C.C.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Br)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCSCC1 JVZFVEPBKPMYAV-UHFFFAOYSA-N 0.000 description 1
- MMVHSWGRHVFIGX-UHFFFAOYSA-N C.C.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCC1 Chemical compound C.C.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCC1 MMVHSWGRHVFIGX-UHFFFAOYSA-N 0.000 description 1
- MABRCBTYGSZFDU-UHFFFAOYSA-N C.C.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2Cl)C=C1.N=C(NO)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Br)C=C2)C=C1 Chemical compound C.C.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2Cl)C=C1.N=C(NO)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Br)C=C2)C=C1 MABRCBTYGSZFDU-UHFFFAOYSA-N 0.000 description 1
- NBGXWAIBNZCMFT-UHFFFAOYSA-N C.C.CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1 Chemical compound C.C.CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1 NBGXWAIBNZCMFT-UHFFFAOYSA-N 0.000 description 1
- UJPMIJSAJRUNDR-UHFFFAOYSA-N C.C.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Br)C=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Br)C=C2)C=C1)N1CCCCC1 Chemical compound C.C.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Br)C=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Br)C=C2)C=C1)N1CCCCC1 UJPMIJSAJRUNDR-UHFFFAOYSA-N 0.000 description 1
- KCHPMYCEKCVXJH-UHFFFAOYSA-N C.C.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Br)C=C2)C=C1)N1CCOCC1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Br)C=C2)C=C1 Chemical compound C.C.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Br)C=C2)C=C1)N1CCOCC1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Br)C=C2)C=C1 KCHPMYCEKCVXJH-UHFFFAOYSA-N 0.000 description 1
- OGPQLUOGWNFODV-UHFFFAOYSA-N C.C.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCOCC1.N=C(NO)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1 Chemical compound C.C.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCOCC1.N=C(NO)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1 OGPQLUOGWNFODV-UHFFFAOYSA-N 0.000 description 1
- VPISDVHXXGSTLM-UHFFFAOYSA-N C.C.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2Cl)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2Cl)C=C1)N1CCCCC1 Chemical compound C.C.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2Cl)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2Cl)C=C1)N1CCCCC1 VPISDVHXXGSTLM-UHFFFAOYSA-N 0.000 description 1
- CUQDHLDZCISHLJ-ZCZNPZJMSA-N C.CCCCNS(=O)(=O)C1=CC=CC=C1C1=CC=C(N)C=C1.CCCCNS(=O)(=O)C1=CC=CC=C1C1=CC=C(N)C=C1.O=BP.O=BP.O=CC(F)(F)F.[H]/C(C(=O)NC1=CC=C(C2=CC=CC=C2S(=O)(=O)NCCCC)C=C1)=C(\[H])C(=O)OC.[H]/C(C(=O)NC1=CC=C(OC)C=C1)=C(\[H])C(=O)NC1=CC=C(C2=CC=CC=C2S(=O)(=O)NCCCC)C=C1.[H]/C(C(=O)NC1=CC=C(OC)C=C1)=C(\[H])C(=O)NC1=CC=C(C2=CC=CC=C2S(N)(=O)=O)C=C1.[H]/C(C(=O)O)=C(\[H])C(=O)NC1=CC=C(OC)C=C1.[H]/C(C(=O)O)=C(\[H])C(=O)OC Chemical compound C.CCCCNS(=O)(=O)C1=CC=CC=C1C1=CC=C(N)C=C1.CCCCNS(=O)(=O)C1=CC=CC=C1C1=CC=C(N)C=C1.O=BP.O=BP.O=CC(F)(F)F.[H]/C(C(=O)NC1=CC=C(C2=CC=CC=C2S(=O)(=O)NCCCC)C=C1)=C(\[H])C(=O)OC.[H]/C(C(=O)NC1=CC=C(OC)C=C1)=C(\[H])C(=O)NC1=CC=C(C2=CC=CC=C2S(=O)(=O)NCCCC)C=C1.[H]/C(C(=O)NC1=CC=C(OC)C=C1)=C(\[H])C(=O)NC1=CC=C(C2=CC=CC=C2S(N)(=O)=O)C=C1.[H]/C(C(=O)O)=C(\[H])C(=O)NC1=CC=C(OC)C=C1.[H]/C(C(=O)O)=C(\[H])C(=O)OC CUQDHLDZCISHLJ-ZCZNPZJMSA-N 0.000 description 1
- ZSNFLUVQTNTJIF-GDEFNOCQSA-N C/C(=N/C#N)N(C)C.CC1=CC=CC=C1.CC1=CC=CS1.CN(C(=N)N)C1COC1.CN(C)CCN(C)/C(=N/C#N)N(C)C.CN(C)CCN1CCCC1.CN(C)CCN1CCCCC1.CN(C)CCN1CCCCC1.CN(C)CCN1CCOCC1.CNCCN(C)C1=NCCN1.CNCCN(C)C1=NCCN1C.CNCCNC1=NCCN1.COCCN.COCCN/C(N)=N/C#N.COCCN/C(N)=N/[N+](=O)[O-].COCCNC(=N)N.COCCNC(=N)N(C)C Chemical compound C/C(=N/C#N)N(C)C.CC1=CC=CC=C1.CC1=CC=CS1.CN(C(=N)N)C1COC1.CN(C)CCN(C)/C(=N/C#N)N(C)C.CN(C)CCN1CCCC1.CN(C)CCN1CCCCC1.CN(C)CCN1CCCCC1.CN(C)CCN1CCOCC1.CNCCN(C)C1=NCCN1.CNCCN(C)C1=NCCN1C.CNCCNC1=NCCN1.COCCN.COCCN/C(N)=N/C#N.COCCN/C(N)=N/[N+](=O)[O-].COCCNC(=N)N.COCCNC(=N)N(C)C ZSNFLUVQTNTJIF-GDEFNOCQSA-N 0.000 description 1
- ZOWZOKCOFQRVAF-GDEFNOCQSA-N C/C(=N/C#N)N(C)C.CC1=CC=CC=C1.CC1=CC=CS1.CN(C(=N)N)C1COC1.CN(C)CCN(C)/C(=N/C#N)N(C)C.CN(C)CCN1CCCC1.CN(C)CCN1CCCCC1.CN(C)CCN1CCCCC1.CN(C)CCN1CCOCC1.CNCCN(C)C1=NCCN1.CNCCN(C)C1=NCCN1C.CNCCNC1=NCCN1.COCCN/C(N)=N/C#N.COCCN/C(N)=N/[N+](=O)[O-].COCCNC(=N)N.COCCNC(=N)N(C)C.NC1COC1 Chemical compound C/C(=N/C#N)N(C)C.CC1=CC=CC=C1.CC1=CC=CS1.CN(C(=N)N)C1COC1.CN(C)CCN(C)/C(=N/C#N)N(C)C.CN(C)CCN1CCCC1.CN(C)CCN1CCCCC1.CN(C)CCN1CCCCC1.CN(C)CCN1CCOCC1.CNCCN(C)C1=NCCN1.CNCCN(C)C1=NCCN1C.CNCCNC1=NCCN1.COCCN/C(N)=N/C#N.COCCN/C(N)=N/[N+](=O)[O-].COCCNC(=N)N.COCCNC(=N)N(C)C.NC1COC1 ZOWZOKCOFQRVAF-GDEFNOCQSA-N 0.000 description 1
- OAHFOEOBEWEMMA-FKKZVNILSA-N C/C(=N\O)N(C)C.C/C(=N\O)N1CCCC1.C/N=C(\C)N(C)C.C/N=C(\C)N1CCCC1.C/N=C(\C)NC(C)(C)C.CC(=N)N.CC(=N)N(C)C.CC(=N)N1C(C)CCC1C.CC(=N)N1CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCC1C.CC(=N)N1CCCC1C(=O)O.CC(=N)N1CCCCC1.CC(=N)N1CCCCCC1.CC(=N)N1CCCN(C)CC1.CC(=N)N1CCCNCC1.CC(=N)N1CCN(C)CC1.CC(=N)N1CCNCC1.CC(=N)N1CCOCC1.CC(=N)N1CCS(=O)(=O)CC1.CC(=N)N1CCS(=O)CC1.CC(=N)N1CCSCC1.CC(=N)NN.CC(=N)NO.CCN(CC)C(C)=N.CCNC(C)=N.CNC(C)=N.CNNC(C)=N.COC(=O)C1CCCN1C(C)=N.CONC(C)=N Chemical compound C/C(=N\O)N(C)C.C/C(=N\O)N1CCCC1.C/N=C(\C)N(C)C.C/N=C(\C)N1CCCC1.C/N=C(\C)NC(C)(C)C.CC(=N)N.CC(=N)N(C)C.CC(=N)N1C(C)CCC1C.CC(=N)N1CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCC1C.CC(=N)N1CCCC1C(=O)O.CC(=N)N1CCCCC1.CC(=N)N1CCCCCC1.CC(=N)N1CCCN(C)CC1.CC(=N)N1CCCNCC1.CC(=N)N1CCN(C)CC1.CC(=N)N1CCNCC1.CC(=N)N1CCOCC1.CC(=N)N1CCS(=O)(=O)CC1.CC(=N)N1CCS(=O)CC1.CC(=N)N1CCSCC1.CC(=N)NN.CC(=N)NO.CCN(CC)C(C)=N.CCNC(C)=N.CNC(C)=N.CNNC(C)=N.COC(=O)C1CCCN1C(C)=N.CONC(C)=N OAHFOEOBEWEMMA-FKKZVNILSA-N 0.000 description 1
- JTDGIGQVHMELCZ-NYZLDFTISA-N C/C(=N\O)N(C)C.C/C(=N\O)N1CCCC1.C/N=C(\C)N(C)C.C/N=C(\C)N1CCCC1.CC(=N)N.CC(=N)N(C)C.CC(=N)N1C(C)CCC1C.CC(=N)N1CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCC1C.CC(=N)N1CCCC1C(=O)O.CC(=N)N1CCCCC1.CC(=N)N1CCCCCC1.CC(=N)N1CCCN(C)CC1.CC(=N)N1CCCNCC1.CC(=N)N1CCN(C)CC1.CC(=N)N1CCNCC1.CC(=N)N1CCOCC1.CC(=N)N1CCS(=O)(=O)CC1.CC(=N)N1CCS(=O)CC1.CC(=N)N1CCSCC1.CC(=N)NN.CC(=N)NO.CC(C)=N.CC1=NCCCN1.CC1=NCCCN1C.CC1=NCCN1.CC1=NCCN1C.CCN(CC)C(C)=N.CCN1CCCN=C1C.CCN1CCN=C1C.CCNC(C)=N.CNC(C)=N.CNNC(C)=N.COC(=O)C1CCCN1C(C)=N.CONC(C)=N Chemical compound C/C(=N\O)N(C)C.C/C(=N\O)N1CCCC1.C/N=C(\C)N(C)C.C/N=C(\C)N1CCCC1.CC(=N)N.CC(=N)N(C)C.CC(=N)N1C(C)CCC1C.CC(=N)N1CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCC1C.CC(=N)N1CCCC1C(=O)O.CC(=N)N1CCCCC1.CC(=N)N1CCCCCC1.CC(=N)N1CCCN(C)CC1.CC(=N)N1CCCNCC1.CC(=N)N1CCN(C)CC1.CC(=N)N1CCNCC1.CC(=N)N1CCOCC1.CC(=N)N1CCS(=O)(=O)CC1.CC(=N)N1CCS(=O)CC1.CC(=N)N1CCSCC1.CC(=N)NN.CC(=N)NO.CC(C)=N.CC1=NCCCN1.CC1=NCCCN1C.CC1=NCCN1.CC1=NCCN1C.CCN(CC)C(C)=N.CCN1CCCN=C1C.CCN1CCN=C1C.CCNC(C)=N.CNC(C)=N.CNNC(C)=N.COC(=O)C1CCCN1C(C)=N.CONC(C)=N JTDGIGQVHMELCZ-NYZLDFTISA-N 0.000 description 1
- SJWPDXGMHJIROS-QQJDZUIHSA-N C/C=C\C.CC1=C(C)C2=C(C=CC=C2)N1.CC1=C(C)C2=C(C=CC=C2)S1.CC1=C(C)C2=C(CCC2)S1.CC1=C(C)CCC1.CC1=C(C)CCCC1.CC1=C(C)COC1.CC1=C(C)NC=C1.CC1=C(C)NC=N1.CC1=C(C)NN=C1.CC1=C(C)NN=C1.CC1=C(C)O=CN1.CC1=C(C)OC=C1.CC1=C(C)SC(C2=CC=CC=C2)=C1.CC1=C(C)SC=C1.CC1=C(C)SC=C1.CC1=C(C)SC=N1.CC1=C(C)SC=N1.CC1=CC2=C(C=C1C)NC=N2.CC1=CC2=C(C=CC=C2)C=C1C.CC1=CC2=C(C=CC=C2)N1C.CC1=CC2=C(C=NC=C2)C=C1C.CC1=CC=CC=C1C.CC1=CC=CN=C1C.CC1=CC=CN=C1C.CC1=CC=NC=C1C.CC1=CC=NC=C1C.CC1=CC=NN1C.CC1=CN(C)N=C1.CC1=CN=CN=C1C.CC1=CN=CN=C1C.CC1=CNN=C1C.CC1=CNN=C1C.CC1=CNO=C1C.CC1=CON=C1C.CC1=CSC=C1C.CC1=NC=CN=C1C.CC1CC1C.CC1CCC1C.CC1CCCC1C.CC1CCCCC1C Chemical compound C/C=C\C.CC1=C(C)C2=C(C=CC=C2)N1.CC1=C(C)C2=C(C=CC=C2)S1.CC1=C(C)C2=C(CCC2)S1.CC1=C(C)CCC1.CC1=C(C)CCCC1.CC1=C(C)COC1.CC1=C(C)NC=C1.CC1=C(C)NC=N1.CC1=C(C)NN=C1.CC1=C(C)NN=C1.CC1=C(C)O=CN1.CC1=C(C)OC=C1.CC1=C(C)SC(C2=CC=CC=C2)=C1.CC1=C(C)SC=C1.CC1=C(C)SC=C1.CC1=C(C)SC=N1.CC1=C(C)SC=N1.CC1=CC2=C(C=C1C)NC=N2.CC1=CC2=C(C=CC=C2)C=C1C.CC1=CC2=C(C=CC=C2)N1C.CC1=CC2=C(C=NC=C2)C=C1C.CC1=CC=CC=C1C.CC1=CC=CN=C1C.CC1=CC=CN=C1C.CC1=CC=NC=C1C.CC1=CC=NC=C1C.CC1=CC=NN1C.CC1=CN(C)N=C1.CC1=CN=CN=C1C.CC1=CN=CN=C1C.CC1=CNN=C1C.CC1=CNN=C1C.CC1=CNO=C1C.CC1=CON=C1C.CC1=CSC=C1C.CC1=NC=CN=C1C.CC1CC1C.CC1CCC1C.CC1CCCC1C.CC1CCCCC1C SJWPDXGMHJIROS-QQJDZUIHSA-N 0.000 description 1
- GSMDNSNFSCQVET-UAJBKYHUSA-N C/C=C\C.CC1=C(C)C2=C(C=CC=C2)N1.CC1=C(C)C2=C(C=CC=C2)S1.CC1=C(C)C2=C(CCC2)S1.CC1=C(C)CCC1.CC1=C(C)CCCC1.CC1=C(C)NC=C1.CC1=C(C)NC=N1.CC1=C(C)NN=C1.CC1=C(C)NN=C1.CC1=C(C)O=CN1.CC1=C(C)OC=C1.CC1=C(C)SC(C2=CC=CC=C2)=C1.CC1=C(C)SC=C1.CC1=C(C)SC=C1.CC1=C(C)SC=N1.CC1=C(C)SC=N1.CC1=C2C=CC=CC2=NN1C.CC1=CC2=C(C=C1C)NC=N2.CC1=CC2=C(C=CC=C2)C=C1C.CC1=CC2=C(C=CC=C2)N1C.CC1=CC2=C(C=NC=C2)C=C1C.CC1=CC=CC=C1C.CC1=CC=CN=C1C.CC1=CC=CN=C1C.CC1=CC=NC=C1C.CC1=CC=NC=C1C.CC1=CC=NN1C.CC1=CN(C)N=C1.CC1=CN=CN=C1C.CC1=CN=CN=C1C.CC1=CNN=C1C.CC1=CNN=C1C.CC1=CNO=C1C.CC1=CON=C1C.CC1=CSC=C1C.CC1=NC=CN=C1C.CC1CC1C Chemical compound C/C=C\C.CC1=C(C)C2=C(C=CC=C2)N1.CC1=C(C)C2=C(C=CC=C2)S1.CC1=C(C)C2=C(CCC2)S1.CC1=C(C)CCC1.CC1=C(C)CCCC1.CC1=C(C)NC=C1.CC1=C(C)NC=N1.CC1=C(C)NN=C1.CC1=C(C)NN=C1.CC1=C(C)O=CN1.CC1=C(C)OC=C1.CC1=C(C)SC(C2=CC=CC=C2)=C1.CC1=C(C)SC=C1.CC1=C(C)SC=C1.CC1=C(C)SC=N1.CC1=C(C)SC=N1.CC1=C2C=CC=CC2=NN1C.CC1=CC2=C(C=C1C)NC=N2.CC1=CC2=C(C=CC=C2)C=C1C.CC1=CC2=C(C=CC=C2)N1C.CC1=CC2=C(C=NC=C2)C=C1C.CC1=CC=CC=C1C.CC1=CC=CN=C1C.CC1=CC=CN=C1C.CC1=CC=NC=C1C.CC1=CC=NC=C1C.CC1=CC=NN1C.CC1=CN(C)N=C1.CC1=CN=CN=C1C.CC1=CN=CN=C1C.CC1=CNN=C1C.CC1=CNN=C1C.CC1=CNO=C1C.CC1=CON=C1C.CC1=CSC=C1C.CC1=NC=CN=C1C.CC1CC1C GSMDNSNFSCQVET-UAJBKYHUSA-N 0.000 description 1
- GUHXJLPGRXTFEB-OAXWKQDWSA-N C/N=C(/C)N1CCCC1.C/N=C(\C)N(C)C.CC(=N)N.CC(=N)N(C)C.CC(=N)N1C(C)CCC1C.CC(=N)N1CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCC1C.CC(=N)N1CCCC1C(=O)O.CC(=N)N1CCCN(C)CC1.CC(=N)N1CCCNCC1.CC(=N)N1CCN(C)CC1.CC(=N)N1CCNCC1.CC(=N)N1CCOCC1.CC(=N)N1CCS(=O)(=O)CC1.CC(=N)N1CCS(=O)CC1.CC(=N)N1CCSCC1.CC(=N)NC1CCCC1.CC(=N)NN.CC(=N)NO.CC(=O)N(C)C.CC(=O)N1CCCC1.CC(C)=N.CC(N)=O.CC1=NCCCN1.CC1=NCCCN1.CC1=NCCCN1C.CC1=NCCN1.CC1=NCCN1C.CCN(CC)C(C)=N.CCN1CCN=C1C.CCNC(C)=N.CNC(C)=N.CNNC(C)=N.CONC(C)=N Chemical compound C/N=C(/C)N1CCCC1.C/N=C(\C)N(C)C.CC(=N)N.CC(=N)N(C)C.CC(=N)N1C(C)CCC1C.CC(=N)N1CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCC1C.CC(=N)N1CCCC1C(=O)O.CC(=N)N1CCCN(C)CC1.CC(=N)N1CCCNCC1.CC(=N)N1CCN(C)CC1.CC(=N)N1CCNCC1.CC(=N)N1CCOCC1.CC(=N)N1CCS(=O)(=O)CC1.CC(=N)N1CCS(=O)CC1.CC(=N)N1CCSCC1.CC(=N)NC1CCCC1.CC(=N)NN.CC(=N)NO.CC(=O)N(C)C.CC(=O)N1CCCC1.CC(C)=N.CC(N)=O.CC1=NCCCN1.CC1=NCCCN1.CC1=NCCCN1C.CC1=NCCN1.CC1=NCCN1C.CCN(CC)C(C)=N.CCN1CCN=C1C.CCNC(C)=N.CNC(C)=N.CNNC(C)=N.CONC(C)=N GUHXJLPGRXTFEB-OAXWKQDWSA-N 0.000 description 1
- UVHXYTHUALANIR-UHFFFAOYSA-N C/N=C(/NC)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(Cl)C=C2OC)C=C1 Chemical compound C/N=C(/NC)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(Cl)C=C2OC)C=C1 UVHXYTHUALANIR-UHFFFAOYSA-N 0.000 description 1
- DKSQNUQDTCMFLM-NROBKIFHSA-N C/N=C(\C)N(C)C.C/N=C(\C)N1CC1.C/N=C(\C)N1CCC1.C/N=C(\C)N1CCCC1.C/N=C(\C)N1CCCCC1.CC(=N)N.CC(=N)N1CC1.CC(=N)N1CCC(C(=O)O)CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCCC1.CC1=C(CN(C)C)C=C(F)C=C1.CC1=C(CN(C)C)C=[N+](O)C=C1.CC1=CC=C(F)C=C1S(C)(=O)=O.CC1=CC=C(F)C=C1S(N)(=O)=O.CC1=CC=CC=C1C(=N)N(C)C.CC1=CC=CC=C1CN(C)C.CC1=CC=CC=C1S(C)(=O)=O.CC1=CC=CC=C1S(N)(=O)=O.CC1=CC=N(O)C(CN(C)C)=C1.CC1=CC=NC(CN(C)C)=C1.CC1=CC=NC(CN(C)C)=N1.CC1=CC=NC(N)=C1.CC1=CC=NC(N)=N1.CC1=CC=[N+](O)C=C1.CC1=NCCN1C.CCN(C)C(C)=N.CCOC(=O)C1CCN(C(C)=N)CC1.CN(C)C=N.CN1CCCN(C)CC1 Chemical compound C/N=C(\C)N(C)C.C/N=C(\C)N1CC1.C/N=C(\C)N1CCC1.C/N=C(\C)N1CCCC1.C/N=C(\C)N1CCCCC1.CC(=N)N.CC(=N)N1CC1.CC(=N)N1CCC(C(=O)O)CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCCC1.CC1=C(CN(C)C)C=C(F)C=C1.CC1=C(CN(C)C)C=[N+](O)C=C1.CC1=CC=C(F)C=C1S(C)(=O)=O.CC1=CC=C(F)C=C1S(N)(=O)=O.CC1=CC=CC=C1C(=N)N(C)C.CC1=CC=CC=C1CN(C)C.CC1=CC=CC=C1S(C)(=O)=O.CC1=CC=CC=C1S(N)(=O)=O.CC1=CC=N(O)C(CN(C)C)=C1.CC1=CC=NC(CN(C)C)=C1.CC1=CC=NC(CN(C)C)=N1.CC1=CC=NC(N)=C1.CC1=CC=NC(N)=N1.CC1=CC=[N+](O)C=C1.CC1=NCCN1C.CCN(C)C(C)=N.CCOC(=O)C1CCN(C(C)=N)CC1.CN(C)C=N.CN1CCCN(C)CC1 DKSQNUQDTCMFLM-NROBKIFHSA-N 0.000 description 1
- LXWIKKIZFKVERC-KDUBQDSXSA-N C/N=C(\C)N(C)C.C/N=C(\C)N1CCCC1.CC(=N)N.CC(=N)N(C)C.CC(=N)N1CCCC1.CC(=N)N1CCN(C)CC1.CC(=N)NO.CC(=O)N1CCCC1.CC1=C(C(=N)N(C)C)C=CC=C1.CC1=C(C(=N)N)C=CC=C1.CC1=C(C(N)=O)C=CC=C1.CC1=C(CN(C)C)C=CC=C1.CC1=C(CN)C=CC=C1.CC1=C(CN2CCCC2)C=CC=C1.CC1=C(S(C)(=O)=O)C=CC=C1.CC1=C(S(N)(=O)=O)C=CC=C1.CC1=CC(C(N)=O)=CC=C1.CC1=CC(C(N)=O)=NC=C1.CC1=CC(CN(C)C)=CC=C1.CC1=CC(CN)=CC=C1.CC1=CC(N)=NC=C1.CC1=CC(O)=NC=C1.CC1=CC=CC=C1CNC1=NCCN1.CC1=CC=CC=C1CNC1=NCCN1C.CC1=NCCN1C.CCN(C)C.CCN1CCN(C)CC1.CCN1CCOCC1.CN1CCCNCC1.CN1CCN(C(=N)N)CC1.CN1CCOCC1.CNC(C)=N.CONC(C)=N Chemical compound C/N=C(\C)N(C)C.C/N=C(\C)N1CCCC1.CC(=N)N.CC(=N)N(C)C.CC(=N)N1CCCC1.CC(=N)N1CCN(C)CC1.CC(=N)NO.CC(=O)N1CCCC1.CC1=C(C(=N)N(C)C)C=CC=C1.CC1=C(C(=N)N)C=CC=C1.CC1=C(C(N)=O)C=CC=C1.CC1=C(CN(C)C)C=CC=C1.CC1=C(CN)C=CC=C1.CC1=C(CN2CCCC2)C=CC=C1.CC1=C(S(C)(=O)=O)C=CC=C1.CC1=C(S(N)(=O)=O)C=CC=C1.CC1=CC(C(N)=O)=CC=C1.CC1=CC(C(N)=O)=NC=C1.CC1=CC(CN(C)C)=CC=C1.CC1=CC(CN)=CC=C1.CC1=CC(N)=NC=C1.CC1=CC(O)=NC=C1.CC1=CC=CC=C1CNC1=NCCN1.CC1=CC=CC=C1CNC1=NCCN1C.CC1=NCCN1C.CCN(C)C.CCN1CCN(C)CC1.CCN1CCOCC1.CN1CCCNCC1.CN1CCN(C(=N)N)CC1.CN1CCOCC1.CNC(C)=N.CONC(C)=N LXWIKKIZFKVERC-KDUBQDSXSA-N 0.000 description 1
- RMLJTGBDFQRCHB-LXKVIBFUSA-N C/N=C(\C)N(C)C.C/N=C(\C)NC.CC(=N)N.CC(=N)N(C)C.CC(=N)N1CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCCCC1.CC(=N)N1CCN(C)CC1.CC(=N)N1CCNCC1.CC(=N)N1CCOCC1.CC(=N)N1CCOCC1.CC(=N)N1CCSCC1.CC(=N)NN.CC(=N)NO.CC1=CC=N(O)C=C1.CC1=CC=NC=C1.CC1=NCCCN1C.CC1=NCCN1C.CCN1CCN=C1C.CN1CCCN(C)CC1.CNNC(C)=N.CONC(C)=N Chemical compound C/N=C(\C)N(C)C.C/N=C(\C)NC.CC(=N)N.CC(=N)N(C)C.CC(=N)N1CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCCCC1.CC(=N)N1CCN(C)CC1.CC(=N)N1CCNCC1.CC(=N)N1CCOCC1.CC(=N)N1CCOCC1.CC(=N)N1CCSCC1.CC(=N)NN.CC(=N)NO.CC1=CC=N(O)C=C1.CC1=CC=NC=C1.CC1=NCCCN1C.CC1=NCCN1C.CCN1CCN=C1C.CN1CCCN(C)CC1.CNNC(C)=N.CONC(C)=N RMLJTGBDFQRCHB-LXKVIBFUSA-N 0.000 description 1
- DLOCIIYRYNHIBL-RPEWYAMFSA-N C/N=C(\C)N(C)C.CC(=N)C1=CC=CC=C1.CC(=N)C1=CC=CS1.CC(=N)C1=CC=NC=C1.CC(=N)C1CC1.CC(=N)C1CCC1.CC(=N)C1CCCC1.CC(=N)C1CCCCC1.CC(=N)N(C)C.CC(=N)N(C)C.CC(=N)N1CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCCC1.CC1=CC=CS1.CC1=NCCC1.CC1=NCCCC1.CC1=NCCS1.CC1CC1.CC1CCC1.CC1CCCC1.CC1CCCCC1.CC1CCN(C)CC1.CC1CCNCC1.CC1CCOCC1.CC1CCSCC1.CCN1CCC(C)CC1.CN.CN.CNC(=N)N(C)C.CNC(C)=N Chemical compound C/N=C(\C)N(C)C.CC(=N)C1=CC=CC=C1.CC(=N)C1=CC=CS1.CC(=N)C1=CC=NC=C1.CC(=N)C1CC1.CC(=N)C1CCC1.CC(=N)C1CCCC1.CC(=N)C1CCCCC1.CC(=N)N(C)C.CC(=N)N(C)C.CC(=N)N1CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCCC1.CC1=CC=CS1.CC1=NCCC1.CC1=NCCCC1.CC1=NCCS1.CC1CC1.CC1CCC1.CC1CCCC1.CC1CCCCC1.CC1CCN(C)CC1.CC1CCNCC1.CC1CCOCC1.CC1CCSCC1.CCN1CCC(C)CC1.CN.CN.CNC(=N)N(C)C.CNC(C)=N DLOCIIYRYNHIBL-RPEWYAMFSA-N 0.000 description 1
- IDWYASFWKLAOAD-CQFYEVFRSA-N C/N=C(\C)N1CC1.C/N=C(\C)N1CCC1.C/N=C(\C)N1CCCC1.C/N=C(\C)N1CCCCC1.CC(=N)N.CC(=N)N(C)C.CC(=N)N1CC1.CC(=N)N1CCC(C(=O)O)CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCCC1.CC1=C(CN(C)C)C=C(F)C=C1.CC1=C(CN(C)C)C=[N+](O)C=C1.CC1=CC=C(F)C=C1S(C)(=O)=O.CC1=CC=C(F)C=C1S(N)(=O)=O.CC1=CC=CC=C1C(=N)N(C)C.CC1=CC=CC=C1CN(C)C.CC1=CC=CC=C1S(C)(=O)=O.CC1=CC=CC=C1S(N)(=O)=O.CC1=CC=N(O)C(CN(C)C)=C1.CC1=CC=NC(CN(C)C)=C1.CC1=CC=NC(CN(C)C)=N1.CC1=CC=NC(N)=C1.CC1=CC=NC(N)=N1.CC1=CC=[N+](O)C=C1.CC1=NCCN1C.CCN(C)C(C)=N.CCOC(=O)C1CCN(C(C)=N)CC1.CN(C)C=N.CN1CCCN(C)CC1 Chemical compound C/N=C(\C)N1CC1.C/N=C(\C)N1CCC1.C/N=C(\C)N1CCCC1.C/N=C(\C)N1CCCCC1.CC(=N)N.CC(=N)N(C)C.CC(=N)N1CC1.CC(=N)N1CCC(C(=O)O)CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCCC1.CC1=C(CN(C)C)C=C(F)C=C1.CC1=C(CN(C)C)C=[N+](O)C=C1.CC1=CC=C(F)C=C1S(C)(=O)=O.CC1=CC=C(F)C=C1S(N)(=O)=O.CC1=CC=CC=C1C(=N)N(C)C.CC1=CC=CC=C1CN(C)C.CC1=CC=CC=C1S(C)(=O)=O.CC1=CC=CC=C1S(N)(=O)=O.CC1=CC=N(O)C(CN(C)C)=C1.CC1=CC=NC(CN(C)C)=C1.CC1=CC=NC(CN(C)C)=N1.CC1=CC=NC(N)=C1.CC1=CC=NC(N)=N1.CC1=CC=[N+](O)C=C1.CC1=NCCN1C.CCN(C)C(C)=N.CCOC(=O)C1CCN(C(C)=N)CC1.CN(C)C=N.CN1CCCN(C)CC1 IDWYASFWKLAOAD-CQFYEVFRSA-N 0.000 description 1
- QBKKFXOFWZHTCP-QEYCDUBXSA-N C/N=C(\C1=CC(Cl)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N(C)C.C/N=C(\C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N(C)C.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCC1 Chemical compound C/N=C(\C1=CC(Cl)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N(C)C.C/N=C(\C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N(C)C.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCC1 QBKKFXOFWZHTCP-QEYCDUBXSA-N 0.000 description 1
- UIONTRDCWAKIGT-APGJPFGKSA-N C/N=C(\C1=CC(Cl)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OC)C=C1)N(C)C.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=C(Cl)C=C(C(=N)N2CCC2)C=C1 Chemical compound C/N=C(\C1=CC(Cl)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OC)C=C1)N(C)C.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=C(Cl)C=C(C(=N)N2CCC2)C=C1 UIONTRDCWAKIGT-APGJPFGKSA-N 0.000 description 1
- BCRTXBMPDSJVAH-IQODEUHDSA-N C/N=C(\C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N(C)C.N=C(C1=CC(Cl)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N1CC1.N=C(C1=CC(Cl)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N1CCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N1CCC1 Chemical compound C/N=C(\C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N(C)C.N=C(C1=CC(Cl)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N1CC1.N=C(C1=CC(Cl)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N1CCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N1CCC1 BCRTXBMPDSJVAH-IQODEUHDSA-N 0.000 description 1
- FFZHIDPURJEZDS-BQPVZQOISA-N C/N=C(\C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OC(F)(F)F)C=C1)N(C)C.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=C(Cl)C=C(C(=N)N2CC2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OC(F)(F)F)C=C1)N1CC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OC(F)(F)F)C=C1)N1CCC1 Chemical compound C/N=C(\C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OC(F)(F)F)C=C1)N(C)C.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=C(Cl)C=C(C(=N)N2CC2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OC(F)(F)F)C=C1)N1CC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OC(F)(F)F)C=C1)N1CCC1 FFZHIDPURJEZDS-BQPVZQOISA-N 0.000 description 1
- MLIPTMJQSGLOBM-SZGYWAGOSA-N C/N=C(\C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OC)C=C1)N(C)C.C/N=C(\C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OC)C=C1)N(C)C.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCC2)C=C1 Chemical compound C/N=C(\C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OC)C=C1)N(C)C.C/N=C(\C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OC)C=C1)N(C)C.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCC2)C=C1 MLIPTMJQSGLOBM-SZGYWAGOSA-N 0.000 description 1
- ZPLWCKPKUQTGFA-GCPPRROUSA-N C/N=C(\C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N(C)C.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N1CC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N1CC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N1CCC1 Chemical compound C/N=C(\C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N(C)C.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N1CC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N1CC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N1CCC1 ZPLWCKPKUQTGFA-GCPPRROUSA-N 0.000 description 1
- HFCJJMJISBMBKI-VQXTZJAHSA-N C/N=C(\C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OC(F)(F)F)C=C2)C=C1)N(C)C.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CC2)C=C1.COC1=CC(OC(F)(F)F)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCC2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OC(F)(F)F)C=C2)C=C1)N1CCC1 Chemical compound C/N=C(\C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OC(F)(F)F)C=C2)C=C1)N(C)C.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CC2)C=C1.COC1=CC(OC(F)(F)F)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCC2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OC(F)(F)F)C=C2)C=C1)N1CCC1 HFCJJMJISBMBKI-VQXTZJAHSA-N 0.000 description 1
- UHPMNPCHSVEYSE-NJNXFGOHSA-N C/N=C(\C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N(C)C Chemical compound C/N=C(\C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N(C)C UHPMNPCHSVEYSE-NJNXFGOHSA-N 0.000 description 1
- XUZQONBPOKDXDR-UHFFFAOYSA-N C=C(C)NC1=NC(C)=CC=N1.CC1=C(C#N)C=NC=C1.CC1=C(C(N)=O)C=NC=C1.CC1=C(CN(C)C)C=NC=C1.CC1=C(CN(C)C)C=NC=N1.CC1=C(CN)C=NC=C1.CC1=C(CN)C=NC=N1.CC1=C(N(C)C)C=NC=C1.CC1=C(N)C=NC=C1.CC1=C(N)C=NC=N1.CC1=C(S(C)(=O)=O)C=NC=C1.CC1=C(S(N)(=O)=O)C=NC=C1.CC1=CC(C(C)(C)N)=CC=C1.CC1=CC(C(C)(C)N)=NC=C1.CC1=CC(C(C)(C)N)=[N+](O)C=C1.CC1=CC(C(N)=O)=CC=C1.CC1=CC(C(N)=O)=NC=C1.CC1=CC(CN)=CC=C1.CC1=CC(N(C)C)=NC=C1.CC1=CC(N)=NC=C1.CC1=CC(O)=NC=C1.CC1=CC=NC(CN)=N1.CC1=CC=NC(N)=N1.CC1=CC=NC=C1.CC1=CC=NC=N1.CC1=CC=[N+](O)C=C1.CCOC1=NC(C)=CC=N1.O.[C-]#[N+]C1=CC=CC(C)=C1.[C-]#[N+]C1=NC=CC(C)=C1 Chemical compound C=C(C)NC1=NC(C)=CC=N1.CC1=C(C#N)C=NC=C1.CC1=C(C(N)=O)C=NC=C1.CC1=C(CN(C)C)C=NC=C1.CC1=C(CN(C)C)C=NC=N1.CC1=C(CN)C=NC=C1.CC1=C(CN)C=NC=N1.CC1=C(N(C)C)C=NC=C1.CC1=C(N)C=NC=C1.CC1=C(N)C=NC=N1.CC1=C(S(C)(=O)=O)C=NC=C1.CC1=C(S(N)(=O)=O)C=NC=C1.CC1=CC(C(C)(C)N)=CC=C1.CC1=CC(C(C)(C)N)=NC=C1.CC1=CC(C(C)(C)N)=[N+](O)C=C1.CC1=CC(C(N)=O)=CC=C1.CC1=CC(C(N)=O)=NC=C1.CC1=CC(CN)=CC=C1.CC1=CC(N(C)C)=NC=C1.CC1=CC(N)=NC=C1.CC1=CC(O)=NC=C1.CC1=CC=NC(CN)=N1.CC1=CC=NC(N)=N1.CC1=CC=NC=C1.CC1=CC=NC=N1.CC1=CC=[N+](O)C=C1.CCOC1=NC(C)=CC=N1.O.[C-]#[N+]C1=CC=CC(C)=C1.[C-]#[N+]C1=NC=CC(C)=C1 XUZQONBPOKDXDR-UHFFFAOYSA-N 0.000 description 1
- NQHWCMCBKHCQFH-UHFFFAOYSA-N C=C.CC.CC1=NCCCN1.CC1=NCCCN1C.CC1=NCCCO1.CC1=NCCCS1.CC1=NCCN1.CC1=NCCN1C.CC1=NCCO1.CC1CCC(C)N1C.CC1CCCN1C.CCC(=N)NC.CCCN(C)C.CCCN1CCCCC1.CCCN1CCN(C)CC1.CCCN1CCOCC1.CCCOC.CCN(C)CC.CCN1CCCN=C1C.CCN1CCN=C1C.CCNC.CN.CN(C)C.CN(C)CCN(C)CCN(C)C.CN1CCCC1C(=O)O.CN1CCCN(C)CC1.CN1CCCNCC1.CN1CCN(C(=O)OC(C)(C)C)CC1.CN1CCN(C)CC1.CN1CCNCC1.CN1CCS(=O)(=O)CC1.CN1CCS(=O)CC1.CN1CCSCC1.CNC.CNC(=N)C(F)(F)F.CNC(=N)N.CNC(C)=N.CNN.CNNC.CNO.CNOC.COCCN(C)CCN(C)C.COCCN(C)CCOC Chemical compound C=C.CC.CC1=NCCCN1.CC1=NCCCN1C.CC1=NCCCO1.CC1=NCCCS1.CC1=NCCN1.CC1=NCCN1C.CC1=NCCO1.CC1CCC(C)N1C.CC1CCCN1C.CCC(=N)NC.CCCN(C)C.CCCN1CCCCC1.CCCN1CCN(C)CC1.CCCN1CCOCC1.CCCOC.CCN(C)CC.CCN1CCCN=C1C.CCN1CCN=C1C.CCNC.CN.CN(C)C.CN(C)CCN(C)CCN(C)C.CN1CCCC1C(=O)O.CN1CCCN(C)CC1.CN1CCCNCC1.CN1CCN(C(=O)OC(C)(C)C)CC1.CN1CCN(C)CC1.CN1CCNCC1.CN1CCS(=O)(=O)CC1.CN1CCS(=O)CC1.CN1CCSCC1.CNC.CNC(=N)C(F)(F)F.CNC(=N)N.CNC(C)=N.CNN.CNNC.CNO.CNOC.COCCN(C)CCN(C)C.COCCN(C)CCOC NQHWCMCBKHCQFH-UHFFFAOYSA-N 0.000 description 1
- LYSONXXMKQHXED-UHFFFAOYSA-N CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCOCC1.N=C(N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.NN=Cl(F)(Cl)(N=O)=NN=O.O=NN=Cl(F)(Cl)(N=O)=NN=O.[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH] Chemical compound CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCOCC1.N=C(N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.NN=Cl(F)(Cl)(N=O)=NN=O.O=NN=Cl(F)(Cl)(N=O)=NN=O.[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH] LYSONXXMKQHXED-UHFFFAOYSA-N 0.000 description 1
- OKRRLNNUWLNHBO-UHFFFAOYSA-N CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(Cl)C=C2)C=C1)N1CCOCC1.N=C(NO)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(Cl)C=C2)C=C1.O=NN=Br(Cl)(N=O)=NN=O.O=NN=Br(Cl)(N=O)=NN=O.[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH] Chemical compound CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC#CC.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(Cl)C=C2)C=C1)N1CCOCC1.N=C(NO)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(Cl)C=C2)C=C1.O=NN=Br(Cl)(N=O)=NN=O.O=NN=Br(Cl)(N=O)=NN=O.[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH].[HH] OKRRLNNUWLNHBO-UHFFFAOYSA-N 0.000 description 1
- OWICRYXFYHBZGN-UHFFFAOYSA-N CC(=N)C1=CC(C)=CC=C1F.CC1=C(CN)C(Br)=CC(N)=C1.CC1=C(F)C=CC(C(=N)N)=C1.CC1=C(N)C=C2C=CN=C(N)C2=C1.CC1=C(O)C=CC(C(=N)N)=C1.CC1=C([N+](=O)[O-])C=C2C=CN=C(N)C2=C1.CC1=CC(Br)=CC(N)=C1.CC1=CC=C2C=CN=C(N)C2=C1.CC1=CC=CC(C(=N)N)=C1.CC1=CC=CC(C(=N)NN)=C1.CC1=CC=CC(C(N)=O)=C1.CC1=CC=CC(CN)=C1.CC1=CC=CC(N)=C1.CONC(=N)C1=CC(C)=CC=C1.COOSC1=C(C)C=C2C(=C1)C=CN=C2N.[C-]#[N+]C1=C(C)C=C2C(=C1)C=CN=C2N Chemical compound CC(=N)C1=CC(C)=CC=C1F.CC1=C(CN)C(Br)=CC(N)=C1.CC1=C(F)C=CC(C(=N)N)=C1.CC1=C(N)C=C2C=CN=C(N)C2=C1.CC1=C(O)C=CC(C(=N)N)=C1.CC1=C([N+](=O)[O-])C=C2C=CN=C(N)C2=C1.CC1=CC(Br)=CC(N)=C1.CC1=CC=C2C=CN=C(N)C2=C1.CC1=CC=CC(C(=N)N)=C1.CC1=CC=CC(C(=N)NN)=C1.CC1=CC=CC(C(N)=O)=C1.CC1=CC=CC(CN)=C1.CC1=CC=CC(N)=C1.CONC(=N)C1=CC(C)=CC=C1.COOSC1=C(C)C=C2C(=C1)C=CN=C2N.[C-]#[N+]C1=C(C)C=C2C(=C1)C=CN=C2N OWICRYXFYHBZGN-UHFFFAOYSA-N 0.000 description 1
- NXWGXTNMTPXSDV-UHFFFAOYSA-L CC(=N)N(C)CC(=N)N([Rb])[RaH].CC(=N)N(C)CC(=O)N([Rb])[RaH].CC(=N)N(C)CC(=O)N([Rb])[RaH].CC(=N)N(C)CC(=O)N([Rb])[RaH].CC(=N)N(C)CC(=O)O[Rb].CC(=N)N(C)CC(=O)S[Rb].CC(=N)N(C)CN(C)[Re].CC(=N)N([Rb])[RaH].CC(=O)N([Rb])[RaH].CCC(=O)N([Rb])[RaH].COCN(C)C(C)=N.CSCN(C)C(C)=N.[C-]#[N+]CN(C)C(C)=N Chemical compound CC(=N)N(C)CC(=N)N([Rb])[RaH].CC(=N)N(C)CC(=O)N([Rb])[RaH].CC(=N)N(C)CC(=O)N([Rb])[RaH].CC(=N)N(C)CC(=O)N([Rb])[RaH].CC(=N)N(C)CC(=O)O[Rb].CC(=N)N(C)CC(=O)S[Rb].CC(=N)N(C)CN(C)[Re].CC(=N)N([Rb])[RaH].CC(=O)N([Rb])[RaH].CCC(=O)N([Rb])[RaH].COCN(C)C(C)=N.CSCN(C)C(C)=N.[C-]#[N+]CN(C)C(C)=N NXWGXTNMTPXSDV-UHFFFAOYSA-L 0.000 description 1
- AQSRJZSYPYUNAM-UHFFFAOYSA-N CC(=N)N.CC(=N)N(C)C.CC(=N)N1CC1.CC(=N)N1CCC(C(=O)O)CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCCC1.CC1=CC=CC=C1S(C)(=O)=O.CC1=CC=CC=C1S(N)(=O)=O.CC1=NCCN1C.CCN(C)C(C)=N.CCOC(=O)C1CCN(C(C)=N)CC1 Chemical compound CC(=N)N.CC(=N)N(C)C.CC(=N)N1CC1.CC(=N)N1CCC(C(=O)O)CC1.CC(=N)N1CCC1.CC(=N)N1CCCC1.CC(=N)N1CCCCC1.CC1=CC=CC=C1S(C)(=O)=O.CC1=CC=CC=C1S(N)(=O)=O.CC1=NCCN1C.CCN(C)C(C)=N.CCOC(=O)C1CCN(C(C)=N)CC1 AQSRJZSYPYUNAM-UHFFFAOYSA-N 0.000 description 1
- SFLYDRCPIHDGLG-UHFFFAOYSA-N CC(=N)N.CC(=N)N(C)C.CC(=N)N1CCCC1.CC(=O)N(C)CCS(C)(=O)=O.CC(=O)N1CCCCC1.CC(=O)N1CCN(C)CC1.CC(=O)N1CCOCC1.CC(=O)N1CCS(=O)(=O)CC1.CC(=O)NCCS(C)(=O)=O.CC1=NCCN1C.CCNC1=NCCN1.CCNC1=NCCN1C.CN(C)CCN1CCN(C)CC1.CN(C)CCN1CCS(=O)(=O)CC1.CN1CCCCC1.CN1CCN(C)CC1.CN1CCOCC1.CN1CCS(=O)(=O)CC1.CNC1=NCCN1.CNC1=NCCN1.CNCCN1CCN(C)CC1.CNCCN1CCOCC1.CNCCN1CCS(=O)(=O)CC1.COCCN1CCN(C)CC1.COCCN1CCOCC1.COCCN1CCS(=O)(=O)CC1.COCCNC(C)=O Chemical compound CC(=N)N.CC(=N)N(C)C.CC(=N)N1CCCC1.CC(=O)N(C)CCS(C)(=O)=O.CC(=O)N1CCCCC1.CC(=O)N1CCN(C)CC1.CC(=O)N1CCOCC1.CC(=O)N1CCS(=O)(=O)CC1.CC(=O)NCCS(C)(=O)=O.CC1=NCCN1C.CCNC1=NCCN1.CCNC1=NCCN1C.CN(C)CCN1CCN(C)CC1.CN(C)CCN1CCS(=O)(=O)CC1.CN1CCCCC1.CN1CCN(C)CC1.CN1CCOCC1.CN1CCS(=O)(=O)CC1.CNC1=NCCN1.CNC1=NCCN1.CNCCN1CCN(C)CC1.CNCCN1CCOCC1.CNCCN1CCS(=O)(=O)CC1.COCCN1CCN(C)CC1.COCCN1CCOCC1.COCCN1CCS(=O)(=O)CC1.COCCNC(C)=O SFLYDRCPIHDGLG-UHFFFAOYSA-N 0.000 description 1
- NAHYYZFSRVYOBM-UHFFFAOYSA-N CC(=N)N1CCC(C(=O)O)CC1.CC(=N)N1CCCC1.CC(=N)N1CCCCC1.CC1=NCCN1C.COC(=O)C1CCN(C(C)=N)CC1 Chemical compound CC(=N)N1CCC(C(=O)O)CC1.CC(=N)N1CCCC1.CC(=N)N1CCCCC1.CC1=NCCN1C.COC(=O)C1CCN(C(C)=N)CC1 NAHYYZFSRVYOBM-UHFFFAOYSA-N 0.000 description 1
- WJJPQIOUKZEUIY-UHFFFAOYSA-N CC(=N)N1CCC(C)CC1.CC(=N)N1CCN(C)CC1.CC1CCN(C(=N)N)CC1.CCN(C)C.CCN(C)C1=NCCN1.CCN(C)C1=NCCN1C.CCN(C)C1=NCCO1.CCN1CCCCC1.CCN1CCN(C)CC1.CCN1CCNCC1.CCN1CCOCC1.CN1CCCCC1.CN1CCCNCC1.CN1CCN(C(=N)N)CC1.CN1CCN(C)CC1.CN1CCNCC1.CN1CCOCC1.CNC1=NCCN1.CNC1=NCCN1C.COC1CCN(C(=N)N)CC1.COC1CCN(C(C)=N)CC1.[H]N(CC)C1=NCCN1.[H]N(CC)C1=NCCN1C.[H]N(CC)C1=NCCO1 Chemical compound CC(=N)N1CCC(C)CC1.CC(=N)N1CCN(C)CC1.CC1CCN(C(=N)N)CC1.CCN(C)C.CCN(C)C1=NCCN1.CCN(C)C1=NCCN1C.CCN(C)C1=NCCO1.CCN1CCCCC1.CCN1CCN(C)CC1.CCN1CCNCC1.CCN1CCOCC1.CN1CCCCC1.CN1CCCNCC1.CN1CCN(C(=N)N)CC1.CN1CCN(C)CC1.CN1CCNCC1.CN1CCOCC1.CNC1=NCCN1.CNC1=NCCN1C.COC1CCN(C(=N)N)CC1.COC1CCN(C(C)=N)CC1.[H]N(CC)C1=NCCN1.[H]N(CC)C1=NCCN1C.[H]N(CC)C1=NCCO1 WJJPQIOUKZEUIY-UHFFFAOYSA-N 0.000 description 1
- IQBOPKXKKIQCDR-UHFFFAOYSA-N CC(=N)N1CCC(C)CC1.CC1=C(C#N)C=NC=N1.CC1=C(C(=N)N(C)C)C=CC=C1.CC1=C(C(N)=O)C=NC=C1.CC1=C(CN)C=NC=N1.CC1=C(CN)C=NC=N1.CC1=C(N)C=NC=N1.CC1=C(S(C)(=O)=O)C=NC=C1.CC1=C(S(C)(=O)=O)C=NC=N1.CC1=C(S(N)(=O)=O)C=NC=C1.CC1=CC(C(N)=O)=NC=N1.CC1=CC(CN)=NC=N1.CC1=CC(N(C)C)=NC=N1.CC1=CC(N)=NC=N1.CC1=CC=CN=N1.CC1=CC=NC(CN)=N1.CC1=CC=NC(N)=N1.CC1=CC=NC=N1.CC1=CC=NN=C1.CC1=CN=C(N)N1.CC1=CN=CN1.CC1=NCCC1.CC1=NCCCC1.CC1=NCCCO1.CC1=NCCO1.CC1=NCCS1.CC1=NN=NN1.CC1CCN(C(=N)N)CC1.CC1CCN(C)CC1.CCOC1=NC(C)=CC=N1.CN1C=CC=N1.CN1C=CN=C1.CN1C=CN=C1N.[C-]#[N+]C1=NC=NC(C)=C1 Chemical compound CC(=N)N1CCC(C)CC1.CC1=C(C#N)C=NC=N1.CC1=C(C(=N)N(C)C)C=CC=C1.CC1=C(C(N)=O)C=NC=C1.CC1=C(CN)C=NC=N1.CC1=C(CN)C=NC=N1.CC1=C(N)C=NC=N1.CC1=C(S(C)(=O)=O)C=NC=C1.CC1=C(S(C)(=O)=O)C=NC=N1.CC1=C(S(N)(=O)=O)C=NC=C1.CC1=CC(C(N)=O)=NC=N1.CC1=CC(CN)=NC=N1.CC1=CC(N(C)C)=NC=N1.CC1=CC(N)=NC=N1.CC1=CC=CN=N1.CC1=CC=NC(CN)=N1.CC1=CC=NC(N)=N1.CC1=CC=NC=N1.CC1=CC=NN=C1.CC1=CN=C(N)N1.CC1=CN=CN1.CC1=NCCC1.CC1=NCCCC1.CC1=NCCCO1.CC1=NCCO1.CC1=NCCS1.CC1=NN=NN1.CC1CCN(C(=N)N)CC1.CC1CCN(C)CC1.CCOC1=NC(C)=CC=N1.CN1C=CC=N1.CN1C=CN=C1.CN1C=CN=C1N.[C-]#[N+]C1=NC=NC(C)=C1 IQBOPKXKKIQCDR-UHFFFAOYSA-N 0.000 description 1
- OHZRDAKQZBYDJO-UHFFFAOYSA-N CC(=N)N1CCC(C)CC1.CC1=CC2=C(C=C1)C=C(Cl)O2.CC1=CC2=C(C=C1)C=C(Cl)S2.CC1=CC2=C(C=C1)CN(C)CC2.CC1=CC2=C(C=C1)C[N+](C)(C)CC2.CC1=CC2=C(C=C1)NCCC2.CC1=CC2=C(C[N+](C)(C)CC2)S1.CC1=CC2=C(N)N=CC=C2N1.CC1=CC2=CC(C(=N)N(C)C)=CC=C2N1.CC1=CC2=CC(C(=N)N(C)C)=CC=C2S1.CC1=CC2=CC(C3=NCCN3C)=CC=C2N1.CC1=CC2=CC(Cl)=CC=C2N1.CC1=CC2=CC(Cl)=CC=C2S1.CC1=CC2=CC=NC(N)=C2C=C1.CC1CCN(C(=N)N)CC1.CC1CCN(C)CC1.CN1CC1.CN1CCC1.CN1CCCC1.CN1CCCCC1.CN1CCCCCC1.CN1CCOCC1 Chemical compound CC(=N)N1CCC(C)CC1.CC1=CC2=C(C=C1)C=C(Cl)O2.CC1=CC2=C(C=C1)C=C(Cl)S2.CC1=CC2=C(C=C1)CN(C)CC2.CC1=CC2=C(C=C1)C[N+](C)(C)CC2.CC1=CC2=C(C=C1)NCCC2.CC1=CC2=C(C[N+](C)(C)CC2)S1.CC1=CC2=C(N)N=CC=C2N1.CC1=CC2=CC(C(=N)N(C)C)=CC=C2N1.CC1=CC2=CC(C(=N)N(C)C)=CC=C2S1.CC1=CC2=CC(C3=NCCN3C)=CC=C2N1.CC1=CC2=CC(Cl)=CC=C2N1.CC1=CC2=CC(Cl)=CC=C2S1.CC1=CC2=CC=NC(N)=C2C=C1.CC1CCN(C(=N)N)CC1.CC1CCN(C)CC1.CN1CC1.CN1CCC1.CN1CCCC1.CN1CCCCC1.CN1CCCCCC1.CN1CCOCC1 OHZRDAKQZBYDJO-UHFFFAOYSA-N 0.000 description 1
- IXSHIJWRBINTKF-UHFFFAOYSA-N CC(=N)N1CCC(C)CC1.CC1=CC=CC(CN)=C1.CC1=CC=NC(C#N)=C1.CC1=CC=NC(C(N)=O)=C1.CC1=CC=NC(CN)=C1.CC1=CC=NC=C1.CC1=CN=C(N)N1.CC1=CN=CN1.CC1=NC=NC(C#N)=C1.CC1=NC=NC(C(N)=O)=C1.CC1=NC=NC(CN)=C1S(C)(=O)=O.CC1=NC=NC(N(C)C)=C1.CC1=NC=NC(N)=C1.CC1=NC=NC=C1C#N.CC1=NC=NC=C1S(C)(=O)=O.CC1=NCCC1.CC1=NCCCC1.CC1=NCCCN1.CC1=NCCCN1C.CC1=NCCCO1.CC1=NCCCS1.CC1=NCCN1.CC1=NCCN1C.CC1=NCCO1.CC1=NCCS1.CC1=NN=NN1.CC1CCN(C(=N)N)CC1.CC1CCN(C)CC1.CCN(C)CC.CCN1CCCN=C1C.CCN1CCN=C1C.CCNC.CN.CN(C)C.CN1C=CN=C1.CN1C=CN=C1.CN1C=CN=C1N.CN1CCCC1.CN1CCCN(C)CC1.CNC.CNC(=N)N.CNC(C)(C)C.CNC(C)=N.CNN.CNNC.CNO.CNOC Chemical compound CC(=N)N1CCC(C)CC1.CC1=CC=CC(CN)=C1.CC1=CC=NC(C#N)=C1.CC1=CC=NC(C(N)=O)=C1.CC1=CC=NC(CN)=C1.CC1=CC=NC=C1.CC1=CN=C(N)N1.CC1=CN=CN1.CC1=NC=NC(C#N)=C1.CC1=NC=NC(C(N)=O)=C1.CC1=NC=NC(CN)=C1S(C)(=O)=O.CC1=NC=NC(N(C)C)=C1.CC1=NC=NC(N)=C1.CC1=NC=NC=C1C#N.CC1=NC=NC=C1S(C)(=O)=O.CC1=NCCC1.CC1=NCCCC1.CC1=NCCCN1.CC1=NCCCN1C.CC1=NCCCO1.CC1=NCCCS1.CC1=NCCN1.CC1=NCCN1C.CC1=NCCO1.CC1=NCCS1.CC1=NN=NN1.CC1CCN(C(=N)N)CC1.CC1CCN(C)CC1.CCN(C)CC.CCN1CCCN=C1C.CCN1CCN=C1C.CCNC.CN.CN(C)C.CN1C=CN=C1.CN1C=CN=C1.CN1C=CN=C1N.CN1CCCC1.CN1CCCN(C)CC1.CNC.CNC(=N)N.CNC(C)(C)C.CNC(C)=N.CNN.CNNC.CNO.CNOC IXSHIJWRBINTKF-UHFFFAOYSA-N 0.000 description 1
- ZJGUPROEEIYVTB-UHFFFAOYSA-N CC(=N)N1CCC(C)CC1.CC1CCN(C(=N)N)CC1.CCN(C)C1=NCCN1.CCN(C)C1=NCCN1.CCN(C)C1=NCCN1C.CCN(C)C1=NCCO1.CNC1=NCCN1.CNC1=NCCN1C.COC1CCN(C(=N)N)CC1.COC1CCN(C(C)=N)CC1.[H]N(CC)C1=NCCN1C.[H]N(CC)C1=NCCO1 Chemical compound CC(=N)N1CCC(C)CC1.CC1CCN(C(=N)N)CC1.CCN(C)C1=NCCN1.CCN(C)C1=NCCN1.CCN(C)C1=NCCN1C.CCN(C)C1=NCCO1.CNC1=NCCN1.CNC1=NCCN1C.COC1CCN(C(=N)N)CC1.COC1CCN(C(C)=N)CC1.[H]N(CC)C1=NCCN1C.[H]N(CC)C1=NCCO1 ZJGUPROEEIYVTB-UHFFFAOYSA-N 0.000 description 1
- JMBJYHDGIVOSPK-UHFFFAOYSA-N CC(=O)N(C)CCN(C)C.CC1=NCCC1.CC1=NCCN1.CCNC(C)=N.CN(C)CCN1CCOCC1.CNC1=NCCC1.COCCN(C)C(C)=O Chemical compound CC(=O)N(C)CCN(C)C.CC1=NCCC1.CC1=NCCN1.CCNC(C)=N.CN(C)CCN1CCOCC1.CNC1=NCCC1.COCCN(C)C(C)=O JMBJYHDGIVOSPK-UHFFFAOYSA-N 0.000 description 1
- PHNZQHGKDYROOO-UHFFFAOYSA-N CC(=O)N1CC(C)=C(C)C1.CC(=O)N1CCC(C)=C(C)C1.CC1=C(C)CN(C(=O)N(C)C)C1.CC1=C(C)CN(C(=O)N(C)C)CC1.CC1=C(C)CN(S(=O)(=O)N(C)C)C1.CC1=C(C)CN(S(=O)(=O)N(C)C)CC1.CC1=C(C)CN(S(C)(=O)=O)C1.CC1=C(C)CN(S(C)(=O)=O)CC1.CC1=C(C)CS(=O)(=O)C1.CC1=C(C)C[N-](C)C1.CC1=C(C)C[N-](C)CC1.CC1=C(C)C[N-](CC2=CC=CC=C2)CC1 Chemical compound CC(=O)N1CC(C)=C(C)C1.CC(=O)N1CCC(C)=C(C)C1.CC1=C(C)CN(C(=O)N(C)C)C1.CC1=C(C)CN(C(=O)N(C)C)CC1.CC1=C(C)CN(S(=O)(=O)N(C)C)C1.CC1=C(C)CN(S(=O)(=O)N(C)C)CC1.CC1=C(C)CN(S(C)(=O)=O)C1.CC1=C(C)CN(S(C)(=O)=O)CC1.CC1=C(C)CS(=O)(=O)C1.CC1=C(C)C[N-](C)C1.CC1=C(C)C[N-](C)CC1.CC1=C(C)C[N-](CC2=CC=CC=C2)CC1 PHNZQHGKDYROOO-UHFFFAOYSA-N 0.000 description 1
- RFZZRDZKOODKNJ-UHFFFAOYSA-N CC(=O)N1CCC(C)=C(C)C1.CC1=C(C)CN(C(=O)N(C)C)CC1.CC1=C(C)CN(S(=O)(=O)N(C)C)C1.CC1=C(C)CN(S(=O)(=O)N(C)C)CC1.CC1=C(C)CN(S(C)(=O)=O)CC1.CC1=C(C)C[N-](C)CC1.CC1=C(C)C[N-](CC2=CC=CC=C2)CC1 Chemical compound CC(=O)N1CCC(C)=C(C)C1.CC1=C(C)CN(C(=O)N(C)C)CC1.CC1=C(C)CN(S(=O)(=O)N(C)C)C1.CC1=C(C)CN(S(=O)(=O)N(C)C)CC1.CC1=C(C)CN(S(C)(=O)=O)CC1.CC1=C(C)C[N-](C)CC1.CC1=C(C)C[N-](CC2=CC=CC=C2)CC1 RFZZRDZKOODKNJ-UHFFFAOYSA-N 0.000 description 1
- HPELHESPFSJISY-UHFFFAOYSA-N CC(=O)NC1=CC(C(=O)NC2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)=C(OC2=CC(C(=N)N)=CC=C2)C=C1 Chemical compound CC(=O)NC1=CC(C(=O)NC2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)=C(OC2=CC(C(=N)N)=CC=C2)C=C1 HPELHESPFSJISY-UHFFFAOYSA-N 0.000 description 1
- MZACHOJLGWOECX-UHFFFAOYSA-N CC(=O)NC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)C=C1 Chemical compound CC(=O)NC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)C=C1 MZACHOJLGWOECX-UHFFFAOYSA-N 0.000 description 1
- YWQWOLGKDSZRQV-UHFFFAOYSA-N CC(=O)NC1=CC(OC2=CC=CC(C(=N)N)=C2)=C(C(=O)NC2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)C=C1.CN(C)C1=CC(OC2=CC=CC(C(=N)N)=C2)=C(C(=O)NC2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)C=C1.CNC1=CC(OC2=CC=CC(C(=N)N)=C2)=C(C(=O)NC2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)C=C1.CS(=O)(=O)NC1=CC(OC2=CC=CC(C(=N)N)=C2)=C(C(=O)NC2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)C=C1 Chemical compound CC(=O)NC1=CC(OC2=CC=CC(C(=N)N)=C2)=C(C(=O)NC2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)C=C1.CN(C)C1=CC(OC2=CC=CC(C(=N)N)=C2)=C(C(=O)NC2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)C=C1.CNC1=CC(OC2=CC=CC(C(=N)N)=C2)=C(C(=O)NC2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)C=C1.CS(=O)(=O)NC1=CC(OC2=CC=CC(C(=N)N)=C2)=C(C(=O)NC2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)C=C1 YWQWOLGKDSZRQV-UHFFFAOYSA-N 0.000 description 1
- FJKAKDOBAQGOGX-UHFFFAOYSA-N CC(C)(C)OC(=O)N1CCN(CC2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=N4)C=C(Cl)C=C3)C=C2)CC1.CN(CC1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)C1=CC=NC=N1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CN2C=CN=C2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CN2C=CN=N2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CN2C=NN=C2)C=C1 Chemical compound CC(C)(C)OC(=O)N1CCN(CC2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=N4)C=C(Cl)C=C3)C=C2)CC1.CN(CC1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)C1=CC=NC=N1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CN2C=CN=C2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CN2C=CN=N2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CN2C=NN=C2)C=C1 FJKAKDOBAQGOGX-UHFFFAOYSA-N 0.000 description 1
- KXTCQEUEUYGHLJ-UHFFFAOYSA-N CC.CC(=O)N(C)CCN(C)C.CC(=O)N1CCCCC1.CC1=NCCC1.CC1=NCCN1.CCNC(C)=N.CNC1=NCCC1.COCCN(C)C(C)=O.NCCN1CCOCC1 Chemical compound CC.CC(=O)N(C)CCN(C)C.CC(=O)N1CCCCC1.CC1=NCCC1.CC1=NCCN1.CCNC(C)=N.CNC1=NCCC1.COCCN(C)C(C)=O.NCCN1CCOCC1 KXTCQEUEUYGHLJ-UHFFFAOYSA-N 0.000 description 1
- HKKSDYHQOQNSHG-UHFFFAOYSA-N CC.CC1=C(CN(C)C2=NCCN2)C=CC=C1.CC1=C(CN(C)C2=NCCN2C)C=CC=C1.CC1=C(CNC2=NCCN2)C=CC=C1.CC1=C(CNC2=NCCN2C)C=CC=C1.CC1=C(NC2=NCCN2)C=CC=C1.CC1=C(NC2=NCCN2C)C=CC=C1.CC1=C(NC2=NCCO2)C=CC=C1.CC1=CC(C(N)=O)=NC=C1.CC1=CC(CN)=CC=C1.CC1=CC(CN)=NC=C1.CC1=CC=NC=C1.CC1=NCCCN1.CC1=NCCCN1C.CC1=NCCCS1.CC1=NCCN1.CC1=NCCN1C.CC1CCC(C)N1C.CC1CCCN1C.CCN(C)CC.CCN1CCCN=C1C.CCN1CCN=C1C.CCNC.CN.CN(C)C.CN1CC1.CN1CCC1.CN1CCCC1.CN1CCCC1C(=O)O.CN1CCCCC1.CN1CCCCCC1.CN1CCCN(C)CC1.CN1CCCNCC1.CN1CCN(C)CC1.CN1CCNCC1.CN1CCOCC1.CN1CCS(=O)(=O)CC1.CN1CCSCC1.CNC.CNC(=N)N.CNC(C)=N.CNN.CNNC.CNO.CNOC.[C-]#[N+]C1=NC=CC(C)=C1 Chemical compound CC.CC1=C(CN(C)C2=NCCN2)C=CC=C1.CC1=C(CN(C)C2=NCCN2C)C=CC=C1.CC1=C(CNC2=NCCN2)C=CC=C1.CC1=C(CNC2=NCCN2C)C=CC=C1.CC1=C(NC2=NCCN2)C=CC=C1.CC1=C(NC2=NCCN2C)C=CC=C1.CC1=C(NC2=NCCO2)C=CC=C1.CC1=CC(C(N)=O)=NC=C1.CC1=CC(CN)=CC=C1.CC1=CC(CN)=NC=C1.CC1=CC=NC=C1.CC1=NCCCN1.CC1=NCCCN1C.CC1=NCCCS1.CC1=NCCN1.CC1=NCCN1C.CC1CCC(C)N1C.CC1CCCN1C.CCN(C)CC.CCN1CCCN=C1C.CCN1CCN=C1C.CCNC.CN.CN(C)C.CN1CC1.CN1CCC1.CN1CCCC1.CN1CCCC1C(=O)O.CN1CCCCC1.CN1CCCCCC1.CN1CCCN(C)CC1.CN1CCCNCC1.CN1CCN(C)CC1.CN1CCNCC1.CN1CCOCC1.CN1CCS(=O)(=O)CC1.CN1CCSCC1.CNC.CNC(=N)N.CNC(C)=N.CNN.CNNC.CNO.CNOC.[C-]#[N+]C1=NC=CC(C)=C1 HKKSDYHQOQNSHG-UHFFFAOYSA-N 0.000 description 1
- VXHYZKKKMGHUIG-UHFFFAOYSA-N CC1=C(C#N)C=CC=C1.CC1=C(C#N)C=NC=C1.CC1=C(C(=N)N)C=CC=C1.CC1=C(C(N)=O)C=CC=C1.CC1=C(CN(C)C)C=CC=C1.CC1=C(CN(C)C)C=NC=C1.CC1=C(CN)C=CC=C1.CC1=C(CN)C=NC=C1.CC1=C(CN2CCCC2)C=CC=C1.CC1=C(CN2CCCCC2)C=CC=C1.CC1=C(CN2CCOCC2)C=CC=C1.CC1=C(N(C)C)C=NC=C1.CC1=C(N)C=NC=C1.CC1=C(S(C)(=O)=O)C=CC=C1.CC1=C(S(N)(=O)=O)C=CC=C1.CC1=CC(C(C)(C)N)=NC=C1.CC1=CC(C(N)=O)=CC=C1.CC1=CC(N(C)C)=NC=C1.CC1=CC(N)=NC=C1.CC1=CC=CC=C1.CNS(=O)(=O)C1=C(C)C=CC=C1.[C-]#[N+]C1=CC=CC(C)=C1 Chemical compound CC1=C(C#N)C=CC=C1.CC1=C(C#N)C=NC=C1.CC1=C(C(=N)N)C=CC=C1.CC1=C(C(N)=O)C=CC=C1.CC1=C(CN(C)C)C=CC=C1.CC1=C(CN(C)C)C=NC=C1.CC1=C(CN)C=CC=C1.CC1=C(CN)C=NC=C1.CC1=C(CN2CCCC2)C=CC=C1.CC1=C(CN2CCCCC2)C=CC=C1.CC1=C(CN2CCOCC2)C=CC=C1.CC1=C(N(C)C)C=NC=C1.CC1=C(N)C=NC=C1.CC1=C(S(C)(=O)=O)C=CC=C1.CC1=C(S(N)(=O)=O)C=CC=C1.CC1=CC(C(C)(C)N)=NC=C1.CC1=CC(C(N)=O)=CC=C1.CC1=CC(N(C)C)=NC=C1.CC1=CC(N)=NC=C1.CC1=CC=CC=C1.CNS(=O)(=O)C1=C(C)C=CC=C1.[C-]#[N+]C1=CC=CC(C)=C1 VXHYZKKKMGHUIG-UHFFFAOYSA-N 0.000 description 1
- XKFJDOYXHRJIDK-UHFFFAOYSA-N CC1=C(C(N)=O)C=C(Cl)C=C1.CC1=C(F)C=C(Br)C=C1.CC1=C(F)C=C(F)C=C1.CC1=C(SOON)C=C(Cl)C=C1.CC1=C(SOON)C=CC=C1.CC1=C([N+](=O)[O-])C=C(Br)C=C1.CC1=C([N+](=O)[O-])C=C(Cl)C=C1.CC1=C([N+](=O)[O-])C=CC=C1.CC1=CC(F)=C(Cl)C=C1.CC1=CC=C(Br)C=C1.CC1=CC=C(Cl)C=C1.CC1=CC=C(F)C=C1.CC1=CC=NC(N)=C1.CC1=CN=C(C)C=C1.CC1=CN=NC(N)=C1.CC1=NC=C(Br)C=C1.CC1=NC=C(Br)C=N1.CC1=NC=C(Cl)C=C1.CC1=NC=C(Cl)C=N1.CC1=NC=C(F)C=C1.CC1=NC=C(F)C=N1.COC(=O)C1=C(C)C=CC(Br)=C1.COC(=O)C1=C(C)C=CC(Cl)=C1.COC1=CN=C(C)C=C1.COOSC1=C(C)C=CC(Br)=C1.COOSC1=C(C)C=CC(Cl)=C1.COOSC1=C(C)C=CC=C1.[C-]#[N+]C1=C(C)C=CC(Cl)=C1 Chemical compound CC1=C(C(N)=O)C=C(Cl)C=C1.CC1=C(F)C=C(Br)C=C1.CC1=C(F)C=C(F)C=C1.CC1=C(SOON)C=C(Cl)C=C1.CC1=C(SOON)C=CC=C1.CC1=C([N+](=O)[O-])C=C(Br)C=C1.CC1=C([N+](=O)[O-])C=C(Cl)C=C1.CC1=C([N+](=O)[O-])C=CC=C1.CC1=CC(F)=C(Cl)C=C1.CC1=CC=C(Br)C=C1.CC1=CC=C(Cl)C=C1.CC1=CC=C(F)C=C1.CC1=CC=NC(N)=C1.CC1=CN=C(C)C=C1.CC1=CN=NC(N)=C1.CC1=NC=C(Br)C=C1.CC1=NC=C(Br)C=N1.CC1=NC=C(Cl)C=C1.CC1=NC=C(Cl)C=N1.CC1=NC=C(F)C=C1.CC1=NC=C(F)C=N1.COC(=O)C1=C(C)C=CC(Br)=C1.COC(=O)C1=C(C)C=CC(Cl)=C1.COC1=CN=C(C)C=C1.COOSC1=C(C)C=CC(Br)=C1.COOSC1=C(C)C=CC(Cl)=C1.COOSC1=C(C)C=CC=C1.[C-]#[N+]C1=C(C)C=CC(Cl)=C1 XKFJDOYXHRJIDK-UHFFFAOYSA-N 0.000 description 1
- CGAMUUYWRBCRPB-UHFFFAOYSA-N CC1=C(C(N)=O)C=C(Cl)C=C1.CC1=C(SOON)C=C(Cl)C=C1.CC1=C(SOON)C=CC=C1.CC1=C([N+](=O)[O-])C=C(Br)C=C1.CC1=C([N+](=O)[O-])C=C(Cl)C=C1.CC1=C([N+](=O)[O-])C=CC=C1.CC1=CC(Cl)=C(F)C=C1.CC1=CC(Cl)=CC(Cl)=C1.CC1=CC(N)=NC=C1.CC1=CC(N)=NN=C1.CC1=CC=C(Br)C=C1.CC1=CC=C(Br)C=C1F.CC1=CC=C(Br)C=N1.CC1=CC=C(Cl)C(F)=C1.CC1=CC=C(Cl)C=C1.CC1=CC=C(Cl)C=N1.CC1=CC=C(F)C=C1.CC1=CC=C(F)C=C1F.CC1=CC=C(F)C=N1.CC1=CC=C(N)N=C1.CC1=CC=C(O)C=C1.CC1=CC=NC=C1.CC1=CC=NN=C1.CC1=NC=C(Br)C=N1.CC1=NC=C(Cl)C=N1.CC1=NC=C(F)C=N1.COC1=C(Cl)C=CC(C)=C1.COC1=CC=C(C)C=C1.COOSC1=C(C)C=CC(Br)=C1.COOSC1=C(C)C=CC(Cl)=C1.COOSC1=C(C)C=CC=C1.[C-]#[N+]C1=C(C)C=CC(Cl)=C1 Chemical compound CC1=C(C(N)=O)C=C(Cl)C=C1.CC1=C(SOON)C=C(Cl)C=C1.CC1=C(SOON)C=CC=C1.CC1=C([N+](=O)[O-])C=C(Br)C=C1.CC1=C([N+](=O)[O-])C=C(Cl)C=C1.CC1=C([N+](=O)[O-])C=CC=C1.CC1=CC(Cl)=C(F)C=C1.CC1=CC(Cl)=CC(Cl)=C1.CC1=CC(N)=NC=C1.CC1=CC(N)=NN=C1.CC1=CC=C(Br)C=C1.CC1=CC=C(Br)C=C1F.CC1=CC=C(Br)C=N1.CC1=CC=C(Cl)C(F)=C1.CC1=CC=C(Cl)C=C1.CC1=CC=C(Cl)C=N1.CC1=CC=C(F)C=C1.CC1=CC=C(F)C=C1F.CC1=CC=C(F)C=N1.CC1=CC=C(N)N=C1.CC1=CC=C(O)C=C1.CC1=CC=NC=C1.CC1=CC=NN=C1.CC1=NC=C(Br)C=N1.CC1=NC=C(Cl)C=N1.CC1=NC=C(F)C=N1.COC1=C(Cl)C=CC(C)=C1.COC1=CC=C(C)C=C1.COOSC1=C(C)C=CC(Br)=C1.COOSC1=C(C)C=CC(Cl)=C1.COOSC1=C(C)C=CC=C1.[C-]#[N+]C1=C(C)C=CC(Cl)=C1 CGAMUUYWRBCRPB-UHFFFAOYSA-N 0.000 description 1
- URUSUGUAASHJOJ-UHFFFAOYSA-N CC1=C(C(N)=O)C=C(Cl)C=C1.CC1=C(SOON)C=C(Cl)C=C1.CC1=C(SOON)C=CC=C1.CC1=C([N+](=O)[O-])C=C(Br)C=C1.CC1=C([N+](=O)[O-])C=C(Cl)C=C1.CC1=C([N+](=O)[O-])C=CC=C1.CC1=CC=C(Br)C=C1.CC1=CC=C(Br)C=C1F.CC1=CC=C(Br)C=N1.CC1=CC=C(C)N=C1.CC1=CC=C(Cl)C(F)=C1.CC1=CC=C(Cl)C=C1.CC1=CC=C(Cl)C=N1.CC1=CC=C(F)C=C1.CC1=CC=C(F)C=C1F.CC1=CC=C(F)C=N1.CC1=NC=C(Br)C=N1.CC1=NC=C(Cl)C=N1.CC1=NC=C(F)C=N1.COC(=O)C1=C(C)C=CC(Br)=C1.COC(=O)C1=C(C)C=CC(Cl)=C1.COC1=CC=C(C)C=C1.COC1=CC=C(C)N=C1.COOSC1=C(C)C=CC(Br)=C1.COOSC1=C(C)C=CC(Cl)=C1.COOSC1=C(C)C=CC=C1.[C-]#[N+]C1=C(C)C=CC(Cl)=C1 Chemical compound CC1=C(C(N)=O)C=C(Cl)C=C1.CC1=C(SOON)C=C(Cl)C=C1.CC1=C(SOON)C=CC=C1.CC1=C([N+](=O)[O-])C=C(Br)C=C1.CC1=C([N+](=O)[O-])C=C(Cl)C=C1.CC1=C([N+](=O)[O-])C=CC=C1.CC1=CC=C(Br)C=C1.CC1=CC=C(Br)C=C1F.CC1=CC=C(Br)C=N1.CC1=CC=C(C)N=C1.CC1=CC=C(Cl)C(F)=C1.CC1=CC=C(Cl)C=C1.CC1=CC=C(Cl)C=N1.CC1=CC=C(F)C=C1.CC1=CC=C(F)C=C1F.CC1=CC=C(F)C=N1.CC1=NC=C(Br)C=N1.CC1=NC=C(Cl)C=N1.CC1=NC=C(F)C=N1.COC(=O)C1=C(C)C=CC(Br)=C1.COC(=O)C1=C(C)C=CC(Cl)=C1.COC1=CC=C(C)C=C1.COC1=CC=C(C)N=C1.COOSC1=C(C)C=CC(Br)=C1.COOSC1=C(C)C=CC(Cl)=C1.COOSC1=C(C)C=CC=C1.[C-]#[N+]C1=C(C)C=CC(Cl)=C1 URUSUGUAASHJOJ-UHFFFAOYSA-N 0.000 description 1
- WNFKLDYEPZSNQT-UHFFFAOYSA-N CC1=C(C(N)=O)C=C(Cl)C=C1F.CC1=C(F)C=C(Cl)C=C1F.CC1=C(N)C=C(Cl)C=C1F.CC1=C(SOON)C=C(Cl)C=C1F.CC1=CC(N)=C(Cl)C=C1F.CC1=CC=C(Cl)C(CN)=C1.CC1=CC=C(Cl)C(N)=C1.CC1=CC=C(Cl)C=C1C(N)=O.CC1=CC=C(Cl)C=C1N.CC1=CC=C(Cl)C=C1[N+](=O)[O-].COC1=CC=C(C)C=C1N.COOSC1=C(C)C(F)=CC(Cl)=C1.[C-]#[N+]C1=C(C)C(F)=CC(Cl)=C1.[C-]#[N+]C1=CC(Cl)=CC=C1C Chemical compound CC1=C(C(N)=O)C=C(Cl)C=C1F.CC1=C(F)C=C(Cl)C=C1F.CC1=C(N)C=C(Cl)C=C1F.CC1=C(SOON)C=C(Cl)C=C1F.CC1=CC(N)=C(Cl)C=C1F.CC1=CC=C(Cl)C(CN)=C1.CC1=CC=C(Cl)C(N)=C1.CC1=CC=C(Cl)C=C1C(N)=O.CC1=CC=C(Cl)C=C1N.CC1=CC=C(Cl)C=C1[N+](=O)[O-].COC1=CC=C(C)C=C1N.COOSC1=C(C)C(F)=CC(Cl)=C1.[C-]#[N+]C1=C(C)C(F)=CC(Cl)=C1.[C-]#[N+]C1=CC(Cl)=CC=C1C WNFKLDYEPZSNQT-UHFFFAOYSA-N 0.000 description 1
- IFRLPFBGTJEJLM-UHFFFAOYSA-N CC1=C(C(N)=O)C=C(Cl)C=N1.CC1=C(F)C=C(Cl)C=N1.CC1=C(F)C=C(Cl)N=C1.CC1=C(F)C=C(F)C=N1.CC1=C(F)C=CC=N1.CC1=C(SOON)C=C(Cl)C=N1.CC1=CC(F)=C(C)N=C1.CC1=CC(N)=NC=C1.CC1=CC=C(Cl)N=C1.COOSC1=C(C)N=CC(Cl)=C1.[C-]#[N+]C1=C(C)N=CC(Cl)=C1 Chemical compound CC1=C(C(N)=O)C=C(Cl)C=N1.CC1=C(F)C=C(Cl)C=N1.CC1=C(F)C=C(Cl)N=C1.CC1=C(F)C=C(F)C=N1.CC1=C(F)C=CC=N1.CC1=C(SOON)C=C(Cl)C=N1.CC1=CC(F)=C(C)N=C1.CC1=CC(N)=NC=C1.CC1=CC=C(Cl)N=C1.COOSC1=C(C)N=CC(Cl)=C1.[C-]#[N+]C1=C(C)N=CC(Cl)=C1 IFRLPFBGTJEJLM-UHFFFAOYSA-N 0.000 description 1
- GKSWRSARCJISCY-UHFFFAOYSA-N CC1=C(C(N)=O)C=CC(C(=N)N)=C1.CC1=C(C(N)=O)C=CC(CN)=C1.CC1=C(F)C=CC(C(=N)N)=C1.CC1=C(N)C=CC(C(=N)N)=C1.CC1=C(N)C=CC(CN)=C1.CC1=C(O)C=CC(C(=N)N)=C1.CC1=C(O)C=CC(CN)=C1.CC1=C(SOON)C=CC(C(=N)N)=C1.CC1=C(SOON)C=CC(CN)=C1.CC1=CC(F)=CC(C(=N)N)=C1.CC1=CC=CC(C(=N)N)=C1.CC1=CC=CC(C(=N)NO)=C1.CC1=CC=CC(C(N)=O)=C1.CONC(=N)C1=CC(C)=CC=C1.COOSC1=C(C)C=C(C(=N)N)C=C1.COOSC1=C(C)C=C(CN)C=C1.[C-]#[N+]C1=C(C)C=C(C(=N)N)C=C1.[C-]#[N+]C1=C(C)C=C(CN)C=C1 Chemical compound CC1=C(C(N)=O)C=CC(C(=N)N)=C1.CC1=C(C(N)=O)C=CC(CN)=C1.CC1=C(F)C=CC(C(=N)N)=C1.CC1=C(N)C=CC(C(=N)N)=C1.CC1=C(N)C=CC(CN)=C1.CC1=C(O)C=CC(C(=N)N)=C1.CC1=C(O)C=CC(CN)=C1.CC1=C(SOON)C=CC(C(=N)N)=C1.CC1=C(SOON)C=CC(CN)=C1.CC1=CC(F)=CC(C(=N)N)=C1.CC1=CC=CC(C(=N)N)=C1.CC1=CC=CC(C(=N)NO)=C1.CC1=CC=CC(C(N)=O)=C1.CONC(=N)C1=CC(C)=CC=C1.COOSC1=C(C)C=C(C(=N)N)C=C1.COOSC1=C(C)C=C(CN)C=C1.[C-]#[N+]C1=C(C)C=C(C(=N)N)C=C1.[C-]#[N+]C1=C(C)C=C(CN)C=C1 GKSWRSARCJISCY-UHFFFAOYSA-N 0.000 description 1
- IBZRGTOTOPBHJX-UHFFFAOYSA-N CC1=C(C(N)=O)C=CC(C(=N)N)=C1.CC1=C(C(N)=O)C=CC(CN)=C1.CC1=C(F)C=CC(C(=N)N)=C1.CC1=C(O)C=CC(CN)=C1.CC1=C(SOON)C=CC(C(=N)N)=C1.CC1=C(SOON)C=CC(CN)=C1.CC1=CC=CC(C(N)=O)=C1.COOSC1=C(C)C=C(C(=N)N)C=C1.COOSC1=C(C)C=C(CN)C=C1.[C-]#[N+]C1=C(C)C=C(CN)C=C1 Chemical compound CC1=C(C(N)=O)C=CC(C(=N)N)=C1.CC1=C(C(N)=O)C=CC(CN)=C1.CC1=C(F)C=CC(C(=N)N)=C1.CC1=C(O)C=CC(CN)=C1.CC1=C(SOON)C=CC(C(=N)N)=C1.CC1=C(SOON)C=CC(CN)=C1.CC1=CC=CC(C(N)=O)=C1.COOSC1=C(C)C=C(C(=N)N)C=C1.COOSC1=C(C)C=C(CN)C=C1.[C-]#[N+]C1=C(C)C=C(CN)C=C1 IBZRGTOTOPBHJX-UHFFFAOYSA-N 0.000 description 1
- FXJJPFFKWLXUNA-UHFFFAOYSA-N CC1=C(C(N)=O)C=CC(CN)=C1.CC1=C(CN)C(Br)=CC(N)=C1.CC1=C(F)C=CC(C(=N)N)=C1.CC1=C(O)C=C2C=CN=C(N)C2=C1.CC1=C(O)C=CC(C(=N)N)=C1.CC1=C(SOON)C=CC(N)=C1.CC1=C([N+](=O)[O-])C(Br)=CC(N)=C1.CC1=C([N+](=O)[O-])C=C2C=CC(Br)=CC2=C1.CC1=CC(Br)=CC(N)=C1.CC1=CC=C(F)C(C(=N)N)=C1.CC1=CC=CC(C(=N)N)=C1.CC1=CC=CC(C(=N)NN)=C1.CC1=CC=CC(C(=N)NO)=C1.CC1=CC=CC(C(N)=O)=C1.CC1=CC=CC(CN)=C1.CC1=CC=CC(N)=C1.CONC(=N)C1=CC(C)=CC=C1.COOSC1=C(C)C=C(C(N)=O)C=C1.[C-]#[N+]C1=C(C)C=C(C(=N)N)C=C1 Chemical compound CC1=C(C(N)=O)C=CC(CN)=C1.CC1=C(CN)C(Br)=CC(N)=C1.CC1=C(F)C=CC(C(=N)N)=C1.CC1=C(O)C=C2C=CN=C(N)C2=C1.CC1=C(O)C=CC(C(=N)N)=C1.CC1=C(SOON)C=CC(N)=C1.CC1=C([N+](=O)[O-])C(Br)=CC(N)=C1.CC1=C([N+](=O)[O-])C=C2C=CC(Br)=CC2=C1.CC1=CC(Br)=CC(N)=C1.CC1=CC=C(F)C(C(=N)N)=C1.CC1=CC=CC(C(=N)N)=C1.CC1=CC=CC(C(=N)NN)=C1.CC1=CC=CC(C(=N)NO)=C1.CC1=CC=CC(C(N)=O)=C1.CC1=CC=CC(CN)=C1.CC1=CC=CC(N)=C1.CONC(=N)C1=CC(C)=CC=C1.COOSC1=C(C)C=C(C(N)=O)C=C1.[C-]#[N+]C1=C(C)C=C(C(=N)N)C=C1 FXJJPFFKWLXUNA-UHFFFAOYSA-N 0.000 description 1
- MVMFXHJECCAYPT-UHFFFAOYSA-N CC1=C(C(N)=O)C=CC(CN)=C1.CC1=C(N)C=CC(CN)=C1.CC1=C(O)C=CC(CN)=C1.CC1=C(SOON)C=CC(CN)=C1.CC1=CC=C2C=CN=C(N)C2=C1.CC1=CC=CC(C(N)=O)=C1.COOSC1=C(C)C=C(CN)C=C1.[C-]#[N+]C1=C(C)C=C(CN)C=C1 Chemical compound CC1=C(C(N)=O)C=CC(CN)=C1.CC1=C(N)C=CC(CN)=C1.CC1=C(O)C=CC(CN)=C1.CC1=C(SOON)C=CC(CN)=C1.CC1=CC=C2C=CN=C(N)C2=C1.CC1=CC=CC(C(N)=O)=C1.COOSC1=C(C)C=C(CN)C=C1.[C-]#[N+]C1=C(C)C=C(CN)C=C1 MVMFXHJECCAYPT-UHFFFAOYSA-N 0.000 description 1
- OLIFKNWLHXZKLG-UHFFFAOYSA-N CC1=C(C(N)=O)C=CC(CN)=C1.CC1=C(O)C=C2C=CN=C(N)C2=C1.CC1=C(SOON)C=C2C=CC(Br)=CC2=C1.CC1=C(SOON)C=CC(N)=C1.CC1=C([N+](=O)[O-])C(Br)=CC(N)=C1.CC1=C([N+](=O)[O-])C=C2C=CC(Br)=CC2=C1.CC1=CC(Cl)=CC(Cl)=C1.CC1=CC=C(Br)C(Cl)=C1.CC1=CC=C(Cl)C(Cl)=C1.CC1=CC=C(F)C(Cl)=C1.CC1=CC=C(O)C=C1.CC1=CC=C2C(=C1)NN=C2Cl.CC1=CC=C2C=CNC2=C1.CC1=CC=C2C=NNC2=C1.CC1=CC=CC(C(=N)NO)=C1.CC1=CC=NC=C1.CC1=CN=C(N)C=C1.CC1=CN=NC=C1.COC1=CC(C)=CC=C1Cl.COC1=CC=C(C)C=C1Cl.COOSC1=C(C)C=C(C(N)=O)C=C1.COOSC1=C(C)C=C2C=C(Br)C=CC2=C1.[C-]#[N+]C1=C(C)C=C(C(=N)N)C=C1 Chemical compound CC1=C(C(N)=O)C=CC(CN)=C1.CC1=C(O)C=C2C=CN=C(N)C2=C1.CC1=C(SOON)C=C2C=CC(Br)=CC2=C1.CC1=C(SOON)C=CC(N)=C1.CC1=C([N+](=O)[O-])C(Br)=CC(N)=C1.CC1=C([N+](=O)[O-])C=C2C=CC(Br)=CC2=C1.CC1=CC(Cl)=CC(Cl)=C1.CC1=CC=C(Br)C(Cl)=C1.CC1=CC=C(Cl)C(Cl)=C1.CC1=CC=C(F)C(Cl)=C1.CC1=CC=C(O)C=C1.CC1=CC=C2C(=C1)NN=C2Cl.CC1=CC=C2C=CNC2=C1.CC1=CC=C2C=NNC2=C1.CC1=CC=CC(C(=N)NO)=C1.CC1=CC=NC=C1.CC1=CN=C(N)C=C1.CC1=CN=NC=C1.COC1=CC(C)=CC=C1Cl.COC1=CC=C(C)C=C1Cl.COOSC1=C(C)C=C(C(N)=O)C=C1.COOSC1=C(C)C=C2C=C(Br)C=CC2=C1.[C-]#[N+]C1=C(C)C=C(C(=N)N)C=C1 OLIFKNWLHXZKLG-UHFFFAOYSA-N 0.000 description 1
- YRBYHZZACDXXKE-UHFFFAOYSA-N CC1=C(C(N)=O)C=CC=C1.CC1=C(CN)C(N)=CC(Cl)=C1.CC1=C(F)C=CC=C1.CC1=C(N)C=CC=C1.CC1=C(SOON)C=CC=C1.CC1=C([N+](=O)[O-])C=CC=C1.CC1=CC(Cl)=C(Cl)C=C1.CC1=CC(N)=C(Cl)C=C1.CC1=CC(N)=C(Cl)C=C1F.CC1=CC(N)=CC(Cl)=C1.CC1=CC=CC=C1.COC1=C(Cl)C=C(C)C=C1.COC1=C(Cl)C=CC(C)=C1.COC1=C(N)C=C(C)C=C1.COOSC1=C(C)C=CC=C1.[C-]#[N+]C1=C(C)C=CC=C1 Chemical compound CC1=C(C(N)=O)C=CC=C1.CC1=C(CN)C(N)=CC(Cl)=C1.CC1=C(F)C=CC=C1.CC1=C(N)C=CC=C1.CC1=C(SOON)C=CC=C1.CC1=C([N+](=O)[O-])C=CC=C1.CC1=CC(Cl)=C(Cl)C=C1.CC1=CC(N)=C(Cl)C=C1.CC1=CC(N)=C(Cl)C=C1F.CC1=CC(N)=CC(Cl)=C1.CC1=CC=CC=C1.COC1=C(Cl)C=C(C)C=C1.COC1=C(Cl)C=CC(C)=C1.COC1=C(N)C=C(C)C=C1.COOSC1=C(C)C=CC=C1.[C-]#[N+]C1=C(C)C=CC=C1 YRBYHZZACDXXKE-UHFFFAOYSA-N 0.000 description 1
- ARPWIGFKMVWHQO-UHFFFAOYSA-N CC1=C(C)C2=C(C=CC=C2)N1.CC1=C(C)C2=C(C=CC=C2)S1.CC1=C(C)C2=C(CCC2)S1.CC1=C(C)N(C)C2=C1C=CC=C2.CC1=CC2=C(C=CC=C2)C=C1C Chemical compound CC1=C(C)C2=C(C=CC=C2)N1.CC1=C(C)C2=C(C=CC=C2)S1.CC1=C(C)C2=C(CCC2)S1.CC1=C(C)N(C)C2=C1C=CC=C2.CC1=CC2=C(C=CC=C2)C=C1C ARPWIGFKMVWHQO-UHFFFAOYSA-N 0.000 description 1
- RYLYMSQFHSWFBO-UHFFFAOYSA-N CC1=C(C)C2=C(C=CC=C2)N1.CC1=C(C)C2=C(C=CC=C2)S1.CC1=C(C)C2=C(CCC2)S1.CC1=CC2=C(C=CC=C2)C=C1C.CC1=CC2=C(C=CC=C2)N1C Chemical compound CC1=C(C)C2=C(C=CC=C2)N1.CC1=C(C)C2=C(C=CC=C2)S1.CC1=C(C)C2=C(CCC2)S1.CC1=CC2=C(C=CC=C2)C=C1C.CC1=CC2=C(C=CC=C2)N1C RYLYMSQFHSWFBO-UHFFFAOYSA-N 0.000 description 1
- SCHNJRDEGBKETG-UHFFFAOYSA-N CC1=C(C)SC=C1.CC1=C(C)SC=C1.CC1=CSC=C1C Chemical compound CC1=C(C)SC=C1.CC1=C(C)SC=C1.CC1=CSC=C1C SCHNJRDEGBKETG-UHFFFAOYSA-N 0.000 description 1
- IEZAFDXBZLMZQN-UHFFFAOYSA-N CC1=C(C2=CC=C(C(=O)Cl)C=C2)C=CC=C1.CC1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC(C#N)=CC=C4)SC=C3)C=C2)C=CC=C1.COC(=O)C1=C(N)C=CS1.COC(=O)C1=C(NC(=O)C2=CC=C(C3=C(C)C=CC=C3)C=C2)C=CS1.C[Al](C)C.N#CC1=CC=CC(N)=C1.N=C(N)C1=CC=CC(NC(=O)C2=C(NC(=O)C3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=CS2)=C1 Chemical compound CC1=C(C2=CC=C(C(=O)Cl)C=C2)C=CC=C1.CC1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC(C#N)=CC=C4)SC=C3)C=C2)C=CC=C1.COC(=O)C1=C(N)C=CS1.COC(=O)C1=C(NC(=O)C2=CC=C(C3=C(C)C=CC=C3)C=C2)C=CS1.C[Al](C)C.N#CC1=CC=CC(N)=C1.N=C(N)C1=CC=CC(NC(=O)C2=C(NC(=O)C3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=CS2)=C1 IEZAFDXBZLMZQN-UHFFFAOYSA-N 0.000 description 1
- JPBWWAFCYJHMTP-UHFFFAOYSA-N CC1=C(CN)C=NC=N1.CC1=C(N(C)C)C=NC=N1.CC1=C(N)C=NC=N1.CC1=CC=CC=C1CNC1=NCCN1.CC1=CC=CC=C1CNC1=NCCN1C.CC1=CC=NN=C1.CC1=CN=C(N)N1.CC1=CN=CN1.CC1=NC=CN1.CC1=NN=C(N)C=C1.CC1=NN=C(O)C=C1.CC1=NN=CC=C1.CC1=NN=NN1.CCOC1=NC(C)=CC=N1.CN1C=CN=C1 Chemical compound CC1=C(CN)C=NC=N1.CC1=C(N(C)C)C=NC=N1.CC1=C(N)C=NC=N1.CC1=CC=CC=C1CNC1=NCCN1.CC1=CC=CC=C1CNC1=NCCN1C.CC1=CC=NN=C1.CC1=CN=C(N)N1.CC1=CN=CN1.CC1=NC=CN1.CC1=NN=C(N)C=C1.CC1=NN=C(O)C=C1.CC1=NN=CC=C1.CC1=NN=NN1.CCOC1=NC(C)=CC=N1.CN1C=CN=C1 JPBWWAFCYJHMTP-UHFFFAOYSA-N 0.000 description 1
- OGJVUJZJPFRCMC-UHFFFAOYSA-N CC1=C(CN)C=NC=N1.CC1=C(N(C)C)C=NC=N1.CC1=C(N)C=NC=N1.CC1=CC=CC=C1CNC1=NCCN1.CC1=CC=CC=C1CNC1=NCCN1C.CC1=CC=NN=C1.CC1=CN=CN1.CC1=NC=CN1.CC1=NN=C(N)C=C1.CC1=NN=C(O)C=C1.CC1=NN=CC=C1.CC1=NN=NN1.CCOC1=NC(C)=CC=N1.CN1=CC=N(N)C1.CN1C=CN=C1 Chemical compound CC1=C(CN)C=NC=N1.CC1=C(N(C)C)C=NC=N1.CC1=C(N)C=NC=N1.CC1=CC=CC=C1CNC1=NCCN1.CC1=CC=CC=C1CNC1=NCCN1C.CC1=CC=NN=C1.CC1=CN=CN1.CC1=NC=CN1.CC1=NN=C(N)C=C1.CC1=NN=C(O)C=C1.CC1=NN=CC=C1.CC1=NN=NN1.CCOC1=NC(C)=CC=N1.CN1=CC=N(N)C1.CN1C=CN=C1 OGJVUJZJPFRCMC-UHFFFAOYSA-N 0.000 description 1
- NCBYLOFIOBQLOI-UHFFFAOYSA-N CC1=C(F)C(Cl)=CC=N1.CC1=C(F)C=C(Cl)C(N)=N1.CC1=C(F)C=NC(N)=C1.CC1=C(N)C=CC(C(=N)N)=C1.CC1=C(O)C=CC(C(=N)N)=C1.CC1=CC(Cl)=NC(N)=C1.CC1=CC(Cl)=NC=C1.CC1=CC=C(Cl)C(N)=N1.CC1=CC=CC(C(=N)N)=C1.CC1=CC=NC(N)=N1.[C-]#[N+]C1=C(C)C=C(C(=N)N)C=C1 Chemical compound CC1=C(F)C(Cl)=CC=N1.CC1=C(F)C=C(Cl)C(N)=N1.CC1=C(F)C=NC(N)=C1.CC1=C(N)C=CC(C(=N)N)=C1.CC1=C(O)C=CC(C(=N)N)=C1.CC1=CC(Cl)=NC(N)=C1.CC1=CC(Cl)=NC=C1.CC1=CC=C(Cl)C(N)=N1.CC1=CC=CC(C(=N)N)=C1.CC1=CC=NC(N)=N1.[C-]#[N+]C1=C(C)C=C(C(=N)N)C=C1 NCBYLOFIOBQLOI-UHFFFAOYSA-N 0.000 description 1
- OGFQADZNEGIDOP-UHFFFAOYSA-N CC1=C(F)C=C(C2=CC=[N+]([O-])C=C2)C=C1.CC1=CC2=C(C=C1)C=C(Cl)S2.CC1=CC2=C(C=C1)CN(C)CC2.CC1=CC2=C(C=C1)C[N+](C)(C)CC2.CC1=CC2=C(C=C1)NCCC2.CC1=CC2=C(CN(C)CC2)S1.CC1=CC2=C(C[N+](C)(C)CC2)S1.CC1=CC2=C(N)N=CC=C2N1.CC1=CC2=C(N)N=CC=C2S1.CC1=CC2=CC(N)=NC=C2C=C1.CC1=CC2=CC=NC(N)=C2C=C1.CC1=CC2=CN=CC=C2N1.CC1=CC2=CN=CC=C2S1.CC1=CC2=NC=CC=C2N1.CC1=CC2=NC=NC(N)=C2C=C1.CC1=CC=C(C2=CC(N)=NC=C2)C=C1.CC1=CC=C(C2=CC=NC=C2)C=C1.CC1=CC=C(C2=CC=[N+]([O-])C=C2)C=C1.CC1=CN=C(C2=CC=[N+](O)C=C2)N=C1 Chemical compound CC1=C(F)C=C(C2=CC=[N+]([O-])C=C2)C=C1.CC1=CC2=C(C=C1)C=C(Cl)S2.CC1=CC2=C(C=C1)CN(C)CC2.CC1=CC2=C(C=C1)C[N+](C)(C)CC2.CC1=CC2=C(C=C1)NCCC2.CC1=CC2=C(CN(C)CC2)S1.CC1=CC2=C(C[N+](C)(C)CC2)S1.CC1=CC2=C(N)N=CC=C2N1.CC1=CC2=C(N)N=CC=C2S1.CC1=CC2=CC(N)=NC=C2C=C1.CC1=CC2=CC=NC(N)=C2C=C1.CC1=CC2=CN=CC=C2N1.CC1=CC2=CN=CC=C2S1.CC1=CC2=NC=CC=C2N1.CC1=CC2=NC=NC(N)=C2C=C1.CC1=CC=C(C2=CC(N)=NC=C2)C=C1.CC1=CC=C(C2=CC=NC=C2)C=C1.CC1=CC=C(C2=CC=[N+]([O-])C=C2)C=C1.CC1=CN=C(C2=CC=[N+](O)C=C2)N=C1 OGFQADZNEGIDOP-UHFFFAOYSA-N 0.000 description 1
- IYNAKUDXVFISSG-UHFFFAOYSA-N CC1=C(F)C=C(Cl)C(N)=C1.CC1=CC2=C(C=CC=C2)N1.CC1=CC2=C(C=CN=C2)N1.CC1=CC=C(Br)C=C1.CC1=CC=C(Cl)C(N)=C1.CC1=CC=C(Cl)C=C1.CC1=CC=C(F)C=C1.CC1=NC=C(Br)C=C1.CC1=NC=C(Cl)C(F)=C1.CC1=NC=C(Cl)C(N)=C1.CC1=NC=C(Cl)C=C1.CC1=NC=C(F)C(Cl)=C1.CC1=NC=C(N)C=C1.CC1=NC=C2C=CC=CC2=C1.CC1=NC=C2C=CN=C(N)C2=C1.CC1=NC=C2C=CNC2=C1.CC1=NC=C2NC=CC2=C1.CC1=NC=CC(N)=C1.CC1=NC=NC(N)=C1.COC1=CN=C(C)C=C1 Chemical compound CC1=C(F)C=C(Cl)C(N)=C1.CC1=CC2=C(C=CC=C2)N1.CC1=CC2=C(C=CN=C2)N1.CC1=CC=C(Br)C=C1.CC1=CC=C(Cl)C(N)=C1.CC1=CC=C(Cl)C=C1.CC1=CC=C(F)C=C1.CC1=NC=C(Br)C=C1.CC1=NC=C(Cl)C(F)=C1.CC1=NC=C(Cl)C(N)=C1.CC1=NC=C(Cl)C=C1.CC1=NC=C(F)C(Cl)=C1.CC1=NC=C(N)C=C1.CC1=NC=C2C=CC=CC2=C1.CC1=NC=C2C=CN=C(N)C2=C1.CC1=NC=C2C=CNC2=C1.CC1=NC=C2NC=CC2=C1.CC1=NC=CC(N)=C1.CC1=NC=NC(N)=C1.COC1=CN=C(C)C=C1 IYNAKUDXVFISSG-UHFFFAOYSA-N 0.000 description 1
- YIWLSUZGAAKWBL-UHFFFAOYSA-N CC1=C(N)C=C2C=CN=C(N)C2=C1.CC1=C(N)C=C2C=CN=C(N)C2=C1.CC1=C(O)C=C2C=CN=C(N)C2=C1.CC1=C(SOON)C=C2C=CN=C(N)C2=C1.CC1=C([N+](=O)[O-])C=C2C=CN=C(N)C2=C1.COOSC1=C(C)C=C2C(=C1)C=CN=C2N Chemical compound CC1=C(N)C=C2C=CN=C(N)C2=C1.CC1=C(N)C=C2C=CN=C(N)C2=C1.CC1=C(O)C=C2C=CN=C(N)C2=C1.CC1=C(SOON)C=C2C=CN=C(N)C2=C1.CC1=C([N+](=O)[O-])C=C2C=CN=C(N)C2=C1.COOSC1=C(C)C=C2C(=C1)C=CN=C2N YIWLSUZGAAKWBL-UHFFFAOYSA-N 0.000 description 1
- CNVRJBYLTCPEQX-UHFFFAOYSA-N CC1=C(N)C=C2C=CN=C(N)C2=C1.CC1=C(SOON)C=C2C=CC(Br)=CC2=C1.CC1=C([N+](=O)[O-])C=C2C=CN=C(N)C2=C1.CC1=CC(Cl)=C(Br)C=C1.CC1=CC(Cl)=C(Cl)C=C1.CC1=CC(Cl)=C(F)C=C1.CC1=CC(Cl)=C(F)C=C1.CC1=CC(Cl)=CC(Cl)=C1.CC1=CC(N)=NC=C1.CC1=CC(N)=NN=C1.CC1=CC2=C(C=C1)C(Cl)=NN2.CC1=CC2=C(C=C1)C=NN2.CC1=CC2=C(C=CN2)C=C1.CC1=CC=C(N)N=C1.CC1=CC=C(O)C=C1.CC1=CC=C2C=CN=C(N)C2=C1.CC1=CC=NC=C1.CC1=CC=NN=C1.COC1=C(Cl)C=C(C)C=C1.COC1=C(Cl)C=CC(C)=C1.COOSC1=C(C)C=C2C(=C1)C=CN=C2N.COOSC1=C(C)C=C2C=C(Br)C=CC2=C1.[C-]#[N+]C1=C(C)C=C2C(=C1)C=CN=C2N Chemical compound CC1=C(N)C=C2C=CN=C(N)C2=C1.CC1=C(SOON)C=C2C=CC(Br)=CC2=C1.CC1=C([N+](=O)[O-])C=C2C=CN=C(N)C2=C1.CC1=CC(Cl)=C(Br)C=C1.CC1=CC(Cl)=C(Cl)C=C1.CC1=CC(Cl)=C(F)C=C1.CC1=CC(Cl)=C(F)C=C1.CC1=CC(Cl)=CC(Cl)=C1.CC1=CC(N)=NC=C1.CC1=CC(N)=NN=C1.CC1=CC2=C(C=C1)C(Cl)=NN2.CC1=CC2=C(C=C1)C=NN2.CC1=CC2=C(C=CN2)C=C1.CC1=CC=C(N)N=C1.CC1=CC=C(O)C=C1.CC1=CC=C2C=CN=C(N)C2=C1.CC1=CC=NC=C1.CC1=CC=NN=C1.COC1=C(Cl)C=C(C)C=C1.COC1=C(Cl)C=CC(C)=C1.COOSC1=C(C)C=C2C(=C1)C=CN=C2N.COOSC1=C(C)C=C2C=C(Br)C=CC2=C1.[C-]#[N+]C1=C(C)C=C2C(=C1)C=CN=C2N CNVRJBYLTCPEQX-UHFFFAOYSA-N 0.000 description 1
- OZECKYQXKKXKLK-UHFFFAOYSA-N CC1=C(N)C=CC(CN)=C1.CC1=CC(F)=CC(C(=N)N)=C1.CC1=CC=C(F)C(C(=N)N)=C1.CC1=CC=C2C=CN=CC2=C1.CC1=CC=CC(C(=N)NN)=C1.CC1=CC=CC(C(=N)NO)=C1.CONC(=N)C1=CC(C)=CC=C1 Chemical compound CC1=C(N)C=CC(CN)=C1.CC1=CC(F)=CC(C(=N)N)=C1.CC1=CC=C(F)C(C(=N)N)=C1.CC1=CC=C2C=CN=CC2=C1.CC1=CC=CC(C(=N)NN)=C1.CC1=CC=CC(C(=N)NO)=C1.CONC(=N)C1=CC(C)=CC=C1 OZECKYQXKKXKLK-UHFFFAOYSA-N 0.000 description 1
- DKLVGOOVBNVVOT-UHFFFAOYSA-N CC1=CC(C(=O)NC2=CC=C(Cl)C=C2C(=O)NC2=NC=C(Cl)C=C2)=CC=C1 Chemical compound CC1=CC(C(=O)NC2=CC=C(Cl)C=C2C(=O)NC2=NC=C(Cl)C=C2)=CC=C1 DKLVGOOVBNVVOT-UHFFFAOYSA-N 0.000 description 1
- MBZBLMSALZMHRA-UHFFFAOYSA-N CC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N(C)C)C=C2)C=C1 Chemical compound CC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N(C)C)C=C2)C=C1 MBZBLMSALZMHRA-UHFFFAOYSA-N 0.000 description 1
- GXEOEKOYKYRXFR-UHFFFAOYSA-N CC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NCC2=CC=C(C(=N)N3CCCC3)C=C2)C=C1 Chemical compound CC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NCC2=CC=C(C(=N)N3CCCC3)C=C2)C=C1 GXEOEKOYKYRXFR-UHFFFAOYSA-N 0.000 description 1
- LQTHVPXTYCTYBI-UHFFFAOYSA-N CC1=CC(C(=O)NC2=NC=C(Cl)C=C2)=C(NC(=O)C2=CC=C(C(=N)N3CCCC3)C=C2)C=C1.CC1=CC(C(=O)NC2=NC=C(Cl)C=C2)=C(NC(=O)C2=CC=C(C(=N)N3CCCCC3)C=C2)C=C1.CC1=CC(C(=O)NC2=NC=C(Cl)C=C2)=C(NC(=O)C2=CC=C(C3=NCCN3C)C=C2)C=C1 Chemical compound CC1=CC(C(=O)NC2=NC=C(Cl)C=C2)=C(NC(=O)C2=CC=C(C(=N)N3CCCC3)C=C2)C=C1.CC1=CC(C(=O)NC2=NC=C(Cl)C=C2)=C(NC(=O)C2=CC=C(C(=N)N3CCCCC3)C=C2)C=C1.CC1=CC(C(=O)NC2=NC=C(Cl)C=C2)=C(NC(=O)C2=CC=C(C3=NCCN3C)C=C2)C=C1 LQTHVPXTYCTYBI-UHFFFAOYSA-N 0.000 description 1
- IIRCSWQGHBXAEP-UHFFFAOYSA-N CC1=CC(C(=O)NC2=NC=C(Cl)C=C2)=C(NC(=O)C2=CC=C(C3=NCCN3C)C=C2)C=C1 Chemical compound CC1=CC(C(=O)NC2=NC=C(Cl)C=C2)=C(NC(=O)C2=CC=C(C3=NCCN3C)C=C2)C=C1 IIRCSWQGHBXAEP-UHFFFAOYSA-N 0.000 description 1
- UFSACVYWYVKXPB-UHFFFAOYSA-N CC1=CC(CN)=C(Cl)C=C1.CC1=CC(CN)=CC=C1.CC1=CC(N)=C(Cl)C=C1.CC1=CC=C(Cl)C=C1.CC1=CC=C(Cl)C=C1N.CC1=CC=C(Cl)N=C1.CC1=CC=C(N)N=C1.CC1=CC=C2C=CNC2=C1.CC1=CC=CC=C1.CC1=CN=C(C)C(F)=C1.CC1=NC2=C(C=C1)NC=C2.CC1=NC=C(Cl)C(F)=C1.CC1=NC=C(Cl)C=C1.CC1=NC=C(Cl)C=C1C(N)=O.CC1=NC=C(Cl)C=C1F.CC1=NC=C(Cl)C=C1F.CC1=NC=C(Cl)C=C1N.CC1=NC=C(Cl)C=C1SOON.CC1=NC=C(F)C=C1F.CC1=NC=C2C=CNC2=C1.COC1=CC=C(C)C=C1.COC1=CC=C(C)C=C1N.COOSC1=CC(Cl)=CN=C1C.[C-]#[N+]C1=CC(Cl)=CN=C1C Chemical compound CC1=CC(CN)=C(Cl)C=C1.CC1=CC(CN)=CC=C1.CC1=CC(N)=C(Cl)C=C1.CC1=CC=C(Cl)C=C1.CC1=CC=C(Cl)C=C1N.CC1=CC=C(Cl)N=C1.CC1=CC=C(N)N=C1.CC1=CC=C2C=CNC2=C1.CC1=CC=CC=C1.CC1=CN=C(C)C(F)=C1.CC1=NC2=C(C=C1)NC=C2.CC1=NC=C(Cl)C(F)=C1.CC1=NC=C(Cl)C=C1.CC1=NC=C(Cl)C=C1C(N)=O.CC1=NC=C(Cl)C=C1F.CC1=NC=C(Cl)C=C1F.CC1=NC=C(Cl)C=C1N.CC1=NC=C(Cl)C=C1SOON.CC1=NC=C(F)C=C1F.CC1=NC=C2C=CNC2=C1.COC1=CC=C(C)C=C1.COC1=CC=C(C)C=C1N.COOSC1=CC(Cl)=CN=C1C.[C-]#[N+]C1=CC(Cl)=CN=C1C UFSACVYWYVKXPB-UHFFFAOYSA-N 0.000 description 1
- VVBKFEOSIWNZTM-UHFFFAOYSA-N CC1=CC(Cl)=C(Br)C=C1.CC1=CC(Cl)=C(Cl)C=C1.CC1=CC(Cl)=C(F)C=C1.CC1=CC2=C(C=C1)C(Cl)=NN2.CC1=CC2=C(C=C1)C=NN2.CC1=CC2=C(C=CN2)C=C1.CC1=CC=C(C)N=C1.CC1=CC=CC=N1.COC1=C(Cl)C=C(C)C=C1.COC1=CC=C(C)N=C1 Chemical compound CC1=CC(Cl)=C(Br)C=C1.CC1=CC(Cl)=C(Cl)C=C1.CC1=CC(Cl)=C(F)C=C1.CC1=CC2=C(C=C1)C(Cl)=NN2.CC1=CC2=C(C=C1)C=NN2.CC1=CC2=C(C=CN2)C=C1.CC1=CC=C(C)N=C1.CC1=CC=CC=N1.COC1=C(Cl)C=C(C)C=C1.COC1=CC=C(C)N=C1 VVBKFEOSIWNZTM-UHFFFAOYSA-N 0.000 description 1
- LJZPCWFFVBGDSJ-UHFFFAOYSA-N CC1=CC(Cl)=C(C)C(F)=C1.CC1=CC(Cl)=C(C)C=C1.CC1=CC(F)=C(C)C(F)=C1.CC1=CC(F)=C(C)C=C1.CC1=CC(F)=C(C)C=C1.CC1=CC(F)=C(C)C=C1F.CC1=CC=C(C)C(Br)=C1.CC1=CC=C(C)C(C(=O)O)=C1.CC1=CC=C(C)C(C)=C1.CC1=CC=C(C)C=C1.CC1=CC=C(C)N=C1.CC1=CC=C(C)N=C1.CC1=CN(C)C(C)=C1.CC1=CN=C(C)C=N1.CC1=CN=C(C)N=C1.CC1=CN=C(C)N=C1.CC1=CNC(C)=C1.CC1=COC(C)=C1.CC1=CSC(C)=C1.CC1=CSC(C)=C1.CC1=CSC(C)=C1Cl.CC1=CSC(C)=N1.CC1CCC(C)CC1.CC1CCN(C)CC1.CC1CCN(C)CC1.CN1CCN(C)CC1.COC1=CC(C)=CC=C1C Chemical compound CC1=CC(Cl)=C(C)C(F)=C1.CC1=CC(Cl)=C(C)C=C1.CC1=CC(F)=C(C)C(F)=C1.CC1=CC(F)=C(C)C=C1.CC1=CC(F)=C(C)C=C1.CC1=CC(F)=C(C)C=C1F.CC1=CC=C(C)C(Br)=C1.CC1=CC=C(C)C(C(=O)O)=C1.CC1=CC=C(C)C(C)=C1.CC1=CC=C(C)C=C1.CC1=CC=C(C)N=C1.CC1=CC=C(C)N=C1.CC1=CN(C)C(C)=C1.CC1=CN=C(C)C=N1.CC1=CN=C(C)N=C1.CC1=CN=C(C)N=C1.CC1=CNC(C)=C1.CC1=COC(C)=C1.CC1=CSC(C)=C1.CC1=CSC(C)=C1.CC1=CSC(C)=C1Cl.CC1=CSC(C)=N1.CC1CCC(C)CC1.CC1CCN(C)CC1.CC1CCN(C)CC1.CN1CCN(C)CC1.COC1=CC(C)=CC=C1C LJZPCWFFVBGDSJ-UHFFFAOYSA-N 0.000 description 1
- HMZRTWGETAYJJO-UHFFFAOYSA-N CC1=CC(Cl)=C(C)C(F)=C1.CC1=CC(F)=C(C)C(F)=C1.CC1=CC(F)=C(C)C=C1F.CC1=CC=C(C)C(Br)=C1.CC1=CC=C(C)C(C)=C1.CC1=CC=C(C)C(Cl)=C1.CC1=CC=C(C)C(F)=C1.CC1=CC=C(C)C(F)=C1.CC1=CC=C(C)C=C1.CC1=CC=C(C)N=C1.CC1=CN(C)C(C)=C1.CC1=CN=C(C)N=C1.CC1=COC(C)=C1.CC1=CSC(C)=C1.CC1=CSC(C)=C1Cl Chemical compound CC1=CC(Cl)=C(C)C(F)=C1.CC1=CC(F)=C(C)C(F)=C1.CC1=CC(F)=C(C)C=C1F.CC1=CC=C(C)C(Br)=C1.CC1=CC=C(C)C(C)=C1.CC1=CC=C(C)C(Cl)=C1.CC1=CC=C(C)C(F)=C1.CC1=CC=C(C)C(F)=C1.CC1=CC=C(C)C=C1.CC1=CC=C(C)N=C1.CC1=CN(C)C(C)=C1.CC1=CN=C(C)N=C1.CC1=COC(C)=C1.CC1=CSC(C)=C1.CC1=CSC(C)=C1Cl HMZRTWGETAYJJO-UHFFFAOYSA-N 0.000 description 1
- NAUAOVQBALBTNU-UHFFFAOYSA-N CC1=CC(Cl)=C(F)C=C1.CC1=CC(Cl)=CC(Cl)=C1.CC1=CC(Cl)=CC(N)=C1.CC1=CC(F)=C(Cl)C=C1N.CC1=CC=CC(CN)=C1.CC1=CC=CC(N)=C1.CC1=CC=CC=C1.CC1=CC=NC=C1 Chemical compound CC1=CC(Cl)=C(F)C=C1.CC1=CC(Cl)=CC(Cl)=C1.CC1=CC(Cl)=CC(N)=C1.CC1=CC(F)=C(Cl)C=C1N.CC1=CC=CC(CN)=C1.CC1=CC=CC(N)=C1.CC1=CC=CC=C1.CC1=CC=NC=C1 NAUAOVQBALBTNU-UHFFFAOYSA-N 0.000 description 1
- GAJYBPLIIQSLGW-UHFFFAOYSA-N CC1=CC(Cl)=C(F)C=N1.CC1=CC(F)=C(Cl)C=N1.CC1=CC(N)=C(Cl)C=N1.CC1=CC(N)=CC=N1.CC1=CC(N)=NC=N1.CC1=CC(N)=NN=C1.CC1=CC=C(Br)C=N1.CC1=CC=C(Cl)C=N1.CC1=CC=C(Cl)N=C1.CC1=CC=C(F)C=N1.CC1=CC=C(N)C=N1.CC1=CC=C(N)N=C1.CC1=CC=C(O)C=N1.CC1=CC=NN=C1.CC1=NC=C(Br)C=N1.CC1=NC=C(Cl)C=N1.CC1=NC=C(F)C=N1.COC1=CC2=CC=C(C)C=C2C=C1.COC1=CC=C(C)N=C1 Chemical compound CC1=CC(Cl)=C(F)C=N1.CC1=CC(F)=C(Cl)C=N1.CC1=CC(N)=C(Cl)C=N1.CC1=CC(N)=CC=N1.CC1=CC(N)=NC=N1.CC1=CC(N)=NN=C1.CC1=CC=C(Br)C=N1.CC1=CC=C(Cl)C=N1.CC1=CC=C(Cl)N=C1.CC1=CC=C(F)C=N1.CC1=CC=C(N)C=N1.CC1=CC=C(N)N=C1.CC1=CC=C(O)C=N1.CC1=CC=NN=C1.CC1=NC=C(Br)C=N1.CC1=NC=C(Cl)C=N1.CC1=NC=C(F)C=N1.COC1=CC2=CC=C(C)C=C2C=C1.COC1=CC=C(C)N=C1 GAJYBPLIIQSLGW-UHFFFAOYSA-N 0.000 description 1
- URFPIQOGFSXYIJ-UHFFFAOYSA-N CC1=CC(OC2=CC(C(=N)N)=CC=C2)=C(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)C=C1.COC1=CC(OC2=CC(C(=N)N)=CC=C2)=C(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)C=C1.N=C(N)C1=CC=CC(OC2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=CC(Br)=C2)=C1.N=C(N)C1=CC=CC(OC2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=CC(Cl)=C2)=C1 Chemical compound CC1=CC(OC2=CC(C(=N)N)=CC=C2)=C(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)C=C1.COC1=CC(OC2=CC(C(=N)N)=CC=C2)=C(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)C=C1.N=C(N)C1=CC=CC(OC2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=CC(Br)=C2)=C1.N=C(N)C1=CC=CC(OC2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=CC(Cl)=C2)=C1 URFPIQOGFSXYIJ-UHFFFAOYSA-N 0.000 description 1
- LSDYUUICOLESQZ-UHFFFAOYSA-N CC1=CC2=C(/C=C(Cl)\C=C/2)O1.CC1=CC2=C(/C=C(Cl)\C=C/2)S1.CC1=CC2=C(/C=C\C(Cl)=C/2)N1.CC1=CC2=C(/C=C\C(Cl)=C/2)S1.CC1=CC=C2C=CC(Cl)=CC2=C1.COC1=CC2=CC(C)=CC=C2C=C1 Chemical compound CC1=CC2=C(/C=C(Cl)\C=C/2)O1.CC1=CC2=C(/C=C(Cl)\C=C/2)S1.CC1=CC2=C(/C=C\C(Cl)=C/2)N1.CC1=CC2=C(/C=C\C(Cl)=C/2)S1.CC1=CC=C2C=CC(Cl)=CC2=C1.COC1=CC2=CC(C)=CC=C2C=C1 LSDYUUICOLESQZ-UHFFFAOYSA-N 0.000 description 1
- UTDZNERJJAMMMD-UHFFFAOYSA-N CC1=CC2=C(/C=C\C=C/2)N1.CC1=CC=C2C=C(Cl)C=CC2=C1.CC1=CC=C2C=CC(Br)=CC2=C1.CC1=CC=C2NC(Cl)=CC2=C1.CC1=CC=C2NC(Cl)=CC2=C1.CC1=CC=C2NC=CC2=C1.CC1=CC=C2NN=CC2=C1.CC1=NC2=C(C=C1)NC=C2.CC1=NC=C2C=CC=CC2=C1.CC1=NC=C2C=CN=C(N)C2=C1.CC1=NC=C2C=CNC2=C1.CC1=NC=C2NC=CC2=C1 Chemical compound CC1=CC2=C(/C=C\C=C/2)N1.CC1=CC=C2C=C(Cl)C=CC2=C1.CC1=CC=C2C=CC(Br)=CC2=C1.CC1=CC=C2NC(Cl)=CC2=C1.CC1=CC=C2NC(Cl)=CC2=C1.CC1=CC=C2NC=CC2=C1.CC1=CC=C2NN=CC2=C1.CC1=NC2=C(C=C1)NC=C2.CC1=NC=C2C=CC=CC2=C1.CC1=NC=C2C=CN=C(N)C2=C1.CC1=NC=C2C=CNC2=C1.CC1=NC=C2NC=CC2=C1 UTDZNERJJAMMMD-UHFFFAOYSA-N 0.000 description 1
- PXIRDAQRPIWBLS-UHFFFAOYSA-N CC1=CC2=CC(C(=N)N(C)C)=CC=C2N1.CC1=CC2=CC(C(=N)N(C)C)=CC=C2S1.CC1=CC2=CC(C3=NCCN3C)=CC=C2N1.CC1=CC2=CC(C3=NCCN3C)=CC=C2S1.CC1=CC2=CC(Cl)=CC=C2N1.CC1=CC2=CC(Cl)=CC=C2S1.CC1=NC=C(C2=CC=[N+]([O-])C=C2)C=N1 Chemical compound CC1=CC2=CC(C(=N)N(C)C)=CC=C2N1.CC1=CC2=CC(C(=N)N(C)C)=CC=C2S1.CC1=CC2=CC(C3=NCCN3C)=CC=C2N1.CC1=CC2=CC(C3=NCCN3C)=CC=C2S1.CC1=CC2=CC(Cl)=CC=C2N1.CC1=CC2=CC(Cl)=CC=C2S1.CC1=NC=C(C2=CC=[N+]([O-])C=C2)C=N1 PXIRDAQRPIWBLS-UHFFFAOYSA-N 0.000 description 1
- WJDMCRWILYLUOJ-UHFFFAOYSA-N CC1=CC=C(Br)C=C1.CC1=CC=C(Br)C=C1F.CC1=CC=C(Cl)C(F)=C1.CC1=CC=C(Cl)C=C1.CC1=CC=C(Cl)C=C1F.CC1=CC=C(Cl)C=C1SOON.CC1=CC=C(F)C=C1.CC1=CC=C(F)C=C1F.CC1=CC=C(N)C=C1.CC1=CC=C(N)C=C1F.CC1=CC=C(O)C=C1.COC1=CC=C(C)C(F)=C1.COC1=CC=C(C)C=C1.COOSC1=CC(Cl)=CC=C1C Chemical compound CC1=CC=C(Br)C=C1.CC1=CC=C(Br)C=C1F.CC1=CC=C(Cl)C(F)=C1.CC1=CC=C(Cl)C=C1.CC1=CC=C(Cl)C=C1F.CC1=CC=C(Cl)C=C1SOON.CC1=CC=C(F)C=C1.CC1=CC=C(F)C=C1F.CC1=CC=C(N)C=C1.CC1=CC=C(N)C=C1F.CC1=CC=C(O)C=C1.COC1=CC=C(C)C(F)=C1.COC1=CC=C(C)C=C1.COOSC1=CC(Cl)=CC=C1C WJDMCRWILYLUOJ-UHFFFAOYSA-N 0.000 description 1
- SAYGYRNHBIKDDN-UHFFFAOYSA-N CC1=CC=C(C(=O)N(C)C2=CC=C(Cl)C=C2C(=O)NC2=NC=C(Cl)C=C2)C=C1.CC1=CC=C(C(=O)NC2=CC=C(Cl)C=C2C(=O)N(C)C2=CC=C(Cl)C=C2)C=C1 Chemical compound CC1=CC=C(C(=O)N(C)C2=CC=C(Cl)C=C2C(=O)NC2=NC=C(Cl)C=C2)C=C1.CC1=CC=C(C(=O)NC2=CC=C(Cl)C=C2C(=O)N(C)C2=CC=C(Cl)C=C2)C=C1 SAYGYRNHBIKDDN-UHFFFAOYSA-N 0.000 description 1
- LDFVNMLTOADCLP-UHFFFAOYSA-N CC1=CC=C(C(=O)NC2=CC=C(Cl)C=C2C(=O)NC2=CC=C(Cl)C=N2)C=N1.CC1=CC=C(C(=O)NC2=CC=C(Cl)C=C2C(=O)NC2=NC=C(Cl)C=C2)N=C1.CC1=CC=C(C(=O)NC2=NC=C(Cl)C=C2C(=O)NC2=CC=C(Cl)C=C2)C=C1.CC1=CC=C(C(=O)NC2=NC=C(Cl)C=C2C(=O)NC2=NC=C(Cl)C=C2)C=C1.CC1=CC=C(C(=O)NC2=NC=CN=C2C(=O)NC2=NC=C(Cl)C=C2)C=C1.CC1=CN=C(C(=O)NC2=CC=C(Cl)C=C2C(=O)NC2=CC=C(Cl)C=C2)N=C1.CC1=NC=C(C(=O)NC2=CC=C(Cl)C=C2C(=O)NC2=CC=C(Cl)C=N2)C=N1 Chemical compound CC1=CC=C(C(=O)NC2=CC=C(Cl)C=C2C(=O)NC2=CC=C(Cl)C=N2)C=N1.CC1=CC=C(C(=O)NC2=CC=C(Cl)C=C2C(=O)NC2=NC=C(Cl)C=C2)N=C1.CC1=CC=C(C(=O)NC2=NC=C(Cl)C=C2C(=O)NC2=CC=C(Cl)C=C2)C=C1.CC1=CC=C(C(=O)NC2=NC=C(Cl)C=C2C(=O)NC2=NC=C(Cl)C=C2)C=C1.CC1=CC=C(C(=O)NC2=NC=CN=C2C(=O)NC2=NC=C(Cl)C=C2)C=C1.CC1=CN=C(C(=O)NC2=CC=C(Cl)C=C2C(=O)NC2=CC=C(Cl)C=C2)N=C1.CC1=NC=C(C(=O)NC2=CC=C(Cl)C=C2C(=O)NC2=CC=C(Cl)C=N2)C=N1 LDFVNMLTOADCLP-UHFFFAOYSA-N 0.000 description 1
- SGOGMTYACCESJO-UHFFFAOYSA-N CC1=CC=C(C)C(Br)=C1.CC1=CC=C(C)C(C)=C1.CC1=CC=C(C)C(Cl)=C1.CC1=CC=C(C)C(F)=C1.CC1=CC=C(C)C(F)=C1.CC1=CC=C(C)C=C1.CC1=CC=C(C)N=C1.CC1=CC=C(C)N=C1.CC1=CN=C(C)C=N1.CC1=CN=C(C)N=C1.CC1=CN=C(C)N=C1.COC1=CC(C)=CC=C1C Chemical compound CC1=CC=C(C)C(Br)=C1.CC1=CC=C(C)C(C)=C1.CC1=CC=C(C)C(Cl)=C1.CC1=CC=C(C)C(F)=C1.CC1=CC=C(C)C(F)=C1.CC1=CC=C(C)C=C1.CC1=CC=C(C)N=C1.CC1=CC=C(C)N=C1.CC1=CN=C(C)C=N1.CC1=CN=C(C)N=C1.CC1=CN=C(C)N=C1.COC1=CC(C)=CC=C1C SGOGMTYACCESJO-UHFFFAOYSA-N 0.000 description 1
- GUMKPURKBXWJSA-UHFFFAOYSA-N CC1=CC=C(N)C=C1.CC1=CC=CC=C1C1=CC=C(C(=O)O)C=C1.CC1=CN=C(NC(=O)C2=C(NC(=O)C3=CC=C(C4=CC=CC=C4C)C=C3)C=CC=C2)C=C1.COC(=O)C1=C(N)C=CC=C1.COC(=O)C1=C(N)C=CC=C1.COC(=O)C1=C(NC(=O)C2=CC=C(C3=CC=CC=C3C)C=C2)C=CC=C1.C[Al](C)C.N.N.N.N.O=P(Cl)(Cl)(Cl)(Cl)Cl Chemical compound CC1=CC=C(N)C=C1.CC1=CC=CC=C1C1=CC=C(C(=O)O)C=C1.CC1=CN=C(NC(=O)C2=C(NC(=O)C3=CC=C(C4=CC=CC=C4C)C=C3)C=CC=C2)C=C1.COC(=O)C1=C(N)C=CC=C1.COC(=O)C1=C(N)C=CC=C1.COC(=O)C1=C(NC(=O)C2=CC=C(C3=CC=CC=C3C)C=C2)C=CC=C1.C[Al](C)C.N.N.N.N.O=P(Cl)(Cl)(Cl)(Cl)Cl GUMKPURKBXWJSA-UHFFFAOYSA-N 0.000 description 1
- OLWSVDBDDMGETK-UHFFFAOYSA-N CC1=CC=C(NC(=O)C2=C(NC(=O)C3=CC=C(C(=N)N4CCCCC4)C=C3)C(OCC(=O)O)=CC(Cl)=C2)N=C1.CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OCC(=O)O)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OCC(=O)O)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OCC(=O)O)C=C1)N1CCCC1 Chemical compound CC1=CC=C(NC(=O)C2=C(NC(=O)C3=CC=C(C(=N)N4CCCCC4)C=C3)C(OCC(=O)O)=CC(Cl)=C2)N=C1.CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OCC(=O)O)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OCC(=O)O)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OCC(=O)O)C=C1)N1CCCC1 OLWSVDBDDMGETK-UHFFFAOYSA-N 0.000 description 1
- OFGSZEQSBZPZMK-UHFFFAOYSA-N CC1=CC=C(NC(=O)C2=C(NC(=O)C3=CC=C(C(=N)N4CCCCCCC4)C=C3)C=CC=C2)N=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCSCC1 Chemical compound CC1=CC=C(NC(=O)C2=C(NC(=O)C3=CC=C(C(=N)N4CCCCCCC4)C=C3)C=CC=C2)N=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCSCC1 OFGSZEQSBZPZMK-UHFFFAOYSA-N 0.000 description 1
- SHAGXPSGZYLPAM-UHFFFAOYSA-N CC1=CC=C(NC(=O)C2=C(NC(=O)C3=CC=C(C4=NCCN4C)C=C3)C(O)=CC=C2)N=C1 Chemical compound CC1=CC=C(NC(=O)C2=C(NC(=O)C3=CC=C(C4=NCCN4C)C=C3)C(O)=CC=C2)N=C1 SHAGXPSGZYLPAM-UHFFFAOYSA-N 0.000 description 1
- AAXKZZMSCBNXNY-UHFFFAOYSA-N CC1=CC=C(NC(=O)C2=C(NC(=O)C3=CC=C(C4=NCCN4C)C=C3)C=CC=C2)C=C1 Chemical compound CC1=CC=C(NC(=O)C2=C(NC(=O)C3=CC=C(C4=NCCN4C)C=C3)C=CC=C2)C=C1 AAXKZZMSCBNXNY-UHFFFAOYSA-N 0.000 description 1
- ZNKQJEBYDCPKMV-UHFFFAOYSA-N CC1=CC=C(NC(=O)C2=C(NC(=O)C3=CC=C(C4=NCCN4C)C=C3)C=CC=C2)C=C1.CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1)N1CCCCC1 Chemical compound CC1=CC=C(NC(=O)C2=C(NC(=O)C3=CC=C(C4=NCCN4C)C=C3)C=CC=C2)C=C1.CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1)N1CCCCC1 ZNKQJEBYDCPKMV-UHFFFAOYSA-N 0.000 description 1
- TXSJOONEWSHCEG-UHFFFAOYSA-N CC1=CC=C(NC(=O)C2=C(OCC3=CC=C(C(=N)N4CCCC4)C=C3)C=CC(Cl)=C2)N=C1 Chemical compound CC1=CC=C(NC(=O)C2=C(OCC3=CC=C(C(=N)N4CCCC4)C=C3)C=CC(Cl)=C2)N=C1 TXSJOONEWSHCEG-UHFFFAOYSA-N 0.000 description 1
- ZBOBLPPBVSVNFX-UHFFFAOYSA-N CC1=CC=C(NC(=O)C2=C(OCC3=CC=C(C(=N)N4CCCCC4)C=C3)C=CC(Cl)=C2)N=C1.CN(C)C(=N)C1=CC=C(COC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1.CN1CCN=C1C1=CC=C(COC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1.N=C(C1=CC=C(COC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N1CCC1 Chemical compound CC1=CC=C(NC(=O)C2=C(OCC3=CC=C(C(=N)N4CCCCC4)C=C3)C=CC(Cl)=C2)N=C1.CN(C)C(=N)C1=CC=C(COC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1.CN1CCN=C1C1=CC=C(COC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1.N=C(C1=CC=C(COC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N1CCC1 ZBOBLPPBVSVNFX-UHFFFAOYSA-N 0.000 description 1
- OPRNBVWQDWMVQZ-UHFFFAOYSA-N CC1=CC=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1.CC1=CC=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.CC1=CC=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 Chemical compound CC1=CC=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1.CC1=CC=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.CC1=CC=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 OPRNBVWQDWMVQZ-UHFFFAOYSA-N 0.000 description 1
- CIQVMFVJZJNXPK-UHFFFAOYSA-N CC1=CC=CC=C1CN(C)C1=NCCN1.CC1=CC=CC=C1CNC1=NCCN1C.CC1=CC=CC=C1CNC1=NCCN1C.CC1=CC=CC=C1NC1=NCCN1.CC1=CC=CC=C1NC1=NCCN1C.CC1=CC=CC=C1NC1=NCCO1.CC1CCC(C)N1C.CC1CCCN1C.CN1CC1.CN1CCC1.CN1CCCC1C(=O)O.CN1CCCCC1.CN1CCCCCC1.CN1CCCNCC1.CN1CCN(C)CC1.CN1CCNCC1.CN1CCOCC1.CN1CCS(=O)(=O)CC1.CN1CCSCC1 Chemical compound CC1=CC=CC=C1CN(C)C1=NCCN1.CC1=CC=CC=C1CNC1=NCCN1C.CC1=CC=CC=C1CNC1=NCCN1C.CC1=CC=CC=C1NC1=NCCN1.CC1=CC=CC=C1NC1=NCCN1C.CC1=CC=CC=C1NC1=NCCO1.CC1CCC(C)N1C.CC1CCCN1C.CN1CC1.CN1CCC1.CN1CCCC1C(=O)O.CN1CCCCC1.CN1CCCCCC1.CN1CCCNCC1.CN1CCN(C)CC1.CN1CCNCC1.CN1CCOCC1.CN1CCS(=O)(=O)CC1.CN1CCSCC1 CIQVMFVJZJNXPK-UHFFFAOYSA-N 0.000 description 1
- DDLSVJROXXLLDN-UHFFFAOYSA-N CC1=CN=C(C(=O)NC2=CC=C(Cl)C=C2C(=O)NC2=CC=C(Cl)C=N2)N=C1.COC1=C(NC(=O)C2=NC=C(C)C=N2)C(C(=O)NC2=CC=C(Cl)C=N2)=CC(Cl)=C1 Chemical compound CC1=CN=C(C(=O)NC2=CC=C(Cl)C=C2C(=O)NC2=CC=C(Cl)C=N2)N=C1.COC1=C(NC(=O)C2=NC=C(C)C=N2)C(C(=O)NC2=CC=C(Cl)C=N2)=CC(Cl)=C1 DDLSVJROXXLLDN-UHFFFAOYSA-N 0.000 description 1
- WTRPZMWQLSFEHB-UHFFFAOYSA-N CC1=CSC(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)CCN(S(C)(=O)=O)C2)=C1Cl.CC1=CSC(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)CN(S(C)(=O)=O)CC2)=C1Cl Chemical compound CC1=CSC(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)CCN(S(C)(=O)=O)C2)=C1Cl.CC1=CSC(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)CN(S(C)(=O)=O)CC2)=C1Cl WTRPZMWQLSFEHB-UHFFFAOYSA-N 0.000 description 1
- RWNHOHQOYFEXOE-UHFFFAOYSA-N CC1=CSC(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)CCN(S(C)(=O)=O)C2)=C1Cl.CC1=CSC(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)CN(S(C)(=O)=O)CC2)=C1Cl Chemical compound CC1=CSC(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)CCN(S(C)(=O)=O)C2)=C1Cl.CC1=CSC(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)CN(S(C)(=O)=O)CC2)=C1Cl RWNHOHQOYFEXOE-UHFFFAOYSA-N 0.000 description 1
- FIQMLVUTJNEUQL-UHFFFAOYSA-N CC1=NCCCN1.CC1=NCCCN1C.CC1=NCCCN1C.CC1=NCCN1.CC1=NCCN1C.CCN1CCN=C1C.CN1CCCN(C)CC1 Chemical compound CC1=NCCCN1.CC1=NCCCN1C.CC1=NCCCN1C.CC1=NCCN1.CC1=NCCN1C.CCN1CCN=C1C.CN1CCCN(C)CC1 FIQMLVUTJNEUQL-UHFFFAOYSA-N 0.000 description 1
- VBCRXBCHLGFBBA-UHFFFAOYSA-N CC1=NCCCN1.CC1=NCCCN1C.CC1=NCCN1.CC1=NCCN1C.CCN1CCCN=C1C.CCN1CCN=C1C Chemical compound CC1=NCCCN1.CC1=NCCCN1C.CC1=NCCN1.CC1=NCCN1C.CCN1CCCN=C1C.CCN1CCN=C1C VBCRXBCHLGFBBA-UHFFFAOYSA-N 0.000 description 1
- FDJQQKXYQFYOTP-UHFFFAOYSA-N CC1CCCN1C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCCCC1 Chemical compound CC1CCCN1C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCCCC1 FDJQQKXYQFYOTP-UHFFFAOYSA-N 0.000 description 1
- WYLPCRGALLXCTL-UHFFFAOYSA-N CC1CCCN1C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1)N1CCCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1)N1CCOCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1)N1CCSCC1 Chemical compound CC1CCCN1C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1)N1CCCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1)N1CCOCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1)N1CCSCC1 WYLPCRGALLXCTL-UHFFFAOYSA-N 0.000 description 1
- YPYJGSQROTWEHE-UHFFFAOYSA-N CC1CCCN1C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1 Chemical compound CC1CCCN1C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1 YPYJGSQROTWEHE-UHFFFAOYSA-N 0.000 description 1
- VVNUXLCZIDJIIH-UHFFFAOYSA-N CCN(C)/C(=N\C#N)NC1CCCCC1.CCN(C)/C(N)=N\C#N.CCN(C)C(=N)C1CC1.CCN(C)C(=N)N(C)C.CCN(C)C(=N)N(C)C.CCN(C)C(=N)N(C)C.CCN(C)C(=N)NC.CCNC(=N)N(C)C.CCNC(=N)NC Chemical compound CCN(C)/C(=N\C#N)NC1CCCCC1.CCN(C)/C(N)=N\C#N.CCN(C)C(=N)C1CC1.CCN(C)C(=N)N(C)C.CCN(C)C(=N)N(C)C.CCN(C)C(=N)N(C)C.CCN(C)C(=N)NC.CCNC(=N)N(C)C.CCNC(=N)NC VVNUXLCZIDJIIH-UHFFFAOYSA-N 0.000 description 1
- ZQDFVVDXKWNOPY-UHFFFAOYSA-N CCN(C)C(=N)C1CC1.CCN(C)C(=N)C1CC1.CCN(C)C(=N)N.CCN(C)C(=N)N.CCN(C)C(=N)NC1CCCCC1.CCNC(=N)N.CCNC(=N)N(C)C.CCNC(=N)NC.CCNC(=N)NC1CCCCC1.CN(C)C(=N)C1=CC=CS1.CN(C)C(=N)C1CC1.CN(C)C(=N)N(C)C.CN/C(N)=N\[N+](=O)[O-].CN/C(N)=N\[N+](=O)[O-].CNC(=N)C1=CC=CC=C1.CNC(=N)C1=CC=CS1.CNC(=N)C1=CC=NC=C1.CNC(=N)C1CC1.CNC(=N)CCC(F)(F)F.CNC(=N)N.CNC(=N)N(C)C.CNC(=N)N(C)C.CNC(=N)NC.CNC(=N)NC.CNC(C)=N.CNC(C)=N.CNC1=NC=CN1.CNC1=NCCC1.CNC1=NCCN1.CNC1=NCCN1C Chemical compound CCN(C)C(=N)C1CC1.CCN(C)C(=N)C1CC1.CCN(C)C(=N)N.CCN(C)C(=N)N.CCN(C)C(=N)NC1CCCCC1.CCNC(=N)N.CCNC(=N)N(C)C.CCNC(=N)NC.CCNC(=N)NC1CCCCC1.CN(C)C(=N)C1=CC=CS1.CN(C)C(=N)C1CC1.CN(C)C(=N)N(C)C.CN/C(N)=N\[N+](=O)[O-].CN/C(N)=N\[N+](=O)[O-].CNC(=N)C1=CC=CC=C1.CNC(=N)C1=CC=CS1.CNC(=N)C1=CC=NC=C1.CNC(=N)C1CC1.CNC(=N)CCC(F)(F)F.CNC(=N)N.CNC(=N)N(C)C.CNC(=N)N(C)C.CNC(=N)NC.CNC(=N)NC.CNC(C)=N.CNC(C)=N.CNC1=NC=CN1.CNC1=NCCC1.CNC1=NCCN1.CNC1=NCCN1C ZQDFVVDXKWNOPY-UHFFFAOYSA-N 0.000 description 1
- AWDFPJHMDRECNO-UHFFFAOYSA-N CCN(C)C.CCN(C)C(=O)NC.CCN(C)C(=S)NC1=CC=CC=C1.CCN(C)C1=NCCC1.CCN(C)C1=NCCN1.CCN(C)C1=NCCN1C.CCN(C)C1=NCCO1.CCN(C)C1=NCCS1.CCN(C)C1CCCCC1.CCN(C)C1CCN(C)CC1.CCN(C)CCN(C)C.CCN(C)CCN1CCCCC1.CCN(C)CCN1CCN(C)CC1.CCN(C)CCN1CCOCC1.CCN(C)CCOC.CCN1CCCCC1.CCN1CCN(C)CC1.CCN1CCOCC1.CN(C)CCN(C)CCN(C)C.COCCN(C)CCN(C)C Chemical compound CCN(C)C.CCN(C)C(=O)NC.CCN(C)C(=S)NC1=CC=CC=C1.CCN(C)C1=NCCC1.CCN(C)C1=NCCN1.CCN(C)C1=NCCN1C.CCN(C)C1=NCCO1.CCN(C)C1=NCCS1.CCN(C)C1CCCCC1.CCN(C)C1CCN(C)CC1.CCN(C)CCN(C)C.CCN(C)CCN1CCCCC1.CCN(C)CCN1CCN(C)CC1.CCN(C)CCN1CCOCC1.CCN(C)CCOC.CCN1CCCCC1.CCN1CCN(C)CC1.CCN1CCOCC1.CN(C)CCN(C)CCN(C)C.COCCN(C)CCN(C)C AWDFPJHMDRECNO-UHFFFAOYSA-N 0.000 description 1
- XVHQOBNVVNURLA-UHFFFAOYSA-N CCN(C)C1=CC=NC=C1.CCN(C)C1=NC(N)=CC=N1.CCN(C)C1=NC(N)=NC=C1.CCN1C=CC(N)=N1.CCN1C=CC=N1.CCN1C=CN=C1.CCN1C=CN=C1NC.CCN1C=CN=N1.CCN1C=NCC1.CCN1C=NN=C1.CCN1CCCN=C1N.CCN1CCN=C1N.CCN1N=CC=N1 Chemical compound CCN(C)C1=CC=NC=C1.CCN(C)C1=NC(N)=CC=N1.CCN(C)C1=NC(N)=NC=C1.CCN1C=CC(N)=N1.CCN1C=CC=N1.CCN1C=CN=C1.CCN1C=CN=C1NC.CCN1C=CN=N1.CCN1C=NCC1.CCN1C=NN=C1.CCN1CCCN=C1N.CCN1CCN=C1N.CCN1N=CC=N1 XVHQOBNVVNURLA-UHFFFAOYSA-N 0.000 description 1
- BBFBTAYDUVSZHI-UHFFFAOYSA-N CCN(C)C1=NCCC1.CCN(C)C1=NCCCC1.CCN(C)C1=NCCCN1.CCN(C)C1=NCCCN1C.CCN(C)C1=NCCCO1.CCN(C)C1=NCCCS1.CCN(C)C1=NCCN1.CCN(C)C1=NCCN1C.CCN(C)C1=NCCO1.CCN(C)C1=NCCS1.CCN1C=CC(N)=C1.CCN1CCN(C)CC1.CCN1CCNC1=N Chemical compound CCN(C)C1=NCCC1.CCN(C)C1=NCCCC1.CCN(C)C1=NCCCN1.CCN(C)C1=NCCCN1C.CCN(C)C1=NCCCO1.CCN(C)C1=NCCCS1.CCN(C)C1=NCCN1.CCN(C)C1=NCCN1C.CCN(C)C1=NCCO1.CCN(C)C1=NCCS1.CCN1C=CC(N)=C1.CCN1CCN(C)CC1.CCN1CCNC1=N BBFBTAYDUVSZHI-UHFFFAOYSA-N 0.000 description 1
- ZOHRTTMDJLCBRG-UHFFFAOYSA-N CCN(C)C1=NCCC1.CCN(C)C1=NCCCC1.CCN(C)C1=NCCCN1.CCN(C)C1=NCCCN1C.CCN(C)C1=NCCCO1.CCN(C)C1=NCCCS1.CCN(C)C1=NCCN1.CCN(C)C1=NCCN1C.CCN(C)C1=NCCO1.CCN(C)C1=NCCS1.CCN1C=CC(N)=N1.CCN1CCN(C)CC1.CCN1CCNC1=N Chemical compound CCN(C)C1=NCCC1.CCN(C)C1=NCCCC1.CCN(C)C1=NCCCN1.CCN(C)C1=NCCCN1C.CCN(C)C1=NCCCO1.CCN(C)C1=NCCCS1.CCN(C)C1=NCCN1.CCN(C)C1=NCCN1C.CCN(C)C1=NCCO1.CCN(C)C1=NCCS1.CCN1C=CC(N)=N1.CCN1CCN(C)CC1.CCN1CCNC1=N ZOHRTTMDJLCBRG-UHFFFAOYSA-N 0.000 description 1
- SXJJMQPXMVJKOQ-UHFFFAOYSA-N CCN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.CN1CCCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Br)C=N2)C=CC=C1)C1=CC=C(C2=NCCCN2)C=C1 Chemical compound CCN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.CN1CCCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Br)C=N2)C=CC=C1)C1=CC=C(C2=NCCCN2)C=C1 SXJJMQPXMVJKOQ-UHFFFAOYSA-N 0.000 description 1
- YJALZZJPIZMAGE-UHFFFAOYSA-N CCN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1.CN1CCCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=CC=C1)C1=CC=C(C2=NCCCN2)C=C1 Chemical compound CCN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1.CN1CCCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=CC=C1)C1=CC=C(C2=NCCCN2)C=C1 YJALZZJPIZMAGE-UHFFFAOYSA-N 0.000 description 1
- PVPMUFZKRFXOBH-UHFFFAOYSA-N CCNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1.N=C(NC1CCCC1)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1 Chemical compound CCNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1.N=C(NC1CCCC1)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1 PVPMUFZKRFXOBH-UHFFFAOYSA-N 0.000 description 1
- HHSMJZIEKLAJCF-UHFFFAOYSA-N CCOC(=O)COC1=C(CC)C=C(C(=N)N)C=C1.CCOC(=O)COC1=C(OC)C=C(C(=N)N)C=C1.COC1=C(CC(N)=O)C=CC(C(=N)N)=C1.COC1=C(CS(=O)(=O)O)C=CC(C(=N)N)=C1.COC1=C(CS(C)(=O)=O)C=CC(C(=N)N)=C1.COC1=C(CS(N)(=O)=O)C=CC(C(=N)N)=C1.COC1=CC=C2C=CN=C(N)C2=C1.COC1=CC=CC(C(=N)NO)=C1 Chemical compound CCOC(=O)COC1=C(CC)C=C(C(=N)N)C=C1.CCOC(=O)COC1=C(OC)C=C(C(=N)N)C=C1.COC1=C(CC(N)=O)C=CC(C(=N)N)=C1.COC1=C(CS(=O)(=O)O)C=CC(C(=N)N)=C1.COC1=C(CS(C)(=O)=O)C=CC(C(=N)N)=C1.COC1=C(CS(N)(=O)=O)C=CC(C(=N)N)=C1.COC1=CC=C2C=CN=C(N)C2=C1.COC1=CC=CC(C(=N)NO)=C1 HHSMJZIEKLAJCF-UHFFFAOYSA-N 0.000 description 1
- HCZQMPIOEZHNRB-UHFFFAOYSA-N CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=C(F)C=C(C(=N)N(C)C)C=C2)C=C1 Chemical compound CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=C(F)C=C(C(=N)N(C)C)C=C2)C=C1 HCZQMPIOEZHNRB-UHFFFAOYSA-N 0.000 description 1
- TUEHJRZRHMBCJL-UHFFFAOYSA-N CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=C(F)C=C(C(=N)N)C=C2)C=C1 Chemical compound CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=C(F)C=C(C(=N)N)C=C2)C=C1 TUEHJRZRHMBCJL-UHFFFAOYSA-N 0.000 description 1
- YXFGUZZCUOQLSU-UHFFFAOYSA-N CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=C(F)C=C(C(=N)N3CCC3)C=C2)C=C1 Chemical compound CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=C(F)C=C(C(=N)N3CCC3)C=C2)C=C1 YXFGUZZCUOQLSU-UHFFFAOYSA-N 0.000 description 1
- JQKDEJGRPMWTAX-UHFFFAOYSA-N CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=C(F)C=C(C(=N)N3CCCC3)C=C2)C=C1 Chemical compound CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=C(F)C=C(C(=N)N3CCCC3)C=C2)C=C1 JQKDEJGRPMWTAX-UHFFFAOYSA-N 0.000 description 1
- MTTTZVLUPUHSFL-UHFFFAOYSA-N CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=C(F)C=C(C(=N)N3CCCCC3)C=C2)C=C1 Chemical compound CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=C(F)C=C(C(=N)N3CCCCC3)C=C2)C=C1 MTTTZVLUPUHSFL-UHFFFAOYSA-N 0.000 description 1
- JITBHWXQCWARKL-UHFFFAOYSA-N CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N(C)C)C=C2)C=C1 Chemical compound CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N(C)C)C=C2)C=C1 JITBHWXQCWARKL-UHFFFAOYSA-N 0.000 description 1
- MCUYDQPESFXMNV-UHFFFAOYSA-N CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N)C=C2)C=C1 Chemical compound CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N)C=C2)C=C1 MCUYDQPESFXMNV-UHFFFAOYSA-N 0.000 description 1
- QYZJMVRIVTZAMZ-UHFFFAOYSA-N CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCC3)C=C2)C=C1 Chemical compound CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCC3)C=C2)C=C1 QYZJMVRIVTZAMZ-UHFFFAOYSA-N 0.000 description 1
- UTBVMGGLXKRNSS-UHFFFAOYSA-N CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCCC3)C=C2)C=C1 Chemical compound CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCCC3)C=C2)C=C1 UTBVMGGLXKRNSS-UHFFFAOYSA-N 0.000 description 1
- AVMQUGFACYHRMW-UHFFFAOYSA-N CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCCCC3)C=C2)C=C1 Chemical compound CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCCCC3)C=C2)C=C1 AVMQUGFACYHRMW-UHFFFAOYSA-N 0.000 description 1
- XGKCFXFTNNBGSI-UHFFFAOYSA-N CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C3=NCCN3C)C=C2)C=C1 Chemical compound CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C3=NCCN3C)C=C2)C=C1 XGKCFXFTNNBGSI-UHFFFAOYSA-N 0.000 description 1
- UWVYBDIHQDLBPQ-UHFFFAOYSA-N CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C3=NCCN3C)C=C2F)C=C1 Chemical compound CCOC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C3=NCCN3C)C=C2F)C=C1 UWVYBDIHQDLBPQ-UHFFFAOYSA-N 0.000 description 1
- YIGNCUUNWCGIAS-UHFFFAOYSA-N CCOC(=O)COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1.CCOC(=O)COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.CCOC(=O)COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.CCOC(=O)COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 Chemical compound CCOC(=O)COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1.CCOC(=O)COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.CCOC(=O)COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.CCOC(=O)COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 YIGNCUUNWCGIAS-UHFFFAOYSA-N 0.000 description 1
- MNNARXWEYRXSSN-UHFFFAOYSA-N CCc(cc1)cc(C(CCc(cc2)ncc2Cl)=O)c1NC(c(ccc(C1=NCCN1C)c1)c1F)=O Chemical compound CCc(cc1)cc(C(CCc(cc2)ncc2Cl)=O)c1NC(c(ccc(C1=NCCN1C)c1)c1F)=O MNNARXWEYRXSSN-UHFFFAOYSA-N 0.000 description 1
- YHTJIPDQOMZNBM-UHFFFAOYSA-N CN(C)C(=N)C1=C(C2=CC=C(C(=O)NC3=CC=CC=C3C(=O)NC3=NC=C(Cl)C=C3)C=C2)C=CC=C1.CN(C)C(=N)C1=CC=C(C(=O)NC2=CC=CC=C2C(=O)NC2=NC=C(Cl)C=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=CC=CC=C2C(=O)NC2=NC=C(Cl)C=C2)C=C1.N=C(N)C1=C(C2=CC=C(C(=O)NC3=CC=CC=C3C(=O)NC3=NC=C(Cl)C=C3)C=C2)C=CC=C1.O=C(NC1=CC=CC=C1C(=O)NC1=NC=C(Cl)C=C1)C1=CC=C(C(=O)N2CCCC2)C=C1.O=C(NC1=CC=CC=C1C(=O)NC1=NC=C(Cl)C=C1)C1=CC=C(N2CCOCC2)C=C1 Chemical compound CN(C)C(=N)C1=C(C2=CC=C(C(=O)NC3=CC=CC=C3C(=O)NC3=NC=C(Cl)C=C3)C=C2)C=CC=C1.CN(C)C(=N)C1=CC=C(C(=O)NC2=CC=CC=C2C(=O)NC2=NC=C(Cl)C=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=CC=CC=C2C(=O)NC2=NC=C(Cl)C=C2)C=C1.N=C(N)C1=C(C2=CC=C(C(=O)NC3=CC=CC=C3C(=O)NC3=NC=C(Cl)C=C3)C=C2)C=CC=C1.O=C(NC1=CC=CC=C1C(=O)NC1=NC=C(Cl)C=C1)C1=CC=C(C(=O)N2CCCC2)C=C1.O=C(NC1=CC=CC=C1C(=O)NC1=NC=C(Cl)C=C1)C1=CC=C(N2CCOCC2)C=C1 YHTJIPDQOMZNBM-UHFFFAOYSA-N 0.000 description 1
- QFPHZYUZZGOXFY-UHFFFAOYSA-N CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1.CNC(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1 Chemical compound CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1.CNC(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1 QFPHZYUZZGOXFY-UHFFFAOYSA-N 0.000 description 1
- QPKLNXHLDAPCJY-UHFFFAOYSA-N CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.CNC(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1 Chemical compound CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.CNC(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1 QPKLNXHLDAPCJY-UHFFFAOYSA-N 0.000 description 1
- YBNBHDMLKWBAMC-UHFFFAOYSA-N CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCCC1 Chemical compound CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCCC1 YBNBHDMLKWBAMC-UHFFFAOYSA-N 0.000 description 1
- UVTUTODAHWYZPG-UHFFFAOYSA-N CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCCC1 Chemical compound CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCCC1 UVTUTODAHWYZPG-UHFFFAOYSA-N 0.000 description 1
- SHBYCTXGGKVJTK-UHFFFAOYSA-N CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2N(C)C)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCC2)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCCC2)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C2=NCCN2C)C=C1 Chemical compound CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2N(C)C)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCC2)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCCC2)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C2=NCCN2C)C=C1 SHBYCTXGGKVJTK-UHFFFAOYSA-N 0.000 description 1
- FJHKEOCMCVSKOM-UHFFFAOYSA-N CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1.CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1)N1CC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1)N1CCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1)N1CCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1)N1CCCCC1 Chemical compound CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1.CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1)N1CC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1)N1CCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1)N1CCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1)N1CCCCC1 FJHKEOCMCVSKOM-UHFFFAOYSA-N 0.000 description 1
- HFEROVWXSHODEJ-UHFFFAOYSA-N CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1.CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1)N1CC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1)N1CCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1)N1CCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1)N1CCCCC1 Chemical compound CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1.CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1)N1CC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1)N1CCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1)N1CCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1)N1CCCCC1 HFEROVWXSHODEJ-UHFFFAOYSA-N 0.000 description 1
- FVIPVWKPCJWYLB-UHFFFAOYSA-N CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1 Chemical compound CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1 FVIPVWKPCJWYLB-UHFFFAOYSA-N 0.000 description 1
- WEUOHYVXPBHZSH-UHFFFAOYSA-N CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1 Chemical compound CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1 WEUOHYVXPBHZSH-UHFFFAOYSA-N 0.000 description 1
- UVAUFOLBRSHIST-UHFFFAOYSA-N CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCOCC1 Chemical compound CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCOCC1 UVAUFOLBRSHIST-UHFFFAOYSA-N 0.000 description 1
- VINKZPRFHYGZTI-UHFFFAOYSA-N CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.CNC(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1)N1CCCC1.N=C(N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1 Chemical compound CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.CNC(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1)N1CCCC1.N=C(N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1 VINKZPRFHYGZTI-UHFFFAOYSA-N 0.000 description 1
- MPOFDIMJNOXSHH-UHFFFAOYSA-N CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.CNC(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Br)C=N4)SC=C3)C=C2)C=CC=C1.NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=N4)SC=C3)C=C2)C=CC=C1 Chemical compound CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.CNC(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Br)C=N4)SC=C3)C=C2)C=CC=C1.NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=N4)SC=C3)C=C2)C=CC=C1 MPOFDIMJNOXSHH-UHFFFAOYSA-N 0.000 description 1
- YUIXRLAQGAYMLE-UHFFFAOYSA-N CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1 Chemical compound CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1 YUIXRLAQGAYMLE-UHFFFAOYSA-N 0.000 description 1
- XXTWHUBBQVBBIX-UHFFFAOYSA-N CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCCC1 Chemical compound CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCCC1 XXTWHUBBQVBBIX-UHFFFAOYSA-N 0.000 description 1
- BMIIERWKEVDWKN-UHFFFAOYSA-N CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2N(C)C)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCC2)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCCC2)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C2=NCCN2C)C=C1 Chemical compound CN(C)C(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2N(C)C)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCC2)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCCC2)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C2=NCCN2C)C=C1 BMIIERWKEVDWKN-UHFFFAOYSA-N 0.000 description 1
- GOGOZXPYDKXPCP-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=C(C(N)=O)C=C(Cl)C=C3)C=NN2C)C=C1.CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=C(O)C=C(Cl)C=C3)C=NN2C)C=C1.COOSC1=C(NC(=O)C2=C(NC(=O)C3=CC=C(C(=N)N(C)C)C=C3)N(C)N=C2)C=CC(Cl)=C1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=C(C(N)=O)C=C(Cl)C=C3)C=NN2C)C=C1.CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=C(O)C=C(Cl)C=C3)C=NN2C)C=C1.COOSC1=C(NC(=O)C2=C(NC(=O)C3=CC=C(C(=N)N(C)C)C=C3)N(C)N=C2)C=CC(Cl)=C1 GOGOZXPYDKXPCP-UHFFFAOYSA-N 0.000 description 1
- JGZSCZRSVIMKGO-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=C(F)C=C(Cl)C=C3)C=CC=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=C(F)C=C(Cl)C=C3)C=CC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=C(F)C=C(Cl)C=C3)C=CC=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=C(F)C=C(Cl)C=C3)C=CC=C2)C=C1)N1CCCCC1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=C(F)C=C(Cl)C=C3)C=CC=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=C(F)C=C(Cl)C=C3)C=CC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=C(F)C=C(Cl)C=C3)C=CC=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=C(F)C=C(Cl)C=C3)C=CC=C2)C=C1)N1CCCCC1 JGZSCZRSVIMKGO-UHFFFAOYSA-N 0.000 description 1
- OLJHLFALPSCDJF-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=C(F)C=C(Cl)C=C3)C=NN2C)C=C1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=C(F)C=C(Cl)C=C3)C=NN2C)C=C1 OLJHLFALPSCDJF-UHFFFAOYSA-N 0.000 description 1
- FINVDQVNEHXNDG-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C(F)=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C(F)=C2)C=C1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C(F)=C2)C=C1.N=C(NO)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C(F)=C2)C=C1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C(F)=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C(F)=C2)C=C1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C(F)=C2)C=C1.N=C(NO)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C(F)=C2)C=C1 FINVDQVNEHXNDG-UHFFFAOYSA-N 0.000 description 1
- KHVDGWUKIYYNIU-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1.CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Br)C=N2)C=C(F)C=C1)C1=CC=C(C2=NCCN2)C=C1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1.CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Br)C=N2)C=C(F)C=C1)C1=CC=C(C2=NCCN2)C=C1 KHVDGWUKIYYNIU-UHFFFAOYSA-N 0.000 description 1
- BYSHBACJZCSIGC-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.CN1CCN(C(=N)C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Br)C=N4)C=CC=C3)C=C2)CC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCOCC1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.CN1CCN(C(=N)C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Br)C=N4)C=CC=C3)C=C2)CC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCOCC1 BYSHBACJZCSIGC-UHFFFAOYSA-N 0.000 description 1
- PZJULDOEVASNFI-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCCC1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCCC1 PZJULDOEVASNFI-UHFFFAOYSA-N 0.000 description 1
- KAEUCVHOAUMZCA-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCCC1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCCC1 KAEUCVHOAUMZCA-UHFFFAOYSA-N 0.000 description 1
- YDANAZDRFYRABP-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2N(C)C)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(Cl)C=C2N(C)C)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 YDANAZDRFYRABP-UHFFFAOYSA-N 0.000 description 1
- VEWDUPJXRRTZHD-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1)N1CC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1)N1CCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1)N1CCCCC1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1)N1CC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1)N1CCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1)N1CCCCC1 VEWDUPJXRRTZHD-UHFFFAOYSA-N 0.000 description 1
- URGSIKWJLPXPIR-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1)N1CC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1)N1CCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1)N1CCCCC1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1)N1CC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1)N1CCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=CC=C2)C=C1)N1CCCCC1 URGSIKWJLPXPIR-UHFFFAOYSA-N 0.000 description 1
- BRDDBZQHMIPDPT-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=NN2C)C=C1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=NN2C)C=C1 BRDDBZQHMIPDPT-UHFFFAOYSA-N 0.000 description 1
- HNUIDMLMHBOUTG-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2N(C)C)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2N(C)C)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 HNUIDMLMHBOUTG-UHFFFAOYSA-N 0.000 description 1
- GLUJWKNMSLOIND-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2N2CCOCC2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2N2CCOCC2)C=C1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2N2CCOCC2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2N2CCOCC2)C=C1 GLUJWKNMSLOIND-UHFFFAOYSA-N 0.000 description 1
- JITBRWSABYJDSF-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2O)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2O)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2O)C=C1)N1CCCCC1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2O)C=C1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2O)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2O)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2O)C=C1)N1CCCCC1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2O)C=C1 JITBRWSABYJDSF-UHFFFAOYSA-N 0.000 description 1
- XBJIODIGMFLUJD-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1)N1CCCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCCC1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1)N1CCCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCCC1 XBJIODIGMFLUJD-UHFFFAOYSA-N 0.000 description 1
- YRKQCBSGTTWMFF-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(N(C)C)C=C2)C=C1.COC1=CC(N(C)C)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(N(C)C)C=C2)C=C1.COC1=CC(N(C)C)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 YRKQCBSGTTWMFF-UHFFFAOYSA-N 0.000 description 1
- SHPLZBCWEPFFFA-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(N3CCOCC3)C=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(N3CCOCC3)C=C2)C=C1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(N3CCOCC3)C=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(N3CCOCC3)C=C2)C=C1 SHPLZBCWEPFFFA-UHFFFAOYSA-N 0.000 description 1
- VCQUPPLFOBCIHW-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1 VCQUPPLFOBCIHW-UHFFFAOYSA-N 0.000 description 1
- OAAVBJQEFYRXDS-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC([N+](=O)[O-])=C2)C=C1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC([N+](=O)[O-])=C2)C=C1 OAAVBJQEFYRXDS-UHFFFAOYSA-N 0.000 description 1
- RYBYBIPTYZISNI-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCCNCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCNCC1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCCNCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCNCC1 RYBYBIPTYZISNI-UHFFFAOYSA-N 0.000 description 1
- HCOYUSHHGXLAMS-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2O)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2O)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2O)C=C1)N1CCOCC1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2O)C=C1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2O)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2O)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2O)C=C1)N1CCOCC1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2O)C=C1 HCOYUSHHGXLAMS-UHFFFAOYSA-N 0.000 description 1
- VRUIADVEOBJNQO-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1)N1CCCC1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1)N1CCCC1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1 VRUIADVEOBJNQO-UHFFFAOYSA-N 0.000 description 1
- QTPIXVBFHQLPNC-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(C(F)(F)F)C=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(C(F)(F)F)C=C2)C=C1.COC1=CC(NC(=O)C2=CC=C(C3=NCCN3C)C=C2)=C(C(=O)NC2=NC=C(Br)C=C2)C=C1OC Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(C(F)(F)F)C=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(C(F)(F)F)C=C2)C=C1.COC1=CC(NC(=O)C2=CC=C(C3=NCCN3C)C=C2)=C(C(=O)NC2=NC=C(Br)C=C2)C=C1OC QTPIXVBFHQLPNC-UHFFFAOYSA-N 0.000 description 1
- KJAZPPJXQFETBD-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(Cl)C=C2)C=C1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(Cl)C=C2)C=C1 KJAZPPJXQFETBD-UHFFFAOYSA-N 0.000 description 1
- UWGGLQAXOABHKS-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCCC1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1)N1CCCCC1 UWGGLQAXOABHKS-UHFFFAOYSA-N 0.000 description 1
- PKBIRTVXVJUDOD-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2N(C)C)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2N(C)C)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 PKBIRTVXVJUDOD-UHFFFAOYSA-N 0.000 description 1
- GFVZLQXQFWOWKK-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(F)C(F)=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(F)C(F)=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(F)C(F)=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(F)C(F)=C2)C=C1)N1CCCCC1 Chemical compound CN(C)C(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(F)C(F)=C2)C=C1.CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(F)C(F)=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(F)C(F)=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(F)C(F)=C2)C=C1)N1CCCCC1 GFVZLQXQFWOWKK-UHFFFAOYSA-N 0.000 description 1
- QMPYHGYSCBQTBP-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1.CN1CCN=C1C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1.N=C(C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N1CCC1.N=C(C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N1CCCCC1 Chemical compound CN(C)C(=N)C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1.CN1CCN=C1C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1.N=C(C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N1CCC1.N=C(C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)N1CCCCC1 QMPYHGYSCBQTBP-UHFFFAOYSA-N 0.000 description 1
- FQLLSKNCRXTALL-UHFFFAOYSA-N CN(C)C(=N)C1=CC=C(NCC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1.N=C(C1=CC=C(NCC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCC1.N=C(C1=CC=C(NCC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCCCC1.N=C(C1=CC=C(NCC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCOCC1 Chemical compound CN(C)C(=N)C1=CC=C(NCC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1.N=C(C1=CC=C(NCC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCC1.N=C(C1=CC=C(NCC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCCCC1.N=C(C1=CC=C(NCC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCOCC1 FQLLSKNCRXTALL-UHFFFAOYSA-N 0.000 description 1
- UTRUPKWFJOXBCK-UHFFFAOYSA-N CN(C)C(=N)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CC=C2)C=C1 Chemical compound CN(C)C(=N)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CC=C2)C=C1 UTRUPKWFJOXBCK-UHFFFAOYSA-N 0.000 description 1
- BYHCQLXHAXZWGG-UHFFFAOYSA-N CN(C)C(c1ccc(C(Nc(ccc(I)c2)c2C(Nc(cc2)ncc2[I]2CC2)=O)=O)c(F)c1)=N Chemical compound CN(C)C(c1ccc(C(Nc(ccc(I)c2)c2C(Nc(cc2)ncc2[I]2CC2)=O)=O)c(F)c1)=N BYHCQLXHAXZWGG-UHFFFAOYSA-N 0.000 description 1
- VLYVKBYFVDGQAU-UHFFFAOYSA-N CN(C)C.CN(C)CCN(C)C.CN(C)CCN1CCCC1.CN(C)CCN1CCCCC1.CN(C)CCN1CCOCC1.CN(C)CCN1CCS(=O)(=O)CC1.CN1CCCC1.CN1CCCCC1.CN1CCN(C)CC1.CN1CCOCC1.COCCN(C)C.COCCN(C)C.COCCN(C)CCOC.COCCN1CCCC1.COCCN1CCCCC1.COCCN1CCOCC1.COCCN1CCS(=O)(=O)CC1.COCCOC.COCCS(C)(=O)=O Chemical compound CN(C)C.CN(C)CCN(C)C.CN(C)CCN1CCCC1.CN(C)CCN1CCCCC1.CN(C)CCN1CCOCC1.CN(C)CCN1CCS(=O)(=O)CC1.CN1CCCC1.CN1CCCCC1.CN1CCN(C)CC1.CN1CCOCC1.COCCN(C)C.COCCN(C)C.COCCN(C)CCOC.COCCN1CCCC1.COCCN1CCCCC1.COCCN1CCOCC1.COCCN1CCS(=O)(=O)CC1.COCCOC.COCCS(C)(=O)=O VLYVKBYFVDGQAU-UHFFFAOYSA-N 0.000 description 1
- ZEEQXRHRYPVRTN-UHFFFAOYSA-N CN(C)C1=CC(C(=O)NC2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)=C(OC2=CC=CC(C(=N)N)=C2)C=C1 Chemical compound CN(C)C1=CC(C(=O)NC2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)=C(OC2=CC=CC(C(=N)N)=C2)C=C1 ZEEQXRHRYPVRTN-UHFFFAOYSA-N 0.000 description 1
- KIQLHZPDGJHQFR-UHFFFAOYSA-N CN(C)C1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCCC3)C=C2)C=C1.CN(C)C1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCCCC3)C=C2)C=C1 Chemical compound CN(C)C1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCCC3)C=C2)C=C1.CN(C)C1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCCCC3)C=C2)C=C1 KIQLHZPDGJHQFR-UHFFFAOYSA-N 0.000 description 1
- CVMWBHNEUMSPOL-UHFFFAOYSA-N CN(C)C1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1 Chemical compound CN(C)C1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.CN(C)C1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1 CVMWBHNEUMSPOL-UHFFFAOYSA-N 0.000 description 1
- QCGIQWLUVIWBBC-UHFFFAOYSA-N CN(C)CC1=C(C2=CC=C(C(=O)NC3=CC=CC=C3C(=O)NC3=NC=C(Cl)C=C3)C=C2)C=CC=C1.NCC1=C(C2=CC=C(C(=O)NC3=CC=CC=C3C(=O)NC3=NC=C(Cl)C=C3)C=C2)C=CC=C1.NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=CC=CC=C3C(=O)NC3=NC=C(Cl)C=C3)C=C2)C=CC=C1.NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=CC=CC=C3NC(=O)C3=NC=C(Cl)C=C3)C=C2)C=CC=C1.NS(=O)(=O)C1=C(C2=CC=C(NC(=O)C3=CC=CC=C3C(=O)NC3=NC=C(Cl)C=C3)C=C2)C=CC=C1.NS(=O)(=O)C1=C(C2=CC=C(NC(=O)C3=CC=CC=C3NC(=O)C3=NC=C(Cl)C=C3)C=C2)C=CC=C1 Chemical compound CN(C)CC1=C(C2=CC=C(C(=O)NC3=CC=CC=C3C(=O)NC3=NC=C(Cl)C=C3)C=C2)C=CC=C1.NCC1=C(C2=CC=C(C(=O)NC3=CC=CC=C3C(=O)NC3=NC=C(Cl)C=C3)C=C2)C=CC=C1.NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=CC=CC=C3C(=O)NC3=NC=C(Cl)C=C3)C=C2)C=CC=C1.NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=CC=CC=C3NC(=O)C3=NC=C(Cl)C=C3)C=C2)C=CC=C1.NS(=O)(=O)C1=C(C2=CC=C(NC(=O)C3=CC=CC=C3C(=O)NC3=NC=C(Cl)C=C3)C=C2)C=CC=C1.NS(=O)(=O)C1=C(C2=CC=C(NC(=O)C3=CC=CC=C3NC(=O)C3=NC=C(Cl)C=C3)C=C2)C=CC=C1 QCGIQWLUVIWBBC-UHFFFAOYSA-N 0.000 description 1
- KQICCYXXTYGXDU-UHFFFAOYSA-N CN(CC1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)C1=CC=CC=N1.CN(CC1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)C1=NCCN1.CN1CCN=C1N(C)CC1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1.CN1CCN=C1NCC1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CNC2=NCCN2)C=C1 Chemical compound CN(CC1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)C1=CC=CC=N1.CN(CC1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1)C1=NCCN1.CN1CCN=C1N(C)CC1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1.CN1CCN=C1NCC1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CNC2=NCCN2)C=C1 KQICCYXXTYGXDU-UHFFFAOYSA-N 0.000 description 1
- VQBOBKQYEQQALA-UHFFFAOYSA-N CN1C(c(cc2)cc(F)c2C(Nc(ccc(F)c2)c2C(Nc(cc2)ncc2Br)=O)=O)=NCC1 Chemical compound CN1C(c(cc2)cc(F)c2C(Nc(ccc(F)c2)c2C(Nc(cc2)ncc2Br)=O)=O)=NCC1 VQBOBKQYEQQALA-UHFFFAOYSA-N 0.000 description 1
- FWCIVTXRKDWGNH-UHFFFAOYSA-N CN1CCCCC1.CN1CCCCC1.CN1CCN(C)CC1.CN1CCNCC1 Chemical compound CN1CCCCC1.CN1CCCCC1.CN1CCN(C)CC1.CN1CCNCC1 FWCIVTXRKDWGNH-UHFFFAOYSA-N 0.000 description 1
- CAPNTVYBHBEKNH-UHFFFAOYSA-N CN1CCCN(C(=N)C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=N4)C=CC=C3)C=C2)CC1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1 Chemical compound CN1CCCN(C(=N)C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=N4)C=CC=C3)C=C2)CC1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1 CAPNTVYBHBEKNH-UHFFFAOYSA-N 0.000 description 1
- NHSVYWZKKRHEJN-UHFFFAOYSA-N CN1CCCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1)N1CCCCCC1.N=C(NO)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1 Chemical compound CN1CCCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1)N1CCCCCC1.N=C(NO)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1 NHSVYWZKKRHEJN-UHFFFAOYSA-N 0.000 description 1
- BECYWMPOBWHXMA-UHFFFAOYSA-N CN1CCCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1 Chemical compound CN1CCCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1 BECYWMPOBWHXMA-UHFFFAOYSA-N 0.000 description 1
- TWYICXFZWHEIDT-UHFFFAOYSA-N CN1CCN(C(=N)C2=CC(F)=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=N4)C=C(F)C=C3)C=C2)CC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCS(=O)(=O)CC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCSCC1 Chemical compound CN1CCN(C(=N)C2=CC(F)=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=N4)C=C(F)C=C3)C=C2)CC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCS(=O)(=O)CC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCSCC1 TWYICXFZWHEIDT-UHFFFAOYSA-N 0.000 description 1
- SDJWAESWYPBMPD-UHFFFAOYSA-N CN1CCN(C(=N)C2=CC(F)=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=N4)C=CC=C3)C=C2)CC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCSCC1.N=C(N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1 Chemical compound CN1CCN(C(=N)C2=CC(F)=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=N4)C=CC=C3)C=C2)CC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCSCC1.N=C(N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1 SDJWAESWYPBMPD-UHFFFAOYSA-N 0.000 description 1
- ZLUXREWBLCVHDJ-UHFFFAOYSA-N CN1CCN(C(=N)C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Br)C=N4)C=C(F)C=C3)C=C2)CC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1)N1CCOCC1 Chemical compound CN1CCN(C(=N)C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Br)C=N4)C=C(F)C=C3)C=C2)CC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1)N1CCOCC1 ZLUXREWBLCVHDJ-UHFFFAOYSA-N 0.000 description 1
- JXADQKNAJNOIHV-UHFFFAOYSA-N CN1CCN(C(=N)C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=C4)C=NN3C)C=C2)CC1 Chemical compound CN1CCN(C(=N)C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=C4)C=NN3C)C=C2)CC1 JXADQKNAJNOIHV-UHFFFAOYSA-N 0.000 description 1
- VQYMLZYTBPMCNI-UHFFFAOYSA-N CN1CCN(C(=N)C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=N4)C=NN3C)C=C2)CC1 Chemical compound CN1CCN(C(=N)C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=N4)C=NN3C)C=C2)CC1 VQYMLZYTBPMCNI-UHFFFAOYSA-N 0.000 description 1
- LOQFWJQCKWJAPD-UHFFFAOYSA-N CN1CCN(C(=N)C2=CC=C(CNC3=C(C(=O)NC4=CC=C(Cl)C=N4)C=CC=C3)C=C2)CC1.N=C(C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCCCCC1.N=C(C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCNCC1.N=C(C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCSCC1 Chemical compound CN1CCN(C(=N)C2=CC=C(CNC3=C(C(=O)NC4=CC=C(Cl)C=N4)C=CC=C3)C=C2)CC1.N=C(C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCCCCC1.N=C(C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCNCC1.N=C(C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCSCC1 LOQFWJQCKWJAPD-UHFFFAOYSA-N 0.000 description 1
- WIWDJWUWHPRFSL-UHFFFAOYSA-N CN1CCN(C2=CC(C(=O)NC3=CC=C(Br)C=N3)=C(NC(=O)C3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=C2)CC1 Chemical compound CN1CCN(C2=CC(C(=O)NC3=CC=C(Br)C=N3)=C(NC(=O)C3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=C2)CC1 WIWDJWUWHPRFSL-UHFFFAOYSA-N 0.000 description 1
- FYLNCGHAQLRJSD-UHFFFAOYSA-N CN1CCN(CC2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=N4)C=C(Cl)C=C3)C=C2)CC1.NC1=NC=CN1CC1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CN2C=NN=N2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CN2CCNCC2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CN2CCSCC2)C=C1 Chemical compound CN1CCN(CC2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=N4)C=C(Cl)C=C3)C=C2)CC1.NC1=NC=CN1CC1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CN2C=NN=N2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CN2CCNCC2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CN2CCSCC2)C=C1 FYLNCGHAQLRJSD-UHFFFAOYSA-N 0.000 description 1
- FNCCDSQXNRXBJI-UHFFFAOYSA-N CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1)N1CCC1 Chemical compound CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1)N1CCC1 FNCCDSQXNRXBJI-UHFFFAOYSA-N 0.000 description 1
- JBLITAKBVUQPFX-UHFFFAOYSA-N CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCOCC1 Chemical compound CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCOCC1 JBLITAKBVUQPFX-UHFFFAOYSA-N 0.000 description 1
- RIEMNBCQTHHNBE-UHFFFAOYSA-N CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1 Chemical compound CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1 RIEMNBCQTHHNBE-UHFFFAOYSA-N 0.000 description 1
- DHMVUKSCMGLGMF-UHFFFAOYSA-N CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCCC1.N=C(N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1 Chemical compound CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCCC1.N=C(N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1 DHMVUKSCMGLGMF-UHFFFAOYSA-N 0.000 description 1
- DVDGIBDBRCLSND-UHFFFAOYSA-N CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1)N1CCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1)N1CCCCC1.N=C(N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1 Chemical compound CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1)N1CCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1)N1CCCCC1.N=C(N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1 DVDGIBDBRCLSND-UHFFFAOYSA-N 0.000 description 1
- WFPAMIIYAZDEMR-UHFFFAOYSA-N CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1)N1CCCCC1 Chemical compound CN1CCN=C1C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)SC=C2)C=C1)N1CCCCC1 WFPAMIIYAZDEMR-UHFFFAOYSA-N 0.000 description 1
- VGKLAFJAZLEECD-UHFFFAOYSA-N CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1 Chemical compound CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1 VGKLAFJAZLEECD-UHFFFAOYSA-N 0.000 description 1
- MJHPGZPNWOVNLX-UHFFFAOYSA-N CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)SC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)SC=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)SC=C2)C=C1)N1CCCCC1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)SC=C2)C=C1 Chemical compound CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)SC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)SC=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)SC=C2)C=C1)N1CCCCC1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)SC=C2)C=C1 MJHPGZPNWOVNLX-UHFFFAOYSA-N 0.000 description 1
- IEAQEPIBEOKEDZ-UHFFFAOYSA-N CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=NN2C)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCNCC2)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCOCC2)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCSCC2)C=C1 Chemical compound CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=NN2C)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCNCC2)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCOCC2)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCSCC2)C=C1 IEAQEPIBEOKEDZ-UHFFFAOYSA-N 0.000 description 1
- RFONTHXEGKHJFY-UHFFFAOYSA-N CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1 Chemical compound CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1 RFONTHXEGKHJFY-UHFFFAOYSA-N 0.000 description 1
- PBRGWLMYIBXDKW-UHFFFAOYSA-N CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2O)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2O)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2O)C=C1)N1CCCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2O)C=C1)N1CCOCC1 Chemical compound CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2O)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2O)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2O)C=C1)N1CCCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2O)C=C1)N1CCOCC1 PBRGWLMYIBXDKW-UHFFFAOYSA-N 0.000 description 1
- YYFPWLYTERZXTC-UHFFFAOYSA-N CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CS2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)SC=C2)C=C1)N1CCOCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)SC=C2)C=C1)N1CCSCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CS2)C=C1)N1CCCC1 Chemical compound CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CS2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)SC=C2)C=C1)N1CCOCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)SC=C2)C=C1)N1CCSCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CS2)C=C1)N1CCCC1 YYFPWLYTERZXTC-UHFFFAOYSA-N 0.000 description 1
- WISHKRDBVMTJAW-UHFFFAOYSA-N CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=NN2C)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCNCC2)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCOCC2)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCSCC2)C=C1 Chemical compound CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=NN2C)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCNCC2)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCOCC2)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCSCC2)C=C1 WISHKRDBVMTJAW-UHFFFAOYSA-N 0.000 description 1
- CGEJQQMDMLZCLP-UHFFFAOYSA-N CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)SC=C2)C=C1 Chemical compound CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)SC=C2)C=C1 CGEJQQMDMLZCLP-UHFFFAOYSA-N 0.000 description 1
- LKPZPGHQDNNARH-UHFFFAOYSA-N CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1 Chemical compound CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1 LKPZPGHQDNNARH-UHFFFAOYSA-N 0.000 description 1
- AEAWUYAERVKLEH-UHFFFAOYSA-N CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CS2)C=C1 Chemical compound CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CS2)C=C1 AEAWUYAERVKLEH-UHFFFAOYSA-N 0.000 description 1
- KEKHBBGENFWNMG-UHFFFAOYSA-N CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1 Chemical compound CN1CCN=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1 KEKHBBGENFWNMG-UHFFFAOYSA-N 0.000 description 1
- RRPXBXZZNMHGMS-UHFFFAOYSA-N CN1CCN=C1C1=CC=C(NCC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1 Chemical compound CN1CCN=C1C1=CC=C(NCC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1 RRPXBXZZNMHGMS-UHFFFAOYSA-N 0.000 description 1
- SXHQRQCDJATNJH-UHFFFAOYSA-N CN1CCN=C1NCC1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CC=C2)C=C1 Chemical compound CN1CCN=C1NCC1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CC=C2)C=C1 SXHQRQCDJATNJH-UHFFFAOYSA-N 0.000 description 1
- WAYPRAVIUGAUCO-FIZFVFNCSA-N CN1CCN=C1[2H]C(=O)NC1=CC=C(Cl)C=C1C(=O)NC1=NC=C(Cl)C=C1 Chemical compound CN1CCN=C1[2H]C(=O)NC1=CC=C(Cl)C=C1C(=O)NC1=NC=C(Cl)C=C1 WAYPRAVIUGAUCO-FIZFVFNCSA-N 0.000 description 1
- IXNAHZVOBBOUBP-UHFFFAOYSA-N CN1N=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CC2)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCC2)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=NN2C)C=C1 Chemical compound CN1N=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CC2)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCC2)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=NN2C)C=C1 IXNAHZVOBBOUBP-UHFFFAOYSA-N 0.000 description 1
- UNLPBUKKJNVTEG-UHFFFAOYSA-N CN1N=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CC2)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCC2)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=NN2C)C=C1 Chemical compound CN1N=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CC2)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCC2)C=C1.CN1N=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=NN2C)C=C1 UNLPBUKKJNVTEG-UHFFFAOYSA-N 0.000 description 1
- GGHKKFHZFDOXOA-UHFFFAOYSA-N CNC(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OC)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=C(F)C=C(C(=N)N(C)C)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=C(F)C=C(C(=N)N)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCC2)C=C1 Chemical compound CNC(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OC)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=C(F)C=C(C(=N)N(C)C)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=C(F)C=C(C(=N)N)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCC2)C=C1 GGHKKFHZFDOXOA-UHFFFAOYSA-N 0.000 description 1
- SDRKGHXRARYNCC-UHFFFAOYSA-N CNC(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1.N/C(=N/O)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCC1 Chemical compound CNC(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1.N/C(=N/O)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCC1 SDRKGHXRARYNCC-UHFFFAOYSA-N 0.000 description 1
- QCJBAUBTYUPAEM-UHFFFAOYSA-N CNC(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1.N/C(=N/O)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1 Chemical compound CNC(=N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1.N/C(=N/O)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1 QCJBAUBTYUPAEM-UHFFFAOYSA-N 0.000 description 1
- OYTIJOQMAYMDBR-UHFFFAOYSA-N CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(OC)C=C2)C=C1 Chemical compound CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(OC)C=C2)C=C1 OYTIJOQMAYMDBR-UHFFFAOYSA-N 0.000 description 1
- VYCHDJOLENELGQ-UHFFFAOYSA-N CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCCCCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCSCC1 Chemical compound CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCCCCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCSCC1 VYCHDJOLENELGQ-UHFFFAOYSA-N 0.000 description 1
- JROUXDZEAJPGHP-UHFFFAOYSA-N CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(C)C=N3)C=C(Cl)C=C2OC)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Br)C=N2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1 Chemical compound CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(C)C=N3)C=C(Cl)C=C2OC)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Br)C=N2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1 JROUXDZEAJPGHP-UHFFFAOYSA-N 0.000 description 1
- UPKOENOCEGZTBO-UHFFFAOYSA-N CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(C)C=N3)C=CC=C2O)C=C1 Chemical compound CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(C)C=N3)C=CC=C2O)C=C1 UPKOENOCEGZTBO-UHFFFAOYSA-N 0.000 description 1
- MMTZJJOBBNPLTM-UHFFFAOYSA-N CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2O)C=C1 Chemical compound CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2O)C=C1 MMTZJJOBBNPLTM-UHFFFAOYSA-N 0.000 description 1
- FMDNBJMHZMMJSO-UHFFFAOYSA-N CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OC)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1 Chemical compound CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2OC)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1 FMDNBJMHZMMJSO-UHFFFAOYSA-N 0.000 description 1
- ROZHQUJDVVDJHG-UHFFFAOYSA-N CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1.N/C(=N/O)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCS(=O)(=O)CC1 Chemical compound CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1.N/C(=N/O)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCS(=O)(=O)CC1 ROZHQUJDVVDJHG-UHFFFAOYSA-N 0.000 description 1
- RZNYEDGWOJXLTN-UHFFFAOYSA-N CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OC)C=C2)C(F)=C1 Chemical compound CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OC)C=C2)C(F)=C1 RZNYEDGWOJXLTN-UHFFFAOYSA-N 0.000 description 1
- WPIREPCQDPQVJB-UHFFFAOYSA-N CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OC)C=C2)C=C1 Chemical compound CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OC)C=C2)C=C1 WPIREPCQDPQVJB-UHFFFAOYSA-N 0.000 description 1
- VGHPDJBFEFEWGU-UHFFFAOYSA-N CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2O)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2O)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2O)C=C1)N1CCCCC1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2O)C=C1 Chemical compound CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2O)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2O)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2O)C=C1)N1CCCCC1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2O)C=C1 VGHPDJBFEFEWGU-UHFFFAOYSA-N 0.000 description 1
- QEWIYJRDJDUYMB-UHFFFAOYSA-N CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2OC)C=C1.COC1=CC=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1 Chemical compound CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2OC)C=C1.COC1=CC=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1 QEWIYJRDJDUYMB-UHFFFAOYSA-N 0.000 description 1
- NWDBEWGRRZQSSC-UHFFFAOYSA-N CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(F)C(F)=C2)C=C1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(F)C(F)=C2)C=C1.N=C(NO)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(F)C(F)=C2)C=C1 Chemical compound CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(F)C(F)=C2)C=C1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(F)C(F)=C2)C=C1.N=C(NO)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(F)C(F)=C2)C=C1 NWDBEWGRRZQSSC-UHFFFAOYSA-N 0.000 description 1
- DZVLGIZGVNUEFH-UHFFFAOYSA-N CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1 Chemical compound CNC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1 DZVLGIZGVNUEFH-UHFFFAOYSA-N 0.000 description 1
- OSACAUROBUBBOM-UHFFFAOYSA-N CNC(=N)C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1.N=C(C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCCC1.N=C(C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCOCC1.N=C(N)C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1 Chemical compound CNC(=N)C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1.N=C(C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCCC1.N=C(C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCOCC1.N=C(N)C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1 OSACAUROBUBBOM-UHFFFAOYSA-N 0.000 description 1
- UMKHWHDCTRBHBL-UHFFFAOYSA-N CNC(c(cc1)ccc1C(Nc(c(O)ccc1)c1C(Nc(nc1)ccc1Cl)=O)=O)=N Chemical compound CNC(c(cc1)ccc1C(Nc(c(O)ccc1)c1C(Nc(nc1)ccc1Cl)=O)=O)=N UMKHWHDCTRBHBL-UHFFFAOYSA-N 0.000 description 1
- FZDIUCGRMAUDIW-UHFFFAOYSA-N CNC1=CC=CC(C(=N)N)=C1.CNC1=CC=CC(C(N)=O)=C1.CNC1=CC=CC(CN)=C1.COC1=C(C#N)C=CC(C(=N)N)=C1.COC1=C(C(N)=O)C=CC(C(=N)N)=C1.COC1=C(F)C=CC(C(=N)N)=C1.COC1=C(N)C=CC(C(=N)N)=C1.COC1=C(O)C=CC(C(=N)N)=C1.COC1=C(OCC(=O)O)C=CC(C(=N)N)=C1.COC1=C(S(C)(=O)=O)C=CC(C(=N)N)=C1.COC1=C(S(N)(=O)=O)C=CC(C(=N)N)=C1.COC1=C([N+](=O)[O-])C=CC(C(=N)N)=C1.COC1=CC=C(F)C(C(=N)N)=C1.COC1=CC=CC(C(=N)N)=C1.COC1=CC=CC(C(N)=O)=C1.COC1=CC=CC(CN)=C1 Chemical compound CNC1=CC=CC(C(=N)N)=C1.CNC1=CC=CC(C(N)=O)=C1.CNC1=CC=CC(CN)=C1.COC1=C(C#N)C=CC(C(=N)N)=C1.COC1=C(C(N)=O)C=CC(C(=N)N)=C1.COC1=C(F)C=CC(C(=N)N)=C1.COC1=C(N)C=CC(C(=N)N)=C1.COC1=C(O)C=CC(C(=N)N)=C1.COC1=C(OCC(=O)O)C=CC(C(=N)N)=C1.COC1=C(S(C)(=O)=O)C=CC(C(=N)N)=C1.COC1=C(S(N)(=O)=O)C=CC(C(=N)N)=C1.COC1=C([N+](=O)[O-])C=CC(C(=N)N)=C1.COC1=CC=C(F)C(C(=N)N)=C1.COC1=CC=CC(C(=N)N)=C1.COC1=CC=CC(C(N)=O)=C1.COC1=CC=CC(CN)=C1 FZDIUCGRMAUDIW-UHFFFAOYSA-N 0.000 description 1
- OQMCCJRBJLFQAT-UHFFFAOYSA-N CNCC1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1.NCC1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CN2CCC2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CN2CCCCC2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CN2CCOCC2)C=C1 Chemical compound CNCC1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1.NCC1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CN2CCC2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CN2CCCCC2)C=C1.O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CN2CCOCC2)C=C1 OQMCCJRBJLFQAT-UHFFFAOYSA-N 0.000 description 1
- JDYAJNSVWOEJKI-UHFFFAOYSA-N COC(=O)C1=C(NC(=O)C2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(C)(=O)=O)C=C3)C=CC=C2)C=CC(Br)=C1 Chemical compound COC(=O)C1=C(NC(=O)C2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(C)(=O)=O)C=C3)C=CC=C2)C=CC(Br)=C1 JDYAJNSVWOEJKI-UHFFFAOYSA-N 0.000 description 1
- FCHUOKTZYIIQIG-UHFFFAOYSA-N COC(=O)C1=CC(NC(=O)C2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)=C(OC2=CC=CC(C(=N)NO)=C2)C=C1.N=C(NO)C1=CC(OC2=C(C(=O)NC3=C(F)C=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=C([N+](=O)[O-])C=C2)=CC=C1.N=C(NO)C1=CC(OC2=C(C(=O)NC3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=C([N+](=O)[O-])C=C2)=CC=C1.N=C(NO)C1=CC(OC2=C(NC(=O)C3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=CC=C2)=CC=C1 Chemical compound COC(=O)C1=CC(NC(=O)C2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)=C(OC2=CC=CC(C(=N)NO)=C2)C=C1.N=C(NO)C1=CC(OC2=C(C(=O)NC3=C(F)C=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=C([N+](=O)[O-])C=C2)=CC=C1.N=C(NO)C1=CC(OC2=C(C(=O)NC3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=C([N+](=O)[O-])C=C2)=CC=C1.N=C(NO)C1=CC(OC2=C(NC(=O)C3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=CC=C2)=CC=C1 FCHUOKTZYIIQIG-UHFFFAOYSA-N 0.000 description 1
- WDWINFCNLACNAP-UHFFFAOYSA-N COC(=O)C1=CC(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)=C(OC2=CC(C(=N)N)=CC=C2)C=C1 Chemical compound COC(=O)C1=CC(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)=C(OC2=CC(C(=N)N)=CC=C2)C=C1 WDWINFCNLACNAP-UHFFFAOYSA-N 0.000 description 1
- ZUKNYRWWURNYRT-UHFFFAOYSA-N COC(=O)C1=CC2=CC(Br)=CC=C2C=C1OC1=C(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)C=CC=C1 Chemical compound COC(=O)C1=CC2=CC(Br)=CC=C2C=C1OC1=C(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)C=CC=C1 ZUKNYRWWURNYRT-UHFFFAOYSA-N 0.000 description 1
- CIFQXAYKMGIQNN-UHFFFAOYSA-N COC(=O)C1=CC2=CC=CC=C2C=C1OC1=C(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)C=CC=C1 Chemical compound COC(=O)C1=CC2=CC=CC=C2C=C1OC1=C(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)C=CC=C1 CIFQXAYKMGIQNN-UHFFFAOYSA-N 0.000 description 1
- OQISFVCHBSJCIR-UHFFFAOYSA-N COC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N(C)C)C=C2)C=C1 Chemical compound COC(=O)COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N(C)C)C=C2)C=C1 OQISFVCHBSJCIR-UHFFFAOYSA-N 0.000 description 1
- NWIGPBDMFNUUDD-UHFFFAOYSA-N COC1=CC(Br)=CC(C(=O)NC2=CC=C(Br)C=N2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1.COC1=CC(Br)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1 Chemical compound COC1=CC(Br)=CC(C(=O)NC2=CC=C(Br)C=N2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1.COC1=CC(Br)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1 NWIGPBDMFNUUDD-UHFFFAOYSA-N 0.000 description 1
- MGNLNWVSWNELMX-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Br)C=N2)=C(NC(=O)C2=CC=C(/C(N)=N/O)C=C2)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Br)C=N2)=C(NC(=O)C2=CC=C(/C(N)=N/O)C=C2)C=C1 MGNLNWVSWNELMX-UHFFFAOYSA-N 0.000 description 1
- PUFYOWCAOMJHMX-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Br)C=N2)=C(NC(=O)C2=CC=C(C(=N)N(C)C)C=C2)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Br)C=N2)=C(NC(=O)C2=CC=C(C(=N)N(C)C)C=C2)C=C1 PUFYOWCAOMJHMX-UHFFFAOYSA-N 0.000 description 1
- NDLROAMCEWQKHQ-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Br)C=N2)=C(NC(=O)C2=CC=C(C(=N)N)C=C2)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Br)C=N2)=C(NC(=O)C2=CC=C(C(=N)N)C=C2)C=C1 NDLROAMCEWQKHQ-UHFFFAOYSA-N 0.000 description 1
- JDOAUDDQLSQFKD-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Br)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCCC3)C=C2)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Br)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCCC3)C=C2)C=C1 JDOAUDDQLSQFKD-UHFFFAOYSA-N 0.000 description 1
- NTZHDHUSDFNUFI-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Br)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCCCC3)C=C2)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Br)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCCCC3)C=C2)C=C1 NTZHDHUSDFNUFI-UHFFFAOYSA-N 0.000 description 1
- GYMAGHIVWQXXFC-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Br)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCOCC3)C=C2)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Br)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCOCC3)C=C2)C=C1 GYMAGHIVWQXXFC-UHFFFAOYSA-N 0.000 description 1
- AELWKAWYFANERE-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Br)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCSCC3)C=C2)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Br)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCSCC3)C=C2)C=C1 AELWKAWYFANERE-UHFFFAOYSA-N 0.000 description 1
- XPKJZPJJAIEOGD-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Br)C=N2)=C(NC(=O)C2=CC=C(C3=NCCN3C)C=C2)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Br)C=N2)=C(NC(=O)C2=CC=C(C3=NCCN3C)C=C2)C=C1 XPKJZPJJAIEOGD-UHFFFAOYSA-N 0.000 description 1
- YCIHCUJPIDBJHR-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=C(F)C=C(C(=N)N(C)C)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=C(F)C=C(C(=N)N3CC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=C(F)C=C(C(=N)N3CCC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=C(F)C=C(C(=N)N3CCCC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=C(F)C=C(C(=N)N3CCCCC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=C(F)C=C(C3=NCCN3C)C=C2)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=C(F)C=C(C(=N)N(C)C)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=C(F)C=C(C(=N)N3CC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=C(F)C=C(C(=N)N3CCC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=C(F)C=C(C(=N)N3CCCC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=C(F)C=C(C(=N)N3CCCCC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=C(F)C=C(C3=NCCN3C)C=C2)C=C1 YCIHCUJPIDBJHR-UHFFFAOYSA-N 0.000 description 1
- REFDAMWNNZBVTP-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=CC=C(C(=N)N(C)C)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=CC=C(C(=N)N3CC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=CC=C(C(=N)N3CCC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=CC=C(C(=N)N3CCCC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=CC=C(C(=N)N3CCCCC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=CC=C(C3=NCCN3C)C=C2)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=CC=C(C(=N)N(C)C)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=CC=C(C(=N)N3CC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=CC=C(C(=N)N3CCC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=CC=C(C(=N)N3CCCC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=CC=C(C(=N)N3CCCCC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=C2)=C(NC(=O)C2=CC=C(C3=NCCN3C)C=C2)C=C1 REFDAMWNNZBVTP-UHFFFAOYSA-N 0.000 description 1
- FNNKPVLTGFPEKB-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(/C(N)=N/O)C=C2F)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(/C(N)=N/O)C=C2F)C=C1 FNNKPVLTGFPEKB-UHFFFAOYSA-N 0.000 description 1
- XHOLNRLADUSQLD-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N(C)C)C=C2)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N(C)C)C=C2)C=C1 XHOLNRLADUSQLD-UHFFFAOYSA-N 0.000 description 1
- DFINKIAMPKTVTB-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N(C)C)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCC3)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OC(F)(F)F)C=C2)C=C1)N1CC1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N(C)C)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCC3)C=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OC(F)(F)F)C=C2)C=C1)N1CC1 DFINKIAMPKTVTB-UHFFFAOYSA-N 0.000 description 1
- BVAOXAZADJUBBQ-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N)C=C2)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N)C=C2)C=C1 BVAOXAZADJUBBQ-UHFFFAOYSA-N 0.000 description 1
- ITYYYRDELOWBQJ-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N)C=C2F)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N)C=C2F)C=C1 ITYYYRDELOWBQJ-UHFFFAOYSA-N 0.000 description 1
- NNYXJLZYNNBUQP-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCCC3)C=C2)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCCC3)C=C2)C=C1 NNYXJLZYNNBUQP-UHFFFAOYSA-N 0.000 description 1
- DTYFURWOGSOPHB-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCCC3)C=C2F)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCCC3)C=C2F)C=C1 DTYFURWOGSOPHB-UHFFFAOYSA-N 0.000 description 1
- OUOFPLQBQGPAFR-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCCCC3)C=C2)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCCCC3)C=C2)C=C1 OUOFPLQBQGPAFR-UHFFFAOYSA-N 0.000 description 1
- OLJDNRCYPYUOIL-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCCCC3)C=C2F)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCCCC3)C=C2F)C=C1 OLJDNRCYPYUOIL-UHFFFAOYSA-N 0.000 description 1
- XIEDFNQLMXPITM-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCOCC3)C=C2)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCOCC3)C=C2)C=C1 XIEDFNQLMXPITM-UHFFFAOYSA-N 0.000 description 1
- CEXGQNDSCXNCLS-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCOCC3)C=C2F)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCOCC3)C=C2F)C=C1 CEXGQNDSCXNCLS-UHFFFAOYSA-N 0.000 description 1
- PFJWTDXTWDDNCB-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCSCC3)C=C2)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCSCC3)C=C2)C=C1 PFJWTDXTWDDNCB-UHFFFAOYSA-N 0.000 description 1
- RXCWSTXIYQYHPX-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCSCC3)C=C2F)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C(=N)N3CCSCC3)C=C2F)C=C1 RXCWSTXIYQYHPX-UHFFFAOYSA-N 0.000 description 1
- BOAASANGNHDEFD-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C3=NCCN3C)C=C2)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C3=NCCN3C)C=C2)C=C1 BOAASANGNHDEFD-UHFFFAOYSA-N 0.000 description 1
- ZTZPWERJUCDBEJ-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C3=NCCN3C)C=C2F)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C3=NCCN3C)C=C2F)C=C1 ZTZPWERJUCDBEJ-UHFFFAOYSA-N 0.000 description 1
- BBDBINVTVFHHLQ-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NCC2=CC=C(C(=N)N(C)C)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NCC2=CC=C(C(=N)N3CCC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NCC2=CC=C(C(=N)N3CCCCC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NCC2=CC=C(C3=NCCN3C)C=C2)C=C1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NCC2=CC=C(C(=N)N(C)C)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NCC2=CC=C(C(=N)N3CCC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NCC2=CC=C(C(=N)N3CCCCC3)C=C2)C=C1.COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NCC2=CC=C(C3=NCCN3C)C=C2)C=C1 BBDBINVTVFHHLQ-UHFFFAOYSA-N 0.000 description 1
- NLGXCJJDMOSYPQ-UHFFFAOYSA-N COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NCC2=CC=C(C(=N)N3CCCC3)C=C2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NCC1=CC=C(C(=N)N2CCCC2)C=C1.N=C(C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCCC1 Chemical compound COC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NCC2=CC=C(C(=N)N3CCCC3)C=C2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NCC1=CC=C(C(=N)N2CCCC2)C=C1.N=C(C1=CC=C(CNC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCCC1 NLGXCJJDMOSYPQ-UHFFFAOYSA-N 0.000 description 1
- FPQUEPQOUQSGMG-UHFFFAOYSA-N COC1=CC(C(=O)NC2=NC=C(Br)C=C2)=C(NC(=O)C2=CC=C(C3=NCCN3C)C=C2)C(OC)=C1OC Chemical compound COC1=CC(C(=O)NC2=NC=C(Br)C=C2)=C(NC(=O)C2=CC=C(C3=NCCN3C)C=C2)C(OC)=C1OC FPQUEPQOUQSGMG-UHFFFAOYSA-N 0.000 description 1
- VXCXLSAWAXMSRB-UHFFFAOYSA-N COC1=CC(C)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N(C)C)C=C1 Chemical compound COC1=CC(C)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N(C)C)C=C1 VXCXLSAWAXMSRB-UHFFFAOYSA-N 0.000 description 1
- ZWPDSNLWSTZBHI-UHFFFAOYSA-N COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Br)C=N2)=C1NC(=O)C1=CC=C(C(=N)N)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Br)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Br)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Br)C=N2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 Chemical compound COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Br)C=N2)=C1NC(=O)C1=CC=C(C(=N)N)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Br)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Br)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Br)C=N2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 ZWPDSNLWSTZBHI-UHFFFAOYSA-N 0.000 description 1
- LZWHQOVQHDFJOI-UHFFFAOYSA-N COC1=CC(Cl)=CC(C(=O)NC2=CC=C(C)C=N2)=C1NC(=O)C1=C(F)C=C(C2=NCCN2C)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCCC2)C=C1 Chemical compound COC1=CC(Cl)=CC(C(=O)NC2=CC=C(C)C=N2)=C1NC(=O)C1=C(F)C=C(C2=NCCN2C)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCCC2)C=C1 LZWHQOVQHDFJOI-UHFFFAOYSA-N 0.000 description 1
- WONCYJPLAHPCLX-UHFFFAOYSA-N COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N(C)C)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C2=NCCN2C)C=C1 Chemical compound COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N(C)C)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C2=NCCN2C)C=C1 WONCYJPLAHPCLX-UHFFFAOYSA-N 0.000 description 1
- RVSCVQBNLLOZRP-UHFFFAOYSA-N COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N(C)C)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C2=NCCN2C)C=C1 Chemical compound COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N(C)C)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C2=NCCN2C)C=C1 RVSCVQBNLLOZRP-UHFFFAOYSA-N 0.000 description 1
- DESZVTSCKWCWFD-UHFFFAOYSA-N COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 Chemical compound COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 DESZVTSCKWCWFD-UHFFFAOYSA-N 0.000 description 1
- SYVLVSUFZMUTNB-UHFFFAOYSA-N COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 Chemical compound COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 SYVLVSUFZMUTNB-UHFFFAOYSA-N 0.000 description 1
- WWLKVXPXEJZVGJ-UHFFFAOYSA-N COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1 Chemical compound COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1 WWLKVXPXEJZVGJ-UHFFFAOYSA-N 0.000 description 1
- JUBVMHJUFKTRBV-UHFFFAOYSA-N COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCOCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 Chemical compound COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCOCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 JUBVMHJUFKTRBV-UHFFFAOYSA-N 0.000 description 1
- OIZVEXTYZJOYTG-UHFFFAOYSA-N COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NCC1=CC=C(C(=N)N(C)C)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NCC1=CC=C(C(=N)N2CCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NCC1=CC=C(C(=N)N2CCCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NCC1=CC=C(C2=NCCN2C)C=C1 Chemical compound COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NCC1=CC=C(C(=N)N(C)C)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NCC1=CC=C(C(=N)N2CCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NCC1=CC=C(C(=N)N2CCCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NCC1=CC=C(C2=NCCN2C)C=C1 OIZVEXTYZJOYTG-UHFFFAOYSA-N 0.000 description 1
- XXCQNCDEMDQNDL-UHFFFAOYSA-N COC1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N(C)C)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C2=NCCN2C)C=C1 Chemical compound COC1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N(C)C)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C(=N)N2CCCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=C(F)C=C(C2=NCCN2C)C=C1 XXCQNCDEMDQNDL-UHFFFAOYSA-N 0.000 description 1
- KNLPEJOGKJDCQE-UHFFFAOYSA-N COC1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 Chemical compound COC1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.COC1=CC(Cl)=CC(C(=O)NC2=NC=C(Cl)C=C2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 KNLPEJOGKJDCQE-UHFFFAOYSA-N 0.000 description 1
- QYVNMVWOTPVASW-UHFFFAOYSA-N COC1=CC(NC(=O)C2=CC=C(C(=N)N(C)C)C=C2)=C(C(=O)NC2=NC=C(Br)C=C2)C=C1OC Chemical compound COC1=CC(NC(=O)C2=CC=C(C(=N)N(C)C)C=C2)=C(C(=O)NC2=NC=C(Br)C=C2)C=C1OC QYVNMVWOTPVASW-UHFFFAOYSA-N 0.000 description 1
- KAYCNJGOJHDPMX-UHFFFAOYSA-N COC1=CC(NC(=O)C2=CC=C(C(=N)N)C=C2)=C(C(=O)NC2=CC=C(Br)C=N2)C=C1OC Chemical compound COC1=CC(NC(=O)C2=CC=C(C(=N)N)C=C2)=C(C(=O)NC2=CC=C(Br)C=N2)C=C1OC KAYCNJGOJHDPMX-UHFFFAOYSA-N 0.000 description 1
- BZDJZUOKAKCSKV-UHFFFAOYSA-N COC1=CC=CC(C(=O)NC2=CC=C(C)C=N2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1 Chemical compound COC1=CC=CC(C(=O)NC2=CC=C(C)C=N2)=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1 BZDJZUOKAKCSKV-UHFFFAOYSA-N 0.000 description 1
- GAPGARQDMOXYGT-UHFFFAOYSA-N COC1=CC=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N)C=C1.COC1=CC=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1 Chemical compound COC1=CC=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N)C=C1.COC1=CC=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCC2)C=C1 GAPGARQDMOXYGT-UHFFFAOYSA-N 0.000 description 1
- FDGNMYPHNADRCR-UHFFFAOYSA-N COC1=CC=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.COC1=CC=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 Chemical compound COC1=CC=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C(=N)N2CCCCC2)C=C1.COC1=CC=CC(C(=O)NC2=CC=C(Cl)C=N2)=C1NC(=O)C1=CC=C(C2=NCCN2C)C=C1 FDGNMYPHNADRCR-UHFFFAOYSA-N 0.000 description 1
- XVIOMYSSLOPYHI-UHFFFAOYSA-N CONC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCCC1.N=C(NO)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1 Chemical compound CONC(=N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCCC1.N=C(NO)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1 XVIOMYSSLOPYHI-UHFFFAOYSA-N 0.000 description 1
- WSSASXSAQMURJO-UHFFFAOYSA-N CS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC(C(=N)N)=CC=C4)SC=C3)C=C2)C=CC=C1.N=C(N)C1=CC(OC2=CC=CC=C2NC(=O)C2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)=C(O)C=C1.NC1=NC=CC2=CC=C(OC3=CC=CC=C3NC(=O)C3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=C21.NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=CC=CC=C3OC3=CC=C4C=C(Br)C=CC4=C3)C=C2)C=CC=C1 Chemical compound CS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC(C(=N)N)=CC=C4)SC=C3)C=C2)C=CC=C1.N=C(N)C1=CC(OC2=CC=CC=C2NC(=O)C2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)=C(O)C=C1.NC1=NC=CC2=CC=C(OC3=CC=CC=C3NC(=O)C3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=C21.NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=CC=CC=C3OC3=CC=C4C=C(Br)C=CC4=C3)C=C2)C=CC=C1 WSSASXSAQMURJO-UHFFFAOYSA-N 0.000 description 1
- CQRMUVINBNNXIQ-UHFFFAOYSA-N CS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=CC=CC=C3C(=O)NC3=CC(C(=N)N)=CC=C3)C(F)=C2)C=CC=C1.CS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=CC=CC=C3NC(=O)C3=CC(C(=N)N)=CC=C3)C=C2)C=CC=C1.CS(=O)(=O)C1=C(C2=CC=C(NC(=O)C3=CC=CC=C3C(=O)NC3=CC(C(=N)N)=CC=C3)C(F)=C2)C=CC=C1.CS(=O)(=O)C1=C(C2=CC=C(NC(=O)C3=CC=CC=C3NC(=O)C3=CC(C(=N)N)=CC=C3)C=C2)C=CC=C1 Chemical compound CS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=CC=CC=C3C(=O)NC3=CC(C(=N)N)=CC=C3)C(F)=C2)C=CC=C1.CS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=CC=CC=C3NC(=O)C3=CC(C(=N)N)=CC=C3)C=C2)C=CC=C1.CS(=O)(=O)C1=C(C2=CC=C(NC(=O)C3=CC=CC=C3C(=O)NC3=CC(C(=N)N)=CC=C3)C(F)=C2)C=CC=C1.CS(=O)(=O)C1=C(C2=CC=C(NC(=O)C3=CC=CC=C3NC(=O)C3=CC(C(=N)N)=CC=C3)C=C2)C=CC=C1 CQRMUVINBNNXIQ-UHFFFAOYSA-N 0.000 description 1
- JUIJBGGCQHMUCG-UHFFFAOYSA-N CS(=O)(=O)C1=C(C2=CC=C(NC(=O)C3=CC=CC=C3OC3=CC=CC(C(=N)N)=C3)C(F)=C2)C=CC=C1.N=C(N)C1=CC(OC2=CC=CC=C2C(=O)NC2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)=CC=C1.N=C(N)C1=CC(OC2=CC=CC=C2NC(=O)C2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)=CC=C1.NCC1=CC(OC2=CC=CC=C2C(=O)NC2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)=CC=C1 Chemical compound CS(=O)(=O)C1=C(C2=CC=C(NC(=O)C3=CC=CC=C3OC3=CC=CC(C(=N)N)=C3)C(F)=C2)C=CC=C1.N=C(N)C1=CC(OC2=CC=CC=C2C(=O)NC2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)=CC=C1.N=C(N)C1=CC(OC2=CC=CC=C2NC(=O)C2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)=CC=C1.NCC1=CC(OC2=CC=CC=C2C(=O)NC2=CC=C(C3=C(S(N)(=O)=O)C=CC=C3)C=C2)=CC=C1 JUIJBGGCQHMUCG-UHFFFAOYSA-N 0.000 description 1
- NRTNLSLCHHNPNM-UHFFFAOYSA-N CS(=O)(=O)C1=CC(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)=C(OC2=CC(C(=N)N)=CC=C2)C=C1 Chemical compound CS(=O)(=O)C1=CC(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)=C(OC2=CC(C(=N)N)=CC=C2)C=C1 NRTNLSLCHHNPNM-UHFFFAOYSA-N 0.000 description 1
- YQEZGLKKLAOYBK-UHFFFAOYSA-N CS(=O)(=O)C1=CC(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)=C(OC2=CC3=C(N)N=CC=C3C=C2)C=C1 Chemical compound CS(=O)(=O)C1=CC(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)=C(OC2=CC3=C(N)N=CC=C3C=C2)C=C1 YQEZGLKKLAOYBK-UHFFFAOYSA-N 0.000 description 1
- IRBRYLCPRDDYMP-UHFFFAOYSA-N CS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=C([N+](=O)[O-])C=C(Br)C=C3)C=CC=C2)C=C1 Chemical compound CS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=C([N+](=O)[O-])C=C(Br)C=C3)C=CC=C2)C=C1 IRBRYLCPRDDYMP-UHFFFAOYSA-N 0.000 description 1
- UJMYIUYGJLADJS-UHFFFAOYSA-N CS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1 Chemical compound CS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1 UJMYIUYGJLADJS-UHFFFAOYSA-N 0.000 description 1
- YXEFIQZOVVLEJI-UHFFFAOYSA-N CS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1 Chemical compound CS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1 YXEFIQZOVVLEJI-UHFFFAOYSA-N 0.000 description 1
- NSQRNVJINADEIA-UHFFFAOYSA-N CS(=O)(=O)NC1=CC(C(=O)NC2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)=C(OC2=CC(C(=N)N)=CC=C2)C=C1 Chemical compound CS(=O)(=O)NC1=CC(C(=O)NC2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)=C(OC2=CC(C(=N)N)=CC=C2)C=C1 NSQRNVJINADEIA-UHFFFAOYSA-N 0.000 description 1
- PSLCSOVHXFHTIW-UHFFFAOYSA-N CS(=O)(=O)NC1=CC(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)=C(C(=O)NC2=CC=C(Br)C=N2)C=C1 Chemical compound CS(=O)(=O)NC1=CC(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)=C(C(=O)NC2=CC=C(Br)C=N2)C=C1 PSLCSOVHXFHTIW-UHFFFAOYSA-N 0.000 description 1
- BEAIWOQOBGBGFM-UHFFFAOYSA-N Cc1c(C)[s]c2c1CCC2 Chemical compound Cc1c(C)[s]c2c1CCC2 BEAIWOQOBGBGFM-UHFFFAOYSA-N 0.000 description 1
- ACHMHHCOSAKQSS-UHFFFAOYSA-N Cc1c(C)[s]c2ccccc12 Chemical compound Cc1c(C)[s]c2ccccc12 ACHMHHCOSAKQSS-UHFFFAOYSA-N 0.000 description 1
- OLMKETZTNACYRL-UHFFFAOYSA-N N/C(/c(cc1)ccc1C(Nc(ccc(F)c1)c1C(Nc(cc1)ncc1Br)=O)=O)=N/O Chemical compound N/C(/c(cc1)ccc1C(Nc(ccc(F)c1)c1C(Nc(cc1)ncc1Br)=O)=O)=N/O OLMKETZTNACYRL-UHFFFAOYSA-N 0.000 description 1
- NGXLVFDEUYNVRH-UHFFFAOYSA-N N/C(=N/O)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1.N/C(=N/O)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.N=C(N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1 Chemical compound N/C(=N/O)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1.N/C(=N/O)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1.N=C(N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1 NGXLVFDEUYNVRH-UHFFFAOYSA-N 0.000 description 1
- RMMMYMSWJCYZFY-UHFFFAOYSA-N N/C(=N/O)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1 Chemical compound N/C(=N/O)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1.N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1 RMMMYMSWJCYZFY-UHFFFAOYSA-N 0.000 description 1
- ANTXQWUHCVTXLC-UHFFFAOYSA-N N/C(=N/O)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(O)C=C2)C=C1 Chemical compound N/C(=N/O)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(O)C=C2)C=C1 ANTXQWUHCVTXLC-UHFFFAOYSA-N 0.000 description 1
- IOAOLRJWYUEVBW-UHFFFAOYSA-N N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1)N1CCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1)N1CCSCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCSCC1 Chemical compound N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1)N1CCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1)N1CCSCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCSCC1 IOAOLRJWYUEVBW-UHFFFAOYSA-N 0.000 description 1
- BEGSUNNEZGUHLU-UHFFFAOYSA-N N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1)N1CCCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1)N1CCOCC1.N=C(N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1 Chemical compound N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1)N1CCCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1)N1CCOCC1.N=C(N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C=C2)C=C1 BEGSUNNEZGUHLU-UHFFFAOYSA-N 0.000 description 1
- GUBPTUHGXPBEPB-UHFFFAOYSA-N N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCOCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCSCC1 Chemical compound N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCOCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCSCC1 GUBPTUHGXPBEPB-UHFFFAOYSA-N 0.000 description 1
- SCLNFWAEVZSISH-UHFFFAOYSA-N N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1)N1CCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1)N1CCCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=C3)C=C(F)C=C2)C=C1)N1CCC1 Chemical compound N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1)N1CCCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=C3)C=C(F)C=C2)C=C1)N1CCCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=C3)C=C(F)C=C2)C=C1)N1CCC1 SCLNFWAEVZSISH-UHFFFAOYSA-N 0.000 description 1
- UZMWJLXXHZYQQE-UHFFFAOYSA-N N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCC1 Chemical compound N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCC1.N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCC1 UZMWJLXXHZYQQE-UHFFFAOYSA-N 0.000 description 1
- BOHAGNCBKFZSHS-UHFFFAOYSA-N N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1)N1CCC1 Chemical compound N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1)N1CCC1 BOHAGNCBKFZSHS-UHFFFAOYSA-N 0.000 description 1
- QVJVVBRAPYWDTP-UHFFFAOYSA-N N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1)N1CCCC1 Chemical compound N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1)N1CCCC1 QVJVVBRAPYWDTP-UHFFFAOYSA-N 0.000 description 1
- UDMLJQOSUJXLHW-UHFFFAOYSA-N N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1)N1CCCCC1 Chemical compound N=C(C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1)N1CCCCC1 UDMLJQOSUJXLHW-UHFFFAOYSA-N 0.000 description 1
- TUNHJGFMMVVYOL-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C(F)=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C(F)=C2)C=C1)N1CCCCC1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C(F)=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(F)C(F)=C2)C=C1)N1CCCCC1 TUNHJGFMMVVYOL-UHFFFAOYSA-N 0.000 description 1
- UXLPNIFXGAGSJY-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCCNCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCNCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCCCC1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCCNCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1)N1CCNCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCCCC1 UXLPNIFXGAGSJY-UHFFFAOYSA-N 0.000 description 1
- PPEZBVQCWVQMKS-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2N2CCOCC2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2N2CCOCC2)C=C1)N1CCCCC1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2N2CCOCC2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(Cl)C=C2N2CCOCC2)C=C1)N1CCCCC1 PPEZBVQCWVQMKS-UHFFFAOYSA-N 0.000 description 1
- PGXOMUJZFGBOET-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCOCC1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(F)C=C2)C=C1)N1CCOCC1 PGXOMUJZFGBOET-UHFFFAOYSA-N 0.000 description 1
- LYFRHBLNNRNIJH-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(N3CCOCC3)C=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(N3CCOCC3)C=C2)C=C1)N1CCCCC1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(N3CCOCC3)C=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(N3CCOCC3)C=C2)C=C1)N1CCCCC1 LYFRHBLNNRNIJH-UHFFFAOYSA-N 0.000 description 1
- AQDPAIPHEYHKER-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1)N1CCC1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1)N1CCC1 AQDPAIPHEYHKER-UHFFFAOYSA-N 0.000 description 1
- DVJWHLILEQDYSJ-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1)N1CCCC1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1)N1CCCC1 DVJWHLILEQDYSJ-UHFFFAOYSA-N 0.000 description 1
- IACXIAOZIVWOPR-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1)N1CCCCC1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1)N1CCCCC1 IACXIAOZIVWOPR-UHFFFAOYSA-N 0.000 description 1
- NOKAWDIHDJAGNI-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCOCC1.N=C(NO)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCCC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCOCC1.N=C(NO)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1 NOKAWDIHDJAGNI-UHFFFAOYSA-N 0.000 description 1
- FWPPOXPGFWDMIU-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCS(=O)(=O)CC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCS(=O)CC1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCS(=O)(=O)CC1.N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCS(=O)CC1 FWPPOXPGFWDMIU-UHFFFAOYSA-N 0.000 description 1
- NIOKCFVEYFFHHR-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(F)C=C3)C=CC=C2)C=C1)N1CCCC1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(F)C=C3)C=CC=C2)C=C1)N1CCCC1 NIOKCFVEYFFHHR-UHFFFAOYSA-N 0.000 description 1
- UEKHGTQZPJJRNN-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(F)C=C3)C=CC=C2)C=C1)N1CCCCC1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(F)C=C3)C=CC=C2)C=C1)N1CCCCC1 UEKHGTQZPJJRNN-UHFFFAOYSA-N 0.000 description 1
- PATDLDZRDVHTOT-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)SC=C2)C=C1)N1CCCC1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)SC=C2)C=C1)N1CCCC1 PATDLDZRDVHTOT-UHFFFAOYSA-N 0.000 description 1
- PQGRIPSVFIAEAB-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)SC=C2)C=C1)N1CCCCC1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)SC=C2)C=C1)N1CCCCC1 PQGRIPSVFIAEAB-UHFFFAOYSA-N 0.000 description 1
- RUHCJEOHUZNYRC-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)SC=C2)C=C1)N1CCOCC1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)SC=C2)C=C1)N1CCOCC1 RUHCJEOHUZNYRC-UHFFFAOYSA-N 0.000 description 1
- KBCAMZSPLNVUPM-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)SC=C2)C=C1)N1CCSCC1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)SC=C2)C=C1)N1CCSCC1 KBCAMZSPLNVUPM-UHFFFAOYSA-N 0.000 description 1
- TXVFFWCOSKKKFY-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CS2)C=C1)N1CCCC1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CS2)C=C1)N1CCCC1 TXVFFWCOSKKKFY-UHFFFAOYSA-N 0.000 description 1
- IQYJCJHONUOPRQ-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1)N1CCCC1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1)N1CCCC1 IQYJCJHONUOPRQ-UHFFFAOYSA-N 0.000 description 1
- HMFFMLLFCFDYNO-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1)N1CCCCCC1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1)N1CCCCCC1 HMFFMLLFCFDYNO-UHFFFAOYSA-N 0.000 description 1
- OIOKXKQABSCVNY-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1)N1CCOCC1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1)N1CCOCC1 OIOKXKQABSCVNY-UHFFFAOYSA-N 0.000 description 1
- FFXWDCJTQXEGJS-UHFFFAOYSA-N N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1)N1CCSCC1 Chemical compound N=C(C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1)N1CCSCC1 FFXWDCJTQXEGJS-UHFFFAOYSA-N 0.000 description 1
- KVIDZBAMACRNHF-UHFFFAOYSA-N N=C(C1=CC=C(NCC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCCC1 Chemical compound N=C(C1=CC=C(NCC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1)N1CCCC1 KVIDZBAMACRNHF-UHFFFAOYSA-N 0.000 description 1
- VAHWHGXLCNFVCR-UHFFFAOYSA-N N=C(N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1 Chemical compound N=C(N)C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1 VAHWHGXLCNFVCR-UHFFFAOYSA-N 0.000 description 1
- VQEGUTHDTBWMLI-UHFFFAOYSA-N N=C(N)C1=CC(OC2=C(C(=O)NC3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=CC(Br)=C2)=CC=C1.N=C(N)C1=CC(OC2=C(C(=O)NC3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=CC(Cl)=C2)=CC=C1.N=C(N)C1=CC(OC2=C(C(=O)NC3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=CC(N)=C2)=CC=C1.N=C(N)C1=CC(OC2=C(C(=O)NC3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=CC([N+](=O)[O-])=C2)=CC=C1 Chemical compound N=C(N)C1=CC(OC2=C(C(=O)NC3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=CC(Br)=C2)=CC=C1.N=C(N)C1=CC(OC2=C(C(=O)NC3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=CC(Cl)=C2)=CC=C1.N=C(N)C1=CC(OC2=C(C(=O)NC3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=CC(N)=C2)=CC=C1.N=C(N)C1=CC(OC2=C(C(=O)NC3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=CC([N+](=O)[O-])=C2)=CC=C1 VQEGUTHDTBWMLI-UHFFFAOYSA-N 0.000 description 1
- YUBXDPJPXIHSCQ-UHFFFAOYSA-N N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1 Chemical compound N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1 YUBXDPJPXIHSCQ-UHFFFAOYSA-N 0.000 description 1
- DUVIALUKSVHAGZ-UHFFFAOYSA-N N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(O)C=C2)C=C1 Chemical compound N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(O)C=C2)C=C1 DUVIALUKSVHAGZ-UHFFFAOYSA-N 0.000 description 1
- WITGOXJMPIAMLX-UHFFFAOYSA-N N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1 Chemical compound N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)O)C=C2)C=C1 WITGOXJMPIAMLX-UHFFFAOYSA-N 0.000 description 1
- FPXZPOJVQGESEM-UHFFFAOYSA-N N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)SC=C2)C=C1 Chemical compound N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)SC=C2)C=C1 FPXZPOJVQGESEM-UHFFFAOYSA-N 0.000 description 1
- LCXLMWUGOXTGNC-UHFFFAOYSA-N N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1 Chemical compound N=C(N)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1 LCXLMWUGOXTGNC-UHFFFAOYSA-N 0.000 description 1
- SPHABVOQQZPEIZ-UHFFFAOYSA-N N=C(N)C1=CC=CC(C(=O)NC2=C(NC(=O)C3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=CC=C2)=C1 Chemical compound N=C(N)C1=CC=CC(C(=O)NC2=C(NC(=O)C3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=CC=C2)=C1 SPHABVOQQZPEIZ-UHFFFAOYSA-N 0.000 description 1
- INYJNALPRWWYPW-UHFFFAOYSA-N N=C(N)C1=CC=CC(NC(=O)C2=C(NC(=O)C3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=CS2)=C1 Chemical compound N=C(N)C1=CC=CC(NC(=O)C2=C(NC(=O)C3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=CS2)=C1 INYJNALPRWWYPW-UHFFFAOYSA-N 0.000 description 1
- LTPHGAMJGVWJFM-UHFFFAOYSA-N N=C(N)C1=CC=CC(NC2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=CC=C2)=C1 Chemical compound N=C(N)C1=CC=CC(NC2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=CC=C2)=C1 LTPHGAMJGVWJFM-UHFFFAOYSA-N 0.000 description 1
- YVMKGNIOENIXEQ-UHFFFAOYSA-N N=C(N)C1=CC=CC(OC2=C(C(=O)NC3=C(F)C=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=C(N)C=C2)=C1 Chemical compound N=C(N)C1=CC=CC(OC2=C(C(=O)NC3=C(F)C=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=C(N)C=C2)=C1 YVMKGNIOENIXEQ-UHFFFAOYSA-N 0.000 description 1
- QJPXTGLCOWMDIX-UHFFFAOYSA-N N=C(N)C1=CC=CC(OC2=C(C(=O)NC3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=C(Br)C=C2)=C1 Chemical compound N=C(N)C1=CC=CC(OC2=C(C(=O)NC3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=C(Br)C=C2)=C1 QJPXTGLCOWMDIX-UHFFFAOYSA-N 0.000 description 1
- GIMYYUSGVPGFHW-UHFFFAOYSA-N N=C(N)C1=CC=CC(OC2=C(C(=O)NC3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=C(Cl)C=C2)=C1 Chemical compound N=C(N)C1=CC=CC(OC2=C(C(=O)NC3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=C(Cl)C=C2)=C1 GIMYYUSGVPGFHW-UHFFFAOYSA-N 0.000 description 1
- SYOTWQBTJARFEQ-UHFFFAOYSA-N N=C(N)C1=CC=CC(OC2=C(C(=O)NC3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=C(N)C=C2)=C1 Chemical compound N=C(N)C1=CC=CC(OC2=C(C(=O)NC3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=C(N)C=C2)=C1 SYOTWQBTJARFEQ-UHFFFAOYSA-N 0.000 description 1
- WWLITDFYKJYGEL-UHFFFAOYSA-N N=C(N)C1=CC=CC(OC2=C(C(=O)NC3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=C([N+](=O)[O-])C=C2)=C1 Chemical compound N=C(N)C1=CC=CC(OC2=C(C(=O)NC3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=C([N+](=O)[O-])C=C2)=C1 WWLITDFYKJYGEL-UHFFFAOYSA-N 0.000 description 1
- FUKVFZYOSCTXRM-UHFFFAOYSA-N N=C(N)C1=CC=CC(OC2=C(C(=O)NC3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=CC=C2)=C1 Chemical compound N=C(N)C1=CC=CC(OC2=C(C(=O)NC3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=CC=C2)=C1 FUKVFZYOSCTXRM-UHFFFAOYSA-N 0.000 description 1
- KWPUGQUTZXLRNF-UHFFFAOYSA-N N=C(N)C1=CC=CC(OC2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=C(C(=O)O)C=C2)=C1 Chemical compound N=C(N)C1=CC=CC(OC2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=C(C(=O)O)C=C2)=C1 KWPUGQUTZXLRNF-UHFFFAOYSA-N 0.000 description 1
- FCEQWUOAIRVQKP-UHFFFAOYSA-N N=C(N)C1=CC=CC(OC2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=C(C(F)(F)F)C=C2)=C1 Chemical compound N=C(N)C1=CC=CC(OC2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=C(C(F)(F)F)C=C2)=C1 FCEQWUOAIRVQKP-UHFFFAOYSA-N 0.000 description 1
- QINMQXZJWXQDLT-UHFFFAOYSA-N N=C(N)C1=CC=CC(OC2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=CC(O)=C2)=C1 Chemical compound N=C(N)C1=CC=CC(OC2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=CC(O)=C2)=C1 QINMQXZJWXQDLT-UHFFFAOYSA-N 0.000 description 1
- DUVLBBOBLCROSD-UHFFFAOYSA-N N=C(N)C1=CC=CC(OC2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=CC=C2)=C1 Chemical compound N=C(N)C1=CC=CC(OC2=C(NC(=O)C3=CC=C(C4=CC=CC=C4S(N)(=O)=O)C=C3)C=CC=C2)=C1 DUVLBBOBLCROSD-UHFFFAOYSA-N 0.000 description 1
- PBVFSKOICDHBEK-UHFFFAOYSA-N N=C(N)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CC=C2)C=C1 Chemical compound N=C(N)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CC=C2)C=C1 PBVFSKOICDHBEK-UHFFFAOYSA-N 0.000 description 1
- IPUXEGATWZQWJX-UHFFFAOYSA-N N=C(NO)C1=CC(OC2=C(C(=O)NC3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=C(Br)C=C2)=CC=C1.N=C(NO)C1=CC(OC2=C(C(=O)NC3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=C(Cl)C=C2)=CC=C1 Chemical compound N=C(NO)C1=CC(OC2=C(C(=O)NC3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=C(Br)C=C2)=CC=C1.N=C(NO)C1=CC(OC2=C(C(=O)NC3=CC=C(C4=C(S(N)(=O)=O)C=CC=C4)C=C3)C=C(Cl)C=C2)=CC=C1 IPUXEGATWZQWJX-UHFFFAOYSA-N 0.000 description 1
- DWPFMRGKVGIWKU-UHFFFAOYSA-N N=C(NO)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1 Chemical compound N=C(NO)C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)SC=C2)C=C1 DWPFMRGKVGIWKU-UHFFFAOYSA-N 0.000 description 1
- AHGWIXSZYMYNQH-UHFFFAOYSA-N N=C(NO)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CC=C2)C=C1 Chemical compound N=C(NO)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CC=C2)C=C1 AHGWIXSZYMYNQH-UHFFFAOYSA-N 0.000 description 1
- GGIWLDWGDSYCRV-UHFFFAOYSA-N N=C(c(cc1)cc(F)c1C(Nc(ccc(I)c1)c1C(Nc(cc1)ncc1Br)=O)=O)N1CCCCC1 Chemical compound N=C(c(cc1)cc(F)c1C(Nc(ccc(I)c1)c1C(Nc(cc1)ncc1Br)=O)=O)N1CCCCC1 GGIWLDWGDSYCRV-UHFFFAOYSA-N 0.000 description 1
- SYGCUXNKKNGTNO-UHFFFAOYSA-N N=C(c(cc1)cc([Tl])c1C(Cc(ccc(Cl)c1)c1C(Nc(nc1)ccc1Cl)=O)=O)NO Chemical compound N=C(c(cc1)cc([Tl])c1C(Cc(ccc(Cl)c1)c1C(Nc(nc1)ccc1Cl)=O)=O)NO SYGCUXNKKNGTNO-UHFFFAOYSA-N 0.000 description 1
- VXFRAUIOYJILDP-UHFFFAOYSA-N N=C(c(cc1)ccc1C(Nc(c(C(Nc(nc1)ccc1Cl)=O)c1)ccc1OCC([O])=O)=O)N1CCCC1 Chemical compound N=C(c(cc1)ccc1C(Nc(c(C(Nc(nc1)ccc1Cl)=O)c1)ccc1OCC([O])=O)=O)N1CCCC1 VXFRAUIOYJILDP-UHFFFAOYSA-N 0.000 description 1
- JQWPVEAYMTUSRV-UHFFFAOYSA-N N=C(c(cc1)ccc1C(Nc(cccc1)c1C(Nc(nc1)ccc1Cl)=O)=O)N(CC1)CCS1(=O)=O Chemical compound N=C(c(cc1)ccc1C(Nc(cccc1)c1C(Nc(nc1)ccc1Cl)=O)=O)N(CC1)CCS1(=O)=O JQWPVEAYMTUSRV-UHFFFAOYSA-N 0.000 description 1
- YUJNXIAOYLWAFM-UHFFFAOYSA-N NC(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CC=C2)C=C1 Chemical compound NC(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CC=C2)C=C1 YUJNXIAOYLWAFM-UHFFFAOYSA-N 0.000 description 1
- NHUIVMTWCUNKOP-UHFFFAOYSA-N NC(c(cc1)ccc1C(Nc(ccc(F)c1)c1C(Nc(cc1)ncc1Br)=O)=O)=N Chemical compound NC(c(cc1)ccc1C(Nc(ccc(F)c1)c1C(Nc(cc1)ncc1Br)=O)=O)=N NHUIVMTWCUNKOP-UHFFFAOYSA-N 0.000 description 1
- YGTCMTRNZLRMBT-UHFFFAOYSA-N NC1=C(C(=O)NC2=NC=C(Br)C=C2)C=C(Cl)C=C1 Chemical compound NC1=C(C(=O)NC2=NC=C(Br)C=C2)C=C(Cl)C=C1 YGTCMTRNZLRMBT-UHFFFAOYSA-N 0.000 description 1
- FMMFHGVZUPFPTH-UHFFFAOYSA-N NC1=C2C=C(OC3=C(NC(=O)C4=CC=C(C5=CC=CC=C5S(N)(=O)=O)C=C4)C=C(C(F)(F)F)C=C3)C=CC2=CC=N1 Chemical compound NC1=C2C=C(OC3=C(NC(=O)C4=CC=C(C5=CC=CC=C5S(N)(=O)=O)C=C4)C=C(C(F)(F)F)C=C3)C=CC2=CC=N1 FMMFHGVZUPFPTH-UHFFFAOYSA-N 0.000 description 1
- IJWFYXNPCKRWPH-UHFFFAOYSA-N NC1=C2C=C(OC3=C(NC(=O)C4=CC=C(C5=CC=CC=C5S(N)(=O)=O)C=C4)C=C(F)C=C3)C=CC2=CC=N1 Chemical compound NC1=C2C=C(OC3=C(NC(=O)C4=CC=C(C5=CC=CC=C5S(N)(=O)=O)C=C4)C=C(F)C=C3)C=CC2=CC=N1 IJWFYXNPCKRWPH-UHFFFAOYSA-N 0.000 description 1
- JUWFUGYOXYMLNK-UHFFFAOYSA-N NC1=C2C=C(OC3=C(NC(=O)C4=CC=C(C5=CC=CC=C5S(N)(=O)=O)C=C4)C=CC=C3)C=CC2=CC=N1 Chemical compound NC1=C2C=C(OC3=C(NC(=O)C4=CC=C(C5=CC=CC=C5S(N)(=O)=O)C=C4)C=CC=C3)C=CC2=CC=N1 JUWFUGYOXYMLNK-UHFFFAOYSA-N 0.000 description 1
- BCTHOXQGNCNZPW-UHFFFAOYSA-N NC1=CC(C(=O)NC2=CC=C(Br)C=N2)=C(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)C=C1 Chemical compound NC1=CC(C(=O)NC2=CC=C(Br)C=N2)=C(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)C=C1 BCTHOXQGNCNZPW-UHFFFAOYSA-N 0.000 description 1
- ZCSWLBURSBZESH-UHFFFAOYSA-N NC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)C=C1 Chemical compound NC1=CC(C(=O)NC2=CC=C(Cl)C=N2)=C(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)C=C1 ZCSWLBURSBZESH-UHFFFAOYSA-N 0.000 description 1
- PBIBDSDWIMZYDT-UHFFFAOYSA-N NC1=CC(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)=C(C(=O)NC2=CC=C(Br)C=N2)C=C1 Chemical compound NC1=CC(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)=C(C(=O)NC2=CC=C(Br)C=N2)C=C1 PBIBDSDWIMZYDT-UHFFFAOYSA-N 0.000 description 1
- JVALKLBLUHGHNP-UHFFFAOYSA-N NC1=CC(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)=C(C(=O)NC2=CC=C(Cl)C=N2)C=C1 Chemical compound NC1=CC(NC(=O)C2=CC=C(C3=CC=CC=C3S(N)(=O)=O)C=C2)=C(C(=O)NC2=CC=C(Cl)C=N2)C=C1 JVALKLBLUHGHNP-UHFFFAOYSA-N 0.000 description 1
- LPCINVCZMCLEGM-UHFFFAOYSA-N NCC1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CC=C2)C=C1 Chemical compound NCC1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=CC=C2)C=C1 LPCINVCZMCLEGM-UHFFFAOYSA-N 0.000 description 1
- KUVZGGJBACVUAA-UHFFFAOYSA-N NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Br)C=N4)C=C(F)C(F)=C3)C=C2)C=CC=C1 Chemical compound NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Br)C=N4)C=C(F)C(F)=C3)C=C2)C=CC=C1 KUVZGGJBACVUAA-UHFFFAOYSA-N 0.000 description 1
- UJGBTBNZYNOGJH-UHFFFAOYSA-N NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Br)C=N4)C=C(F)C=C3)C=C2)C=CC=C1 Chemical compound NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Br)C=N4)C=C(F)C=C3)C=C2)C=CC=C1 UJGBTBNZYNOGJH-UHFFFAOYSA-N 0.000 description 1
- QVHCSJQCOXBFFY-UHFFFAOYSA-N NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=N4)C=C(F)C(F)=C3)C=C2)C=CC=C1 Chemical compound NS(=O)(=O)C1=C(C2=CC=C(C(=O)NC3=C(C(=O)NC4=CC=C(Cl)C=N4)C=C(F)C(F)=C3)C=C2)C=CC=C1 QVHCSJQCOXBFFY-UHFFFAOYSA-N 0.000 description 1
- DSXXUHVGPRCTRC-UHFFFAOYSA-N NS(=O)(=O)C1=C(C2=CC=C(NC(=O)NC3=C(C(=O)NC4=CC=C(Br)C=N4)C=C(F)C=C3)C=C2)C=CC=C1 Chemical compound NS(=O)(=O)C1=C(C2=CC=C(NC(=O)NC3=C(C(=O)NC4=CC=C(Br)C=N4)C=C(F)C=C3)C=C2)C=CC=C1 DSXXUHVGPRCTRC-UHFFFAOYSA-N 0.000 description 1
- HLUMWMDWGMLTRX-UHFFFAOYSA-N NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C([N+](=O)[O-])C=C2)C=C1.NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C([N+](=O)[O-])C=C2)C=C1.NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC([N+](=O)[O-])=C2)C=C1 Chemical compound NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C([N+](=O)[O-])C=C2)C=C1.NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C([N+](=O)[O-])C=C2)C=C1.NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC([N+](=O)[O-])=C2)C=C1 HLUMWMDWGMLTRX-UHFFFAOYSA-N 0.000 description 1
- CEGMMABBTNUSIO-UHFFFAOYSA-N NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1 Chemical compound NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=C2)C=C1 CEGMMABBTNUSIO-UHFFFAOYSA-N 0.000 description 1
- KJBCXSVAAZPYKS-UHFFFAOYSA-N NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=N2)C=C1 Chemical compound NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CC=N2)C=C1 KJBCXSVAAZPYKS-UHFFFAOYSA-N 0.000 description 1
- YHCVPWGUVHTBRY-UHFFFAOYSA-N NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CN=C2)C=C1.NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=NC=C2)C=C1.NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CN=C2)C=C1.NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=NC=C2)C=C1 Chemical compound NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=CN=C2)C=C1.NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=NC=C2)C=C1.NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CN=C2)C=C1.NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=NC=C2)C=C1 YHCVPWGUVHTBRY-UHFFFAOYSA-N 0.000 description 1
- MDSPRVILVNUAGV-UHFFFAOYSA-N NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)N=CC=C2)C=C1 Chemical compound NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)N=CC=C2)C=C1 MDSPRVILVNUAGV-UHFFFAOYSA-N 0.000 description 1
- GKCSOJWDPJQQEU-UHFFFAOYSA-N NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1 Chemical compound NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=C2)C=C1 GKCSOJWDPJQQEU-UHFFFAOYSA-N 0.000 description 1
- NRYPQCIFWFTPKX-UHFFFAOYSA-N NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=N2)C=C1 Chemical compound NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=CC=N2)C=C1 NRYPQCIFWFTPKX-UHFFFAOYSA-N 0.000 description 1
- PJABDQSLGPZVHV-UHFFFAOYSA-N NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)N=CC=C2)C=C1 Chemical compound NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)N=CC=C2)C=C1 PJABDQSLGPZVHV-UHFFFAOYSA-N 0.000 description 1
- QTULBZARQYTKPU-UHFFFAOYSA-N NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1 Chemical compound NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1 QTULBZARQYTKPU-UHFFFAOYSA-N 0.000 description 1
- IKPWMNXHZAWVPL-UHFFFAOYSA-N NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(OC3=CC4=CC=C(Br)C=C4C=C3)C=CC=C2)C=C1 Chemical compound NS(=O)(=O)C1=CC=CC=C1C1=CC=C(C(=O)NC2=C(OC3=CC4=CC=C(Br)C=C4C=C3)C=CC=C2)C=C1 IKPWMNXHZAWVPL-UHFFFAOYSA-N 0.000 description 1
- ZHVPTERSBUMMHK-UHFFFAOYSA-N Nc1cc(cccc2)c2cc1O Chemical compound Nc1cc(cccc2)c2cc1O ZHVPTERSBUMMHK-UHFFFAOYSA-N 0.000 description 1
- PIQXBELMAOEHCF-UHFFFAOYSA-N O=C(NC1=C(C(=O)NC2=CC=C(Br)C=N2)C=CC=C1)C1=CC=C(C2=NCCN2)C=C1 Chemical compound O=C(NC1=C(C(=O)NC2=CC=C(Br)C=N2)C=CC=C1)C1=CC=C(C2=NCCN2)C=C1 PIQXBELMAOEHCF-UHFFFAOYSA-N 0.000 description 1
- DZESXFZAOQEIBS-UHFFFAOYSA-N O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CN2CCCC2)C=C1 Chemical compound O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=C(Cl)C=C1)C1=CC=C(CN2CCCC2)C=C1 DZESXFZAOQEIBS-UHFFFAOYSA-N 0.000 description 1
- WQLOWAOWARASDB-UHFFFAOYSA-N O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=CC=C1)C1=CC=C(C2=NCCN2)C=C1 Chemical compound O=C(NC1=C(C(=O)NC2=CC=C(Cl)C=N2)C=CC=C1)C1=CC=C(C2=NCCN2)C=C1 WQLOWAOWARASDB-UHFFFAOYSA-N 0.000 description 1
- OMDJZGIGSOMJKA-UHFFFAOYSA-N O=C(NC1=C(C(=O)NC2=NC=C(Cl)C=C2)C=CC=C1)C1=CC=C(CNC2=NCCN2)C=C1 Chemical compound O=C(NC1=C(C(=O)NC2=NC=C(Cl)C=C2)C=CC=C1)C1=CC=C(CNC2=NCCN2)C=C1 OMDJZGIGSOMJKA-UHFFFAOYSA-N 0.000 description 1
- XMTKTCHWAFEJDJ-UHFFFAOYSA-N [C-]#[N+]C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1 Chemical compound [C-]#[N+]C1=CC(F)=C(C(=O)NC2=C(C(=O)NC3=NC=C(Cl)C=C3)C=C(Cl)C=C2)C=C1 XMTKTCHWAFEJDJ-UHFFFAOYSA-N 0.000 description 1
- MTXHGHSYGORCNJ-UHFFFAOYSA-N [C-]#[N+]C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(OC)C(OC)=C2)C=C1 Chemical compound [C-]#[N+]C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(OC)C(OC)=C2)C=C1 MTXHGHSYGORCNJ-UHFFFAOYSA-N 0.000 description 1
- FYXQMJQWZDTKHV-UHFFFAOYSA-N [C-]#[N+]C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(OC)C=C2)C=C1 Chemical compound [C-]#[N+]C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Br)C=N3)C=C(OC)C=C2)C=C1 FYXQMJQWZDTKHV-UHFFFAOYSA-N 0.000 description 1
- VFQZHFCTQAKZPQ-UHFFFAOYSA-N [C-]#[N+]C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(O)C=C2)C=C1 Chemical compound [C-]#[N+]C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(O)C=C2)C=C1 VFQZHFCTQAKZPQ-UHFFFAOYSA-N 0.000 description 1
- NBQIGLLYESIOHP-UHFFFAOYSA-N [C-]#[N+]C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OC)C=C2)C=C1 Chemical compound [C-]#[N+]C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OC)C=C2)C=C1 NBQIGLLYESIOHP-UHFFFAOYSA-N 0.000 description 1
- GANIXRZQPNTBPU-UHFFFAOYSA-N [C-]#[N+]C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)OCC)C=C2)C=C1 Chemical compound [C-]#[N+]C1=CC=C(C(=O)NC2=C(C(=O)NC3=CC=C(Cl)C=N3)C=C(OCC(=O)OCC)C=C2)C=C1 GANIXRZQPNTBPU-UHFFFAOYSA-N 0.000 description 1
- VFGMEXLHQVVOAL-UHFFFAOYSA-N [C-]#[N+]C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(Cl)C=C2)C=C1 Chemical compound [C-]#[N+]C1=CC=C(C(=O)NC2=C(C(=O)NC3=NC=C(Br)C=C3)C=C(Cl)C=C2)C=C1 VFGMEXLHQVVOAL-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/60—Salicylic acid; Derivatives thereof
- A61K31/612—Salicylic acid; Derivatives thereof having the hydroxy group in position 2 esterified, e.g. salicylsulfuric acid
- A61K31/616—Salicylic acid; Derivatives thereof having the hydroxy group in position 2 esterified, e.g. salicylsulfuric acid by carboxylic acids, e.g. acetylsalicylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4858—Organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4866—Organic macromolecular compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/28—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a non-condensed six-membered aromatic ring of the carbon skeleton
- C07C237/42—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a non-condensed six-membered aromatic ring of the carbon skeleton having nitrogen atoms of amino groups bound to the carbon skeleton of the acid part, further acylated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/49—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C255/58—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton containing cyano groups and singly-bound nitrogen atoms, not being further bound to other hetero atoms, bound to the carbon skeleton
- C07C255/60—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton containing cyano groups and singly-bound nitrogen atoms, not being further bound to other hetero atoms, bound to the carbon skeleton at least one of the singly-bound nitrogen atoms being acylated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C257/00—Compounds containing carboxyl groups, the doubly-bound oxygen atom of a carboxyl group being replaced by a doubly-bound nitrogen atom, this nitrogen atom not being further bound to an oxygen atom, e.g. imino-ethers, amidines
- C07C257/10—Compounds containing carboxyl groups, the doubly-bound oxygen atom of a carboxyl group being replaced by a doubly-bound nitrogen atom, this nitrogen atom not being further bound to an oxygen atom, e.g. imino-ethers, amidines with replacement of the other oxygen atom of the carboxyl group by nitrogen atoms, e.g. amidines
- C07C257/18—Compounds containing carboxyl groups, the doubly-bound oxygen atom of a carboxyl group being replaced by a doubly-bound nitrogen atom, this nitrogen atom not being further bound to an oxygen atom, e.g. imino-ethers, amidines with replacement of the other oxygen atom of the carboxyl group by nitrogen atoms, e.g. amidines having carbon atoms of amidino groups bound to carbon atoms of six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C259/00—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups
- C07C259/12—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups with replacement of the other oxygen atom of the carboxyl group by nitrogen atoms, e.g. N-hydroxyamidines
- C07C259/18—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups with replacement of the other oxygen atom of the carboxyl group by nitrogen atoms, e.g. N-hydroxyamidines having carbon atoms of hydroxamidine groups bound to carbon atoms of six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/15—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings
- C07C311/16—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to hydrogen atoms or to an acyclic carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/30—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/45—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups at least one of the singly-bound nitrogen atoms being part of any of the groups, X being a hetero atom, Y being any atom, e.g. N-acylaminosulfonamides
- C07C311/46—Y being a hydrogen or a carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/26—Sulfones; Sulfoxides having sulfone or sulfoxide groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C317/32—Sulfones; Sulfoxides having sulfone or sulfoxide groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton with sulfone or sulfoxide groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton
- C07C317/34—Sulfones; Sulfoxides having sulfone or sulfoxide groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton with sulfone or sulfoxide groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton having sulfone or sulfoxide groups and amino groups bound to carbon atoms of six-membered aromatic rings being part of the same non-condensed ring or of a condensed ring system containing that ring
- C07C317/38—Sulfones; Sulfoxides having sulfone or sulfoxide groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton with sulfone or sulfoxide groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton having sulfone or sulfoxide groups and amino groups bound to carbon atoms of six-membered aromatic rings being part of the same non-condensed ring or of a condensed ring system containing that ring with the nitrogen atom of at least one amino group being part of any of the groups, X being a hetero atom, Y being any atom, e.g. N-acylaminosulfones
- C07C317/40—Y being a hydrogen or a carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/44—Sulfones; Sulfoxides having sulfone or sulfoxide groups and carboxyl groups bound to the same carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/75—Amino or imino radicals, acylated by carboxylic or carbonic acids, or by sulfur or nitrogen analogues thereof, e.g. carbamates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
- C07D213/82—Amides; Imides in position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/22—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the nitrogen-containing ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/56—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/64—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms, e.g. histidine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/12—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly or doubly bound nitrogen atoms
- C07D295/125—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly or doubly bound nitrogen atoms with the ring nitrogen atoms and the substituent nitrogen atoms attached to the same carbon chain, which is not interrupted by carbocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/38—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- R 1e is a member selected from the group of —F, —Cl, —Br, —OH, -Me and —OMe.
- the invention provides compound of formula Ib, as described above, having the following stricture:
- the invention provides a compound of formula Ib, as described above, having the following structure:
- each G group is substituted by 0-4 R 1d groups and each such R 1d group is independently selected from the group consisting of:
- amidino groups illustrated above as substituents for the cyclized amine heterocyclic ring are instead form an acyclic amidino A-Q group and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.
- Dosage formulations of the compounds of this invention to be used for therapeutic administration must be sterile. Sterility is readily accomplished by filtration through sterile membranes such as 0.2 micron membranes, or by other conventional methods. Formulations typically will be stored in lyophilized form or as an aqueous solution.
- the pH of the preparations of this invention typically will be 3-11, more preferably 5-9 and most preferably 7-8. It will be understood that use of certain of the foregoing excipients, carriers, or stabilizers will result in the formation of cyclic polypeptide salts.
- While the preferred route of administration is by injection, other methods of administration are also anticipated such as orally, intravenously (bolus and/or infusion), subcutaneously, intramuscularly, colonically, rectally, nasally, transdermally or intraperitoneally, employing a variety of dosage forms such as suppositories, implanted pellets or small cylinders, aerosols, oral dosage formulations and topical formulations such as ointments, drops and dermal patches.
- the compounds of this invention are desirably incorporated into shaped articles such as implants which may employ inert materials such as biodegradable polymers or synthetic silicones, for example, Silastic, silicone rubber or other polymers commercially available.
- Diagnostic applications of the compounds of this invention will typically utilize formulations such as solution or suspension.
- the compounds of this invention may be utilized in compositions such as tablets, capsules or elixirs for oral administration, suppositories, sterile solutions or suspensions or injectable administration, and the like, or incorporated into shaped articles.
- Subjects in need of treatment (typically mammalian) using the compounds of this invention can be administered dosages that will provide optimal efficacy.
- the dose and method of administration will vary from subject to subject and be dependent upon such factors as the type of mammal being treated, its sex, weight, diet, concurrent medication, overall clinical condition, the particular compounds employed, the specific use for which these compounds are employed, and other factors which those skilled in the medical arts will recognize.
- dissolution or suspension of the active compound in a vehicle such as an oil or a synthetic fatty vehicle like ethyl oleate, or into a liposome may be desired.
- a vehicle such as an oil or a synthetic fatty vehicle like ethyl oleate
- Buffers, preservatives, antioxidants and the like can be incorporated according to accepted pharmaceutical practice.
- Step 2 To a solution of 4-bromo-2-nitroaniline (43.4 mg, 0.2 mmol, 2.0 equiv) in 5 ml of methylene chloride treated with AlMe 3 (2M in hexane, 0.3 mL, 6 equiv) for 30 min was added methyl 2-(4-[(2-methylsulfonyl)phenyl]phenylcarbonyl)aminobenzoate (46.6 mg, 1 equiv). The mixture was stirred at rt overnight, quenched with saturated aqueous potassium sodium tartrate. The organic layer was dried over MgSO 4 , filtered and evaporated.
- Step 2 To A solution of 2-amino-5-bromopridine (135 mg, 4.0 equiv) in 5 mL of methylene chloride treated with AlMe 3 (2M in hexane, 1 mL, 10 equiv) for 30 min was added methyl 2-(4-[(2-t-butylaminosulfonyl)phenyl]phenylcarbonyl)amino-4-nitrobenzoate (100 mg, 0.2 mmol, 1 equiv). The mixture was stirred at rt overnight, quenched with saturated aqueous potassium sodium tartrate.
- Step 1 To a solution of 5-methyl-2-nitrobenzoic acid (1 g, 5.52 mmol) in dichloromethane (5 ml) was added oxalyl chloride (0.964 ml, 11.04 mmol) and a few drops of dimethylformamide. The mixture was stirred at r.t. for 2 hrs. After the evaporation of the solvent, the residue was dissolved in dichloromethane (5 ml). 2-amino-5-chloropyridine (852 mg, 6.62 mmol) and pyridine (1.34 ml, 16.56 mmol) were added to the solution. The mixture was stirred at r.t. overnight.
- Step 3 A mixture of N-(5-bromo-2-pyridinyl)-(2-(4-hydroxyamidinophenylcarbonyl)amino)5-fluorophenylcarboxamide (0.56 g, 1.19 mmol, 1.0 equiv) and zinc dust (0.39 g, 5.0 equiv), in 10 mL of acetic acid was stirred at rt for 45 min. The volatile was filtered and evaporated.
- Example 284 To a solution of Example 284 (10 mg) in MeOH (1 mL) was added 50 ⁇ L of 1N aq. LiOH solution. The mixture was stirred for 1 hour and purified by RP-HPLC to give the title compound. MS found for C 24 H 22 ClN 5 O 5 (M+H) + : 496.
- Step 1 A solution of methyl 2-amino-5-nitrobenzoate (1 equiv) and 4-cyanobenzoic acid (1 equiv) in pyridine was treated with POCl 3 (1.1 equiv) for 1 h. The resulting mixture was quenched by slow addition of water, and extracted with EtOAc. The organic layer was dried over MgSO 4 , filtered and flash chromatographied to give the desired product.
- Step 2 A solution of 2-amino-5-bromopridine (45 mg, 4.0 equiv) in 5 mL of methylene chloride treated with AlMe 3 (2M in hexane, 0.65 mL, 20 equiv) for 30 min was added the compound obtained in step 1 (0.064 mmol, 1 equiv). The mixture was stirred at rt overnight, quenched with saturated aqueous potassium sodium tartrate. The organic layer was dried over MgSO 4 , filtered, evaporated and purified by column chromatography to give the desired product.
- Step 3 The product obtained in step 2 was subjected to standard Pinner conditions to give the title compound after HPLC (C18 reversed phase, eluting with 0.5% TFA in H 2 O/CH 3 CN). MS (M+H) + : 467.
- Example 297 (1 equiv) in CH 2 Cl 2 was treated with BBr 3 (4 equiv) overnight, quenched with ice water. HPLC (C18 reversed phase, eluting with 0.5% TFA in H 2 O/CH 3 CN) gave the title compound. MS (M+H) + : 438.
- Step 1 A mixture of 4-cyanobenzaldehyde (1 equiv), 4-chloro-2-(5-chloro-2-pyridinyl)amino-carbonyl aniline (1 equiv) and glacial acetic acid (10 equiv) in CH 2 Cl 2 was stirred at rt for 30 min. NaBH(OAc) 3 (3 equiv) was added at once and the mixture was stirred overnight. The reaction was quenched with water and the organic layer was washed with brine and dried over Na 2 SO 4 . Column separation over silica gel gave the desired product.
- Step 2 A solution of the compound obtained in step 3 (1S mg) in anhydrous pyridine (10 mL) and triethyl amine (2 mL) was saturated with hydrogen sulfide gas at 0° C. The mixture was stirred at rt overnight. After concentration, the residue was dissolved in anhydrous acetone (10 mL) and iodomethane (1 mL) was added. The mixture was refluxed for 2 hrs. After concentration, the residue was dissolved in anhydrous methanol (5 mL) and a solution of pyrrolidine 0.5 mL) and acetic acid (0.5 mL) in anhydrous methanol (5 ml) was added. The mixture was refluxed for 15 min. After concentrated, the crude residue was purified by RP-HPLC to give target. MS (M+H) 435.
- Step 3 A solution of the compound obtained in step 2 (IS mg) in anhydrous pyridine (10 mL) and triethyl amine (2 mL) was saturated with hydrogen sulfide gas at 0° C. The mixture was stirred at rt overnight. After concentration, the residue was dissolved in anhydrous acetone (10 mL) and iodomethane (1 mL) was added. The mixture was refluxed for 2 hrs. After concentration, the residue was dissolved in anhydrous methanol (5 mL) and a solution of pyrrolidine (0.5 mL) and acetic acid (0.5 mL) in anhydrous methanol (5 ml) was added. The mixture was refluxed for 15 min. After concentrated, the crude residue was purified by RP-HPLC to give target. MS (M+H) 434.
Abstract
Description
A-Q-D-E-G-J-X (I)
where:
- A is selected from:
- (a) C1-C6-alkyl;
- (b) C3-C8-cycloalkyl;
- (c) —N(R1,R2), N(R1,R2)—C(═NR3)—, N(R1,R2)—C(═NR3)—N(R4)—, R1—C(═NR3)—, R1—C(═NR3)—N(R4)—;
- (d) phenyl, which is independently substituted with 0-2 R substitutents;
- (e) naphthyl, which is independently substituted with 0-2 R substituents;
and - (f) a monocyclic or fused bicyclic heterocyclic ring system having from 5 to 10 ring atoms, wherein 1-4 ring atoms of the ring system are selected from N, O and S, and wherein the ring system may be substituted with 0-2 R substituents;
- R is selected from:
- H, halo, —CN, —CO2R1, —C(═O)—N(R1, R2), —(CH2)m—CO2R1, —(CH2)m—C(═O)—N(R1, R2), —NO2, —SO2N(R1, R2), —SO2R1, —(CH2)m—NR1R2, —(CH2)m—C(═NR3)—R1, —(CH2)m—C(═NR3)—N(R1,R2), —(CH2)m—N(R4)—C(═NR3)—N(R1,R2), —(CH2)mNR1— group appended to a 3 to 6 membered heterocyclic ring containing from 1-4 heteroatoms selected from N, O and S, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CF3, —OR2, and a 5-6 membered heterocyclic system containing from 1-4 heteroatoms selected from N, O and S, wherein from 1-4 hydrogen atoms on the heterocyclic system may be independently replaced with a member selected from the group consisting of halo, —C1-C4-alkyl, —C1-4alkyl-CN, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl and —NO2;
- m is an integer of 0-2;
- R1, R2, R3 and R4 are independently selected from the group consisting of:
- H, —OR5, —N(—R5, —R6), —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —C0-4alkylphenyl and —C0-4alkylnaphthyl, wherein from 1-4 hydrogen atoms on the ring atoms of the phenyl and naphthyl moieties may be independently replaced with a member selected from the group consisting of halo, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CN, and —NO2; or
- R1 and R2, or R2 and R3 taken together can form a 3-8 membered cycloalkyl or a heterocyclic ring system, wherein the heterocyclic ring system may have from 3 to 10 ring atoms, with 1 to 2 rings being in the ring system and contain from 1-4 heteroatoms selected from N, O and S, wherein from 1-4 hydrogen atoms on the heterocyclic ring system may be independently replaced with a member selected from the group consisting of halo, C1-C4-alkyl, —CN—C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl and —NO2;
- R5 and R6 are independently selected from the group consisting of:
- H, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —C0-4alkylphenyl and —C0-4alkylnaphthyl, wherein from 1-4 hydrogen atoms on the ring atoms of the phenyl and naphthyl moieties may be independently replaced with a member selected from the group consisting of halo, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CN, and —NO2; or
- R5 and R6 taken together can form a 3-8 membered cycloalkyl or a heterocyclic ring system, wherein the heterocyclic ring system may have from 3 to 10 ring atoms, with 1 to 2 rings being in the ring system and contain from 1-4 heteroatoms selected from N, O and S, wherein from 1-4 hydrogen atoms on the heterocyclic ring system may be independently replaced with a member selected from the group consisting of halo, —C1-C4-alkyl, —CN—C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl and —NO2;
- Q is a member selected from the group consisting of:
- a direct link, —CH2—, —C(═O)—, —O—, —N(R7)—, —N(R7)CH2—, —CH2N(R7)—, —C(═NR7)—, —C(═O)—N(R7)—, —N(R7)—C(═O)—, —S—, —SO—, —SO2—, —SO2—N(R7)— and —N(R7)—SO2—;
- R7 is selected from:
- H, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —C0-4alkylphenyl and —C0-4alkylnaphthyl, wherein from 1-4 hydrogen atoms on the ring atoms of the phenyl and naphthyl moieties may be independently replaced with a member selected from the group consisting of halo, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CN, and —NO2;
- D is a direct link or is a member selected from the group consisting of:
- (a) phenyl, which is independently substituted with 0-2 R1a substituents;
- (b) naphthyl, which is independently substituted with 0-2 R1a substituents; and
- (c) a monocyclic or fused bicyclic heterocyclic ring system having from 5 to 10 ring atoms, wherein 1-4 ring atoms of the ring system are selected from N, O and S, and wherein the ring system may be substituted from 0-2 R1a substituents;
- R1a is selected from:
- halo, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CN, —NO2, —(CH2)nNR2aR3a, —(CH2)nCO2R2a, —(CH2)nCONR2aR3a, —SO2NR2aR3a, —SO2R2a, —CF3, —OR2a, and a 5-6 membered aromatic heterocyclic system containing from 1-4 heteroatoms selected from N, O and S, wherein from 1-4 hydrogen atoms on the aromatic heterocyclic system may be independently replaced with a member selected from the group consisting of halo, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CN and —NO2,
- R2a and R3a are independently selected from the group consisting of:
- H, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —C0-4alkylphenyl and —C0-4alkylnaphthyl, wherein from 1-4 hydrogen atoms on the ring atoms of the phenyl and naphthyl moieties may be independently replaced with a member selected from the group consisting of halo, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CN and —NO2;
- n is an integer of 0-2;
- E is a direct link or a member selected from the group consisting of:
- —C1-2-alkyl-, —O—, —S—, —SO—, —SO2—, —C0-1-alkyl-C(═O), —C0-1-alkyl-C(═O)—N(—R8)—C0-1-alkyl-, —C0-1-alkyl-N(—R8)—C(═O)—C0-1-alkyl-, —N(—R8)—C(═O)—N(—R8)— and —C0-1-alkyl-N(—R8)—;
- R8 is a member selected from the group consisting of:
- H; —C1-4-alkyl; —C0-4-alkylaryl; —C0-4-alkyl-heteroaryl; —C1-4-alkyl-C(═O)—OH, —C1-4-alkyl-C(═O)—O—C1-4-alkyl, and —C1-4-alkyl-C(═O)—N(—R2b, —R3b);
- R2b and R3b are each a member independently selected from the group consisting of:
- H, —C1-4-alkyl, —C0-4-alkyl-aryl; —C0-4-alkyl-heterocyclic group, and R2b and R3b together with the N atom to which they are attached can form a 5-8 membered heterocyclic ring containing 1-4 heteroatoms selected from N, O and S, wherein the heterocyclic ring may be substituted with 0-2 R1c groups;
- R1c is a member selected from the group consisting of:
- Halo; —C1-4-alkyl; —CN, —NO2; —C(═O)—N(—R2c, —R3c); —C(═O)—OR2c; —(CH2)q—N(—R2c, —R3c); —SO2—N(—R2c, —R3c); —SO2R2c; —CF3 and —(CH2)q—OR2c;
- R2c and R3c are each independently a member selected from the group consisting of:
- H; —C1-4-alkyl and —C1-4-alkyl-aryl;
- q is an integer of 0-2;
- G is a member selected from the group consisting of:
- (a) C2-alkenyl or C3-8-cycloalkenyl, wherein the alkenyl and cycloalkenyl attachment points are the alkenyl carbon atoms and wherein the —C2-alkenyl or —C3-8-cycloalkenyl are substituted with 0-4 R1d groups;
- (b) a phenylene group wherein the ring carbon atoms of the phenylene group are substituted with 0-4 R1d groups;
- (c) a 3-8 membered a saturated, partially unsaturated or aromatic monocyclic-heterocyclic ring system containing 1-4 heteroatoms selected from N, O and S, wherein 0-2 ring atoms of the heterocyclic ring may be substituted with 0-4 R1d groups; and,
- (d) an 8-10 membered fused heterocyclic bicyclic ring system, containing 1-4 heteroatoms selected from N, O and S, wherein 0-2 ring atoms of the fused bicyclic ring system may be substituted with 0-4 R1d groups;
- R1d is a member selected from the group consisting of:
- H, halo; C1-6-alkyl, carbocylic aryl, —CN; —NO2; —(CH2)0-6—NR2dR3d; —SO2NR2dR3d; —SO2R2d; —CF3; —(CH2)0-6—OR2d; —OH, —OC1-6alkyl, —O—(CH2)1-6OR2d; —O—(CH2)1-6—C(═O)—O—R2d; —O—(CH2)1-6—C(═O)—N(R2d,R3d); —N(R5a)—(CH2)1-6—OR2d; —N(R5a)—(CH2)1-6—N(R2d,R3d); —C(═O)—N(R2d,R3d); —N(R5a)—(CH2)1-6—C(═O)—N(R2d,R3d); —N(—(CH2)1-6—OR2d)2; —N(R5a)—(CH2)1-6—OR2d; —N(R5a)—C(═O)—R2d; —N(R5a)—SO2—R2d; —(CH2)0-6—C(═O)—O—R2d; —(CH2)0-6—C(═O)—N(R2d,R3d); —(CH2)0-6—C(═NR2d)—O—R2d; —(CH2)0-6—N(R5a)C(═NR2d)—N(R3d,R4d); a —(CH2)0-6—N(R3d)C5-6 membered saturated, partially unsaturated or aromatic heterocyclic ring containing 1-4 heteroatoms selected from N, O and S, and a —(CH2)0-6-5-6 membered saturated, partially unsaturated or aromatic heterocyclic ring containing 1-4 heteroatoms selected from N, O and S;
- R5a, R2d, R3d and R4d are each independently a member selected from the group consisting of:
- H, C1-6-alkyl and C1-6-alkylaryl, —CN; —NO2; carbocylic aryl, —CN; —NO2; or
- R2d and R3d taken together with the N atoms they are independently attached form a 5-7 membered saturated, partially unsaturated or aromatic heterocyclic ring; or
- R3d and R4d taken together with the N atom to which they are attached form a 5-8 membered saturated, partially unsaturated or aromatic heterocyclic ring containing 1-4 heteroatoms selected from N, O and S;
- J is a direct link or is a member selected from the group consisting of:
- —N(—R9)—C(═O)—; —C(═O)—N(—R9)—; —O—; —S—; —SO—; —SO2—; —CH2—; —N(—R9)—; and —N(—R9)—SO2—;
- R9 is a member selected from the group consisting of:
- H; —C1-4-alkyl; —C0-4-alkyl-carbocyclic aryl; —(CH2)0-4-5-6 membered saturated, partially unsaturated or aromatic heterocyclic ring containing 1-4 heteroatoms selected from N, O and S; —(CH2)1-6—C(═O)—O—C1-4-alkyl; and —(CH2)1-6—C(═O)—N(R6a,R6b);
- R6a and R6b are each a member independently selected from the group consisting of:
- H and —C1-6-alkyl;
- X is a member selected from the group consisting of:
- (a) phenyl substituted with 0-3 R1e groups;
- (b) naphthyl substituted with 0-3 R1e groups and
- (c) a 6-membered aromatic heterocyclic ring system containing 1-3 N atoms and having 0-3 ring atoms substituted with 0-3 R1e groups; and
- (d) an 8-10 membered fused aromatic heterocyclic bicyclic ring system containing 1-4 heteroatoms selected from N, O and S and 0-3 ring atoms of the fused heterocyclic bicyclic ring system are substituted with 0-3 R1e groups;
- R1e is a member independently selected from the group consisting of:
- Halo; CF3; —C1-4-alkyl; carbocyclic aryl; —C0-2-alkyl-CN; —O—R2e; —C0-2-alkyl-C(═O)—O—R2e; —C0-2-alkyl-C(═O)—N(R2e, R3e); —C0-2-alkyl-NO2; —C0-2-alkyl-N(R2e, R3e); —C0-2-alkyl-SO2—N(R2e, R3e); —C0-2-alkyl-SO2—R2e; trihaloalkyl; —O—C0-2alkyl-O—R2e; —C0-2-alkyl-O—R2e; —O—C1-4-alkyl-C(═O)—N(R2e, R3e); —O—C1-4-alkyl-C(═O)—O—R2e; —C0-2-alkyl-N(R2e)—C(═O)—R3e; —C0-2-alkyl-N(—R2e)—SO2—R3e; —CH2—N(R2e)—C(═O)—R3e; —CH2—N(R2e)—SO2—R3e; —(CH2)0-6NR2eR3e; —C(═O)—N(R2e,R3e); —N(—(CH2)1-6—OR2e)2; —N(R10)—(CH2)1-6—OR2e; —N(R10)—C(═O)—R2e; —N(R10)—SO2—R2e; —C(═N(R10))—N(R2e,R3e); and a —(CH2)0-6-5-6 membered saturated, partially unsaturated or aromatic heterocyclic ring containing 14 heteroatoms selected from N, O and S;
- R10, R2e and R3w are each independently a member selected from the group consisting of:
- H; —C1-4-alkyl; —C0-2-alkyl-O—R1g; —C0-2-alkyl-N(—R1g, —R2g); —C1-4-alkyl-carbocyclic aryl; —C1-4-alkyl-heterocyclic; and R10 and R2e, or R2e and R3e together with the N atom to which they are attached can form 5-8 membered heterocyclic ring containing 1-4 heteroatoms selected from N, O and S which can be substituted with 0-2 R1g groups;
- R1g and R2g are independently a member selected from the group of:
- H; halo; —C1-4-alkyl, a carbocyclic aryl group; a saturated, partially unsaturated or aromatic heterocyclic group; —CN; —C(═O)—N(R3g)R4g; —C(═O)—OR3g; —NO2; —(CH2)p—NR3gR4g; —SO2NR3gR4g; —SO2R3g; —CF3; and —(CH2)pOR3g;
- p is an integer of 0-2;
- R3g and R4g are each independently selected from the group consisting of:
- H; C1-4-alkyl and —C0-4-alkyl-carbocyclic aryl;
- and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.
A-Q-D-E-G-J-X (I)
where:
- A is selected from:
- (a) C1-C6-alkyl;
- (b) C3-C8-cycloalkyl;
- (c) —N(R1,R2), N(R1,R2)—C(═NR3)—, N(R1,R2)—C(═NR3)—N(R4), R1—C(═NR3)—, R1—C(═NR3)—N(R4)—;
- (d) phenyl, which is independently substituted with 0-2 R substituents;
- (e) naphthyl, which is independently substituted with 0-2 R substituents;
and - (f) a monocyclic or fused bicyclic heterocyclic ring system having from 5 to 10 ring atoms, wherein 1-4 ring atoms of the ring system are selected from N, O and S, and wherein the ring system may be substituted with 0-2 R substituents;
- R is selected from:
- H, halo, —CN, —CO2R1, —C(═O)—N(R1, R2), —(CH2)m—CO2R1, —(CH2)m—C(═O)—N(R1, R2), —NO2, —SO2N(R1, R2), —SO2R1, —(CH2)mNR1R2; —(CH2)m—C(═NR3)—R1, —(CH2)m—C(═NR3)—N(R1,R2), (CH2)m—N(R4)—C(═NR3)—N(R1,R2), —(CH2)mNR1—C3-6heterocyclics, C1-4alkyl, C2-6alkenyl, C2-6alkynyl, C3-8cycloalkyl, C0-4alkylC3-8cycloalkyl, —CF3, —OR2, and a 5-6 membered heterocyclic system containing from 1-4 heteroatoms selected from N, O and S, wherein from 1-4 hydrogen atoms on the heterocyclic system may be independently replaced with a member selected from the group consisting of halo, C1-C4-alkyl, CN—C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl and —NO2;
- m is an integer of 0-2;
- R1, R2, R3 and R4 are independently selected from the group consisting of:
- H, —OR5, —N(—R5, —R6), —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —C0-4alkylphenyl and —C0-4alkylnaphthyl, wherein from 1-4 hydrogen atoms on the ring atoms of the phenyl and naphthyl moieties may be independently replaced with a member selected from the group consisting of halo, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CN, and —NO2; or
- R1 and R2, or R2 and R3 taken together can form a 3-8 membered cycloalkyl or a heterocyclic ring system, wherein the heterocyclic ring system may have from 3 to 10 ring atoms, with 1 to 2 rings being in the ring system and contain from 1-4 heteroatoms selected from N, O and S, wherein from 1-4 hydrogen atoms on the heterocyclic ring system may be independently replaced with a member selected from the group consisting of halo, C1-C4-alkyl, —CN—C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl and —NO2;
- R5 and R6 are independently selected from the group consisting of:
- H, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —C0-4alkylphenyl and —C0-4alkylnaphthyl, wherein from 1-4 hydrogen atoms on the ring atoms of the phenyl and naphthyl moieties may be independently replaced with a member selected from the group consisting of halo, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CN, and —NO2; or
- R5 and R6 taken together can form a 3-8 membered cycloalkyl or a heterocyclic ring system, wherein the heterocyclic ring system may have from 3 to 10 ring atoms, with 1 to 2 rings being in the ring system and contain from 1-4 heteroatoms selected from N, O and S, wherein from 1-4 hydrogen atoms on the heterocyclic ring system may be independently replaced with a member selected from the group consisting of halo, C1-C4-alkyl, —CN—C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl and —NO2;
- Q is a member selected from the group consisting of:
- a direct link, —CH2—, —C(═O)—, —O—, —N(R7)—, —N(R7)CH2—, —CH2N(R7)—, —C(═NR7)—, —C(═O)—N(R7)—, —N(R7)—C(═O)—, —S—, —SO—, —SO2—, —SO2—N(R7)— and —N(R7)—SO2—;
- R7 is selected from:
- H, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —C0-4alkylphenyl and —C0-4alkylnaphthyl, wherein from 1-4 hydrogen atoms on the ring atoms of the phenyl and naphthyl moieties may be independently replaced with a member selected from the group consisting of halo, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CN, and —NO2;
- D is a direct link or is a member selected from the group consisting of:
- (a) phenyl, which is independently substituted with 0-2 R1a substituents;
- (b) naphthyl, which is independently substituted with 0-2 R12 substituents; and
- (c) a monocyclic or fused bicyclic heterocyclic ring system having from 5 to 10 ring atoms, wherein 1-4 ring atoms of the ring system are selected from N, O and S, and wherein the ring system may be substituted from 0-2 R1a substituents;
- R1a is selected from:
- halo, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CN, —NO2, —(CH2)nNR2aR3a, —(CH2)nCO2R2a, —(CH2)CONR2aR3a, —SO2NR2aR3a, —SO2R2a, —CF3, —OR2a, and a 5-6 membered aromatic heterocyclic system containing from 1-4 heteroatoms selected from N, O and S, wherein from 1-4 hydrogen atoms on the aromatic heterocyclic system may be independently replaced with a member selected from the group consisting of halo, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CN and —NO2;
- R2a and R3a are independently selected from the group consisting of:
- H, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —C0-4alkylphenyl and —C0-4alkylnaphthyl, wherein from 1-4 hydrogen atoms on the ring atoms of the phenyl and naphthyl moieties may be independently replaced with a member selected from the group consisting of halo, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CN and —NO2;
- n is an integer of 0-2;
- E is a direct link or a member selected from the group consisting of:
- —C1-2-alkyl-, —O—, —S—, —SO—, —SO2—, —CO0-1-alkyl-C(═O)—, —C0-1-alkyl-C(═O)—N(—R8)—C0-1-alkyl-, —C0-1-alkyl-N(—R8)—C(═O)—C0-1-alkyl-, —N(—R8)—C(═O)—N(—R8)— and —C0-1-alkyl-N(—R8)—;
- R8 is a member selected from the group consisting of:
- H; —C1-4-alkyl; —C0-4-alkylaryl; —C0-4-alkyl-heteroaryl; —C1-4-alkyl-C(═O)—OH, —C1-4-alkyl-C(═O)—O—C1-4-alkyl, and —C1-4-alkyl-C(═O)—N(—R2b, —R3b);
- R2b and R3b are each a member independently selected from the group consisting of:
- H, —C1-4-alkyl, —C0-4-alkyl-aryl; —C0-4-alkyl-heterocyclic group, and R2b and R3b together with the N atom to which they are attached can form a 5-8 membered heterocyclic ring containing 1-4 heteroatoms selected from N, O and S, wherein the heterocyclic ring may be substituted with 0-2 R1c groups;
- R1c is a member selected from the group consisting of:
- Halo; —C1-4-alkyl; —CN, —NO2; —C(═O)—N(—R2c, —R3c); —C(═O)OR2c; —(CH2)q—N(—R2c, —R3c); —SO2—N(—R2c, —R3c); —SO2R2c; —CF3 and —(CH2)q—OR2c;
- R2c and R3c are each independently a member selected from the group consisting of:
- H; —C1-4-alkyl and —C1-4-alkyl-aryl;
- q is an integer of 0-2;
- G is a member selected from the group consisting of:
- (a) C2-alkenyl or C3-8-cycloalkenyl, wherein the alkenyl and cycloalkenyl attachment points are the alkenyl carbon atoms and wherein C2-alkenyl or C3-8-cycloalkenyl are substituted with 0-4 R1d groups;
- (b) a phenylene group wherein the ring carbon atoms of the phenylene group are substituted with 0-4 R1d groups;
- (c) a 3-8 membered a saturated, partially unsaturated or aromatic monocyclic-heterocyclic ring system containing 1-4 heteroatoms selected from N, O and S, wherein 0-4 ring atoms of the heterocyclic ring may be substituted with 0-4 R1d groups; and,
- (d) an 8-10 membered fused heterocyclic bicyclic ring system, containing 1-4 heteroatoms selected from N, O and S, wherein 0-4 ring atoms of the fused bicyclic ring system may be substituted with 0-4 R1d groups;
- R1d is a member selected from the group consisting of:
- H, halo; C1-6alkyl, carbocylic aryl, —CN; —NO2; —(CH2)0-6—NR2dR3d; —SO2 NR2dR3d; —SO2R2d; —CF3; —(CH2)0-6—OR2d; —OH, —OC1-6alkyl, —O—(CH2)1-6OR2d; —O—(CH2)1-6—C(═O)—O—R2d; —O—(CH2)1-6—C(═O)—N(R2dR3d); —N(R5a)—(CH2)1-6—OR2d; —N(R5a)—(CH2)1-6—N(R2d,R3d); —C(═O)—N(R2d,R3d); —N(R5a)—(CH2)1-6—C(═O)—N(R2dR3d); —N(—(CH2)1-6—OR2d)2; —N(R5a)—(CH2)1-6OR2d; —N(R5a)—C(═O)—R2d; —N(R5a)—SO2—R2d; —(CH2)0-6—C(═O)—O—R2d; —(C2)0-6—C(═O)—N(R2d,R3d); —(CH2)0-6—C(═NR2d)—N(R3d,R4d); —(CH2)0-6—N(R5a)C(═NR2d)—N(R3d,R4d); and —(CH2)0-6—N(—R3d)— group attached directly by its nitrogen atom to a carbon atom of a 5 to 6 membered saturated, partially unsaturated or aromatic heterocyclic ring containing 1-4 heteroatoms selected from N, O and S, and a —(CH2)0-6— group attached to a 5-6 membered saturated, partially unsaturated or aromatic heterocyclic ring containing 1-4 heteroatoms selected from N, O and S;
- R5a, R2d, R3d and R4d are each independently a member selected from the group consisting of:
- H, C1-6-alkyl and C1-6-alkylaryl, —CN; —NO2; carbocylic aryl, —CN; —NO2; or
- R2d and R3d taken together with the N atoms there are independently attached form a 5-7 membered saturated, partially unsaturated or aromatic heterocyclic ring; or
- R3d and R4d taken together with the N atom to which they are attached form a 5-8 membered saturated, partially unsaturated or aromatic heterocyclic ring containing 1-4 heteroatoms selected from N, O and S;
- J is a direct link or is a member selected from the group consisting of:
- —N(—R9)—C(═O)—; —C(═O)—N(—R9)—; —O—; —S—; —SO—; —SO2—; —CH2—; —N(—R9)—; and —N(—R9)—SO2—;
- R9 is a member selected from the group consisting of:
- H; —C1-4-alkyl; —C0-4-alkyl-carbocyclic aryl; —(CH2)0-4-5-6 membered saturated, partially unsaturated or aromatic heterocyclic ring containing 1-4 heteroatoms selected from N, O and S; —(CH2)1-6—C(═O)—O—C1-4-alkyl; and —(CH2)1-6—C(═O)—N(R6a,R6b);
- R6a and R6b are each a member independently selected from the group consisting of:
- H and —C1-6alkyl;
- X is a member selected from the group consisting of:
- (a) phenyl substituted with 0-3 R1e groups;
- (b) naphthyl substituted with 0-3 R1e groups;
- (c) a 6-membered aromatic heterocyclic ring system containing 1-3 N atoms and having 0-3 ring atoms substituted with 0-3 R1e groups; and
- (d) an 8-10 membered fused aromatic heterocyclic bicyclic ring system containing 1-4 heteroatoms selected from N, O and S and 0-3 ring atoms of the fused heterocyclic bicyclic ring system are substituted with 0-3 R1e groups;
- R1e is a member independently selected from the group consisting of:
- Halo; CF3; —C1-4-alkyl; carbocyclic aryl; —C0-2-alkyl-CN; —O—R2e; —C0-2-alkyl-C(═O)—O—R2e; —C0-2-alkyl-C(═O)—N(R2e, R3e); —C0-2-alkyl-NO2; —C0-2-alkyl-N(R2e, R3e); —C0-2-alkyl-SO2—N(R2e, R3e); —C0-2-alkyl-SO2—R2e; trihaloalkyl; —O—C0-2-alkyl-O—R2e; —C0-2-alkyl-O—R2e; —O—C1-4-alkyl-C(═O)—N(R2e, R3e); —O—C1-4-alkyl-C(═O)—O—R2e; —C0-2-alkyl-N(R2e)—C(═O)—R3e; —C0-2-alkyl-N(—R2e)—SO2—R3e; —CH2—N(R2e)—C(═O)—R3e; —CH2—N(R2e)—SO2—R3e; —(CH2)0-6NR2eR3e; —C(═O)—N(R2e,R3e); —N(—(CH2)1-6—OR2e)2; —N(R10)—(CH2)1-6—OR2e; —N(R10)—C(═O)—R2e; —N(R10)—SO2—R2e; —C(═N(R10))—N(R2e,R3e); and a —(CH2)0-6-5-6 membered saturated, partially unsaturated or aromatic heterocyclic ring containing 1-4 heteroatoms selected from N, O and S;
- R10, R2e and R3e are each independently a member selected from the group consisting of:
- H; —C1-4alkyl; —C0-2-alkyl-O—R1g; —C0-2-alkyl-N(—R1g, —R2g); —C1-4-alkyl-carbocyclic aryl; —C1-4-alkyl-heterocyclic; and R10 and R2e, or R2e and R3e together with the N atom to which they are attached can form 5-8 membered heterocyclic ring containing 1-4 heteroatoms selected from N, O and S which can be substituted with 0-2 R1g groups;
- R1g and R2g are independently a member selected from the group of:
- H; halo; —C1-4-alkyl, a carbocyclic aryl group; a saturated, partially unsaturated or aromatic heterocyclic group; —CN; —C(═O)—N(R3g)R4g; —C(═O)—OR3g; —NO2; —(CH2)p—NR3gR4g; —SO2NR3gR4g; —SO2R3g; —CF3; and —(CH2)pOR3g;
- p is an integer of 0-2; and
- R3g and R4g are each independently selected from the group consisting of:
- H; C1-4-alkyl and —C0-4-alkyl-carbocyclic aryl;
A-Q-D-E-G-J-X (Ia)
where:
- A is selected from:
- (a) C1-C6-alkyl;
- (b) C3-C8-cycloalkyl;
- (c) —N(R1,R2), N(R1,R2)—C(═NR3)—, N(R1,R2)—C(═NR3)—N(R4)—, R1—C(═NR3)—, R1—C(═NR3)—N(R4)—;
- (d) phenyl, which is independently substituted with 0-2 R substituents;
- (e) naphthyl, which is independently substituted with 0-2 R substituents;
and - (f) monocyclic or fused bicyclic heterocyclic ring system having from 5 to 10 ring atoms, wherein 1-4 ring atoms of the ring system are selected from N, O and S, and wherein the ring system may be substituted with 0-2 R substituents;
- R is selected from:
- H, halo, —CN, —CO2R1, —C(═O)—N(R1, R2), —(CH2)m—CO2R1, —(CH2)m—C(═O)—N(R1, R2), —NO2, —SO2N(R1, R2), —SO2R1, —(CH2)mNR1R2, —(CH2)m—C(═NR3)—R1; —(CH2)m—C(═NR3)—N(R1,R2), —(CH2)m—N(R4)—C(═NR3)—N(R1,R2), (CH2)mNR1— group attached to a 3-6 membered heterocylic ring having from 1 to 3 heteroatoms selected from the group consisting of N, O and S, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CF3, —OR2, and a 5-6 membered heterocyclic aromatic or partially saturated system, including imidazoline, containing from 1-4 heteroatoms selected from N, O and S, wherein from 1-4 hydrogen atoms on the heterocyclic system may be independently replaced with a member selected from the group consisting of halo, -methyl, —C2-C4-alkyl, —CN, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl and —NO2;
- m is an integer of 0-2;
- R1, R2, R3 and R4 are independently selected from the group consisting of:
- H, —OR5, —N(—R5, —R6), —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —C0-4alkylphenyl and —C0-4alkylnaphthyl, wherein from 1-4 hydrogen atoms on the ring atoms of the phenyl and naphthyl moieties may be independently replaced with a member selected from the group consisting of halo, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CN, and —NO2; or
- R1 and R2, or R2 and R3 taken together can form a 3-8 membered cycloalkyl or a heterocyclic ring system, wherein the heterocyclic ring system may have from 3 to 10 ring atoms, with 1 to 2 rings being in the ring system and contain from 1-4 heteroatoms selected from N, O and S, wherein from 1-4 hydrogen atoms on the heterocyclic ring system may be independently replaced with a member selected from the group consisting of halo, C1-C4-alkyl, —CN—C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl and —NO2;
- R5 and R6 are independently selected from the group consisting of:
- H, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —C0-4alkylphenyl and —C0-4alkylnaphthyl, wherein from 1-4 hydrogen atoms on the ring atoms of the phenyl and naphthyl moieties may be independently replaced with a member selected from the group consisting of halo, —C1-14alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CN, and —NO2; or
- R5 and R6 taken together can form a 3-8 membered cycloalkyl or a heterocyclic ring system, wherein the heterocyclic ring system may have from 3 to 10 ring atoms, with 1 to 2 rings being in the ring system and contain from 1-4 heteroatoms selected from N, O and S, wherein from 1-4 hydrogen atoms on the heterocyclic ring system may be independently replaced with a member selected from the group consisting of halo, C1-C4-alkyl, —CN—C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl and —NO2;
- Q is a member selected from the group consisting of:
- a direct link, —CH2—, —C(═O)—, —O—, —NH—, —NMe-, —NHCH2—, —NMeCH2—, —CH2NH—, —C(═NH)—, —C(═O)—NH—, —NH—C(═O)—, —CH2NMe-, —C(—NMe)-;
- D is a direct link or is a member selected from the group consisting of:
-
- a monocyclic or fused bicyclic heterocyclic ring system having from 5 to 10 ring atoms, wherein 1-4 ring atoms of the ring system are selected from N, O and S, and wherein the ring system may be substituted from 0-2 R1a substituents;
- R1a is selected from:
- halo, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CN, —NO2, —(CH2)nNR2aR3a, —(CH2)nCO2R2a, (CH2)nCONR2aR3a, —SO2NR2aR3a, —SO2R2a, —CF3, —OR2a, and a 5-6 membered aromatic heterocyclic system containing from 1-4 heteroatoms selected from N, O and S, wherein from 1-4 hydrogen atoms on the aromatic heterocyclic system may be independently replaced with a member selected from the group consisting of halo, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CN and —NO2;
- R2a and R3a are independently selected from the group consisting of:
- H, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —C0-4alkylphenyl and —C0-4alkylnaphthyl, wherein from 1-4 hydrogen atoms on the ring atoms of the phenyl and naphthyl moieties may be independently replaced with a member selected from the group consisting of halo, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CN and —NO2;
- n is an integer of 0-2;
- E is a member selected from the group consisting of:
- a direct link, —O—, —NH—, —CH2NH—, —NHCH2—, —NMe-, —NH—C(═O)—NH—, —C(═O)—NH—, —NH—C(═O)—;
- G is a member selected from the group consisting of:
- (a) a C2-alkenyl group or a C3-8-cycloalkenyl group, wherein the alkenyl group and cycloalkenyl group attachment points are the alkenyl carbon atoms and wherein the C2-alkenyl group or C3-8-cycloalkenyl group is substituted with 0-4 R1d groups;
- (b) a phenylene group wherein the ring carbon atoms of the phenylene group are substituted with 0-4 R1d groups;
- (c) a 3-8 membered a saturated, partially unsaturated or aromatic monocyclic-heterocyclic ring system containing 1-4 heteroatoms selected from N, O and S, wherein 0-4 ring atoms of the heterocyclic ring may be substituted with 0-4 R1d groups; and,
- (d) an 8-10 membered fused heterocyclic bicyclic ring system, containing 1-4 heteroatoms selected from N, O and S, wherein 0-4 ring atoms of the fused bicyclic ring system may be substituted with 0-4 R1d groups;
- R1d is a member selected from the group consisting of:
- H, halo; C1-6alkyl, carbocylic aryl, —CN; —NO2; —(CH2)0-6NR2dR3d; —SO2NR2dR3d; —SO2R2d; —CF3; —(CH2)0-6—OR2d; —OH, —OC1-6alkyl, —O—(CH2)1-6OR2d; —O—(CH2)1-6—C(═O)—O—R2d; —O—(CH2)1-6—C(═O)—N(R2d,R3d); —N(R5a)—(CH2)1-6—OR2d; —N(R5a)—(CH2)1-6—N(R2d,R3d); —C(═O)—N(R2d,R3d); —N(R5a)—(CH2)1-6—C(═O)—N(R2d,R3d); —N(—(CH2)1-6—OR2d)2; —N(R5a)—(CH2)1-6—OR2d; —N(R5a)—C(═O)—R2d; —N(R5a)—SO2R2d; —(CH2)0-6—C(═O)—O—R2d; —(CH2)0-6C(═O)—N(R2d,R3d); —(CH2)0-6—C(═NR2d)—N(R3d,R4d); —(CH2)0-6—N(R5a)C(═NR2d)—N(R3d,R4d); and a —(CH2)0-6—N(R3d) group which is attached via the nitrogen atom to a carbon atom of a 5 to 6 membered saturated, partially unsaturated or aromatic heterocyclic ring containing 1-4 heteroatoms selected from N, O and S, and a —(CH2)0-6— group attached to a 5-6 membered saturated, partially unsaturated or aromatic heterocyclic ring containing 1-4 heteroatoms selected from N, O and S;
- R5a, R2d, R3d and R4d are each independently a member selected from the group consisting of:
- H, C1-6-alkyl and C1-4-alkylaryl, —CN; —NO2; carbocylic aryl, —CN; —NO2; or
- R2d and R3d taken together with the N atoms there are independently attached form a 5-7 membered saturated, partially unsaturated or aromatic heterocyclic ring; or
- R3d and R4d taken together with the N atom to which they are attached form a 5-8 membered saturated, partially unsaturated or aromatic heterocyclic ring containing 1-4 heteroatoms selected from N, O and S;
- J is a member selected from the group consisting of:
- a direct link, —O—, —NH—, —NMe-, —C(═O)—NH—, —NH—C(═O)—;
- X is a member selected from the group consisting of:
- (a) phenyl substituted with 0-3 R1e groups;
- (b) naphthyl substituted with 0-3 R1e groups and
- (c) a 6-membered aromatic heterocyclic ring system containing 1-3 N atoms and having 0-3 ring atoms substituted with 0-3 R1e groups; and
- (d) an 8-10 membered fused aromatic heterocyclic bicyclic ring system containing 1-4 heteroatoms selected from N, O and S and 0-3 ring atoms of the fused heterocyclic bicyclic ring system are substituted with 0-3 R1e groups;
- R1e is a member independently selected from the group consisting of:
- Halo; CF3; —C1-4-alkyl; carbocyclic aryl; —C0-2-alkyl-CN; —O—R2e; —C0-2-alkyl-C(═O)—O—R2e; —C0-2-alkyl-C(═O)—N(R2e, R3e); —C0-2-alkyl-NO2; —C0-2-alkyl-N(R2e, R3e); —C0-2-alkyl-SO2—N(R2e, R3e); —C0-2-alkyl-SO2—R2e; trihaloalkyl; —O—C0-2-alkyl-O—R2e; —C0-2-alkyl-O—R2e; —O—C1-4-alkyl-C(═O)—N(R2e, R3e); —O—C1-4-alkyl-C(═O)—O—R2e; —C0-2-alkyl-N(R2e)—C(═O)—R3e; —C0-2-alkyl-N(—R2e)SO2—R3e; —CH2—N(R2e)—C(═O)—R3e; —CH2—N(R2e)—SO2—R3e; —(CH2)0-6NR2eR3e; —C(═O)—N(R2e,R3e); —N(—(CH2)1-6—OR2e)2; —N(R10)—(CH2)1-6—OR2e; —N(R10)—C(═O)—R2e; —N(R10)—SO2—R2e; —C(═N(R10))—N(R2e,R3e); and a —(CH2)0-6-5-6 membered saturated, partially unsaturated or aromatic heterocyclic ring containing 1-4 heteroatoms selected from N, O and S;
- R10, R2e and R3e are each independently a member selected from the group consisting of:
- H; —C1-4-alkyl; —C0-2-alkyl-O—R1g; —C0-2-alkyl-N(—R1g, —R2g); —C1-4-alkyl-carbocyclic aryl; —C1-4-alkyl-heterocyclic; and R10 and R2e, or R2e and R3e together with the N atom to which they are attached can form 5-8 membered heterocyclic ring containing 1-4 heteroatoms selected from N, O and S which can be substituted with 0-2 R1g groups;
- R1g and R2g are independently a member selected from the group of:
- H; halo; —C1-4-alkyl, a carbocyclic aryl group; a saturated, partially unsaturated or aromatic heterocyclic group; —CN; —C(═O)—N(R3g,R4g); —C(═O)—OR3g; —NO2; —(CH2)p—NR3gR4g; —SO2NR3gR4g; —SO2R3g; —CF3; and —(CH2)pOR3g;
- p is an integer of 0-2; and
- R3g and R4g are each independently selected from the group consisting of:
- H; C1-4-alkyl and —C0-4-alkyl-carbocyclic aryl;
A-Q-D-E-G-J-X (Ib)
where:
- A is selected from:
- (a) C1-C6-alkyl;
- (b) C3-C8-cycloalkyl;
- (c) —N(R1,R2), N(R1,R2)—C(═NR3)—, N(R1,R2)—C(═NR3)—N(R4)—, R1—C(═NR3)—, R1—C(═NR3)—N(R4)—;
- (d) phenyl, which is independently substituted with 0-2 R substituents;
- (e) naphthyl, which is independently substituted with 0-2 R substituents;
- (f) a monocyclic or fused bicyclic ring system having from 5 to 10 ring atoms, wherein 1-4 ring atoms of the ring system are selected from N, O and S, and wherein the ring system may be substituted with 0-2 R substituents;
- R is selected from:
- H, halo, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CF3, —CN, —(CH2)m—CO2R1, —(CH2)m—C(═O)—N(R1, R2), —(CH2)m—C(═S)—N(R1, R2), —NO2, —(CH2)m—SO2N(R1, R2), —(CH2)m—SO2R1, —(CH2)mNR1R2, —(CH2)mOR1, —(CH2)m—C(═NR3)—R1, —(CH2)m—C(═NR1)—N(R1,R2), —(CH2)m—N(R4)—C(═NR3)—N(R1,R2), and a 3-8 membered cyclic system containing from 1-4 heteroatoms selected from N, O and S, wherein from 1-4 hydrogen atoms on the heterocyclic ring system may be independently replaced with a member selected from the group consisting of halo, C1-C4-alkyl, —CN—C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl and —NO2;
- m is an integer of 0-2;
- R1, R2, R3 and R4 are independently selected from the group consisting of:
- H, —(CH2)0-4OR5, —(CH2)0-4—CO2R5, —(CH2)0-4N(—R5, —R6), —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —C0-4alkylaryl and —C0-4alkylheteroaryl, and a 3-8 membered cyclic system containing from 1-4 heteroatoms selected from N, O and S, wherein from 1-4 hydrogen atoms on the heterocyclic ring system may be independently replaced with a member selected from the group consisting of halo, C1-C4-alkyl, —CN—C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl and —NO2; or
- R1 and R2, or R2 and R3 taken together can form a 3-8 membered cycloalkyl or a heterocyclic ring system, wherein the heterocyclic ring system may have from 3 to 10 ring atoms, with 1 to 2 rings being in the ring system and contain from 1-4 heteroatoms selected from N, O and S, where the hydrogen atoms on the heterocyclic ring system may be independently replaced with a member selected from the group consisting of halo, C1-C4-alkyl, —CN, —CO2R5, —OH, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl and —NO2;
- R5 and R6 are independently selected from the group consisting of:
- H, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —C0-4alkylaryl and —C0-4alkylheteroaryl, wherein from 1-4 hydrogen atoms on the ring atoms of the phenyl and naphthyl moieties may be independently replaced with a member selected from the group consisting of halo, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CN, and —NO2; or
- R5 and R6 taken together can form a 3-8 membered cycloalkyl or a heterocyclic ring system, wherein the heterocyclic ring system may have from 3 to 10 ring atoms, with 1 to 2 rings being in the ring system and contain from 1-4 heteroatoms selected from N, O and S, wherein from 1-4 hydrogen atoms on the heterocyclic ring system may be independently replaced with a member selected from the group consisting of halo, —C1-C4-alkyl, —CN—C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl and —NO2;
- Q is a member selected from the group consisting of:
- R7 is selected from:
- H; —C1-4-alkyl; —C0-4-alkylaryl; —C0-4-alkyl-heteroaryl; —C1-4-alkyl-O—C1-4-alkyl, —C1-4-alkyl-N(—C1-4-alkyl, —C1-4-alkyl); —C1-4-alkyl-C(═O)—O—C1-4-alkyl, and —C1-4-alkyl-C(═O)—N(—C1-4-alkyl, —C1-4-alkyl);
- D is a direct link or is a member selected from the group consisting of:
- (a) phenyl, which is independently substituted with 0-2 R1a substituents;
- (b) naphthyl, which is independently substituted with 0-2 R1a substituents; and
- (c) a monocyclic or fused bicyclic heterocyclic ring system having from 5 to 10 ring atoms, wherein 14 ring atoms of the ring system are selected from N, O and S, and wherein the ring system may be substituted from 0-2 R1a substituents;
- R1a is selected from:
- halo, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CN, —NO2, —(CH2)nOR2a, —(CH2)nNR2aR3a, —(CH2)nCO2R2a, —(CH2)nCONR2aR3a, —SO2NR2aR3a, —SO2R2a, —CF3, and a 5-6 membered aromatic heterocyclic system containing from 1-4 heteroatoms selected from N, O and S, wherein from 1-4 hydrogen atoms on the aromatic heterocyclic system may be independently replaced with a member selected from the group consisting of halo, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CN and —NO2:
- R2a and R3a are independently selected from the group consisting of:
- H, —C4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —C0-4alkylaryl and —C0-4alkylheteroaryl, wherein from 1-4 hydrogen atoms on the ring atoms of the phenyl and naphthyl moieties may be independently replaced with a member selected from the group consisting of halo, —C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl, —CN and —NO2;
- n is an integer of 0-2;
- E is a direct link or a member selected from the group consisting of:
- R8 is a member selected from the group consisting of:
- H; —C1-4-alkyl; —C0-4-alkylaryl; —C0-4-alkyl-heteroaryl; —C1-4-alkyl-OR2b, —C1-4-alkyl-N(—R2b, —R3b); —C1-4-alkyl-C(═O)—OR2b; —C1-4-alkyl-C(═O)—N(—R2b, —R3b); —C0-4-alkyl-C(═O)—R2b; and —C0-4-alkyl-SO2—R2b;
- R2b and R3b are each a member independently selected from the group consisting of:
- H, —C1-4-alkyl, —C1-4-alkyl-CO2—C0-4-alkyl, —C0-4-alkyl-aryl; —C0-4-alkyl-heterocyclic group, and R2b and R3b together with the N atom to which they are attached can form a 5-8 membered heterocyclic ring containing 1-4 heteroatoms selected from N, O and S, wherein the heterocyclic ring may be substituted with 0-2 R1c groups;
- R1c is a member selected from the group consisting of:
- Halo; —C1-4-alkyl; —CN, —NO2; —C(═O)—N(—R2, —R3); —C(═O)—OR2c; —(CH2)q—N(—R2c, —R3c); —SO2—N(—R2c, —R3c); —SO2R2c; —CF3 and —(CH2)q—OR2c;
- R2c and R3c are each independently a member selected from the group consisting of:
- H; —C1-4-alkyl and —C1-4alkyl-aryl;
- q is an integer of 0-2;
- G is a member selected from the group consisting of:
- (a) C2-alkenyl or C3-8-cycloalkenyl, wherein the alkenyl and cycloalkenyl attachment points are the alkenyl carbon atoms and wherein the —C2-alkenyl or —C3-8-cycloalkenyl are substituted with 0-4 R1J groups;
- (b) a phenylene group wherein the ring carbon atoms of the phenylene group are substituted with 0-4 R1d groups;
- (c) a 3-8 membered a saturated, partially unsaturated or aromatic monocyclic ring system containing 1-4 heteroatoms selected from N, O and S, wherein 0-2 ring atoms of the heterocyclic ring may be substituted with 0-4 R1d groups; and,
- (d) an 8-10 membered fused cyclic system, containing 0-4 heteroatoms selected from N, O and S, wherein 0-2 ring atoms of the fused bicyclic ring system may be substituted with 0-4 R1d groups;
- R1d is a member selected from the group consisting of:
- H, halo; —CF3; —OCF3, —OCF2H, —OCFH2, —OCH2CF3, —OCF2CF3, C1-6-alkyl, carbocylic aryl, —CN; —NO2; —(CH2)0-6—NR2dR3d; —(CH2)0-6—OR2d; —OH, —OC1-6alkyl, —O—(CH2)1-6OR2d; —O—(CH2)1-6—NR2dR2d; —N(R5a)—(CH2)1-6—OR2d; —N(R5a)—(CH2)1-6—N(R2d,R3d); —(CH2)0-6—C(═O)—O—R2d; —(CH2)0-6—C(═O)—N(R2d,R3d); —O—(CH2)1-6—C(═O)—O—R2d; —O—(CH2)1-6—C(═O)—N(R2d,R3d); —N(R5a)—(CH2)1-6—C(═O)—O—R2d; —N(R5a)—(CH2)1-6—C(═O)—N(R2d,R3d); —N(—(CH2)1-6—OR2d)2; —N(—(CH2)1-6—N(R2d,R3d))2; —(CH2)0-6—SO2NR2dR3d; —(CH2)0-6—SO2R2d; —(CH2)0-6—N(R5a)—C(═O)—R2d; —(CH2)0-6—N(R5a)—SO2—R2d, —(CH2)0-6—C(═NR2d)—N(R3d,R4d); —(CH2)0-6—N(R5a)C(═NR2d)—N(R3d,R4d); —(CH2)0-6—N(R5a)C(═NR2d)—R4d; —O—(CH2)1-6—SO2NR2dR3d; —O—(CH2)1-6—SO2R2d; —O—(CH2)1-6—N(R5a)—C(═O)—R2d; —O—(CH2)1-6—N(R5a)—SO2—R2d, —O—(CH2)1-6—C(═NR2d)—N(R3d,R4d); —O—(CH2)1-6—N(R5a)C(═NR2d)—N(R3dR4d); —O—(CH2)1-6—N(R5a)C(═NR2d)—R4d; —N(R5d)—(CH2)1-6—SO2NR2dR3d; —N(R5d)—(CH2)1-6—SO2R2d; —N(R5d)—(CH2)1-6—N(R5a)—C(═O)—R2d; —N(R5d)—(CH2)1-6—N(R5a)—SO2—R2d, —N(R5d)—(CH2)1-6—C(═NR2d)—N(R3d,R4d); —N(R5d)—(CH2)1-6—N(R5a)C(═NR2d)—N(R3d,R4d); —N(R5d)—(CH2)1-6—N(R5a)C(═NR2d)—R4d; and a 3-8 membered cyclic system containing from 1-4 heteroatoms selected from N, O and S, wherein from 1-4 hydrogen atoms on the heterocyclic ring system may be independently replaced with a member selected from the group consisting of halo, C1-C4-alkyl, —CN—C1-4alkyl, —C2-6alkenyl, —C2-6alkynyl, —C3-8cycloalkyl, —C0-4alkylC3-8cycloalkyl and —NO2;
- R5a, R2d, R3d, R4d and R5d are each independently a member selected from the group consisting of:
- H, C1-6-alkyl and C1-6-alkylaryl, —CN; —NO2; or
- R2d and R3d, or R3d and R4d taken together with the N atoms they are independently attached form a 3-8 membered saturated, partially unsaturated or aromatic heterocyclic ring;
- J is a direct link or is a member selected from the group consisting of:
- R9 is a member selected from the group consisting of:
- H; —C1-4-alkyl; —C0-4-alkylaryl; —C0-4-alkyl-heteroaryl; —C1-4-alkyl-OR6a, —C1-4-alkyl-N(—R6a, —R6b); —C1-4-alkyl-C(═O)—OR6a, and —C1-4-alkyl-C(═O)—N(R6a, R6b);
- R6a and R6b are each a member independently selected from the group consisting of:
- H and —C1-6-alkyl;
- X is a member selected from the group consisting of:
- (a) phenyl substituted with 0-3 R1e groups;
- (b) naphthyl substituted with 0-3 R1e groups and
- (c) a 6-membered aromatic heterocyclic ring system containing 1-3 N atoms and having 0-3 ring atoms substituted with 0-3 R1e groups; and
- (d) an 8-10 membered fused bicyclic ring system containing 1-4 heteroatoms selected from N, O and S and 0-3 ring atoms of the fused heterocyclic bicyclic ring system are substituted with 0-3 R1e groups;
- R1e is a member independently selected from the group consisting of:
- Halo; CF3; —C1-4-alkyl; carbocyclic aryl; —C0-2-alkyl-CN; —O—R2e; —C0-2-alkyl-C(═O)—O—R2e; —C0-2-alkyl-C(═O)—N(R2e, R3e); —C0-2-alkyl-NO2; —C0-2-alkyl-N(R2e, R3e); —C0-2-alkyl-SO2—N(R2e, R3e); —C0-2-alkyl-SO2—R2e; trihaloalkyl; —O—C0-2-alkyl-O—R2e; —C0-2-alkyl-O—R2e; —O—C1-4-alkyl-C(═O)—N(R2e, R3e); —O—C1-4-alkyl-C(═O)—O—R2e; —C0-2-alkyl-N(R2e)—C(═O)—R3e; —C0-2-alkyl-N(—R2e)—SO2—R2e; —CH2—N(R2e)—C(═O)—R3e; —CH2—N(R2e)—SO2—R3e; —(CH2)0-6—NR2eR3e; —C(═O)—N(R2e,R3e); —N(—(CH2)1-6—OR2e)2; —N(R10)—(CH2)1-6—OR2e; —N(R10)—C(═O)—R2e; —N(R10)—SO2—R2e; —C(═N(R10))—N(R2e,R3e); and a —(CH2)0-6-5-6 membered saturated, partially unsaturated or aromatic heterocyclic ring containing 1-4 heteroatoms selected from N, O and S;
- R10, R2e and R3e are each independently a member selected from the group consisting of:
- H; —C1-4-alkyl; —C0-2-alkyl-O—R1g; —C0-2-alkyl-N(—R1g, —R2g); —C1-4-alkyl-carbocyclic aryl; —C1-4-alkyl-heterocyclic; and R10 and R2e, or R2e and R3e together with the N atom to which they are attached can form 5-8 membered heterocyclic ring containing 1-4 heteroatoms selected from N, O and S which can be substituted with 0-2 R1g groups;
- R1g and R2g are independently a member selected from the group of:
- H; halo; —C1-4-alkyl, a carbocyclic aryl group; a saturated, partially unsaturated or aromatic heterocyclic group; —CN; —C(—O)—N(R3g)R4g; —C(═O)—OR3g; —NO2; —(CH2)p—NR3gR4g; —SO2NR3gR4g; —SO2R3g; —CF3; and —(CH2)pOR3g;
- p is an integer of 0-2;
- R3g and R4g are each independently selected from the group consisting of:
- H; C1-4-alkyl and —C0-4-alkyl-carbocyclic aryl;
A-Q-D-E-G-J-X (Ic)
where:
- A is a member selected from the group consisting of:



- Q is a member selected from the group consisting of:
- D is a direct link or is a member selected from the group consisting of:

- E is a member selected from the group consisting of:
- a direct link, —CH2NH—, —C(═O)—NH—, —NH—C(═O)—;
- G is a member selected from the group consisting of:


- G is substituted by 0-4 R1d groups and each R1d group is independently selected from the group consisting of:
- H, —CH3, —CF3, —Cl, —F, —Br, —NH2, —NMe2, —OH, —OMe, —NHSO2Me, —NO2, —CN, —C(═O)—OMe, —CO2H, —CONH2, —SO2NH2, —SO2CH3, —NHC(═O)Me, —C(═O)N(-Me)2, —CH2NH2, —CH2N(-Me)2, —CH2OH, —OCH2CO2H, —OCH2C(═O)—OMe, —OCH2C(═O)—NH2 and —OCH2C(═O)N(-Me)2.


- J is a member selected from the group consisting of:
- a direct link, —O—, —NH—, —C(═O)—NH— and —NH—C(═O)—;
- X is a member selected from the group consisting of:




where:
- R1a is a member selected from the group consisting of:
- H, —F, —Cl and —Br;
- R1e is a member selected from the group consisting of:
- H, —F, —Cl, —Br, —OMe, —OH, -Me, —CF3 and —CH2NH2; and
- A-Q is a member selected from the group consisting of:



where:
- R1a is a member selected from the group consisting of:
- H, —F, —Cl and —Br;
- R1e is a member selected from the group consisting of:
- H, —F, —Cl, —Br, —OMe, —OH, -Me, —CF3 and —CH2NH2; and
- A-Q is a member selected from the group consisting of:


where:
- R1a is a member selected from the group consisting of:
- H, —F, —Cl and —Br;
- R1e is a member selected from the group consisting of:
- H, —F, —Cl, —Br, —OMe, —OH, -Me, —CF3 and —CH2NH2;
- A-Q is a member selected from the group consisting of:


where:
- R1e is a member selected from the group consisting of:
- H, —F, —Cl, —Br, —OMe, —OH, -Me, —CF3 and —CH2NH2;
- A-Q is a member selected from the group consisting of


- D is a member selected from the group consisting of:


where:
- J is a member selected from the group consisting of:
- —NHC(═O)—, —C(═O)NH—;
- X is a member selected from the group consisting of:



wherein:
- R is a member selected from the group of:
- —SO2—NH2 and —SO2Me;
- R1a is a member selected from the group of:
- H, —F, —Cl and Br;
- E is a member selected from the group consisting of:
- —NHC(═O)— and —C(═O)NH—;
- R1d1, R1d2, and R1d4 are independently a member selected from the group of:
- H, —F, —Cl, —Br, -Me, —NO2, —OH, —OMe, —NH2, —NHAc, —NHSO2Me, —CH2OH and —CH2NH2;
- R1d3 is a member selected from the group of:
- H, —CH3, —CF3, —Cl, —F, —Br, —NH2, —N(-Me)2, —OH, —OMe, —NHSO2Me, —NO2, —CN, —C(═O)—OMe, —CO2H, —C(═O)—NH2, —SO2NH2, —SO2CH3, —NHC(═O)-Me, —C(═O)—N(-Me)2, —CH2NH2, —CH2—N(-Me)2, —CH2OH, —OCH2CO2H, —OCH2C(═O)—OMe, —OCH2C(═O)—NH2, and —OCH2C(═O)—N(-Me)2,


- R1e is a member selected from the group of:
- F, —Cl, —Br, —OH, -Me and —OMe,
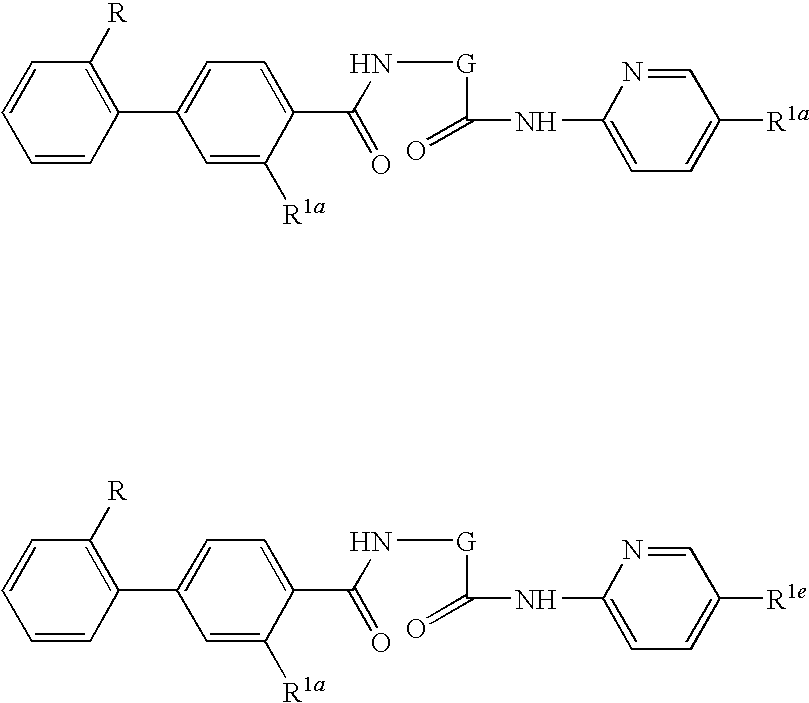
wherein:
- R is a member selected from the group consisting of:
- —SO2NH2, —SO2Me;
- R1a is a member selected from the group consisting of:
- H, —F, —Cl and Br;
- R1e is a member selected from the group consisting of:
- H, —F, —Cl, —Br, —OMe, —OH, -Me, —CF3 and —CH2NH2; and
- G is a member selected from the group consisting of:


wherein each G group may be substituted by 0-4 R1d groups and each such R1d group is independently selected from the group consisting of:
-
- H, —CH3, —CF3, —Cl, —F, —Br, —NH2, —N(-Me)2, —OH, —OMe, —NHSO2Me, —NO2, —CN, —C(═O)—OMe, —CO2H, —C(═O)—NH2, —SO2NH2, —SO2CH3, —NH—C(═O)-Me, —C(═O)—N(-Me)2, —CH2NH2, —CH2—N(-Me)2, —CH2OH, —OCH2CO2H, —OCH2CO2Me, —OCH2C(═O)—NH2, —OCH2C(═O)—N(-Me)2.



wherein:
- J-X are collectively a member selected from the group consisting of:



wherein:
- R is a member selected from the group of:
- —SO2NH2, and —SO2Me;
- R1a is a member selected from the group of:
- H, —F, —Cl and Br;
- E is a member selected from the group consisting of:
- —NHC(═O)— and —C(═O)NH—;
- J is a member selected from the group consisting of:
- —NHC(═O)— and —C(═O)NH—, O;
- R1d1, R1d2, and R1d4 are independently a member selected from the group of:
- H, —F, —Cl, —Br, -Me, —NO2, —OH, —OMe, —NH2, —NHAc, —NHSO2Me, —CH2OH, —CH2NH2;
- R1d3 is a member selected from the group of:
- H, —CH3, —CF3, —Cl, —F, —Br, —NH2, —N(-Me)2, —OH, —OMe, —NHSO2Me, —NO2, —CN, —CO2Me, —CO2H, —C(═O)—NH2, —SO2NH2, —SO2CH3, —NHC(═O)-Me, —C(═O)—N(-Me)2, —CH2NH2, —CH2—N(-Me)2, —CH2OH, —OCH2CO2H, —OCH2C(═O)—OMe, —OCH2C(═O)—NH2, —OCH2C(═O)—N(-Me)2


- R13 is a member selected from the group of:
- F, —Cl, —Br, —OH, -Me and —OMe;


wherein:
- R is a member selected from the group of:
- —SO2—NH2, and —SO2Me;
- R1a is a member selected from the group of:
- H, —F, —Cl and Br;
- R1d is a member selected from the group consisting of:
- —H, —CH3, —CF3, —CN, —SO2NH2 and —SO2CH3; and
- R1e is a member selected from the group of:
- —Cl and —Br;

wherein:
- A-Q taken together are a member selected from the group consisting of:


wherein:
- A-Q is a member selected from the group of:

- R1a is a member selected from the group of:
- H, —F, —Cl and Br;
- R1d1, R1d2, and R1d4 are independently a member selected from the group of:
- H, —F, —Cl, —Br, -Me, —NO2, —OH, —OMe, —NH2, —NHAc, —NHSO2Me, —CH2OH, —CH2NH2
- R1d3 is a member selected from the group of:


- R1e is a member selected from the group of:
- F, —Cl, —Br, —OH, -Me and —OMe;

and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.
In formula VI:






and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.
In formula VII:

- where Z′ is as described above;
- R1a is a member selected from the group of H, —F, —Cl and Br;
- R1d2 and R1d4 are each H;
- R1d1 is R1d3 are each independently a member selected from the group of H, —Cl, —F, —Br, —OH and —OMe;
- R1e is a member selected from the group of —F, —Cl, —Br, —OH, -Me and —OMe.
Examples of suitable compounds of formula VII, as described above, include, but are not limited to:





















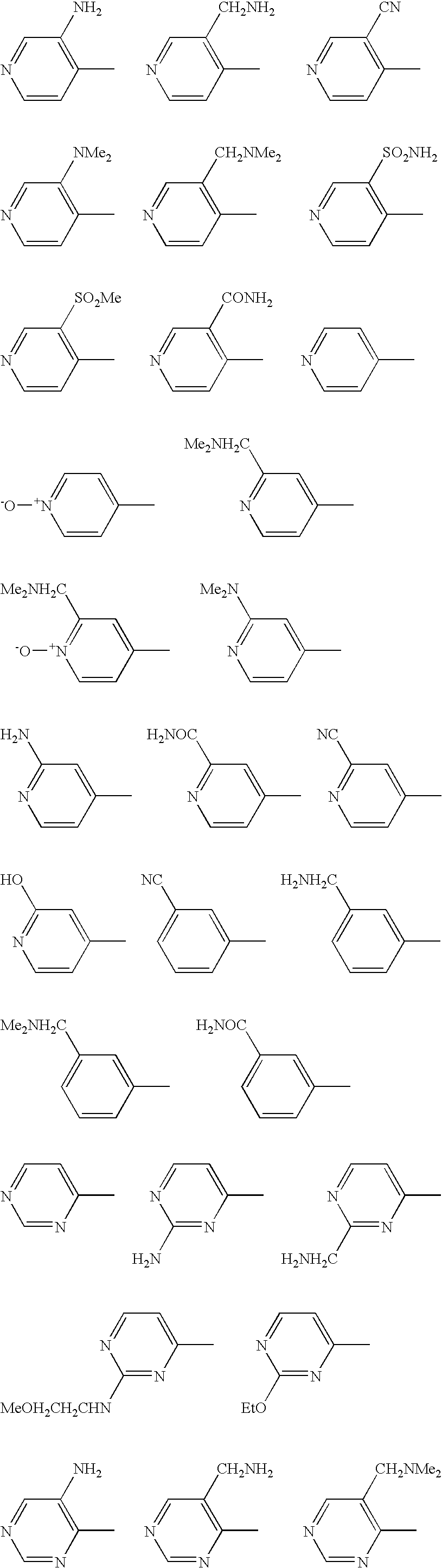




- Q is a member selected from the group consisting of:
- a direct link, —CH2—, —C(═O)—, —NH—, —N(Me)—, —NHCH2—, —N(Me)CH2—, —C(═NH)—, —C(═NMe)-;
- D is a direct link or is a member selected from the group consisting of:

- E is a member selected from the group consisting of:
- a direct link, —CH2NH—, —NHCH2—, —CH2O—, —OCH2—, —CH2NH—, —CONH—, —NHCO—, —CONMe-, —NMeCO—;
- G is a member selected from the group consisting of:


- G is substituted by 0-4 R1d groups and each R1d group is independently selected from the group consisting of:
- H, -Me, —F, —Cl, —Br, aryl, heteroaryl, —NH2, —NMe2, —NHMe, —NHSO2Me, —NHCOMe, —CH3, —CF3, —OH, —OCH3, —SCH3, —OCF3, —OCH2F, —OCHF2, —OCH2CF3, —OCF2CF3, —NO2, —CN, —CO2H, —CO2Me, —CO2Et, —CONH2, —CONHMe, —CONMe2, —SO2NH2, —SO2CH3, —SO2NMe2, —CH2OH, —CH2NH2, —CH2NHMe, —CH2NMe2, —OCH2CO2H, —OCH2CO2Me, —OCH2CO2Et, —OCH2CONH2, —OCH2CONMe2, —OCH2CONHMe, —OCH2CH2OMe, —OCH2CH2OEt, —OCH2CH2NH2, —OCH2CH2NHMe, —OCH2CH2NMe2, —NHCH2CH2OMe, —SCH2CH2OMe, —SO2CH2CH2OMe, —OCH2CH2SO2Me, —NHCH2CH2NHMe, —NHCH2CH2NMe2, —N(CH2CH2OH)2, —N(CH2CH2OMe)2, —NHCH2CO2H, —NHCH2CO2Et, —NHCH2CO2Et, —NHCH2CONH2, —NHCH2CONMe2, —NHCH2CONHMe, —N(CH3)CH2CO2H, —N(CH3)CH2CO2Et, —(NMe)CH2COOH, —N(Me)CH2CONH2, —N(Me)CH2CH2NMe2, —N(Me)CH2CH2OMe, —NHCH2CH2OMe,



- J is a member selected from the group consisting of:
- a direct link, —SO2—, —CO—, —O—, —NH—, —C(═O)—NH— and —NH—C(═O)—;
- X is a member selected from the group consisting of:













and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

where:
- R1a is a member selected from the group consisting of:
- H, —F, —Cl and —Br;
- R1e is a member selected from the group consisting of:
- H, —F, —Cl, —Br, —OMe, —OH, -Me, —CF3 and —CH2NH2; and
- A-Q is a member selected from the group consisting of:


and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- R is a member selected from the group consisting of:
- —SO2Me, —SO2NH2, —CH2NH2, —CH2N(CH3)2;
- R1a is a member selected from the group consisting of:
- H, —F;
- R1d1 is a member selected from the group consisting of:
- H, -Me, —F, —Cl, —Br, aryl, heteroaryl, —NH2, —NMe2, —NHMe, —NHSO2Me, —NHCOMe, —CH3, —CF3, —OH, —OCH3, —SCH3, —OCF3, —OCH2F, —OCHF2, —OCH2CF3, —OCF2CF3, —NO2, —CN, —CO2H, —CO2Me, —CO2Et, —CONH2, —CONHMe, —CONMe2, —SO2NH2, —SO2CH3, —SO2NMe2, —CH2OH, —CH2NH2, —CH2NHMe, —CH2NMe2, —OCH2CO2H, —OCH2CO2Me, —OCH2CO2Et, —OCH2CONH2, —OCH2CONMe2, —OCH2CONHMe, —OCH2CH2OMe, —OCH2CH2OEt, —OCH2CH2NH2, —OCH2CH2NHMe, —OCH2CH2NMe2, —NHCH2CH2OMe, —SCH2CH2OMe, —SO2CH2CH2OMe, —OCH2CH2SO2Me, —NHCH2CH2NHMe, —NHCH2CH2NMe2, —N(CH2CH2OH)2, —N(CH2CH2OMe)2, —NHCH2CO2H, —NH2CO2Et, —NHCH2CO2Et, —NHCH2CONH2, —NHCH2CONMe2, —NHCH2CONHMe, —N(CH3)CH2CO2H, —N(CH3)CH2CO2Et, —(NMe)CH2COOH, —N(Me)CH2CONH2, —N(Me)CH2CH2NMe2, —N(Me)CH2CH2OMe, —NHCH2CH2OMe,



and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- A-Q is a member selected from the group consisting of:

- R1a is a member selected from the group consisting of:
- H, —F;
- R1d1 is a member selected from the group consisting of:
- H, -Me, —F, —Cl, —Br, aryl, heteroaryl, —NH2, —NMe2, —NHMe, —NHSO2Me, —NHCOMe, —CH3, —CF3, —OH, —OCH3, —SCH3, —OCF3, —OCH2F, —OCHF2, —OCH2CF3, —OCF2CF3, —NO2, —CN, —CO2H, —CO2Me, —CO2Et, —CONH2, —CONHMe, —CONMe2, —SO2NH2, —SO2CH3, —SO2NMe2, —CH2OH, —CH2NH2, —CH2NHMe, —CH2NMe2, —OCH2CO2H, —OCH2CO2Me, —OCH2CO2Et, —OCH2CONH2, —OCH2CONMe2, —OCH2CONHMe, —OCH2CH2OMe, —OCH2CH2OEt, —OCH2CH2NH2, —OCH2CH2NHMe, —OCH2CH2NMe2, —NHCH2CH2OMe, —SCH2CH2OMe, —SO2CH2CH2OMe, —OCH2CH2SO2Me, —NHCH2CH2NHMe, —NHCH2CH2NMe2, —N(CH2CH2OH)2, —N(CH2CH2OMe)2, —NHCH2CO2H, —NHCH2CO2Et, —NHCH2CO2Et, —NHCH2CONH2, —NHCH2CONMe2, —NHCH2CONHMe, —N(CH3)CH2CO2H, —N(CH3)CH2CO2Et, —(NMe)CH2COOH, —N(Me)CH2CONH2, —N(Me)CH2CH2NMe2, —N(Me)CH2CH2OMe, —NHCH2CH2OMe,



and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof

wherein:
- A-Q is a member selected from the group consisting of:

- R1a is a member selected from the group consisting of:
- H, —F;
- R1e is a member selected from the group consisting of:
- H, —F, —SO2Me, —SO2NH2, —CN, —CONH2, —CH2NH2, —CH2NMe2;
- R1d3 is a member selected from the group consisting of:
- H, -Me, —F, —Cl, —Br, aryl, heteroaryl, —NH2, —NMe2, —NHMe, —NHSO2Me, —NHCOMe, —CH3, —CF3, —OH, —OCH3, —SCH3, —OCF3, —OCH2F, —OCHF2, —OCH2CF3, —OCF2CF3, —NO2, —CN, —CO2H, —CO2Me, —CO2Et, —CONH2, —CONHMe, —CONMe2, —SO2NH2, —SO2CH3, —SO2NMe2, —CH2OH, —CH2NH2, —CH2NHMe, —CH2NMe2, —OCH2CO2H, —OCH2CO2Me, —OCH2CO2Et, —OCH2CONH2, —OCH2CONMe2, —OCH2CONHMe, —OCH2CH2OMe, —OCH2CH2OEt, —OCH2CH2NH2, —OCH2CH2NHMe, —OCH2CH2NMe2, —NHCH2CH2OMe, —SCH2CH2OMe, —SO2CH2CH2OMe, —OCH2CH2SO2Me, —NHCH2CH2NHMe, —NHCH2CH2NMe2, —N(CH2CH2OH)2, —N(CH2CH2OMe)2, —NHCH2CO2H, —NHCH2CO2Et, —NHCH2CO2Et, —NHCH2CONH2, —NHCH2CONMe2, —NHCH2CONHMe, —N(CH3)CH2CO2H, —N(CH3)CH2CO2Et, —(NMe)CH2COOH, —N(Me)CH2CONH2, —N(Me)CH2CH2NMe2, —N(Me)CH2CH2OMe, —NHCH2CH2OMe,



and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- A-Q is a member selected from the group consisting of:

- R1a is a member selected from the group consisting of:
- H, —F;
- R1e is a member selected from the group consisting of:
- —Cl, —Br;
- R1d3 is a member selected from the group consisting of:
- H, F, Cl, Br, —OCH3, —OCF3, —OCH2F, —OCHF2, —OCH2CF3, —OCF2CF3;
and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.
- H, F, Cl, Br, —OCH3, —OCF3, —OCH2F, —OCHF2, —OCH2CF3, —OCF2CF3;

wherein:
- A-Q is a member selected from the group consisting of:

- R1a is a member selected from the group consisting of:
- H, —F;
- R1d3 is a member selected from the group consisting of:
- H, —F, —Cl, —Br, —OCH3, —OCF3, —OCH2F, —OCHF2, —OCH2CF3, —OCF2CF3, —NHSO2Me, —NHAc, —SO2Me, —SO2NH2;
- X is a member selected from the group consisting of:

and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- A-Q is a member selected from the group consisting of:


- R1a is a member selected from the group consisting of:
- H, —F;
and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.
- H, —F;

wherein:
- R1a is a member selected from the group consisting of:
- H, —F;
- R1d1 is a member selected from the group consisting of:
- H, —OMe;
- R1d3 is a member selected from the group consisting of:
- H, —F, —Cl, —Br, —OMe, —OCF3;
- R1e is a member selected from the group consisting of:
- H, —F, —Cl, —Br;
- A-Q is a member selected from the group consisting of:

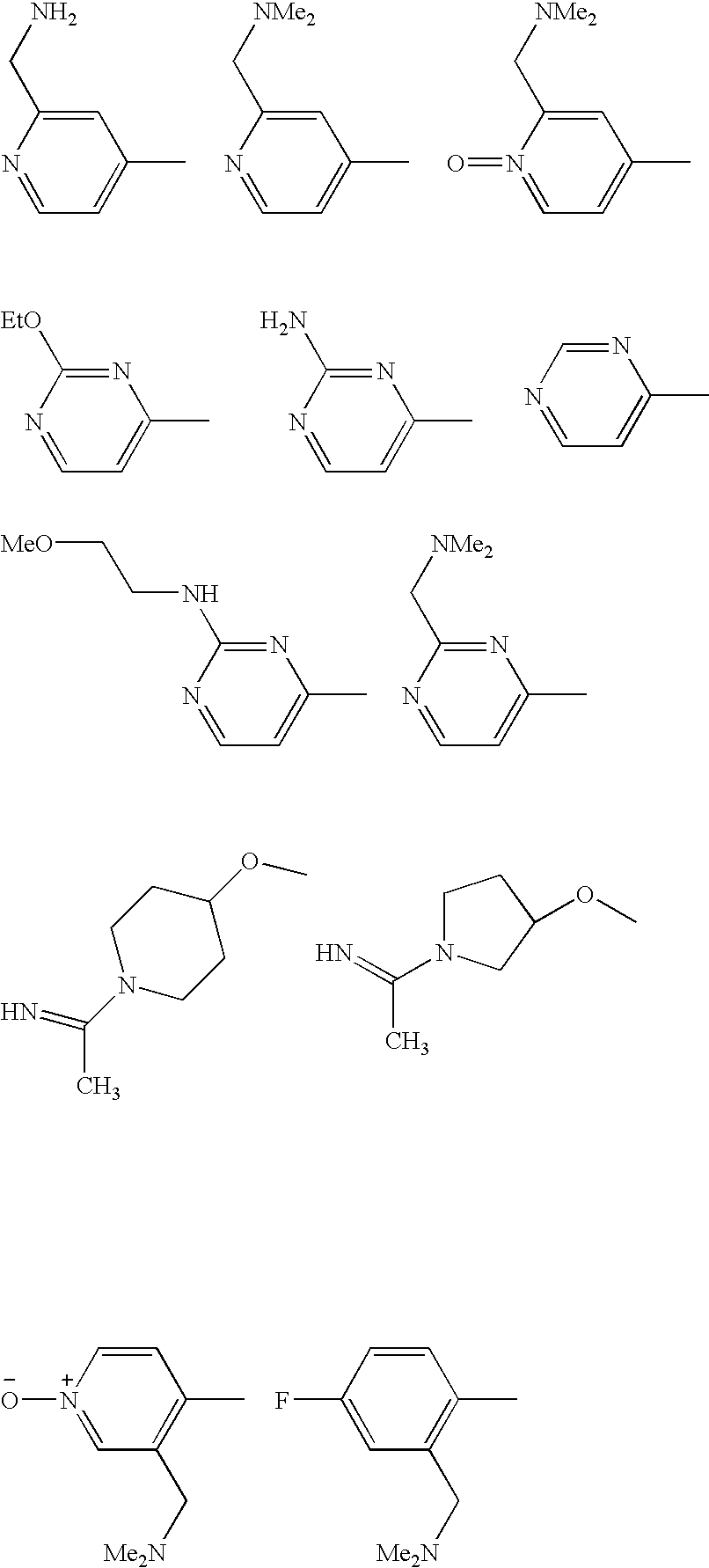
and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- D is a member selected from the group consisting of:

and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- R1d1 is H or —OMe;
- A-Q-D is a member selected from the group consisting of:


and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- R1a is H or F;
- R1d1 is H or —OMe;
- A-Q is a member selected from the group consisting of:


and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- R1a is H or F;
- R1d1 is H or —OMe;
- A-Q is a member selected from the group consisting of:

- R1a is a member selected from the group consisting of: H, —F,
and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- R1a is H or F;
- R1d1 is H or —OMe; and
- A-Q is a member selected from the group consisting of:

and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- A-Q is a member selected from the group consisting of:


and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- R1a is H or F;
- R1d1 is H or —OMe;
- A-Q is a member selected from the group consisting of:


- and X is a member selected from the group consisting of:

and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- R1a is H or F;
- A-Q is a member selected from the group consisting of:


- R1d1 is a member selected from the group consisting of:
- H, —F, —Cl, —Br, aryl, heteroaryl, —NH2, —NMe2, —NHMe, —NHSO2Me, —NHCOMe, —CH3, —CF3, —OH, —OCH3, —SCH3, —OCF3, —OCH2F, —OCHF2, —OCH2CF3, —OCF2CF3, —NO2, —CN, —CO2H, —CO2Me, —CO2Et, —CONH2, —CONHMe, —CONMe2, —SO2NH2, —SO2CH3, —SO2NMe2, —CH2OH, —CH2NH2, —CH2NHMe, —CH2NMe2, —OCH2CO2H, —OCH2CO2Me, —OCH2CO2Et, —OCH2CONH2, —OCH2CONMe2, —OCH2CONHMe, —OCH2CH2OMe, —OCH2CH2OEt, —OCH2CH2NH2, —OCH2CH2NHMe, —OCH2CH2NMe2, —NHCH2CH2OMe, —SCH2CH2OMe, —SO2CH2CH2OMe, —OCH2CH2SO2Me, —NHCH2CH2NHMe, —NHCH2CH2NMe2, —N(CH2CH2OH)2, —N(CH2CH2OMe)2, —NHCH2CO2H, —NHCH2CO2Et, —NHCH2CO2Et, —NHCH2CONH2, —NHCH2CONMe2, —NHCH2CONHMe, —N(CH3)CH2CO2H, —N(CH3)CH2,CO2Et, —(NMe)CH2COOH, —N(Me)CH2CONH2, —N(Me)CH2CH2NMe2, —N(Me)CH2CH2OMe, —NHCH2CH2OMe,



- R1d3 is a member selected from the group consisting of:
- H, —F, —Cl, —Br, —OCH3, —OCF3, —OCH2F, —OCHF2, —OCH2CF3, —OCF2CF3; and
- X is a member selected from the group consisting of:


and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- A-Q is a member selected from the group consisting of:


and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.


wherein:
- A-Q is a member selected from the group consisting of:


and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- R1a is H or F;
- A-Q is a member selected from the group consisting of:


- R1d1 is a member selected from the group consisting of: H, OMe, Cl, F, OCF3,

and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- R1a is H or F;
- A-Q is a member selected from the group consisting of:

- R1d1 is a member selected from the group consisting of:
- H, —F, —Cl, —Br, —OMe, —OCF3, —OH, —NMe2, —OCH2CO2Et, —OCH2CO2H;
- R1d3 is a member selected from the group consisting of:
- H, —F, —Cl, —Br, —OMe, —OCF3, —OH, —NMe2, —OCH2CO2Et, —OCH2CO2H, —OCF2H, —OCFH2, —OCF2CF3, —OCH2CH3,

and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- R1a is H or F;
- A-Q is a member selected from the group consisting of:

- R1d1 is a member selected from the group consisting of:
- H, —F, —Cl, —Br, —OMe, —OCF3, —OH, —NMe2, —OCH2CO2Et, —OCH2CO2H

- R1d3 is a member selected from the group consisting of:
- H, —F, —Cl, —Br, —OMe, —OCF3, —OH, —NMe2, —OCH2CO2Et, —OCH2CO2H, —OCF2H, —OCFH2, —OCF2CF3, —OCH2CH3,
and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.
- H, —F, —Cl, —Br, —OMe, —OCF3, —OH, —NMe2, —OCH2CO2Et, —OCH2CO2H, —OCF2H, —OCFH2, —OCF2CF3, —OCH2CH3,

wherein:
- R1a is H or F;
- R1d1 is selected from H, —OMe, —NMe2,

- R1d3 is Cl or Br;
- A-Q is a member selected from the group consisting of:

and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- R1a is H or F;
- R1d1 is selected from H, —OMe, —NMe2,

- R1d3 is Cl or Br;
- A-Q is a member selected from the group consisting of:

and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- R1a is H or F;
- R1d1 is selected from H, —OMe, —NMe2,

- A-Q is a member selected from the group consisting of:

and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- R1a is H or F;
- R1d1 is selected from H, —OMe, —NMe2,

- A-Q is a member selected from the group consisting of:

and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- R1a is H or F;
- R1d1 is selected from H, —OMe, —NMe2,

- A-Q is a member selected from the group consisting of:

and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- R1a is H or F;
- R1d1 is selected from H, —OMe, —NMe2,

- A-Q is a member selected from the group consisting of:

and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- A-Q is a member selected from the group consisting of:


- R1a is a member selected from the group consisting of:
- H, —F, —Cl and Br;
- R1e is a member selected from the group consisting of:
- H, —F, —Cl, —Br, —OMe, —OH, -Me, —CF3 and —CH2NH2; and
- G is a member selected from the group consisting of:


wherein each G group is substituted by 0-4 R1d groups and each such R1d group is independently selected from the group consisting of:
-
- H, -Me, —F, —Cl, —Br, aryl, heteroaryl, —NH2, —NMe2, —NHMe, —NHSO2Me, —NHCOMe, —CH3, —CF3, —OH, —OCH3, —SCH3, —OCF3, —OCH2F, —OCHF2, —OCH2CF3, —OCF2CF3, —NO2, —CN, —CO2H, —CO2Me, —CO2Et, —CONH2, —CONHMe, —CONMe2, —SO2NH2, —SO2CH3, —SO2 NMe2, —CH2OH, —CH2NH2, —CH2NHMe, —CH2NMe2, —OCH2CO2H, —OCH2CO2Me, —OCH2CO2Et, —OCH2CONH2, —OCH2CONMe2, —OCH2CONHMe, —OCH2CH2OMe, —OCH2CH2OEt, —OCH2CH2NH2, —OCH2CH2NHMe, —OCH2CH2NMe2, —NHCH2CH2OMe, —SCH2CH2OMe, —SO2CH2CH2OMe, —OCH2CH2SO2Me, —NHCH2CH2NHMe, —NHCH2CH2NMe2, —N(CH2CH2OH)2, —N(CH2CH2OMe)2, —NHCH2CO2H, —NHCH2CO2Et, —NHCH2CO2Et, —NHCH2CONH2, —NHCH2CONMe2, —NHCH2CONHMe, —N(CH3)CH2CO2H, —N(CH3)CH2CO2Et, —(NMe)CH2COOH, —N(Me)CH2CONH2, —N(Me)CH2CH2NMe2, —N(Me)CH2CH2OMe, —NHCH2CH2OMe,



and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:


- R1a is a member selected from the group of:
- H, —F, —Cl, —Br;
- R1d is a member selected from the group of:
- H, —F, —Cl, —Br, —OMe;
- R1e1 is a member selected from the group of:
- H, —F, —Cl, —Br, —NH2, —CH2NH2, —OMe, —OH, —CN, —SO2Me, —SO2NH2; and
- R1e2 is a member selected from the group of:
- H, —F, —Cl, —Br, —NH2,
and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.
- H, —F, —Cl, —Br, —NH2,

wherein:
- A-Q is a member selected from the group of:


- R1a is a member selected from the group of:
- H, —F, —Cl and Br; and
- R1d is a member selected from the group of:
- H, —F, —Cl, —Br, —OMe,
and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.
- H, —F, —Cl, —Br, —OMe,

wherein:
- A-Q is a member selected from the group of:

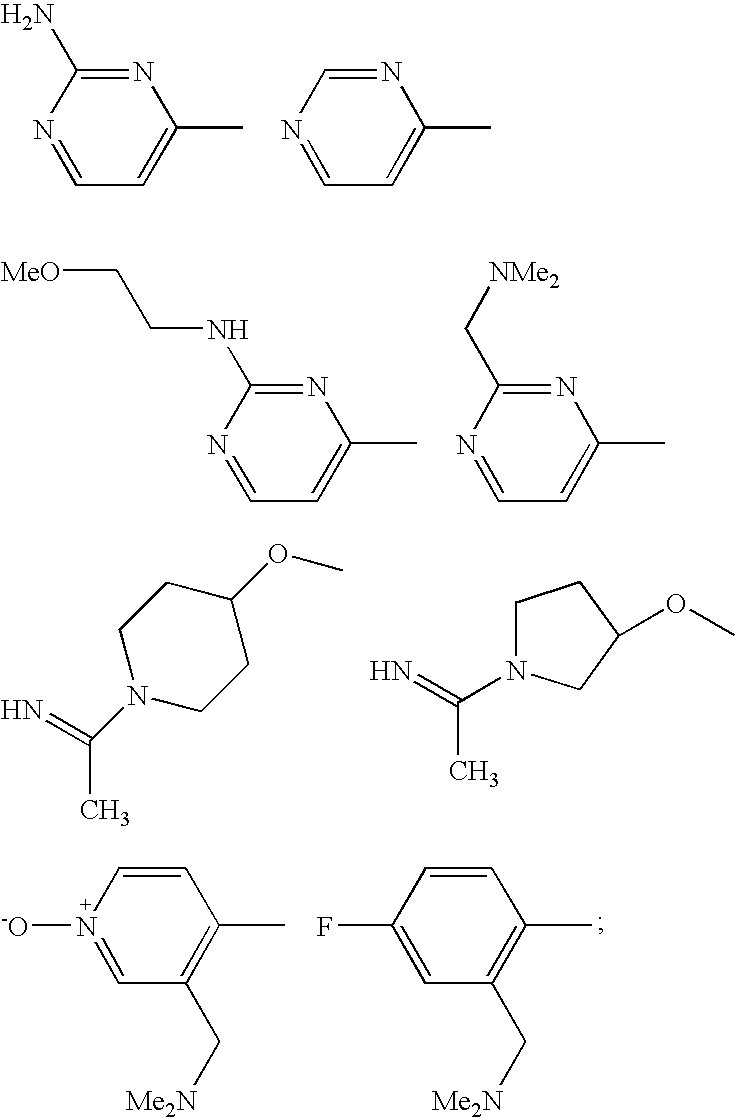
- R1a is a member selected from the group of:
- H, —F, —Cl and Br; and
- R1e is a member selected from the group of:
- H, —F, —Cl, —Br, —NH2, —CH2NH2, —OMe, —OH, —CN, —SO2Me, —SO2NH2.
and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.
- H, —F, —Cl, —Br, —NH2, —CH2NH2, —OMe, —OH, —CN, —SO2Me, —SO2NH2.


wherein:
- R is a member selected from the group of:
- —SO2NH2, —SO2Me, —CH2NMe2;
- R1a is a member selected from the group of:
- H, —F, —Cl, —Br;
- R1d is a member selected from the group of:
- H, —F, —Cl, —Br, —CN, CF3, —CH3, —SO2NH2, —SO2Me; and
- R1e is a member selected from the group of:
- —Cl, —Br,
and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.
- —Cl, —Br,

wherein:
- A-Q taken together are a member selected from the group consisting of:

and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- A-Q is a member selected from the group consisting of:


- R1a is a member selected from the group consisting of:
- H, —F, —Cl and Br;
- G is a member selected from the group consisting of:


wherein each G group is substituted by 0-4 R1d groups and each such R1d group is independently selected from the group consisting of:
-
- H, -Me, —F, —Cl, —Br, aryl, heteroaryl, —NH2, —NMe2, —NH Me, —NHSO2Me, —NHCOMe, —CH3, —CF3, —OH, —OCH3, —SCH3, —OCF3, —OCH2F, —OCHF2, —OCH2CF3, —OCF2CF3, —NO2, —CN, —CO2H, —CO2Me, —CO2Et, —CONH2, —CONHMe, —CONMe2, —SO2NH2, —SO2CH3, —SO2NMe2, —CH2OH, —CH2NH2, —CH2NHMe, —CH2NMe2, —OCH2CO2H, —OCH2CO2Me, —OCH2CO2Et, —OCH2CONH2, —OCH2CONMe2, —OCH2CONHMe, —OCH2CH2OMe, —OCH2CH2OEt, —OCH2CH2NH2, —OCH2CH2NHMe, —OCH2CH12NMe2, —NHCH2CH2OMe, —SCH2CH2OMe, —SO2CH2CH2OMe, —OCH2CH2SO2Me, —NHCH2CH2NHMe, —NHCH2CH2NMe2, —N(CH2CH2OH)2, —N(CH2CH2OMe)2, —NHCH2CO2H, —NHCH2CO2Et, —NHCH2CO2Et, —NHCH2CONH2, —NHCH2CONMe2, —NHCH2CONHMe, —N(CH3)CH2CO2H, —N(CH3)CH2CO2Et, —(NMe)CH2COOH, —N(Me)CH2CONH2, —N(Me)CH2CH2NMe2, —N(Me)CH2CH2OMe, —NHCH2CH2OMe,



- J is a member selected from the group consisting of:
- —CONH—, —NHCO—, —O—, —NH—, —NMe-, —CONMe-, —NMeCO—; and
- X is a member selected from the group consisting of:


and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:
- A-Q is a member selected from the group of:


- R1a is a member selected from the group of:
- H, —F, —Cl, —Br;
- R1d1, R1d2, R1d3 and R1d4 is independently a member selected from the group of:
- H, —F, —Cl, —Br, —NO2, —NH2, —NHMe, —NMe2, —NHAc, —NHSO2Me, —SO2Me, —CO2H, —CO2Me, —OH, —OMe, —N(Me)CO2H, —N(Me)CO2Et and

- R1e is a member selected from the group of:
- H, —OH,
and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.
- H, —OH,

- R is a member selected from the group of:
- —SO2Me, —SO2NH2, —CH2NH2, —CH2N(CH3)2;
- R1a is a member selected from the group of:
- H, —F;
- R1d2 and R1d3 is independently a member selected from the group of:
- H, —F, —Cl, —Br, —NO2; —NH2, —NHMe, —NMe2, —NHAc, —NHSO2Me, —SO2Me, —CO2H, —CO2Me, —OH, —OMe; and
- R1e is a member selected from the group of:
- H, —OH,
and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.
- H, —OH,

wherein:
- R is a member selected from the group of:
- —SO2Me, —SO2NH2, —CH2NH2, —CH2N(CH3)2;
- R1a is a member selected from the group of:
- H, —F;
- R1d2 and R1d3 is independently a member selected from the group of:
- H, —F, —Cl, —Br, —NO2, —NH2, —NHMe, —NMe2, —NHAc, —NHSO2Me, —SO2Me, —CO2H, —CO2Me, —OH, —OMe; and
- R1e is a member selected from the group of:
- H, —OH,
and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.
- H, —OH,

- R is a member selected from the group of:
- —SO2Me, —SO2NH2, —CH2NH2, —CH2N(CF3)2;
- R1a is a member selected from the group of:
- H, —F;
- R1d1 and R1d2 is independently a member selected from the group of:
- H, —F, —Cl, —Br, —OMe; and
- R1e is a member selected from the group of:
- H, —OH,
and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.
- H, —OH,

wherein:


and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:


and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.








wherein
- R1d3 is a member selected from the group consisting of:
- H, —F, —Cl, —Br, —OMe, —OCF3, —OCF2H, and —OCF2H;
and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:


and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein:


and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.








wherein
- R1d3 is a member selected from the group consisting of:
- H, —F, —Cl, —Br, —OMe, —OCF3, —OCF2H, and —OCF2H,
and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.

wherein each of Ra, Rb, Rc, Rd and Re is independently a member selected from the group consisting of C1-C8 alkyl, C2-C8 alkenyl, C1-C8 acyl and C1-C8 acyl C1-C8 alkyl ester, and the Ra and Rb groups together with the nitrogen atom to which they are both attached may be cyclized to form a C3-C8 heterocylic ring having from 1 to 4 additional hetero ring atoms selected from O, N and S,
and all pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives thereof.













































































































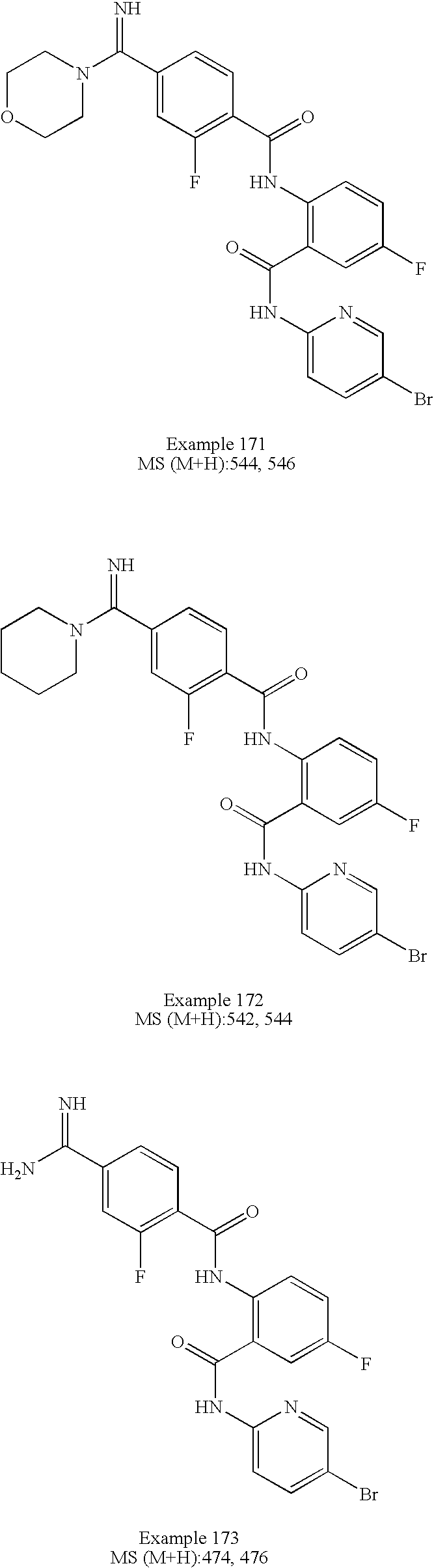























































































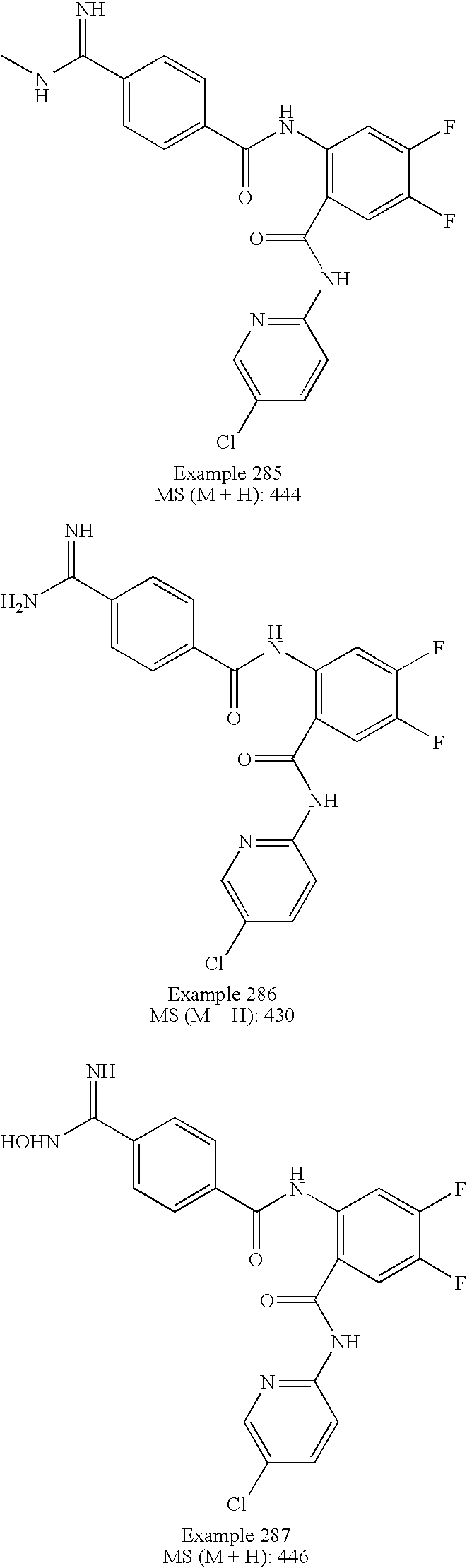
























































































































































Claims (10)
A-Q-D-E-G-J-X (Ic)





























Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11/924,480 US7727981B2 (en) | 2000-02-29 | 2007-10-25 | Benzamides and related inhibitors of factor Xa |
| US12/756,037 US8063036B2 (en) | 2000-02-29 | 2010-04-07 | Benzamides and related inhibitors of factor Xa |
| US13/247,937 US8518977B2 (en) | 2000-02-29 | 2011-09-28 | Benzamides and related inhibitors of factor XA |
| US13/612,597 US8691847B2 (en) | 2000-02-29 | 2012-09-12 | Benzamides and related inhibitors of factor Xa |
| US14/175,087 US9108922B2 (en) | 2000-02-29 | 2014-02-07 | Benzamides and related inhibitors of factor Xa |
| US14/742,465 US9629831B2 (en) | 2000-02-29 | 2015-06-17 | Benzamides and related inhibitors of factor XA |
| US15/466,530 US10179124B2 (en) | 2000-02-29 | 2017-03-22 | Benzamides and related inhibitors of factor Xa |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US18574600P | 2000-02-29 | 2000-02-29 | |
| US09/663,420 US6844367B1 (en) | 1999-09-17 | 2000-09-15 | Benzamides and related inhibitors of factor Xa |
| US09/794,225 US6376515B2 (en) | 2000-02-29 | 2001-02-28 | Benzamides and related inhibitors of factor Xa |
| US10/126,976 US20030162690A1 (en) | 2000-02-29 | 2002-04-22 | Benzamides and related inhibitors of factor Xa |
| US10/687,334 US6835739B2 (en) | 2000-02-29 | 2003-10-15 | Benzamides and related inhibitors of factor Xa |
| US10/942,733 US7314874B2 (en) | 2000-02-29 | 2004-09-15 | Benzamides and related inhibitors of factor Xa |
| US11/442,060 US7342013B2 (en) | 2000-02-29 | 2006-05-26 | Benzamides and related inhibitors of factor Xa |
| US11/924,480 US7727981B2 (en) | 2000-02-29 | 2007-10-25 | Benzamides and related inhibitors of factor Xa |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/442,060 Continuation US7342013B2 (en) | 2000-02-29 | 2006-05-26 | Benzamides and related inhibitors of factor Xa |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/756,037 Continuation US8063036B2 (en) | 2000-02-29 | 2010-04-07 | Benzamides and related inhibitors of factor Xa |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| US20090131411A1 US20090131411A1 (en) | 2009-05-21 |
| US7727981B2 true US7727981B2 (en) | 2010-06-01 |
Family
ID=26881428
Family Applications (13)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/794,225 Expired - Lifetime US6376515B2 (en) | 2000-02-29 | 2001-02-28 | Benzamides and related inhibitors of factor Xa |
| US10/126,976 Abandoned US20030162690A1 (en) | 2000-02-29 | 2002-04-22 | Benzamides and related inhibitors of factor Xa |
| US10/687,334 Expired - Lifetime US6835739B2 (en) | 2000-02-29 | 2003-10-15 | Benzamides and related inhibitors of factor Xa |
| US10/942,733 Expired - Fee Related US7314874B2 (en) | 2000-02-29 | 2004-09-15 | Benzamides and related inhibitors of factor Xa |
| US11/442,060 Expired - Lifetime US7342013B2 (en) | 2000-02-29 | 2006-05-26 | Benzamides and related inhibitors of factor Xa |
| US11/488,455 Expired - Fee Related US7727982B2 (en) | 2000-02-29 | 2006-07-17 | Benzamides and related inhibitors of factor Xa |
| US11/924,480 Expired - Fee Related US7727981B2 (en) | 2000-02-29 | 2007-10-25 | Benzamides and related inhibitors of factor Xa |
| US12/756,037 Expired - Fee Related US8063036B2 (en) | 2000-02-29 | 2010-04-07 | Benzamides and related inhibitors of factor Xa |
| US13/247,937 Expired - Fee Related US8518977B2 (en) | 2000-02-29 | 2011-09-28 | Benzamides and related inhibitors of factor XA |
| US13/612,597 Expired - Fee Related US8691847B2 (en) | 2000-02-29 | 2012-09-12 | Benzamides and related inhibitors of factor Xa |
| US14/175,087 Expired - Fee Related US9108922B2 (en) | 2000-02-29 | 2014-02-07 | Benzamides and related inhibitors of factor Xa |
| US14/742,465 Expired - Fee Related US9629831B2 (en) | 2000-02-29 | 2015-06-17 | Benzamides and related inhibitors of factor XA |
| US15/466,530 Expired - Fee Related US10179124B2 (en) | 2000-02-29 | 2017-03-22 | Benzamides and related inhibitors of factor Xa |
Family Applications Before (6)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/794,225 Expired - Lifetime US6376515B2 (en) | 2000-02-29 | 2001-02-28 | Benzamides and related inhibitors of factor Xa |
| US10/126,976 Abandoned US20030162690A1 (en) | 2000-02-29 | 2002-04-22 | Benzamides and related inhibitors of factor Xa |
| US10/687,334 Expired - Lifetime US6835739B2 (en) | 2000-02-29 | 2003-10-15 | Benzamides and related inhibitors of factor Xa |
| US10/942,733 Expired - Fee Related US7314874B2 (en) | 2000-02-29 | 2004-09-15 | Benzamides and related inhibitors of factor Xa |
| US11/442,060 Expired - Lifetime US7342013B2 (en) | 2000-02-29 | 2006-05-26 | Benzamides and related inhibitors of factor Xa |
| US11/488,455 Expired - Fee Related US7727982B2 (en) | 2000-02-29 | 2006-07-17 | Benzamides and related inhibitors of factor Xa |
Family Applications After (6)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/756,037 Expired - Fee Related US8063036B2 (en) | 2000-02-29 | 2010-04-07 | Benzamides and related inhibitors of factor Xa |
| US13/247,937 Expired - Fee Related US8518977B2 (en) | 2000-02-29 | 2011-09-28 | Benzamides and related inhibitors of factor XA |
| US13/612,597 Expired - Fee Related US8691847B2 (en) | 2000-02-29 | 2012-09-12 | Benzamides and related inhibitors of factor Xa |
| US14/175,087 Expired - Fee Related US9108922B2 (en) | 2000-02-29 | 2014-02-07 | Benzamides and related inhibitors of factor Xa |
| US14/742,465 Expired - Fee Related US9629831B2 (en) | 2000-02-29 | 2015-06-17 | Benzamides and related inhibitors of factor XA |
| US15/466,530 Expired - Fee Related US10179124B2 (en) | 2000-02-29 | 2017-03-22 | Benzamides and related inhibitors of factor Xa |
Country Status (10)
| Country | Link |
|---|---|
| US (13) | US6376515B2 (en) |
| EP (1) | EP1259485B1 (en) |
| AT (1) | ATE311366T1 (en) |
| AU (2) | AU2001250783A1 (en) |
| CA (1) | CA2401778C (en) |
| DE (1) | DE60115394T2 (en) |
| DK (1) | DK1259485T3 (en) |
| ES (1) | ES2254385T3 (en) |
| HK (1) | HK1051539A1 (en) |
| WO (2) | WO2001064642A2 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100234352A1 (en) * | 2004-06-18 | 2010-09-16 | Millennium Pharmaceuticals, Inc. | Factor xa inhibitors |
| US20100249117A1 (en) * | 2006-05-05 | 2010-09-30 | Millennium Pharmaceuticals, Inc. | Factor xa inhibitors |
| US20110033459A1 (en) * | 2007-05-02 | 2011-02-10 | Portola Pharmaceuticals, Inc. | Combination therapy with a compound acting as a platelet adp receptor inhibitor |
| US20110152530A1 (en) * | 2009-12-17 | 2011-06-23 | Millennium Pharmaceuticals, Inc. | Methods of preparing factor xa inhibitors and salts thereof |
| US20110178135A1 (en) * | 2009-12-17 | 2011-07-21 | Millennium Pharmaceuticals, Inc. | Salts and crystalline forms of a factor xa inhibitor |
| US8377974B2 (en) | 2004-06-18 | 2013-02-19 | Millennium Pharmaceuticals, Inc. | Factor Xa inhibitors |
| US8394964B2 (en) | 2009-12-17 | 2013-03-12 | Millennium Pharmaceuticals, Inc. | Methods of synthesizing factor Xa inhibitors |
| US8455440B2 (en) | 2007-04-13 | 2013-06-04 | Millennium Pharmaceuticals, Inc. | Combination anticoagulant therapy with a compound that acts as a factor Xa inhibitor |
| US8518977B2 (en) | 2000-02-29 | 2013-08-27 | Millennium Pharmaceuticals, Inc. | Benzamides and related inhibitors of factor XA |
| US8524907B2 (en) | 2006-11-02 | 2013-09-03 | Millennium Pharmaceuticals, Inc. | Methods of synthesizing pharmaceutical salts of a factor Xa inhibitor |
| US9200268B2 (en) | 2012-12-27 | 2015-12-01 | Portola Pharmaceuticals, Inc. | Compounds and methods for purification of serine proteases |
Families Citing this family (209)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6822267B1 (en) * | 1997-08-20 | 2004-11-23 | Advantest Corporation | Signal transmission circuit, CMOS semiconductor device, and circuit board |
| AU776053B2 (en) | 2000-03-31 | 2004-08-26 | Astellas Pharma Inc. | Diazepan derivatives or salts thereof |
| TWI290136B (en) | 2000-04-05 | 2007-11-21 | Daiichi Seiyaku Co | Ethylenediamine derivatives |
| US7160878B2 (en) | 2000-07-27 | 2007-01-09 | Eli Lilly And Company | Substituted heterocyclic amides |
| PL204898B1 (en) | 2000-11-22 | 2010-02-26 | Astellas Pharma Inc | Substituted benzene derivatives or salts thereof |
| WO2002064567A2 (en) * | 2000-11-28 | 2002-08-22 | Eli Lilly And Company | Substituted carboxamides as inhibitors of factor xa |
| US7067539B2 (en) | 2001-02-08 | 2006-06-27 | Schering Corporation | Cannabinoid receptor ligands |
| US7507767B2 (en) | 2001-02-08 | 2009-03-24 | Schering Corporation | Cannabinoid receptor ligands |
| US7312235B2 (en) * | 2001-03-30 | 2007-12-25 | Millennium Pharmaceuticals, Inc. | Benzamide inhibitors of factor Xa |
| WO2003000657A1 (en) | 2001-06-20 | 2003-01-03 | Daiichi Pharmaceutical Co., Ltd. | Diamine derivatives |
| SE0102764D0 (en) | 2001-08-17 | 2001-08-17 | Astrazeneca Ab | Compounds |
| AUPR738301A0 (en) * | 2001-08-30 | 2001-09-20 | Starpharma Limited | Chemotherapeutic agents |
| JP3795044B2 (en) | 2001-09-14 | 2006-07-12 | メシルジーン、インコーポレイテッド | Inhibitors of histone deacetylase |
| US6897220B2 (en) * | 2001-09-14 | 2005-05-24 | Methylgene, Inc. | Inhibitors of histone deacetylase |
| US7868204B2 (en) | 2001-09-14 | 2011-01-11 | Methylgene Inc. | Inhibitors of histone deacetylase |
| AR037352A1 (en) | 2001-11-14 | 2004-11-03 | Schering Corp | LIGHTING COMPOUNDS OF CANNABINOID RECEPTORS, A PHARMACEUTICAL COMPOSITION, AND THE USE OF SUCH COMPOUNDS FOR THE MANUFACTURE OF MEDICINES |
| EP1314733A1 (en) | 2001-11-22 | 2003-05-28 | Aventis Pharma Deutschland GmbH | Indole-2-carboxamides as factor Xa inhibitors |
| US7030141B2 (en) * | 2001-11-29 | 2006-04-18 | Christopher Franklin Bigge | Inhibitors of factor Xa and other serine proteases involved in the coagulation cascade |
| AU2002350172A1 (en) * | 2001-12-07 | 2003-06-23 | Eli Lilly And Company | Substituted heterocyclic carboxamides with antithrombotic activity |
| US20050026929A1 (en) * | 2002-04-23 | 2005-02-03 | Axys Pharmaceuticals, Inc. | Novel phenyl derivatives as inducers of apoptosis |
| US7217732B2 (en) | 2002-06-19 | 2007-05-15 | Schering Corporation | Cannabinoid receptor agonists |
| TW200409637A (en) | 2002-06-26 | 2004-06-16 | Glaxo Group Ltd | Compounds |
| GB0215293D0 (en) | 2002-07-03 | 2002-08-14 | Rega Foundation | Viral inhibitors |
| US7307088B2 (en) * | 2002-07-09 | 2007-12-11 | Amgen Inc. | Substituted anthranilic amide derivatives and methods of use |
| JP2006505559A (en) | 2002-10-18 | 2006-02-16 | エフ.ホフマン−ラ ロシュ アーゲー | 4-piperazinylbenzenesulfonylindole with 5-HT6 receptor affinity |
| GB0226930D0 (en) | 2002-11-19 | 2002-12-24 | Astrazeneca Ab | Chemical compounds |
| DK1569912T3 (en) | 2002-12-03 | 2015-06-29 | Pharmacyclics Inc | 2- (2-hydroxybiphenyl-3-yl) -1h-benzoimidazole-5-carboxamidine derivatives as factor VIIa inhibitors. |
| TWI299664B (en) | 2003-01-06 | 2008-08-11 | Osi Pharm Inc | (2-carboxamido)(3-amino)thiophene compounds |
| US7696225B2 (en) | 2003-01-06 | 2010-04-13 | Osi Pharmaceuticals, Inc. | (2-carboxamido)(3-Amino) thiophene compounds |
| RU2332412C2 (en) | 2003-02-07 | 2008-08-27 | Дайити Фармасьютикал Ко., Лтд. | Pyrazole derivatives |
| TW200505902A (en) | 2003-03-20 | 2005-02-16 | Schering Corp | Cannabinoid receptor ligands |
| US7557102B2 (en) * | 2003-04-03 | 2009-07-07 | Merck Patent Gmbh | Pyrazolidine-1,2-dicarboxyldiphenylamide derivatives as coagulation factor Xa inhibitors for the treatment of thromboses |
| MXPA05010444A (en) * | 2003-04-03 | 2005-11-04 | Merck Patent Gmbh | Pyrrolidino-1, 2-dicarboxy -1-(phenylamide) -2-(4-(3-oxo- morpholino -4-yl)- phenylamide) derivatives and related compounds for use as inhibitors of coagulation factor xa in the treatment of thrombo-embolic diseases. |
| TWI372050B (en) | 2003-07-03 | 2012-09-11 | Astex Therapeutics Ltd | (morpholin-4-ylmethyl-1h-benzimidazol-2-yl)-1h-pyrazoles |
| NZ544756A (en) | 2003-07-22 | 2009-09-25 | Astex Therapeutics Ltd | 3,4-Disubstituted 1H-pyrazole compounds and their use as cyclin dependent kinases (CDK) and glycogen synthase kinase-3 (GSK-3) modulators |
| CN1882569B (en) | 2003-09-19 | 2010-09-29 | 阿斯利康(瑞典)有限公司 | Quinazoline derivatives |
| KR101153335B1 (en) | 2003-09-24 | 2012-07-05 | 메틸진 인코포레이티드 | Inhibitors of histone deacetylase |
| SE0302573D0 (en) * | 2003-09-26 | 2003-09-26 | Astrazeneca Ab | Benzimidazole derivatives, compositions containing them, preparation thereof and uses thereof |
| EP1670783A2 (en) * | 2003-09-30 | 2006-06-21 | Eli Lilly And Company | Antithrombotic aromatic ethers |
| TW200526631A (en) | 2003-10-07 | 2005-08-16 | Renovis Inc | Amide derivatives as ion-channel ligands and pharmaceutical compositions and methods of using the same |
| ES2338881T3 (en) | 2003-10-09 | 2010-05-13 | Millennium Pharmaceuticals, Inc. | BENZAMIDS REPLACED WITH THIOETER AS INHIBITORS OF THE XA FACTOR. |
| EP1706403B9 (en) * | 2003-12-22 | 2012-07-25 | K.U.Leuven Research & Development | Imidazo[4,5-c]pyridine compounds and methods of antiviral treatment |
| CA2548374C (en) | 2003-12-23 | 2014-05-27 | Astex Therapeutics Limited | Pyrazole derivatives as protein kinase modulators |
| WO2005073193A1 (en) * | 2004-01-23 | 2005-08-11 | Amgen Inc. | Vanilloid receptor ligands and their use in treatments |
| AU2005214137B2 (en) * | 2004-02-18 | 2008-05-29 | Astrazeneca Ab | Compounds |
| ATE426597T1 (en) * | 2004-02-18 | 2009-04-15 | Astrazeneca Ab | BENZAMIDE DERIVATIVES AND THE USE THEREOF AS GLUCOCINASE ACTIVATE AGENTS |
| US7253204B2 (en) | 2004-03-26 | 2007-08-07 | Methylgene Inc. | Inhibitors of histone deacetylase |
| US7625949B2 (en) | 2004-04-23 | 2009-12-01 | Roche Palo Alto Llc | Methods for treating retroviral infections |
| US7166738B2 (en) | 2004-04-23 | 2007-01-23 | Roche Palo Alto Llc | Non-nucleoside reverse transcriptase inhibitors |
| TW200600086A (en) * | 2004-06-05 | 2006-01-01 | Astrazeneca Ab | Chemical compound |
| PE20060417A1 (en) | 2004-08-03 | 2006-06-13 | Wyeth Corp | INDAZOLES AS LXRS MODULATORS |
| KR101269869B1 (en) * | 2004-09-24 | 2013-06-07 | 네오메드 인스티튜트 | Benzimidazole derivatives, compositions containing them, preparation thereof and uses thereof |
| GB0422556D0 (en) * | 2004-10-11 | 2004-11-10 | Syngenta Participations Ag | Novel insecticides |
| GB0423043D0 (en) * | 2004-10-16 | 2004-11-17 | Astrazeneca Ab | Compounds |
| WO2006040527A1 (en) | 2004-10-16 | 2006-04-20 | Astrazeneca Ab | Process for making phenoxy benzamide compounds |
| GB0423044D0 (en) * | 2004-10-16 | 2004-11-17 | Astrazeneca Ab | Compounds |
| EP1809619A1 (en) | 2004-10-21 | 2007-07-25 | Transtech Pharma, Inc. | Bissulfonamide compounds as agonists of galr1, compositions, and methods of use |
| MX2007005113A (en) | 2004-11-03 | 2007-06-26 | Hoffmann La Roche | Dicarboxamide derivatives and their use as factor xa inhibitors. |
| WO2006057845A1 (en) | 2004-11-24 | 2006-06-01 | Eli Lilly And Company | Aromatic ether derivatives useful as thrombin inhibitors |
| WO2006057868A1 (en) * | 2004-11-29 | 2006-06-01 | Eli Lilly And Company | Antithrombotic diamides |
| JP2008524246A (en) | 2004-12-16 | 2008-07-10 | タケダ サン ディエゴ インコーポレイテッド | Histone deacetylase inhibitor |
| ES2324794T3 (en) * | 2004-12-21 | 2009-08-14 | Gilead Sciences Inc | IMIDAZO (4,5-C) COMPONENT PYRIDINE AND ANTIVIRAL TREATMENT METHOD. |
| BRPI0519759A2 (en) | 2004-12-30 | 2009-03-10 | Astex Therapeutics Ltd | pharmaceutical compositions |
| EP1846372B1 (en) | 2005-01-07 | 2014-04-16 | Synta Pharmaceuticals Corp. | Compounds for inflammation and immune-related uses |
| AR054425A1 (en) | 2005-01-21 | 2007-06-27 | Astex Therapeutics Ltd | PIPERIDIN ADDITION SALTS 4-IL-ACID AMID 4- (2,6-DICLORO-BENZOILAMINO) 1H-PIRAZOL-3-CARBOXILICO. |
| BRPI0606480A (en) | 2005-01-21 | 2008-03-11 | Astex Therapeutics Ltd | pharmaceutical compounds |
| KR101346886B1 (en) | 2005-01-21 | 2014-01-02 | 아스텍스 테라퓨틱스 리미티드 | Pharmaceutical compounds |
| US8404718B2 (en) | 2005-01-21 | 2013-03-26 | Astex Therapeutics Limited | Combinations of pyrazole kinase inhibitors |
| WO2006083003A1 (en) * | 2005-02-02 | 2006-08-10 | Ajinomoto Co., Inc. | Novel benzamidine compound |
| US7576099B2 (en) | 2005-02-28 | 2009-08-18 | Renovis, Inc. | Amide derivatives as ion-channel ligands and pharmaceutical compositions and methods of using the same |
| CA2600409C (en) | 2005-03-10 | 2011-07-05 | Pfizer Inc. | Substituted n-sulfonylaminophenylethyl-2-phenoxy acetamide compounds |
| US7264402B2 (en) * | 2005-03-10 | 2007-09-04 | Corning Cable Systems Llc | Multi-fiber optic receptacle and plug assembly |
| JP2008540574A (en) | 2005-05-11 | 2008-11-20 | タケダ サン ディエゴ インコーポレイテッド | Histone deacetylase inhibitor |
| EP1891069A1 (en) * | 2005-05-24 | 2008-02-27 | AstraZeneca AB | 2-phenyl substituted imidazol [4,5b]pyridine/ pyrazine and purine derivatives as glucokinase modulators |
| TW200714597A (en) * | 2005-05-27 | 2007-04-16 | Astrazeneca Ab | Chemical compounds |
| US8343953B2 (en) | 2005-06-22 | 2013-01-01 | Astex Therapeutics Limited | Pharmaceutical compounds |
| US8541461B2 (en) | 2005-06-23 | 2013-09-24 | Astex Therapeutics Limited | Pharmaceutical combinations comprising pyrazole derivatives as protein kinase modulators |
| US20080234273A1 (en) * | 2005-07-09 | 2008-09-25 | Mckerrecher Darren | Heteroaryl Benzamide Derivatives for Use as Glk Activators in the Treatment of Diabetes |
| US20110053910A1 (en) * | 2005-07-09 | 2011-03-03 | Mckerrecher Darren | 2 -heterocyclyloxybenzoyl amino heterocyclyl compounds as modulators of glucokinase for the treatment of type 2 diabetes |
| US7642259B2 (en) * | 2005-07-09 | 2010-01-05 | Astrazeneca Ab | Heteroaryl benzamide derivatives for use as GLK activators in the treatment of diabetes |
| BRPI0613429A2 (en) * | 2005-07-14 | 2009-02-10 | Takeda San Diego Inc | histone deacetylase inhibitors |
| US9202182B2 (en) * | 2005-08-11 | 2015-12-01 | International Business Machines Corporation | Method and system for analyzing business architecture |
| NZ566276A (en) | 2005-09-16 | 2011-03-31 | Arrow Therapeutics Ltd | Biphenyl derivatives and their use in treating hepatitis C |
| EP1940781A1 (en) | 2005-10-19 | 2008-07-09 | F.Hoffmann-La Roche Ag | Phenyl-acetamide nnrt inhibitors |
| PE20110070A1 (en) * | 2005-11-08 | 2011-03-04 | Millennium Pharm Inc | PROCEDURE FOR THE PREPARATION OF THE COMPOUND [2 - ({4 - [(DIMETHYLAMINE) IMINOMETIL] PHENYL} CARBONYLAMINE) -5-METOXYLAMINE] -N- (5-CHLORO (2-PYRIDYL)) CARBOXAMIDE |
| TW200738621A (en) | 2005-11-28 | 2007-10-16 | Astrazeneca Ab | Chemical process |
| US8399442B2 (en) | 2005-12-30 | 2013-03-19 | Astex Therapeutics Limited | Pharmaceutical compounds |
| EP1976835A2 (en) * | 2006-01-13 | 2008-10-08 | Takeda San Diego, Inc. | Histone deacetylase inhibitors |
| US8168658B2 (en) | 2006-02-28 | 2012-05-01 | Merck Sharp & Dohme Corp. | Inhibitors of histone deacetylase |
| TW200745049A (en) * | 2006-03-23 | 2007-12-16 | Astrazeneca Ab | New crystalline forms |
| US20090186810A1 (en) * | 2006-03-27 | 2009-07-23 | Portola Pharmaceuticals, Inc. | Potassium channel modulators and platelet procoagulant activity |
| JP2009531385A (en) | 2006-03-31 | 2009-09-03 | グラクソ グループ リミテッド | Piperazine derivatives as growth hormone secretagogue (GHS) receptor agonists |
| CA2648804C (en) * | 2006-04-07 | 2014-05-27 | Methylgene Inc. | Benzamide derivatives as inhibitors of histone deacetylase |
| TW200808769A (en) * | 2006-04-18 | 2008-02-16 | Astrazeneca Ab | Therapeutic compounds |
| WO2008001115A2 (en) | 2006-06-29 | 2008-01-03 | Astex Therapeutics Limited | Pharmaceutical combinations of 1-cyclopropyl-3- [3- (5-m0rphoolin-4-ylmethyl-1h-benzoimidazol-2-yl) -lh-1-pyrazol- 4-yl] -urea |
| EP2043635A2 (en) * | 2006-06-29 | 2009-04-08 | Astex Therapeutics Limited | Pharmaceutical combinations |
| ATE454384T1 (en) * | 2006-07-07 | 2010-01-15 | Gilead Sciences Inc | NEW PYRIDAZINE COMPOUND AND USE THEREOF |
| TW200825063A (en) * | 2006-10-23 | 2008-06-16 | Astrazeneca Ab | Chemical compounds |
| SA07280576B1 (en) * | 2006-10-26 | 2011-06-22 | استرازينيكا ايه بي | Benzoyl amino heterocyclyl compounds as glucokinase (GLK) activators |
| KR20090064478A (en) | 2006-11-13 | 2009-06-18 | 화이자 프로덕츠 인크. | Diaryl, dipyridinyl and aryl-pyridinyl derivatives and uses thereof |
| RU2452484C2 (en) * | 2006-12-08 | 2012-06-10 | Милленниум Фармасьютикалз, Инк. | Standard drug preparations and methods of treating thrombosis by oral administration of factor xa inhibitor |
| UY30822A1 (en) * | 2006-12-21 | 2008-07-31 | Astrazeneca Ab | NEW CRYSTAL FORM OF 3 - {[5-AZETIDIN-1-YLCABONYL) PYRAZIN-2-YL] OXY} -5- [1-METHYLETHYLOXY] -N-1H-PYRAZOL-3-YLBENZAMIDA, COMPOSITIONS CONTAINING AND PREPARATION, PROCESSING AND PREPARATION APPLICATIONS |
| AU2008205093A1 (en) * | 2007-01-05 | 2008-07-17 | Millennium Pharmaceuticals, Inc. | Factor Xa inhibitors |
| UY30892A1 (en) | 2007-02-07 | 2008-09-02 | Smithkline Beckman Corp | AKT ACTIVITY INHIBITORS |
| US8618324B2 (en) * | 2007-03-13 | 2013-12-31 | Board Of Regents, The University Of Texas System | Composition and method for making oligo-benzamide compounds |
| GB0704932D0 (en) | 2007-03-14 | 2007-04-25 | Astex Therapeutics Ltd | Pharmaceutical compounds |
| UA99466C2 (en) | 2007-07-06 | 2012-08-27 | Гилиад Сайенсиз, Инк. | Crystalline pyridazine compound |
| EP2915564B1 (en) * | 2007-09-28 | 2020-11-04 | Portola Pharmaceuticals, Inc. | Antidotes for factor XA inhibitors and methods of using the same |
| UA103319C2 (en) | 2008-05-06 | 2013-10-10 | Глаксосмитклайн Ллк | Thiazole- and oxazole-benzene sulfonamide compounds |
| EA201100097A1 (en) | 2008-08-04 | 2011-10-31 | Астразенека Аб | PYRAZOLO [3,4] PYRIMIDIN-4-ILA DERIVATIVES AND THEIR APPLICATIONS FOR TREATING DIABETES AND OBESITY |
| JP5709316B2 (en) | 2008-11-14 | 2015-04-30 | ポートラ ファーマシューティカルズ, インコーポレイテッド | Methods for using antidotes in combination with antidotes for factor Xa inhibitors and blood coagulants |
| US8609711B2 (en) * | 2009-01-30 | 2013-12-17 | Glaxosmithkline Llc | Crystalline N-{(1S)-2-amino-1-[(3-fluorophenyl)methyl]ethyl}-5-chloro-4-(4-chloro-1-methyl-1H-pyrazol-5-yl)-2-thiophenecarboxamic hydrochloride |
| GB0902406D0 (en) * | 2009-02-13 | 2009-04-01 | Astrazeneca Ab | Crystalline polymorphic form |
| GB0902434D0 (en) * | 2009-02-13 | 2009-04-01 | Astrazeneca Ab | Chemical process |
| AU2010223919B2 (en) | 2009-03-13 | 2016-03-31 | Les Laboratoires Servier | Methods and compositions for cell-proliferation-related disorders |
| PT2414517T (en) | 2009-03-30 | 2016-12-27 | Portola Pharm Inc | Antidotes for factor xa inhibitors and methods of using the same |
| EP2417123A2 (en) | 2009-04-06 | 2012-02-15 | Agios Pharmaceuticals, Inc. | Therapeutic compositions and related methods of use |
| WO2010116176A1 (en) * | 2009-04-09 | 2010-10-14 | Astrazeneca Ab | Pyrazolo [4, 5-e] pyrimidine derivative and its use to treat diabetes and obesity |
| AR076220A1 (en) | 2009-04-09 | 2011-05-26 | Astrazeneca Ab | DERIVATIVES OF PIRAZOL [4,5 - E] PYRIMIDINE |
| CA2766882C (en) | 2009-06-29 | 2016-11-22 | Agios Pharmaceuticals, Inc. | Therapeutic compounds and compositions |
| ES2605801T3 (en) * | 2009-07-15 | 2017-03-16 | Portola Pharmaceuticals, Inc. | Unit dose formulation of antidote for factor Xa inhibitors for use in the prevention of bleeding |
| US20110086853A1 (en) * | 2009-10-08 | 2011-04-14 | William Brown | Therapeutic Compounds |
| ES2594402T3 (en) | 2009-10-21 | 2016-12-20 | Agios Pharmaceuticals, Inc. | Methods and compositions for disorders related to cell proliferation |
| EA015918B1 (en) * | 2010-03-03 | 2011-12-30 | Дмитрий Геннадьевич ТОВБИН | URETHANES, UREAS, AMIDES AND RELATED INHIBITORS OF Xa FACTORS |
| EP2575453B1 (en) | 2010-05-28 | 2018-08-01 | The Board of Regents of the University of Texas System | Oligo-benzamide compounds and their use |
| WO2012006477A1 (en) | 2010-07-07 | 2012-01-12 | Ardelyx, Inc. | Compounds and methods for inhibiting phosphate transport |
| JP5823514B2 (en) | 2010-07-07 | 2015-11-25 | アーデリクス,インコーポレーテッド | Compounds and methods for inhibiting phosphate transport |
| WO2012006473A1 (en) * | 2010-07-07 | 2012-01-12 | Ardelyx, Inc. | Compounds and methods for inhibiting phosphate transport |
| JP5827327B2 (en) | 2010-07-07 | 2015-12-02 | アーデリクス,インコーポレーテッド | Compounds and methods for inhibiting phosphate transport |
| TW201240664A (en) | 2010-09-01 | 2012-10-16 | Portola Pharm Inc | Methods and formulations of treating thrombosis with betrixaban and a P-glycoprotein inhibitor |
| AR082804A1 (en) | 2010-09-01 | 2013-01-09 | Portola Pharm Inc | CRYSTAL FORMS OF AN XA FACTOR INHIBITOR |
| ES2564952T3 (en) | 2010-12-17 | 2016-03-30 | Agios Pharmaceuticals, Inc. | New derivatives of N- (4- (azetidine-1-carbonyl) phenyl) - (hetero-) arylsulfonamide as modulators of pyruvate kinase M2 (PKM2) |
| MX336022B (en) | 2010-12-21 | 2016-01-06 | Agios Pharmaceuticals Inc | Bicyclic pkm2 activators. |
| TWI549947B (en) | 2010-12-29 | 2016-09-21 | 阿吉歐斯製藥公司 | Therapeutic compounds and compositions |
| US20140050743A1 (en) | 2011-01-19 | 2014-02-20 | Bayer Intellectual Property Gmbh | Binding proteins to inhibitors of coagulation factors |
| GB201104267D0 (en) | 2011-03-14 | 2011-04-27 | Cancer Rec Tech Ltd | Pyrrolopyridineamino derivatives |
| CN114432312A (en) | 2011-05-03 | 2022-05-06 | 安吉奥斯医药品有限公司 | Pyruvate kinase activators for therapy |
| WO2012151452A1 (en) | 2011-05-03 | 2012-11-08 | Agios Pharmaceuticals, Inc | Pyruvate kinase activators for use in therapy |
| BR112013028430A2 (en) | 2011-05-04 | 2017-08-01 | Merck Sharp & Dohme | compound, pharmaceutical composition, method for treating a spleen tyrosine kinase (syk) mediated disease or condition, and use of a therapeutically efficient amount of the compound |
| CN102827170A (en) | 2011-06-17 | 2012-12-19 | 安吉奥斯医药品有限公司 | Active treatment compositions and use method thereof |
| CN102827073A (en) | 2011-06-17 | 2012-12-19 | 安吉奥斯医药品有限公司 | Therapeutically active compositions and application methods thereof |
| US20150224091A1 (en) | 2011-08-31 | 2015-08-13 | Portola Pharmaceuticals, Inc. | Prevention and treatment of thrombosis in medically ill patients |
| MY167798A (en) | 2011-10-03 | 2018-09-26 | Respivert Ltd | 1-pyrazolyl-3- (4- ( (2 -anilinopyrimidin- 4 - yl) oxy) napththalen- 1 - yl) ureas as p38 map kinase inhibitors |
| EP2578582A1 (en) | 2011-10-03 | 2013-04-10 | Respivert Limited | 1-Pyrazolyl-3-(4-((2-anilinopyrimidin-4-yl)oxy)napththalen-1-yl)ureas as p38 MAP kinase inhibitors |
| US8835493B2 (en) | 2011-11-23 | 2014-09-16 | Board Of Regents, The University Of Texas System | Oligo-benzamide compounds for use in treating cancers |
| US8754124B2 (en) | 2011-11-23 | 2014-06-17 | Board Of Regents, The University Of Texas System | Oligo-benzamide compounds and their use in treating cancers |
| US9474779B2 (en) | 2012-01-19 | 2016-10-25 | Agios Pharmaceuticals, Inc. | Therapeutically active compositions and their methods of use |
| PT2841428T (en) | 2012-04-24 | 2018-11-29 | Vertex Pharma | Dna-pk inhibitors |
| GB201214750D0 (en) | 2012-08-17 | 2012-10-03 | Respivert Ltd | Compounds |
| GB201215357D0 (en) | 2012-08-29 | 2012-10-10 | Respivert Ltd | Compounds |
| GB201216018D0 (en) | 2012-09-07 | 2012-10-24 | Cancer Rec Tech Ltd | Pharmacologically active compounds |
| WO2014053968A1 (en) | 2012-10-04 | 2014-04-10 | Pfizer Limited | Pyrrolo[3,2-c]pyridine tropomyosin-related kinase inhibitors |
| US20140346397A1 (en) | 2012-12-27 | 2014-11-27 | Portola Pharmaceuticals, Inc. | Compounds and methods for purification of serine proteases |
| FR3000895B1 (en) | 2013-01-11 | 2017-02-24 | Laboratoire Francais Du Fractionnement Et Des Biotechnologies Sa | USE OF ANTIDOTES OF COAGULATION INHIBITORS INDICATED IN THE PREVENTION OR TREATMENT OF THROMBOEMBOLIC PATHOLOGIES |
| US9340557B2 (en) | 2013-03-12 | 2016-05-17 | Vertex Pharmaceuticals Incorporated | Substituted quinoxaline DNA-PK inhibitors |
| EP2970190A1 (en) | 2013-03-14 | 2016-01-20 | Respivert Limited | Kinase inhibitors |
| US10376510B2 (en) | 2013-07-11 | 2019-08-13 | Agios Pharmaceuticals, Inc. | 2,4- or 4,6-diaminopyrimidine compounds as IDH2 mutants inhibitors for the treatment of cancer |
| US9579324B2 (en) | 2013-07-11 | 2017-02-28 | Agios Pharmaceuticals, Inc | Therapeutically active compounds and their methods of use |
| WO2015003355A2 (en) | 2013-07-11 | 2015-01-15 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| WO2015003360A2 (en) | 2013-07-11 | 2015-01-15 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US20150031627A1 (en) | 2013-07-25 | 2015-01-29 | Agios Pharmaceuticals, Inc | Therapeutically active compounds and their methods of use |
| EA201300843A1 (en) * | 2013-08-01 | 2015-04-30 | Арсений Валерьевич Айбушев | CARBAMOILPHENYL DERIVATIVES FOR STOPPING, PREVENTION AND PREVENTION OF BLEEDING OR STRENGTHENING OF THE HEMOSTASIS SYSTEM |
| GB201314452D0 (en) | 2013-08-13 | 2013-09-25 | Ostara Biomedical Ltd | Embryo implantation |
| DK3054936T5 (en) | 2013-10-10 | 2024-03-18 | Eastern Virginia Medical School | 4-((2-HYDROXY-3-METHOXYBENZYL)AMINO) BENZENESULFONAMIDE DERIVATIVES AS 12-LIPOXYGENASE INHIBITORS |
| LT3424920T (en) | 2013-10-17 | 2020-08-10 | Vertex Pharmaceuticals Incorporated | Co-crystals of (s)-n-methyl-8-(1-((2`-methyl-[4,5`-bipyrimidin]-6-yl)amino)propan-2-yl)quinoline-4-carboxamide and deuterated derivatives thereof as dna-pk inhibitors |
| US9636298B2 (en) | 2014-01-17 | 2017-05-02 | Methylgene Inc. | Prodrugs of compounds that enhance antifungal activity and compositions of said prodrugs |
| AU2015216957B2 (en) | 2014-02-14 | 2019-04-04 | Respivert Limited | Aromatic heterocyclic compounds as antiinflammatory compounds |
| MX2016011810A (en) | 2014-03-14 | 2017-04-27 | Agios Pharmaceuticals Inc | Pharmaceutical compositions of therapeutically active compounds. |
| DE102014108210A1 (en) | 2014-06-11 | 2015-12-17 | Dietrich Gulba | rodenticide |
| WO2016029061A1 (en) | 2014-08-20 | 2016-02-25 | Portola Pharmaceuticals, Inc. | Lyophilized formulations for factor xa antidote |
| GB201501302D0 (en) | 2015-01-27 | 2015-03-11 | Ostara Biomedical Ltd | Embryo implantation |
| EP3307271B1 (en) | 2015-06-11 | 2023-09-13 | Agios Pharmaceuticals, Inc. | Methods of using pyruvate kinase activators |
| WO2017012502A1 (en) | 2015-07-17 | 2017-01-26 | Sunshine Lake Pharma Co., Ltd. | Substituted quinazoline compounds and preparation and uses thereof |
| GB201514021D0 (en) | 2015-08-07 | 2015-09-23 | Arner Elias Set Jeno | Novel Pyridines and their use in the treatment of cancer |
| GB201517523D0 (en) | 2015-10-05 | 2015-11-18 | Ostara Biomedical Ltd | Methods and compositions for managing reproduction |
| MA42999A (en) | 2015-10-15 | 2018-08-22 | Agios Pharmaceuticals Inc | POLYTHERAPY FOR THE TREATMENT OF MALIGNITIES |
| JP7033061B2 (en) | 2015-10-15 | 2022-03-09 | アジオス ファーマシューティカルズ, インコーポレイテッド | Combination therapy to treat malignant tumors |
| WO2017091757A1 (en) | 2015-11-24 | 2017-06-01 | Portola Pharmaceuticals, Inc. | Isotopically enriched betrixaban |
| CA3012031C (en) | 2016-01-22 | 2024-03-26 | Janssen Pharmaceutica Nv | 6-membered heteroaromatic substituted cyanoindoline derivatives as nik inhibitors |
| TWI739783B (en) | 2016-01-22 | 2021-09-21 | 比利時商健生藥品公司 | New substituted cyanoindoline derivatives as nik inhibitors |
| CN108883160B (en) | 2016-02-24 | 2022-06-28 | 博尔托拉制药公司 | Lyophilized formulations of factor XA antidotes |
| RU2698202C2 (en) * | 2016-06-01 | 2019-08-23 | Закрытое акционерное общество "ФАРМА ВАМ" | METHOD FOR PRODUCING DIRECT FACTOR Xa INHIBITOR |
| CA3027457A1 (en) | 2016-06-17 | 2017-12-21 | Portola Pharmaceuticals, Inc. | Preparation of factor xa derivatives |
| WO2018002217A1 (en) | 2016-06-30 | 2018-01-04 | Janssen Pharmaceutica Nv | Heteroaromatic derivatives as nik inhibitors |
| US20190119299A1 (en) | 2016-06-30 | 2019-04-25 | Janssen Pharmaceutica Nv | Cyanoindoline derivatives as nik inhibitors |
| ES2885003T3 (en) | 2016-09-02 | 2021-12-13 | Cyclerion Therapeutics Inc | Bicyclic fused SGC stimulators |
| KR20190062485A (en) | 2016-09-27 | 2019-06-05 | 버텍스 파마슈티칼스 인코포레이티드 | Methods of treating cancer using a combination of DNA-damaging agents and DNA-PK inhibitors |
| WO2018069936A1 (en) | 2016-10-13 | 2018-04-19 | Mylan Laboratories Limited | Polymorphs and solid dispersion of betrixaban and methods for the preparation thereof |
| BR112019016233A2 (en) | 2017-02-07 | 2020-04-07 | Oblique Therapeutics Ab | heterocyclyl sulfonyl substituted pyridines and their use in cancer treatment |
| CN110382465A (en) | 2017-02-07 | 2019-10-25 | 欧比力克治疗公司 | Sulfinyl pyridine and its purposes in cancer treatment |
| RU2019128063A (en) | 2017-02-07 | 2021-03-09 | Облик Терапьютикс Аб | HYDROCARBONSULPHONYL SUBSTITUTED PYRIDINS AND THEIR APPLICATION IN THE TREATMENT OF CANCER |
| US11028067B2 (en) | 2017-02-07 | 2021-06-08 | Oblique Therapeutics Ab | Heteroarylsulfonyl-substituted pyridines and their use in the treatment of cancer |
| WO2018229796A2 (en) | 2017-06-14 | 2018-12-20 | Mylan Laboratories Limited | A process for betrixaban hydrochloride and betrixaban maleate salt |
| KR20200026987A (en) | 2017-07-11 | 2020-03-11 | 버텍스 파마슈티칼스 인코포레이티드 | Carboxamides as Modulators of Sodium Channels |
| SG11202001684PA (en) | 2017-10-06 | 2020-03-30 | Forma Therapeutics Inc | Inhibiting ubiquitin specific peptidase 30 |
| SG10202011632RA (en) | 2017-10-27 | 2021-01-28 | Boehringer Ingelheim Int | Pyridine carbonyl derivatives and therapeutic uses thereof as trpc6 inhibitors |
| CA3098283C (en) | 2018-04-26 | 2023-05-23 | Pfizer Inc. | 2-amino-pyridine or 2-amino-pyrimidine derivatives as cyclin dependent kinase inhibitors |
| GB201807014D0 (en) | 2018-04-30 | 2018-06-13 | Univ Leeds Innovations Ltd | Factor xlla inhibitors |
| US10980788B2 (en) | 2018-06-08 | 2021-04-20 | Agios Pharmaceuticals, Inc. | Therapy for treating malignancies |
| GB201810581D0 (en) | 2018-06-28 | 2018-08-15 | Ctxt Pty Ltd | Compounds |
| FI3860989T3 (en) | 2018-10-05 | 2023-05-25 | Forma Therapeutics Inc | Fused pyrrolines which act as ubiquitin-specific protease 30 (usp30) inhibitors |
| CA3143666A1 (en) | 2019-06-18 | 2020-12-24 | Pfizer Inc. | Benzisoxazole sulfonamide derivatives |
| UY38979A (en) | 2019-12-06 | 2021-07-30 | Vertex Pharma | TETRAHYDROFURANS REPLACED AS SODIUM CHANNEL MODULATORS |
| TW202322824A (en) | 2020-02-18 | 2023-06-16 | 美商基利科學股份有限公司 | Antiviral compounds |
| WO2022211680A1 (en) * | 2021-03-30 | 2022-10-06 | Общество С Ограниченной Ответственностью "Фармадиол" | Process for producing the anticoagulant n-(5-chloropyridin-2-yl)-2-({4-[ethanimidoyl(methyl)amino]benzoyl}amino)-5-methylbenzamide hydrochloride |
| EP4070658A1 (en) | 2021-04-06 | 2022-10-12 | BIORoxx GmbH | Use of anticoagulant active compounds as rodenticide |
| AU2022256476A1 (en) | 2021-04-16 | 2023-10-12 | Gilead Sciences, Inc. | Methods of preparing carbanucleosides using amides |
| GB202107722D0 (en) | 2021-05-28 | 2021-07-14 | Lunac Therapeutics Ltd | Factor XIIA Inhibitors |
| AR126073A1 (en) | 2021-06-04 | 2023-09-06 | Vertex Pharma | N-(HYDROXYALKYL(HETERO)ARYL)TETRAHYDROFURAN CARBOXAMIDES AS SODIUM CHANNEL MODULATORS |
Citations (54)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2095619A (en) | 1937-10-12 | Aryl oxides | ||
| CH520657A (en) | 1961-09-11 | 1972-03-31 | Wander Ag Dr A | Bis-anilides with basic substituents - having chemotherapeutic activity |
| EP0077534A2 (en) | 1981-10-15 | 1983-04-27 | Chugai Seiyaku Kabushiki Kaisha | Benzamide derivatives, a process for preparing the same, and pharmaceutical compositions containing the same |
| JPS59181257A (en) | 1983-03-31 | 1984-10-15 | Chugai Pharmaceut Co Ltd | Ureidobenzamide derivative |
| US4514416A (en) | 1981-02-27 | 1985-04-30 | Torii & Co. Ltd. | Amidine compound, process for producing same and anti-complement agent comprising same |
| US4588587A (en) | 1983-03-01 | 1986-05-13 | Pennsylvania Hospital | Method of treatment to inhibit metastasis |
| GB2220206A (en) | 1988-06-29 | 1990-01-04 | Otsuka Pharma Co Ltd | Dialkoxyphosphinoylmethyl-benzamide compounds |
| US4912001A (en) | 1987-12-29 | 1990-03-27 | Dainichi Seika Color & Chemicals Mfg. Co., Ltd. | Electrophotographic photoreceptor comprising azo compound and coupler |
| JPH0418675A (en) | 1990-05-11 | 1992-01-22 | Ricoh Co Ltd | Circuit diagram input device |
| EP0540051A1 (en) | 1991-10-31 | 1993-05-05 | Daiichi Pharmaceutical Co., Ltd. | Aromatic amidine derivatives and salts thereof |
| WO1994013693A1 (en) | 1992-12-15 | 1994-06-23 | Corvas International, Inc. | NOVEL INHIBITORS OF FACTOR Xa |
| WO1996028427A1 (en) | 1995-03-10 | 1996-09-19 | Berlex Laboratories, Inc. | Benzamidine derivatives their preparation and their use as anti-coagulants |
| US5569768A (en) | 1992-06-03 | 1996-10-29 | Eli Lilly And Company | Angiotensin II antagonists |
| WO1997021437A1 (en) | 1995-12-08 | 1997-06-19 | Berlex Laboratories, Inc. | Naphthyl-substituted benzimidazole derivatives as anticoagulants |
| WO1997029067A1 (en) | 1996-02-12 | 1997-08-14 | Berlex Laboratories, Inc. | Benzamidine derivatives substituted by amino acid and hydroxy acid derivatives and their use as anti-coagulants |
| EP0798295A1 (en) | 1994-12-02 | 1997-10-01 | Yamanouchi Pharmaceutical Co. Ltd. | Novel amidinonaphthyl derivative or salt thereof |
| WO1998006694A1 (en) | 1996-08-16 | 1998-02-19 | Du Pont Pharmaceuticals Company | Amidinophenyl-pyrrolidines, -pyrrolines, and -isoxazolidines and derivatives thereof |
| WO1998009630A1 (en) | 1996-09-09 | 1998-03-12 | Smithkline Beecham Corporation | Compounds and methods |
| WO1998028282A2 (en) | 1996-12-23 | 1998-07-02 | Du Pont Pharmaceuticals Company | OXYGEN OR SULFUR CONTAINING 5-MEMBERED HETEROAROMATICS AS FACTOR Xa INHIBITORS |
| WO1998028269A1 (en) | 1996-12-23 | 1998-07-02 | Du Pont Pharmaceuticals Company | NITROGEN CONTAINING HETEROAROMATICS AS FACTOR Xa INHIBITORS |
| WO1998057934A1 (en) | 1997-06-19 | 1998-12-23 | The Du Pont Merck Pharmaceutical Company | (AMIDINO)6-MEMBERED AROMATICS AS FACTOR Xa INHIBITORS |
| WO1999000127A1 (en) | 1997-06-26 | 1999-01-07 | Eli Lilly And Company | Antithrombotic agents |
| WO1999000128A1 (en) | 1997-06-26 | 1999-01-07 | Eli Lilly And Company | Antithrombotic agents |
| WO1999000126A1 (en) | 1997-06-26 | 1999-01-07 | Eli Lilly And Company | Antithrombotic agents |
| WO1999000121A1 (en) | 1997-06-26 | 1999-01-07 | Eli Lilly And Company | Antithrombotic agents |
| US5872115A (en) | 1995-05-05 | 1999-02-16 | Grelan Pharmaceutical Co. Ltd. | 2-ureido-benzamide derivatives |
| WO1999007379A1 (en) * | 1997-08-11 | 1999-02-18 | Boehringer Ingelheim Pharmaceuticals, Inc. | 5,6-HETEROARYL-DIPYRIDO[2,3-b:3',2'-f]AZEPINES AND THEIR USE IN THE PREVENTION OR TREATMENT OF HIV INFECTION |
| WO1999010316A1 (en) | 1997-08-27 | 1999-03-04 | Kissei Pharmaceutical Co., Ltd. | 3-amidinoaniline derivatives, activated blood coagulation factor x inhibitors, and intermediates for producing both |
| WO1999032477A1 (en) | 1997-12-19 | 1999-07-01 | Schering Aktiengesellschaft | Ortho-anthranilamide derivatives as anti-coagulants |
| EP0937711A1 (en) | 1998-02-18 | 1999-08-25 | Roche Diagnostics GmbH | Thiobenzamides, process for their preparation and medicaments containing them |
| JPH11302177A (en) | 1998-04-27 | 1999-11-02 | Otsuka Pharmaceut Factory Inc | Medicine for treating nephritis |
| WO2000034238A1 (en) | 1998-12-09 | 2000-06-15 | American Home Products Corporation | Thiourea inhibitors of herpes viruses |
| WO2000034237A2 (en) | 1998-12-09 | 2000-06-15 | American Home Products Corporation | Acetamide and substituted acetamide-containing thiourea inhibitors of herpes viruses |
| WO2000034261A2 (en) | 1998-12-09 | 2000-06-15 | American Home Products Corporation | Heterocyclic carboxamide-containing thiourea inhibitors of herpes viruses containing a substituted phenylenediamine group |
| WO2000034258A2 (en) | 1998-12-09 | 2000-06-15 | American Home Products Corporation | Heterocyclic carboxamide-containing thiourea inhibitors of herpes viruses containing phenylenediamine group |
| WO2000034269A1 (en) | 1998-12-09 | 2000-06-15 | American Home Products Corporation | Thiourea inhibitors of herpes viruses |
| WO2000034268A1 (en) | 1998-12-09 | 2000-06-15 | American Home Products Corporation | Thiourea inhibitors of herpes viruses |
| WO2000034260A2 (en) | 1998-12-09 | 2000-06-15 | American Home Products Corporation | Alpha-methylbenzyl-containing thiourea inhibitors of herpes viruses containing a phenylenediamine group |
| WO2000039118A1 (en) | 1998-12-23 | 2000-07-06 | Eli Lilly And Company | Aromatic amides |
| US6140351A (en) | 1997-12-19 | 2000-10-31 | Berlex Laboratories, Inc. | Ortho-anthranilamide derivatives as anti-coagulants |
| US6376515B2 (en) | 2000-02-29 | 2002-04-23 | Cor Therapeutics, Inc. | Benzamides and related inhibitors of factor Xa |
| US20030069250A1 (en) | 2001-03-30 | 2003-04-10 | Bing-Yan Zhu | Benzamide inhibitors of factor Xa |
| US6632815B2 (en) | 1999-09-17 | 2003-10-14 | Millennium Pharmaceuticals, Inc. | Inhibitors of factor Xa |
| US6686368B1 (en) | 1999-09-17 | 2004-02-03 | Millennium Pahrmaceuticals, Inc. | Inhibitors of factor Xa |
| US6844367B1 (en) | 1999-09-17 | 2005-01-18 | Millennium Pharmaceuticals, Inc. | Benzamides and related inhibitors of factor Xa |
| US7022695B2 (en) | 2003-10-09 | 2006-04-04 | Millennium Pharmaceuticals, Inc. | Thioether-substituted benzamides as inhibitors of Factor Xa |
| US20060100193A1 (en) | 2004-06-18 | 2006-05-11 | Millennium Pharmaceuticals, Inc. | Factor Xa inhibitors |
| US20070112039A1 (en) | 2005-11-08 | 2007-05-17 | Millennium Pharmaceuticals, Inc. | Novel pharmaceutical salts and polymorphs of a factor Xa inhibitor |
| US20070185092A1 (en) | 2004-06-18 | 2007-08-09 | Millennium Pharmaceuticals, Inc. | FACTOR Xa INHIBITORS |
| US20070259924A1 (en) | 2006-05-05 | 2007-11-08 | Millennium Pharmaceuticals, Inc. | Factor xa inhibitors |
| US20080153876A1 (en) | 2006-12-08 | 2008-06-26 | Millennium Pharmaceuticals, Inc. | Unit dose formulations and methods of treating thrombosis with an oral factor Xa inhibitor |
| US20080254036A1 (en) | 2007-04-13 | 2008-10-16 | Millennium Pharmaceuticals, Inc. | Combination anticoagulant therapy with a compound that acts as a factor xa inhibitor |
| US20080279845A1 (en) | 2007-05-02 | 2008-11-13 | Portola Pharmaceuticals, Inc. | Combination therapy with a compound acting as a platelet adp receptor inhibitor |
| US20080293704A1 (en) | 2007-01-05 | 2008-11-27 | Millennium Pharmaceuticals, Inc. | FACTOR Xa INHIBITORS |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1047245A (en) * | 1963-02-15 | |||
| JPS6033389B2 (en) * | 1979-02-22 | 1985-08-02 | 日産化学工業株式会社 | Heterocyclic ether phenoxy fatty acid derivative, its production method, and herbicide containing the derivative |
| JPS648675A (en) | 1987-06-30 | 1989-01-12 | Mitsubishi Electric Corp | Formation of schottky gate |
| GB9816837D0 (en) * | 1998-08-04 | 1998-09-30 | Zeneca Ltd | Amide derivatives |
| US5610335A (en) | 1993-05-26 | 1997-03-11 | Cornell Research Foundation | Microelectromechanical lateral accelerometer |
| MX226123B (en) | 1999-09-17 | 2005-02-07 | Millennium Pharm Inc | BENZAMIDES AND RELATED INHIBITORS OF FACTOR Xa. |
| AU776053B2 (en) * | 2000-03-31 | 2004-08-26 | Astellas Pharma Inc. | Diazepan derivatives or salts thereof |
| US6373515B1 (en) * | 2000-08-09 | 2002-04-16 | Hewlett-Packard Company | Variable resolution transition placement device |
| ES2236548T3 (en) * | 2001-07-31 | 2005-07-16 | Wayne State University | DERIVATIVES OF QUINOLINA AND ITS USE AS ANTITUMOR AGENTS. |
| US20090186810A1 (en) * | 2006-03-27 | 2009-07-23 | Portola Pharmaceuticals, Inc. | Potassium channel modulators and platelet procoagulant activity |
-
2001
- 2001-02-28 DE DE60115394T patent/DE60115394T2/en not_active Expired - Lifetime
- 2001-02-28 EP EP01918257A patent/EP1259485B1/en not_active Expired - Lifetime
- 2001-02-28 CA CA2401778A patent/CA2401778C/en not_active Expired - Lifetime
- 2001-02-28 AU AU2001250783A patent/AU2001250783A1/en not_active Abandoned
- 2001-02-28 US US09/794,225 patent/US6376515B2/en not_active Expired - Lifetime
- 2001-02-28 ES ES01918257T patent/ES2254385T3/en not_active Expired - Lifetime
- 2001-02-28 WO PCT/US2001/006247 patent/WO2001064642A2/en active Search and Examination
- 2001-02-28 DK DK01918257T patent/DK1259485T3/en active
- 2001-02-28 AU AU2001245353A patent/AU2001245353A1/en not_active Abandoned
- 2001-02-28 AT AT01918257T patent/ATE311366T1/en active
- 2001-02-28 WO PCT/US2001/006255 patent/WO2001064643A2/en active IP Right Grant
-
2002
- 2002-04-22 US US10/126,976 patent/US20030162690A1/en not_active Abandoned
-
2003
- 2003-05-27 HK HK03103788A patent/HK1051539A1/en not_active IP Right Cessation
- 2003-10-15 US US10/687,334 patent/US6835739B2/en not_active Expired - Lifetime
-
2004
- 2004-09-15 US US10/942,733 patent/US7314874B2/en not_active Expired - Fee Related
-
2006
- 2006-05-26 US US11/442,060 patent/US7342013B2/en not_active Expired - Lifetime
- 2006-07-17 US US11/488,455 patent/US7727982B2/en not_active Expired - Fee Related
-
2007
- 2007-10-25 US US11/924,480 patent/US7727981B2/en not_active Expired - Fee Related
-
2010
- 2010-04-07 US US12/756,037 patent/US8063036B2/en not_active Expired - Fee Related
-
2011
- 2011-09-28 US US13/247,937 patent/US8518977B2/en not_active Expired - Fee Related
-
2012
- 2012-09-12 US US13/612,597 patent/US8691847B2/en not_active Expired - Fee Related
-
2014
- 2014-02-07 US US14/175,087 patent/US9108922B2/en not_active Expired - Fee Related
-
2015
- 2015-06-17 US US14/742,465 patent/US9629831B2/en not_active Expired - Fee Related
-
2017
- 2017-03-22 US US15/466,530 patent/US10179124B2/en not_active Expired - Fee Related
Patent Citations (63)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2095619A (en) | 1937-10-12 | Aryl oxides | ||
| CH520657A (en) | 1961-09-11 | 1972-03-31 | Wander Ag Dr A | Bis-anilides with basic substituents - having chemotherapeutic activity |
| US4514416A (en) | 1981-02-27 | 1985-04-30 | Torii & Co. Ltd. | Amidine compound, process for producing same and anti-complement agent comprising same |
| EP0077534A2 (en) | 1981-10-15 | 1983-04-27 | Chugai Seiyaku Kabushiki Kaisha | Benzamide derivatives, a process for preparing the same, and pharmaceutical compositions containing the same |
| US4588587A (en) | 1983-03-01 | 1986-05-13 | Pennsylvania Hospital | Method of treatment to inhibit metastasis |
| JPS59181257A (en) | 1983-03-31 | 1984-10-15 | Chugai Pharmaceut Co Ltd | Ureidobenzamide derivative |
| US4912001A (en) | 1987-12-29 | 1990-03-27 | Dainichi Seika Color & Chemicals Mfg. Co., Ltd. | Electrophotographic photoreceptor comprising azo compound and coupler |
| GB2220206A (en) | 1988-06-29 | 1990-01-04 | Otsuka Pharma Co Ltd | Dialkoxyphosphinoylmethyl-benzamide compounds |
| US4971957A (en) | 1988-06-29 | 1990-11-20 | Otsuka Pharmaceutical Factory, Inc. | Carboxamide compounds, processes for preparing the same and a pharmaceutical composition containing the same |
| JPH0418675A (en) | 1990-05-11 | 1992-01-22 | Ricoh Co Ltd | Circuit diagram input device |
| US5576343A (en) | 1991-10-31 | 1996-11-19 | Daiichi Pharmaceutical Co., Ltd. | Aromatic amidine derivatives and salts thereof |
| EP0540051A1 (en) | 1991-10-31 | 1993-05-05 | Daiichi Pharmaceutical Co., Ltd. | Aromatic amidine derivatives and salts thereof |
| US5569768A (en) | 1992-06-03 | 1996-10-29 | Eli Lilly And Company | Angiotensin II antagonists |
| WO1994013693A1 (en) | 1992-12-15 | 1994-06-23 | Corvas International, Inc. | NOVEL INHIBITORS OF FACTOR Xa |
| EP0798295A1 (en) | 1994-12-02 | 1997-10-01 | Yamanouchi Pharmaceutical Co. Ltd. | Novel amidinonaphthyl derivative or salt thereof |
| WO1996028427A1 (en) | 1995-03-10 | 1996-09-19 | Berlex Laboratories, Inc. | Benzamidine derivatives their preparation and their use as anti-coagulants |
| US5872115A (en) | 1995-05-05 | 1999-02-16 | Grelan Pharmaceutical Co. Ltd. | 2-ureido-benzamide derivatives |
| WO1997021437A1 (en) | 1995-12-08 | 1997-06-19 | Berlex Laboratories, Inc. | Naphthyl-substituted benzimidazole derivatives as anticoagulants |
| WO1997029067A1 (en) | 1996-02-12 | 1997-08-14 | Berlex Laboratories, Inc. | Benzamidine derivatives substituted by amino acid and hydroxy acid derivatives and their use as anti-coagulants |
| WO1998006694A1 (en) | 1996-08-16 | 1998-02-19 | Du Pont Pharmaceuticals Company | Amidinophenyl-pyrrolidines, -pyrrolines, and -isoxazolidines and derivatives thereof |
| WO1998009630A1 (en) | 1996-09-09 | 1998-03-12 | Smithkline Beecham Corporation | Compounds and methods |
| WO1998028269A1 (en) | 1996-12-23 | 1998-07-02 | Du Pont Pharmaceuticals Company | NITROGEN CONTAINING HETEROAROMATICS AS FACTOR Xa INHIBITORS |
| WO1998028282A2 (en) | 1996-12-23 | 1998-07-02 | Du Pont Pharmaceuticals Company | OXYGEN OR SULFUR CONTAINING 5-MEMBERED HETEROAROMATICS AS FACTOR Xa INHIBITORS |
| WO1998057934A1 (en) | 1997-06-19 | 1998-12-23 | The Du Pont Merck Pharmaceutical Company | (AMIDINO)6-MEMBERED AROMATICS AS FACTOR Xa INHIBITORS |
| WO1999000127A1 (en) | 1997-06-26 | 1999-01-07 | Eli Lilly And Company | Antithrombotic agents |
| WO1999000128A1 (en) | 1997-06-26 | 1999-01-07 | Eli Lilly And Company | Antithrombotic agents |
| WO1999000126A1 (en) | 1997-06-26 | 1999-01-07 | Eli Lilly And Company | Antithrombotic agents |
| WO1999000121A1 (en) | 1997-06-26 | 1999-01-07 | Eli Lilly And Company | Antithrombotic agents |
| WO1999007379A1 (en) * | 1997-08-11 | 1999-02-18 | Boehringer Ingelheim Pharmaceuticals, Inc. | 5,6-HETEROARYL-DIPYRIDO[2,3-b:3',2'-f]AZEPINES AND THEIR USE IN THE PREVENTION OR TREATMENT OF HIV INFECTION |
| WO1999010316A1 (en) | 1997-08-27 | 1999-03-04 | Kissei Pharmaceutical Co., Ltd. | 3-amidinoaniline derivatives, activated blood coagulation factor x inhibitors, and intermediates for producing both |
| WO1999032477A1 (en) | 1997-12-19 | 1999-07-01 | Schering Aktiengesellschaft | Ortho-anthranilamide derivatives as anti-coagulants |
| US6140351A (en) | 1997-12-19 | 2000-10-31 | Berlex Laboratories, Inc. | Ortho-anthranilamide derivatives as anti-coagulants |
| EP0937711A1 (en) | 1998-02-18 | 1999-08-25 | Roche Diagnostics GmbH | Thiobenzamides, process for their preparation and medicaments containing them |
| WO1999042439A1 (en) | 1998-02-18 | 1999-08-26 | Roche Diagnostics Gmbh | Novel thiobenzamides |
| JPH11302177A (en) | 1998-04-27 | 1999-11-02 | Otsuka Pharmaceut Factory Inc | Medicine for treating nephritis |
| WO2000034238A1 (en) | 1998-12-09 | 2000-06-15 | American Home Products Corporation | Thiourea inhibitors of herpes viruses |
| WO2000034237A2 (en) | 1998-12-09 | 2000-06-15 | American Home Products Corporation | Acetamide and substituted acetamide-containing thiourea inhibitors of herpes viruses |
| WO2000034261A2 (en) | 1998-12-09 | 2000-06-15 | American Home Products Corporation | Heterocyclic carboxamide-containing thiourea inhibitors of herpes viruses containing a substituted phenylenediamine group |
| WO2000034258A2 (en) | 1998-12-09 | 2000-06-15 | American Home Products Corporation | Heterocyclic carboxamide-containing thiourea inhibitors of herpes viruses containing phenylenediamine group |
| WO2000034269A1 (en) | 1998-12-09 | 2000-06-15 | American Home Products Corporation | Thiourea inhibitors of herpes viruses |
| WO2000034268A1 (en) | 1998-12-09 | 2000-06-15 | American Home Products Corporation | Thiourea inhibitors of herpes viruses |
| WO2000034260A2 (en) | 1998-12-09 | 2000-06-15 | American Home Products Corporation | Alpha-methylbenzyl-containing thiourea inhibitors of herpes viruses containing a phenylenediamine group |
| WO2000039118A1 (en) | 1998-12-23 | 2000-07-06 | Eli Lilly And Company | Aromatic amides |
| US6720317B1 (en) | 1999-09-17 | 2004-04-13 | Millennium Pharmaceuticals, Inc. | Inhibitors of factor Xa |
| US7285565B2 (en) | 1999-09-17 | 2007-10-23 | Millennium Pharmaceuticals, Inc. | Benzamides and related inhibitors of factor Xa |
| US6632815B2 (en) | 1999-09-17 | 2003-10-14 | Millennium Pharmaceuticals, Inc. | Inhibitors of factor Xa |
| US6686368B1 (en) | 1999-09-17 | 2004-02-03 | Millennium Pahrmaceuticals, Inc. | Inhibitors of factor Xa |
| US6844367B1 (en) | 1999-09-17 | 2005-01-18 | Millennium Pharmaceuticals, Inc. | Benzamides and related inhibitors of factor Xa |
| US6376515B2 (en) | 2000-02-29 | 2002-04-23 | Cor Therapeutics, Inc. | Benzamides and related inhibitors of factor Xa |
| US6835739B2 (en) | 2000-02-29 | 2004-12-28 | Millennium Pharmaceuticals, Inc. | Benzamides and related inhibitors of factor Xa |
| US7342013B2 (en) | 2000-02-29 | 2008-03-11 | Millennium Pharmaceuticals, Inc. | Benzamides and related inhibitors of factor Xa |
| US7314874B2 (en) | 2000-02-29 | 2008-01-01 | Millennium Pharmaceuticals, Inc. | Benzamides and related inhibitors of factor Xa |
| US7312235B2 (en) | 2001-03-30 | 2007-12-25 | Millennium Pharmaceuticals, Inc. | Benzamide inhibitors of factor Xa |
| US20030069250A1 (en) | 2001-03-30 | 2003-04-10 | Bing-Yan Zhu | Benzamide inhibitors of factor Xa |
| US7022695B2 (en) | 2003-10-09 | 2006-04-04 | Millennium Pharmaceuticals, Inc. | Thioether-substituted benzamides as inhibitors of Factor Xa |
| US20070185092A1 (en) | 2004-06-18 | 2007-08-09 | Millennium Pharmaceuticals, Inc. | FACTOR Xa INHIBITORS |
| US20060100193A1 (en) | 2004-06-18 | 2006-05-11 | Millennium Pharmaceuticals, Inc. | Factor Xa inhibitors |
| US20070112039A1 (en) | 2005-11-08 | 2007-05-17 | Millennium Pharmaceuticals, Inc. | Novel pharmaceutical salts and polymorphs of a factor Xa inhibitor |
| US20070259924A1 (en) | 2006-05-05 | 2007-11-08 | Millennium Pharmaceuticals, Inc. | Factor xa inhibitors |
| US20080153876A1 (en) | 2006-12-08 | 2008-06-26 | Millennium Pharmaceuticals, Inc. | Unit dose formulations and methods of treating thrombosis with an oral factor Xa inhibitor |
| US20080293704A1 (en) | 2007-01-05 | 2008-11-27 | Millennium Pharmaceuticals, Inc. | FACTOR Xa INHIBITORS |
| US20080254036A1 (en) | 2007-04-13 | 2008-10-16 | Millennium Pharmaceuticals, Inc. | Combination anticoagulant therapy with a compound that acts as a factor xa inhibitor |
| US20080279845A1 (en) | 2007-05-02 | 2008-11-13 | Portola Pharmaceuticals, Inc. | Combination therapy with a compound acting as a platelet adp receptor inhibitor |
Non-Patent Citations (46)
| Title |
|---|
| "Dictionary of Organic Compounds, 5th Ed., vol. 5" Chapman and Hall, New York, NY, US, compounds T-00160, T-00161, T-00162, p. 5119, 1982. |
| Berge, S.M., et al. "Pharmaceutical Salts" J. Pharmac. Sci. 66:1-19, 1997. |
| Bing-Yan Zhu, et al., Annual Reports in Medicinal Chemistry, 35, pp. 83-102, 2000. |
| Cheng, Y., et al., "Relationship between the Inhibition Constant (KI) and the Concentration of Inhibitor which Causes 50 Percent Inhibition (I50) of an Enzymatic Reaction" Biochem. Pharmacol. 22(23):3099-3108, 1973. |
| Claeson, G. "Synthetic peptides and peptidomimetics as substrates and inhibitors of thrombin and other proteases in the blood coagulation system", Blood Coag. FIBRINOL. 5:411-436, 1994. |
| David K. Herron, et al., "1-2-Dibenzamidobenze Inhibitors of Human Factor XA", J. Med. Chem., vol. 43, pp. 859-872, 2000. |
| El-Nagy, S., Asian J. Chem. 2(4):368-78, 1990. |
| Elodi, et al. "Optimization of Conditions for the Catalytic Effect of the Factor IXa-factor VIII complex: probable role of the complex in the amplification of blood coagulation" Thromb. Res. 15:617-629, 1979. |
| El-Zanfally, S. Egypt. J. Pharm. Sci. vol. 17(1), 29-34, 1976. |
| Finlayson, K. et al., "[3H]dofetilide binding to HERG transfected membranes: A Potential High Throughout Preclinical Screen" European J. Pharmacol. 430:147, 2001. |
| Gresele et al. "Novel approaches to the treatment of thrombosis." Trends in Pharmacological Sciences, 23(1), 25-32, Jan. 2002. |
| Hamilton, A., "Intra-and Intermolecular Hydrogen Bonding Control of Supermolecular Structure" NATO ASI Ser. C. (Computation Approaches in Supramolecular Chemistry), vol. 426, pp, 101-108, 1994. |
| Hauptmann, J. et al., "Comparison of the Anticoagulant and Antithrombolic Effects of Synthetic Thrombin and Factor Xa Inhibitors", Thromb. Haemost. 63:220-223, 1990. |
| Haverkamp, W. et al. "The potential for QT prolongation and pro-arrhythmia by non-anti-arrhythmic drugs: Clinical and regulatory implications. Report on a Policy Conference of the European Society of Cardiology" Cardiovasc. Res. 47:219-33, 2000. |
| Herbert J. Sipe, Jr., et al., "An Improved Synthesis of Aryl Sulfones", Synthesis, No. 3, pp. 283-284 1984. |
| Hey, D. J. Chem. Soc. 16, 1513-18, 1967. |
| Hitomi Suzuki, et al., "Selective Reduction with Lithium Aluminum Hydride/Diphosphorus Tetraiodide. A Mild Conversion of Aromatic Ketones to Parent Hydrocarbons", Chemistry Letters, pp. 909-910, 1983. |
| Hitomi, Y., et al. "Inhibitory Effect of New Synthetic Protease Inhibitor (FUT-175) on the Coagulation System" Heamostasis 15:164-168 (1985). |
| John D. Young, et al., "Interannular Interactions in Para-Substituted Diphenylmethane Anion Radicals", J. Am. Chem. Soc., 94:25, pp. 8790-8794, Dec. 1972. |
| Kam, C. et al., "Mechanism Based Isocoumarin Inhibitors for Trypsin and Blood Coagulation Serine Protease: New Anticoagulants" Biochemistry 27:2547-2557 (1988). |
| Kulkarni, J. Indian Chem. Soc. 61(8):720-721, 1984. |
| Kuzmic, P. et al., "High-throughput Screening of Enzyme Inhibitors: Automatic Determination of Tight-Binding Inhibition Constants" Anal. Biochem 281:62-67 (2000). |
| Kuzmic, P. et al., "High-throughput Screening of Enzyme Inhibitors: Automatic Determination of Tight-Binding Inhibition Constants" Anal. Biochem 281:62-67 (2000). |
| Michael R. Wiley, et al., Structure-Based Design of Potent, Amidine-Derived Inhibitors of Factor Xa: Evaluation of Selectively, Anticoagulant Activity, and Antithrombotic Activity, J. Med. Chem., vol. 43, pp. 883-899, 2000. |
| Netzer, R. et al., "Screening Lead Compounds for QT Interval Prolongation" Drug Discovery Today, 6:78-84 (2001). |
| Nutt, E. et al., "The Amino Acid Sequence of Antistasin, a Potent Inhibitor of Factor XA Reveals a Repeated Internal Structure" J. Biol. Chem. 263:10162-10167 (1988). |
| Pearlstein, R. et al., "Characterization of HERG Potassium Channel Inhibition Using CoMSiA 3D QSAR and Homology Modeling Approaches" Bioorg. Med. Chem. Lett. 13:1829-35 (2003). |
| R. Kahn, et al., "Addition von Maleinsaure-anhydrid von Polene", Berichte Der Deutschen Chemischen Gelsellschaft, vol. 63, 1930, pp. 2662-2679. |
| Redfern, W. et al., "Relationships between preclinical Cardiac Electrophysiology, Clinical QT Interval Prolongation and Torsade de Pointes for a Broad Range of Drugs: Evidence for a Provisional Safety Margin in Drug Development" Cardiovasc. Res. 58:32-45 (2003). |
| Richard Kuhn et al, "Addition von Maleinsaure-anhydrid an Polyene (Uber konjugierte Doppelbindugen, XIV" Berichte Der Deutschen Chemischen Gesellschaft, vol. 63, pp. 2662-2679, 1930. |
| St. Goldschmidt and P. Modderman, "Biphenyl Derivatives II Basic 4-Biphenyl Compounds", Recueil, 69, pp. 1109-1117, 1950. |
| Sturzebecher, J. et al., "Synthetic Inhibitors of Bovine Factor Xa and Thrombin. Comparison of Their Anticoagulant Efficiency", Thromb. Res. 54:245-252 (1989). |
| Takashi Keumi et al., "2-(Trifluoromethylsulfonyloxy)pyridine as a Reagent for the Ketone Synthesis from Carboxylic Acids and Aromatic Hydrocarbons", Bull. Chem. Soc. Japan., vol. 61, pp. 455-459, 1988. |
| Tan, et al., "Factor X inhibitors." Expert Opin. Investig. Drugs, 12(5), 799-804, 2003. |
| Tidwell, R. et al., "Strategies for Anticoagulation with Synthetic Protease Inhibitors. Xa Inhibitors Versus Thrombin Inhibitors" Thromb. Res. 19:339-349 (1980). |
| Turner, A. et al., "p-Amidino Esters as Irreversible Inhibitors of Factor IXa and Xa and Thrombin" Biochemistry 25:4929-4935 (1986). |
| Turple, Am. J. Health Syst. Pharm., 60.(22 Suppl. 7), S20-4, Nov. 15, 2003. |
| U.S. Appl. No. 11/924,480, filed Oct. 25, 2007, Bing-Yan et al. |
| U.S. Appl. No. 12/166,944, filed Jul. 2, 2008, Yonghong Song et al. |
| U.S. Appl. No. 12/280,531, filed Mar. 26, 2007, Bevers et al. |
| W. E. Bachmann, et al., Reduction by Magnesium +Magnesium Halide, XIII. The reaction Between Epoxy Ketones and Grignard Reagents, Journal of the American Chemical Society, vol. 56, No. 7, pp. 1559-1560, 1934. |
| W.F. Cockburn, et al., "Molecular Rearrangement of Tertiary Amines. Part I," J. Chem. Soc. No. 8, pp. 3340-3346 1960. |
| Waxman, L. et al., "Tick Anticoagulant Peptide (TAP) is a novel Inhibitor of Blood Coagulation Factor Xa" Science 248:593-596 (1990). |
| West, Anthony R., Solid State Chemistry and its Applications, Wiley, New York, 1988, pp. 358 & 365. * |
| Ying K. lee, et al., "N2-Aroylanthranilamide Inhibitors of Human Factor Xa", J. Med. Chem., vol. 43, pp. 873-882, 2000. |
| Zhou, Z. et al. "Properties of HERG Channels Stably Expressed in HEK293 Cells Studied at Physiological Temperature" Biophys. J. 74:230-241 (1998). |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9629831B2 (en) | 2000-02-29 | 2017-04-25 | Millennium Pharmaceuticals, Inc. | Benzamides and related inhibitors of factor XA |
| US9108922B2 (en) | 2000-02-29 | 2015-08-18 | Millennium Pharmaceuticals, Inc. | Benzamides and related inhibitors of factor Xa |
| US8691847B2 (en) | 2000-02-29 | 2014-04-08 | Millennium Pharmaceuticals, Inc. | Benzamides and related inhibitors of factor Xa |
| US8518977B2 (en) | 2000-02-29 | 2013-08-27 | Millennium Pharmaceuticals, Inc. | Benzamides and related inhibitors of factor XA |
| US10179124B2 (en) | 2000-02-29 | 2019-01-15 | Millennium Pharmaceuticals, Inc. | Benzamides and related inhibitors of factor Xa |
| US8377974B2 (en) | 2004-06-18 | 2013-02-19 | Millennium Pharmaceuticals, Inc. | Factor Xa inhibitors |
| US20100234352A1 (en) * | 2004-06-18 | 2010-09-16 | Millennium Pharmaceuticals, Inc. | Factor xa inhibitors |
| US8063077B2 (en) | 2006-05-05 | 2011-11-22 | Millennium Pharmaceuticals, Inc. | Factor Xa inhibitors |
| US8349873B2 (en) | 2006-05-05 | 2013-01-08 | Millennium Pharmaceuticals, Inc. | Factor XA inhibitors |
| US20110118244A2 (en) * | 2006-05-05 | 2011-05-19 | Millennium Pharmaceuticals, Inc. | Factor xa inhibitors |
| US20100249117A1 (en) * | 2006-05-05 | 2010-09-30 | Millennium Pharmaceuticals, Inc. | Factor xa inhibitors |
| US8524907B2 (en) | 2006-11-02 | 2013-09-03 | Millennium Pharmaceuticals, Inc. | Methods of synthesizing pharmaceutical salts of a factor Xa inhibitor |
| US9221758B2 (en) | 2006-11-02 | 2015-12-29 | Millennium Pharmaceuticals, Inc. | Methods of synthesizing pharmaceutical salts of a factor Xa inhibitor |
| US8455440B2 (en) | 2007-04-13 | 2013-06-04 | Millennium Pharmaceuticals, Inc. | Combination anticoagulant therapy with a compound that acts as a factor Xa inhibitor |
| US8946219B2 (en) | 2007-05-02 | 2015-02-03 | Portola Pharmaceuticals, Inc. | Combination therapy with a compound acting as a platelet ADP receptor inhibitor |
| US20110033459A1 (en) * | 2007-05-02 | 2011-02-10 | Portola Pharmaceuticals, Inc. | Combination therapy with a compound acting as a platelet adp receptor inhibitor |
| US20110178135A1 (en) * | 2009-12-17 | 2011-07-21 | Millennium Pharmaceuticals, Inc. | Salts and crystalline forms of a factor xa inhibitor |
| US8987463B2 (en) | 2009-12-17 | 2015-03-24 | Millennium Pharmaceuticals, Inc. | Methods of synthesizing factor Xa inhibitors |
| US8742120B2 (en) | 2009-12-17 | 2014-06-03 | Millennium Pharmaceuticals, Inc. | Methods of preparing factor xa inhibitors and salts thereof |
| US8530501B2 (en) | 2009-12-17 | 2013-09-10 | Millennium Pharmaceuticals, Inc. | Salts and crystalline forms of a factor Xa inhibitor |
| US8394964B2 (en) | 2009-12-17 | 2013-03-12 | Millennium Pharmaceuticals, Inc. | Methods of synthesizing factor Xa inhibitors |
| US20110152530A1 (en) * | 2009-12-17 | 2011-06-23 | Millennium Pharmaceuticals, Inc. | Methods of preparing factor xa inhibitors and salts thereof |
| US9200268B2 (en) | 2012-12-27 | 2015-12-01 | Portola Pharmaceuticals, Inc. | Compounds and methods for purification of serine proteases |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10179124B2 (en) | Benzamides and related inhibitors of factor Xa | |
| US7285565B2 (en) | Benzamides and related inhibitors of factor Xa | |
| KR100760434B1 (en) | Benzamides And Related Inhibitors Of Factor Xa | |
| US6632815B2 (en) | Inhibitors of factor Xa | |
| US7312235B2 (en) | Benzamide inhibitors of factor Xa | |
| US6720317B1 (en) | Inhibitors of factor Xa | |
| US20040077690A1 (en) | Quaternary amidino based inhibitors of factor xa |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| FEPP | Fee payment procedure |
Free format text: PAYOR NUMBER ASSIGNED (ORIGINAL EVENT CODE: ASPN); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY |
|
| STCF | Information on status: patent grant |
Free format text: PATENTED CASE |
|
| CC | Certificate of correction | ||
| AS | Assignment |
Owner name: COR THERAPEUTICS, INC., CALIFORNIA Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:ZHU, BING-YAN;ZHANG, PENGLIE;WANG, LINGYAN;AND OTHERS;SIGNING DATES FROM 20010302 TO 20010414;REEL/FRAME:029502/0737 Owner name: MILLENNIUM PHARMACEUTICALS, INC., MASSACHUSETTS Free format text: MERGER;ASSIGNOR:COR THERAPEUTICS, INC.;REEL/FRAME:029504/0084 Effective date: 20020212 |
|
| FPAY | Fee payment |
Year of fee payment: 4 |
|
| MAFP | Maintenance fee payment |
Free format text: PAYMENT OF MAINTENANCE FEE, 8TH YEAR, LARGE ENTITY (ORIGINAL EVENT CODE: M1552) Year of fee payment: 8 |
|
| FEPP | Fee payment procedure |
Free format text: MAINTENANCE FEE REMINDER MAILED (ORIGINAL EVENT CODE: REM.); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY |
|
| LAPS | Lapse for failure to pay maintenance fees |
Free format text: PATENT EXPIRED FOR FAILURE TO PAY MAINTENANCE FEES (ORIGINAL EVENT CODE: EXP.); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY |
|
| STCH | Information on status: patent discontinuation |
Free format text: PATENT EXPIRED DUE TO NONPAYMENT OF MAINTENANCE FEES UNDER 37 CFR 1.362 |
|
| FP | Lapsed due to failure to pay maintenance fee |
Effective date: 20220601 |