US6756497B1 - Benzoic acid derivatives and processes for the preparation thereof - Google Patents
Benzoic acid derivatives and processes for the preparation thereof Download PDFInfo
- Publication number
- US6756497B1 US6756497B1 US09/720,825 US72082501A US6756497B1 US 6756497 B1 US6756497 B1 US 6756497B1 US 72082501 A US72082501 A US 72082501A US 6756497 B1 US6756497 B1 US 6756497B1
- Authority
- US
- United States
- Prior art keywords
- group
- formula
- preparation
- methyl
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 238000002360 preparation method Methods 0.000 title claims abstract description 43
- 238000000034 method Methods 0.000 title claims abstract description 32
- 150000001558 benzoic acid derivatives Chemical class 0.000 title claims abstract description 11
- 150000001875 compounds Chemical class 0.000 claims abstract description 68
- -1 alkali metal alkoxide Chemical class 0.000 claims description 128
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 51
- 239000002904 solvent Substances 0.000 claims description 50
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 45
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 43
- 239000002585 base Substances 0.000 claims description 19
- IYFXXIUALYIKQR-UHFFFAOYSA-N 3-(cyclohepten-1-yl)cyclohept-2-en-1-one Chemical class O=C1CCCCC(C=2CCCCCC=2)=C1 IYFXXIUALYIKQR-UHFFFAOYSA-N 0.000 claims description 18
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 16
- 229920006395 saturated elastomer Polymers 0.000 claims description 15
- 229910052739 hydrogen Inorganic materials 0.000 claims description 13
- 239000001257 hydrogen Substances 0.000 claims description 13
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 claims description 12
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 claims description 12
- 125000000623 heterocyclic group Chemical group 0.000 claims description 12
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 11
- 125000005842 heteroatom Chemical group 0.000 claims description 11
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 10
- 229910052760 oxygen Inorganic materials 0.000 claims description 9
- QYCGBAJADAGLLK-UHFFFAOYSA-N 1-(cyclohepten-1-yl)cycloheptene Chemical class C1CCCCC=C1C1=CCCCCC1 QYCGBAJADAGLLK-UHFFFAOYSA-N 0.000 claims description 8
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 8
- 229910052757 nitrogen Inorganic materials 0.000 claims description 8
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 7
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 claims description 7
- 125000002373 5 membered heterocyclic group Chemical group 0.000 claims description 6
- 229910052799 carbon Inorganic materials 0.000 claims description 6
- 125000004432 carbon atom Chemical group C* 0.000 claims description 6
- 125000004070 6 membered heterocyclic group Chemical group 0.000 claims description 5
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 5
- 125000004765 (C1-C4) haloalkyl group Chemical group 0.000 claims description 4
- WPYMKLBDIGXBTP-UHFFFAOYSA-N Benzoic acid Natural products OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 claims description 4
- 229910052783 alkali metal Inorganic materials 0.000 claims description 4
- 150000008044 alkali metal hydroxides Chemical class 0.000 claims description 4
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 claims description 4
- 125000005530 alkylenedioxy group Chemical group 0.000 claims description 4
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 claims description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 3
- 239000000460 chlorine Substances 0.000 claims description 3
- 229910052801 chlorine Inorganic materials 0.000 claims description 3
- 230000007062 hydrolysis Effects 0.000 claims description 3
- 238000006460 hydrolysis reaction Methods 0.000 claims description 3
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 2
- 239000005711 Benzoic acid Substances 0.000 claims 3
- 235000010233 benzoic acid Nutrition 0.000 claims 3
- 125000003158 alcohol group Chemical group 0.000 claims 1
- 238000006243 chemical reaction Methods 0.000 abstract description 54
- 239000000543 intermediate Substances 0.000 abstract description 6
- 239000003905 agrochemical Substances 0.000 abstract description 4
- 229940079593 drug Drugs 0.000 abstract description 4
- 239000003814 drug Substances 0.000 abstract description 4
- 230000002363 herbicidal effect Effects 0.000 abstract description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 72
- 239000000243 solution Substances 0.000 description 43
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 42
- 235000019441 ethanol Nutrition 0.000 description 29
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 28
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 27
- 0 CC1(C)C(Cl)=C(Cl)C(Cl)=C1Cl.[1*]C1=C(C)C=C(Cl)C(Cl)=C1C.[1*]C1=C(C)C=CC(C)=C1C.[1*]C1C(C)C2(Cl)C(=O)C1(Cl)C(Cl)=C2Cl.[1*]C1C(C)C2(Cl)C(Cl)=C(Cl)C1(Cl)C2(C)C.[1*]C=CC Chemical compound CC1(C)C(Cl)=C(Cl)C(Cl)=C1Cl.[1*]C1=C(C)C=C(Cl)C(Cl)=C1C.[1*]C1=C(C)C=CC(C)=C1C.[1*]C1C(C)C2(Cl)C(=O)C1(Cl)C(Cl)=C2Cl.[1*]C1C(C)C2(Cl)C(Cl)=C(Cl)C1(Cl)C2(C)C.[1*]C=CC 0.000 description 22
- 239000013078 crystal Substances 0.000 description 22
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 21
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 20
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 20
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 20
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 19
- 238000002844 melting Methods 0.000 description 19
- 230000008018 melting Effects 0.000 description 19
- 239000012044 organic layer Substances 0.000 description 19
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 18
- LSDPWZHWYPCBBB-UHFFFAOYSA-N Methanethiol Chemical compound SC LSDPWZHWYPCBBB-UHFFFAOYSA-N 0.000 description 16
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 15
- 239000005457 ice water Substances 0.000 description 15
- 150000003839 salts Chemical class 0.000 description 15
- 238000010898 silica gel chromatography Methods 0.000 description 15
- 239000012043 crude product Substances 0.000 description 13
- ZSWFCLXCOIISFI-UHFFFAOYSA-N cyclopentadiene Chemical class C1C=CC=C1 ZSWFCLXCOIISFI-UHFFFAOYSA-N 0.000 description 13
- 238000010828 elution Methods 0.000 description 12
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 10
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 10
- 239000000203 mixture Substances 0.000 description 10
- 229910000027 potassium carbonate Inorganic materials 0.000 description 10
- 235000011181 potassium carbonates Nutrition 0.000 description 10
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 10
- 235000017557 sodium bicarbonate Nutrition 0.000 description 10
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 9
- 150000001298 alcohols Chemical class 0.000 description 9
- 238000001914 filtration Methods 0.000 description 9
- 238000006722 reduction reaction Methods 0.000 description 9
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 8
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 8
- 238000001816 cooling Methods 0.000 description 7
- 229910052751 metal Inorganic materials 0.000 description 7
- 239000002184 metal Substances 0.000 description 7
- 239000012046 mixed solvent Substances 0.000 description 7
- 238000003756 stirring Methods 0.000 description 7
- UHSMEJQTFMHABA-UHFFFAOYSA-N 1,2,3,4-tetrachloro-5,5-dimethoxycyclopenta-1,3-diene Chemical compound COC1(OC)C(Cl)=C(Cl)C(Cl)=C1Cl UHSMEJQTFMHABA-UHFFFAOYSA-N 0.000 description 6
- IOHPVZBSOKLVMN-UHFFFAOYSA-N 2-(2-phenylethyl)benzoic acid Chemical compound OC(=O)C1=CC=CC=C1CCC1=CC=CC=C1 IOHPVZBSOKLVMN-UHFFFAOYSA-N 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 6
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 6
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 6
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 6
- VPHHJAOJUJHJKD-UHFFFAOYSA-N 3,4-dichlorobenzoic acid Chemical class OC(=O)C1=CC=C(Cl)C(Cl)=C1 VPHHJAOJUJHJKD-UHFFFAOYSA-N 0.000 description 5
- CANBLQWGAVGDEP-UHFFFAOYSA-N ethyl 4,5-dichloro-2-methyl-3-(3-methyl-1,2-oxazol-5-yl)benzoate Chemical compound CCOC(=O)C1=CC(Cl)=C(Cl)C(C=2ON=C(C)C=2)=C1C CANBLQWGAVGDEP-UHFFFAOYSA-N 0.000 description 5
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 5
- 125000004433 nitrogen atom Chemical group N* 0.000 description 5
- 238000007254 oxidation reaction Methods 0.000 description 5
- 125000004430 oxygen atom Chemical group O* 0.000 description 5
- 125000004434 sulfur atom Chemical group 0.000 description 5
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 5
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 4
- GOJIQJJMZNNSCR-UHFFFAOYSA-N C1=CC=NC=C1.C1=CN=CN=C1.C1=CNN=C1.C1=COC=N1.C1=CON=C1.C1=CSC=N1.C1=NOCC1.CC.CC.CC.CC.CC.CC.CC.CC.CC.CC.CC.CC.CC.CC.CC.CC.CC.CC1OCCCO1.CC1OCCO1 Chemical compound C1=CC=NC=C1.C1=CN=CN=C1.C1=CNN=C1.C1=COC=N1.C1=CON=C1.C1=CSC=N1.C1=NOCC1.CC.CC.CC.CC.CC.CC.CC.CC.CC.CC.CC.CC.CC.CC.CC.CC.CC.CC1OCCCO1.CC1OCCO1 GOJIQJJMZNNSCR-UHFFFAOYSA-N 0.000 description 4
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 4
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 4
- KFSLWBXXFJQRDL-UHFFFAOYSA-N Peracetic acid Chemical compound CC(=O)OO KFSLWBXXFJQRDL-UHFFFAOYSA-N 0.000 description 4
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 238000009835 boiling Methods 0.000 description 4
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 4
- 150000002170 ethers Chemical class 0.000 description 4
- 239000012442 inert solvent Substances 0.000 description 4
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 4
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 4
- 239000010410 layer Substances 0.000 description 4
- MWYNZHNSSDDVEM-UHFFFAOYSA-N o-ethyl 5-chloro-2,4-dimethyl-3-(3-methyl-1,2-oxazol-5-yl)benzenecarbothioate Chemical compound CCOC(=S)C1=CC(Cl)=C(C)C(C=2ON=C(C)C=2)=C1C MWYNZHNSSDDVEM-UHFFFAOYSA-N 0.000 description 4
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 229910052708 sodium Inorganic materials 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 4
- WPWHSFAFEBZWBB-UHFFFAOYSA-N 1-butyl radical Chemical compound [CH2]CCC WPWHSFAFEBZWBB-UHFFFAOYSA-N 0.000 description 3
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 150000004703 alkoxides Chemical class 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- CTBUQELNDXASFA-UHFFFAOYSA-N ethyl 5-chloro-2-methyl-3-(3-methyl-1,2-oxazol-5-yl)-4-methylsulfonylbenzoate Chemical compound CCOC(=O)C1=CC(Cl)=C(S(C)(=O)=O)C(C=2ON=C(C)C=2)=C1C CTBUQELNDXASFA-UHFFFAOYSA-N 0.000 description 3
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 3
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 3
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 description 3
- 235000017550 sodium carbonate Nutrition 0.000 description 3
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 3
- 125000001424 substituent group Chemical group 0.000 description 3
- 125000006002 1,1-difluoroethyl group Chemical group 0.000 description 2
- 125000004776 1-fluoroethyl group Chemical group [H]C([H])([H])C([H])(F)* 0.000 description 2
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 2
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- YNJSNEKCXVFDKW-UHFFFAOYSA-N 3-(5-amino-1h-indol-3-yl)-2-azaniumylpropanoate Chemical compound C1=C(N)C=C2C(CC(N)C(O)=O)=CNC2=C1 YNJSNEKCXVFDKW-UHFFFAOYSA-N 0.000 description 2
- YFCGUICCZOJPGV-UHFFFAOYSA-N 3-methyl-5-prop-1-enyl-1,2-oxazole Chemical compound CC=CC1=CC(C)=NO1 YFCGUICCZOJPGV-UHFFFAOYSA-N 0.000 description 2
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 2
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 2
- KDDQRKBRJSGMQE-UHFFFAOYSA-N 4-thiazolyl Chemical group [C]1=CSC=N1 KDDQRKBRJSGMQE-UHFFFAOYSA-N 0.000 description 2
- ZTQGXYHKRGOXQS-UHFFFAOYSA-N C.CC=CC1=CC(C)=NO1.COC1(OC)C(Cl)=C(Cl)C(Cl)=C1Cl.COC1(OC)C2(Cl)C(Cl)=C(Cl)C1(Cl)C(C1=CC(C)=NO1)C2C Chemical compound C.CC=CC1=CC(C)=NO1.COC1(OC)C(Cl)=C(Cl)C(Cl)=C1Cl.COC1(OC)C2(Cl)C(Cl)=C(Cl)C1(Cl)C(C1=CC(C)=NO1)C2C ZTQGXYHKRGOXQS-UHFFFAOYSA-N 0.000 description 2
- 238000005698 Diels-Alder reaction Methods 0.000 description 2
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 2
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical class C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 239000007868 Raney catalyst Substances 0.000 description 2
- 229910000564 Raney nickel Inorganic materials 0.000 description 2
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 2
- 239000005708 Sodium hypochlorite Substances 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- OCBFFGCSTGGPSQ-UHFFFAOYSA-N [CH2]CC Chemical compound [CH2]CC OCBFFGCSTGGPSQ-UHFFFAOYSA-N 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 2
- FFBHFFJDDLITSX-UHFFFAOYSA-N benzyl N-[2-hydroxy-4-(3-oxomorpholin-4-yl)phenyl]carbamate Chemical compound OC1=C(NC(=O)OCC2=CC=CC=C2)C=CC(=C1)N1CCOCC1=O FFBHFFJDDLITSX-UHFFFAOYSA-N 0.000 description 2
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 239000003638 chemical reducing agent Substances 0.000 description 2
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 2
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 2
- 238000006298 dechlorination reaction Methods 0.000 description 2
- 125000004772 dichloromethyl group Chemical group [H]C(Cl)(Cl)* 0.000 description 2
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 2
- DNJIEGIFACGWOD-UHFFFAOYSA-N ethanethiol Chemical compound CCS DNJIEGIFACGWOD-UHFFFAOYSA-N 0.000 description 2
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 2
- 150000008282 halocarbons Chemical class 0.000 description 2
- OTTZHAVKAVGASB-UHFFFAOYSA-N hept-2-ene Chemical compound CCCCC=CC OTTZHAVKAVGASB-UHFFFAOYSA-N 0.000 description 2
- 239000012456 homogeneous solution Substances 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- 238000007327 hydrogenolysis reaction Methods 0.000 description 2
- 150000004679 hydroxides Chemical class 0.000 description 2
- WQYVRQLZKVEZGA-UHFFFAOYSA-N hypochlorite Chemical class Cl[O-] WQYVRQLZKVEZGA-UHFFFAOYSA-N 0.000 description 2
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 2
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000004284 isoxazol-3-yl group Chemical group [H]C1=C([H])C(*)=NO1 0.000 description 2
- 125000004499 isoxazol-5-yl group Chemical group O1N=CC=C1* 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- 239000012280 lithium aluminium hydride Substances 0.000 description 2
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 2
- 235000019341 magnesium sulphate Nutrition 0.000 description 2
- SCBJGSXTGZCKQF-UHFFFAOYSA-N methyl 4,5-dichloro-2-methyl-3-(3-methyl-1,2-oxazol-5-yl)benzoate Chemical compound COC(=O)C1=CC(Cl)=C(Cl)C(C=2ON=C(C)C=2)=C1C SCBJGSXTGZCKQF-UHFFFAOYSA-N 0.000 description 2
- MERKVXRKYFANAY-UHFFFAOYSA-N methyl 4,5-dichloro-3-(4,5-dihydro-1,2-oxazol-3-yl)-2-methylbenzoate Chemical compound COC(=O)C1=CC(Cl)=C(Cl)C(C=2CCON=2)=C1C MERKVXRKYFANAY-UHFFFAOYSA-N 0.000 description 2
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 2
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 2
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- LAOHMOGCDZDXBR-UHFFFAOYSA-N o-ethyl 2,4-dimethyl-3-(3-methyl-1,2-oxazol-5-yl)benzenecarbothioate Chemical compound CCOC(=S)C1=CC=C(C)C(C=2ON=C(C)C=2)=C1C LAOHMOGCDZDXBR-UHFFFAOYSA-N 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 150000002894 organic compounds Chemical class 0.000 description 2
- 125000004287 oxazol-2-yl group Chemical group [H]C1=C([H])N=C(*)O1 0.000 description 2
- 125000003145 oxazol-4-yl group Chemical group O1C=NC(=C1)* 0.000 description 2
- 125000004304 oxazol-5-yl group Chemical group O1C=NC=C1* 0.000 description 2
- 239000007800 oxidant agent Substances 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 2
- 150000002978 peroxides Chemical class 0.000 description 2
- 239000011736 potassium bicarbonate Substances 0.000 description 2
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 2
- 235000015497 potassium bicarbonate Nutrition 0.000 description 2
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 2
- 229940086066 potassium hydrogencarbonate Drugs 0.000 description 2
- SATVIFGJTRRDQU-UHFFFAOYSA-N potassium hypochlorite Chemical compound [K+].Cl[O-] SATVIFGJTRRDQU-UHFFFAOYSA-N 0.000 description 2
- 125000004289 pyrazol-3-yl group Chemical group [H]N1N=C(*)C([H])=C1[H] 0.000 description 2
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 2
- 125000004527 pyrimidin-4-yl group Chemical group N1=CN=C(C=C1)* 0.000 description 2
- 125000004528 pyrimidin-5-yl group Chemical group N1=CN=CC(=C1)* 0.000 description 2
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical compound [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 description 2
- KXCAEQNNTZANTK-UHFFFAOYSA-N stannane Chemical compound [SnH4] KXCAEQNNTZANTK-UHFFFAOYSA-N 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 229910000083 tin tetrahydride Inorganic materials 0.000 description 2
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 2
- UAYWVJHJZHQCIE-UHFFFAOYSA-L zinc iodide Chemical compound I[Zn]I UAYWVJHJZHQCIE-UHFFFAOYSA-L 0.000 description 2
- DSHVOLRBCATFNZ-UHFFFAOYSA-N 1,2,3,4-tetrachloro-6-methyl-5-(3-methyl-1,2-oxazol-5-yl)bicyclo[2.2.1]hept-2-en-7-one Chemical compound O=C1C2(Cl)C(Cl)=C(Cl)C1(Cl)C(C)C2C1=CC(C)=NO1 DSHVOLRBCATFNZ-UHFFFAOYSA-N 0.000 description 1
- 125000001724 1,2,3-oxadiazol-4-yl group Chemical group [H]C1=C(*)N=NO1 0.000 description 1
- 125000004503 1,2,3-oxadiazol-5-yl group Chemical group O1N=NC=C1* 0.000 description 1
- 125000004512 1,2,3-thiadiazol-4-yl group Chemical group S1N=NC(=C1)* 0.000 description 1
- 125000001359 1,2,3-triazol-4-yl group Chemical group [H]N1N=NC([*])=C1[H] 0.000 description 1
- 125000001506 1,2,3-triazol-5-yl group Chemical group [H]N1N=NC([H])=C1[*] 0.000 description 1
- 125000001766 1,2,4-oxadiazol-3-yl group Chemical group [H]C1=NC(*)=NO1 0.000 description 1
- 125000004505 1,2,4-oxadiazol-5-yl group Chemical group O1N=CN=C1* 0.000 description 1
- 125000004515 1,2,4-thiadiazol-3-yl group Chemical group S1N=C(N=C1)* 0.000 description 1
- 125000004516 1,2,4-thiadiazol-5-yl group Chemical group S1N=CN=C1* 0.000 description 1
- 125000001305 1,2,4-triazol-3-yl group Chemical group [H]N1N=C([*])N=C1[H] 0.000 description 1
- 125000001414 1,2,4-triazol-5-yl group Chemical group [H]N1N=C([H])N=C1[*] 0.000 description 1
- 125000004507 1,2,5-oxadiazol-3-yl group Chemical group O1N=C(C=N1)* 0.000 description 1
- 125000004518 1,2,5-thiadiazol-3-yl group Chemical group S1N=C(C=N1)* 0.000 description 1
- XBYRMPXUBGMOJC-UHFFFAOYSA-N 1,2-dihydropyrazol-3-one Chemical class OC=1C=CNN=1 XBYRMPXUBGMOJC-UHFFFAOYSA-N 0.000 description 1
- 125000004509 1,3,4-oxadiazol-2-yl group Chemical group O1C(=NN=C1)* 0.000 description 1
- 125000004521 1,3,4-thiadiazol-2-yl group Chemical group S1C(=NN=C1)* 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- OCJBOOLMMGQPQU-UHFFFAOYSA-N 1,4-dichlorobenzene Chemical compound ClC1=CC=C(Cl)C=C1 OCJBOOLMMGQPQU-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 1
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000004278 2-oxazolin-2-yl group Chemical group [H]C1([H])OC(*)=NC1([H])[H] 0.000 description 1
- 125000004485 2-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])([H])C1([H])* 0.000 description 1
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- ZTTDTLQZRZUFQV-UHFFFAOYSA-N 3,4-dichloro-6-methyl-3-(3-methyl-1,2-oxazol-5-yl)cyclohexa-1,5-diene-1-carboxylic acid Chemical compound O1N=C(C)C=C1C1(Cl)C(Cl)C=C(C)C(C(O)=O)=C1 ZTTDTLQZRZUFQV-UHFFFAOYSA-N 0.000 description 1
- UYGMPZSLJYVODL-NSCUHMNNSA-N 3-[(e)-prop-1-enyl]-1h-pyrrole Chemical compound C\C=C\C=1C=CNC=1 UYGMPZSLJYVODL-NSCUHMNNSA-N 0.000 description 1
- OCWWBKGUCHLVKS-NSCUHMNNSA-N 3-[(e)-prop-1-enyl]-4,5-dihydro-1,2-oxazole Chemical compound C\C=C\C1=NOCC1 OCWWBKGUCHLVKS-NSCUHMNNSA-N 0.000 description 1
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- UQBRHGWHQPYGGJ-UHFFFAOYSA-N 3-methyl-5-(1,2,3,4-tetrachloro-7,7-dimethoxy-6-methyl-5-bicyclo[2.2.1]hept-2-enyl)-1,2-oxazole Chemical compound ClC1=C(Cl)C2(Cl)C(OC)(OC)C1(Cl)C(C)C2C1=CC(C)=NO1 UQBRHGWHQPYGGJ-UHFFFAOYSA-N 0.000 description 1
- YFCGUICCZOJPGV-ONEGZZNKSA-N 3-methyl-5-[(e)-prop-1-enyl]-1,2-oxazole Chemical compound C\C=C\C1=CC(C)=NO1 YFCGUICCZOJPGV-ONEGZZNKSA-N 0.000 description 1
- 125000004575 3-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001397 3-pyrrolyl group Chemical group [H]N1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000004487 4-tetrahydropyranyl group Chemical group [H]C1([H])OC([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- ZZXAZUPHEDIDMX-UHFFFAOYSA-N 5-prop-1-enyl-1,2-oxazole Chemical compound CC=CC1=CC=NO1 ZZXAZUPHEDIDMX-UHFFFAOYSA-N 0.000 description 1
- HIADDRJDIIBQMJ-UHFFFAOYSA-N 5-prop-1-enyl-1,3-oxazole Chemical compound CC=CC1=CN=CO1 HIADDRJDIIBQMJ-UHFFFAOYSA-N 0.000 description 1
- CWDWFSXUQODZGW-UHFFFAOYSA-N 5-thiazolyl Chemical group [C]1=CN=CS1 CWDWFSXUQODZGW-UHFFFAOYSA-N 0.000 description 1
- BIZPDNYASWPKNH-UHFFFAOYSA-N C.C.CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(Cl)C(Cl)=C1.CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(SC)C(Cl)=C1.CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(SC)C=C1 Chemical compound C.C.CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(Cl)C(Cl)=C1.CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(SC)C(Cl)=C1.CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(SC)C=C1 BIZPDNYASWPKNH-UHFFFAOYSA-N 0.000 description 1
- NCUMTAXBQDMWOJ-UHFFFAOYSA-N C.C.COC(=O)C1=C(C)C(C2=NOCC2)=C(Cl)C(Cl)=C1.COC(=O)C1=C(C)C(C2=NOCC2)=C(SC)C(Cl)=C1.COC(=O)C1=C(C)C(C2=NOCC2)=C(SC)C=C1 Chemical compound C.C.COC(=O)C1=C(C)C(C2=NOCC2)=C(Cl)C(Cl)=C1.COC(=O)C1=C(C)C(C2=NOCC2)=C(SC)C(Cl)=C1.COC(=O)C1=C(C)C(C2=NOCC2)=C(SC)C=C1 NCUMTAXBQDMWOJ-UHFFFAOYSA-N 0.000 description 1
- ZTQGXYHKRGOXQS-NKZKMTPJSA-N C.C/C=C/C1=CC(C)=NO1.COC1(OC)C(Cl)=C(Cl)C(Cl)=C1Cl.COC1(OC)C2(Cl)C(Cl)=C(Cl)C1(Cl)C(C1=CC(C)=NO1)C2C Chemical compound C.C/C=C/C1=CC(C)=NO1.COC1(OC)C(Cl)=C(Cl)C(Cl)=C1Cl.COC1(OC)C2(Cl)C(Cl)=C(Cl)C1(Cl)C(C1=CC(C)=NO1)C2C ZTQGXYHKRGOXQS-NKZKMTPJSA-N 0.000 description 1
- IWYRTJRBEBKGOO-QIICRJKOSA-N C.C/C=C/C1=NOCC1.COC1(OC)C(Cl)=C(Cl)C(Cl)=C1Cl.COC1(OC)C2(Cl)C(Cl)=C(Cl)C1(Cl)C(C1=NOCC1)C2C Chemical compound C.C/C=C/C1=NOCC1.COC1(OC)C(Cl)=C(Cl)C(Cl)=C1Cl.COC1(OC)C2(Cl)C(Cl)=C(Cl)C1(Cl)C(C1=NOCC1)C2C IWYRTJRBEBKGOO-QIICRJKOSA-N 0.000 description 1
- ZTQGXYHKRGOXQS-DVACKJPTSA-N C.C/C=C\C1=CC(C)=NO1.COC1(OC)C(Cl)=C(Cl)C(Cl)=C1Cl.COC1(OC)C2(Cl)C(Cl)=C(Cl)C1(Cl)C(C1=CC(C)=NO1)C2C Chemical compound C.C/C=C\C1=CC(C)=NO1.COC1(OC)C(Cl)=C(Cl)C(Cl)=C1Cl.COC1(OC)C2(Cl)C(Cl)=C(Cl)C1(Cl)C(C1=CC(C)=NO1)C2C ZTQGXYHKRGOXQS-DVACKJPTSA-N 0.000 description 1
- IWYRTJRBEBKGOO-GDNVVUIBSA-N C.C/C=C\C1=NOCC1.COC1(OC)C(Cl)=C(Cl)C(Cl)=C1Cl.COC1(OC)C2(Cl)C(Cl)=C(Cl)C1(Cl)C(C1=NOCC1)C2C Chemical compound C.C/C=C\C1=NOCC1.COC1(OC)C(Cl)=C(Cl)C(Cl)=C1Cl.COC1(OC)C2(Cl)C(Cl)=C(Cl)C1(Cl)C(C1=NOCC1)C2C IWYRTJRBEBKGOO-GDNVVUIBSA-N 0.000 description 1
- BLGCXDDSEIPDCJ-UHFFFAOYSA-N CC1(C)C(Cl)=C(Cl)C(Cl)C1Cl Chemical compound CC1(C)C(Cl)=C(Cl)C(Cl)C1Cl BLGCXDDSEIPDCJ-UHFFFAOYSA-N 0.000 description 1
- ZLMQAQPXIQNLBF-UHFFFAOYSA-M CC1=C(C(=O)O)C=C(Cl)C(Cl)=C1C1=NOCC1.CC1C(C2=NOCC2)C2(Cl)C(=O)C1(Cl)C(Cl)=C2Cl.O.O[Na] Chemical compound CC1=C(C(=O)O)C=C(Cl)C(Cl)=C1C1=NOCC1.CC1C(C2=NOCC2)C2(Cl)C(=O)C1(Cl)C(Cl)=C2Cl.O.O[Na] ZLMQAQPXIQNLBF-UHFFFAOYSA-M 0.000 description 1
- IDXCHUXNWGJRIZ-UHFFFAOYSA-N CC1=NOC(C2=C(Cl)C(Cl)=CC(C(=O)O)=C2C)=C1.CC1=NOC(C2C(C)C3(Cl)C(=O)C2(Cl)C(Cl)=C3Cl)=C1 Chemical compound CC1=NOC(C2=C(Cl)C(Cl)=CC(C(=O)O)=C2C)=C1.CC1=NOC(C2C(C)C3(Cl)C(=O)C2(Cl)C(Cl)=C3Cl)=C1 IDXCHUXNWGJRIZ-UHFFFAOYSA-N 0.000 description 1
- YUKBACQIJHAVOX-UHFFFAOYSA-N CC1=NOC(C2C(C)C3(Cl)C(=O)C2(Cl)C(Cl)=C3Cl)=C1.CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(Cl)C(Cl)=C1.CCO[Na] Chemical compound CC1=NOC(C2C(C)C3(Cl)C(=O)C2(Cl)C(Cl)=C3Cl)=C1.CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(Cl)C(Cl)=C1.CCO[Na] YUKBACQIJHAVOX-UHFFFAOYSA-N 0.000 description 1
- HJKOZDBKOJXGIF-UHFFFAOYSA-N CC1=NOC(C2C(C)C3(Cl)C(=O)C2(Cl)C(Cl)=C3Cl)=C1.COC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(Cl)C(Cl)=C1.COC1(OC)C2(Cl)C(Cl)=C(Cl)C1(Cl)C(C1=CC(C)=NO1)C2C.CO[Na] Chemical compound CC1=NOC(C2C(C)C3(Cl)C(=O)C2(Cl)C(Cl)=C3Cl)=C1.COC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(Cl)C(Cl)=C1.COC1(OC)C2(Cl)C(Cl)=C(Cl)C1(Cl)C(C1=CC(C)=NO1)C2C.CO[Na] HJKOZDBKOJXGIF-UHFFFAOYSA-N 0.000 description 1
- BAUVMEYBQTWXEU-UHFFFAOYSA-N CC1C(C2=NOCC2)C2(Cl)C(=O)C1(Cl)C(Cl)=C2Cl.CO.COC(=O)C1=C(C)C(C2=NOCC2)=C(Cl)C(Cl)=C1.COC1(OC)C2(Cl)C(Cl)=C(Cl)C1(Cl)C(C1=NOCC1)C2C.CO[Na] Chemical compound CC1C(C2=NOCC2)C2(Cl)C(=O)C1(Cl)C(Cl)=C2Cl.CO.COC(=O)C1=C(C)C(C2=NOCC2)=C(Cl)C(Cl)=C1.COC1(OC)C2(Cl)C(Cl)=C(Cl)C1(Cl)C(C1=NOCC1)C2C.CO[Na] BAUVMEYBQTWXEU-UHFFFAOYSA-N 0.000 description 1
- FFOFKCJXCCFTJN-UHFFFAOYSA-N CCCCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(Cl)C(Cl)=C1.CCCCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(SC)C=C1.CO Chemical compound CCCCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(Cl)C(Cl)=C1.CCCCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(SC)C=C1.CO FFOFKCJXCCFTJN-UHFFFAOYSA-N 0.000 description 1
- XYGKPECLBFWMTB-UHFFFAOYSA-N CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(Cl)C(Cl)=C1.CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(SC)C=C1 Chemical compound CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(Cl)C(Cl)=C1.CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(SC)C=C1 XYGKPECLBFWMTB-UHFFFAOYSA-N 0.000 description 1
- YOTCYYFLQMRKCH-UHFFFAOYSA-M CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(Cl)C(Cl)=C1.CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(SC)C=C1.O=COO[K].[KH] Chemical compound CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(Cl)C(Cl)=C1.CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(SC)C=C1.O=COO[K].[KH] YOTCYYFLQMRKCH-UHFFFAOYSA-M 0.000 description 1
- GLVKEZAKJXOFPV-UHFFFAOYSA-N CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(S(C)(=O)=O)C(Cl)=C1.CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(S(C)(=O)=O)C=C1 Chemical compound CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(S(C)(=O)=O)C(Cl)=C1.CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(S(C)(=O)=O)C=C1 GLVKEZAKJXOFPV-UHFFFAOYSA-N 0.000 description 1
- QYZBZTGFPXILRE-UHFFFAOYSA-N CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(S(C)(=O)=O)C(Cl)=C1.CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(SC)C(Cl)=C1 Chemical compound CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(S(C)(=O)=O)C(Cl)=C1.CCOC(=O)C1=C(C)C(C2=CC(C)=NO2)=C(SC)C(Cl)=C1 QYZBZTGFPXILRE-UHFFFAOYSA-N 0.000 description 1
- MTRRMMYWDKMIBK-UHFFFAOYSA-N CCOC(=O)C1=C(C)C(C2=NOCC2)=C(Cl)C(Cl)=C1.CCOC(=O)C1=C(C)C(C2=NOCC2)=C(SC)C=C1 Chemical compound CCOC(=O)C1=C(C)C(C2=NOCC2)=C(Cl)C(Cl)=C1.CCOC(=O)C1=C(C)C(C2=NOCC2)=C(SC)C=C1 MTRRMMYWDKMIBK-UHFFFAOYSA-N 0.000 description 1
- VHZLBGCBSIBGCL-UHFFFAOYSA-N CCOC(c(c(C)c1-c2cc(C)n[o]2)cc(Cl)c1SC)=O Chemical compound CCOC(c(c(C)c1-c2cc(C)n[o]2)cc(Cl)c1SC)=O VHZLBGCBSIBGCL-UHFFFAOYSA-N 0.000 description 1
- DEJDAMZXAKNXNQ-UHFFFAOYSA-N CCOC(c(c(C)c1-c2cc(C)n[o]2)ccc1SC)=O Chemical compound CCOC(c(c(C)c1-c2cc(C)n[o]2)ccc1SC)=O DEJDAMZXAKNXNQ-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- RWSOTUBLDIXVET-UHFFFAOYSA-N Dihydrogen sulfide Chemical compound S RWSOTUBLDIXVET-UHFFFAOYSA-N 0.000 description 1
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 229910000288 alkali metal carbonate Inorganic materials 0.000 description 1
- 150000008041 alkali metal carbonates Chemical class 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 125000004106 butoxy group Chemical group [*]OC([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 1
- 239000000920 calcium hydroxide Substances 0.000 description 1
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 1
- 150000007942 carboxylates Chemical group 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- QSKWJTXWJJOJFP-UHFFFAOYSA-N chloroform;ethoxyethane Chemical compound ClC(Cl)Cl.CCOCC QSKWJTXWJJOJFP-UHFFFAOYSA-N 0.000 description 1
- ARUKYTASOALXFG-UHFFFAOYSA-N cycloheptylcycloheptane Chemical group C1CCCCCC1C1CCCCCC1 ARUKYTASOALXFG-UHFFFAOYSA-N 0.000 description 1
- 238000005695 dehalogenation reaction Methods 0.000 description 1
- 229940117389 dichlorobenzene Drugs 0.000 description 1
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- 229920001971 elastomer Polymers 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- SMLJGJXWRHBLJO-UHFFFAOYSA-N ethyl 2-methyl-3-(3-methyl-1,2-oxazol-5-yl)-4-methylsulfonylbenzoate Chemical compound CCOC(=O)C1=CC=C(S(C)(=O)=O)C(C=2ON=C(C)C=2)=C1C SMLJGJXWRHBLJO-UHFFFAOYSA-N 0.000 description 1
- WTRZQJZDYUQXEV-UHFFFAOYSA-N ethyl 4,5-dichloro-3-(4,5-dihydro-1,2-oxazol-3-yl)-2-methylbenzoate Chemical compound CCOC(=O)C1=CC(Cl)=C(Cl)C(C=2CCON=2)=C1C WTRZQJZDYUQXEV-UHFFFAOYSA-N 0.000 description 1
- QDQPFZXOCVKCDU-UHFFFAOYSA-N ethyl 4-ethylsulfanyl-2-methyl-3-(3-methyl-1,2-oxazol-5-yl)benzoate Chemical compound CCOC(=O)C1=CC=C(SCC)C(C=2ON=C(C)C=2)=C1C QDQPFZXOCVKCDU-UHFFFAOYSA-N 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical group 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 239000004009 herbicide Substances 0.000 description 1
- 229910000037 hydrogen sulfide Inorganic materials 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 125000003037 imidazol-2-yl group Chemical group [H]N1C([*])=NC([H])=C1[H] 0.000 description 1
- 125000002140 imidazol-4-yl group Chemical group [H]N1C([H])=NC([*])=C1[H] 0.000 description 1
- 125000000336 imidazol-5-yl group Chemical group [H]N1C([H])=NC([H])=C1[*] 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000001678 irradiating effect Effects 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001793 isothiazol-3-yl group Chemical group [H]C1=C([H])C(*)=NS1 0.000 description 1
- 125000004500 isothiazol-4-yl group Chemical group S1N=CC(=C1)* 0.000 description 1
- 125000004501 isothiazol-5-yl group Chemical group S1N=CC=C1* 0.000 description 1
- 125000004498 isoxazol-4-yl group Chemical group O1N=CC(=C1)* 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- YJJBEQYJSLXZBL-UHFFFAOYSA-N methyl 2-methyl-3-(3-methyl-1,2-oxazol-5-yl)-4-methylsulfanylbenzoate Chemical compound COC(=O)C1=CC=C(SC)C(C=2ON=C(C)C=2)=C1C YJJBEQYJSLXZBL-UHFFFAOYSA-N 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- 125000004312 morpholin-2-yl group Chemical group [H]N1C([H])([H])C([H])([H])OC([H])(*)C1([H])[H] 0.000 description 1
- 125000004572 morpholin-3-yl group Chemical group N1C(COCC1)* 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- ANAMHDXMWXXKFJ-UHFFFAOYSA-N o-ethyl 3-(4,5-dihydro-1,2-oxazol-3-yl)-2,4-dimethylbenzenecarbothioate Chemical compound CCOC(=S)C1=CC=C(C)C(C=2CCON=2)=C1C ANAMHDXMWXXKFJ-UHFFFAOYSA-N 0.000 description 1
- AHZDONALLYJBOA-UHFFFAOYSA-N o-methyl 3-(4,5-dihydro-1,2-oxazol-3-yl)-2,4-dimethylbenzenecarbothioate Chemical compound COC(=S)C1=CC=C(C)C(C=2CCON=2)=C1C AHZDONALLYJBOA-UHFFFAOYSA-N 0.000 description 1
- HPTDZXIWGIVWIG-UHFFFAOYSA-N o-methyl 5-chloro-3-(4,5-dihydro-1,2-oxazol-3-yl)-2,4-dimethylbenzenecarbothioate Chemical compound COC(=S)C1=CC(Cl)=C(C)C(C=2CCON=2)=C1C HPTDZXIWGIVWIG-UHFFFAOYSA-N 0.000 description 1
- DFUKYOQAHMXNNH-UHFFFAOYSA-N o-tert-butyl 2,4-dimethyl-3-(3-methyl-1,2-oxazol-5-yl)benzenecarbothioate Chemical compound O1N=C(C)C=C1C1=C(C)C=CC(C(=S)OC(C)(C)C)=C1C DFUKYOQAHMXNNH-UHFFFAOYSA-N 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 125000004483 piperidin-3-yl group Chemical group N1CC(CCC1)* 0.000 description 1
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 125000004307 pyrazin-2-yl group Chemical group [H]C1=C([H])N=C(*)C([H])=N1 0.000 description 1
- 125000004497 pyrazol-5-yl group Chemical group N1N=CC=C1* 0.000 description 1
- 125000002206 pyridazin-3-yl group Chemical group [H]C1=C([H])C([H])=C(*)N=N1 0.000 description 1
- 125000004940 pyridazin-4-yl group Chemical group N1=NC=C(C=C1)* 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000000630 rising effect Effects 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- XKDPXDDIHAYASJ-UHFFFAOYSA-N tert-butyl 4,5-dichloro-2-methyl-3-(3-methyl-1,2-oxazol-5-yl)benzoate Chemical compound O1N=C(C)C=C1C1=C(C)C(C(=O)OC(C)(C)C)=CC(Cl)=C1Cl XKDPXDDIHAYASJ-UHFFFAOYSA-N 0.000 description 1
- 125000004299 tetrazol-5-yl group Chemical group [H]N1N=NC(*)=N1 0.000 description 1
- 125000000437 thiazol-2-yl group Chemical group [H]C1=C([H])N=C(*)S1 0.000 description 1
- 125000004495 thiazol-4-yl group Chemical group S1C=NC(=C1)* 0.000 description 1
- 125000004496 thiazol-5-yl group Chemical group S1C=NC=C1* 0.000 description 1
- 238000011282 treatment Methods 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/54—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/55—Acids; Esters
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/26—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/04—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/06—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members
- C07D261/08—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/30—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D263/32—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/22—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D277/30—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/10—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 not condensed with other rings
- C07D317/14—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 not condensed with other rings with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D317/30—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D319/00—Heterocyclic compounds containing six-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D319/04—1,3-Dioxanes; Hydrogenated 1,3-dioxanes
- C07D319/06—1,3-Dioxanes; Hydrogenated 1,3-dioxanes not condensed with other rings
Definitions
- the present invention relates to benzoic acid derivatives useful as intermediates for the preparation of drugs and agricultural chemicals, particularly compounds having herbicidal activity, and to processes for the preparation thereof.
- Known methods of dehalogenation of aromatic halogenated compounds include, for example, catalytic hydrogenolysis using palladium-carbon or Raney nickel as a catalyst, a method of using metal and metal salts such as lithium or sodium, hydrogenolytic reduction with tin hydride, reduction with metal-hydrogen complex compounds such as lithium aluminum hydride, and electrolytic reduction, which are described in Shin Jikkenkagakukouza, Vol. 14, Syntheses and Reactions of Organic Compounds [I] pages 22-30 (edited by the Chemical Society of Japan, published by Maruzen Co., Ltd.).
- Substituted benzoic acid compounds such as 4-alkylthiobenzoic acid derivatives, are important as intermediates for the preparation of agricultural chemicals and drugs. It has been desired to develop easy and industrially advantageous processes for the preparation of the said benzoic acid derivatives.
- the present invention is directed to
- R 1 is hydrogen or C 1-4 alkyl
- R 2 is hydrogen or C 1-6 alkyl
- Q is an optionally substituted, saturated or unsaturated, 5- or 6-membered heterocyclic group containing 1 to 4 N, O or S atoms and combining with the benzene ring via a carbon atom);
- R 1 are as defined above, and X is chlorine or C 1-4 alkoxy and two X's may join to form a C 2-3 alkylenedioxy group optionally substituted with C 1-4 alkyl), and of acting a base and water or alcohol on a bicycloheptenone derivative of the said Formula (2) in an appropriate solvent;
- R 1 , R 2 and Q are as defined above and R 4 is C 1-6 alkyl
- R 4 is C 1-6 alkyl
- R 1 is hydrogen, or C 1-4 alkyl such as methyl, ethyl, propyl, isopropyl n-butyl isobutyl s-butyl or t-butyl;
- a R 2 is hydrogen, or C 1-6 alkyl such as methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl, s-butyl t-butyl, n-pentyl, isopentyl, neopentyl, n-hexyl or isohexyl;
- X is halogen such as chlorine, or C 1-4 alkoxy such as methoxy, ethoxy, propoxy, isopropoxy or butoxy;
- Two X's may join to form a C 2-3 alkylenedioxy group, such as ethylenedioxy or trimethylenedioxy;
- the said C 2-3 alkylenedioxy group may be substituted with C 1-4 alkyl such as methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl, s-butyl and t-butyl; and
- Q is an optionally substituted, saturated or unsaturate 5- or 6-membered heterocyclic group containing 1 to 4 N, O or S atoms and combining with the benzene or bicycloheptane ring via a carbon atom.
- hetero rings include, for example, 5-membered heterocyclic groups containing 1 to 4 N, O or S atoms, such as
- These groups may have one or more, same or different, substituents at arbitrary positions of the hetero rings.
- substituents include, for example, C 1-4 alkyl such as methyl, ethyl, propyl isopropyl, n-butyl, isobutyl, s-butyl and t-butyl, and C 1-4 haloalkyl such as chloromethyl, dichloromethyl, trichloromethyl, fluoromethyl, difluoromethyl, trifluoromethyl, 1-fluoroethyl, 1,1-difluoroethyl, 2,2,2-trifluoroethyl and pentafluoroethyl.
- Q is more preferably one of the following groups represented by Q-1 to Q-9.
- R 3 is, for example, C 1-4 alkyl such as methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl, s-butyl or t-butyl, or C 1-4 haloalkyl such as chloromethyl, dichloromethyl, trichloromethyl, fluoromethyl, difluoromethyl, trifluoromethyl, 1-fluoroethyl, 1,1-difluoroethyl, 2,2,2-trifluoroethyl or pentafluoroethyl, and n is 0 or an integer of 1 or 2.
- C 1-4 alkyl such as methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl, s-butyl or t-butyl
- C 1-4 haloalkyl such as chloromethyl, dichloromethyl, trichloromethyl, fluoromethyl, di
- R 4 is C 1-6 alkyl such as methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl s-butyl, t-butyl, n-pentyl, isopentyl, neopentyl, n-hexyl or isohexyl.
- the compounds of the present invention represented by the said Formulae (2) and (3), may have stereoisomers, depending on substituents at positions 5 and 6. They are all covered by the present invention.
- the compounds of the present invention may be produced by the following processes:
- a bicycloheptenone derivative (2) of Formula (2) is reacted with a base and water or alcohol in an appropriate solvent to give a compound of Formula (1).
- Bases used for this reaction include, for example, alkali metal alcolates such as sodium methylate, sodium ethylate and potassium t-butoxide; alkali metal hydroxides such as sodium hydroxide and potassium hydroxide; alkaline earth metal hydroxides such as calcium hydroxide; alkali metal carbonates such as sodium carbonate, potassium carbonate and sodium hydrogen carbonate; and alkaline earth metal carbonates.
- alkali metal alcolates such as sodium methylate, sodium ethylate and potassium t-butoxide
- alkali metal hydroxides such as sodium hydroxide and potassium hydroxide
- alkaline earth metal hydroxides such as calcium hydroxide
- alkali metal carbonates such as sodium carbonate, potassium carbonate and sodium hydrogen carbonate
- alkaline earth metal carbonates alkaline earth metal carbonates.
- An amount of a base used is preferably 2 to 5 equivalents to 1 mole of a bicycloheptenone derivative (2).
- Solvents able to be used for the reaction include alcohols such as methanol, ethanol, propanol, isopropanol, butanol and t-butanol; ethers such as diethyl ether and tetrahydrofuran (THF); hydrocarbons such as benzene and toluene; acetonitrile, dimethylformamide (DMF), water, toluene, benzene and the like, or mixtures of 2 or more of these solvents.
- alcohols such as methanol, ethanol, propanol, isopropanol, butanol and t-butanol
- ethers such as diethyl ether and tetrahydrofuran (THF)
- hydrocarbons such as benzene and toluene
- acetonitrile, dimethylformamide (DMF) water, toluene, benzene and the like, or mixtures of 2 or more of these solvent
- alcoholic solvents that is, alcohols, or mixed solvents of alcohol and other solvents such as water and alcohol or alcohol and ether.
- an ester (COOR 2 ) having a portion corresponding to an alcohol (R 2 OH) used or the alkoxide portion of a metal alkoxide (MOR 2 ) used can be obtained; for example, a methyl ester is obtained with the use of methyl alcohol and an ethyl ester from ethyl alcohol.
- More preferred combinations of a base and a solvent include, for example, sodium methoxide and methanol (or a mixed solvent of methanol and other solvents), sodium ethoxide and ethanol (or a mixed solvent of ethanol and other solvents), sodium hydroxide and alcohol (or a mixed solvent of alcohol and other solvents), potassium hydroxide and alcohol (or a mixed solvent of alcohol and other solvents), and potassium t-butoxide and butanol.
- Solvents used together with water are favorably alcohols and ethers.
- bases the aforementioned hydroxides or carbonates are preferably used.
- Preferred reaction temperatures are between ⁇ 10° C. and the boiling point of solvents used.
- a bicycloheptenone derivative (2) can be obtained by hydrolysis of a bicycloheptene derivative (3) with an acid such as hydrochloric acid or sulfuric acid.
- a compound (2) can be obtained by hydrolyzing a compound (3) without using a solvent or with a solvent including alcohols such as methanol, ethanol and t-butanol, ethers such as diethyl ether and tetrahydrofuran (THF) or aromatic hydrocarbons such as benzene and toluene, or a mixed solvent of two or more of these solvents, at temperature between ⁇ 10° C. and the boiling point of solvents used.
- a solvent including alcohols such as methanol, ethanol and t-butanol, ethers such as diethyl ether and tetrahydrofuran (THF) or aromatic hydrocarbons such as benzene and toluene, or a mixed solvent of two or more of these solvents, at temperature between ⁇ 10° C. and the boiling point of solvents used.
- a target benzoic acid derivative can be prepared in the same way as that described in Process 1 above.
- the Diels-Alder reaction can be carried out according to methods described, for example, in Tetrahedron, 42, 1741-1744 (1986) or J. Org. Chem., 26, 2066-2072 (1961).
- cyclopentadienes and ethylene derivatives are reacted while heating.
- a molar ratio of the ethylene derivatives used in the reaction is 0.5 to 10 times in equivalent, preferably 1 to 3 equivalents, to 1 mole of cyclopentadienes.
- the reaction is carried out at temperature between room temperature and 25° C., more preferably between 70° C. and 200° C.
- solvents may be used.
- solvents used include aromatic hydrocarbons such as benzene, toluene, xylene, chlorobenzene and dichlorobenzene; alcohols such as ethanol, n-propyl alcohol, ethylene glycol, 1,3-butanediol and ethylene glycol monomethyl ether; ethers such as dimethoxyethane, dioxane and diethylene glycol dimethyl ether, amides such as N,N-dimethylformamide, N,N-dimethylacetamide, N-methylpyrolidone and N,N-dimethylimidazoline; hydrocarbons containing sulfur such as dimethylsulfoxide and sulfolane, and water.
- the reaction proceeds more smoothly by adding a polymerization inhibitor such as hydroquinone in the presence of a base such as sodium hydrogen carbonate, potassium hydrogen carbonate, sodium carbonate or potassium carbonate.
- a polymerization inhibitor such as hydroquinone
- a base such as sodium hydrogen carbonate, potassium hydrogen carbonate, sodium carbonate or potassium carbonate.
- a compound of Formula (5) a starting material, can be prepared according to methods described, for example, in the following papers:
- a 4,5-dichlorobenzoic acid derivative of Formula (1) is reacted with an excessive amount of alkane thiol of Formula R 4 SH (R 4 is as defined above) in an appropriate inert solvent in the presence of a base, to give a 4-alkylthiobenzoic acid derivative of Formula (6).
- the reaction proceeds more smoothly under the irradiation of light (of a specified wavelength).
- Various light sources can be used, including sunlight, fluorescent lights, mercury lamps, arc lamps and incandescent lamps. It is preferable to carry out the reaction under inert gas atmosphere, after the reaction system is sufficiently degassed.
- the reaction may sometimes proceed more smoothly by adding water of 0.1 to 5 times in equivalent to the amount of 4,5-dichlorobenzoic acid derivative (1).
- solvents used for this reaction are amides, such as dimethylformamide DMF), N,N-dimethylacetamide and N-methylpyrolidone.
- amides such as dimethylformamide DMF
- N,N-dimethylacetamide and N-methylpyrolidone.
- solvents used are not restricted to them.
- bases used include hydroxides such as sodium hydroxide and potassium hydroxide; carbonates such as sodium carbonate, sodium hydrogen carbonate, potassium carbonate and potassium hydrogen carbonate; and metal alcolates such as sodium methylate, sodium etheylate and potassium t-butoxide.
- a salt of alkane thiol such as sodium or potassium salt of alkane thiol, prepared from an alkane thiol and a base beforehand, can be used for the reaction.
- Amounts of base and alkane thiol used are preferably 2 to 20 equivalents to that of 4,5-dichlorobenzoic acid derivative (1).
- Benzoic acid derivatives of Formula (6) can be produced by isolation of Compound (7) followed by reduction, as shown in the following reaction scheme:
- the same alkane thiol (R 4 SH) as that used in the previous reaction can be used as a reducing agent.
- other reducing agents such as hydrogen sulfide and aromatic thiols can be employed.
- a compound of Formula (6) can be derived to a corresponding SO 2 R 4 compound (8) by oxidation reaction of the SR 4 group.
- This oxidation reaction may be carried out using an oxidizing agent including peroxides such as hydrogen peroxide, peracetic acid, perbenzoic acid and m-chloroperbenzoic acid, and hypochlorites such as sodium hypochlorite and potassium hypochlorite, in an inert solvent including water, alcohols such as methanol and ethanol organic acids such as acetic acid, and halogenated hydrocarbons such as dichloromethane, chloroform and carbon tetrachloride.
- the reaction proceeds smoothly in the temperature range from 0° C. to the boiling point of the solvent used.
- a SO 2 R 4 compound (8) can be obtained by oxidation of a compound of the said Formula (7) and then a reduction reaction for dechlorination.
- a compound of Formula (9) can be produced by oxidation of a compound of Formula (7).
- This oxidation reaction may be carried out using an oxidizing agent including peroxides such as hydrogen peroxide, peracetic acid, perbenzoic acid and m-chloroperbenzoic acid, and hypochlorites such as sodium hypochlorite and potassium hypochlorite, in an inert solvent including water, alcohols such as methanol and ethanol, organic acids such as acetic acid, and halogenated hydrocarbons such as dichloromethane, chloroform and carbon tetrachloride.
- peroxides such as hydrogen peroxide, peracetic acid, perbenzoic acid and m-chloroperbenzoic acid
- hypochlorites such as sodium hypochlorite and potassium hypochlorite
- an inert solvent including water, alcohols such as methanol and ethanol, organic acids such as acetic acid, and halogenated hydrocarbons such as dichloromethane
- the next dechlorination of a compound of Formula (9) is carried out by such a method as catalytic hydrogenolysis using Raney nickel as a catalyst, method of using metal and metal salts such as lithium and sodium hydrogenolytic reduction with tin hydride, reduction with a metal-hydrogen complex compound such as lithium aluminum hydride, or electrolytic reduction, described in Shin Jikkenkagakukouza Vol. 14, Syntheses and Reactions of Organic Compounds [I] pages 22-30 (edited by the Chemical Society of Japan, published by Maruzen Co., Ltd.).
- the obtained viscous liquid was purified on silica gel column chromatography (elution with n-hexane/ethyl acetate) to give 0.024 g of the title compound as white crystals. Melting point 86-88° C.
- the obtained viscous liquid was purified on silica gel column chromatography (elution with n-hexane/ethyl acetate) to give 0.42 g of the title compound as white crystals. Melting point: 76-77° C.
- the product was again purified on silica gel column chromatography to give the title compound with melting point of 102-103° C. as white crystals.
- Table 3-1 Compounds 3-1 to 3-400, where R 4 is CH 3 , that are derived from Compounds Nos. from 1-1 to 1-400 in Table 1.
- Table 3-2 Compounds 3-401 to 3-800, where R 4 is C 2 H 5 , that are derived from Compounds Nos. from 1-1 to 1-400 in Table 1.
- Table 3-3 Compounds 3-801 to 3-1200, where R 4 is i-C 3 H 7 , that are derived from Compounds Nos. from 1-1 to 1-400 in Table 1.
- Table 3-4 Compounds 3-1201 to 3-1600, where R 4 is t-C 4 H 9 , that are derived from Compounds Nos. from 1-1 to 1-400 in Table 1.
- benzoic acid derivatives useful as intermediates for the preparation of agricultural chemicals and drugs can be produced simply, easily and industrially advantageously in a short process, using inexpensive cyclopentadiene derivatives and ethylene derivatives substituted with hetero rings as starting materials.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Benzoic acid derivatives useful as intermediates for the preparation of drugs and agricultural chemicals, particularly compounds having herbicidal activity; and easy and economical processes for the preparation of the same. The processes are specifically those represented by reaction formula for the preparation of compounds represented by general formulae (1) and (6). 
Description
The present invention relates to benzoic acid derivatives useful as intermediates for the preparation of drugs and agricultural chemicals, particularly compounds having herbicidal activity, and to processes for the preparation thereof.
A method (A) of obtaining benzoic acid derivatives by reactions represented by the following reaction scheme, 
between bicycloheptenone derivatives substituted with carboxylates and alcolates or the like is described in Tetrahedron, 42, 1741 (1986) and J. Org. Chem., 26, 2066 (1961).
However, there are no reports on reactions of bicycloheptenone derivatives substituted with hetero rings.
Known methods of dehalogenation of aromatic halogenated compounds include, for example, catalytic hydrogenolysis using palladium-carbon or Raney nickel as a catalyst, a method of using metal and metal salts such as lithium or sodium, hydrogenolytic reduction with tin hydride, reduction with metal-hydrogen complex compounds such as lithium aluminum hydride, and electrolytic reduction, which are described in Shin Jikkenkagakukouza, Vol. 14, Syntheses and Reactions of Organic Compounds [I] pages 22-30 (edited by the Chemical Society of Japan, published by Maruzen Co., Ltd.).
Substituted benzoic acid compounds, such as 4-alkylthiobenzoic acid derivatives, are important as intermediates for the preparation of agricultural chemicals and drugs. It has been desired to develop easy and industrially advantageous processes for the preparation of the said benzoic acid derivatives.
The present invention is directed to
(a) a benzoic acid derivative represented by Formula (1) 
(wherein R1 is hydrogen or C1-4 alkyl,
R2 is hydrogen or C1-6 alkyl, and
Q is an optionally substituted, saturated or unsaturated, 5- or 6-membered heterocyclic group containing 1 to 4 N, O or S atoms and combining with the benzene ring via a carbon atom);
(b) a process for the preparation of a benzoic acid derivative of the said Formula (1), characterized by acting a base on a bicycloheptenone derivative of Formula (2) 
(wherein R1 and Q are as defined above), in an appropriate solvent;
(c) a process for the preparation of a benzoic acid derivative of the said Formula (1), characterized by consisting of a stage of preparing a bicycloheptenone derivative of the said Formula (2) by hydrolysis of a bicycloheptene derivative of Formula (3) 
(wherein R1 are as defined above, and X is chlorine or C1-4 alkoxy and two X's may join to form a C2-3 alkylenedioxy group optionally substituted with C1-4 alkyl), and of acting a base and water or alcohol on a bicycloheptenone derivative of the said Formula (2) in an appropriate solvent;
(d) a process for the preparation of a bicycloheptene derivative of the said Formula (3), characterized by reacting a cyclopentadiene derivative of Formula (4) 
(wherein X is as defined above) with an ethylene derivative substituted with a hetero ring, of Formula (5) 
(wherein R1 and Q are defined above);
(e) a bicycloheptenone derivative of the said formula (2); and
(f) a process for the preparation of a benzoic acid derivative of Formula (6) 
(wherein R1, R2 and Q are as defined above and R4 is C1-6 alkyl), characterized by reacting a 4,5-dichlorobenzoic acid derivative of the said Formula (1), with a mercaptan of Formula R4SH (wherein R4 is as defined above) and a base or with a salt of mercaptan of Formula R4SH (wherein R4 is as defined above)
In the definitions of the compounds of the said Formulae (1) and (2), which are the compounds of the present invention, the compounds of Formula (3) of their precursors and the compounds of Formula (6),
R1 is hydrogen, or C1-4 alkyl such as methyl, ethyl, propyl, isopropyl n-butyl isobutyl s-butyl or t-butyl;
a R2 is hydrogen, or C1-6 alkyl such as methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl, s-butyl t-butyl, n-pentyl, isopentyl, neopentyl, n-hexyl or isohexyl;
X is halogen such as chlorine, or C1-4 alkoxy such as methoxy, ethoxy, propoxy, isopropoxy or butoxy;
Two X's may join to form a C2-3 alkylenedioxy group, such as ethylenedioxy or trimethylenedioxy;
Further, the said C2-3 alkylenedioxy group may be substituted with C1-4 alkyl such as methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl, s-butyl and t-butyl; and
Q is an optionally substituted, saturated or unsaturate 5- or 6-membered heterocyclic group containing 1 to 4 N, O or S atoms and combining with the benzene or bicycloheptane ring via a carbon atom.
Such hetero rings include, for example, 5-membered heterocyclic groups containing 1 to 4 N, O or S atoms, such as
2-furyl, 3-furyl,
2-thienyl, 3-thienyl,
2,3-dihydrofuran-2-yl, 2,3-dihydrofuran-3-yl, 2,3-dihydrofuran4-yl,2,3-dihydrofuran-5-yl, 2,5-dihydrofuran-2-yl, 2,5-dihydrofuran-3-yl,
2,3-dihydrothiophen-2-yl, 2,3-dihydrothiophen-3-yl, 2,3-dihydrothiophen-4-yl, 2,3-dihydrothiophen-5-yl, 2,5-dihydrothiophen-2-yl, 2,5-dihydrothiophen-3-yl,
pyrrol-2-yl, pyrrol-3-yl,
imidazol-2-yl, imidazol-4-yl, imidazol-5-yl,
2-imidazolin-2-yl, 2-imidazolin-4-yl, 2-imidazolin-5-yl,
pyrazol-3-yl, pyrozol-4-yl, pyrazol-5-yl,
oxazol-2-yl, oxazol-4-yl, oxazol-5-yl,
isoxazol-3-yl, isoxazol-4-yl, isoxazol-5-yl.
1,2,4-oxadiazol-3-yl, 1,2,4-oxadiazol-5-yl, 1,3,4-oxadiazol-2-yl, 1,2,3-oxadiazol-4-yl, 1,2,3-oxadiazol-5-yl, 1,2,5-oxadiazol-3-yl,
4-thiazolyl, 4-thiazolyl, 5-thiazolyl,
isothiazol-3-yl, isothiazol-4-yl, isothiazol-5-yl,
1,2,4-thiadiazol-3-yl, 1,2,4-thiadiazol-5-yl, 1,3,4-thiadiazol-2-yl, 1,2,3-thiadiazol-4-yl, 1,2,3-thiadiazol-5-yl, 1,2,5-thiadiazol-3-yl,
1,2,4-triazol-3-yl, 1,2,4-triazol-5-yl, 1,3,4-triazol-2-yl, 1,2,3-triazol-4-yl, 1,2,3-triazol-5-yl, tetrazol-5-yl,
2-pyrrolin-1-yl, 2-pyrrolin-2-yl, 2-pyrrolin-3-yl, 2-pyrrolin4-yl, 2-pyrrolin-5-yl,
2-oxazolin-2-yl, 2-oxazolin-4-yl, 2-oxazolin-5-yl, 3-oxazolin-2-yl, 3-oxazolin-4-yl, 3-oxazolin-5-yl, 4-oxazolin-2-yl, 4-oxazolin-4-yl, 4oxazolin-5-yl,
2-isoxazolin-3-yl, 2-isoxazolin-4-yl, 2-isoxazolin-5-yl,
3-isoxazolin-3-yl, 3-isoxazolin-4-yl, 3-isoxazolin-5-yl,
4-isoxazolin-3-yl, 4-isoxazolin-4-yl, 4-isoxazolin-5-yl,
2-thiazolin-2-yl, 4-thiazolin-4-yl, 4-thiazolin-5-yl,
2-isothiazolin-3-yl, 2-isothiazolin-4-yl, 2-isothiazolin-5-yl,
3-isothiazolin-3-yl, 3-isothiazolin-4-yl, 3-isothiazolin-5-yl,
4-isothiazolin-3-yl, 4-isothiazolin-4-yl, 4-isothiazolin-5-yl,
1-pyrazolin-3-yl, 1-pyrazolin-4-yl, 1-pyrazolin-5-yl,
2-pyrazolin-3-yl, 2-pyrazolin-4-yl, 2-pyrazolin-5-yl,
3-pyrazolin-3-yl, 3-pyrazolin-4-yl and 3-pyrazolin-5-yl; saturated 5-membered heterocyclic groups containing 1 to 4 N, O or S atoms, such as
2-pyrrolidinyl, 3-pyrrolidinyl,
2-tetrahydrofuranyl, 3-tetrahydrofuranyl,
2-tetrahydrothienyl, 3-tetrahydrothienyl,
2-oxazolidinyl, 4-oxazolidinyl, 5-oxazolidinyl,
3-isoxazolidinyl, 4-isoxazolidinyl, 5-isoxazolidinyl,
2-thiazolidinyl, 4-thiazolidinyl, 5-thiazolidinyl,
3-isothiazolidinyl, 4-isothiazolidinyl, 5-isothiazolidinyl,
2-imidazolidinyl, 4-imidazolidinyl,
1,2,4-oxadiazolidin-3-yl, 1,2,4-oxadiazolidin-5-yl, 1,3,4-oxadiazolidin-2-yl,
1,2,4-thiadiazolidin-3-yl, 1,2,4-thiadiazolidin-5-yl, 1,3,4-thiadiazolidin-2-yl,
1,3,4-triazolidin-2-yl,
1,3-dioxolan-2-yl, 1,3-dioxolan-4-yl,
1,3-dithiolan-2-yl, 1,3-dithiolan-4-yl, and
1,3-oxathiolan-2-yl; and
6-membered heterocyclic groups containing 1 to 4 N, O or S atoms, such as
2-pyridyl, 3-pyridyl, 4-pyridyl,
3-pyridazinyl, 4-pyridazinyl,
2-pyrimidinyl, 4-pyrimidinyl, 5-pyrimidinyl,
pyrazin-2-yl,
2H-pyran-3-yl, 2H-thiopyran-3-yl,
2-piperidinyl, 3-piperidinyl, 4-piperidinyl
2-piperadinyl,
2-morpholinyl, 3-morpholinyl,
5,6-dihydro-4H-1,3-thiazin-2-yl,
2-tetrahydropyranyl, 3-tetrahydropyranyl, 4-tetrahydropyranyl,
2-tetrahydrothiopyranyl, 3-tetrahydrothiopyranyl and 4-tetrahydrothiopyranyl.
These groups may have one or more, same or different, substituents at arbitrary positions of the hetero rings. Such substituents include, for example, C1-4 alkyl such as methyl, ethyl, propyl isopropyl, n-butyl, isobutyl, s-butyl and t-butyl, and C1-4 haloalkyl such as chloromethyl, dichloromethyl, trichloromethyl, fluoromethyl, difluoromethyl, trifluoromethyl, 1-fluoroethyl, 1,1-difluoroethyl, 2,2,2-trifluoroethyl and pentafluoroethyl.
Q is more preferably one of the following groups represented by Q-1 to Q-9. 
In the above formulae, R3 is, for example, C1-4 alkyl such as methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl, s-butyl or t-butyl, or C1-4 haloalkyl such as chloromethyl, dichloromethyl, trichloromethyl, fluoromethyl, difluoromethyl, trifluoromethyl, 1-fluoroethyl, 1,1-difluoroethyl, 2,2,2-trifluoroethyl or pentafluoroethyl, and n is 0 or an integer of 1 or 2.
Further, in the said Formula (6), R4 is C1-6 alkyl such as methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl s-butyl, t-butyl, n-pentyl, isopentyl, neopentyl, n-hexyl or isohexyl.
The compounds of the present invention, represented by the said Formulae (2) and (3), may have stereoisomers, depending on substituents at positions 5 and 6. They are all covered by the present invention.
The compounds of the present invention may be produced by the following processes:
(Process 1) Process for the Preparation of a 4,5-dichlorobenzoic Acid Derivative from a Bicycloheptenone Derivative 
(wherein R1, R2 and Q are as defined above).
A bicycloheptenone derivative (2) of Formula (2) is reacted with a base and water or alcohol in an appropriate solvent to give a compound of Formula (1).
Bases used for this reaction include, for example, alkali metal alcolates such as sodium methylate, sodium ethylate and potassium t-butoxide; alkali metal hydroxides such as sodium hydroxide and potassium hydroxide; alkaline earth metal hydroxides such as calcium hydroxide; alkali metal carbonates such as sodium carbonate, potassium carbonate and sodium hydrogen carbonate; and alkaline earth metal carbonates.
An amount of a base used is preferably 2 to 5 equivalents to 1 mole of a bicycloheptenone derivative (2).
Solvents able to be used for the reaction include alcohols such as methanol, ethanol, propanol, isopropanol, butanol and t-butanol; ethers such as diethyl ether and tetrahydrofuran (THF); hydrocarbons such as benzene and toluene; acetonitrile, dimethylformamide (DMF), water, toluene, benzene and the like, or mixtures of 2 or more of these solvents.
In the aforementioned reaction, it is particularly preferable to use alcoholic solvents, that is, alcohols, or mixed solvents of alcohol and other solvents such as water and alcohol or alcohol and ether.
In the above reaction, an ester (COOR2) having a portion corresponding to an alcohol (R2OH) used or the alkoxide portion of a metal alkoxide (MOR2) used can be obtained; for example, a methyl ester is obtained with the use of methyl alcohol and an ethyl ester from ethyl alcohol.
More preferred combinations of a base and a solvent include, for example, sodium methoxide and methanol (or a mixed solvent of methanol and other solvents), sodium ethoxide and ethanol (or a mixed solvent of ethanol and other solvents), sodium hydroxide and alcohol (or a mixed solvent of alcohol and other solvents), potassium hydroxide and alcohol (or a mixed solvent of alcohol and other solvents), and potassium t-butoxide and butanol.
In the case of the use of mixed solvents or aqueous solvents, carboxylic acids (R2=H) can be obtained. Solvents used together with water are favorably alcohols and ethers. As for bases, the aforementioned hydroxides or carbonates are preferably used.
In the case of the use of metal alkoxides, it is preferable to use corresponding alcohols, as described above. It is of course possible to use other alcohols.
Preferred reaction temperatures are between −10° C. and the boiling point of solvents used.
(Process 2) 
(wherein R1, R2, Q and X are as defined above.)
A bicycloheptenone derivative (2) can be obtained by hydrolysis of a bicycloheptene derivative (3) with an acid such as hydrochloric acid or sulfuric acid.
That is, a compound (2) can be obtained by hydrolyzing a compound (3) without using a solvent or with a solvent including alcohols such as methanol, ethanol and t-butanol, ethers such as diethyl ether and tetrahydrofuran (THF) or aromatic hydrocarbons such as benzene and toluene, or a mixed solvent of two or more of these solvents, at temperature between −10° C. and the boiling point of solvents used.
After this, a target benzoic acid derivative can be prepared in the same way as that described in Process 1 above.
(Process 3) 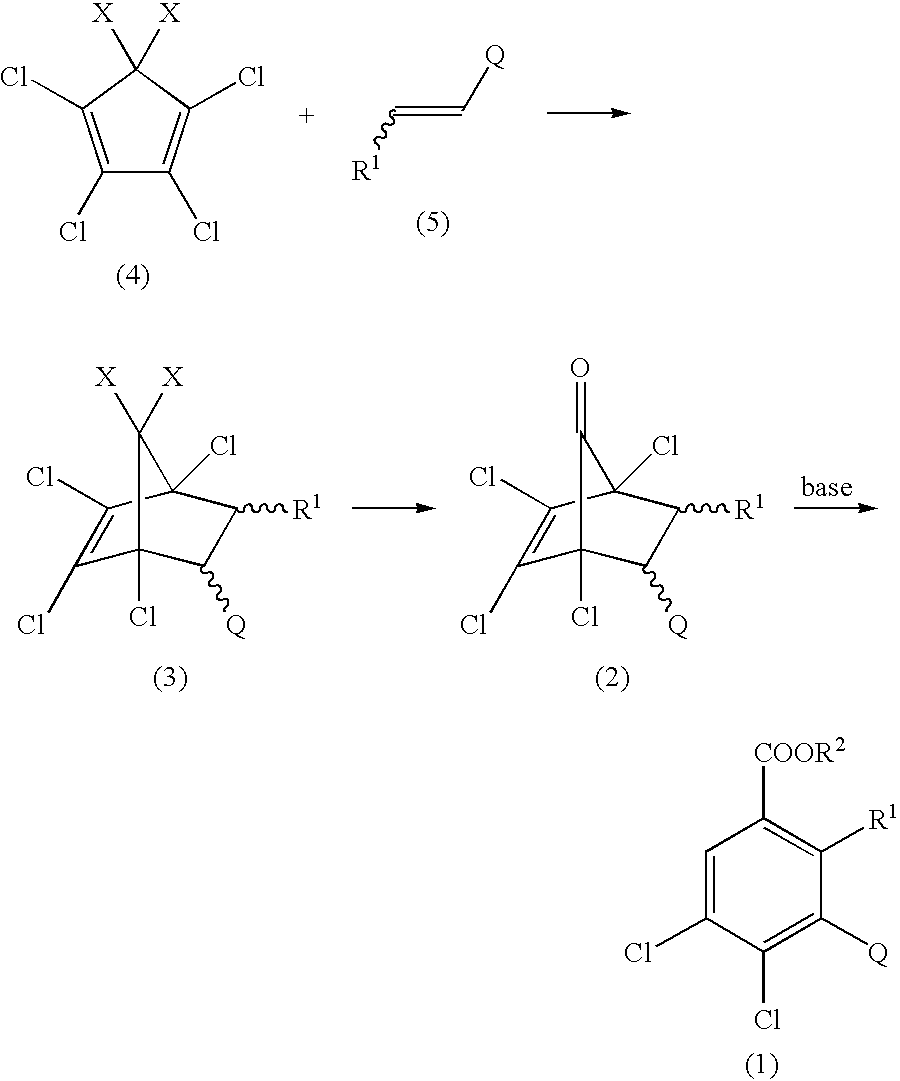
(wherein R1, R2, Q and X are as defined above.)
A Diels-Alder reaction of a cyclopentadiene derivative (4) and an ethylene derivative substituted with a hetero ring (5) gives a bicycloheptene derivative (3).
The Diels-Alder reaction can be carried out according to methods described, for example, in Tetrahedron, 42, 1741-1744 (1986) or J. Org. Chem., 26, 2066-2072 (1961).
In the above reaction, cyclopentadienes and ethylene derivatives are reacted while heating. A molar ratio of the ethylene derivatives used in the reaction is 0.5 to 10 times in equivalent, preferably 1 to 3 equivalents, to 1 mole of cyclopentadienes. The reaction is carried out at temperature between room temperature and 25° C., more preferably between 70° C. and 200° C.
Although this reaction is usually carried out without solvents, solvents may be used. Examples of solvents used include aromatic hydrocarbons such as benzene, toluene, xylene, chlorobenzene and dichlorobenzene; alcohols such as ethanol, n-propyl alcohol, ethylene glycol, 1,3-butanediol and ethylene glycol monomethyl ether; ethers such as dimethoxyethane, dioxane and diethylene glycol dimethyl ether, amides such as N,N-dimethylformamide, N,N-dimethylacetamide, N-methylpyrolidone and N,N-dimethylimidazoline; hydrocarbons containing sulfur such as dimethylsulfoxide and sulfolane, and water.
The reaction proceeds more smoothly by adding a polymerization inhibitor such as hydroquinone in the presence of a base such as sodium hydrogen carbonate, potassium hydrogen carbonate, sodium carbonate or potassium carbonate.
After this, a target benzoic acid derivative can be produced in the same way as that in Process 2.
A compound of Formula (5), a starting material, can be prepared according to methods described, for example, in the following papers:
Chim. Ind. (Milan), 52 (1), 56 (1977): Preparation of 3-(1-propenyl)-1H-pyrrole;
Zh. Organ. Khim., 2, 417-423 (1966): Preparation of 5-(1-propenyl)-isoxazole;
J. Org. Chem., 62, 3671-3677 (1997): Preparation of 3(1-propenyl)-2-isoxazoline;
Heterocycles, 22 (11), 2475-2478 (1984): Preparation of 4(1-propenyl)pyridine; and
Heterocycles, 29(1), 103-114 (1989): Preparation of 5-(1-propenyl)-oxazole.
(Process 4) 
(wherein R1, R2, R4 and Q are as defined above.)
In the process, a 4,5-dichlorobenzoic acid derivative of Formula (1) is reacted with an excessive amount of alkane thiol of Formula R4SH (R4 is as defined above) in an appropriate inert solvent in the presence of a base, to give a 4-alkylthiobenzoic acid derivative of Formula (6).
While doing so, the reaction proceeds more smoothly under the irradiation of light (of a specified wavelength). Various light sources can be used, including sunlight, fluorescent lights, mercury lamps, arc lamps and incandescent lamps. It is preferable to carry out the reaction under inert gas atmosphere, after the reaction system is sufficiently degassed.
The reaction may sometimes proceed more smoothly by adding water of 0.1 to 5 times in equivalent to the amount of 4,5-dichlorobenzoic acid derivative (1).
Particularly preferred solvents used for this reaction are amides, such as dimethylformamide DMF), N,N-dimethylacetamide and N-methylpyrolidone. However, solvents used are not restricted to them.
Examples of bases used include hydroxides such as sodium hydroxide and potassium hydroxide; carbonates such as sodium carbonate, sodium hydrogen carbonate, potassium carbonate and potassium hydrogen carbonate; and metal alcolates such as sodium methylate, sodium etheylate and potassium t-butoxide. In this case, a salt of alkane thiol, such as sodium or potassium salt of alkane thiol, prepared from an alkane thiol and a base beforehand, can be used for the reaction.
Amounts of base and alkane thiol used are preferably 2 to 20 equivalents to that of 4,5-dichlorobenzoic acid derivative (1).
Benzoic acid derivatives of Formula (6) can be produced by isolation of Compound (7) followed by reduction, as shown in the following reaction scheme: 
To produce a compound of Formula (7), an equivalent of a compound of Formula (1) is reacted with about 1 to 3 equivalents of an alkane thiol of Formula R4SH (R4 is as defined above) in an inert solvent in the presence of an appropriate base.
In the reduction reaction from a compound of Formula (7) to a compound of Formula (6), the same alkane thiol (R4SH) as that used in the previous reaction can be used as a reducing agent. Further, other reducing agents such as hydrogen sulfide and aromatic thiols can be employed.
A compound of Formula (6) can be derived to a corresponding SO2R4 compound (8) by oxidation reaction of the SR4 group. This oxidation reaction may be carried out using an oxidizing agent including peroxides such as hydrogen peroxide, peracetic acid, perbenzoic acid and m-chloroperbenzoic acid, and hypochlorites such as sodium hypochlorite and potassium hypochlorite, in an inert solvent including water, alcohols such as methanol and ethanol organic acids such as acetic acid, and halogenated hydrocarbons such as dichloromethane, chloroform and carbon tetrachloride. The reaction proceeds smoothly in the temperature range from 0° C. to the boiling point of the solvent used.
Further, a SO2R4 compound (8) can be obtained by oxidation of a compound of the said Formula (7) and then a reduction reaction for dechlorination. 
(wherein R1, R2, R4 and Q are as defined above.)
A compound of Formula (9) can be produced by oxidation of a compound of Formula (7). This oxidation reaction may be carried out using an oxidizing agent including peroxides such as hydrogen peroxide, peracetic acid, perbenzoic acid and m-chloroperbenzoic acid, and hypochlorites such as sodium hypochlorite and potassium hypochlorite, in an inert solvent including water, alcohols such as methanol and ethanol, organic acids such as acetic acid, and halogenated hydrocarbons such as dichloromethane, chloroform and carbon tetrachloride. The reaction proceeds smoothly in the temperature range from 0° C. to the boiling point of the solvent used.
The next dechlorination of a compound of Formula (9) is carried out by such a method as catalytic hydrogenolysis using Raney nickel as a catalyst, method of using metal and metal salts such as lithium and sodium hydrogenolytic reduction with tin hydride, reduction with a metal-hydrogen complex compound such as lithium aluminum hydride, or electrolytic reduction, described in Shin Jikkenkagakukouza Vol. 14, Syntheses and Reactions of Organic Compounds [I] pages 22-30 (edited by the Chemical Society of Japan, published by Maruzen Co., Ltd.).
The compounds of the present invention, intermediates and others can be obtained with usual post-treatments after the completion of the reactions.
The structures of the compounds of the present invention, intermediates and others were determined by IR, NMR, MS and other means.
The present invention is described in more detail in reference to Examples and Reference Examples.
In Examples, of the compounds of the present invention, (1S, 4R, 5R, 6S) and (1R, 4S, 5S, 6R) isomers are represented as “trans”, and (1S, 4R, 5R, 6R) and (1R, 4S, 5S, 6S) isomers as “cis”.

0.11 g of 6-methyl-5-(3-methylisoxazol-5-yl)-1,2,3,4-tetrachlorobicyclo[2.2.1]hept-2en-7-one (a mixture of trans:cis=1:1) was dissolved in 1 ml of ethanol, and 0.06 g of sodium ethylate was added, while cooling with ice. The resulting solution was stirred for an hour at room temperature. The reaction solution was poured into 50 ml of ice-water, and extracted with 30 ml of ethyl acetate twice. The organic layer was washed with saturated salt water and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure. The obtained viscous liquid was purified on silica gel column chromatography (elution with n-hexane/ethyl acetate) to give 0.024 g of the title compound as white crystals. Melting point 86-88° C.

To 0.99 g of 6-methyl-cis-5-(3-methylisoxazol-5-yl)-7,7-dimethoxy-1,2,3,4-tetrachlorobicyclo[2.2]hept-2-ene was added 6.02 g of concentrated sulfuric acid, while cooling with ice. After returning to room temperature, the mixture was stirred for an hour. The resulting homogeneous solution was left undisturbed at room temperature for 15 hours. The reaction solution was poured into 20 ml of ice-water, and extracted with 20 ml of ethyl acetate twice. The organic layer was washed with aqueous sodium bicarbonate twice and then with saturated salt water, and dried over anhydrous magnesium sulfate. The magnesium sulfate was removed and the solvent was concentrated under reduced pressure to give 0.79 g of a crude product of 6-methyl-cis-5-(3-methylisoxazol-5-yl)-1,2,3,4-tetrachlorobicyclo[2.2.1]hept-2-en-7-one as a yellow sticky substance.
0.94 g of the obtained crude 6-methyl-cis-5-(3-methylisoxazol-5-yl)-1,2,3,4 tetrachlorobicyclo[2.2.1.]hept-2-en-7-one was dissolved in 10 ml of methanol, and 1.44 g of a 28% methanol solution of sodium methylate was dropped over 5 minutes, while cooling with ice. The resulting solution was stirred for 4 hours at room temperature, poured into 50 ml of ice-water, and extracted with 30 ml of ethyl acetate twice. The organic layer was washed with saturated salt water and dried over anhydrous magnesium sulfate. The solvent was concentrated under reduced pressure. The obtained viscous liquid was purified on silica gel column chromatography (elution with n-hexane/ethyl acetate) to give 0.42 g of the title compound as white crystals. Melting point: 76-77° C.

A solution of 156.9 g of 6-methyl-cis-5-(3-methylisoxazol-5-yl1,2,3,4-tetrachlorobicyclo[2.2.1]hept-2-en-7-one in 1150 ml of THF was dropped into a solution of 92 g of sodium hydroxide in 1150 ml of water at temperature between −3° C. and 0° C. over 1.5 hours. After the completion of the dropping, the cooling bath was removed. The reaction solution was stirred for 1.5 hours, while gradually rising the temperature to room temperature. After the reaction was completed, 500 ml of toluene was added to the reaction solution, and the aqueous layer was separated. Further, 500 ml of 1N aqueous solution of sodium hydroxide was added to the organic layer, and the aqueous layer was separated. The aqueous layers were combined, and 210 ml of concentrated hydrochloric acid was added to precipitate crystals. The crystals were separated by filtration, dissolved in 300 ml of ethyl acetate, and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure to give 95.0 g of the title compound as white crystals. Melting point: 212-213° C.

To 1.0 g of 6-methyl-cis-5-(2-isoxazolin-3-yl)-7,7-dimethoxy-1,2,3,4-tetrachlorobicyclo[2.2.1]hept-2-ene was added 6.3 g of concentrated sulfuric acid, while cooling with ice. After returning to room temperature, the mixture was stirred for an hour. The resulting homogeneous solution was left undisturbed at room temperature for 15 hours. The reaction solution was poured into 20 ml of ice-water and extracted with 20 ml of ethyl acetate twice. The organic layer was washed with aqueous sodium bicarbonate twice and then with saturated salt water, and dried over anhydrous magnesium sulfate. The magnesium sulfate was removed and the solvent was concentrated under reduced pressure to give 0.94 g of a crude product of 6-methyl-cis-5-(2-isoxazolin-3-yl)-1,2,3,4-tetrachlorobicyclo[2.2.1]hept-2-en-7-one as a yellow sticky substance.
0.94 g of the obtained crude product was dissolved in 10 ml of methanol, and 1.5 g of a 28% methanol solution of sodium methylate was dropped over 5 minutes, while cooling with ice. The resulting solution was stirred at room temperature for 2 hours. The reaction solution was poured into 50 ml of ice-water, and extracted with 30 ml of ethyl acetate twice. The organic layer was washed with saturated salt water and dried over anhydrous magnesium sulfate. The solvent was concentrated under reduced pressure. The obtained viscous liquid was purified on silica gel column chromatography (with development solvents: n-hexane/ethyl acetate) to give 0.71 g of the title compound as white crystals. Melting point: 74-76° C.
The above procedure was repeated to produce ethyl 4,5-dichloro-2-methyl-3-(2-isoxazolin-3-yl)benzoate. Melting point: 96-97° C.

1.6 g of a crude product of 6-methyl-cis-5-(2-isoxazolin-3-yl)-1,2,3,4-tetrachlorobicyclo[2.2.1]hept-2-en-7-one, obtained in the same manner as that in Example 4, was dissolved in 5 ml of tetrahydrofuran and 5 ml of water, and 14.6 g of 1N aqueous solution of sodium hydroxide was dropped over 5 minutes, while cooling with ice. The resulting solution was stirred at room temperature for 2 hours. The reaction solution was poured into 50 ml of ice-water, acidified in pH with hydrochloric acid, and extracted with 30 ml of ethyl acetate twice. The organic layer was washed with saturated salt water, and dried over anhydrous magnesium sulfate. The solvent was concentrated under reduced pressure. The obtained crystals were washed with n-hexane to give 0.6 g of the title compound as white crystals. Melting point: 203-206° C.

2.15 g of 5,5-dimethoxy-1,2,3,4-tetrachlorocyclopenta-1,3-diene, 0.81 g of sodium hydrogen carbonate, 1.00 g of 1-(3-methylisoxazol-5-yl)-1-propene (an equivalent mixture of trans and cis isomers), 0.40 g of zinc iodide and 0.10 g of hydroquinone were mixed and heated with stirring at 150° C. for 8 hours. The reaction solution was cooled and 100 ml of water was added. The resulting solution was extracted with 200 ml of ethyl acetate. Insoluble matter present was removed by filtration. The organic layer was washed with saturated salt water and dried over anhydrous magnesium sulfate. The solvent was concentrated under reduced pressure. n-Hexane was added to the obtained crude product. The precipitated crystals were separated by filtration to give 0.92 g of the title compound. Melting point: 100-101° C.
Further, the above filtrate was purified on silica gel column chromatography (elution with benzene) to give 0.74 g of the tide compound.

4.30 g of 5,5-dimethoxy-1,2,3,4-tetrachlorocyclopenta-1,3-diene, 1.00 g of 1-(3-methylisoxazol-5-yl)-1-propene (an equivalent mixture of trans and cis isomers) and 0.18 g of hydroquinone were added to 20 ml of 1,3-butanediol. The resulting mixture was heated at reflux for 7 hours. The reaction solution was cooled, poured into ice-water, and extracted with ethyl acetate. The organic layer was washed with saturated salt water, and dried over anhydrous magnesium sulfate. The solvent was concentrated under reduced pressure. n-Hexane was added to the obtained crude product. The separated viscous oil was removed. The n-hexane layer was concentrated under reduced pressure. The obtained crude product was purified on silica gel column chromatography (elution with benzene) to give 1.41 g of the title compound as white crystals. Melting point: 100-101° C.

1.07 g of 5,5-dimethoxy-1,2,3,4-tetrachlorocyclopenta-1,3-diene, 0.34 g of sodium hydrogen carbonate, 0.50g of trans-1-(3-methylisoxazol-5-yl)-1-propene and 0.05 g of hydroquinone were mixed and heated with stirring at 180° C. for 7 hours. The reaction solution was cooled and ethyl acetate was added. Insoluble matter was removed by filtration. Then the solvent was concentrated under reduced pressure. The obtained crude product was purified on silica gel column chromatography (elution with benzene) to give 0.70 g of the title compound as a mixture of isomers.

64.39 g of 5,5-dimethoxy-1,2,3,4-tetrachlorocyclopenta-1,3-diene, 14.07 g of sodium hydrogen carbonate, 20.00 g of cis-1-(3-methylisoxazol-5-yl)-1-propene and 2.00 g of hydroquinone were mixed and heated with stirring at 120° C. for 6 hours. The reaction solution was cooled and 500 ml of water was added. The resulting solution was extracted with ethyl acetate three times. Insoluble matter present was removed by filtration. The organic layer was washed with saturated salt water and dried over anhydrous magnesium sulfate.
The solvent was concentrated under reduced pressure. n-Hexane was added to the obtained crude product. The precipitated crystals were separated by filtration to give 45.81 g of the title compound. Melting point: 100-101° C.
Further, the filtrate after the filtration of the crystals was purified on silica gel column chromatography (elution with benzene) to give 4.50 g of the title compound.
1H-NMR data of isomers (CDCl3, δppm) cis isomer
0.83 (d, 3H), 2.30 (s, 3H), 3.08 (m, 1H), 3.58 (s, 3H), 3.66 (s, 3H), 4.17 (d, 1H), 6.02 (s, 1H) trans isomer-1
1.40 (d, 3H), 2.30 (s, 3H), 2.37 (m, 1H), 3.58 (s, 3H), 3.62 (d, 1H), 3.66 (s, 3H), 6.00 (s, 1H) trans isomer-2
1.09 (d, 3H), 2.34 (s, 3H), 2.84 (d, 1H), 3.07 (m, 1H), 3.50 (s, 3H), 3.57 (s, 3H), 6.19 (s, 1H)

2.14 g of 5,5-dimethoxy-1,2,3,4-tetrachlorocyclopenta-1,3-diene, 0.46 g of sodium hydrogen carbonate and 0.6 g of cis-1-(2-isoxazolin-3-yl)-1-propene were mixed and heated with stirring at 120° C. for 6 hours. The reaction solution was cooled to room temperature and ice-water was added. The resulting solution was extracted with 200 ml of ethyl acetate. The organic layer was washed with water and dried over anhydrous magnesium sulfate. The solvent was concentrated under reduced pressure. The obtained oily product was purified on silica gel column chromatography (elution with n-hexane/ethyl acetate) to give 1.0 g of the title compound. Melting point: 98-100° C.

2.62 g of 5,5-dimethoxy-1,2,3,4-tetrachlorocyclopenta-1,3-diene, 1.1 g of sodium hydrogen carbonate and 1.0 g of trans-1-(2-isoxazolin-3-yl)-1-propene were mixed and heated with stirring at 200° C. for 20 hours. The reaction solution was cooled to room temperature, poured into ice-water, and extracted with ethyl acetate. The organic layer was washed with water and dried over anhydrous magnesium sulfate. The solvent was concentrated under reduced pressure. The obtained oily product was purified on silica gel column chromatography (elution with n-hexane/ethyl acetate) to give 1.8 g of the title compound. nD 19.9 1.5087

0.50 g of ethyl 4,5-dichloro-2-methyl-3-(3-methylisoxazol-5-yl)benzoate and 4.40 g of potassium carbonate were added to 15 ml of DMF, and a solution of 1.53 g of methyl mercaptan dissolved in 5 ml of DMF was dropped at room temperature. The resulting solution was further stirred for 19.5 hours. Then, the reaction solution was poured into ice-water and extracted with ethyl acetate. The organic layer was washed with water and then with saturated salt water, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained crude product was purified on silica gel column chromatography to give 0.42 g of the title compound as white crystals. Melting point: 72-74° C.

4.48 g of ethyl 4,5-dichloro-2-methyl-3-(3-methylisoxazol-5-yl)benzoate and 3.00 g of potassium carbonate were added to 100 ml of DMF, and 2.1 g of methyl mercaptan gas was blown into the reaction system at room temperature. The resulting solution was further stirred at room temperature for 15 hours. The reaction solution was poured into ice-water and extracted with ethyl acetate. The organic layer was washed with water and then with saturated salt water, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to give 4.40 g of ethyl 5-chloro-2-methyl-3-(3-methylisoxazol-5-yl)-4-methylthiobenzoate as white crystals. Melting point: 79-82° C.
0.50 g of the obtained ethyl 5-chloro-2-methyl-3-(3-methylisoxazol-5-yl)-4-methylthiobenzoate and 1.06 g of potassium carbonate were added to 15 ml of DMF, and further a solution of 0.36 g of methyl mercaptan dissolved in 5 ml of DMF was dropped at room temperature. The resulting solution was stirred at room temperature for 23 hours. The same procedure as that in Example 1 was repeated to give 0.29 g of the title compound as white crystals. 0.08 g of ethyl 5-chloro-2-methyl-3-(3-methylisoxazol-5-yl)-4-methylthiobenzoate was recovered.

10 ml of degassed DMF was placed in a colorless, transparent glass container, and 420 mg of methyl mercaptan was blown in to dissolve. Then 600 mg of potassium carbonate and 500 mg of methyl 4,5-dichloro-3-(2-isoxazolin-3-yl)-2-methylbenzoate were added one by one. The resulting solution was stirred with a Teflon magnetic stirrer at room temperature with light shielded under the nitrogen atmosphere. After stirring it for an hour, it was confirmed by NMR that methyl 5-chloro-3-(2-isoxazolin-3-yl)-2-methyl-4-methylthiobenzoate was a major product. Succeedingly, the reaction system was stirred at 40° C. for 2 hours, while irradiating with white fluorescent light. The reaction solution was poured into ice-water, and extracted with ether-chloroform (5:1) and then with ether. The organic layers were washed with water, dried over sodium sulfate, and concentrated under reduced pressure. The obtained residue was purified on silica gel column chromatography (elution with n-hexane/ethyl acetate=95/5) to give 424 mg of the title compound as white crystals. Melting point: 109-111° C.

27 mg of water was added to 10 ml of DMF, and 360 mg of methyl mercaptan was blown into the resulting solution. Then the reaction system was purged by nitrogen. To the resulting solution were added 518 mg of potassium carbonate and 513 mg of t-butyl 4,5-dichloro-2-methyl-3-(3-methylisoxazol-5-yl)benzoate to stir at room temperature for 20 hours. The reaction solution was poured into 25 ml of ice-water and extracted with 25 ml of ether. The organic layer was washed with water and dried over anhydrous sodium sulfate. The solvent was distilled off under reduced pressure. The obtained crude product was purified on silica gel column chromatography (elution with n-hexane/ethyl acetate=95/5) to give 484 mg of the title compound with 83% purity as a white solid. (The purity was determined by NMR spectra.) The product was again purified on silica gel column chromatography to give the title compound with melting point of 102-103° C. as white crystals.

Into a 10-ml round-bottomed flask purged by nitrogen were placed 7.9 mg of water, 1.1 ml of DMF and a solution of 105 mg of methyl mercaptan in 1.5 ml of DMF, and then 151 mg of potassium carbonate and 132 mg of ethyl 4,5-dichloro-3-(2-isoxazol-3-yl)-2-methylbenzoate were added. The DMF used here was purged by nitrogen beforehand. The resulting mixture was stirred with a magnetic stirrer at room temperature for 3 hours and 45 minutes under the irradiation of 40W fluorescent light. The reaction solution was poured into 20 ml of ice-water, and extracted with 20 ml of diethyl ether. The organic layer was washed with water, and dried over anhydrous sodium sulfate. The solvent was distilled off under reduced pressure. The obtained crude product was purified on silica gel column chromatography (elution with n-hexane/ethyl acetate=95/5) to give 87.8 mg of the title compound as white crystals. Melting point: 93˜94° C.

Into a 250-ml round-bottomed flask were placed 0.311 g of ethyl mercaptan, 0.018 g of water and 6 ml of DMF, and then 0.346 g of potassium carbonate and 0.314 g of ethyl 4,5-dichloro-2-methyl-3-(3-methylisoxazol-5-yl)benzoate were adder The resulting mixture was stirred with a magnetic stirrer at room temperature for 6 hours under the irradiation of 40W fluorescent light. The reaction solution was poured into 25 ml of ice-water, and extracted with 25 ml of diethyl ether. The organic layer was washed with water, and dried over anhydrous sodium sulfate. The solvent was distilled off under reduced pressure. The obtained crude product was purified on silica gel column chromatography (elution with n-hexane/ethyl acetate=95/5) to give 0.132 g of the title compound as white crystals. Melting point: 39-40° C.
The above procedure was repeated to produce methyl 4-methylthio-2-methyl-3-(3-methylisoxazol-5-yl)benzoate by reacting methyl 4,5-dichloro-2-methyl-3-(3-methylisoxazol-5-yl)benzoate and methyl mercaptan. Melting point: 94-95° C.
Examples of the preparation of the aforementioned 4-SO2R4 compounds (8) are described in reference to Reference Examples.
Preparation of ethyl 2-methyl-3-(3-methylisoxazol-5-yl)-4-methylsulfonylbenzoate 
0.30 g of ethyl 5-chloro-2-methyl-3-(3-methylisoxazol-5-yl)-4-methylsulfonylbenzoate was dissolved in 20 ml of ethanol. After the reaction system was purged by nitrogen, 0.02 g of 5% palladium-carbon was added. The system was purged by hydrogen. A rubber ball filled with hydrogen was attached and the solution was stirred at room temperature for 3 hours. The palladium-carbon was removed from the reaction solution by filtration. The solvent was distilled off under reduced pressure to give 0.28 g of the title compound as white crystals. Melting point: 83-86° C.
Preparation of ethyl 5-chloro-2-methyl-3-(3-methylisoxazol-5-yl)-4-methylsulfonylbenzoate 
4.40 g of ethyl-5-chloro-2-methyl-3-(3-methylisoxazol-5-yl)-4-methylthiobenzoate was dissolved in 50 ml of chloroform, and 12.70 g of 55% m-chloroperbenzoic acid was added at room temperature. The resulting solution was stirred at room temperature for 19 hours. Precipitated crystals were separated from the reaction solution by filtration. The filtrate was washed with 80 ml of a 4% aqueous solution of sodium hydroxide. The organic layer was dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure to give 4.49 g of the title compound as white crystals. Melting point: 126-129° C.
Representative examples of the compounds of Formulae (1) and (2) of the present invention, including those in the above examples, are shown in Tables 1 and 2. The abbreviations of Q's in the tables have the following meanings:
Q1: isoxazol-5-yl, Q2: 3-methylisoxazol-5-yl, Q3: isoxazol-3-yl, Q4: 2-isoxazolin-3-yl, Q5: oxazol-2-yl, Q6: oxazol-4-yl, Q7: oxazol-5-yl, Q8: thiazol-2-yl, Q9: thiazol-4-yl, Q10: thiazol-5-yl, Q11: pyrazol-3-yl, Q12: 1-methyl-pyrazol-3-yl, Q13: 2-pyridyl: Q14: 3-pyridyl, Q15: 4-pyridyl, Q16: pyrimidin-2-yl, Q17: pyrimidin-4-yl, Q18: pyrimidin-5-yl, Q19: 1,3-dioxoran-2-yl, and Q20: 1,3-dioxan-2-yl
| TABLE 1 | |
| (1) | |
 |
|
| Compound No. | Q | ||
| Table 1-1 | |||
| R1: CH3 | |||
| R2: H | |||
| 1-1 | Q1 | ||
| 1-2 | Q2 | ||
| 1-3 | Q3 | ||
| 1-4 | Q4 | ||
| 1-5 | Q5 | ||
| 1-6 | Q6 | ||
| 1-7 | Q7 | ||
| 1-8 | Q8 | ||
| 1-9 | Q9 | ||
| 1-10 | Q10 | ||
| 1-11 | Q11 | ||
| 1-12 | Q12 | ||
| 1-13 | Q13 | ||
| 1-14 | Q14 | ||
| 1-15 | Q15 | ||
| 1-16 | Q16 | ||
| 1-17 | Q17 | ||
| 1-18 | Q18 | ||
| 1-19 | Q19 | ||
| 1-20 | Q20 | ||
| Table 1-2 | |||
| R1: CH3 | |||
| R2: CH3 | |||
| 1-21 | Q1 | ||
| 1-22 | Q2 | ||
| 1-23 | Q3 | ||
| 1-24 | Q4 | ||
| 1-25 | Q5 | ||
| 1-26 | Q6 | ||
| 1-27 | Q7 | ||
| 1-28 | Q8 | ||
| 1-29 | Q9 | ||
| 1-30 | Q10 | ||
| 1-31 | Q11 | ||
| 1-32 | Q12 | ||
| 1-33 | Q13 | ||
| 1-34 | Q14 | ||
| 1-35 | Q15 | ||
| 1-36 | Q16 | ||
| 1-37 | Q17 | ||
| 1-38 | Q18 | ||
| 1-39 | Q19 | ||
| 1-40 | Q20 | ||
| Table 1-3 | |||
| R1: CH3 | |||
| R2: C2H5 | |||
| 1-41 | Q1 | ||
| 1-42 | Q2 | ||
| 1-43 | Q3 | ||
| 1-44 | Q4 | ||
| 1-45 | Q5 | ||
| 1-46 | Q6 | ||
| 1-47 | Q7 | ||
| 1-48 | Q8 | ||
| 1-49 | Q9 | ||
| 1-50 | Q10 | ||
| 1-51 | Q11 | ||
| 1-52 | Q12 | ||
| 1-53 | Q13 | ||
| 1-54 | Q14 | ||
| 1-55 | Q15 | ||
| 1-56 | Q16 | ||
| 1-57 | Q17 | ||
| 1-58 | Q18 | ||
| 1-59 | Q19 | ||
| 1-60 | Q20 | ||
| Table 1-4 | |||
| R1: CH3 | |||
| R2: n-C3H7 | |||
| 1-61 | Q1 | ||
| 1-62 | Q2 | ||
| 1-63 | Q3 | ||
| 1-64 | Q4 | ||
| 1-65 | Q5 | ||
| 1-66 | Q6 | ||
| 1-67 | Q7 | ||
| 1-68 | Q8 | ||
| 1-69 | Q9 | ||
| 1-70 | Q10 | ||
| 1-71 | Q11 | ||
| 1-72 | Q12 | ||
| 1-73 | Q13 | ||
| 1-74 | Q14 | ||
| 1-75 | Q15 | ||
| 1-76 | Q16 | ||
| 1-77 | Q17 | ||
| 1-78 | Q18 | ||
| 1-79 | Q19 | ||
| 1-80 | Q20 | ||
| Table 1-5 | |||
| R1: CH3 | |||
| R2: i-C3H7 | |||
| 1-81 | Q1 | ||
| 1-82 | Q2 | ||
| 1-83 | Q3 | ||
| 1-84 | Q4 | ||
| 1-85 | Q5 | ||
| 1-86 | Q6 | ||
| 1-87 | Q7 | ||
| 1-88 | Q8 | ||
| 1-89 | Q9 | ||
| 1-90 | Q10 | ||
| 1-91 | Q11 | ||
| 1-92 | Q12 | ||
| 1-93 | Q13 | ||
| 1-94 | Q14 | ||
| 1-95 | Q15 | ||
| 1-96 | Q16 | ||
| 1-97 | Q17 | ||
| 1-98 | Q18 | ||
| 1-99 | Q19 | ||
| 1-100 | Q20 | ||
| Table 1-6 | |||
| R1: CH3 | |||
| R2: n-C4H9 | |||
| 1-101 | Q1 | ||
| 1-102 | Q2 | ||
| 1-103 | Q3 | ||
| 1-104 | Q4 | ||
| 1-105 | Q5 | ||
| 1-106 | Q6 | ||
| 1-107 | Q7 | ||
| 1-108 | Q8 | ||
| 1-109 | Q9 | ||
| 1-110 | Q10 | ||
| 1-111 | Q11 | ||
| 1-112 | Q12 | ||
| 1-113 | Q13 | ||
| 1-114 | Q14 | ||
| 1-115 | Q15 | ||
| 1-116 | Q16 | ||
| 1-117 | Q17 | ||
| 1-118 | Q18 | ||
| 1-119 | Q19 | ||
| 1-120 | Q20 | ||
| Table 1-7 | |||
| R1: CH3 | |||
| R2: n-C4H9 a | |||
| 1-121 | Q1 | ||
| 1-122 | Q2 | ||
| 1-123 | Q3 | ||
| 1-124 | Q4 | ||
| 1-125 | Q5 | ||
| 1-126 | Q6 | ||
| 1-127 | Q7 | ||
| 1-128 | Q8 | ||
| 1-129 | Q9 | ||
| 1-130 | Q10 | ||
| 1-131 | Q11 | ||
| 1-132 | Q12 | ||
| 1-133 | Q13 | ||
| 1-134 | Q14 | ||
| 1-135 | Q15 | ||
| 1-136 | Q16 | ||
| 1-137 | Q17 | ||
| 1-138 | Q18 | ||
| 1-139 | Q19 | ||
| 1-140 | Q20 | ||
| Table 1-8 | |||
| R1: CH3 | |||
| R2: t-C4H9 | |||
| 1-141 | Q1 | ||
| 1-142 | Q2 | ||
| 1-143 | Q3 | ||
| 1-144 | Q4 | ||
| 1-145 | Q5 | ||
| 1-146 | Q6 | ||
| 1-147 | Q7 | ||
| 1-148 | Q8 | ||
| 1-149 | Q9 | ||
| 1-150 | Q10 | ||
| 1-151 | Q11 | ||
| 1-152 | Q12 | ||
| 1-153 | Q13 | ||
| 1-154 | Q14 | ||
| 1-155 | Q15 | ||
| 1-156 | Q16 | ||
| 1-157 | Q17 | ||
| 1-158 | Q18 | ||
| 1-159 | Q19 | ||
| 1-160 | Q20 | ||
| Table 1-9 | |||
| R1: CH3 | |||
| R2: i-C5H11 | |||
| 1-161 | Q1 | ||
| 1-162 | Q2 | ||
| 1-163 | Q3 | ||
| 1-164 | Q4 | ||
| 1-165 | Q5 | ||
| 1-166 | Q6 | ||
| 1-167 | Q7 | ||
| 1-168 | Q8 | ||
| 1-169 | Q9 | ||
| 1-170 | Q10 | ||
| 1-171 | Q11 | ||
| 1-172 | Q12 | ||
| 1-173 | Q13 | ||
| 1-174 | Q14 | ||
| 1-175 | Q15 | ||
| 1-176 | Q16 | ||
| 1-177 | Q17 | ||
| 1-178 | Q18 | ||
| 1-179 | Q19 | ||
| 1-180 | Q20 | ||
| Table 1-10 | |||
| R1: CH3 | |||
| R2: t-C5H11 | |||
| 1-181 | Q1 | ||
| 1-182 | Q2 | ||
| 1-183 | Q3 | ||
| 1-184 | Q4 | ||
| 1-185 | Q5 | ||
| 1-186 | Q6 | ||
| 1-187 | Q7 | ||
| 1-188 | Q8 | ||
| 1-189 | Q9 | ||
| 1-190 | Q10 | ||
| 1-191 | Q11 | ||
| 1-192 | Q12 | ||
| 1-193 | Q13 | ||
| 1-194 | Q14 | ||
| 1-195 | Q15 | ||
| 1-196 | Q16 | ||
| 1-197 | Q17 | ||
| 1-198 | Q18 | ||
| 1-199 | Q19 | ||
| 1-200 | Q20 | ||
| Table 1-11 | |||
| R1: C2H5 | |||
| R2: H | |||
| 1-201 | Q1 | ||
| 1-202 | Q2 | ||
| 1-203 | Q3 | ||
| 1-204 | Q4 | ||
| 1-205 | Q5 | ||
| 1-206 | Q6 | ||
| 1-207 | Q7 | ||
| 1-208 | Q8 | ||
| 1-209 | Q9 | ||
| 1-210 | Q10 | ||
| 1-211 | Q11 | ||
| 1-212 | Q12 | ||
| 1-213 | Q13 | ||
| 1-214 | Q14 | ||
| 1-215 | Q15 | ||
| 1-216 | Q16 | ||
| 1-217 | Q17 | ||
| 1-218 | Q18 | ||
| 1-219 | Q19 | ||
| 1-220 | Q20 | ||
| Table 1-12 | |||
| R1: C2H5 | |||
| R2: CH3 | |||
| 1-221 | Q1 | ||
| 1-222 | Q2 | ||
| 1-223 | Q3 | ||
| 1-224 | Q4 | ||
| 1-225 | Q5 | ||
| 1-226 | Q6 | ||
| 1-227 | Q7 | ||
| 1-228 | Q8 | ||
| 1-229 | Q9 | ||
| 1-230 | Q10 | ||
| 1-231 | Q11 | ||
| 1-232 | Q12 | ||
| 1-233 | Q13 | ||
| 1-234 | Q14 | ||
| 1-235 | Q15 | ||
| 1-236 | Q16 | ||
| 1-237 | Q17 | ||
| 1-238 | Q18 | ||
| 1-239 | Q19 | ||
| 1-240 | Q20 | ||
| Table 1-13 | |||
| R1: C2H5 | |||
| R2: C2H5 | |||
| 1-241 | Q1 | ||
| 1-242 | Q2 | ||
| 1-243 | Q3 | ||
| 1-244 | Q4 | ||
| 1-245 | Q5 | ||
| 1-246 | Q6 | ||
| 1-247 | Q7 | ||
| 1-248 | Q8 | ||
| 1-249 | Q9 | ||
| 1-250 | Q10 | ||
| 1-251 | Q11 | ||
| 1-252 | Q12 | ||
| 1-253 | Q13 | ||
| 1-254 | Q14 | ||
| 1-255 | Q15 | ||
| 1-256 | Q16 | ||
| 1-257 | Q17 | ||
| 1-258 | Q18 | ||
| 1-259 | Q19 | ||
| 1-260 | Q20 | ||
| Table 1-14 | |||
| R1: C2H5 | |||
| R2: n-C3H7 | |||
| 1-261 | Q1 | ||
| 1-262 | Q2 | ||
| 1-263 | Q3 | ||
| 1-264 | Q4 | ||
| 1-265 | Q5 | ||
| 1-266 | Q6 | ||
| 1-267 | Q7 | ||
| 1-268 | Q8 | ||
| 1-269 | Q9 | ||
| 1-270 | Q10 | ||
| 1-271 | Q11 | ||
| 1-272 | Q12 | ||
| 1-273 | Q13 | ||
| 1-274 | Q14 | ||
| 1-275 | Q15 | ||
| 1-276 | Q16 | ||
| 1-277 | Q17 | ||
| 1-278 | Q18 | ||
| 1-279 | Q19 | ||
| 1-280 | Q20 | ||
| Table 1-15 | |||
| R1: C2H5 | |||
| R2: i-C3H7 | |||
| 1-281 | Q1 | ||
| 1-282 | Q2 | ||
| 1-283 | Q3 | ||
| 1-284 | Q4 | ||
| 1-285 | Q5 | ||
| 1-286 | Q6 | ||
| 1-287 | Q7 | ||
| 1-288 | Q8 | ||
| 1-289 | Q9 | ||
| 1-290 | Q10 | ||
| 1-291 | Q11 | ||
| 1-292 | Q12 | ||
| 1-293 | Q13 | ||
| 1-294 | Q14 | ||
| 1-295 | Q15 | ||
| 1-296 | Q16 | ||
| 1-297 | Q17 | ||
| 1-298 | Q18 | ||
| 1-299 | Q19 | ||
| 1-300 | Q20 | ||
| Table 1-16 | |||
| R1: C2H5 | |||
| R2: n-C4H9 | |||
| 1-301 | Q1 | ||
| 1-302 | Q2 | ||
| 1-303 | Q3 | ||
| 1-304 | Q4 | ||
| 1-305 | Q5 | ||
| 1-306 | Q6 | ||
| 1-307 | Q7 | ||
| 1-308 | Q8 | ||
| 1-309 | Q9 | ||
| 1-310 | Q10 | ||
| 1-311 | Q11 | ||
| 1-312 | Q12 | ||
| 1-313 | Q13 | ||
| 1-314 | Q14 | ||
| 1-315 | Q15 | ||
| 1-316 | Q16 | ||
| 1-317 | Q17 | ||
| 1-318 | Q18 | ||
| 1-319 | Q19 | ||
| 1-320 | Q20 | ||
| Table 1-17 | |||
| R1: C2H5 | |||
| R2: s-C4H9 | |||
| 1-321 | Q1 | ||
| 1-322 | Q2 | ||
| 1-323 | Q3 | ||
| 1-324 | Q4 | ||
| 1-325 | Q5 | ||
| 1-326 | Q6 | ||
| 1-327 | Q7 | ||
| 1-328 | Q8 | ||
| 1-329 | Q9 | ||
| 1-330 | Q10 | ||
| 1-331 | Q11 | ||
| 1-332 | Q12 | ||
| 1-333 | Q13 | ||
| 1-334 | Q14 | ||
| 1-335 | Q15 | ||
| 1-336 | Q16 | ||
| 1-337 | Q17 | ||
| 1-338 | Q18 | ||
| 1-339 | Q19 | ||
| 1-340 | Q20 | ||
| Table 1-18 | |||
| R1: C2H5 | |||
| R2: t-C4H9 | |||
| 1-341 | Q1 | ||
| 1-342 | Q2 | ||
| 1-343 | Q3 | ||
| 1-344 | Q4 | ||
| 1-345 | Q5 | ||
| 1-346 | Q6 | ||
| 1-347 | Q7 | ||
| 1-348 | Q8 | ||
| 1-349 | Q9 | ||
| 1-350 | Q10 | ||
| 1-351 | Q11 | ||
| 1-352 | Q12 | ||
| 1-353 | Q13 | ||
| 1-354 | Q14 | ||
| 1-355 | Q15 | ||
| 1-356 | Q16 | ||
| 1-357 | Q17 | ||
| 1-358 | Q18 | ||
| 1-359 | Q19 | ||
| 1-360 | Q20 | ||
| Table 1-19 | |||
| R1: C2H5 | |||
| R2: i-C5H11 | |||
| 1-361 | Q1 | ||
| 1-362 | Q2 | ||
| 1-363 | Q3 | ||
| 1-364 | Q4 | ||
| 1-365 | Q5 | ||
| 1-366 | Q6 | ||
| 1-367 | Q7 | ||
| 1-368 | Q8 | ||
| 1-369 | Q9 | ||
| 1-370 | Q10 | ||
| 1-371 | Q11 | ||
| 1-372 | Q12 | ||
| 1-373 | Q13 | ||
| 1-374 | Q14 | ||
| 1-375 | Q15 | ||
| 1-376 | Q16 | ||
| 1-377 | Q17 | ||
| 1-378 | Q18 | ||
| 1-379 | Q19 | ||
| 1-380 | Q20 | ||
| Table 1-20 | |||
| R1: C2H5 | |||
| R2: t-C5H11 | |||
| 1-381 | Q1 | ||
| 1-382 | Q2 | ||
| 1-383 | Q3 | ||
| 1-384 | Q4 | ||
| 1-385 | Q5 | ||
| 1-386 | Q6 | ||
| 1-387 | Q7 | ||
| 1-388 | Q8 | ||
| 1-389 | Q9 | ||
| 1-390 | Q10 | ||
| 1-391 | Q11 | ||
| 1-392 | Q12 | ||
| 1-393 | Q13 | ||
| 1-394 | Q14 | ||
| 1-395 | Q15 | ||
| 1-396 | Q16 | ||
| 1-397 | Q17 | ||
| 1-398 | Q18 | ||
| 1-399 | Q19 | ||
| 1-400 | Q20 | ||
| TABLE 2 | |
| (2) | |
 |
|
| Compound No. | Q | ||
| Table 2-1 | |||
| R1: CH3 | |||
| 2-1 | Q1 | ||
| 2-2 | Q2 | ||
| 2-3 | Q3 | ||
| 2-4 | Q4 | ||
| 2-5 | Q5 | ||
| 2-6 | Q6 | ||
| 2-7 | Q7 | ||
| 2-8 | Q8 | ||
| 2-9 | Q9 | ||
| 2-10 | Q10 | ||
| 2-11 | Q11 | ||
| 2-12 | Q12 | ||
| 2-13 | Q13 | ||
| 2-14 | Q14 | ||
| 2-15 | Q15 | ||
| 2-16 | Q16 | ||
| 2-17 | Q17 | ||
| 2-18 | Q18 | ||
| 2-19 | Q19 | ||
| 2-20 | Q20 | ||
| Table 2-2 | |||
| R1: C2H5 | |||
| 2-21 | Q1 | ||
| 2-22 | Q2 | ||
| 2-23 | Q3 | ||
| 2-24 | Q4 | ||
| 2-25 | Q5 | ||
| 2-26 | Q6 | ||
| 2-27 | Q7 | ||
| 2-28 | Q8 | ||
| 2-29 | Q9 | ||
| 2-30 | Q10 | ||
| 2-31 | Q11 | ||
| 2-32 | Q12 | ||
| 2-33 | Q13 | ||
| 2-34 | Q14 | ||
| 2-35 | Q15 | ||
| 2-36 | Q16 | ||
| 2-37 | Q17 | ||
| 2-38 | Q18 | ||
| 2-39 | Q19 | ||
| 2-40 | Q20 | ||
| TABLE 3 | |
| (6) | |
 |
|
| Table 3-1 |
| Compounds 3-1 to 3-400, where R4 is CH3, that are derived from |
| Compounds Nos. from 1-1 to 1-400 in Table 1. |
| Table 3-2 |
| Compounds 3-401 to 3-800, where R4 is C2H5, that are derived from |
| Compounds Nos. from 1-1 to 1-400 in Table 1. |
| Table 3-3 |
| Compounds 3-801 to 3-1200, where R4 is i-C3H7, that are derived from |
| Compounds Nos. from 1-1 to 1-400 in Table 1. |
| Table 3-4 |
| Compounds 3-1201 to 3-1600, where R4 is t-C4H9, that are derived from |
| Compounds Nos. from 1-1 to 1-400 in Table 1. |
As described above, according to the present invention, benzoic acid derivatives useful as intermediates for the preparation of agricultural chemicals and drugs, particularly compounds having herbicidal activity, can be produced simply, easily and industrially advantageously in a short process, using inexpensive cyclopentadiene derivatives and ethylene derivatives substituted with hetero rings as starting materials.
For example, reactions of compounds of Formula (6), which are produced according to the present invention, with 3-hydroxypyrazoles give herbicides disclosed in WO 96/26206, WO 97/41118 and others.
Claims (5)
1. A process for the preparation of a substituted benzoic acid of Formula (1) 
wherein R1is hydrogen or C1-4 alkyl, R2 is hydrogen or C1-6 alkyl, and Q is an optionally substituted, saturated or unsaturated, 5- or 6-membered heterocyclic group wherein said heterocyclic group is selected from the group consisting of:
a) a five member heterocyclic ring having at least one heteroatom which is selected from the group consisting of N, O, or S, and where the heterocyclic group is linked to the benzene ring via a carbon atom; or
b) a six member heterocyclic ring having at least one heteroatom which is selected from the group consisting of N, O, or S, and where the heterocyclic group is linked to the benzene ring via a carbon atom;
which comprises reacting a substituted bicycloheptenone of Formula (2) 
wherein R1 is as defined above and Q 
is an optionally substituted, saturated or unsaturated, 5- or 6-membered heterocyclic group wherein said heterocyclic group is selected from the group consisting of:
a) a five member heterocyclic ring having at least one heteroatom which is selected from the group consisting of N, O, or S, and where the heterocyclic group is linked to the benzene ring via a carbon atom; or
b) a six member heterocyclic ring having at least one heteroatom which is selected from the group consisting of N, O, or S, and where the heterocyclic group is linked to the benzene ring via a carbon atom;
with a base and water or alcohol of Formula R2OH, wherein R2 is hydrogen or C1-6 alkyl, in an appropriate solvent, and wherein the base is an alkali metal alkoxide, all alkali metal hydroxide or an alkaline earth metal hydroxide, the alcohol is methanol, ethanol, propanol, isopropanol, butanol or t-butanol, and the solvent is methanol, ethanol, propanol, isopropanol, butanol or t-butanol, or THF.
2. A process for the preparation of a substituted benzoic acid of Formula (1) 
wherein R1 is hydrogen or C1-4 alkyl, R2 is hydrogen or C1-6 alkyl, and Q is selected from the group consisting of Q-1, Q-2, Q-3, Q-4, Q-5, Q-6, Q-7, Q-8 and Q-9 
wherein R3 is C1-4 alkyl or C1-4 haloalkyl, and n is 0 or an integer of 1 or 2, which comprises having a stage for the preparation of a substituted bicycloheptenone of Formula (2) 
wherein R1 and Q are as defined above by hydrolysis of a substituted bicycloheptene of Formula (3) 
wherein R1 and Q are as defined above, and X is chlorine or C1-4 alkoxy; and the two X's may join together to form a C2-3 alkylenedioxy group optionally substituted with C1-4 alkyl, and a stage of reacting the substituted bicycloheptenone of the above Formula (2) with a base and water or alcohol in an appropriate solvent, and wherein the base is an alkali metal alkoxide, an alkali metal hydroxide or an alkaline earth metal hydroxide, the alcohol is methanol, ethanol, propanol, isopropanol, butanol or t-butanol, and the solvent is methanol, ethanol, propanol, isopropanol, butanol or t-butanol, or THF.
3. A process for the preparation of a substituted benzoic acid of the above Formula (1) according to claim 1 , in which the base is one or more compounds selected from the group consisting of alkali metal alkoxides, alkali metal hydroxides and alkaline earth metal hydroxides.
4. A process according to claim 1 , in which the solvent is an alcohol or a solvent containing an alcohol.
5. A process according to claim 2 , in which the hetero ring represented by Q is a group selected from the following group consisting of Q-1, Q-2, Q-3, Q-4, Q-5, Q-6, Q-7, Q-8 and Q-9 
wherein R3 is C1-4 alkyl or C1-4 haloalkyl, and n is 0 or an integer of 1 or 2.
Applications Claiming Priority (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP19912598 | 1998-07-14 | ||
| JP19912698 | 1998-07-14 | ||
| JP10-199125 | 1998-07-14 | ||
| JP11-064856 | 1999-03-11 | ||
| JP6485699 | 1999-03-11 | ||
| JP12293999 | 1999-04-28 | ||
| JP11-122939 | 1999-04-28 | ||
| JP10-199126 | 1999-04-28 | ||
| PCT/JP1999/003773 WO2000003988A1 (en) | 1998-07-14 | 1999-07-13 | Novel benzoic acid derivatives and processes for the preparation thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| US6756497B1 true US6756497B1 (en) | 2004-06-29 |
Family
ID=27464496
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/720,825 Expired - Fee Related US6756497B1 (en) | 1998-07-14 | 1999-07-13 | Benzoic acid derivatives and processes for the preparation thereof |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US6756497B1 (en) |
| EP (1) | EP1097925B1 (en) |
| DE (1) | DE69933222T2 (en) |
| WO (1) | WO2000003988A1 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9398768B2 (en) | 2012-04-27 | 2016-07-26 | Basf Se | Substituted N-(tetrazol-5-yl)- and N-(triazol-5-yl)pyridin-3-yl-carboxamide compounds and their use as herbicides |
| US9902704B2 (en) | 2013-10-10 | 2018-02-27 | Basf Se | Substituted N-(tetrazol-5-yl)- and N-(triazol-5-yl)arylcarboxamide compounds and their use as herbicides |
| US9926284B2 (en) | 2013-07-18 | 2018-03-27 | Basf Se | Substituted N-(1,2,4-triazol-3-yl)Arylcarboxamide compounds and their use as herbicides |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4804615B2 (en) * | 2000-09-12 | 2011-11-02 | 日本曹達株式会社 | Method for producing bicycloheptene derivative |
| AR088857A1 (en) | 2011-11-14 | 2014-07-10 | Basf Se | 1,2,5-OXADIAZOL SUBSTITUTED COMPOUNDS AND ITS USE AS HERBICIDES |
| CN104428297A (en) | 2012-04-27 | 2015-03-18 | 巴斯夫欧洲公司 | Substituted N-(tetrazol-5-yl)- and N-(triazol-5-yl)arylcarboxamide compounds and their use as herbicides |
| JP6426714B2 (en) | 2013-05-15 | 2018-11-21 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | Substituted N- (tetrazol-5-yl)-and N- (triazol-5-yl) arylcarboxamide compounds and their use as herbicides |
| WO2014184058A1 (en) | 2013-05-15 | 2014-11-20 | Basf Se | Substituted 1,2,5-oxadiazole compounds and their use as herbicides |
| WO2015052173A1 (en) | 2013-10-10 | 2015-04-16 | Basf Se | Tetrazole and triazole compounds and their use as herbicides |
| WO2015052178A1 (en) | 2013-10-10 | 2015-04-16 | Basf Se | 1,2,5-oxadiazole compounds and their use as herbicides |
| EP2907807A1 (en) | 2014-02-18 | 2015-08-19 | Basf Se | Benzamide compounds and their use as herbicides |
| CN108473444A (en) | 2015-12-17 | 2018-08-31 | 巴斯夫欧洲公司 | Benzamide compounds and its purposes as herbicide |
| WO2018219935A1 (en) | 2017-05-30 | 2018-12-06 | Basf Se | Benzamide compounds and their use as herbicides |
| AU2018276360A1 (en) | 2017-05-30 | 2019-12-19 | Basf Se | Benzamide compounds and their use as herbicides II |
| AR112112A1 (en) | 2017-06-20 | 2019-09-18 | Basf Se | BENZAMIDE COMPOUNDS AND THEIR USE AS HERBICIDES |
| AR112342A1 (en) | 2017-07-21 | 2019-10-16 | Basf Se | BENZAMIDE COMPOUNDS AND THEIR USE AS HERBICIDES |
| WO2019122345A1 (en) | 2017-12-22 | 2019-06-27 | Basf Se | Benzamide compounds and their use as herbicides |
| WO2019162309A1 (en) | 2018-02-21 | 2019-08-29 | Basf Se | Benzamide compounds and their use as herbicides |
| WO2019162308A1 (en) | 2018-02-21 | 2019-08-29 | Basf Se | Benzamide compounds and their use as herbicides |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3625977A (en) | 1968-10-04 | 1971-12-07 | Eastman Kodak Co | 2-acetal-7-ketal-5-norbornene and 2-acetal-7-ketalnorbornane compounds |
| US4643757A (en) * | 1985-05-20 | 1987-02-17 | Nissan Chemical Industries, Ltd. | Herbicidal 4-benzoyl-1-methyl-5-hydroxypyrazoles |
| US4966998A (en) | 1985-03-07 | 1990-10-30 | Ici Americas Inc. | Trisubstituted benzoic acid intermediates |
| WO1996026206A1 (en) * | 1995-02-24 | 1996-08-29 | Basf Aktiengesellschaft | Pyrazol-4-yl-benzoyl derivatives and their use as herbicides |
| US5834402A (en) * | 1995-02-24 | 1998-11-10 | Basf Aktiengesellschaft | Isoxazolylbenzoyl derivatives |
| US5948917A (en) * | 1996-03-26 | 1999-09-07 | Nippon Soda Co., Ltd. | 3-(isoxazol-5-yl)-substituted benzoic acid derivative and method for production thereof |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4986845A (en) * | 1988-07-15 | 1991-01-22 | Nissan Chemical Industries Ltd. | Pyrazole derivatives and herbicides containing them |
-
1999
- 1999-07-13 US US09/720,825 patent/US6756497B1/en not_active Expired - Fee Related
- 1999-07-13 EP EP99929810A patent/EP1097925B1/en not_active Expired - Lifetime
- 1999-07-13 WO PCT/JP1999/003773 patent/WO2000003988A1/en not_active Ceased
- 1999-07-13 DE DE69933222T patent/DE69933222T2/en not_active Expired - Fee Related
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3625977A (en) | 1968-10-04 | 1971-12-07 | Eastman Kodak Co | 2-acetal-7-ketal-5-norbornene and 2-acetal-7-ketalnorbornane compounds |
| US4966998A (en) | 1985-03-07 | 1990-10-30 | Ici Americas Inc. | Trisubstituted benzoic acid intermediates |
| US4643757A (en) * | 1985-05-20 | 1987-02-17 | Nissan Chemical Industries, Ltd. | Herbicidal 4-benzoyl-1-methyl-5-hydroxypyrazoles |
| WO1996026206A1 (en) * | 1995-02-24 | 1996-08-29 | Basf Aktiengesellschaft | Pyrazol-4-yl-benzoyl derivatives and their use as herbicides |
| US5834402A (en) * | 1995-02-24 | 1998-11-10 | Basf Aktiengesellschaft | Isoxazolylbenzoyl derivatives |
| US5948917A (en) * | 1996-03-26 | 1999-09-07 | Nippon Soda Co., Ltd. | 3-(isoxazol-5-yl)-substituted benzoic acid derivative and method for production thereof |
Non-Patent Citations (5)
| Title |
|---|
| Hoch, a study of the acid and bse hydrolysis of bridged ketones, Nov. 1960, J. of Organic Chem., 26, 2066-71.* * |
| Hoch; A Study of the Acid and Base Hydrolysis of Bridged Ketones Derived from Diels-Alder Adducts of 1,2,3,4-Tetrachloro-5,5-dimethoxycyclopentadiene, Chem. & Ind., 110 (1958), vol. 26 p. 2066-2072. |
| Mark; Nonstereospecific Diels-Alder Reactions. I. Reaction of Hexachlorocyclopentadiene with 1,2-Disubstituted Ethylenes. J. Org. Chem., vol. 39, No. 21, p. 3179-3181 (1974). |
| Mitchell et al, an approach utilizing a diels-alder route to 1,2,3-trisubstituted benzenes, Apr. 1985, Tetrahedron, 42, 1741-42.* * |
| Mitchell et al.; Cis-dimethyldihydropyrene synhesis. An Approach Utilizing a Diels-Alder Route to 1,2,3-Trisubstituted Benzenes.; Tetrahedron, vol. 42, No. 6, p. 1741-1744 (1986). |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9398768B2 (en) | 2012-04-27 | 2016-07-26 | Basf Se | Substituted N-(tetrazol-5-yl)- and N-(triazol-5-yl)pyridin-3-yl-carboxamide compounds and their use as herbicides |
| US9926284B2 (en) | 2013-07-18 | 2018-03-27 | Basf Se | Substituted N-(1,2,4-triazol-3-yl)Arylcarboxamide compounds and their use as herbicides |
| US9902704B2 (en) | 2013-10-10 | 2018-02-27 | Basf Se | Substituted N-(tetrazol-5-yl)- and N-(triazol-5-yl)arylcarboxamide compounds and their use as herbicides |
Also Published As
| Publication number | Publication date |
|---|---|
| DE69933222T2 (en) | 2007-01-04 |
| WO2000003988A1 (en) | 2000-01-27 |
| DE69933222D1 (en) | 2006-10-26 |
| EP1097925A4 (en) | 2001-12-19 |
| EP1097925B1 (en) | 2006-09-13 |
| EP1097925A1 (en) | 2001-05-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6756497B1 (en) | Benzoic acid derivatives and processes for the preparation thereof | |
| AU2020343788B2 (en) | Piperonylic acid derivative and application thereof | |
| EP1541561B1 (en) | Pyrazole derivatives and process for the production thereof | |
| ES2674813T3 (en) | Heterocyclic compound containing partially saturated nitrogen | |
| WO2021127643A1 (en) | Fluoroalkyl-oxadiazoles and uses thereof | |
| AU2006311797A1 (en) | Aminopyrimidines useful as kinase inhibitors | |
| AU2004225977A1 (en) | Thiazoles useful as inhibitors of protein kinases | |
| IL147651A (en) | Isoxazoline derivative and herbicide containing the same as the active ingredient | |
| WO2003064397A1 (en) | Indazole compounds useful as protein kinase inhibitors | |
| AU2003212833A1 (en) | Indazole compounds useful as protein kinase inhibitors | |
| EA010628B1 (en) | 5-phenylpyrimidines, method for production and use thereof | |
| CA3149137A1 (en) | Process and intermediates for the preparation of pyroxasulfone | |
| CN115066419B (en) | Preparation of Substituted 3-Aryl-5-trifluoromethyl-1,2,4-oxadiazoles | |
| KR102946998B1 (en) | Preparation of substituted 3-aryl-5-trifluoromethyl-1,2,4-oxadiazole | |
| UA73735C2 (en) | A method for the preparation of substituted pirimidines (variants) | |
| JP4377992B2 (en) | Process for producing aromatic thioether compound | |
| ES2315441T3 (en) | PROCESS FOR THE PREPARATION OF CYCLIC DICETONES. | |
| JP4592129B2 (en) | Novel benzoic acid derivatives and process for producing the same | |
| TW202146395A (en) | 6-(fluoroalkyl)-3,4-dihydro-2h-pyran-5-carboxylic acid ester derivative, method for producing derivative, method for producing 2-(fluoroalkyl)nicotinic acid ester derivative, and method for producing 2-(fluoroalkyl)nicotinic acid derivative | |
| EP0934255B1 (en) | Production of pyridazine herbicides | |
| JP4804615B2 (en) | Method for producing bicycloheptene derivative | |
| JP4600859B2 (en) | Heterocyclic substituted thiophenol compounds, intermediates for production thereof, and production methods | |
| WO2026041700A1 (en) | Process for preparing oxazole compounds | |
| WO2001053271A1 (en) | Imidazole compounds and their use as adenosine deaminase inhibitors | |
| AU2013201630B2 (en) | Aminopyrimidines useful as kinase inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: NIPPON SODA CO., LTD., JAPAN Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:NAKAGAWA, YUUKI;YAMAGUCHI, MASAO;ADACHI, HIROYUKI;AND OTHERS;REEL/FRAME:011504/0598;SIGNING DATES FROM 20001212 TO 20001218 |
|
| FEPP | Fee payment procedure |
Free format text: PAYOR NUMBER ASSIGNED (ORIGINAL EVENT CODE: ASPN); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY |
|
| REMI | Maintenance fee reminder mailed | ||
| LAPS | Lapse for failure to pay maintenance fees | ||
| STCH | Information on status: patent discontinuation |
Free format text: PATENT EXPIRED DUE TO NONPAYMENT OF MAINTENANCE FEES UNDER 37 CFR 1.362 |
|
| FP | Lapsed due to failure to pay maintenance fee |
Effective date: 20080629 |