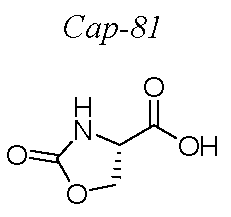
KR101624365B1 - C형 간염 바이러스 억제제 - Google Patents
C형 간염 바이러스 억제제 Download PDFInfo
- Publication number
- KR101624365B1 KR101624365B1 KR1020107020306A KR20107020306A KR101624365B1 KR 101624365 B1 KR101624365 B1 KR 101624365B1 KR 1020107020306 A KR1020107020306 A KR 1020107020306A KR 20107020306 A KR20107020306 A KR 20107020306A KR 101624365 B1 KR101624365 B1 KR 101624365B1
- Authority
- KR
- South Korea
- Prior art keywords
- alkyl
- mmol
- aryl
- heterocyclyl
- halo
- Prior art date
Links
- 0 C(C1*CC1)C1*CCC1 Chemical compound C(C1*CC1)C1*CCC1 0.000 description 11
- JVWZSHDIPVXYQE-UHFFFAOYSA-N CC(C(O)=O)(c1ccccc1)N(C)C Chemical compound CC(C(O)=O)(c1ccccc1)N(C)C JVWZSHDIPVXYQE-UHFFFAOYSA-N 0.000 description 1
- VXIIZQXOIDYWBS-QUFPSDLASA-N CC(C)(C)OC(N(C(C1)[C@H]1C1)[C@@H]1C(O)=O)=O Chemical compound CC(C)(C)OC(N(C(C1)[C@H]1C1)[C@@H]1C(O)=O)=O VXIIZQXOIDYWBS-QUFPSDLASA-N 0.000 description 1
- PDCUJLHHLCAIAN-DNKZHYAASA-N CC(C)(C)OC(N(CCC1)C1c1ncc(-c(cc2)ccc2-c2ccc(-c(nc3[C@H](CCCC4)N4C(OC(C)(C)C)=O)c(CCCC4)[n]3O)c4c2)[nH]1)=O Chemical compound CC(C)(C)OC(N(CCC1)C1c1ncc(-c(cc2)ccc2-c2ccc(-c(nc3[C@H](CCCC4)N4C(OC(C)(C)C)=O)c(CCCC4)[n]3O)c4c2)[nH]1)=O PDCUJLHHLCAIAN-DNKZHYAASA-N 0.000 description 1
- WWYQPVXUIRJIMN-HEVIKAOCSA-N CC(C)(C)OC(N(CCC1)[C@@H]1c1ncc(-c(cc2)ccc2-c2ccc(-c3c(CCCC4)[nH]c([C@H](CCCC5)N5C(OC(C)(C)C)=O)n3)c4c2)[nH]1)=O Chemical compound CC(C)(C)OC(N(CCC1)[C@@H]1c1ncc(-c(cc2)ccc2-c2ccc(-c3c(CCCC4)[nH]c([C@H](CCCC5)N5C(OC(C)(C)C)=O)n3)c4c2)[nH]1)=O WWYQPVXUIRJIMN-HEVIKAOCSA-N 0.000 description 1
- SIGJBDNBLCTRPL-SCSAIBSYSA-N CC(C)([C@@H](C(O)=O)NC(OC)=O)O Chemical compound CC(C)([C@@H](C(O)=O)NC(OC)=O)O SIGJBDNBLCTRPL-SCSAIBSYSA-N 0.000 description 1
- VNWSHSMCDQYRJL-LEKBNUHKSA-N CC(C)[C@@H](C(N(C(C1C2)C1N=C)[C@@H]2c1ncc(-c(c(F)c2)ccc2-c2cc3ccc4[nH]c([C@H](CC5C6C5)N6OC(c5ccccc5)=O)nc4c3cc2)[nH]1)=O)NC(OC)=O Chemical compound CC(C)[C@@H](C(N(C(C1C2)C1N=C)[C@@H]2c1ncc(-c(c(F)c2)ccc2-c2cc3ccc4[nH]c([C@H](CC5C6C5)N6OC(c5ccccc5)=O)nc4c3cc2)[nH]1)=O)NC(OC)=O VNWSHSMCDQYRJL-LEKBNUHKSA-N 0.000 description 1
- SPTVPPUZZFHDTO-CPCREDONSA-N CC(C)[C@@H](C(N(CCC1)[C@@H]1c1nc2ccc(cc(cc3)-c4cc5ccc6[nH]c([C@H]7NCCCC7)nc6c5cc4)c3c2[nH]1)=O)NC(OC)=O Chemical compound CC(C)[C@@H](C(N(CCC1)[C@@H]1c1nc2ccc(cc(cc3)-c4cc5ccc6[nH]c([C@H]7NCCCC7)nc6c5cc4)c3c2[nH]1)=O)NC(OC)=O SPTVPPUZZFHDTO-CPCREDONSA-N 0.000 description 1
- HQQGFVVRYLEWDY-MTHGYUFKSA-N CC(C)[C@@H](C(N([C@@H](CC1C2)c3nc4ccc(cc(cc5)-c6cc7ccc8[nH]c([C@H](CC[C@@H]9C%10C9)N%10OC(c9ccccc9)=O)nc8c7cc6)c5c4[nH]3)C12C=C)=O)NC(OC)=O Chemical compound CC(C)[C@@H](C(N([C@@H](CC1C2)c3nc4ccc(cc(cc5)-c6cc7ccc8[nH]c([C@H](CC[C@@H]9C%10C9)N%10OC(c9ccccc9)=O)nc8c7cc6)c5c4[nH]3)C12C=C)=O)NC(OC)=O HQQGFVVRYLEWDY-MTHGYUFKSA-N 0.000 description 1
- LQCMMXGKEGWUIM-UHFFFAOYSA-N CC(c(c(O)c1)ccc1Br)=O Chemical compound CC(c(c(O)c1)ccc1Br)=O LQCMMXGKEGWUIM-UHFFFAOYSA-N 0.000 description 1
- ZFJVQDOOKDWBQX-UHFFFAOYSA-N CCCCCC[ClH+] Chemical compound CCCCCC[ClH+] ZFJVQDOOKDWBQX-UHFFFAOYSA-N 0.000 description 1
- YQYOETRYLLWJMC-NNAABLSCSA-N CCN(CC)[C@@H](C(N(C(C1)C1C1)[C@@H]1c1ncc(-c(c(F)c2)ccc2-c2cc3ccc4[nH]c([C@H](C[C@@H]5C6CC5)N6C([C@@H](c5ccccc5)N(CC)CC)=O)nc4c3cc2)[nH]1)=O)c1ccccc1 Chemical compound CCN(CC)[C@@H](C(N(C(C1)C1C1)[C@@H]1c1ncc(-c(c(F)c2)ccc2-c2cc3ccc4[nH]c([C@H](C[C@@H]5C6CC5)N6C([C@@H](c5ccccc5)N(CC)CC)=O)nc4c3cc2)[nH]1)=O)c1ccccc1 YQYOETRYLLWJMC-NNAABLSCSA-N 0.000 description 1
- ZKXOJPCWOHKOQR-UHFFFAOYSA-N CCOc1c(cc(nc2C#N)Cl)c2ccc1 Chemical compound CCOc1c(cc(nc2C#N)Cl)c2ccc1 ZKXOJPCWOHKOQR-UHFFFAOYSA-N 0.000 description 1
- ANQBUBVGJORSTE-UHFFFAOYSA-N CN(C)C1(C(O)=O)c2ccccc2CC1 Chemical compound CN(C)C1(C(O)=O)c2ccccc2CC1 ANQBUBVGJORSTE-UHFFFAOYSA-N 0.000 description 1
- SKCUMFJFAWFXDL-UHFFFAOYSA-N CN(C)C1(Cc2ccccc2C1)C(O)=O Chemical compound CN(C)C1(Cc2ccccc2C1)C(O)=O SKCUMFJFAWFXDL-UHFFFAOYSA-N 0.000 description 1
- MLOBRLOZPSSKKO-SECBINFHSA-N CN(C)[C@@H](C(O)=O)c1ccccc1 Chemical compound CN(C)[C@@H](C(O)=O)c1ccccc1 MLOBRLOZPSSKKO-SECBINFHSA-N 0.000 description 1
- NLVXWIBDIWBXLM-UHFFFAOYSA-N CN(CCC#N)Cc1cccc(Br)c1 Chemical compound CN(CCC#N)Cc1cccc(Br)c1 NLVXWIBDIWBXLM-UHFFFAOYSA-N 0.000 description 1
- WGUNWFNMFQNCGQ-UHFFFAOYSA-N COC(NC(C1)C1(C1CCOCC1)C(O)=O)=O Chemical compound COC(NC(C1)C1(C1CCOCC1)C(O)=O)=O WGUNWFNMFQNCGQ-UHFFFAOYSA-N 0.000 description 1
- XQEHBQMNCVYKKI-ZBZPLXCBSA-N COC(N[C@@H](C(N(C(C1)C1C1)[C@@H]1c1nc2ccc(cc(cc3)-c4cc5ccc6[nH]c([C@H](CC[C@@H]7C8C7)N8C([C@@H](c7ccccc7)NC(OC)=O)=O)nc6c5cc4)c3c2[nH]1)=O)c1ccccc1)=O Chemical compound COC(N[C@@H](C(N(C(C1)C1C1)[C@@H]1c1nc2ccc(cc(cc3)-c4cc5ccc6[nH]c([C@H](CC[C@@H]7C8C7)N8C([C@@H](c7ccccc7)NC(OC)=O)=O)nc6c5cc4)c3c2[nH]1)=O)c1ccccc1)=O XQEHBQMNCVYKKI-ZBZPLXCBSA-N 0.000 description 1
- QCYOIFVBYZNUNW-BYPYZUCNSA-N C[C@@H](C(O)=O)N(C)C Chemical compound C[C@@H](C(O)=O)N(C)C QCYOIFVBYZNUNW-BYPYZUCNSA-N 0.000 description 1
- OOIJOKDKOOJZJS-BYPYZUCNSA-N C[C@@H](C(O)=O)NC(N(C)C)=O Chemical compound C[C@@H](C(O)=O)NC(N(C)C)=O OOIJOKDKOOJZJS-BYPYZUCNSA-N 0.000 description 1
- YGYLYUIRSJSFJS-QMMMGPOBSA-N C[C@@H](C(OCc1ccccc1)=O)N Chemical compound C[C@@H](C(OCc1ccccc1)=O)N YGYLYUIRSJSFJS-QMMMGPOBSA-N 0.000 description 1
- VOGHVBZAMPRNEG-IBGZPJMESA-N O=C(c1ccccc1)ON(CCC1)[C@@H]1c1nc(CCc2cc(Br)ccc2-2)c-2[nH]1 Chemical compound O=C(c1ccccc1)ON(CCC1)[C@@H]1c1nc(CCc2cc(Br)ccc2-2)c-2[nH]1 VOGHVBZAMPRNEG-IBGZPJMESA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/044—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/044—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
- C07D491/052—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring the oxygen-containing ring being six-membered
Abstract
본 개시내용은 하기 구조 I의 화합물, 조성물 및 C형 간염 바이러스 (HCV) 감염의 치료를 위한 방법에 관한 것이다. 또한, 본 발명은 상기 화합물을 함유하는 제약 조성물 및 HCV 감염의 치료에 있어서의 이들 화합물의 사용 방법에 관한 것이다.
<화학식 I>
<화학식 I>
Description
<관련 출원의 교차 참조>
본원은 2008년 2월 13일자로 출원된 미국 가출원 일련 번호 61/028,277호의 이익을 청구한다.
본 개시내용은 일반적으로 항바이러스성 화합물에 관한 것이고, 더욱 구체적으로는 C형 간염 바이러스 (HCV)에 의해 코딩되는 NS5A 단백질의 기능을 억제할 수 있는 화합물, 이러한 화합물을 포함하는 조성물, 및 NS5A 단백질의 기능을 억제하는 방법에 관한 것이다.
HCV는 세계적으로 1억 7천만 명 - 인간 면역결핍 바이러스 제1형에 감염된 숫자의 대략 5배 - 이 감염된 것으로 추산되는 주요 인간 병원체이다. 이들 HCV에 감염된 개체 중 높은 비율에서 간경변 및 간세포성 암종을 비롯한 심각한 진행성 간 질환이 발생한다.
현재, 가장 효과적인 HCV 치료법은 환자의 40%에서 지속적인 효능을 이끌어내는 알파-인터페론 및 리바비린의 조합물을 이용하는 것이다. 최근의 임상 결과는 페길화된 알파-인터페론이 단일요법으로서 비-변성된 알파-인터페론보다 우수함을 증명하였다. 그러나, 페길화된 알파-인터페론 및 리바비린의 조합물을 포함하는 실험적 치료 레지멘으로도, 높은 비율의 환자에서 바이러스 부하를 지속적으로 감소시키지는 못했다. 따라서, HCV 감염의 치료에 효과적인 치료제를 개발해야 할 분명하고도 절실한 필요성이 존재한다.
HCV는 양성 가닥 RNA 바이러스이다. 추정되는 아미노산 서열 및 5' 비-번역 영역에서의 광범위한 유사성의 비교를 기준으로, HCV는 플라비비리다에(Flaviviridae) 과의 별개의 속으로 분류된다. 플라비비리다에 과의 모든 구성원은 중단되지 않은 단일 오픈 리딩 프레임의 번역을 통해 모든 공지된 바이러스-특이적 단백질을 코딩하는 양성 가닥 RNA 게놈을 함유하는 외피 비리온을 갖는다.
HCV 게놈 전체에 걸쳐 뉴클레오티드 및 코딩된 아미노산 서열 내에서 상당한 이종성이 발견된다. 6개 이상의 주요 유전자형이 특성화되어 있고, 50개 초과의 아형이 기재되어 있다. HCV의 주요 유전자형은 세계적으로 그 분포가 상이하며, 발병학 및 치료법에서의 유전자형의 가능한 효과에 대한 다수의 연구에도 불구하고, HCV의 유전적 이종성의 임상적 중요성은 파악되지 못한 채로 남아있다.
단일 가닥 HCV RNA 게놈은 대략 9500개 뉴클레오티드의 길이이고, 약 3000개 아미노산의 단일한 거대 다중단백질을 코딩하는 단일 오픈 리딩 프레임 (ORF)을 갖는다. 감염된 세포에서, 상기 다중단백질은 세포성 및 바이러스성 프로테아제에 의해 다수의 부위에서 절단되어 구조적 및 비-구조적 (NS) 단백질을 생성한다. HCV의 경우, 성숙한 비-구조적 단백질 (NS2, NS3, NS4A, NS4B, NS5A 및 NS5B)의 발생은 2개의 바이러스성 프로테아제에 의해 행해진다. 첫 번째 것은 메탈로프로테아제인 것으로 여겨지며, 이는 NS2-NS3 접합부를 절단하고; 두 번째 것은 NS3의 N-말단 영역 내에 함유된 세린 프로테아제 (본원에서 NS3 프로테아제로도 지칭됨)로서, 이는 NS3-NS4A 절단 부위에서는 시스로, 나머지 NS4A-NS4B, NS4B-NS5A, NS5A-NS5B 부위에 대해서는 트랜스로 NS3의 하류의 모든 후속 절단을 매개한다. NS4A 단백질은 NS3 프로테아제에 대한 보조인자로서 작용하고, NS3 및 다른 바이러스성 레플리카제 성분의 막 국지화를 보조할 수 있는 복합 기능을 수행하는 것으로 보인다. NS3 단백질 및 NS4A의 복합체 형성은 모든 부위에서 단백질분해 효율을 증진시키므로 프로세싱 사건에 필수적인 것으로 여겨진다. NS3 단백질은 또한 뉴클레오시드 트리포스파타제 및 RNA 헬리카제 활성을 나타낸다. NS5B (본원에서 HCV 폴리머라제로도 지칭됨)는 HCV의 복제에 관여하는 RNA-의존성 RNA 폴리머라제이다.
HCV 바이러스성 복제를 선택적으로 억제하는, HCV-감염 환자의 치료에 유용한 화합물이 요망된다. 특히, NS5A 단백질의 기능을 억제하는데 효과적인 화합물이 요망된다. HCV NS5A 단백질은, 예를 들어 문헌 [Tan, S.-L., Katzel, M.G. Virology 2001, 284, 1-12]; 및 [Park, K.-J.; Choi, S.-H, J. Biological Chemistry 2003]에 기재되어 있다.
제1 측면에서, 본 개시내용은 하기 화학식 I의 화합물 또는 그의 제약상 허용되는 염을 제공한다:
<화학식 I>

상기 식 중,
u 및 u'는 독립적으로 0, 1, 2 또는 3이고;
D 및 D'는 각각 독립적으로 NR5, O 및 S로부터 선택되고; 여기서 각각의 R5는 독립적으로 수소, 알콕시카르보닐, 알킬, 아릴알콕시카르보닐, 카르복시, 할로알킬, 히드록시, (NRaRb)카르보닐 및 트리알킬실릴알콕시알킬로부터 선택되고;
각각의 R1 및 R1'는 독립적으로 알콕시, 알콕시알킬, 알콕시카르보닐, 알킬, 아릴알콕시카르보닐, 카르복시, 포르밀, 할로, 할로알킬, 히드록시, 히드록시알킬, -NRaRb, (NRaRb)알킬 및 (NRaRb)카르보닐로부터 선택되고;
R2는 수소, 알콕시카르보닐, 알킬, 아릴알콕시카르보닐, 카르복시, 할로알킬 및 (NRaRb)카르보닐로부터 선택되고;
R3은 수소, 알콕시, 알콕시알킬, 알콕시카르보닐, 알킬, 아릴알콕시카르보닐, 카르복시, 포르밀, 할로, 할로알킬, 히드록시, 히드록시알킬, -NRaRb, (NRaRb)알킬 및 (NRaRb)카르보닐로부터 선택되거나; 또는
R2 및 R3은, 이들이 부착되어 있는 탄소 원자와 함께, 질소, 산소 및 황으로부터 독립적으로 선택되는 1 또는 2개의 헤테로원자를 임의로 함유하는 5원 내지 8원 방향족 또는 비-방향족 고리를 형성하고; 여기서 상기 5원 내지 8원 고리는 알콕시, 알콕시알킬, 알콕시카르보닐, 알킬, 알킬술포닐, 아릴, 아릴알킬, 아릴술포닐, 카르복시, 포르밀, 할로, 할로알콕시, 할로알킬, 히드록시, 히드록시알킬, -NRaRb, (NRaRb)알킬, (NRaRb)카르보닐, 옥소 및 스피로사이클로부터 독립적으로 선택되는 1, 2 또는 3개의 치환기로 임의로 치환되고;
R2' 및 R3'는, 이들이 부착되어 있는 탄소 원자와 함께, 질소, 산소 및 황으로부터 독립적으로 선택되는 1 또는 2개의 헤테로원자를 임의로 함유하는 5원 내지 8원 방향족 또는 비-방향족 고리를 형성하고; 여기서 상기 5원 내지 8원 고리는 알콕시, 알콕시알킬, 알콕시카르보닐, 알킬, 알킬술포닐, 아릴, 아릴알킬, 아릴술포닐, 카르복시, 포르밀, 할로, 할로알콕시, 할로알킬, 히드록시, 히드록시알킬, -NRaRb, (NRaRb)알킬, (NRaRb)카르보닐, 옥소 및 스피로사이클로부터 독립적으로 선택되는 1, 2 또는 3개의 치환기로 임의로 치환되고;
R4 및 R4'는 각각 독립적으로

여기서
각각의 m은 독립적으로 0, 1 또는 2이고;
각각의 s는 독립적으로 0, 1, 2, 3 또는 4이고;
각각의 X는 독립적으로 O, S, S(O), SO2, CH2, CHR6 및 C(R6)2로부터 선택되되; 단, n이 0인 경우, X는 CH2, CHR6 및 C(R6)2로부터 선택되고;
각각의 R6은 독립적으로 알콕시, 알킬, 아릴, 할로, 할로알킬, 히드록시 및 -NRaRb로부터 선택되고, 여기서 상기 알킬은 인접한 탄소 원자와 함께, 융합된 3원 내지 6원 고리를 임의로 형성할 수 있고, 이때 상기 3원 내지 6원 고리는 1 또는 2개의 알킬기로 임의로 치환되고;
각각의 R7은 독립적으로 수소 및 R11-C(O)- 및 R11-C(S)-로부터 선택되고;
R8은 수소 및 알킬로부터 선택되고;
R9 및 R10은 각각 독립적으로 수소, 알케닐, 알콕시알킬, 알킬, 할로알킬 및 (NRaRb)알킬로부터 선택되거나; 또는
R9 및 R10은, 이들이 부착되어 있는 탄소 원자와 함께, NRz, O 및 S로부터 선택되는 1 또는 2개의 헤테로원자를 임의로 함유하는 5원 또는 6원 포화 고리를 형성하고; 여기서 Rz는 수소 및 알킬로부터 선택되고;
각각의 R11은 독립적으로 알콕시, 알콕시알킬, 알콕시카르보닐, 알콕시카르보닐알킬, 알킬, 알킬카르보닐알킬, 아릴, 아릴알케닐, 아릴알콕시, 아릴알킬, 아릴옥시알킬, 시클로알킬, (시클로알킬)알케닐, (시클로알킬)알킬, 시클로알킬옥시알킬, 할로알킬, 헤테로시클릴, 헤테로시클릴알케닐, 헤테로시클릴알콕시, 헤테로시클릴알킬, 헤테로시클릴옥시알킬, 히드록시알킬, -NRcRd, (NRcRd)알케닐, (NRcRd)알킬 및 (NRcRd)카르보닐로부터 선택된다.
제1 측면의 제1 실시양태에서, 본 개시내용은 D 및 D'가 각각 NR5인 화학식 I의 화합물 또는 그의 제약상 허용되는 염을 제공한다. 제1 측면의 제2 실시양태에서, 각각의 R5는 독립적으로 수소 및 히드록시로부터 선택된다.
제1 측면의 제3 실시양태에서, 본 개시내용은 u 및 u'가 각각 0인 화학식 I의 화합물 또는 그의 제약상 허용되는 염을 제공한다.
제1 측면의 제4 실시양태에서, 본 개시내용은 R2가 수소 및 할로알킬로부터 선택되는 화학식 I의 화합물 또는 그의 제약상 허용되는 염을 제공한다.
제1 측면의 제5 실시양태에서, 본 개시내용은 R3이 수소 및 할로로부터 선택되는 화학식 I의 화합물 또는 그의 제약상 허용되는 염을 제공한다.
제1 측면의 제6 실시양태에서, 본 개시내용은 R2 및 R3이, 이들이 부착되어 있는 탄소 원자와 함께, 6원 또는 7원 카르보시클릭 고리를 형성하는 화학식 I의 화합물 또는 그의 제약상 허용되는 염을 제공한다.
제1 측면의 제7 실시양태에서, 본 개시내용은 R2' 및 R3'가, 이들이 부착되어 있는 탄소 원자와 함께, 산소, 질소 및 황으로부터 선택되는 1개의 헤테로원자를 임의로 함유하는 6원 내지 8원 고리를 형성하고; 여기서 상기 고리가 1 또는 2개의 알킬기로 임의로 치환되는 화학식 I의 화합물 또는 그의 제약상 허용되는 염을 제공한다.
제2 측면에서, 본 개시내용은 하기 화학식 II의 화합물 또는 그의 제약상 허용되는 염을 제공한다:
<화학식 II>

상기 식 중,
R2는 수소 및 할로알킬로부터 선택되고;
R3은 수소 및 할로로부터 선택되거나; 또는
R2 및 R3은, 이들이 부착되어 있는 탄소 원자와 함께, 5원 또는 6원 방향족 또는 비-방향족 고리 카르보시클릭 고리를 형성하고;
R2' 및 R3'는, 이들이 부착되어 있는 탄소 원자와 함께, 산소, 질소 및 황으로부터 선택되는 1개의 헤테로원자를 임의로 함유하는 6원 내지 8원 방향족 또는 비-방향족 고리를 형성하고; 여기서 상기 고리는 1 또는 2개의 알킬기로 임의로 치환되고;
R4 및 R4'는 각각 독립적으로

여기서
각각의 s는 0 또는 2이고;
각각의 R6은 독립적으로 알킬 및 할로로부터 선택되고, 여기서 상기 알킬은 인접한 탄소 원자와 함께, 융합된 3원 고리를 형성하고;
각각의 R7은 독립적으로 수소 및 R11-C(O)-로부터 선택되고;
R8은 수소 및 알킬로부터 선택되고;
R9 및 R10은 각각 독립적으로 수소 및 알킬로부터 선택되고;
각각의 R11은 독립적으로 알킬, 아릴알콕시, 아릴알킬 및 (NRcRd)알킬로부터 선택된다.
제3 측면에서, 본 개시내용은 화학식 I의 화합물 또는 그의 제약상 허용되는 염, 및 제약상 허용되는 담체를 포함하는 조성물을 제공한다. 제3 측면의 제1 실시양태에서, 상기 조성물은 항-HCV 활성을 갖는 1 또는 2종의 추가 화합물을 포함한다. 제2 실시양태에서, 추가 화합물 중 1종 이상은 인터페론 또는 리바비린이다. 제3 실시양태에서 인터페론은 인터페론 알파 2B, 페길화된 인터페론 알파, 컨센서스 인터페론, 인터페론 알파 2A 및 림프아구성 인터페론 타우로부터 선택된다.
제4 실시양태에서 본 개시내용은 화학식 I의 화합물 또는 그의 제약상 허용되는 염, 제약상 허용되는 담체, 및 항-HCV 활성을 갖는 1 또는 2종의 추가 화합물을 포함하는 조성물을 제공하며, 여기서 추가 화합물 중 1종 이상은 인터류킨 2, 인터류킨 6, 인터류킨 12, 제1형 헬퍼 T 세포 반응의 발생을 증진시키는 화합물, 간섭 RNA, 안티-센스 RNA, 이미퀴모드(Imiqimod), 리바비린, 이노신 5'-모노포스페이트 데히드로게나제 억제제, 아만타딘 및 리만타딘으로부터 선택된다.
제5 실시양태에서 본 개시내용은 화학식 I의 화합물 또는 그의 제약상 허용되는 염, 제약상 허용되는 담체, 및 항-HCV 활성을 갖는 1 또는 2종의 추가 화합물을 포함하는 조성물을 제공하며, 여기서 추가 화합물 중 1종 이상은 HCV 감염의 치료를 위해 HCV 메탈로프로테아제, HCV 세린 프로테아제, HCV 폴리머라제, HCV 헬리카제, HCV NS4B 단백질, HCV 진입, HCV 조립, HCV 방출, HCV NS5A 단백질 및 IMPDH로부터 선택되는 표적의 기능을 억제하는데 효과적이다.
제4 측면에서 본 개시내용은 치료적 유효량의 화학식 I의 화합물 또는 그의 제약상 허용되는 염을 환자에게 투여하는 것을 포함하는, 환자에서의 HCV 감염의 치료 방법을 제공한다. 제4 측면의 제1 실시양태에서 상기 방법은 항-HCV 활성을 갖는 1 또는 2종의 추가 화합물을 화학식 I의 화합물 또는 그의 제약상 허용되는 염의 투여 전에, 후에 또는 투여와 동시에 투여하는 것을 더 포함한다. 제2 실시양태에서 추가 화합물 중 1종 이상은 인터페론 또는 리바비린이다. 제3 실시양태에서 인터페론은 인터페론 알파 2B, 페길화된 인터페론 알파, 컨센서스 인터페론, 인터페론 알파 2A 및 림프아구성 인터페론 타우로부터 선택된다.
제4 측면의 제4 실시양태에서 본 개시내용은 치료적 유효량의 화학식 I의 화합물 또는 그의 제약상 허용되는 염, 및 항-HCV 활성을 갖는 1 또는 2종의 추가 화합물을 화학식 I의 화합물 또는 그의 제약상 허용되는 염의 투여 전에, 후에 또는 투여와 동시에 환자에게 투여하는 것을 포함하는, 환자에서의 HCV 감염의 치료 방법을 제공하며, 여기서 추가 화합물 중 1종 이상은 인터류킨 2, 인터류킨 6, 인터류킨 12, 제1형 헬퍼 T 세포 반응의 발생을 증진시키는 화합물, 간섭 RNA, 안티-센스 RNA, 이미퀴모드, 리바비린, 이노신 5'-모노포스페이트 데히드로게나제 억제제, 아만타딘 및 리만타딘으로부터 선택된다.
제4 측면의 제5 실시양태에서 본 개시내용은 치료적 유효량의 화학식 I의 화합물 또는 그의 제약상 허용되는 염, 및 항-HCV 활성을 갖는 1 또는 2종의 추가 화합물을 화학식 I의 화합물 또는 그의 제약상 허용되는 염의 투여 전에, 후에 또는 투여와 동시에 투여 환자에게 투여하는 것을 포함하는, 환자에서의 HCV 감염의 치료 방법을 제공하며, 여기서 추가 화합물 중 1종 이상은 HCV 감염의 치료를 위해 HCV 메탈로프로테아제, HCV 세린 프로테아제, HCV 폴리머라제, HCV 헬리카제, HCV NS4B 단백질, HCV 진입, HCV 조립, HCV 방출, HCV NS5A 단백질 및 IMPDH로부터 선택되는 표적의 기능을 억제하는데 효과적이다.
본 개시내용의 다른 실시양태는 본원에 개시된 둘 이상의 실시양태 및/또는 측면의 적절한 조합을 포함할 수 있다.
개시내용의 다른 실시양태 및 측면은 이하에 제공된 기재에 따라 명확해질 것이다.
본 개시내용의 화합물은 또한 호변이성질체로서 존재하며, 따라서, 본 개시내용은 모든 호변이성질체 형태를 또한 포함한다.
본원에서 본 개시내용의 기재는 화학 결합의 법칙 및 원칙과 일치하는 것으로 해석되어야 한다.
본 개시내용에 포함되는 화합물은 적합하게는 약제로서 사용하기에 안정한 것으로 이해되어야 한다.
분자 내 특정 위치에서의 임의의 치환기 또는 가변기 (예를 들어, R1, R2, R5, R6 등)의 정의는 상기 분자 내 다른 위치에서의 그의 정의와 독립적인 것으로 의도된다. 예를 들어, u가 2인 경우, 2개의 R1 기 각각은 동일하거나 상이할 수 있다.
명세서에서 언급된 모든 특허, 특허 출원 및 참고 문헌은 그의 전문이 본원에 참고로 도입된다. 불일치되는 경우, 정의를 비롯하여, 본 개시내용이 우선할 것이다.
본 명세서에 사용된 하기 용어는 지시된 바와 같은 의미를 갖는다:
본원에 사용된 단수형 및 정관사는 문맥상 명확하게 다르게 지시되지 않는 한, 복수 형태를 포함한다.
달리 언급되지 않는 한, 본 개시내용의 모든 아릴, 시클로알킬 및 헤테로시클릴기는 그의 개별적 정의 각각에 기재된 바와 같이 치환될 수 있다. 예를 들어, 아릴알킬기의 아릴 부분은 용어 '아릴'의 정의에 기재된 바와 같이 치환될 수 있다.
본원에 사용된 용어 "알케닐"은 1개 이상의 탄소-탄소 이중 결합을 함유하는, 2개 내지 6개 탄소 원자의 직쇄 또는 분지쇄 기를 지칭한다.
본원에 사용된 용어 "알케닐옥시"는 산소 원자를 통해 모 분자 부분에 부착된 알케닐기를 지칭한다.
본원에 사용된 용어 "알케닐옥시카르보닐"은 카르보닐기를 통해 모 분자 부분에 부착된 알케닐옥시기를 지칭한다.
본원에 사용된 용어 "알콕시"는 산소 원자를 통해 모 분자 부분에 부착된 알킬기를 지칭한다.
본원에 사용된 용어 "알콕시알킬"은 1개, 2개 또는 3개의 알콕시기로 치환된 알킬기를 지칭한다.
본원에 사용된 용어 "알콕시알킬카르보닐"은 카르보닐기를 통해 모 분자 부분에 부착된 알콕시알킬기를 지칭한다.
본원에 사용된 용어 "알콕시카르보닐"은 카르보닐기를 통해 모 분자 부분에 부착된 알콕시기를 지칭한다.
본원에 사용된 용어 "알콕시카르보닐알킬"은 1개, 2개 또는 3개의 알콕시카르보닐기로 치환된 알킬기를 지칭한다.
본원에 사용된 용어 "알킬"은 1개 내지 6개의 탄소 원자를 함유하는 직쇄 또는 분지쇄의 포화된 탄화수소로부터 유도된 기를 지칭한다. 본 개시내용의 화합물에서, m이 1 또는 2이고; X가 CHR6이고, R6이 알킬인 경우, 각각의 알킬은 인접한 탄소 원자와 함께 3원 내지 6원의 융합된 고리를 임의로 형성하여 하기 제시된 구조 중 하나를 제공할 수 있다:

(식 중, z는 1, 2, 3 또는 4이고, w는 0, 1 또는 2이고, R50은 알킬임). w가 2인 경우, 2개의 R50 알킬기는 동일하거나 상이할 수 있다.
본원에 사용된 용어 "알킬카르보닐"은 카르보닐기를 통해 모 분자 부분에 부착된 알킬기를 지칭한다.
본원에 사용된 용어 "알킬카르보닐알킬"은 1개, 2개 또는 3개의 알킬카르보닐기로 치환된 알킬기를 지칭한다.
본원에 사용된 용어 "알킬카르보닐옥시"는 산소 원자를 통해 모 분자 부분에 부착된 알킬카르보닐기를 지칭한다.
본원에 사용된 용어 "알킬술파닐"은 황 원자를 통해 모 분자 부분에 부착된 알킬기를 지칭한다.
본원에 사용된 용어 "알킬술포닐"은 술포닐기를 통해 모 분자 부분에 부착된 알킬기를 지칭한다.
본원에 사용된 용어 "아릴"은 페닐기, 또는 고리 중 하나 또는 둘 다가 페닐기인 융합된 바이시클릭 고리계를 지칭한다. 융합된 바이시클릭 고리계는 4원 내지 6원의 방향족 또는 비-방향족 카르보시클릭 고리에 융합된 페닐기로 이루어진다. 본 개시내용의 아릴기는 기 내의 임의의 치환가능한 탄소 원자를 통해 모 분자 부분에 부착될 수 있다. 아릴기의 대표적인 예에는 인다닐, 인데닐, 나프틸, 페닐 및 테트라히드로나프틸이 포함되지만 이에 한정되는 것은 아니다. 본 개시내용의 아릴기는 알콕시, 알콕시알킬, 알콕시카르보닐, 알킬, 알킬카르보닐, 제2 아릴기, 아릴알콕시, 아릴알킬, 아릴카르보닐, 시아노, 할로, 할로알콕시, 할로알킬, 헤테로시클릴, 헤테로시클릴알킬, 헤테로시클릴카르보닐, 히드록시, 히드록시알킬, 니트로, -NRxRy, (NRxRy)알킬, 옥소 및 -P(O)OR2로부터 독립적으로 선택되는 1개, 2개, 3개, 4개 또는 5개의 치환기로 임의로 치환되며, 여기서 각각의 R은 수소 및 알킬로부터 독립적으로 선택되고; 아릴알킬 및 헤테로시클릴알킬의 알킬 부분은 비치환되고, 제2 아릴기, 아릴알킬의 아릴 부분, 아릴카르보닐의 아릴 부분, 헤테로시클릴, 및 헤테로시클릴알킬 및 헤테로시클릴카르보닐의 헤테로시클릴 부분은 알콕시, 알킬, 시아노, 할로, 할로알콕시, 할로알킬 및 니트로로부터 독립적으로 선택되는 1개, 2개 또는 3개의 치환기로 임의로 추가 치환된다.
본원에 사용된 용어 "아릴알케닐"은 1개, 2개 또는 3개의 아릴기로 치환된 알케닐기를 지칭한다.
본원에 사용된 용어 "아릴알콕시"는 알콕시기를 통해 모 분자 부분에 부착된 아릴기를 지칭한다.
본원에 사용된 용어 "아릴알콕시알킬"은 1개, 2개 또는 3개의 아릴알콕시기로 치환된 알킬기를 지칭한다.
본원에 사용된 용어 "아릴알콕시알킬카르보닐"은 카르보닐기를 통해 모 분자 부분에 부착된 아릴알콕시알킬기를 지칭한다.
본원에 사용된 용어 "아릴알콕시카르보닐"은 카르보닐기를 통해 모 분자 부분에 부착된 아릴알콕시기를 지칭한다.
본원에 사용된 용어 "아릴알킬"은 1개, 2개 또는 3개의 아릴기로 치환된 알킬기를 지칭한다. 아릴알킬의 알킬 부분은 알콕시, 알킬카르보닐옥시, 할로, 할로알콕시, 할로알킬, 헤테로시클릴, 히드록시 및 -NRcRd로부터 독립적으로 선택되는 1개 또는 2개의 추가 기로 임의로 치환되며, 여기서 헤테로시클릴은 알콕시, 알킬, 비치환된 아릴, 비치환된 아릴알콕시, 비치환된 아릴알콕시카르보닐, 할로, 할로알콕시, 할로알킬, 히드록시 및 -NRxRy로부터 독립적으로 선택되는 1개 또는 2개의 치환기로 임의로 추가 치환된다.
본원에 사용된 용어 "아릴알킬카르보닐"은 카르보닐기를 통해 모 분자 부분에 부착된 아릴알킬기를 지칭한다.
본원에 사용된 용어 "아릴카르보닐"은 카르보닐기를 통해 모 분자 부분에 부착된 아릴기를 지칭한다.
본원에 사용된 용어 "아릴옥시"는 산소 원자를 통해 모 분자 부분에 부착된 아릴기를 지칭한다.
본원에 사용된 용어 "아릴옥시알킬"은 1개, 2개 또는 3개의 아릴옥시기로 치환된 알킬기를 지칭한다.
본원에 사용된 용어 "아릴옥시카르보닐"은 카르보닐기를 통해 모 분자 부분에 부착된 아릴옥시기를 지칭한다.
본원에 사용된 용어 "아릴술포닐"은 술포닐기를 통해 모 분자 부분에 부착된 아릴기를 지칭한다.
본원에 사용된 용어 "Cap" 및 "cap"은 말단 질소-함유 고리, 즉, 화학식 I의 화합물의 피롤리딘 고리의 질소 원자 상에 위치한 기를 지칭한다. "Cap" 또는 "cap"은 말단 질소-함유 고리에 기를 부가하는 데 사용되는 시약 또는 최종 생성물 내의 단편, 즉, "Cap-51", 또는 실시예 5에서 발견된 Cap-51 단편"을 지칭할 수 있음을 이해하여야 한다.
본원에 사용된 용어 "카르보닐"은 -C(O)-를 지칭한다.
본원에 사용된 용어 "카르복시"는 -CO2H를 지칭한다.
본원에 사용된 용어 "시아노"는 -CN을 지칭한다.
본원에 사용된 용어 "시클로알킬"은 3개 내지 7개의 탄소 원자 및 0개의 헤테로원자를 갖는 포화된 모노시클릭 탄화수소 고리계를 지칭한다. 시클로알킬기의 대표적인 예에는 시클로프로필, 시클로펜틸 및 시클로헥실이 포함되지만 이에 한정되는 것은 아니다. 본 개시내용의 시클로알킬기는 알콕시, 알킬, 아릴, 시아노, 할로, 할로알콕시, 할로알킬, 헤테로시클릴, 히드록시, 히드록시알킬, 니트로 및 -NRxRy로부터 독립적으로 선택되는 1개, 2개, 3개, 4개 또는 5개의 치환기로 임의로 치환되며, 여기서 아릴 및 헤테로시클릴은 알콕시, 알킬, 시아노, 할로, 할로알콕시, 할로알킬, 히드록시 및 니트로로부터 독립적으로 선택되는 1개, 2개 또는 3개의 치환기로 임의로 추가 치환된다.
본원에 사용된 용어 "(시클로알킬)알케닐"은 1개, 2개 또는 3개의 시클로알킬기로 치환된 알케닐기를 지칭한다.
본원에 사용된 용어 "(시클로알킬)알킬"은 1개, 2개 또는 3개의 시클로알킬기로 치환된 알킬기를 지칭한다. (시클로알킬)알킬의 알킬 부분은 히드록시 및 -NRcRd로부터 독립적으로 선택되는 1개 또는 2개의 기로 임의로 추가 치환된다.
본원에 사용된 용어 "시클로알킬옥시"는 산소 원자를 통해 모 분자 부분에 부착된 시클로알킬기를 지칭한다.
본원에 사용된 용어 "시클로알킬옥시알킬"은 1개, 2개 또는 3개의 시클로알킬옥시기로 치환된 알킬기를 지칭한다.
본원에 사용된 용어 "시클로알킬술포닐"은 술포닐기를 통해 모 분자 부분에 부착된 시클로알킬기를 지칭한다.
본원에 사용된 용어 "포르밀"은 -CHO를 지칭한다.
본원에 사용된 용어 "할로" 및 "할로겐"은 F, Cl, Br 또는 I를 지칭한다.
본원에 사용된 용어 "할로알콕시"는 산소 원자를 통해 모 분자 부분에 부착된 할로알킬기를 지칭한다.
본원에 사용된 용어 "할로알콕시카르보닐"은 카르보닐기를 통해 모 분자 부분에 부착된 할로알콕시기를 지칭한다.
본원에 사용된 용어 "할로알킬"은 1개, 2개, 3개 또는 4개의 할로겐 원자에 의해 치환된 알킬기를 지칭한다.
본원에 사용된 용어 "헤테로시클릴"은 질소, 산소 및 황으로부터 독립적으로 선택되는 1개, 2개, 3개 또는 4개의 헤테로원자를 함유하는 4원, 5원, 6원 또는 7원의 고리를 지칭한다. 4원의 고리는 0개의 이중 결합을 갖고, 5원의 고리는 0개 내지 2개의 이중 결합을 갖고, 6원 및 7원의 고리는 0개 내지 3개의 이중 결합을 갖는다. 용어 "헤테로시클릴"은 또한, 헤테로시클릴 고리가 또다른 모노시클릭 헤테로시클릴기, 또는 4원 내지 6원의 방향족 또는 비-방향족 카르보시클릭 고리에 융합된 바이시클릭 기뿐만 아니라 가교된 바이시클릭 기, 예컨대 7-아자바이시클로[2.2.1]헵트-7-일, 2-아자바이시클로[2.2.2]옥-2-틸 및 2-아자바이시클로[2.2.2]옥-3-틸을 포함한다. 본 개시내용의 헤테로시클릴기는 기 내의 임의의 탄소 원자 또는 질소 원자를 통해 모 분자 부분에 부착될 수 있다. 헤테로시클릴기의 예에는 벤조티에닐, 푸릴, 이미다졸릴, 인돌리닐, 인돌릴, 이소티아졸릴, 이속사졸릴, 모르폴리닐, 옥사졸릴, 피페라지닐, 피페리디닐, 피라졸릴, 피리디닐, 피롤리디닐, 피롤로피리디닐, 피롤릴, 티아졸릴, 티에닐, 티오모르폴리닐, 7-아자바이시클로[2.2.1]헵트-7-일, 2-아자바이시클로[2.2.2]옥-2-틸 및 2-아자바이시클로[2.2.2]옥-3-틸이 포함되지만 이에 한정되는 것은 아니다. 본 개시내용의 헤테로시클릴기는 알콕시, 알콕시알킬, 알콕시카르보닐, 알킬, 알킬카르보닐, 아릴, 아릴알킬, 아릴카르보닐, 시아노, 할로, 할로알콕시, 할로알킬, 제2 헤테로시클릴기, 헤테로시클릴알킬, 헤테로시클릴카르보닐, 히드록시, 히드록시알킬, 니트로, -NRxRy, (NRxRy)알킬 및 옥소로부터 독립적으로 선택되는 1개, 2개, 3개, 4개 또는 5개의 치환기로 임의로 치환되며, 여기서 아릴알킬 및 헤테로시클릴알킬의 알킬 부분은 비치환되고, 아릴, 아릴알킬의 아릴 부분, 아릴카르보닐의 아릴 부분, 제2 헤테로시클릴기, 및 헤테로시클릴알킬 및 헤테로시클릴카르보닐의 헤테로시클릴 부분은 알콕시, 알킬, 시아노, 할로, 할로알콕시, 할로알킬 및 니트로로부터 독립적으로 선택되는 1개, 2개 또는 3개의 치환기로 임의로 추가 치환된다.
본원에 사용된 용어 "헤테로시클릴알케닐"은 1개, 2개 또는 3개의 헤테로시클릴기로 치환된 알케닐기를 지칭한다.
본원에 사용된 용어 "헤테로시클릴알콕시"는 알콕시기를 통해 모 분자 부분에 부착된 헤테로시클릴기를 지칭한다.
본원에 사용된 용어 "헤테로시클릴알콕시카르보닐"은 카르보닐기를 통해 모 분자 부분에 부착된 헤테로시클릴알콕시기를 지칭한다.
본원에 사용된 용어 "헤테로시클릴알킬"은 1개, 2개 또는 3개의 헤테로시클릴기로 치환된 알킬기를 지칭한다. 헤테로시클릴알킬의 알킬 부분은 알콕시, 알킬카르보닐옥시, 아릴, 할로, 할로알콕시, 할로알킬, 히드록시 및 -NRcRd로부터 독립적으로 선택되는 1개 또는 2개의 추가 기로 임의로 추가 치환되며, 여기서 아릴은 알콕시, 알킬, 비치환된 아릴, 비치환된 아릴알콕시, 비치환된 아릴알콕시카르보닐, 할로, 할로알콕시, 할로알킬, 히드록시 및 -NRxRy로부터 독립적으로 선택되는 1개 또는 2개의 치환기로 임의로 추가 치환된다.
본원에 사용된 용어 "헤테로시클릴알킬카르보닐"은 카르보닐기를 통해 모 분자 부분에 부착된 헤테로시클릴알킬기를 지칭한다.
본원에 사용된 용어 "헤테로시클릴카르보닐"은 카르보닐기를 통해 모 분자 부분에 부착된 헤테로시클릴기를 지칭한다.
본원에 사용된 용어 "헤테로시클릴옥시"는 산소 원자를 통해 모 분자 부분에 부착된 헤테로시클릴기를 지칭한다.
본원에 사용된 용어 "헤테로시클릴옥시알킬"은 1개, 2개 또는 3개의 헤테로시클릴옥시기로 치환된 알킬기를 지칭한다.
본원에 사용된 용어 "헤테로시클릴옥시카르보닐"은 카르보닐기를 통해 모 분자 부분에 부착된 헤테로시클릴옥시기를 지칭한다.
본원에 사용된 용어 "히드록시"는 -OH를 지칭한다.
본원에 사용된 용어 "히드록시알킬"은 1개, 2개 또는 3개의 히드록시기로 치환된 알킬기를 지칭한다.
본원에 사용된 용어 "히드록시알킬카르보닐"은 카르보닐기를 통해 모 분자 부분에 부착된 히드록시알킬기를 지칭한다.
본원에 사용된 용어 "니트로"는 -NO2를 지칭한다.
본원에 사용된 용어 "-NRaRb"는 질소 원자를 통해 모 분자 부분에 부착된 2개의 기 Ra 및 Rb를 지칭한다. Ra 및 Rb는 수소, 알케닐 및 알킬로부터 독립적으로 선택된다.
본원에 사용된 용어 "(NRaRb)알킬"은 1개, 2개 또는 3개의 -NRaRb기로 치환된 알킬기를 지칭한다.
본원에 사용된 용어 "(NRaRb)카르보닐"은 카르보닐기를 통해 모 분자 부분에 부착된 -NRaRb기를 지칭한다.
본원에 사용된 용어 "-NRcRd"는 질소 원자를 통해 모 분자 부분에 부착된 2개의 기 Rc 및 Rd를 지칭한다. Rc 및 Rd는 수소, 알케닐옥시카르보닐, 알콕시알킬카르보닐, 알콕시카르보닐, 알킬, 알킬카르보닐, 알킬술포닐, 아릴, 아릴알콕시카르보닐, 아릴알킬, 아릴알킬카르보닐, 아릴카르보닐, 아릴옥시카르보닐, 아릴술포닐, 시클로알킬, 시클로알킬술포닐, 포르밀, 할로알콕시카르보닐, 헤테로시클릴, 헤테로시클릴알콕시카르보닐, 헤테로시클릴알킬, 헤테로시클릴알킬카르보닐, 헤테로시클릴카르보닐, 헤테로시클릴옥시카르보닐, 히드록시알킬카르보닐, (NReRf)알킬, (NReRf)알킬카르보닐, (NReRf)카르보닐, (NReRf)술포닐, -C(NCN)OR' 및 -C(NCN)NRxRy로부터 독립적으로 선택되며, 여기서 R'는 알킬 및 비치환된 페닐로부터 선택되고, 아릴알킬, 아릴알킬카르보닐, 헤테로시클릴알킬 및 헤테로시클릴알킬카르보닐의 알킬 부분은 1개의 -NReRf기로 임의로 추가 치환되고; 아릴, 및 아릴알콕시카르보닐, 아릴알킬, 아릴알킬카르보닐, 아릴카르보닐, 아릴옥시카르보닐 및 아릴술포닐의 아릴 부분, 헤테로시클릴, 및 헤테로시클릴알콕시카르보닐, 헤테로시클릴알킬, 헤테로시클릴알킬카르보닐, 헤테로시클릴카르보닐 및 헤테로시클릴옥시카르보닐의 헤테로시클릴 부분은 알콕시, 알킬, 시아노, 할로, 할로알콕시, 할로알킬 및 니트로로부터 독립적으로 선택되는 1개, 2개 또는 3개의 치환기로 임의로 추가 치환된다.
본원에 사용된 용어 "(NRcRd)알케닐"은 1개, 2개 또는 3개의 -NRcRd기로 치환된 알케닐기를 지칭한다.
본원에 사용된 용어 "(NRcRd)알킬"은 1개, 2개 또는 3개의 -NRcRd기로 치환된 알킬기를 지칭한다. (NRcRd)알킬의 알킬 부분은 알콕시, 알콕시알킬카르보닐, 알콕시카르보닐, 알킬술파닐, 아릴알콕시알킬카르보닐, 카르복시, 헤테로시클릴, 헤테로시클릴카르보닐, 히드록시 및 (NReRf)카르보닐로부터 선택되는 1개 또는 2개의 추가 기로 임의로 추가 치환되며, 여기서 헤테로시클릴은 알콕시, 알킬, 시아노, 할로, 할로알콕시, 할로알킬 및 니트로로부터 독립적으로 선택되는 1개, 2개, 3개, 4개 또는 5개의 치환기로 임의로 추가 치환된다.
본원에 사용된 용어 "(NRcRd)카르보닐"은 카르보닐기를 통해 모 분자 부분에 부착된 -NRcRd기를 지칭한다.
본원에 사용된 용어 "-NReRf"는 질소 원자를 통해 모 분자 부분에 부착된 2개의 기 Re 및 Rf를 지칭한다. Re 및 Rf는 수소, 알킬, 비치환된 아릴, 비치환된 아릴알킬, 비치환된 시클로알킬, 비치환된 (시클로알킬)알킬, 비치환된 헤테로시클릴, 비치환된 헤테로시클릴알킬, (NRxRy)알킬 및 (NRxRy)카르보닐로부터 독립적으로 선택된다.
본원에 사용된 용어 "(NReRf)알킬"은 1개, 2개 또는 3개의 -NReRf기로 치환된 알킬기를 지칭한다.
본원에 사용된 용어 "(NReRf)알킬카르보닐"은 카르보닐기를 통해 모 분자 부분에 부착된 (NReRf)알킬기를 지칭한다.
본원에 사용된 용어 "(NReRf)카르보닐"은 카르보닐기를 통해 모 분자 부분에 부착된 -NReRf기를 지칭한다.
본원에 사용된 용어 "(NReRf)술포닐"은 술포닐기를 통해 모 분자 부분에 부착된 -NReRf기를 지칭한다.
본원에 사용된 용어 "-NRxRy"는 질소 원자를 통해 모 분자 부분에 부착된 2개의 기 Rx 및 Ry를 지칭한다. Rx 및 Ry는 수소, 알콕시카르보닐, 알킬, 알킬카르보닐, 비치환된 아릴, 비치환된 아릴알콕시카르보닐, 비치환된 아릴알킬, 비치환된 시클로알킬, 비치환된 헤테로시클릴 및 (NRx'Ry')카르보닐로부터 독립적으로 선택되며, 여기서 Rx' 및 Ry'는 수소 및 알킬로부터 독립적으로 선택된다.
본원에 사용된 용어 "(NRxRy)알킬"은 1개, 2개 또는 3개의 -NRxRy기로 치환된 알킬기를 지칭한다.
본원에 사용된 용어 "(NRxRy)카르보닐"은 카르보닐기를 통해 모 분자 부분에 부착된 -NRxRy기를 지칭한다.
본원에 사용된 용어 "옥소"는 =O를 지칭한다.
본원에 사용된 용어 "술포닐"은 -SO2-를 지칭한다.
본원에 사용된 용어 "트리알킬실릴"은 -SiR3 (여기서, R은 알킬임)을 지칭한다. R 기는 동일하거나 상이할 수 있다.
본원에 사용된 용어 "트리알킬실릴알킬"은 1개, 2개 또는 3개의 트리알킬실릴기로 치환된 알킬기를 지칭한다.
본원에 사용된 용어 "트리알킬실릴알콕시"는 산소 원자를 통해 모 분자 부분에 부착된 트리알킬실릴알킬기를 지칭한다.
본원에 사용된 용어 "트리알킬실릴알콕시알킬"은 1개, 2개 또는 3개의 트리알킬실릴알콕시기로 치환된 알킬기를 지칭한다.
비대칭 중심이 본 개시내용의 화합물에 존재한다. 이러한 중심은 키랄 탄소 원자 주위의 치환기의 배위에 따라 기호 "R" 또는 "S"로 표시된다. 본 개시내용은 NS5A를 억제하는 능력을 갖는, 모든 입체화학적 이성질체 형태 또는 이들의 혼합물을 포함하는 것으로 이해되어야 한다. 화합물의 개별적 입체이성질체는 키랄 중심을 함유하는 시판중인 출발 물질로부터 합성하여 제조할 수 있거나, 또는 거울상이성질체 생성물의 혼합물을 제조한 다음 분리, 예컨대 부분입체이성질체의 혼합물로의 전환에 이은 분리 또는 재결정화, 크로마토그래피 기술에 의하거나, 또는 키랄 크로마토그래피 컬럼 상에서 거울상이성질체를 직접 분리함으로써 제조할 수 있다. 특정 입체화학의 출발 화합물은 시판중이거나, 또는 당업계에 공지된 기술에 의해 제조된 다음 분할될 수 있다.
본 개시내용의 특정 화합물은 또한, 분리될 수 있는 상이한 안정한 입체구조 형태로 존재할 수 있다. 예를 들어 입체 장해 또는 고리 변형 때문인, 비대칭 단일 결합에 대한 제한된 회전으로 인한 비틀림 비대칭은 상이한 형태이성질체의 분리를 가능케 할 수 있다. 본 개시내용은 상기 화합물의 각 형태이성질체 및 이들의 혼합물을 포함한다.
용어 "본 개시내용의 화합물" 및 등가의 표현은 화학식 I의 화합물, 및 그의 제약상 허용되는 거울상이성질체, 부분입체이성질체 및 염을 포함하는 것으로 한다. 유사하게, 중간체에 대한 언급은 문맥상 허용되는 경우에 그의 염을 포함하는 것으로 한다.
본 개시내용의 화합물은 제약상 허용되는 염으로서 존재할 수 있다. 본원에 사용된 용어 "제약상 허용되는 염"은 안전 의료 평가의 범위 내에서, 합리적인 유익/유해 비율에 상응하도록, 과도한 독성, 자극, 알레르기성 반응, 또는 기타 문제점 또는 합병증 없이 환자의 조직과의 접촉에서 사용하기에 적합하고, 그의 의도되는 용도에 대해 효과적인, 수용성 또는 지용성이거나, 또는 수분산성 또는 지분산성인 본 개시내용의 화합물의 염 또는 쯔비터이온 형태를 나타낸다. 염은 화합물의 최종 단리 및 정제 동안 제조할 수 있거나, 또는 별도로 적합한 질소 원자를 적합한 산과 반응시켜 제조할 수 있다. 대표적인 산 부가염은 아세테이트, 아디페이트, 알기네이트, 시트레이트, 아스파르테이트, 벤조에이트, 벤젠술포네이트, 바이술페이트, 부티레이트, 캄포레이트, 캄포르술포네이트; 디글루코네이트, 디히드로브로마이드, 디히드로클로라이드, 디히드로요오다이드, 글리세로포스페이트, 헤미술페이트, 헵타노에이트, 헥사노에이트, 포르메이트, 푸마레이트, 히드로클로라이드, 히드로브로마이드, 히드로요오다이드, 2-히드록시에탄술포네이트, 락테이트, 말레에이트, 메시틸렌술포네이트, 메탄술포네이트, 나프틸렌술포네이트, 니코티네이트, 2-나프탈렌술포네이트, 옥살레이트, 팔모에이트, 펙티네이트, 퍼술페이트, 3-페닐프로프리오네이트, 피크레이트, 피발레이트, 프로피오네이트, 숙시네이트, 타르트레이트, 트리클로로아세테이트, 트리플루오로아세테이트, 포스페이트, 글루타메이트, 바이카르보네이트, 파라-톨루엔술포네이트 및 운데카노에이트를 포함한다. 제약상 허용되는 부가염을 형성하는데 사용될 수 있는 산의 예에는 염산, 브롬화수소산, 황산 및 인산과 같은 무기산, 및 옥살산, 말레산, 숙신산 및 시트르산과 같은 유기산이 포함된다.
염기성 부가염은 화합물의 최종 단리 및 정제 동안 카르복시기를 적합한 염기, 예컨대 금속 양이온의 수산화물, 탄산염 또는 중탄산염, 또는 암모니아 또는 1급, 2급 또는 3급 유기 아민과 반응시켜 제조할 수 있다. 제약상 허용되는 염의 양이온에는 리튬, 나트륨, 칼륨, 칼슘, 마그네슘 및 알루미늄, 및 무독성 4급 아민 양이온, 예컨대 암모늄, 테트라메틸암모늄, 테트라에틸암모늄, 메틸아민, 디메틸아민, 트리메틸아민, 트리에틸아민, 디에틸아민, 에틸아민, 트리부틸아민, 피리딘, N,N-디메틸아닐린, N-메틸피페리딘, N-메틸모르폴린, 디시클로헥실아민, 프로카인, 디벤질아민, N,N-디벤질페네틸아민 및 N,N'-디벤질에틸렌디아민이 포함된다. 염기 부가염의 형성에 유용한 다른 대표적인 유기 아민에는 에틸렌디아민, 에탄올아민, 디에탄올아민, 피페리딘 및 피페라진이 포함된다.
치료법에서 사용하기 위해, 치료적 유효량의 화학식 I의 화합물, 및 그의 제약상 허용되는 염을 정제되지 않은 화학물질로서 투여하는 것이 가능한 경우, 활성 성분은 제약 조성물로서 제공될 수 있다. 따라서, 본 개시내용은 추가로, 치료적 유효량의 화학식 I의 화합물 또는 그의 제약상 허용되는 염, 및 1종 이상의 제약상 허용되는 담체, 희석제 또는 부형제를 포함하는 제약 조성물을 제공한다. 본원에 사용된 용어 "치료적 유효량"은 환자에 대한 의미있는 이점, 예를 들어 바이러스 부하의 감소를 나타내기에 충분한 각 활성 성분의 총량을 지칭한다. 단독으로 투여되는 개별적인 활성 성분에 적용되는 경우, 상기 용어는 활성 성분만의 양을 지칭한다. 조합물에 적용되는 경우, 상기 용어는 조합되어 투여되거나, 순차적으로 또는 동시에 투여되든지에 상관없이, 치료 효과를 야기하는 활성 성분들의 총량을 지칭한다. 화학식 I의 화합물 및 그의 제약상 허용되는 염은 상기 기재된 바와 같다. 담체(들), 희석제(들) 또는 부형제(들)은 제제의 다른 성분과 상용적이고 그의 수용자에게 유해하지 않다는 의미에서 허용가능해야 한다. 본 개시내용의 또다른 측면에 따라, 화학식 I의 화합물 또는 그의 제약상 허용되는 염을 1종 이상의 제약상 허용되는 담체, 희석제 또는 부형제와 혼합시키는 것을 포함하는, 제약 제제의 제조 방법이 또한 제공된다. 본원에 사용된 용어 "제약상 허용되는"은 안전 의료 평가의 범위 내에서, 합리적인 유익/유해 비율에 상응하도록, 과도한 독성, 자극, 알레르기성 반응, 또는 기타 문제점 또는 합병증 없이 환자의 조직과의 접촉에서 사용하기에 적합하고, 그의 의도되는 용도에 대해 효과적인 화합물, 물질, 조성물 및/또는 투여 형태를 지칭한다.
제약 제제는 단위 투여 당 소정량의 활성 성분을 함유하는 단위 투여 형태로 제공될 수 있다. 일일 체중 kg 당 약 0.01 내지 약 250 mg ("mg/kg"), 바람직하게는 일일 체중 kg 당 약 0.05 내지 약 100 mg의 본 개시내용의 화합물의 투여량 수준이 HCV 매개 질환의 예방 및 치료를 위한 단일요법에서 전형적이다. 전형적으로, 본 개시내용의 제약 조성물은 일일 약 1회 내지 약 5회, 또는 다르게는 연속 주입으로서 투여될 것이다. 이러한 투여는 장기 또는 단기 치료법으로 사용될 수 있다. 단일 투여 형태를 제조하기 위해 담체 물질과 조합될 수 있는 활성 성분의 양은 치료될 증상, 증상의 중증도, 투여 시간, 투여 경로, 사용되는 화합물의 배설 속도, 치료의 지속기간, 및 환자의 연령, 성별, 체중 및 상태에 따라 달라질 것이다. 바람직한 단위 투여 제제는 본원에서 상기 언급된 바와 같은 일일 투여량 또는 하위-투여량, 또는 그의 적절한 분율의 활성 성분을 함유하는 것이다. 치료는 화합물의 최적 투여량보다 실질적으로 적은 투여량으로 개시될 수 있다. 그 후, 주어진 상황 하에서 최적의 효과에 도달할 때까지 투여량을 소량씩 증가시킨다. 통상적으로는, 일반적으로 어떠한 해롭거나 유해한 부작용을 일으키지 않으면서 항바이러스적으로 효과적인 결과를 가져오는 농도 수준으로 화합물을 투여하는 것이 가장 바람직하다.
본 개시내용의 조성물이 본 개시내용의 화합물 및 1종 이상의 추가 치료제 또는 예방제의 조합물을 포함하는 경우, 화합물 및 추가 작용제 둘다는 보통, 단일요법 레지멘으로 통상적으로 투여되는 투여량의 약 10 내지 150%, 보다 바람직하게는 약 10 내지 80%의 투여량 수준으로 존재한다.
제약 제제는 임의의 적절한 경로, 예를 들어 경구 (협측 또는 설하 포함), 직장, 비내, 국소 (협측, 설하 또는 경피 포함), 질, 또는 비경구 (피하, 피내, 근육내, 관절내, 활액내, 흉골내, 수막강내, 병변내, 정맥내, 또는 진피내 주사 또는 주입 포함) 경로에 의한 투여에 대해 적합할 수 있다. 이러한 제제는 약학 분야에 공지된 임의의 방법에 의해, 예를 들어 활성 성분을 담체(들) 또는 부형제(들)과 회합시켜 제조할 수 있다. 경구 투여 또는 주사에 의한 투여가 바람직하다.
경구 투여에 대해 적합화된 제약 제제는 개별적 단위, 예컨대 캡슐제 또는 정제; 분말제 또는 과립제; 수성 또는 비-수성 액체 중의 용액제 또는 현탁액제; 식용 폼(foam) 또는 휩(whip); 또는 수중유형 액체 에멀젼제 또는 유중수형 에멀젼제로서 존재할 수 있다.
예를 들어, 정제 또는 캡슐제 형태로의 경구 투여에 대해, 활성 약물 성분은 제약상 허용되는 무독성의 경구용 불활성 담체, 예컨대 에탄올, 글리세롤, 물 등과 조합될 수 있다. 분말제는 화합물을 적합하게 미세한 크기로 분쇄하고, 유사하게 분쇄된 제약 담체, 예컨대 식용 탄수화물 (예를 들어, 전분 또는 만니톨로서)과 혼합시켜 제조한다. 향미제, 보존제, 분산화제 및 착색제가 또한 존재할 수 있다.
캡슐제는 상기 기재된 바와 같이 분말 혼합물을 제조한 다음, 성형된 젤라틴 외피에 충전함으로써 제조된다. 충전 작업 전에, 콜로이드 실리카, 활석, 마그네슘 스테아레이트, 칼슘 스테아레이트 또는 고체 폴리에틸렌 글리콜과 같은 활택제 및 윤활제가 분말 혼합물에 첨가될 수 있다. 한천-한천, 탄산칼슘 또는 탄산나트륨과 같은 붕해제 또는 가용화제를 또한 첨가하여 캡슐제를 복용했을 때 약제의 이용률을 개선시킬 수 있다.
또한, 바람직하거나 필요한 경우, 적합한 결합제, 윤활제, 붕해제 및 착색제가 상기 혼합물에 또한 혼입될 수 있다. 적합한 결합제에는 전분, 젤라틴, 천연 당, 예컨대 글루코스 또는 베타-락토스, 옥수수 감미료, 천연 및 합성 고무, 예컨대 아카시아, 트라가칸트 또는 나트륨 알기네이트, 카르복시메틸셀룰로스, 폴리에틸렌 글리콜 등이 포함된다. 상기 투여 형태에서 사용되는 윤활제에는 나트륨 올레에이트, 염화나트륨 등이 포함된다. 붕해제에는 전분, 메틸 셀룰로스, 한천, 벤토나이트, 크산탄 고무 등이 포함되지만 이에 한정되는 것은 아니다. 정제는, 예를 들어 분말 혼합물을 제조하고, 과립화하거나 슬러그화하고, 윤활제 및 붕해제를 첨가하고, 정제로 압착함으로써 제제화된다. 분말 혼합물은 적합하게 분쇄된 화합물을 상기 기재된 바와 같은 희석제 또는 베이스, 및 임의로는 결합제, 예컨대 카르복시메틸셀룰로스, 알기네이트, 젤라틴, 또는 폴리비닐 피롤리돈, 용액 지연제, 예컨대 파라핀, 재흡수 촉진제, 예컨대 4차 염 및/또는 흡수제, 예컨대 벤토나이트, 카올린 또는 인산이칼슘과 혼합시켜 제조한다. 분말 혼합물은 결합제, 예컨대 시럽, 전분 페이스트, 아라비아 점액, 또는 셀룰로스 또는 중합체 물질의 용액으로 습윤시키고, 스크린을 통해 통과시켜 과립화할 수 있다. 과립화에 대한 별법으로, 분말 혼합물을 타정기에 통과시킬 수 있으며, 그 결과는 과립으로 부수어지는 불완전하게 형성된 슬러그이다. 정제 형성 다이에 점착되는 것을 방지하기 위해, 스테아르산, 스테아레이트 염, 활석 또는 광유를 첨가함으로써 과립을 윤활시킬 수 있다. 이어서, 윤활된 혼합물을 정제로 압착한다. 본 개시내용의 화합물은 또한, 불활성의 자유 유동 담체와 조합하여, 과립화 또는 슬러그화 단계를 거치지 않고 직접적으로 정제로 압착시킬 수 있다. 셸락의 밀봉 코팅, 당 또는 중합체 물질의 코팅, 및 왁스의 광택 코팅으로 이루어지는 투명 또는 불투명 보호 코팅이 제공될 수 있다. 다른 단위 투여형과 구별하기 위해서 이들 코팅에 염료를 첨가할 수 있다.
용액제, 시럽제 및 엘릭시르제와 같은 경구 유동액은 제시된 양에 소정량의 화합물이 함유되어 있는 투여 단위 형태로 제조될 수 있다. 시럽제는 적합하게 가미된 수용액에 화합물을 용해시킴으로써 제조될 수 있는 반면, 엘릭시르제는 무독성 비히클의 사용을 통해 제조된다. 가용화제 및 유화제, 예컨대 에톡실화 이소스테아릴 알콜 및 폴리옥시에틸렌 소르비톨 에테르, 보존제, 풍미 첨가제, 예컨대 페퍼민트 오일 또는 천연 감미료, 또는 사카린 또는 기타 인공 감미료 등이 또한 첨가될 수 있다.
적절한 경우, 경구 투여용 투여 단위 제제는 마이크로캡슐화될 수 있다. 제제는 또한, 예를 들어 미립자 물질을 중합체, 왁스 등으로 코팅하거나 이에 매립함으로써 방출이 연장되거나 또는 지속되도록 제조할 수 있다.
화학식 I의 화합물 및 그의 제약상 허용되는 염은 또한 리포솜 전달 시스템, 예컨대 소형 단층 소포, 대형 단층 소포 및 다층 소포 형태로 투여될 수 있다. 리포솜은 다양한 인지질, 예컨대 콜레스테롤, 스테아릴아민 또는 포스파티딜콜린으로부터 형성될 수 있다.
화학식 I의 화합물 및 그의 제약상 허용되는 염은 또한, 화합물 분자가 결합된 개별적 담체로서의 모노클로날 항체를 사용함으로써 전달될 수도 있다. 화합물은 또한 표적화될 수 있는 약물 담체로서의 가용성 중합체와 결합될 수 있다. 이러한 중합체에는 폴리비닐피롤리돈, 피란 공중합체, 폴리히드록시프로필메타크릴아미드페놀, 폴리히드록시에틸아스파르트아미드페놀, 또는 팔리토일 잔기로 치환된 폴리에틸렌옥시드폴리리신이 포함될 수 있다. 또한, 화합물은 약물의 제어된 방출을 달성하는데 유용한 생물분해성 중합체 부류, 예를 들어 폴리락트산, 폴렙실론 카프로락톤, 폴리히드록시 부티르산, 폴리오르토에스테르, 폴리아세탈, 폴리디히드로피란, 폴리시아노아크릴레이트, 및 히드로겔의 가교된 또는 양친매성의 블록 공중합체에 결합될 수 있다.
경피 투여에 대해 적합화된 제약 제제는 연장된 기간 동안 수용자의 상피에 밀접하게 접촉된 채 유지되도록 의도된 별개의 패치로서 제공될 수 있다. 예를 들어, 활성 성분은 문헌 [Pharmaceutical Research 1986, 3(6), 318]에 개괄적으로 기재된 바와 같이 이온이동법(iontophoresis)에 의해 패치로부터 전달될 수 있다.
국소 투여에 적합화된 제약 제제는 연고, 크림, 현탁액제, 로션, 분말제, 용액제, 페이스트, 겔, 스프레이, 에어로졸 또는 오일로서 제제화될 수 있다.
직장 투여에 적합화된 제약 제제는 좌제 또는 관장제로서 제공될 수 있다.
담체가 고체인 비내 투여에 적합화된 제약 제제는 코로 들이쉬는 방식으로, 즉, 코에 밀착시켜 유지한 분말제-함유 용기로부터 콧구멍을 통해 빠르게 흡입함으로써 투여되는, 예를 들어 20 내지 500 마이크로미터 범위의 입도를 갖는 조대 분말제를 포함한다. 비내 스프레이 또는 점비제로서 투여하기 위한, 담체가 액체인 적합한 제제는 활성 성분의 수용성 또는 오일 용액제를 포함한다.
흡입에 의한 투여에 적합화된 제약 제제는 다양한 유형의 계량식 가압 에어로졸, 분무기 또는 취입기에 의해 생성될 수 있는 미세한 입자 분진 또는 미스트를 포함한다.
질내 투여에 대해 적합화된 제약 제제는 질좌제, 탐폰, 크림, 겔, 페이스트, 폼 또는 스프레이 제제로서 제공될 수 있다.
비경구 투여에 대해 적합화된 제약 제제는 항산화제, 완충제, 정균제, 및 제제를 의도된 수용자의 혈액과 등장성이 되도록 하는 용질을 함유할 수 있는 수성 및 비-수성 멸균 주사액제; 및 현탁화제 및 농후화제를 포함할 수 있는 수성 및 비-수성 멸균 현탁액제를 포함한다. 제제는 단위-투여 또는 다중-투여 용기, 예를 들어 밀봉된 앰플 및 바이알 안에 제공될 수 있고, 사용 직전에 멸균 액체 담체, 예를 들어 주사용수를 첨가하기만 하면 되는 동결건조된 상태로 저장될 수 있다. 임시처방 주사액제 및 현탁액제는 멸균 분말제, 과립제 및 정제로부터 제조될 수 있다.
상기에 구체적으로 언급된 성분 이외에, 제제는 해당 제제의 유형과 관련된 분야에서 통상적인 다른 작용제를 포함할 수 있는 것으로 이해되어야 하고, 예를 들어 경구 투여에 적합한 것은 향미제를 포함할 수 있다.
용어 "환자"는 인간 및 다른 포유동물 모두를 포함한다.
용어 "치료하는"은 (i) 질환, 장애 및/또는 증상에 걸리기 쉬울 수 있지만 아직 걸린 것으로 진단되지는 않은 환자에서 상기 질환, 장애 또는 증상이 발생하는 것을 방지하는 것; (ii) 질환, 장애 또는 증상을 억제, 즉, 그의 진행을 저지하는 것; 및 (iii) 질환, 장애 또는 증상을 경감, 즉, 질환, 장애 및/또는 증상의 감퇴를 야기하는 것을 지칭한다.
본 개시내용의 화합물은 또한 시클로스포린, 예를 들어 시클로스포린 A와 함께 투여될 수 있다. 시클로스포린 A는 임상 실험에서 HCV에 대해 활성인 것으로 입증되었다 (문헌 [Hepatology 2003, 38, 1282]; [Biochem. Biophys. Res. Commun. 2004, 313, 42]; [J. Gastroenterol. 2003, 38, 567]).
하기 표 1은 본 개시내용의 화합물과 함께 투여될 수 있는 몇몇 예시적인 화합물을 열거한다. 본 개시내용의 화합물은 조합요법에서 다른 항-HCV 활성 화합물과 함께 공동으로 또는 개별적으로, 또는 화합물들을 조성물 내에 화합시킴으로써 투여될 수 있다.




본 개시내용의 화합물은 또한 실험실 시약으로서 사용될 수 있다. 화합물은 바이러스 복제 분석법의 고안, 동물 분석 시스템의 정당성 확인, 및 HCV 질환 메카니즘의 지식을 더욱 증진시키기 위한 구조 생물학 연구에 대해 조사 도구를 제공하는데 도움이 될 수 있다. 추가로, 본 개시내용의 화합물은, 예를 들어 경쟁적 억제에 의해, 다른 항바이러스성 화합물의 결합 부위를 확립하거나 결정하는데 유용하다.
본 개시내용의 화합물은 또한 물질의 바이러스 오염을 치료하거나 예방하기 위해 사용될 수 있으며, 따라서, 예를 들어 혈액, 조직, 수술 기구 및 의류, 실험실 기구 및 의류, 및 채혈 또는 수혈 장치 및 재료와 접촉하는 실험실 또는 병원 직원 또는 환자의 바이러스 감염의 위험을 낮출 수 있다.
본 개시내용은, 합성 과정에 의하거나, 또는 인간 또는 동물 신체내 (생체내)에서 발생하는 과정 또는 시험관내에서 발생하는 과정을 비롯한 대사 과정에 의해 제조되는 경우 화학식 I을 갖는 화합물을 포함하는 것으로 의도된다.
구체적으로 하기의 예시적 반응식 및 실시예를 비롯한, 본원에 사용된 약어는 당업자에게 익히 공지되어 있다. 사용된 몇몇 약어들은 다음과 같다: TFA = 트리플루오로아세트산; Ph = 페닐; tBu 또는 t-Bu = tert-부틸; DMSO = 디메틸술폭시드; DMF = N,N-디메틸포름아미드; EtOH = 에탄올; Boc 또는 BOC = tert-부톡시카르보닐; THF = 테트라히드로푸란; Et2O = 디에틸 에테르; Et = 에틸; MeOH = 메탄올; EtOAc 및 EtOAC = 에틸 아세테이트; RT = 실온 또는 체류 시간 (문맥에서 결정함); Rt 또는 tR = 체류 시간; h = 시간; sat'd = 포화; PCC = 피리디늄 클로로크로메이트; TBDPS = tert-부틸디페닐실릴; DMAP = 4-디메틸아미노피리딘; TBAF = 테트라부틸암모늄 플루오라이드; Et3N 또는 TEA = 트리에틸아민; min = 분; OAc = 아세테이트; Cbz = 카르보벤질옥시; SEM = 2-트리메틸실릴에톡시메톡시; AIBN = 아조비스이소부티로니트릴; HATU = O-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트; DDQ = 2,3-디클로로-5,6-디시아노-1,4-벤조퀴논; dppf = 1,1'-비스(디페닐포스피노)페로센; 및 iPr2NEt 또는 DIPEA 또는 DIEA = 디이소프로필에틸아민.
본 개시내용의 화합물 및 방법은 본 개시내용의 화합물을 제조할 수 있는 방법들을 예시하는 하기 합성 반응식과 연계하여 보다 잘 이해될 것이다. 출발 물질은 상업적 공급원으로부터 입수할 수 있거나, 또는 당업자에게 공지된 잘 확립된 문헌 방법에 의해 제조할 수 있다. 상기 정의된 화합물은 하기 나타낸 합성에서 적절한 반응물 및 작용제의 치환에 의해 합성될 수 있음이 당업자에게 쉽게 명백할 것이다. 선택적 보호 및 탈보호 단계, 및 그 단계들 자체의 순위는 하기 합성을 성공적으로 완료하기 위한 변수들의 특성에 따라, 순서를 다르게 하여 수행할 수 있음이 당업자에게 또한 쉽게 명백할 것이다. 변수들은 하기에 다르게 언급되지 않는 한, 상기 정의된 바와 같다.
반응식 1: 치환된
페닐글리신
유도체
치환된 페닐글리신 유도체는 하기에 나타낸 수많은 방법에 의해 제조할 수 있다. 페닐글리신 t-부틸 에스테르는 산성 매질에서 적절한 알데히드 및 환원제, 예컨대 나트륨 시아노보로히드라이드를 사용하여 환원성 알킬화시킬 수 있다 (경로 A). t-부틸 에스테르의 가수분해는 강산, 예컨대 HCl 또는 트리플루오로아세트산을 사용하여 달성할 수 있다. 별법으로, 페닐글리신은 알킬 할라이드, 예컨대 에틸 요오다이드 및 염기, 예컨대 중탄산나트륨 또는 탄산칼륨을 사용하여 알킬화시킬 수 있다 (경로 B). 경로 C는 경로 A에서와 같은 페닐글리신의 환원성 알킬화에 이어서 환원제 및 산의 존재하에 다른 알데히드, 예컨대 포름알데히드를 사용한 2차 환원성 알킬화를 예시한다. 경로 D는 상응하는 만델산 유사체를 통한 치환된 페닐글리신의 합성을 예시한다. 2급 알콜의 유능한 이탈기로의 전환은 p-톨루엔술포닐 클로라이드를 사용하여 달성할 수 있다. 적절한 아민으로 토실레이트기를 치환한 다음 벤질 에스테르를 환원성 제거하여 치환된 페닐글리신 유도체를 제공할 수 있다. 경로 E에서, 라세미 치환된 페닐글리신 유도체는 거울상이성질체적으로 순수한 키랄 보조제, 예컨대 (+)-1-페닐에탄올, (-)-1-페닐에탄올, 에반(Evan) 옥사졸리디논, 또는 거울상이성질체적으로 순수한 판토락톤 (이에 한정되지 않음)으로의 에스테르화에 의해 분할된다. 부분입체이성질체들의 분리는 크로마토그래피 (실리카겔, HPLC, 결정화 등)에 이어서 키랄 보조제를 제거하여 달성함으로써 거울상이성질체적으로 순수한 페닐글리신 유도체를 제공한다. 경로 H는 경로 E와 교차하는 합성 순서를 예시하며, 여기서 상기 언급된 키랄 보조제는 아민 첨가 전에 위치한다. 별법으로, 아릴아세트산의 에스테르는 브로모늄 이온 공급원, 예컨대 브롬, N-브로모숙신이미드 또는 CBr4를 사용하여 브롬화시킬 수 있다. 생성된 벤질성 브로마이드는 3급 아민 염기, 예컨대 트리에틸아민 또는 휴닉 염기의 존재하에, 다양한 일치환 또는 이치환된 아민으로 대체시킬 수 있다. 저온에서 수산화리튬으로, 또는 승온에서 6 N HCl로의 처리를 통해 메틸 에스테르를 가수분해시켜 치환된 페닐글리신 유도체를 제공한다. 또다른 방법은 경로 G로 제시된다. 글리신 유사체는 팔라듐 (0) 공급원, 예컨대 팔라듐 비스(트리부틸포스핀) 및 염기, 예컨대 인산칼륨의 존재하에, 다양한 아릴 할라이드를 사용하여 유도체화시킬 수 있다. 이어서, 생성된 에스테르를 염기 또는 산으로 처리하여 가수분해시킬 수 있다. 페닐글리신 유도체를 제조하기 위한 잘 알려진 기타 방법들이 당업계에 존재하고, 이는 본 기재내용의 목적하는 화합물을 제공하기 위해 보정될 수 있는 것으로 이해된다. 또한, 최종 페닐글리신 유도체를 분취용 HPLC를 통해 98%ee 초과의 거울상이성질체 순도로 정제할 수 있음을 이해해야 한다.

반응식 2:
아실화된
아미노산 유도체
본 개시내용의 또다른 실시양태에서, 아실화된 페닐글리신 유도체는 하기 예시된 바와 같이 제조할 수 있다. 카르복실산이 쉽게 제거되는 에스테르로서 보호된 페닐글리신 유도체는, 염기, 예컨대 트리에틸아민의 존재하에 산 클로라이드로 아실화시켜 상응하는 아미드를 제공할 수 있다 (경로 A). 경로 B는 적절한 클로로포르메이트를 사용한 출발 페닐글리신 유도체의 아실화를 예시하고, 경로 C는 적절한 이소시아네이트 또는 카르바모일 클로라이드와의 반응을 나타낸다. 경로 A 내지 C에서 제시된 3개의 중간체 각각은 당업자에게 공지된 방법으로 탈보호시킬 수 있다 (즉, 강산, 예컨대 HCl 또는 트리플루오로아세트산으로 t-부틸 에스테르의 처리).

반응식 3
아미노-치환된 페닐아세트산은 클로로메틸페닐아세트산을 과량의 아민으로 처리하여 제조할 수 있다.

화합물 분석 조건
워터스 마이크로매스(Waters Micromass) ZQ MS 시스템과 연결된 시마주(Shimadzu) LC 시스템 상에서 순도 평가 및 저분해능 질량 분석을 수행하였다. 체류 시간은 기계 간에 다소 다를 수 있다는 것을 주의하여야 한다. 체류 시간 (RT)을 측정하는데 이용되는 LC 조건은 다음과 같다:
일반적
cap
의 합성
달리 언급되지 않는 한, 추가적인 LC 조건은 본 섹션에 적용가능하다.
조건-
MS
-
W1
컬럼 = 엑스테라(XTERRA) 3.0×50 mm S7
시작 %B = 0
최종 %B = 100
구배 시간 = 2 분
중지 시간 = 3 분
유속 = 5 ml/분
파장 = 220 nm
용매 A = 10% 메탄올/90% H2O 중 0.1% TFA
용매 B = 90% 메탄올/10% H2O 중 0.1% TFA
조건-
MS
-
W2
컬럼 = 엑스테라 3.0×50 mm S7
시작 %B = 0
최종 %B = 100
구배 시간 = 3 분
중지 시간 = 4 분
유속 = 4 ml/분
파장 = 220 nm
용매 A = 10% 메탄올/90% H2O 중 0.1% TFA
용매 B = 90% 메탄올/10% H2O 중 0.1% TFA
조건- MS - W5
컬럼 = 엑스테라 3.0×50 mm S7
시작 %B = 0
최종 %B = 30
구배 시간 = 2 분
중지 시간 = 3 분
유속 = 5 ml/분
파장 = 220 nm
용매 A = 10% 메탄올/90% H2O 중 0.1% TFA
용매 B = 90% 메탄올/10% H2O 중 0.1% TFA
조건-
D1
컬럼 = 엑스테라 C18 3.0×50 mm S7
시작 %B = 0
최종 %B = 100
구배 시간 = 3 분
중지 시간 = 4 분
유속 = 4 ml/분
파장 = 220 nm
용매 A = 10% 메탄올/90% H2O 중 0.1% TFA
용매 B = 90% 메탄올/10% H2O 중 0.1% TFA
조건-
D2
컬럼 = 페노메넥스-루나(Phenomenex-Luna) 4.6×50 mm S10
시작 %B = 0
최종 %B = 100
구배 시간 = 3 분
중지 시간 = 4 분
유속 = 4 ml/분
파장 = 220 nm
용매 A = 10% 메탄올/90% H2O 중 0.1% TFA
용매 B = 90% 메탄올/10% H2O 중 0.1% TFA
조건-
M3
컬럼 = 엑스테라 C18 3.0×50 mm S7
시작 %B = 0
최종 %B = 40
구배 시간 = 2 분
중지 시간 = 3 분
유속 = 5 ml/분
파장 = 220 nm
용매 A = 10% 메탄올/90% H2O 중 0.1% TFA
용매 B = 90% 메탄올/10% H2O 중 0.1% TFA
조건 I
컬럼 = 페노메넥스-루나 3.0×50 mm S10
시작 %B = 0
최종 %B = 100
구배 시간 = 2 분
중지 시간 = 3 분
유속 = 4 ml/분
파장 = 220 nm
용매 A = 10% 메탄올/90% H2O 중 0.1% TFA
용매 B = 90% 메탄올/10% H2O 중 0.1% TFA
조건 II
컬럼 = 페노메넥스-루나 4.6×50 mm S10
시작 %B = 0
최종 %B = 100
구배 시간 = 2 분
중지 시간 = 3 분
유속 = 5 ml/분
파장 = 220 nm
용매 A = 10% 메탄올/90% H2O 중 0.1% TFA
용매 B = 90% 메탄올/10% H2O 중 0.1% TFA
조건 III
컬럼 = 엑스테라 C18 3.0×50mm S7
시작 %B = 0
최종 %B = 100
구배 시간 = 3 분
중지 시간 = 4 분
유속 = 4 ml/분
파장 = 220 nm
용매 A = 10% 메탄올/90% H2O 중 0.1% TFA
용매 B = 90% 메탄올/10% H2O 중 0.1% TFA

메탄올 (10 mL) 중 10% Pd/C (2.0 g)의 현탁액을 (R)-2-페닐글리신 (10 g, 66.2 mmol), 포름알데히드 (물 중 37 중량% 33 mL), 1 N HCl (30 mL) 및 메탄올 (30 mL)의 혼합물에 첨가하고, H2 (60 psi)에 3 시간 동안 노출시켰다. 반응 혼합물을 규조토 (셀라이트(Celite)®)를 통해 여과하고, 여과물을 진공하에 농축시켰다. 생성된 조 물질을 이소프로판올로부터 재결정화시켜, Cap-1의 HCl 염을 백색 침상체로서 수득하였다 (4.0 g).


NaBH3CN (6.22 g, 94 mmol)을 (R)-2-페닐글리신 (6.02 g, 39.8 mmol) 및 메탄올 (100 mL)의 냉각된 (얼음/물) 혼합물에 몇분에 걸쳐 조금씩 첨가하고, 5 분 동안 교반하였다. 아세트알데히드 (10 mL)를 10 분에 걸쳐 적가하고, 동일한 냉각 온도에서 45 분 동안, 및 주위 온도에서 약 6.5 시간 동안 교반을 계속하였다. 반응 혼합물을 다시 빙수조로 냉각시키고, 물 (3 mL)로 처리한 후, 혼합물의 pH가 약 1.5 내지 2.0이 될 때까지 약 45 분에 걸쳐 진한 HCl을 적가하여 켄칭시켰다. 냉각조를 제거하고, 혼합물의 pH가 약 1.5 내지 2.0을 유지하도록 진한 HCl을 첨가하면서 교반을 계속하였다. 반응 혼합물을 밤새 교반하고 여과하여, 백색 현탁액을 제거하고, 여과물을 진공하에 농축시켰다. 조 물질을 에탄올로부터 재결정화시켜, Cap-2의 HCl 염을 빛나는 백색 고체로서 두번의 수집물로 수득하였다 (수집물-1: 4.16 g; 수집물-2: 2.19 g).


아세트알데히드 (5.0 mL, 89.1 mmol), 및 메탄올/H2O (4 mL/1 mL) 중 10% Pd/C (720 mg)의 현탁액을 (R)-2-페닐글리신 (3.096 g, 20.48 mmol), 1 N HCl (30 mL) 및 메탄올 (40 mL)의 냉각된 (약 15℃) 혼합물에 순차적으로 첨가하였다. 냉각조를 제거하고, 반응 혼합물을 H2 풍선하에 17 시간 동안 교반하였다. 추가의 아세트알데히드 (10 mL, 178.2 mmol)를 첨가하고, H2 분위기 하에 24 시간 동안 교반을 계속하였다 [주: H2의 공급은 반응 전반에 걸쳐 필요에 따라 보충함]. 반응 혼합물을 규조토 (셀라이트®)를 통해 여과하고, 여과물을 진공하에 농축시켰다. 생성된 조 물질을 이소프로판올로부터 재결정화시켜, (R)-2-(에틸아미노)-2-페닐아세트산의 HCl 염을 빛나는 백색 고체로서 수득하였다 (2.846 g).

메탄올/H2O (3 mL/1 mL) 중 10% Pd/C (536 mg)의 현탁액을 (R)-2-(에틸아미노)-2-페닐아세트산/HCl (1.492 g, 6.918 mmol), 포름알데히드 (물 중 37 중량% 20 mL), 1 N HCl (20 mL) 및 메탄올 (23 mL)의 혼합물에 첨가하였다. 반응 혼합물을 H2 풍선하에 약 72 시간 동안 교반하고, 여기서 H2 공급은 필요에 따라 보충하였다. 반응 혼합물을 규조토 (셀라이트®)를 통해 여과하고, 여과물을 진공하에 농축시켰다. 생성된 조 물질을 이소프로판올 (50 mL)로부터 재결정화시켜, Cap-3의 HCl 염을 백색 고체로서 수득하였다 (985 mg).


ClCO2Me (3.2 mL, 41.4 mmol)를 (R)-tert-부틸 2-아미노-2-페닐아세테이트/HCl (9.877 g, 40.52 mmol) 및 디이소프로필에틸아민 (14.2 mL, 81.52 mmol)의 냉각된 (얼음/물) THF (410 mL) 반-용액에 6 분에 걸쳐 적가하고, 유사한 온도에서 5.5 시간 동안 교반하였다. 휘발성 성분을 진공하에 제거하고, 잔류물을 물 (100 mL) 및 에틸 아세테이트 (200 mL) 사이에 분배하였다. 유기층을 1 N HCl (25 mL) 및 NaHCO3 포화 용액 (30 mL)으로 세척하고, 건조시키고 (MgSO4), 여과하고, 진공하에 농축시켰다. 생성된 무색 오일을 헥산으로부터 연화처리하고, 여과하고, 헥산 (100 mL)으로 세척하여, (R)-tert-부틸 2-(메톡시카르보닐아미노)-2-페닐아세테이트를 백색 고체로서 수득하였다 (7.7 g).

TFA (16 mL)를 상기 생성물의 냉각된 (얼음/물) CH2Cl2 (160 mL) 용액에 7 분에 걸쳐 적가하고, 냉각조를 제거하고, 반응 혼합물을 20 시간 동안 교반하였다. 탈보호가 여전히 완료되지 않았기 때문에, 추가의 TFA (1.0 mL)를 첨가하고, 추가 2 시간 동안 교반을 계속하였다. 휘발성 성분을 진공하에 제거하고, 생성된 오일 잔류물을 디에틸 에테르 (15 mL) 및 헥산 (12 mL)으로 처리하여, 침전물을 수득하였다. 침전물을 여과하고, 디에틸 에테르/헥산 (약 1:3 비; 30 mL)으로 세척하고, 진공하에 건조시켜, Cap-4를 솜털모양 백색 고체로서 수득하였다 (5.57 g).


에탄올 (40 mL) 중 (R)-2-페닐글리신 (1.0 g, 6.62 mmol), 1,4-디브로모부탄 (1.57 g, 7.27 mmol) 및 Na2CO3 (2.10 g, 19.8 mmol)의 혼합물을 100℃에서 21 시간 동안 가열하였다. 반응 혼합물을 주위 온도로 냉각시키고 여과하고, 여과물을 진공하에 농축시켰다. 잔류물을 에탄올 중에 용해시키고, 1 N HCl을 사용하여 pH 3 내지 4로 산성화시키고, 휘발성 성분을 진공하에 제거하였다. 생성된 조 물질을 역상 HPLC (물/메탄올/TFA)로 정제하여, Cap-5의 TFA 염을 반-점성 백색 포말체로서 수득하였다 (1.0 g).


Cap-6의 TFA 염을 Cap-5의 제조 방법을 이용하여 (R)-2-페닐글리신 및 1-브로모-2-(2-브로모에톡시)에탄으로부터 합성하였다.


p-톨루엔술포닐 클로라이드 (8.65 g, 45.4 mmol)의 CH2Cl2 (200 mL) 용액을 -5℃ 내지 0℃로 온도를 유지하면서 (S)-벤질 2-히드록시-2-페닐아세테이트 (10.0 g, 41.3 mmol), 트리에틸아민 (5.75 mL, 41.3 mmol) 및 4-디메틸아미노피리딘 (0.504 g, 4.13 mmol)의 냉각된 (-5℃) CH2Cl2 (200 mL) 용액에 적가하였다. 반응물을 0℃에서 9 시간 동안 교반한 후, 냉동고 (-25℃)에서 14 시간 동안 저장하였다. 이것을 주위 온도로 해동하고, 물 (200 mL), 1 N HCl (100 mL) 및 염수 (100 mL)로 세척하고, 건조시키고 (MgSO4), 여과하고, 진공하에 농축시켜, 정치시 고체화되는 점성 오일로서 벤질 2-페닐-2-(토실옥시)아세테이트를 수득하였다 (16.5 g). 생성물의 키랄 보전성은 확인하지 않고, 생성물을 추가 정제 없이 다음 단계에 사용하였다.

벤질 2-페닐-2-(토실옥시)아세테이트 (6.0 g, 15.1 mmol), 1-메틸피페라진 (3.36 mL, 30.3 mmol) 및 N,N-디이소프로필에틸아민 (13.2 mL, 75.8 mmol)의 THF (75 mL) 용액을 65℃에서 7 시간 동안 가열하였다. 반응물을 주위 온도로 냉각시키고, 휘발성 성분을 진공하에 제거하였다. 잔류물을 에틸아세테이트 및 물 사이에 분배하고, 유기층을 물 및 염수로 세척하고, 건조시키고 (MgSO4), 여과하고, 진공하에 농축시켰다. 생성된 조 물질을 플래시 크로마토그래피 (실리카겔, 에틸 아세테이트)로 정제하여, 벤질 2-(4-메틸피페라진-1-일)-2-페닐아세테이트를 오렌지색을 띠는 갈색 점성 오일로서 수득하였다 (4.56 g). 키랄 HPLC 분석 (키랄셀(Chiralcel) OD-H)은 샘플이 38.2 대 58.7 비율의 거울상이성질체들의 혼합물임을 나타내었다. 거울상이성질체들의 분리는 하기와 같이 수행하였다: 생성물을 에탄올/헵탄 (1:1) 120 mL 중에 용해시키고, 75 mL/분으로 85:15 헵탄/에탄올로 용리시키는 키랄 HPLC 컬럼 (키랄셀 OJ, 5 cm ID×50 cm L, 20 μm) 상에서 주입하고 (5 mL/주입), 220 nm에서 모니터링하였다. 거울상이성질체-1 (1.474 g) 및 거울상이성질체-2 (2.2149 g)를 점성 오일로서 회수하였다.

벤질 2-(4-메틸피페라진-1-일)-2-페닐아세테이트 (1.0 g, 3.1 mmol)의 어느 하나의 거울상이성질체의 메탄올 (10 mL) 용액을 메탄올 (5.0 mL) 중 10% Pd/C (120 mg)의 현탁액에 첨가하였다. 반응 혼합물을 주의깊게 모니터링하면서 50 분 미만 동안 수소 풍선에 노출시켰다. 반응의 완료 직후, 촉매를 규조토 (셀라이트®)를 통해 여과하고, 여과물을 진공하에 농축시켜, 페닐아세트산으로 오염된 Cap-7을 황갈색 포말체로서 수득하였다 (867.6 mg; 질량이 이론적 수율을 초과함). 생성물을 추가 정제 없이 다음 단계에 사용하였다.

SN2 치환 단계에 적절한 아민 (즉, Cap-8에 대해서는 4-히드록시피페리딘 및 Cap-9에 대해서는 (S)-3-플루오로피롤리딘)을 사용하고 하기 기재된 바와 같은 각 입체이성질체 중간체의 분리를 위해 변형된 조건을 사용하여, Cap-8 및 Cap-9의 합성을 Cap-7의 합성에 따라 수행하다.

중간체 벤질 2-(4-히드록시피페리딘-1-일)-2-페닐 아세테이트의 거울상이성질체 분리를 하기 조건을 이용하여 수행하였다: 화합물 (500 mg)을 에탄올/헵탄 (5 mL/45 mL) 중에 용해시켰다. 생성된 용액을 10 mL/분으로 80:20 헵탄/에탄올로 용리시키는 키랄 HPLC 컬럼 (키랄셀 OJ, 2 cm ID×25 cm L, 10 μm) 상에 주입하고 (5 mL/주입), 220 nm에서 모니터링하여, 186.3 mg의 거울상이성질체-1 및 209.1 mg의 거울상이성질체-2를 담황색 점성 오일로서 수득하였다. 상기 벤질 에스테르를 Cap-7의 제조에 따라 수소화분해시켜 Cap-8을 수득하였다:


중간체 벤질 2-((S)-3-플루오로피롤리딘-1-일)-2-페닐아세테이트의 부분입체이성질체 분리를 하기 조건을 이용하여 수행하였다: 에스테르 (220 mg)를 10 bar 압력, 70 mL/분 유속 및 35℃ 온도에서 0.1% TFA를 함유하는 95% CO2/5% 메탄올로 용리시키는 키랄 HPLC 컬럼 (키랄셀 OJ-H, 0.46 cm ID×25 cm L, 5 μm) 상에서 분리하였다. 개별 입체이성질체에 대한 HPLC 용리액을 농축시키고, 잔류물을 CH2Cl2 (20 mL) 중에 용해시키고, 수성 매질 (물 10 mL + NaHCO3 포화 용액 1 mL)로 세척하였다. 유기상을 건조시키고 (MgSO4), 여과하고, 진공하에 농축시켜, 92.5 mg의 분획-1 및 59.6 mg의 분획-2를 수득하였다. 이들 벤질 에스테르를 Cap-7의 제조에 따라 가수소분해시켜 Cap 9a 및 9b를 제조하였다.


메탄올 (15 mL) 중 D-프롤린 (2.0 g, 17 mmol) 및 포름알데히드 (물 중 37 중량% 2.0 mL)의 용액에 메탄올 (5 mL) 중 10% Pd/C (500 mg)의 현탁액을 첨가하였다. 혼합물을 수소 풍선하에 23 시간 동안 교반하였다. 반응 혼합물을 규조토 (셀라이트®)를 통해 여과하고, 진공하에 농축시켜, Cap-10을 회백색 고체로서 수득하였다 (2.15 g).


메탄올 (20 mL) 중 (2S,4R)-4-플루오로피롤리딘-2-카르복실산 (0.50 g, 3.8 mmol), 포름알데히드 (물 중 37 중량% 0.5 mL), 12 N HCl (0.25 mL) 및 10% Pd/C (50 mg)의 혼합물을 수소 풍선하에 19 시간 동안 교반하였다. 반응 혼합물을 규조토 (셀라이트®)를 통해 여과하고, 여과물을 진공하에 농축시켰다. 잔류물을 이소프로판올로부터 재결정화시켜, Cap-11의 HCl 염을 백색 고체로서 수득하였다 (337.7 mg).


L-알라닌 (2.0 g, 22.5 mmol)을 10% 탄산나트륨 수용액 (50 mL) 중에 용해시키고, 메틸 클로로포르메이트 (4.0 mL)의 THF (50 mL) 용액을 상기 수용액에 첨가하였다. 반응 혼합물을 주위 조건하에 4.5 시간 동안 교반하고, 진공하에 농축시켰다. 생성된 백색 고체를 물 중에 용해시키고, 1 N HCl을 사용하여 pH 약 2 내지 3으로 산성화시켰다. 생성된 용액을 에틸 아세테이트 (3×100 mL)로 추출하고, 합한 유기상을 건조시키고 (Na2SO4), 여과하고 진공하에 농축시켜, 무색 오일을 수득하였다 (2.58 g). 상기 물질 500 mg을 역상 HPLC (H2O/메탄올/TFA)로 정제하여, 150 mg의 Cap-12를 무색 오일로서 수득하였다.

메탄올 (30 mL) 중 L-알라닌 (2.5 g, 28 mmol), 포름알데히드 (8.4 g, 37 중량%), 1 N HCl (30 mL) 및 10% Pd/C (500 mg)의 혼합물을 수소 대기 (50 psi)하에 5 시간 동안 교반하였다. 반응 혼합물을 규조토 (셀라이트®)를 통해 여과하고, 여과물을 진공하에 농축시켜, 진공하에 정치시 고체화되는 오일로서 Cap-13의 HCl 염을 수득하였다 (4.4 g; 질량이 이론적 수율을 초과함). 생성물을 추가 정제 없이 사용하였다.

단계 1: (R)-(-)-D-페닐글리신 tert-부틸 에스테르 (3.00 g, 12.3 mmol), NaBH3CN (0.773 g, 12.3 mmol), KOH (0.690 g, 12.3 mmol) 및 아세트산 (0.352 mL, 6.15 mmol)의 혼합물을 메탄올 중에서 0℃에서 교반하였다. 이 혼합물에 글루타르산 디알데히드 (2.23 mL, 12.3 mmol)를 5 분에 걸쳐 적가하였다. 반응 혼합물을 그대로 교반하면서 주위 온도로 가온하고, 동일한 온도에서 16 시간 동안 교반을 계속하였다. 용매를 후속적으로 제거하고, 잔류물을 10% 수성 NaOH 및 에틸 아세테이트로 분배하였다. 유기상을 분리하고, 건조시키고 (MgSO4), 여과하고, 농축 건조시켜, 투명 오일을 수득하였다. 이 물질을 역상 분취용 HPLC (프라임스피어(Primesphere) C-18, 30×100 mm; CH3CN-H2O-0.1% TFA)로 정제하여, 중간체 에스테르 (2.70 g, 56%)를 투명 오일로서 수득하였다.

단계 2: 디클로로메탄 (10 mL) 중 중간체 에스테르 (1.12 g, 2.88 mmol)의 교반 용액에 TFA (3 mL)를 첨가하였다. 반응 혼합물을 주위 온도에서 4 시간 동안 교반한 후, 이것을 농축 건조시켜 담황색 오일을 수득하였다. 오일을 역상 분취용 HPLC (프라임스피어 C-18, 30×100 mm; CH3CN-H2O-0.1% TFA)를 사용하여 정제하였다. 적절한 분획을 합하고, 진공하에 농축 건조시켰다. 이어서, 잔류물을 최소량의 메탄올 중에 용해시키고, MCX LP 추출 카트리지 (2×6 g)에 적용시켰다. 카트리지를 메탄올 (40 mL)로 헹군 후, 목적하는 화합물을 메탄올 (50 mL) 중 2 M 암모니아를 사용하여 용리시켰다. 생성물 함유 분획을 합하고 농축시키고, 잔류물을 물 중에 용해시켰다. 이 용액을 동결건조시켜 표제 화합물 (0.492 g, 78%)을 담황색 고체로서 수득하였다.


단계 1; (S)-1-페닐에틸 2-브로모-2-페닐아세테이트: 무수 디클로로메탄 (100 mL) 중 α-브로모페닐아세트산 (10.75 g, 0.050 mol), (S)-(-)-1-페닐에탄올 (7.94 g, 0.065 mol) 및 DMAP (0.61 g, 5.0 mmol)의 혼합물에 고체 EDCI (12.46 g, 0.065 mol)를 모두 한번에 첨가하였다. 생성된 용액을 실온에서 Ar하에 18 시간 동안 교반한 후, 이것을 에틸 아세테이트로 희석시키고, 세척하고 (H2O×2, 염수), 건조시키고 (Na2SO4), 여과하고, 농축시켜 옅은 황색 오일을 수득하였다. 이 오일을 플래시 크로마토그래피 (SiO2/헥산-에틸 아세테이트, 4:1)로 정제하여, 표제 화합물 (11.64 g, 73%)을 백색 고체로서 수득하였다.

단계 2; (S)-1-페닐에틸 (R)-2-(4-히드록시-4-메틸피페리딘-1-일)-2-페닐아세테이트: THF (8 mL) 중 (S)-1-페닐에틸 2-브로모-2-페닐아세테이트 (0.464 g, 1.45 mmol)의 용액에 트리에틸아민 (0.61 mL, 4.35 mmol)에 이어 테트라부틸암모늄 요오다이드 (0.215 g, 0.58 mmol)를 첨가하였다. 반응 혼합물을 실온에서 5 분 동안 교반한 후, THF (2 mL) 중 4-메틸-4-히드록시피페리딘 (0.251 g, 2.18 mmol)의 용액을 첨가하였다. 혼합물을 실온에서 1 시간 동안 교반한 후, 이것을 55 내지 60℃ (오일조 온도)에서 4 시간 동안 가열하였다. 이어서, 냉각된 반응 혼합물을 에틸 아세테이트 (30 mL)로 희석시키고, 세척하고 (H2O×2, 염수), 건조시키고 (MgSO4), 여과하고, 농축시켰다. 잔류물을 실리카겔 크로마토그래피 (0→60% 에틸 아세테이트-헥산)로 정제하여, 먼저 표제 화합물의 (S,R)-이성질체 (0.306 g, 60%)를 백색 고체로서, 이어서 상응하는 (S,S)-이성질체 (0.120 g, 23%)를 또한 백색 고체로서 수득하였다.

단계 3; (R)-2-(4-히드록시-4-메틸피페리딘-1-일)-2-페닐아세트산: 디클로로메탄 (3 mL) 중 (S)-1-페닐에틸 (R)-2-(4-히드록시-4-메틸피페리딘-1-일)-2-페닐아세테이트 (0.185 g, 0.52 mmol)의 용액에 트리플루오로아세트산 (1 mL)을 첨가하고, 혼합물을 실온에서 2 시간 동안 교반하였다. 휘발성 물질을 후속적으로 진공하에 제거하고, 잔류물을 역상 분취용 HPLC (프라임스피어 C-18, 20×100 mm; CH3CN-H2O-0.1% TFA)로 정제하여, 표제 화합물 (TFA 염)을 옅은 청색을 띠는 고체로서 수득하였다 (0.128 g, 98%).

단계 1; (S)-1-페닐에틸 2-(2-플루오로페닐)아세테이트: CH2Cl2 (100 mL) 중 2-플루오로페닐아세트산 (5.45 g, 35.4 mmol), (S)-1-페닐에탄올 (5.62 g, 46.0 mmol), EDCI (8.82 g, 46.0 mmol) 및 DMAP (0.561 g, 4.60 mmol)의 혼합물을 실온에서 12 시간 동안 교반하였다. 이어서, 용매를 농축시키고, 잔류물을 H2O-에틸 아세테이트로 분배하였다. 상들을 분리하고, 수성층을 에틸 아세테이트 (2×)로 역추출하였다. 합한 유기상을 세척하고 (H2O, 염수), 건조시키고 (Na2SO4), 여과하고, 진공하에 농축시켰다. 잔류물을 실리카겔 크로마토그래피 (바이오티지(Biotage)/0→20% 에틸 아세테이트-헥산)로 정제하여, 표제 화합물을 무색 오일로서 수득하였다 (8.38 g, 92%).
단계 2; (R)-((S)-1-페닐에틸) 2-(2-플루오로페닐)-2-(피페리딘-1-일)아세테이트: 0℃에서 THF (1200 mL) 중 (S)-1-페닐에틸 2-(2-플루오로페닐)아세테이트 (5.00 g, 19.4 mmol)의 용액에 DBU (6.19 g, 40.7 mmol)를 첨가하고, 용액을 30 분 동안 교반하면서 실온으로 가온하였다. 이어서, 용액을 -78℃로 냉각시키고, THF (100 mL) 중 CBr4 (13.5 g, 40.7 mmol)의 용액을 첨가하고, 혼합물을 -10℃로 가온하고, 이 온도에서 2 시간 동안 교반하였다. 반응 혼합물을 포화 수성 NH4Cl로 켄칭시키고, 층을 분리하였다. 수성층을 에틸 아세테이트 (2×)로 역추출하고, 합한 유기상을 세척하고 (H2O, 염수), 건조시키고 (Na2SO4), 여과하고, 진공하에 농축시켰다. 잔류물에 피페리딘 (5.73 mL, 58.1 mmol)을 첨가하고, 용액을 실온에서 24 시간 동안 교반하였다. 이어서, 휘발성 물질을 진공하에 농축시키고, 잔류물을 실리카겔 크로마토그래피 (바이오티지/0→30% 디에틸 에테르-헥산)로 정제하여, 미반응 출발 물질 (2.53 g, 51%)과 함께 부분입체이성질체들의 순수한 혼합물 (1H NMR에 의해 2:1 비)을 황색 오일로서 수득하였다 (2.07 g, 31%). 부분입체이성질체 혼합물을 추가 크로마토그래피 (바이오티지/0→10% 디에틸 에테르-톨루엔)하여, 표제 화합물을 무색 오일로서 수득하였다 (0.737 g, 11%).

단계 3; (R)-2-(2-플루오로페닐)-2-(피페리딘-1-일)아세트산: 에탄올 (30 mL) 중 (R)-((S)-1-페닐에틸) 2-(2-플루오로페닐)-2-(피페리딘-1-일)아세테이트 (0.737 g, 2.16 mmol) 및 20% Pd(OH)2/C (0.070 g)의 혼합물을 실온 및 대기압 (H2 풍선)에서 2 시간 동안 수소화시켰다. 용액을 Ar으로 퍼징하고, 규조토 (셀라이트®)를 통해 여과하고, 진공하에 농축시켰다. 이로써 표제 화합물을 무색 고체로서 수득하였다 (0.503 g, 98%).


단계 1; (S)-1-페닐에틸 (R)-2-(4-히드록시-4-페닐피페리딘-1-일)-2-페닐아세테이트: THF (25 mL) 중 (S)-1-페닐에틸 2-브로모-2-페닐아세테이트 (1.50 g, 4.70 mmol)의 용액에 트리에틸아민 (1.31 mL, 9.42 mmol)에 이어 테트라부틸암모늄 요오다이드 (0.347 g, 0.94 mmol)를 첨가하였다. 반응 혼합물을 실온에서 5 분 동안 교반한 후, THF (5 mL) 중 4-페닐-4-히드록시피페리딘 (1.00 g, 5.64 mmol)의 용액을 첨가하였다. 혼합물을 16 시간 동안 교반한 후, 이것을 에틸 아세테이트 (100 mL)로 희석시키고, 세척하고 (H2O×2, 염수), 건조시키고 (MgSO4), 여과하고, 농축시켰다. 잔류물을 실리카겔 컬럼 (0→60% 에틸 아세테이트-헥산) 상에서 정제하여, 부분입체이성질체들의 대략 2:1 혼합물을 수득하였다 (1H NMR로 판단). 이들 이성질체의 분리를 초임계 유체 크로마토그래피 (키랄셀 OJ-H, 30×250 mm; 35℃에서 CO2 중 20% 에탄올)를 사용하여 수행하여, 먼저 표제 화합물의 (R)-이성질체 (0.534 g, 27%)를 황색 오일로서, 이어서 상응하는 (S)-이성질체 (0.271 g, 14%)를 또한 황색 오일로서 수득하였다.

하기의 에스테르를 유사한 방식으로 제조하였다:



체류 시간 측정을 위한 키랄 SFC 조건
조건 I
컬럼: 키랄팩(Chiralpak) AD-H 컬럼, 4.62×50 mm, 5 μm
용매: 90% CO2 - 10% 메탄올 + 0.1% DEA
온도: 35℃
압력: 150 bar
유속: 2.0 mL/분
220 nm에서의 UV 모니터링
주입: 1.0 mg/메탄올 3 mL
조건 II
컬럼: 키랄셀 OD-H 컬럼, 4.62×50 mm, 5 μm
용매: 90% CO2 - 10% 메탄올 + 0.1% DEA
온도: 35℃
압력: 150 bar
유속: 2.0 mL/분
220 nm에서의 UV 모니터링
주입: 1.0 mg/메탄올 mL
Cap 17, 단계 2; (R)-2-(4-히드록시-4-페닐피페리딘-1-일)-2-페닐아세트산: 디클로로메탄 (5 mL) 중 (S)-1-페닐에틸 (R)-2-(4-히드록시-4-페닐피페리딘-1-일)-2-페닐아세테이트 (0.350 g, 0.84 mmol)의 용액에 트리플루오로아세트산 (1 mL)을 첨가하고, 혼합물을 실온에서 2 시간 동안 교반하였다. 휘발성 물질을 후속적으로 진공하에 제거하고, 잔류물을 역상 분취용 HPLC (프라임스피어 C-18, 20×100 mm; CH3CN-H2O-0.1% TFA)로 정제하여, 표제 화합물 (TFA 염)을 백색 고체로서 수득하였다 (0.230 g, 88%).
하기의 카르복실산을 유사한 방식으로 광학적으로 순수한 형태로 제조하였다:

체류 시간 측정을 위한 LCMS 조건
조건 I
컬럼: 페노메넥스-루나 4.6×50 mm S10
시작 %B = 0
최종 %B = 100
구배 시간 = 4 분
유속 = 4 mL/분
파장 = 220
용매 A = 10% 메탄올 - 90% H2O - 0.1% TFA
용매 B = 90% 메탄올 - 10% H2O - 0.1% TFA
조건 II
컬럼: 워터스-선파이어(Waters-Sunfire) 4.6×50 mm S5
시작 %B = 0
최종 %B = 100
구배 시간 = 2 분
유속 = 4 mL/분
파장 = 220
용매 A = 10% 메탄올 - 90% H2O - 0.1% TFA
용매 B = 90% 메탄올 - 10% H2O - 0.1% TFA
조건 III
컬럼: 페노메넥스 10μ 3.0×50 mm
시작 %B = 0
최종 %B = 100
구배 시간 = 2 분
유속 = 4 mL/분
파장 = 220
용매 A = 10% 메탄올 - 90% H2O - 0.1% TFA
용매 B = 90% 메탄올 - 10% H2O - 0.1% TFA

단계 1; (R,S)-에틸 2-(4-피리딜)-2-브로모아세테이트: 0℃에서 아르곤하에 건조 THF (150 mL) 중 에틸 4-피리딜아세테이트 (1.00 g, 6.05 mmol)의 용액에 DBU (0.99 mL, 6.66 mmol)를 첨가하였다. 반응 혼합물을 30 분에 걸쳐 실온으로 가온한 후, 이것을 -78℃로 냉각시켰다. 이 혼합물에 CBr4 (2.21 g, 6.66 mmol)를 첨가하고, -78℃에서 2 시간 동안 교반을 계속하였다. 이어서, 반응 혼합물을 포화 수성 NH4Cl로 켄칭시키고, 상들을 분리하였다. 유기상을 세척하고 (염수), 건조시키고 (Na2SO4), 여과하고, 진공하에 농축시켰다. 생성된 황색 오일을 플래시 크로마토그래피 (SiO2/헥산-에틸 아세테이트, 1:1)로 즉시 정제하여, 표제 화합물 (1.40 g, 95%)을 다소 불안정한 황색 오일로서 수득하였다.

단계 2; (R,S)-에틸 2-(4-피리딜)-2-(N,N-디메틸아미노)아세테이트: DMF (10 mL) 중 (R,S)-에틸 2-(4-피리딜)-2-브로모아세테이트 (1.40 g, 8.48 mmol)의 용액에 실온에서 디메틸아민 (THF 중 2 M, 8.5 mL, 17.0 mmol)을 첨가하였다. 반응의 완료 후 (박층 크로마토그래피로 판단), 휘발성 물질을 진공하에 제거하고, 잔류물을 플래시 크로마토그래피 (바이오티지, 40+M SiO2 컬럼; 50%→100% 에틸 아세테이트-헥산)로 정제하여, 표제 화합물 (0.539 g, 31%)을 담황색 오일로서 수득하였다.

단계 3; (R,S)-2-(4-피리딜)-2-(N,N-디메틸아미노)아세트산: THF-메탄올-H2O (1:1:1, 6 mL)의 혼합물 중 (R,S)-에틸 2-(4-피리딜)-2-(N,N-디메틸아미노)아세테이트 (0.200 g, 0.960 mmol)의 용액에 분말화 LiOH (0.120 g, 4.99 mmol)를 실온에서 첨가하였다. 용액을 3 시간 동안 교반한 후, 이것을 1 N HCl을 사용하여 pH 6으로 산성화시켰다. 수성상을 에틸 아세테이트로 세척한 후, 이것을 동결건조시켜, 표제 화합물의 디히드로클로라이드를 황색 고체로서 수득하였다 (LiCl 함유). 생성물을 후속 단계에 그대로 사용하였다.
하기의 실시예를 상기 기재된 방법을 사용하여 유사한 방식으로 제조하였다:



단계 1; (R,S)-에틸 2-(퀴놀린-3-일)-2-(N,N-디메틸아미노)-아세테이트: 에틸 N,N-디메틸아미노아세테이트 (0.462 g, 3.54 mmol), K3PO4 (1.90 g, 8.95 mmol), Pd(t-Bu3P)2 (0.090 g, 0.176 mmol) 및 톨루엔 (10 mL)의 혼합물을 Ar 버블 스트림으로 15 분 동안 탈기시켰다. 이어서, 반응 혼합물을 100℃에서 12 시간 동안 가열한 후, 이것을 실온으로 냉각시키고, H2O에 부었다. 혼합물을 에틸 아세테이트 (2×)로 추출하고, 합한 유기상을 세척하고 (H2O, 염수), 건조시키고 (Na2SO4), 여과하고, 진공하에 농축시켰다. 잔류물을 역상 분취용 HPLC (프라임스피어 C-18, 30×100 mm; CH3CN-H2O-5 mM NH4OAc)로 먼저 정제한 후, 플래시 크로마토그래피 (SiO2/헥산-에틸 아세테이트, 1:1)로 정제하여, 표제 화합물 (0.128 g, 17%)을 오렌지색 오일로서 수득하였다.

단계 2; (R,S) 2-(퀴놀린-3-일)-2-(N,N-디메틸아미노)아세트산: (R,S)-에틸 2-(퀴놀린-3-일)-2-(N,N-디메틸아미노)아세테이트 (0.122 g, 0.472 mmol) 및 6 M HCl (3 mL)의 혼합물을 100℃에서 12 시간 동안 가열하였다. 용매를 진공하에 제거하여, 표제 화합물의 디히드로클로라이드 (0.169 g, >100%)를 담황색 포말체로서 수득하였다. 비정제 물질을 추가 정제 없이 후속 단계에 사용하였다. 

단계 1; (R)-((S)-1-페닐에틸) 2-(디메틸아미노)-2-(2-플루오로페닐)아세테이트 및 (S)-((S)-1-페닐에틸) 2-(디메틸아미노)-2-(2-플루오로페닐)아세테이트: CH2Cl2 (40 mL) 중 (RS)-2-(디메틸아미노)-2-(2-플루오로페닐)아세트산 (2.60 g, 13.19 mmol), DMAP (0.209 g, 1.71 mmol) 및 (S)-1-페닐에탄올 (2.09 g, 17.15 mmol)의 혼합물에 EDCI (3.29 g, 17.15 mmol)를 첨가하고, 혼합물을 실온에서 12 시간 동안 교반하였다. 이어서, 용매를 진공하에 제거하고, 잔류물을 에틸 아세테이트-H2O로 분배하였다. 층을 분리하고, 수성층을 에틸 아세테이트 (2×)로 역추출하고, 합한 유기상을 세척하고 (H2O, 염수), 건조시키고 (Na2SO4), 여과하고, 진공하에 농축시켰다. 잔류물을 실리카겔 크로마토그래피 (바이오티지/0→50% 디에틸 에테르-헥산)로 정제하였다. 이어서, 생성된 순수한 부분입체이성질체 혼합물을 역상 분취용 HPLC (프라임스피어 C-18, 30×100 mm; CH3CN-H2O-0.1% TFA)에 의해 분리하여, 먼저 (S)-1-페네틸 (R)-2-(디메틸아미노)-2-(2-플루오로페닐)아세테이트 (0.501 g, 13%) 및 이어서 (S)-1-페네틸 (S)-2-(디메틸아미노)-2-(2-플루오로페닐)-아세테이트 (0.727 g, 18%)를 둘 다 그들의 TFA 염으로서 수득하였다.

단계 2; (R)-2-(디메틸아미노)-2-(2-플루오로페닐)아세트산: 에탄올 (30 mL) 중 (R)-((S)-1-페닐에틸) 2-(디메틸아미노)-2-(2-플루오로페닐)아세테이트 TFA 염 (1.25 g, 3.01 mmol) 및 20% Pd(OH)2/C (0.125 g)의 혼합물을 실온 및 대기압 (H2 풍선)에서 4 시간 동안 수소화시켰다. 이어서, 용액을 Ar으로 퍼징하고, 규조토 (셀라이트®)를 통해 여과하고, 진공하에 농축시켰다. 이로써 표제 화합물을 무색 고체로서 수득하였다 (0.503 g, 98%).

유사한 방식으로 (S)-((S)-1-페닐에틸) 2-(디메틸아미노)-2-(2-플루오로페닐)아세테이트 TFA 염으로부터 S-이성질체를 수득할 수 있었다.

(R)-(2-클로로페닐)글리신 (0.300 g, 1.62 mmol), 포름알데히드 (35% 수용액, 0.80 mL, 3.23 mmol) 및 20% Pd(OH)2/C (0.050 g)의 혼합물을 실온 및 대기압 (H2 풍선)에서 4 시간 동안 수소화시켰다. 이어서, 용액을 Ar으로 퍼징하고, 규조토 (셀라이트®)를 통해 여과하고, 진공하에 농축시켰다. 잔류물을 역상 분취용 HPLC (프라임스피어 C-18, 30×100 mm; CH3CN-H2O-0.1% TFA)로 정제하여, 표제 화합물 (R)-2-(디메틸아미노)-2-(2-클로로페닐)아세트산의 TFA 염을 무색 오일로서 수득하였다 (0.290 g, 55%).


H2O (5.5 mL) 중 (R)-(2-클로로페닐)글리신 (1.00 g, 5.38 mmol) 및 NaOH (0.862 g, 21.6 mmol)의 빙냉 용액에 메틸 클로로포르메이트 (1.00 mL, 13.5 mmol)를 적가하였다. 상기 혼합물을 0℃에서 1 시간 동안 교반한 후, 이것을 진한 HCl (2.5 mL)을 첨가하여 산성화시켰다. 혼합물을 에틸 아세테이트 (2×)로 추출하고, 합한 유기상을 세척하고 (H2O, 염수), 건조시키고 (Na2SO4), 여과하고, 진공하에 농축시켜, 표제 화합물 (R)-2-(메톡시카르보닐아미노)-2-(2-클로로페닐)아세트산을 황색-오렌지색 포말체로서 수득하였다 (1.31 g, 96%).


THF (20 mL) 중 2-(2-(클로로메틸)페닐)아세트산 (2.00 g, 10.8 mmol)의 현탁액에 모르폴린 (1.89 g, 21.7 mmol)을 첨가하고, 용액을 실온에서 3 시간 동안 교반하였다. 이어서, 반응 혼합물을 에틸 아세테이트로 희석시키고, H2O (2×)로 추출하였다. 수성상을 동결건조시키고, 잔류물을 실리카겔 크로마토그래피 (바이오티지/0→10% 메탄올-CH2Cl2)로 정제하여, 표제 화합물 2-(2-(모르폴리노메틸)페닐)아세트산을 무색 고체로서 수득하였다 (2.22 g, 87%).

하기의 실시예를 Cap-41에 대해 기재된 방법을 사용하여 유사하게 제조하였다:


HMDS (1.85 mL, 8.77 mmol)를 CH2Cl2 (10 mL) 중 (R)-2-아미노-2-페닐아세트산 p-톨루엔술포네이트 (2.83 g, 8.77 mmol)의 현탁액에 첨가하고, 혼합물을 실온에서 30 분 동안 교반하였다. 메틸 이소시아네이트 (0.5 g, 8.77 mmol)를 30 분 동안 교반을 계속하면서 한번에 첨가하였다. 반응물을 H2O (5 mL)를 첨가하여 켄칭시키고, 생성된 침전물을 여과하고, H2O 및 n-헥산으로 세척하고, 진공하에 건조시켰다. (R)-2-(3-메틸우레이도)-2-페닐아세트산 (1.5 g; 82%)을 백색 고체로서 회수하였고, 이것을 추가 정제 없이 사용하였다.


Cap-45a에 기재된 방법에 따라 목적하는 생성물을 제조하였다.


단계 1; (R)-tert-부틸 2-(3,3-디메틸우레이도)-2-페닐아세테이트: DMF (40 mL) 중 (R)-tert-부틸-2-아미노-2-페닐아세테이트 (1.0 g, 4.10 mmol) 및 휴닉 염기 (1.79 mL, 10.25 mmol)의 교반 용액에 디메틸카르바모일 클로라이드 (0.38 mL, 4.18 mmol)를 10 분에 걸쳐 적가하였다. 실온에서 3 시간 동안 교반한 후, 반응물을 감압하에 농축시키고, 생성된 잔류물을 에틸 아세테이트 중에 용해시켰다. 유기 층을 H2O, 1 N 수성 HCl 및 염수로 세척하고, 건조시키고 (MgSO4), 여과하고, 감압하에 농축시켰다. (R)-tert-부틸 2-(3,3-디메틸우레이도)-2-페닐아세테이트를 백색 고체로서 수득하였고 (0.86 g; 75%), 이를 추가 정제 없이 사용하였다.

단계 2; (R)-2-(3,3-디메틸우레이도)-2-페닐아세트산: CH2Cl2 (250 mL) 중 ((R)-tert-부틸 2-(3,3-디메틸우레이도)-2-페닐아세테이트 (0.86 g, 3.10 mmol)의 교반 용액에 TFA (15 mL)를 적가하고, 생성된 용액을 실온에서 3 시간 동안 교반하였다. 이어서, EtOAC:헥산 (5:20)의 혼합물을 사용하여 상기 용액으로부터 목적하는 생성물을 침전시키고, 여과해내고, 감압하에 건조시켰다. (R)-2-(3,3-디메틸우레이도)-2-페닐아세트산을 백색 고체로서 단리시켰고 (0.59 g, 86%), 이를 추가 정제 없이 사용하였다.


단계 1; (R)-tert-부틸 2-(3-시클로펜틸우레이도)-2-페닐아세테이트: DMF (15 mL) 중 (R)-2-아미노-2-페닐아세트산 히드로클로라이드 (1.0 g, 4.10 mmol) 및 휴닉 염기 (1.0 mL, 6.15 mmol)의 교반 용액에 시클로펜틸 이소시아네이트 (0.46 mL, 4.10 mmol)를 10 분에 걸쳐 적가하였다. 실온에서 3 시간 동안 교반한 후, 반응물을 감압하에 농축시키고, 생성된 잔류물을 에틸 아세테이트 중에 용해시켰다. 유기 층을 H2O 및 염수로 세척하고, 건조시키고 (MgSO4), 여과하고, 감압하에 농축시켰다. (R)-tert-부틸 2-(3-시클로펜틸우레이도)-2-페닐아세테이트를 불투명 오일로서 수득하였고 (1.32 g; 100%), 이를 추가 정제 없이 사용하였다.

단계 2; (R)-2-(3-시클로펜틸우레이도)-2-페닐아세트산: CH2Cl2 (25 mL) 중 (R)-tert-부틸 2-(3-시클로펜틸우레이도)-2-페닐아세테이트 (1.31 g, 4.10 mmol)의 교반 용액에 TFA (4 mL) 및 트리에틸실란 (1.64 mL; 10.3 mmol)을 적가하고, 생성된 용액을 실온에서 6 시간 동안 교반하였다. 휘발성 성분을 감압하에 제거하고, 조 생성물을 에틸 아세테이트/펜탄에서 재결정화시켜, (R)-2-(3-시클로펜틸우레이도)-2-페닐아세트산을 백색 고체로서 수득하였다 (0.69 g, 64%).


포름산 (91 mL) 중 2-(벤질아미노)아세트산 (2.0 g, 12.1 mmol)의 교반 용액에 포름알데히드 (6.94 mL, 93.2 mmol)를 첨가하였다. 70℃에서 5 시간 후, 반응 혼합물을 감압하에 20 mL로 농축시켰고, 백색 고체가 침전되었다. 여과 후, 모액을 수집하고, 감압하에 추가로 농축시켜, 조 생성물을 수득하였다. 역상 분취용 HPLC (엑스테라 30×100 mm, 220 nm에서 검출, 유속 35 mL/분, 8 분에 걸쳐 0→35% B; A = 90% 물, 10% 메탄올, 0.1% TFA, B = 10% 물, 90% 메탄올, 0.1% TFA)로 정제하여, 표제 화합물 2-(벤질(메틸)-아미노)아세트산을 그의 TFA 염 (723 mg, 33%)으로서 무색 왁스로서 수득하였다.


물 (30 mL) 중 3-메틸-2-(메틸아미노)부탄산 (0.50 g, 3.81 mmol)의 교반 용액에 K2CO3 (2.63 g, 19.1 mmol) 및 벤질 클로라이드 (1.32 g, 11.4 mmol)를 첨가하였다. 반응 혼합물을 주위 온도에서 18 시간 동안 교반하였다. 반응 혼합물을 에틸 아세테이트 (30 mL×2)로 추출하였고, 수성 층을 감압하에 농축시켜, 조 생성물을 수득하였고, 이것을 역상 분취용 HPLC (엑스테라 30×100 mm, 220 nm에서 검출, 유속 40 mL/분, 6 분에 걸쳐 20→80% B; A = 90% 물, 10% 메탄올, 0.1% TFA, B = 10% 물, 90% 메탄올, 0.1% TFA)로 정제하여, 2-(벤질(메틸)아미노)-3-메틸부탄산, TFA 염 (126 mg, 19%)을 무색 왁스로서 수득하였다.


Na2CO3 (1.83 g, 17.2 mmol)을 L-발린 (3.9 g, 33.29 mmol)의 NaOH (1 M/H2O 33 mL, 33 mmol) 용액에 첨가하고, 생성된 용액을 빙수조로 냉각시켰다. 메틸 클로로포르메이트 (2.8 mL, 36.1 mmol)를 15 분에 걸쳐 적가하였고, 냉각조를 제거하고, 반응 혼합물을 주위 온도에서 3.25 시간 동안 교반하였다. 반응 혼합물을 에테르 (50 mL, 3×)로 세척하고, 수성 상을 빙수조로 냉각시키고, 진한 HCl을 사용하여 pH 1 내지 2의 영역으로 산성화시키고, CH2Cl2 (50 mL, 3×)로 추출하였다. 유기 상을 건조시키고 (MgSO4), 진공하에 증발시켜, Cap-51을 백색 고체로서 수득하였다 (6 g).


Cap-51의 합성에 대해 기재된 절차에 따라 L-알라닌으로부터 Cap-52를 합성하였다. 특징규명의 목적을 위해, 조 물질의 일부를 역상 HPLC (H2O/메탄올/TFA)로 정제하여, Cap-52를 무색 점성 오일로서 수득하였다.
Cap-51의 합성에 대해 기재된 절차에 따라, 만일 존재하는 경우에는 명시된 변형법으로, 적절한 출발 물질로부터 Cap-53 내지 Cap-64를 제조하였다.




메틸 클로로포르메이트 (0.65 mL, 8.39 mmol)를 Na2CO3 (0.449 g, 4.23 mmol), NaOH (1 M/H2O 8.2 mL, 8.2 mmol) 및 (S)-2-아미노-3-히드록시-3-메틸부탄산 (1.04 g, 7.81 mmol)의 냉각된 (빙수) 혼합물에 5 분에 걸쳐 적가하였다. 반응 혼합물을 45 분 동안 교반한 후, 냉각조를 제거하고, 추가의 3.75 시간 동안 교반을 계속하였다. 반응 혼합물을 CH2Cl2로 세척하고, 수성 상을 빙수조로 냉각시키고, 진한 HCl을 사용하여 pH 1 내지 2 영역으로 산성화시켰다. 휘발성 성분을 진공하에 제거하고, 잔류물을 MeOH/CH2Cl2의 2:1 혼합물 (15 mL) 중에 용해시키고 여과하고, 여과물을 회전증발시켜, Cap-65를 백색 반-점성 포말체로서 수득하였다 (1.236 g).

Cap-65의 합성에 대해 기재된 절차를 사용하여 시판 공급되는 적절한 출발 물질로부터 Cap-66 및 -67을 제조하였다.





메틸 클로로포르메이트 (0.38 ml, 4.9 mmol)를 1 N NaOH (수성) (9.0 ml, 9.0 mmol), 1 M NaHCO3 (수성) (9.0 ml, 9.0 mmol), L-아스파르트산 β-벤질 에스테르 (1.0 g, 4.5 mmol) 및 디옥산 (9 ml)의 혼합물에 적가하였다. 반응 혼합물을 주위 조건에서 3 시간 동안 교반한 후, 에틸 아세테이트 (50 ml, 3×)로 세척하였다. 수성 층을 12 N HCl을 사용하여 pH 약 1 내지 2로 산성화시키고, 에틸 아세테이트 (3×50 ml)로 추출하였다. 합한 유기 층을 염수로 세척하고, 건조시키고 (Na2SO4), 여과하고, 진공하에 농축시켜, Cap-68을 담황색 오일로서 수득하였다 (1.37 g; 질량이 이론적 수율을 초과하고, 생성물은 추가 정제 없이 사용함).


NaCNBH3 (2.416 g, 36.5 mmol)을 알라닌 (1.338 g, 15.0 mmol)의 냉각된 (약 15℃) 물 (17 mL)/MeOH (10 mL) 용액에 배치로 첨가하였다. 몇분 후에, 아세트알데히드 (4.0 mL, 71.3 mmol)를 4 분에 걸쳐 적가하고, 냉각조를 제거하고, 반응 혼합물을 주위 조건에서 6 시간 동안 교반하였다. 추가의 아세트알데히드 (4.0 mL)를 첨가하고, 반응물을 2 시간 동안 교반하였다. pH가 약 1.5에 도달할 때까지 진한 HCl을 반응 혼합물에 서서히 첨가하고, 생성된 혼합물을 40℃에서 1 시간 동안 가열하였다. 대부분의 휘발성 성분을 진공하에 제거하고, 잔류물을 도웩스(Dowex)® 50WX8-100 이온 교환 수지 (컬럼을 물로 세척하고, 화합물을 묽은 NH4OH (NH4OH 18 ml 및 물 282 ml를 혼합하여 제조함)로 용리시킴)로 정제하여, Cap-69 (2.0 g)를 연질의 회백색 흡습성 고체로서 수득하였다.

Cap-69의 합성에 대해 기재된 절차에 따라, 적절한 출발 물질을 사용하여 Cap-70 내지 Cap-74x를 제조하였다.




NaBH3CN (1.6 g, 25.5 mmol)을 H-D-Ser-OBzl HCl (2.0 g, 8.6 mmol)의 냉각된 (얼음/물 조) 물 (25 ml)/메탄올 (15 ml) 용액에 첨가하였다. 아세트알데히드 (1.5 ml, 12.5 mmol)를 5 분에 걸쳐 적가하고, 냉각조를 제거하고, 반응 혼합물을 주위 조건에서 2 시간 동안 교반하였다. 반응물을 12 N HCl로 조심스럽게 켄칭시키고, 진공하에 농축시켰다. 잔류물을 물 중에 용해시키고, 역상 HPLC (MeOH/H2O/TFA)로 정제하여, (R)-벤질 2-(디에틸아미노)-3-히드록시프로파노에이트의 TFA 염을 무색 점성 오일로서 수득하였다 (1.9 g).

Cap
-75
NaH (0.0727 g, 1.82 mmol, 60%)를 상기 제조된 TFA 염 (R)-벤질 2-(디에틸아미노)-3-히드록시프로파노에이트 (0.3019 g, 0.8264 mmol)의 냉각된 (빙수) THF (3.0 mL) 용액에 첨가하고, 혼합물을 15 분 동안 교반하였다. 메틸 요오다이드 (56 μL, 0.90 mmol)를 첨가하고, 조를 주위 조건으로 해동시키면서 18 시간 동안 교반을 계속하였다. 반응물을 물로 켄칭시키고, MeOH 예비-컨디셔닝된 MCX (6 g) 카트리지 상에 로딩하고, 메탄올로 세척하고, 이어서 화합물을 2 N NH3/메탄올로 용리시켰다. 휘발성 성분을 진공하에 제거하여, (R)-2-(디에틸아미노)-3-히드록시프로판산으로 오염된 Cap-75를 황색 반-고체로서 수득하였다 (100 mg). 생성물을 추가 정제 없이 그대로 사용하였다.

NaCNBH3 (1.60 g, 24.2 mmol)을 (S)-4-아미노-2-(tert-부톡시카르보닐아미노)부탄산 (2.17 g, 9.94 mmol)의 냉각된 (약 15℃) 물/MeOH (각각 12 mL) 용액에 배치로 첨가하였다. 몇분 후에, 아세트알데히드 (2.7 mL, 48.1 mmol)를 2 분에 걸쳐 적가하고, 냉각조를 제거하고, 반응 혼합물을 주위 조건에서 3.5 시간 동안 교반하였다. 추가의 아세트알데히드 (2.7 mL, 48.1 mmol)를 첨가하고, 반응물을 20.5 시간 동안 교반하였다. 대부분의 MeOH 성분을 진공하에 제거하고, 남아있는 혼합물을 pH가 약 1.0에 도달할 때까지 진한 HCl로 처리한 후, 40℃에서 2 시간 동안 가열하였다. 휘발성 성분을 진공하에 제거하고, 잔류물을 4 M HCl/디옥산 (20 mL)으로 처리하고, 주위 조건에서 7.5 시간 동안 교반하였다. 휘발성 성분을 진공하에 제거하고, 잔류물을 도웩스® 50WX8-100 이온 교환 수지 (컬럼을 물로 세척하고, 화합물을 묽은 NH4OH (NH4OH 18 ml 및 물 282 ml로부터 제조함)로 용리시킴)로 정제하여, 중간체 (S)-2-아미노-4-(디에틸아미노)부탄산을 회백색 고체로서 수득하였다 (1.73 g).
메틸 클로로포르메이트 (0.36 mL, 4.65 mmol)를 Na2CO3 (0.243 g, 2.29 mmol), NaOH (1 M/H2O 4.6 mL, 4.6 mmol) 및 상기 생성물 (802.4 mg)의 냉각된 (빙수) 혼합물에 11 분에 걸쳐 적가하였다. 반응 혼합물을 55 분 동안 교반한 후, 냉각조를 제거하고, 추가의 5.25 시간 동안 교반을 계속하였다. 반응 혼합물을 동일한 부피의 물로 희석시키고, CH2Cl2 (30 mL, 2×)로 세척하고, 수성 상을 빙수조로 냉각시키고, 진한 HCl을 사용하여 pH 2의 영역으로 산성화시켰다. 이어서, 휘발성 성분을 진공하에 제거하고, 조 물질을 MCX 수지 (6.0 g; 컬럼을 물로 세척하고, 샘플을 2.0 M NH3/MeOH로 용리시킴)로 유리 염기화시켜, 불순물이 섞인 Cap-76을 회백색 고체로서 수득하였다 (704 mg).


SN2 치환 단계에 대해 7-아자바이시클로[2.2.1]헵탄을 사용하고, 하기 조건을 사용하여 중간체 벤질 2-(7-아자바이시클로[2.2.1]헵탄-7-일)-2-페닐아세테이트의 거울상이성질체 분리를 수행함으로써, Cap-7에 기재된 절차에 따라 Cap-77의 합성을 수행하였다: 중간체 (303.7 mg)를 에탄올 중에 용해시키고, 생성된 용액을 키랄 HPLC 컬럼 (키랄셀 AD-H 컬럼, 30×250 mm, 5 um) 상에 주입하고, 35℃의 온도에서 70 mL/분의 90% CO2-10% EtOH로 용리시켜, 124.5 mg의 거울상이성질체-1 및 133.8 mg의 거울상이성질체-2를 수득하였다. 이들 벤질 에스테르를 Cap-7의 제조에 따라 가수소분해하여 Cap-77을 수득하였다.


NaCNBH3 (0.5828 g, 9.27 mmol)을 MeOH (10 mL) 중 (1-에톡시시클로프로폭시)트리메틸실란 (1.640 g, 9.40 mmol) 및 (R)-2-(에틸아미노)-2-페닐아세트산 (Cap-3의 합성에서의 중간체; 0.9923 mg, 4.60 mmol)의 HCl 염의 혼합물에 첨가하고, 반-불균질 혼합물을 오일조로 50℃에서 20 시간 동안 가열하였다. 추가의 (1-에톡시시클로프로폭시)트리메틸실란 (150 mg, 0.86 mmol) 및 NaCNBH3 (52 mg, 0.827 mmol)을 첨가하고, 반응 혼합물을 추가의 3.5 시간 동안 가열하였다. 이어서, 이것을 주위 온도로 냉각시키고, 진한 HCl을 사용하여 pH 약 2의 영역으로 산성화시키고, 혼합물을 여과하고, 여과물을 회전증발시켰다. 생성된 조 물질을 i-PrOH (6 mL) 중에 용해시키고, 가열하여 용해시키고, 비-용해된 부분을 여과해내고, 여과물을 진공하에 농축시켰다. 생성된 조 물질의 약 1/3을 역상 HPLC (H2O/MeOH/TFA)로 정제하여, Cap-78의 TFA 염을 무색 점성 오일로서 수득하였다 (353 mg).


반응 혼합물이 청색 색조를 얻을 때까지, 오존을 Cap-55 (369 mg, 2.13 mmol)의 냉각된 (-78℃) CH2Cl2 (5.0 mL) 용액을 통해 약 50 분 동안 버블링하였다. Me2S (10 피펫 방울)를 첨가하고, 반응 혼합물을 35 분 동안 교반하였다. -78℃ 조를 -10℃ 조로 대체하고, 추가의 30 분 동안 교반을 계속한 후, 휘발성 성분을 진공하에 제거하여, 무색 점성 오일을 수득하였다.
NaBH3CN (149 mg, 2.25 mmol)을 상기 조 물질 및 모르폴린 (500 μL, 5.72 mmol)의 MeOH (5.0 mL) 용액에 첨가하고, 혼합물을 주위 조건에서 4 시간 동안 교반하였다. 이것을 빙수 온도로 냉각시키고, 진한 HCl로 처리하여, 그의 pH가 약 2.0이 되도록 하였으며, 이후 2.5 시간 동안 교반하였다. 휘발성 성분을 진공하에 제거하고, 잔류물을 MCX 수지 (MeOH 세척; 2.0 N NH3/MeOH 용리) 및 역상 HPLC (H2O/MeOH/TFA)의 조합에 의해 정제하여, 확인되지 않은 양의 모르폴린을 함유하는 Cap-79를 수득하였다.
모르폴린 오염물을 소모시키기 위해, 상기 물질을 CH2Cl2 (1.5 mL) 중에 용해시키고, Et3N (0.27 mL, 1.94 mmol)에 이어 아세트산 무수물 (0.10 mL, 1.06 mmol)로 처리하고, 주위 조건에서 18 시간 동안 교반하였다. THF (1.0 mL) 및 H2O (0.5 mL)를 첨가하고, 1.5 시간 동안 교반을 계속하였다. 휘발성 성분을 진공하에 제거하고, 생성된 잔류물을 MCX 수지 (MeOH 세척; 2.0 N NH3/MeOH 용리)를 통해 통과시켜, 불순물이 섞인 Cap-79를 갈색 점성 오일로서 수득하였고, 이것을 추가 정제 없이 다음 단계에 사용하였다.

SOCl2 (6.60 mL, 90.5 mmol)를 (S)-3-아미노-4-(벤질옥시)-4-옥소부탄산 (10.04 g, 44.98 mmol) 및 MeOH (300 mL)의 냉각된 (빙수) 혼합물에 15 분에 걸쳐 적가하고, 냉각조를 제거하고, 반응 혼합물을 주위 조건에서 29 시간 동안 교반하였다. 대부분의 휘발성 성분을 진공하에 제거하고, 잔류물을 EtOAc (150 mL) 및 NaHCO3 포화 용액 사이에 조심스럽게 분배하였다. 수성 상을 EtOAc (150 mL, 2×)로 추출하였고, 합한 유기 상을 건조시키고 (MgSO4), 여과하고, 진공하에 농축시켜, (S)-1-벤질 4-메틸 2-아미노숙시네이트를 무색 오일로서 수득하였다 (9.706 g).

Pb(NO3)2 (6.06 g, 18.3 mmol)를 (S)-1-벤질 4-메틸 2-아미노숙시네이트 (4.50 g, 19.0 mmol), 9-브로모-9-페닐-9H-플루오렌 (6.44 g, 20.0 mmol) 및 Et3N (3.0 mL, 21.5 mmol)의 CH2Cl2 (80 mL) 용액에 1 분에 걸쳐 첨가하고, 불균질 혼합물을 주위 조건에서 48 시간 동안 교반하였다. 혼합물을 여과하고, 여과물을 MgSO4로 처리하고, 다시 여과하고, 최종 여과물을 농축시켰다. 생성된 조 물질을 바이오티지 정제 (350 g 실리카겔, CH2Cl2 용리)에 적용하여, (S)-1-벤질 4-메틸 2-(9-페닐-9H-플루오렌-9-일아미노)숙시네이트를 고점성 무색 오일로서 수득하였다 (7.93 g).

LiHMDS (1.0 M/THF 9.2 mL, 9.2 mmol)를 (S)-1-벤질 4-메틸 2-(9-페닐-9H-플루오렌-9-일아미노)숙시네이트 (3.907 g, 8.18 mmol)의 냉각된 (-78℃) THF (50 mL) 용액에 10 분에 걸쳐 적가하고, 약 1 시간 동안 교반하였다. MeI (0.57 mL, 9.2 mmol)를 8 분에 걸쳐 혼합물에 적가하고, 냉각조를 실온으로 해동시키면서 16.5 시간 동안 교반을 계속하였다. NH4Cl 포화 용액 (5 mL)으로 켄칭시킨 이후, 대부분의 유기 성분을 진공하에 제거하고, 잔류물을 CH2Cl2 (100 mL) 및 물 (40 mL) 사이에 분배하였다. 유기 층을 건조시키고 (MgSO4), 여과하고, 진공하에 농축시키고, 생성된 조 물질을 바이오티지 (350 g 실리카겔; 25% EtOAc/헥산)로 정제하여, 약 1.0:0.65 비 (1H NMR)의 1-벤질 4-메틸 3-메틸-2-(9-페닐-9H-플루오렌-9-일아미노)숙시네이트의 2S/3S 및 2S/3R 부분입체이성질체의 혼합물 3.65 g을 수득하였다. 우세한 이성질체의 입체화학은 이 시점에서 측정하지 않았으며, 혼합물을 분리 없이 다음 단계로 전달하였다.

디이소부틸알루미늄 히드라이드 (헥산 중 1.0 M 20.57 ml, 20.57 mmol)를 상기 제조된 (2S)-1-벤질 4-메틸 3-메틸-2-(9-페닐-9H-플루오렌-9-일아미노)숙시네이트 (3.37 g, 6.86 mmol)의 냉각된 (-78℃) THF (120 mL) 용액에 10 분에 걸쳐 적가하고, -78℃에서 20 시간 동안 교반하였다. 반응 혼합물을 냉각조로부터 제거하고, 교반하면서 약 1 M H3PO4/H2O (250 mL)에 신속히 붓고, 혼합물을 에테르 (100 mL, 2×)로 추출하였다. 합한 유기 상을 염수로 세척하고, 건조시키고 (MgSO4), 여과하고, 진공하에 농축시켰다. 조 물질의 실리카겔 메쉬를 제조하고, 크로마토그래피 (25% EtOAc/헥산; 중력 용리)에 적용하여, 벤질 알콜로 오염된 (2S,3S)-벤질 4-히드록시-3-메틸-2-(9-페닐-9H-플루오렌-9-일아미노)부타노에이트 1.1 g을 무색 점성 오일로서, 및 (2S,3R) 입체이성질체를 함유하는 (2S,3R)-벤질 4-히드록시-3-메틸-2-(9-페닐-9H-플루오렌-9-일아미노)부타노에이트를 불순물로서 수득하였다. 후자 샘플을 다시 동일한 컬럼 크로마토그래피 정제하여, 정제된 물질 750 mg을 백색 포말체로서 수득하였다. [주의: 상기 조건하에 (2S,3S) 이성질체가 (2S,3R) 이성질체의 이전에 용리함].

DIBAL-환원 생성물의 상대적 입체화학 지정은 하기 프로토콜을 사용하여 각 이성질체로부터 제조된 락톤 유도체에 대해 수행되는 NOE 연구를 기초로 이루어졌다: LiHMDS (1.0 M/THF 50 μL, 0.05 mmol)를 (2S,3S)-벤질 4-히드록시-3-메틸-2-(9-페닐-9H-플루오렌-9-일아미노)부타노에이트 (62.7 mg, 0.135 mmol)의 냉각 (빙수)된 THF (2.0 mL) 용액에 첨가하고, 반응 혼합물을 유사한 온도에서 약 2 시간 동안 교반하였다. 휘발성 성분을 진공하에 제거하고, 잔류물을 CH2Cl2 (30 mL), 물 (20 mL) 및 NH4Cl 포화 수용액 (1 mL) 사이에 분배하였다. 유기 층을 건조시키고 (MgSO4), 여과하고, 진공하에 농축시키고, 생성된 조 물질을 바이오티지 정제 (40 g 실리카겔; 10-15% EtOAc/헥산)하여, (3S,4S)-4-메틸-3-(9-페닐-9H-플루오렌-9-일아미노)디히드로푸란-2(3H)-온을 무색 고체 필름으로서 수득하였다 (28.1 mg). (2S,3R)-벤질 4-히드록시-3-메틸-2-(9-페닐-9H-플루오렌-9-일아미노)부타노에이트를 (3S,4R)-4-메틸-3-(9-페닐-9H-플루오렌-9-일아미노)디히드로푸란-2(3H)-온과 유사하게 만들었다.

TBDMS-Cl (48 mg, 0.312 mmol)에 이어 이미다졸 (28.8 mg, 0.423 mmol)을 (2S,3S)-벤질 4-히드록시-3-메틸-2-(9-페닐-9H-플루오렌-9-일아미노)부타노에이트 (119.5 mg, 0.258 mmol)의 CH2Cl2 (3 ml) 용액에 첨가하고, 혼합물을 주위 조건에서 14.25 시간 동안 교반하였다. 이어서, 반응 혼합물을 CH2Cl2 (30 mL)로 희석시키고, 물 (15 mL)로 세척하고, 유기 층을 건조시키고 (MgSO4), 여과하고, 진공하에 농축시켰다. 생성된 조 물질을 바이오티지 (40 g 실리카겔; 5% EtOAc/헥산)로 정제하여, TBDMS계 불순물로 오염된 (2S,3S)-벤질 4-(tert-부틸디메틸실릴옥시)-3-메틸-2-(9-페닐-9H-플루오렌-9-일아미노)부타노에이트를 무색 점성 오일로서 수득하였다 (124.4 mg). (2S,3R)-벤질 4-히드록시-3-메틸-2-(9-페닐-9H-플루오렌-9-일아미노)부타노에이트를 (2S,3R)-벤질 4-(tert-부틸디메틸실릴옥시)-3-메틸-2-(9-페닐-9H-플루오렌-9-일아미노)부타노에이트와 유사하게 만들었다.

수소 풍선을 EtOAc (16 mL) 중 (2S,3S)-벤질 4-(tert-부틸디메틸실릴옥시)-3-메틸-2-(9-페닐-9H-플루오렌-9-일아미노)부타노에이트 (836 mg, 1.447 mmol) 및 10% Pd/C (213 mg)의 혼합물에 부착하고, 혼합물을 실온에서 약 21 시간 동안 교반하였고, 이때 필요에 따라 풍선을 H2로 재충전시켰다. 반응 혼합물을 CH2Cl2로 희석시키고, 규조토 패드 (셀라이트-545®)를 통해 여과하고, 패드를 EtOAc (200 mL), EtOAc/MeOH (1:1 혼합물, 200 mL) 및 MeOH (750 mL)로 세척하였다. 합한 유기 상을 농축시키고, 생성된 조 물질로부터 실리카겔 메쉬를 제조하고, 플래시 크로마토그래피 (EtOAc/i-PrOH/H2O의 8:2:1 혼합물)에 적용하여, (2S,3S)-2-아미노-4-(tert-부틸디메틸실릴옥시)-3-메틸부탄산을 솜털모양 백색 고체로서 수득하였다 (325 mg). (2S,3R)-벤질 4-(tert-부틸디메틸실릴옥시)-3-메틸-2-(9-페닐-9H-플루오렌-9-일아미노)부타노에이트를 (2S,3R)-2-아미노-4-(tert-부틸디메틸실릴옥시)-3-메틸부탄산과 유사하게 만들었다.

물 (1 mL) 및 NaOH (1.0 M/H2O 0.18 mL, 0.18 mmol)를 (2S,3S)-2-아미노-4-(tert-부틸디메틸실릴옥시)-3-메틸부탄산 (41.9 mg, 0.169 mmol) 및 Na2CO3 (11.9 mg, 0.112 mmol)의 혼합물에 첨가하고, 약 1 분 동안 초음파처리하여 반응물을 용해시켰다. 이어서, 혼합물을 빙수조로 냉각시키고, 메틸 클로로포르메이트 (0.02 mL, 0.259 mmol)를 30 초에 걸쳐 첨가하고, 유사한 온도에서 40 분 동안 및 이어서 주위 온도에서 2.7 시간 동안 격렬한 교반을 계속하였다. 반응 혼합물을 물 (5 mL)로 희석시키고, 빙수조로 냉각시키고, 1.0 N HCl 수용액 (약 0.23 mL)으로 적하 처리하였다. 혼합물을 물 (10 mL)로 추가로 희석시키고, CH2Cl2 (15 mL, 2×)로 추출하였다. 합한 유기 상을 건조시키고 (MgSO4), 여과하고, 진공하에 농축시켜, Cap-80a를 회백색 고체로서 수득하였다. (2S,3R)-2-아미노-4-(tert-부틸디메틸실릴옥시)-3-메틸부탄산을 Cap-80b와 유사하게 만들었다.

조 생성물을 추가 정제 없이 사용하였다.
문헌 [Falb et al. Synthetic Communications 1993, 23, 2839]에 기재된 프로토콜에 따라 제조하였다.
Cap
-82 내지
Cap
-85
Cap-51 또는 Cap-13에 대해 기재된 걸차에 따라 적절한 출발 물질로부터 Cap-82 내지 Cap-85를 합성하였다. 샘플은 그의 거울상이성질체 (즉, 각각 Cap-4, Cap-13, Cap-51 및 Cap-52)의 스펙트럼 프로파일과 유사한 스펙트럼 프로파일을 나타내었다.

H2O (15 mL) 중 O-메틸-L-트레오닌 (3.0 g, 22.55 mmol), NaOH (0.902 g, 22.55 mmol)의 혼합물에 ClCO2Me (1.74 mL, 22.55 mmol)를 0℃에서 적가하였다. 혼합물을 12 시간 동안 교반하고, 1 N HCl을 사용하여 pH 1로 산성화시켰다. 수성 상을 EtOAc (2×250 mL), 및 CH2Cl2 (250 mL) 중 10% MeOH로 추출하고, 합한 유기 상을 진공하에 농축시켜, 무색 오일 (4.18 g, 97%)을 수득하였고, 이것은 후속 단계에서 사용하기에 충분한 순도였다.


H2O (15 mL) 중 L-호모세린 (2.0 g, 9.79 mmol), Na2CO3 (2.08 g, 19.59 mmol)의 혼합물에 ClCO2Me (0.76 mL, 9.79 mmol)를 0℃에서 적가하였다. 혼합물을 48 시간 동안 교반하고, 1 N HCl을 사용하여 pH 1로 산성화시켰다. 수성 상을 EtOAc (2×250 mL)로 추출하고, 합한 유기 상을 진공하에 농축시켜 무색 고체 (0.719 g, 28%)를 수득하였고, 이것은 후속 단계에서 사용하기에 충분한 순도였다.


DMSO (10 mL) 중 L-발린 (1.0 g, 8.54 mmol), 3-브로모피리딘 (1.8 mL, 18.7 mmol), K2CO3 (2.45 g, 17.7 mmol) 및 CuI (169 mg, 0.887 mmol)의 혼합물을 100℃에서 12 시간 동안 가열하였다. 반응 혼합물을 실온으로 냉각시키고, H2O (약 150 mL)에 붓고, EtOAc (×2)로 세척하였다. 유기 층을 소량의 H2O로 추출하고, 합한 수성 상을 6 N HCl을 사용하여 pH 약 2로 산성화시켰다. 부피를 약 1/3으로 감소시키고, 양이온 교환 수지 (스트라타(Strata)) 20 g을 첨가하였다. 슬러리를 20 분 동안 정치시키고, 양이온 교환 수지 (스트라타) 패드 (약 25 g)에 로딩하였다. 패드를 H2O (200 mL), MeOH (200 mL) 및 이후 NH3 (MeOH 중 3 M, 2×200 mL)으로 세척하였다. 적절한 분획을 진공하에 농축시키고, 잔류물 (약 1.1 g)을 H2O 중에 용해시키고, 동결시키고, 동결건조시켰다. 표제 화합물을 포말체로서 수득하였다 (1.02 g, 62%).


DMSO (10 mL) 중 L-발린 (1.0 g, 8.54 mmol), 5-브로모피리미딘 (4.03 g, 17.0 mmol), K2CO3 (2.40 g, 17.4 mmol) 및 CuI (179 mg, 0.94 mmol)의 혼합물을 100℃에서 12 시간 동안 가열하였다. 반응 혼합물을 실온으로 냉각시키고, H2O (약 150 mL)에 붓고, EtOAc (×2)로 세척하였다. 유기 층을 소량의 H2O로 추출하고, 합한 수성 상을 6 N HCl을 사용하여 pH 약 2로 산성화시켰다. 부피를 약 1/3로 감소시키고, 양이온 교환 수지 (스트라타) 20 g을 첨가하였다. 슬러리를 20 분 동안 정치시키고, 양이온 교환 수지 (스트라타) 패드 (약 25 g)에 로딩하였다. 패드를 H2O (200 mL), MeOH (200 mL) 및 이후 NH3 (MeOH 중 3 M, 2×200 mL)으로 세척하였다. 적절한 분획을 진공하에 농축시키고, 잔류물 (약 1.1 g)을 H2O 중에 용해시키고, 동결시키고, 동결건조시켰다. 표제 화합물을 포말체로서 수득하였다 (1.02 g, 62%). 1H NMR (400 MHz, CD3OD)은 혼합물이 발린을 함유한다는 것을 나타내었으며, 순도는 추정할 수 없었다. 물질을 후속 반응에서 그대로 사용하였다.

Cap-1의 제조에 대해 기재된 방법에 따라 Cap-90을 제조하였다. 조 물질을 후속 단계에서 그대로 사용하였다.
달리 명시되지 않는 한, cap 51의 제조에 사용된 방법에 따라 하기 Cap을 제조하였다.






Cap
-117 내지
Cap
-123
Cap-117 내지 Cap-123의 제조를 위하여 Boc 아미노산을 시판 공급원으로부터 입수하였고, CH2Cl2 중 25% TFA로 처리하여 탈보호시켰다. LCMS에 의해 판단된 바에 따라 반응이 완료된 후, 용매를 진공하에 제거하고, Cap-51에 대해 기재된 절차에 따라, 상응하는 아미노산의 TFA 염을 메틸 클로로포르메이트로 카르바모일화하였다.



Cap-51에 대한 절차에 따라 L-트레오닌 tert-부틸 에스테르의 히드로클로라이드 염을 카르바모일화하였다. 조 반응 혼합물을 1 N HCl을 사용하여 pH 약 1로 산성화시키고, 혼합물을 EtOAc (2×50 mL)로 추출하였다. 합한 유기 상을 진공하에 농축시켜, 정치시 고체화되는 무색 오일을 수득하였다. 수성 층을 진공하에 농축시키고, 생성된 생성물 및 무기 염의 혼합물을 EtOAc-CH2Cl2-MeOH (1:1:0.1)로 연화처리한 후, 유기 상을 진공하에 농축시켜 무색 오일을 수득하였고, 이것은 LCMS에 의해 목적하는 생성물인 것으로 나타났다. 두 수집물 모두를 합하여 고체 0.52 g을 수득하였다.


메탄올 (15 mL) 중 Pd(OH)2 (20%, 100 mg), 수성 포름알데히드 (37 중량%, 4 ml), 아세트산 (0.5 mL)의 현탁액에 (S)-4-아미노-2-(tert-부톡시카르보닐아미노)부탄산 (1 g, 4.48 mmol)을 첨가하였다. 반응물을 수소로 여러번 퍼징하고, 수소 풍선하에 실온에서 밤새 교반하였다. 반응 혼합물을 규조토 패드 (셀라이트®)를 통해 여과하고, 휘발성 성분을 진공하에 제거하였다. 생성된 조 물질을 그대로 다음 단계에 사용하였다.

이 절차는 Cap-51의 제조에 사용된 절차의 변형이다. 0℃에서 THF (10 mL) 및 H2O (10 mL) 중 3-메틸-L-히스티딘 (0.80 g, 4.70 mmol)의 현탁액에 NaHCO3 (0.88 g, 10.5 mmol)을 첨가하였다. 생성된 혼합물을 ClCO2Me (0.40 mL, 5.20 mmol)로 처리하고, 혼합물을 0℃에서 교반하였다. 약 2 시간 동안 교반한 후, LCMS는 출발 물질이 남아있지 않음을 나타내었다. 반응물을 6 N HCl을 사용하여 pH 2로 산성화시켰다.
용매를 진공하에 제거하고, 잔류물을 CH2Cl2 중 20% MeOH 20 mL에 현탁시켰다. 혼합물을 여과하고 농축시켜, 담황색 포말체를 수득하였다 (1.21 g). LCMS 및 1H NMR은 물질이 메틸 에스테르 및 목적하는 생성물의 9:1 혼합물임을 나타내었다. 이 물질을 THF (10 mL) 및 H2O (10 mL) 중에 용해시키고, 0℃로 냉각시키고, LiOH (249.1 mg, 10.4 mmol)를 첨가하였다. 약 1 시간 교반한 후, LCMS는 에스테르가 남아있지 않음을 나타내었다. 이에 따라, 혼합물을 6 N HCl로 산성화시키고, 용매를 진공하에 제거하였다. LCMS 및 1H NMR에서 에스테르의 부재를 확인하였다. 표제 화합물을 무기 염으로 오염된 HCl 염으로서 수득하였다 (1.91 g, >100%). 화합물을 추가 정제 없이 후속 단계에서 그대로 사용하였다.


(S)-2-아미노-3-(1-메틸-1H-이미다졸-4-일)프로판산 (1.11 g, 6.56 mmol), NaHCO3 (1.21 g, 14.4 mmol) 및 ClCO2Me (0.56 mL, 7.28 mmol)로부터 출발하여 상기 Cap-126에 대한 방법에 따라 Cap-127을 제조하였다. 표제 화합물을 무기 염으로 오염된 HCl 염 (1.79 g, >100%)으로서 수득하였다. LCMS 및 1H NMR은 약 5%의 메틸 에스테르의 존재를 나타내었다. 조 혼합물을 추가 정제 없이 그대로 사용하였다.


단계 1. (S)-벤질 2-(tert-부톡시카르보닐아미노)펜트-4-이노에이트 (cj-27b)의 제조.

0℃에서 CH2Cl2 (100 mL) 중 cj-27a (1.01 g, 4.74 mmol), DMAP (58 mg, 0.475 mmol) 및 iPr2NEt (1.7 mL, 9.8 mmol)의 용액에 Cbz-Cl (0.68 mL, 4.83 mmol)을 첨가하였다. 용액을 0℃에서 4 시간 동안 교반하고, 세척하고 (1 N KHSO4, 염수), 건조시키고 (Na2SO4), 여과하고, 진공하에 농축시켰다. 잔류물을 플래시 컬럼 크로마토그래피 (TLC 6:1 헥스:EtOAc)로 정제하여, 표제 화합물 (1.30 g, 91%)을 무색 오일로서 수득하였다.

단계 2. (S)-벤질 3-(1-벤질-1H-1,2,3-트리아졸-4-일)-2-(tert-부톡시카르보닐아미노)프로파노에이트 (cj-28)의 제조.

실온에서 DMF-H2O (5 mL, 4:1) 중 (S)-벤질 2-(tert-부톡시카르보닐아미노)펜트-4-이노에이트 (0.50 g, 1.65 mmol), 나트륨 아스코르베이트 (0.036 g, 0.18 mmol), CuSO4-5H2O (0.022 g, 0.09 mmol) 및 NaN3 (0.13 g, 2.1 mmol)의 혼합물에 BnBr (0.24 mL, 2.02 mmol)을 첨가하고, 혼합물을 65℃로 가온하였다. 5 시간 후, LCMS는 낮은 전환을 나타내었다. 추가 분량의 NaN3 (100 mg)을 첨가하고, 12 시간 동안 가열을 계속하였다. 반응물을 EtOAc 및 H2O에 붓고, 진탕시켰다. 층을 분리하고, 수성 층을 EtOAc로 3회 추출하고, 합한 유기 상을 세척하고 (H2O×3, 염수), 건조시키고 (Na2SO4), 여과하고, 농축시켰다. 잔류물을 플래시 (바이오티지, 40+M CH2Cl2 중 0→5% MeOH; TLC, CH2Cl2 중 3% MeOH)로 정제하여, 정치시 고체화되는 담황색 오일을 수득하였다 (748.3 mg, 104%). NMR은 목적하는 생성물과 일치하였으나, DMF의 존재를 시사한다. 물질을 추가 정제 없이 그대로 사용하였다.

단계 2. (S)-벤질 3-(1-벤질-1H-1,2,3-트리아졸-4-일)-2-(메톡시카르보닐아미노)프로파노에이트 (cj-29)의 제조.

CH2Cl2 중 (S)-벤질 3-(1-벤질-1H-1,2,3-트리아졸-4-일)-2-(tert-부톡시카르보닐아미노)프로파노에이트 (0.52 g, 1.15 mmol)의 용액에 TFA (4 mL)를 첨가하였다. 혼합물을 실온에서 2 시간 동안 교반하였다. 혼합물을 진공하에 농축시켜, 정치시 고체화되는 무색 오일을 수득하였다. 이 물질을 THF-H2O 중에 용해시키고, 0℃로 냉각시켰다. 고체 NaHCO3 (0.25 g, 3.00 mmol)에 이어 ClCO2Me (0.25 mL, 3.25 mmol)를 첨가하였다. 1.5 시간 동안 교반한 후, 혼합물을 6 N HCl을 사용하여 pH 약 2로 산성화시킨 후, H2O-EtOAc에 부었다. 층을 분리하고, 수성 상을 EtOAc로 2회 추출하였다. 합한 유기 층을 세척하고 (H2O, 염수), 건조시키고 (Na2SO4), 여과하고, 진공하에 농축시켜, 펌프에서 정치시 고체화되는 무색 오일 (505.8 mg, 111%, NMR은 미확인 불순물의 존재를 시사함)을 수득하였다. 물질을 추가 정제 없이 그대로 사용하였다.

단계 4. (S)-2-(메톡시카르보닐아미노)-3-(1H-1,2,3-트리아졸-4-일)프로판산 (Cap-128)의 제조.

(S)-벤질 3-(1-벤질-1H-1,2,3-트리아졸-4-일)-2-(메톡시카르보닐아미노)프로파노에이트 (502 mg, 1.11 mmol)를 12 시간 동안 대기압에서 MeOH (5 mL) 중 Pd-C (82 mg)의 존재하에 수소화시켰다. 혼합물을 규조토 (셀라이트®)를 통해 여과하고, 진공하에 농축시켰다. (S)-2-(메톡시카르보닐아미노)-3-(1H-1,2,3-트리아졸-4-일)프로판산 (266 mg, 111%)을 무색 검으로서 수득하였고, 이것은 약 10%의 메틸 에스테르로 오염되어 있었다. 물질을 추가 정제 없이 그대로 사용하였다.


단계 1. (S)-2-(벤질옥시카르보닐아미노)-3-(1H-피라졸-1-일)프로판산 (cj-31)의 제조.

CH3CN (12 mL) 중 (S)-벤질 2-옥소옥세탄-3-일카르바메이트 (0.67 g, 3.03 mmol) 및 피라졸 (0.22 g, 3.29 mmol)의 현탁액을 50℃에서 24 시간 동안 가열하였다. 혼합물을 밤새 실온으로 냉각시키고, 고체를 여과하여, (S)-2-(벤질옥시카르보닐아미노)-3-(1H-피라졸-1-일)프로판산을 수득하였다 (330.1 mg). 여과물을 진공하에 농축시킨 후, 소량의 CH3CN (약 4 mL)으로 연화처리하여, 제2 수집물을 수득하였다 (43.5 mg). 총 수율 370.4 mg (44%). 융점 165.5-168℃. 문헌 상의 융점 168.5-169.5; 문헌 [Vederas et al. J. Am. Chem. Soc. 1985, 107, 7105].

단계 2. (S)-2-(메톡시카르보닐아미노)-3-(1H-피라졸-1-일)프로판산 (Cap-129)의 제조.

(S)-2-(벤질옥시카르보닐아미노)-3-(1H-피라졸-1-일)프로판산 (0.20 g, 0.70 mmol)을 2 시간 동안 대기압에서 MeOH (5 mL) 중 Pd-C (45 mg)의 존재하에 수소화시켰다. 생성물은 MeOH 중에 불용성인 것으로 나타났고, 이에 따라 반응 혼합물을 H2O 5 mL 및 몇 방울의 6 N HCl로 희석시켰다. 균질한 용액을 규조토 (셀라이트®)를 통해 여과하고, MeOH를 진공하에 제거하였다. 나머지 용액을 동결시키고, 동결건조시켜, 황색 포말체를 수득하였다 (188.9 mg). 이 물질을 THF-H2O (1:1, 10 mL) 중에 현탁시킨 후, 0℃로 냉각시켰다. 저온 혼합물에 NaHCO3 (146.0 mg, 1.74 mmol)을 조심스럽게 첨가하였다 (CO2의 방출). 기체 방출이 중지된 이후 (약 15분), ClCO2Me (0.06 mL, 0.78 mmol)를 적가하였다. 혼합물을 2 시간 동안 교반하고, 6 N HCl을 사용하여 pH 약 2로 산성화시키고, EtOAc에 부었다. 층을 분리하고, 수성 상을 EtOAC로 추출하였다 (×5). 합한 유기 층을 세척하고 (염수), 건조시키고 (Na2SO4), 여과하고, 농축시켜, 표제 화합물을 무색 고체로서 수득하였다 (117.8 mg, 79%).


문헌 [Calmes, M.; Daunis, J.; Jacquier, R.; Verducci, J. Tetrahedron, 1987, 43(10), 2285]에서 주어진 절차와 유사하게, 시판 공급되는 (R)-페닐글리신의 아실화에 의해 Cap-130을 제조하였다.

단계 a: 디메틸카르바모일 클로라이드 (0.92 mL, 10 mmol)를 THF (50 mL) 중 (S)-벤질 2-아미노-3-메틸부타노에이트 히드로클로라이드 (2.44 g; 10 mmol) 및 휴닉 염기 (3.67 mL, 21 mmol)의 용액에 서서히 첨가하였다. 생성된 백색 현탁액을 실온에서 밤새 교반하고 (16 시간), 감압하에 농축시켰다. 잔류물을 에틸 아세테이트 및 물 사이에 분배하였다. 유기층을 염수로 세척하고, 건조시키고 (MgSO4), 여과하고, 감압하에 농축시켰다. 생성된 황색 오일을 에틸 아세테이트:헥산 (1:1)으로 용리시키는 플래시 크로마토그래피로 정제하였다. 수집된 분획을 진공하에 농축시켜, 투명 오일 2.35 g (85%)을 수득하였다.
단계 b: 상기 제조된 중간체 (2.35 g; 8.45 mmol)의 MeOH (50 mL) 용액에 Pd/C (10%; 200 mg)를 첨가하고, 생성된 검정색 현탁액을 N2로 플러슁하고 (3×), H2 1 atm 하에 두었다. 혼합물을 실온에서 밤새 교반하고, 미세섬유 필터를 통해 여과하여, 촉매를 제거하였다. 이어서, 생성된 투명 용액을 감압하에 농축시켜, 1.43 g (89%)의 Cap-131을 백색 포말체로서 수득하였고, 이것을 추가 정제 없이 사용하였다.


Cap-132를 Cap-131에 대해 기재한 방법에 따라 (S)-벤질 2-아미노프로파노에이트 히드로클로라이드로부터 제조하였다.


Cap-133을 Cap-47에 대해 기재한 방법에 따라 (S)-tert-부틸 2-아미노-3-메틸부타노에이트 히드로클로라이드 및 2-플루오로에틸 클로로포르메이트로부터 제조하였다.


Cap-134를 Cap-51에 대해 기재한 방법에 따라 (S)-디에틸 알라닌 및 메틸 클로로포르메이트로부터 제조하였다.


메탄올 (5 mL) 중 D-2-아미노-(4-플루오로페닐)아세트산 (338 mg, 2.00 mmol), 디에틸에테르 중 1 N HCl (2.0 mL, 2.0 mmol) 및 포르말린 (37%, 1 mL)의 용액을 10% 탄소상 팔라듐 (60 mg) 상에서 16 시간 동안 25℃에서 풍선 수소화시켰다. 이어서, 혼합물을 셀라이트를 통해 여과하여, Cap-135의 HCl 염을 백색 포말체로서 수득하였다 (316 mg, 80%).


무수 디에틸 에테르 (50 mL) 중 1-벤질-1H-이미다졸 (1.58 g, 10.0 mmol)의 냉각된 (-50℃) 현탁액에 질소하에 n-부틸 리튬 (헥산 중 2.5 M, 4.0 mL, 10.0 mmol)을 적가하였다. 20 분 동안 -50℃에서 교반한 후, 건조 이산화탄소 (드라이어라이트(Drierite)를 통과한 것)를 반응 혼합물에 10 분 동안 버블링한 후, 이것을 25℃까지 가온하였다. 반응 혼합물에 이산화탄소를 첨가했을 때 형성된 중질 침전물을 여과하여, 흡습성 백색 고체를 수득하였고, 이것을 물 (7 mL) 중에 용해시키고, pH = 3으로 산성화시키고, 냉각시키고, 스크래칭(scratching)으로 결정화를 유도하였다. 이 침전물을 여과하여 백색 고체를 수득하고, 이것을 메탄올 중에 현탁시키고, 1 N HCl/디에틸 에테르 (4 mL)로 처리하고, 진공하에 농축시켰다. 잔류물을 물 (5 mL)로부터 동결건조시켜, Cap-136의 HCl 염을 백색 고체로서 수득하였다 (817 mg, 40%).



무수 디옥산 (10 mL) 중 1-클로로-3-시아노이소퀴놀린 (188 mg, 1.00 mmol; WO 2003/099274에서의 절차에 따라 제조) (188 mg, 1.00 mmol), 세슘 플루오라이드 (303.8 mg, 2.00 mmol), 비스(트리-tert-부틸포스핀)팔라듐 디클로라이드 (10 mg, 0.02 mmol) 및 2-(트리부틸스탄닐)푸란 (378 μL, 1.20 mmol)의 현탁액을 질소하에 80℃에서 16 시간 동안 가열한 후, 25℃로 냉각시키고, 1 시간 동안 격렬히 교반하면서 칼륨 플루오라이드 포화 수용액으로 처리하였다. 혼합물을 에틸 아세테이트 및 물 사이에 분배하고, 유기상을 분리하고, 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 농축시켰다. 잔류물을 실리카겔 상에서 정제 (0%→30% 에틸 아세테이트/헥산으로 용리)하여, Cap-137, 단계 a를 백색 고체로서 수득하였고, 이것을 그대로 사용하였다 (230 mg, 105%).

사염화탄소 (1 mL), 아세토니트릴 (1 mL) 및 물 (1.5 mL) 중 Cap 137, 단계 a (110 mg, 0.50 mmol) 및 과요오드화나트륨 (438 mg, 2.05 mmol)의 현탁액에 루테늄 트리클로라이드 히드레이트 (2 mg, 0.011 mmol)를 첨가하였다. 혼합물을 25℃에서 2 시간 동안 교반한 후, 디클로로메탄 및 물 사이에 분배하였다. 수성층을 분리하고, 디클로로메탄으로 2회 추출하고, 합한 디클로로메탄 추출물을 Na2SO4 상에서 건조시키고, 여과하고, 농축시켰다. 잔류물을 헥산으로 연화처리하여, Cap-137 (55 mg, 55%)을 회색을 띠는 색의 고체로서 수득하였다.

Cap
138 내지 158
합성 계획. 방법 A.



무수 테트라히드로푸란 (20 mL) 중 5-히드록스이소퀴놀린 (WO 2003/099274에서의 절차에 따라 제조) (2.0 g, 13.8 mmol) 및 트리페닐포스핀 (4.3 g, 16.5 mmol)의 교반 현탁액에 무수 메탄올 (0.8 mL) 및 디에틸 아조디카르복실레이트 (3.0 mL, 16.5 mmol)를 조금씩 첨가하였다. 혼합물을 실온에서 20 시간 동안 교반한 후, 에틸 아세테이트로 희석시키고, 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 농축시켰다. 잔류물을 실리카겔 상에 예비 흡수시키고, 정제 (40% 에틸 아세테이트/헥산으로 용리)하여, Cap-138, 단계 a를 담황색 고체로서 수득하였다 (1.00 g, 45%).


무수 디클로로메탄 (50 mL) 중 Cap 138, 단계 a (2.34 g, 14.7 mmol)의 교반 용액에 실온에서 메타-클로로퍼벤조산 (77%, 3.42 g, 19.8 mmol)을 한번에 첨가하였다. 20 시간 동안 교반한 후, 분말화 탄산칼륨 (2.0 g)을 첨가하고, 혼합물을 실온에서 1 시간 동안 교반한 후, 이것을 여과하고 농축시켜, Cap-138, 단계 b를 옅은 황색 고체로서 수득하였고, 이것은 후속과정에 전달되기에 충분히 순수하였다 (2.15 g, 83.3%).


건조 아세토니트릴 (20 mL) 중 Cap 138, 단계 b (0.70 g, 4.00 mmol) 및 트리에틸아민 (1.1 mL, 8.00 mmol)의 교반 용액에 실온에서 질소하에 트리메틸실릴시아니드 (1.60 mL, 12.00 mmol)를 첨가하였다. 혼합물을 75℃에서 20 시간 동안 가열한 후, 실온으로 냉각시키고, 에틸 아세테이트로 희석시키고, 중탄산나트륨 포화 용액 및 염수로 세척한 후, Na2SO4 상에서 건조시키고, 용매를 농축시켰다. 잔류물을 실리카겔 상에서 플래시 크로마토그래피 (5% 에틸 아세테이트/헥산→25% 에틸 아세테이트/헥산으로 용리)하여, 백색 결정성 고체로서의 Cap-138, 단계 c (498.7 mg)를, 여과물로부터 회수된 추가의 Cap-138, 단계 c 223 mg과 함께 수득하였다.


Cap-138, 단계 c (0.45 g, 2.44 mmol)를 5 N 수산화나트륨 용액 (10 mL)으로 처리하고, 생성된 현탁액을 85℃에서 4 시간 동안 가열하고, 25℃로 냉각시키고, 디클로로메탄으로 희석시키고, 1 N 염산으로 산성화시켰다. 유기상을 분리하고, 염수로 세척하고, Na2SO4 상에서 건조시키고, ¼ 부피로 농축시키고, 여과하여, Cap-138을 황색 고체로서 수득하였다 (0.44 g, 88.9%).

합성 계획. 방법 B (문헌 [Tetrahedron Letters, 2001, 42, 6707]에서 유래됨).



무수 톨루엔 (6 mL) 중 1-클로로-6-메톡시이소퀴놀린 (1.2 g, 6.2 mmol; WO 2003/099274에서의 절차에 따라 제조), 시안화칼륨 (0.40 g, 6.2 mmol), 1,5-비스(디페닐포스피노)펜탄 (0.27 g, 0.62 mmol) 및 팔라듐(II) 아세테이트 (70 mg, 0.31 mmol)의 아르곤-탈기된 현탁액을 함유하는 두꺼운 벽의 스크류-탑 바이알에 에 N,N,N',N'-테트라메틸에틸렌디아민 (0.29 mL, 2.48 mmol)을 첨가하였다. 바이알을 밀봉하고, 150℃에서 22 시간 동안 가열한 후, 25℃로 냉각시켰다. 반응 혼합물을 에틸 아세테이트로 희석시키고, 물과 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 농축시켰다. 잔류물을 실리카겔 상에서 5% 에틸 아세테이트/헥산→25% 에틸 아세테이트/헥산으로 용리시키며 정제하여, Cap-139, 단계 a를 백색 고체로서 수득하였다 (669.7 mg).


Cap 138에 대해 기재된 절차에 따라 5 N NaOH를 사용하여, Cap-139, 단계 a의 염기성 가수분해로부터 Cap-139를 제조하였다.



무수 디메틸아세트아미드 (2 mL) 중 1,3-디클로로-5-에톡시이소퀴놀린 (482 mg, 2.00 mmol; WO 2005/051410에서의 절차에 따라 제조), 팔라듐(II) 아세테이트 (9 mg, 0.04 mmol), 탄산나트륨 (223 mg, 2.10 mmol) 및 1,5-비스(디페닐포스피노)펜탄 (35 mg, 0.08 mmol)의 격렬히 교반된 혼합물에 25℃에서 질소하에 N,N,N',N'-테트라메틸에틸렌디아민 (60 mL, 0.40 mmol)을 첨가하였다. 10 분 후, 혼합물을 150℃로 가열한 후, 아세톤 시아노히드린의 원액 용액 (DMA 4.34 mL 중 아세톤 시아노히드린 457 μL로부터 제조)을 1 mL 분량으로 18 시간에 걸쳐 시린지 펌프를 사용하여 첨가하였다. 이어서, 혼합물을 에틸 아세테이트 및 물 사이에 분배하고, 유기층을 분리하고, 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 농축시켰다. 잔류물을 실리카겔 상에서 10% 에틸 아세테이트/헥산→40% 에틸 아세테이트/헥산으로 용리시켜 정제하여, Cap-140, 단계 a를 황색 고체로서 수득하였다 (160 mg, 34%).

하기 기재된 Cap 141의 제조 절차에 기재된 바와 같이 12 N HCl을 사용하는 Cap-140, 단계 a의 산 가수분해에 의해 Cap-140을 제조하였다.


Cap-140, 단계 a (상기 참조)의 제조 절차에 기재된 바와 같이 하여 1-브로모-3-플루오로이소퀴놀린 (문헌 [J. Med. Chem. 1970, 13, 613]에 개괄된 절차을 이용하여 3-아미노-1-브로모이소퀴놀린으로부터 제조)으로부터 Cap-141, 단계 a를 제조하였다.


Cap-141, 단계 a (83 mg, 0.48 mmol)를 12 N HCl (3 mL)로 처리하고, 생성된 슬러리를 80℃에서 16 시간 동안 가열한 후, 실온으로 냉각시키고, 물 (3 mL)로 희석시켰다. 혼합물을 10 분 동안 교반한 다음, 여과하여 Cap-141을 회백색 고체로서 수득하였다 (44.1 mg, 47.8%). 여과물을 디클로로메탄으로 희석시키고, 염수로 세척하고, Na2SO4 상에서 건조시키고, 농축시켜, 추가의 Cap-141을 수득하였고, 이것은 후속과정에 바로 전달되기에 충분히 순수하였다 (29.30 mg, 31.8%).



Cap-138, 단계 b 및 c의 제조를 위한 2-단계 절차에 기재된 바와 같이 하여 4-브로모이소퀴놀린 N-옥시드로부터 Cap-142, 단계 a를 제조하였다.

무수 톨루엔 (1 mL) 중 Cap-142, 단계 a (116 mg, 0.50 mmol), 삼염기성 인산칼륨 (170 mg, 0.80 mmol), 팔라듐(II) 아세테이트 (3.4 mg, 0.015 mmol) 및 2-(디시클로헥실포스피노)바이페닐 (11 mg, 0.03 mmol)의 아르곤-탈기된 현탁액에 모르폴린 (61 μL, 0.70 mmol)을 첨가하였다. 혼합물을 100℃에서 16 시간 동안 가열하고, 25℃로 냉각시키고, 규조토 (셀라이트®)를 통해 여과하였다. 잔류물을 실리카겔 상에서 10%→70% 에틸 아세테이트/헥산으로 용리시키며 정제하여, Cap-142, 단계 b (38 mg, 32%)를 황색 고체로서 수득하였고, 이것을 후속과정으로 바로 전달하였다.


Cap 138에 대한 절차에 기재된 바와 같이 5 N 수산화나트륨을 사용하여 Cap-142, 단계 b로부터 Cap-142를 제조하였다.



무수 디메틸포름아미드 (10 mL) 중 3-아미노-1-브로모이소퀴놀린 (444 mg, 2.00 mmol)의 교반 용액에 수소화나트륨 (60%, 세척되지 않음, 96 mg, 2.4 mmol)을 한번에 첨가하였다. 혼합물을 25℃에서 5 분 동안 교반한 후, 2-브로모에틸 에테르 (90%, 250 μL, 2.00 mmol)를 첨가하였다. 혼합물을 25℃에서 5 시간 동안 및 75℃에서 72 시간 동안 더 교반한 후, 이것을 25℃로 냉각시키고, 염화암모늄 포화 용액으로 켄칭시키고, 에틸 아세테이트로 희석시켰다. 유기층을 분리하고, 물 및 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 농축시켰다. 잔류물을 실리카겔 상에서 0%→70% 에틸 아세테이트/헥산으로 용리시켜 정제하여, Cap-143, 단계 a를 황색 고체로서 수득하였다 (180 mg, 31%).


무수 테트라히드로푸란 (5 mL) 중 Cap-143, 단계 a (154 mg, 0.527 mmol)의 저온 (-60℃) 용액에 헥산 중 n-부틸리튬의 용액 (2.5 M, 0.25 mL, 0.633 mmol)을 첨가하였다. 10 분 후, 건조 이산화탄소를 반응 혼합물에 10 분 동안 버블링한 후, 이것을 1 N HCl로 켄칭시키고, 25℃로 가온하였다. 이어서, 혼합물을 디클로로메탄 (3×30 mL)으로 추출하고, 합한 유기 추출물을 진공하에 농축시켰다. 잔류물을 역상 HPLC (MeOH/물/TFA)로 정제하여, Cap-143을 수득하였다 (16 mg, 12%).


1,3-디클로로이소퀴놀린 (2.75 g, 13.89 mmol)을 발연 질산 (10 mL) 및 진한 황산 (10 mL)의 저온 (0℃) 용액에 소량씩 첨가하였다. 혼합물을 0℃에서 0.5 시간 동안 교반한 후, 점차적으로 25℃로 가온하고, 여기서 이것을 16 시간 동안 교반하였다. 이어서, 부순 얼음 및 물을 함유하는 비이커에 상기 혼합물을 붓고, 생성된 현탁액을 1 시간 동안 0℃에서 교반한 후, 이것을 여과하여, Cap-144, 단계 a (2.73 g, 81%)를 황색 고체로서 수득하였고, 이것을 바로 사용하였다.

Cap-144, 단계 a (0.30 g, 1.23 mmol)를 메탄올 (60 mL) 중에 용해시키고, 산화백금 (30 mg)으로 처리하고, 현탁액을 7 psi H2에서 1.5 시간 동안 파아(Parr) 수소화시켰다. 이어서, 포르말린 (5 mL) 및 추가의 산화백금 (30 mg)을 첨가하고, 현탁액을 45 psi H2에서 13 시간 동안 다시 파아 수소화시켰다. 이어서, 이것을 규조토 (셀라이트®)를 통해 흡인 여과하고, ¼ 부피로 농축시켰다. 생성된 침전물을 흡인 여과하여, 표제 화합물을 황색 고체로서 수득하였고, 이것을 실리카겔 상에서 헥산 중 5% 에틸 아세테이트→헥산 중 25% 에틸 아세테이트로 용리시키며 플래시 크로마토그래피하여, Cap-144, 단계 b (231 mg, 78%)를 옅은 황색 고체로서 수득하였다.


Cap-139, 단계 a의 제조에 대해 기재된 절차에 따라 Cap-144, 단계 b로부터 Cap-144, 단계 c를 제조하였다.


Cap-141에 대해 기재된 절차에 따라 Cap-144를 제조하였다.

Cap
-145 내지
Cap
-162
하기 개괄된 바와 같이 달리 명시되지 않는 한, Cap-138 (방법 A) 또는 Cap-139 (방법 B)의 제조에 대해 기재된 절차에 따라 적절한 1-클로로이소퀴놀린으로부터 Cap-145 내지 Cap-162를 제조하였다.








디에틸에테르 (25 ml) 중 2-케토부티르산 (1.0 g, 9.8 mmol)의 용액에 페닐마그네슘 브로마이드 (22 ml, THF 중 1 M)를 적가하였다. 반응물을 약 25℃에서 질소하에 17.5 시간 동안 교반하였다. 반응물을 1 N HCl로 산성화시키고, 생성물을 에틸 아세테이트 (3×100 ml)로 추출하였다. 합한 유기층을 물에 이어 염수로 세척하고, MgSO4 상에서 건조시켰다. 진공하에 농축 후, 백색 고체를 수득하였다. 고체를 헥산/에틸 아세테이트로부터 재결정화시켜, Cap-163을 백색 침상체로서 수득하였다 (883.5 mg).


MeOH (40 mL) 중 2-아미노-2-페닐부티르산 (1.5 g, 8.4 mmol), 포름알데히드 (14 mL, 물 중 37%), 1 N HCl (10 mL) 및 10% Pd/C (0.5 mg)의 혼합물을 파아 용기 내에서 42 시간 동안 H2 50 psi에 노출시켰다. 반응물을 셀라이트 상에서 여과하고, 진공하에 농축시키고, 잔류물을 MeOH (36 mL) 중에 용해시키고, 생성물을 역상 HPLC (MeOH/H2O/TFA)로 정제하여, Cap-164의 TFA 염을 백색 고체로서 수득하였다 (1.7 g).


1,2-디클로로에탄 (7 ml) 중 2-아미노-2-인단카르복실산 (258.6 mg, 1.46 mmol) 및 포름산 (0.6 ml, 15.9 mmol)의 혼합물에 포름알데히드 (0.6 ml, 물 중 37%)를 첨가하였다. 혼합물을 약 25℃에서 15 분 동안에 교반한 후, 70℃에서 8 시간 동안 가열하였다. 휘발성 성분을 진공하에 제거하고, 잔류물을 DMF (14 mL) 중에 용해시키고, 역상 HPLC (MeOH/H2O/TFA)로 정제하여, Cap-165의 TFA 염을 점성 오일로서 수득하였다 (120.2 mg).


반-분취용 키랄셀 OJ 컬럼, 20×250 mm, 10 μm를 사용하여 85:15 헵탄/에탄올 혼합물로 10 ml/분의 용리 속도에서 25 분 동안 용리시켜 벤질 에스테르 중간체를 분리한 것을 제외하고는, Cap-7a 및 Cap-7b의 합성에 대해 기재한 방법에 따라, Cap-166a 및 -166b를 (1S,4S)-(+)-2-메틸-2,5-디아자바이시클로[2.2.1]헵탄 (2HBr)으로부터 제조하였다.


20% TFA/CH2Cl2 중 라세미체 Boc-1,3-디히드로-2H-이소인돌 카르복실산 (1.0g, 3.8 mmol)의 용액을 약 25℃에서 4 시간 동안 교반하였다. 모든 휘발성 성분을 진공하에 제거하였다. MeOH 중 생성된 조 물질, 포름알데히드 (15 mL, 물 중 37%), 1 N HCl (10 mL) 및 10% Pd/C (10 mg)의 혼합물을 파아 용기 내에서 23 시간 동안 H2 (40 PSI)에 노출시켰다. 반응 혼합물을 셀라이트 상에서 여과하고, 진공하에 농축시켜, Cap-167을 황색 포말체로서 수득하였다 (873.5 mg).


Cap-167의 제조에 대해 기재된 절차에 따라, 라세미체 Cap-168을 라세미체 Boc-아미노인단-1-카르복실산으로부터 제조하였다. 조 물질을 그대로 사용하였다.

MeOH (60 mL) 중 2-아미노-2-페닐프로판산 히드로클로라이드 (5.0 g, 2.5 mmol), 포름알데히드 (15 ml, 물 중 37%), 1 N HCl (15 ml) 및 10% Pd/C (1.32 g)의 혼합물을 파아 용기 내에 두고, 수소 (55 PSI)하에서 4 일 동안 진탕시켰다. 반응 혼합물을 셀라이트 상에서 여과하고, 진공하에 농축시켰다. 잔류물을 MeOH 중에 용해시키고, 역상 분취용-HPLC (MeOH/물/TFA)로 정제하여, Cap-169의 TFA 염을 점성 반-고체로서 수득하였다 (2.1 g).


물 (15 ml) 중 (S)-2-아미노-2-(테트라히드로-2H-피란-4-일)아세트산 (505 mg; 3.18 mmol; 아스타테크(Astatech)로부터 입수)에 탄산나트륨 (673 mg; 6.35 mmol)을 첨가하고, 생성된 혼합물을 0℃로 냉각시킨 후, 메틸 클로로포르메이트 (0.26 ml; 3.33 mmol)를 5 분에 걸쳐 적가하였다. 조를 주위 온도로 해동시키면서 반응물을 18 시간 동안 교반하였다. 이어서, 반응 혼합물을 1 N HCl 및 에틸 아세테이트 사이에 분배하였다. 유기층을 제거하고, 수성층을 2개의 추가 분량의 에틸 아세테이트로 더 추출하였다. 합한 유기층을 염수로 세척하고, 황산마그네슘 상에서 건조시키고, 여과하고, 진공하에 농축시켜, Cap-170을 무색 잔류물로서 수득하였다.


에틸 아세테이트 (7 ml) 및 CH2Cl2 (4.00 ml) 중 메틸 2-(벤질옥시카르보닐아미노)-2-(옥세탄-3-일리덴)아세테이트 (200 mg, 0.721 mmol; 문헌 [Il Farmaco (2001), 56, 609-613])의 용액을 10 분 동안 질소 버블링에 의해 탈기시켰다. 이어서, 디메틸 디카르보네이트 (0.116 ml, 1.082 mmol) 및 Pd/C (20 mg, 0.019 mmol)를 첨가하고, 반응 혼합물에 수소 풍선을 장착하고, 주위 온도에서 밤새 교반하였고, 이때 TLC (95:5 CH2Cl2/MeOH: Ce(NH4)2SO4 1 g, 암모늄 몰리브데이트 6 g, 황산 6 ml 및 물 100 ml로 만들어진 착색제로 가시화됨)는 완전한 전환을 나타내었다. 반응물을 셀라이트를 통해 여과하고 농축시켰다. 잔류물을 바이오티지®를 통해 정제하였다 (25 개의 샘플렛에 상에 디클로로메탄으로 로딩; 25S 컬럼 상에서 디클로로메탄 (3CV)에 이어 250 ml에 걸친 0→5% MeOH/디클로로메탄으로 용리시킨 후, 5% MeOH/디클로로메탄 (250 ml)에서 유지시킴; 분획 9 ml). 목적하는 물질을 함유하는 분획을 수거하고 농축시켜, 메틸 2-(메톡시카르보닐아미노)-2-(옥세탄-3-일)아세테이트 120 mg (81%)을 무색 오일로서 수득하였다.

THF (2 mL) 및 물 (0.5 mL) 중 메틸 2-(메톡시카르보닐아미노)-2-(옥세탄-3-일)아세테이트 (50 mg, 0.246 mmol)에 수산화리튬 일수화물 (10.33 mg, 0.246 mmol)을 첨가하였다. 생성된 용액을 밤새 주위 온도에서 교반하였다. TLC (1:1 EA/Hex; 하네시안(Hanessian) 착색제 [Ce(NH4)2SO4 1 g, 암모늄 몰리브데이트 6 g, 황산 6 ml 및 물 100 ml])는 약 10%의 출발 물질이 남아있음을 나타내었다. LiOH 추가 3 mg을 첨가하고, 밤새 교반하였고, 이때 TLC는 출발 물질이 남아있지 않음을 나타내었다. 진공하에 농축시키고, 고진공하에 밤새 두어, 리튬 2-(메톡시카르보닐아미노)-2-(옥세탄-3-일)아세테이트 55 mg을 무색 고체로서 수득하였다.

실시예
본 개시내용은 이제 그의 범주를 제한하는 것으로 의도되지는 않는 특정 실시양태와 관련하여 기재될 것이다. 반면, 본 개시내용은 특허청구범위의 범주 내에 포함될 수 있는 모든 변형, 변경 및 등가물을 포함한다. 따라서, 특정 실시양태를 포함하는 하기 실시예는 본 개시내용의 한 실시를 예시할 것이며, 실시예는 특정 실시양태의 예시 목적을 위한 것이고, 그의 절차 및 구상 측면의 가장 유용하고 쉽게 이해되는 기재로 여겨지는 것을 제공하기 위해 제시되는 것으로 이해된다.
달리 언급되지 않는다면, 용액 백분율은 부피에 대한 중량 관계를 표현하며, 용액 비는 부피에 대한 부피 관계를 표현한다. 핵 자기 공명 (NMR) 스펙트럼은 브루커(Bruker) 300, 400 또는 500 MHz 분광계로 기록하였으며, 화학적 이동 (δ)은 백만 당 부로 보고되었다. 스틸(Still)의 플래쉬 크로마토그래피 기법에 따라 실리카겔 (SiO2) 상에서 플래쉬 크로마토그래피를 수행하였다 (문헌 [J. Org. Chem. 1978, 43, 2923]).
워터스 마이크로매스 ZQ MS 시스템과 연결된 시마주 LC 시스템에서 순도 평가 및 저분해능 질량 분석을 수행하였다. 체류 시간은 기계 사이에 약간 달라질 수 있음을 주목하여야 한다. 체류 시간 (RT)을 측정하는데 이용되는 LC 조건은 다음과 같다:
체류 시간은 하기의 LC/MS 조건에 따라 측정하였다:
조건 1
컬럼 = 페노메넥스-루나 3.0×50 mm S10
시작 %B = 0
최종 %B = 100
구배 시간 = 2 분
중지 시간 = 3 분
유속 = 4 ml/분
파장 = 220 nm
용매 A = 10% 메탄올/90%H2O 중 0.1% TFA
용매 B = 90% 메탄올/10% H2O 중 0.1% TFA
조건 2
컬럼 = 페노메넥스-루나 4.6×50 mm S10
시작 %B = 0
최종 %B = 100
구배 시간 = 3 분
중지 시간 = 3 분
유속 = 4 ml/분
파장 = 220 nm
용매 A = 10% 메탄올/90%H2O 중 0.1% TFA
용매 B = 90% 메탄올/10% H2O 중 0.1% TFA
조건 3
컬럼 = LCMS. 게미니(GEMINI) C-18 4.6 □ 50 mm
시작 %B = 0
최종 %B = 100
구배 시간 = 3 분
중지 시간 = 4 분
유속 = 4 ml/분
파장 = 220 nm
용매 A = 10% 아세토니트릴/90%H2O 중 0.1% NH4OAc
용매 B = 90% 아세토니트릴/10%H2O 중 0.1% NH4OAc
조건 4
컬럼 = 페노메넥스-루나 3.0×50 mm S10
시작 %B = 0
최종 %B = 100
구배 시간 = 4 분
중지 시간 = 5 분
유속 = 4 ml/분
파장 = 220 nm
용매 A = 10% 메탄올/90%H2O 중 0.1% TFA
용매 B = 90% 메탄올/10% H2O 중 0.1% TFA
조건 5
컬럼 30 mm × = 루나 C18 4.6
시작 %B = 0
최종 %B = 100
구배 시간 = 2 분
중지 시간 = 3 분
유속 = 5 ml/분
파장 = 220 nm
용매 A = 10% 아세토니트릴/90% H2O 중 0.1% NH4OAc
용매 B = 90% 아세토니트릴/10% H2O 중 0.1% NH4OAc
합성 경로 1.

참조: (위티그(Wittig)/환원/고리화) 문헌 [J.Med.Chem. (2005) 48, 7351-7362].

THF (80 mL) 중 칼륨 tert-부톡시드의 1 M 용액을 질소하에 24℃에서 무수 DMSO (20 mL) 중 (3-카르복시프로필)트리페닐포스포늄 브로마이드 (17 g, 40 mmol)에 적가하고, 상기 용액을 30 분 교반한 후에, 3-브로모벤즈알데히드 (4.7 mL, 40 mmol)를 첨가하였다. 몇 분 후에, 침전물이 관찰되고, DMSO 추가 20 mL를 첨가하여 용매화를 보조하고, 반응물을 18 시간 교반하였다. 용액을 물 (120 mL)에 붓고, 클로로포름으로 세척하였다. 수성층을 진한 HCl로 산성화시키고, 클로로포름으로 추출하였다 (3×250 mL). 유기상을 농축시키고, 65 (M) 바이오티지® 실리카겔 컬럼; 2 L에 걸친 15→65% B (A = 헥산; B = EtOAc)의 구배 용리에 적용하여, 실시예 1, (E)-5-(3-브로모페닐)펜트-4-에노산 8.2 g (82%)을 수득하였다.



실시예 1, (E)-5-(3-브로모페닐)펜트-4-에노산 (4 g, 15.8 mmol)을 무수 에탄올 (200 mL) 중에 용해시키고, 질소로 플러슁한 후에, 탄소 상 5% 황화백금 (2.5 g)을 첨가하였다. 용액을 대기압에서 수소로 플러슁하고, 5 시간 교반하였다. 촉매를 규조토 (셀라이트®)를 통한 여과에 의해 제거하고, 용매를 회전 증발에 의해 즉시 제거하여 (에스테르화를 최소화시키기 위한 것임), 실시예 2, 5-(3-브로모페닐)펜탄산 4 g (99%)을 수득하였고, 이것을 추가 정제없이 후속 단계에 사용하였다.


참조: (알킬화/가수분해) 문헌 [J.Am.Chem.Soc. (1946) 68, 1468-1470].

휴닉 염기 (26.5 mL, 0.15 mol)를 무수 DMF (400 mL) 중 3-브로모벤질 브로마이드 (38 g, 0.15 mol) 및 N-메틸-β-알라닌 니트릴 (14.2 mL, 0.15 mol)에 첨가하였다. 용액을 16 시간 교반하고, 진공하에 농축시켜 거의 건조시키고, 에테르/EtOAc로 연화처리하고, 여과하였다. [두번째 반응은 12 g에 대해 반복하였다]. 합한 여과물을 회전 증발에 의해 농축하고, 잔류물을 65 (M) 바이오티지® 실리카겔 컬럼에 충전시키고 (CH2Cl2); 구획 1:1.3 L에 걸친 5→15% B; 구획 2: 6.75 L에 걸친 15%→100% B (A = 헥산; B = EtOAc)의 구배 용리에 적용하여, 실시예 4, 3-((3-브로모벤질)(메틸)아미노)-프로판니트릴 44.4 g (88%)을 수득하였다.



1 L 스크류 캡 압력 용기에서 6 N HCl을 실시예 4, 3-((3-브로모벤질)(메틸)아미노)-프로판니트릴 (44.4 g, 0.175 mol)에 첨가하고, 밀봉된 용액을 90℃에서 40 시간 동안 가열하였다. 냉각시킨 후에, 용액을 진공하에 ¼ 부피로 농축시켰다. 여과, 모액의 농축 및 여과 (2x)로 정량 수율의 실시예 5, 3-((N-(3-브로모벤질)-N-메틸)아미노)프로판산을 백색 고체로서 수득하였다 (HCl 염).



실시예 2, 5-(3-브로모페닐)펜탄산 (4 g, 15.6 mmol)을 폴리인산 (15 g) 중에 녹이고, 승화로 인한 생성물 손실을 방지하기 위해, 캡핑된 150 mL 압력 용기에서 8 시간 동안 140℃로 가열하였다. 반응 혼합물을 물 150 mL 및 CH2Cl2 (600 mL) 사이에 분배하였다. [CH2Cl2의 비등을 방지하기 위한 주의가 필요하다.] 유기상을 물 및 염수로 세척하고 농축시켰다. 조 생성물을 40 (S) 바이오티지® 실리카겔 컬럼 및 5→60% (EtOAc/Hex)로 구배 용리에 적용하여, 실시예 3, 2-브로모-6,7,8,9-테트라히드로-5H-벤조[7]안눌렌-5-온 1.7 g (40%)을 수득하였다.


참조: (고리화) 문헌 [JACS 1962 27, 70-76].

티오닐 클로라이드 (13.2 mL, 181 mmol)를 질소 분위기하에 1 L 둥근-바닥 플라스크 내의 실시예 2a, 6-(3-브로모페닐)헥산산 (16 g, 59.2 mmol)에 (순수) 첨가하였다. 혼합물을 2 시간 동안 60℃로 가온시키고, 과량의 시약을 진공하에 제거하고, 잔류물을 공비 조건 (벤젠 3x)에 적용하였다. 산 클로라이드를 이황화탄소 (550 mL) 중에 용해시키고, 환류에서 가열된 이황화탄소 (1315 mL) 중 삼염화알루미늄 (26.3 g, 198 mmol)의 용액에 캐뉼레이터를 꽂았다. 반응물을 20 시간 교반하고 냉각시키고 경사분리하고 농축시키고, 생성된 고체를 디에틸 에테르/THF (1:1, 1 L) 및 1 N HCl (500 mL) 중에서 1.5 시간 동안 교반하였다. 유기층을 여과하고, 물 및 염수로 세척하여, 실시예 3c, 2-브로모-7,8,9,10-테트라히드로벤조[8]안눌렌-5(6H)-온 10.1 g (66%)을 무색 오일로서 수득하였다.

참조: 문헌 [J.Org.Chem. USSR (Engl.Transl.) (1985) p 2201-2205].

마그네슘 (0.636 g, 26.2 mmol)을 질소하에 THF (100 mL) 중 4-브로모-1,1,1-트리플루오로부탄 (5 g, 26.2 mmol)의 용액에 첨가하였다. 용액을 18 시간 동안 24℃에서 교반하고, 그리냐드(Grignard) 시약을 캐뉼라를 통해 THF (50 mL) 중 4-브로모벤즈알데히드 (4.85 g, 26.2 mmol)의 용액에 -78℃에서 질소하에 전달하였다. 빙조를 제거하고, 반응물을 가온시키고, 18 시간 교반하고, Et2O (1 부피)로 희석하고, 포화 NH4Cl 용액으로 켄칭하고, 염수로 세척하였다. 농축하고, CH2Cl2 중에 녹이고, 40 M 바이오티지® 실리카겔 카트리지에 충전시켰다. 1 L에 걸친 15%→100% B (A/B 헥산/EtOAc)로 구배 용리를 수행하여, 1-(4-브로모페닐)-5,5,5-트리플루오로펜탄-1-올 6.4 g (82%)을 수득하였다.
PCC (9.29 g, 43.1 mmol)를 SiO2 9 g와 혼합하고, 막자와 막자사발로 분쇄한 후에, 디클로로메탄 (350 mL) 중에 용해시킨 1-(4-브로모페닐)-5,5,5-트리플루오로펜탄-1-올 (6.4 g, 21.54 mmol)의 용액에 한 번에 첨가하였다. 반응물을 4 시간 교반하고, 규조토 (셀라이트®)를 통해 여과하고, 농축시키고, 160 g 톰슨(Thomson)® 실리카겔 컬럼에 적용하였다. 용리: 1.5 L에 걸친 10→60% B (A/B 헥산/EtOAc)로 용리하여, 실시예 5b, 1-(4-브로모페닐)-5,5,5-트리플루오로펜탄-1-온 6.1 g (86%)을 수득하였다.


참조: (이미다졸 합성) 문헌 [Bioorg. Med. Chem. Lett. 2002 1009-1011].

실시예 3, 2-브로모-6,7,8,9-테트라히드로-5H-벤조[7]안눌렌-5-온 (1.5 g, 5.9 mmol)을 2:1 Et2O/THF (120 mL) 중에 용해시키고, Et2O 중 1 N HCl (9 mL)을 첨가하였다. 용액을 0℃로 냉각시킨 후에, 이소-아밀니트리트 (1.2 mL, 9 mmol)를 첨가하고, 반응물을 24℃에서 18 시간 교반하고, 농축시키고, 25 (M) 바이오티지® 실리카겔 컬럼에 적용하였다. 1 L에 걸친 15→100% B (A = 헥산; B = EtOAc)의 구배 용리에 적용하고, 실시예 6, (E)-2-브로모-6-(히드록시이미노)-6,7,8,9-테트라히드로-5H-벤조[7]안눌렌-5-온 1 g (64%)을 수득하였다.


진한 28% 수산화암모늄 용액 (12 mL)을 메탄올 (35 mL) 중 실시예 6, (E)-2-브로모-6-(히드록시이미노)-6,7,8,9-테트라히드로-5H-벤조[7]-안눌렌-5-온 (1 g, 3.7 mmol) 및 N-Boc-L-프롤리날 (850 mg, 4.3 mmol)의 용액에 첨가하고, 반응 혼합물을 24℃에서 18 시간 교반하고, 부분적으로 농축시켜 메탄올을 제거하고, 수성 잔류물을 CH2Cl2로 추출하였다. 유기상을 물로 세척하고 농축시키고, 조 생성물을 40 (S) 바이오티지® 실리카겔 컬럼에 충전시키고 (CH2Cl2); 구획 1. 300 mL에 걸친 15%→30% B; 구획 2. 700 mL에 걸친 30%→100% B (A = 1:1 헥산/CH2Cl2; B = EtOAc)의 구배 용리에 적용하여, 실시예 7, 700 mg (44%)을 수득하였다.



트리에틸 포스파이트 (0.78 mL, 4.7 mmol)를 DMF (2 mL) 중 실시예 7 (700 mg, 1.57 mmol)의 용액에 첨가하고, 용액을 80℃에서 18 시간 동안 질소 분위기하에 가열하였다. 반응 혼합물을 에틸 아세테이트 (100 mL) 중에 녹이고, 물 및 염수로 세척하였다. 농축 후 조 생성물을 40 (S) 바이오티지® 실리카겔 컬럼에 적용하고, 구획 1. 300 mL에 걸친 5%→15% B; 구획 2. 600 mL에 걸친 15%→100% B (A = CH2Cl2; B = EtOAc)의 구배 용리에 적용하여, 실시예 8, 675 mg (100%)을 수득하였다.


합성 경로 2.


디클로로메탄 (50 mL) 중 (S)-5-(히드록시메틸)피롤리딘-2-온 (10 g, 87 mmol)의 용액에 tert-부틸클로로디페닐실란 (25.6 g, 93 mmol), Et3N (12.1 mL, 87 mmol) 및 DMAP (1.06 g, 8.7 mmol)를 첨가하였다. 출발 피롤리디논이 완전히 소비될 때까지 혼합물을 실온에서 교반하고, 그 후 이것을 디클로로메탄 (50 mL)로 희석하고, 물 (50 mL)로 세척하였다. 유기층을 건조시키고 (Na2SO4), 여과하고, 진공하에 농축시키고, 조 물질을 플래쉬 크로마토그래피 (실리카겔; 에틸 아세테이트/헥산 30→100%)에 적용하여, 실릴 에테르를 무색 오일로서 수득하였다 (22.7 g, 74% 수율).

디-tert-부틸 디카르보네이트 (38.5 g, 177 mmol)를 실릴 에테르 (31.2 g, 88.3 mmol), Et3N (8.93 g, 88 mmol) 및 DMAP (1.08 g, 8.83 mmol)의 디클로로메탄 (200 mL)의 용액에 10 분에 걸쳐 고체로서 조금씩 나누어 첨가하고, 18 시간 동안 24℃에서 교반하였다. 대부분의 휘발성 물질을 진공하에 제거하고, 조 물질을 20% 에틸 아세테이트/헥산 중에 녹이고, 실리카겔 1.3 L를 함유한 2 L 깔대기에 적용하고, 그 후에 3 L의 20% 에틸 아세테이트/헥산 및 2 L의 50% 에틸 아세테이트로 용리하였다. 회전 증발기에서 목적하는 구획을 농축하여, 고체의 백색 슬러리를 형성하였고, 이를 여과하고, 헥산으로 세척하고 진공하에 건조시켜, 카르바메이트 M.1을 백색 고체로서 수득하였다 (32.65 g, 82% 수율).


온도계 및 질소 유입구가 장착된 3-목 플라스크를 카르바메이트 M.1 (10.05 g, 22.16 mmol) 및 톨루엔 (36 mL)으로 충전시키고, -55℃ 냉각조에서 저하시켰다. 혼합물의 내부 온도가 -50℃에 도달했을 때, 리튬 트리에틸보로히드라이드 (1.0 M/테트라히드로푸란 23 mL, 23.00 mmol)를 30 분에 걸쳐 적가하고, -50℃ 내지 -45℃의 내부 온도를 유지하면서, 혼합물을 35 분 동안 교반하였다. 휴닉 염기 (16.5 mL, 94 mmol)를 10 분에 걸쳐 적가하였다. 이어서, DMAP (34 mg, 0.278 mmol)를 한 배치로 첨가한 후에, -50℃ 내지 -45℃의 내부 온도를 유지하면서, 15 분에 걸쳐 트리플루오로아세트산 무수물 (3.6 mL, 25.5 mmol)을 첨가하였다. 냉각조를 10 분 후에 제거하고, 반응 혼합물이 주위 온도로 상승되도록 하면서, 이것을 14 시간 동안 교반하였다. 이것을 톨루엔 (15 mL)으로 희석시키고, 빙수조를 이용하여 냉각시키고, 5 분에 걸쳐 물 (55 mL)로 서서히 처리하였다. 상들을 분리하고, 유기층을 물 (50 mL, 2×)로 세척하고, 진공하에 농축시켰다. 조 물질을 플래쉬 크로마토그래피 (실리카겔; 5% 에틸 아세테이트/헥산)로 정제하여, 디히드로피롤 M.2를 무색 점성 오일로서 수득하였다 (7.947 g, 82% 수율). 하기의 HPLC 조건 하에서 Rt = 2.41 분: 2 분에 걸친 100% A:0% B→0% A:100% B (A = 0.1% TFA + 1:9 메탄올/물; B = 0.1% TFA + 9:1 메탄올/물)의 용매 구배 및 1 분 동안 유지; 220 nm에서의 검출; 페노메넥스-루나 3.0×50 mm S10 컬럼.


디에틸아연 (톨루엔 중 약 1.1 M, 19 mL, 20.9 mmol)을 디히드로피롤 M.2 (3.94 g, 9.0 mmol)의 냉각된 (-30℃) 톨루엔 (27 mL) 용액에 15 분에 걸쳐 적가하였다. 클로로요오도메탄 (구리 상에서 안정화됨; 3.0 mL, 41.2 mmol)을 10 분에 걸쳐 적가하고, 냉각조 온도를 -25℃에서 1 시간 동안 및 -25℃ 내지 -21℃에서 18.5 시간 동안 유지하면서 교반하였다. 반응 혼합물을 공기에 개방하고, 50% 포화 NaHCO3 용액 (40 mL)을 서서히 첨가하여 켄칭한 다음, 냉각조로부터 제거하고, 주위 온도에서 20 분 동안 교반하였다. 이것을 필터지를 통해 여과하고, 백색 케이크를 톨루엔 50 mL로 세척하였다. 여과물의 유기상을 분리하고, 물 (40 mL, 2x)로 세척하고, 건조시키고 (MgSO4), 여과하고 진공하에 농축시켰다. 조 물질을 바이오티지® 시스템 (350 g 실리카겔; 샘플을 7% 에틸 아세테이트/헥산과 함께 로딩하고; 7→20% 에틸 아세테이트/헥산으로 용리함)을 이용하여 정제하여, 메타노피롤리딘 (M.3이 우세함)을 무색 점성 오일로서 수득하였다 (3.69 g, 90.7%). [주: 정확한 시스/트랜스-이성질체 비율은 이 단계에서 측정하지 않았다]. 하기의 HPLC 조건하에서 Rt = 2.39 분: 2 분에 걸친 100% A:0% B→0% A:100% B (A = 0.1% TFA + 1:9 메탄올/물; B = 0.1% TFA + 9:1 메탄올/물)의 용매 구배, 및 1 분 동안 유지; 220 nm에서의 검출; 페노메넥스-루나 3.0×50 mm S10 컬럼.


TBAF (테트라히드로푸란 중 1.0 M, 7.27 mL, 7.27 mmol)를 실릴 에테르 M.3 (3.13 g, 6.93 mmol)의 테트라히드로푸란 (30 mL) 용액에 5 분에 걸쳐 적가하고, 혼합물을 주위 온도에서 4.75 시간 동안 교반하였다. 포화 염화암모늄 용액 (5 mL)을 첨가한 후에, 대부분의 휘발성 물질을 진공하에 제거하고, 잔류물을 디클로로메탄 (70 mL) 및 50% 포화 염화암모늄 용액 (30 mL) 사이에 분배하였다. 수성상을 디클로로메탄 (30 mL)으로 추출하고, 합한 유기상을 건조시키고 (MgSO4), 여과하고, 진공하에 농축시킨 다음, 고진공에 밤새 노출시켰다. 조 물질을 바이오티지® (실리카겔; 40→50% 에틸 아세테이트/헥산)를 이용하여 정제하여, 미량의 하위의 Rf 반점으로 오염된 알콜의 혼합물을 무색 오일로서 수득하였다 (1.39 g, 약 94% 수율). [주: 정확한 시스/트랜스 이성질체 비율은 이 단계에서 측정하지 않았다.]

물 (31 mL) 중 과요오드산나트륨 (6.46 g, 30.2 mmol)의 반-용액을 아세토니트릴 (20 mL) 및 사염화탄소 (20 mL) 중 알콜 (2.15 g, 10.08 mmol)의 용액에 첨가하였다. 삼염화루테늄 (0.044 g, 0.212 mmol)을 즉시 첨가하고, 불균일 반응 혼합물을 75 분 동안 격렬하게 교반하였다. 반응 혼합물을 물 (60 mL)로 희석하고, 디클로로메탄 (50 mL, 3x)으로 추출하였다. 합한 유기상을 메탄올 1 mL로 처리하고, 약 5 분 동안 정치시킨 다음, 규조토 (셀라이트®)를 통해 여과하였다. 패드를 디클로로메탄 (50 mL)으로 세척하고, 여과물을 진공하에 농축시켜, 밝은 차콜-색상의 고체를 수득하였다. 조 물질을 가열하면서 에틸 아세테이트 (약 10 mL) 중에 용해시키고, 시딩하면서 주위 온도에서 정치시켰다. 냉각 상태에서 약 15 분 후에, 빠른 결정 형성이 관찰되었다. 약 1 시간 후에, 헥산 (약 6 mL)을 첨가하고, 혼합물을 밤새 냉장시켰다 (추가 물질의 침전은 나타나지 않음). 혼합물을 여과하고, 얼음/물-냉각된 헥산/에틸 아세테이트 (2:1 비율; 20 mL)로 세척하고, 고 진공하에 건조시켜, 산 M.4 (회백색 결정, 1.222 g)의 제1 수집물을 수득하였다. 모액을 진공하에 농축시키고, 가열하면서 에틸 아세테이트 약 3 mL 중에 용해시킨 잔류물을 주위 온도에서 1 시간 동안 정치시킨 다음, 헥산 3 mL를 첨가하고, 냉장고에서 약 15 시간 동안 저장하였다. 합친 수율 59%를 위해 산 M.4의 제2 수집물을 유사하게 회수하였다 (회색 결정, 0.133 g). 하기 HPLC 조건 하에서 Rt = 1.48 분: 3 분에 걸친 100% A:0% B→0% A:100% B (A = 0.1% TFA + 1:9 메탄올/물; B = 0.1% TFA + 9:1 메탄올/물)의 용매 구배; 220 nm에서의 검출; 페노메넥스-루나 3.0×50 mm S10 컬럼. 제1 수집물에 대한 MP (분해) = 147.5-149.5℃.

합성 경로 3.

참조: (브롬화) 문헌 [JACS (1952) 74, 6263].
참조: (대체/고리화) 문헌 [J.Med.Chem. (2001) 44, 2990].
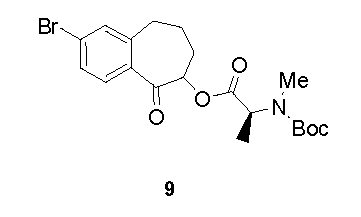
실시예 3, 2-브로모-6,7,8,9-테트라히드로-5H-벤조[7]안눌렌-5-온 (1.5 g, 5.9 mmol)을 Et2O (25 mL) 중에 용해시키고, 0℃로 냉각시키고, 브롬 (0.35 mL, 6.6 mmol)을 적가하였다. TLC가 반응 완료를 나타낼 때까지 (2 내지 18 시간; 규모 의존적), 용액을 24℃에서 교반하였다. 용매를 회전 증발에 의해 제거하고, 조 생성물을 25 (M) 바이오티지® 실리카겔 컬럼에 적용하였다. 500 mL에 걸친 50→100% B (A = 헥산; B = 10% EtOAc/hex)의 구배 용리에 적용하여, 2,6-디브로모-6,7,8,9-테트라히드로-5H-벤조[7]안눌렌-5-온 700 mg, 및 디브로마이드를 함유한 제2 분획 (1:1) 1.5 g을 수득하였다. RT = 2.2 분 (조건 1).
2,6-디브로모-6,7,8,9-테트라히드로-5H-벤조[7]안눌렌-5-온 (700 mg, 2.22 mmol) 및 N-메틸-L-알라닌 (493 mg, 2.4 mmol)을 아세토니트릴 (8 mL) 중에 용해시키고, 휴닉 염기 1.1 mL를 적가하고, 반응물을 55℃에서 18 시간 교반하였다. 용매를 회전 증발에 의해 제거하고, 잔류물 EtOAc 중에 녹이고, 0.1 N HCl 용액, 포화 NaHCO3 용액 및 염수로 세척하고 농축하여, 실시예 9, (2S)-2-브로모-5-옥소-6,7,8,9-테트라히드로-5H-벤조[7]안눌렌-6-일 2-(tert-부톡시카르보닐(메틸)아미노)프로파노에이트 960 mg (98%)을 수득하였고, 이것을 추가 정제없이 후속 단계에 사용하였다.



실시예 9, (2S)-2-브로모-5-옥소-6,7,8,9-테트라히드로-5H-벤조[7]안눌렌-6-일 2-(tert-부톡시카르보닐(메틸)아미노)프로파노에이트 (960 mg, 2.2 mmol)를 크실렌 (20 mL) 중에 녹이고, 100 mL 스크류캡 압력 용기에 넣었다. 암모늄 아세테이트 (1.7 g, 10 당량)를 첨가하고, 혼합물을 120℃ (오일조 온도)에서 4 시간 동안 가열하였다. 냉각된 혼합물을 에틸 아세테이트 (150 mL) 및 포화 NaHCO3 용액으로 희석하고, 유기상을 농축시켰다. 조 생성물을 25 (M) 바이오티지® 실리카겔 카트리지에 적용하였다 (CH2Cl2). 구획 1. 300 mL 동안 15% B 유지; 구획 2. 600 mL에 걸친 10→100% B (A = 헥산; B = EtOAc)의 구배 용리에 적용하여, 실시예 10, 150 mg (16%)을 수득하였다. 주: 크로마토그래피 동안 화합물의 손실이 발생하였다 (수율은 더 높아야 함).


합성 경로 4.




합성 경로 5.






합성 경로 6.

세척되지 않은 60% 수소화나트륨 (54 mg, 1.35 mmol)을 무수 DMF (10 mL) 중 실시예 10a.3 (565 mg, 1.35 mmol)의 교반 용액에 질소하에 한 번에 첨가하였다. 혼합물을 5 분 교반한 후에, SEM-Cl (0.24 mL, 1.35 mmol)을 첨가하고, 3 시간 동안 교반하고, 포화 염화암모늄 (1 mL)으로 켄칭하고, EtOAc (50 mL)로 희석하고, 유기상을 포화 NaHCO3 용액 및 염수로 세척하였다. 수성상을 EtOAc로 2회 더 추출하고, 초기 유기 추출물과 합한 후에 건조시켰다. 적용된 잔류물을 농축시키고, 이것을 40 (S) 바이오티지® 실리카겔 카트리지에 적용하였다 (CH2Cl2). 구획 1. 75 mL 동안 15% B 유지; 구획 2. 750 mL에 걸친 15%→50% B; 구획 3: 750 mL에 걸친 50→100%B (A = hex; B = EtOAc)의 구배 용리를 적용하여, 위치이성질체 생성물 실시예 11, 497 mg (67 %)을 수득하였다.


디옥산 중 저온 4N HCl (0.871 mL, 3.49 mmol)을 MeOH (20 mL) 중 실시예 10a.3 (1R,3S,5R)-tert-부틸 3-(7-브로모-4,5-디히드로-1H-나프토[1,2-d]이미다졸-2-일)-2-아자바이시클로[3.1.0]헥산-2-카르복실레이트 (1.5 g, 3.49 mmol)의 용액에 첨가하고, 반응물을 2 시간 동안 실온에서 교반하였다. 농축시켜 황갈색 고체를 수득하였고, 이것을 디옥산 (20 mL)/물 (20 mL) 중에 녹이고, 0℃로 냉각시켰다. Na2CO3 (0.369 g, 3.49 mmol) 및 CBz-Cl (0.498 mL, 3.49 mmol)을 첨가하고, 반응 혼합물이 실온으로 가온될 때까지 5 시간에 걸쳐 이를 교반하고, 그 후에 EtOAc 및 포화 NaHCO3 용액 사이에 분배하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고 농축시켰다. 조 생성물을 110 g 톰슨(Thompson)® 실리카겔 카트리지에 충전시키고 (CH2Cl2), 1.5 L에 걸쳐 20%→100% B (A/B = 헥산/EtOAc)로 용리하였다. 다양한 순도의 3개 구획으로부터 실시예 11a (1R,3S,5R)-벤질 3-(7-브로모-4,5-디히드로-1H-나프토[1,2-d]이미다졸-2-일)-2-아자바이시클로[3.1.0]헥산-2-카르복실레이트의 합한 수율은 1.4 g (75%)이었다.


합성 경로 7.

참조: 문헌 [J.Chem.Soc. Perkin Trans 2 (1993) p 1305].

AIBN (98 mg, 0.59 mmol)을 CCl4 (50 ml) 중 4-브로모-1,2-디메틸벤젠 (11 g, 59.4 mmol) 및 새로 결정화된 N-브로모숙신이미드 (21.1 g, 119 mmol)에 첨가하였다. 반응 혼합물을 2 시간 동안 질소하에 환류에서 가열하였다. 실온으로 냉각시킨 후에, 용액을 여과하고, 회전 증발로 농축하여, 대략 (75%) 4-브로모-1,2-비스(브로모메틸)벤젠을 3-브로모-1,2-비스(브로모메틸)벤젠과 함께 함유한 혼합물 21 g을 수득하였다. [주: 출발 물질은 25% 3-브로모-1,2-디메틸벤젠으로 오염되었다]. RT = 2.2/2.3 분 (조건 1).
4- 및 3-브로모-1,2-비스(브로모메틸)벤젠의 조 혼합물 (21 g)을 CH2Cl2 (313 ml) 중에 녹이고, 디메틸 3-옥소펜탄디오에이트 (12.8 g, 73.5 mmol)를 첨가하고, 용액을 물 (250 ml) 중 KHCO3 (64 g, 0.64 mol)의 20% 용액으로 희석하였다. 상 전이 촉매인 벤질트리메틸암모늄 클로라이드 (1.7 g, 9.19 mmol)를 첨가하고, 반응 혼합물을 질소하에 50℃에서 18 시간 교반하였다. 유기상의 분리 후에, 수성상을 CH2Cl2로 추출하고, 합한 유기층을 염수로 세척하였다. 농축하여 디메틸 2-브로모-7-옥소-6,7,8,9-테트라히드로-5H-벤조[7]안눌렌-6,8-디카르복실레이트를 함유한 착체 혼합물 (위치이성질체 및 에스테르 부분입체이성질체 둘다) 23 g을 수득하였고, 이것을 정제없이 후속 단계에서 사용하였다.

조 디메틸 2-브로모-7-옥소-6,7,8,9-테트라히드로-5H-벤조[7]안눌렌-6,8-디카르복실레이트 (약 8 g, 22.5 mmol 함유)의 혼합물 23 g을 환류 응축기가 장착된 1 L 둥근-바닥 플라스크에 넣고, 에탄올 (250 ml) 중에 용해시켰다. 물 (250 ml) 중에 용해시킨 KOH (8.8 g, 0.23 mol)의 용액을 첨가하였고, 환류에서 16 시간 동안 가열함에 따라 초기 황갈색 현탁액이 흑색으로 변하였다. 반응 혼합물을 EtOAc (1 부피)에 붓고, 유기상을 분리하였다. 염수 (1/4 부피)를 수성층에 첨가하고, 이것을 EtOAc로 추출하고, 합한 유기층을 염수로 세척하였다. 회전 증발에 의해 농축한 후에, 잔류물을 40 (M) 바이오티지® 실리카겔 컬럼 (하위의 컬럼으로부터 진공을 적용하여 흡수를 보조함)에 충전시키고 (CH2Cl2), 10-100% B (A = 헥산; B = EtOAc)의 구배 용리에 적용하여, 브로마이드 4:1 혼합물을 수득하였고, 이것은 실시예 12, 2-브로모-8,9-디히드로-5H-벤조[7]안눌렌-7(6H)-온이 우세하고, 1-브로모-8,9-디히드로-5H-벤조[7]안눌렌-7(6H)-온, 8.3 g (61%)을 함유하였다. 혼합물을 추가 정제없이 후속 단계에 사용하였지만, 실시예 12의 순수한 샘플은 혼합물을 -5℃에서 3 주 동안 정치시켜 결정화하여 수득하였다.


브롬 (1.4 mL, 26.8 mmol)을 디에틸 에테르 (250 mL) 중 2- 및 1-브로모-8,9-디히드로-5H-벤조[7]안눌렌-7(6H)-온 (6.4 g, 26.8 mmol)의 각각 4:1 혼합물의 용액에 첨가하였다. 반응물을 4 시간 동안 교반하고 농축시키고, 조 α-브로모 케톤을 아세토니트릴 (200 mL) 중에 녹였다. 나트륨 아지드 (1.7 g, 26.8 mmol)를 첨가하고, 반응물을 16 시간 교반한 후에 농축시켰다. 잔류물을 EtOAc 및 포화 NaHCO3 용액 사이에 분배하고, 유기층을 물 및 염수로 세척하고 농축시켰다. RT = 2.0 분 (넓은 피크, 조건 1).
조 α-케토 아지드를 CH3OH (350 mL) 중에 용해시키고, 주석 (II) 클로라이드 이수화물 (13.2 g, 69.6 mmol)을 첨가하고, 반응 혼합물을 3.5 시간 동안 65℃에서 교반하고, 회전 증발로 농축하고, 고 진공하에 18 시간 동안 건조시켰다.
조 α-아미노 케톤을 DMF (300 mL) 중에 녹이고, 휴닉 염기 (24.41 g, 189 mmol), (S)-1-(tert-부톡시카르보닐)피롤리딘-2-카르복실산 (4.07 g, 18.89 mmol) 및 HATU (7.18 g, 18.89 mmol)를 격렬하게 교반하면서 한 번에 첨가하였다. 8 시간 후 반응물을 진공하에 1/3 부피로 농축시키고, EtOAc (1 L) 및 0.1 N HCl (150 mL) 사이에 분배하였다. 주석 염을 규조토 (셀라이트®)를 통한 여과에 의해 제거하고, 유기층을 염수로 세척하고 농축시켰다. 조 생성물을 40M 바이오티지® 실리카겔 카트리지 (2회 진행으로 분할함)에 충전시켰다 (CH2Cl2). 1.5 L에 걸쳐 15%→100% B (A = 헥산; B = 에틸 아세테이트)의 구배 용리에 적용하여, 실시예 13a.1 및 13b.1 (3.8 g, 35.7% 합한 수율) 각각을 부분입체이성질체의 혼합물로서 수득하였다.

암모늄 아세테이트 (4.4 g, 73.1 mmol)를 크실렌 (75 mL) 중 아미드 13a.1 및 13b.1 (3.3 g, 7.31 mmol)의 용액에 첨가하고, 140℃에서 4 시간 동안 가열된 압력 용기 내에서 교반하였다. 냉각시킨 후에, 반응 혼합물을 EtOAc (500 mL)로 희석하고, 포화 NaHCO3 및 염수로 세척하고, 유기상을 농축시켰다. 조 생성물을 40S 바이오티지® 실리카겔 카트리지에 충전시켰다 (CH2Cl2). 750 mL에 걸친 15%→100% B (A = CH2Cl2; B = EtOAc)의 구배 용리에 적용하여, 혼합물 2.49 g을 수득하였고, 이것을 제2 크로마토그래피; 40S 바이오티지® 실리카겔 카트리지, 1 L에 걸친 5%→80% B (A = 1:1 Hex/CH2Cl2; B = 10% CH3OH/EtOAc)의 용리에 적용하여, 실시예 13a.2 및 13b.2의 혼합물 1.1 g (34.8%)을 제공하였다. 생성물 샘플을 반-분취용 정상 상 HPLC (키랄셀 OD 컬럼, 20×250 mm, 10 um; 10 ml/분으로 98:2 헵탄/EtOH; UV 220/254 nm)에 적용하고, 35 내지 36 분에 용리되는 실시예 13a.2를 수집하였다.

합성 경로 8.

실시예 14

N,N-디이소프로필에틸아민 (18 mL, 103.3 mmol)을 DMF (105 mL) 중 N-Boc-L-프롤린 (7.139 g, 33.17 mmol), HATU (13.324 g, 35.04 mmol), 2-아미노-1-(4-브로모-페닐)에타논의 HCl 염 (8.127 g, 32.44 mmol)의 불균일 혼합물에 15 분에 걸쳐 적가하고, 주변 조건에서 55 분 동안 교반하였다. DMF를 진공하에 제거하고, 생성된 잔류물을 에틸 아세테이트 (300 mL) 및 물 (200 mL) 사이에 분배하였다. 유기층을 물 (200 mL) 및 염수로 세척하고, 건조시키고 (MgSO4), 여과하고 농축시켰다. 실리카겔 메쉬를 잔류물로부터 제조하고, 플래쉬 크로마토그래피 (실리카겔; 50→60% 에틸 아세테이트/헥산)에 적용하여, 실시예 14, (S)-tert-부틸 2-(2-(4-브로모페닐)-2-옥소에틸카르바모일)피롤리딘-1-카르복실레이트를 백색 고체로서 제공하였다 (12.8 g).


실시예 14c

실시예 14c; 단계 1:
아세틸 클로라이드 (10.2 mL, 144 mmol)를 디클로로메탄 (100 mL) 중 3-브로모페놀 (25 g, 144 mmol) 및 피리딘 (10.8 mL, 144 mmol)의 냉각 (빙조) 용액에 1 시간에 걸쳐 적하 방식으로 첨가하고, 반응물을 실온에서 18 시간 교반하였다. 물 (150 mL)을 첨가하고, 유기층을 디클로로메탄으로 추출하였다. 합한 유기층을 2.5 N NaHSO4, 3 N NaOH, 물 및 염수로 세척하고, Na2SO4 상에서 건조시키고 여과하였다. 농축하여 3-브로모페닐 아세테이트를 핑크색 액체로서 수득하였다 (28 g, 96%).

실시예 14c; 단계 2:

3-브로모페닐 아세테이트 (36 g, 167 mmol) 및 무수 AlCl3 (33.5 g, 251 mmol)을 140 내지 150℃에서 2 시간 동안 가열하였다. 냉각시킨 후, 5% HCl 용액 (100 ml)을 첨가하고, 고체 물질이 용해될 때까지 혼합물을 가열하였다 (증기조). 갈색 오일을 분리하고, 디클로로메탄 내로 추출하였다. 추출물에 5N NaOH (300 mL) 및 충분한 물을 첨가하여, 생성된 침전물을 용해시켰다. 수성층을 분리하고, 산성화시키고 (pH 2.0), 에틸 아세테이트로 추출하였다. 추출물을 Na2SO4 상에서 건조시키고 여과하고 농축시켜, 1-(4-브로모-2-히드록시페닐)에타논을 결정으로 수득하였다 (35.8 g, 99%).

실시예 14c; 단계 3:

무수 벤젠 (800 mL) 중 1-(4-브로모-2-히드록시페닐)에타논 (35.8 g, 167 mmol)에 N,N-디메틸포름아미드 디메틸아세탈 (44 mL, 333 mmol)을 첨가하고, 용액을 환류에서 4 시간 동안 가열하고 농축 건조시켰다. 생성된 잔류물을 디클로로메탄 (300 mL) 중에 용해시키고, SiO2 상에 여과하였다. 유기층을 농축시켜, (E)-1-(4-브로모-2-히드록시페닐)-3-(디메틸아미노)프로프-2-엔-1-온을 담황색 고체로서 수득하였다 (31.9 g, 71%).

실시예 14c; 단계 4:

진한 HCl (100 mL)을 디클로로메탄 (900 mL) 중 (E)-1-(4-브로모-2-히드록시페닐)-3-(디메틸아미노)프로프-2-엔-1-온 (31.9 g, 118 mmol)의 용액에 첨가하고, 반응 혼합물을 격렬하게 교반하면서 환류에서 40 분 동안 가열하였다. 실온으로 냉각시킨 후에, 유기층을 분리하고, 수성층을 디클로로메탄으로 추출하였다. 합한 유기층을 3 M 탄산칼륨 용액 및 물로 세척하고, Na2SO4 상에서 건조시켰다. 여과 및 농축하여 7-브로모-4H-크로멘-4-온을 옅은 황색 고체로서 수득하였다.
실시예 14c; 단계 5:

무수 THF (500 mL) 중 7-브로모-4H-크로멘-4-온 (26 g, 116 mmol)의 용액을 질소하에 1 시간 동안 -80℃로 냉각시키고, 디이소부틸알루미늄 히드라이드 (톨루엔 중 2 M) 173 mL를 30 분에 걸쳐 첨가하였다. 반응물을 동일한 온도에서 30 분 동안 교반하고, SiO2 (52 g)/물 (52 mL) 현탁액으로 켄칭하고, 0℃로 가온시켰다. 용액을 여과하고, SiO2를 EtOAc로 세척하고, 합한 여과물을 농축 건조시켰다. 잔류물을 CHCl3 (400 mL) 중에 용해시키고, 1 N NaOH (300 mL)로 세척하고, Na2SO4 상에서 건조시키고 농축시켰다. SiO2 (60-120 메쉬) 상의 크로마토그래피에 의해 EtOAc/Pet 에테르 (구배 용리 0→15%)로 정제하여, 7-브로모크로만-4-온 18.1 g (69%)을 옅은 황색 고체로서 수득하였다.
실시예 14c; 단계 6:

H2O (30 mL) 중 NaOAc (3.78 g, 46.2 mmol)를 EtOH (70 ml) 중 7-브로모 4-크로마논 (3.5 g, 15.4 mmol) 및 히드록실-아민 히드로클로라이드 (1.75 g, 23.1 mmol)의 용액에 첨가하였다. 반응물을 2 시간 동안 환류하에 가열하고, 실온으로 냉각시키고 농축시켰다. 잔류물을 H2O로 희석하고, 침전물을 여과하여, 침상형 결정으로서 수득되는 7-브로모크로만-4-온 옥심 3.5 g (98%)을 수득하였다.

실시예 14c; 단계 7:

p-톨루엔술폰산 무수물 (5 g, 16 mmol)을 디클로로메탄 (125 ml) 중 7-브로모크로만-4-온 옥심 (3.5 g, 14.46 mmol) 및 트리에틸아민 (2.4 ml, 17.35 mmol)의 용액에 첨가하였다. 혼합물을 실온에서 3 시간 동안 교반하고, 물 및 염수로 세척하고 건조시켰다 (Na2SO4). 감압하에 여과 및 농축하여, 7-브로모크로만-4-온-O-p-톨루엔술폭심 5.5 g (96 %)을 회백색 고체로서 수득하였고, 이것을 추가 정제없이 사용하였다.

실시예 14c; 단계 8:

에탄올 (35 ml) 중 칼륨 에톡시드 (1.27 g, 15.11 mmol)의 용액을 톨루엔 (60 ml) 중 7-브로모크로만-4-온-O-p-톨루엔술폭심 (5.7 g, 14.3 mmol)의 용액에 첨가하고, 혼합물을 실온에서 15 시간 동안 교반하였다. 침전물 (칼륨 토실레이트)을 여과에 의해 제거하고, 디에틸 에테르로 세척하였다. 합한 여과물에 메탄올 중 3 N HCl (20 ml)을 첨가하고, 용액을 실온에서 2 시간 교반하여, HCl 염을 형성하였다. 용매를 제거한 후, 잔류물을 디에틸 에테르로 연화처리하였다. 생성된 고체를 여과하고 에테르로 세척하고 건조시켜, 7-브로모-3-아미노-크로만-4-온 히드로클로라이드 2.8 g (80 %)을 회백색 분말로서 수득하였다.



크실렌 (155 mL) 중 실시예 14, (S)-tert-부틸 2-(2-(4-브로모페닐)-2-옥소에틸카르바모일)-피롤리딘-1-카르복실레이트 (12.8 g, 31.12 mmol) 및 NH4OAc (12.0 g, 155.7 mmol)의 혼합물을 140℃에서 2 시간 동안 밀봉 튜브에서 가열하였다. 휘발성 성분을 진공하에 제거하고, 잔류물을 에틸 아세테이트 및 물 사이에 조심스럽게 분배하고, 이에 따라 충분한 포화 NaHCO3 용액을 첨가하여, 2상 시스템을 진단시킨 후에 수성상의 pH가 약간 염기성이 되게 하였다. 층을 분리하고, 수성층을 추가의 에틸 아세테이트로 추출하였다. 합한 유기상을 염수로 세척하고, 건조시키고 (MgSO4), 여과하고 농축시켰다. 생성된 물질을 에틸 아세테이트/헥산으로 재결정하여, 실시예 15, (S)-tert-부틸 2-(5-(4-브로모페닐)-1H-이미다졸-2-일)피롤리딘-1-카르복실레이트 5.85 g의 제2 수집물을 제공하였다. 모액을 진공하에 농축시키고, 플래쉬 크로마토그래피 (실리카겔; 30% 에틸 아세테이트/헥산)에 적용하여, 추가 2.23 g을 제공하였다.


합성 경로 9.

Pd(Ph3P)4 (469 mg, 0.406 mmol)를 실시예 15, (S)-tert-부틸 2-(5-(4-브로모페닐)-1H-이미다졸-2-일)피롤리딘-1-카르복실레이트 (4 g, 10.22 mmol), 비스(피나콜레이토)디보론 (5.4 g, 21.35 mmol), 칼륨 아세테이트 (2.6 g, 26.21 mmol) 및 1,4-디옥산 (80 mL)의 혼합물을 함유한 스크류캡 압력 튜브에 첨가하였다. 반응 플라스크를 질소로 퍼징하고 캡핑하고 16 시간 동안 가열하였다 (오일조 80℃). 반응 혼합물을 여과하고, 여과물을 진공하에 농축시켰다. 조 물질을 CH2Cl2 (150 mL) 및 수성 매질 (50 mL 물 + 10 mL 포화 NaHCO3 용액) 사이에 조심스럽게 분배하였다. 수성층을 CH2Cl2로 추출하고, 합한 유기상을 건조시키고 (MgSO4), 여과하고 진공하에 농축시켰다. 생성된 물질을 플래쉬 크로마토그래피 (샘플은 용리 용매; 20→35% 에틸 아세테이트/CH2Cl2로 로딩함)로 정제하여, 피나콜로 오염된 실시예 16, (S)-tert-부틸 2-(5-(4-(4,4,5,5-테트라메틸-1,3,2-디옥사보롤란-2-일)페닐)-1H-이미다졸-2-일)피롤리딘-1-카르복실레이트를 회백색 조밀 고체로서 제공하였고; 실시예 16 대 피나콜의 상대 몰비율은 약 10:1이었다 (1H NMR). 샘플을 고진공에 약 2.5 일 노출시킨 후에 3.9 g을 칭량하였다.





실시예 8 (950 mg, 2.19 mmol), 실시예 16, (S)-tert-부틸 2-(5-(4-(4,4,5,5-테트라메틸-1,3,2-디옥사보롤란-2-일)페닐)-1H-이미다졸-2-일)피롤리딘-1-카르복실레이트 (965 mg, 2.2 mmol) 및 NaHCO3 (462 mg, 5.5 mmol)을 1,2-디메톡시에탄 (25 mL) 중에 용해시키고, 물 (4 mL)을 첨가하였다. 반응 혼합물을 배출시키고, 질소로 플러슁하고 (3x), Pd(Ph3P)4 (127 mg, 0.11 mmol)를 첨가하고, 혼합물을 8 시간 동안 캡핑된 100 mL 압력 용기에서 (80℃의 오일조) 가열하였다. 추가의 촉매 (35 mg)를 첨가하고, 16 시간 동안 계속 가열하였다. 냉각시킨 후에, 용액을 EtOAc/물에 분배하고, 유기층을 염수로 세척하였다. 조 생성물을 40 (S) 바이오티지® 실리카겔 컬럼에 적용하고 (CH2Cl2), 1 L에 걸쳐 20→100% B (A = CH2Cl2; B = EtOAc)의 구배 용리에 적용하여, 실시예 17, 680 mg (49%)을 수득하였다.


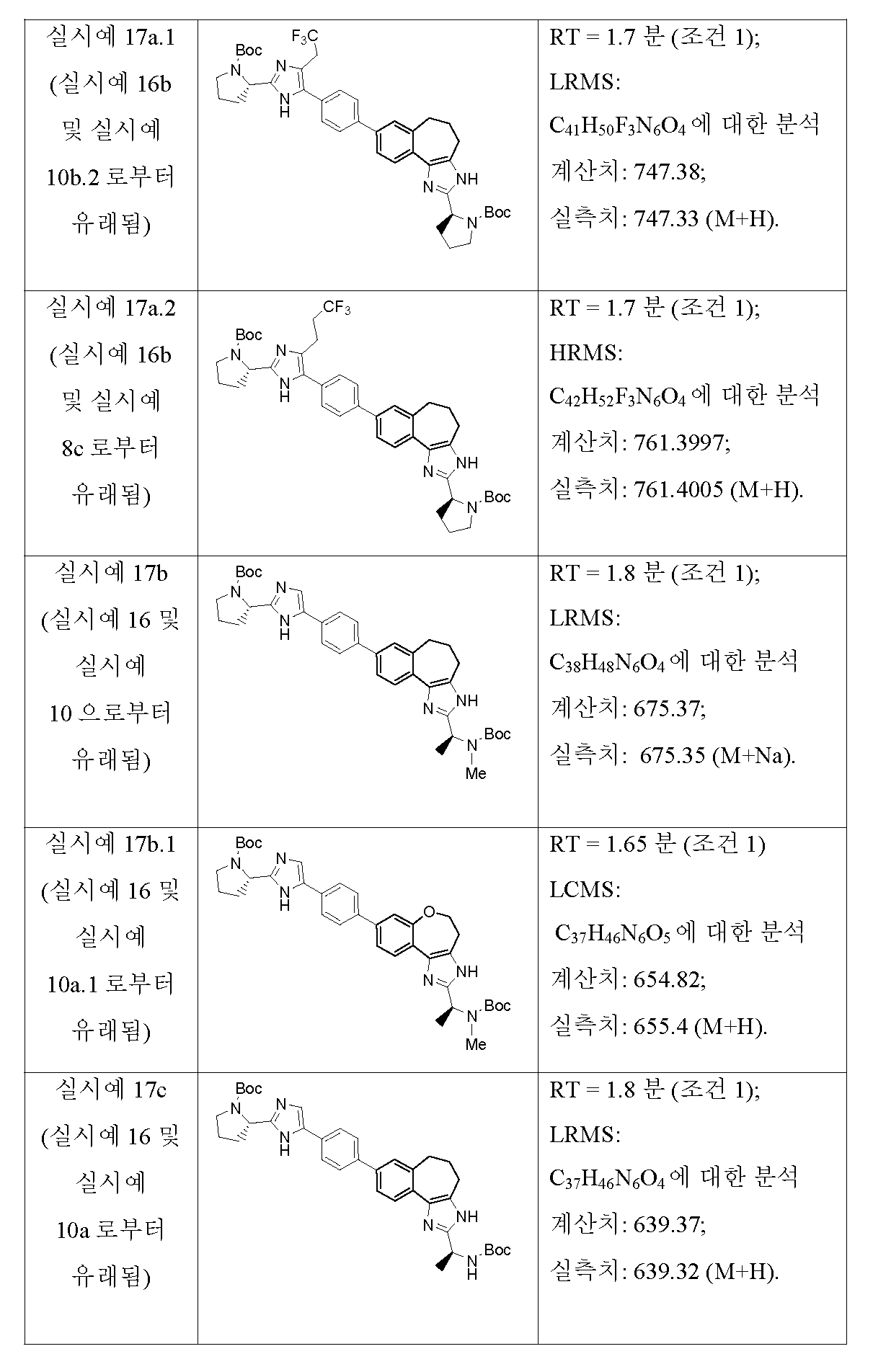






합성 경로 10.

DDQ (116 mg, 0.51 mmol)를 벤젠 (5 mL) 중 실시예 17r (S)-tert-부틸 2-(7-(4-(5-((S)-1-(tert-부톡시카르보닐)피롤리딘-2-일)-1H-이미다졸-2-일)페닐)-4,5-디히드로-1H-나프토[1,2d]이미다졸-2-일)피롤리딘-1-카르복실레이트 (332 mg, 0.51 mmol)의 용액에 질소 분위기 하에 첨가하고, 어두운 갈색 혼합물을 2 시간 동안 환류에서 가열하였다. 용매를 진공하에 제거하고, 잔류물을 25 (M) 바이오티지® 실리카겔 컬럼에 충전시켰다 (CH2Cl2). 구획 1; 720 mL에 걸친 50→100% B (A = CH2Cl2; B = CH2Cl2 중 20% CH3OH)의 구배 용리, 구획 2; 720 mL에 걸친 0→50% B (A = CH2Cl2; B = CH3OH)의 구배 용리에 적용하여, 실시예 18, 261 mg (79%)을 수득하였다.

합성 경로 10a.

활성화된 MnO2 (77 mg, 0.889 mmol)를 무수 CH2Cl2 (2 mL) 중 실시예 17r.2 (200 mg, 0.296 mmol)의 교반 용액에 한 번에 첨가하고, 현탁물을 4 시간 동안 교반하였다. 추가의 MnO2 (300 mg)를 첨가하고, 혼합물을 16 시간 교반하였다. LCMS가 잔류하는 출발 물질을 나타내지 않을 때까지, 이 순서를 반복하였다. 반응 혼합물을 규조토 (셀라이트®)를 통해 여과하고 농축시키고, 고 진공하에 1 시간 동안 건조시켜, 실시예 18.1 (185.8 mg, 92%)을 황색빛-오렌지색 고체로서 수득하였다.





합성 경로 11

디클로로메탄과의 [1,1'-비스(디페닐포스피노)페로센)디클로로팔라듐(ii)착체 (11.2 mg, 0.014 mmol)를 스크류탑 압력 용기에서 아르곤 분위기 하에 DMSO (2.5 mL) 중 실시예 18a (150 mg, 0.274 mmol), 비스-피나콜 디보란 (34.8 mg, 0.137 mmol), dppf (7.61 mg, 0.014 mmol) 및 K2CO3 (114 mg, 0.823 mmol)의 교반 용액에 첨가하였다. 용액을 아르곤으로 철저하게 플러슁하고 밀봉하고, 80℃에서 예열된 오일조에서 침지시키고, 18 시간 교반하였다. 혼합물을 냉각시키고, EtOAc로 희석하고, 포화 NaHCO3 용액 및 염수로 세척하고, 건조시키고 (Na2SO4) 여과하였다. 용매를 진공하에 제거하고, 잔류물을 25 (M) 바이오티지® 실리카겔 컬럼에 충전시키고 (CH2Cl2): 구획 1. 75 mL 동안 10% B; 구획 2. 1.8 L에 걸친 10→100% B (A = 헥산; B = EtOAc)의 구배 용리에 적용하여, 실시예 19, 88.7 mg (33%)을 수득하였다.

합성 경로 12

트리에틸포스파이트 (0.257 mL, 1.48 mmol)를 DMF (5 mL) 중 실시예 17q (340 mg, 0.489 mmol)에 첨가하고, 80℃에서 16 시간 동안 교반하였다. LCMS가 반응 완료를 나타낼 때까지, 추가의 트리에틸포스파이트 (0.3 mL 및 0.6 mL)를 8 시간 간격으로 첨가하였다. 용매를 진공하에 제거하고, 잔류물을 25 (M) 바이오티지® 실리카겔 컬럼에 충전시켰다 (CH2Cl2). 구획 1. 300 mL 동안 0% B; 구획 2. 1440 mL에 걸친 0→50% B의 구배 용리; 구획 3. 600 mL에 걸친 50→100% B (A = CH2Cl2; B = CH2Cl2 중 20% CH3OH)의 구배 용리에 적용하여, 실시예 20, 286 mg (82%)을 수득하였다.

합성 경로 13

실시예 17 (411 mg, 0.62 mmol)을 CH3OH (20 mL) 중에 용해시키고, HCl/디옥산 (4 N, 100 mL)을 첨가하고, 반응물을 4 시간 교반하였다. 용매를 진공하에 제거하고, 테트라 HCl 염을 고진공에 18 시간 동안 노출시켜, 실시예 21, 350 mg (94%)을 HCl 염으로서 수득하였고, 이것을 추가 정제없이 사용하였다.









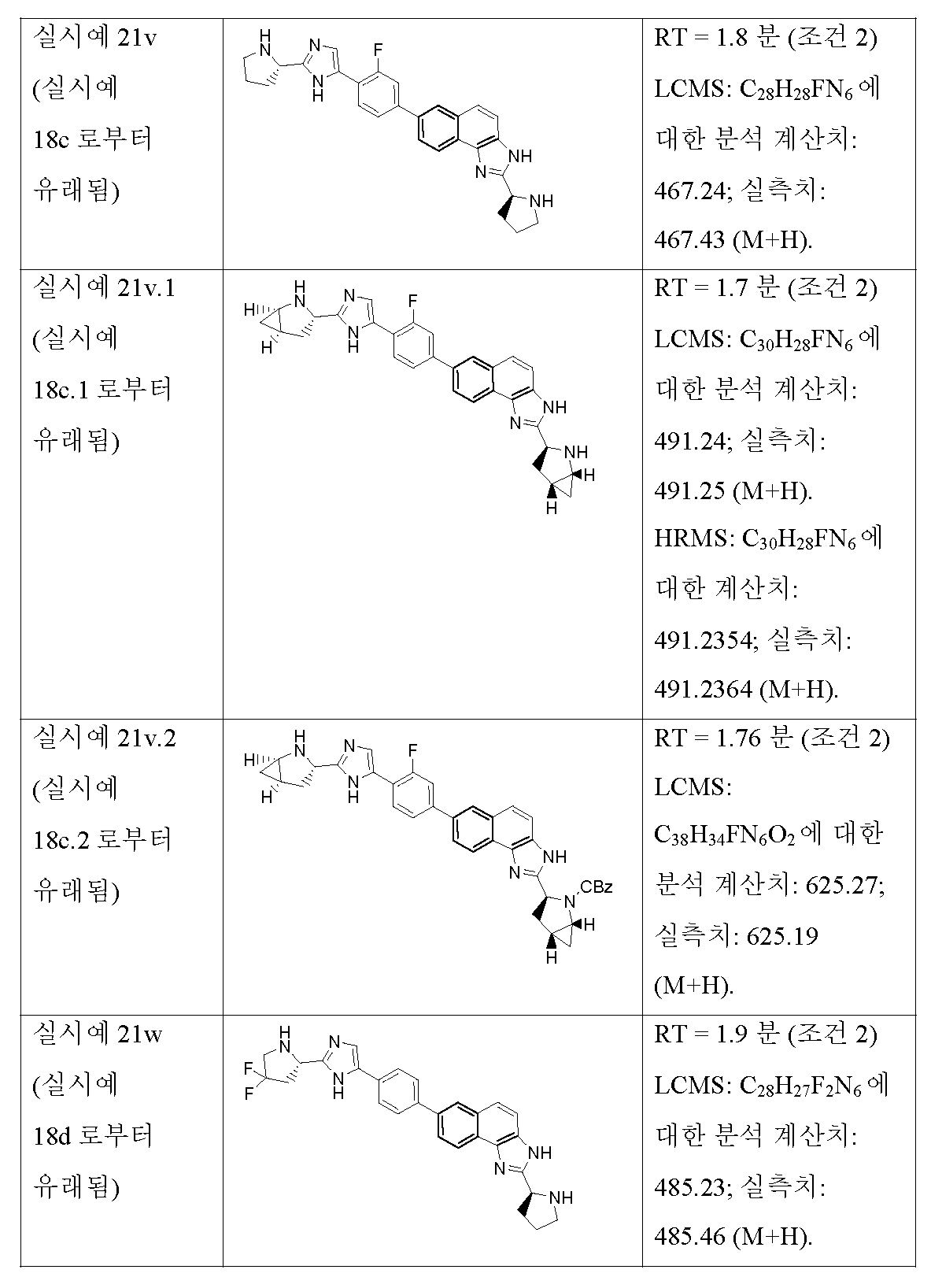

합성 경로 14

HATU (88 mg, 0.33 mmol)를 첨가하고, DMF (3.5 mL) 중 실시예 21 (87.5 mg, 0.145 mmol), Cap-51: N-메톡시카르보닐-L-발린 (63.3 mg, 0.36 mmol) 및 휴닉 염기 (0.23 mL, 1.25 mmol)의 용액을 빠르게 교반하였다. 반응 혼합물을 16 시간 교반한 후에, CH3OH (1 부피)로 희석하고, 반-분취용 HPLC (각각 2 mL로 4회 주입; 다이나맥스(Dynamax) 60A 분취용 C8 컬럼; 30 분에 걸쳐 25%→100% B; 유속 = 20 ml/분; 파장 = 220 nm; 용매 A = 0.1% TFA + 10% 메탄올/90% H2O; 용매 B = 0.1% TFA + 90% 메탄올/10% H2O)에 직접 적용하여, 실시예 22 (TFA 염)를 수득하였다.






























합성 경로 15 (비대칭 Cap 유사체)

CH3OH (5 mL) 및 물 (0.3 mL) 중 실시예 22m (149 mg, 0.197 mmol) 및 K2CO3 (27 mg)의 용액을 질소로 플러슁하고, 10% 팔라듐/탄소 (30 mg)를 첨가하였다. 반응물을 수소로 플러슁하고, 6 시간 교반하고, 규조토 (셀라이트®)를 통해 여과하고, 여과물을 농축시키고, 고진공하에 건조시켜, 실시예 23, 122 mg (100%)을 수득하였다.


합성 경로 16 (비대칭 Cap 유사체)

1 당량의 HATU 및 4 당량의 휴닉 염기를 사용하는 것만을 제외하고는, 실시예 22를 제조하기 위한 합성 경로 14에 기재된 바와 같이 실시예 23에 캡핑을 수행하여, 실시예 24를 제조하였다:





생물학적 활성
본 개시내용에서는 HCV 레플리콘(Replicon) 검정이 이용되었고, 이것은 본원과 공동 소유의 PCT/US2006/022197 및 문헌 [O'Boyle et. al. Antimicrob Agents Chemother. 2005 Apr;49(4):1346-53]에 기재된 바와 같이 준비하여 수행하고 검증하였다.
HCV 1b-377-neo 레플리콘 세포를 사용하여, 본원에 기재된 화합물류 및 NS5A에 Y2065H 돌연변이를 함유하여 화합물 A에 대해 내성인 세포 (출원 PCT/US2006/022197에 기재됨)를 시험하였다. 시험된 화합물은 돌연변이를 함유하는 세포에 대한 억제 활성이 야생형 세포에 대한 것보다 10배 넘게 덜한 것으로 측정되었고, 이는 상기 2종의 화합물류 사이의 관련 작용 메카니즘을 나타낸다. 따라서, 본 개시내용의 화합물은 HCV NS5A 단백질의 기능을 억제하는데 효과적일 수 있고, 출원 PCT/US2006/022197 및 공동 소유의 WO/04014852에 이전에 기재된 바와 같이 조합시에 효과적인 것으로 이해된다. 추가로, 본 개시내용의 화합물은 HCV 1b 유전자형에 대하여 효과적일 수 있다. 또한, 본 개시내용의 화합물이 HCV의 다중 유전자형을 억제할 수 있다는 것을 이해해야 한다. 표 2는 HCV 1b 유전자형에 대한 본 개시내용의 대표적인 화합물의 EC50 값을 보여준다. 한 실시양태에서, 본 개시내용의 화합물은 1a, 1b, 2a, 2b, 3a, 4a 및 5a 유전자형에 대해 활성이다. HCV 1b에 대한 EC50 범위는 다음과 같다: A = >100 nM; B = 1 내지 99 nM; C = 101 내지 999 pM; 및 D = 1 내지 100 pM.
본 개시내용의 화합물은 NS5A 억제 이외의 또는 NS5A 억제와 다른 메카니즘으로 HCV를 억제할 수 있다. 한 실시양태에서 본 개시내용의 화합물은 HCV 레플리콘을 억제하고, 또다른 실시양태에서는 본 개시내용의 화합물이 NS5A를 억제한다.




본 개시내용이 전술한 예시적인 실시예로 제한되지 않으며, 본 발명의 본질적인 사상에서 벗어나지 않으면서 다른 특정 형태로 구현될 수 있음이 당업자에게 명백할 것이다. 따라서, 실시예는 모든 면에서 예시적이고 비-제한적인 것으로 고려되며, 전술한 실시예가 아니라 첨부된 청구의 범위를 참조해야 하며, 이에 따라 청구범위의 등가물의 의미 및 범위 내에 속하는 모든 변화가 본원에 포함되는 것으로 의도되는 것이 바람직하다.
본 개시내용의 화합물은 NS5A 억제 이외의 메카니즘 또는 NS5A 억제와 다른 메카니즘으로 HCV를 억제할 수 있다. 한 실시양태에서 본 개시내용의 화합물은 HCV 레플리콘을 억제하고, 또다른 실시양태에서는 본 개시내용의 화합물이 NS5A를 억제한다. 본 개시내용의 화합물은 다중 유전자형의 HCV를 억제할 수 있다.
Claims (22)
- 하기 화학식 I의 화합물 또는 그의 제약상 허용되는 염:
<화학식 I>

상기 식 중,
u 및 u'는 0이고;
D 및 D'는 각각 독립적으로 NR5로부터 선택되고; 여기서 각각의 R5는 독립적으로 수소, 히드록시, 및 트리-C1-C6 알킬실릴-C1-C6 알콕시-C1-C6 알킬로부터 선택되고;
각각의 R1 및 R1'는 독립적으로 C1-C6 알콕시, C1-C6 알콕시-C1-C6 알킬, C1-C6 알콕시카르보닐, C1-C6 알킬, C6-C12 아릴-C1-C6 알콕시카르보닐, 카르복시, 포르밀, 할로, 할로-C1-C6 알킬, 히드록시, 히드록시-C1-C6 알킬, -NRaRb, (NRaRb)-C1-C6 알킬 및 (NRaRb)카르보닐로부터 선택되고;
R2는 수소, C1-C6 알콕시카르보닐, C1-C6 알킬, C6-C12 아릴-C1-C6 알콕시카르보닐, 카르복시, 할로-C1-C6 알킬 및 (NRaRb)카르보닐로부터 선택되고;
R3은 수소, C1-C6 알콕시, C1-C6 알콕시-C1-C6 알킬, C1-C6 알콕시카르보닐, C1-C6 알킬, C6-C12 아릴-C1-C6 알콕시카르보닐, 카르복시, 포르밀, 할로, 할로-C1-C6 알킬, 히드록시, 히드록시-C1-C6 알킬, -NRaRb, (NRaRb)-C1-C6 알킬 및 (NRaRb)카르보닐로부터 선택되거나; 또는
R2 및 R3은, 이들이 부착되어 있는 탄소 원자와 함께, 질소, 산소 및 황으로부터 독립적으로 선택되는 1 또는 2개의 헤테로원자를 임의로 함유하는 5원 내지 8원 방향족 또는 비-방향족 고리를 형성하고; 여기서 상기 5원 내지 8원 고리는 C1-C6 알콕시, C1-C6 알콕시-C1-C6 알킬, C1-C6 알콕시카르보닐, C1-C6 알킬, C1-C6 알킬술포닐, C6-C12 아릴, C6-C12 아릴-C1-C6 알킬, C6-C12 아릴술포닐, 카르복시, 포르밀, 할로, 할로-C1-C6 알콕시, 할로-C1-C6 알킬, 히드록시, 히드록시-C1-C6 알킬, -NRaRb, (NRaRb)C1-C6 알킬, (NRaRb)카르보닐, 및 옥소로부터 독립적으로 선택되는 1, 2 또는 3개의 치환기로 임의로 치환되고;
R2' 및 R3'는, 이들이 부착되어 있는 탄소 원자와 함께, 질소, 산소 및 황으로부터 독립적으로 선택되는 1 또는 2개의 헤테로원자를 임의로 함유하는 5원 내지 8원 방향족 또는 비-방향족 고리를 형성하고; 여기서 상기 5원 내지 8원 고리는 C1-C6 알콕시, C1-C6 알콕시-C1-C6 알킬, C1-C6 알콕시카르보닐, C1-C6 알킬, C1-C6 알킬술포닐, C6-C12 아릴, C6-C12 아릴-C1-C6 알킬, C6-C12 아릴술포닐, 카르복시, 포르밀, 할로, 할로-C1-C6 알콕시, 할로-C1-C6 알킬, 히드록시, 히드록시-C1-C6 알킬, -NRaRb, (NRaRb)-C1-C6 알킬, (NRaRb)카르보닐, 및 옥소로부터 독립적으로 선택되는 1, 2 또는 3개의 치환기로 임의로 치환되고;
R4 및 R4'는 각각 독립적으로
으로부터 선택되고;
여기서
각각의 m은 독립적으로 0, 1 또는 2이고;
각각의 s는 독립적으로 0, 1, 2, 3 또는 4이고;
각각의 X는 독립적으로 O, S, S(O), SO2, CH2, CHR6 및 C(R6)2로부터 선택되되; 단, m이 0인 경우, X는 CH2, CHR6 및 C(R6)2로부터 선택되고;
각각의 R6은 독립적으로 C1-C6 알콕시, C1-C6 알킬, C6-C12 아릴, 할로, 할로-C1-C6 알킬, 히드록시 및 -NRaRb로부터 선택되고, 여기서 상기 C1-C6 알킬은 인접한 탄소 원자와 함께, 융합된 3원 내지 6원 고리를 임의로 형성할 수 있고, 이때 상기 3원 내지 6원 고리는 1 또는 2개의 C1-C6 알킬기로 임의로 치환되고;
각각의 R7은 독립적으로 수소 및 R11-C(O)- 및 R11-C(S)-로부터 선택되고;
R8은 수소 및 C1-C6 알킬로부터 선택되고;
R9 및 R10은 각각 독립적으로 수소, C2-C6 알케닐, C1-C6 알콕시-C1-C6 알킬, C1-C6 알킬, 할로-C1-C6 알킬 및 (NRaRb)-C1-C6 알킬로부터 선택되거나; 또는
R9 및 R10은, 이들이 부착되어 있는 탄소 원자와 함께, NRz, O 및 S로부터 선택되는 1 또는 2개의 헤테로원자를 임의로 함유하는 5원 또는 6원 포화 고리를 형성하고; 여기서 Rz는 수소 및 C1-C6 알킬로부터 선택되고;
각각의 R11은 독립적으로 C1-C6 알콕시, C1-C6 알콕시-C1-C6 알킬, C1-C6 알콕시카르보닐, C1-C6 알콕시카르보닐-C1-C6 알킬, C1-C6 알킬, C1-C6 알킬카르보닐-C1-C6 알킬, C6-C12 아릴, C6-C12 아릴-C2-C6 알케닐, C6-C12 아릴-C1-C6 알콕시, C6-C12 아릴-C1-C6 알킬, C6-C12 아릴옥시-C1-C6 알킬, C3-C7 시클로알킬, (C3-C7 시클로알킬)-C2-C6 알케닐, (C3-C7 시클로알킬)-C1-C6 알킬, C3-C7 시클로알킬옥시-C1-C6 알킬, 할로-C1-C6 알킬, 헤테로시클릴, 헤테로시클릴-C2-C6 알케닐, 헤테로시클릴-C1-C6 알콕시, 헤테로시클릴-C1-C6 알킬, 헤테로시클릴옥시-C1-C6 알킬, 히드록시-C1-C6 알킬, -NRcRd, (NRcRd)-C2-C6 알케닐, (NRcRd)-C1-C6 알킬 및 (NRcRd)카르보닐로부터 선택되고;
여기서, Ra 및 Rb는 수소, C2-C6 알케닐 및 C1-C6 알킬로부터 독립적으로 선택되고;
Rc 및 Rd는 수소, C2-C6 알케닐옥시카르보닐, C1-C6 알콕시-C1-C6 알킬카르보닐, C1-C6 알콕시카르보닐, C1-C6 알킬, C1-C6 알킬카르보닐, C1-C6 알킬술포닐, C6-C12 아릴, C6-C12 아릴-C1-C6 알콕시카르보닐, C6-C12 아릴-C1-C6 알킬, C6-C12 아릴-C1-C6 알킬카르보닐, C6-C12 아릴카르보닐, C6-C12 아릴옥시카르보닐, C6-C12 아릴술포닐, C3-C7 시클로알킬, C3-C7 시클로알킬술포닐, 포르밀, 할로-C1-C6 알콕시카르보닐, 헤테로시클릴, 헤테로시클릴-C1-C6 알콕시카르보닐, 헤테로시클릴-C1-C6 알킬, 헤테로시클릴-C1-C6 알킬카르보닐, 헤테로시클릴카르보닐, 헤테로시클릴옥시카르보닐, 히드록시-C1-C6 알킬카르보닐, (NReRf)-C1-C6 알킬, (NReRf)-C1-C6 알킬카르보닐, (NReRf)카르보닐, (NReRf)술포닐, -C(NCN)OR' 및 -C(NCN)NRxRy로부터 독립적으로 선택되며, 여기서 R'는 C1-C6 알킬 및 비치환된 페닐로부터 선택되고, C6-C12 아릴-C1-C6 알킬, C6-C12 아릴-C1-C6 알킬카르보닐, 헤테로시클릴-C1-C6 알킬 및 헤테로시클릴-C1-C6 알킬카르보닐의 C1-C6 알킬 부분은 1개의 -NReRf기로 임의로 추가 치환되고; C6-C12 아릴, 및 C6-C12 아릴-C1-C6 알콕시카르보닐, C6-C12 아릴-C1-C6 알킬, C6-C12 아릴-C1-C6 알킬카르보닐, C6-C12 아릴카르보닐, C6-C12 아릴옥시카르보닐 및 C6-C12 아릴술포닐의 C6-C12 아릴 부분, 헤테로시클릴, 및 헤테로시클릴-C1-C6 알콕시카르보닐, 헤테로시클릴-C1-C6 알킬, 헤테로시클릴-C1-C6 알킬카르보닐, 헤테로시클릴카르보닐 및 헤테로시클릴옥시카르보닐의 헤테로시클릴 부분은 C1-C6 알콕시, C1-C6 알킬, 시아노, 할로, 할로-C1-C6 알콕시, 할로-C1-C6 알킬 및 니트로로부터 독립적으로 선택되는 1개, 2개 또는 3개의 치환기로 임의로 추가 치환되고;
Re 및 Rf는 수소, C1-C6 알킬, 비치환된 C6-C12 아릴, 비치환된 C6-C12 아릴-C1-C6 알킬, 비치환된 C3-C7 시클로알킬, 비치환된 (C3-C7 시클로알킬)-C1-C6 알킬, 비치환된 헤테로시클릴, 비치환된 헤테로시클릴-C1-C6 알킬, (NRxRy)-C1-C6 알킬 및 (NRxRy)카르보닐로부터 독립적으로 선택되고;
Rx 및 Ry는 수소, C1-C6 알콕시카르보닐, C1-C6 알킬, C1-C6 알킬카르보닐, 비치환된 C6-C12 아릴, 비치환된 C6-C12 아릴-C1-C6 알콕시카르보닐, 비치환된 C6-C12 아릴-C1-C6 알킬, 비치환된 C3-C7 시클로알킬, 비치환된 헤테로시클릴 및 (NRx'Ry')카르보닐로부터 독립적으로 선택되며, 여기서 Rx' 및 Ry'는 수소 및 C1-C6 알킬로부터 독립적으로 선택되고;
여기서 헤테로시클릴은 질소, 산소 및 황으로부터 독립적으로 선택되는 1개, 2개, 3개 또는 4개의 헤테로원자를 함유하는 4원, 5원, 6원 또는 7원의 고리이다. - 삭제
- 제1항에 있어서, 각각의 R5가 독립적으로 수소 및 히드록시로부터 선택되는 화합물 또는 그의 제약상 허용되는 염.
- 삭제
- 제1항에 있어서, R2가 수소 및 할로-C1-C6 알킬로부터 선택되는 화합물 또는 그의 제약상 허용되는 염.
- 제1항에 있어서, R3이 수소 및 할로로부터 선택되는 화합물 또는 그의 제약상 허용되는 염.
- 제1항에 있어서, R2 및 R3이, 이들이 부착되어 있는 탄소 원자와 함께, 6원 또는 7원 카르보시클릭 고리를 형성하는 화합물 또는 그의 제약상 허용되는 염.
- 제1항에 있어서, R2' 및 R3'가, 이들이 부착되어 있는 탄소 원자와 함께, 산소, 질소 및 황으로부터 선택되는 1개의 헤테로원자를 임의로 함유하는 6원 내지 8원 고리를 형성하고; 여기서 상기 고리가 1 또는 2개의 C1-C6 알킬기로 임의로 치환되는 화합물 또는 그의 제약상 허용되는 염.
- 삭제
-






















로부터 선택되는 화합물 또는 그의 제약상 허용되는 염. - 제1항의 화합물 또는 그의 제약상 허용되는 염, 및 제약상 허용되는 담체를 포함하는, HCV 감염의 치료를 위한 제약 조성물.
- 제11항에 있어서, 항-HCV 활성을 갖는 1 또는 2종의 추가 화합물을 더 포함하는 제약 조성물.
- 제12항에 있어서, 추가 화합물 중 1종 이상이 인터페론 또는 리바비린인 제약 조성물.
- 제13항에 있어서, 인터페론이 인터페론 알파 2B, 페길화된 인터페론 알파, 컨센서스 인터페론, 인터페론 알파 2A 및 림프아구성 인터페론 타우로부터 선택되는 것인 제약 조성물.
- 치료적 유효량의 제1항의 화합물 또는 그의 제약상 허용되는 염을 포함하는, 환자에서의 HCV 감염의 치료를 위한 제약 조성물.
- 제15항에 있어서, 항-HCV 활성을 갖는 1 또는 2종의 추가 화합물과 조합되며 상기 추가 화합물은 제1항의 화합물 또는 그의 제약상 허용되는 염의 투여 전에, 후에 또는 투여와 동시에 투여되는 것인, HCV 감염의 치료를 위한 제약 조성물.
- 제16항에 있어서, 추가 화합물 중 1종 이상이 인터페론 또는 리바비린인 제약 조성물.
- 제17항에 있어서, 인터페론이 인터페론 알파 2B, 페길화된 인터페론 알파, 컨센서스 인터페론, 인터페론 알파 2A 및 림프아구성 인터페론 타우로부터 선택되는 것인 제약 조성물.
- 삭제
- 삭제
- 삭제
- 삭제
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US2827708P | 2008-02-13 | 2008-02-13 | |
| US61/028,277 | 2008-02-13 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| KR20100124290A KR20100124290A (ko) | 2010-11-26 |
| KR101624365B1 true KR101624365B1 (ko) | 2016-05-25 |
Family
ID=40581591
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020107020306A KR101624365B1 (ko) | 2008-02-13 | 2009-02-06 | C형 간염 바이러스 억제제 |
Country Status (28)
| Country | Link |
|---|---|
| US (1) | US8147818B2 (ko) |
| EP (1) | EP2245027B1 (ko) |
| JP (1) | JP5455933B2 (ko) |
| KR (1) | KR101624365B1 (ko) |
| CN (1) | CN102143959B (ko) |
| AR (1) | AR070366A1 (ko) |
| AT (1) | ATE555109T1 (ko) |
| AU (1) | AU2009214997B2 (ko) |
| BR (1) | BRPI0908240A2 (ko) |
| CA (1) | CA2715559A1 (ko) |
| CL (1) | CL2009000324A1 (ko) |
| CO (1) | CO6290682A2 (ko) |
| CY (1) | CY1112999T1 (ko) |
| DK (1) | DK2245027T3 (ko) |
| EA (1) | EA018313B1 (ko) |
| ES (1) | ES2384911T3 (ko) |
| HK (1) | HK1143812A1 (ko) |
| HR (1) | HRP20120523T1 (ko) |
| IL (1) | IL207459A (ko) |
| MX (1) | MX2010008749A (ko) |
| NZ (1) | NZ587322A (ko) |
| PE (1) | PE20091381A1 (ko) |
| PL (1) | PL2245027T3 (ko) |
| PT (1) | PT2245027E (ko) |
| SI (1) | SI2245027T1 (ko) |
| TW (1) | TWI458482B (ko) |
| WO (1) | WO2009102633A1 (ko) |
| ZA (1) | ZA201005484B (ko) |
Families Citing this family (109)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8303944B2 (en) * | 2006-08-11 | 2012-11-06 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7906655B2 (en) | 2008-08-07 | 2011-03-15 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8383094B2 (en) | 2008-10-01 | 2013-02-26 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| CA2740195A1 (en) | 2008-12-23 | 2010-07-01 | Abbott Laboratories | Anti-viral compounds |
| CA2740193A1 (en) | 2008-12-23 | 2010-07-01 | Abbott Laboratories | Anti-viral compounds |
| US8242156B2 (en) * | 2009-02-17 | 2012-08-14 | Enanta Pharmaceuticals, Inc. | Linked dibenzimidazole derivatives |
| TWI438200B (zh) | 2009-02-17 | 2014-05-21 | 必治妥美雅史谷比公司 | C型肝炎病毒抑制劑 |
| US8188132B2 (en) | 2009-02-17 | 2012-05-29 | Enanta Pharmaceuticals, Inc. | Linked dibenzimidazole derivatives |
| US8394968B2 (en) | 2009-02-17 | 2013-03-12 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| UY32462A (es) * | 2009-02-23 | 2010-09-30 | Arrow Therapeutics Ltd | Derivados de bifenilo novedosos para el tratamiento de infección por virus de hepatitis c 644 |
| US8673954B2 (en) | 2009-02-27 | 2014-03-18 | Enanta Pharmaceuticals, Inc. | Benzimidazole derivatives |
| NZ595280A (en) | 2009-02-27 | 2013-11-29 | Enanta Pharm Inc | Hepatitis c virus inhibitors |
| US9765087B2 (en) | 2009-02-27 | 2017-09-19 | Enanta Pharmaceuticals, Inc. | Benzimidazole derivatives |
| US8101643B2 (en) * | 2009-02-27 | 2012-01-24 | Enanta Pharmaceuticals, Inc. | Benzimidazole derivatives |
| US8507522B2 (en) | 2009-03-06 | 2013-08-13 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8796466B2 (en) * | 2009-03-30 | 2014-08-05 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| TW201038559A (en) | 2009-04-09 | 2010-11-01 | Bristol Myers Squibb Co | Hepatitis C virus inhibitors |
| US8143414B2 (en) | 2009-04-13 | 2012-03-27 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| JP5734956B2 (ja) * | 2009-04-15 | 2015-06-17 | アッヴィ・インコーポレイテッド | 抗ウィルス化合物 |
| EP2430015B1 (en) | 2009-05-12 | 2015-06-17 | Merck Sharp & Dohme Corp. | Fused tricyclic compounds useful for the treatment of viral diseases |
| US8088368B2 (en) | 2009-05-13 | 2012-01-03 | Gilead Sciences, Inc. | Antiviral compounds |
| US8211928B2 (en) | 2009-05-29 | 2012-07-03 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8138215B2 (en) | 2009-05-29 | 2012-03-20 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2010138790A1 (en) | 2009-05-29 | 2010-12-02 | Schering Corporation | Antiviral compounds composed of three aligned aryl moieties to treat diseases such as hepatitis c |
| EP2435424B1 (en) | 2009-05-29 | 2015-01-21 | Merck Sharp & Dohme Corp. | Antiviral compounds composed of three linked aryl moieties to treat diseases such as hepatitis c |
| US8716454B2 (en) | 2009-06-11 | 2014-05-06 | Abbvie Inc. | Solid compositions |
| US9394279B2 (en) | 2009-06-11 | 2016-07-19 | Abbvie Inc. | Anti-viral compounds |
| US8937150B2 (en) | 2009-06-11 | 2015-01-20 | Abbvie Inc. | Anti-viral compounds |
| RS53856B1 (en) * | 2009-06-11 | 2015-08-31 | Abbvie Bahamas Ltd. | HETEROCYCLIC COMPOUNDS AS HEPATITIS C VIRUS INHIBITORS |
| US8221737B2 (en) | 2009-06-16 | 2012-07-17 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8609648B2 (en) | 2009-07-02 | 2013-12-17 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8354419B2 (en) | 2009-07-16 | 2013-01-15 | Vertex Pharmaceuticals Incorporated | Benzimidazole analogues for the treatment or prevention of flavivirus infections |
| CA2771327A1 (en) | 2009-09-04 | 2011-03-10 | Glaxosmithkline Llc | Chemical compounds |
| US8759332B2 (en) * | 2009-09-11 | 2014-06-24 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| WO2011031904A1 (en) * | 2009-09-11 | 2011-03-17 | Enanta Pharmaceuticals, Inc | Hepatitis c virus inhibitors |
| US8927709B2 (en) * | 2009-09-11 | 2015-01-06 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8703938B2 (en) * | 2009-09-11 | 2014-04-22 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8815928B2 (en) | 2009-09-11 | 2014-08-26 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| WO2011031934A1 (en) * | 2009-09-11 | 2011-03-17 | Enanta Pharmaceuticals, Inc. | Hepatitis c virus inhibitors |
| US8822700B2 (en) * | 2009-09-11 | 2014-09-02 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US20110269956A1 (en) | 2009-11-11 | 2011-11-03 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US20110274648A1 (en) | 2009-11-11 | 2011-11-10 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US20110281910A1 (en) | 2009-11-12 | 2011-11-17 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| JP2013512246A (ja) * | 2009-11-25 | 2013-04-11 | メルク・シャープ・アンド・ドーム・コーポレーション | ウイルス疾患治療に有用な縮合型三環式化合物およびその誘導体 |
| US20110137633A1 (en) * | 2009-12-03 | 2011-06-09 | Abbott Laboratories | Anti-viral compounds and methods of identifying the same |
| EP2512480A4 (en) | 2009-12-14 | 2013-05-15 | Enanta Pharm Inc | HEPATITIS C-VIRUS HEMMER |
| US8377980B2 (en) | 2009-12-16 | 2013-02-19 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| TW201136942A (en) * | 2009-12-18 | 2011-11-01 | Idenix Pharmaceuticals Inc | 5,5-fused arylene or heteroarylene hepatitis C virus inhibitors |
| CN104530079B (zh) * | 2009-12-18 | 2017-10-20 | 北京凯因科技股份有限公司 | C型肝炎病毒复制的新型抑制剂 |
| US20130156731A1 (en) * | 2009-12-22 | 2013-06-20 | Kevin X. Chen | Fused tricyclic compounds and methods of use thereof for the treatment of viral diseas |
| US8362020B2 (en) | 2009-12-30 | 2013-01-29 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8933110B2 (en) | 2010-01-25 | 2015-01-13 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| PE20130244A1 (es) | 2010-01-25 | 2013-03-10 | Enanta Pharm Inc | Inhibidores del virus de la hepatitis c |
| US8178531B2 (en) * | 2010-02-23 | 2012-05-15 | Enanta Pharmaceuticals, Inc. | Antiviral agents |
| US8623814B2 (en) * | 2010-02-23 | 2014-01-07 | Enanta Pharmaceuticals, Inc. | Antiviral agents |
| EA201201235A1 (ru) * | 2010-03-04 | 2013-04-30 | Энанта Фармасьютиклз, Инк. | Комбинации фармацевтических агентов в качестве ингибиторов репликации hcv |
| JP2013522202A (ja) * | 2010-03-09 | 2013-06-13 | メルク・シャープ・エンド・ドーム・コーポレイション | 縮合三環式シリル化合物およびウイルス疾患の治療のためのその使用方法 |
| CN102869657A (zh) | 2010-03-24 | 2013-01-09 | 沃泰克斯药物股份有限公司 | 用于治疗或预防黄病毒感染的类似物 |
| WO2011127350A1 (en) | 2010-04-09 | 2011-10-13 | Enanta Pharmaceuticals, Inc. | Hepatitis c virus inhibitors |
| EP2575819A4 (en) | 2010-06-04 | 2013-11-27 | Enanta Pharm Inc | INHIBITORS OF HEPATITIS C VIRUS |
| NZ605440A (en) | 2010-06-10 | 2014-05-30 | Abbvie Bahamas Ltd | Solid compositions comprising an hcv inhibitor |
| EP2585448A1 (en) | 2010-06-28 | 2013-05-01 | Vertex Pharmaceuticals Incorporated | Compounds and methods for the treatment or prevention of flavivirus infections |
| CA2803248A1 (en) | 2010-06-28 | 2012-01-12 | Vertex Pharmaceuticals Incorporated | Compounds and methods for the treatment or prevention of flavivirus infections |
| JP5596861B2 (ja) * | 2010-08-04 | 2014-09-24 | ブリストル−マイヤーズ スクイブ カンパニー | C型肝炎ウイルス阻害剤 |
| EP2603080A4 (en) | 2010-08-12 | 2014-01-22 | Enanta Pharm Inc | HEPATITIS C-VIRUS HEMMER |
| US20120195857A1 (en) | 2010-08-12 | 2012-08-02 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| AU2011292040A1 (en) | 2010-08-17 | 2013-03-07 | Vertex Pharmaceuticals Incorporated | Compounds and methods for the treatment or prevention of Flaviviridae viral infections |
| EP2616461A4 (en) * | 2010-08-26 | 2014-03-26 | Rfs Pharma Llc | POWERFUL AND SELECTIVE INHIBITORS OF HEPATITIS C VIRUS |
| CA2815537A1 (en) | 2010-10-26 | 2012-05-03 | Presidio Pharmaceuticals, Inc. | Inhibitors of hepatitis c virus |
| SG190785A1 (en) * | 2010-11-17 | 2013-07-31 | Gilead Sciences Inc | Antiviral compounds |
| US9173887B2 (en) | 2010-12-22 | 2015-11-03 | Abbvie Inc. | Hepatitis C inhibitors and uses thereof |
| US8552047B2 (en) | 2011-02-07 | 2013-10-08 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US20120252721A1 (en) | 2011-03-31 | 2012-10-04 | Idenix Pharmaceuticals, Inc. | Methods for treating drug-resistant hepatitis c virus infection with a 5,5-fused arylene or heteroarylene hepatitis c virus inhibitor |
| US9546160B2 (en) * | 2011-05-12 | 2017-01-17 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US10201584B1 (en) | 2011-05-17 | 2019-02-12 | Abbvie Inc. | Compositions and methods for treating HCV |
| EP2709455A4 (en) | 2011-05-18 | 2014-11-05 | Enanta Pharm Inc | PROCESSES FOR THE PREPARATION OF 5-AZASPIRO [2.4] HEPTANE-6-CARBOXYLIC ACID AND ITS DERIVATIVES |
| WO2012175581A1 (en) | 2011-06-24 | 2012-12-27 | F. Hoffmann-La Roche Ag | Antiviral compounds |
| WO2013016492A1 (en) | 2011-07-26 | 2013-01-31 | Vertex Pharmaceuticals Incorporated | Thiophene compounds |
| WO2013016499A1 (en) | 2011-07-26 | 2013-01-31 | Vertex Pharmaceuticals Incorporated | Methods for preparation of thiophene compounds |
| WO2013021344A1 (en) | 2011-08-08 | 2013-02-14 | Lupin Limited | Imidazole derivatives as antiviral agents |
| SG10201606446XA (en) | 2011-08-08 | 2016-09-29 | Lupin Ltd | Antiviral compounds with a fused tricyclic ring |
| WO2013030750A1 (en) | 2011-09-01 | 2013-03-07 | Lupin Limited | Antiviral compounds |
| HUE036588T2 (hu) | 2011-09-16 | 2018-07-30 | Gilead Pharmasset Llc | Eljárások HCV kezelésére |
| MD4521C1 (ro) | 2011-11-16 | 2018-05-31 | Gilead Pharmasset Llc. | Compuşi antivirali pe bază de imidazolil-dihidroizocromeno-naftoimidazoli condensaţi |
| US9034832B2 (en) | 2011-12-29 | 2015-05-19 | Abbvie Inc. | Solid compositions |
| US9326973B2 (en) | 2012-01-13 | 2016-05-03 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| NZ628515A (en) | 2012-02-10 | 2016-06-24 | Lupin Ltd | Antiviral compounds with a dibenzooxaheterocycle moiety |
| NZ630805A (en) | 2012-03-22 | 2016-01-29 | Alios Biopharma Inc | Pharmaceutical combinations comprising a thionucleotide analog |
| SI2850075T1 (sl) | 2012-04-25 | 2017-06-30 | Theravance Biopharma R&D Ip, Llc | Piperazin-piperidinske spojine kot inhibitorji virusa hepatitisa C |
| US9066944B2 (en) | 2012-04-25 | 2015-06-30 | Theravance Biopharma R&D Ip, Llc | Hepatitis C virus inhibitors |
| US20130309196A1 (en) * | 2012-05-16 | 2013-11-21 | Gilead Sciences, Inc. | Antiviral compounds |
| MY172166A (en) | 2013-01-31 | 2019-11-15 | Gilead Pharmasset Llc | Combination formulation of two antiviral compounds |
| US20150065439A1 (en) | 2013-02-28 | 2015-03-05 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions |
| US11484534B2 (en) | 2013-03-14 | 2022-11-01 | Abbvie Inc. | Methods for treating HCV |
| US20150023913A1 (en) | 2013-07-02 | 2015-01-22 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US9717712B2 (en) | 2013-07-02 | 2017-08-01 | Bristol-Myers Squibb Company | Combinations comprising tricyclohexadecahexaene derivatives for use in the treatment of hepatitis C virus |
| WO2015009744A1 (en) | 2013-07-17 | 2015-01-22 | Bristol-Myers Squibb Company | Combinations comprising biphenyl derivatives for use in the treatment of hcv |
| EP4005560A1 (en) | 2013-08-27 | 2022-06-01 | Gilead Pharmasset LLC | Combination formulation of two antiviral compounds |
| WO2015042375A1 (en) | 2013-09-20 | 2015-03-26 | Idenix Pharmaceuticals, Inc. | Hepatitis c virus inhibitors |
| WO2015103490A1 (en) | 2014-01-03 | 2015-07-09 | Abbvie, Inc. | Solid antiviral dosage forms |
| WO2015184644A1 (zh) * | 2014-06-06 | 2015-12-10 | 爱博新药研发(上海)有限公司 | 抑制丙肝病毒的化合物、药物组合物及其应用 |
| TWI721947B (zh) | 2014-06-11 | 2021-03-21 | 美商基利法瑪席特有限責任公司 | 抗病毒化合物的固態形式 |
| CN105968101B (zh) * | 2015-03-12 | 2019-03-01 | 广东东阳光药业有限公司 | 作为丙型肝炎抑制剂的化合物及其在药物中的应用 |
| US10617675B2 (en) | 2015-08-06 | 2020-04-14 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2019160882A1 (en) | 2018-02-13 | 2019-08-22 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| US10899735B2 (en) | 2018-04-19 | 2021-01-26 | Gilead Sciences, Inc. | PD-1/PD-L1 inhibitors |
| ES2962674T3 (es) | 2018-07-13 | 2024-03-20 | Gilead Sciences Inc | Inhibidores PD-1/PD-L1 |
| WO2020086556A1 (en) | 2018-10-24 | 2020-04-30 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| KR20210098504A (ko) | 2018-12-04 | 2021-08-10 | 브리스톨-마이어스 스큅 컴퍼니 | 다중 동위원소체 반응 모니터링에 의한 샘플내 보정 곡선을 사용한 분석 방법 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006020082A1 (en) | 2004-08-09 | 2006-02-23 | Bristol-Myers Squibb Company | Inhibitors of hcv replication |
| WO2006133326A1 (en) | 2005-06-06 | 2006-12-14 | Bristol-Myers Squibb Company | Inhibitors of hcv replication |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5654451B1 (en) * | 1993-01-14 | 2000-02-22 | Magainin Pharma | Amino acids and peptides having modified c-terminals and modified n-terminals |
| CA2153987A1 (en) | 1993-01-14 | 1994-07-21 | U. Prasad Kari | Amino acids and peptides having modified terminals |
| GB2438802A (en) | 2005-02-28 | 2007-12-05 | Univ Rockefeller | Structure of the hepatitis C virus NS5A protein |
| NZ566276A (en) | 2005-09-16 | 2011-03-31 | Arrow Therapeutics Ltd | Biphenyl derivatives and their use in treating hepatitis C |
| WO2007058384A1 (en) | 2005-11-17 | 2007-05-24 | Osaka University | Method of suppressing replication of hepatitis c virus, inhibitor of replication of the virus and method of screening for the same |
| SG133452A1 (en) | 2005-12-30 | 2007-07-30 | Novartis Ag | Peptide deformylase inhibitors for treatment of mycobacterial and other parasitic diseases |
| MX2008014990A (es) | 2006-05-30 | 2008-12-09 | Arrow Therapeutics Ltd | Derivados de bifenilo y su uso en el tratamiento de hepatitis c. |
| US8303944B2 (en) * | 2006-08-11 | 2012-11-06 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7745636B2 (en) * | 2006-08-11 | 2010-06-29 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7659270B2 (en) * | 2006-08-11 | 2010-02-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7759495B2 (en) * | 2006-08-11 | 2010-07-20 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8329159B2 (en) | 2006-08-11 | 2012-12-11 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| TWI399380B (zh) | 2006-12-20 | 2013-06-21 | Abbott Lab | 抗病毒化合物 |
| US7741347B2 (en) | 2007-05-17 | 2010-06-22 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8629171B2 (en) | 2007-08-08 | 2014-01-14 | Bristol-Myers Squibb Company | Crystalline form of methyl ((1S)-1-((25)-2-(5-(4'-(2-((25)-1((2S)-2-((methoxycarbonyl)amino)-3-methylbutanoyl)-2-pyrrolidinyl)-1H-imidazol-2-yl)-1-pyrrolidinyl)carbonyl)-2-methylpropyl)carbamate dihydrochloride salt |
| US7728027B2 (en) | 2007-08-08 | 2010-06-01 | Bristol-Myers Squibb Company | Process for synthesizing compounds useful for treating hepatitis C |
-
2009
- 2009-01-23 US US12/358,587 patent/US8147818B2/en active Active
- 2009-02-06 PL PL09709849T patent/PL2245027T3/pl unknown
- 2009-02-06 WO PCT/US2009/033380 patent/WO2009102633A1/en active Application Filing
- 2009-02-06 DK DK09709849.5T patent/DK2245027T3/da active
- 2009-02-06 PT PT09709849T patent/PT2245027E/pt unknown
- 2009-02-06 ES ES09709849T patent/ES2384911T3/es active Active
- 2009-02-06 SI SI200930300T patent/SI2245027T1/sl unknown
- 2009-02-06 BR BRPI0908240A patent/BRPI0908240A2/pt not_active IP Right Cessation
- 2009-02-06 JP JP2010546841A patent/JP5455933B2/ja not_active Expired - Fee Related
- 2009-02-06 AU AU2009214997A patent/AU2009214997B2/en not_active Ceased
- 2009-02-06 NZ NZ587322A patent/NZ587322A/en not_active IP Right Cessation
- 2009-02-06 EA EA201001273A patent/EA018313B1/ru not_active IP Right Cessation
- 2009-02-06 MX MX2010008749A patent/MX2010008749A/es active IP Right Grant
- 2009-02-06 CN CN200980112993.9A patent/CN102143959B/zh not_active Expired - Fee Related
- 2009-02-06 KR KR1020107020306A patent/KR101624365B1/ko not_active IP Right Cessation
- 2009-02-06 EP EP09709849A patent/EP2245027B1/en active Active
- 2009-02-06 AT AT09709849T patent/ATE555109T1/de active
- 2009-02-06 CA CA2715559A patent/CA2715559A1/en not_active Abandoned
- 2009-02-11 PE PE2009000200A patent/PE20091381A1/es not_active Application Discontinuation
- 2009-02-12 AR ARP090100503A patent/AR070366A1/es unknown
- 2009-02-12 CL CL2009000324A patent/CL2009000324A1/es unknown
- 2009-02-13 TW TW098104731A patent/TWI458482B/zh not_active IP Right Cessation
-
2010
- 2010-07-30 ZA ZA2010/05484A patent/ZA201005484B/en unknown
- 2010-08-08 IL IL207459A patent/IL207459A/en not_active IP Right Cessation
- 2010-08-11 CO CO10098657A patent/CO6290682A2/es active IP Right Grant
- 2010-11-08 HK HK10110404.7A patent/HK1143812A1/xx not_active IP Right Cessation
-
2012
- 2012-06-21 HR HRP20120523AT patent/HRP20120523T1/hr unknown
- 2012-07-20 CY CY20121100642T patent/CY1112999T1/el unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006020082A1 (en) | 2004-08-09 | 2006-02-23 | Bristol-Myers Squibb Company | Inhibitors of hcv replication |
| WO2006133326A1 (en) | 2005-06-06 | 2006-12-14 | Bristol-Myers Squibb Company | Inhibitors of hcv replication |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101624365B1 (ko) | C형 간염 바이러스 억제제 | |
| KR101614964B1 (ko) | C형 간염 바이러스 억제제로서 사용하기 위한 형태적으로 제한된 바이페닐 유도체 | |
| JP5611959B2 (ja) | C型肝炎ウイルス阻害剤 | |
| JP5558556B2 (ja) | C型肝炎ウイルス阻害剤 | |
| KR101468765B1 (ko) | C형 간염 바이러스 억제제 | |
| KR101492672B1 (ko) | C형 간염 바이러스 억제제로서의 이미다졸릴 바이페닐 이미다졸 | |
| JP5599792B2 (ja) | C型肝炎ウイルス阻害剤としてのbi−1h−ベンズアミダゾール | |
| JP5612661B2 (ja) | C型肝炎ウイルス阻害剤 | |
| JP5558557B2 (ja) | C型肝炎ウイルス阻害剤 | |
| JP5612612B2 (ja) | C型肝炎ウイルス阻害剤 | |
| KR20100123717A (ko) | C형 간염 바이러스 억제제로서의 헤테로시클릭 유도체 | |
| KR20120107991A (ko) | C형 간염 바이러스 억제제 | |
| JP2010527373A (ja) | C型肝炎ウイルス阻害剤 | |
| KR20140045903A (ko) | C형 간염 바이러스 억제제 | |
| EP2435425A1 (en) | Hepatitis c virus inhibitors | |
| KR20120034603A (ko) | C형 간염 바이러스 억제제 | |
| JP2012528166A (ja) | C型肝炎ウイルス阻害剤 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A201 | Request for examination | ||
| E902 | Notification of reason for refusal | ||
| E701 | Decision to grant or registration of patent right | ||
| GRNT | Written decision to grant | ||
| LAPS | Lapse due to unpaid annual fee |