JP2011052019A - 血管破壊のターゲッティングに使用するための組成物および方法 - Google Patents
血管破壊のターゲッティングに使用するための組成物および方法 Download PDFInfo
- Publication number
- JP2011052019A JP2011052019A JP2010278674A JP2010278674A JP2011052019A JP 2011052019 A JP2011052019 A JP 2011052019A JP 2010278674 A JP2010278674 A JP 2010278674A JP 2010278674 A JP2010278674 A JP 2010278674A JP 2011052019 A JP2011052019 A JP 2011052019A
- Authority
- JP
- Japan
- Prior art keywords
- cytotoxic
- cells
- phosphate
- prodrug
- vascular
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images









Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/661—Phosphorus acids or esters thereof not having P—C bonds, e.g. fosfosal, dichlorvos, malathion or mevinphos
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/661—Phosphorus acids or esters thereof not having P—C bonds, e.g. fosfosal, dichlorvos, malathion or mevinphos
- A61K31/6615—Compounds having two or more esterified phosphorus acid groups, e.g. inositol triphosphate, phytic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/664—Amides of phosphorus acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
Abstract
【解決手段】微細血管ホスファターゼに対する基質特異性を有するリン酸塩プロドラッグ形態へ処方された細胞毒性試薬を充分な量で投薬することによる、腫瘍または悪性でない血管過多を有する温血動物を治療方法であり、それは細胞毒性が低いプロドラッグ形態は細胞毒性が高い脱ホスホリル化された形態へ変換されるので、他の正常な組織よりも微細血管が優先的に破壊される。
【選択図】なし
Description
本出願は、同時継続出願である1999年、2月18日に出願された出願番号60/120,478の米国仮特許出願の優先権の利益を主張する。
本発明は、ヒトを含む温血動物における標的化された血管破壊を達成するための方法および組成物、さらにそのような使用が可能であることを確認するための手法に関するものである。
二つの薬剤モデルが現れており、すなわち一方の薬剤は腫瘍において新しい血管の形成を妨げるものであり(抗血管新生)、また他方は、腫瘍への栄養を制限し、および/または血管内皮細胞の管腔表面の、免疫療法としてのガン薬剤に対する不浸透性を制限する方法によって血管破壊を標的とするものである(New England Journal of Medicine 339:473-474,1998)。
抗血管新生モデルは、基本的に細胞分裂抑制的なアプローチであり、血管新生因子は一般的に腫瘍により生産され、例えば血管内皮増殖因子(VEGF)、および血小板由来増殖因子があり、これらは、新たな血管の成長を妨げるために、例えば天然ポリペプチドであるアンジオスタチンおよびエンドスタチンのような抗血管新生化合物によって、ブロックされる(The Cancer Journal Scientific American 4(4):209-216,1998; Cell 88:277-285,1997)。
他方、血管破壊モデルとは、細胞毒性的アプローチであり、腫瘍血管が、低酸素または直接作用的化学療法による腫瘍細胞の細胞毒性を高めるために、細胞毒性の標的とされるものである。
それらは特徴として、インビトロでの細胞の細胞毒性がnM〜μMの範囲であるIC50を有するが、しばしばそれらは、インビボでは正常な組織上の腫瘍を殺傷するための乏しい特異性を示し、そのような薬剤の例としては、コンブレタスタチン類、タキソール(および他のタキサン類)、ビンブラスチン(および他のビンカアルカロイド)、コルヒチノイド類、ドラスタチン類、ポドフィロトキシン類、ステガナシン類、アンフェチニル類、フラボノイド類、リゾキシン類、クラシン類A、エポチロン類AおよびB、ウェルウィスタチン類、フェンスタチン類、2−ストリルキナゾリン−4(3H)−オン類、スチルベン類、2−アリール−1,8−ナフチリジン−4(1H)−オン類、5,6−ジハイドロインドロ(2,1−a)イソキノリン類、2,3−ベンゾ(b)チオフェン類、2,3−置換ベンゾ(b)フラン類、2,3−置換インドール類および2−メトキシエストラジオール類がある(Journal of Med. Chem. 41(16):3022-3032,1998; Journal Med. Chem.34(8):2579-2588, 1991; Anticancer Drugs 4(1):19-25,1993; Pharm. Res.8(6):766-781,1991; Expermentia 45(2):209-211,1989; Med. Res. Rev. 16:2067,1996;Tetrahedron Lett. 34:1035,1993; Mol. Pharmacol.49:288,1996;J.Med.Chem.41:1688-1695,1998;J.Med.Chem.33:1721,1990;J.Med.Chem.34:2579,1991;J.Md.Chem.40:3049,1997;Med.Chem.40:3525,1997; Bioorg.Med.Chem.Lett.9:1081-1086,1999; International (PCT) Application No.US98/04380; U.S.Provisional Patent Application No.60/154,639) 。
一般にチューブリン結合薬剤は、腫瘍血流で効果を発揮できるが、効果的な投薬量であるとしばしば他の普通の組織に対して毒性を示し、また特に腫瘍に対して毒性を示すわけではない(Br. J. Cancer 74(Suppl.27):586-88,1996)。
この臨床開発課題を克服するために首尾よく実施されるひとつのアプローチとしては、生物適応性のある水溶性プロドラッグがあり、例えばコンブレタスタチンA4およびタキソールのリン酸塩誘導体があり、それは水不溶性形態へもどる代謝変換を許容するものである(Anticancer Drug Des. 13 (3) : 183-191, 1998; U.S. patent No.5,561,122; Bioorganic Med. Chem. Lett. 3:1766, 1993 ; Bioorganic Med. Chem.Lett. 3:1357, 1993) 。
プロドラッグとは前駆体であり、それはインビボで代謝活性を経て活性薬剤となる。
前記リン酸塩誘導体について更に言及すると、その概念は、哺乳動物のアルカリホスファターゼのような非特異的ホスファターゼは、リン酸塩プロドラッグを脱ホスホリル化して、本来の生物学的に活性な形態にすることが可能である、といことである。
この先行技術は、水不溶性薬剤をより多くの最大吸収量、および水可溶性であり生物学的に適応性のある形態へ処方することによる生物学的利用能の状態での治療目的のために、どのようにして温血動物に投薬するのかを教授する(Krogsgaard-Larsen, P. and Bundegaard, H.,eds., A textbook of Drug Design and Drug Development,Harvard Academic Publishers, p. 148, 1991) 。
一般にリン酸塩プロドラッグが、アルカリホスファターゼの基質として利用できるが、それは如何なることであれ血管ターゲッティングと何らかの関係があるということは、このことからは当業者にとって明らかではなかった。
しかしながら、プロドラッグそれ自身が血管ターゲッティングの能力があるという事実の理解および正しい認識がなかったにもかかわらず、コンブレタスタチンA4のリン酸塩プロドラッグに関する報告データは、優先的な血管毒性の原理を開示している。
すなわち先行技術は、A4およびA4でないプロドラッグは二つの形態間での血管毒性の違いはないものと仮定することによって血管毒性の能力がある、ということを教授するものである。
上記の明白でないところは、A4リン酸塩プロドラッグおよび他のタキソールリン酸塩プロドラッグは、細胞毒性チューブリン結合形態へのホスファターゼ変換の作用を受けやすいので、促進されるにもかかわらず、この酵素は微細血管において高められ、従って、それらを細胞毒性に対してターゲッティングする、という事実によって実証される。
発明の目的は、ヒト(ただし、これに限定されない)を含む温血動物における、ガン、カポジ肉腫、および、その他悪性でない血管増殖疾患、例えば黄斑変性、乾癬、および再狭窄、および一般に血管増殖によって特徴付けられる炎症性疾患の治療のための微細血管破壊モデルをターゲッティングするのに有用な組成物および方法を提供することにある。
慣例的にまたは望ましくは、“その他の細胞”とは線維芽細胞、例えば通常のヒト線維芽細胞であればよい。

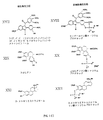
XはO、NH、またはSであり;
YはO、NH、S、O- 、NH- またはS- であり;
ZはOまたはSであり;
R2 およびR3 のそれぞれは、アルキル基、H、1価または2価の陽イオン塩、またはアンモニウム陽イオン塩であり、またR2 およびR3 は、同種または異種であってよく;および
R1 は、Xを含む基である少なくとも一つの基(Ra と表す)を有する化合物を表す構造式R1 −Ra によって定義され、該Ra は、非特異的血管内皮ホスファターゼの基質として利用できるリン酸塩または他の塩を形成することができ、またそれ故に、相対的に非細胞毒性のリン酸塩形態から細胞毒性のR1 −Ra 形態へ変換される。
特別な観点として、それは、すでに予め記載されている、コンブレタスタチン類、タキサン類、ビンカアルカロイド類、コルヒチノイド類、ドラスタチン類、ポドフィロトキシン類、ステガナシン類、アンフェチニル類、フラボノイド類、リゾキシン類、クラシン類A、エポチロン類AおよびB、ウェルウィスタチン類、フェンスタチン類、2−ストリルキナゾリン−4(3H)−オン類、スチルベン類、2−アリール−1,8−ナフチリジン−4(1H)−オン類、5,6−ジハイドロインドロ(2,1−a)イソキノリン類、2,3−ベンゾ(b)チオフェン類、2,3−置換ベンゾ(b)フラン類、2,3−置換インドール類および2−メトキシエストラジオール類のような既知のチューブリン結合薬剤類から選択されればよい。
また更なる特別な意味において、このチューブリンバインダーは、コンブレタスタチン類(コンブレタスタチンA4以外)、コルヒチン、および2−メトキシエストラジオールからなる群より選ばれる化合物であってもよい。
ホスファターゼ(例えば、アルカリ性)が高レベルにあることは、微細血管の通常の生理学の一部分であるが、それは血液凝固機構と共に、アルカリアルカリホスファターゼ活性により生成されるカルシウム沈殿物が、傷回復過程において助力するからである。
本発明は、リン酸塩または他の適当なプロドラッグ構造は、アルカリホスファターゼのようなホスファターゼの基質であるが、それらは微細血管毒性をターゲッティングするのに有用である、という発見を包含するものである。
この目的に適するホスファターゼの典型例には、リン酸化分子が細胞質膜を通過して吸収することが少ないので、外質細胞に位置することが要求される。
外質に位置する知られる脱ホスホリル化酵素には、非特異的アルカリホスファターゼ類、ATPアーゼ、ADPアーゼ、5’−ヌクレオチダーゼ、およびプリンヌクレオシドホスホリラーゼがある。
ホスファターゼが介する脱ホスホリル化による細胞毒性薬剤をターゲッティングするためのもう一つの必要な特性は、それらは基質としてのリン酸塩プロドラッグの幅広さが利用できる、ということである。
この点については、アルカリホスファターゼは、血管内皮細胞に対する毒性に選択的に到達するための魅力的な標的である。
本発明はまた、以下の項目を提供する。
(項目1)動物に、コンブレタスタチンA4リン酸二ナトリウム以外のプロドラッグを含む、血管系が増殖している場所で標的化された血管破壊を達成するために効果的な量で投薬することを含み、該プロドラッグは、実質上、非細胞毒性であるが、血管増殖の箇所で高められたレベルで選択的に誘導される内皮酵素の作用によって、実質上、細胞毒性の薬物に変換される、血管増殖疾患を有する温血動物を治療する方法。
(項目2)該プロドラッグは、以下の一般構造式を有する化合物の部類の範囲内のリン酸塩であり、
XはO、NH、またはSであり;
YはO、NH、S、O - 、NH + またはS - であり;
ZはOまたはSであり;
R 2 およびR 3 のそれぞれは、アルキル基、H、1価または2価の陽イオン塩、またはアンモニウム陽イオン塩であり、またR 2 およびR 3 は、同種または異種であってよく;および
R 1 は、Xを含む基である少なくとも一つの基(R a と表す)を有する化合物を表す構造式R 1 −R a によって定義され、該R a は、非特異的血管内皮ホスファターゼの基質として利用できるリン酸塩または他の塩を形成することができ、またそれ故に、相対的に非細胞毒性のリン酸塩形態から細胞毒性のR 1 −R a 形態へ変換される、
項目1に記載の方法。
(項目3)構造式R 1 −R a によって表される化合物は、チューブリンバインダーである項目2に記載の方法。
(項目4)該チューブリンバインダーは、コンブレタスタチン類、タキサン類、ビンカアルカロイド類、コルヒチノイド類、ドラスタチン類、ポドフィロトキシン類、ステガナシン類、アンフェチニル類、フラボノイド類、リゾキシン類、クラシン類A、エポチロン類AおよびB、ウェルウィスタチン類、フェンスタチン類、2−ストリルキナゾリン−4(3H)−オン類、スチルベン類、2−アリール−1,8−ナフチリジン−4(1H)−オン類、5,6−ジハイドロインドロ(2,1−a)イソキノリン類、2,3−ベンゾ(b)チオフェン類、2,3−置換ベンゾ(b)フラン類、2,3−置換インドール類および2−メトキシエストラジオール類からなる群より選択される化合物である項目3に記載の方法。
(項目5)該動物は、血管増殖の場所で微細血管細胞を有し、該動物は、また、悪性でない他の細胞を有し、および該実質上、細胞毒性の薬物は、該微細血管細胞に対するよりも該悪性でない他の細胞に対しては、実質上、より毒性でない、項目1に記載の方法。
(項目6)該プロドラッグは、多くて約3時間以内に内皮酵素の作用によって、実質上、細胞毒性の薬物に変換される項目5に記載の方法。
(項目7)該プロドラッグは、多くて約3時間以内に内皮酵素の作用によって、実質上、細胞毒性の薬物に変換される項目1に記載の方法。
(項目8)動物に、血管系が増殖している場所で標的化された血管破壊を達成するために効果的なプロドラッグの量で、投薬することを含み、該プロドラッグは、実質上、非細胞毒性であるが、血管増殖の箇所で高められたレベルで選択的に誘導される内皮酵素の作用によって、実質上、細胞毒性の薬物へ変換され得る、悪性でない血管増殖疾患を有する温血動物を治療する方法。
(項目9)該プロドラッグは、一般構造式を有する化合物の部類の範囲内のリン酸塩であり、
XはO、NH、またはSであり;
YはO、NH、S、O - 、NH - またはS - であり;
ZはOまたはSであり;
R 2 およびR 3 のそれぞれは、アルキル基、H、1価または2価の陽イオン塩、またはアンモニウム陽イオン塩であり、またR 2 およびR 3 は、同種または異種であってよく;および
R 1 は、Xを含む基である少なくとも一つの基(R a と表す)を有する化合物を表す構造式R 1 −R a によって定義され、該R a は、非特異的血管内皮ホスファターゼの基質として利用できるリン酸塩または他の塩を形成することができ、またそれ故に、相対的に非細胞毒性のリン酸塩形態から細胞毒性のR 1 −R a 形態へ変換される、
項目8に記載の方法。
(項目10)構造式R 1 −R a によって表される化合物は、チューブリンバインダーである項目9に記載の方法。
(項目11)該チューブリンバインダーは、コンブレタスタチン類、タキサン類、ビンカアルカロイド類、コルヒチノイド類、ドラスタチン類、ポドフィロトキシン類、ステガナシン類、アンフェチニル類、フラボノイド類、リゾキシン類、クラシン類A、エポチロン類AおよびB、ウェルウィスタチン類、フェンスタチン類、2−ストリルキナゾリン−4(3H)−オン類、スチルベン類、2−アリール−1,8−ナフチリジン−4(1H)−オン類、5,6−ジハイドロインドロ(2,1−a)イソキノリン類、2,3−ベンゾ(b)チオフェン類、2,3−置換ベンゾ(b)フラン類、2,3−置換インドール類および2−メトキシエストラジオール類からなる群より選択される化合物である項目10に記載の方法。
(項目12)該動物は、血管増殖の場所で微細血管細胞を有し、該動物は、また、悪性でない他の細胞を有し、および実質上、細胞毒性の薬物は、該微細血管細胞に対するよりも該悪性でない他の細胞に対しては、実質上、毒性でない、項目8に記載の方法。
(項目13)該プロドラッグは、多くて約3時間以内に内皮酵素の作用によって、実質上、細胞毒性試薬に変換される項目12に記載の方法。
(項目14)該プロドラッグは、多くて約3時間以内に内皮酵素の作用によって、実質上、細胞毒性試薬に変換される項目8に記載の方法。
(項目15)コンブレタスタチンA4、パンクラチスタチンおよびタキソールのリン酸塩プロドラッグ以外のプロドラッグを含む、血管系が増殖している場所で標的化された血管破壊を達成するための、血管増殖疾患を有する温血動物を治療するための組成物であって、該プロドラッグは実質上、非細胞毒性であるが、血管増殖の箇所で高められたレベルで選択的に誘導される内皮酵素の作用によって、実質上、細胞毒性の薬物に変換可能である、組成物。
(項目16)該プロドラッグは、以下の一般構造式を有する化合物の部類の範囲内のリン酸塩であり、
XはO、NH、またはSであり;
YはO、NH、S、O - 、NH - またはS - であり;
ZはOまたはSであり;
R 2 およびR 3 のそれぞれは、アルキル基、H、1価または2価の陽イオン塩、またはアンモニウム陽イオン塩であり、またR 2 およびR 3 は、同種または異種であってよく;および
R 1 は、Xを含む基である少なくとも一つの基(R a と表す)を有する化合物を表す構造式R 1 −R a によって定義され、該R a は、非特異的血管内皮ホスファターゼの基質として利用できるリン酸塩または他の塩を形成することができ、またそれ故に、相対的に非細胞毒性のリン酸塩形態から細胞毒性のR 1 −R a 形態へ変換される、
項目15に記載の組成物。
(項目17)構造式R 1 −R a によって表される化合物は、チューブリンバインダーである項目16に記載の定義される組成物。
(項目18)該チューブリンバインダーは、コンブレタスタチン類、タキサン類、ビンカアルカロイド類、コルヒチノイド類、ドラスタチン類、ポドフィロトキシン類、ステガナシン類、アンフェチニル類、フラボノイド類、リゾキシン類、クラシン類A、エポチロン類AおよびB、ウェルウィスタチン類、フェンスタチン類、2−ストリルキナゾリン−4(3H)−オン類、スチルベン類、2−アリール−1,8−ナフチリジン−4(1H)−オン類、5,6−ジハイドロインドロ(2,1−a)イソキノリン類、2,3−ベンゾ(b)チオフェン類、2,3−置換ベンゾ(b)フラン類、2,3−置換インドール類および2−メトキシエストラジオール類からなる群より選択される化合物である項目17に記載の組成物。
(項目19)該動物は、血管増殖の場所で微細血管細胞を有し、該動物は、また、悪性でない他の細胞を有し、および該実質上、細胞毒性の薬物は、該微細血管細胞に対するよりも該悪性でない他の細胞に対しては、実質上、より毒性でない、項目15に記載の組成物。
(項目20)該プロドラッグは、多くて約3時間以内に内皮酵素の作用によって、実質上、細胞毒性の薬物に変換される項目19に記載の組成物。
(項目21)該プロドラッグは、多くて約3時間以内に内皮酵素の作用によって、実質上、細胞毒性の薬物に変換される項目15に記載の方法。
(項目22)上記方法および組成物での使用に適するプロドラッグを確認するための手法であり、コンブレタスタチンA4リン酸二ナトリウム以外のプロドラッグの存在下、制限時間内で、増殖内皮細胞および悪性でない他の細胞を培養する工程;その後それぞれの培養物において、増殖内皮細胞の培養物が、他の細胞の培養物よりも著しく大きな細胞毒性効果を示すかどうかを測定し;および、もしそのようであれば、プロドラッグ、および血管増殖の箇所で高められたレベルで選択的に誘導される内皮酵素の存在下での上記他の細胞の培養が、最初の培養における他の細胞の細胞毒性効果と比較して、酵素存在下の他の細胞に関して細胞毒性が高められ、そのような方法および組成物としてのプロドラッグの適正を示唆する、ことからなる手法。
(項目23)該プロドラッグは、一般構造式を有する化合物の部類の範囲内のリン酸塩であり、
XはO、NH、またはSであり;
YはO、NH、S、O - 、NH - またはS - であり;
ZはOまたはSであり;
R 2 およびR 3 のそれぞれは、アルキル基、H、1価または2価の陽イオン塩、またはアンモニウム陽イオン塩であり、またR 2 およびR 3 は、同種または異種であってよく;および
R 1 は、Xを含む基である少なくとも一つの基(R a と表す)を有する化合物を表す構造式R 1 −R a によって定義され、該R a は、非特異的血管内皮ホスファターゼの基質として利用できるリン酸塩または他の塩を形成することができ、またそれ故に、相対的に非細胞毒性のリン酸塩形態から細胞毒性のR 1 −R a 形態へ変換される、
項目22に記載の手法。
(項目24)構造式R 1 −R a によって表される化合物は、チューブリンバインダーである項目23に記載の手法。
(項目25)該チューブリンバインダーは、コンブレタスタチン類、タキサン類、ビンカアルカロイド類、コルヒチノイド類、ドラスタチン類、ポドフィロトキシン類、ステガナシン類、アンフェチニル類、フラボノイド類、リゾキシン類、クラシン類A、エポチロン類AおよびB、ウェルウィスタチン類、フェンスタチン類、2−ストリルキナゾリン−4(3H)−オン類、スチルベン類、2−アリール−1,8−ナフチリジン−4(1H)−オン類、5,6−ジハイドロインドロ(2,1−a)イソキノリン類、2,3−ベンゾ(b)チオフェン類、2,3−置換ベンゾ(b)フラン類、2,3−置換インドール類および2−メトキシエストラジオール類からなる群より選択される化合物である項目24に記載の手法。
(項目26)該悪性でない他の細胞は、線維芽細胞である項目22に記載の手法。
本発明の更なる特徴および利点は、付随する図面と共に、以下、説明する詳細な説明によって明らかにされよう。
本発明は、腫瘍または悪性でない血管過多を有する温血動物に、細胞毒性が低いプロドラッグ形態が細胞毒性が高い脱ホスホリル化された形態へと変換されることで、微細血管が他の正常な組織よりも優先的に破壊されるために、微細血管ホスファターゼに対する基質特異性を有するプロドラッグ形態に処方される細胞毒性薬剤を十分な量で投薬することからなる、リン酸塩プロドラッグの使用を包含するものである。
血管ターゲッティングのために好まれる細胞毒性の薬剤の例としては、チューブリン結合薬剤があるが、それらはリン酸塩プロドラッグ処方によって、水不溶性から水可溶性へ、チューブリン結合薬剤から非チューブリン結合薬剤へ、および細胞毒性から非細胞毒性へ変換され得るからである(Anti-Cancer Drug Design 13: 183-191, 1998) 。
リン酸塩プロドラッグの形態による微細細胞毒性をターゲッティングするための多様性の例としては、図1〜図3(FIG.1A〜FIG.1C) に示されており、またそれらは、例えば、コンブレタスタチン類、タキサン類、ビンカアルカロイド類、コルヒチノイド類、ドラスタチン類、ポドフィロトキシン類、ステガナシン類、アンフェチニル類、フラボノイド類、リゾキシン類、クラシン類A、エポチロン類AおよびB、ウェルウィスタチン類、フェンスタチン類、2−ストリルキナゾリン−4(3H)−オン類、スチルベン類、2−アリール−1,8−ナフチリジン−4(1H)−オン類、5,6−ジハイドロインドロ(2,1−a)イソキノリン類、2,3−ベンゾ(b)チオフェン類、2,3−置換ベンゾ(b)フラン類、2,3−置換インドール類および2−メトキシエストラジオール類のような、以前、既に掲載された既知のチューブリン結合薬剤から選択された。
図1〜図3(FIG.1A〜FIG.1C)に掲載される化合物は、リン酸塩を生産するための化学反応、および細胞毒性の薬物を非細胞毒性リン酸塩プロドラッグ構造への更なる変換の能力を与える芳香族水酸基またはアミノ基のいずれか一方を有するという構造的要件を満たすものである。
血管毒性をターゲッティングするために必要な他の特徴は:
1.チューブリン結合薬剤類または他の細胞毒性薬剤類(例えば、パンクラチスタチンはチューブリンポリマーに結合することは報告されていない)は、細胞毒性(チューブリン結合)形態の場合には、ヒト微細血管細胞、および線維芽細胞のような他の正常ヒト細胞共に、同様の毒性のレベルを誘導しなければならない、または、二者択一的に、チューブリン結合形態は、微細血管細胞よりも正常細胞に対する細胞毒性が本質的に少なくなければならない。
もしこれがそのような場合でなく、線維芽細胞(即ち、正常細胞)が細胞毒性形態に対して微細血管よりも、より影響を受け易すいのならば、非細胞毒性プロドラッグ形態の場合では、たとえ線維芽細胞には細胞毒性形態を活性化するためのホスファターゼが少なくても、結果として、線維芽細胞において細胞毒性を誘導することを必要とされることは少ないであろう。
最終的な結論は、プロドラッグは正常細胞でなく微細血管に対してよりいっそう毒性であるのは、それらの高められた、細胞毒性形態を生じさせるアルカリホスファターゼ活性のためである、ということである。
2.潜在的なリン酸塩プロドラッグのチューブリン結合または細胞毒性形態は、プロドラッグ形態では細胞毒性であってはならず、それは結果的に1〜3時間、好ましくは、1〜2時間以内で細胞毒性形態へ変換される必要がある。
チューブリン結合薬剤は、数時間以内に周辺の血液循環によって除かれる。
そこで、インビボで血管破壊をターゲッティングすることを効果的に行なうためにはリン酸塩プロドラッグの構造は、優先的な細胞の毒性を誘導するための微細血管におけるホスファターゼによって1〜3時間以内に細胞毒性形態へ変換されなければならない。
したがって、チューブリンに結合するための反応速度は、だいたい1〜3時間以内に結合が完了しなければならない。
R1 は、フェノール性水酸基、または芳香族アミノ基、または、いかなる他の適当な水酸基またはアミノ基である少なくとも一つの基(Ra と表す)を含む化合物を表す構造式R1 −Ra によって定義され、該Ra は、非特異的血管内皮ホスファターゼの基質として利用できるR2−R3ホスフェート金属もしくはアミン塩もしくはリン酸エステルを形成でき、およびそれ故に、相対的に非細胞毒性のリン酸塩形態から細胞毒性の水酸基またはアミノ基形態へ変換される。
GMPで製造されるコンブレタスタチンA4リン酸二ナトリウムは、OXiGENE, Inc. (ボストン)から購入され、細胞培地に添加するための生理食塩水に溶解される。
アルカリホスファターゼは、Sigma(P-6774) から緩衝液として購入され、直接、細胞培地に添加された。
商業的に入手できる4種のヒト細胞系統は、CO2 が5%、湿度80%、37℃の条件で下に示される培地において増殖された。
1.HL60ヒト白血病細胞、10%子牛血清で栄養補強されたRPMI1640で培養されたプロ−アポトーシス細胞系統。
2.K562ヒト白血病細胞、10%子牛血清で強化されたRPMI1640で培養されたアポトーシス耐性細胞系統。
3.ヒト新生児微細血管内皮細胞(HMVEC)、培地131(medium 131)+微小血管成長補足剤(MVGS)+付着因子(AF)=500ml+25ml(AFは2〜3ml/T−25フラスコ;すべての試薬はCascade Biologics,Inc., ポートランド、オレゴン)で培養された。
4.ヒト新生児皮膚線維芽細胞 (HDF)、培地106(medium 106)+低血清成長補足剤(LSGS)=500ml+10ml (Cascade Biologics,Inc.)で培養された。
これは指数関数的増殖段階になり、また細胞生存能力は、トリパンブルー排除法によって95%以上となった。
この試験は、若干修正が加えられたシュバイツアー等によって報告された説明(Ext. Haemetol. 21: 573−578、1993)に基づくものである。
簡単に説明すると、4.2×103 /ml の濃度でHL60、K562、HDF、HMVEC細胞は、96ウェルの平底微生物培養平板で、ウェルあたり190μlの量に、異なる濃度のコンブレタスタチンA4リン酸塩ナトリウムまたは他のチューブリン結合薬剤およびそれらのプロドラッグまたは10μlの分量で加えられるアルカリホスファターゼの単位量(ユニット)を加えて培養された。
上述した標準培養条件下での5日間のインキュベーションの後、コロニー(>40細胞)は、倒立光学顕微鏡によって数えられ、またはMTT試験によって見積もられた。
IC50値は、薬剤濃度に対する百分率の曲線から得られた。
血管内皮細胞に対する毒性を標的とするための、A4プロドラッグをA4に変換することの重要性を立証するために計画された3種類の実験がある。
HL60、K562、HDFおよびHMVEC細胞は96ウェル平板で、示される濃度(図4(FIG.2A)および図5(FIG.2B))で、5日間、A4プロドラッグの存在下で、または2時間接触の後、薬剤を含む培地は取り除かれ、新しい培地が添加されて培養され、そして更に5日間細胞は培養された。
クローン原性増殖は、5日間のインキュベーションの後、すべての処置に関して記録された。
HMVECおよびHDFは、最初に800細胞/ウェルを含む、96ウェルマイクロタイター平板で培養された。
その細胞は、1時間、示されるコンブレタスタチンA4プロドラッグ±1ユニットのアルカリホスファターゼの存在下で、培養された。
培地は取り除かれ、細胞は洗浄され、そして新しい培地が添加され、そして細胞は更に5日間、インキュベートされた。
クローン原性増殖はMTT試験によって見積もられた。
HMVECは、最初に800細胞/ウェルを含む、96ウェルマイクロタイター平板で培養された。
その細胞は、1時間、示されるコンブレタスタチンA4プロドラッグ±示されるユニットのアルカリホスファターゼの存在下で、培養された。
培地は取り除かれ、その細胞は培地で洗浄され、そしてその培養物は更に新しい培地で5日間さらにインキュベートされた。
クローン原性増殖はMTT試験によって見積もられた。
5日目にクローン原性細胞毒性が見積もられる前に、HMVEC、HDF、HL60、およびK562細胞は、2時間(図4(FIG.2A))または5日間(図5(FIG.2B))、コンブレタスタチンA4リン酸二ナトリウムに対して接触された。
IC50値は、5日間の1.5〜2.5nMでの接触後のすべての細胞類で似し、接触が2時間に制限された時、HMVECだけがIC50細胞毒性を示した。
投与量応答細胞毒性は、1ユニットのアルカリホスファターゼの存在下または存在無しの条件下で、様々さ濃度のコンブレタスタチンA4リン酸二ナトリウムに対する1時間の接触の後、見積もられた。
アルカリホスファターゼの添加されないHDFの細胞毒性は無かったが、アルカリホスファターゼが加えられた場合、A4プロドラッグの細胞毒性はHMVECとHDFでは同じであった。
HMVECおよびHDFは、1時間、様々な示される濃度のコンブレタスタチンA4リン酸二ナトリウム+示され添加されるアルカリホスファターゼの存在下で培養された。
データは明らかに、より高いA4プロドラッグの濃度では、特に、細胞毒性に関するアルカリホスファターゼのさらなる依存性を示した。
実施例1は、コンブレタスタチンA4プロドラッグのようなチューブリン結合薬剤への、血管内皮細胞の優先的な細胞毒性についての接触時間の重要性を開示する。
もしクローン原性試験が、5日間、HMVEC、HDF、K562、およびHL60細胞を処理するために、コンブレタスタチンA4リン酸二ナトリウム(プロドラッグ)の濃度を増加させた条件で構成されると、すべての細胞系統はおよそ1.5〜2.5nMのIC50値は類似するだろう(図5(FIG.2B))。
これらのデータは、もし接触時間が充分長ければ、それらの起源に関係なく、ヒト細胞系統の毒性には本質的な違いが無いということを教授する。
しかし、A4プロドラッグは、他のチューブリン結合薬剤と同様、インビボでは数時間内に周辺の血管循環によって取り除かれ、他のチューブリン結合薬剤はこの能力を有することが示されていないが、またこれらの条件下では、A4プロドラッグは、腫瘍における増殖する内皮細胞に対する優先的な毒性を示した(Cancer Res. 57(10):1892-1834, 1997)。
したがって、我々は、A4プロドラッグに対する様々な細胞系統の接触を2〜3時間に制限し、A4を有する培地を取り除き、それを新しい培地に取替え、そして更に5日間培養を続けた。
これらの条件は、HMVECは、HDF、K562、およびHL60細胞と比較してA4プロドラッグ誘導細胞毒性に対してまさに影響を受け易いこと、を示した(図4(FIG.2A))。
これらのデータは、(i) インビトロ細胞モデルでは、A4プロドラッグのようなチューブリン結合薬剤による血管内皮細胞毒性の選択的な誘導を実施するのに使用することが可能であり、(ii)これは、インビボでの薬物動力学で調節された接触の制限をまねるインビトロでの条件下でのみ起こる、および(iii)細胞毒性を調節するチューブリン結合パラメータまたは代謝相違のどちらか、あるいは両方は、A4プロドラッグの血管内皮細胞に対する選択的細胞毒性の原因である、ということを教授する。
コンブレタスタチン類は、天然で、発生する、A−,B−,C−およびD−系統の構造からなるチューブリン結合薬剤のファミリーである(U.S.patents Nos.4,940,726;4,996,237;5,409,953; および5,569,786)。
実施例2は、インビトロでのHDF、HMVECおよびHL60の培養におけるこれらの化合物の選択物によって誘導されるクローン原性毒性のIC50値を比較する。
その化合物は、DMSO(例えば、<0.5%)中の微小培地に添加され、そして毒性は5〜7日後の培養物においてMTT試験によって評価された。
表1のデータは、コンブレタスタチン類似体が、様々な類似体と、評価される様々なヒト細胞種との間の、それらの総合的なクローン原性毒性において、かなり変化したことを示す。
A4は、試験されたすべての細胞において、最も毒性なチューブリン結合の機構を有しており、そしてそれは細胞種の間のクローン原性毒性についての優先性がないことを示した。
しかし、他のコンブレタスタチン類の細胞毒性は一般的に、大きい毒性から小さい毒性の順にクローン原性毒性について、
HL−60>HDF>HMVEC
と並べられよう。
これらのデータは、リン酸塩プロドラッグを、抗有糸分裂毒性の血管ターゲッティングにおいて使用することができるかどうかについて、正常細胞に対する毒性はHMVECに対するよも大きくない、という特性を有するためのチューブリン結合薬剤の必要条件を、確立するものである。
コンブレタスタチン類は、親切にもアリゾナ州立大学のG.R.Pettit教授により提供された。HDF=ヒト二倍体線維芽細胞;HMVEC=ヒト微細血管内皮細胞;HL−60=ヒト骨髄性白血病細胞
様々な種類のチューブリン結合薬剤によって誘導されるクローン原性毒性についての接触時間の効果が、図10(FIG.5)に示されている。
タキソール、タキソテール、ビンクリスチン、およびコンブレタスタチンA1およびA4は、1時間目と6時間目に、HMVECおよびHDFの微小培養物に加えられ、食塩水で洗浄され、およびMTT試験によるクローン原性毒性を評価する前に3日以上完全培地でのインキュベーションが継続された。
この実施例におけるデータは、様々なチューブリン結合薬剤の結合速度は、インビボでの接触と同様の条件(即ち、1時間)下での、それらの細胞毒性に影響を与えること、を示す。
例えば、タキソール、タキソテールおよびコンブレタスタチンA1は、1時間接触後HMVECに対する最大毒性を誘導せず、6時間要求したが、また加えて、速度論的に調節された細胞毒性の応答の程度は、またHMVECと比較してHDFでは異なった。
この場合、リン酸塩プロドラッグは微細血管毒性を標的とすることが可能である。なぜならばそれらは、正常細胞と比較して、プロドラッグをその細胞毒性形態へ変換する、アルカリホスファターゼが増加したからである。
ストレス障害および侵襲性腫瘍細胞の存在の双方は、アルカリホスファターゼのレベルを50倍にするために、微細血管を誘導することができる(J. Invest. Dermatol. 109(4): 597-603, 1997; FEBS lett.350(1):99-103, 1994 )。
アルカリホスファターゼは、細胞膜内に存在し、また血液循環では、リン酸塩を含む化合物を加水分解して、有機分子部分からリン酸塩部分を分離しまたは取り除くことが出来る。
アルカリホスファターゼを高めることによる、それら自身の損傷を修復するための微細血管の生理学的要求は、傷の箇所におけるカルシウム沈殿物の増加された沈殿物を導く、通常の傷治療過程の一部分である。
この代謝の特異性の結論は、リン酸塩に修飾された細胞毒性チューブリン結合薬剤(例えば、A4プロドラッグ)は、やはり、アルカリホスファターゼの基質である、と言う事であろう。
この過程は、また、その結果として、微細血管のチューブリン結合薬剤に対する高められた細胞毒性の感度を導き、チューブリンに結合し、細胞毒性である脱ホスホリル化された形態に対して、ホスホリル化された形態ではチューブリンに結合せず、細胞毒性ではない。
この実施例はこの事実が確かであることを示す。
インビトロにおいて、2時間、高められたA4プロドラッグの濃度に、アルカリホスファターゼが1単位存在あるいは存在しない状態で、培地に接触されたHDFおよびHMVECは、アルカリホスファターゼが添加されていないHMVECに対する高い選択的な細胞毒性の程度を示すが、HDFはアルカリホスファターゼの存在下でのA4プロドラッグに対してHMVECと同一の細胞毒性となる(図6(FIG.3A)および図7(FIG.3B))。
血管破壊をターゲッティングすることは、直接的に、HMVECにおける高レベルのアルカリホスファターゼの存在に依存し、およびその他の正常細胞およびHDFのような腫瘍細胞においては、それを欠くと、結論付けられた。
従って、この実施例は微細血管の増殖破壊をターゲッティングするための方法を教授し、それによって、チューブリン結合化合物のような細胞毒性薬剤は、例えば、血管内皮細胞においてのみ大量に存在し、細胞毒性形態へ戻すアルカリホスファターゼによって選択的に代謝されることが可能であり、細胞毒性を誘導できない、フェノール性ヒドロキシリン酸塩の形態をとることにより、プロドラッグ形態へ変換される。
実施例5では、さらに、実施例4において説明された開示を立証し、証明する。
ここでは、A4プロドラッグの細胞毒性を基準としてアルカリホスファターゼの投薬量の依存を明らかにするという実験計画が計画された。
そのデータは、明白に、どれぐらいのアルカリホスファターゼの量が、HMVECおよびHDF(図8(FIG.4A)および図9(FIG.4B))双方に対するコンブレタスタチンA4リン酸二ナトリウムのクローン原性細胞毒性を決定することを示す。
その結果は、高レベルのアルカリホスファターゼが加えられると、毒性は双方の細胞系統で等しくなるが、直接的に低レベルのアルカリホスファターゼを添加無しない、または添加後のA4プロドラッグに対するクローン原性毒性を、HMVECは直接的に発現するのであるが、HDFがA4プロドラッグによって殺傷され得る前に多くのアルカリホスファターゼを加えなければならない、ことを教授する。
表2に示される化合物は、どのようにして毒性が細胞毒性形態を、増殖している微細血管内皮細胞において他の正常細胞よりも50倍以上の濃度となっている、アルカリホスファターゼのような細胞性ホスファターゼによって細胞毒性形態へ戻されるという変換がなされるまでは結果として細胞毒性はない、リン酸塩プロドラッグへ変換することによって微細血管細胞へ標的とされ得るのか、の例を示す。
一般に、チューブリン結合薬剤はリン酸塩形態ではチューブリンに結合できないので、それらは、血管ターゲッティングのための細胞毒性機構として、好まれる細胞毒性機構を示す。
その化合物のすべてについて、1時間微小培地中で接触させた後、毒性の評価がなされ、そしてさらに培養物を5日間インキュベーションした後、MTT試験によって細胞毒性の試験が行なわれた。
これらの条件下では、チューブリン結合速度は、正常な増殖性HDFおよびHMVECの双方における毒性をもたらすのに充分速いものであった。
表2に報告されるデータは、
(i) HMVECではそうでないが、HDFにおいてプロドラッグに対する高いIC50値によって示されるHMVECに対する毒性の作用がない間、リン酸塩プロドラッグは、一般に、正常HDFに毒性を与えない、(ii)もし細胞毒性薬剤がHMVECに対するよりもHDFに対する毒性が大きいのなら、そしてたとえプロドラッグがHDFに毒性を与えないとしても、それはHDFとHMVECの間の本質的な毒性における違いを埋め合わせることは出来なく、(iii)コンブレタスタチンA1ピペラジンリン酸塩は、細胞毒性からHDFを保護することにおいて,唯一わずかに効果的であるので、ホスファターゼプロドラッグのすべての金属またはアミン塩は、同等に効果的ではなく、および(iv)パンクラチスタチンはチューブリンに結合することは知られていないが、他の細胞毒性機構を有する化合物もまたホスファターゼ機構によって標的とされ得る、
ことを立証する。
総括として、これらのデータは、もし、1時間の接触の内に、リン酸塩プロドラッグをその細胞毒性形態へ代謝するのに充分なアルカリホスファターゼを殆ど有しない正常細胞に対する保護があるのならば、細胞毒性薬剤はリン酸塩プロドラッグ構造による微細血管細胞破壊を標的とすることができる、ということを示す(即ちインビボの条件を模倣する)。
病原性の眼に関する血管新生を模擬実験するために、液体ヒドロペルオキシド(LHP)を30μgの投薬量で、ウサギの眼の角膜内に注射で投薬することによって、眼に関する新生血管形成を行なった。
7日〜14日後、LHPが注射された眼において、眼の血管が形成された。
実験対象は二つのグループに分けられ、一方のグループは、コンブレタスタチンA4リン酸二ナトリウムが、5日間、1日に一度40mg/kgの投薬量で静脈注射によって投薬され、もう一方は、コンブレタスタチンA4リン酸二ナトリウムを含まない賦形剤として、その他のグループに同じ期間に、水が静脈注射によって投与された。
双方の眼のグループは、7日後に検査された。コンブレタスタチンA4リン酸二ナトリウムで治療されたグループにおいて、40%もしくはそれ以上の血管の縮小が確認されたが、他のグループでは確認されなかった。
Claims (1)
- 明細書中に記載の発明。
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12047899P | 1999-02-18 | 1999-02-18 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2000599398A Division JP2002537262A (ja) | 1999-02-18 | 2000-02-16 | 血管破壊のターゲッティングに使用するための組成物および方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2011052019A true JP2011052019A (ja) | 2011-03-17 |
Family
ID=22390566
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2000599398A Pending JP2002537262A (ja) | 1999-02-18 | 2000-02-16 | 血管破壊のターゲッティングに使用するための組成物および方法 |
| JP2010278674A Pending JP2011052019A (ja) | 1999-02-18 | 2010-12-14 | 血管破壊のターゲッティングに使用するための組成物および方法 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2000599398A Pending JP2002537262A (ja) | 1999-02-18 | 2000-02-16 | 血管破壊のターゲッティングに使用するための組成物および方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (2) | US6538038B1 (ja) |
| EP (2) | EP2289521A3 (ja) |
| JP (2) | JP2002537262A (ja) |
| AU (1) | AU776511B2 (ja) |
| CA (1) | CA2358925C (ja) |
| IL (1) | IL144508A0 (ja) |
| WO (1) | WO2000048606A1 (ja) |
Families Citing this family (52)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6867305B2 (en) * | 1996-12-03 | 2005-03-15 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto and analogues thereof |
| CA2273083C (en) | 1996-12-03 | 2012-09-18 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto, analogues and uses thereof |
| US20050043376A1 (en) * | 1996-12-03 | 2005-02-24 | Danishefsky Samuel J. | Synthesis of epothilones, intermediates thereto, analogues and uses thereof |
| US6204388B1 (en) * | 1996-12-03 | 2001-03-20 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto and analogues thereof |
| GB9714249D0 (en) | 1997-07-08 | 1997-09-10 | Angiogene Pharm Ltd | Vascular damaging agents |
| GB9900334D0 (en) | 1999-01-07 | 1999-02-24 | Angiogene Pharm Ltd | Tricylic vascular damaging agents |
| JP2002537262A (ja) * | 1999-02-18 | 2002-11-05 | オキシジェン,インコーポレイティド | 血管破壊のターゲッティングに使用するための組成物および方法 |
| US20020058286A1 (en) * | 1999-02-24 | 2002-05-16 | Danishefsky Samuel J. | Synthesis of epothilones, intermediates thereto and analogues thereof |
| JP4187441B2 (ja) * | 1999-09-17 | 2008-11-26 | ベイラー・ユニバーシテイ | インドール含有およびコンブレタスタチン関連の抗有糸分裂および抗チューブリン重合薬剤 |
| US6849656B1 (en) | 1999-09-17 | 2005-02-01 | Baylor University | Indole-containing and combretastatin-related anti-mitotic and anti-tubulin polymerization agents |
| IL150978A0 (en) * | 2000-02-18 | 2003-02-12 | New York Medical College | Nitroacridine/tumor inhibitor compositions |
| DE60129536T2 (de) * | 2000-03-10 | 2008-06-19 | Baylor University, Waco | Tubulin bindende liganden |
| US7514067B2 (en) * | 2000-04-25 | 2009-04-07 | President And Fellows Of Harvard College | Methods for tumor diagnosis and therapy |
| DK1278758T3 (da) * | 2000-04-27 | 2012-01-23 | Univ Arizona | Combretastatin A-1-phosphat- og combretastatin B-1-phosphat-prodrugs |
| WO2001092224A1 (en) | 2000-05-31 | 2001-12-06 | Astrazeneca Ab | Indole derivatives with vascular damaging activity |
| US6720323B2 (en) | 2000-07-07 | 2004-04-13 | Angiogene Pharmaceuticals Limited | Colchinol derivatives as angiogenesis inhibitors |
| GB0019944D0 (en) * | 2000-08-15 | 2000-09-27 | Angiogene Pharm Ltd | Compositions with vascular damaging activity |
| EP1559718A1 (en) * | 2000-09-14 | 2005-08-03 | Bristol-Myers Squibb Company | Combretastatin A-4 phosphate prodrug salts with nitrogen-containing compounds |
| AU2002246827B2 (en) * | 2000-12-22 | 2008-02-21 | Bristol-Myers Squibb Company | Methods for modulating tumor growth and metastasis |
| AU2002246756A1 (en) | 2000-12-26 | 2002-08-06 | Bristol-Myers Squibb Company | Use of combretastatin a4 and its prodrugs as an immune enhancing therapy |
| JP2004536847A (ja) * | 2001-07-13 | 2004-12-09 | オキシジーン, インコーポレイテッド | 眼の疾患の処置のためのチューブリン結合薬剤を投与するための組成物および方法 |
| US20030181531A1 (en) * | 2003-02-11 | 2003-09-25 | David Sherris | Compositions and methods of administering tubulin binding agents for the treatment of ocular diseases |
| US20050065595A1 (en) * | 2001-09-03 | 2005-03-24 | David Chaplin | Implants containing combretastatin a-4 |
| GB0125532D0 (en) * | 2001-10-24 | 2001-12-12 | Burton Michael | Enzyme activity indicators |
| EP1438281A4 (en) * | 2001-10-26 | 2006-04-26 | Oxigene Inc | FUNCTIONALIZED STYLENE DERIVATIVES AS IMPROVED MEDIUM-TERM MEDICAL DEVICES |
| US7528165B2 (en) | 2001-12-13 | 2009-05-05 | National Health Research Institutes | Indole compounds |
| TWI317634B (en) | 2001-12-13 | 2009-12-01 | Nat Health Research Institutes | Aroyl indoles compounds |
| US7632955B2 (en) | 2001-12-13 | 2009-12-15 | National Health Research Institutes | Indole compounds |
| AU2003214462A1 (en) | 2002-04-03 | 2003-10-13 | Astrazeneca Ab | Indole derivatives having anti-angiogenetic activity |
| US20040127467A1 (en) * | 2002-04-17 | 2004-07-01 | Pettit George R | Synthesis of pancratistatin prodrugs |
| US7358236B1 (en) * | 2002-06-21 | 2008-04-15 | Oxigene, Inc. | Control of acute hypertension and cardiotoxicity in patients treated with vascular targeting agents |
| DK1506203T3 (da) * | 2002-08-23 | 2007-05-14 | Sloan Kettering Inst Cancer | Syntese af epothiloner, mellemprodukter deraf, analoger deraf og anvendelser deraf |
| US6921769B2 (en) | 2002-08-23 | 2005-07-26 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto and analogues thereof |
| US7649006B2 (en) | 2002-08-23 | 2010-01-19 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto and analogues thereof |
| DE60324196D1 (de) | 2002-12-09 | 2008-11-27 | Univ Arizona | Narcistatin-prodrugs |
| KR100616498B1 (ko) * | 2003-07-26 | 2006-08-25 | 주식회사 하이닉스반도체 | 폴리/텅스텐 게이트 전극을 갖는 반도체 소자의 제조방법 |
| DE60336646D1 (de) | 2003-08-07 | 2011-05-19 | Nat Health Research Institutes | Indol-Verbindungen als Inhibitoren der Tubulin-Polymerisation zur Behandlung von angiogenesisbezogenen Erkrankungen |
| US20050261246A1 (en) * | 2004-03-05 | 2005-11-24 | The Arizona Board Of Regents | Methods for treating inflammatory and autoimmune diseases |
| AU2005273961A1 (en) * | 2004-08-11 | 2006-02-23 | Williamsburg Holdings Llc | Noncardiotoxic pharmaceutical compounds |
| US7456289B2 (en) | 2004-12-31 | 2008-11-25 | National Health Research Institutes | Anti-tumor compounds |
| EP1919882A2 (en) * | 2005-02-17 | 2008-05-14 | Synta Pharmaceuticals Corporation | Isoxazole combretastatin derivatives for the treatment of proliferative disorders |
| JP5354775B2 (ja) * | 2005-06-14 | 2013-11-27 | ベイラー ユニバーシティ | チューブリン結合活性を有するコンブレタスタチンアナログ |
| WO2007008848A2 (en) * | 2005-07-07 | 2007-01-18 | Seattle Genetics, Inc. | Monomethylvaline compounds having phenylalanine carboxy modifications at the c-terminus |
| US20070077200A1 (en) * | 2005-09-30 | 2007-04-05 | Baker Clark R | Method and system for controlled maintenance of hypoxia for therapeutic or diagnostic purposes |
| EP1984333B1 (en) | 2006-02-03 | 2012-04-25 | Bionomics Limited | Substituted benzofurans, benzothiophenes, benzoselenophenes and indoles and their use as tubulin polymerisation inhibitors |
| US20080214509A1 (en) * | 2007-03-02 | 2008-09-04 | Robert Kerbel | Methods for enhancing the efficacy of vascular disrupting agents |
| ES2529434T3 (es) | 2007-11-21 | 2015-02-20 | Oxigene, Inc. | Método para tratar neoplasias hematopoyéticas |
| US20090209496A1 (en) * | 2008-02-15 | 2009-08-20 | David Chaplin | Methods and compositions for enhancing the efficacy of rtk inhibitors |
| WO2009129333A1 (en) * | 2008-04-15 | 2009-10-22 | Oxigene, Inc. | Methods for enhancing the efficacy of vascular disrupting agents |
| BR112013023722A2 (pt) * | 2011-03-15 | 2016-11-22 | Univ Utah Res Found | métodos para prever o risco de um indivíduo ter ou desenvolver uma coença, e para diagnosticar um indivíduo com uma doença, kit, modelo de animal não humano, e, sistema para identificar lóbulos coriocapilares aberrantes |
| US11419934B2 (en) | 2015-08-18 | 2022-08-23 | Oncotelic Therapeutics, Inc. | Use of VDAS to enhance immunomodulating therapies against tumors |
| CA3037989A1 (en) * | 2016-09-29 | 2018-04-05 | Janssen Sciences Ireland Unlimited Company | Pyrimidine prodrugs for the treatment of viral infections and further diseases |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63233939A (ja) * | 1987-01-06 | 1988-09-29 | アリゾナ ボード オブ リーゼンツ | コンブレタスタチン類 |
| US5561122A (en) * | 1994-12-22 | 1996-10-01 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Combretastatin A-4 prodrug |
| JPH08337589A (ja) * | 1995-06-06 | 1996-12-24 | Bristol Myers Squibb Co | パクリタキセル誘導体のプロドラッグ |
| WO1998051669A1 (en) * | 1997-05-15 | 1998-11-19 | Vion Pharmaceuticals, Inc. | Prodrug forms of ribonucleotide reductase inhibitors 3-ap and 3-amp |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4996237A (en) | 1987-01-06 | 1991-02-26 | Arizona Board Of Regents | Combretastatin A-4 |
| US4940726A (en) | 1989-04-26 | 1990-07-10 | Arizona Board Of Regents | Cell growth inhibitory macrocyclic lactones denominated Combretastatin D-1 and Combretastatin D-2 |
| GB9106177D0 (en) * | 1991-03-22 | 1991-05-08 | Aston Molecules Ltd | Substituted diphenylethylenes and analogues or derivatives thereof |
| US5529989A (en) * | 1995-01-09 | 1996-06-25 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Pancratistatin prodrug |
| JP2002537262A (ja) * | 1999-02-18 | 2002-11-05 | オキシジェン,インコーポレイティド | 血管破壊のターゲッティングに使用するための組成物および方法 |
| WO2016190184A1 (ja) | 2015-05-28 | 2016-12-01 | オリンパス株式会社 | 内視鏡対物光学系 |
| US9905368B2 (en) | 2015-08-04 | 2018-02-27 | Avx Corporation | Multiple leadwires using carrier wire for low ESR electrolytic capacitors |
-
2000
- 2000-02-16 JP JP2000599398A patent/JP2002537262A/ja active Pending
- 2000-02-16 AU AU35973/00A patent/AU776511B2/en not_active Ceased
- 2000-02-16 EP EP10075411A patent/EP2289521A3/en not_active Withdrawn
- 2000-02-16 CA CA2358925A patent/CA2358925C/en not_active Expired - Fee Related
- 2000-02-16 WO PCT/US2000/003996 patent/WO2000048606A1/en not_active Application Discontinuation
- 2000-02-16 EP EP00914606A patent/EP1152764A4/en not_active Ceased
- 2000-02-16 US US09/505,402 patent/US6538038B1/en not_active Expired - Lifetime
-
2001
- 2001-02-16 IL IL14450801A patent/IL144508A0/xx unknown
-
2002
- 2002-08-14 US US10/218,833 patent/US6956054B2/en not_active Expired - Fee Related
-
2010
- 2010-12-14 JP JP2010278674A patent/JP2011052019A/ja active Pending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63233939A (ja) * | 1987-01-06 | 1988-09-29 | アリゾナ ボード オブ リーゼンツ | コンブレタスタチン類 |
| US5561122A (en) * | 1994-12-22 | 1996-10-01 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Combretastatin A-4 prodrug |
| JPH08337589A (ja) * | 1995-06-06 | 1996-12-24 | Bristol Myers Squibb Co | パクリタキセル誘導体のプロドラッグ |
| WO1998051669A1 (en) * | 1997-05-15 | 1998-11-19 | Vion Pharmaceuticals, Inc. | Prodrug forms of ribonucleotide reductase inhibitors 3-ap and 3-amp |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2000048606A9 (en) | 2001-10-11 |
| US6538038B1 (en) | 2003-03-25 |
| CA2358925C (en) | 2010-04-20 |
| WO2000048606A1 (en) | 2000-08-24 |
| EP2289521A3 (en) | 2011-10-05 |
| AU776511B2 (en) | 2004-09-09 |
| AU3597300A (en) | 2000-09-04 |
| JP2002537262A (ja) | 2002-11-05 |
| US6956054B2 (en) | 2005-10-18 |
| EP1152764A1 (en) | 2001-11-14 |
| IL144508A0 (en) | 2002-05-23 |
| EP1152764A4 (en) | 2005-03-23 |
| EP2289521A2 (en) | 2011-03-02 |
| CA2358925A1 (en) | 2000-08-24 |
| US20030109500A1 (en) | 2003-06-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2011052019A (ja) | 血管破壊のターゲッティングに使用するための組成物および方法 | |
| JP2002537262A5 (ja) | ||
| Freeman et al. | The source of choline for acetylcholine synthesis in brain | |
| US20060128633A1 (en) | Colchinol derivatives as vascular damaging agents | |
| AU2007246023B2 (en) | Use of compounds of the formula A-R-X or their pharmaceutically acceptable salts for producing a pharmaceutical preparation | |
| CA2362281C (en) | Combinations for the treatment of diseases involving angiogenesis | |
| SK13912002A3 (sk) | Farmaceutické prostriedky na vyvolanie účinku, ktorý poškodzuje cievy | |
| WO2002094286A1 (fr) | Inhibiteurs de metastases cancereuses contenant des derives d'acide phosphatidique carbacycliques | |
| Gu et al. | Inhibiting myosin light chain kinase retards the growth of mammary and prostate cancer cells | |
| US20060100179A1 (en) | Compositions and methods for use in targeting vascular destruction | |
| CA2541869C (en) | Compositions and methods for use in targeting vascular destruction | |
| EP1547603A2 (en) | Compositions and methods for use in targeting vascular destruction | |
| AU2004201471B2 (en) | Compositions and Methods for Use in Targeting Vascular Destruction | |
| US20140045797A1 (en) | Bisphosphonate-Prostatic Acid Phosphatase Inhibitor Conjugates To Treat Prostate Cancer Bone Metastasis | |
| MXPA01008291A (en) | Compositions and methods for use in targeting vascular destruction | |
| JPH10511677A (ja) | イノシトール三燐酸の薬剤調製への使用 | |
| US20060135625A1 (en) | Method of administering split doses of a vascular targeting agent | |
| RU2317825C1 (ru) | Способ лечения злокачественных опухолей у животных | |
| Pace-Asciak | Novel eicosanoid pathways: The discovery of prostacyclin/6-keto prostaglandin F 1α and the hepoxilinsand the hepoxilins | |
| ES2344814T3 (es) | Analogos de lipoxina como nuevos inhibidores de la angiogenesis. | |
| Leo | The study of cell motility and plasticity in cancer: the role of the crosstalk between BM-MSCs and tumor in osteosarcoma progression and Claisened Hexafluoro as potential inhibitor of amoeboid motility in metastatic melanoma | |
| WO2023220734A3 (en) | Bisphosphonate lipids, lipid nanoparticle compositions comprising the same, and methods of use thereof for targeted delivery | |
| JP2020063209A (ja) | 細胞周期停止剤および抗腫瘍剤 | |
| Yang et al. | Cytotoxic Effects of Transforming Growth Factor-α Conjugated with Cytotoxin Saporin on Proliferating Vascular Smooth Muscle Cells | |
| 정혜원 | Anti-tumor effect and radiation response modification by tyrosine kinase inhibitor imatinib mesylate on pancreatic cancer cells |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20111201 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130128 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130426 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130502 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130527 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130530 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130620 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20131016 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20140109 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20140115 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20140213 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20140218 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20140314 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20140319 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20140703 |