DESCRIPCIÓN
Anticuerpos humanos contra PD-1
Campo de la invención
La presente invención se refiere a anticuerpos humanos y fragmentos de unión a antígeno de anticuerpos humanos que se unen específicamente al receptor inmunomodulador proteína de muerte programada 1 (PD-1), y métodos terapéuticos y diagnósticos para usar esos anticuerpos.
Exposición de la técnica relacionada
La muerte programada-1 (PD-1) (también llamada CD279) es un receptor de proteína de 288 aminoácidos expresado en linfocitos T y linfocitos B activados, linfocitos citolíticos naturales y monocitos. PD-1 es un miembro de la familia CD28/CTLA-4 (antígeno de linfocito T citotóxico)/ICOS (coestimulador inducible) de receptores coinhibidores de linfocitos T (Chen et al. 2013, Nat. Rev. Immunol. 13: 227-242). La función principal de PD-1 es atenuar la respuesta inmunitaria (Riley 2009, Immunol. Rev. 229: 114-125). PD-1 tiene dos ligandos, el ligando de PD 1 (PD-L1) y Pd -L2. PD-L1 (CD274, B7H1) se expresa ampliamente en tejidos linfoides y no linfoides, tales como linfocitos T CD4 y CD8, células del linaje de los macrófagos, tejidos periféricos, así como en células tumorales, células infectadas por virus y células de tejido autoinmunitario. PD-L2 (CD273, B7-DC) tiene una expresión más restringida que PD-L1, expresándose en células dendríticas activadas y macrófagos (Dong et al. 1999, Nature Med.). PD-L1 se expresa en la mayoría de los cánceres humanos, incluyendo melanoma, glioma, cáncer de pulmón no microcítico, carcinoma de células escamosas de cabeza y cuello, leucemia, cáncer pancreático, carcinoma de células renales y carcinoma hepatocelular y puede ser inducible en casi todos los tipos de cáncer (Zou and Chen 2008, Nat. Rev. Immunol. 8: 467 77). La unión de PD-1 a sus ligandos da como resultado una disminución de la proliferación de linfocitos T y la secreción de citocinas, lo que compromete las respuestas inmunitarias humorales y celulares en enfermedades tales como el cáncer, infecciones víricas y enfermedades autoinmunitarias. El bloqueo de la unión de PD-1 a la inmunosupresión inversa se ha estudiado en inmunoterapia autoinmunitaria, vírica y tumoral (Ribas 2012, NEJM 366: 2517-2519; Watanabe et al. 2012, Clin. Dev. Immunol. Volume 2012, ID Artículo: 269756; Wang et al. 2013, J. Viral Hep. 20: 27-39).
Las moléculas coestimuladoras y coinhibidoras de linfocitos T (denominadas colectivamente moléculas de coseñalización) desempeñan un papel crucial en la regulación de la activación de los linfocitos T, diferenciación de subconjuntos, la función efectora y la supervivencia (Chen et al. 2013, Nature Rev. Immunol. 13: 227-242). Tras el reconocimiento de los complejos péptido-MHC afines en las células presentadoras de antígeno por el receptor de linfocitos T, los receptores de coseñalización se localizan conjuntamente con los receptores de linfocitos T en la sinapsis inmunitaria, donde se sinergizan con la señalización de TCR para promover o inhibir la activación y función de los linfocitos T (Flies et al. 2011, Yale J. Biol. Med. 84: 409-421). La respuesta inmunitaria definitiva está regulada por un equilibrio entre las señales coestimuladoras y coinhibidoras ("puntos de control inmunitario") (Pardoll 2012, Nature 12: 252-264). PD-1 funciona como uno de esos "puntos de control inmunitario" para mediar la tolerancia de los linfocitos T periféricos y para evitar la autoinmunidad. PD-1 se une a PD-L1 o PD-L2 e inhibe la activación de los linfocitos T. Las infecciones virales crónicas y los tumores aprovechan la capacidad de PD1 para inhibir la activación de los linfocitos T para evadir la respuesta inmunitaria. En las infecciones víricas crónicas, PD-1 se expresa en gran medida en los linfocitos T específicos del virus y estos linfocitos T se "agotan" con la pérdida de las funciones efectoras y la capacidad proliferativa (Freeman 2008, p Na S 105: 10275-10276). PD-L1 se expresa en una amplia variedad de tumores y los estudios en modelos animales han demostrado que PD-L1 en tumores inhibe la activación de linfocitos T y la lisis de células tumorales y puede conducir a una mayor muerte de linfocitos T específicos de tumores. El PD-1: El sistema PD-L1 también juega un papel importante en el desarrollo de linfocitos T reguladores (Treg) inducidos y en el mantenimiento de la función Treg (Francisco et al. 2010, Immunol. Rev. 236: 219-242).
Dado que PD-1 juega un papel importante en la autoinmunidad, la inmunidad tumoral y la inmunidad infecciosa, es una diana ideal para la inmunoterapia. Se ha estudiado el bloqueo de PD-1 con antagonistas, incluyendo anticuerpos monoclonales, en tratamientos de cáncer e infecciones víricas crónicas (Sheridan 2012, Nature Biotechnology 30: 729 730).
Los anticuerpos monoclonales contra PD-1 son conocidos en la técnica y se han descrito, por ejemplo, en las patentes de los Estados Unidos número 8008449, 8168757, 20110008369, 20130017199, 20130022595 y en los documentos WO2006121168, WO20091154335, WO2012145493, WO2013014668, WO2009101611, EP2262837 y EP2504028.
Breve sumario de la invención
La presente invención proporciona anticuerpos y fragmentos de unión a antígeno de los mismos como se define en las reivindicaciones que se unen a PD-1. Los anticuerpos de la presente invención son útiles, entre otros, para dirigirse a linfocitos T que expresan PD-1, y para modular la actividad de PD-1. En determinadas realizaciones, los anticuerpos de la invención son útiles para inhibir o neutralizar la actividad de PD-1 y/o para estimular la activación de linfocitos T, por ejemplo, en circunstancias en las que la destrucción mediada por linfocitos T es beneficiosa o deseable. En
aspectos alternativos de la divulgación, los anticuerpos potencian la unión y/o la actividad de PD-1 y pueden usarse para inhibir la activación de linfocitos T. Los anticuerpos anti-PD-1 de la invención, o porciones de unión a antígeno de los mismos, pueden incluirse como parte de una molécula de unión a antígeno multiespecífica, por ejemplo, para modular la respuesta inmunitaria y/o para dirigir los anticuerpos a un tipo celular específico, tal como una célula tumoral, una célula de tejido autoinmunitaria o una célula infectada por virus. Los anticuerpos son útiles en el tratamiento de una enfermedad o trastorno tal como cáncer, infecciones víricas y enfermedades autoinmunitarias.
Los anticuerpos de la invención pueden ser de longitud completa (por ejemplo, un anticuerpo IgG1 o IgG4) o pueden comprender únicamente una porción de unión a antígeno (por ejemplo, un Fab, F(ab')2 o fragmento scFv), y pueden modificarse para afectar a la funcionalidad, por ejemplo, para eliminar las funciones efectoras residuales (Reddy et al., 2000, J. Immunol. 164:1925-1933). En determinadas realizaciones, los anticuerpos pueden ser biespecíficos.
En un primer aspecto, la presente invención proporciona anticuerpos monoclonales recombinantes aislados o fragmentos de unión a antígeno de los mismos como se define en las reivindicaciones que se unen específicamente a PD-1. En determinadas realizaciones, los anticuerpos son totalmente humanos. Los anticuerpos anti-PD-1 ilustrativos de la presente divulgación se enumeran en las Tablas 1 - 3 del presente documento. La Tabla 1 presenta los identificadores de secuencias de aminoácidos de las regiones variables de cadena pesada (HCVR), regiones variables de cadena ligera (LCVR), regiones determinantes de la complementariedad de cadena pesada (HCDR1, HCDR2 y HCDR3) y regiones determinantes de la complementariedad de cadena ligera (LCDR1, LCDR2 y LCDR3) de los anticuerpos anti-PD-1 ilustrativos. La Tabla 2 presenta los identificadores de secuencia de ácido nucleico de las HCVR, LCVR, HCDR1, HCDR2, HCDR3, LCDR1, LCDR2 y LCDR3 de los anticuerpos anti-PD-1 ilustrativos. La Tabla 3 muestra los identificadores de secuencia de aminoácidos de las secuencias de cadena pesada y cadena ligera de anticuerpos anti-PD-1 ilustrativos.
La presente divulgación proporciona anticuerpos o fragmentos de unión a antígeno de los mismos, que comprenden una HCVR que comprende una secuencia de aminoácidos seleccionada de cualquiera de las secuencias de aminoácidos de HCVR enumeradas en la Tabla 1, o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia con la misma.
La presente divulgación también proporciona anticuerpos o fragmentos de unión a antígeno de los mismos, que comprenden una LCVR que comprende una secuencia de aminoácidos seleccionada de cualquiera de las secuencias de aminoácidos de LCVR enumeradas en la Tabla 1, o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia con la misma.
La presente divulgación también proporciona anticuerpos o fragmentos de unión a antígeno de los mismos, que comprenden un par de secuencias de aminoácidos HCVR y LCVR (HCVR/LCVR) que comprende cualquiera de las secuencias de aminoácidos de HCVR enumeradas en la Tabla 1 junto con cualquiera de las secuencias de aminoácidos de LCVR enumeradas en la Tabla 1. De acuerdo con determinados aspectos, la presente divulgación proporciona anticuerpos o fragmentos de unión a antígeno de los mismos, que comprenden un par de secuencias de aminoácidos HCVR/LCVR contenidas dentro de cualquiera de los anticuerpos anti-PD-1 ilustrativos enumerados en la Tabla 1. En determinados aspectos, el par de secuencias de aminoácidos HCVR/LCVR se selecciona del grupo que consiste en SEQ ID NO: 2/10, 18/26, 34/42, 50/58, 66/74, 82/90, 98/106, 114/122, 130/138, 146/154, 162/170, 178/186, 194/202, 210/202, 218/202, 226/202, 234/202, 242/202, 250/202, 258/202, 266/202, 274/202, 282/202, 290/202, 298/186, 306/186 y 314/186. En determinados aspectos, el par de secuencias de aminoácidos HCVR/LCVR se selecciona de uno de SEQ ID NO: 130/138 (por ejemplo, H2M7795N), 162/170 (por ejemplo, H2M7798N), 234/202 (por ejemplo, H4xH9048P), o 314/186 (por ejemplo, H4xH9008P). En la invención, el anticuerpo o fragmento de unión a antígeno del mismo comprende un par de secuencias de aminoácidos HCVR/LCVR de las SEQ ID NO: 162/170.
La presente divulgación también proporciona anticuerpos o fragmentos de unión a antígeno de los mismos, que comprenden una CDR1 de cadena pesada (HCDR1) que comprende una secuencia de aminoácidos seleccionada de cualquiera de las secuencias de aminoácidos de HCDR1 enumeradas en la Tabla 1 o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia.
La presente divulgación también proporciona anticuerpos o fragmentos de unión a antígeno de los mismos, que comprenden una CDR2 de cadena pesada (HCDR2) que comprende una secuencia de aminoácidos seleccionada de cualquiera de las secuencias de aminoácidos de HCDR2 enumeradas en la Tabla 1 o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia.
La presente divulgación también proporciona anticuerpos o fragmentos de unión a antígeno de los mismos, que comprenden una CDR3 de cadena pesada (HCDR3) que comprende una secuencia de aminoácidos seleccionada de cualquiera de las secuencias de aminoácidos de HCDR3 enumeradas en la Tabla 1 o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia.
La presente divulgación también proporciona anticuerpos o fragmentos de unión a antígeno de los mismos, que comprenden una CDR1 de cadena ligera (LCDR1) que comprende una secuencia de aminoácidos seleccionada de cualquiera de las secuencias de aminoácidos de LCDR1 enumeradas en la Tabla 1 o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia.
La presente divulgación también proporciona anticuerpos o fragmentos de unión a antígeno de los mismos, que comprenden una CDR2 de cadena ligera (LCDR2) que comprende una secuencia de aminoácidos seleccionada de cualquiera de las secuencias de aminoácidos de LCDR2 enumeradas en la Tabla 1 o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia.
La presente divulgación también proporciona anticuerpos o fragmentos de unión a antígeno de los mismos, que comprenden una CDR3 de cadena ligera (LCDR3) que comprende una secuencia de aminoácidos seleccionada de cualquiera de las secuencias de aminoácidos de LCDR3 enumeradas en la Tabla 1 o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia.
La presente divulgación también proporciona anticuerpos o fragmentos de unión a antígeno de los mismos, que comprenden un par de secuencias de aminoácidos HCDR3 y LCDR3 (HCDR3/LCDR3) que comprende cualquiera de las secuencias de aminoácidos de HCDR3 enumeradas en la Tabla 1 junto con cualquiera de las secuencias de aminoácidos de LCDR3 enumeradas en la Tabla 1. De acuerdo con determinados aspectos, la presente divulgación proporciona anticuerpos o fragmentos de unión a antígeno de los mismos, que comprenden un par de secuencias de aminoácidos HCDR3/LCDR3 contenidas dentro de cualquiera de los anticuerpos anti-PD-1 ilustrativos enumerados en la Tabla 1. En determinados aspectos, el par de secuencias de aminoácidos HCDR3/LCDR3 se selecciona del grupo que consiste en las SEQ ID NO: 136/144 (por ejemplo, H2M7795N), 168/176 (por ejemplo, H2M7798N), 240/208 (por ejemplo, H4xH9048P), y 320/192 (por ejemplo, H4xH9008P).
La presente divulgación proporciona anticuerpos o fragmentos de unión a antígeno de los mismos, que comprenden una cadena pesada que comprende una secuencia de aminoácidos seleccionada de cualquiera de las secuencias de aminoácidos de HC enumeradas en la Tabla 3 o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia con la misma.
La presente divulgación también proporciona anticuerpos o fragmentos de unión a antígeno de los mismos, que comprenden una cadena ligera que comprende una secuencia de aminoácidos seleccionada de cualquiera de las secuencias de aminoácidos de LC enumeradas en la Tabla 3 o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia con la misma.
La presente divulgación también proporciona anticuerpos o fragmentos de unión a antígeno de los mismos, que comprenden un par de secuencias de aminoácidos HC y LC (HC/LC) que comprende cualquiera de las secuencias de aminoácidos de HC enumeradas en la Tabla 3 junto con cualquiera de las secuencias de aminoácidos de LC enumeradas en la Tabla 3. De acuerdo con determinados aspectos, la presente divulgación proporciona anticuerpos o fragmentos de unión a antígeno de los mismos, que comprenden un par de secuencias de aminoácidos HC/LC contenidas dentro de cualquiera de los anticuerpos anti-PD-1 ilustrativos enumerados en la Tabla 3. En determinados aspectos, el par de secuencias de aminoácidos HC/LC se selecciona del grupo que consiste en SEQ ID NO: 330/331, 332/333, 334/335, y 336/337. En algunas realizaciones, el anticuerpo o fragmento de unión a antígeno del mismo comprende un par de secuencias de aminoácidos HC/LC de las SEQ ID NO: 330/331.
La presente divulgación también proporciona anticuerpos o fragmentos de unión a antígeno de los mismos, que comprenden un conjunto de seis CDR (es decir, HCDR1-HCDR2-HCDR3-LCDR1-LCDR2-LCDR3) contenidas en cualquiera de los anticuerpos anti-PD-1 ilustrativos enumerados en la Tabla 1. En determinados aspectos, el par de secuencias de aminoácidos HCDR1-HCDR2-HCDR3-LCDR1-LCDR2-LCDR3 se selecciona del grupo que consiste en las SEQ ID NO: 132-134-136-140-142-144 (por ejemplo, H2M7795N); 164-166-168-172-174-176 (por ejemplo, H2M7798N); 236-238-240-204-206-208 (por ejemplo, H4xH9048P); y 316-318-320-188-190-192 (por ejemplo, H4xH9008P).
En un aspecto relacionado, la presente divulgación proporciona anticuerpos o fragmentos de unión a antígeno de los mismos, que comprenden un conjunto de seis CDR (es decir, HCDR1-HCDR2-HCDR3-LCDR1-LCDR2-LCDR3) contenidas dentro de un par de secuencias de aminoácidos HCVR/LCVR como se define mediante cualquiera de los anticuerpos anti-PD-1 ilustrativos enumerados en la Tabla 1. Por ejemplo, la presente divulgación incluye anticuerpos o fragmentos de unión a antígeno de los mismos, que comprenden el conjunto de secuencias de aminoácidos HCDR1-HCDR2-HCDR3-LCDR1-LCDR2-LCDR3 contenidas dentro de un par de secuencias de aminoácidos HCVR/LCVR seleccionado del grupo que consiste en las SEQ ID NO: 130/138 (por ejemplo, H2M7795N); 162/170 (por ejemplo, H2M7798N); 234/202 (por ejemplo, H4xH9048P); y 314/186 (por ejemplo, H4xH9008P). Los métodos y técnicas para identificar las CDR dentro de las secuencias de aminoácidos de HCVR y LCVR son bien conocidos en la materia y
pueden utilizarse para identificar las CDR dentro de las secuencias de aminoácidos de HCVR y/o LCVR específicas que se divulgan en el presente documento. Las convenciones ilustrativas que pueden utilizarse para identificar los límites de las CDR incluyen, por ejemplo, la definición de Kabat, la definición de Chothia y la definición de AbM. En términos generales, la definición de Kabat se basa en la variabilidad de secuencia, la definición de Chothia se basa en la ubicación de las regiones de bucles estructurales, y la definición de AbM es un término medio entre las estrategias de Kabat y Chothia. Véanse, por ejemplo, Kabat, "Sequences of Proteins of Immunological Interest", National Institutes of Health, Bethesda, Md. (1991); Al-Lazikani et al., J. Mol. Biol. 273:927-948 (1997); y Martin et al., Proc. Natl. Acad. Sci. USA 86:9268-9272 (1989). También hay disponibles bases de datos públicas para identificar secuencias de CDR dentro de un anticuerpo.
La presente invención incluye anticuerpos anti-PD-1 que tienen un patrón de glucosilación modificado. En algunas realizaciones, puede ser útil la modificación para eliminar sitios de glucosilación indeseables, o un anticuerpo que carezca de una fracción de fucosa presente en la cadena de oligosacárido, por ejemplo, para aumentar la función de citotoxicidad celular dependiente de anticuerpos (ADCC) (véase, Shield et al., (2002) JBC 277:26733). En otras solicitudes, puede hacerse modificación de la galactosilación para modificar la citotoxicidad dependiente del complemento (CDC).
La presente divulgación también proporciona anticuerpos y fragmentos de unión a antígeno de los mismos que compiten por la unión específica a PD-1 con un anticuerpo o fragmento de unión a antígeno del mismo que comprende las CDR de una HCVR y las CDR de una LCVR, en donde la HCVR y la LCVR tienen cada una una secuencia de aminoácidos seleccionada de las secuencias de HCVR y LCVR enumeradas en la Tabla 1.
La presente divulgación también proporciona anticuerpos aislados y fragmentos de unión a antígeno de los mismos que bloquean la unión de PD-1 a PD-L1 o PD-L2. En algunos aspectos, el anticuerpo o fragmento de unión a antígeno del mismo que bloquea la unión de PD-1 a PD-L1 puede unirse al mismo epítopo en PD-1 que PD-L1 o puede unirse a un epítopo diferente en PD-1 que PD-L1.
En aspectos alternativos, la presente divulgación proporciona anticuerpos y fragmentos de unión a antígeno de los mismos que estimulan la unión de PD-1 a PD-L1. En determinados aspectos, la presente divulgación proporciona anticuerpos aislados, o fragmentos de unión a antígeno de los mismos que se unen a PD-1, en donde los anticuerpos o fragmentos de unión a antígeno de los mismos potencian la unión de PD-1 a PD-L1. En algunos aspectos, los anticuerpos aislados o fragmentos de unión a antígeno de los mismos comprenden las CDR de una HCVR, en donde cada HCVR tiene una secuencia de aminoácidos seleccionada del grupo que consiste en las SEQ ID NO: 2, 98 y 250; y las CDR de una LCVR, en donde cada LCVR tiene una secuencia de aminoácidos seleccionada del grupo que consiste en las SEQ ID NO: 10, 106 y 202. En algunos aspectos, los anticuerpos aislados o fragmentos de unión a antígeno de los mismos comprenden un par de secuencias de aminoácidos de HCVR/LCVR seleccionadas del grupo que consiste en las SEQ ID NO: 2/10 (por ejemplo, H1M7789N), 98/106 (por ejemplo, H2M7791N), y 250/202 (por ejemplo, H4H9068P2).
La presente divulgación también proporciona anticuerpos y fragmentos de unión a antígeno de los mismos que se unen específicamente a PD-1 del ser humano o de otras especies. En determinados aspectos, los anticuerpos pueden unirse a PD-1 humano y/o a PD-1 de mono cinomolgo.
La presente divulgación también proporciona anticuerpos y fragmentos de unión a antígeno de los mismos que tienen competencia cruzada por la unión a PD-1 con un anticuerpo de referencia o fragmento de unión a antígeno del mismo que comprende las CDR de una HCVR y las CDR de una LCVR, en donde la HCVR y la LCVR tienen cada una una secuencia de aminoácidos seleccionada de las secuencias de HCVR y LCVR enumeradas en la Tabla 1.
En una realización, la divulgación proporciona un anticuerpo aislado o un fragmento de unión a antígeno que tiene una 0 más de las siguientes características: (a) bloquea la unión de PD-1 a PD-L1 o a PD-L2; (b) se une específicamente a PD-1 humano y/o a PD-1 de mono cinomolgo; (c) bloquea la regulación negativa de los linfocitos T inducida por PD-1 y rescata la señalización de los linfocitos T; (d) suprime el crecimiento tumoral y aumenta la supervivencia en sujetos con cáncer de colon; (e) inhibe la proliferación de linfocitos T en un ensayo de reacción de linfocitos mixtos (MLR); y (f) aumenta la secreción de IL-2 y/o interferón-gamma en un ensayo MLR.
En algunos aspectos, el anticuerpo o fragmento de unión a antígeno del mismo puede unirse específicamente a PD-1 de una manera agonista, es decir, puede potenciar o estimular la unión y/o actividad de PD-1; en otros aspectos, el anticuerpo puede unirse específicamente a PD-1 de una manera antagonista, es decir, puede bloquear la unión de PD-1 a su ligando.
En determinadas realizaciones, los anticuerpos o fragmentos de unión a antígeno de la presente invención son biespecíficos y comprenden una primera especificidad de unión a PD-1 y una segunda especificidad de unión a un segundo epítopo diana. El segundo epítopo diana puede ser otro epítopo en PD-1 o en una proteína diferente. En determinadas realizaciones, el epítopo diana puede estar en una célula diferente incluyendo un linfocito T diferente, un linfocito B, una célula tumoral, una célula de tejido autoinmunitaria o una célula infectada por virus.
En un segundo aspecto, la presente invención proporciona moléculas de ácido nucleico que codifican anticuerpos anti-PD-1 o porciones de los mismos de la invención. La presente divulgación proporciona moléculas de ácido nucleico que codifican cualquiera de las secuencias de aminoácidos de HCVR enumeradas en la Tabla 1; en determinados aspectos, la molécula de ácido nucleico comprende una secuencia de polinucleótidos seleccionada de cualquiera de las secuencias de ácido nucleico de HCVR enumeradas en la Tabla 2, o una secuencia sustancialmente similar a la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia con la misma.
La presente divulgación también proporciona moléculas de ácido nucleico que codifican cualquiera de las secuencias de aminoácidos de LCVR enumeradas en la Tabla 1; en determinados aspectos, la molécula de ácido nucleico comprende una secuencia de polinucleótidos seleccionada de cualquiera de las secuencias de ácido nucleico de LCVR enumeradas en la Tabla 2, o una secuencia sustancialmente similar a la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia con la misma.
La presente divulgación también proporciona moléculas de ácido nucleico que codifican cualquiera de las secuencias de aminoácidos de HCDR1 enumeradas en la Tabla 1; en determinados aspectos, la molécula de ácido nucleico comprende una secuencia de polinucleótidos seleccionada de cualquiera de las secuencias de ácido nucleico de HCDR1 enumeradas en la Tabla 2, o una secuencia sustancialmente similar a la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia con la misma.
La presente divulgación también proporciona moléculas de ácido nucleico que codifican cualquiera de las secuencias de aminoácidos de HCDR2 enumeradas en la Tabla 1; en determinados aspectos, la molécula de ácido nucleico comprende una secuencia de polinucleótidos seleccionada de cualquiera de las secuencias de ácido nucleico de HCDR2 enumeradas en la Tabla 2, o una secuencia sustancialmente similar a la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia con la misma.
La presente divulgación también proporciona moléculas de ácido nucleico que codifican cualquiera de las secuencias de aminoácidos de HCDR3 enumeradas en la Tabla 1; en determinados aspectos, la molécula de ácido nucleico comprende una secuencia de polinucleótidos seleccionada de cualquiera de las secuencias de ácido nucleico de HCDR3 enumeradas en la Tabla 2, o una secuencia sustancialmente similar a la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia con la misma.
La presente divulgación también proporciona moléculas de ácido nucleico que codifican cualquiera de las secuencias de aminoácidos de LCDR1 enumeradas en la Tabla 1; en determinados aspectos, la molécula de ácido nucleico comprende una secuencia de polinucleótidos seleccionada de cualquiera de las secuencias de ácido nucleico de LCDR1 enumeradas en la Tabla 2, o una secuencia sustancialmente similar a la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia con la misma.
La presente divulgación también proporciona moléculas de ácido nucleico que codifican cualquiera de las secuencias de aminoácidos de LCDR2 enumeradas en la Tabla 1; en determinados aspectos, la molécula de ácido nucleico comprende una secuencia de polinucleótidos seleccionada de cualquiera de las secuencias de ácido nucleico de LCDR2 enumeradas en la Tabla 2, o una secuencia sustancialmente similar a la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia con la misma.
La presente divulgación también proporciona moléculas de ácido nucleico que codifican cualquiera de las secuencias de aminoácidos de LCDR3 enumeradas en la Tabla 1; en determinados aspectos, la molécula de ácido nucleico comprende una secuencia de polinucleótidos seleccionada de cualquiera de las secuencias de ácido nucleico de LCDR3 enumeradas en la Tabla 2, o una secuencia sustancialmente similar a la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia con la misma.
La presente divulgación también proporciona moléculas de ácido nucleico que codifican una HCVR, en donde la HCVR comprende un conjunto de tres CDR (es decir, HCDR1-HCDR2-HCDR3), en donde el conjunto de secuencias de aminoácidos de HCDR1-HCDR2-HCDR3 es como se define por cualquiera de los anticuerpos anti-PD-1 ilustrativos enumerados en la Tabla 1.
La presente divulgación también proporciona moléculas de ácido nucleico que codifican una LCVR, en donde la LCVR comprende un conjunto de tres CDR (es decir, LCDR1-LCDR2-LCDR3), en donde el conjunto de secuencias de aminoácidos de LCDR1-LCDR2-LCDR3 es como se define por cualquiera de los anticuerpos anti-PD-1 ilustrativos enumerados en la Tabla 1.
La presente divulgación también proporciona moléculas de ácido nucleico que codifican tanto una HCVR como una LCVR, en donde la HCVR comprende una secuencia de aminoácidos de cualquiera de las secuencias de aminoácidos de HCVR enumeradas en la Tabla 1, y en donde la LCVR comprende una secuencia de aminoácidos de cualquiera de las secuencias de aminoácidos de LCVR enumeradas en la Tabla 1. En determinados aspectos, la molécula de ácido nucleico comprende una secuencia de polinucleótidos seleccionada de cualquiera de las secuencias de ácidos nucleicos de HCVR enumeradas en la Tabla 2, o una secuencia sustancialmente similar de la misma que tiene al
menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia con la misma, y una secuencia de polinucleótidos seleccionada de cualquiera de las secuencias de ácido nucleico de LCVR enumeradas en la Tabla 2, o una secuencia sustancialmente similar a la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia con la misma. En ciertos casos de acuerdo con este aspecto de la divulgación, la molécula de ácido nucleico codifica una HCVR y una LCVR, en donde la HCVR y la LCVR proceden las dos del mismo anticuerpo anti-PD-1 enumerado en la Tabla 1.
La presente divulgación proporciona moléculas de ácido nucleico que codifican cualquiera de las secuencias de aminoácidos de cadena pesada enumeradas en la Tabla 3. La presente divulgación también proporciona moléculas de ácido nucleico que codifican cualquiera de las secuencias de aminoácidos de cadena ligera enumeradas en la Tabla 3.
La presente divulgación también proporciona moléculas de ácido nucleico que codifican tanto una cadena pesada (HC) como una cadena ligera (LC), en donde la HC comprende una secuencia de aminoácidos de cualquiera de las secuencias de aminoácidos de HC enumeradas en la Tabla 3, y en donde la LC comprende una secuencia de aminoácidos de cualquiera de las secuencias de aminoácidos de LC enumeradas en la Tabla 3.
En un aspecto relacionado, la presente invención proporciona vectores de expresión recombinante que comprenden moléculas de la invención. La presente divulgación incluye vectores de expresión recombinantes que comprenden cualquiera de las moléculas de ácido nucleico mencionadas anteriormente, es decir, moléculas de ácido nucleico que codifican cualquiera de las secuencias de HCVR, LCVR y/o CDR como se exponen en la Tabla 1. La presente divulgación también proporciona vectores de expresión recombinantes capaces de expresar un polipéptido que comprende una cadena pesada o ligera de un anticuerpo anti-PD-1. Por ejemplo, la presente divulgación incluye vectores de expresión recombinantes que comprenden cualquiera de las moléculas de ácido nucleico mencionadas anteriormente, es decir, moléculas de ácido nucleico que codifican cualquiera de las secuencias de cadena pesada o cadena ligera como se exponen en la Tabla 3. Dentro del alcance de la presente invención también se incluyen células hospedadoras en donde se han introducido dichos vectores de la invención, así como métodos para producir los anticuerpos o porciones de los mismos de la invención mediante el cultivo de las células hospedadoras de la invención en condiciones que permiten la producción de los anticuerpos o fragmentos de anticuerpo, y la recuperación de los anticuerpos y fragmentos de anticuerpo así producidos.
En un aspecto adicional, la presente divulgación proporciona moléculas de unión a antígeno multiespecíficas y fragmentos de unión a antígeno de las mismas que comprenden una primera especificidad de unión a antígeno que se une específicamente a PD-1 y una segunda especificidad de unión a antígeno que se une específicamente a un antígeno seleccionado del grupo que consiste en un antígeno específico de una célula tumoral, un antígeno específico de un tejido autoinmunitario, un antígeno específico de células infectadas, un coinhibidor de linfocitos T, un receptor de linfocitos T, un receptor Fc, PD-L1 y PD-1. En determinados aspectos, la primera especificidad de unión a antígeno puede comprender tres CDR derivadas de una HCVR con una secuencia de aminoácidos seleccionada de las secuencias de HCVR de la Tabla 1 y tres CDR derivadas de una LCVR con una secuencia de aminoácidos seleccionada de las secuencias de LCVR de la Tabla 1. En un aspecto, la primera especificidad de unión a antígeno puede comprender el dominio extracelular de PD-L1. La segunda especificidad de unión a antígeno puede dirigirse a un antígeno en la misma célula que PD-1 o en una célula diferente del mismo tipo de tejido o de un tipo de tejido diferente. Por ejemplo, la molécula multiespecífica de unión a antígeno puede unirse a un linfocito T en donde la primera especificidad de unión a antígeno puede unirse específicamente a PD-1 y la segunda especificidad de unión a antígeno puede unirse a un receptor de un linfocito T en el linfocito T. Como alternativa, en otro aspecto, la primera especificidad de unión a antígeno puede unirse específicamente a PD-1 en un linfocito T y la segunda especificidad de unión a antígeno puede dirigirse a un antígeno/receptor en un linfocito B o un macrófago o célula presentadora de antígeno. En determinados aspectos, la segunda especificidad de unión a antígeno puede estar dirigida a un antígeno asociado con un tejido autoinmunitario. En un aspecto, la primera especificidad de unión a antígeno puede comprender un dominio extracelular de PD-L1 y la segunda especificidad de unión a antígeno puede unirse a otro epítopo en PD-1. En determinados aspectos, la primera especificidad de unión a antígeno se une a PD-1 con una afinidad menor, por ejemplo, con una Kd de más de 10'7 M, más de 10'6 M, más de 10'5 M, o más de 10'4 M.
En un tercer aspecto, la invención proporciona una composición farmacéutica que comprende un anticuerpo humano recombinante o fragmento del mismo de la invención que se une específicamente a PD-1 y un vehículo farmacéuticamente aceptable. En un aspecto relacionado, la invención presenta una composición que es una combinación de un anticuerpo anti-PD-1 de la invención y un segundo agente terapéutico. En una realización, el segundo agente terapéutico es cualquier agente que se combine de forma ventajosa con un anticuerpo anti-PD-1. Los agentes ilustrativos que pueden combinarse de forma ventajosa con un anticuerpo anti-PD-1 incluyen, sin limitación, otros agentes que se unen y/o modulan la señalización de PD-1 (incluyendo otros anticuerpos o fragmentos de unión a antígeno de los mismos, etc.) y/o agentes que no se unen directamente a PD-1 pero que, sin embargo, modulan la activación de las células inmunitarias. En cualquier parte del presente documento se divulgan terapias de combinación y coformulaciones adicionales que implican los anticuerpos anti-PD-1 de la presente invención.
En un aspecto adicional, la divulgación proporciona métodos para modular la respuesta inmunitaria en un sujeto, comprendiendo el método administrar una cantidad terapéuticamente eficaz de un anticuerpo anti-PD-1 o fragmento
de unión a antígeno del mismo de la invención al sujeto que lo necesita. En determinados aspectos, la divulgación proporciona métodos para potenciar la respuesta inmunitaria en un sujeto, comprendiendo los métodos administrar al sujeto una cantidad eficaz de un anticuerpo o fragmento del mismo de la invención que se une a PD-1 y bloquea la unión de PD-1 a PD-L1. En un aspecto, la divulgación proporciona un método para estimular o potenciar la estimulación de los linfocitos T en un sujeto. En un aspecto, la divulgación proporciona métodos para inhibir un linfocito T regulador (Treg) en un sujeto, comprendiendo los métodos administrar una cantidad terapéuticamente eficaz de un anticuerpo bloqueante o fragmento de unión a antígeno del mismo de la invención al sujeto que lo necesita. En determinados aspectos, el sujeto que lo necesita puede padecer una enfermedad o trastorno tal como cáncer o infección vírica. En aspectos alternativos, la divulgación proporciona métodos para inhibir o suprimir la activación de linfocitos T en un sujeto, comprendiendo los métodos administrar una cantidad terapéuticamente eficaz de un anticuerpo activante o fragmento del mismo de la divulgación al sujeto que lo necesita. En un aspecto, el sujeto puede padecer una enfermedad autoinmunitaria o trastorno.
En otro aspecto, la divulgación proporciona métodos terapéuticos para el tratamiento de una enfermedad o trastorno tal como cáncer, enfermedad autoinmunitaria o infección vírica en un sujeto usando un anticuerpo anti-PD-1 o porción de unión a antígeno de un anticuerpo de la divulgación, en donde los métodos terapéuticos comprenden administrar una cantidad terapéuticamente eficaz de una composición farmacéutica que comprende un anticuerpo o fragmento de un anticuerpo de la divulgación a un sujeto que lo necesita. El trastorno tratado es cualquier enfermedad o afección que se mejora, alivia, inhibe o previene mediante la estimulación o inhibición de la actividad o señalización de PD-1. En determinados aspectos, el anticuerpo o fragmento de unión a antígeno del mismo de la invención se administra en combinación con un segundo agente terapéutico al sujeto que lo necesita. El segundo agente terapéutico puede seleccionarse del grupo que consiste en un anticuerpo contra otro coinhibidor de linfocitos T, un anticuerpo contra un antígeno de célula tumoral, un anticuerpo contra un receptor de linfocitos T, un anticuerpo contra un receptor de Fc, un anticuerpo contra un epítopo de una célula infectada por virus, un anticuerpo contra un antígeno de tejido autoinmunitario, un anticuerpo contra PD-L1, un agente citotóxico, un fármaco contra el cáncer, un fármaco antivírico, un fármaco antiinflamatorio (por ejemplo, corticoesteroides), agente quimioterapéutico, radioterapia, un inmunosupresor y cualquier otro fármaco o terapia conocido en la técnica. En determinados aspectos, el segundo agente terapéutico puede ser un agente que ayude a contrarrestar o reducir cualquier posible efecto o efectos secundarios asociados con un anticuerpo o fragmento de unión a antígeno del mismo de la invención, si ocurriera dicho efecto o efectos.
En determinadas realizaciones, la presente divulgación proporciona métodos para suprimir el crecimiento tumoral. En determinados aspectos, la presente divulgación proporciona métodos para aumentar la supervivencia de los pacientes con cáncer. Los ejemplos de cáncer incluyen, aunque no de forma limitativa, cáncer primario y/o recurrente, incluyendo cáncer de cerebro (por ejemplo, glioblastoma multiforme), cáncer de pulmón (por ejemplo, cáncer de pulmón no microcítico), carcinoma de células escamosas de cabeza y cuello, carcinoma de células renales, melanoma, mieloma múltiple, cáncer de próstata y cáncer de colon. Los métodos comprenden administrar una composición farmacéutica que comprende una cantidad terapéuticamente eficaz de un anticuerpo anti-PD-1 de la presente invención en combinación con un segundo agente terapéutico seleccionado del grupo que consiste en un antagonista del factor de crecimiento endotelial vascular (VEGF) (por ejemplo, aflibercept, bevacizumab), un inhibidor de la angiopoyetina-2 (Ang2) (por ejemplo, un anticuerpo anti-Ang2 tal como nesvacumab), un inhibidor del gen de activación de linfocitos 3 (LAG-3), un inhibidor del antígeno de linfocitos T citotóxicos 4 (CTLA-4) (por ejemplo, ipilimumab), un agente quimioterapéutico y radioterapia. En otra parte del presente documento se describen ejemplos adicionales de terapias/agentes terapéuticos adicionales que se pueden usar en combinación con un anticuerpo anti-PD-1 de la invención para usar en el tratamiento del cáncer.
El anticuerpo o fragmento del mismo se puede administrar por vía subcutánea, intravenosa, intradérmica, intraperitoneal, oral, intramuscular o intracraneal. El anticuerpo o fragmento del mismo se puede administrar a una dosis de aproximadamente 0,1 mg/kg de peso corporal a aproximadamente 100 mg/kg de peso corporal del sujeto.
La presente divulgación también incluye el uso de un anticuerpo anti-PD-1 o fragmento de unión a antígeno del mismo de la invención en la fabricación de un medicamento para el tratamiento de una enfermedad o trastorno que se beneficiaría del bloqueo o mejora de la unión y/o señalización de PD-1.
Otras realizaciones resultarán evidentes tras la revisión de la descripción detallada adjunta.
Breve descripción de las figuras
La Figura 1 es un esquema del bioensayo de PD-1 basado en luciferasa descrito en el Ejemplo 8 del presente documento. Panel A: Células Jurkat inactivas; Panel B: Las células Jurkat se activan mediante la agrupación del receptor de linfocitos T (TCR) a través del anticuerpo biespecífico CD3xCD20; Panel C: La activación de PD-1 atenúa la respuesta en células Jurkat activadas; Panel D: El bloqueo de PD-1 rescata la respuesta en células Jurkat activadas.
La Figura 2 ilustra el crecimiento tumoral y los resultados de supervivencia para ratones implantados con linfocitos tumorales de colon-26 en el día 0 y tratados con las combinaciones indicadas de moléculas mediante inyección en los días 3, 6, 10, 13 y 19 ("modelo de tumor de tratamiento temprano"). El gráfico muestra el volumen tumoral (en
mm3) para los diferentes grupos experimentales en varios puntos temporales después de la implantación. Las flechas hacia arriba a lo largo del eje X indican el momento de las inyecciones de tratamiento. "mlgG2a" es control de isotipo de IgG2; "Fc" es el control de Fc humano; "Trampa de VEGf" es aflibercept; "anti-PD-1" es anti-PD-1 de ratón clon RPMI-14; "anti-PD-L1" es un anticuerpo monoclonal anti-PD-L1 como se describe en otra parte del presente documento.
La Figura 3 ilustra el crecimiento tumoral y los resultados de supervivencia para ratones implantados con linfocitos tumorales de colon-26 en el día 0 y tratados con las combinaciones indicadas de moléculas mediante inyección en los días 3, 6, 10, 13 y 19 ("modelo de tumor de tratamiento temprano"). El gráfico muestra el volumen tumoral (en mm3) de ratones individuales en cada grupo experimental en el día 28 después de la implantación. "mlgG2a" es control de isotipo de IgG2; "Fc" es el control de Fc humano; "Trampa de VEGF" es aflibercept; "anti-PD-1" es anti-PD-1 de ratón clon RPMI-14; "anti-PD-L1" es un anticuerpo monoclonal anti-PD-L1 como se describe en otra parte del presente documento.
Descripción detallada
Antes de describir los presentes métodos, se ha de entender que la presente invención no se limita a los métodos ni a las condiciones experimentales descritos en particular, ya que dichos métodos y condiciones pueden variar. También debe entenderse que la terminología usada en el presente documento únicamente tiene el fin de describir realizaciones particulares, y no se pretende que sea limitante, ya que el alcance de la presente invención estará limitado únicamente por las reivindicaciones adjuntas.
A menos que se definan de otra manera, todos los términos técnicos y científicos usados en el presente documento tienen el mismo significado que el entendido comúnmente por un experto habitual en la materia a la cual pertenece la presente invención. Aunque puede usarse cualquier método y material similar o equivalente a los que se describen en el presente documento en la práctica o ensayo de la presente invención, a continuación, se describen métodos y materiales preferidos.
El término "PD-1" se refiere a la proteína de muerte programada-1, un coinhibidor de linfocitos T, también conocido como CD279. La secuencia de aminoácidos de PD-1 de longitud completa se proporciona en GenBank con número de registro NP_005009.2 y también se hace referencia a ella en el presente documento como SEQ ID NO: 327. El término "PD-1" también incluye variantes de la proteína PD-1 que tienen la secuencia de aminoácidos de las SEQ ID NO: 321, 322, 323, o 324. El término "PD-1" incluye PD-1 recombinante o un fragmento de la misma. El término también abarca PD-1 o un fragmento de la misma acoplado a, por ejemplo, una etiqueta de histidina, Fc de ratón o humano, o una secuencia de señal tal como ROR1. Por ejemplo, el término incluye secuencias ilustradas por las SEQ ID NO: 323 o 324, que comprenden un Fc de ratón (mlgG2a) o Fc humano (hlgG1) en el extremo C-terminal, acoplado a los restos de aminoácidos 25-170 de PD-1 de longitud completa con un cambio C93S. Las variantes de proteínas, como se ilustra en la SEQ ID NO: 321, comprenden una etiqueta de histidina en el extremo C-terminal, acoplada a los restos de aminoácidos 25-170 de PD-1 de longitud completa. A menos que se especifique que proviene de una especie no humana, el término "PD-1" significa PD-1 humana.
PD-1 es un miembro de la familia de coinhibidores de linfocitos T CD28/CTLA-4/ICOS. PD-1 es una proteína de 288 aminoácidos con un dominio N-terminal extracelular que es similar a IgV, un dominio transmembrana y un dominio intracelular que contiene un motivo inhibidor inmunorreceptor basado en tirosina (ITIM) y un motivo intercambiable inmunorreceptor basado en tirosina (ITSM) (Chattopadhyay et al. 2009, Immunol. Rev.). El receptor de PD-1 tiene dos ligandos, el ligando de PD 1 (PD-L1) y PD-L2.
El término "PD-L1" se refiere al ligando del receptor de PD-1 también conocido como CD274 y B7H1. La secuencia de aminoácidos de PD-L1 de longitud completa se proporciona en GenBank con número de registro NP_054862.1 y también se hace referencia a ella en el presente documento como SEQ ID NO: 328. El término también abarca PD-L1 o un fragmento del mismo acoplado a, por ejemplo, una etiqueta de histidina, Fc de ratón o humano, o una secuencia de señal tal como ROR1. Por ejemplo, el término incluye secuencias ilustradas por las SEQ ID NO: 325 o 326, que comprenden un Fc de ratón (mlgG2a) o Fc humano (hlgG1) en el extremo C-terminal, acoplado a los restos de aminoácidos 19-239 de PD-L1 de longitud completa. PD-L1 es una proteína de 290 aminoácidos con un dominio extracelular similar a IgV, un dominio transmembrana y un dominio intracelular altamente conservado de aproximadamente 30 aminoácidos. PD-L1 se expresa constitutivamente en muchas células, tales como las células presentadoras de antígeno (por ejemplo, células dendríticas, macrófagos y linfocitos B) y en células hematopoyéticas y no hematopoyéticas (por ejemplo, células endoteliales vasculares, islotes pancreáticos y sitios de privilegio inmunitario). PD-L1 también se expresa en una amplia variedad de tumores, células infectadas por virus y tejido autoinmunitario, y es un componente del medio inmunosupresor (Ribas 2012, NEJM 366: 2517-2519).
Como se utiliza en el presente documento, el término "coinhibidor de linfocitos T" se refiere a un ligando y/o receptor que modula la respuesta inmunitaria a través de la activación o supresión de linfocitos T. La expresión "coinhibidor de linfocitos T", también conocido como molécula de coseñalización de linfocitos T, incluye, pero sin limitación, proteína del gen de activación de linfocitos 3 (LAG-3, también conocida como CD223), antígeno de linfocitos T citotóxicos 4 (CTLA-4), atenuador de linfocitos B y T (BTLA), CD-28, 2B4, LY108, inmunoglobulina de linfocitos T y mucina 3 (TIM3), inmunorreceptor de linfocitos T con inmunoglobulina e ITIM (TIGIT; también conocido como VSIG9), receptor tipo
inmunoglobulina asociado a leucocitos 1 (LAIR1; también conocido como CD305), coestimulador de linfocitos T inducible (ICOS; también conocido como CD278), dominio V de Ig supresor de la activación de linfocitos T (VISTA) y CD160.
Como se utiliza en el presente documento, la expresión "receptor de Fc" se refiere a la proteína receptora de superficie que se encuentra en las células inmunitarias, incluidos los linfocitos B, linfocitos citolíticos naturales, macrófagos, basófilos, neutrófilos y mastocitos, que tiene una especificidad de unión para la región Fc de un anticuerpo. La expresión "receptor Fc" incluye, pero sin limitación, un receptor Fcy [por ejemplo, FcyRI (CD64), FcyRIIA (CD32), FcyRIIB (CD32), FcyRIIIA (CD16a) y FcyRIIIB (CD16b)], receptor Fca (por ejemplo, FcaRl o CD89) y receptor Fc£ [por ejemplo, FceRI, y FceRII (Cd 23)].
El término "anticuerpo", como se utiliza en el presente documento, pretende hacer referencia a moléculas de inmunoglobulina compuestas por cuatro cadenas polipeptídicas, dos cadenas pesadas (H) y dos cadenas ligeras (L) interconectadas por enlaces disulfuro (es decir, "moléculas de anticuerpo completo"), así como multímeros de las mismas (por ejemplo, IgM) o fragmentos de unión a antígeno de las mismas. Cada cadena pesada está compuesta por una región variable de la cadena pesada ("HCVR" o "Vh") y una región constante de la cadena pesada (comprendida por los dominios Ch1, Ch2 y Ch3). Cada cadena ligera está compuesta por una región variable de la cadena ligera ("LCVR o "Vl") y una región constante de la cadena ligera (Cl). Las regiones Vh y Vl pueden subdividirse además en regiones de hipervariabilidad, denominadas regiones determinantes de la complementariedad (CDR), intercaladas con regiones que están más conservadas, denominadas regiones marco (FR). Cada Vh y Vl está compuesta por tres CDR y cuatro FR, dispuestas desde el extremo amino hasta el extremo carboxilo en el siguiente orden: FR1, CDR1, FR2, Cd R2, FR3, Cd R3, FR4. En determinados aspectos de la divulgación, las FR del anticuerpo (o fragmento de unión a antígeno del mismo) pueden ser idénticas a las secuencias de la línea germinal humana, o pueden modificarse natural o artificialmente. Una secuencia consenso de aminoácidos puede definirse basándose en un análisis paralelo de dos o más CDR.
También es posible la sustitución de uno o más restos de CDR o la omisión de una o más CDR. Se han descrito anticuerpos en la bibliografía científica en los que se puede prescindir de una o dos CDR para la unión. Padlan et al. (1995 FASEB J. 9:133-139) analizaron las regiones de contacto entre los anticuerpos y sus antígenos, basándose en estructuras cristalinas publicadas, y concluyeron que solo aproximadamente de un quinto a un tercio de los restos de CDR realmente entran en contacto con el antígeno. Padlan también descubrió muchos anticuerpos en los que una o dos CDR no tenían aminoácidos en contacto con un antígeno (véase también, Vajdos et al. 2002 J Mol Biol 320:415-428).
Los restos de CDR que no entran en contacto con el antígeno se pueden identificar basándose en estudios anteriores (por ejemplo, los restos H60-H65 en CDRH2 a menudo no son necesarios), de regiones de CDR de Kabat que se encuentran fuera de CDR de Chothia, por modelización molecular y/o empíricamente. Si se omite una CDR o uno o más restos de la misma, generalmente se sustituye con un aminoácido que ocupa la posición correspondiente en otra secuencia de anticuerpo humano o un consenso de dichas secuencias. Las posiciones para la sustitución dentro de CDR y los aminoácidos a sustituir también pueden seleccionarse empíricamente. Las sustituciones empíricas pueden ser sustituciones conservadoras o no conservadoras.
Los anticuerpos monoclonales anti-PD-1 totalmente humanos divulgados en el presente documento pueden comprender una o más sustituciones, inserciones y/o deleciones de aminoácidos en las regiones marco y/o CDR de los dominios variables de la cadena pesada y ligera en comparación con las secuencias de la línea germinal correspondientes. Dichas mutaciones pueden determinarse fácilmente comparando las secuencias de aminoácidos que se divulgan en el presente documento con secuencias de la línea germinal disponibles en, por ejemplo, bases de datos de secuencias de anticuerpos públicas. La presente divulgación incluye anticuerpos y fragmentos de unión a antígeno de los mismos, que se obtienen a partir de cualquiera de las secuencias de aminoácidos que se divulgan en el presente documento, en donde uno o más aminoácidos dentro de una o más regiones marco y/o CDR están mutados en el resto o restos correspondientes de la secuencia de la línea germinal de la que deriva el anticuerpo, o en el resto o restos correspondientes de otra secuencia de la línea germinal humana, o en una sustitución de aminoácidos conservadora del resto o restos de la línea germinal correspondientes (dichos cambios de secuencia se denominan en el presente documento colectivamente "mutaciones de la línea germinal"). Un experto habitual en la materia, partiendo de las secuencias de la región variable de cadena pesada y ligera que se divulgan en el presente documento, puede producir fácilmente numerosos anticuerpos y fragmentos de unión a antígeno que comprenden una o más mutaciones de la línea germinal individuales o combinaciones de las mismas. En determinados aspectos, todos los restos de la región marco y/o CDR dentro de los dominios Vh y/o Vl mutan de vuelta a los restos encontrados en la secuencia de la línea germinal original de la que se obtuvo el anticuerpo. En otros aspectos, únicamente determinados restos mutan de vuelta a la secuencia de la línea germinal original, por ejemplo, únicamente los restos mutados encontrados dentro de los primeros 8 aminoácidos de FR1 o dentro de los últimos 8 aminoácidos de FR4, o únicamente los restos mutados encontrados dentro de CDR1, CDR2 o CDR3. En otros aspectos, uno o más del resto o restos marco y/o CDR están mutados en el resto o restos correspondientes de una secuencia de línea germinal diferente (es decir, una secuencia de línea germinal que es diferente de la secuencia de línea germinal de la cual se obtuvo originalmente el anticuerpo). Asimismo, los anticuerpos de la presente divulgación pueden contener cualquier combinación de dos o más mutaciones de la línea germinal dentro de las regiones marco y/o CDR, por ejemplo, en
donde ciertos restos individuales están mutados en el resto correspondiente de una secuencia de la línea germinal particular mientras que otros restos determinados que difieren de la secuencia de la línea germinal original se mantienen o están mutados en el resto correspondiente de una secuencia de la línea germinal diferente. Una vez obtenidos, los anticuerpos y fragmentos de unión a antígeno que contienen una o más mutaciones de la línea germinal pueden ensayarse fácilmente con respecto a una o más propiedades deseadas tales como, especificidad de unión mejorada, mayor afinidad de unión, propiedades biológicas antagonista o agonista mejoradas o potenciadas (según sea el caso), menor inmunogenia, etc. Los anticuerpos y fragmentos de unión a antígeno obtenidos de esta manera general están incluidos en la presente divulgación.
La presente divulgación también incluye anticuerpos monoclonales anti-PD-1 totalmente humanos que comprenden variantes de cualquiera de las secuencias de aminoácidos de HCVR, LCVR, y/o CDR divulgadas en el presente documento que tienen una o más sustituciones conservadoras. Por ejemplo, la presente divulgación incluye anticuerpos anti-PD-1 que tienen secuencias de aminoácidos de HCVR, LCVR, y/o CDR con, por ejemplo, 10 o menos, 8 o menos, 6 o menos, 4 o menos, etc., sustituciones conservadoras de aminoácidos respecto a cualquiera de las secuencias de aminoácidos de HCVR, LCVR, y/o CDR divulgadas en el presente documento.
La expresión "anticuerpo humano", como se utiliza en el presente documento, pretende incluir anticuerpos que tienen regiones variables y constantes derivadas de secuencias de inmunoglobulina de la línea germinal humana. Los mAb humanos de la divulgación pueden incluir restos de aminoácidos no codificados por las secuencias de inmunoglobulinas de la línea germinal humana (por ejemplo, mutaciones introducidas por mutagénesis aleatoria y específicas de sitio in vitro o mediante mutación somática in vivo), por ejemplo, en las c Dr y en particular CDR3. Sin embargo, la expresión "anticuerpo humano", como se utiliza en el presente documento, no pretende incluir mAb en los cuales las secuencias CDR derivadas de la línea germinal de otra especie de mamífero (por ejemplo, ratón), se han injertado en secuencias FR humanas. La expresión incluye anticuerpos producidos de forma recombinante en un mamífero no humano o en células de un mamífero no humano. La expresión no pretende incluir anticuerpos aislados de o generados en un sujeto humano.
El término "recombinante", como se utiliza en el presente documento, se refiere a anticuerpos o fragmentos de unión a antígeno de los mismos de la invención creados, expresados, aislados u obtenidos mediante tecnologías o métodos conocidos en la técnica como tecnología de ADN recombinante que incluye, por ejemplo, corte y empalme de ADN y expresión transgénica. La expresión se refiere a anticuerpos expresados en un mamífero no humano (incluyendo mamíferos no humanos transgénicos, por ejemplo, ratones transgénicos) o en un sistema de expresión celular (por ejemplo, células CHO) o aislados de una biblioteca de anticuerpos humanos combinatorios recombinantes.
La expresión "moléculas de unión a antígeno multiespecíficas", como se usa en el presente documento, se refiere a moléculas de unión a antígeno biespecíficas, triespecíficas o multiespecíficas y a fragmentos de unión a antígeno de las mismas. Las moléculas de unión a antígeno multiespecíficas pueden ser específicas de diferentes epítopos de un polipéptido diana o pueden contener dominios de unión a antígeno específicos de los epítopos de más de un polipéptido diana. Una molécula de unión a antígeno multiespecífica puede ser un solo polipéptido multifuncional o puede ser un complejo multimérico de dos o más polipéptidos que se asocian covalente o no covalentemente entre sí. La expresión "moléculas de unión a antígeno multiespecíficas" incluye anticuerpos de la presente invención que pueden unirse o coexpresarse con otra molécula funcional, por ejemplo, otro péptido o proteína. Por ejemplo, un anticuerpo o fragmento del mismo puede unirse de forma funcional (por ejemplo, por acoplamiento químico, fusión genética, asociación no covalente o de otro modo) a una o más entidades moleculares distintas, tales como una proteína o fragmento de la misma para producir una molécula de unión a antígeno biespecífica o multiespecífica con una segunda especificidad de unión. De acuerdo con la presente invención, la expresión "moléculas de unión a antígeno multiespecíficas" incluye también anticuerpos biespecíficos, triespecíficos o multiespecíficos o fragmentos de unión a antígeno de los mismos. En determinadas realizaciones, un anticuerpo de la presente invención se une funcionalmente a otro anticuerpo o fragmento de unión a antígeno del mismo para producir un anticuerpo biespecífico con una segunda especificidad de unión. Los anticuerpos biespecíficos y multiespecíficos de la presente invención se describen en otra parte del presente documento.
La expresión "se une específicamente", o "se une específicamente a", o similares, significa que un anticuerpo o fragmento de unión a antígeno del mismo forma un complejo con un antígeno que es relativamente estable en condiciones fisiológicas. La unión específica puede caracterizarse por una constante de disociación en el equilibrio de al menos aproximadamente 1 x 10'8 M o menos (por ejemplo, una Kd menor indica una unión más estrecha). Los métodos para determinar si dos moléculas se unen específicamente son bien conocidos en la técnica e incluyen, por ejemplo, diálisis en equilibrio, resonancia de plasmón superficial y similares. Como se describe en el presente documento, los anticuerpos se han identificado mediante resonancia de plasmón superficial, por ejemplo, BIACORE™, que se une específicamente a PD-1. Además, los anticuerpos multiespecíficos, que se unen a un dominio en PD-1 y uno o más antígenos adicionales o uno biespecífico que se une a dos regiones diferentes de PD-1 se consideran, no obstante, anticuerpos que se "unen específicamente", como se usan en el presente documento.
La expresión anticuerpo "de alta afinidad" se refiere a aquellos mAb que tienen una afinidad de unión a PD-1, expresada como Kd, de al menos 10'7 M; preferentemente 10'8 M; más preferentemente de 10'9 M, incluso más preferentemente de 10'10 M, incluso más preferentemente de 10'11 M, según se mide mediante resonancia de plasmón
superficial, por ejemplo, BIACORE™ o ELISA de afinidad en solución.
Por la expresión "disociación lenta", "Koff' o "kd" se entiende un anticuerpo que se disocia de PD-1, con una constante de velocidad de 1 x 10-3 s-1 o menos, preferentemente 1 x 10-4 s-1 o menos, tal como se determina mediante resonancia de plasmón superficial, por ejemplo, BIACORE™.
Las expresiones "porción de unión a antígeno" de un anticuerpo, "fragmento de unión a antígeno" de un anticuerpo y similares, como se utiliza en el presente documento, incluyen cualquier polipéptido o glucoproteína de origen natural, obtenible enzimáticamente, sintético o modificado por ingeniería genética que se une específicamente a un antígeno para formar un complejo. Las expresiones "fragmento de unión a antígeno" de un anticuerpo o "fragmento de anticuerpo", como se utiliza en el presente documento, se refieren a uno o más fragmentos de un anticuerpo que conservan la capacidad de unirse a PD-1.
En realizaciones específicas, un anticuerpo o fragmentos de anticuerpo de la invención pueden estar conjugados con una fracción tal como un ligando o una fracción terapéutica ("inmunoconjugado"), tal como un antibiótico, un segundo anticuerpo anti-PD-1, o un anticuerpo contra otro antígeno, tal como un antígeno específico de tumor, un antígeno de tejido autoinmunitario, un antígeno de célula infectada por virus, un receptor Fc, un receptor de linfocitos T o un coinhibidor de linfocitos T, o una inmunotoxina, o cualquier otra fracción terapéutica útil para tratar una enfermedad o afección que incluye cáncer, enfermedad autoinmunitaria o infección vírica crónica.
Un "anticuerpo aislado", como se utiliza en el presente documento, pretende hacer referencia a un anticuerpo que está sustancialmente libre de otros anticuerpos (Abs) que tienen diferentes especificidades antigénicas (por ejemplo, un anticuerpo aislado que se une específicamente a PD-1 o un fragmento del mismo, está sustancialmente libre de Abs que se unen específicamente a antígenos diferentes a PD-1.
Un "anticuerpo de bloqueo" o un "anticuerpo neutralizante", como se usa en el presente documento (o un "anticuerpo que neutraliza la actividad de PD-1" o "anticuerpo antagonista"), pretende hacer referencia a un anticuerpo cuya unión a PD-1 da como resultado la inhibición de al menos una actividad biológica de PD-1. Los anticuerpos de la invención previenen o bloquean la unión de PD-1 a PD-L1.
Un "anticuerpo activador" o un "anticuerpo potenciador", como se usa en el presente documento (o un "anticuerpo agonista"), pretende hacer referencia a un anticuerpo cuya unión a PD-1 da como resultado el aumento o la estimulación de al menos una actividad biológica de PD-1. Por ejemplo, un anticuerpo de la divulgación puede aumentar la unión de PD-1 a PD-L1.
La expresión "resonancia de plasmón superficial", como se utiliza en el presente documento, se refiere a un fenómeno óptico que permite el análisis de interacciones biomoleculares en tiempo real mediante la detección de alteraciones en concentraciones de proteína dentro de una matriz biodetectora, por ejemplo usando el sistema BIACORE™ (Pharmacia Biosensor a B, Uppsala, Suecia y Piscataway, N.J.).
El término "Kd", como se utiliza en el presente documento, pretende hacer referencia a la constante de disociación en equilibrio de una interacción particular de anticuerpo-antígeno.
El término "epítopo" se refiere a un determinante antigénico que interactúa con un sitio de unión a antígeno específico en la región variable de una molécula de anticuerpo conocida como parátopo. Un único antígeno puede tener más de un epítopo. Por tanto, diferentes anticuerpos pueden unirse a diferentes áreas en un antígeno o puede tener diferentes efectos biológicos. El término "epítopo" se refiere también a un lugar sobre un antígeno al que responden linfocitos B y/o T. También se refiere a una región de un antígeno a la que se une un anticuerpo. Los epítopos pueden definirse como estructurales o funcionales. Los epítopos funcionales son generalmente un subconjunto de los epítopos estructurales y tienen aquellos restos que contribuyen directamente a la afinidad de la interacción. Los epítopos también pueden ser conformacionales, es decir, compuestos de aminoácidos no lineales. En determinados casos, los epítopos pueden incluir determinantes que son agrupaciones superficiales químicamente activas de moléculas tales como aminoácidos, cadenas laterales de azúcares, grupos fosforilo o grupos sulfonilo y, en determinados casos, pueden tener características estructurales tridimensionales específicas y/o características de carga específicas.
La expresión "identidad sustancial" o "sustancialmente idéntico", cuando hace referencia a un ácido nucleico o fragmento del mismo, indica que, cuando se alinean de forma óptima con inserciones o eliminaciones de nucleótidos adecuadas con otro ácido nucleico (o su cadena complementaria), existe identidad de secuencia de nucleótidos en al menos aproximadamente un 90 % y, más preferentemente, al menos aproximadamente un 95 %, 96 %, 97 %, 98 % o 99 % de las bases nucleotídicas, según se mide mediante cualquier algoritmo bien conocido de identidad de secuencia, tales como FASTA, BLAST o GAP, como se analiza a continuación. Una molécula de ácido nucleico que tiene identidad sustancial con una molécula de ácido nucleico de referencia puede, en determinados casos, codificar un polipéptido que tiene la misma secuencia de aminoácidos o una sustancialmente similar al polipéptido codificado por la molécula de ácido nucleico de referencia.
Aplicada a polipéptidos, la expresión "similitud sustancial" o "sustancialmente similar" significa que dos secuencias
peptídicas, cuando se alinean de forma óptima, tal como mediante los programas GAP o BESTFIT usando ponderaciones de hueco predeterminadas, comparten al menos un 90 % de identidad de secuencia, incluso más preferentemente al menos el 95 %, 98 % o 99 % de identidad de secuencia. Preferentemente, las posiciones de los restos, que no son idénticas, difieren en sustituciones de aminoácidos conservadoras. Una "sustitución de aminoácido conservadora" es una en donde un resto de aminoácido está sustituido por otro resto de aminoácido que tiene una cadena lateral (grupo R) con propiedades químicas similares (por ejemplo, carga o hidrofobia). En general, una sustitución de aminoácidos conservadora no cambiará sustancialmente las propiedades funcionales de una proteína. En los casos donde dos o más secuencias de aminoácidos difieren entre sí por sustituciones conservadoras, el porcentaje o grado de similitud puede ajustarse a la alza para corregir la naturaleza conservadora de la sustitución. Los medios para hacer este ajuste son bien conocidos por los expertos en la materia. Véase, por ejemplo, Pearson (1994) Methods Mol. Biol. 24: 307-331. Los ejemplos de grupos de aminoácidos que tienen cadenas laterales con propiedades químicas similares incluyen 1) cadenas laterales alifáticas: glicina, alanina, valina, leucina e isoleucina; 2) cadenas laterales alifáticas-hidroxilo: serina y treonina; 3) cadenas laterales que contienen amida: asparagina y glutamina; 4) cadenas laterales aromáticas: fenilalanina, tirosina y triptófano; 5) cadenas laterales básicas: lisina, arginina e histidina; 6) cadenas laterales ácidas: aspartato y glutamato, y 7) cadenas laterales que contienen azufre: cisteína y metionina. Son grupos de sustitución conservadora de aminoácidos preferidos: valina-leucina-isoleucina, fenilalanina-tirosina, lisina-arginina, alanina-valina, glutamato-aspartato y asparagina-glutamina. Como alternativa, un remplazo conservador es cualquier cambio que tenga un valor positivo en la matriz de probabilidad logarítmica PAM250 divulgada en Gonnet et al. (1992) Science 256: 1443 45. Un remplazo "moderadamente conservador" es cualquier cambio que tiene un valor no negativo en la matriz de probabilidad logarítmica PAM250.
La similitud de secuencia para polipéptidos se mide normalmente usando un programa informático de análisis de secuencia. El programa informático de análisis de proteínas empareja secuencias similares usando medidas de similitud asignadas a diversas sustituciones, eliminaciones y otras modificaciones, incluyendo sustituciones de aminoácidos conservadoras. Por ejemplo, el programa informático GCG contiene programas tales como GAP y BESTFIT que pueden usarse con parámetros por defecto para determinar la homología de secuencia o la identidad de secuencia entre polipéptidos muy relacionados, tales como polipéptidos homólogos de diferentes especies de organismos o entre una proteína de tipo silvestre y una muteína de la misma. Véase, por ejemplo, GCG Versión 6.1. Las secuencias polipeptídicas también pueden compararse usando FASTA con parámetros por defecto o recomendados; un programa en GCG Versión 6.1. FASTA (por ejemplo, FASTA2 y FASTA3) proporciona alineaciones y porcentajes de identidad de secuencia de las regiones del mejor solapamiento entre las secuencias de consulta y de búsqueda (Pearson (2000) supra). Otro algoritmo preferido cuando se compara una secuencia de la invención con una base de datos que contiene una gran cantidad de secuencias de diferentes organismos es el programa informático BLAST, especialmente BLASTP o TBLASTN, usando parámetros por defecto. Véanse, por ejemplo, Altschul et al. (1990) J. Mol. Biol. 215: 403-410 y (1997) Nucleic Acids Res. 25:3389-3402.
Por la expresión "cantidad terapéuticamente eficaz" se entiende una cantidad que produce el efecto deseado para el que se administra. La cantidad exacta dependerá del propósito del tratamiento, y un experto en la materia podrá determinarla usando técnicas conocidas (véase, por ejemplo, Lloyd (1999) The Art, Science and Technology of Pharmaceutical Compounding).
Como se utiliza en el presente documento, el término "sujeto" se refiere a un animal, preferentemente un mamífero, que necesita mejora, prevención y/o tratamiento de una enfermedad o trastorno tal como una infección vírica, cáncer o enfermedad autoinmunitaria.
Como se utiliza en el presente documento, "fármaco contra el cáncer" significa cualquier agente útil para tratar el cáncer que incluye, aunque no de forma limitativa, citotoxinas y agentes tales como antimetabolitos, agentes alquilantes, antraciclinas, antibióticos, agentes antimitóticos, procarbazina, hidroxiurea, asparaginasa, corticoesteroides, mitotano (O,P'-(DDD)), biológicos (por ejemplo, anticuerpos e interferones) y agentes radioactivos. Como se utiliza en el presente documento, "una citotoxina o agente citotóxico", también se refiere a un agente quimioterapéutico y significa cualquier agente que es perjudicial para las células. Los ejemplos incluyen Taxol® (paclitaxel), temozolamida, citocalasina B, gramicidina D, bromuro de etidio, emetina, cisplatino, mitomicina, etopósido, tenopósido, vincristina, vinblastina, colchicina, doxorrubicina, daunorrubicina, dihidroxiantracindiona, mitoxantrona, mitramicina, actinomicina D, 1-deshidrotestosterona, glucocorticoides, procaína, tetracaína, lidocaína, propanolol y puromicina y análogos u homólogos de los mismos.
Como se utiliza en el presente documento, la expresión "fármaco antivírico" se refiere a cualquier fármaco o terapia usado para tratar, prevenir o mejorar una infección vírica en un sujeto hospedador. La expresión "fármaco antivírico" incluye, pero sin limitación, zidovudina, lamivudina, abacavir, ribavirina, lopinavir, efavirenz, cobicistat, tenofovir, rilpivirina, analgésicos y corticoesteroides. En el contexto de la presente invención, las infecciones crónicas incluyen infecciones a largo plazo o crónicas causadas por virus incluyendo, aunque no de forma limitativa, virus de la inmunodeficiencia humana (VIH), virus de la hepatitis B (VHB), virus de la hepatitis C (VHC), virus del papiloma humano (VPH), virus de la coriomeningitis linfocítica (VCML) y virus de la inmunodeficiencia de los simios (VIS).
Los anticuerpos y fragmentos de unión a antígeno de la presente invención se unen específicamente a PD-1 y modulan la interacción de PD-1 con PD-L1. Los anticuerpos anti-PD-1 de la divulgación pueden unirse a PD-1 con alta o baja
afinidad. Los anticuerpos de la presente invención son anticuerpos bloqueantes, en donde los anticuerpos se unen a PD-1 y bloquean la interacción de PD-1 con PD-L1. Los anticuerpos bloqueantes de la invención bloquean la unión de PD-1 a PD-L1 y estimulan o mejoran la activación de los linfocitos T. En algunas realizaciones, los anticuerpos bloqueantes pueden ser útiles para estimular o potenciar la respuesta inmunitaria y/o para tratar a un sujeto que padece cáncer o una infección vírica crónica. Los anticuerpos, cuando se administran a un sujeto que los necesita, pueden reducir la infección crónica por un virus tal como el VIH, VCML o VHB en el sujeto. Pueden usarse para inhibir el crecimiento de células tumorales en un sujeto. Pueden usarse solos o como terapia complementaria con otras fracciones terapéuticas o modalidades conocidas en la técnica para tratar el cáncer o una infección vírica.
En otros aspectos, los anticuerpos de la presente divulgación pueden ser anticuerpos activadores, en donde los anticuerpos pueden unirse a PD-1 y potenciar la interacción de PD-1 y PD-L1. En algunos aspectos, los anticuerpos activadores pueden potenciar la unión de PD-1 a PD-L1 y/o inhibir o suprimir la activación de linfocitos T. Los anticuerpos activadores de la presente divulgación pueden ser útiles para inhibir la respuesta inmunitaria en un sujeto y/o para tratar una enfermedad autoinmunitaria.
En determinadas realizaciones, los anticuerpos anti-PD-1 pueden ser moléculas de unión a antígeno multiespecíficas, en donde estas comprenden una primera especificidad de unión a PD-1 de la invención y una segunda especificidad de unión a un antígeno seleccionado del grupo que consiste en otro coinhibidor de linfocitos T, un antígeno de tejido autoinmunitario, receptor de linfocitos T, receptor Fc, receptor de linfocitos T, PD-L1, y un epítopo diferente de PD-1.
En determinados aspectos, los anticuerpos de la divulgación se obtienen de ratones inmunizados con un inmunógeno primario, tal como un PD-1 de longitud completa [Véase número de registro de GenBank NP_005009.2 (SEQ ID NO: 327)] o con una forma recombinante de PD-1 o fragmentos de PD-1 humanos modificados (SEQ ID NO: 321, 323 o 324) o con fragmentos de PD-1 de mono cinomolgo modificados (SEQ ID NO: 322), seguido de inmunización con un inmunógeno secundario o con un fragmento inmunogénicamente activo de PD-1.
El inmunógeno puede ser un fragmento biológicamente activo y/o inmunogénico de PD-1 o ADN que codifica el fragmento activo del mismo. El fragmento puede derivarse del dominio N-terminal o C-terminal de PD-1. En determinados aspectos de la divulgación, el inmunógeno es un fragmento de PD-1 que abarca los restos de aminoácidos 25-170 de la SEQ ID NO: 327 con un cambio C93S.
Los péptidos pueden modificarse para incluir la adición o sustitución de ciertos restos para marcar o con fines de conjugación con moléculas vehículo, tales como, KLH. Por ejemplo, puede añadirse una cisteína en el extremo N terminal o C terminal de un péptido, o puede añadirse una secuencia enlazadora para preparar el péptido para conjugarlo con, por ejemplo, KLH para inmunización.
La secuencia de aminoácidos de longitud completa de PD-1 humano de longitud completa se muestra como la SEQ ID NO: 327.
En determinados aspectos, los anticuerpos que se unen específicamente a PD-1 se pueden preparar usando fragmentos de las regiones antes mencionadas, o péptidos que se extienden más allá de las regiones designadas en aproximadamente 5 a aproximadamente 20 restos de aminoácidos de cualquiera, o ambos, el extremo N o C terminal de las regiones descritas en el presente documento. En determinados aspectos, se puede usar cualquier combinación de las regiones mencionadas anteriormente o fragmentos de las mismas en la preparación de anticuerpos específicos de PD-1. En determinados aspectos, se puede usar una cualquiera o más de las regiones de PD-1 indicadas anteriormente, o fragmentos de las mismas, para preparar anticuerpos monoespecíficos, biespecíficos o multiespecíficos.
Los anticuerpos anti-PD-1 de la presente invención pueden unirse y neutralizar la actividad de PD-1, según lo determinado en ensayos in vitro o in vivo. La capacidad de los anticuerpos de la invención para unirse y neutralizar la actividad de PD-1 puede medirse usando cualquier método estándar conocido por los expertos en la materia, incluyendo ensayos de unión o ensayos de actividad, como se describe en el presente documento.
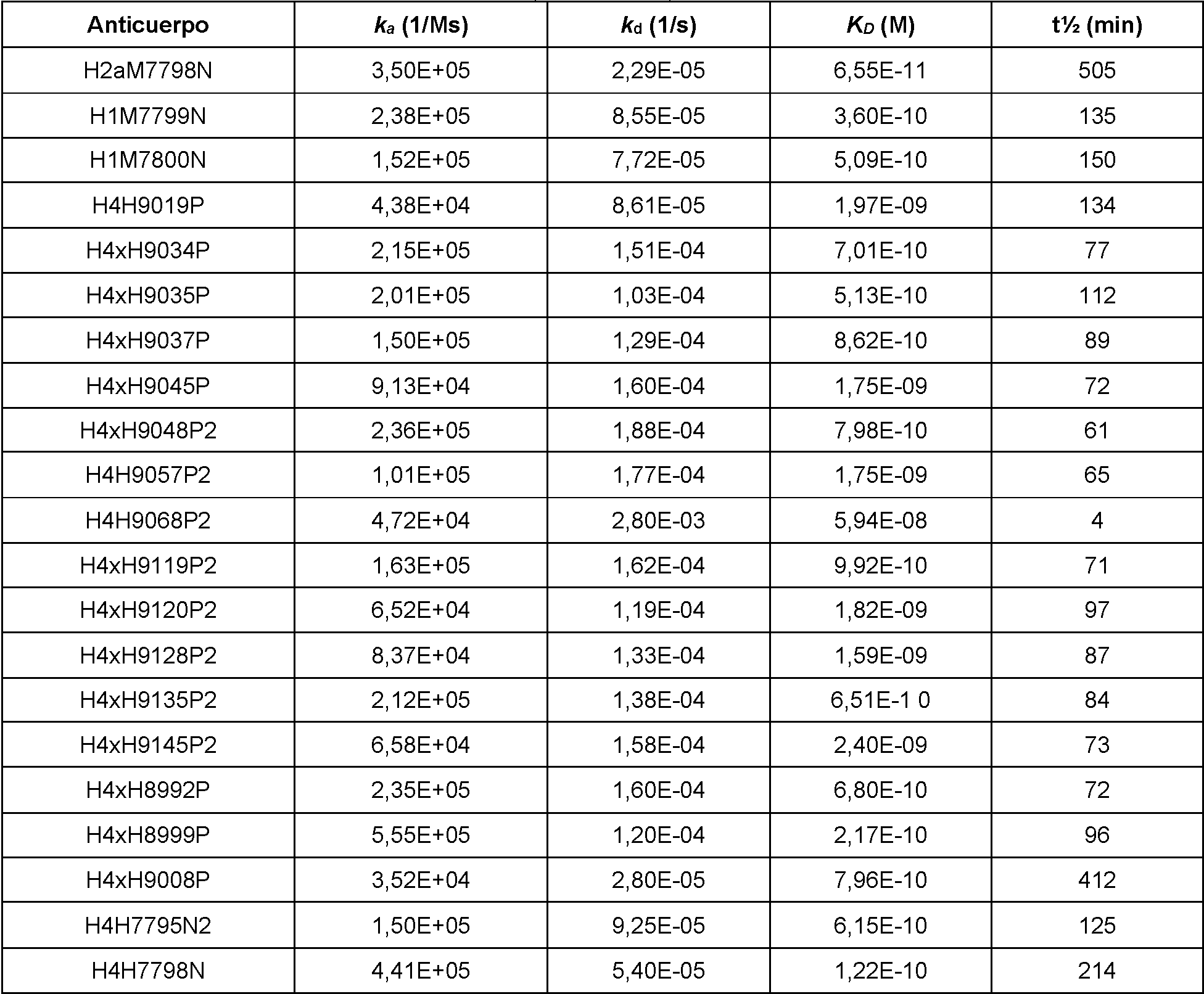
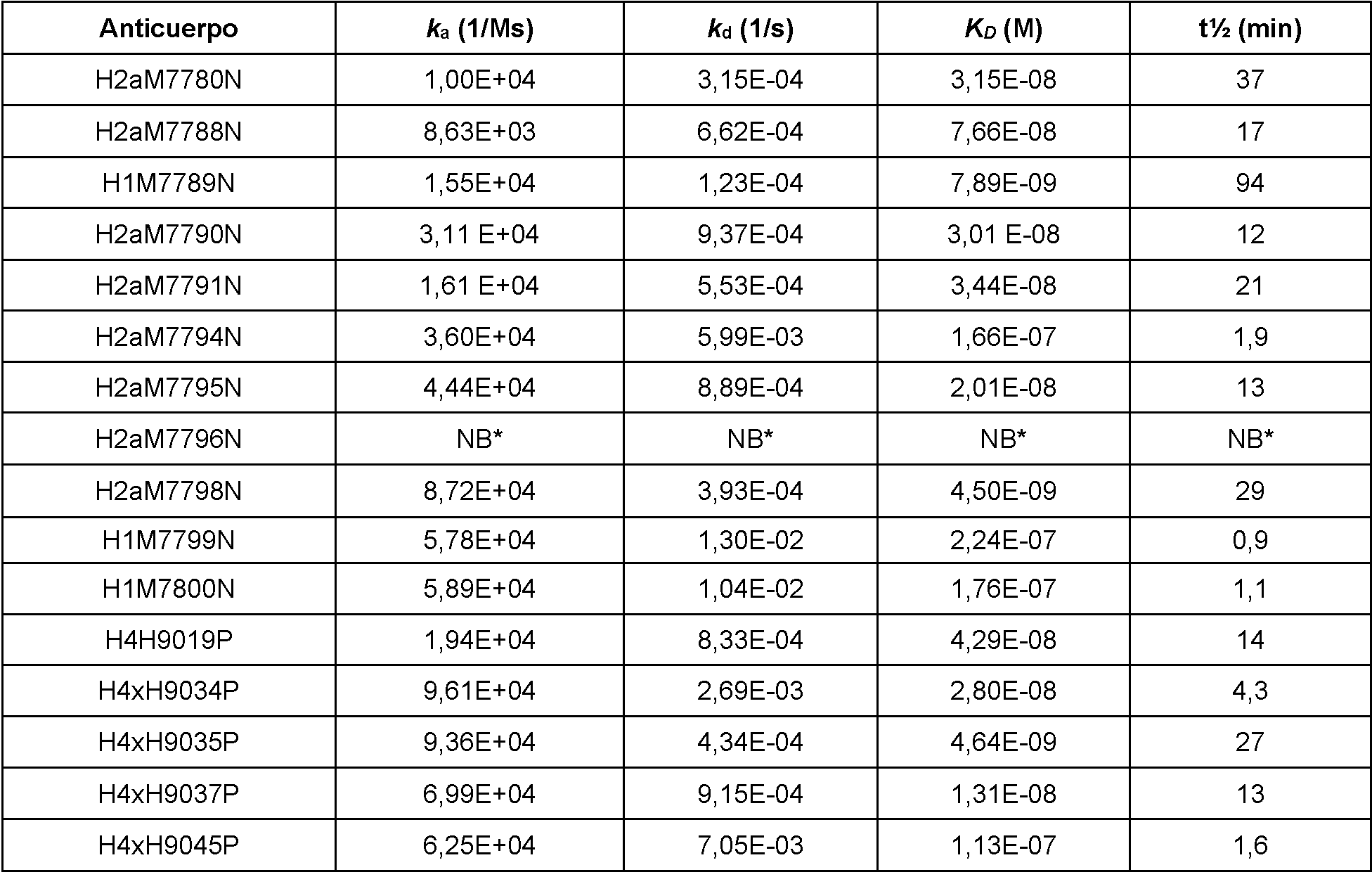
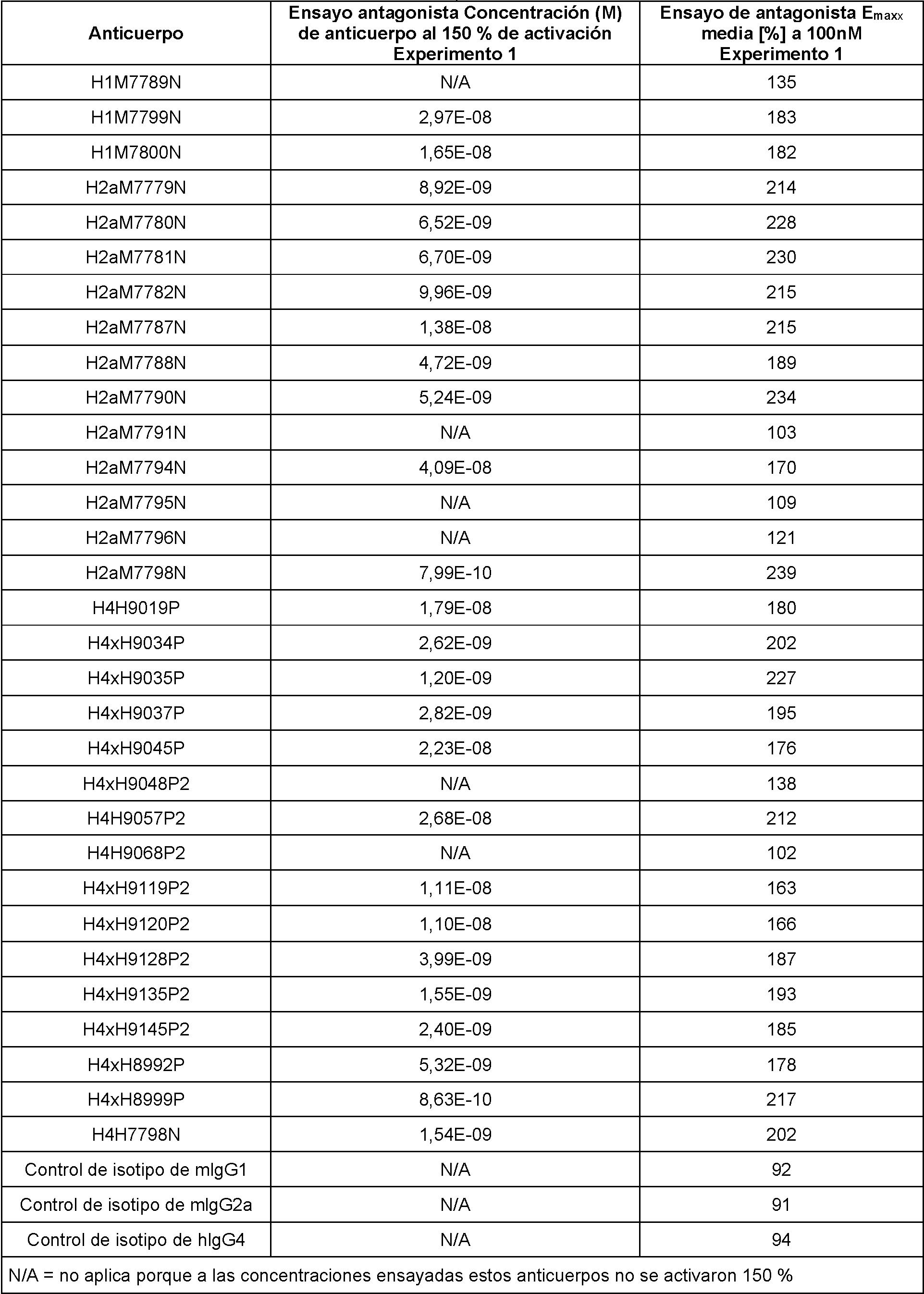
Configuraciones no limitantes, ilustrativas de ensayos in vitro para medir la actividad de unión se ilustran en los Ejemplos del presente documento. En el Ejemplo 3, las afinidades de unión y las constantes cinéticas de los anticuerpos anti-PD-1 humanos para PD-1 humano y PD-1 de mono cinomolgo se determinaron mediante resonancia de plasmón superficial y las mediciones se realizaron en un instrumento Biacore 4000 o T200. En los Ejemplos 4 y 5, se usaron ensayos de bloqueo para determinar la capacidad de los anticuerpos anti-PD-1 para bloquear la capacidad de unión de PD-1 a PD-L1 in vitro. En el Ejemplo 6, se utilizaron ensayos de bloqueo para determinar la competencia cruzada entre anticuerpos anti-PD-1. El ejemplo 7 describe la unión de los anticuerpos a células que sobreexpresan PD-1. En el Ejemplo 8, se usó un ensayo de luciferasa para determinar la capacidad de los anticuerpos anti-PD-1 para antagonizar la señalización de PD-1/PD-L1 en linfocitos T.
Los anticuerpos de la presente invención pueden potenciar o estimular la activación de los linfocitos T in vitro y en un sujeto con cáncer o en un sujeto infectado con un virus tal como el VCML. En determinadas realizaciones, los anticuerpos de la presente invención se utilizan en combinación con un segundo agente activo terapéutico, tal como
un anticuerpo contra un segundo coinhibidor de linfocitos T, para potenciar la respuesta inmunitaria e inhibir el crecimiento tumoral en un sujeto.
Los anticuerpos específicos de PD-1 pueden no contener marcadores o fracciones adicionales, o pueden contener un marcador o fracción N-terminal o C-terminal. En una realización, el marcador o fracción es biotina. En un ensayo de unión, la ubicación de un marcador (si lo hay) puede determinar la orientación del péptido con respecto a la superficie sobre la que se une el péptido. Por ejemplo, si una superficie está recubierta con avidina, un péptido que contiene una biotina N-terminal se orientará de manera que la porción C-terminal del péptido estará distal a la superficie. En una realización, el marcador puede ser un radionúclido, un tinte fluorescente o un marcador detectable por RM. En determinados aspectos, dichos anticuerpos marcados pueden usarse en ensayos de diagnóstico que incluyen ensayos de formación de imágenes.
Fragmentos de anticuerpos de unión a antígeno
A menos que se indique específicamente de otra manera, el término "anticuerpo", como se utiliza en el presente documento, debe entenderse que abarca moléculas de anticuerpos que comprenden dos cadenas pesadas de inmunoglobulina y dos cadenas ligeras de inmunoglobulina (es decir, "moléculas de anticuerpo completo") así como sus fragmentos de unión a antígeno. Las expresiones "porción de unión a antígeno" de un anticuerpo, "fragmento de unión a antígeno" de un anticuerpo y similares, como se utiliza en el presente documento, incluyen cualquier polipéptido o glucoproteína de origen natural, obtenible enzimáticamente, sintético o modificado por ingeniería genética que se une específicamente a un antígeno para formar un complejo. Las expresiones "fragmento de unión a antígeno" de un anticuerpo o "fragmento de anticuerpo", como se utiliza en el presente documento, se refieren a uno o más fragmentos de un anticuerpo que conservan la capacidad de unirse específicamente a PD-1. Un fragmento de anticuerpo puede incluir un fragmento Fab, un fragmento F(ab')2, un fragmento Fv, un fragmento dAb, un fragmento que contiene una CDR, o una CDR aislada. En determinadas realizaciones, la expresión "fragmento de unión a antígeno" se refiere a un fragmento de polipéptido de una molécula de unión a antígeno multiespecífica. En dichos aspectos de la divulgación, la expresión "fragmento de unión a antígeno" incluye, por ejemplo, un dominio extracelular de PD-L1 que se une específicamente a PD-1. Pueden obtenerse fragmentos de unión a antígeno de un anticuerpo, por ejemplo, a partir de moléculas de anticuerpo completo, usando cualquier técnica convencional adecuada tal como digestión proteolítica o técnicas de ingeniería genética recombinante que implican la manipulación y expresión de ADN que codifica dominios variables, y opcionalmente) constantes, de anticuerpo. Dicho a Dn se conoce y/o se puede adquirir fácilmente de, por ejemplo, fuentes comerciales, bibliotecas de ADN (incluyendo, por ejemplo, fagotecas de anticuerpos), o puede sintetizarse. El ADN puede secuenciarse y manipularse químicamente o usando técnicas de biología molecular, por ejemplo, para disponer uno o más dominios variables y/o constantes en una configuración adecuada, o para introducir codones, crear restos de cisteína, modificar, añadir o suprimir aminoácidos, etc.
Los ejemplos no limitantes de fragmentos de unión a antígeno incluyen: (i) fragmentos Fab; (ii) fragmentos F(ab')2; (iii) fragmentos Fd; (iv) fragmentos Fv; (v) moléculas Fv monocatenarias (scFv); (vi) fragmentos dAb; y (vii) unidades de reconocimiento mínimo que consisten en los restos de aminoácidos que imitan la región hipervariable de un anticuerpo (por ejemplo, una región determinante de complementariedad (CDR) aislada, tal como un péptido CDR3) o un péptido FR3-CDR3-FR4 restringido. Otras moléculas modificadas por ingeniería genética, tales como anticuerpos específicos de dominio, anticuerpos de dominio único, anticuerpos de dominio suprimido, anticuerpos quiméricos, anticuerpos injertados con CDR, diacuerpos, triacuerpos, tetracuerpos, minicuerpos, nanocuerpos (por ejemplo, nanocuerpos monovalentes, nanocuerpos bivalentes, etc.), agentes inmunofarmacéuticos modulares pequeños (SMIP) y dominios IgNAR variables de tiburón, también se incluyen dentro de la expresión "fragmento de unión a antígeno", como se usan en el presente documento.
Un fragmento de unión a antígeno de un anticuerpo normalmente comprenderá al menos un dominio variable. El dominio variable puede ser de cualquier tamaño o composición de aminoácidos y generalmente comprenderá al menos una CDR, que está adyacente o en marco con una o más secuencias marco. En fragmentos de unión a antígeno que tienen un dominio Vh asociado a un dominio Vl, los dominios Vh y Vl pueden situarse uno con respecto al otro en cualquier disposición adecuada. Por ejemplo, la región variable puede ser dimérica y contener los dímeros Vh - Vh, Vh - Vl o Vl - Vl. Como alternativa, el fragmento de unión a antígeno de un anticuerpo puede contener un dominio Vh o Vl monomérico.
En determinados aspectos, un fragmento de unión a antígeno de un anticuerpo puede contener al menos un dominio variable unido covalentemente con al menos un dominio constante. Configuraciones no limitantes, ilustrativas, de dominios variables y constantes que pueden encontrarse dentro de un fragmento de unión a antígeno de un anticuerpo de la presente divulgación incluyen: (i) Vh -Ch1; (ii) Vh -Ch2; (iii) Vh -Ch3; (iv) Vh -Ch1-Ch2; (v) Vh -Ch1-Ch2-Ch3; (vi) Vh -Ch2-Ch3; (vii) Vh -Cl; (viii) Vl -Ch1; (ix) Vl -Ch2; (x) Vl -Ch3; (xi) Vl -Ch1-Ch2; (xii) Vl -Ch1-Ch2-Ch3; (xiii) Vl -Ch2-Ch3; y (xiv) Vl -Cl. En cualquier configuración de dominios variables y constantes, incluyendo cualquiera de las configuraciones de ejemplo enumeradas anteriormente, los dominios variables y constantes pueden estar unidos directamente entre sí o pueden estar unidos mediante una región bisagra o enlazadora completa o parcial. Una región bisagra puede consistir en al menos 2 (por ejemplo, 5, 10, 15, 20, 40, 60 o más) aminoácidos, lo que da como resultado una unión flexible o semiflexible entre dominios variables y/o constantes adyacentes en una única molécula polipeptídica. Además, un fragmento de unión a antígeno de un anticuerpo de la presente divulgación puede
comprender un homo-dímero o hetero-dímero (u otro multímero) de cualquiera de las configuraciones de dominio variable y constante enumeradas anteriormente en asociación no covalente entre sí y/o con uno o más dominios Vh o Vl monoméricos (p.ej., mediante enlace(s) disulfuro).
Como ocurre con las moléculas de anticuerpo completas, los fragmentos de unión a antígeno pueden ser monoespecíficos o multiespecíficos (por ejemplo, biespecíficos). Un fragmento de unión a antígeno multiespecífico de un anticuerpo normalmente comprenderá al menos dos dominios variables diferentes, en donde cada dominio variable puede unirse específicamente con un antígeno distinto o con un epítopo diferente en el mismo antígeno. Cualquier formato de anticuerpo multiespecífico, incluyendo los formatos de anticuerpo biespecífico de ejemplo que se divulgan en el presente documento, puede adaptarse para su uso en el contexto de un fragmento de unión a antígeno de un anticuerpo de la presente invención usando técnicas rutinarias disponibles en la técnica.
Preparación de anticuerpos humanos
Los métodos para generar anticuerpos humanos en ratones transgénicos se conocen en la técnica. Cualquiera de dichos métodos conocidos puede usarse en el contexto de la presente divulgación para preparar anticuerpos humanos que se unan específicamente a PD-1.
Puede usarse un inmunógeno que comprenda uno cualquiera de los siguientes para generar anticuerpos contra PD-1. En determinados aspectos, los anticuerpos de la divulgación se obtienen de ratones inmunizados con un PD-1 nativo, de longitud completa (véase el número de registro GenBank NP_005009.2) (SEQ ID NO: 327), o con un péptido PD-1 recombinante. Como alternativa, PD-1 o un fragmento del mismo puede producirse usando técnicas bioquímicas estándar y modificarse (SEQ ID NO: 321 - 324) y usarse como inmunógeno.
En determinados aspectos, el inmunógeno puede ser un péptido del extremo N terminal o C terminal de PD-1. En un aspecto, el inmunógeno es el dominio extracelular o el dominio de PD-1 similar a IgV. En determinados aspectos de la divulgación, el inmunógeno es un fragmento de PD-1 que va desde aproximadamente los restos de aminoácidos 25-170 de la SEQ ID NO: 327 con un cambio C93S.
En algunos aspectos, el inmunógeno puede ser un péptido PD-1 recombinante expresado en E. coli o en cualquier otra célula eucariota o de mamífero tales como células de ovario de hámster chino (CHO).
En determinados aspectos, los anticuerpos que se unen específicamente a PD-1 se pueden preparar usando fragmentos de las regiones antes mencionadas, o péptidos que se extienden más allá de las regiones designadas en aproximadamente 5 a aproximadamente 20 restos de aminoácidos de cualquiera, o ambos, el extremo N o C terminal de las regiones descritas en el presente documento. En determinados aspectos, se puede usar cualquier combinación de las regiones mencionadas anteriormente o fragmentos de las mismas en la preparación de anticuerpos específicos de PD-1.
Usando la tecnología VELOCIMMUNE® (véase, por ejemplo, el documento US 6.596.541, Regeneron Pharmaceuticals, VELOCIMMUNE®) o cualquier otro método conocido para generar anticuerpos monoclonales, inicialmente se aíslan anticuerpos quiméricos de alta afinidad contra PD-1 que tienen una región variable humana y una región constante de ratón. La tecnología VELOCIMMUNE® implica la generación de un ratón transgénico que tiene un genoma que comprende regiones variables de la cadena pesada y ligera humanas unidas operativamente a loci endógenos de regiones constantes de ratón de manera que el ratón produce un anticuerpo que comprende una región variable humana y una región constante de ratón en respuesta a la estimulación antigénica. El ADN que codifica las regiones variables de las cadenas pesada y ligera del anticuerpo se aísla y se une operativamente al ADN que codifica las regiones constantes de la cadena pesada y ligera humana. A continuación, el ADN se expresa en una célula capaz de expresar el anticuerpo totalmente humano.
Bioequivalentes
Los anticuerpos anti-PD-1 y fragmentos de anticuerpo de la presente divulgación abarcan proteínas que tienen secuencias de aminoácidos que varían respecto a las de los anticuerpos descritos, pero que conservan la capacidad de unión a PD-1. Dichos anticuerpos variantes y fragmentos de anticuerpo comprenden una o más adiciones, eliminaciones o sustituciones de aminoácidos cuando se comparan con la secuencia precursora, pero muestran actividad biológica que es esencialmente equivalente a la de los anticuerpos descritos. De forma análoga, las secuencias de ADN que codifican el anticuerpo de la presente divulgación abarcan secuencias que comprenden una o más adiciones, eliminaciones o sustituciones de nucleótidos en comparación con la secuencia divulgada, pero que codifican un anticuerpo o fragmento de anticuerpo que es esencialmente bioequivalente a un anticuerpo o fragmento de anticuerpo de la invención.
Dos proteínas de unión a antígeno, o anticuerpos, se consideran bioequivalentes si, por ejemplo, son equivalentes farmacéuticos o alternativas farmacéuticas cuya velocidad y grado de absorción no muestran una diferencia significativa cuando se administran a la misma dosis molar en condiciones experimentales similares, bien en una sola dosis o en múltiples dosis. Algunos anticuerpos se considerarán equivalentes o alternativas farmacéuticas si son
equivalentes en el grado de su absorción pero no en su velocidad de absorción y aún pueden considerarse bioequivalentes porque dichas diferencias en la velocidad de absorción son intencionadas y se reflejan en el marcaje, no son esenciales para obtener las concentraciones eficaces del fármaco en el organismo en, por ejemplo, el uso crónico, y se consideran médicamente insignificantes para el producto farmacológico particular estudiado.
En un aspecto, dos proteínas de unión a antígeno son bioequivalentes si no hay diferencias clínicamente importantes en su seguridad, pureza, o potencia.
En un aspecto, dos proteínas de unión a antígeno son bioequivalentes si un paciente puede cambiarse una o más veces entre el producto de referencia y el producto biológico sin un aumento esperado en el riesgo de efectos adversos, incluyendo un cambio clínicamente significativo en la inmunogenia, o eficacia disminuida, en comparación con la terapia continuada sin dicho cambio.
En un aspecto, dos proteínas de unión a antígeno son bioequivalentes si ambas actúan mediante un mecanismo o mecanismos comunes de acción para la condición o condiciones de uso, en la medida en que dichos mecanismos sean conocidos.
La bioequivalencia puede demostrarse por métodos in vivo y/o in vitro. Las medidas de bioequivalencia incluyen, por ejemplo, (a) un ensayo in vivo en seres humanos u otros mamíferos, en el que se mide la concentración del anticuerpo o sus metabolitos en sangre, plasma, suero u otro líquido biológico como una función del tiempo; (b) un ensayo in vitro que se ha correlacionado con y es razonablemente predictivo de datos de biodisponibilidad in vivo en seres humanos; (c) un ensayo in vivo en seres humanos u otros mamíferos en el que se mide el efecto farmacológico agudo adecuado del anticuerpo (o su diana) como una función del tiempo; y (d) en un ensayo clínico bien controlado que establece la seguridad, eficacia o biodisponibilidad o bioequivalencia de un anticuerpo.
Las variantes bioequivalentes de los anticuerpos de la invención pueden construirse, por ejemplo, generando diversas sustituciones de restos o secuencias o eliminando secuencias o restos terminales o internos no necesarios para la actividad biológica. Por ejemplo, pueden eliminarse restos de cisteína no esenciales para la actividad biológica, o remplazarse con otros aminoácidos, para evitar la formación de puentes disulfuro intramoleculares innecesarios o incorrectos tras la renaturalización. En otros contextos, los anticuerpos bioequivalentes pueden incluir variantes de anticuerpo que comprenden cambios de aminoácidos, los cuales modifican las características de glucosilación de los anticuerpos, por ejemplo, mutaciones que eliminan o suprimen la glucosilación.
Anticuerpos anti-PD-1 que comprenden variantes de Fc
De acuerdo con determinadas realizaciones de la presente invención, se proporcionan anticuerpos anti-PD-1 que comprenden un dominio Fc que comprende una o más mutaciones que mejoran o reducen la unión del anticuerpo al receptor FcRn, por ejemplo, a pH ácido en comparación con pH neutro. Por ejemplo, la presente invención incluye anticuerpos anti-PD-1 que comprenden una mutación en la región Ch2 o Ch3 del dominio Fc, en los que la mutación o mutaciones aumentan la afinidad del dominio Fc por FcRn en un entorno ácido (por ejemplo, en un endosoma en el que el pH varía de aproximadamente 5,5 a aproximadamente 6,0). Dichas mutaciones pueden dar como resultado un aumento de la semivida en suero del anticuerpo cuando se administra a un animal. Los ejemplos no limitantes de dichas modificaciones de Fc incluyen, por ejemplo, una modificación en la posición 250 (por ejemplo, E o Q); 250 y 428 (por ejemplo, L o F); 252 (por ejemplo, L/Y/F/W o T), 254 (por ejemplo, S o T) y 256 (por ejemplo, S/R/Q/E/D o T); o una modificación en la posición 428 y/o 433 (por ejemplo, H/L/R/S/P/Q o K) y/o 434 (por ejemplo, A, W, H, F o Y [N434A, N434W, N434H, N434F o N434Y]); o una modificación en la posición 250 y/o 428; o una modificación en la posición 307 o 308 (por ejemplo, 308F, V308F) y 434. En una realización, la modificación comprende una modificación 428L (por ejemplo, M428L) y 434S (por ejemplo, N434S); una modificación 428L, 2591 (por ejemplo, V2591) y 308F (por ejemplo, V308F); una modificación 433K (por ejemplo, H433K) y en 434 (por ejemplo, 434Y); una modificación en 252, 254, y 256 (por ejemplo, 252Y, 254T y 256E); una modificación 250Q y 428L (por ejemplo,T250Q y M428L); y una modificación en 307 y/o 308 (por ejemplo, 308F o 308P). En otra realización más, la modificación comprende una modificación 265A (por ejemplo, D265A) y/o una modificación 297A (por ejemplo, N297A).
Por ejemplo, la presente invención incluye anticuerpos anti-PD-1 que comprenden un dominio Fc que comprende uno o más pares o grupos de mutaciones seleccionadas del grupo que consiste en: 250Q y 248L (por ejemplo, T250Q y M248L); 252Y, 254T y 256E (por ejemplo, M252Y, S254T y T256E); 428L y 434S (por ejemplo, M428L y N434S); 257I y 3111 (por ejemplo, P257I y Q311I); 257I y 434H (por ejemplo, P257I y N434H); 376V y 434H (por ejemplo, D376V y N434H); 307A, 380A y 434A (por ejemplo, T307A, E380A y N434A); y 433K y 434F (por ejemplo, H433K y N434F). En una realización, la presente invención incluye anticuerpos anti-PD-1 que comprenden un dominio Fc que comprende una mutación S108P en la región bisagra de IgG4 para promover la estabilización del dímero. Todas las posibles combinaciones de las mutaciones del dominio Fc anteriores, y otras mutaciones dentro de los dominios variables del anticuerpo divulgados en el presente documento, se contemplan.
La presente invención también incluye anticuerpos anti-PD-1 que comprenden una región constante de cadena pesada (Ch) quimérica, en donde la región Ch quimérica comprende segmentos procedentes de las regiones Ch de más de un isotipo de inmunoglobulina. Por ejemplo, los anticuerpos de la invención pueden comprender una región Ch
quimérica que comprende parte o todo el dominio Ch2 derivado de una molécula de IgG1 humana, IgG2 humana o IgG4 humana, en combinación con parte o la totalidad de un dominio Ch3 derivado de una molécula de IgG1 humana, IgG2 humana o IgG4 humana. De acuerdo con determinadas realizaciones, los anticuerpos de la invención comprenden una región Ch quimérica que tiene una región bisagra quimérica. Por ejemplo, una bisagra quimérica puede comprender una secuencia de aminoácidos de "bisagra superior" (restos de aminoácidos de las posiciones 216 a 227 de acuerdo con la numeración EU) derivada de una región bisagra de una IgG1 humana, una IgG2 humana o una IgG4 humana, en combinación con una secuencia "bisagra inferior" (restos de aminoácidos de las posiciones 228 a 236 de acuerdo con la numeración EU) derivada de una región bisagra de una IgG1 humana, una IgG2 humana o una IgG4 humana. De acuerdo con determinadas realizaciones, la región bisagra quimérica comprende restos de aminoácidos procedentes de una bisagra superior de IgG1 humana o IgG4 humana y restos de aminoácidos procedentes de una bisagra inferior de IgG2 humana. Un anticuerpo que comprende una región Ch quimérica como se describe en el presente documento puede, en determinadas realizaciones, presentar funciones efectoras de Fc modificadas sin afectar negativamente a las propiedades terapéuticas o farmacocinéticas del anticuerpo. (Véase, por ejemplo, USSN. 14/170.166, presentada el 31 de enero de 2014).
Características biológicas de los anticuerpos
En general, los anticuerpos de la presente divulgación funcionan uniéndose a PD-1. La presente invención incluye anticuerpos anti-PD-1 y fragmentos de unión a antígeno de los mismos que unen moléculas de PD-1 monomérico o dimérico solubles con alta afinidad. Por ejemplo, la presente divulgación incluye anticuerpos y fragmentos de unión a antígeno de anticuerpos que se unen a PD-1 monomérico (por ejemplo, a 25 °C o a 37 °C) con una Kd de menos de aproximadamente 50 nM según se mide por resonancia de plasmón superficial, por ejemplo, usando el formato de ensayo como se define en el Ejemplo 3 del presente documento. En determinados aspectos, los anticuerpos o fragmentos de unión a antígeno de los mismos se unen a PD-1 monomérico con una Kd de menos de aproximadamente 40 nM, menos de aproximadamente 30 nM, menos de aproximadamente 20 nM, menos de aproximadamente 10 nM o menos de aproximadamente 5 nM, menos de aproximadamente 2 nM o menos de aproximadamente 1 nM, según se mide mediante resonancia de plasmón superficial, por ejemplo, usando el formato de ensayo como se define en el Ejemplo 3 en el presente documento, o un ensayo sustancialmente similar.
La presente divulgación incluye también anticuerpos y fragmentos de unión a antígeno de los mismos que se unen a PD-1 dimérico (por ejemplo, a 25 °C o a 37 °C) con una Kd de menos de aproximadamente 400 pM según se mide por resonancia de plasmón superficial, por ejemplo, usando el formato de ensayo como se define en el Ejemplo 3 del presente documento. En determinados aspectos, los anticuerpos o fragmentos de unión a antígeno de los mismos se unen a PD-1 dimérico con una Kd de menos de aproximadamente 300 pM, menos de aproximadamente 250 pM, menos de aproximadamente 200 pM, menos de aproximadamente 100 pM, o menos de aproximadamente 50 pM, según se mide mediante resonancia de plasmón superficial, por ejemplo, usando el formato de ensayo como se define en el Ejemplo 3 en el presente documento, o un ensayo sustancialmente similar.
La presente divulgación incluye también anticuerpos y fragmentos de unión a antígeno de los mismos que se unen a PD-1 de mono cinomolgo (Macaca fascicularis) (por ejemplo, a 25 °C o a 37 °C) con una Kd de menos de aproximadamente 35 nM según se mide por resonancia de plasmón superficial, por ejemplo, usando el formato de ensayo como se define en el Ejemplo 3 del presente documento. En determinados aspectos, los anticuerpos o fragmentos de unión a antígeno de los mismos se unen a PD-1 de mono cinomolgo con una Kd de menos de aproximadamente 30 nM, menos de aproximadamente 20 nM, menos de aproximadamente 15 nM, menos de aproximadamente 10 nM o menos de aproximadamente 5 nM, según se mide mediante resonancia de plasmón superficial, por ejemplo, usando el formato de ensayo como se define en el Ejemplo 3 en el presente documento, o un ensayo sustancialmente similar.
La presente divulgación también incluye anticuerpos y fragmentos de unión a antígeno de los mismos que se unen a PD-1 con una semivida disociativa (t1^ ) superior a aproximadamente 1,1 minutos, medida por resonancia de plasmón superficial a 25 °C o 37 °C, por ejemplo, usando un formato de ensayo como se define en el ejemplo 3 en el presente documento, o un ensayo sustancialmente similar. En determinados aspectos, los anticuerpos o fragmentos de unión a antígeno de la presente divulgación se unen a PD-1 con una t1^ superior a aproximadamente 5 minutos, superior a aproximadamente 10 minutos, superior a aproximadamente 30 minutos, superior a aproximadamente 50 minutos, superior a aproximadamente 60 minutos, superior a aproximadamente 70 minutos, superior a aproximadamente 80 minutos, superior a aproximadamente 90 minutos, superior a aproximadamente 100 minutos, superior a aproximadamente 200 minutos, superior a aproximadamente 300 minutos, superior a aproximadamente 400 minutos, superior a aproximadamente 500 minutos, superior a aproximadamente 600 minutos, superior a aproximadamente 700 minutos, superior a aproximadamente 800 minutos, superior a aproximadamente 900 minutos, superior a aproximadamente 1000 minutos o superior a aproximadamente 1200 minutos, medido por resonancia de plasmón superficial a 25 °C o 37 °C, por ejemplo, usando un formato de ensayo como se define en el Ejemplo 3 en el presente documento (p.ej., formato de captura de mAb o captura de antígeno), o un ensayo sustancialmente similar.
La presente divulgación también incluye anticuerpos o fragmentos de unión a antígeno de los mismos que bloquean la unión de PD-1 a PD-L1 con una CI50 de menos de aproximadamente 3 nM determinado usando un ensayo de inmunoensayo basado en ELISA, por ejemplo, como se muestra en el Ejemplo 4, o un ensayo sustancialmente similar.
La presente divulgación también incluye anticuerpos y fragmentos de unión a antígeno de los mismos que se unen a PD-1 y mejoran la unión de PD-1 a p D-L1.
En algunos aspectos, los anticuerpos de la presente divulgación pueden unirse al dominio de PD-1 o a un fragmento del dominio. En algunos aspectos, los anticuerpos de la presente divulgación pueden unirse a más de un dominio (anticuerpos de reactividad cruzada). En determinados aspectos, los anticuerpos de la presente divulgación pueden unirse a un epítopo ubicado en el dominio extracelular que comprende los restos de aminoácidos 21-171 de PD-1 (SEQ ID NO: 327). En un aspecto, los anticuerpos pueden unirse a un epítopo que comprende uno o más aminoácidos seleccionados del grupo que consiste en los restos de aminoácidos 1-146 de la SEQ ID NO: 321 - 324.
En determinados aspectos, los anticuerpos de la presente divulgación pueden funcionar bloqueando o inhibiendo la actividad de unión de PD-L1 asociada con PD-1 al unirse a cualquier otra región o fragmento de la proteína de longitud completa, cuya secuencia de aminoácidos que se muestra en la SEQ ID NO: 327. En determinados aspectos, los anticuerpos pueden atenuar o modular la interacción entre PD-1 y PD-L1.
En determinadas realizaciones, los anticuerpos de la presente invención pueden ser anticuerpos biespecíficos. Los anticuerpos biespecíficos de la invención pueden unirse a un epítopo en un dominio y también pueden unirse a un segundo epítopo en un dominio de PD-1 diferente. En determinadas realizaciones, los anticuerpos biespecíficos de la invención pueden unirse a dos epítopos diferentes en el mismo dominio. En una realización, la molécula de unión a antígeno multiespecífica comprende una primera especificidad de unión en la que la primera especificidad de unión comprende el dominio extracelular o fragmento del mismo de PD-L1; y una segunda especificidad de unión a otro epítopo de PD-1.
En un aspecto, la divulgación proporciona un anticuerpo monoclonal totalmente humano aislado o un fragmento de unión a antígeno del mismo que se une a PD-1, en donde el anticuerpo o fragmento del mismo presenta una o más de las siguientes características: (i) comprende una HCVR que tiene una secuencia de aminoácidos seleccionada del grupo que consiste en las SEQ ID NO: 2, 18, 34, 50, 66, 82, 98, 114, 130, 146, 162, 178, 194, 210, 218, 226, 234, 242, 250, 258, 266, 274, 282, 290, 298, 306 y 314, o una secuencia sustancialmente similar a la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia; (ii) comprende una LCVR que tiene una secuencia de aminoácidos seleccionada del grupo que consiste en la SEQ ID NO: 10, 26, 42, 58, 74, 90, 106, 122, 138, 154, 170, 186, y 202, o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia; (iii) comprende un dominio HCDR3 que tiene una secuencia de aminoácidos seleccionada del grupo que consiste en la SEQ ID NO: 8, 24, 40, 56, 72, 88, 104, 120, 136, 152, 168, 184, 200, 216, 224, 232, 240, 248, 256, 264, 272, 280, 288, 296, 304, 312 y 320, o una secuencia sustancialmente similar a la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia; y un dominio LCDR3 que tiene una secuencia de aminoácidos seleccionada del grupo que consiste en la SEQ ID NO: 16, 32, 48, 64, 80, 96, 112, 128, 144, 160, 176, 192, y 208, o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia; (iv) comprende un dominio HCDR1 que tiene una secuencia de aminoácidos seleccionada del grupo que consiste en la SEQ ID NO: 4, 20, 36, 52, 68, 84, 100, 116, 132, 148, 164, 180, 196, 212, 220, 228, 236, 244, 252, 260, 268, 276, 284, 292, 300, 308 y 316, o una secuencia sustancialmente similar a la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia; un dominio HCDR2 que tiene una secuencia de aminoácidos seleccionada del grupo que consiste en la SEQ ID NO: 6, 22, 38, 54, 70, 86, 102, 118, 134, 150, 166, 182, 198, 214, 222, 230, 238, 246, 254, 262, 270, 278, 286, 294, 302, 310 y 318, o una secuencia sustancialmente similar a la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia; un dominio LCDR1 que tiene una secuencia de aminoácidos seleccionada del grupo que consiste en la SEQ ID NO: 12, 28, 44, 60, 76, 92, 108, 124, 140, 156, 172, 188, y 204, o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia; y un dominio LCDR2 que tiene una secuencia de aminoácidos seleccionada del grupo que consiste en la SEQ ID NO: 14, 30, 46, 62, 78, 94, 110, 126, 142, 158, 174, 190, y 206, o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia; (v) es una molécula de unión a antígeno multiespecífica que comprende una primera especificidad de unión a PD-1 y una segunda especificidad de unión a un antígeno seleccionado del grupo que consiste en PD-1, un antígeno específico de tumores, un antígeno específico de tejido autoinmunitario, un antígeno de célula infectada por virus, un coinhibidor de linfocitos T diferente, receptor de linfocitos T y un receptor Fc; (vi) se une a PD-1 humano con una Kd de aproximadamente 28 pM a aproximadamente 1,5 pM; (vii) se une a PD-1 de mono cinomolgo con una Kd de aproximadamente 3 nM a aproximadamente 7,5 pM; (viii) bloquea o potencia la unión de PD-1 a PD-L1 con una CI50 < aproximadamente 3,3 nM; (ix) bloquea la regulación negativa de linfocitos T inducida por PD-1 y/o rescata la señalización de linfocitos T en un ensayo de indicador de luciferasa de linfocitos T/APC; (x) estimula la proliferación y actividad de linfocitos T en un ensayo de reacción de linfocitos mixtos (MLR); (xi) induce la producción de IL-2 y/o IFNy en un ensayo MLR; y (xii) suprime el crecimiento tumoral y aumenta la supervivencia en sujetos con cáncer.
En un aspecto, la divulgación proporciona un anticuerpo monoclonal totalmente humano aislado o un fragmento de unión a antígeno del mismo que bloquea la unión de PD-1 a PD-L1, en donde el anticuerpo o fragmento del mismo presenta una o más de las siguientes características: (i) comprende una HCVR que tiene una secuencia de
aminoácidos seleccionada del grupo que consiste en la SEQ ID NO: 130, 162, 234 y 314, o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia; (ii) comprende una LCVR que tiene una secuencia de aminoácidos seleccionada del grupo que consiste en la SEQ ID NO: 138, 170, 186 y 202, o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia; (iii) comprende un dominio HCDR3 que tiene una secuencia de aminoácidos seleccionada del grupo que consiste en la SEQ ID NO: 136, 168, 240, y 320, o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia; y un dominio LCDR3 que tiene una secuencia de aminoácidos seleccionada del grupo que consiste en la SEQ ID NO: 144, 176, 192, y 208, o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia; (iv) comprende un dominio HCDR1 que tiene una secuencia de aminoácidos seleccionada del grupo que consiste en la SEQ ID NO: 132, 164, 236, y 316, o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia; y un dominio HCDR2 que tiene una secuencia de aminoácidos seleccionada del grupo que consiste en la SEQ ID NO: 134, 166, 238, y 318, o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia; y un dominio LCDR1 que tiene una secuencia de aminoácidos seleccionada del grupo que consiste en la SEQ ID NO: 140, 172, 188, y 204, o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia; y un dominio LCDR2 que tiene una secuencia de aminoácidos seleccionada del grupo que consiste en la SEQ ID NO: 142, 174, 190, y 206, o una secuencia sustancialmente similar de la misma que tiene al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 % de identidad de secuencia; (v) es una molécula de unión a antígeno multiespecífica que comprende una primera especificidad de unión a PD-1 y una segunda especificidad de unión a un antígeno seleccionado del grupo que consiste en un epítopo diferente de PD-1, un antígeno específico de tumores, un antígeno específico de tejido autoinmunitario, un antígeno de célula infectada por virus, un coinhibidor de linfocitos T diferente, receptor de linfocitos T y un receptor Fc; (vi) unión a PD-1 humano con una Kd á de 10-9M; (vi) unión a PD-1 de mono cinomolgo con una Kd á de 10-8M; (viii) bloquea la unión de PD-1 a PD-L1 con una CI50 á 10'10M; (ix) bloquea la regulación negativa de linfocitos T inducida por PD-1 y/o rescata la señalización de linfocitos T en un ensayo de indicador de luciferasa de linfocitos T/APC; (x) estimula la proliferación y actividad de linfocitos T en un ensayo de reacción de linfocitos mixtos (MLR); (xi) induce la producción de IL-2 y/o IFNy en un ensayo MLR; y (xii) suprime el crecimiento tumoral y aumenta la supervivencia en sujetos con cáncer.
Los anticuerpos de la presente divulgación pueden poseer una o más de las características biológicas mencionadas anteriormente, o cualquier combinación de las mismas. Otras características biológicas de los anticuerpos de la presente invención serán evidentes para un experto en la materia a partir de una revisión de la presente divulgación que incluye los Ejemplos de trabajo del presente documento.
Selectividad de especie y reactividad cruzada de especie
De acuerdo con determinados aspectos de la divulgación, los anticuerpos anti-PD-1 se unen a PD-1 humano, pero no a PD-1 de otras especies. Como alternativa, los anticuerpos anti-PD-1 de la divulgación, en determinados aspectos, se unen a PD-1 humano y a PD-1 de una o más especies no humanas. Por ejemplo, los anticuerpos anti-PD-1 de la divulgación pueden unirse a PD-1 humano y pueden unirse o no, según sea el caso, a uno o más de PD-1 de ratón, rata, cobaya, hámster, jerbo, cerdo, gato, perro, conejo, cabra, oveja, vaca, caballo, camello, cinomolgo, tití, mono Rhesus o chimpancé. En determinados aspectos, los anticuerpos anti-PD-1 de la divulgación pueden unirse a PD-1 humano y de mono cinomolgo con las mismas afinidades o con diferentes afinidades, pero no se unen a PD-1 de rata y de ratón.
Mapeo de epítopos y tecnologías relacionadas
La presente divulgación incluye anticuerpos anti-PD-1 que interactúan con uno o más aminoácidos que se encuentran dentro de uno o más dominios de la molécula de PD-1, incluidos, por ejemplo, dominio extracelular (similar a IgV), un dominio transmembrana y un dominio intracelular que contiene el motivo inhibidor inmunorreceptor basado en tirosina (ITIM) y un motivo intercambiable inmunorreceptor basado en tirosina (ITSM). El epítopo al que se unen los anticuerpos puede consistir en una sola secuencia contigua de 3 o más (por ejemplo, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 o más) aminoácidos localizados dentro de cualquiera de los dominios de la molécula de PD-1 anteriormente mencionados (por ejemplo, un epítopo lineal en un dominio). Como alternativa, el epítopo puede consistir en una pluralidad de aminoácidos (o secuencias de aminoácidos) no contiguos dentro de uno o ambos de los dominios anteriormente mencionados de la molécula de PD-1 (por ejemplo, un epítopo conformacional).
Pueden usarse diversas técnicas conocidas por los expertos en la materia para determinar si un anticuerpo "interactúa con uno o más aminoácidos" dentro de un polipéptido o proteína. Las técnicas de ejemplo incluyen, por ejemplo, ensayos de retrobloqueo rutinario, tales como los descritos en Antibodies, Harlow y Lane (Cold Spring Harbor Press, Cold Spring Harbor, NY). Otros métodos incluyen análisis mutacionales de barrido de alanina, análisis de hibridación de péptidos (Reineke (2004) Methods Mol. Biol. 248: 443-63), análisis de escisión de péptidos, estudios cristalográficos y análisis por RMN. Además, pueden emplearse métodos tales como escisión de epítopos, extracción de epítopos y
modificación química de antígenos (Tomer (2000) Prot. Sci. 9: 487-496). Otro método que puede usarse para identificar los aminoácidos dentro de un polipéptido con el que interactúa un anticuerpo es el intercambio de hidrógeno/deuterio detectado por espectrometría de masas. En términos generales, el método de intercambio hidrógeno/deuterio implica el marcaje con deuterio de la proteína de interés, seguido de la unión del anticuerpo a la proteína marcada con deuterio. A continuación, el complejo proteína/anticuerpo se transfiere al agua y los protones intercambiables dentro de los aminoácidos que están protegidos por el complejo del anticuerpo experimentan un intercambio inverso de deuterio a hidrógeno a una velocidad más lenta que los protones intercambiables dentro de los aminoácidos que no forman parte de la interfaz. Como resultado, los aminoácidos que forman parte de la interfaz proteína/anticuerpo pueden retener deuterio y, por lo tanto, presentan una masa relativamente mayor en comparación con los aminoácidos no incluidos en la interfaz. Después de la disociación del anticuerpo, la proteína diana se somete a escisión por proteasa y análisis de espectrometría de masas, revelando de esta manera los restos marcados con deuterio que corresponden a los aminoácidos específicos con los que interactúa el anticuerpo. Véanse, por ejemplo, Ehring (1999) Analytical Biochemistry 267: 252-259; Engen y Smith (2001) Anal. Chem. 73: 256A-265A.
El término "epítopo"se refiere a un lugar sobre un antígeno al que responden linfocitos B y/o T. Los epítopos de linfocitos B pueden formarse tanto a partir de aminoácidos contiguos como a partir de aminoácidos no contiguos yuxtapuestos mediante el plegamiento terciario de una proteína. Los epítopos formados a partir de aminoácidos contiguos normalmente se mantienen al tratarlos con disolventes desnaturalizantes, mientras que los epítopos formados por plegamiento terciario normalmente se pierden al tratarlos con disolventes desnaturalizantes. Un epítopo normalmente incluye al menos 3, y más habitualmente, al menos 5 u 8-10 aminoácidos en una conformación espacial única.
El perfilado asistido por modificación (MAP), también conocido como perfilado de anticuerpos basado en la estructura del antígeno (ASAP) es un método que categoriza grandes números de anticuerpos monoclonales (mAb) dirigidos contra el mismo antígeno de acuerdo con las similitudes del perfil de unión de cada anticuerpo a superficies de antígenos modificadas química o enzimáticamente (Véase el documento US 2004/0101920). Cada categoría puede reflejar un epítopo único diferente o parcialmente superpuesto con el epítopo representado por otra categoría. Esta tecnología permite el filtrado rápido de anticuerpos genéticamente idénticos, de modo que la caracterización puede centrarse en anticuerpos genéticamente distintos. Cuando se aplica al cribado de hibridomas, MAP puede facilitar la identificación de clones de hibridomas raros que producen mAb que tienen las características deseadas. MAP se puede usar para clasificar los anticuerpos de la invención en grupos de anticuerpos que se unen a diferentes epítopos.
En determinados aspectos, los anticuerpos anti-PD-1 o fragmentos de unión a antígeno de los mismos se unen a un epítopo dentro de una o más de las regiones ilustradas en la PD-1, ya sea en forma natural, como se ilustra en la SEQ ID NO: 327, o producida de forma recombinante, como se ilustra en las SEQ ID NO: 321-324, o en un fragmento del mismo. En algunos aspectos, los anticuerpos de la divulgación se unen a una región extracelular que comprende uno 0 más aminoácidos seleccionados del grupo que consiste en los restos de aminoácidos 21-171 de PD-1. En algunos aspectos, los anticuerpos de la divulgación se unen a una región extracelular que comprende uno o más aminoácidos seleccionados del grupo que consiste en los restos de aminoácidos 1-146 de PD-1 del mono cinomolgo, como se ilustra por la SEQ ID NO: 322.
En determinados aspectos, los anticuerpos de la divulgación, como se muestra en la Tabla 1, interactúan con al menos una secuencia de aminoácidos seleccionada del grupo que consiste en restos de aminoácidos que varían desde aproximadamente la posición 21 hasta aproximadamente la posición 136 de la SEQ ID NO: 327; o restos de aminoácidos que varían desde aproximadamente la posición 136 hasta aproximadamente la posición 171 de la SEQ ID NO: 327. Estas regiones están parcialmente ilustradas en las SEQ ID No : 321 - 324.
La presente divulgación incluye anticuerpos anti-PD-1 que se unen al mismo epítopo, o una porción del epítopo, como cualquiera de los anticuerpos ilustrativos específicos descritos en el presente documento en la Tabla 1, o un anticuerpo que tiene las secuencias CDR de cualquiera de los anticuerpos ilustrativos descritos en la Tabla 1. De forma análoga, la presente divulgación también incluye anticuerpos anti-PD-1 que compiten por unirse a PD-1 o un fragmento de PD-1 con cualquiera de los anticuerpos ilustrativos específicos descritos en el presente documento en la Tabla 1, o un anticuerpo que tiene las secuencias CDR de cualquiera de los anticuerpos ilustrativos descritos en la Tabla 1. Por ejemplo, la presente divulgación incluye anticuerpos anti-PD-1 que compiten de forma cruzada para unirse a PD-1 con uno o más anticuerpos como se define en el Ejemplo 6 en el presente documento (por ejemplo, H2aM7788N, H4xH8992P, H4xH8999P, H1M7799N, H2aM7780N, H1M7800N, H2aM7794N, H2aM7798N, H4xH9145P2, H4H9057P2, H4xH9120P2, H4xH9128P2, H4H9019P, H4xH9119P2, H4xH9135P2, H4xH9034P, H2aM7790N, H4xH9035P, H4xH9037P, H4xH9045P y H2aM7795N).
Puede determinarse fácilmente si un anticuerpo se une al mismo epítopo que, o compite por la unión con, un anticuerpo anti-PD-1 de referencia usando métodos rutinarios conocidos en la técnica. Por ejemplo, para determinar si un anticuerpo de ensayo se une al mismo epítopo que un anticuerpo anti-PD-1 de referencia de la invención, se permite que el anticuerpo de referencia se una a una proteína o péptido PD-1 en condiciones de saturación. A continuación, se evalúa la capacidad de un anticuerpo de ensayo de unirse a la molécula de PD-1. Si el anticuerpo de ensayo puede unirse a PD-1 después de la unión por saturación con el anticuerpo anti-PD-1 de referencia, puede concluirse que el anticuerpo de ensayo se une a un epítopo diferente del anticuerpo anti-PD-1 de referencia. Por otro lado, si el
anticuerpo de ensayo no puede unirse a la proteína de PD-1 después de la unión por saturación con el anticuerpo anti-PD-1 de referencia, entonces el anticuerpo de ensayo puede unirse al mismo epítopo al que se une el anticuerpo anti-PD-1 de referencia de la invención.
Para determinar si un anticuerpo compite por la unión con un anticuerpo anti-PD-1 de referencia, se realiza la metodología de unión descrita anteriormente en dos orientaciones: En una primera orientación, se permite que el anticuerpo de referencia se una a una proteína PD-1 en condiciones de saturación seguido por evaluación de la unión del anticuerpo de ensayo a la molécula de PD-1. En una segunda orientación, se permite que el anticuerpo de ensayo se una a una molécula PD-1 en condiciones de saturación seguido por evaluación de la unión del anticuerpo de referencia a la molécula de PD-1. Si, en ambas orientaciones, únicamente el primer anticuerpo (de saturación) puede unirse a la molécula de PD-1, entonces se concluye que el anticuerpo de ensayo y el anticuerpo de referencia compiten por la unión a PD-1. Como apreciará un experto en la materia, un anticuerpo que compite por la unión con un anticuerpo de referencia puede que no se una necesariamente al epítopo idéntico al anticuerpo de referencia, sino que puede bloquear estéricamente la unión del anticuerpo de referencia mediante la unión a un epítopo solapante o adyacente.
Dos anticuerpos se unen al mismo epítopo o epítopos solapantes si cada uno inhibe (bloquea) competitivamente la unión del otro al antígeno. Es decir, un exceso en un factor de 1, 5, 10, 20 o 100 de un anticuerpo inhibe la unión del otro en al menos un 50 %, pero preferentemente en un 75 %, 90 % o incluso un 99 %, según se mide en un ensayo de unión competitiva (véase, por ejemplo, Junghans et al., Cancer Res. 1990 50:1495-1502). Como alternativa, dos anticuerpos tienen el mismo epítopo si esencialmente todas las mutaciones de aminoácido en el antígeno que reducen o eliminan la unión de un anticuerpo reducen o eliminan la unión del otro. Dos anticuerpos tienen epítopos solapantes si algunas mutaciones de aminoácido que reducen o eliminan la unión de un anticuerpo reducen o eliminan la unión del otro.
A continuación, puede realizarse experimentación de rutina adicional (por ejemplo, mutación de péptidos y análisis de unión) para confirmar si la ausencia observada de unión del anticuerpo de ensayo se debe, de hecho, a la unión al mismo epítopo que el anticuerpo de referencia o si el bloqueo estérico (u otro fenómeno) es responsable de la ausencia de la unión observada. Los experimentos de este tipo pueden realizarse usando ELISA, RIA, resonancia del plasmón superficial, citometría de flujo o cualquier otro ensayo cuantitativo o cualitativo de unión de anticuerpos disponible en la técnica.
Inmunoconjugados
La invención abarca un anticuerpo monoclonal anti-PD-1 humano de la invención conjugado con una fracción terapéutica ("inmunoconjugado"), tal como una citotoxina o un agente quimioterapéutico para tratar el cáncer. Como se utiliza en el presente documento, el término "inmunoconjugado" se refiere a un anticuerpo que está unido química o biológicamente a una citotoxina, un agente radiactivo, una citocina, un interferón, una diana o fracción indicadora, una enzima, una toxina, un péptido o proteína o un agente terapéutico. El anticuerpo se puede unir a la citotoxina, agente radiactivo, citocina, interferón, diana o fracción indicadora, enzima, toxina, péptido o agente terapéutico en cualquier lugar a lo largo de la molécula siempre que sea capaz de unirse a su diana. Los ejemplos de inmunoconjugados incluyen conjugados anticuerpo-fármaco y proteínas de fusión anticuerpo-toxina. En una realización, el agente puede ser un segundo anticuerpo diferente contra PD-1. En determinadas realizaciones, el anticuerpo puede conjugarse con un agente específico de una célula tumoral o una célula infectada por virus. El tipo de fracción terapéutica que se puede conjugar con el anticuerpo anti-PD-1 tendrá en cuenta la afección a tratar y el efecto terapéutico deseado a lograr. En la técnica se conocen ejemplos de agentes adecuados para formar inmunoconjugados; véase, por ejemplo, el documento WO 05/103081.
Anticuerpos multiespecíficos
Los anticuerpos de la presente invención pueden ser monoespecíficos, biespecíficos, o multiespecíficos. Los anticuerpos multiespecíficos pueden ser específicos para diferentes epítopos de un polipéptido diana o pueden contener dominios de unión a antígeno específicos para más de un polipéptido diana. Véanse, por ejemplo, Tutt et al., 1991, J. Immunol. 147:60-69; Kufer et al., 2004, Trends Biotechnol. 22:238-244.
En un aspecto, la presente divulgación incluye moléculas de unión a antígeno multiespecíficas o fragmentos de unión a antígeno de las mismas en las que una especificidad de una inmunoglobulina es específica del dominio extracelular de PD-1, o un fragmento del mismo, y la otra especificidad de la inmunoglobulina es específica para unirse fuera del dominio extracelular de PD-1, o una segunda diana terapéutica, o está conjugada con un resto terapéutico. En determinados aspectos, la primera especificidad de unión a antígeno puede comprender PD-L1 o PD-L2, o un fragmento del mismo. En determinados aspectos de la divulgación, una especificidad de una inmunoglobulina es específica de un epítopo que comprende los restos de aminoácidos 21-171 de PD-1 (SEQ ID NO: 327) o un fragmento del mismo, y la otra especificidad de la inmunoglobulina es específica de un segundo antígeno diana. El segundo antígeno diana puede estar en la misma célula que PD-1 o en una célula diferente. En un aspecto, la segunda célula diana está en una célula inmunitaria distinta de un linfocito T, tal como un linfocito B, célula presentadora de antígenos, monocito, macrófago o célula dendrítica. En algún aspecto, el segundo antígeno diana puede estar presente en una célula tumoral o una célula de tejido autoinmunitario o en una célula infectada por virus.
En otro aspecto, la divulgación proporciona moléculas de unión a antígeno multiespecíficas o fragmentos de unión a antígeno de las mismas que comprenden una primera especificidad de unión a antígeno que se une a PD-1 y una segunda especificidad de unión a antígeno que se une a un receptor de linfocitos T, un receptor de linfocitos B o un receptor Fc. En un aspecto relacionado, la divulgación proporciona moléculas de unión a antígeno multiespecíficas o fragmentos de unión a antígeno de las mismas que comprenden una primera especificidad de unión a antígeno que se une a PD-1 y una segunda especificidad de unión a antígeno que se une a un coinhibidor de linfocitos T diferente, tal como LAG-3, CTLA-4, BTLA, CD-28, 2B4, LY108, TIGIT, TIM3, LAIR1, ICOS y CD160.
En otro aspecto, la divulgación proporciona moléculas de unión a antígeno multiespecíficas o fragmentos de unión a antígeno de las mismas que comprenden una primera especificidad de unión a antígeno que se une a PD-1 y una segunda especificidad de unión a antígeno que se une a un antígeno específico de tejido autoinmunitario. En determinados aspectos, los anticuerpos pueden ser anticuerpos activantes o agonistas.
Cualquiera de las moléculas de unión a antígeno multiespecíficas de la invención, o variantes de las mismas, puede construirse usando técnicas de biología molecular estándar (por ejemplo, tecnología de ADN recombinante y expresión de proteínas), como sabrá un experto habitual en la materia.
En algunas realizaciones, los anticuerpos específicos de PD-1 se generan en un formato biespecífico (un "biespecífico") en el que las regiones variables que se unen a dominios distintos de PD-1 se unen para conferir especificidad de dominio doble dentro de una sola molécula de unión. Los biespecíficos adecuadamente diseñados pueden mejorar la eficacia inhibidora general de PD-1 al aumentar tanto la especificidad como la avidez de unión. Las regiones variables con especificidad para dominios individuales, (por ejemplo, segmentos del dominio N-terminal), o que pueden unirse a diferentes regiones dentro de un dominio, se emparejan en un armazón estructural que permite que cada región se una simultáneamente a los epítopos separados, o a diferentes regiones dentro de un dominio. En un ejemplo de un biespecífico, las regiones variables de la cadena pesada (Vh) de un ligante con especificidad para un dominio se recombinan con regiones variables de la cadena ligera (Vl) a partir de una serie de ligantes con especificidad para un segundo dominio para identificar parejas Vl no afines que se pueden emparejar con una Vh original sin alterar la especificidad original para esa Vh. De esta forma, un único segmento Vl (por ejemplo, Vl1) se puede combinar con dos dominios Vh diferentes (por ejemplo, Vh1 y Vh2) para generar un biespecífico comprendido por dos "brazos" de unión " (Vh1-Vl1 y Vh2-Vl1). El uso de un solo segmento de Vl reduce la complejidad del sistema y, por lo tanto, simplifica y aumenta la eficiencia en los procesos de clonación, expresión y purificación utilizados para generar el biespecífico (véase, por ejemplo, USSN13/022759 y US2010/0331527).
Como alternativa, los anticuerpos que se unen a más de un dominio y una segunda diana, tales como, aunque no de forma limitativa, por ejemplo, un segundo anticuerpo anti-PD-1 diferente, pueden prepararse en un formato biespecífico usando técnicas descritas en el presente documento, u otras técnicas conocidas por los expertos en la materia. Las regiones variables de anticuerpos que se unen a regiones distintas pueden unirse junto con regiones variables que se unen a sitios relevantes en, por ejemplo, el dominio extracelular de PD-1, para conferir especificidad de antígeno doble dentro de una única molécula de unión. Los biespecíficos adecuadamente diseñados de esta naturaleza cumplen una doble función. Las regiones variables con especificidad para el dominio extracelular se combinan con una región variable con especificidad del exterior del dominio extracelular y se emparejan en un armazón estructural que permite que cada región variable se una a los antígenos separados.
Un formato de anticuerpo biespecífico de ejemplo que puede usarse en el contexto de la presente invención implica el uso de un primer dominio CH3 de inmunoglobulina (Ig) y un segundo dominio CH3 de Ig, en donde el primer y el segundo dominio Ch3 de Ig difieren entre sí en al menos un aminoácido, y en donde al menos una diferencia de aminoácido reduce la unión del anticuerpo biespecífico a la proteína A en comparación con un anticuerpo biespecífico que carece de la diferencia de aminoácido. En una realización, el primer dominio Ch3 de Ig está unido a proteína A y el segundo dominio Ch3 de Ig contiene una mutación que reduce o anula la unión a proteína A tal como una modificación H95R (por numeración de exones IMGT; H435R por numeración EU). El segundo Ch3 puede comprender además una modificación Y96F (por IMGT; Y436F por EU). Otras modificaciones adicionales que pueden encontrarse dentro del segundo Ch3 incluyen: D16E, L18M, N44S, K52N, V57M y V82I (por IMGT; D356E, L358M, N384S, K392N, V397M y V422I por EU) en el caso de anticuerpos IgG1; N44S, K52N y V82I (IMGT; N384S, K392N y V422I por EU) en el caso de anticuerpos IgG2; y Q15R, N44S, K52N, V57M, R69K, E79Q y V82I (por IMGT; Q355R, N384S, K392N, V397M, R409K, E419Q y V422I por EU) en el caso de anticuerpos IgG4. Dentro del alcance de la presente invención se contemplan variaciones en el formato de anticuerpo biespecífico descrito anteriormente.
Otros formatos biespecíficos ilustrativos que pueden usarse en el contexto de la presente invención incluyen, sin limitación, por ejemplo, formatos biespecíficos basados en scFv o de diacuerpo, fusiones IgG-scFv, dominio variable doble (DVD)-lg, cuadroma, "botón en ojal", cadena ligera común (por ejemplo, cadena ligera común con "botón en ojal", etc.), CrossMab, CrossFab, (SEED)cuerpo, cremallera de leucina, Duocuerpo, lgG1/lgG2, Fab de acción doble (DAF)-IgG y formatos biespecíficos de Mab2 (véase, e.g., Klein et al. 2012, mAbs 4:6, 1-11, y las referencias citadas en el mismo, para una revisión de los formatos anteriores). También se pueden construir anticuerpos biespecíficos usando conjugación de péptido/ácido nucleico, por ejemplo, en los que se usan aminoácidos no naturales con reactividad química ortogonal para generar conjugados de anticuerpo-oligonucleótido específicos de sitio que después
se autoensamblan en complejos multiméricos con composición, valencia y geometría definidas. (Véase, por ejemplo, Kazane et al., J. Am. Chem. Soc. [Publicación electrónica: 4 de diciembre de 2012]).
Administración terapéutica y formulaciones
La invención proporciona composiciones terapéuticas que comprenden los anticuerpos anti-PD-1 o fragmentos de unión a antígeno de los mismos de la presente invención. Las composiciones terapéuticas de acuerdo con la invención se administrarán con vehículos, excipientes y otros agentes adecuados que se incorporan en las formulaciones para proporcionar una mejor transferencia, administración, tolerancia y similares. Puede encontrarse una multitud de formulaciones adecuadas en el formulario conocido para todos los químicos farmacéuticos: Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA. Estas formulaciones incluyen, por ejemplo, polvos, pastas, pomadas, gelatinas, ceras, aceites, lípidos, vesículas que contienen lípidos (catiónicos o aniónicos) (tales como LIPOFECTIN™), conjugados de ADN, pastas de absorción anhidras, emulsiones de aceite en agua y agua en aceite, emulsiones de Carbowax (polietilenglicoles de diversos pesos moleculares), geles semisólidos y mezclas semisólidas que contienen Carbowax. Véase también Powell et al. "Compendium of excipients for parenteral formulations" PDA (1998) J Pharm Sci Technol 52:238-311.
La dosis de anticuerpo puede variar dependiendo de la edad y el tamaño de un sujeto a recibir la administración, la enfermedad diana, afecciones, vía de administración y similares. Cuando un anticuerpo de la presente invención se usa para tratar una enfermedad o trastorno en un paciente adulto, o para prevenir dicha enfermedad, es ventajoso administrar el anticuerpo de la presente invención, normalmente en una única dosis de aproximadamente 0,1 a aproximadamente 60 mg/kg de peso corporal, más preferentemente de aproximadamente 5 a aproximadamente 60, de aproximadamente 10 a aproximadamente 50, o de aproximadamente 20 a aproximadamente 50 mg/kg de peso corporal. Dependiendo de la intensidad de la afección, pueden ajustarse la frecuencia y la duración del tratamiento. En determinados aspectos, el anticuerpo o fragmento de unión a antígeno del mismo de la invención puede administrarse como una dosis inicial de al menos aproximadamente 0,1 mg a aproximadamente 800 mg, aproximadamente 1 a aproximadamente 500 mg, aproximadamente 5 a aproximadamente 300 mg, o aproximadamente 10 a aproximadamente 200 mg, hasta aproximadamente 100 mg, o hasta aproximadamente 50 mg. En determinados aspectos, la dosis inicial puede ir seguida de la administración de una segunda o una pluralidad de dosis posteriores del anticuerpo o fragmento de unión a antígeno del mismo en una cantidad que puede ser aproximadamente la misma o menor que la de la dosis inicial, en donde las dosis posteriores están separadas por al menos 1 día a 3 días; al menos una semana, al menos 2 semanas; al menos 3 semanas; al menos 4 semanas; al menos 5 semanas; al menos 6 semanas; al menos 7 semanas; al menos 8 semanas; al menos 9 semanas; al menos 10 semanas; al menos 12 semanas; o al menos 14 semanas.
Se conocen diversos sistemas de administración y pueden usarse para administrar la composición farmacéutica de la invención, por ejemplo, encapsulación en liposomas, micropartículas, microcápsulas, células recombinantes capaces de expresar los virus mutantes, endocitosis mediada por receptor (véase, por ejemplo, Wu et al. (1987) J. Biol. Chem.
262:4429-4432). Los métodos de introducción incluyen, aunque no de forma limitativa, las vías intradérmica, transdérmica, intramuscular, intraperitoneal, intravenosa, subcutánea, intranasal, epidural y oral. La composición puede administrarse por cualquier vía conveniente, por ejemplo, por infusión o inyección en bolo, por absorción a través de revestimientos epiteliales o mucocutáneos (por ejemplo, mucosa oral, mucosa rectal e intestinal, etc.) y puede administrarse junto con otros agentes biológicamente activos. La administración puede ser sistémica o local. La composición farmacéutica también puede administrarse en una vesícula, en particular un liposoma (véase, por ejemplo, Langer (1990) Science 249:1527-1533).
El uso de nanopartículas para administrar los anticuerpos de la presente invención también se contempla en el presente documento. Las nanopartículas conjugadas con anticuerpo pueden usarse tanto para aplicaciones terapéuticas como de diagnóstico. Las nanopartículas conjugadas con anticuerpo y los métodos de preparación y uso se describen en detalle por Arruebo, M., et al. 2009 ("Antibody-conjugated nanoparticles for biomedical applications" en J. Nanomat. Volume 2009, Artículo ID 439389, 24 páginas), doi: 10.1155/2009/439389). Las nanopartículas se pueden desarrollar y conjugar con anticuerpos contenidos en composiciones farmacéuticas para dirigirse a células tumorales diana o células de tejido autoinmunitario o células infectadas por virus. También se han descrito nanopartículas para la administración de fármacos en, por ejemplo, los documentos EP 8257740 o EP 8246995.
En determinadas situaciones, la composición farmacéutica puede suministrarse en un sistema de liberación controlada. En una realización, se puede usar una bomba. En otra realización, pueden usarse materiales poliméricos. En otra realización más, puede ubicarse un sistema de liberación controlada en las proximidades de la diana de la composición, siendo necesaria, por lo tanto, solo una fracción de la dosis sistémica.
Las preparaciones inyectables pueden incluir formas farmacéuticas para inyecciones intravenosas, subcutáneas, intracutáneas, intracraneales, intraperitoneales e intramusculares, infusiones por goteo, etc. Estas preparaciones inyectables pueden prepararse por métodos conocidos públicamente. Por ejemplo, las preparaciones inyectables pueden prepararse, por ejemplo, disolviendo, suspendiendo o emulsionando el anticuerpo o su sal descrita anteriormente en un medio acuoso estéril o un medio oleoso utilizado convencionalmente para inyecciones. Como medio acuoso para inyecciones, existen, por ejemplo, solución salina fisiológica, una solución isotónica que contiene
glucosa y otros auxiliares, etc., que pueden usarse en combinación con un agente solubilizante apropiado tal como un alcohol (por ejemplo, etanol), un polialcohol (por ejemplo, propilenglicol, polietilenglicol), un tensioactivo no iónico [por ejemplo, polisorbato 80, HCO-50 (aducto de polioxietileno (50 mol) de aceite de ricino hidrogenado)], etc. Como medio oleoso, se emplean, por ejemplo, aceite de sésamo, aceite de soja, etc., que pueden usarse en combinación con un agente solubilizante tal como benzoato de bencilo, alcohol bencílico, etc. La inyección preparada de este modo se carga preferentemente en una ampolla adecuada.
Una composición farmacéutica de la presente invención puede administrarse por vía subcutánea o por vía intravenosa con una aguja y una jeringa convencionales. Además, con respecto a la administración subcutánea, un dispositivo inyector de pluma tiene fácilmente aplicaciones en la administración de una composición farmacéutica de la presente invención. Dicho dispositivo inyector de pluma puede ser reutilizable o desechable. Un dispositivo inyector de pluma reutilizable utiliza generalmente un cartucho reemplazable que contiene una composición farmacéutica. Una vez que se ha administrado toda la composición farmacéutica dentro del cartucho y que el cartucho está vacío, el cartucho vacío puede desecharse fácilmente y remplazarse con un nuevo cartucho que contiene la composición farmacéutica. El dispositivo inyector de pluma puede entonces reutilizarse. En un dispositivo inyector de pluma desechable, no hay cartucho reemplazable. Más bien, el dispositivo inyector de pluma desechable viene precargado con la composición farmacéutica mantenida en un depósito dentro del dispositivo. Una vez que el depósito se vacía de la composición farmacéutica, el dispositivo completo se desecha.
Numerosos dispositivos inyectores de pluma y autoinyectores reutilizables tienen aplicaciones en la administración subcutánea de una composición farmacéutica de la presente invención. Los ejemplos incluyen, aunque ciertamente sin limitación, AUTOPEN™ (Owen Mumford, Inc., Woodstock, RU), pluma DiSETRONIC™ (Disetronic Medical Systems, Bergdorf, Suiza), pluma HUMALOG MIX 75/25™, pluma HUMALOG™, pluma HUMALIN 70/30™ (Eli Lilly y Co., Indianápolis, IN), NOVOPEN™1 I, II y III (Novo Nordisk, Copenhague, Dinamarca), NOVOPEN JUNIOR™ (Novo Nordisk, Copenhague, Dinamarca), pluma BD™ (Becton Dickinson, Franklin Lakes, NJ), OPTIPEN™, OPTIPEN PRO™, OpT iPEN STARLET™, y OpTI-CLIK™ (Sanofi-Aventis, Frankfurt, Alemania), por nombrar solo algunos. Los ejemplos de dispositivos inyectores de pluma desechables que tienen aplicaciones en la administración subcutánea de una composición farmacéutica de la presente invención incluyen, aunque ciertamente sin limitación, la pluma SOLOSTAR™ (Sanofi-Aventis), la FLEXPEN™ (Novo Nordisk) y la KWIKPEN™ (Eli Lilly), el autoinyector SURECLICK ™ (Amgen, Thousand Oaks, CA), el PENLET ™ (Haselmeier, Stuttgart, Alemania), el EPIPEN (Dey, L.P.) y la pluma HUMIRA ™ (Abbott Labs, Abbott Park, IL), por nombrar solo algunos.
Ventajosamente, las composiciones farmacéuticas para su uso oral o parenteral descritas anteriormente se preparan en formas farmacéuticas en una dosis unitaria adecuada para ajustarse a una dosis de los principios activos. Dichas formas farmacéuticas en una dosis unitaria incluyen, por ejemplo, comprimidos, pastillas, cápsulas, inyecciones (ampollas), supositorios, etc. La cantidad del anticuerpo contenida generalmente es de aproximadamente 5 a aproximadamente 500 mg por forma farmacéutica en una dosis unitaria; especialmente en forma de inyección, se prefiere que el anticuerpo esté contenido en aproximadamente 5 a aproximadamente 100 mg y en aproximadamente 10 a aproximadamente 250 mg para las otras formas farmacéuticas.
Usos terapéuticos de los anticuerpos
Los anticuerpos de la invención son útiles, entre otros, para el tratamiento, prevención y/o mejora de cualquier enfermedad o trastorno asociado o mediado por la expresión, señalización o actividad de PD-1, o que se puede tratar bloqueando la interacción entre PD-1 y un ligando de PD-1 (por ejemplo, PD-L1 o PD-L2) o inhibiendo de otro modo la actividad y/o señalización de PD-1. Por ejemplo, la presente divulgación proporciona métodos para el tratamiento del cáncer (inhibición del crecimiento tumoral), infecciones víricas crónicas y/o enfermedad autoinmunitaria mediante la administración de un anticuerpo anti-PD-1 (o composición farmacéutica que comprende un anticuerpo anti-PD-1) como se describe en el presente documento a un paciente que necesita dicho tratamiento. Los anticuerpos de la presente divulgación son útiles en el tratamiento, prevención y/o mejora de una enfermedad o trastorno o afección tal como cáncer, enfermedad autoinmunitaria o una infección vírica y/o para mejorar al menos un síntoma asociado con dicha enfermedad, trastorno o afección. En el contexto de los métodos de tratamiento descritos en el presente documento, el anticuerpo anti-PD-1 se puede administrar como monoterapia (es decir, como único agente terapéutico) o en combinación con uno o más agentes terapéuticos adicionales (ejemplos de los cuales se describen en otra parte del presente documento).
En algunos aspectos de la divulgación, los anticuerpos descritos en el presente documento son útiles para tratar sujetos que padecen cáncer primario o recurrente, incluyendo, aunque no de forma limitativa, carcinoma de células renales, cáncer colorrectal, cáncer de pulmón no microcítico, cáncer de cerebro (por ejemplo, glioblastoma multiforme), carcinoma de células escamosas de cabeza y cuello, cáncer gástrico, cáncer de próstata, cáncer de ovario, cáncer de riñón, cáncer de mama, mieloma múltiple y melanoma.
Los anticuerpos pueden usarse para tratar los síntomas del cáncer en etapa temprana o tardía. En un aspecto, se puede usar un anticuerpo o fragmento del mismo de la invención para tratar el cáncer metastásico. Los anticuerpos son útiles para reducir o inhibir o evitar el crecimiento tumoral tanto de tumores sólidos como de cánceres sanguíneos. En determinados aspectos, el tratamiento con un anticuerpo o fragmento de unión a antígeno del mismo de la invención
conduce a más del 50 % de regresión, más del 60 % de regresión, más del 70 % de regresión, más del 80 % de regresión o más del 90 % de regresión de un tumor en un sujeto. En determinados aspectos, los anticuerpos pueden usarse para prevenir la recaída de un tumor. En determinados aspectos, los anticuerpos son útiles para prolongar la supervivencia general en un sujeto con cáncer. En algunos aspectos, los anticuerpos son útiles para reducir la toxicidad debida a la quimioterapia o la radioterapia a la vez que mantienen la supervivencia a largo plazo en un paciente que padece cáncer.
En determinados aspectos, los anticuerpos de la invención son útiles para tratar sujetos que padecen una infección vírica crónica. En algunos aspectos, los anticuerpos de la invención son útiles para disminuir los títulos víricos en el hospedador y/o rescatar los linfocitos T agotados. En determinados aspectos, se puede usar un anticuerpo o fragmento del mismo de la invención para tratar la infección vírica crónica por el virus de la coriomeningitis linfocítica (VCML). En algunos aspectos, un anticuerpo o fragmento de unión a antígeno del mismo de la invención puede administrarse a una dosis terapéutica a un paciente con una infección por el virus de la inmunodeficiencia humana (VIH) o el virus del papiloma humano (VPH) o el virus de la hepatitis B/C (VHB/VHC). En un aspecto relacionado, un anticuerpo o fragmento de unión a antígeno del mismo de la invención puede usarse para tratar una infección por el virus de la inmunodeficiencia de simios (VIS) en un sujeto simio tal como el mono cinomolgo.
En determinados aspectos, se puede administrar un anticuerpo bloqueante de la presente invención en una cantidad terapéuticamente eficaz a un sujeto que padece un cáncer o una infección vírica.
En determinados aspectos, los anticuerpos de la divulgación son útiles para tratar una enfermedad autoinmunitaria, incluyendo pero sin limitación, alopecia areata, hepatitis autoinmunitaria, enfermedad celíaca, enfermedad de Graves, síndrome de Guillain-Barré, enfermedad de Hashimoto, anemia hemolítica, enfermedad inflamatoria intestinal, miopatías inflamatorias, esclerosis múltiple, cirrosis biliar primaria, psoriasis, artritis reumatoide, esclerodermia, síndrome de Sjogren, lupus eritematoso sistémico, vitíligo, pancreatitis autoinmunitaria, urticaria autoinmunitaria, púrpura trombocitopénica autoinmunitaria, enfermedad de Crohn, diabetes tipo I, fascitis eosinofílica, enterogastritis eosinofílica, síndrome de Goodpasture, miastenia grave, artritis psoriásica, fiebre reumática, colitis ulcerosa, vasculitis y granulomatosis de Wegener. En determinados aspectos, se puede usar un anticuerpo activador de la divulgación para tratar a un sujeto que padece una enfermedad autoinmunitaria.
Se pueden administrar uno o más anticuerpos de la presente invención para aliviar o prevenir o disminuir la gravedad de uno o más de los síntomas o afecciones de la enfermedad o trastorno.
También se contempla en el presente documento utilizar uno o más anticuerpos de la presente divulgación de forma profiláctica en pacientes con riesgo de desarrollar una enfermedad o trastorno tal como cáncer, enfermedad autoinmunitaria e infección vírica crónica.
En un aspecto adicional de la divulgación, los presentes anticuerpos se utilizan para la preparación de una composición farmacéutica para el tratamiento de pacientes que padecen cáncer, enfermedad autoinmunitaria o infección vírica. En otro aspecto de la divulgación, los presentes anticuerpos se usan como terapia adjunta con cualquier otro agente o cualquier otra terapia conocida por los expertos en la técnica útil para tratar el cáncer, enfermedad autoinmunitaria o infección vírica.
Terapias y formulaciones de combinación
Las terapias de combinación pueden incluir un anticuerpo anti-PD-1 de la invención y cualquier agente terapéutico adicional que pueda combinarse de manera ventajosa con un anticuerpo de la invención, o con un fragmento biológicamente activo de un anticuerpo de la invención.
Los anticuerpos de la presente invención pueden combinarse sinérgicamente con uno o más fármacos contra el cáncer o terapia usada para tratar el cáncer, incluyendo, por ejemplo, carcinoma de células renales, cáncer colorrectal, glioblastoma multiforme, carcinoma de células escamosas de cabeza y cuello, cáncer de pulmón no microcítico, cáncer de colon, cáncer de ovario, adenocarcinoma, cáncer de próstata, glioma, y melanoma. En el presente documento se contempla usar anticuerpos anti-PD-1 de la invención en combinación con terapias inmunoestimuladoras y/o inmunosupresoras para inhibir el crecimiento tumoral y/o mejorar la supervivencia de pacientes con cáncer. Las terapias inmunoestimuladoras incluyen terapias inmunoestimuladoras directas para aumentar la actividad de las células inmunitarias "liberando el freno" sobre las células inmunitarias suprimidas o "pisando el acelerador" para activar una respuesta inmunitaria. Los ejemplos incluyen el direccionamiento a otros receptores de puntos de control, vacunación y adyuvantes. Las modalidades de inmunosupresión pueden aumentar la antigenicidad del tumor al promover la muerte celular inmunogénica, la inflamación o tener otros efectos indirectos que promueven una respuesta inmunitaria antitumoral. Los ejemplos incluyen radiación, quimioterapia, agentes anti-angiogénicos y cirugía.
En diversas realizaciones, se pueden usar uno o más anticuerpos de la presente invención en combinación con un anticuerpo contra PD-L1, un segundo anticuerpo contra PD-1 (por ejemplo, nivolumab), un inhibidor de LAG-3, un inhibidor de CTLA-4 (por ejemplo, ipilimumab), un inhibidor de TlM3, un inhibidor de BTLA, un inhibidor de TIGIT, un inhibidor de CD47, un antagonista de otro coinhibidor o ligando de linfocitos T (por ejemplo, un anticuerpo contra CD28, 2B4, LY108, LAIR1, ICOS, CD160 o VISTA), un inhibidor de la indolamina-2,3-dioxigenasa (IDO), un antagonista del factor de crecimiento endotelial vascular (VEGF) [por ejemplo, una "trampa de VEGF" tal como aflibercept u otra proteína de fusión inhibidora de VEGF como se establece en el documento US 7.087.411 o un anticuerpo anti-VEGF o fragmento de unión a antígeno del mismo (por ejemplo, bevacizumab, o ranibizumab) o un inhibidor de cinasa de molécula pequeña del receptor de VEGF (por ejemplo, sunitinib, sorafenib o pazopanib)], un inhibidor de Ang2 (por ejemplo, nesvacumab), un inhibidor del factor de crecimiento transformante beta (TGFp), un inhibidor del receptor del factor de crecimiento epidérmico (EGFR) (por ejemplo, erlotinib, cetuximab), un agonista de un receptor coestimulador (por ejemplo, un agonista de la proteína relacionada con TNFR inducida por glucocorticoides), un anticuerpo contra un antígeno específico de tumores (por ejemplo, CA9, CA125, antígeno 3 asociado a melanoma (MAGE3), antígeno carcinoembrionario (CEA), vimentina, tumor-M2-PK, antígeno específico de la próstata (AEP), mucina-1, MART-1, y CA19-9), una vacuna (por ejemplo, el bacilo de Calmette-Guerin, una vacuna contra el cáncer), un adyuvante para aumentar la presentación de antígenos (por ejemplo, factor estimulante de colonias de granulocitos-macrófagos), un anticuerpo biespecífico (por ejemplo, un anticuerpo biespecífico CD3xCD20, un anticuerpo biespecífico PSMAxCD3), una citotoxina, un agente quimioterapéutico (por ejemplo, dacarbazina, temozolomida, ciclofosfamida, docetaxel, doxorrubicina, daunorrubicina, cisplatino, carboplatino, gemcitabina, metotrexato, mitoxantrona, oxaliplatino, paclitaxel y vincristina), ciclofosfamida, radioterapia, un inhibidor de IL-6R (por ejemplo, sarilumab), un inhibidor de lL-4R (por ejemplo, dupilumab), un inhibidor de la IL-10, una citocina tal como IL-2, IL-7, IL-21 y IL-15, un conjugado anticuerpofármaco (ADC) (por ejemplo, el ADC anti-CD19-DM4 y el ADC anti-DS6-DM4), corticoesteroides y fármacos antiinflamatorios no esteroideos), un suplemento dietético, tal como los antioxidantes o cualquier cuidado paliativo para tratar el cáncer. En determinadas realizaciones, los anticuerpos anti-PD-1 de la presente invención pueden usarse en combinación con vacunas contra el cáncer, incluidas vacunas de células dendríticas, virus oncolíticos, vacunas contra células tumorales, etc. para aumentar la respuesta antitumoral. Ejemplos de vacunas contra el cáncer que se pueden usar en combinación con anticuerpos anti-pD-1 de la presente invención incluyen la vacuna MAGE3 para el melanoma y el cáncer de vejiga, la vacuna MUC1 para el cáncer de mama, EGFRv3 (por ejemplo, rindopepimut) para el cáncer de cerebro (incluido el glioblastoma multiforme) o ALVAC-CEA (para los cánceres CEA+).
En determinados aspectos, los anticuerpos anti-PD-1 de la invención pueden administrarse en combinación con radioterapia en métodos para generar respuestas antitumorales duraderas a largo plazo y/o mejorar la supervivencia de pacientes con cáncer. En algunos aspectos, los anticuerpos anti-PD-1 de la invención pueden administrarse antes de, concomitantemente o después de administrar radioterapia a un paciente con cáncer. Por ejemplo, la radioterapia se puede administrar en una o más dosis a las lesiones tumorales seguido de la administración de una o más dosis de anticuerpos anti-PD-1 de la invención. En algunos aspectos, la radioterapia se puede administrar localmente a una lesión tumoral para potenciar la inmunogenicidad local del tumor de un paciente (radiación adyuvante) y/o para destruir las células tumorales (radiación ablativa) seguida de la administración sistémica de un anticuerpo anti-PD-1. de la invención. Por ejemplo, se puede administrar radiación intracraneal a un paciente con cáncer de cerebro (por ejemplo, glioblastoma multiforme) en combinación con la administración sistémica de un anticuerpo anti-PD-1 de la invención. En determinados aspectos, los anticuerpos anti-PD-1 de la invención pueden administrarse en combinación con radioterapia y un agente quimioterapéutico (por ejemplo, temozolomida) o un antagonista de VEGF (por ejemplo, aflibercept).
En determinados aspectos, los anticuerpos anti-PD-1 de la invención se pueden administrar en combinación con uno o más fármacos antivíricos para tratar la infección vírica crónica causada por el VCML, VIH, VPH, VHB o VHC. Ejemplos de fármacos antivíricos incluyen, aunque no de forma limitativa, zidovudina, lamivudina, abacavir, ribavirina, lopinavir, efavirenz, cobicistat, tenofovir, rilpivirina y corticosteroides. En algunos aspectos, los anticuerpos anti-PD-1 de la invención pueden administrarse en combinación con un inhibidor de LAG3, un inhibidor de CTLA-4 o cualquier antagonista de otro coinhibidor de linfocitos T para tratar una infección vírica crónica.
En determinados aspectos, los anticuerpos anti-PD-1 de la divulgación pueden combinarse con un anticuerpo contra un receptor Fc en células inmunitarias para el tratamiento de una enfermedad autoinmunitaria. En un aspecto, un anticuerpo o fragmento del mismo de la divulgación se administra en combinación con un anticuerpo o proteína de unión a antígeno dirigido a un antígeno específico de tejido autoinmunitario. En determinados aspectos, un anticuerpo o fragmento de unión a antígeno del mismo de la divulgación se administra en combinación con un anticuerpo o proteína de unión a antígeno dirigido a un receptor de linfocitos T o un receptor de linfocitos B, incluyendo pero sin limitación, Fca (por ejemplo, CD89), Fcy (por ejemplo, CD64, CD32, CD16a y CD16b), CD19, etc. Los anticuerpos o fragmentos de los mismos de la divulgación pueden usarse en combinación con cualquier fármaco o terapia conocida en la técnica (por ejemplo, corticosteroides y otros inmunosupresores) para tratar una enfermedad o trastorno autoinmunitario que incluye, pero sin limitación, alopecia areata, hepatitis autoinmunitaria, enfermedad celíaca, enfermedad de Graves, síndrome de Guillain-Barré, enfermedad de Hashimoto, anemia hemolítica, enfermedad inflamatoria intestinal, miopatías inflamatorias, esclerosis múltiple, cirrosis biliar primaria, psoriasis, artritis reumatoide, esclerodermia, síndrome de Sjogren, lupus eritematoso sistémico, vitíligo, pancreatitis autoinmunitaria, urticaria autoinmunitaria, púrpura trombocitopénica autoinmunitaria, enfermedad de Crohn, diabetes tipo I, fascitis eosinofílica, enterogastritis eosinofílica, síndrome de Goodpasture, miastenia grave, artritis psoriásica, fiebre reumática, colitis ulcerosa, vasculitis y granulomatosis de Wegener.
El agente o agentes/componente o componentes terapéuticamente activos adicionales pueden administrarse antes de, simultáneamente con o después de la administración del anticuerpo anti-PD-1 de la presente invención. Para los
fines de la presente divulgación, dichos regímenes de administración se consideran la administración de un anticuerpo anti-PD-1 "en combinación con" un segundo componente terapéuticamente activo.
El componente o componentes terapéuticamente activos adicionales se pueden administrar a un sujeto antes de la administración de un anticuerpo anti-PD-1 de la presente invención. Por ejemplo, se puede considerar que un primer componente se administra "antes" de un segundo componente si el primer componente se administra 1 semana antes,
72 horas antes, 60 horas antes, 48 horas antes, 36 horas antes, 24 horas antes, 12 horas antes, 6 horas antes, 5 horas antes, 4 horas antes, 3 horas antes, 2 horas antes, 1 hora antes, 30 minutos antes, 15 minutos antes, 10 minutos antes, 5 minutos antes, o menos de 1 minuto antes de la administración del segundo componente. En otros aspectos, el componente o componentes terapéuticamente activos adicionales se pueden administrar a un sujeto después de la administración de un anticuerpo anti-PD-1 de la presente invención. Por ejemplo, se puede considerar que un primer componente se administra "después" de un segundo componente si el primer componente se administra 1 minuto después, 5 minutos después, 10 minutos después, 15 minutos después, 30 minutos después, 1 hora después, 2 horas después, 3 horas después, 4 horas después, 5 horas después, 6 horas después, 12 horas después, 24 horas después,
36 horas después, 48 horas después, 60 horas después, 72 horas después de la administración del segundo componente. En otros aspectos más, el componente o componentes terapéuticamente activos adicionales se pueden administrar a un sujeto concurrente con la administración de un anticuerpo anti-PD-1 de la presente invención. La administración "concurrente", para los fines de la presente invención, incluye, por ejemplo, la administración de un anticuerpo anti-PD-1 y un componente terapéuticamente activo adicional a un sujeto en una forma de dosificación única (por ejemplo, coformulado), o en formas de dosificación separadas administradas al sujeto en aproximadamente
30 minutos o menos entre sí. Si se administran en formas de dosificación separadas, cada forma de dosificación puede administrarse por la misma vía (por ejemplo, tanto el anticuerpo anti- PD-1 como el componente terapéuticamente activo adicional se pueden administrar por vía intravenosa, subcutánea, etc.); como alternativa, cada forma de dosificación puede administrarse por una vía diferente (por ejemplo, el anticuerpo anti-PD-1 se puede administrar por vía intravenosa, y el componente terapéuticamente activo se puede administrar por vía subcutánea). En cualquier caso, la administración de los componentes en una única forma de dosificación, en formas de dosificación separadas por la misma vía, o en formas de dosificación separadas por vías diferentes, se consideran "administración concurrente", para los fines de la presente divulgación. Para los fines de la presente divulgación, la administración de un anticuerpo anti-PD-1 "antes de", "concurrente con", o "después" (como se definen esos términos en el presente documento anteriormente) de la administración de un componente terapéuticamente activo adicional se considera la administración de un anticuerpo anti-PD-1 "en combinación con" un componente terapéuticamente activo adicional).
La presente invención incluye composiciones farmacéuticas en las que un anticuerpo anti-PD-1 de la presente invención se formula conjuntamente con uno o más del componente o los componentes terapéuticamente activos adicionales como se describe en otra parte del presente documento utilizando una variedad de combinaciones de dosificación.
En aspectos ilustrativos en donde se administra un anticuerpo anti-PD-1 de la invención en combinación con un antagonista de VEGF (por ejemplo, una trampa de VEGF tal como aflibercept), incluida la administración de coformulaciones que comprenden un anticuerpo anti-PD-1 y un antagonista de VEGF, los componentes individuales pueden administrarse a un sujeto y/o coformularse usando una variedad de combinaciones de dosificación. Por ejemplo, el anticuerpo anti-PD-1 puede administrarse a un sujeto y/o estar contenido en una coformulación en una cantidad seleccionada del grupo que consiste en 0,01 mg, 0,02 mg, 0,03 mg, 0,04 mg, 0,05 mg, 0,1 mg, 0,2 mg, 0,3 mg, 0,4 mg, 0,5 mg, 0,6 mg, 0,7 mg, 0,8 mg, 0,9 mg, 1,0 mg, 1,5 mg, 2,0 mg, 2,5 mg, 3,0 mg, 3,5 mg, 4,0 mg, 4,5 5,0 mg, 6,0 mg, 7,0 mg, 8,0 mg, 9,0 mg, y 10,0 mg; y el antagonista de VEGF (por ejemplo, una trampa de VEGF tal como aflibercept) puede administrarse al sujeto y/o estar contenida en una coformulación en una cantidad seleccionada del grupo que consiste en 0,1 mg, 0,2 mg, 0,3 mg, 0,4 mg, 0,5 mg, 0,6 mg, 0,7 mg, 0,8 mg, 0,9 mg, 1,0 mg, 1,1 mg, 1,2 mg, 1,3 mg, 1,4 mg, 1,5 mg, 1,6 mg, 1,7 mg, 1,8 mg, 1,9 mg, 2,0 mg, 2,1 mg, 2,2 mg, 2,3 mg, 2,4 2,5 mg, 2,6 mg, 2,7 mg, 2,8 mg, 2,9 mg y 3,0 mg. Las combinaciones/coformulaciones se pueden administrar a un sujeto de acuerdo con cualquiera de los regímenes de administración divulgados en otra parte del presente documento, incluyendo, por ejemplo, dos veces a la semana, una vez cada semana, una vez cada 2 semanas, una vez cada 3 semanas, una vez al mes, una vez cada 2 meses, una vez cada 3 meses, una vez cada 4 meses, una vez cada 5 meses, una vez cada 6 meses, etc.
Regímenes de administración
De acuerdo con ciertos aspectos de la presente divulgación, se pueden administrar múltiples dosis de un anticuerpo anti-PD-1 (o una composición farmacéutica que comprende una combinación de un anticuerpo anti-PD-1 y cualquiera de los agentes terapéuticamente activos adicionales mencionados en el presente documento) a un sujeto durante un transcurso de tiempo definido. Los métodos de acuerdo con este aspecto de la divulgación comprenden administrar secuencialmente a un sujeto múltiples dosis de un anticuerpo anti-PD-1 de la invención. Como se utiliza en el presente documento, "administrar secuencialmente" significa que cada dosis de anticuerpo anti-PD-1 se administra al sujeto en un diferente punto temporal, por ejemplo, en días diferentes separados por un intervalo predeterminado (por ejemplo, horas, días, semanas o meses). La presente divulgación incluye métodos que comprenden administrar secuencialmente al paciente una dosis inicial única de un anticuerpo anti-PD-1, seguida por una o más dosis secundarias del anticuerpo anti-PD-1, y opcionalmente seguidas por una o más dosis terciarias del anticuerpo anti
PD-1. El anticuerpo anti-PD-1 se puede administrar a una dosis entre 0,1 mg/kg y 100 mg/kg.
Las expresiones "dosis inicial", "dosis secundarias", y "dosis terciarias", se refieren a la secuencia temporal de administración del anticuerpo anti-PD-1 de la invención. Por tanto, la "dosis inicial" es la dosis que se administra al inicio del régimen de tratamiento (también mencionada como la "dosis basal"); las "dosis secundarias" son las dosis que se administran después de la dosis inicial; y las "dosis terciarias" son las dosis que se administran después de las dosis secundarias. Las dosis inicial, secundarias y terciarias pueden contener todas la misma cantidad de anticuerpo anti-PD-1, pero generalmente pueden diferir entre sí en términos de frecuencia de administración. En determinadas realizaciones, sin embargo, la cantidad de anticuerpo anti-PD-1 contenida en las dosis inicial, secundarias y/o terciarias varían entre sí (p.ej., ajustadas hacia arriba o hacia abajo según corresponda) durante el transcurso del tratamiento. En determinados aspectos, dos o más (por ejemplo, 2, 3, 4 o 5) dosis se administran al comienzo del régimen de tratamiento como "dosis de carga" seguidas de dosis posteriores que se administran con menos frecuencia (por ejemplo, "dosis de mantenimiento").
En ciertos aspectos ilustrativos de la presente divulgación, cada dosis secundaria y/o terciaria se administra de 1 a 26 (por ejemplo, 1, 1 / , 2, 2 / , 3, 3 / , 4, 41/ 2, 5, 51/ 2, 6, 61/ 2, 7, 71/ 2, 8, 81/ 2, 9, 91/ 2, 10, 101/ 2, 11, 11 /, 12, 121/ 2, 13, 131/ 2, 14, 141/ 2, 15, 15 /, 16, 16 /, 17, 17 /, 18, 18 /, 19, 19 /, 20, 20 /, 21, 21 / , 22, 22 /, 23, 23 /, 24, 24 /, 25, 25 /, 26, 26 / o más) semanas después de la dosis inmediatamente anterior. La expresión "la dosis inmediatamente anterior", como se utiliza en el presente documento, significa, en una secuencia de administraciones múltiples, la dosis de anticuerpo anti-PD-1 que se administra a un paciente antes de la administración de la siguiente dosis en la secuencia sin dosis intermedias.
Los métodos de acuerdo con este aspecto de la divulgación pueden comprender administrar a un paciente cualquier cantidad de dosis secundarias y/o terciarias de un anticuerpo anti-PD-1. Por ejemplo, en determinados aspectos, solo se administra una dosis secundaria única al paciente. En otros aspectos, dos o más (por ejemplo, 2, 3, 4, 5, 6, 7, 8 o más) dosis secundarias se administran al paciente. De forma análoga, en determinados aspectos, solo se administra una dosis terciaria única al paciente. En otros aspectos, dos o más (por ejemplo, 2, 3, 4, 5, 6, 7, 8 o más) dosis terciarias se administran al paciente.
En aspectos que implican múltiples dosis secundarias, cada dosis secundaria puede administrarse a la misma frecuencia que las otras dosis secundarias. Por ejemplo, cada dosis secundaria puede administrarse al paciente de 1 a 2 semanas o de 1 a 2 meses después de la dosis inmediatamente anterior. De forma similar, en aspectos que implican múltiples dosis terciarias, cada dosis terciaria puede administrarse a la misma frecuencia que las otras dosis terciarias. Por ejemplo, cada dosis terciaria puede administrarse al paciente de 2 a 12 semanas después de la dosis inmediatamente anterior. En determinados aspectos de la divulgación, la frecuencia con la que se administran las dosis secundarias y/o terciarias a un paciente puede variar en el transcurso del régimen de tratamiento. La frecuencia de la administración también puede ajustarla un médico durante el transcurso del tratamiento, dependiendo de las necesidades del paciente individual después de la exploración clínica.
La presente divulgación incluye la administración de regímenes en los que se administran de 2 a 6 dosis de carga a un paciente en una primera frecuencia (por ejemplo, una vez a la semana, una vez cada dos semanas, una vez cada tres semanas, una vez al mes, una vez cada dos meses, etc.), seguido de la administración de dos o más dosis de mantenimiento al paciente de forma menos frecuente. Por ejemplo, de acuerdo con este aspecto de la divulgación, si las dosis de carga se administran con una frecuencia de, por ejemplo, una vez al mes (por ejemplo, dos, tres, cuatro o más dosis de carga administradas una vez al mes), entonces las dosis de mantenimiento pueden administrarse al paciente una vez cada cinco semanas, una vez cada seis semanas, una vez cada siete semanas, una vez cada ocho semanas, una vez cada diez semanas, una vez cada doce semanas, etc.).
Usos de diagnóstico de los anticuerpos
Los anticuerpos anti-PD-1 de la presente invención pueden usarse para detectar y/o medir PD-1 en una muestra, por ejemplo, con fines de diagnóstico. Algunos aspectos contemplan el uso de uno o más anticuerpos de la presente divulgación en ensayos para detectar una enfermedad o trastorno, tal como cáncer, enfermedad autoinmunitaria o infección vírica crónica. Los ensayos de diagnóstico ilustrativos para PD-1 pueden comprender, por ejemplo, poner en contacto una muestra, obtenida de un paciente, con un anticuerpo anti-PD-1 de la invención, en donde el anticuerpo anti-PD-1 está marcado con un marcador detectable o molécula indicadora o se usa como un ligando de captura para aislar selectivamente PD-1 de las muestras del paciente. Como alternativa, puede usarse un anticuerpo anti-PD-1 no marcado en aplicaciones de diagnóstico en combinación con un anticuerpo secundario que está marcado de forma detectable por sí mismo. El marcador detectable o molécula indicadora puede ser un radioisótopo, tal como 3H, 14C, 32P, 35S o 125I; una fracción fluorescente o quimioluminiscente tal como isotiocianato de fluoresceína o rodamina; o una enzima tal como una fosfatasa alcalina, p-galactosidasa, peroxidasa de rábano picante o luciferasa. Los ensayos ilustrativos específicos que pueden usarse para detectar o medir PD-1 en una muestra incluyen el ensayo inmunoabsorbente ligado a enzimas (ELISA), radioinmunoensayo (RIA) y clasificación de células activadas por fluorescencia (FACS).
Las m ue s tra s que pueden usa rse en e n sa yo s de d ia g n ó s tico de PD-1 de a cu e rd o con la p re sen te d ivu lg a c ió n inc luyen
cualquier muestra de tejido o fluido obtenible de un paciente, que contiene cantidades detectables de proteína PD-1, o fragmentos de la misma, en condiciones normales o patológicas. Generalmente, se medirán los niveles de PD-1 en una muestra particular obtenida de un paciente sano (por ejemplo, un paciente no afectado por cáncer o por una enfermedad autoinmunitaria) para establecer un nivel basal, o estándar, nivel de PD-1. Este nivel basal de PD-1 a continuación puede compararse con los niveles de PD-1 medidos en muestras obtenidas de individuos de los que se sospecha que tienen una afección relacionada con el cáncer, o síntomas asociados con dicha afección.
Los anticuerpos específicos de PD-1 pueden no contener marcadores o fracciones adicionales, o pueden contener un marcador o fracción N-terminal o C-terminal. En una realización, el marcador o fracción es biotina. En un ensayo de unión, la ubicación de un marcador (si lo hay) puede determinar la orientación del péptido con respecto a la superficie sobre la que se une el péptido. Por ejemplo, si una superficie está recubierta con avidina, un péptido que contiene una biotina N-terminal se orientará de manera que la porción C-terminal del péptido estará distal a la superficie.
Los aspectos de la divulgación se refieren al uso de los anticuerpos divulgados como marcadores para predecir el pronóstico de cáncer o un trastorno autoinmunitario en pacientes. Los anticuerpos de la presente invención pueden usarse en ensayos de diagnóstico para evaluar el pronóstico de la enfermedad de cáncer en un paciente y predecir la supervivencia.
Ejemplos
Los siguientes ejemplos se presentan para proporcionar a los expertos en la materia una divulgación y descripción completa de cómo preparar y usar los métodos y las composiciones de la invención, y no se pretende que limiten el alcance de lo que los inventores consideran su invención. Se han realizado esfuerzos para garantizar la exactitud con respecto a los números utilizados (por ejemplo, cantidades, temperatura, etc.) pero deben tenerse en cuenta algunos errores y desviaciones experimentales. A menos que se indique lo contrario, las partes son partes en peso, el peso molecular es peso molecular promedio, la temperatura está en grados centígrados, la temperatura ambiente es de aproximadamente 25 °C y la presión es o está cerca de la atmosférica.
Ejemplo 1: Generación de anticuerpos humanos contra PD-1
Se generaron anticuerpos humanos contra PD-1 usando un fragmento de PD-1 que varía entre aproximadamente los aminoácidos 25-170 del n.° de registro GenBank NP_005009.2 (SEQ ID NO: 327) con un cambio de C93S. El inmunógeno se administró directamente, con un adyuvante para estimular la respuesta inmunitaria, a un ratón VELOCIMMUNE® que comprende ADN que codifica las regiones variables de cadena pesada y cadena ligera kappa de inmunoglobulina humana. La respuesta inmunitaria de anticuerpos se controló mediante un inmunoensayo específico de PD-1. Cuando se consiguió una respuesta inmunitaria deseada, se recogieron los esplenocitos y se fusionaron con células de mieloma de ratón para conservar su viabilidad y formar estirpes celulares de hibridoma. Las líneas celulares de hibridoma se cribaron y seleccionaron para identificar líneas celulares que producían anticuerpos específicos de PD-1. Utilizando esta técnica y el inmunógeno descrito anteriormente, se obtuvieron varios anticuerpos quiméricos anti-PD-1 (es decir, anticuerpos que poseen dominios variables humanos y dominios constantes de ratón); los anticuerpos de ejemplo generados de esta manera se designaron como H1M7789N, H1M7799N, H1M7800N, H2M7780N, H2M7788N, H2M7790N, H2M7791N, H2M7794N, H2M7795N, H2M7796N y H2M7798N.
También se aislaron anticuerpos anti-PD-1 directamente de linfocitos B positivos a antígeno sin fusión a células de mieloma, tal como se describe en el documento U.S. 2007/0280945A1, incorporado específicamente en el presente documento por referencia en su totalidad. Usando este método, se obtuvieron varios anticuerpos anti-PD-1 completamente humanos (es decir, anticuerpos que poseen dominios variables humanos y dominios constantes humanos); los anticuerpos de ejemplo generados de esta manera se denominaron del siguiente modo: H4H9019P, H4xH9034P2, H4xH9035P2, H4xH9037P2, H4xH9045P2, H4xH9048P2, H4H9057P2, H4H9068P2, H4xH9119P2, H4xH9120P2, H4xH9128P2, H4xH9135P2, H4xH9145P2, H4xH8992P, H4xH8999P y H4xH9008P.
Determinadas propiedades biológicas de los anticuerpos de ejemplo generados de acuerdo con los métodos de este Ejemplo se describen en detalle en los Ejemplos que se exponen a continuación.
Ejemplo 2: Secuencias de aminoácidos y nucleótidos de la región variable de la cadena pesada y ligera
La Tabla 1 muestra los identificadores de la secuencia de aminoácidos de las regiones variables de la cadena pesada y ligera y las CDR de los anticuerpos anti-PD-1 seleccionados de la invención. Los identificadores de secuencia de ácido nucleico correspondientes se exponen en la Tabla 2.
Tabla 1: Identificadores de secuencia de aminoácidos
Tabla 2: Identificadores de secuencia de ácido nucleico
continuación
Los anticuerpos se denominan normalmente en el presente documento de acuerdo con la siguiente nomenclatura: prefijo Fc (por ejemplo, "H4xH", "H1M", "H2M", etc.), seguido de un identificador numérico (por ejemplo, "7789", "7799", etc., como se muestra en la Tabla 1), seguido de un sufijo "P", "P2", "N", o "B". Por tanto, de acuerdo con esta nomenclatura, un anticuerpo puede denominarse en el presente documento, por ejemplo, "H1H7789N", "H1M7799N", "H2M7780N", etc. Los prefijos H4xH, H1M, H2M y H2aM en las denominaciones de anticuerpo usadas en el presente documento indican el isotipo de la región Fc particular del anticuerpo. Por ejemplo, un anticuerpo "H4xH" tiene un Fc de IgG4 humana con 2 o más cambios de aminoácidos como se divulga en el documento US20100331527, un anticuerpo "H1M" tiene un Fc de IgG1 de ratón, y un anticuerpo "H2M" tiene un Fc de IgG2 de ratón (isotipo a o b) (todas las regiones variables son completamente humanas como se indica por la primera "H" en la designación del anticuerpo). Como apreciará un experto en la materia, un anticuerpo que tiene un isotipo de Fc particular se puede convertir en un anticuerpo con un isotipo de Fc diferente (por ejemplo, un anticuerpo con una Fc de IgG1 de ratón se puede convertir en un anticuerpo con una IgG4 humana, etc.), pero en cualquier caso, los dominios variables (incluyendo las CDR), que se indican mediante los identificadores numéricos que se muestran en la Tabla 1, seguirán siendo los mismos, y se espera que las propiedades de unión sean idénticas o sustancialmente similares independientemente de la naturaleza del dominio Fc.
En determinadas realizaciones, los anticuerpos seleccionados con un Fc de IgG1 de ratón se convirtieron en anticuerpos con Fc de IgG4 humana. En una realización, el dominio Fc de IgG4 comprende una mutación de serina a prolina en la región bisagra (S108P) para promover la estabilización del dímero. La Tabla 3 muestra los identificadores de secuencia de aminoácidos de las secuencias de la cadena pesada y la cadena ligera de anticuerpos anti-PD-1 con Fc de IgG4 seleccionados.
Tabla 3
Cada secuencia de cadena pesada en la Tabla 3 comprendía una región variable (Vh o HCVR; que comprende HCDR1, HCDR2 y HCDR3) y una región constante (que comprende los dominios Ch 1, Ch2 y Ch3). Cada secuencia de cadena ligera en la Tabla 3 comprendía una región variable (Vl o LCVR; que comprende LCDR1, LCDR2 y LCDR3) y una región constante (Cl). SEQ ID NO: 330 que comprendía una HCVR que comprende los aminoácidos 1 - 117 y una región constante que comprende los aminoácidos 118 - 444. SEQ ID NO: 331 que comprendía una LCVR que comprende los aminoácidos 1 - 107 y una región constante que comprende los aminoácidos 108 - 214. SEQ ID NO: 332 que comprendía una HCVR que comprende los aminoácidos 1 - 122 y una región constante que comprende los aminoácidos 123 - 449. SEQ ID NO: 333 que comprendía una LCVR que comprende los aminoácidos 1 - 107 y una región constante que comprende los aminoácidos 108 - 214. SEQ ID NO: 334 que comprendía una HCVR que comprende los aminoácidos 1 - 119 y una región constante que comprende los aminoácidos 120 - 446. SEQ ID NO: 335 que comprendía una LCVR que comprende los aminoácidos 1 - 108 y una región constante que comprende los aminoácidos 109 - 215. SEQ ID NO: 336 que comprendía una HCVR que comprende los aminoácidos 1 - 121 y una región constante que comprende los aminoácidos 122 - 448. SEQ ID NO: 337 que comprendía una LCVR que comprende los aminoácidos 1 - 108 y una región constante que comprende los aminoácidos 109 - 215.
Ejemplo 3: Unión de anticuerpos a PD-1 determinada mediante resonancia de plasmón superficial
Las constantes de velocidad de asociación de unión y tasa de disociación (ka y kd, respectivamente), las constantes de disociación y las semividas de disociación en el equilibrio (Kd y t1^ , respectivamente) para la unión del antígeno a anticuerpos anti-PD1 purificados se determinaron usando un ensayo de biosensor de resonancia de plasmón superficial en tiempo real en un instrumento Biacore 4000 o Biacore T200. La superficie del sensor Biacore se derivatizó con un anticuerpo policlonal anti-ratón de conejo (GE, N.° BR-1008-38) o con un anticuerpo monoclonal anti-Fc humano de ratón (GE, N.° BR-1008-39) para capturar aproximadamente 100-900 RU de anticuerpos monoclonales anti-PD-1, expresados con un Fc de ratón o un Fc humano, respectivamente. Los reactivos PD-1 ensayados para la unión a los anticuerpos anti-PD-1 incluyeron PD-1 humano recombinante expresado con una etiqueta myc-mychexahistidina C-terminal (hPD-1-MMH; SEQ ID NO: 321), PD-1 de mono cinomolgo recombinante expresado con una etiqueta myc-myc-hexahistidina C-terminal (MfPD-1-MMH; SEQ ID NO: 322), dímero de PD-1 humano recombinante expresado con una etiqueta Fc de IgG2a de ratón C-terminal (hPD-1-mFc; SEQ ID NO: 323) o con un Fc de IgG1 humana C-terminal (hPD1-hFc; SEQ ID NO: 324), y PD-1 de mono con mFc (SEQ ID n O: 329). Se inyectaron diferentes concentraciones de reactivos PD-1 que variaban de 200 nM a 3,7 nM sobre la superficie capturada del anticuerpo monoclonal anti-PD-1 a un caudal de 30 pl/min en Biacore 4000 o de 50 pl/min en Biacore T200. La unión de los reactivos PD-1 a los anticuerpos monoclonales capturados se controló durante 3 a 5 minutos mientras que su disociación de los anticuerpos se controló durante 7 a 10 minutos en tampón de análisis HBST (HEPES 0,01 M pH 7,4, NaCI 0,15 M, EDTA 3 mM, tensioactivo P20 al 0,05 % v/v. Los experimentos se realizaron a 25 °C y 37 °C. Las constantes de velocidad de asociación cinética (ka) y disociación cinética (kd) se determinaron procesando y ajustando los datos a un modelo de unión 1:1 usando el software de ajuste de curvas Scrubber 2.0c. Las constantes en equilibrio de disociación de unión (KD) y las semividas disociativas (ty2) se calcularon a continuación a partir de las constantes de velocidad cinética como: Kd (M) = kd / ka y t!^ (min) = [In2/(60*kd)]. Los parámetros de la cinética de unión para diferentes anticuerpos monoclonales anti-PD-1 que se unen a diferentes reactivos PD-1 a 25 °C y 37 °C se tabulan en las Tablas 4-11.
Tabla 4: Parámetros de la cinética de unión de anticuerpos monoclonales anti-PD-1 que se unen a PD-1-MMH humano a 25 °C.
continuación
Tabla 5: Parámetros de la cinética de unión de anticuerpos monoclonales anti-PD-1 que se unen a PD-1-MMH humano a 37 °C.
continuación
Tabla 6: Parámetros de la cinética de unión de anticuerpos monoclonales anti-PD-1 que se unen al dímero de PD-1
- - - - °
continuación
Tabla 7: Parámetros de la cinética de unión de anticuerpos monoclonales anti-PD-1 que se unen al dímero de PD-1 h m n PD-1-mF h m n PD-1-hF h m n 7 ° .
continuación
Tabla 8: Parámetros de la cinética de unión de anticuerpos monoclonales anti-PD-1 que se unen a MfPD-1-MMH a
25 °C.
continuación
Tabla 9: Parámetros de la cinética de unión de anticuerpos monoclonales anti-PD-1 que se unen a MfPD-1-MMH a
37 °C
continuación
Tabla 10: Parámetros de la cinética de unión de anticuerpos monoclonales anti-PD-1 que se unen al dímero de PD-1 de mono PD-1-mFc de mono a 25 °C
Tabla 11: Parámetros de la cinética de unión de anticuerpos monoclonales anti-PD-1 que se unen al dímero de PD-1
- - °
Como se muestra en la Tabla 4, a 25 °C, 28 de los 29 anticuerpos anti-PD-1 de la invención se unieron a hPD-1-MMH con valores KD que van de 2,1 nM a 291 nM. Un anticuerpo, H4H9068P2, no demostró ninguna unión medible a hPD-1-MMH a 25 °C. Como se muestra en la Tabla 5, a 37 °C, 26 de los 29 anticuerpos anti-PD-1 de la invención se unieron a hPD-1-MMH con valores KD que van de 3,79 nM a 1,51 pM. Tres anticuerpos de la invención no demostraron ninguna unión concluyente a hPD-1-MMH a 37 °C. Como se muestra en la Tabla 6, a 25 °C, los 29 anticuerpos anti-PD-1 de la invención se unieron a las proteínas del dímero de hPD-1 con valores KD que van de 65,5 pM a 59,4 nM. Como se muestra en la Tabla 7, a 37 °C, los 27 anticuerpos anti-PD-1 de la invención se unieron a las proteínas del dímero de hPD-1 con valores KD que van de 3,09 pM a 551 nM. Como se muestra en la Tabla 8, a 25 °C, 27 de los 29 anticuerpos anti-PD-1 de la invención se unieron a MfPD-1-MMH con valores KD que van de 3,09 nM a 551 nM. Dos anticuerpos de la invención no demostraron ninguna unión concluyente a MfPD-1-MMH a 25 °C. Como se muestra en la Tabla 9, a 37 °C, 25 de los 29 anticuerpos anti-PD-1 de la invención se unieron a MfPD-1-MMH con valores KDque van de 7,00 nM a 7,54 pM. Cuatro anticuerpos de la invención no demostraron ninguna unión concluyente a MfPD-1-MMH a 37 °C. Como se muestra en la Tabla 10, a 25 °C, 18 de los anticuerpos anti-PD-1 ensayados de la invención se unieron al dímero de MfPD-1 con valores KDque van de 137 pM a 54,2 nM. Como se muestra en la Tabla 11, a 37 °C, 18 de los anticuerpos anti-PD-1 ensayados de la invención se unieron al dímero de MfPD-1 con valores KDque van de 49 pM a 86,3 nM.
Ejemplo 4: Bloqueo de la unión de PD-1 a PD-L1 determinado por ELISA
La capacidad de los anticuerpos anti-PD-1 para bloquear la unión de PD-1 humano a su ligando, el receptor de PD-L1, se midió utilizando tres formatos de ELISA sándwich de competición. Las proteínas PD-L1 humanas dimérica, compuestas por una porción del dominio extracelular de PD-L1 humano expresado con una etiqueta Fc humana C-terminal (hPD-L1-hFc; SEQ ID: 325) o una etiqueta Fc de ratón C-terminal (hPD-L1-mFc; Se Q ID: 326), o PD-L2 humana dimérica, compuesta por una región extracelular de PD-L2 humana producida con una etiqueta Fc humano C-terminal (hPD-L2-hFc; R&D Systems, N.° 1224- PL) se recubrieron por separado a una concentración de 2 pg/ml en PBS en una placa de microtitulación de 96 pocillos durante la noche a 4 °C. Los sitios de unión inespecíficos se
bloquearon posteriormente usando una solución al 0,5 % (p/v) de BSA en PBS. En un primer formato de competición, se añadió una concentración constante de 1,5 nM de una proteína PD-1 humana dimérica, compuesta por un dominio extracelular de PD-1 humana expresada con una etiqueta Fc humano C-terminal (hPD-1-mFc; s Eq ID: 323) a diluciones seriadas de anticuerpos anti-PD-1 o anticuerpos de control de isotipo de modo que las concentraciones finales de anticuerpos variaran de 0 a 200 nM. En un segundo formato de competición, se añadió una concentración constante de 200 pM de proteína PD-1 humana biotinilada dimérica, compuesta por el dominio extracelular de PD-1 humana que se expresó con una etiqueta Fc humana C-terminal (biot-hPD-1-hFc; SEQ ID: 323) se añadió de forma similar a diluciones en serie de anticuerpos anti-PD-1 o un control de isotipo a concentraciones finales de anticuerpo que van de 0 a 50 nM. En un tercer formato de competición, se añadió de forma similar una concentración constante de 100 pM de proteína dimérica hPD-1-mFc a diluciones en serie de anticuerpos anti-PD-1 o un control de isotipo a concentraciones finales de anticuerpo que varían de 0 a 100 nM. Estos complejos anticuerpo-proteína se incubaron a continuación durante 1 hora a temperatura ambiente (TA). Los complejos anticuerpo-proteína con 1,5 nM constante de hPD-1-mFc se transfirieron a placas de microvaloración recubiertas con hPD-L1-hFc, los complejos anticuerpoproteína con 200 pM constante de biot-hPD-1-hFc se transfirieron a placas recubiertas con hPD-L1-mFc, y los complejos anticuerpo-proteína con 100 pM constante de hPD-1-mFc se transfirieron a placas de microvaloración recubiertas con hPD-L2-hFc. Después de incubar durante 1 hora a TA, se lavaron los pocillos y se detectó el hPD-1-mFc unido a la placa con un anticuerpo policlonal anti-mFc conjugado con peroxidasa de rábano picante (HRP) (Jackson ImmunoResearch Inc., N.° 115-035-164 ), y se detectó biot-hPD-1-hFc unido a la placa con estreptavidina conjugada con HRP (Thermo Scientific, N.° N200). Las muestras se desarrollaron con una solución TMB (BD Biosciences, N.° 51-2606KC y N.° 51-2607KC) para producir una reacción colorimétrica y a continuación el desarrollo del color se estabilizó mediante la adición de ácido sulfúrico 1 M antes de medir la absorbancia a 450 nm en un lector de placas Victor X5. El análisis de los datos se realizó usando un modelo de respuesta a dosis sigmoideo dentro del programa informático Prism™ (GraphPad). El valor de CI50 calculado, definido como la concentración de anticuerpo requerida para reducir el 50 % de la unión de PD-1 humano a PD-L1 o PD-L2 humano, se usó como indicador de la potencia de bloqueo. El porcentaje de bloqueo máximo se calculó como una medida de la capacidad de los anticuerpos para bloquear completamente la unión de PD-1 humano a PD-L1 o PD-L2 humano en la placa determinado a partir de la curva de dosis. Este porcentaje de bloqueo máximo se calculó restando del 100 % la proporción de la reducción en la señal observada en presencia de la concentración más alta probada para cada anticuerpo respecto a la diferencia entre la señal observada para una muestra de PD-1 humano que no contiene anticuerpo anti-PD-1 (0 % de bloqueo) y la señal de fondo del anticuerpo secundario conjugado con HRP solo (100 % de bloqueo).
El porcentaje máximo de bloqueo y los valores de CI50 calculados para los anticuerpos que bloquean más del 35 % de la señal de unión a hPD-1 se muestran en las Tablas 12 - 14. Los anticuerpos que mostraron una disminución en la señal de unión de hPD-1 del 35 % o menos se definieron como no bloqueantes. Los anticuerpos que mostraron un aumento del 35 % o más en la señal de unión de PD-1 humano se caracterizaron como no bloqueantes/potenciadores. La base teórica del ensayo, definida como la concentración mínima de anticuerpo teóricamente necesaria para ocupar el 50 % de los sitios de unión de PD-1 humano en el ensayo, es 0,75 nM para el formato que usa 1,5 nM constante de hPD-1-mFc, 100 pM para el formato que usa 200 pM constante de biot-hPD-1-hFc y 50 pM para el formato que usa 100 pM constante de hPD-1-mFc, lo que indica que los valores de CI50 inferiores calculados no representan unión al sitio de unión al anticuerpo. Por esta razón, los anticuerpos con valores de CI50 calculados de menos de 0,75 nM en el ensayo con hPD-1-mFc constante y recubrimiento de hPD-L1, de menos de 100 pM en el ensayo con biot-hPD-1-hFc constante y recubrimiento de hPD-L1, y menos de 50 pM en el ensayo con hPD-1-mFc constantes y recubrimiento de hPD-L2 se indican en las Tablas 12 - 14 como <7,5E-10M, <1,0E-10M y <5,0E-11M, respectivamente.
Tabla 12: ELISA de blo ueo de la unión de PD-1 humano a PD-L1 humano mediante anticuer os anti-PD-1
continuación
Tabla 13: ELISA de bloqueo de la unión de PD-1 humano biotinilado a PD-L1 humano mediante anticuerpos anti-
PD-1
continuación
Tabla 14: ELISA de blo ueo de la unión de PD-1 humano a PD-L2 humano mediante anticuer os anti-PD-1
o m o se ind ica en la T a b la 12, en el p rim e r fo rm a to de ensayo , 23 de los 27 a n ticu e rp o s an ti-P D -1 b loq uea ron 1,5 nM
de hPD-1-mFc de la unión a hPD-L1-hFc con valores de CI50 que van de 190 pM a 3,3 nM con el porcentaje de bloqueo máximo que va del 67 % al 100 %. Un anticuerpo, H2aM7796N, se identificó como no bloqueante. Se identificaron tres anticuerpos anti-PD-1 (H4H9068P2, H1M7789N y H2aM7791N) como no bloqueantes/potenciadores.
Como se muestra en la Tabla 13, en el segundo formato de ensayo, 23 de los 27 anticuerpos anti-PD-1 bloquearon 200 pM de biothPD-1-hFc de la unión a hPD-L1-mFc con valores de CI50 que van de 59 pM a 1,3 nM con el porcentaje de bloqueo máximo que va del 60 % al 101 %. Un anticuerpo, H1M7789N, se identificó como no bloqueante. Se identificaron tres anticuerpos anti-PD-1 (H4H9057P2, H4H9068P2 y H2aM7791N) como no bloqueantes/potenciadores.
En el tercer formato de ensayo como se muestra en la Tabla 14, se probaron cuatro anticuerpos anti-PD-1 de la invención y un control de isotipo. Los 4 anticuerpos anti-PD-1 de la invención bloquearon 100 pM (concentración fija) de hPD-1-mFc de la unión a hPD-L2-hFc que recubre la placa con valores de CI50 que varían de 0,13 nM a 1,3 nM y con un porcentaje de bloqueo máximo que varía del 94 % al 100 %.
Ejemplo 5: Bloqueo de la unión de PD-1 a PD-L1 determinado mediante un ensayo de biosensor y resonancia de plasmón superficial
Se estudió la inhibición de la unión de PD-1 humano a PD-L1 humano por diferentes anticuerpos monoclonales anti-PD-1, ya sea utilizando un ensayo de interferometría de biocapa en tiempo real en un instrumento biosensor Octet Red96 (Fortebio Inc.) o utilizando un ensayo de biosensor de resonancia de plasmón superificial en tiempo real en un instrumento Biacore 3000.
Se realizaron estudios de inhibición de anticuerpos monoclonales anti-PD-1 expresados con un Fc de ratón en un instrumento Octet Red 96. En primer lugar, se incubaron 100 nM de un PD-1 humano recombinante expresado con una etiqueta Fc de IgG2a de ratón C-terminal (hPD-1-mFc; SEQ ID NO: 323) con 500 nM de cada anticuerpo monoclonal anti-PD-1 durante al menos 1 hora antes de realizar el ensayo de inhibición. Se capturaron alrededor de 0,8 nm a 1,2 nm de PD-L1 humano recombinante expresado con una etiqueta Fc de IgG1 humana C-terminal (hPD-L1-hFc; SEQ ID NO: 325) usando un biosensor Octet de captura de Fc de anti-IgG humana. Los biosensores Octet recubiertos con hPD-L1-hFc se sumergieron a continuación en pocillos que contenían la mezcla de hPD-1-mFc y diferentes anticuerpos monoclonales anti-PD-1. Todo el experimento se realizó a 25 °C en tampón Octet HBST (0,01 M HEPES pH 7,4, NaCl 0,15 M, EDTA 3 mM, Tensioactivo P20 al 0,05 % v/v, 0,1 mg/ml de BSA) con la placa en agitación a una velocidad de 1000 rpm. Los biosensores se lavaron en tampón Octet HBST entre cada etapa del experimento. Las respuestas de unión en tiempo real se controlaron durante todo el transcurso del experimento y se registró la respuesta de unión al final de cada etapa. La unión de hPD-1-mFc al hPD-L1-hFc capturado se comparó en presencia y ausencia de diferentes anticuerpos monoclonales anti-PD-1 y se usó para determinar el comportamiento de bloqueo de los anticuerpos ensayados como se muestra en la Tabla 15.
Tabla 15: Inhibición de la unión de PD-L1 humano a PD-1 mediante anticuerpos monoclonales anti-PD-1 ex resados con Fc de ratón medido en un instrumento Octet Red 96
C o m o se m ue s tra en la T a b la 15, 9 de los 11 a n ticu e rp o s an ti-P D -1 e n sa ya d o s en el in s tru m e n to O c te t Red 96
demostraron un fuerte bloqueo de hPD-1-mFc de la unión a hPD-L1-hFc desde el 86 % hasta el bloqueo completo de la unión. Un anticuerpo anti-PD-1 (H1M7789N) ensayado mostró un bloqueo más débil de la unión de hPD-1-mFc a hPD-L1-hFc con un bloqueo del 29 %. Un anticuerpo (H2aM7791N) ensayado demostró la capacidad de mejorar la unión de hPD-1-mFc a hPD-L1-hFc.
A continuación, se realizaron estudios de inhibición de anticuerpos monoclonales anti-PD-1 expresados con un Fc humano en un instrumento Biacore 3000. En primer lugar, se incubaron 100 nM de un PD-1 humano recombinante expresado con una etiqueta Fc de IgG1 humana C-terminal (hPD-1-hFc; SEQ ID: 324) con 500 nM de cada anticuerpo monoclonal anti-PD-1 durante al menos 2 horas antes de realizar el ensayo de inhibición. En primer lugar, se derivatizó una superficie de sensor CM5 Biacore con anticuerpo policlonal anti-ratón de conejo (Catálogo GE N.° BR-1008-38) usando química estándar EDC-NHS. Se capturaron alrededor de 730 RU de PD-L1 humano recombinante expresado con una etiqueta Fc de IgG2a de ratón C-terminal (hPD-L1.mFc; SEQ ID: 326) seguido a continuación de la inyección de 100 nM de hPD-1.hFc en presencia y ausencia de diferentes anticuerpos monoclonales anti-PD-1 a un caudal de 25 pl/min durante 3 minutos. Todo el experimento se realizó a 25 °C en tampón de análisis que comprendía HEPES 0,01 M pH 7,4, NaCl 0,15 M, EDTA 3 mM, tensioactivo Tween-20 al 0,05 % v/v (tampón de análisis HBS-ET). Las respuestas de unión en tiempo real se controlaron durante todo el transcurso del experimento y se registró la respuesta de unión al final de cada etapa. La unión de hPD-1-hFc al hPD-L1-mFc capturado se comparó en presencia y ausencia de diferentes anticuerpos monoclonales anti-PD-1 y se usó para determinar el comportamiento de bloqueo de los anticuerpos ensayados como se muestra en la Tabla 16.
Tabla 16: Inhibición de la unión de PD-L1 humano a PD-1 mediante anticuerpos monoclonales anti-PD-1 ex resados con Fc humano medido en un instrumento Biacore 3000
Como se muestra en la Tabla 16, 18 de los 20 anticuerpos anti-PD-1 de la invención ensayados en el instrumento Biacore 3000 demostraron un fuerte bloqueo de la unión de hPD-1-hFc a hPD-L1-mFc con un bloqueo que varía de 96% a 100%. Un anticuerpo demostró la capacidad de mejorar la unión de hPD-1-hFc a hPD-L1-mFc. En este estudio,
uno de los anticuerpos ensayados de la invención (H4H9057P2) demostró una unión de fondo no específica a la superficie de captura del Fc anti-ratón.
Ejemplo 6: Competencia cruzada en Octet entre anticuerpos anti-PD-1
La competencia de unión entre anticuerpos monoclonales anti-PD-1 se determinó usando un ensayo de interferometría de biocapa sin marcaje en tiempo real en un biosensor Octet RED384 (Pall ForteBio Corp.). Todo el experimento se realizó a 25 °C en tampón HEPES 0,01 M pH 7,4, NaCl 0,15 M, EDTA 3 mM, Tensioactivo Tween-20 al 0,05 % v/v, 0,1 mg/ml de BSA (tampón Octet HBS-ET) con la placa en agitación a una velocidad de 1000 rpm. Para evaluar si 2 anticuerpos podían competir entre sí para unirse a sus respectivos epítopos en un PD-1 humano expresado de manera recombinante con una etiqueta myc-myc-hexahistidina C-terminal (hPD-1-MMH; SEQ ID: 321), se capturaron alrededor de 0,1 nM de hPD-1-MMH primero en puntas de biosensor Octet recubiertas con anticuerpo anti-Penta-His (Pall ForteBio Corp., N.° 18-5079) sumergiendo las puntas durante 5 minutos en pocillos que contenían una solución de 50 |jg/ml de hPD-1-MMH. Las puntas del biosensor capturadas con antígeno se saturaron a continuación con el primer anticuerpo monoclonal anti-PD-1 (posteriormente denominado mAb-1) sumergiéndolo en pocillos que contenían 50 jg/m l de solución de mAb-1 durante 5 minutos. Las puntas del biosensor se sumergieron a continuación en pocillos que contenían una solución de 50 jg/m l de un segundo anticuerpo monoclonal anti-PD-1 (posteriormente denominado mAb-2). Las puntas del biosensor se lavaron en tampón Octet HBS-ET entre cada etapa del experimento. La respuesta de unión en tiempo real se controló durante todo el transcurso del experimento y se registró la respuesta de unión al final de cada etapa. Se comparó la respuesta de la unión de mAb-2 a hPD-1-MMH formando un complejo previo con mAb-1 y se determinó el comportamiento competitivo/no competitivo de diferentes anticuerpos monoclonales anti-PD-1. Los resultados se resumen en la Tabla 17 (* No se enumeran los mAb2 de autocompetencia).
T l 17: m n i r z nr r n i r ni-PD-1 l i n
(continuación)
(continuación)
Se determinó una segunda competencia de unión entre un panel de anticuerpos monoclonales anti-PD-1 seleccionados usando un ensayo de interferometría de biocapa sin marcaje en tiempo real en un biosensor Octet HTX (Pall ForteBio Corp.). Todo el experimento se realizó a 25 °C en tampón HEPES 0,01 M pH 7,4, NaCl 0,15 M, EDTA 3 mM, Tensioactivo Tween-20 al 0,05 % v/v, 0,1 mg/ml de BSA (tampón Octet HBS-ET) con la placa en agitación a una velocidad de 1000 rpm. Para evaluar si 2 anticuerpos podían competir entre sí para unirse a sus respectivos epítopos en el hPD-1-MMH, se capturaron alrededor de 0,25 nM de hPD-1-MMH primero en puntas de biosensor Octet recubiertas con anticuerpo anti-Penta-His (ForteBio Inc., N.° 18-5079) sumergiendo las puntas durante 150 segundos en pocillos que contenían una solución de 10 pg/ml de hPD-1-MMH. Las puntas del biosensor capturadas con antígeno se saturaron a continuación con un primer anticuerpo monoclonal anti-PD-1 (posteriormente denominado mAb-1) sumergiéndolo en pocillos que contenían 100 pg/ml de solución de mAb-1 durante 5 minutos. Las puntas del biosensor se sumergieron continuación en pocillos que contenían una solución de 100 pg/ml de un segundo anticuerpo monoclonal anti-PD-1 (posteriormente denominado mAb-2) durante 4 minutos. Todos los biosensores se lavaron en tampón Octet HBS-ET entre cada etapa del experimento. La respuesta de unión en tiempo real se controló durante todo el transcurso del experimento y se registró la respuesta de unión al final de cada etapa como se muestra en la Figura 2. Se comparó la respuesta de la unión de mAb-2 a hPD-1-MMH formando un complejo previo con mAb-1 y se determinó el comportamiento competitivo/no competitivo de diferentes anticuerpos monoclonales anti-PD-1. Los resultados se resumen en la Tabla 18 (*No se enumeran los mAb2 de autocompetencia).
T l 1 : m n i r z nr r n i r ni-PD-1 l i n
En las condiciones experimentales divulgadas en este ejemplo, H4H7795N2 mostraba competencia cruzada con H4H7798N; H4H7798N mostraba competencia cruzada con H4H7795N2 y H4H9008P; H4H9008P mostraba competencia cruzada con H4H7798N y H4H9068P2; H4H9068P2 mostraba competencia cruzada con H4H9008P y H4H9048P2.
Ejemplo 7: Unión de anticuerpos a células que sobreexpresan PD-1
La un ión de a n ticu e rp o s an ti-P D -1 a una línea c e lu la r de riñón e m b rio n a rio hu m ano (H E K 293 ; A T C C , N .° C R L -1573 )
transfectada de manera estable con PD-1 humano de longitud completa (aminoácidos 1 a 289 con número de registro NP_005009.2) (HEK293/hPD-1) se determinó mediante FACS.
Para el ensayo, las células adherentes se separaron usando tripsina o tampón de disociación sin enzimas y se bloquearon con medio completo. Las células se centrifugaron y se resuspendieron a una concentración de 2,5-6 x1oA6 células/ml en PBS frío que contiene FBS al 2 %. A continuación, se incubaron células HEK293 parentales y HEK293/hPD-1 durante 15-30 min en hielo con 100 nM de cada anticuerpo anti-PD-1. Los anticuerpos no unidos se eliminaron lavando con D-PBS que contenía FBS al 2 %, y las células se incubaron posteriormente con un F(ab')2 secundario conjugado con aloficocianina que reconocía Fc humano (Jackson ImmunoResearch, N.° 109-136-170) o Fc de ratón (Jackson ImmunoResearch, #115-136-146) durante 15-30 minutos en hielo. Las células se lavaron con D-PBS que contenía FBS al 2 % para eliminar el F(ab')2 secundario no unido y las mediciones de fluorescencia se adquirieron utilizando un citómetro de flujo HyperCyte (IntelliCyt, Inc.) o un citómetro de flujo Accuri (BD Biosciences). Los datos se analizaron utilizando el software FlowJo (Tree Star).
Tabla 19: Unión or FACS de anticuer os anti-PD-1 a células HEK293/hPD-1 células HEK293 arentales
C o m o se m ue s tra en la T a b la 19, 25 de los 27 a n ticu e rp o s an ti-P D -1 de la inven c ión m ostra ro n una fu e rte un ión a las
células HEK293/hPD-1 en comparación con la unión a la línea HEK293 parental. Dos anticuerpos de la invención (H2aM7795N y H4H9068P2) se unieron más débilmente a las células que expresan PD-1 humano en comparación con los otros anticuerpos probados.
Para caracterizar adicionalmente los anticuerpos anti-PD1 de la invención, la unión dependiente de la dosis a una línea celular de riñón embrionario humano (HEK293; ATCC, N.° CRL-1573) transfectada de manera estable con PD-1 humano de longitud completa (aminoácidos 1 a 289 con número de registro NP_005009.2) (HEK293/hPD-1) se determinó mediante FACS.
Para el ensayo, las células adherentes se separaron usando tripsina y se bloquearon con medio completo. Las células se centrifugaron y se resuspendieron a una concentración de 6 x10A6 células/ml en tampón de tinción (FBS al 1 % FBS en PBS). Para determinar la CE50 y la Emax de los anticuerpos anti-PD1, se incubaron 90 pl de suspensión celular durante 30 minutos en hielo con una dilución en serie de anticuerpos anti-PD-1 y los controles se diluyeron hasta una concentración final que oscilaba de 5 pM a 100 nM (no se incluyó ninguna muestra de mAb como control negativo) en tampón de tinción. A continuación, las células se centrifugaron y los sedimentos se lavaron una vez con tampón de tinción para eliminar los anticuerpos no unidos. Posteriormente, las células se incubaron durante 30 minutos en hielo con un F(ab')2 secundario conjugado con aloficocianina que reconoce Fc humano (Jackson ImmunoResearch, N.° 109-136-170) o Fc de ratón (Jackson ImmunoResearch, N.° 115-136-071). Las células se centrifugaron y los sedimentos se lavaron una vez con tampón de tinción para eliminar el F(ab')2 secundario no unido y a continuación se fijaron durante la noche con una dilución 1:1 de Cytofix (BD Biosciences, N.° 554655) y tampón de tinción. El día siguiente, las células se centrifugaron y los sedimentos se lavaron una vez con tampón de tinción, se resuspendieron y se filtraron. Las medidas de fluorescencia se adquirieron en un citómetro Hypercyt® y se analizaron en ForeCyt™ (intelliCyt; Albuquerque, NM) para determinar las intensidades medias de fluorescencia (MFI). Los valores de CE50 se calcularon a partir de una ecuación logística de cuatro parámetros sobre una curva de respuesta de 11 puntos utilizando GraphPad Prism. La Emax para cada anticuerpo se definió como la unión a la dosis de anticuerpo más alta (100 nM) ensayada.
Tabla 20: FACS de endiente^ de dosis de la unión de anticuer os anti-PD-1 a células HEK293/hPD-1
continuación
Tabla 21: FACS de endiente de dosis de la unión de anticuer os anti-PD-1 a células HEK293/hPD-1
Como se muestra en la Tabla 20, 25 de 28 anticuerpos anti-PD1 de la invención mostraron unión dependiente de la dosis a células HEK293/hPD-1 con valores de CE50 que van de 33,18 pM a 2,59 nM y valores de Emax que varían de 37.789 a 11,368 MFI. Tres anticuerpos anti-PD1 de la invención no demostraron una unión fuerte a las células HEK293/hPD-1 y, por lo tanto, no se pudo determinar un valor de CE50. Ninguno de los controles de isotipo demostró unión medible en este ensayo.
Como se muestra en la Tabla 21,3 de 6 anticuerpos anti-PD1 de la invención mostraron unión dependiente de la dosis a células HEK293/hPD-1 con valores de CE50 que van de 509 pM a 4,81 nM y valores de Emax que van de 39.774 a 14.111 MFI. Tres anticuerpos de la invención ensayados se unieron a células HEK293/hPD-1, pero no alcanzaron una meseta. Por consiguiente, no se pudieron determinar sus valores de CE50 y sus valores de CE50 se consideran no concluyentes. Ninguno de los controles de isotipo demostró unión medible en este ensayo.
Ejemplo 8: Bloqueo de la regulación negativa de linfocitos T inducida por PD-1 en un ensayo de indicador de luciferasa en linfocitos/APC
La activación de los linfocitos T se logra estimulando los receptores de los linfocitos T (TcR) que reconocen péptidos específicos presentados por las proteínas del complejo principal de histocompatibilidad de clase I o II en las células presentadoras de antígeno (APC). Los TcR activados a su vez inician una cascada de eventos de señalización que pueden ser controlados por genes indicadores impulsados por factores de transcripción como la proteína activadora 1 (AP-1), el factor nuclear de linfocitos T activados (NFAT) o el factor nuclear potenciador de la cadena ligera kappa de linfocitos B activados (NFKb). La respuesta de los linfocitos T se modula mediante el acoplamiento de correceptores expresados de forma constitutiva o inducible en los linfocitos T. Uno de esos receptores es el PD-1, un regulador negativo de la actividad de los linfocitos T. PD-1 interactúa con su ligando, PD-L1, que se expresa en las células diana, incluidas las APC o las células cancerosas, y actúa para enviar señales inhibitorias al reclutar fosfatasas en el
signalosoma de TcR, lo que da como resultado la supresión de la señalización positiva.
La capacidad de los anticuerpos anti-PD-1 para antagonizar la señalización mediada por PD-1/PD-L1 a través del receptor PD-1 en líneas de linfocitos T humanos se evaluó usando un ensayo basado en células in vitro mostrado en la Figura 1. El bioensayo se desarrolló para medir la señalización de linfocitos T inducida por la interacción entre APC y linfocitos T utilizando un cultivo mixto derivado de dos líneas de células de mamíferos: células Jurkat (una línea de linfocitos T inmortalizada) y células Raji (una línea de linfocitos B). Para el primer componente del bioensayo, las células Jurkat Clón E6-1 (ATCC, N.° TIB-152) se transdujeron con Cignal Lenti AP-1 Luc Reporter (Qiagen -Sabiosciences, N.° CLS-011L) según las instrucciones del fabricante. El lentivirus codifica el gen de luciferasa de luciérnaga bajo el control de un promotor mínimo de CMV, repeticiones en tándem del elemento de respuesta transcripcional inducible por TPA (TRE) y un gen de resistencia a puromicina. La línea celular Jurkat diseñada fue posteriormente transducida con una quimera de PD-1 que comprende el dominio extracelular de PD-1 humano (aminoácidos de 1 a 170 de PD1 humano; número de registro NP_005009.2) y los dominios transmembrana y citoplásmico de CD300a humano (aminoácidos de 181 a 299 de CD300a humano; número de registro NP_009192.2). La línea celular estable resultante (Jurkat/AP1-Luc/hPD1-hCD300a) se seleccionó y mantuvo en RPMI/FBS al 10 %/penicilina/estreptomicina/glutamina suplementada con 500 pg/ml de G418 1 pg/ml de puromicina.
Para el segundo componente del bioensayo, se transdujeron células Raji (ATCC, N.° CCL-86) con el gen PD-L1 humano (aminoácidos 1-290 del número de registro NP_054862.1) que se había clonado en un sistema de vector lentiviral (pLEX) (Thermo Scientific Biosystems, N.° OHS4735). Las células Raji, positivas para PD-L1 (Raji/hPD-L1) se aislaron mediante FACS usando un anticuerpo PD-L1 y se mantuvieron en Iscove/FBS al 10 %/penicilina/estreptomicina/glutamina suplementada con 1 pg/ml de puromicina.
Para simular la interacción APC/linfocito T, se utilizó un anticuerpo biespecífico compuesto por un brazo Fab que se une a CD3 en linfocitos T y el otro brazo Fab que se une a CD20 en células Raji (anticuerpo biespecífico CD3xCD20; por ejemplo, como se divulga en el documento US20140088295). La presencia de la molécula biespecífica en el ensayo da como resultado la activación de los linfocitos T y APC al unir las subunidades CD3 de los linfocitos T con CD20 expresado endógenamente en las células Raji. Se ha demostrado que la ligación de CD3 con anticuerpos anti-CD3 conduce a la activación de los linfocitos T. En este bioensayo, los anticuerpos que bloquean la interacción PD1/PD-L1 rescatan la actividad de los linfocitos T desactivando la señalización inhibidora y, posteriormente, conduciendo a un aumento de la activación de AP1-Luc.
En el bioensayo basado en luciferasa, se usó RPMI1640 suplementado con FBS al 10 % y penicilina/estreptomicina/glutamina como medio de ensayo para preparar suspensiones celulares y diluciones de anticuerpos para llevar a cabo el cribado de anticuerpos monoclonales (mAb) anti-PD1. El día del cribado, se determinaron los valores de CE50 de los mAb anti-PD1, en presencia de una concentración fija de anticuerpo biespecífico CD3xCD20 (30 pM), así como la CE50 del anticuerpo biespecífico solo. En el siguiente orden, se añadieron células y reactivos a placas de fondo plano y blanco de 96 pocillos. Para las determinaciones de la CE50 de los mAb anti-PD1, primero se preparó una concentración fija de anticuerpo biespecífico CD3xCD20 (30 pM final) y se añadió a los pocillos de la placa de microvaloración. A continuación, se añadieron diluciones en serie de 12 puntos de mAb anti-PD1 y controles (concentraciones finales en el intervalo de 1,7 pM a 100 nM; más pocillos con medio de ensayo solo). Para la determinación del anticuerpo biespecífico (solo) CE50 , se añadió el anticuerpo biespecífico, a concentraciones finales que varían de 0,17 pM a 10 nM (más pocillos con medio de ensayo solo), a los pocillos de la placa de microvaloración. Posteriormente, se preparó una suspensión celular de 2,5 x 10A6/ml de células Raji/hPD-L1 y se añadieron 20 pl por pocillo (número final de células (pocillo 5 x 10A4 células). Las placas se dejaron a temperatura ambiente (15-20 minutos), mientras se preparaba una suspensión de 2,5 x 10A6/ml de Jurkat/AP1-Luc/hPD1(ecto)-hCD300a (TM-Cyto). Se añadieron 20 pl de la suspensión Jurkat (número final de células/pocillo 5x10A4 células) por pocillo. Las placas que contenían el cocultivo se incubaron durante 5 a 6 horas a 37 °C/5 % de CO2. Las muestras se analizaron por duplicado y a continuación se detectó la actividad luciferasa después de la adición de reactivo ONE-Glo™ (Promega, # E6051) y se midieron las unidades de luz relativa (RLU) en un luminómetro Victor.
Los valores de RLU para cada anticuerpo seleccionado se normalizaron estableciendo las condiciones de ensayo con concentración fija (30 pM) del anticuerpo biespecífico CD3/CD20, pero sin anticuerpo anti-PD-1 hasta el 100 %. Esta condición corresponde a la respuesta máxima de AP1-Luc provocada por la molécula biespecífica en presencia de la señal inhibidora de PD-1/PD-L1. Tras la adición del anticuerpo anti-PD-1, se suprime la señal inhibidora y la estimulación aumentada se muestra aquí como Emax, el porcentaje de aumento de la señal en presencia de la dosis de anticuerpo más alta probada (100 nM). Para comparar la potencia de los anticuerpos anti-PD1 ensayados, se determinó la concentración de anticuerpo a la que el valor de RLU normalizado alcanzó el 150 % de activación a partir de una ecuación logística de cuatro parámetros sobre una curva de respuesta de 12 puntos utilizando GraphPad Prism. Los resultados se resumen en la Tabla 22 y la Tabla 23, respectivamente.
Tabla 22: Anticuerpo anti-PD1 que bloquea la inhibición dependiente de PD-1/PD-L1 de la señalización de AP1-Luc en el Ex erimento 1
Tabla 23: Anticuerpo anti-PD1 que bloquea la inhibición dependiente de PD-1/PD-L1 de la señalización de AP1-Luc en el Ex erimento 2
Como se muestra en la Tabla 22, 25 de los 31 anticuerpos anti-PD-1 de la invención ensayados bloquearon la inhibición de PD-1/PD-L1 con valores de Emax que van de 239 a 163. Seis de los 31 anticuerpos anti-PD-1 de la invención no demostraron un bloqueo sustancial de la interacción PD1/PD-L1 cuando se ensayaron en este ensayo.
Como se muestra en la Tabla 23, 2 de los 4 anticuerpos anti-PD-1 de la invención ensayados bloquearon la inhibición de PD-1/PD-L1 con valores de Emax de 150 y 343 %, respectivamente. Dos de los 4 anticuerpos anti-PD-1 de la invención no demostraron un bloqueo sustancial de la interacción PD1/PD-L1 cuando se ensayaron en este ensayo.
Ejemplo 9: Eficacia in vivo de los anticuerpos anti-PD-1
Para determinar el efecto de un número selecto de anticuerpos anti-PD-1 de la invención en un modelo in vivo relevante, se llevaron a cabo tres estudios de crecimiento tumoral MC38.ova, que implican la inyección subcutánea de células tumorales y que comenzaron en días diferentes, en ratones que eran homocigotos para la expresión del dominio extracelular de PD-1 humano en lugar del dominio extracelular de PD-1 de ratón (ratones PD-1 Humln) en un 75 % de C57/BI6/25 % de 129 cepas de fondo.
Para los estudios, los ratones se dividieron uniformemente según el peso corporal en 5 grupos de tratamiento o control para el Estudio 1 (5 ratones por grupo), 8 grupos de tratamiento o control para el Estudio 2 (5 ratones por grupo) y 5 grupos de tratamiento o control para el Estudio 3 (7 ratones por grupo). En el día 0, los ratones fueron anestesiados por inhalación de isoflurano y a continuación recibieron una inyección por vía subcutánea en el flanco derecho de 5 x 105 células MC38.ova en suspensión de 100 pl de DMEM en el Estudio 1 o 1 x 106 células MC38.ova en suspensión de 100 pl de DMEM en el Estudio 2 y el Estudio 3. Para el estudio 1, se inyectaron por vía intraperitoneal los grupos de tratamiento con 200 pg de uno de los tres anticuerpos anti-PD-1 de la invención, o un anticuerpo control de isotipo con especificidad irrelevante los días 3, 7, 10, 14 y 17 del experimento, mientras que un grupo de ratones se dejó sin tratar. Para el estudio 2, los grupos de tratamiento se inyectaron por vía intraperitoneal con uno de los tres anticuerpos anti-PD-1 de la invención a 10 mg/kg o 5 mg/kg por dosis, un anticuerpo de la invención (H4H7795N2) a 10 mg/kg por dosis, o un anticuerpo de control de isotipo con especificidad irrelevante a 10 mg/kg los días 3, 7, 10, 14 y 17 del experimento. Para el estudio 3, los grupos de tratamiento se inyectaron por vía intraperitoneal con uno de los dos anticuerpos anti-PD-1 de la invención a 5 mg/kg o 2,5 mg/kg por/dosis, o un anticuerpo de control de isotipo con especificidad irrelevante a 5 mg/kg los días 3, 7, 10, 14 y 17 del experimento. El protocolo experimental de dosificación y tratamiento para grupos de ratones se muestra en la Tabla 24.
continuación
Para los estudios, se registraron los volúmenes tumorales promedio determinados por medidas de calibre y el porcentaje de supervivencia en el día 14 o 17 y el día 23 o 24 de cada experimento para cada grupo de tratamiento. Además, también se evaluó el número de ratones sin tumor al final del estudio (día 42 para el estudio 1 y día 31 para el estudio 2 y el estudio 3). Los resultados, expresados como la media del volumen tumoral (mm3) (± DE), el porcentaje de supervivencia y el número de ratones sin tumor se muestran en la Tabla 23 para el Estudio 1, la Tabla 3 para el Estudio 2 y la Tabla 4 para el Estudio 3.
Tabla 25: Media del volumen tumoral, porcentaje de supervivencia y número de ratones sin tumor en cada grupo de tratamiento del tumor in vivo Estudio 1
Como se muestra en la Tabla 25 para el Estudio 1, los ratones tratados con un anticuerpo de la invención, H4H7798N no desarrollaron ningún tumor detectable durante el transcurso del estudio. Los ratones tratados con H4H9008P exhibieron un volumen tumoral reducido sostenido en comparación con los controles en los días 17 y 24 del estudio con 3 de 5 ratones o 4 de 5 ratones sin tumores al final del experimento, respectivamente. En cambio, el tratamiento con uno de los anticuerpos anti-PD1, H4H7795N2, no demostró una eficacia significativa en la reducción del volumen tumoral en este estudio en comparación con los controles. En el día 23 del estudio, 1 de cada 5 ratones murió en el grupo H4H7795N2, y 2 de cada 5 ratones murieron en el grupo de tratamiento de control de isotipo. En el grupo sin tratamiento y en el grupo de control de isotipo, algunos ratones mostraron una regresión espontánea de los tumores (1 de cada 5 ratones y 2 de cada 5 ratones, respectivamente).
Como se muestra en la Tabla 26 para el Estudio 2, los ratones tratados con un anticuerpo de la invención, H4H7798N a 10 mg/kg no desarrollaron ningún tumor detectable durante el transcurso del estudio. Los grupos de ratones tratados con 10 mg/kg de H4H9008P o H4H9048P2 mostraron un volumen tumoral sustancialmente reducido en comparación con los controles en los días 17 y 24 del estudio. Cuatro de cada 5 ratones en cada grupo tratados con 10 mg/kg de H4H9008P o H4H9048P2 no tenían tumores el día 31, mientras que en el grupo de tratamiento de control de isotipo solo 1 de cada 5 animales no tenía tumores como resultado de la regresión espontánea del tumor. Un anticuerpo ensayado a 10 mg/kg, H4H7795N2, demostró un volumen tumoral sustancialmente reducido en comparación con los controles en los días 17 y 24 del estudio, pero este anticuerpo fue el anticuerpo anti-PD1 menos eficaz y solo 2 de cada 5 ratones sobrevivieron al final del experimento.
Se observó una respuesta dependiente de la dosis en la supresión tumoral a las dosis ensayadas (5 mg/kg y 10 mg/kg) en los grupos tratados con H4H7798N, H4H9008P y H4H9048P2. La terapia con H4H7798N o H4H9008P a 5 mg/kg fue menos eficaz, con 4 de 5 ratones sin tumor al final del experimento el día 21, mientras que 5 de 5 ratones permanecieron sin tumores en ambos grupos de dosis de 10 mg/kg de H4H7798N y H4H9008P.
La prueba de Dunnett en comparaciones múltiples ANOVA bidireccional reveló que las diferencias en el crecimiento tumoral entre el grupo tratado con anticuerpo de control de isotipo con 10 mg/kg como referencia y los grupos tratados con 10 mg/kg con H4H7798N, H4H9008P o H4H9048P2 fueron estadísticamente significativas con valor de p <0,005. Las diferencias en el crecimiento tumoral entre el grupo tratado con anticuerpo de control de isotipo con 10 mg/kg como referencia y los grupos tratados con 5 mg/kg con H4H7798N, H4H9008P o H4H9048P2 también fueron estadísticamente significativas con un valor de p < 0,05.
Tabla 27: Media del volumen tumoral, porcentaje de supervivencia y número de ratones sin tumor en cada grupo de tratamiento del tumor in vivo Estudio 3
Como se muestra en la Tabla 27 para el Estudio 3, 6 de cada 7 ratones tratados con un anticuerpo de la invención, H4H7798N, u otro anticuerpo de la invención, H4H9008P, con 5 mg/kg no tenían tumor al final del experimento, mientras que en el grupo de control de isotipo no había ningún animal sin tumor. Un ratón portador de tumores en el grupo de control de IgG4 murió el día 17 posterior a la implantación. Solo 4 de los 7 ratones tratados con H4H9008P a una dosis de 2,5 mg/kg permanecían sin tumores al final del experimento. La diferencia en los volúmenes tumorales el día 21 entre los anticuerpos anti-PD-1 ensayados y un grupo de control de isotipo fue estadísticamente significativa según lo determinado por ANOVA unidireccional con la prueba posterior de comparación múltiple de Dunnett con p < 0,01. Los cuatro anticuerpos anti-PD-1 fueron igualmente más eficaces con la dosis de 5 mg/kg que con la dosis de 2,5 mg/kg.
Ejemplo 10: Efectos antitumorales de una combinación de un anticuerpo anti-PD-1 y un antagonista de VEGF en un modelo de tum or de tratamiento temprano en ratón
Se desarrolló un modelo de tumor de tratamiento temprano para probar la eficacia de una combinación de un anticuerpo anti-PD-1 y un antagonista de VEGF. En este modelo, la terapia de combinación se administra poco después de la implantación del tumor. El experimento también usó un anticuerpo anti-PD-L1 solo y en combinación con el antagonista de VEGF. El anticuerpo anti-PD-1 usado en este experimento fue el clon "RPMI-14" anti-PD-1 de ratón con IgG2b de rata (Bio X Cell, West Lebanon, NH). El antagonista de VEGF usado en este experimento fue aflibercept (una molécula quimérica basada en el receptor de VEGF, también conocida como "trampa de VEGF" o "VEGFR1R2-FcAC1(a)", cuya descripción completa se proporciona en otra parte del presente documento). El anticuerpo anti-PD-L1 utilizado en este experimento fue un anticuerpo monoclonal anti-PD-L1 con secuencias Vh/Vl de anticuerpo "YW243.55S70" de acuerdo con el documento US20100203056A1 (Genentech, Inc.), con IgG2a de ratón y que mostró reactividad cruzada con PD-L1 de ratón.
P ara este m ode lo e xp e rim e n ta l, se im p lan ta ron po r v ía su b cu tá n e a 1,0 x 106 de cé lu la s tu m o ra le s de co lo n -26 en
ratones BALB/c el día 0. Comenzando el Día 3, antes del establecimiento de tumores medibles, los ratones se trataron con una de las terapias mono o de combinación, o combinación de control, como se expone en la Tabla 28.
Tabla 28: Dosificación ex erimental ru os de tratamiento
Las diversas terapias se administraron en cinco puntos temporales diferentes durante un período de dos semanas (es decir, inyecciones en el Día 3, Día 6, Día 10, Día 13 y Día 19).
Los animales de cada grupo de terapia se evaluaron en términos de incidencia tumoral, volumen tumoral, mediana del tiempo de supervivencia y número de animales sin tumor el Día 50. La extensión del crecimiento tumoral se resume en la Figura 2 (curvas de crecimiento tumoral) y la Figura 3 (volumen tumoral en el día 28). Los resultados también se resumen en la Tabla 29.
T l 2 : R n in m r n r r mi n
El crecimiento tumoral se redujo sustancialmente en los animales tratados con la combinación de trampa de VEGF anticuerpo anti-PD-1 en comparación con los regímenes de tratamiento que implican cualquier agente terapéutico solo (véanse las Figuras 2 y 3). Asimismo, la supervivencia aumentó sustancialmente en el grupo de anticuerpo trampa de VEGF anti-PD-1, sobreviviendo el 70 % de los animales hasta al menos el día 50 después de la implantación del tumor. Por el contrario, para los grupos de anti-PD-1 y trampa de VEGF en monoterapia, la supervivencia hasta el día 50 fue sólo del 40 % y 30 % respectivamente (véase la Figura 3 y la Tabla 29).
Ejemplo 11: Estudio de ensayo clín ico de dosificación repetida con anticuerpo anti-PD-1 como terapia única y en combinación con otras terapias contra el cáncer en pacientes con neoplasias malignas avanzadas
Este es un estudio de aumento de la dosis del anticuerpo anti-PD-1, solo o en combinación con radioterapia, ciclofosfamida o ambos en pacientes con neoplasias malignas avanzadas. El anticuerpo anti-PD-1 ilustrativo ("mAb") usado en este Ejemplo comprende HCVR de SEQ ID NO: 162 y LCVR de SEQ ID NO: 170.
Objetivos del estudio
El objetivo principal del estudio es caracterizar la seguridad, tolerabilidad, TLD del mAb administrado por vía intravenosa como monoterapia o en combinación con radiación dirigida (con la intención de que esto sirva como una terapia inmunoestimuladora, en lugar de básicamen una terapia ablativa del tumor), ciclofosfamida en dosis bajas (una terapia que se ha demostrado que inhibe las respuestas de los linfocitos T reguladores), o ambas en pacientes con neoplasias malignas avanzadas.
Los objetivos secundarios del estudio son: (1) determinar una dosis de fase 2 recomendada (RP2D) de mAb como monoterapia y en combinación con otras terapias contra el cáncer (radiación dirigida, ciclofosfamida en dosis bajas o ambas); (2) describir la actividad antitumoral preliminar de mAb, solo y con cada pareja(s) de combinación; (3) caracterizar la FC de mAb como monoterapia y en combinación con otras terapias contra el cáncer (radiación dirigida, ciclofosfamida en dosis bajas o ambas); y (4) evaluar la inmunogenicidad de mAb.
Diseño del estudio
El estudio de seguridad se evaluará en cohortes de aumento de la dosis 3 3 estándar separadas (en monoterapia), combinación con radioterapia, combinación con ciclofosfamida, y combinación con radioterapia más ciclofosfamida). La elección de la terapia combinada con radiación, ciclofosfamida o ambas se basará en la evaluación del investigador de la mejor opción de terapia para un paciente individual en consulta con el patrocinador. Para ser incluido en una cohorte de radioterapia, un paciente debe tener una lesión que pueda irradiarse de manera segura y para la cual la radiación en las dosis paliativas limitadas contempladas, se consideraría médicamente apropiada, y al menos otra lesión adecuada para la evaluación de la respuesta. Se permitirá la inclusión de un paciente solo si hay un hueco disponible en la cohorte para el tratamiento elegido.
Los pacientes se someterán a procedimientos de detección para determinar la elegibilidad dentro de los 28 días anteriores a la administración inicial del mAb. Después de la inclusión de los pacientes en una cohorte de monoterapia con mAb, la inclusión de cohortes posteriores se determinará por la aparición de TLD en cohortes anteriores (es decir, sin TLD en una cohorte de 3 pacientes, o no más de 1 TLD en una cohorte ampliada de 6 pacientes), y la disponibilidad de huecos para pacientes. Los niveles de dosis de monoterapia planificados son 1, 3 o 10 mg/kg administrados por vía intravenosa cada 14 días (2 semanas).
Una vez que uno o ambos períodos de observación de la TLD de la cohorte de monoterapia de 1 mg/kg o 3 mg/kg de mAb en monoterapia se completan sin una TLD en una cohorte de 3 pacientes o con no más de 1 TLD en una cohorte ampliada de 6 pacientes, los pacientes se pueden incluir en una cohorte que combina ciclofosfamida o radioterapia con mAb a ese nivel de dosis de monoterapia. Los pacientes pueden incluirse en una cohorte de combinación de mAb ciclofosfamida/radioterapia una vez que se completen los períodos de observación de TLD tanto para la cohorte para ese nivel de dosis de mAb ciclofosfamida como para la cohorte para ese nivel de dosis de mAb el mismo régimen de radioterapia sin TLD en una cohorte de 3 pacientes, o no más de 1 TLD en una cohorte ampliada de 6 pacientes.
Una vez que se completa el período de observación de TLD de la cohorte de 3 mg/kg de mAb en monoterapia sin TLD en una cohorte de 3 pacientes, o no más de 1 TLD en una cohorte ampliada de 6 pacientes, también se puede incluir una cohorte de 10 mg/kg de mAb en monoterapia.
Las cohortes de mAb de 3 mg/kg y 10 mg/kg en monoterapia se incluirán solo después de que el número requerido de pacientes en la cohorte de dosis de monoterapia previa (es decir, 1 mg/kg y 3 mg/kg, respectivamente) hayan superado el período de observación de TLD de 28 días sin que se demuestre una dosis máxima tolerada (DMT) para ese nivel de dosis. Una cohorte de tratamiento de combinación de 1 mg/kg de mAb se incluirá solo después de completar el período de observación de TLD para la cohorte de 1 mg/kg en monoterapia. Las cohortes de combinación que reciben 3 mg/kg de mAb se incluirán solo cuando el número requerido de pacientes en las respectivas cohortes de combinación de 1 mg/kg de mAb haya superado el período de observación de TLD sin demostrar una DMT. Las cohortes de combinación triple que combinan mAb con ciclofosfamida y un régimen de radiación se incluirán solo cuando el número requerido de pacientes en las dos cohortes de combinación doble correspondientes a ese nivel de dosificación haya superado el período de observación de TLD sin que se demuestre una d Mt .
La Tabla 30 resume las cohortes de aumento de la dosis en las cuales se incluirán los pacientes.
Tabla 30: Posibles cohortes de aumento de la dosis
con tin u a c ió n
U na T LD se de fine com o cu a lq u ie ra de lo s igu ien te : una to x ic id a d no h e m a to ló g ica (p o r e jem p lo , uve ítis o c u a lq u ie r o A A ri) o una to x ic id a d h e m a to ló g ica (p o r e jem p lo , neu tropen ia , tro m b o c ito p e n ia , n e u tro p e n ia febril).
La dos is m áx im a to le ra d a (D M T ) se de fine com o la dos is m ás a lta a la que m enos de un te rc io de una coho rte am p liad a de 6 p a c ien tes e xp e rim e n ta una T LD d u ra n te el p rim e r c ic lo de tra ta m ie n to . P o r tan to , la D M T se de fine co m o el n ive l de dos is in m e d ia ta m e n te po r de b a jo de l n ive l en el que se de tie ne la d o s ifica c ió n de b id o a la apa ric ió n de 2 o m ás T LD en una coh o rte a m p liad a de 6 pac ien tes. Si el a u m en to de la d os is no se de tie ne de b id o a la apa ric ió n de TLD, se co n s id e ra rá que no se ha d e te rm in a do la DM T. Es pos ib le que en este es tu d io no se de fina una DM T, ya sea para un g rupo de m on o te rap ia o para g ru p o s de co m b in a c ió n ind iv idua les . A d ic io n a lm e n te , es pos ib le que las D M T de los m A b puedan d ife rir en tre la m on o te rap ia y cad a rég im en de tra ta m ie n to com b inado .
Duración del estudio
Los pac ie n te s rec ib irán hasta 48 se m a n a s de tra ta m ie n to , d e sp ué s de las cua les habrá un pe ríodo de se g u im ie n to de 24 sem anas. Un pac ie n te rec ib irá tra ta m ie n to hasta que se co m p le te el pe ríodo de tra ta m ie n to de 48 sem anas, o hasta la p rog res ión de la en fe rm e da d , to x ic id a d inacep tab le , re tirada de l c o n se n tim ie n to o cu m p lim ie n to de o tro crite rio de re tirada de l es tud io . D e spués de un m ín im o de 24 se m a n a s de tra ta m ie n to , los p a c ie n te s con resp u e s ta s co m p le ta s (R C ) co n firm a d a s pueden o p ta r p o r in te rru m p ir el tra ta m ie n to y c o n tin u a r con to d a s las e va lu a c io n e s re leva n te s del es tud io (p o r e jem p lo , e va lu a c io n e s de e ficac ia ). D esp u é s de un m ín im o de 24 se m a n a s de tra ta m ie n to , los p a c ien tes con e va lu a c io n e s de la ca rga tu m o ra l de e n fe rm e da d es ta b le (E E ) o respues ta pa rc ia l (R P ) que no han ca m b ia d o du ra n te 3 e va lu a c io n e s tu m o ra le s su ce s iva s ta m b ié n pueden o p ta r po r in te rru m p ir el tra ta m ie n to y c o n tin u a r con to d as las e va lu a c io n e s re leva n te s del es tu d io (p o r e jem p lo , e v a lu a c io n e s de la e ficac ia ).
Población del estudio
La p o b lac ión ob je tivo de este es tu d io co m p re n d e p a c ie n te s con n e o p las ias m a lig na s a va n za d a s que no son ca n d id a to s para la te ra p ia es tánda r, que no desean so m e te rse a una te ra p ia e s tá n d a r o para q u ie n es no se esp e ra que una te ra p ia d isp o n ib le p ro p o rc io n e un b e ne fic io c lín ico ; y p a c ie n te s con n e o p las ias que son in cu ra b le s y no han resp o n d id o o han m os tra d o p ro g re s ió n de l tu m o r a p e sa r de la te ra p ia es tándar.
C rite rio s de inc lus ión : Un pac ie n te d ebe cu m p lir los s ig u ie n tes c rite rio s para se r e le g ib le para su inc lus ión en el es tud io : (1 ) p rog res ión d e m o s tra d a de un tu m o r só lido sin una opc ión te ra p é u tica de re fe re n c ia a lte rn a tiva d ispon ib le ; (2 ) al m enos 1 les ión para la e va lu a c ió n de la respues ta . Los p a c ie n te s a s ig n a d o s a rad io te ra p ia requ ie ren al m enos una les ión a d ic io n a l que pueda irrad ia rse de fo rm a se g u ra sin a fe c ta r a las les io n e s de re fe re n c ia y para la cua l la rad iac ión en las d os is p a lia tivas lim ita d as co n tem p la d a s se co n s id e ra ría m é d ica m e n te a p rop iada ; (3 ) E s tado fu n c io n a l del E aste rn C o o p e ra tive O n co lo g y G roup (E C O G ) < 1; (4) m ás de 18 años; (5) fu n c ió n hepática : a. b ilirru b in a to ta l < 1,5x el lím ite su p e rio r de la no rm a lid a d (LS N ; si ex is ten m e tá s ta s is hepá tica s < 3x LSN), b. tra n s a m in a s a s < 3x LSN (o < 5 ,0 x LSN, si ex is ten m e tá s ta s is hepá ticas), c. fo s fa ta sa a lca lin a (A L P ) < 2 ,5x LSN (o 5 ,0x LSN, si hay m e tás tas is hepáticas); (6) fun c ió n renal: c rea tin in a sé rica < 1 ,5x LSN; (7) recu e n to de n e u tró filo s (A N C ) > 1,5 x 109/l, c. recuen to de p la q u e ta s > 75 x 109/l; (8) ca p a c id ad para p ro p o rc io n a r el c o n se n tim ie n to in fo rm a d o firm ado ; y (9) ca p a c id ad y vo lu n tad de cu m p lir con las v is ita s p rog ra m a d a s , los p lanes de tra ta m ie n to , las p rue b a s de lab o ra to rio y o tros p ro ce d im ie n tos re lac ion a d o s con el es tud io .
C rite rio s de exc lus ión : Un pac ien te que cu m p la c u a lq u ie ra de los s ig u ie n te s c rite rio s se rá e xc lu id o de l es tud io : (1) E v id e n c ia a c tua l o rec ien te (en los ú ltim os 5 años) de e n fe rm e da d a u to in m u n ita ria s ig n ifica tiva que requ irió tra ta m ie n to con tra ta m ie n to s in m u n o su p re so re s s is tém icos , que pueden su g e rir r iesgo de A A ri; (2 ) T ra ta m ie n to p rev io con un agen te que b lo q u ea la v ía P D -1 /P D -L1 ; (3) T ra ta m ie n to p rev io con o tros a g e n te s in m u n o m o d u la d o re s en m enos de 4 se m a n a s o 4 se m iv idas , lo que sea m ayor, a n tes de la p rim e ra d os is de m Ab; (4) Los e je m p lo s de ag e n te s in m u n o m o d u la d o re s inc luyen b lo q u e a d o re s de C T LA -4 , 4 -1 B B (C D 137), O X -40 , va c u n a s te ra p é u tica s o tra ta m ie n to s con c itoc inas; (5 ) M e tá s ta s is ce re b ra le s no tra ta d a s que pueden co n s id e ra rse activas. Los pac ie n te s con m e tá s ta s is ce re b ra le s p re v ia m e n te tra ta d a s pueden p a rtic ip a r s ie m pre que estén e s ta b les (es decir, s in e v id e n c ia de p rog res ión p o r im á ge n e s du ra n te al m enos 4 se m a n a s a n tes de la p rim e ra d os is de l tra ta m ie n to de l es tud io , y cu a lq u ie r s ín tom a n e u ro ló g ico haya vu e lto a los va lo re s in ic ia le s ) y no haya e v id e n c ia de m e tá s ta s is ce re b ra le s nuevas o en aum en to ; (6) D osis de co rtico s te ro id e s in m u n o su p re so re s (> 10 m g de p re d n iso n a al d ía o e q u iva le n te ) en las 4 sem a n a s a n te rio re s a la p rim e ra d os is de m Ab; (7 ) T ro m b o s is v e n o sa p ro funda , em b o lia p u lm o n a r ( in c lu id a e m b o lia p u lm o n a r a s in to m á tica id e n tifica da en las im á ge n e s) u o tro e p iso d io tro m b o e m b ó lic o en los 6 m eses a n te rio re s a la p rim era dos is de m Ab; (8 ) In fecc ión ac tiva que requ ie re te ra p ia , inc lu id a la in fecc ión co n o c id a p o r el v iru s de la in m u n o d e fic ie n c ia hu m a n a o la in fecc ión ac tiva po r el v iru s de la hep a titis B o la hep a titis C; (9 ) A n te c e d e n te s de ne u m o n itis en los ú ltim os 5 años; (10 ) C u a lq u ie r tra ta m ie n to de in ve s tig a c ió n o a n titum o ra l en los 30 d ías a n te rio re s a
la a d m in is tra c ió n in ic ia l de m Ab; (11 ) A n te c e d e n te s de re a cc io n e s a lé rg ica s d o cu m e n ta d a s o re a cc io n e s de h ip e rse n s ib ilid a d a guda a trib u id a s al tra ta m ie n to con te ra p ia s con a n ticu e rp o s en genera l, o a ag e n te s e sp e c ífica m e n te u tiliza d o s en el es tud io ; 12) A le rg ia co n o c id a a la d o x ic ic lin a o te tra c ic lin a (p re ca u c ió n de b id o a la p re senc ia de co m p o n e n te s tra z a en m Ab); (13 ) Lac tan c ia m aterna ; (14 ) P rueba de e m b a ra zo en su e ro pos itiva ; (15 ) A n te ce d e n te s en los ú ltim os 5 años de una n e o p las ia m a ligna invas iva d is tin ta a la tra ta d a en este estud io , con la exce p c ió n de ca rc ino m a de piel de cé lu la s e sca m o sa s o basa l re se ca d o /e x tirp a d o o ca rc ino m a in situ del cue llo u te rino u o tros tu m o re s loca les c o n s id e ra d o s cu ra d o s con tra ta m ie n to loca l; (16 ) P ro b le m a s p s iq u iá trico s ag u do s o c rón ico s que, según la eva lu a c ió n de l inves tig a do r, hacen que el pac ie n te no sea e le g ib le para pa rtic ipa r; y (17 ) A c tiv id a d sexua l co n tin u a d a en h o m bres o m u je res en edad fé rtil que no están d isp u e s to s a u tiliza r m é to d o s a n tico n ce p tivo s a d e cua d o s d u ra n te el es tud io .
Tratamientos del estudio
El m A b se su m in is tra rá co m o un líqu ido en v ia le s e s té rile s de un so lo uso. C ada v ia l co n ten d rá un v o lu m e n su fic ie n te pa ra e x tra e r 10 ml de m A b a una co n ce n tra c ió n de 25 m g/m l. Las in s tru cc io n e s so b re la p repa ra c ió n de la d os is se p rop o rc io n an en los m an u a le s de re fe re n c ia de l es tud io . El m A b se a d m in is tra rá de fo rm a a m b u la to ria com o una in fus ión in tra ve n o sa de 30 m inutos. La d os is de cada pac ie n te d e p e n d e rá del peso co rp o ra l ind iv idua l. La dos is de m A b debe a ju s ta rse en cada c ic lo para ca m b io s en el peso co rp o ra l de >10% . El m A b se a d m in is tra rá so lo y en co m b in a c ió n con rad ia c ió n yo c ic lo fos fam ida .
M on o te rap ia
El m A b se a d m in is tra rá de fo rm a a m b u la to ria m ed ia n te in fus ión in tra ve n o sa du ra n te 30 m in u to s cada 14 d ías du ran te 48 se m a n a s (es decir, los d ías 1, 15 ± 3, 29 ± 3, y 43 ± 3 de un c ic lo de 56 d ías). Los reg ím e n es de m on o te rap ia p la n ifica d os que se a s ig n a rá n pueden inc lu ir: (i) in fus ión IV de 1 m g/kg d u ra n te 30 m in u to s cada 14 d ías du ra n te 48 sem anas; (ii) in fus ión de 3 m g/kg du ra n te 30 m inu tos cada 14 d ías du ra n te 48 sem anas; ( iii) in fus ión de 10 m g/kg d u ra n te 30 m inu tos cada 14 d ías du ra n te 48 sem anas; y (iv ) in fus ión de 0 ,3 m g/kg d u ra n te 30 m inu tos cada 14 d ías d u ra n te 48 se m a n a s (si se de te rm in a que la D M T es in fe rio r a 1 m g/kg).
T e ra p ia de co m b in a c ió n
La rad io te ra p ia co n co m ita n te y la c ic lo fo s fa m id a se a d m in is tra rá n m ed ia n te rece ta m éd ica y su uso, dosis , m od ifica c io n e s de la dosis , reducc iones , o retrasos, a s í com o cu a lq u ie r A A po ten c ia l resu ltan te de su uso, se ras trea rán ju n to con el de l m Ab.
Coadministración de mAb y radiación: El m A b se a d m in is tra rá m ed ia n te in fus ión IV d u ra n te 30 m in u to s cada 14 d ías du ra n te 48 se m a n a s en co m b in a c ió n con rad io te ra p ia desde el d ía 8 hasta el d ía 12. Los reg ím e n es de co m b in a c ió n de m A b y ra d io te ra p ia p la n ifica d o s pueden inclu ir:
• 1 m g/kg de in fus ión de m A b du ra n te 30 m in u to s cada 14 d ías du ra n te 48 se m a n a s m ás rad io te ra p ia 30 G y (6 G y x 5 ve ce s /se m a n a ; a d m in is tra d a 1 se m a n a d e sp ué s de la p rim e ra d os is de m Ab, p re fe re n te m e n te en d ías co n se cu tivo s )
• 1 m g/kg de in fus ión de m A b du ra n te 30 m inu tos cada 14 d ías du ra n te 48 se m a n a s m ás rad io te ra p ia 27 G y (9 G y x 3 ve ce s /sem a n a ; a d m in is tra d a 1 se m a n a d e sp ué s de la p rim e ra d os is de m Ab, p re fe re n te m e n te en d ías no co n se cu tivo s )
• 3 m g/kg de in fus ión de m A b du ra n te 30 m inu tos cada 14 d ías du ra n te 48 se m a n a s m ás rad io te ra p ia 30 G y (6 G y x 5 ve ce s /se m a n a ; a d m in is tra d a 1 se m a n a d e sp ué s de la p rim e ra d os is de m Ab, p re fe re n te m e n te en d ías co n se cu tivo s )
• 3 m g/kg de in fus ión de m A b du ra n te 30 m in u to s cada 14 d ías d u ra n te 48 se m a n a s m ás rad io te ra p ia 27 G y (9 G y x 3 ve ce s /sem a n a ; a d m in is tra d a 1 se m a n a d e sp ué s de la p rim e ra d os is de m Ab, p re fe re n te m e n te en d ías no co n se cu tivo s )
Los p a c ie n te s rec ib irán 30 G y a d m in is tra d o s com o 5 fra cc io n e s de 6 G y a d m in is tra d o s d ia ria m e n te co m e n za n d o 1 se m a n a d e sp ué s de la p rim e ra dos is de m Ab, o 27 G y a d m in is tra d o s com o 3 fra cc io n e s de 9 G y a d m in is tra d o s en d ías a lte rn o s co m e n za n d o 1 se m a n a d e sp ué s de la p rim e ra d os is de m Ab. La les ión se le cc io n a d a para la rad iac ión debe se r una les ión que pueda irra d ia rse de m ane ra se g u ra con irra d ia c ió n foca l sin a fe c ta r a la les ión o les iones de re fe renc ia , y para las cua les la rad iac ión en las dos is p a lia tiva s lim ita d as c o n te m p la d a s se co n s id e ra ría m é d ica m e n te ap rop iada . La d os is ob je tivo pa ra un pac ie n te se basa rá en la as ig n a c ió n de co h o rte s y debe a ju s ta rse a los requ is itos no rm a le s de l te jido , de a cu e rd o con la p rác tica e s tá n d a r de o n co lo g ía rad io te rá p ica . El tra ta m ie n to con el rég im en de dos ifica c ió n e sp e c ifica do po r el p ro to co lo es tá pe rm itid o so lo si se cum p le n los c r ite rio s de te jid o norm a l. Si los c rite rio s de te jid o no rm a l no se pueden cu m p lir en n in g u no de los reg ím e n es de rad io te ra p ia e sp e c ifica d o s en el p ro toco lo , el pac ien te no es e leg ib le pa ra su inc lus ión en una co h o rte de tra ta m ie n to co m b in a d o de rad iac ión en este estud io . Coadministración de mAb y ciclofosfam ida: El m A b se a d m in is tra rá m ed ia n te pe rfus ión in tra ve n o sa d u ra n te 30 m in u to s cada 14 d ías (2 se m a n a s) du ra n te 48 se m a n a s en co m b in a c ió n con c ic lo fo s fa m id a 200 m g /m 2 cada 14 d ías du ra n te 4 dosis. C ada una de las 4 d os is de c ic lo fo s fa m id a se a d m in is tra rá 1 d ía an tes de cada una de las p rim e ras 4
dos is de m A b (d ía s -1, 14, 28 y 42 de l p rim e r c ic lo de 56 d ías).
A u n q u e la c ic lo fo s fa m id a se ha u tiliza d o con éx ito al m ism o tie m p o que o tros fá rm acos , según se ha descrito , la tasa de m e ta b o lism o y la a c tiv idad le u co p én ica de la c ic lo fo s fa m id a a u m e n ta n con la a d m in is tra c ió n c rón ica de a ltas dos is de fe n ob a rb ita l. El tra ta m ie n to con c ic lo fo s fa m id a p ro vo ca una inh ib ic ión m arcada y p e rs is te n te de la a c tiv idad de la co lines te ra sa , lo que po ten c ia el e fec to de l c lo ru ro de su cc in ilco lin a . El rég im en p la n ifica d o de co m b in a c ió n de m A b y c ic lo fo s fa m id a que se a s ig n a rá es:
• C ic lo fo s fa m id a 200 m g /m 2 cada 14 d ías (d ía s -1, 14, 28 y 42 de l p rim e r c ic lo de 56 d ías) para un to ta l de 4 dos is m ás
• 3 m g/kg de in fus ión de m A b du ra n te 30 m inu tos cada 14 d ías d u ra n te 48 se m a n a s (dos is de m on o te rap ia a d m in is tra d a de 3 m g/kg < DM T; si 3 m g/kg > DM T, la d os is se rá 1 m g/kg).
Coadministración de mAb, radiación y ciclofosfam ida: El rég im en p la n ifica d o de co m b in a c ió n de m Ab, rad iac ión y c ic lo fo s fa m id a incluye:
• C ic lo fo s fa m id a 200 m g /m 2 cada 14 d ías (d ía s -1, 14, 28 y 42 de l p rim e r c ic lo de 56 d ías) para un to ta l de 4 dos is m ás
• 27 G y de ra d io te ra p ia (9 G y x 3 ve ce s /sem a n a ; a d m in is tra d a 1 se m a n a d e sp ué s de la p rim e ra d os is de m Ab, p re fe re n te m e n te en d ías no c o n se cu tivo s ) O
• 30 G y de rad io te ra p ia (6 G y x 5 ve ce s /se m a n a ; a d m in is tra d a 1 se m a n a desp ué s de la p rim e ra dos is de m Ab, p re fe re n te m e n te en d ías c o n se cu tivo s ) m ás
• 3 m g/kg de in fus ión de m A b du ra n te 30 m inu tos cada 14 d ías d u ra n te 48 se m a n a s (dos is de m on o te rap ia a d m in is tra d a de 3 m g/kg < DM T; si 3 m g/kg > DM T, la d os is se rá 1 m g/kg )
Variables del estudio
V a ria b le s p rinc ipa les : Las va ria b le s p rinc ipa le s de se g u rid a d inc luyen la in c id e n c ia de TLD , la in c id e n c ia y la g rave d a d de los a co n te c im ie n to s a d ve rso s su rg id o s du ra n te el tra ta m ie n to (A A S T ) y los resu lta d o s de la b o ra to rio a n o rm a le s du ra n te las 48 se m a n a s de tra ta m ie n to .
V a ria b le s s e c u n d a r ia s : Las va ria b le s s e cu n d a ria s c lave inc luyen las s igu ien tes :
• C o n ce n tra c ió n sé rica y fa rm a co c in é tica (FC ) de m Ab
• A c tiv id a d e s a n titu m o ra le s e va lu a d a s según los c rite rio s a d e cu a d o s pa ra la ind icac ión :
o C rite rio s de e va lu a c ió n de la resp u e s ta en tu m o re s só lidos (R E C IS T ), m ed id o s po r to m o g ra fía co m p u ta riza da (TC ) o re so n a n c ia m ag n é tica (R M )
o T am b ié n deben u sa rse o tros c rite rio s de eva lu a c ió n pa ra tu m o re s e sp e c ífico s en los que las m ed ic ion e s R E C IS T no son el es tándar.
o C rite rio s de resp u e s ta re lac ion a d o s con el s is te m a in m un ita rio (C R ri) a p lica d o s a las m e d ic ion e s R E C IS T . En to d os los casos, los C R ri se rán la h e rra m ien ta que rige para d e te rm in a r la p rog res ión de la en fe rm e da d (PE), DE, RC, RP. Los da tos e s tá n d a r de R E C IS T ta m b ié n se recop ila rán con fin e s in fo rm a tivos .
• A n tic u e rp o s an ti-m A b
Procedimientos del estudio
En la se le cc ió n se rea liza rán los s ig u ie n tes p ro ce d im ie n tos con el fin de d e te rm in a r la e leg ib ilid a d para el es tu d io o ca ra c te r iz a r la pob lac ión de re fe renc ia : (i) p -H C G en su e ro (el resu lta d o d ebe s e r < 72 horas an tes de la p rim e ra dosis); (ii) R e co g id a de m a te ria l tu m o ra l a rch ivado : D e spués de que un pac ie n te haya d ado su c o n se n tim ie n to in fo rm ado , se le ped irá al pac ie n te que rea lice las g e s tio ne s n e ce sa ria s para p ro p o rc io n a r las m ue s tra s tu m o ra le s recog idas p re v ia m e n te d ispon ib les ; (iii) RM ce reb ra l: se req u ie re una reso n a n c ia m ag n é tica ce re b ra l en la se lecc ió n si no se rea lizó en los 60 d ías an te rio res ; y (iv ) rad io g ra fía de tó rax: se req u ie re una rad io g ra fía de tó ra x en la se le cc ió n si no se rea lizó en los 60 d ías an te rio res .
P ro ce d im ie n to s de la e ficac ia : S e rea liza rá una to m o g ra fía c o m p u ta r iza d a o reso n a n c ia m ag n é tica para la eva lu a c ió n de l tu m o r en la v is ita de se lecc ió n (de n tro de los 28 d ías a n te rio re s a la in fus ión ) y du ra n te cada c ic lo (a p ro x im a d a m e n te cada 8 se m a n a s) el d ía 56 ± 3, y cu a n do se so sp e che la p rog res ión de la en fe rm edad . A d ic io n a lm e n te , para los p a c ie n te s que no han p ro g re sa do en el es tud io , se rea liza rá una eva lu a c ió n del tu m o r para las v is ita s de se g u im ie n to 3, 5 y 7. U na ve z que se haya d e c id id o u tiliza r una T C o una RM, las e v a lu a c io n e s pos te rio re s se rea liza rán u tiliza n d o la m ism a m oda lidad .
La e va lu a c ió n de la resp u e s ta tu m o ra l se rea liza rá de a cu e rd o con los c rite rio s de resp u e s ta re lac ion a d o s con el s is te m a inm un ita rio (C R ri; N ish ino 2013). Las e va lu a c io n e s de a cu e rd o con los C rite rio s de eva lu a c ió n de la respues ta en tu m o re s só lid o s (R E C IS T ) ve rs ión 1.1 (E ise n h a u e r 2009 ) ta m b ié n se rea liza rán co m o e xp lo ra c ió n de apoyo; sin
embargo, la determinación principal de la progresión de la enfermedad para un paciente individual se realizará de acuerdo con los CRri. Las lesiones medibles seleccionadas como lesiones objetivo para las evaluaciones RECIST también se incluirán como lesiones de referencia para las evaluaciones de los CRri.
Procedimientos de seguridad: Se recogerán las constantes vitales, incluyendo la temperatura, presión arterial en reposo, pulso y respiración. Cuando se programe en la misma visita que otros procedimientos, las constantes vitales deben medirse antes de las evaluaciones de laboratorio clínico, FC o recogida de muestras exploratorias. Durante el ciclo 1, las constantes vitales se registrarán los días de tratamiento antes del tratamiento, al final de la infusión, cada 30 minutos durante las primeras 4 horas posteriores a la infusión y a las 6 y 8 horas posteriores a la administración del fármaco del estudio. En ciclos posteriores, las constantes vitales de los días de tratamiento se evaluarán y documentarán antes de la infusión, cada 30 minutos durante las primeras 2 horas y luego cada hora hasta las 4 horas posteriores a la administración del fármaco del estudio.
Se realizará un examen físico completo o limitado en las visitas. La exploración física completa incluirá un examen de la piel, cabeza, ojos, nariz, garganta, cuello, articulaciones, pulmones, corazón, pulso, abdomen (incluyendo hígado y bazo), ganglios linfáticos y extremidades, así como una breve exploración neurológica. La exploración física limitada incluirá pulmones, corazón, abdomen y piel.
Se realizará un ECG estándar de 12 derivaciones. Cualquier hallazgo de ECG que el investigador considere un cambio clínicamente significativo (empeoramiento) en comparación con el valor inicial se considerará un AA, se registrará y se controlará.
Los ensayos de seguridad inmunitaria consisten en el factor reumatoide (RF), hormona estimulante tiroidea (TSH), proteína C reactiva (PCR) y el título y patrón de anticuerpos antinucleares (ANA). Si, durante el transcurso del estudio, se observa un aumento de 4 veces o más con respecto al valor inicial en RF o ANA o niveles anormales de TSH o CRP, también se pueden realizar las siguientes pruebas: anticuerpos anti-DNA, anticuerpos anti-antígeno A del síndrome de Sjogren (SSA) (Ro), anticuerpos anti-antígeno B del síndrome de Sjogren (SSB) (La), anticuerpos antitiroglobulina, anticuerpos anti-LKM, anticuerpos antifosfolipídicos, anticuerpos anti-células de los islotes, anticuerpos anti-citoplasma de neutrófilos, C3, C4, CH50. El tiempo de tromboplastina parcial activada (aPTT) y el índice internacional normalizado (INR) serán analizados por el laboratorio local del centro.
Seguridad
Un acontecimiento adverso (AA) es cualquier acontecimiento médico no deseado en un paciente al que se le administra un fármaco del estudio que puede tener o no una relación causal con el fármaco del estudio. Por consiguiente, un AA es cualquier signo desfavorable e involuntario (incluido un hallazgo anormal de laboratorio), síntoma o enfermedad asociado que se asocia temporalmente con el uso de un fármaco del estudio, independientemente de si se considera relacionado o no con el fármaco del estudio. Un AA también incluye cualquier empeoramiento (es decir, cualquier cambio clínicamente significativo en la frecuencia y/o intensidad) de una afección preexistente que está temporalmente asociado con el uso del fármaco del estudio. La progresión de la neoplasia maligna subyacente no se considerará un AA si es claramente coherente con el patrón de progresión típico del cáncer subyacente (incluido el curso temporal, los órganos afectados, etc.). Los síntomas clínicos de progresión se pueden notificar como AA si no se puede determinar que el síntoma se debe exclusivamente a la progresión de la neoplasia maligna subyacente o no se ajusta al patrón de progresión esperado para la enfermedad en estudio.
Un acontecimiento adverso grave (AAG) es cualquier acontecimiento médico adverso que a cualquier dosis tiene como resultado la muerte, es potencialmente mortal, requiere hospitalización o prolongación de la hospitalización existente, tiene como resultado una discapacidad/incapacidad persistente o significativa (interrupción sustancial de la capacidad de uno mismo para llevar a cabo las funciones de la vida normal), es una anomalía congénita/defecto congénito.
Se registrará la información del paciente sobre todos los AA y AAG.
Plan estadístico
El aumento de la dosis del estudio se basa en un diseño tradicional 3 3 con 3 a 6 pacientes asignados por nivel de dosis. El número exacto de pacientes incluidos en el estudio dependerá del número de TLD definidas por el protocolo observadas y de la necesidad de ampliar los niveles de dosis definidos actualmente o abrir cohortes adicionales a niveles de dosis más bajos. Después de que se haya producido la inclusión inicial requerida en la siguiente cohorte de aumento de la dosis, la inclusión en cada una de las cohortes previas anteriores por debajo de la DMT para ese tratamiento se ampliará (si no se amplió previamente durante el aumento de la dosis) hasta un total de 6 pacientes.
Los datos se resumirán utilizando únicamente estadísticos descriptivos. En general, los datos se resumirán por niveles de dosis y combinaciones. Los resúmenes y análisis de seguridad se realizarán en el conjunto de análisis de seguridad (CAS). El análisis principal de seguridad se basará en AA surgidos durante el tratamiento (AAST).