EP1707563A2 - Methods and compositions utilizing quinazolinones - Google Patents
Methods and compositions utilizing quinazolinones Download PDFInfo
- Publication number
- EP1707563A2 EP1707563A2 EP06075276A EP06075276A EP1707563A2 EP 1707563 A2 EP1707563 A2 EP 1707563A2 EP 06075276 A EP06075276 A EP 06075276A EP 06075276 A EP06075276 A EP 06075276A EP 1707563 A2 EP1707563 A2 EP 1707563A2
- Authority
- EP
- European Patent Office
- Prior art keywords
- substituted
- alkyl
- chosen
- hydrogen
- aryl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
- 0 C*(C(*)(*)/C(/*1*)=*/CCc(c(*)c(c(*)c2O)I)c2C1=O)[N+](*)[O-] Chemical compound C*(C(*)(*)/C(/*1*)=*/CCc(c(*)c(c(*)c2O)I)c2C1=O)[N+](*)[O-] 0.000 description 7
Images

























































































Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/86—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in position 4
- C07D239/88—Oxygen atoms
- C07D239/90—Oxygen atoms with acyclic radicals attached in position 2 or 3
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/86—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in position 4
- C07D239/88—Oxygen atoms
- C07D239/91—Oxygen atoms with aryl or aralkyl radicals attached in position 2 or 3
Definitions
- This invention relates to quinazolinone derivatives, which are inhibitors of the mitotic kinesin KSP and are useful in the treatment of cellular proliferative diseases, for example cancer, hyperplasias, restenosis, cardiac hypertrophy, immune disorders and inflammation.
- Quinazolinones and derivatives thereof are now known to have a wide variety of biological properties including hypnotic, sedative, analgesic, anticonvulsant, antitussive and anti-inflammatory activities.
- Quinazolinone derivatives for which specific biological uses have been described include U.S. Patent No. 5,147,875 describing 2-(substituted phenyl)-4-oxo quinazolines with bronchodilator activity.
- U.S. Patent Nos. 3,723,432, 3,740,442, and 3,925,548 describe a class of 1 -substituted-4-aryl-2(1H)-quinazolinone derivatives useful as anti-inflammatory agents.
- European patent publication EP 0 056 637 B1 claims a class of 4(3H)-quinazolinone derivatives for the treatment of hypertension.
- European patent publication EP 0 884 319 A1 describes pharmaceutical compositions of quinazolin-4-one derivatives used to treat neurodegenerative, psychotropic, and drug and alcohol induced central and peripheral nervous system disorders.
- Quinazolinones are among a growing number of therapeutic agents used to treat cell proliferative disorders, including cancer.
- PCT WO 96/06616 describes a pharmaceutical composition containing a quinazolinone derivative to inhibit vascular smooth cell proliferation.
- PCT WO 96/19224 uses this same quinazolinone derivative to inhibit mesengial cell proliferation.
- U.S. Patent Nos. 4,981,856, 5,081,124 and 5,280,027 describe the use of quinazolinone derivatives to inhibit thymidylate synthase, the enzyme that catalyzes the methylation of deoxyuridine monophosphate to produce thymidine monophosphate which is required for DNA synthesis.
- Taxanes and vinca alkaloids act on microtubules, which are present in a variety of cellular structures.
- Microtubules are the primary structural element of the mitotic spindle. The mitotic spindle is responsible for distribution of replicate copies of the genome to each of the two daughter cells that result from cell division. It is presumed that disruption of the mitotic spindle by these drugs results in inhibition of cancer cell division, and induction of cancer cell death.
- microtubules form other types of cellular structures, including tracks for intracellular transport in nerve processes. Because these agents do not specifically target mitotic spindles, they have side effects that limit their usefulness.
- Mitotic kinesins are enzymes essential for assembly and function of the mitotic spindle, but are not generally part of other microtubule structures, such as in nerve processes. Mitotic kinesins play essential roles during all phases of mitosis. These enzymes are "molecular motors" that transform energy released by hydrolysis of ATP into mechanical force which drives the directional movement of cellular cargoes along microtubules. The catalytic domain sufficient for this task is a compact structure of approximately 340 amino acids. During mitosis, kinesins organize microtubules into the bipolar structure that is the mitotic spindle.
- Kinesins mediate movement of chromosomes along spindle microtubules, as well as structural changes in the mitotic spindle associated with specific phases of mitosis.
- Experimental perturbation of mitotic kinesin function causes malformation or dysfunction of the mitotic spindle, frequently resulting in cell cycle arrest and cell death.
- KSP belongs to an evolutionarily conserved kinesin subfamily of plus end-directed microtubule motors that assemble into bipolar homotetramers consisting of antiparallel homodimers.
- KSP associates with microtubules of the mitotic spindle.
- Microinjection of antibodies directed against KSP into human cells prevents spindle pole separation during prometaphase, giving rise to monopolar spindles and causing mitotic arrest and induction of programmed cell death.
- KSP and related kinesins in other, non-human, organisms bundle antiparallel microtubules and slide them relative to one another, thus forcing the two spindle poles apart.
- KSP may also mediate in anaphase B spindle elongation and focussing of microtubules at the spindle pole.
- HsEg5 Human KSP (also termed HsEg5) has been described [Blangy, et al., Cell, 83:1159-69 (1995); Whitehead, et al., Arthritis Rheum., 39:1635-42 (1996); Galgio et al., J. Cell Biol., 135:339-414 (1996); Blangy, et al., J Biol. Chem., 272:19418-24 (1997); Blangy, et al., Cell Motil Cytoskeleton, 40:174-82 (1998); Whitehead and Rattner, J.
- Mitotic kinesins are attractive targets for the discovery and development of novel mitotic chemotherapeutics. Accordingly, it is an object of the present invention to provide methods and compositions useful in the inhibition of KSP, a mitotic kinesin.
- compositions and methods that can be used to treat diseases of proliferating cells.
- the compositions are KSP inhibitors, particularly human KSP inhibitors.
- the invention relates to methods for treating cellular proliferative diseases, for treating disorders associated with KSP kinesin activity, and for inhibiting KSP kinesin.
- the methods employ compounds chosen from the group consisting of: wherein:
- Diseases and disorders that respond to therapy with compounds of the invention include cancer, hyperplasia, restenosis, cardiac hypertrophy, immune disorders and inflammation; especially cancer, hyperplasia, restenosis, and cardiac hypertrophy; particularly cancer.
- the invention relates to compounds useful in inhibiting KSP kinesin.
- the compounds have the structures shown above.
- the present invention provides methods of screening for compounds that will bind to a KSP kinesin, for example compounds that will displace or compete with the binding of the compositions of the invention.
- the methods comprise combining a labeled compound of the invention, a KSP kinesin, and at least one candidate agent and determining the binding of the candidate bioactive agent to the KSP kinesin.
- the invention provides methods of screening for modulators of KSP kinesin activity.
- the methods comprise combining a composition of the invention, a KSP kinesin, and at least one candidate agent and determining the effect of the candidate bioactive agent on the KSP kinesin activity.
- the present invention is directed to a class of novel compounds, based on a core quinazolinone structure, that are modulators of mitotic kinesins.
- mitotic kinesins By inhibiting or modulating mitotic kinesins, but not other kinesins (e.g., transport kinesins), specific inhibition of cellular proliferation is accomplished.
- the present invention capitalizes on the finding that perturbation of mitotic kinesin function causes malformation or dysfunction of mitotic spindles, frequently resulting in cell cycle arrest and cell death.
- the methods of inhibiting a human KSP kinesin comprise contacting an inhibitor of the invention with a KSP kinesin, particularly human KSP kinesins, including fragments and variants of KSP.
- the inhibition can be of the ATP hydrolysis activity of the KSP kinesin and/or the mitotic spindle formation activity, such that the mitotic spindles are disrupted. Meiotic spindles may also be disrupted.
- An object of the present invention is to develop inhibitors and modulators of mitotic kinesins, in particular KSP, for the treatment of disorders associated with cell proliferation.
- KSP mitotic kinesins
- An object of the present invention is to develop inhibitors and modulators of mitotic kinesins, in particular KSP, for the treatment of disorders associated with cell proliferation.
- dramatic improvements in the treatment of cancer, one type of cell proliferative disorder have been associated with identification of therapeutic agents acting through novel mechanisms. Examples of this include not only the taxane class of agents that appear to act on microtubule formation, but also the camptothecin class of topoisomerase I inhibitors.
- the compositions and methods described herein can differ in their selectivity and are preferably used to treat diseases of proliferating cells, including, but not limited to cancer, hyperplasias, restenosis, cardiac hypertrophy, immune disorders and inflammation.
- the present invention relates to methods employing quinazolinone amides of formula 1a: quinazolinone sulfonamides of formula 1b and quinazolinone amines of formulae 1c and 1d wherein:
- optionally substituted alkyl means either “alkyl” or “substituted alkyl,” as defined below. It will be understood by those skilled in the art with respect to any group containing one or more substituents that such groups are not intended to introduce any substitution or substitution patterns (e.g., substituted alkyl includes optionally substituted cycloalkyl groups, which in turn are defined as including optionally substituted alkyl groups, potentially ad infinitum ) that are sterically impractical and/or synthetically non-feasible.
- Alkyl is intended to include linear, branched, or cyclic hydrocarbon structures and combinations thereof.
- Lower alkyl refers to alkyl groups of from 1 to 5 carbon atoms. Examples of lower alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, s-and t-butyl and the like.
- Preferred alkyl groups are those of C 20 or below. More preferred alkyl groups are those of C 13 or below.
- Cycloalkyl is a subset of alkyl and includes cyclic hydrocarbon groups of from 3 to 13 carbon atoms.
- cycloalkyl groups include c-propyl, c-butyl, c-pentyl, norbomyl, adamantyl and the like.
- alkyl refers to alkanyl, as well as the unsaturated alkenyl and alkynyl residues; it is intended to include cyclohexylmethyl, vinyl, allyl, isoprenyl and the like.
- Alkylene refers to the same residues as alkyl, but having two points of attachment.
- butyl is meant to include n-butyl, sec-butyl, isobutyl and t-butyl;
- propyl includes n-propyl and isopropyl (or “i-propyl”).
- Alkoxy or alkoxyl refers to groups of from 1 to 8 carbon atoms of a straight, branched, cyclic configuration and combinations thereof attached to the parent structure through an oxygen. Examples include methoxy, ethoxy, propoxy, isopropoxy, cyclopropyloxy, cyclohexyloxy and the like. Lower-alkoxy refers to groups containing one to four carbons.
- Acyl refers to groups of from 1 to 8 carbon atoms of a straight, branched, cyclic configuration, saturated, unsaturated and aromatic and combinations thereof, attached to the parent structure through a carbonyl functionality.
- One or more carbons in the acyl residue may be replaced by nitrogen, oxygen or sulfur as long as the point of attachment to the parent remains at the carbonyl. Examples include acetyl, benzoyl, propionyl, isobutyryl, t-butoxycarbonyl, benzyloxycarbonyl and the like.
- Lower-acyl refers to groups containing one to four carbons.
- Aryl and heteroaryl mean a 5- or 6-membered aromatic or heteroaromatic ring containing 0-3 heteroatoms selected from O, N, or S; a bicyclic 9- or 10-membered aromatic or heteroaromatic ring system containing 0-3 heteroatoms selected from O, N, or S; or a tricyclic 13- or 14-membered aromatic or heteroaromatic ring system containing 0-3 heteroatoms selected from O, N, or S.
- the aromatic 6- to 14-membered carbocyclic rings include, e.g., benzene, naphthalene, indane, tetralin, and fluorene and the 5- to 10-membered aromatic heterocyclic rings include, e.g., imidazole, pyridine, indole, thiophene, benzopyranone, thiazole, furan, benzimidazole, quinoline, isoquinoline, quinoxaline, pyrimidine, pyrazine, tetrazole and pyrazole.
- Alkylaryl refers to a residue in which an aryl moiety is attached to the parent structure via an alkylene residue. Examples are benzyl, phenethyl, phenylvinyl, phenylallyl and the like.
- Oxaalkyl and oxaalkylaryl refer to alkyl and alkylaryl residues in which one or more methylenes have been replaced by oxygen. Examples of oxaalkyl and oxaalkylaryl residues are ethoxyethoxyethyl (3,6-dioxaoctyl), benzyloxymethyl and phenoxymethyl; in general, glycol ethers, such as polyethyleneglycol, are intended to be encompassed by this group.
- Alkylheteroaryl refers to a residue in which a heteroaryl moiety is attached to the parent structure via an alkylene residue. Examples include furanylmethyl, pyridinylmethyl, pyrimidinylethyl and the like.
- Heterocycle means a cycloalkyl or aryl residue in which one to four of the carbons is replaced by a heteroatom such as oxygen, nitrogen or sulfur.
- heterocycles that fall within the scope of the invention include imidazoline, pyrrolidine, pyrazole, pyrrole, indole, quinoline, isoquinoline, tetrahydroisoquinoline, benzofuran, benzodioxan, benzodioxole (commonly referred to as methylenedioxyphenyl, when occurring as a substituent), tetrazole, morpholine, thiazole, pyridine, pyridazine, pyrimidine, thiophene, furan, oxazole, oxazoline, isoxazole, dioxane, tetrahydrofuran and the like.
- N-heterocyclyl refers to a nitrogen-containing heterocycle as a substituent residue.
- the term heterocyclyl encompasses heteroaryl, which is a subset of heterocyclyl.
- Examples of N-heterocyclyl residues include 4-morpholinyl, 4-thiomorpholinyl, 1-piperidinyl, 1-pyrrolidinyl, 3-thiazolidinyl, piperazinyl and 4-(3,4-dihydrobenzoxazinyl).
- substituted heterocyclyl include 4-methyl-1-piperazinyl and 4-benzyl-1-piperidinyl.
- Substituted alkyl, aryl and heteroaryl refer to alkyl, aryl or heteroaryl wherein one or more H atoms are replaced with alkyl, halogen, hydroxy, alkoxy, alkylenedioxy (e.g., methylenedioxy), fluoroalkyl, carboxy (-COOH), carboalkoxy (i.e., acyloxy -O(O)CR), carboxyalkyl (i.e., esters -C(O)OR), carboxamido, sulfonamidoalkyl, sulfonamidoaryl, aminocarbonyl, benzyloxycarbonylamino (CBZ-amino), cyano, carbonyl, nitro, primary-, secondary- and tertiary-amino (e.g., alkylamino and dialkylamino) and aminoaklylene, alkylthio, alkylsulfinyl, alkyl
- substituted alkyl also includes oxaalkyl residues, i.e. alkyl residues in which one or more carbons has been replaced by oxygen.
- Substituted alkylaryl and substituted oxaalkylaryl refer to residues where either or both of the alkylene and aryl moieties are substituted.
- Substituted alkylheteroaryl and substituted oxaalkylheteroaryl residues where either or both of the alkylene and heteroaryl moieties are substituted. It should additionally be noted that certain positions may contain two or even three substitution groups, R, R' and R" (e.g., diethylamino and trifluoromethyl).
- Halogen refers to fluorine, chlorine, bromine or iodine. Fluorine, chlorine and bromine are preferred.
- Dihaloaryl, dihaloalkyl, trihaloaryl, etc. refer to aryl and alkyl substituted with a plurality of halogens, but not necessarily a plurality of the same halogen; thus 4-chloro-3-fluorophenyl is within the scope of dihaloaryl.
- Most of the compounds described herein contain one or more asymmetric centers (e.g. the carbon to which R 2 and R 2 ' are attached) and may thus give rise to enantiomers, diastereomers, and other stereoisomeric forms that may be defined, in terms of absolute stereochemistry, as (R)- or (S)-.
- the present invention is meant to include all such possible isomers, including racemic mixtures, optically pure forms and intermediate mixtures.
- Optically active (R)- and (S)- isomers may be prepared using chiral synthons or chiral reagents, or resolved using conventional techniques.
- the compounds described herein contain olefinic double bonds or other centers of geometric asymmetry, and unless specified otherwise, it is intended that the compounds include both E and Z geometric isomers. Likewise, all tautomeric forms are also intended to be included.
- the R- and S-isomers may be resolved by methods known to those skilled in the art, for example by formation of diastereoisomeric salts or complexes which may be separated, for example, by crystallisation; via formation of diastereoisomeric derivatives which may be separated, for example, by crystallisation, gas-liquid or liquid chromatography; selective reaction of one enantiomer with an enantiomer-specific reagent, for example enzymatic oxidation or reduction, followed by separation of the modified and unmodified enantiomers; or gas-liquid or liquid chromatography in a chiral environment, for example on a chiral support, such as silica with a bound chiral ligand or in the presence of a chiral solvent.
- a chiral support such as silica with a bound chiral ligand or in the presence of a chiral solvent.
- the two adjacent R 2 groups may be fused together to form a ring structure.
- the fused ring structure may contain heteroatoms and may be substituted with one or more substitution groups "R".
- R substitution groups
- certain positions may contain two substitution groups, R and R'.
- R 1 is selected from hydrogen, alkyl, aryl, substituted alkyl, substituted aryl, heteroaryl, substituted heteroaryl, alkylaryl and substituted alkylaryl.
- R 1 is selected from hydrogen, lower alkyl,substituted lower alkyl, benzyl, substituted benzyl, phenyl, naphthyl and substituted phenyl.
- R 1 is chosen from hydrogen, ethyl, propyl, methoxyethyl, naphthyl, phenyl, bromophenyl, chlorophenyl, methoxyphenyl, ethoxyphenyl, tolyl, dimethylphenyl, chorofluorophenyl, methylchlorophenyl, ethylphenyl, phenethyl, benzyl, chlorobenzyl, methylbenzyl, methoxybenzyl, cyanobenzyl, hydroxybenzyl, tetrahydrofuranylmethyl and (ethoxycarbonyl)ethyl.
- R 2 is hydrogen, alkyl,cycloalkyl or substituted alkyl.
- Formulae 1a, 1b, 1c and 1d possess a potentially chiral center at the carbon to which R 2 is attached.
- the R 2 position may comprise two substitution groups, R 2 and R 2 '.
- the R 2 and R 2 ' groups may be the same or different; if different, the composition is chiral.
- preferred embodiments utilize only a single non-hydrogen R 2 .
- the invention contemplates the use of pure enantiomers and mixtures of enantiomers, including racemic mixtures, although the use of the substantially optically pure eutomer will generally be preferred, particularly the R enantiomer.
- R 2 is chosen from hydrogen, lower alkyl and substituted lower alkyl, and R 2 ' is hydrogen.
- R 2 is chosen from hydrogen, methyl, ethyl, propyl (particularly i -propyl), butyl (particularly t-butyl), methylthioethyl, aminobutyl, (CBZ)aminobutyl, cyclohexylmethyl, benzyloxymethyl, methylsulfinylethyl, methylsulfinylmethyl, hydroxymethyl, benzyl and indolylmethyl.
- R enantiomer where R 2 is i -propyl.
- R 3 is selected from chosen from alkyl, substituted alkyl, alkylaryl, heteroaryl, aryl, substituted aryl, substituted oxaalkylaryl, -O-R 15 and -NH-R 15 , and R 15 is chosen from alkyl, aryl and substituted aryl.
- R 3 when R 3 is not -NHR 15 , R 3 is chosen from C 1 -C 13 alkyl; substituted lower alkyl;aryl, including phenyl, biphenyl and naphthyl; substituted aryl, including phenyl substituted with one or more halo, lower alkyl, loweralkoxy, nitro, carboxy, methylenedioxy or trifluoromethyl; benzyl; phenoxymethyl; halophenoxymethyl; phenylvinyl; heteroaryl; heteroaryl substituted with lower alkyl; and benzyloxymethyl.
- R 3 is chosen from ethyl, propyl, chloropropyl, butoxy, heptyl, butyl, octyl, tridecanyl, (ethoxycarbonyl)ethyl, dimethylaminoethyl, dimethylaminomethyl, phenyl, naphthyl, halophenyl, dihalophenyl, cyanophenyl, halo(trifluoromethyl)phenyl, chlorophenoxymethyl, methoxyphenyl, carboxyphenyl, ethylphenyl, tolyl, biphenyl, methylenedioxyphenyl, methylsulfonylphenyl, methoxychlorophenyl, chloronaphthyl, methylhalophenyl, trifluoromethylphenyl, butylphenyl, pentylphenyl, methylnitrophen
- R 15 is chosen from lower alkyl; cyclohexyl; phenyl; and phenyl substituted with halo, lower alkyl, loweralkoxy, or lower alkylthio.
- R 15 is isopropyl, butyl, cyclohexyl, phenyl, bromophenyl, dichlorophenyl, methoxyphenyl, ethylphenyl, tolyl, trifluoromethylphenyl or methylthiophenyl.
- R 4 is chosen from alkyl, aryl, alkylaryl, alkylheteroaryl, substituted alkyl, substituted aryl, and -alkylene-R 16 , and R 16 is chosen from alkoxy, amino, alkylamino, dialkylamino and N-heterocyclyl.
- R 4 is selected from lower alkyl, substituted lower alkyl, cyclohexyl; phenyl substituted with hydroxy, lower alkoxy or lower alkyl; benzyl; heteroarylmethyl; heteroarylethyl; heteroarylpropyl and -alkylene-R 16 , wherein R 16 is amino, lower alkylamino, di(lower alkyl)amino, lower alkoxy, or N-heterocyclyl.
- R 4 is chosen from methyl, ethyl, propyl, butyl, cyclohexyl, carboxyethyl, carboxymethyl, methoxyethyl, hydroxyethyl, hydroxypropyl, dimethylaminoethyl, dimethylaminopropyl, diethylaminoethyl, diethylaminopropyl, aminopropyl, methylaminopropyl, 2,2-dimethyl-3-(dimethylamino)propyl, 1-cyclohexyl-4-(diethylamino)butyl, aminoethyl, aminobutyl, aminopentyl, aminohexyl, aminoethoxyethyl, isopropylaminopropyl, diisopropylaminoethyl, 1-methyl-4-(diethylamino)butyl, (t-Boc)aminopropyl, hydroxyphenyl,
- R 5 , R 6 , R 7 and R 8 are chosen from hydrogen, halo (particularly chloro and fluoro), lower alkyl (particularly methyl), substituted lower alkyl (particularly trifluoromethyl), lower alkoxy (particularly methoxy), and cyano; more preferably from hydrogen and halo.
- R 5 is hydrogen or halo
- R 6 is hydrogen, methyl or halo
- R 7 is hydrogen, halo, alkyl (particularly methyl), alkoxy (particularly methoxy) or cyano
- R 8 is hydrogen or halo.
- Still further preferred are the compounds where only one of R 5 , R 6 , R 7 and R 8 is not hydrogen, especially R 7 .
- R 1 is benzyl or halobenzyl
- R 2 is chosen from ethyl and propyl
- R 2 ' is hydrogen
- R 3 (or R 3' or R 3" ) is substituted phenyl
- R 4 is - (CH 2 ) m OH where m is two or three, or -(CH 2 ) p- R 16 where p is one to three and R 16 is amino, propylamino or azetidinyl
- R 5 is hydrogen
- R 6 is hydrogen
- R 7 is halo
- R 8 is hydrogen.
- R 1 is preferably chosen from hydrogen, lower alkyl, substituted lower alkyl, benzyl, substituted benzyl, phenyl and substituted phenyl;
- R 2 is chosen from hydrogen and lower alkyl and R 2 ' is hydrogen;
- R 3' is chosen from C 1 -C 13 alkyl; phenyl; naphthyl; phenyl substituted with halo, lower alkyl, lower alkoxy, nitro, methylenedioxy, or trifluoromethyl; biphenylyl and heteroaryl; and R 4 is chosen from lower alkyl, cyclohexyl; phenyl substituted with hydroxy, lower alkoxy or lower alkyl; benzyl; heteroarylmethyl; heteroarylethyl; heteroarylpropyl; heteroarylethyl; heteroarylpropyl and -alkylene-R 16 , wherein R 16 is di(lower alkyl)
- R 1 is most preferably chosen from lower alkyl, benzyl, substituted benzyl and substituted phenyl;
- R 2 is hydrogen or lower alkyl;
- R 2 ' is hydrogen;
- R 3 is chosen from substituted phenyl and naphthyl;
- R 4 is -alkylene-R 16 ;
- R 7 is hydrogen, fluoro, methyl or chloro;
- R 5 , R 6 and R 8 are hydrogen; and
- R 16 is chosen from di(lower alkylamino), (lower alkyl)amino, amino, pyrrolidino, piperidino, imidazolyl and morpholino.
- R 1 is preferably chosen from hydrogen, lower alkyl, substituted lower alkyl, benzyl, substituted benzyl, phenyl, naphthyl and substituted phenyl;
- R 2 is chosen from hydrogen, lower alkyl and substituted lower alkyl and R 2 ' is hydrogen;
- R 3" is chosen from C 1 -C 13 alkyl; substituted lower alkyl; phenyl; naphthyl; phenyl substituted with halo, lower alkyl, lower alkoxy, nitro, methylenedioxy, or trifluoromethyl; biphenylyl, benzyl and heterocyclyl; and
- R 4 is chosen from lower alkyl; cyclohexyl; phenyl substituted with hydroxy, lower alkoxy or lower alkyl; benzyl; substituted benzyl; heterocyclyl; heteroarylmethyl; heteroarylethyl; heteroarylpropyl
- R 1 is most preferably chosen from lower alkyl, benzyl, substituted benzyl and substituted phenyl;
- R 2 is hydrogen or lower alkyl;
- R 2 ' is hydrogen;
- R 3" is chosen from substituted phenyl, heterocyclyl and naphthyl;
- R 4 is chosen from subtituted benzyl, heterocyclyl and -alkylene-R 16 ;
- R 6 and R 7 are chosen from hydrogen and halo;
- R 5 and R 8 are hydrogen; and
- R 16 is chosen from di(lower alkylamino), (lower alkyl)amino, amino, pyrrolidinyl, piperidinyl, imidazolyl and morpholinyl.
- R 3" is present (as in 1d) it is most preferably chosen from halophenyl, polyhalophenyl, tolyl, dimethylphenyl, methoxyphenyl, dimethoxyphenyl, cyanophenyl, trifluoromethylphenyl, trifluoromethoxyphenyl, bis(trifluoromethyl)phenyl, carboxyphenyl, t-butylphenyl, methoxycarbonylphenyl, piperidinyl and naphthyl.
- R 1 is alkylaryl or substituted alkylaryl (particularly benzyl or substituted benzyl), R 2 is lower alkyl (particularly i-propyl), R 2 ' is hydrogen, R 3 is substituted aryl (particularly methyl-and/or halo-substituted phenyl), R 4 is substituted alkyl (particularly 3-amino-n-propyl), R 5 , R 6 and R 8 are hydrogen, and R 7 is halo or cyano (particularly chloro), where the stereogenic center to which R 2 and R 2 ' are attached is of the R configuration.
- compositions of the invention are synthesized as outlined below, utilizing techniques well known in the art. For example, as described in Ager et al., J. of Med. Chem., 20:379-386 (1977), hereby incorporated by reference, quinazolinones can be obtained by acid-catalyzed condensation of N-acylanthranilic acids with aromatic primary amines. Other processes for preparing quinazolinones are described in U.S. Patent applications 5,783,577, 5,922,866 and 5,187,167, all of which are incorporated by reference.
- the compositions of the invention may be made as shown in Figures 1, 2, 4 and 5. Compounds of formulae 1d are made in analogous fashion to Figure 1, except that the acyl halide in the final step is replaced by an alkyl halide.
- compositions of the invention find use in a variety of applications.
- mitosis may be altered in a variety of ways; that is, one can affect mitosis either by increasing or decreasing the activity of a component in the mitotic pathway. Stated differently, mitosis may be affected (e.g., disrupted) by disturbing equilibrium, either by inhibiting or activating certain components. Similar approaches may be used to alter meiosis.
- compositions of the invention are used to modulate mitotic spindle formation, thus causing prolonged cell cycle arrest in mitosis.
- modulate herein is meant altering mitotic spindle formation, including increasing and decreasing spindle formation.
- mitotic spindle formation herein is meant organization of microtubules into bipolar structures by mitotic kinesins.
- mitotic spindle dysfunction herein is meant mitotic arrest and monopolar spindle formation.
- compositions of the invention are useful to bind to and/or modulate the activity of a mitotic kinesin, KSP.
- the KSP is human KSP, although KSP kinesins from other organisms may also be used.
- modulate means either increasing or decreasing spindle pole separation, causing malformation, i.e., splaying, of mitotic spindle poles, or otherwise causing morphological perturbation of the mitotic spindle.
- variants and/or fragments of KSP See U.S. Patent Application "Methods of Screening for Modulators of Cell Proliferation and Methods of Diagnosing Cell Proliferation States", filed Oct. 27, 1999 (U.S. Serial Number 09/428,156), hereby incorporated by reference in its entirety.
- other mitotic kinesins may be used in the present invention.
- the compositions of the invention have been shown to have specificity for KSP.
- KSP or a compound according to the invention is non-diffusably bound to an insoluble support having isolated sample receiving areas (e.g., a microtiter plate, an array, etc.).
- the insoluble support may be made of any composition to which the compositions can be bound, is readily separated from soluble material, and is otherwise compatible with the overall method of screening.
- the surface of such supports may be solid or porous and of any convenient shape.
- suitable insoluble supports include microtiter plates, arrays, membranes and beads. These are typically made of glass, plastic (e.g., polystyrene), polysaccharides, nylon or nitrocellulose, TeflonTM, etc.
- Microtiter plates and arrays are especially convenient because a large number of assays can be carried out simultaneously, using small amounts of reagents and samples.
- the particular manner of binding of the composition is not crucial so long as it is compatible with the reagents and overall methods of the invention, maintains the activity of the composition and is nondiffusable.
- Preferred methods of binding include the use of antibodies (which do not sterically block either the ligand binding site or activation sequence when the protein is bound to the support), direct binding to "sticky" or ionic supports, chemical crosslinking, the synthesis of the protein or agent on the surface, etc. Following binding of the protein or agent, excess unbound material is removed by washing. The sample receiving areas may then be blocked through incubation with bovine serum albumin (BSA), casein or other innocuous protein or other moiety.
- BSA bovine serum albumin
- the antimitotic agents of the invention may be used on their own to modulate the activity of a mitotic kinesin, particularly KSP.
- the mitotic agents of the invention are combined with KSP and the activity of KSP is assayed.
- Kinesin activity is known in the art and includes one or more kinesin activities. Kinesin activities include the ability to affect ATP hydrolysis; microtubule binding; gliding and polymerization/depolymerization (effects on microtubule dynamics); binding to other proteins of the spindle; binding to proteins involved in cell-cycle control; serving as a substrate to other enzymes; such as kinases or proteases; and specific kinesin cellular activities such as spindle pole separation.
- ATPase hydrolysis activity assay utilizes 0.3 M PCA (perchloric acid) and malachite green reagent (8.27 mM sodium molybdate II, 0.33 mM malachite green oxalate, and 0.8 mM Triton X-1 00).
- ATPase activity ofkinesin motor domains also can be used to monitor the effects of modulating agents.
- ATPase assays of kinesin are performed in the absence of microtubules.
- the ATPase assays are performed in the presence of microtubules.
- Different types of modulating agents can be detected in the above assays.
- the effect of a modulating agent is independent of the concentration of microtubules and ATP.
- the effect of the agents on kinesin ATPase can be decreased by increasing the concentrations of ATP, microtubules or both.
- the effect of the modulating agent is increased by increasing concentrations of ATP, microtubules or both.
- Agents that modulate the biochemical activity of KSP in vitro may then be screened in vivo.
- Methods for such agents in vivo include assays of cell cycle distnbution, cell viability, or the presence, morphology, activity, distribution, or amount of mitotic spindles.
- Methods for monitoring cell cycle distribution of a cell population, for example, by flow cytometry are well known to those skilled in the art, as are methods for determining cell viability. See for example, U.S. Patent Application "Methods of Screening for Modulators of Cell Proliferation and Methods of Diagnosing Cell Proliferation States," filed Oct. 22, 1999, serial number 09/428,156, hereby incorporated by reference in its entirety.
- compositions of the invention inhibit the KSP kinesin.
- One measure of inhibition is IC 50 , defined as the concentration of the composition at which the activity of KSP is decreased by fifty percent.
- Preferred compositions have IC 50 's of less than about 1 mM, with preferred embodiments having IC 50 's of less than about 100 ⁇ M, with more preferred embodiments having IC 50 's of less than about 10 ⁇ M, with particularly preferred embodiments having IC 50 's of less than about 1 ⁇ M, and especially preferred embodiments having IC 50 's of less than about 100 nM, and with the most preferred embodiments having IC 50 's of less than about 10 nM.
- Measurement of IC 50 is done using an ATPase assay.
- K i Another measure of inhibition is K i .
- the K i or K d is defined as the dissociation rate constant for the interaction of the quinazolinone with KSP.
- Preferred compounds have K i 's of less than about 100 ⁇ M, with preferred embodiments having K i 's of less than about 10 ⁇ M, and particularly preferred embodiments having K i 's of less than about 1 ⁇ M and especially preferred embodiments having K i 's of less than about 100 nM, and with the most preferred embodiments having K i 's of less than about 10 nM.
- the K i for a compound is determined from the IC 50 based on three assumptions.
- V is the observed rate
- V max is the rate of the free enzyme
- I 0 is the inhibitor concentration
- E 0 is the enzyme concentration
- K d is the dissociation constant of the enzyme-inhibitor complex.
- GI 50 defined as the concentration of the compound that results in a decrease in the rate of cell growth by fifty percent.
- Preferred compounds have GI 50 's of less than about 1 mM.
- the level of preferability of embodiments is a function of their GI 50 : those having GI 50 's of less than about 20 ⁇ M are more preferred; those having GI 50 's of 10 ⁇ M more so; those having GI 50 of less than about 1 ⁇ M more so; those having GI 50 's of 100 nM more so; those having GI 50 of less than about 10 nM even more so.
- Measurement of GI 50 is done using a cell proliferation assay.
- compositions of the invention are used to treat cellular proliferation diseases.
- Disease states which can be treated by the methods and compositions provided herein include, but are not limited to, cancer (further discussed below), autoimmune disease, arthritis, graft rejection, inflammatory bowel disease, proliferation induced after medical procedures, including, but not limited to, surgery, angioplasty, and the like. It is appreciated that in some cases the cells may not be in a hyper or hypo proliferation state (abnormal state) and still require treatment. For example, during wound healing, the cells may be proliferating "normally", but proliferation enhancement may be desired.
- cells may be in a "normal" state, but proliferation modulation may be desired to enhance a crop by directly enhancing growth of a crop, or by inhibiting the growth of a plant or organism which adversely affects the crop.
- the invention herein includes application to cells or individuals afflicted or impending affliction with any one of these disorders or states.
- compositions and methods provided herein are particularly deemed useful for the treatment of cancer including solid tumors such as skin, breast, brain, cervical carcinomas, testicular carcinomas, etc. More particularly, cancers that may be treated by the compositions and methods of the invention include, but are not limited to: Cardiac: sarcoma (angiosarcoma, fibrosarcoma, rhabdomyosarcoma, liposarcoma), myxoma, rhabdomyoma, fibroma, lipoma and teratoma; Lung : bronchogenic carcinoma (squamous cell, undifferentiated small cell, undifferentiated large cell, adenocarcinoma), alveolar (bronchiolar) carcinoma, bronchial adenoma, sarcoma, lymphoma, chondromatous hamartoma, mesothelioma; Gastrointestinal: esophagus (squamous cell carcinoma, adenocar
- compositions of the invention are administered to cells.
- administered herein is meant administration of a therapeutically effective dose of the mitotic agents of the invention to a cell either in cell culture or in a patient.
- therapeutically effective dose herein is meant a dose that produces the effects for which it is administered. The exact dose will depend on the purpose of the treatment, and will be ascertainable by one skilled in the art using known techniques. As is known in the art, adjustments for systemic versus localized delivery, age, body weight, general health, sex, diet, time of administration, drug interaction and the severity of the condition may be necessary, and will be ascertainable with routine experimentation by those skilled in the art.
- cells herein is meant almost any cell in which mitosis or meiosis can be altered.
- a "patient” for the purposes of the present invention includes both humans and other animals, particularly mammals, and other organisms. Thus the methods are applicable to both human therapy and veterinary applications.
- the patient is a mammal, and in the most preferred embodiment the patient is human.
- Mitotic agents having the desired pharmacological activity may be administered in a physiologically acceptable carrier to a patient, as described herein.
- the compounds may be formulated in a variety of ways as discussed below.
- the concentration of therapeutically active compound in the formulation may vary from about 0.1-100 wt.%.
- the agents may be administered alone or in combination with other treatments, i.e., radiation, or other chemotherapeutic agents.
- the pharmaceutical compositions are in a water soluble form, such as pharmaceutically acceptable salts, which is meant to include both acid and base addition salts.
- “Pharmaceutically acceptable acid addition salt” refers to those salts that retain the biological effectiveness of the free bases and that are not biologically or otherwise undesirable, formed with inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid and the like, and organic acids such as acetic acid, propionic acid, glycolic acid, pyruvic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, p-toluenesulfonic acid, salicylic acid and the like.
- “Pharmaceutically acceptable base addition salts” include those derived from inorganic bases such as sodium, potassium, lithium, ammonium, calcium, magnesium, iron, zinc, copper, manganese, aluminum salts and the like. Particularly preferred are the ammonium, potassium, sodium, calcium, and magnesium salts.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines and basic ion exchange resins, such as isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine, and ethanolamine.
- compositions can be prepared in various forms, such as granules, tablets, pills, suppositories, capsules, suspensions, salves, lotions and the like.
- Pharmaceutical grade organic or inorganic carriers and/or diluents suitable for oral and topical use can be used to make up compositions containing the therapeutically-active compounds.
- Diluents known to the art include aqueous media, vegetable and animal oils and fats. Stabilizing agents, wetting and emulsifying agents, salts for varying the osmotic pressure or buffers for securing an adequate pH value, and skin penetration enhancers can be used as auxiliary agents.
- compositions may also include one or more of the following: carrier proteins such as serum albumin; buffers; fillers such as microcrystalline cellulose, lactose, corn and other starches; binding agents; sweeteners and other flavoring agents; coloring agents; and polyethylene glycol.
- carrier proteins such as serum albumin
- buffers such as buffers
- fillers such as microcrystalline cellulose, lactose, corn and other starches
- binding agents such as microcrystalline cellulose, lactose, corn
- the administration of the mitotic agents of the present invention can be done in a variety of ways as discussed above, including, but not limited to, orally, subcutaneously, intravenously, intranasally, transdermally, intraperitoneally, intramuscularly, intrapulmonary, vaginally, rectally, or intraocularly.
- the anti-mitotic agents may be directly applied as a solution or spray.
- the KSP is bound to a support, and a compound of the invention (which is a mitotic agent) is added to the assay.
- a compound of the invention which is a mitotic agent
- the compound of the invention is bound to the support and KSP is added.
- Classes of compounds among which novel binding agents may be sought include specific antibodies, non-natural binding agents identified in screens of chemical libraries, peptide analogs, etc. Of particular interest are screening assays for candidate agents that have a low toxicity for human cells.
- assays may be used for this purpose, including labeled in vitro protein-protein binding assays, electrophoretic mobility shift assays, immunoassays for protein binding, functional assays (phosphorylation assays, etc.) and the like.
- the determination of the binding of the mitotic agent to KSP may be done in a number of ways.
- the mitotic agent (the compound of the invention) is labeled, for example, with a fluorescent or radioactive moiety and binding determined directly.
- this may be done by attaching all or a portion of KSP to a solid support, adding a labeled mitotic agent (for example a compound of the invention in which at least one atom has been replaced by a detectable isotope), washing off excess reagent, and determining whether the amount of the label is that present on the solid support.
- a labeled mitotic agent for example a compound of the invention in which at least one atom has been replaced by a detectable isotope
- washing off excess reagent for example a compound of the invention in which at least one atom has been replaced by a detectable isotope
- Various blocking and washing steps may be utilized as is known in the art.
- label herein is meant that the compound is either directly or indirectly labeled with a label which provides a detectable signal, e.g., radioisotope, fluorescent tag, enzyme, antibodies, particles such as magnetic particles, chemiluminescent tag, or specific binding molecules, etc.
- Specific binding molecules include pairs, such as biotin and streptavidin, digoxin and antidigoxin etc.
- the complementary member would normally be labeled with a molecule which provides for detection, in accordance with known procedures, as outlined above.
- the label can directly or indirectly provide a detectable signal.
- the kinesin proteins may be labeled at tyrosine positions using 125 I, or with fluorophores.
- more than one component may be labeled with different labels; using 125 I for the proteins, for example, and a fluorophor for the mitotic agents.
- the compounds of the invention may also be used as competitors to screen for additional drug candidates.
- "Candidate bioactive agent” or “drug candidate” or grammatical equivalents as used herein describe any molecule, e.g., protein, oligopeptide, small organic molecule, polysaccharide, polynucleotide, etc., to be tested for bioactivity. They may be capable of directly or indirectly altering the cellular proliferation phenotype or the expression of a cellular proliferation sequence, including both nucleic acid sequences and protein sequences. In other cases, alteration of cellular proliferation protein binding and/or activity is screened. Screens of this sort may be performed either in the presence or absence of microtubules.
- preferred embodiments exclude molecules already known to bind to that particular protein, for example, polymer structures such as microtubules, and energy sources such as ATP.
- Preferred embodiments of assays herein include candidate agents which do not bind the cellular proliferation protein in its endogenous native state termed herein as "exogenous" agents.
- exogenous agents further exclude antibodies to KSP.
- Candidate agents can encompass numerous chemical classes, though typically they are organic molecules, preferably small organic compounds having a molecular weight of more than 100 and less than about 2,500 daltons.
- Candidate agents comprise functional groups necessary for structural interaction with proteins, particularly hydrogen bonding and lipophilic binding, and typically include at least an amine, carbonyl, hydroxyl, ether, or carboxyl group, preferably at least two of the functional chemical groups.
- the candidate agents often comprise cyclical carbon or heterocyclic structures and/or aromatic or polyaromatic structures substituted with one or more of the above functional groups.
- Candidate agents are also found among biomolecules including peptides, saccharides, fatty acids, steroids, purines, pyrimidines, derivatives, structural analogs or combinations thereof. Particularly preferred are peptides.
- Candidate agents are obtained from a wide variety of sources including libraries of synthetic or natural compounds. For example, numerous means are available for random and directed synthesis of a wide variety of organic compounds and biomolecules, including expression of randomized oligonucleotides. Alternatively, libraries of natural compounds in the form of bacterial, fungal, plant and animal extracts are available or readily produced. Additionally, natural or synthetically produced libraries and compounds are readily modified through conventional chemical, physical and biochemical means. Known pharmacological agents may be subjected to directed or random chemical modifications, such as acylation, alkylation, esterification, amidification to produce structural analogs.
- a second sample comprises a mitotic agent, KSP and a drug candidate. This may be performed in either the presence or absence of microtubules.
- the binding of the drug candidate is determined for both samples, and a change, or difference in binding between the two samples indicates the presence of an agent capable of binding to KSP and potentially modulating its activity. That is, if the binding of the drug candidate is different in the second sample relative to the first sample, the drug candidate is capable of binding to KSP.
- the binding of the candidate agent is determined through the use of competitive binding assays.
- the competitor is a binding moiety known to bind to KSP, such as an antibody, peptide, binding partner, ligand, etc. Under certain circumstances, there may be competitive binding as between the candidate agent and the binding moiety, with the binding moiety displacing the candidate agent.
- the candidate agent is labeled. Either the candidate agent, or the competitor, or both, is added first to KSP for a time sufficient to allow binding, if present. Incubations may be performed at any temperature which facilitates optimal activity, typically between 4 and 40°C.
- Incubation periods are selected for optimum activity, but may also be optimized to facilitate rapid high throughput screening. Typically between 0.1 and 1 hour will be sufficient. Excess reagent is generally removed or washed away. The second component is then added, and the presence or absence of the labeled component is followed, to indicate binding.
- the competitor is added first, followed by the candidate agent.
- Displacement of the competitor is an indication the candidate agent is binding to KSP and thus is capable of binding to, and potentially modulating, the activity of KSP.
- either component can be labeled.
- the presence of label in the wash solution indicates displacement by the agent.
- the candidate agent is labeled, the presence of the label on the support indicates displacement.
- the candidate agent is added first, with incubation and washing, followed by the competitor.
- the absence of binding by the competitor may indicate the candidate agent is bound to KSP with a higher affinity.
- the candidate agent is labeled, the presence of the label on the support, coupled with a lack of competitor binding, may indicate the candidate agent is capable of binding to KSP.
- KSP binding site of KSP. This can be done in a variety of ways. In one embodiment, once KSP has been identified as binding to the mitotic agent, KSP is fragmented or modified and the assays repeated to identify the necessary components for binding.
- Modulation is tested by screening for candidate agents capable of modulating the activity of KSP comprising the steps of combining a candidate agent with KSP, as above, and determining an alteration in the biological activity of KSP.
- the candidate agent should both bind to KSP (although this may not be necessary), and alter its biological or biochemical activity as defined herein.
- the methods include both in vitro screening methods and in vivo screening of cells for alterations in cell cycle distribution, cell viability, or for the presence, morpohology, activity, distribution, or amount of mitotic spindles, as are generally outlined above.
- differential screening may be used to identify drug candidates that bind to the native KSP, but cannot bind to modified KSP.
- Positive controls and negative controls may be used in the assays.
- Preferably all control and test samples are performed in at least triplicate to obtain statistically significant results. Incubation of all samples is for a time sufficient for the binding of the agent to the protein. Following incubation, all samples are washed free of non-specifically bound material and the amount of bound, generally labeled agent determined. For example, where a radiolabel is employed, the samples may be counted in a scintillation counter to determine the amount of bound compound.
- reagents may be included in the screening assays. These include reagents like salts, neutral proteins, e.g., albumin, detergents, etc which may be used to facilitate optimal protein-protein binding and/or reduce non-specific or background interactions. Also reagents that otherwise improve the efficiency of the assay, such as protease inhibitors, nuclease inhibitors, anti-microbial agents, etc., may be used. The mixture of components may be added in any order that provides for the requisite binding.
- Step 1 N-butyryl anthranilic acid.
- Step 2 2-Propyl-3,1-[4H]benzoxazin-4-one.
- Step 4 2-(1'-bromopropyl)-3-benzylquinazolin-4-one.
- Step 5 2-[1'-(N,N-dimethylethylenediamino)propyl]-3-benzylquinazolin-4-one.
- Step 6 2-[1'-(N-4-fluorobenzoyl)-(N,N-dimethylethylenediamino)propyl]-3-benzylquinazolin-4-one .
- a stock solution of compound 5 (1.822 g, 5.0 mmol) was prepared in HPLC grade CHCl 3 (0.5 mL).
- a third solution of triethylamine (2.0 mL of 0.5 M) was prepared in HPLC grade 1,2-dichlorethane.
- a 100 ⁇ L aliquot of each solution was pipetted into a glass reaction vessel using a Beckman Biomet 2000 automated liquid dispenser.
- reaction mixture was shaken using a mechanical shaker, sonicated in an ultrasonic water bath, and then incubated overnight at room temperature.
- the mixture was diluted in CHCl 3 (300 ⁇ L) and washed with 5% aqueous NaHCO 3 and water.
- the solvent was removed in vacuo to provide compound 6 (65%).
- the purity of the compound was analyzed by TLC eluted with CH 2 Cl 2 -ethanol-concentrated aqueous NH 3 , 100:10:1.
- Filtrations were performed using Whatman/Polyfiltronics 24 well, 10 mL filtration blocks. Evaporation of volatile materials from the array was performed with a Labconco Vortex-Evaporator or by sweeping with a 4 x 6 nitrogen manifold.
- Step 2 The resin from Step 1 was swelled in 3 mL of DCM. DIEA (0.130 mL, 0.72 mmol) and 4-bromobenzyl bromide (0.12 g, 0.48 mmol) were added. The vial was sealed and shaken overnight. LCMS analysis of a cleaved aliquot revealed an approximate 1:1 mixture of starting material and product. Another 0.130 mL of DIEA and 0.12 g of 4-bromobenzyl bromide were added and the mixture was shaken at 70 °C for 8 h. The resin was filtered, washed (as above), and dried under vacuum.
- Step 3 The resin from Step 2 was twice shaken for 30 min with 5:5:90 TFA:TES:DCM and filtered. The filtrates were combined and evaporated, yielding 140 mg of an orange oil. This material was purified by reverse phase preparative HPLC (acetonitrile-water gradient) to provide 27 mg (17% for 3 steps) of the mono-TFA salt.
- STEP 1 1,2-Diaminoethane trityl resin (Novabiochem, 0.95 mmol/g) (200 g, 1.9 mmol) and 1,3-Diaminopropane trityl resin (Novabiochem, 1.14 mmol/g) (2.0 g, 2.28 mmol) were each placed in different 10 mL polypropylene fritted tubes (Bio-Rad). To each were added 4 mL of DMF, 4 mL of chloroform, 3 eq. of DIEA (1.0 mL and 1.2 mL, respectively) and 2 eq.
- the R enantiomer of B can be crystallized selectively by heating a mixture of B with 1.1 equivalents of D -tartaric acid in a mixture of isopropanol and methanol and then letting the mixture return to room temperature.
- Racemic intermediate B (1.5 g), dissolved in 100 mL of boiling isopropanol, was mixed with 0.8 g of D-tartaric acid in 100 mL of boiling methanol. The mixture was allowed to slowly reach room temperature. After standing overnight, the solid was removed by filtration and rinsed with ethyl acetate and hexanes, and allowed to air dry. The dried solid (0.8 g) was then dissolved in a boiling mixture of 50 mL of isopropanol and 50 mL of methanol and allowed to slowly cool to room temperature. After standing overnight, the resulting solid was removed by filtration and rinsed with ethyl acetate and hexanes, and allowed to air dry.
- FACS analysis to determine cell cycle stage by measuring DNA content was performed as follows. Skov-3 cells (human ovarian cancer) were split 1:10 for plating in 10cm dishes and grown to subconfluence with RPMI 1640 medium containing 5% fetal bovine serum (FBS). The cells were then treated with either 10nM paclitaxel, 400nM quinazolinone 1, 200nM quinazolinone2, or 0.25% DMSO (vehicle for compounds) for 24 hours. Cells were then rinsed off the plates with PBS containing 5mM EDTA, pelleted, washed once in PBS containing 1% FCS, and then fixed overnight in 85% ethanol at 4°C.
- Skov-3 cells human ovarian cancer
- RNAse ribonuclease
- human tumor cell lines Skov-3 (ovarian), HeLa (cervical), and A549 (lung) were plated in 96-well plates at densities of 4,000 cells per well (SKOV-3 & HeLa) or 8,000 cells per well (A549), allowed to adhere for 24 hours, and treated with various concentrations of the quinazolinone compounds for 24 hours.
- Cells were fixed in 4% formaldehyde and stained with anti-tubulin antibodies (subsequently recognized using fluorescently-labeled secondary antibody) and Hoechst dye (which stains DNA).
- a Gi 50 was calculated by plotting the concentration of compound in ⁇ M vs the percentage of cell growth of cell growth in treated wells.
- Solution 1 consists of 3 mM phosphoenolpyruvate potassium salt (Sigma P-7127), 2 mM ATP (Sigma A-3377), 1 mM IDTT (Sigma D-9779), 5 ⁇ M paclitaxel (Sigma T-7402), 10 ppm antifoam 289 (Sigma A-8436), 25 mM Pipes/KOH pH 6.8 (Sigma P6757), 2 mM MgCl2 (VWR JT400301), and 1 mM EGTA (Sigma E3889).
- Solution 1 consists of 3 mM phosphoenolpyruvate potassium salt (Sigma P-7127), 2 mM ATP (Sigma A-3377), 1 mM IDTT (Sigma D-9779), 5 ⁇ M paclitaxel (Sigma T-7402), 10 ppm antifoam 289 (Sigma A-8436), 25 mM Pipes/KOH pH 6.8 (Sigma P6757), 2
- Solution 2 consists of 1 mM NADH (Sigma N8129), 0.2 mg/ml BSA (Sigma A7906), pyruvate kinase 7U/ml, L-lactate dehydrogenase 10 U/ml (Sigma P0294), 100 nM KSP motor domain, 50 ⁇ g/ml microtubules, 1 mM DTT (Sigma D9779), 5 ⁇ M paclitaxel (Sigma T-7402), 10 ppm antifoam 289 (Sigma A-8436), 25 mM Pipes/KOH pH 6.8 (Sigma P6757), 2 mM MgC12 (VWR JT4003-01), and 1 mM EGTA (Sigma E3889).
- the quinazolinone compounds inhibit growth in a variety of cell lines, including cell lines (MCF-7/ADR-RES, HCT1 5) that express P-glycoprotein (also known as Multidrug Resistance, or MDR + ), which conveys resistance to other chemotherapeutic drugs, such as pacilitaxel. Therefore, the quinazolinones are anti-mitotics that inhibit cell proliferation, and are not subject to resistance by overexpression of MDR + by drug-resistant tumor lines.
- GI 50 values for the quinazolinone compounds tested ranged from 200 nM to greater than the highest concentration tested. By this we mean that although most of the compounds that inhibited KSP activity biochemically did inhibit cell proliferation, for some, at the highest concentration tested (generally about 20 ⁇ M), cell growth was inhibited less than 50%. Many of the compounds have GI 50 values less than 10 ⁇ M, and several have GI 50 values less than 1 ⁇ M.
- Antiproliferative compounds that have been successfully applied in the clinic to treatment of cancer have GI 50 's that vary greatly.
- paclitaxel GI 50 is 4 nM
- doxorubicin is 63 nM
- 5-fluorouracil is 1 ⁇ M
- hydroxyurea is 500 ⁇ M (data provided by National Cancer Institute, Developmental Therapeutic Program, http://dtp.nci.nih.gov/). Therefore, compounds that inhibit cellular proliferation at virtually any concentration may be useful.
- compounds will have GI 50 values of less than 1 mM. More preferably, compounds will have GI 50 values of less than 20 ⁇ M. Even more preferably, compounds will have GI 50 values of less than 10 ⁇ M. Further reduction in GI 50 values may also be desirable, including compounds with GI 50 values of less than 1 ⁇ M.
- mice Female nude mice weighing approximately 20 g were implanted s.c. by trocar with fragments of human tumor carcinomas harvested from s.c. growing tumors in nude mice host. When the tumors were approximately 77 mg in size, the animals were pair matched into treatment and control groups. Each group containted 8 tumored mice, each of which was ear-tagged and followed individually throughout the experiment. Initial doses (10 mL/kg of a 66 mM Citrate buffer, pH 5.0/0.9% Saline / 10% Tween 80 formulation of each test compound having a maximum concentration of 5 mg/mL) were given on Day 1 following pair matching, dosing at the levels and schedules indicated.
- mice were weighed twice weekly, and tumor measurements were taken by calipers twice weekly, starting on Day 1. These tumor measurements were converted to mg tumor weight by a well-known formula, W 2 x L/2. The experiment was terminated when the control group tumor size reached an average of 1 gram. Upon termination, the mice were weighted, sacrificed and their tumors excised. Tumors were weighted and the mean treated tumor weight per group was calculated. In this model, the change in mean treated tumor weight / the change in mean control tumor weight x 100% ( ⁇ T/ ⁇ C) was subtracted from 100% to give the tumor growth inhibition (TGI) for each group.
- TGI tumor growth inhibition
Abstract
Description
- This invention relates to quinazolinone derivatives, which are inhibitors of the mitotic kinesin KSP and are useful in the treatment of cellular proliferative diseases, for example cancer, hyperplasias, restenosis, cardiac hypertrophy, immune disorders and inflammation.
- Interest in the medicinal chemistry of quinazoline derivatives was stimulated in the early 1950's with the elucidation of the structure of a quinazoline alkaloid, 3-[β-keto-gamma-(3-hydroxy-2-piperidyl)-propyl]-4-quinazolone, from an Asian plant known for its antimalarial properties. In a quest to find additional antimalarial agents, various substituted quinazolines have been synthesized. Of particular import was the synthesis of the derivative 2-methyl-3-o-tolyl-4-(3H)-quinazolinone. This compound, known by the name methaqualone, though ineffective against protozoa, was found to be a potent hypnotic.
- Since the introduction of methaqualone and its discovery as a hypnotic, the pharmacological activity of quinazolinones and related compounds has been investigated. Quinazolinones and derivatives thereof are now known to have a wide variety of biological properties including hypnotic, sedative, analgesic, anticonvulsant, antitussive and anti-inflammatory activities.
- Quinazolinone derivatives for which specific biological uses have been described include U.S. Patent No. 5,147,875 describing 2-(substituted phenyl)-4-oxo quinazolines with bronchodilator activity. U.S. Patent Nos. 3,723,432, 3,740,442, and 3,925,548 describe a class of 1 -substituted-4-aryl-2(1H)-quinazolinone derivatives useful as anti-inflammatory agents. European
patent publication EP 0 056 637 B1 claims a class of 4(3H)-quinazolinone derivatives for the treatment of hypertension. Europeanpatent publication EP 0 884 319 A1 describes pharmaceutical compositions of quinazolin-4-one derivatives used to treat neurodegenerative, psychotropic, and drug and alcohol induced central and peripheral nervous system disorders. - Quinazolinones are among a growing number of therapeutic agents used to treat cell proliferative disorders, including cancer. For example, PCT WO 96/06616 describes a pharmaceutical composition containing a quinazolinone derivative to inhibit vascular smooth cell proliferation. PCT WO 96/19224 uses this same quinazolinone derivative to inhibit mesengial cell proliferation. U.S. Patent Nos. 4,981,856, 5,081,124 and 5,280,027 describe the use of quinazolinone derivatives to inhibit thymidylate synthase, the enzyme that catalyzes the methylation of deoxyuridine monophosphate to produce thymidine monophosphate which is required for DNA synthesis. U.S. Patent Nos. 5,747,498 and 5,773,476 describe quinazolinone derivatives used to treat cancers characterized by over-activity or inappropriate activity of tyrosine receptor kinases. U.S. Patent No. 5,037,829 claims (1H-azol-1-ylmethyl) substituted quinazoline compositions to treat carcinomas which occur in epithelial cells. PCT WO 98/34613 describes a composition containing a quinazolinone derivative useful for attenuating neovascularization and for treating malignancies. U.S. Patent 5,187,167 describes pharmaceutical compositions comprising quinazolin-4-one derivatives which possess anti-tumor activity.
- Other therapeutic agents used to treat cancer include the taxanes and vinca alkaloids. Taxanes and vinca alkaloids act on microtubules, which are present in a variety of cellular structures. Microtubules are the primary structural element of the mitotic spindle. The mitotic spindle is responsible for distribution of replicate copies of the genome to each of the two daughter cells that result from cell division. It is presumed that disruption of the mitotic spindle by these drugs results in inhibition of cancer cell division, and induction of cancer cell death. However, microtubules form other types of cellular structures, including tracks for intracellular transport in nerve processes. Because these agents do not specifically target mitotic spindles, they have side effects that limit their usefulness.
- Improvements in the specificity of agents used to treat cancer is of considerable interest because of the therapeutic benefits which would be realized if the side effects associated with the administration of these agents could be reduced. Traditionally, dramatic improvements in the treatment of cancer are associated with identification of therapeutic agents acting through novel mechanisms. Examples of this include not only the taxanes, but also the camptothecin class of topoisomerase I inhibitors. From both of these perspectives, mitotic kinesins are attractive targets for new anti-cancer agents.
- Mitotic kinesins are enzymes essential for assembly and function of the mitotic spindle, but are not generally part of other microtubule structures, such as in nerve processes. Mitotic kinesins play essential roles during all phases of mitosis. These enzymes are "molecular motors" that transform energy released by hydrolysis of ATP into mechanical force which drives the directional movement of cellular cargoes along microtubules. The catalytic domain sufficient for this task is a compact structure of approximately 340 amino acids. During mitosis, kinesins organize microtubules into the bipolar structure that is the mitotic spindle. Kinesins mediate movement of chromosomes along spindle microtubules, as well as structural changes in the mitotic spindle associated with specific phases of mitosis. Experimental perturbation of mitotic kinesin function causes malformation or dysfunction of the mitotic spindle, frequently resulting in cell cycle arrest and cell death.
- Among the mitotic kinesins that have been identified is KSP. KSP belongs to an evolutionarily conserved kinesin subfamily of plus end-directed microtubule motors that assemble into bipolar homotetramers consisting of antiparallel homodimers. During mitosis KSP associates with microtubules of the mitotic spindle. Microinjection of antibodies directed against KSP into human cells prevents spindle pole separation during prometaphase, giving rise to monopolar spindles and causing mitotic arrest and induction of programmed cell death. KSP and related kinesins in other, non-human, organisms, bundle antiparallel microtubules and slide them relative to one another, thus forcing the two spindle poles apart. KSP may also mediate in anaphase B spindle elongation and focussing of microtubules at the spindle pole.
- Human KSP (also termed HsEg5) has been described [Blangy, et al., Cell, 83:1159-69 (1995); Whitehead, et al., Arthritis Rheum., 39:1635-42 (1996); Galgio et al., J. Cell Biol., 135:339-414 (1996); Blangy, et al., J Biol. Chem., 272:19418-24 (1997); Blangy, et al., Cell Motil Cytoskeleton, 40:174-82 (1998); Whitehead and Rattner, J. Cell Sci., 111:2551-61 (1998); Kaiser, et al., JBC 274:18925-31 (1999); GenBank accession numbers: X85137, NM004523 and U37426], and a fragment of the KSP gene (TRIP5) has been described [Lee, et al., Mol Endocrinol., 9:243-54 (1995); GenBank accession number L40372]. Xenopus KSP homologs (Eg5), as well as Drosophila KLP61 F/KRP1 30 have been reported.
- Mitotic kinesins are attractive targets for the discovery and development of novel mitotic chemotherapeutics. Accordingly, it is an object of the present invention to provide methods and compositions useful in the inhibition of KSP, a mitotic kinesin.
- In accordance with the objects outlined above, the present invention provides compositions and methods that can be used to treat diseases of proliferating cells. The compositions are KSP inhibitors, particularly human KSP inhibitors.
- In one aspect, the invention relates to methods for treating cellular proliferative diseases, for treating disorders associated with KSP kinesin activity, and for inhibiting KSP kinesin. The methods employ compounds chosen from the group consisting of:


- R1
- is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;
- R2 and R2'
- are independently chosen from hydrogen, alkyl, oxaalkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; or R2 and R2' taken together form a 3- to 7-membered ring;
- R3
- is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, oxaalkyl, oxaalkylaryl, substituted oxaalkylaryl, oxaalkyl heteroaryl, substituted oxaalkylheteroaryl, R15O- and R15-NH-;
- R3'
- is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl and R15-NH-;
- R3"
- is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;
- R4
- is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, and R16-alkylene-;
- R5, R6, R7 and R8
- are independently chosen from hydrogen, alkyl, alkoxy, halogen, fluoroalkyl, nitro, cyano, dialkylamino, alkylsulfonyl, alkylsulfonamido, sulfonamidoalkyl, sulfonamidoaryl, alkylthio, carboxyalkyl, carboxamido, aminocarbonyl, aryl and heretoaryl;
- R15
- is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;
- R16
- is chosen from alkoxy, amino, alkylamino, dialkylamino, N-heterocyclyl and substituted N-heterocyclyl.
- Diseases and disorders that respond to therapy with compounds of the invention include cancer, hyperplasia, restenosis, cardiac hypertrophy, immune disorders and inflammation; especially cancer, hyperplasia, restenosis, and cardiac hypertrophy; particularly cancer.
- In another aspect, the invention relates to compounds useful in inhibiting KSP kinesin. The compounds have the structures shown above.
- In an additional aspect, the present invention provides methods of screening for compounds that will bind to a KSP kinesin, for example compounds that will displace or compete with the binding of the compositions of the invention. The methods comprise combining a labeled compound of the invention, a KSP kinesin, and at least one candidate agent and determining the binding of the candidate bioactive agent to the KSP kinesin.
- In a further aspect, the invention provides methods of screening for modulators of KSP kinesin activity. The methods comprise combining a composition of the invention, a KSP kinesin, and at least one candidate agent and determining the effect of the candidate bioactive agent on the KSP kinesin activity.
-
- Figure 1 depicts a generic synthetic scheme to make compositions of the invention.
- Figure 2 depicts a synthetic route for the synthesis of quinazolinone KSP inhibitors.
- Figure 3 depicts representative chemical structures of quinazolinone KSP inhibitors.
- Figure 4 depicts a synthetic route to substantially pure single enantiomers.
- Figure 5 depicts synthetic routes to sulfonamides (5a), carbamates (5b), ureas (5c) and amines (5d).
- The present invention is directed to a class of novel compounds, based on a core quinazolinone structure, that are modulators of mitotic kinesins. By inhibiting or modulating mitotic kinesins, but not other kinesins (e.g., transport kinesins), specific inhibition of cellular proliferation is accomplished. Thus, the present invention capitalizes on the finding that perturbation of mitotic kinesin function causes malformation or dysfunction of mitotic spindles, frequently resulting in cell cycle arrest and cell death. The methods of inhibiting a human KSP kinesin comprise contacting an inhibitor of the invention with a KSP kinesin, particularly human KSP kinesins, including fragments and variants of KSP. The inhibition can be of the ATP hydrolysis activity of the KSP kinesin and/or the mitotic spindle formation activity, such that the mitotic spindles are disrupted. Meiotic spindles may also be disrupted.
- An object of the present invention is to develop inhibitors and modulators of mitotic kinesins, in particular KSP, for the treatment of disorders associated with cell proliferation. Traditionally, dramatic improvements in the treatment of cancer, one type of cell proliferative disorder, have been associated with identification of therapeutic agents acting through novel mechanisms. Examples of this include not only the taxane class of agents that appear to act on microtubule formation, but also the camptothecin class of topoisomerase I inhibitors. The compositions and methods described herein can differ in their selectivity and are preferably used to treat diseases of proliferating cells, including, but not limited to cancer, hyperplasias, restenosis, cardiac hypertrophy, immune disorders and inflammation.
- Accordingly, the present invention relates to methods employing quinazolinone amides of formula 1a:



- R1
- is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;
- R2 and R2'
- are independently chosen from hydrogen, alkyl, oxaalkyl, aryl, alkylaryl, heteroaryl, allcylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; or R2 and R2' taken together form a 3- to 7-membered ring;
- R3
- is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, oxaalkyl, oxaalkylaryl, substituted oxaalkylaryl, oxaalkylheteroaryl, substituted oxaalkylheteroaryl, R15O- and R15-NH-;
- R3' is
- chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, and R15-NH-;
- R3"
- is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;
- R4
- is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, and R16-alkylene-;
- R5, R6, R7 and R8
- are independently chosen from hydrogen, alkyl, alkoxy, halogen, fluoroalkyl, nitro, cyano, dialkylamino, alkylsulfonyl, alkylsulfonamido, sulfonamidoalkyl, sulfonamidoaryl, alkylthio, carboxyalkyl, carboxamido, aminocarbonyl, aryl and heteroaryl;
- R15
- is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;
- R16
- is chosen from alkoxy, amino, alkylamino, dialkylamino, N-heterocyclyl and substituted N-heterocyclyl.
- All of the compounds falling within the foregoing parent genus and its subgenera are useful as kinesin inhibitors, but not all the compounds are novel. In particular, certain ureas (i.e. compounds in which R3 is R15NH) are disclosed in US patent 5,756,502 as agents which modify cholecystokinin action. The specific exceptions in the claims reflect applicants' intent to avoid claiming subject matter that, while functionally part of the inventive concept, is not patentable to them for reasons having nothing to do with the scope of the invention.
- The term "optional" or "optionally" means that the subsequently described event or circumstance may or may not occur, and that the description includes instances where said event or circumstance occurs and instances in which it does not. For example, "optionally substituted alkyl" means either "alkyl" or "substituted alkyl," as defined below. It will be understood by those skilled in the art with respect to any group containing one or more substituents that such groups are not intended to introduce any substitution or substitution patterns (e.g., substituted alkyl includes optionally substituted cycloalkyl groups, which in turn are defined as including optionally substituted alkyl groups, potentially ad infinitum) that are sterically impractical and/or synthetically non-feasible.
- Alkyl is intended to include linear, branched, or cyclic hydrocarbon structures and combinations thereof. Lower alkyl refers to alkyl groups of from 1 to 5 carbon atoms. Examples of lower alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, s-and t-butyl and the like. Preferred alkyl groups are those of C20 or below. More preferred alkyl groups are those of C13 or below. Cycloalkyl is a subset of alkyl and includes cyclic hydrocarbon groups of from 3 to 13 carbon atoms. Examples of cycloalkyl groups include c-propyl, c-butyl, c-pentyl, norbomyl, adamantyl and the like. In this application, alkyl refers to alkanyl, as well as the unsaturated alkenyl and alkynyl residues; it is intended to include cyclohexylmethyl, vinyl, allyl, isoprenyl and the like. Alkylene refers to the same residues as alkyl, but having two points of attachment. Examples of alkylene include ethylene (-CH2CH2-), ethenylene (-CH2=CH2-), propylene (-CH2CH2CH2-), dimethylpropylene (-CH2C(CH3)2CH2-) and cyclohexylpropylene (-CH2CH2CH(C6H13)-). When an alkyl residue having a specific number of carbons is named, all geometric isomers having that number of carbons are intended to be encompassed; thus, for example, "butyl" is meant to include n-butyl, sec-butyl, isobutyl and t-butyl; "propyl" includes n-propyl and isopropyl (or "i-propyl").
- Alkoxy or alkoxyl refers to groups of from 1 to 8 carbon atoms of a straight, branched, cyclic configuration and combinations thereof attached to the parent structure through an oxygen. Examples include methoxy, ethoxy, propoxy, isopropoxy, cyclopropyloxy, cyclohexyloxy and the like. Lower-alkoxy refers to groups containing one to four carbons.
- Acyl refers to groups of from 1 to 8 carbon atoms of a straight, branched, cyclic configuration, saturated, unsaturated and aromatic and combinations thereof, attached to the parent structure through a carbonyl functionality. One or more carbons in the acyl residue may be replaced by nitrogen, oxygen or sulfur as long as the point of attachment to the parent remains at the carbonyl. Examples include acetyl, benzoyl, propionyl, isobutyryl, t-butoxycarbonyl, benzyloxycarbonyl and the like. Lower-acyl refers to groups containing one to four carbons.
- Aryl and heteroaryl mean a 5- or 6-membered aromatic or heteroaromatic ring containing 0-3 heteroatoms selected from O, N, or S; a bicyclic 9- or 10-membered aromatic or heteroaromatic ring system containing 0-3 heteroatoms selected from O, N, or S; or a tricyclic 13- or 14-membered aromatic or heteroaromatic ring system containing 0-3 heteroatoms selected from O, N, or S. The aromatic 6- to 14-membered carbocyclic rings include, e.g., benzene, naphthalene, indane, tetralin, and fluorene and the 5- to 10-membered aromatic heterocyclic rings include, e.g., imidazole, pyridine, indole, thiophene, benzopyranone, thiazole, furan, benzimidazole, quinoline, isoquinoline, quinoxaline, pyrimidine, pyrazine, tetrazole and pyrazole.
- Alkylaryl refers to a residue in which an aryl moiety is attached to the parent structure via an alkylene residue. Examples are benzyl, phenethyl, phenylvinyl, phenylallyl and the like. Oxaalkyl and oxaalkylaryl refer to alkyl and alkylaryl residues in which one or more methylenes have been replaced by oxygen. Examples of oxaalkyl and oxaalkylaryl residues are ethoxyethoxyethyl (3,6-dioxaoctyl), benzyloxymethyl and phenoxymethyl; in general, glycol ethers, such as polyethyleneglycol, are intended to be encompassed by this group. Alkylheteroaryl refers to a residue in which a heteroaryl moiety is attached to the parent structure via an alkylene residue. Examples include furanylmethyl, pyridinylmethyl, pyrimidinylethyl and the like.
- Heterocycle means a cycloalkyl or aryl residue in which one to four of the carbons is replaced by a heteroatom such as oxygen, nitrogen or sulfur. Examples of heterocycles that fall within the scope of the invention include imidazoline, pyrrolidine, pyrazole, pyrrole, indole, quinoline, isoquinoline, tetrahydroisoquinoline, benzofuran, benzodioxan, benzodioxole (commonly referred to as methylenedioxyphenyl, when occurring as a substituent), tetrazole, morpholine, thiazole, pyridine, pyridazine, pyrimidine, thiophene, furan, oxazole, oxazoline, isoxazole, dioxane, tetrahydrofuran and the like. "N-heterocyclyl" refers to a nitrogen-containing heterocycle as a substituent residue. The term heterocyclyl encompasses heteroaryl, which is a subset of heterocyclyl. Examples of N-heterocyclyl residues include 4-morpholinyl, 4-thiomorpholinyl, 1-piperidinyl, 1-pyrrolidinyl, 3-thiazolidinyl, piperazinyl and 4-(3,4-dihydrobenzoxazinyl). Examples of substituted heterocyclyl include 4-methyl-1-piperazinyl and 4-benzyl-1-piperidinyl.
- Substituted alkyl, aryl and heteroaryl refer to alkyl, aryl or heteroaryl wherein one or more H atoms are replaced with alkyl, halogen, hydroxy, alkoxy, alkylenedioxy (e.g., methylenedioxy), fluoroalkyl, carboxy (-COOH), carboalkoxy (i.e., acyloxy -O(O)CR), carboxyalkyl (i.e., esters -C(O)OR), carboxamido, sulfonamidoalkyl, sulfonamidoaryl, aminocarbonyl, benzyloxycarbonylamino (CBZ-amino), cyano, carbonyl, nitro, primary-, secondary- and tertiary-amino (e.g., alkylamino and dialkylamino) and aminoaklylene, alkylthio, alkylsulfinyl, alkylsulfonyl, alkylsulfonamido, arylthio, arylsulfinyl, arylsulfonyl, amidino, aryl (e.g., phenyl and benzyl), heteroaryl, heterocyclyl, phenoxy, benzyloxy, or heteroaryloxy. For the purposes of the present invention, substituted alkyl also includes oxaalkyl residues, i.e. alkyl residues in which one or more carbons has been replaced by oxygen. Substituted alkylaryl and substituted oxaalkylaryl refer to residues where either or both of the alkylene and aryl moieties are substituted. Substituted alkylheteroaryl and substituted oxaalkylheteroaryl residues where either or both of the alkylene and heteroaryl moieties are substituted. It should additionally be noted that certain positions may contain two or even three substitution groups, R, R' and R" (e.g., diethylamino and trifluoromethyl).
- Halogen refers to fluorine, chlorine, bromine or iodine. Fluorine, chlorine and bromine are preferred. Dihaloaryl, dihaloalkyl, trihaloaryl, etc. refer to aryl and alkyl substituted with a plurality of halogens, but not necessarily a plurality of the same halogen; thus 4-chloro-3-fluorophenyl is within the scope of dihaloaryl.
- Most of the compounds described herein contain one or more asymmetric centers (e.g. the carbon to which R2 and R2' are attached) and may thus give rise to enantiomers, diastereomers, and other stereoisomeric forms that may be defined, in terms of absolute stereochemistry, as (R)- or (S)-. The present invention is meant to include all such possible isomers, including racemic mixtures, optically pure forms and intermediate mixtures. Optically active (R)- and (S)- isomers may be prepared using chiral synthons or chiral reagents, or resolved using conventional techniques. When the compounds described herein contain olefinic double bonds or other centers of geometric asymmetry, and unless specified otherwise, it is intended that the compounds include both E and Z geometric isomers. Likewise, all tautomeric forms are also intended to be included.
- When desired, the R- and S-isomers may be resolved by methods known to those skilled in the art, for example by formation of diastereoisomeric salts or complexes which may be separated, for example, by crystallisation; via formation of diastereoisomeric derivatives which may be separated, for example, by crystallisation, gas-liquid or liquid chromatography; selective reaction of one enantiomer with an enantiomer-specific reagent, for example enzymatic oxidation or reduction, followed by separation of the modified and unmodified enantiomers; or gas-liquid or liquid chromatography in a chiral environment, for example on a chiral support, such as silica with a bound chiral ligand or in the presence of a chiral solvent. It will be appreciated that where the desired enantiomer is converted into another chemical entity by one of the separation procedures described above, a further step may be required to liberate the desired enantiomeric form. Alternatively, specific enantiomer may be synthesized by asymmetric synthesis using optically active reagents, substrates, catalysts or solvents, or by converting on enantiomer to the other by asymmetric transformation. An example of a synthesis from optically active starting materials is shown in Figure 4.
- In one embodiment, as will be appreciated by those in the art, the two adjacent R2 groups may be fused together to form a ring structure. Again, the fused ring structure may contain heteroatoms and may be substituted with one or more substitution groups "R". It should additionally be noted that for cycloalkyl (i.e., saturated ring structures), certain positions may contain two substitution groups, R and R'.
- Considering formulae 1a, 1b, 1c and 1d, but focusing on 1a, in a preferred embodiment R1 is selected from hydrogen, alkyl, aryl, substituted alkyl, substituted aryl, heteroaryl, substituted heteroaryl, alkylaryl and substituted alkylaryl.
- In a more preferred embodiment R1 is selected from hydrogen, lower alkyl,substituted lower alkyl, benzyl, substituted benzyl, phenyl, naphthyl and substituted phenyl.
- In a most preferred embodiment R1 is chosen from hydrogen, ethyl, propyl, methoxyethyl, naphthyl, phenyl, bromophenyl, chlorophenyl, methoxyphenyl, ethoxyphenyl, tolyl, dimethylphenyl, chorofluorophenyl, methylchlorophenyl, ethylphenyl, phenethyl, benzyl, chlorobenzyl, methylbenzyl, methoxybenzyl, cyanobenzyl, hydroxybenzyl, tetrahydrofuranylmethyl and (ethoxycarbonyl)ethyl.
- In a preferred embodiment R2 is hydrogen, alkyl,cycloalkyl or substituted alkyl. As will be appreciated by those in the art, Formulae 1a, 1b, 1c and 1d possess a potentially chiral center at the carbon to which R2 is attached. Thus, the R2 position may comprise two substitution groups, R2 and R2'. The R2 and R2' groups may be the same or different; if different, the composition is chiral. When the R2 and R2' are different, preferred embodiments utilize only a single non-hydrogen R2. The invention contemplates the use of pure enantiomers and mixtures of enantiomers, including racemic mixtures, although the use of the substantially optically pure eutomer will generally be preferred, particularly the R enantiomer.
- In a more preferred embodiment, R2 is chosen from hydrogen, lower alkyl and substituted lower alkyl, and R2' is hydrogen. In a most preferred embodiment R2 is chosen from hydrogen, methyl, ethyl, propyl (particularly i-propyl), butyl (particularly t-butyl), methylthioethyl, aminobutyl, (CBZ)aminobutyl, cyclohexylmethyl, benzyloxymethyl, methylsulfinylethyl, methylsulfinylmethyl, hydroxymethyl, benzyl and indolylmethyl. Especially preferred is the R enantiomer where R2 is i-propyl.
- In a preferred embodiment R3 is selected from chosen from alkyl, substituted alkyl, alkylaryl, heteroaryl, aryl, substituted aryl, substituted oxaalkylaryl, -O-R15 and -NH-R15, and R15 is chosen from alkyl, aryl and substituted aryl.
- In a more preferred embodiment, when R3 is not -NHR15, R3 is chosen from C1-C13 alkyl; substituted lower alkyl;aryl, including phenyl, biphenyl and naphthyl; substituted aryl, including phenyl substituted with one or more halo, lower alkyl, loweralkoxy, nitro, carboxy, methylenedioxy or trifluoromethyl; benzyl; phenoxymethyl; halophenoxymethyl; phenylvinyl; heteroaryl; heteroaryl substituted with lower alkyl; and benzyloxymethyl.
- In a most preferred embodiment, when R3 is not -NHR15, R3 is chosen from ethyl, propyl, chloropropyl, butoxy, heptyl, butyl, octyl, tridecanyl, (ethoxycarbonyl)ethyl, dimethylaminoethyl, dimethylaminomethyl, phenyl, naphthyl, halophenyl, dihalophenyl, cyanophenyl, halo(trifluoromethyl)phenyl, chlorophenoxymethyl, methoxyphenyl, carboxyphenyl, ethylphenyl, tolyl, biphenyl, methylenedioxyphenyl, methylsulfonylphenyl, methoxychlorophenyl, chloronaphthyl, methylhalophenyl, trifluoromethylphenyl, butylphenyl, pentylphenyl, methylnitrophenyl, phenoxymethyl, dimethoxyphenyl, phenylvinyl, nitrochlorophenyl, nitrophenyl, dinitrophenyl, bis(trifluoromethyl)phenyl, benzyloxymethyl, benzyl, furanyl, benzofuranyl, pyridinyl, indolyl, methylpyridinyl, quinolinyl, picolinyl, pyrazolyl, and imidazolyl.
- In a more preferred embodiment, when R3 is -NHR15, R15 is chosen from lower alkyl; cyclohexyl; phenyl; and phenyl substituted with halo, lower alkyl, loweralkoxy, or lower alkylthio.
- In a most preferred embodiment, when R3 is -NHR15, R15 is isopropyl, butyl, cyclohexyl, phenyl, bromophenyl, dichlorophenyl, methoxyphenyl, ethylphenyl, tolyl, trifluoromethylphenyl or methylthiophenyl.
- In a preferred embodiment R4 is chosen from alkyl, aryl, alkylaryl, alkylheteroaryl, substituted alkyl, substituted aryl, and -alkylene-R16, and R16 is chosen from alkoxy, amino, alkylamino, dialkylamino and N-heterocyclyl.
- In a more preferred embodiment, R4 is selected from lower alkyl, substituted lower alkyl, cyclohexyl; phenyl substituted with hydroxy, lower alkoxy or lower alkyl; benzyl; heteroarylmethyl; heteroarylethyl; heteroarylpropyl and -alkylene-R16, wherein R16 is amino, lower alkylamino, di(lower alkyl)amino, lower alkoxy, or N-heterocyclyl.
- In a most preferred embodiment, R4 is chosen from methyl, ethyl, propyl, butyl, cyclohexyl, carboxyethyl, carboxymethyl, methoxyethyl, hydroxyethyl, hydroxypropyl, dimethylaminoethyl, dimethylaminopropyl, diethylaminoethyl, diethylaminopropyl, aminopropyl, methylaminopropyl, 2,2-dimethyl-3-(dimethylamino)propyl, 1-cyclohexyl-4-(diethylamino)butyl, aminoethyl, aminobutyl, aminopentyl, aminohexyl, aminoethoxyethyl, isopropylaminopropyl, diisopropylaminoethyl, 1-methyl-4-(diethylamino)butyl, (t-Boc)aminopropyl, hydroxyphenyl, benzyl, methoxyphenyl, methylinethoxyphenyl, dimethylphenyl, tolyl, ethylphenyl, (oxopyrrolidinyl)propyl, (methoxycarbonyl)ethyl, benzylpiperidinyl, pyridinylethyl, pyridinylmethyl, morpholinylethyl, morpholinylpropyl, piperidinyl, azetidinylmethyl, azetidinylpropyl, pyrrolidinylethyl, pyrrolidinylpropyl, piperidinylmethyl, piperidinylethyl, imidazolylpropyl, imidazolylethyl, (ethylpyrrolidinyl)methyl, (methylpyrrolidinyl)ethyl, (methylpiperidinyl)propyl, (methylpiperazinyl)propyl, furanylmethyl and indolylethyl.
- In other preferred embodiments, R5, R6, R7 and R8 are chosen from hydrogen, halo (particularly chloro and fluoro), lower alkyl (particularly methyl), substituted lower alkyl (particularly trifluoromethyl), lower alkoxy (particularly methoxy), and cyano; more preferably from hydrogen and halo. Further preferred for each of the specific substituents: R5 is hydrogen or halo; R6 is hydrogen, methyl or halo; R7 is hydrogen, halo, alkyl (particularly methyl), alkoxy (particularly methoxy) or cyano; and R8 is hydrogen or halo. Still further preferred are the compounds where only one of R5, R6, R7 and R8 is not hydrogen, especially R7.
- In a particularly preferred subgenus, R1 is benzyl or halobenzyl; R2 is chosen from ethyl and propyl; R2' is hydrogen; R3 (or R3' or R3") is substituted phenyl; R4 is - (CH2)mOH where m is two or three, or -(CH2)p-R16 where p is one to three and R16 is amino, propylamino or azetidinyl; R5 is hydrogen; R6 is hydrogen; R7 is halo; and R8 is hydrogen.
- When considering primarily the sulfonamides of formula 1b, R1 is preferably chosen from hydrogen, lower alkyl, substituted lower alkyl, benzyl, substituted benzyl, phenyl and substituted phenyl; R2 is chosen from hydrogen and lower alkyl and R2' is hydrogen; R3' is chosen from C1-C13 alkyl; phenyl; naphthyl; phenyl substituted with halo, lower alkyl, lower alkoxy, nitro, methylenedioxy, or trifluoromethyl; biphenylyl and heteroaryl; and R4 is chosen from lower alkyl, cyclohexyl; phenyl substituted with hydroxy, lower alkoxy or lower alkyl; benzyl; heteroarylmethyl; heteroarylethyl; heteroarylpropyl; heteroarylethyl; heteroarylpropyl and -alkylene-R16, wherein R16 is di(lower alkyl)amino, (lower alkyl)amino, amino, lower alkoxy, or N-heterocyclyl, particularly pyrrolidino, piperidino or imidazolyl.
- When considering primarily the sulfonamides of formula 1b, R1 is most preferably chosen from lower alkyl, benzyl, substituted benzyl and substituted phenyl; R2 is hydrogen or lower alkyl; R2' is hydrogen; R3 is chosen from substituted phenyl and naphthyl; R4 is -alkylene-R16; R7 is hydrogen, fluoro, methyl or chloro; R5, R6 and R8 are hydrogen; and R16 is chosen from di(lower alkylamino), (lower alkyl)amino, amino, pyrrolidino, piperidino, imidazolyl and morpholino.
- When considering primarily the amines of formulae 1c and 1d, R1 is preferably chosen from hydrogen, lower alkyl, substituted lower alkyl, benzyl, substituted benzyl, phenyl, naphthyl and substituted phenyl; R2 is chosen from hydrogen, lower alkyl and substituted lower alkyl and R2' is hydrogen; R3" is chosen from C1-C13 alkyl; substituted lower alkyl; phenyl; naphthyl; phenyl substituted with halo, lower alkyl, lower alkoxy, nitro, methylenedioxy, or trifluoromethyl; biphenylyl, benzyl and heterocyclyl; and R4 is chosen from lower alkyl; cyclohexyl; phenyl substituted with hydroxy, lower alkoxy or lower alkyl; benzyl; substituted benzyl; heterocyclyl; heteroarylmethyl; heteroarylethyl; heteroarylpropyl and -alkylene-R16, wherein R16 is di(lower alkyl)amino, (lower alkyl)amino, amino, lower alkoxy, or N-heterocyclyl.
- When considering primarily the amines of formulae 1c and 1d, R1 is most preferably chosen from lower alkyl, benzyl, substituted benzyl and substituted phenyl; R2 is hydrogen or lower alkyl; R2' is hydrogen; R3" is chosen from substituted phenyl, heterocyclyl and naphthyl; R4 is chosen from subtituted benzyl, heterocyclyl and -alkylene-R16; R6 and R7 are chosen from hydrogen and halo; R5 and R8 are hydrogen; and R16 is chosen from di(lower alkylamino), (lower alkyl)amino, amino, pyrrolidinyl, piperidinyl, imidazolyl and morpholinyl. When R3" is present (as in 1d) it is most preferably chosen from halophenyl, polyhalophenyl, tolyl, dimethylphenyl, methoxyphenyl, dimethoxyphenyl, cyanophenyl, trifluoromethylphenyl, trifluoromethoxyphenyl, bis(trifluoromethyl)phenyl, carboxyphenyl, t-butylphenyl, methoxycarbonylphenyl, piperidinyl and naphthyl.
- In view of the foregoing, particularly when taken in consideration of the test data presented below, it will be appreciated that preferred for the compounds, pharmaceutical formulations, methods of manufacture and use of the present invention are the following combinations and permutations of substituent groups (sub-grouped, respectively, in increasing order of preference):
- 1. Any of formulae 1a, 1b, 1c or 1d (preferably formulae 1a or 1d) where R1 is hydrogen, lower alkyl, substituted lower alkyl, alkylaryl or substituted alkylaryl (preferably benzyl or substituted benzyl):
- a. Especially where the stereogenic center to which R2 and R2' are attached is of the R configuration, where R2' is hydrogen.
- i. Particularly where R2 is lower alkyl (preferably ethyl, i-proyl, c-propyl or t-butyl).
- 1. Most preferably where R2 is i-propyl.
- i. Particularly where R2 is lower alkyl (preferably ethyl, i-proyl, c-propyl or t-butyl).
- b. Especially those where R4 is substituted alkyl, (preferably a primary-, secondary- or tertiary-amino-substituted lower alkyl).
- i. Most preferably where R4 is primary-amino lower alkyl.
- c. Especially those where R5, R6, R7 and R8 are chosen from hydrogen, halo, lower alkyl (preferably methyl), substituted lower alkyl, lower alkoxy (preferably methoxy), and cyano.
- i. Preferably R5, R6, and R8 are hydrogen.
- 1. More preferably R7 is halo or cyano, especially fluoro or chloro, and most preferably chloro.
- i. Preferably R5, R6, and R8 are hydrogen.
- d. In the case of formulae 1a and 1d, especially those where R3 or R3" is aryl (preferably phenyl), substituted aryl (preferably lower alkyl- or lower alkoxy-substituted phenyl), alkylaryl (preferably benzyl and phenylviny), alkylheteroaryl, oxaalkylaryl (preferably phenoxy lower alkyl), oxaalkylheteroaryl, substituted alkylaryl (preferably substituted benzyl and substituted phenylviny), substituted alkylheteroaryl, substituted oxaalkylaryl (preferably substituted phenoxy lower alkyl), or substituted oxaalkylheteroaryl.
- i. Most preferably those where R3 or R3" is aryl, substituted aryl, lower alkylaryl or substituted lower alkylaryl.
- a. Especially where the stereogenic center to which R2 and R2' are attached is of the R configuration, where R2' is hydrogen.
- 2. Any of formulae 1a, 1b, 1c or 1d, where the stereogenic center to which R2 and R2' are attached is of the R configuration, particularly where R2, is hydrogen:
- a. Especially where R2 is lower alkyl (preferably ethyl, i-proyl, c-propyl or t-butyl).
- i. Most preferably where R2 is i-propyl.
- b. Especially where R4 is substituted alkyl (preferably a primary-, secondary-or tertiary-amino-substituted lower alkyl).
- i. Most preferably where R4 is primary amino-lower alkyl.
- c. Especially where R5, R6, and R8 are hydrogen.
- i. Most preferably where R7 is hydrogen, halo (particularly chloro or fluoro), lower alkyl (particularly methyl), substituted lower alkyl, lower alkoxy (particularly methoxy), or cyano
- 1. Especialy where R7 is chloro.
- i. Most preferably where R7 is hydrogen, halo (particularly chloro or fluoro), lower alkyl (particularly methyl), substituted lower alkyl, lower alkoxy (particularly methoxy), or cyano
- a. Especially where R2 is lower alkyl (preferably ethyl, i-proyl, c-propyl or t-butyl).
- 3. Any of formulae 1a, 1b, 1c or 1d where R7 is hydrogen, halo (preferably chloro or fluoro), lower alkyl (preferably methyl), substituted lower alkyl, lower alkoxy (preferably methoxy), or cyano.
- a. Especially those where R7 is halo or cyano.
- b. Especially those where R5, R6, and R8 are hydrogen.
- i. Most preferably those where R7 is chloro.
- 4. Any of formulae 1b or 1c, where R4 is substituted alkyl (preferably a primary-, secondary- or tertiary-amino-substituted lower alkyl, especially primary-amino lower alkyl).
- a. Especially where the stereogenic center to which R2 and R2' are attached is of the R configuration, particularly where R2' is hydrogen:
- i. Especially where R2 is lower alkyl (preferably ethyl, i-proyl, c-propyl or t-butyl).
- 1. Most preferably where R2 is i-propyl.
- i. Especially where R2 is lower alkyl (preferably ethyl, i-proyl, c-propyl or t-butyl).
- b. Especially where R5, R6, and R8 are hydrogen.
- i. Most preferably where R7 is hydrogen, halo (particularly chloro or fluoro), lower alkyl (particularly methyl), substituted lower alkyl, lower alkoxy (particularly methoxy), or cyano
- 1. Especialy where R7 is chloro.
- i. Most preferably where R7 is hydrogen, halo (particularly chloro or fluoro), lower alkyl (particularly methyl), substituted lower alkyl, lower alkoxy (particularly methoxy), or cyano
- a. Especially where the stereogenic center to which R2 and R2' are attached is of the R configuration, particularly where R2' is hydrogen:
- Most preferred for the compounds, pharmaceutical formulations, methods of manufacture and use of the present invention is formula 1a incorporating the following combinations and permutations of substituent groups (sub-grouped, respectively, in increasing order of preference):
- 1. R1 is alkylaryl or substituted alkylaryl (preferably benzyl or substituted benzyl; most preferably benzyl).
- a. Especially where the stereogenic center to which R2 and R2' are attached is of the R configuration, where R2' is hydrogen.
- i. Particularly those where R2 is lower alkyl (preferably ethyl, i-propyl, c-propyl or t-butyl).
- 1. Most preferably those where R2 is i-propyl.
- i. Particularly those where R2 is lower alkyl (preferably ethyl, i-propyl, c-propyl or t-butyl).
- b. Especially those where R3 is aryl (preferably phenyl), substituted aryl (preferably lower alkyl-, lower alkoxy- and/or halo-substituted phenyl), alkylaryl (preferably benzyl and phenylviny), alkylheteroaryl, oxaalkylaryl (preferably phenoxy lower alkyl), oxaalkylheteroaryl, substituted alkylaryl (preferably substituted benzyl and substituted phenylviny), substituted alkylheteroaryl, substituted oxaalkylaryl (preferably substituted phenoxy lower alkyl), or substituted oxaalkylheteroaryl.
- i. Particularly those where R3 is aryl, substituted aryl, lower alkylaryl, substituted lower alkylaryl, oxa(lower)alkylaryl.
- 1. Most preferably those where R3 is methyl- and/or halo-substituted phenyl.
- i. Particularly those where R3 is aryl, substituted aryl, lower alkylaryl, substituted lower alkylaryl, oxa(lower)alkylaryl.
- c. Especially those where R4 is substituted alkyl (preferably a primary-, secondary- or tertiary-amino-substituted lower alkyl).
- i. Particularly those where R4 is a primary-amino-substituted lower alkyl.
- 1. Most preferably those where R4 is 3-amino-n-propyl.
- i. Particularly those where R4 is a primary-amino-substituted lower alkyl.
- d. Especially those where R5, R6, R7 and R8 are chosen from hydrogen, halo (preferably chloro and fluoro), lower alkyl (preferably methyl), substituted lower alkyl, lower alkoxy (preferably methoxy), and cyano.
- i. Particularly those where R5, R6, R7 and R8 are hydrogen, halo, lower alkyl or cyano.
- 1. Most preferably those where R5, R6 and R8 are hydrogen.
- a. Especially those where R7 is halo or cyano (most preferably chloro).
- 2. Especially those where R7 is halo or cyano (most preferably chloro).
- 1. Most preferably those where R5, R6 and R8 are hydrogen.
- i. Particularly those where R5, R6, R7 and R8 are hydrogen, halo, lower alkyl or cyano.
- a. Especially where the stereogenic center to which R2 and R2' are attached is of the R configuration, where R2' is hydrogen.
- 2. Where the stereogenic center to which R2 and R2' are attached is of the R configuration (preferably where R2' is hydrogen).
- a. Especially those where R2 is lower alkyl (preferably ethyl, i-propyl, c-propyl or t-butyl).
- i. Most preferably those where R2 is i-propyl.
- b. Especially those where R3 is aryl (preferably phenyl), substituted aryl (preferably lower alkyl-, lower alkoxy-, and/or halo-substituted phenyl), alkylaryl (preferably benzyl and phenylviny), alkylheteroaryl, oxaalkylaryl (preferably phenoxy lower alkyl), oxaalkylheteroaryl, substituted alkylaryl (preferably substituted benzyl and substituted phenylviny), substituted alkylheteroaryl, substituted oxaalkylaryl (preferably substituted phenoxy lower alkyl), or substituted oxaalkylheteroaryl.
- i. Particularly those where R3 is aryl, substituted aryl, lower alkylaryl, substituted lower alkylaryl, oxa(lower)alkylaryl.
- 1. Most preferably those where R3 is methyl- and/or halo-substituted phenyl.
- i. Particularly those where R3 is aryl, substituted aryl, lower alkylaryl, substituted lower alkylaryl, oxa(lower)alkylaryl.
- c. Especially those where R4 is substituted alkyl (preferably a primary-, secondary- or tertiary-amino-substituted lower alkyl).
- i. Particularly those where R4 is a primary-amino-substituted lower alkyl.
- 1. Most preferably those where R4 is 3-amino-n-propyl.
- i. Particularly those where R4 is a primary-amino-substituted lower alkyl.
- d. Especially those where R5, R6, R7 and R8 are chosen from hydrogen, halo (preferably chloro and fluoro), lower alkyl (preferably methyl), substituted lower alkyl, lower alkoxy (preferably methoxy), and cyano.
- i. Particularly those where R5, R6, R7 and R8 are hydrogen, halo or lower alkyl.
- 1. Most preferably those where R5, R6 and R8 are hydrogen.
- a. Especially those where R7 is halo or cyano (most preferably chloro).
- 2. Especially those where R7 is halo or cyano (most preferably chloro).
- 1. Most preferably those where R5, R6 and R8 are hydrogen.
- i. Particularly those where R5, R6, R7 and R8 are hydrogen, halo or lower alkyl.
- a. Especially those where R2 is lower alkyl (preferably ethyl, i-propyl, c-propyl or t-butyl).
- 3. R3 is aryl (preferably phenyl), substituted aryl (preferably lower alkyl-, lower alkoxy-, and/or halo-substituted phenyl), alkylaryl (preferably benzyl and phenylviny), alkylheteroaryl, oxaalkylaryl (preferably phenoxy lower alkyl), oxaalkylheteroaryl, substituted alkylaryl (preferably substituted benzyl and substituted phenylviny), substituted alkylheteroaryl, substituted oxaalkylaryl (preferably substituted phenoxy lower alkyl), or substituted oxaalkylheteroaryl.
- a. Especially those where R3 is aryl, substituted aryl, lower alkylaryl, substituted lower alkylaryl, oxa(lower)alkylaryl.
- i. Most preferably those where R3 is methyl- and/or halo-substituted phenyl.
- b. Especially those where R4 is substituted alkyl (preferably a primary-, secondary- or tertiary-amino-substituted lower alkyl).
- i. Particularly those where R4 is a primary-amino-substituted lower alkyl.
- 1. Most preferably those where R4 is 3-amino-n-propyl.
- i. Particularly those where R4 is a primary-amino-substituted lower alkyl.
- c. Especially those where R5, R6, R7 and R8 are chosen from hydrogen, halo (preferably chloro and fluoro), lower alkyl (preferably methyl), substituted lower alkyl, lower alkoxy (preferably methoxy), and cyano.
- i. Particularly those where R5, R6, R7 and R8 are hydrogen, halo, lower alkyl or cyano.
- 1. Most preferably those where R5, R6 and R8 are hydrogen.
- a. Especially those where R7 is halo or cyano (most preferably chloro).
- 2. Especially those where R7 is halo or cyano (most preferably chloro).
- 1. Most preferably those where R5, R6 and R8 are hydrogen.
- i. Particularly those where R5, R6, R7 and R8 are hydrogen, halo, lower alkyl or cyano.
- a. Especially those where R3 is aryl, substituted aryl, lower alkylaryl, substituted lower alkylaryl, oxa(lower)alkylaryl.
- 4. R4 is substituted alkyl (preferably a primary-, secondary- or tertiary-amino-substituted lower alkyl).
- a. Particularly those where R4 is a primary-amino-substituted lower alkyl.
- i. Most preferably those where R4 is 3-amino-n-propyl.
- b. Especially those where R5, R6, R7 and R8 are chosen from hydrogen, halo (preferably chloro and fluoro), lower alkyl (preferably methyl), substituted lower alkyl, lower alkoxy (preferably methoxy), and cyano.
- i. Particularly those where R5, R6, R7 and R8 are hydrogen, halo, lower alkyl or cyano.
- 1. Most preferably those where R5, R6 and Rs are hydrogen.
- a. Especially those where R7 is halo or cyano (most preferably chloro).
- 2. Especially those where R7 is halo or cyano (most preferably chloro).
- 1. Most preferably those where R5, R6 and Rs are hydrogen.
- i. Particularly those where R5, R6, R7 and R8 are hydrogen, halo, lower alkyl or cyano.
- a. Particularly those where R4 is a primary-amino-substituted lower alkyl.
- 5. R5, R6, R7 and R8 are chosen from hydrogen, halo (preferably chloro and fluoro), lower alkyl (preferably methyl), substituted lower alkyl, lower alkoxy (preferably methoxy), and cyano.
- a. Especially those where R5, R6, R7 and R8 are hydrogen, halo, lower alkyl or cyano.
- i. Most preferably those where R5, R6 and R8 are hydrogen.
- 1. Especially those where R7 is halo or cyano (most preferably chloro).
- ii. Especially those where R7 is halo or cyano (most preferably chloro).
- i. Most preferably those where R5, R6 and R8 are hydrogen.
- a. Especially those where R5, R6, R7 and R8 are hydrogen, halo, lower alkyl or cyano.
- Especially preferred for the compounds, pharmaceutical formulations, methods of manufacture and use of the present invention is formula 1a where R1 is alkylaryl or substituted alkylaryl (particularly benzyl or substituted benzyl), R2 is lower alkyl (particularly i-propyl), R2' is hydrogen, R3 is substituted aryl (particularly methyl-and/or halo-substituted phenyl), R4 is substituted alkyl (particularly 3-amino-n-propyl), R5, R6 and R8 are hydrogen, and R7 is halo or cyano (particularly chloro), where the stereogenic center to which R2 and R2' are attached is of the R configuration.
- The compositions of the invention are synthesized as outlined below, utilizing techniques well known in the art. For example, as described in Ager et al., J. of Med. Chem., 20:379-386 (1977), hereby incorporated by reference, quinazolinones can be obtained by acid-catalyzed condensation of N-acylanthranilic acids with aromatic primary amines. Other processes for preparing quinazolinones are described in U.S. Patent applications 5,783,577, 5,922,866 and 5,187,167, all of which are incorporated by reference.
The compositions of the invention may be made as shown in Figures 1, 2, 4 and 5. Compounds of formulae 1d are made in analogous fashion to Figure 1, except that the acyl halide in the final step is replaced by an alkyl halide. - Once made, the compositions of the invention find use in a variety of applications. As will be appreciated by those in the art, mitosis may be altered in a variety of ways; that is, one can affect mitosis either by increasing or decreasing the activity of a component in the mitotic pathway. Stated differently, mitosis may be affected (e.g., disrupted) by disturbing equilibrium, either by inhibiting or activating certain components. Similar approaches may be used to alter meiosis.
- In a preferred embodiment, the compositions of the invention are used to modulate mitotic spindle formation, thus causing prolonged cell cycle arrest in mitosis. By "modulate" herein is meant altering mitotic spindle formation, including increasing and decreasing spindle formation. By "mitotic spindle formation" herein is meant organization of microtubules into bipolar structures by mitotic kinesins. By "mitotic spindle dysfunction" herein is meant mitotic arrest and monopolar spindle formation.
- The compositions of the invention are useful to bind to and/or modulate the activity of a mitotic kinesin, KSP. In a preferred embodiment, the KSP is human KSP, although KSP kinesins from other organisms may also be used. In this context, modulate means either increasing or decreasing spindle pole separation, causing malformation, i.e., splaying, of mitotic spindle poles, or otherwise causing morphological perturbation of the mitotic spindle. Also included within the definition of KSP for these purposes are variants and/or fragments of KSP. See U.S. Patent Application "Methods of Screening for Modulators of Cell Proliferation and Methods of Diagnosing Cell Proliferation States", filed Oct. 27, 1999 (U.S. Serial Number 09/428,156), hereby incorporated by reference in its entirety. In addition, other mitotic kinesins may be used in the present invention. However, the compositions of the invention have been shown to have specificity for KSP.
- For assay of activity, generally either KSP or a compound according to the invention is non-diffusably bound to an insoluble support having isolated sample receiving areas (e.g., a microtiter plate, an array, etc.). The insoluble support may be made of any composition to which the compositions can be bound, is readily separated from soluble material, and is otherwise compatible with the overall method of screening. The surface of such supports may be solid or porous and of any convenient shape. Examples of suitable insoluble supports include microtiter plates, arrays, membranes and beads. These are typically made of glass, plastic (e.g., polystyrene), polysaccharides, nylon or nitrocellulose, Teflon™, etc. Microtiter plates and arrays are especially convenient because a large number of assays can be carried out simultaneously, using small amounts of reagents and samples. The particular manner of binding of the composition is not crucial so long as it is compatible with the reagents and overall methods of the invention, maintains the activity of the composition and is nondiffusable. Preferred methods of binding include the use of antibodies (which do not sterically block either the ligand binding site or activation sequence when the protein is bound to the support), direct binding to "sticky" or ionic supports, chemical crosslinking, the synthesis of the protein or agent on the surface, etc. Following binding of the protein or agent, excess unbound material is removed by washing. The sample receiving areas may then be blocked through incubation with bovine serum albumin (BSA), casein or other innocuous protein or other moiety.
- The antimitotic agents of the invention may be used on their own to modulate the activity of a mitotic kinesin, particularly KSP. In this embodiment, the mitotic agents of the invention are combined with KSP and the activity of KSP is assayed. Kinesin activity is known in the art and includes one or more kinesin activities. Kinesin activities include the ability to affect ATP hydrolysis; microtubule binding; gliding and polymerization/depolymerization (effects on microtubule dynamics); binding to other proteins of the spindle; binding to proteins involved in cell-cycle control; serving as a substrate to other enzymes; such as kinases or proteases; and specific kinesin cellular activities such as spindle pole separation.
- Methods of performing motility assays are well known to those of skill in the art. [See e.g., Hall, et al. (1996), Biophys. J., 71: 3467-3476, Turner et al., 1996, AnaL Biochem. 242 (1):20-5; Gittes et al., 1996, Biophys. J. 70(1): 418-29; Shirakawa et al., 1995, J. Exp. BioL 198: 1809-15; Winkelmann et al., 1995, Biophys. J. 68: 2444-53; Winkelmann et al., 1995, Biophys. J. 68: 72S.]
- Methods known in the art for determining ATPase hydrolysis activity also can be used. Preferably, solution based assays are utilized. U.S. application 09/314,464, filed May 18, 1999, hereby incorporated by reference in its entirety, describes such assays. Alternatively, conventional methods are used. For example, Pi release from kinesin can be quantified. In one preferred embodiment, the ATPase hydrolysis activity assay utilizes 0.3 M PCA (perchloric acid) and malachite green reagent (8.27 mM sodium molybdate II, 0.33 mM malachite green oxalate, and 0.8 mM Triton X-1 00). To perform the assay, 10 µL of reaction is quenched in 90 µL of cold 0.3 M PCA. Phosphate standards are used so data can be converted to mM inorganic phosphate released. When all reactions and standards have been quenched in PCA, 100 µL of malachite green reagent is added to the relevant wells in e.g., a microtiter plate. The mixture is developed for 10-15 minutes and the plate is read at an absorbance of 650 nm. If phosphate standards were used, absorbance readings can be converted to mM Pi and plotted over time. Additionally, ATPase assays known in the art include the luciferase assay.
- ATPase activity ofkinesin motor domains also can be used to monitor the effects of modulating agents. In one embodiment ATPase assays of kinesin are performed in the absence of microtubules. In another embodiment, the ATPase assays are performed in the presence of microtubules. Different types of modulating agents can be detected in the above assays. In a preferred embodiment, the effect of a modulating agent is independent of the concentration of microtubules and ATP. In another embodiment, the effect of the agents on kinesin ATPase can be decreased by increasing the concentrations of ATP, microtubules or both. In yet another embodiment, the effect of the modulating agent is increased by increasing concentrations of ATP, microtubules or both.
- Agents that modulate the biochemical activity of KSP in vitro may then be screened in vivo. Methods for such agents in vivo include assays of cell cycle distnbution, cell viability, or the presence, morphology, activity, distribution, or amount of mitotic spindles. Methods for monitoring cell cycle distribution of a cell population, for example, by flow cytometry, are well known to those skilled in the art, as are methods for determining cell viability. See for example, U.S. Patent Application "Methods of Screening for Modulators of Cell Proliferation and Methods of Diagnosing Cell Proliferation States," filed Oct. 22, 1999, serial number 09/428,156, hereby incorporated by reference in its entirety.
- In addition to the assays described above, microscopic methods for monitoring spindle formation and malformation are well known to those of skill in the art (see, e.g., Whitehead and Rattner (1998), J. Cell Sci. 111:2551-61; Galgio et al, (1996) J. Cell biol., 135:399-414).
- The compositions of the invention inhibit the KSP kinesin. One measure of inhibition is IC50, defined as the concentration of the composition at which the activity of KSP is decreased by fifty percent. Preferred compositions have IC50's of less than about 1 mM, with preferred embodiments having IC50's of less than about 100 µM, with more preferred embodiments having IC50's of less than about 10 µM, with particularly preferred embodiments having IC50's of less than about 1 µM, and especially preferred embodiments having IC50's of less than about 100 nM, and with the most preferred embodiments having IC50's of less than about 10 nM. Measurement of IC50 is done using an ATPase assay.
- Another measure of inhibition is Ki. For compounds with IC50's less than 1 µM, the Ki or Kd is defined as the dissociation rate constant for the interaction of the quinazolinone with KSP. Preferred compounds have Ki's of less than about 100 µM, with preferred embodiments having Ki's of less than about 10 µM, and particularly preferred embodiments having Ki's of less than about 1 µM and especially preferred embodiments having Ki's of less than about 100 nM, and with the most preferred embodiments having Ki's of less than about 10 nM. The Ki for a compound is determined from the IC50 based on three assumptions. First, only one compound molecule binds to the enzyme and there is no cooperativity. Second, the concentrations of active enzyme and the compound tested are known (i.e., there are no significant amounts of impurities or inactive forms in the preparations). Third, the enzymatic rate of the enzyme-inhibitor complex is zero. The rate (i.e., compound concentration) data are fitted to the equation:

- Where V is the observed rate, Vmax is the rate of the free enzyme, I0 is the inhibitor concentration, E0 is the enzyme concentration, and Kd is the dissociation constant of the enzyme-inhibitor complex.
- Another measure of inhibition is GI50, defined as the concentration of the compound that results in a decrease in the rate of cell growth by fifty percent. Preferred compounds have GI50's of less than about 1 mM. The level of preferability of embodiments is a function of their GI50 : those having GI50's of less than about 20 µM are more preferred; those having GI50's of 10 µM more so; those having GI50 of less than about 1 µM more so; those having GI50's of 100 nM more so; those having GI50 of less than about 10 nM even more so. Measurement of GI50 is done using a cell proliferation assay.
- The compositions of the invention are used to treat cellular proliferation diseases. Disease states which can be treated by the methods and compositions provided herein include, but are not limited to, cancer (further discussed below), autoimmune disease, arthritis, graft rejection, inflammatory bowel disease, proliferation induced after medical procedures, including, but not limited to, surgery, angioplasty, and the like. It is appreciated that in some cases the cells may not be in a hyper or hypo proliferation state (abnormal state) and still require treatment. For example, during wound healing, the cells may be proliferating "normally", but proliferation enhancement may be desired. Similarly, as discussed above, in the agriculture arena, cells may be in a "normal" state, but proliferation modulation may be desired to enhance a crop by directly enhancing growth of a crop, or by inhibiting the growth of a plant or organism which adversely affects the crop. Thus, in one embodiment, the invention herein includes application to cells or individuals afflicted or impending affliction with any one of these disorders or states.
- The compositions and methods provided herein are particularly deemed useful for the treatment of cancer including solid tumors such as skin, breast, brain, cervical carcinomas, testicular carcinomas, etc. More particularly, cancers that may be treated by the compositions and methods of the invention include, but are not limited to: Cardiac: sarcoma (angiosarcoma, fibrosarcoma, rhabdomyosarcoma, liposarcoma), myxoma, rhabdomyoma, fibroma, lipoma and teratoma; Lung: bronchogenic carcinoma (squamous cell, undifferentiated small cell, undifferentiated large cell, adenocarcinoma), alveolar (bronchiolar) carcinoma, bronchial adenoma, sarcoma, lymphoma, chondromatous hamartoma, mesothelioma; Gastrointestinal: esophagus (squamous cell carcinoma, adenocarcinoma, leiomyosarcoma, lymphoma), stomach (carcinoma, lymphoma, leiomyosarcoma), pancreas (ductal adenocarcinoma, insulinoma, glucagonoma, gastrinoma, carcinoid tumors, vipoma), small bowel (adenocarcinoma, lymphoma, carcinoid tumors, Karposi's sarcoma, leiomyoma, hemangioma, lipoma, neurofibroma, fibroma), large bowel (adenocarcinoma, tubular adenoma, villous adenoma, hamartoma, leiomyoma); Genitourinary tract: kidney (adenocarcinoma, Wilm's tumor [nephroblastoma], lymphoma, leukemia), bladder and urethra (squamous cell carcinoma, transitional cell carcinoma, adenocarcinoma), prostate (adenocarcinoma, sarcoma), testis (seminoma, teratoma, embryonal carcinoma, teratocarcinoma, choriocarcinoma, sarcoma, interstitial cell carcinoma, fibroma, fibroadenoma, adenomatoid tumors, lipoma); Liver: hepatoma (hepatocellular carcinoma), cholangiocarcinoma, hepatoblastoma, angiosarcoma, hepatocellular adenoma, hemangioma; Bone: osteogenic sarcoma (osteosarcoma), fibrosarcoma, malignant fibrous histiocytoma, chondrosarcoma, Ewing's sarcoma, malignant lymphoma (reticulum cell sarcoma), multiple myeloma, malignant giant cell tumor chordoma, osteochronfroma (osteocartilaginous exostoses), benign chondroma, chondroblastoma, chondromyxofibroma, osteoid osteoma and giant cell tumors; Nervous system: skull (osteoma, hemangioma, granuloma, xanthoma, osteitis deformans), meninges (meningioma, meningiosarcoma, gliomatosis), brain (astrocytoma, medulloblastoma, glioma, ependymoma, germinoma [pinealoma], glioblastoma multiform, oligodendroglioma, schwannoma, retinoblastoma, congenital tumors), spinal cord neurofibroma, meningioma, glioma, sarcoma); Gynecological: uterus (endometrial carcinoma), cervix (cervical carcinoma, pre-tumor cervical dysplasia), ovaries (ovarian carcinoma [serous cystadenocarcinoma, mucinous cystadenocarcinoma, unclassified carcinoma], granulosa-thecal cell tumors, Sertoli-Leydig cell tumors, dysgerminoma, malignant teratoma), vulva (squamous cell carcinoma, intraepithelial carcinoma, adenocarcinoma, fibrosarcoma, melanoma), vagina (clear cell carcinoma, squamous cell carcinoma, botryoid sarcoma (embryonal rhabdomyosarcoma], fallopian tubes (carcinoma); Hematologic: blood (myeloid leukemia [acute and chronic], acute lymphoblastic leukemia, chronic lymphocytic leukemia, myeloproliferative diseases, multiple myeloma, myelodysplastic syndrome), Hodgkin's disease, non-Hodgkin's lymphoma [malignant lymphoma]; Skin: malignant melanoma, basal cell carcinoma, squamous cell carcinoma, Karposi's sarcoma, moles dysplastic nevi, lipoma, angioma, dermatofibroma, keloids, psoriasis; and Adrenal glands: neuroblastoma. Thus, the term "cancerous cell" as provided herein, includes a cell afflicted by any one of the above-identified conditions.
- Accordingly, the compositions of the invention are administered to cells. By "administered" herein is meant administration of a therapeutically effective dose of the mitotic agents of the invention to a cell either in cell culture or in a patient. By "therapeutically effective dose" herein is meant a dose that produces the effects for which it is administered. The exact dose will depend on the purpose of the treatment, and will be ascertainable by one skilled in the art using known techniques. As is known in the art, adjustments for systemic versus localized delivery, age, body weight, general health, sex, diet, time of administration, drug interaction and the severity of the condition may be necessary, and will be ascertainable with routine experimentation by those skilled in the art. By "cells" herein is meant almost any cell in which mitosis or meiosis can be altered.
- A "patient" for the purposes of the present invention includes both humans and other animals, particularly mammals, and other organisms. Thus the methods are applicable to both human therapy and veterinary applications. In the preferred embodiment the patient is a mammal, and in the most preferred embodiment the patient is human.
- Mitotic agents having the desired pharmacological activity may be administered in a physiologically acceptable carrier to a patient, as described herein. Depending upon the manner of introduction, the compounds may be formulated in a variety of ways as discussed below. The concentration of therapeutically active compound in the formulation may vary from about 0.1-100 wt.%. The agents may be administered alone or in combination with other treatments, i.e., radiation, or other chemotherapeutic agents.
- In a preferred embodiment, the pharmaceutical compositions are in a water soluble form, such as pharmaceutically acceptable salts, which is meant to include both acid and base addition salts. "Pharmaceutically acceptable acid addition salt" refers to those salts that retain the biological effectiveness of the free bases and that are not biologically or otherwise undesirable, formed with inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid and the like, and organic acids such as acetic acid, propionic acid, glycolic acid, pyruvic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, p-toluenesulfonic acid, salicylic acid and the like. "Pharmaceutically acceptable base addition salts" include those derived from inorganic bases such as sodium, potassium, lithium, ammonium, calcium, magnesium, iron, zinc, copper, manganese, aluminum salts and the like. Particularly preferred are the ammonium, potassium, sodium, calcium, and magnesium salts. Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines and basic ion exchange resins, such as isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine, and ethanolamine.
- The pharmaceutical compositions can be prepared in various forms, such as granules, tablets, pills, suppositories, capsules, suspensions, salves, lotions and the like. Pharmaceutical grade organic or inorganic carriers and/or diluents suitable for oral and topical use can be used to make up compositions containing the therapeutically-active compounds. Diluents known to the art include aqueous media, vegetable and animal oils and fats. Stabilizing agents, wetting and emulsifying agents, salts for varying the osmotic pressure or buffers for securing an adequate pH value, and skin penetration enhancers can be used as auxiliary agents. The pharmaceutical compositions may also include one or more of the following: carrier proteins such as serum albumin; buffers; fillers such as microcrystalline cellulose, lactose, corn and other starches; binding agents; sweeteners and other flavoring agents; coloring agents; and polyethylene glycol. Additives are well known in the art, and are used in a variety of formulations.
- The administration of the mitotic agents of the present invention can be done in a variety of ways as discussed above, including, but not limited to, orally, subcutaneously, intravenously, intranasally, transdermally, intraperitoneally, intramuscularly, intrapulmonary, vaginally, rectally, or intraocularly. In some instances, for example, in the treatment of wounds and inflammation, the anti-mitotic agents may be directly applied as a solution or spray.
- To employ the compounds of the invention in a method of screening for compounds that bind to KSP kinesin, the KSP is bound to a support, and a compound of the invention (which is a mitotic agent) is added to the assay. Alternatively, the compound of the invention is bound to the support and KSP is added. Classes of compounds among which novel binding agents may be sought include specific antibodies, non-natural binding agents identified in screens of chemical libraries, peptide analogs, etc. Of particular interest are screening assays for candidate agents that have a low toxicity for human cells. A wide variety of assays may be used for this purpose, including labeled in vitro protein-protein binding assays, electrophoretic mobility shift assays, immunoassays for protein binding, functional assays (phosphorylation assays, etc.) and the like.
- The determination of the binding of the mitotic agent to KSP may be done in a number of ways. In a preferred embodiment, the mitotic agent (the compound of the invention) is labeled, for example, with a fluorescent or radioactive moiety and binding determined directly. For example, this may be done by attaching all or a portion of KSP to a solid support, adding a labeled mitotic agent (for example a compound of the invention in which at least one atom has been replaced by a detectable isotope), washing off excess reagent, and determining whether the amount of the label is that present on the solid support. Various blocking and washing steps may be utilized as is known in the art.
- By "labeled" herein is meant that the compound is either directly or indirectly labeled with a label which provides a detectable signal, e.g., radioisotope, fluorescent tag, enzyme, antibodies, particles such as magnetic particles, chemiluminescent tag, or specific binding molecules, etc. Specific binding molecules include pairs, such as biotin and streptavidin, digoxin and antidigoxin etc. For the specific binding members, the complementary member would normally be labeled with a molecule which provides for detection, in accordance with known procedures, as outlined above. The label can directly or indirectly provide a detectable signal.
- In some embodiments, only one of the components is labeled. For example, the kinesin proteins may be labeled at tyrosine positions using 125I, or with fluorophores. Alternatively, more than one component may be labeled with different labels; using 125I for the proteins, for example, and a fluorophor for the mitotic agents.
- The compounds of the invention may also be used as competitors to screen for additional drug candidates. "Candidate bioactive agent" or "drug candidate" or grammatical equivalents as used herein describe any molecule, e.g., protein, oligopeptide, small organic molecule, polysaccharide, polynucleotide, etc., to be tested for bioactivity. They may be capable of directly or indirectly altering the cellular proliferation phenotype or the expression of a cellular proliferation sequence, including both nucleic acid sequences and protein sequences. In other cases, alteration of cellular proliferation protein binding and/or activity is screened. Screens of this sort may be performed either in the presence or absence of microtubules. In the case where protein binding or activity is screened, preferred embodiments exclude molecules already known to bind to that particular protein, for example, polymer structures such as microtubules, and energy sources such as ATP. Preferred embodiments of assays herein include candidate agents which do not bind the cellular proliferation protein in its endogenous native state termed herein as "exogenous" agents. In another preferred embodiment, exogenous agents further exclude antibodies to KSP.
- Candidate agents can encompass numerous chemical classes, though typically they are organic molecules, preferably small organic compounds having a molecular weight of more than 100 and less than about 2,500 daltons. Candidate agents comprise functional groups necessary for structural interaction with proteins, particularly hydrogen bonding and lipophilic binding, and typically include at least an amine, carbonyl, hydroxyl, ether, or carboxyl group, preferably at least two of the functional chemical groups. The candidate agents often comprise cyclical carbon or heterocyclic structures and/or aromatic or polyaromatic structures substituted with one or more of the above functional groups. Candidate agents are also found among biomolecules including peptides, saccharides, fatty acids, steroids, purines, pyrimidines, derivatives, structural analogs or combinations thereof. Particularly preferred are peptides.
- Candidate agents are obtained from a wide variety of sources including libraries of synthetic or natural compounds. For example, numerous means are available for random and directed synthesis of a wide variety of organic compounds and biomolecules, including expression of randomized oligonucleotides. Alternatively, libraries of natural compounds in the form of bacterial, fungal, plant and animal extracts are available or readily produced. Additionally, natural or synthetically produced libraries and compounds are readily modified through conventional chemical, physical and biochemical means. Known pharmacological agents may be subjected to directed or random chemical modifications, such as acylation, alkylation, esterification, amidification to produce structural analogs.
- Competitive screening assays may be done by combining KSP and a drug candidate in a first sample. A second sample comprises a mitotic agent, KSP and a drug candidate. This may be performed in either the presence or absence of microtubules. The binding of the drug candidate is determined for both samples, and a change, or difference in binding between the two samples indicates the presence of an agent capable of binding to KSP and potentially modulating its activity. That is, if the binding of the drug candidate is different in the second sample relative to the first sample, the drug candidate is capable of binding to KSP.
- In a preferred embodiment, the binding of the candidate agent is determined through the use of competitive binding assays. In this embodiment, the competitor is a binding moiety known to bind to KSP, such as an antibody, peptide, binding partner, ligand, etc. Under certain circumstances, there may be competitive binding as between the candidate agent and the binding moiety, with the binding moiety displacing the candidate agent.
- In one embodiment, the candidate agent is labeled. Either the candidate agent, or the competitor, or both, is added first to KSP for a time sufficient to allow binding, if present. Incubations may be performed at any temperature which facilitates optimal activity, typically between 4 and 40°C.
- Incubation periods are selected for optimum activity, but may also be optimized to facilitate rapid high throughput screening. Typically between 0.1 and 1 hour will be sufficient. Excess reagent is generally removed or washed away. The second component is then added, and the presence or absence of the labeled component is followed, to indicate binding.
- In a preferred embodiment, the competitor is added first, followed by the candidate agent. Displacement of the competitor is an indication the candidate agent is binding to KSP and thus is capable of binding to, and potentially modulating, the activity of KSP. In this embodiment, either component can be labeled. Thus, for example, if the competitor is labeled, the presence of label in the wash solution indicates displacement by the agent. Alternatively, if the candidate agent is labeled, the presence of the label on the support indicates displacement.
- In an alternative embodiment, the candidate agent is added first, with incubation and washing, followed by the competitor. The absence of binding by the competitor may indicate the candidate agent is bound to KSP with a higher affinity. Thus, if the candidate agent is labeled, the presence of the label on the support, coupled with a lack of competitor binding, may indicate the candidate agent is capable of binding to KSP.
- It may be of value to identify the binding site of KSP. This can be done in a variety of ways. In one embodiment, once KSP has been identified as binding to the mitotic agent, KSP is fragmented or modified and the assays repeated to identify the necessary components for binding.
- Modulation is tested by screening for candidate agents capable of modulating the activity of KSP comprising the steps of combining a candidate agent with KSP, as above, and determining an alteration in the biological activity of KSP. Thus, in this embodiment, the candidate agent should both bind to KSP (although this may not be necessary), and alter its biological or biochemical activity as defined herein. The methods include both in vitro screening methods and in vivo screening of cells for alterations in cell cycle distribution, cell viability, or for the presence, morpohology, activity, distribution, or amount of mitotic spindles, as are generally outlined above.
- Alternatively, differential screening may be used to identify drug candidates that bind to the native KSP, but cannot bind to modified KSP.
- Positive controls and negative controls may be used in the assays. Preferably all control and test samples are performed in at least triplicate to obtain statistically significant results. Incubation of all samples is for a time sufficient for the binding of the agent to the protein. Following incubation, all samples are washed free of non-specifically bound material and the amount of bound, generally labeled agent determined. For example, where a radiolabel is employed, the samples may be counted in a scintillation counter to determine the amount of bound compound.
- A variety of other reagents may be included in the screening assays. These include reagents like salts, neutral proteins, e.g., albumin, detergents, etc which may be used to facilitate optimal protein-protein binding and/or reduce non-specific or background interactions. Also reagents that otherwise improve the efficiency of the assay, such as protease inhibitors, nuclease inhibitors, anti-microbial agents, etc., may be used. The mixture of components may be added in any order that provides for the requisite binding.
- The following examples serve to more fully describe the manner of using the above-described invention, as well as to set forth the best modes contemplated for carrying out various aspects of the invention. It is understood that these examples in no way serve to limit the true scope of this invention, but rather are presented for illustrative purposes. All references cited herein are incorporated by reference in their entirety.
- The following abbreviations and terms have the indicated meanings throughout:
- Ac
- = acetyl
- BNB
- = 4-bromomethyl-3-nitrobenzoic acid
- Boc
- = t-butyloxy carbonyl
- Bu
- = butyl
- c-
- = cyclo
- CBZ
- = carbobenzoxy = benzyloxycarbonyl
- DBU
- = diazabicyclo[5.4.0]undec-7-ene
- DCM
- = dichloromethane = methylene chloride = CH2Cl2
- DCE
- = dichloroethylene
- DEAD
- = diethyl azodicarboxylate
- DIC
- = diisopropylcarbodiimide
- DIEA
- = N,N-diisopropylethyl amine
- DMAP
- = 4-N,N-dimethylaminopyridine
- DMF
- = N,N-dimethylformamide
- DMSO
- = dimethyl sulfoxide
- DVB
- = 1,4-divinylbenzene
- EEDQ
- = 2-ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline
- Et
- = ethyl
- Fmoc
- = 9-fluorenylmethoxycarbonyl
- GC
- = gas chromatography
- HATU
- = O-(7-Azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HMDS
- = hexamethyldisilazane
- HOAc
- = acetic acid
- HOBt
- = hydroxybenzotriazole
- Me
- = methyl
- mesyl
- = methanesulfonyl
- MTBE
- = methyl t-butyl ether
- NMO
- = N-methylmorpholine oxide
- PEG
- = polyethylene glycol
- Ph
- = phenyl
- PhOH
- = phenol
- PfP
- = pentafluorophenol
- PPTS
- = pyridinium p-toluenesulfonate
- Py
- = pyridine
- PyBroP
- = bromo-tris-pyrrolidino-phosphonium hexafluorophosphate
- rt
- = room temperature
- sat=d
- = saturated
- s-
- = secondary
- t-
- = tertiary
- TBDMS
- = t-butyldimethylsilyl
- TES
- = triethylsilane
- TFA
- = trifluoroacetic acid
- THF
- = tetrahydrofuran
- TMOF
- = trimethyl orthoformate
- TMS
- = trimethylsilyl
- tosyl
- = p-toluenesulfonyl
- Trt .
- = triphenylmethyl
- The general synthesis is shown in Figures 1 and 2.
- To a three-necked, 500 mL round-bottom flask equipped with a thermometer, dropping funnel, and an efficient magnetic stir bar, was added anthranilic acid (1) (0.5 mole, 68.5 g) and dimethyl formamide (250 mL). To this solution was added butyryl chloride (0.55 mole, 57.1 mL) dropwise at such a rate that the temperature of the mixture did not rise above 40°C. The suspension was stirred vigorously at room temperature for at least an additional 3 h. The mixture was poured into water (2000 mL) and stirred for another 1 h. The precipitated product was collected by filtration, washed with cold water, and dried under reduced pressure over P2O5, yielding compound 2 (67.3 g, 65%).
- Compound 2 (51.8 g, 0.25 mole) was dissolved in acetic anhydride (180 mL) in a 500 mL round-bottom flask equipped with a magnetic stir bar, a Claisen-distillation head (with vacuum inlet) and a thermometer. The flask was placed in an oil bath and slowly heated to 170-180°C with vigorous stirring. The acetic acid produced was slowly distilled off under atmospheric pressure. Monitoring the head temperature of the distillation unit was used to follow the progress of the transformation. The reaction mixture was then cooled to 60 °C and the excess of acetic anhydride removed by distillation under reduced pressure (ca. 20 mm Hg). The residue was afterward cooled and the product crystallized. The product was triturated with n-hexane (75 mL) and isolated by filtration to yield 2-propyl-3,1-[4H]benzoxazin-4-one (3) (29.3 g, 62%). The above procedure gave
compound 3 sufficiently pure to use directly in the next step. - Compound 3 (28.4 g, 0.15 mole) and benzylamine (17.5 mL, 0.16 mole) were refluxed in chloroform (50 ml) in a one-neck 250 mL round-bottom flask for 6 h. After complete consumption of
compound 3, the chloroform was evaporated under reduced pressure. Ethylene glycol (100 mL) and NaOH pellets (0.60 g) were added to the residue and the flask equipped with a Claisen-distillation head and a magnetic stir bar. The flask was immersed in an oil bath and reheated to 130-140 °C bath temperature with vigorous stirring and maintained there for 5 h while the water produced was removed by distillation. After completion of the reaction, the clear solution was allowed to cool to room temperature and kept overnight to precipitate the product. The pH of the suspension was adjusted to 7-8 by adding 3% aq. HCl, the crystals were filtered off and washed with cold water, and then recrystallized from isopropanol (or alternatively from acetone) to provide the compound, 2-propyl-3-benzylquinazolin-4-one (compound 4) (28.0 g, 67%). - To a three-neck 250 mL round-bottom flask equipped with a thermometer, dropping funnel, and efficient magnetic stir bar was added compound 4 (27.8 g, 0. 10 mole), anhydrous sodium acetate (10.0 g) and glacial acetic acid (130 mL). Bromine (16.0 g, 0.10 mole) dissolved in acetic acid (10 mL) was added dropwise to the above solution at 40 °C for 1-2 h. After addition was complete, the mixture was poured into water (1500 mL) and stirred for 1-2 h at room temperature. The precipitated product, 2-(1'-bromopropyl)-3-benzylquinazolin-4-one (5) was isolated by filtration, washed with warm water to remove traces of acetic acid, and rinsed with a small amount of isopropanol. Drying yielded compound 5 (33.0 g, 92%).
- Compound 5 (10.7 g, 0.03 mole) and N,N-dimethylethylenediamine (6.6 mL, 0.06 mole) were dissolved in abs. ethanol (60 mL) and heated at reflux for 6 h. After completion of the reaction, the solvent was evaporated under reduced pressure. The residue was dissolved in dichloromethane (150 mL) and washed with 3% aq. NaOH solution (ca. 10-20 mL). The organic layer was dried over MgSO4 and evaporated to dryness under reduced pressure. The remaining oily product was purified by flash chromatography on a short silica gel pad using an eluent of CHCl3-MeOH-aq.NH3, 90:10:0.1, to give the desired compound (5), 2-[1'-(N,N-dimethylethylenediamino)propyl]-3-benzylquinazolin-4-one (6) (6.0 g, 55%).
- A stock solution of compound 5 (1.822 g, 5.0 mmol) was prepared in HPLC grade CHCl3 (0.5 mL). A stock solution of p-flurobenzoyl chloride (160.2 mg, 1 mmol) in
HPLC grade 1,2-dichloroethane (2.0 mL) was prepared in a 2.0 mL volumetric flask. A third solution of triethylamine (2.0 mL of 0.5 M) was prepared inHPLC grade 1,2-dichlorethane. A 100 µL aliquot of each solution was pipetted into a glass reaction vessel using a Beckman Biomet 2000 automated liquid dispenser. The reaction mixture was shaken using a mechanical shaker, sonicated in an ultrasonic water bath, and then incubated overnight at room temperature. The mixture was diluted in CHCl3 (300 µL) and washed with 5% aqueous NaHCO3 and water. The solvent was removed in vacuo to provide compound 6 (65%). The purity of the compound was analyzed by TLC eluted with CH2Cl2-ethanol-concentrated aqueous NH3, 100:10:1. -

- All anhydrous solvents were purchased from Aldrich chemical company in SureSeal® containers. Most reagents were purchase from Aldrich Chemical Company. Abbreviations: DCM, dichloromethane; DIEA, N,N-diisopropylethylamine; DMF, N,N-dimethylformamide; TES, triethylsilane; TFA, trifluoroacetic acid. Array synthesis was conducted in 15 x 75 mm glass round bottom screw-cap vials contained in a 4 x 6 array aluminum synthesis block, sealed with a Teflon-lined rubber membrane. Reagents were added and aqueous extractions performed with single or multichannel pipettors. Filtrations were performed using Whatman/Polyfiltronics 24 well, 10 mL filtration blocks. Evaporation of volatile materials from the array was performed with a Labconco Vortex-Evaporator or by sweeping with a 4 x 6 nitrogen manifold.
- STEP 1: 1,3-Diaminopropane trityl resin (Novabiochem, 1.2 mmol/g) (0.20 g, 0.24 mmol) was weighed into a screw-cap vial and 3 mL of a 1:1 mixture of DMF and chloroform was added. DIEA (0.130 mL, 0.72 mmol) and 2-(1'-bromopropyl)-3-benzylquinazolin-4-one (from Example 1) (0.188 g, 0.48 mmol) were added. The vial was sealed, heated to 70 °C and shaken overnight. The resin was filtered and washed (3 x DCM, 2 x MeOH, 1 x DCM, 2 x ether) and dried under vacuum. A 27 mg aliquot of resin was treated with 5:5:90 TFA:TES:DCM for 15 min and the mixture was filtered and evaporated, resulting in 8 mg (64% yield) of the quinazolinone-diamine intermediate. LCMS analysis showed >80 % purity.
- STEP 2: The resin from
Step 1 was swelled in 3 mL of DCM. DIEA (0.130 mL, 0.72 mmol) and 4-bromobenzyl bromide (0.12 g, 0.48 mmol) were added. The vial was sealed and shaken overnight. LCMS analysis of a cleaved aliquot revealed an approximate 1:1 mixture of starting material and product. Another 0.130 mL of DIEA and 0.12 g of 4-bromobenzyl bromide were added and the mixture was shaken at 70 °C for 8 h. The resin was filtered, washed (as above), and dried under vacuum. - STEP 3: The resin from
Step 2 was twice shaken for 30 min with 5:5:90 TFA:TES:DCM and filtered. The filtrates were combined and evaporated, yielding 140 mg of an orange oil. This material was purified by reverse phase preparative HPLC (acetonitrile-water gradient) to provide 27 mg (17% for 3 steps) of the mono-TFA salt. - STEP 1: 1,2-Diaminoethane trityl resin (Novabiochem, 0.95 mmol/g) (200 g, 1.9 mmol) and 1,3-Diaminopropane trityl resin (Novabiochem, 1.14 mmol/g) (2.0 g, 2.28 mmol) were each placed in different 10 mL polypropylene fritted tubes (Bio-Rad). To each were added 4 mL of DMF, 4 mL of chloroform, 3 eq. of DIEA (1.0 mL and 1.2 mL, respectively) and 2 eq. of 2-(1'-bromopropyl)-3-benzylquinazolin-4-one (from Example 1) (1.5 g and 1.8 g, respectively). The mixtures were shaken at 70 °C overnight. Each mixture was washed (3 x DCM, 2 x MeOH, 1 x DCM, 2 x ether) and dried under vacuum. Analysis of a cleaved aliquot revealed the presence of the appropriate quinazolinone-diamine for each in >90 % purity.
- STEP 2: The quinazolinone ethyl-diamine resin (105 mg, 0.10 mmol) was placed into each of the vials in the first 2 rows of the array, and the quinazolinone propyl-diamine resin (88 mg, 0.10 mmol) was placed into each vial of the last 2 rows of the array. To each vial was added DIEA (0.131 mL, 0.75 mmol). Into each vial of the first 2 rows of the array was added a different amine, and the additions were repeated for the last two rows of the array. The reaction block was shaken at 70 °C overnight. Liquid was removed from each vial by multichannel pipette using fine-pointed gel-well tips, and the resins were washed (2 x DCM, 1 x MeOH, 1 x DCM) and dried under vacuum.
- STEP 3: To each vial of the array was added 2 mL of a 10:5:85 TFA:TES:DCM solution. The reaction block was shaken for 45 min and the mixtures were transferred to a filter block, filtered, and washed twice with 0.75 mL DCM. The solutions were evaporated to yield yellow-to-red oils. These thick oils were triturated twice with ether, dissolved in DCM and treated with 4 M HCl in dioxane to provide the HCl salts (unknown number of salts per compound) as tan-to-white powdery or amorphous solids. Analysis by LCMS showed all to be >75 % pure.
- Six racemic quinazolinones were separated into their enantiomers by chiral chromatography. The chiral chromatography of three of these compounds is described below:
-

- Column - Chiralpak AD, 250 x 4.6 mm (Diacel Inc.). Sample- 0.5 mg/mL in EtOH.
- Conditions - 15 min at 60% EtOH in Hexane,
enantiomer 1 elutes at 4.5 min,enantiomer 2 elutes at 4.9 min. -

- Column - Chiralcel OJ, 250 x 4.6 mm (Diacel Inc.). Sample - 0.5 mg/mL in EtOH. Conditions - 15 min at 10% EtOH in Hexane, (R)-enantiomer elutes at 8.4 min, (S)-enantiomer elutes at 9.6 min.
-
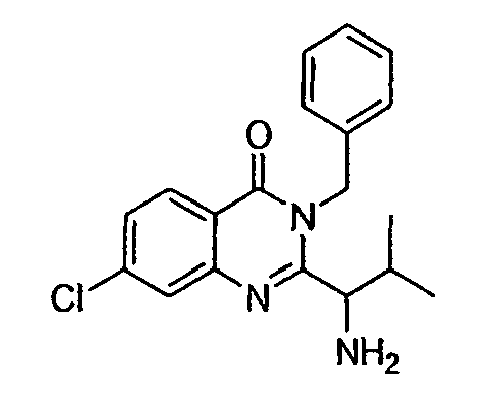
- Column - Chiralpak AD, 250 x 4.6 mm (Diacel Inc.). Sample - 0.5 mg/mL in EtOH. Conditions - 15 min at 70% EtOH in Hexane,
enantiomer 1 elutes at 6.5 min,enantiomer 2 elutes at 8.8 min. - The table below depicts the IC50 activity of the racemate and the enantiomers of three other compounds separated as above. In all three cases, one enantiomer was significantly more potent than the other. By independent chiral synthesis, it appears that the more active enantiomer is the R enantiomer.

- The following two compounds were synthesized as single enantiomers by the route shown in Figure 4. The data indicate that the more active enantiomer is the R enantiomer.

- Intermediate A, prepared in Example 1, can be converted to an intermediate B, which, upon resolution, provides an alternative to the first five steps shown in Figure 4. The process is shown in the scheme below:

- The R enantiomer of B can be crystallized selectively by heating a mixture of B with 1.1 equivalents of D-tartaric acid in a mixture of isopropanol and methanol and then letting the mixture return to room temperature.
- Racemic intermediate B (1.5 g), dissolved in 100 mL of boiling isopropanol, was mixed with 0.8 g of D-tartaric acid in 100 mL of boiling methanol. The mixture was allowed to slowly reach room temperature. After standing overnight, the solid was removed by filtration and rinsed with ethyl acetate and hexanes, and allowed to air dry. The dried solid (0.8 g) was then dissolved in a boiling mixture of 50 mL of isopropanol and 50 mL of methanol and allowed to slowly cool to room temperature. After standing overnight, the resulting solid was removed by filtration and rinsed with ethyl acetate and hexanes, and allowed to air dry. The dried solid was then stirred with saturated sodium bicarbonate for 30 min and extracted with ethyl acetate. The organics were dried (MgSO4), filtered and evaporated to dryness. The resulting clear oil weighed 345 mg. Chiral purity of >95% was determined by conversion of a portion to the S-Mosher amide and examination of the product by 1HNMR.
- The enantiomerically pure compounds below were prepared, according to the remaining steps in Figure 4, from material resulting from the procedure described above using both D- and L-tartaric acid.

- FACS analysis to determine cell cycle stage by measuring DNA content was performed as follows. Skov-3 cells (human ovarian cancer) were split 1:10 for plating in 10cm dishes and grown to subconfluence with RPMI 1640 medium containing 5% fetal bovine serum (FBS). The cells were then treated with either 10nM paclitaxel,
400nM quinazolinone 1, 200nM quinazolinone2, or 0.25% DMSO (vehicle for compounds) for 24 hours. Cells were then rinsed off the plates with PBS containing 5mM EDTA, pelleted, washed once in PBS containing 1% FCS, and then fixed overnight in 85% ethanol at 4°C. Before analysis, the cells were pelleted, washed once, and stained in a solution of 10µg propidium iodide and 250µg of ribonuclease (RNAse) A per milliliter at 37°C for half an hour. Flow cytometry analysis was performed on a Becton-Dickinson FACScan, and data from 10,000 cells per sample was analyzed with Modfit software. The quinazolinone compounds, as well as the known anti-mitotic agent paclitaxel, caused a shift in the population of cells from a GO/G 1 cell cycle stage (2n DNA content) to a G2/M cell cycle stage (4n DNA content). Other compounds of this class were found to have similar effects. - To determine the nature of the G2/M accumulation, human tumor cell lines Skov-3 (ovarian), HeLa (cervical), and A549 (lung) were plated in 96-well plates at densities of 4,000 cells per well (SKOV-3 & HeLa) or 8,000 cells per well (A549), allowed to adhere for 24 hours, and treated with various concentrations of the quinazolinone compounds for 24 hours. Cells were fixed in 4% formaldehyde and stained with anti-tubulin antibodies (subsequently recognized using fluorescently-labeled secondary antibody) and Hoechst dye (which stains DNA).
- Visual inspection revealed that the quinazolinone compounds caused cell cycle arrest in the prometaphase stage of mitosis. DNA was condensed and spindle formation had initiated, but arrested cells uniformly displayed monopolar spindles, indicating that there was an inhibition of spindle pole body separation. Microinjection of anti-KSP antibodies also causes mitotic arrest with arrested cells displaying monopolar spindles.
- Inhibition of Cellular Proliferation in Tumor Cell Lines Treated with Quinazolinone KSP Inhibitors.
- Cells were plated in 96-well plates at densities from 1000-2500 cells/well of a 96-well plate (depending on the cell line) and allowed to adhere/grow for 24 hours. They were then treated with various concentrations of drug for 48 hours. The time at which compounds are added is considered To. A tetrazolium-based assay using the reagent 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) (I.S> Patent No. 5,185,450) (see Promega product catalog #G3580, CellTiter 96® AQueous One Solution Cell Proliferation Assay) was used to determine the number of viable cells at To and the number of cells remaining after 48 hours compound exposure. The number of cells remaining after 48 hours was compared to the number of viable cells at the time of drug addition, allowing for calculation of growth inhibition.
- The growth over 48 hours of cells in control wells that had been treated with vehicle only (0.25% DMSO) is considered 100% growth and the growth of cells in wells with compounds is compared to this. Quinazolinone KSP inhibitors inhibited cell proliferation in human tumor cell lines of the following tumor types: lung (NCI-H460, A549), breast (MDA-MB-231, MCF-7, MCF-7/ADR-RES), colon (HT29, HCT15), ovarian (SKOV-3, OVCAR-3), leukemia (HL-60(TB), K-562), central nervous system (SF-268), renal (A498), osteosarcoma (U2-OS), and cervical (HeLa). In addition, a mouse tumor line (B16, melanoma) was also growth-inhibited in the presence of the quinazolinone compounds.
- A Gi50 was calculated by plotting the concentration of compound in µM vs the percentage of cell growth of cell growth in treated wells. The Gi50 calculated for the compounds is the estimated concentration at which growth is inhibited by 50% compared to control, i.e., the concentration at which:

- All concentrations of compounds are tested in duplicate and controls are averaged over 12 wells. A very similar 96-well plate layout and Gi50 calculation scheme is used by the National Cancer Institute (see Monks, et al., J. NatI. Cancer Inst. 83:757-766 (1991)). However, the method by which the National Cancer Institute quantitates cell number does not use MTS, but instead employs alternative methods.
- Measurement of a composition's IC50 for KSP activity uses an ATPase assay. The following solutions are used:
Solution 1 consists of 3 mM phosphoenolpyruvate potassium salt (Sigma P-7127), 2 mM ATP (Sigma A-3377), 1 mM IDTT (Sigma D-9779), 5 µM paclitaxel (Sigma T-7402), 10 ppm antifoam 289 (Sigma A-8436), 25 mM Pipes/KOH pH 6.8 (Sigma P6757), 2 mM MgCl2 (VWR JT400301), and 1 mM EGTA (Sigma E3889).Solution 2 consists of 1 mM NADH (Sigma N8129), 0.2 mg/ml BSA (Sigma A7906), pyruvate kinase 7U/ml, L-lactate dehydrogenase 10 U/ml (Sigma P0294), 100 nM KSP motor domain, 50 µg/ml microtubules, 1 mM DTT (Sigma D9779), 5 µM paclitaxel (Sigma T-7402), 10 ppm antifoam 289 (Sigma A-8436), 25 mM Pipes/KOH pH 6.8 (Sigma P6757), 2 mM MgC12 (VWR JT4003-01), and 1 mM EGTA (Sigma E3889). Serial dilutions (8-12 two-fold dilutions) of the composition are made in a 96-well microtiter plate (Coming Costar 3695) usingSolution 1. Following serial dilution each well has 50 µl ofSolution 1. The reaction is started by adding 50 µl ofsolution 2 to each well. This may be done with a multichannel pipettor either manually or with automated liquid handling devices. The microtiter plate is then transferred to a microplate absorbance reader and multiple absorbance readings at 340 nm are taken for each well in a kinetic mode. The observed rate of change, which is proportional to the ATPase rate, is then plotted as a function of the compound concentration. For a standard IC50 determination the data acquired is fit by the following four parameter equation using a nonlinear fitting program (e.g., Grafit 4):
- Where y is the observed rate and x the compound concentration.
- The quinazolinone compounds inhibit growth in a variety of cell lines, including cell lines (MCF-7/ADR-RES, HCT1 5) that express P-glycoprotein (also known as Multidrug Resistance, or MDR+), which conveys resistance to other chemotherapeutic drugs, such as pacilitaxel. Therefore, the quinazolinones are anti-mitotics that inhibit cell proliferation, and are not subject to resistance by overexpression of MDR+ by drug-resistant tumor lines.
- Other compounds of this class were found to inhibit cell proliferation, although GI50 values varied. GI50 values for the quinazolinone compounds tested ranged from 200 nM to greater than the highest concentration tested. By this we mean that although most of the compounds that inhibited KSP activity biochemically did inhibit cell proliferation, for some, at the highest concentration tested (generally about 20 µM), cell growth was inhibited less than 50%. Many of the compounds have GI50 values less than 10 µM, and several have GI50 values less than 1 µM. Antiproliferative compounds that have been successfully applied in the clinic to treatment of cancer (cancer chemotherapeutics) have GI50's that vary greatly. For example, in A549 cells, paclitaxel GI50 is 4 nM, doxorubicin is 63 nM, 5-fluorouracil is 1 µM, and hydroxyurea is 500 µM (data provided by National Cancer Institute, Developmental Therapeutic Program, http://dtp.nci.nih.gov/). Therefore, compounds that inhibit cellular proliferation at virtually any concentration may be useful. However, preferably, compounds will have GI50 values of less than 1 mM. More preferably, compounds will have GI50 values of less than 20 µM. Even more preferably, compounds will have GI50 values of less than 10 µM. Further reduction in GI50 values may also be desirable, including compounds with GI50 values of less than 1 µM. Some of the quinazolinone compounds of the invention inhibit cell proliferation with GI50 values from below 200 nM to below 10 nM.
- Female nude mice weighing approximately 20 g were implanted s.c. by trocar with fragments of human tumor carcinomas harvested from s.c. growing tumors in nude mice host. When the tumors were approximately 77 mg in size, the animals were pair matched into treatment and control groups. Each group containted 8 tumored mice, each of which was ear-tagged and followed individually throughout the experiment. Initial doses (10 mL/kg of a 66 mM Citrate buffer, pH 5.0/0.9% Saline / 10% Tween 80 formulation of each test compound having a maximum concentration of 5 mg/mL) were given on
Day 1 following pair matching, dosing at the levels and schedules indicated. - Mice were weighed twice weekly, and tumor measurements were taken by calipers twice weekly, starting on
Day 1. These tumor measurements were converted to mg tumor weight by a well-known formula, W2 x L/2. The experiment was terminated when the control group tumor size reached an average of 1 gram. Upon termination, the mice were weighted, sacrificed and their tumors excised. Tumors were weighted and the mean treated tumor weight per group was calculated. In this model, the change in mean treated tumor weight / the change in mean control tumor weight x 100% (ΔT/ΔC) was subtracted from 100% to give the tumor growth inhibition (TGI) for each group. - Compounds 1-5 (below) were tested by the above-described method, giving the results summarized below in Tables A - D. Other compounds of the present invention show comparable activities when tested by this method.





Table A SKOV3 tumor xenograft Vehicle Compound 1 Taxol Dose & Schedule Daily x 5 80 mg/kg every 3 days x 4 20 mg/kg daily x 5 Route i.v. i.v. i.p. # Mice at Start 8 8 8 Final Tumor Weight (Mean ± SEM) 904.1 ± 126.5 554.3 ± 76.8 90.4 ± 36.0 Tumor Growth Inhibition --- 41.5% 91.7% Mice with Partial Tumor Shrinkage 0 0 4 Mean % Tumor Shrinkage --- --- 27.9% Maximum Weight Loss None None 16.5 % Mortalities 0 1 0 Table B SKOV3 tumor xenograft Vehicle Compound 2 Compound 3Taxol Dose & Schedule Daily x 5 50 mg/kg every 3 days x 4 60 mg/kg daily x 5 20 mg/kg daily x 5 Route i.v. i.v. i.v. i.p. # Mice at Start 8 8 8 8 Final Tumor Weight (Mean ± SEM) 1506.3 ± 227.1 340.8 ± 93.0 806.1 ± 163.8 55.9 ± 29.4 Tumor Growth Inhibition --- 81.5% 48.3% 99.7% Mice with Partial Tumor Shrinkage 0 0 0 7 Mean % Tumor Shrinkage --- --- --- 43.5% Maximum Weight Loss None None 2.76% 7.49 % Mortalities 0 0 1 0 Table C SKOV3 tumor xenograft Vehicle Compound 4 Taxol Dose & Schedule Daily x 5 4 mg/kg weekly x 3 20 mg/kg daily x 5 Route i.v. i.v. i.p. # Mice at Start 8 8 8 Final Tumor Weight (Mean ± SEM) 1191.1 ± 239.6 726.9 ± 147.2 90.6 ± 34.5 Tumor Growth Inhibition --- 40.1% 87.1% Mice with Partial Tumor Shrinkage 0 0 4 Mean % Tumor Shrinkage --- --- 44.4% Maximum Weight Loss None 0.02% 12.26 % Mortalities 1 1 1 Table D SKOV3 tumor xenograft Vehicle Compound 5 Taxol Dose & Schedule Daily x 5 25 mg/kg daily x 5 20 mg/kg daily x 5 Route i.v. i.v. i.p. # Mice at Start 8 8 8 Final Tumor Weight (Mean ± SEM) 1230.4 ± 227.3 405.6 ± 124.8 379.0 ± 154.0 Tumor Growth Inhibition --- 71.0% 73.0% Mice with Partial Tumor Shrinkage 0 1 0 Mean % Tumor Shrinkage --- 56.3% --- Maximum Weight Loss None None 8.77 % Mortalities 0 0 0 - While the present invention has been described with reference to the specific embodiments thereof, it should be understood by those skilled in the art that various changes may be made and equivalents may be substituted without departing from the true spirit and scope of the invention. In addition, many modifications may be made to adapt a particular situation, material, composition of matter, process, process step or steps, to the objective, spirit and scope of the present invention. All such modifications are intended to be within the scope of the claims appended hereto. All patents and publications cited above are hereby incorporated by reference.
Claims (64)
- A method of treating cellular proliferative diseases comprising administering a compound chosen from the group consisting of:
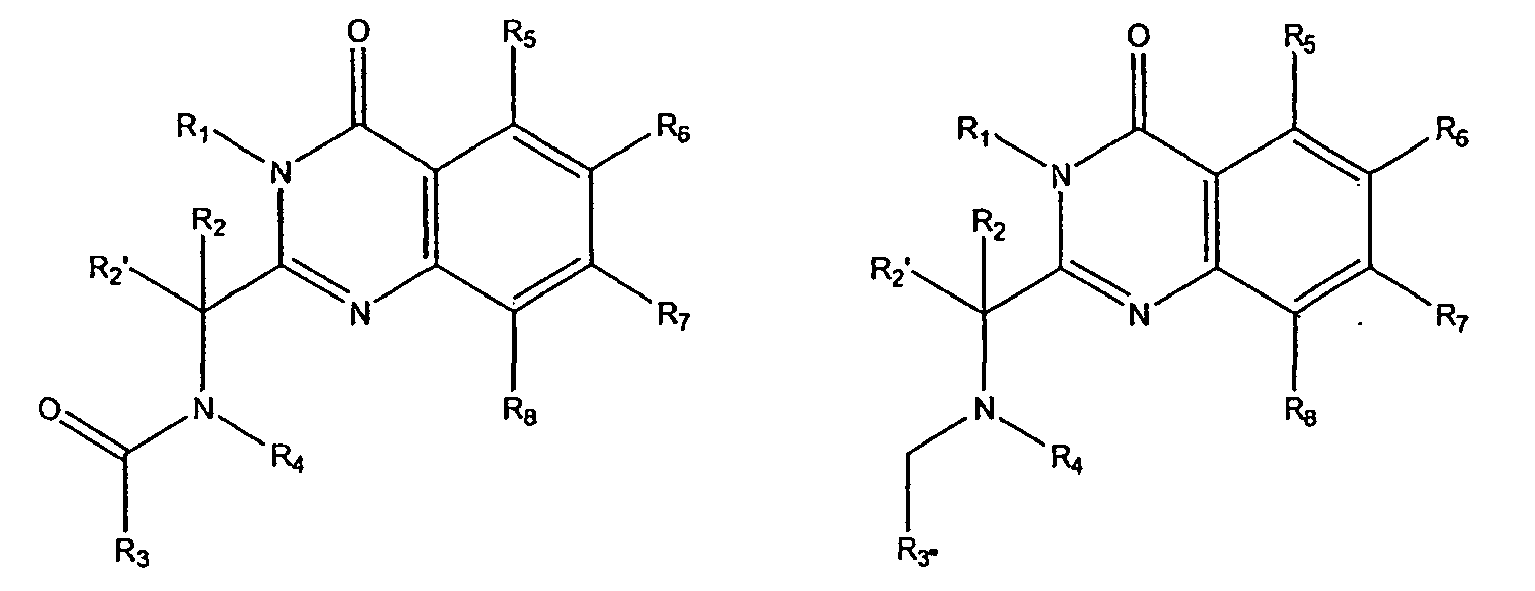
 R1 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R2 and R2' are independently chosen from hydrogen, alkyl, oxaalkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; or R2 and R2' taken together form a 3- to 7-membered ring;R3 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, oxaalkyl, oxaalkylaryl, substituted oxaalkylaryl, oxaalkylheteroaryl, substituted oxaalkylheteroaryl, R15O- and R15-NH-;R3' is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, and R15-NH-;R3" is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R4 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, and R16-alkylene-;R5, R6, R7 and R8 are independently chosen from hydrogen, alkyl, alkoxy, halogen, fluoroalkyl, nitro, cyano, dialkylamino, alkylsulfonyl, alkylsulfonamido, sulfonamidoalkyl, sulfonamidoaryl, alkylthio, carboxyalkyl, carboxamido, aminocarbonyl, aryl and heteroaryl;R15 is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; andR16 is chosen from alkoxy, amino, alkylamino, dialkylamino, N-heterocyclyl and substituted N-heterocyclyl,or a pharmaceutically acceptable salt thereof.
R1 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R2 and R2' are independently chosen from hydrogen, alkyl, oxaalkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; or R2 and R2' taken together form a 3- to 7-membered ring;R3 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, oxaalkyl, oxaalkylaryl, substituted oxaalkylaryl, oxaalkylheteroaryl, substituted oxaalkylheteroaryl, R15O- and R15-NH-;R3' is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, and R15-NH-;R3" is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R4 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, and R16-alkylene-;R5, R6, R7 and R8 are independently chosen from hydrogen, alkyl, alkoxy, halogen, fluoroalkyl, nitro, cyano, dialkylamino, alkylsulfonyl, alkylsulfonamido, sulfonamidoalkyl, sulfonamidoaryl, alkylthio, carboxyalkyl, carboxamido, aminocarbonyl, aryl and heteroaryl;R15 is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; andR16 is chosen from alkoxy, amino, alkylamino, dialkylamino, N-heterocyclyl and substituted N-heterocyclyl,or a pharmaceutically acceptable salt thereof. - A method of treating a disorder associated with KSP kinesin activity comprising administering a compound chosen from the group consisting of:

 R1 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R2 and R2' are independently chosen from hydrogen, alkyl, oxaalkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; or R2 and R2' taken together form a 3- to 7-membered ring;R3 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, oxaalkyl, oxaalkylaryl, substituted oxaalkylaryl, oxaalkylheteroaryl, substituted oxaalkylheteroaryl, R15O-and R15-NH-;R3' is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl and R15-NH-;R3" is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R4 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, and R16-alkylene-;R5, R6, R7 and R8 are independently chosen from hydrogen, alkyl, alkoxy, halogen, fluoroalkyl, nitro, cyano, dialkylamino, alkylsulfonyl, alkylsulfonamido, sulfonamidoalkyl, sulfonamidoaryl, alkylthio, carboxyalkyl, carboxamido, aminocarbonyl, aryl and heteroaryl;R15 is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; andor a pharmaceutically acceptable salt thereof.
R1 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R2 and R2' are independently chosen from hydrogen, alkyl, oxaalkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; or R2 and R2' taken together form a 3- to 7-membered ring;R3 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, oxaalkyl, oxaalkylaryl, substituted oxaalkylaryl, oxaalkylheteroaryl, substituted oxaalkylheteroaryl, R15O-and R15-NH-;R3' is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl and R15-NH-;R3" is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R4 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, and R16-alkylene-;R5, R6, R7 and R8 are independently chosen from hydrogen, alkyl, alkoxy, halogen, fluoroalkyl, nitro, cyano, dialkylamino, alkylsulfonyl, alkylsulfonamido, sulfonamidoalkyl, sulfonamidoaryl, alkylthio, carboxyalkyl, carboxamido, aminocarbonyl, aryl and heteroaryl;R15 is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; andor a pharmaceutically acceptable salt thereof.
R16 is chosen from alkoxy, amino, alkylamino, dialkylamino, N-heterocyclyl and substituted N-heterocyclyl, - A method of inhibiting KSP kinesin comprising contacting KSP kinesin with a compound chosen from the group consisting of:

 R1 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R2 and R2' are independently chosen from hydrogen, alkyl, oxaalkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; or R2 and R2' taken together form a 3- to 7-membered ring;R3 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, oxaalkyl, oxaalkylaryl, substituted oxaalkylaryl, oxaalkylheteroaryl, substituted oxaalkylheteroaryl, R15O-and R15-NH-;R3' is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl and R15-NH-;R3" is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R4 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, and R16-alkylene-;R5, R6, R7 and R8 are independently chosen from hydrogen, alkyl, alkoxy, halogen, fluoroalkyl, nitro, cyano, dialkylamino, alkylsulfonyl, alkylsulfonamido, sulfonamidoalkyl, sulfonamidoaryl, alkylthio, carboxyalkyl, carboxamido, aminocarbonyl, aryl and heteroaryl;R15 is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; andR16 is chosen from alkoxy, amino, alkylamino, dialkylamino, N-heterocyclyl and substituted N-heterocyclyl,or a pharmaceutically acceptable salt thereof.
R1 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R2 and R2' are independently chosen from hydrogen, alkyl, oxaalkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; or R2 and R2' taken together form a 3- to 7-membered ring;R3 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, oxaalkyl, oxaalkylaryl, substituted oxaalkylaryl, oxaalkylheteroaryl, substituted oxaalkylheteroaryl, R15O-and R15-NH-;R3' is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl and R15-NH-;R3" is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R4 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, and R16-alkylene-;R5, R6, R7 and R8 are independently chosen from hydrogen, alkyl, alkoxy, halogen, fluoroalkyl, nitro, cyano, dialkylamino, alkylsulfonyl, alkylsulfonamido, sulfonamidoalkyl, sulfonamidoaryl, alkylthio, carboxyalkyl, carboxamido, aminocarbonyl, aryl and heteroaryl;R15 is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; andR16 is chosen from alkoxy, amino, alkylamino, dialkylamino, N-heterocyclyl and substituted N-heterocyclyl,or a pharmaceutically acceptable salt thereof. - A method according to claim 1, 2 or 3 wherein:R1 is chosen from hydrogen, alkyl, aryl, substituted alkyl, substituted aryl, heteroaryl, substituted heteroaryl, alkylaryl, substituted alkylaryl and substituted alkylheteroaryl;R2 is chosen from hydrogen, alkyl and substituted alkyl;R2' is hydrogen;R3 is chosen from alkyl, substituted alkyl, alkylaryl, heteroaryl, aryl, substituted aryl, substituted hereroaryl, substituted oxaalkylaryl R15O- and R15-NH-;R4 is chosen from alkyl, aryl, alkylaryl, alkylheteroaryl, substituted alkyl, substituted aryl, and R16-alkylene-;R5 is hydrogen;R6, R7 and R8 are independently chosen from hydrogen, halogen, methyl, cyano and trifluoromethyl;R15 is chosen from alkyl, aryl and substituted aryl;R16 is chosen from alkoxy, amino, alkylamino, dialkylamino and N-heterocyclyl.
- A method according to claim 1, 2 or 3 wherein: R2 is chosen from hydrogen, alkyl and substituted alkyl; R2' is hydrogen; and the stereogenic center to which R2 and R2' are attached is of the R configuration.
- A method according to claim 1, 2 or 3 comprising administering a compound of the formula:
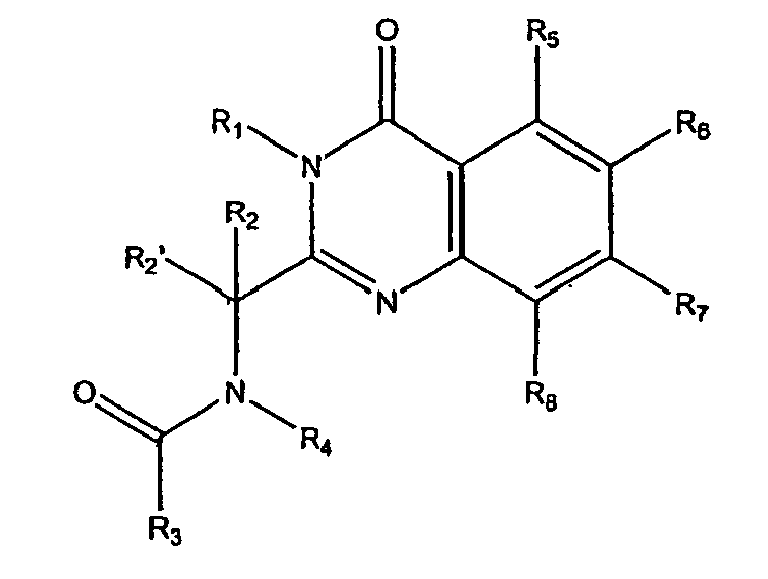
- A method according to claim 6 wherein R1 is chosen from hydrogen, lower alkyl, substituted lower alkyl, aryl, substituted aryl, alkylaryl and substituted alkylaryl.
- A method according to claim 7 wherein R1 is chosen from hydrogen, ethyl, propyl, methoxyethyl, naphthyl, phenyl, bromophenyl, chlorophenyl, methoxyphenyl, ethoxyphenyl, tolyl, dimethylphenyl, chorofluorophenyl, methylchlorophenyl, ethylphenyl, phenethyl, benzyl, chlorobenzyl, methylbenzyl, methoxybenzyl, tetrahydrofuranylmethyl and (ethoxycarbonyl)ethyl.
- A method according to claim 6 wherein R2 is chosen from hydrogen, lower alkyl and substituted lower alkyl; R2' is hydrogen; and the stereogenic center to which R2 and R2' are attached is of the R configuration.
- A method according to claim 9 wherein R2 is chosen from hydrogen, methyl, ethyl, propyl, methylthioethyl, aminobutyl, (CBZ)aminobutyl, cyclohexylmethyl, benzyloxymethyl, methylsulfinylethyl, methylsulfinylmethyl, hydroxymethyl, benzyl and indolylmethyl.
- A method according to claim 6 wherein R3 is chosen from C1-C13 alkyl; substituted lower alkyl; aryl, substituted aryl, alkylaryl, alkylheteroaryl, oxaalkylaryl, oxaalkylheteroaryl, substituted alkylaryl, substituted alkylheteroaryl, substituted oxaalkylaryl, and substituted oxaalkylheteroarlyl.
- A method according to claim 11 wherein R3 is chosen from ethyl, propyl, chloropropyl, butoxy, heptyl, butyl, octyl, tridecanyl, (ethoxycarbonyl)ethyl, dimethylaminoethyl, dimethylaminomethyl, phenyl, naphthyl, halophenyl, dihalophenyl, cyanophenyl, halo(trifluoromethyl)phenyl, chlorophenoxymethyl, methoxyphenyl, carboxyphenyl, ethylphenyl, tolyl, biphenylyl, methylenedioxyphenyl, methylsulfonylphenyl, methoxychlorophenyl, chloronaphthyl, methylhalophenyl, trifluoromethylphenyl, butylphenyl, pentylphenyl, methylnitrophenyl, phenoxymethyl, dimethoxyphenyl, phenylvinyl, nitrochlorophenyl, nitrophenyl, dinitrophenyl, bis(trifluoromethyl)phenyl, benzyloxymethyl, benzyl, furanyl, benzofuranyl, pyridinyl, indolyl, methylpyridinyl, quinolinyl, picolinyl, pyrazolyl, and imidazolyl.
- A method according to claim 6 wherein R3 is R15-NH- and R15 is chosen from lower alkyl; cyclohexyl; phenyl; and phenyl substituted with halo, lower alkyl, loweralkoxy, or lower alkylthio.
- A method according to claim 13 wherein R15 is chosen from isopropyl, butyl, cyclohexyl, phenyl, bromophenyl, dichlorophenyl, methoxyphenyl, ethylphenyl, tolyl, trifluoromethylphenyl and methylthiophenyl.
- A method according to claim 6 wherein R4 is chosen from lower alkyl, substituted lower alkyl, cyclohexyl; phenyl substituted with hydroxy, lower alkoxy or lower alkyl; benzyl; heteroarylmethyl; heteroarylethyl; heteroarylpropyl and R16-alkylene-, wherein R16 is amino, lower alkylamino, di(lower alkyl)amino, lower alkoxy, or N-heterocyclyl.
- A method according to claim 15 wherein R4 is chosen from methyl, ethyl, propyl, butyl, cyclohexyl, carboxyethyl, carboxymethyl, methoxyethyl, hydroxyethyl, hydroxypropyl, dimethylaminoethyl, dimethylaminopropyl, diethylaminoethyl, diethylaminopropyl, aminopropyl, methylaminopropyl, 2,2-dimethyl-3-(dimethylamino)propyl, 1-cyclohexyl-4-(diethylamino)butyl, aminoethyl, aminobutyl, aminopentyl, aminohexyl, aminoethoxyethyl, isopropylaminopropyl, diisopropylaminoethyl, 1-methyl-4-(diethylamino)butyl, (t-Boc)aminopropyl, hydroxyphenyl, benzyl, methoxyphenyl, methylmethoxyphenyl, dimethylphenyl, tolyl, ethylphenyl, (oxopyrrolidinyl)propyl, (methoxycarbonyl)ethyl, benzylpiperidinyl, pyridinylethyl, pyridinylmethyl, morpholinylethyl, morpholinylpropyl, piperidinyl, azetidinylmethyl, azetidinylpropyl, pyrrolidinylethyl, pyrrolidinylpropyl, piperidinylmethyl, piperidinylethyl, imidazolylpropyl, imidazolylethyl, (ethylpyrrolidinyl)methyl, (methylpyrrolidinyl)ethyl, (methylpiperidinyl)propyl, (methylpiperazinyl)propyl, furanylmethyl and indolylethyl.
- A method according to claim 6 wherein
R1 is chosen from lower alkyl, benzyl, substituted benzyl and substituted phenyl;
R2 is chosen from hydrogen, alkyl, substituted lower alkyl and benzyl;
R2' is hydrogen;
R3 is chosen from substituted phenyl and naphthyl;
R4 is chosen from substituted alkyl and R16-alkylene-;
R5 is hydrogen or halo
R6 is hydrogen, methyl or halo;
R7 is hydrogen, halo, lower alkyl, substituted lower alkyl, lower alkoxy or cyano;
R8 is hydrogen or halo; and
R16 is chosen from di(lower alkylamino), (lower alkyl)amino, amino, N-heterocyclyl and substituted N-heterocyclyl. - A method according to claim 1, 2 or 3 wherein
R1 is benzyl or halobenzyl;
R2 is chosen from ethyl and propyl;
R2' is hydrogen;
R3 is substituted phenyl;
R3' is substituted phenyl;
R3" is substituted phenyl;
R4 is -(CH2)mOH or -(CH2)pR16 wherein m is 2 or 3 and p is 1-3;
R5 is hydrogen;
R6 is hydrogen;
R7 is halo;
R8 is hydrogen; and
R16 is chosen from amino, propylamino, and azetidinyl. - A method according to claim 18 wherein the stereogenic center to which R2 and R2' are attached is of the R configuration.
- A method according to claim 1, 2 or 3 comprising administering a compound of formula:

- A method according to claim 20 wherein:R1 is chosen from hydrogen, lower alkyl, substituted lower alkyl, benzyl, substituted benzyl, phenyl, naphthyl and substituted phenyl;R2 is chosen from hydrogen, lower alkyl and substituted lower alkyl and R2' is hydrogen;R3' is chosen from C1-C13 alkyl; phenyl; naphthyl; phenyl substituted with halo, lower alkyl, lower alkoxy, nitro, methylenedioxy, or trifluoromethyl; biphenylyl, benzyl and heteroaryl; andR4 is chosen from lower alkyl, substituted lower alkyl, cyclohexyl; phenyl substituted with hydroxy, lower alkoxy or lower alkyl; benzyl; heteroarylmethyl; heteroarylethyl; heteroarylpropyl and R16-alkylene, whereinR16 is amino, (lower alkyl)amino, di(lower alkyl)amino, lower alkoxy, or N-heterocyclyl.
- A method according to claim 20 wherein
R1 is chosen from lower alkyl, benzyl, substituted benzyl and substituted phenyl;
R2 is hydrogen or lower alkyl;
R2' is hydrogen;
R3' is chosen from substituted phenyl and naphthyl;
R4 is R16-alkylene-, hydroxy lower alkyl or carboxy lower alkyl;
R6 and R7 are chosen from hydrogen and halo;
R5 and R8 are hydrogen;
R16 is chosen from di(lower alkylamino), (lower alkyl)amino, amino, piperidinyl, azetidinyl pyrrolidinyl and morpbolinyl. - A method according to claim 1, 2 or 3 comprising administering a compound of formula:

- A method according to claim 23 wherein:R1 is chosen from hydrogen, lower alkyl, substituted lower alkyl, benzyl, substituted benzyl, phenyl, naphthyl and substituted phenyl;R2 is chosen from hydrogen, lower alkyl and substituted lower alkyl and R2' is hydrogen; andR4 is chosen from lower alkyl, cyclohexyl; phenyl substituted with hydroxy, lower alkoxy or lower alkyl; benzyl; heteroarylmethyl; heteroarylethyl; heteroarylpropyl and R16-alkylene, wherein R16 is di(lower alkyl)amino, alkylamino, amino, lower alkoxy, or N-heterocyclyl.
- A method according to claim 23 wherein
R1 is chosen from lower alkyl, benzyl, substituted benzyl and substituted phenyl;
R2 is hydrogen or lower alkyl;
R2' is hydrogen;
R4 is R16-alkylene-;
R6 and R7 are chosen from hydrogen and halo;
R5 and R8 are hydrogen; and
R16 is chosen from di(lower alkylamino), (lower alkyl)amino, amino, pyrrolidinyl, piperidinyl, imidazolyl and morpholinyl. - A method according to claim 1, 2 or 3 comprising administering a compound of formula:

- A method according to claim 26 wherein:R1 is chosen from hydrogen, lower alkyl, substituted lower alkyl, benzyl, substituted benzyl, phenyl, naphthyl and substituted phenyl;R2 is chosen from hydrogen, lower alkyl and substituted lower alkyl and R2' is hydrogen;R3" is chosen from C1-C13 alkyl; substituted lower alkyl; phenyl; naphthyl; phenyl substituted with halo, lower alkyl, lower alkoxy, nitro, methylenedioxy, or trifluoromethyl; biphenylyl; benzyl and heterocyclyl; andR4 is chosen from lower alkyl, substituted lower alkyl; cyclohexyl; phenyl substituted with hydroxy, lower alkoxy or lower alkyl; benzyl; substituted benzyl; heterocyclyl; heteroarylmethyl; heteroarylethyl; heteroarylpropyl and R16-alkylene, whereinR16 is di(lower alkyl)amino, (lower alkyl)amino, amino, lower alkoxy, or N-heterocyclyl.
- A method according to claim 27 whereinR1 is chosen from lower alkyl, benzyl, substituted benzyl and substituted phenyl;R2 is hydrogen or lower alkyl;R2' is hydrogen;R3" is chosen from substituted phenyl, heterocyclyl and naphthyl;R4 is chosen from subtituted benzyl, heterocyclyl substituted lower alkyl and R16-alkylene-;R6 and R7 are chosen from hydrogen and halo;R5 and R8 are hydrogen;R16 is chosen from di(lower alkylamino), (lower alkyl)amino, amino, pyrrolidinyl, azetidinyl piperidinyl, imidazolyl and morpholinyl.
- A method according to claim 28 whereinR1 is benzyl;R2 is ethyl;R2' is hydrogen;R3" is chosen from halophenyl, polyhalophenyl, tolyl, dimethylphenyl, methoxyphenyl, dimethoxyphenyl, cyanophenyl, trifluoromethylphenyl, trifluoromethoxyphenyl, bis(trifluoromethyl)phenyl, carboxyphenyl, t-butylphenyl, methoxycarbonylphenyl, piperidinyl and naphthyl;R4 is chosen from substituted benzyl, piperidinyl, hydroxy (lower alkyl) and R16-alkylene-;R6 and R7 are chosen from hydrogen and halo;R5 and R8 are hydrogen;R16 is chosen from dimethylamino, amino, pyrrolidinyl and piperidinyl.
- A method according to claim 1 or 2 wherein said disease or disorder is chosen from the group consisting of cancer, hyperplasia, restenosis, and cardiac hypertrophy.
- A compound chosen from the group consisting of:
 R1 is chosen from hydrogen, alkyl, alkylaryl, alkylheteroaryl, substituted alkyl, substituted alkylaryl, and substituted alkylheteroaryl;R2 and R2' are independently chosen from hydrogen, alkyl, oxaalkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; or R2 and R2' taken together form a 3- to 7-membered ring;R3 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, oxaalkyl, oxaalkylaryl, substituted oxaalkylaryl, oxaalkylheteroaryl, substituted oxaalkylheteroaryl, R15O- and R15-NH-;R3" is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R4 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, and R16-alkylene-;R5, R6, R7 and R8 are independently chosen from hydrogen, alkyl, alkoxy, halogen, fluoroalkyl, nitro, cyano, dialkylamino, alkylsulfonyl, alkylsulfonamido, sulfonamidoalkyl, sulfonamidoaryl, alkylthio, carboxyalkyl, carboxamido, aminocarbonyl, aryl and heteroaryl;R15 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; andR16 is chosen from alkoxy, amino, alkylamino, dialkylamino, N-heterocyclyl and substituted N-heterocyclyl,or a pharmaceutically acceptable salt thereof,
R1 is chosen from hydrogen, alkyl, alkylaryl, alkylheteroaryl, substituted alkyl, substituted alkylaryl, and substituted alkylheteroaryl;R2 and R2' are independently chosen from hydrogen, alkyl, oxaalkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; or R2 and R2' taken together form a 3- to 7-membered ring;R3 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, oxaalkyl, oxaalkylaryl, substituted oxaalkylaryl, oxaalkylheteroaryl, substituted oxaalkylheteroaryl, R15O- and R15-NH-;R3" is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R4 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, and R16-alkylene-;R5, R6, R7 and R8 are independently chosen from hydrogen, alkyl, alkoxy, halogen, fluoroalkyl, nitro, cyano, dialkylamino, alkylsulfonyl, alkylsulfonamido, sulfonamidoalkyl, sulfonamidoaryl, alkylthio, carboxyalkyl, carboxamido, aminocarbonyl, aryl and heteroaryl;R15 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; andR16 is chosen from alkoxy, amino, alkylamino, dialkylamino, N-heterocyclyl and substituted N-heterocyclyl,or a pharmaceutically acceptable salt thereof,
with the proviso that when R3 is R15-NH-, both of R2 and R4 must be other than hydrogen. - A compound chosen from the group consisting of:
 R1 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R2 and R2' are independently chosen from hydrogen, alkyl, oxaalkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; or R2 and R2' taken together form a 3- to 7-membered ring;R3' is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl and R15-NH-;R4 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, and R16-alkylene-;R5, R6, R7 and R8 are independently chosen from hydrogen, alkyl, alkoxy, halogen, fluoroalkyl, nitro, cyano, dialkylamino, alkylsulfonyl, alkylsulfonamido, sulfonamidoalkyl, sulfonamidoaryl, alkylthio, carboxyalkyl, carboxamido, aminocarbonyl, aryl and heteroaryl;R15 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; andR16 is chosen from alkoxy, amino, alkylamino, dialkylamino, N-heterocyclyl and substituted N-heterocyclyl,or a pharmaceutically acceptable salt thereof.
R1 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R2 and R2' are independently chosen from hydrogen, alkyl, oxaalkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; or R2 and R2' taken together form a 3- to 7-membered ring;R3' is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl and R15-NH-;R4 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, and R16-alkylene-;R5, R6, R7 and R8 are independently chosen from hydrogen, alkyl, alkoxy, halogen, fluoroalkyl, nitro, cyano, dialkylamino, alkylsulfonyl, alkylsulfonamido, sulfonamidoalkyl, sulfonamidoaryl, alkylthio, carboxyalkyl, carboxamido, aminocarbonyl, aryl and heteroaryl;R15 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; andR16 is chosen from alkoxy, amino, alkylamino, dialkylamino, N-heterocyclyl and substituted N-heterocyclyl,or a pharmaceutically acceptable salt thereof. - A compound or salt according to claim 31 or 32 wherein:R1 is chosen from hydrogen, alkyl, aryl, substituted alkyl, substituted aryl, heteroaryl, substituted heteroaryl, alkylaryl, alkylheteroaryl and substituted alkylaryl;R2 is chosen from hydrogen, alkyl and substituted alkyl; R2' is hydrogen; and the stereogenic center to which R2 and R2' are attached is of the R configuration.R3 is chosen from alkyl, aryl, alkylaryl, heteroaryl, substituted aryl, substituted alkyl, substituted heteroaryl, oxaalkylaryl, substituted oxaalkylaryl, R15O- and R15-NH-;R4 is chosen from alkyl, aryl, alkylaryl, alkylheteroaryl, substituted alkyl, substituted aryl, and R16-alkylene-;R5 is hydrogen;R6, R7 and R8 are independently chosen from hydrogen, halogen, methyl and trifluoromethyl;R15 is chosen from alkyl, aryl and substituted aryl; andR16 is chosen from alkoxy, amino, alkylamino, dialkylamino and N-heterocyclyl.
- A compound according to claim 31 of formula:

- A compound or salt according to claim 34 wherein R1 is chosen from hydrogen, lower alkyl, substituted lower alkyl, alkylaryl or substituted alkylaryl.
- A compound or salt according to claim 35 wherein R1 is chosen from hydrogen, ethyl, propyl, methoxyethyl, phenethyl, benzyl, chlorobenzyl, methylbenzyl, methoxybenzyl, tetrahydrofuranylmethyl and (ethoxycarbonyl)ethyl.
- A compound or salt according to claim 34 wherein R2 is chosen from hydrogen, lower alkyl and substituted lower alkyl; R2' is hydrogen.; and the stereogenic center to which R2 and R2' are attached is of the R configuration.
- A compound or salt according to claim 37 wherein R2 is chosen from ethyl, i-propyl, c-propyl or t-butyl.
- A compound or salt according to claim 34 or 37 wherein R3 is chosen from aryl, substituted aryl, alkylaryl, alkylheteroaryl, oxaalkylaryl, oxaalkylheteroaryl, substituted alkylaryl, substituted alkylheteroaryl, substituted oxaalkylaryl or substituted oxaalkylheteroaryl.
- A compound or salt according to claim 39 wherein R3 is chosen from phenyl, substituted phenyl, benzyl, substituted benzyl, phenylvinyl, substituted phenylvinyl, phenoxy lower alkyl and substituted phenoxy lower alkyl, furanyl, benzofuranyl, pyridinyl, indolyl, methylpyridinyl, quinolinyl, picolinyl, pyrazolyl, and imidazolyl.
- A compound or salt according to claim 40 wherein R3 is chosen from halophenyl, dihalophenyl, cyanophenyl, halo(trifluoromethyl)phenyl, chlorophenoxymethyl, methoxyphenyl, carboxyphenyl, methylphenyl, ethylphenyl, tolyl, biphenylyl, methylenedioxyphenyl, methylsulfonylphenyl, methoxychlorophenyl, methylhalophenyl, trifluoromethylphenyl, butylphenyl, pentylphenyl, methylnitrophenyl, dimethoxyphenyl, phenylvinyl, nitrochlorophenyl, nitrophenyl, dinitrophenyl, bis(trifluoromethyl)phenyl,
- A compound or salt according to claim 37 wherein R3 is R15-NH-where R15 is chosen from isopropyl, butyl, cyclohexyl, phenyl, bromophenyl, dichlorophenyl, methoxyphenyl, ethylphenyl, tolyl, trifluoromethylphenyl and methylthiophenyl.
- A compound or salt according to claim 34, 37 or 39 wherein R4 is chosen from lower alkyl, substituted lower alkyl, and R16-alkylene-, wherein R16 is amino, lower alkylamino, di(lower alkyl)amino, lower alkoxy, or N-heterocyclyl.
- A compound or salt according to claim 34, 37 or 39 wherein R5, R6, R7 and R8 are chosen from hydrogen, halo, lower alkyl, substituted lower alkyl, lower alkoxy and cyano.
- A compound or salt according to claim 44 wherein R5, R6 and R8 are hydrogen.
- A compound or salt according to claim 45 wherein R7 is halo.
- A compound or salt according to claim 34 wherein:R1 is chosen from alkylaryl or substituted alkylaryl;R2 is lower alkyl; R2' is hydrogen; and the stereogenic center to which R2 and R2' are attached is of the R configuration;R3 is substituted aryl;R4 is substituted alkyl;R5 is hydrogen;R6 is hydrogen;R7 is halo or cyano; andR8 is hydrogen.
- The compound or salt according to claim 47 wherein:R1 is benzyl or substituted benzyl;R2 is i-propyl;R3 is methyl- and/or halo-substituted phenyl;R4 is 3-amino-n-propyl;R5 is hydrogen;R6 is hydrogen;R7 is chloro or cyano; andR8 is hydrogen.
- The compound or salt according to claim 47 wherein:R1 is benzyl;R2 is i-propyl;R3 is p-fluorophenyl;R4 is 3-amino-n-propyl;R5 is hydrogen;R6 is hydrogen;R7 is fluoro; andR8 is hydrogen.
- The compound or salt according to claim 47 wherein:R1 is benzyl;R2 is i-propyl;R3 is p-fluorophenyl;R4 is 3-amino-n-propyl;R5 is hydrogen;R6 is hydrogen;R7 is chloro; andR8 is hydrogen.
- The compound or salt according to claim 47 wherein:R1 is benzyl;R2 is i-propyl;R3 is 3-fluoro-4-methylphenyl;R4 is 3-amino-n-propyl;R5 is hydrogen;R6 is hydrogen;R7 is chloro; andR8 is hydrogen.
- The compound or salt according to claim 47 wherein:R1 is benzyl;R2 is i-propyl;R3 is p-methylphenyl;R4 is 3-amino-n-propyl;R5 is hydrogen;R6 is hydrogen;R7 is chloro; andR8 is hydrogen.
- The compound or salt according to claim 47 wherein:R1 is benzyl;R2 is i-propyl;R3 is p-methylphenyl;R4 is 3-amino-n-propyl;R5 is hydrogen;R6 is hydrogen;R7 is cyano; andR8 is hydrogen.
- A compound according to claim 32 of formula:

- A compound according to claim 54 wherein:R1 is chosen from hydrogen, lower alkyl, substituted lower alkyl, benzyl, substituted benzyl, phenyl, naphthyl and substituted phenyl;R2 is chosen from hydrogen, lower alkyl and substituted lower alkyl and R2' is hydrogen;R3' is chosen from C1-C13 alkyl; phenyl; naphthyl; phenyl substituted with halo, lower alkyl, lower alkoxy, nitro, methylenedioxy, or trifluoromethyl; biphenylyl, benzyl and heteroaryl; andR4 is chosen from lower alkyl, substituted lower alkyl, cyclohexyl; phenyl substituted with hydroxy, lower alkoxy or lower alkyl; benzyl; heteroarylmethyl; heteroarylethyl; heteroarylpropyl and R16-alkylene, whereinR16 is amino, (lower alkyl)amino, di(lower alkyl)amino, lower alkoxy, or N-heterocyclyl.
- A compound according to claim 55 wherein:R1 is chosen from lower alkyl, benzyl, substituted benzyl and substituted phenyl;R2 is hydrogen or lower alkyl;R2' is hydrogen;R3 is chosen from substituted phenyl and naphthyl;R4 is R16-alkylene-, hydroxy(lower alkyl) or carboxy (lower alkyl);R7 is hydrogen, fluoro, chloro or methyl;R5, R6 and R8 are hydrogen;R16 is chosen from di(lower alkyl)amino, (lower alkyl)amino, amino, pyrrolidinyl and piperidinyl.
- A compound according to claim 32 of formula:

- A compound or salt according to claim 57 wherein:R1 is chosen from hydrogen, lower alkyl, substituted lower alkyl, benzyl, substituted benzyl, phenyl, naphthyl and substituted phenyl;R2 is chosen from hydrogen, lower alkyl and substituted lower alkyl and R2' is hydrogen; andR4 is chosen from lower alkyl, cyclohexyl; phenyl substituted with hydroxy, lower alkoxy or lower alkyl; benzyl; heteroarylmethyl; heteroarylethyl; heteroarylpropyl and R16-alrylene, wherein R16 is di(lower alkyl)amino, (lower alkyl)amino, amino, lower alkoxy, or N-heterocyclyl.
- A compound or salt according to claim 58 wherein:R1 is chosen from lower alkyl, benzyl, substituted benzyl and substituted phenyl;R2 is hydrogen or lower alkyl;R2' is hydrogen;R4 is R16-alkylene-;R7 is hydrogen, fluoro, chloro or methyl;R5, R6 and R8 are hydrogen;R16 is chosen from di(lower alkylamino), (lower alkyl)amino, amino, pyrrolidinyl, piperidinyl, imidazolyl and morpholinyl.
- A compound according to claim 31 of formula:

- A compound or salt according to claim 60 wherein:R1 is chosen from hydrogen, lower alkyl, substituted lower alkyl, alkylaryl or substituted alkylaryl;R2 is chosen from hydrogen, lower alkyl and substituted lower alkyl; R2' is hydrogen; and the stereogenic center to which R2 and R2' are attached is of the R configuration;R3" is chosen from aryl, substituted aryl, alkylaryl, alkylheteroaryl, oxaalkylaryl, oxaalkylheteroaryl, substituted alkylaryl, substituted alkylheteroaryl, substituted oxaalkylaryl or substituted oxaalkylheteroaryl; andR4 is chosen from lower alkyl, substituted lower alkyl, and R16-alkylene, whereinR16 is di(lower alkyl)amino, (lower alkyl)amino, amino, lower alkoxy, or N-heterocyclyl.
- A compound according to claim 31 wherein said compound is of a formula as defined in Figure 3.
- A method of screening for KSP kinesin modulators comprising:(a) combining a kinesin, a candidate bioactive agent and a compound chosen from the group consisting of:

 R1 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R2 and R2' are independently chosen from hydrogen, alkyl, oxaalkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; or R2 and R2' taken together form a 3- to 7-membered ring;R3 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, oxaalkyl, oxaalkylaryl, substituted oxaalkylaryl, oxaalkylheteroaryl, substituted oxaalkylheteroaryl, R15O- and R15-NH-;R3' is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl and R15-NH-;R3" is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R4 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, and R16-alkylene-;R5, R6, R7 and R8 are independently chosen from hydrogen, alkyl, alkoxy, halogen, fluoroalkyl, nitro, cyano, dialkylamino, alkylsulfonyl, alkylsulfonamido, sulfonamidoalkyl, sulfonamidoaryl, alkylthio, carboxyalkyl, carboxamido, aminocarbonyl, aryl and heteroaryl;R15 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; andR16 is chosen from alkoxy, amino, alkylamino, dialkylamino, N-heterocyclyl and substituted N-heterocyclyl; and(b) determining the effect of said candidate bioactive agent on the activity of said kinesin.
R1 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R2 and R2' are independently chosen from hydrogen, alkyl, oxaalkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; or R2 and R2' taken together form a 3- to 7-membered ring;R3 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, oxaalkyl, oxaalkylaryl, substituted oxaalkylaryl, oxaalkylheteroaryl, substituted oxaalkylheteroaryl, R15O- and R15-NH-;R3' is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl and R15-NH-;R3" is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R4 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, and R16-alkylene-;R5, R6, R7 and R8 are independently chosen from hydrogen, alkyl, alkoxy, halogen, fluoroalkyl, nitro, cyano, dialkylamino, alkylsulfonyl, alkylsulfonamido, sulfonamidoalkyl, sulfonamidoaryl, alkylthio, carboxyalkyl, carboxamido, aminocarbonyl, aryl and heteroaryl;R15 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; andR16 is chosen from alkoxy, amino, alkylamino, dialkylamino, N-heterocyclyl and substituted N-heterocyclyl; and(b) determining the effect of said candidate bioactive agent on the activity of said kinesin. - A method of screening for compounds that bind to KSP kinesin comprising:(a) combining a kinesin, a candidate bioactive agent and a labeled compound chosen from the group consisting of:

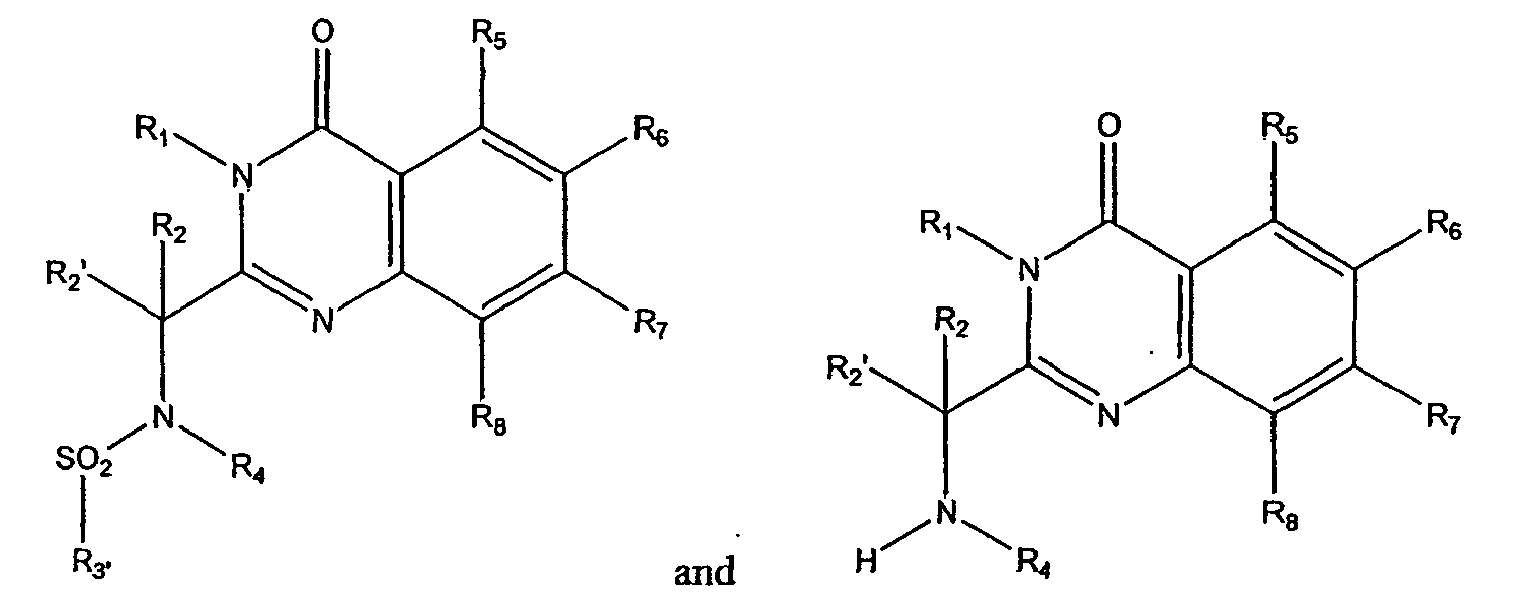 R1 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R2 and R2' are independently chosen from hydrogen, alkyl, oxaalkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; or R2 and R2' taken together form a 3- to 7-membered ring;R3 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, oxaalkyl, oxaalkylaryl, substituted oxaalkylaryl, oxaalkylheteroaryl, substituted oxaalkylheteroaryl, R15O- and R15-NH-;R3, is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl and R15-NH-;R3" is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R4 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, and R16-alkylene-;R5, R6, R7 and R8 are independently chosen from hydrogen, alkyl, alkoxy, halogen, fluoroalkyl, nitro, cyano, dialkylamino, alkylsulfonyl, alkylsulfonamido, sulfonamidoalkyl, sulfonamidoaryl, alkylthio, carboxyalkyl, carboxamido, aminocarbonyl, aryl and heteroaryl;R15 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; andR16 is chosen from alkoxy, amino, alkylamino, dialkylamino, N-heterocyclyl and substituted N-heterocyclyl; and(b) determining the binding of said candidate bioactive agent to said kinesin.
R1 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R2 and R2' are independently chosen from hydrogen, alkyl, oxaalkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; or R2 and R2' taken together form a 3- to 7-membered ring;R3 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, oxaalkyl, oxaalkylaryl, substituted oxaalkylaryl, oxaalkylheteroaryl, substituted oxaalkylheteroaryl, R15O- and R15-NH-;R3, is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl and R15-NH-;R3" is chosen from alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl;R4 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, substituted alkylheteroaryl, and R16-alkylene-;R5, R6, R7 and R8 are independently chosen from hydrogen, alkyl, alkoxy, halogen, fluoroalkyl, nitro, cyano, dialkylamino, alkylsulfonyl, alkylsulfonamido, sulfonamidoalkyl, sulfonamidoaryl, alkylthio, carboxyalkyl, carboxamido, aminocarbonyl, aryl and heteroaryl;R15 is chosen from hydrogen, alkyl, aryl, alkylaryl, heteroaryl, alkylheteroaryl, substituted alkyl, substituted aryl, substituted alkylaryl, substituted heteroaryl, and substituted alkylheteroaryl; andR16 is chosen from alkoxy, amino, alkylamino, dialkylamino, N-heterocyclyl and substituted N-heterocyclyl; and(b) determining the binding of said candidate bioactive agent to said kinesin.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US21310400P | 2000-06-21 | 2000-06-21 | |
| US09/699,047 US6545004B1 (en) | 1999-10-27 | 2000-10-24 | Methods and compositions utilizing quinazolinones |
| EP01932769A EP1296959B1 (en) | 2000-06-21 | 2001-04-27 | Methods and compositions utilizing quinazolinones |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP01932769A Division EP1296959B1 (en) | 2000-06-21 | 2001-04-27 | Methods and compositions utilizing quinazolinones |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP1707563A2 true EP1707563A2 (en) | 2006-10-04 |
| EP1707563A3 EP1707563A3 (en) | 2007-06-06 |
Family
ID=26907773
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP06075276A Withdrawn EP1707563A3 (en) | 2000-06-21 | 2001-04-27 | Methods and compositions utilizing quinazolinones |
| EP01932769A Expired - Lifetime EP1296959B1 (en) | 2000-06-21 | 2001-04-27 | Methods and compositions utilizing quinazolinones |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP01932769A Expired - Lifetime EP1296959B1 (en) | 2000-06-21 | 2001-04-27 | Methods and compositions utilizing quinazolinones |
Country Status (24)
| Country | Link |
|---|---|
| US (7) | US6545004B1 (en) |
| EP (2) | EP1707563A3 (en) |
| JP (1) | JP4854906B2 (en) |
| KR (1) | KR100847788B1 (en) |
| CN (1) | CN1229353C (en) |
| AT (1) | ATE323684T1 (en) |
| AU (3) | AU5927001A (en) |
| BR (1) | BR0111898A (en) |
| CA (1) | CA2413426C (en) |
| CY (1) | CY1105070T1 (en) |
| CZ (1) | CZ303007B6 (en) |
| DE (1) | DE60118921T2 (en) |
| DK (1) | DK1296959T3 (en) |
| ES (1) | ES2262649T3 (en) |
| HK (1) | HK1053837A1 (en) |
| HU (1) | HU229078B1 (en) |
| IL (1) | IL153554A (en) |
| MX (1) | MXPA02012627A (en) |
| NO (1) | NO324440B1 (en) |
| NZ (1) | NZ523233A (en) |
| PL (1) | PL204525B1 (en) |
| PT (1) | PT1296959E (en) |
| SI (1) | SI1296959T1 (en) |
| WO (1) | WO2001098278A1 (en) |
Families Citing this family (121)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4965825A (en) | 1981-11-03 | 1990-10-23 | The Personalized Mass Media Corporation | Signal processing apparatus and methods |
| US7831204B1 (en) | 1981-11-03 | 2010-11-09 | Personalized Media Communications, Llc | Signal processing apparatus and methods |
| JP4831906B2 (en) | 1999-08-27 | 2011-12-07 | ケモセントリックス, インコーポレイテッド | Heterocyclic compounds and methods for modulating CXCR3 function |
| US20070021493A1 (en) * | 1999-09-16 | 2007-01-25 | Curis, Inc. | Mediators of hedgehog signaling pathways, compositions and uses related thereto |
| IL149164A0 (en) * | 1999-10-27 | 2002-11-10 | Cytokinetics Inc | Methods and compositions utilizing quinazolinones |
| US7230000B1 (en) | 1999-10-27 | 2007-06-12 | Cytokinetics, Incorporated | Methods and compositions utilizing quinazolinones |
| US6545004B1 (en) * | 1999-10-27 | 2003-04-08 | Cytokinetics, Inc. | Methods and compositions utilizing quinazolinones |
| US7671200B2 (en) * | 1999-10-27 | 2010-03-02 | Cytokinetics, Inc. | Quinazolinone KSP inhibitors |
| HUP0500880A2 (en) * | 2000-12-11 | 2006-05-29 | Tularik Inc | Cxcr3 antagonists and pharmaceutical compositions thereof |
| EP1360180A1 (en) * | 2001-01-19 | 2003-11-12 | Cytokinetics, Inc. | Phenothiazine kinesin inhibitors |
| US6794379B2 (en) | 2001-06-06 | 2004-09-21 | Tularik Inc. | CXCR3 antagonists |
| CA2450762A1 (en) | 2001-06-22 | 2003-01-03 | Pfizer Products Inc. | Pharmaceutical compositions comprising low-solubility and/or acid-sensitive drugs and neutralized acidic polymers |
| EP2116248A1 (en) * | 2001-09-05 | 2009-11-11 | Minerva Biotechnologies Corporation | Compositions and Methods of Treatment of Cancer |
| AU2002363429B2 (en) | 2001-11-07 | 2008-05-08 | Merck & Co., Inc. | Mitotic kinesin inhibitors |
| US7053216B2 (en) * | 2001-11-19 | 2006-05-30 | Iconix Pharmaceuticals, Inc. | Modulators of Rho C activity |
| DK1847534T3 (en) * | 2001-12-11 | 2011-08-29 | Kyowa Hakko Kirin Co Ltd | Thiadiazoline derivatives for the treatment of cancer |
| CA2475879A1 (en) * | 2002-02-15 | 2003-08-28 | Cytokinetics, Inc. | Synthesis of quinazolinones |
| ATE507210T1 (en) * | 2002-03-07 | 2011-05-15 | X Ceptor Therapeutics Inc | QUINAZOLINONE MODULATORS OF NUCLE RECEPTORS |
| DE60333094D1 (en) * | 2002-04-17 | 2010-08-05 | Cytokinetics Inc | COMPOUNDS, COMPOSITIONS AND METHODS |
| JP2005530785A (en) | 2002-05-09 | 2005-10-13 | サイトキネティクス・インコーポレーテッド | Compounds, compositions and methods |
| RU2004135554A (en) * | 2002-05-09 | 2006-01-20 | Цитокинетикс, Инк. (Us) | Pyrimidinones, compositions based on them and methods for their use |
| WO2003103575A2 (en) * | 2002-05-23 | 2003-12-18 | Cytokinetics, Inc. | Compounds, compositions, and methods |
| WO2003106426A1 (en) * | 2002-06-14 | 2003-12-24 | Cytokinetics, Inc. | Compounds, compositions, and methods |
| EP1539727B1 (en) * | 2002-07-17 | 2009-02-18 | Cytokinetics, Inc. | Compounds, compositions, and methods for treating cellular proliferative diseases |
| US7211580B2 (en) * | 2002-07-23 | 2007-05-01 | Cytokinetics, Incorporated | Compounds, compositions, and methods |
| AU2003262747A1 (en) * | 2002-08-21 | 2004-03-11 | Cytokinetics, Inc. | Compounds, compositions, and methods |
| EP1554265A4 (en) * | 2002-09-13 | 2008-05-07 | Cytokinetics Inc | Compounds, compositions and methods |
| JP2006501306A (en) * | 2002-09-30 | 2006-01-12 | サイトキネティクス・インコーポレーテッド | Compounds, compositions and methods |
| US7358262B2 (en) | 2003-01-29 | 2008-04-15 | Whitehead Institute For Biomedical Research | Identification of genotype-selective anti-tumor agents |
| KR20050107784A (en) * | 2003-03-07 | 2005-11-15 | 아스트라제네카 아베 | Novel fused heterocycles and uses thereof |
| SE0300627D0 (en) * | 2003-03-07 | 2003-03-07 | Astrazeneca Ab | Novel fused heterocycles and uses therof |
| WO2006018628A1 (en) * | 2003-03-07 | 2006-02-23 | Astrazeneca Ab | Enantiomers of selected fused pyrimidones and uses in the treatment and preventi on of cancer |
| WO2004092123A2 (en) * | 2003-04-10 | 2004-10-28 | Microbia, Inc. | Inhibitors of fungal invasion |
| US20070232597A1 (en) * | 2003-04-10 | 2007-10-04 | Xiangping Qian | Compounds, Compositions, and Methods |
| ES2394850T3 (en) | 2003-04-18 | 2013-02-06 | Kyowa Hakko Kirin Co., Ltd. | Mitotic kinesin inhibitor |
| JP2007500213A (en) * | 2003-05-07 | 2007-01-11 | サイトキネティクス・インコーポレーテッド | Compounds, compositions and methods |
| US20070032536A1 (en) * | 2003-05-15 | 2007-02-08 | Xianping Qian | Compounds, compositions and methods |
| US7345046B2 (en) | 2003-05-30 | 2008-03-18 | Chiron Corporation | Heteroaryl-fused pyrimidinyl compounds as anticancer agents |
| EP1632484A4 (en) * | 2003-06-10 | 2008-09-03 | Kyowa Hakko Kogyo Kk | Thiadiazoline derivative |
| WO2004113335A2 (en) * | 2003-06-20 | 2004-12-29 | Chiron Corporation | Pyridino[1,2-a]pyrimidin-4-one compounds as anticancer agents |
| AU2004264533B2 (en) * | 2003-08-15 | 2009-01-22 | Merck Sharp & Dohme Corp. | Mitotic kinesin inhibitors |
| EP1670456A2 (en) * | 2003-10-06 | 2006-06-21 | Cytokinetics, Inc. | Compounds, compositions and methods |
| EP1671957A4 (en) * | 2003-10-10 | 2007-03-14 | Thiadiazoline derivatives | |
| JP2007510652A (en) * | 2003-11-03 | 2007-04-26 | サイトキネティクス・インコーポレーテッド | Pyrimidin-4-one compounds, compositions and methods |
| JP2007510660A (en) * | 2003-11-07 | 2007-04-26 | サイトキネティクス・インコーポレーテッド | Compounds, compositions and methods |
| CA2546932A1 (en) | 2003-11-25 | 2005-06-09 | Chiron Corporation | Quinazolinone compounds as anticancer agents |
| JP2007513154A (en) | 2003-12-08 | 2007-05-24 | サイトキネティクス・インコーポレーテッド | Compounds, compositions and methods |
| US7662581B1 (en) | 2003-12-18 | 2010-02-16 | Novartis Vaccines And Diagnostics, Inc. | Eg5 co-crystals |
| CN1898215A (en) * | 2003-12-19 | 2007-01-17 | 默克公司 | Mitotic kinesin inhibitors |
| JP2007517071A (en) * | 2003-12-19 | 2007-06-28 | メルク エンド カムパニー インコーポレーテッド | Mitotic kinesin inhibitor |
| WO2005061518A1 (en) * | 2003-12-19 | 2005-07-07 | Merck & Co., Inc. | Mitotic kinesin inhibitors |
| MXPA06011236A (en) | 2004-04-02 | 2007-01-16 | Prana Biotechnology Ltd | Neurologically-active compounds. |
| US7618981B2 (en) * | 2004-05-06 | 2009-11-17 | Cytokinetics, Inc. | Imidazopyridinyl-benzamide anti-cancer agents |
| US7504413B2 (en) * | 2004-05-06 | 2009-03-17 | Cytokinetics, Inc. | N-(4-(imidazo[1,2A]pyridin-YL)phenethyl)benzamide inhibitors of the mitotic kinesin CENP-E for treating certain cellular proliferation diseases |
| US7795448B2 (en) * | 2004-05-06 | 2010-09-14 | Cytokinetics, Incorporated | Imidazoyl-benzamide anti-cancer agents |
| PL1753723T3 (en) * | 2004-05-21 | 2009-01-30 | Novartis Vaccines & Diagnostics Inc | Substituted quinoline derivatives as mitotic kinesin inhibitors |
| US20050282834A1 (en) * | 2004-06-09 | 2005-12-22 | Fady Malik | Compositions, devices and methods for treating cardiovascular disease |
| US7576221B2 (en) * | 2004-06-18 | 2009-08-18 | Novartis Vaccines And Diagnostics, Inc. | Substituted imidazole derivatives |
| US7939538B2 (en) * | 2004-06-28 | 2011-05-10 | Amgen Inc. | Compounds, compositions and methods for prevention and treatment of inflammatory and immunoregulatory disorders and diseases |
| US7375102B2 (en) | 2004-06-28 | 2008-05-20 | Amgen Sf, Llc | Tetrahydroquinazolin-4(3H)-one-related and tetrahydropyrido[2,3-D]pyrimidin-4(3H)-one-related compounds, compositions and methods for their use |
| US7271271B2 (en) | 2004-06-28 | 2007-09-18 | Amgen Sf, Llc | Imidazolo-related compounds, compositions and methods for their use |
| UA84954C2 (en) * | 2004-07-22 | 2008-12-10 | Astrazeneca Ab | Fused pyrimidones usefuel in the treatment and the prevention of cancer |
| KR20070046176A (en) * | 2004-08-18 | 2007-05-02 | 아스트라제네카 아베 | Enantiomers of selected fused pyrimidones and uses in the treatment and prevention of cancer |
| US20060041128A1 (en) * | 2004-08-18 | 2006-02-23 | Astrazeneca Ab | Selected fused heterocyclics and uses thereof |
| EP1786265A4 (en) * | 2004-08-30 | 2009-08-19 | Smithkline Beecham Corp | Novel compositions and methods of treatment |
| WO2007053135A1 (en) * | 2004-09-14 | 2007-05-10 | Minerva Biotechnologies Corporation | Methods for diagnosis and treatment of cancer |
| US7449486B2 (en) * | 2004-10-19 | 2008-11-11 | Array Biopharma Inc. | Mitotic kinesin inhibitors and methods of use thereof |
| RU2007118523A (en) * | 2004-10-19 | 2008-11-27 | Новартис Вэксинс Энд Диагностикс Инк. (Us) | DERIVATIVES OF INDOL AND BENZIMIDAZOLE |
| WO2006060737A2 (en) * | 2004-12-03 | 2006-06-08 | Takeda San Diego, Inc. | Mitotic kinesin inhibitors |
| ES2409686T3 (en) | 2005-01-25 | 2013-06-27 | Prolexys Pharmaceuticals, Inc. | Quinoxaline derivatives as antitumor agents |
| US20070161644A1 (en) * | 2005-01-25 | 2007-07-12 | Stockwell Brent R | Erastin analogs and uses thereof |
| TW200714593A (en) * | 2005-03-22 | 2007-04-16 | Kyowa Hakko Kogyo Kk | Agent for treatment of solid tumor |
| WO2006101103A1 (en) * | 2005-03-22 | 2006-09-28 | Kyowa Hakko Kogyo Co., Ltd. | Agent for treatment of hematopoietic tumor |
| DE102005024017A1 (en) * | 2005-05-25 | 2006-11-30 | Merck Patent Gmbh | quinazolinones |
| US7910611B2 (en) * | 2005-06-24 | 2011-03-22 | Kyowa Hakko Kirin Co., Ltd. | Therapeutic agent for restenosis |
| MY147188A (en) * | 2005-08-09 | 2012-11-14 | Novartis Ag | Substituted imidazole compounds as ksp inhibitors |
| AU2006294835B2 (en) * | 2005-09-26 | 2011-05-26 | Merck Sharp & Dohme Corp. | N-(4-oxo-3,4-dihydroquinazolin-2-yl)butanamides as androgen receptor modulators |
| DE102006002065B4 (en) * | 2006-01-16 | 2007-11-29 | Infineon Technologies Austria Ag | Compensation component with reduced and adjustable on-resistance |
| GB0603041D0 (en) | 2006-02-15 | 2006-03-29 | Angeletti P Ist Richerche Bio | Therapeutic compounds |
| WO2007149476A2 (en) * | 2006-06-19 | 2007-12-27 | Trustees Of Columbia University In The City Of New York | Assays for non-apoptotic cell death and uses thereof |
| WO2008014373A2 (en) * | 2006-07-27 | 2008-01-31 | Smithkline Beecham Corporation | Hspa1a as a marker for sensitivity to ksp inhibitors |
| US8173629B2 (en) | 2006-09-22 | 2012-05-08 | Merck Sharp & Dohme Corp. | Method of treatment using fatty acid synthesis inhibitors |
| CN101547922B (en) * | 2006-10-04 | 2012-06-20 | 辉瑞产品公司 | Pyrido[4,3-d]pyrimidin-4(3h)-one derivatives as calcium receptor antagonists |
| US8916552B2 (en) | 2006-10-12 | 2014-12-23 | Astex Therapeutics Limited | Pharmaceutical combinations |
| WO2008044045A1 (en) | 2006-10-12 | 2008-04-17 | Astex Therapeutics Limited | Pharmaceutical combinations |
| US20110218176A1 (en) | 2006-11-01 | 2011-09-08 | Barbara Brooke Jennings-Spring | Compounds, methods, and treatments for abnormal signaling pathways for prenatal and postnatal development |
| CN101558049B (en) * | 2006-11-13 | 2012-08-15 | 诺瓦提斯公司 | Substituted pyrazole and triazole compounds as KSP inhibitors |
| JP2010515687A (en) * | 2007-01-05 | 2010-05-13 | ノバルティス アーゲー | Imidazole derivatives as kinesin spindle protein inhibitors (EG-5) |
| RS58936B1 (en) | 2007-01-10 | 2019-08-30 | Msd Italia Srl | Amide substituted indazoles as poly(adp-ribose)polymerase (parp) inhibitors |
| MX2009009304A (en) | 2007-03-01 | 2009-11-18 | Novartis Ag | Pim kinase inhibitors and methods of their use. |
| WO2008144062A1 (en) | 2007-05-21 | 2008-11-27 | Novartis Ag | Csf-1r inhibitors, compositions, and methods of use |
| KR20100024494A (en) | 2007-06-22 | 2010-03-05 | 아르퀼 인코포레이티드 | Quinazolinone compounds and methods of use thereof |
| WO2009002553A1 (en) * | 2007-06-25 | 2008-12-31 | Prolexys Pharmaceuticals, Inc. | Methods of treating multiple myeloma and resistant cancers |
| EP2170076B1 (en) | 2007-06-27 | 2016-05-18 | Merck Sharp & Dohme Corp. | 4-carboxybenzylamino derivatives as histone deacetylase inhibitors |
| CA2720664A1 (en) * | 2008-04-22 | 2009-11-26 | Centre National De La Recherche Scientifique | Novel compositions which inhibit melanogenesis and uses thereof |
| EP3330377A1 (en) | 2010-08-02 | 2018-06-06 | Sirna Therapeutics, Inc. | Rna interference mediated inhibition of catenin (cadherin-associated protein), beta 1 (ctnnb1) gene expression using short interfering nucleic acid (sina) |
| CN108676800B (en) | 2010-08-17 | 2022-11-11 | 瑟纳治疗公司 | RNA interference-mediated suppression of Hepatitis B Virus (HBV) gene expression using short interfering nucleic acids (siNA) |
| US8883801B2 (en) | 2010-08-23 | 2014-11-11 | Merck Sharp & Dohme Corp. | Substituted pyrazolo[1,5-a]pyrimidines as mTOR inhibitors |
| WO2012036997A1 (en) | 2010-09-16 | 2012-03-22 | Schering Corporation | Fused pyrazole derivatives as novel erk inhibitors |
| US8927559B2 (en) * | 2010-10-11 | 2015-01-06 | Merck Sharp & Dohme Corp. | Quinazolinone-type compounds as CRTH2 antagonists |
| ES2663009T3 (en) | 2010-10-29 | 2018-04-10 | Sirna Therapeutics, Inc. | Inhibition of RNA-mediated gene expression using short interference nucleic acids (ANic) |
| EP2455456A1 (en) | 2010-11-22 | 2012-05-23 | Institut Curie | Use of kinesin inhibitors in HIV infection treatment and a method for screening them |
| WO2012143879A1 (en) | 2011-04-21 | 2012-10-26 | Piramal Healthcare Limited | A crystalline form of a salt of a morpholino sulfonyl indole derivative and a process for its preparation |
| US8940742B2 (en) | 2012-04-10 | 2015-01-27 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US20150299696A1 (en) | 2012-05-02 | 2015-10-22 | Sirna Therapeutics, Inc. | SHORT INTERFERING NUCLEIC ACID (siNA) COMPOSITIONS |
| ES2685568T3 (en) | 2012-12-21 | 2018-10-10 | Gilead Calistoga Llc | Inhibitors of isoquinolinone or quinazolinone phosphatidylinositol 3-kinase |
| PT2941426T (en) | 2012-12-21 | 2018-07-18 | Gilead Calistoga Llc | Substituted pyrimidine aminoalkyl-quinazolones as phosphatidylinositol 3-kinase inhibitors |
| BR112015031475A2 (en) | 2013-06-14 | 2017-07-25 | Gilead Sciences Inc | compound, pharmaceutical composition, method for treating a disease or condition, kit, and use of compound. |
| US20160194368A1 (en) | 2013-09-03 | 2016-07-07 | Moderna Therapeutics, Inc. | Circular polynucleotides |
| PE20160685A1 (en) | 2013-10-04 | 2016-07-23 | Infinity Pharmaceuticals Inc | HETEROCYCLIC COMPOUNDS AND USES OF THEM |
| WO2015051241A1 (en) * | 2013-10-04 | 2015-04-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| MX2016012021A (en) | 2014-03-19 | 2017-04-13 | Infinity Pharmaceuticals Inc | Heterocyclic compounds for use in the treatment of pi3k-gamma mediated disorders. |
| JO3589B1 (en) | 2014-08-06 | 2020-07-05 | Novartis Ag | Protein kinase c inhibitors and methods of their use |
| US9708348B2 (en) | 2014-10-03 | 2017-07-18 | Infinity Pharmaceuticals, Inc. | Trisubstituted bicyclic heterocyclic compounds with kinase activities and uses thereof |
| AU2016322552B2 (en) | 2015-09-14 | 2021-03-25 | Infinity Pharmaceuticals, Inc. | Solid forms of isoquinolinone derivatives, process of making, compositions comprising, and methods of using the same |
| US10759806B2 (en) | 2016-03-17 | 2020-09-01 | Infinity Pharmaceuticals, Inc. | Isotopologues of isoquinolinone and quinazolinone compounds and uses thereof as PI3K kinase inhibitors |
| WO2017214269A1 (en) | 2016-06-08 | 2017-12-14 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| TW201825465A (en) | 2016-09-23 | 2018-07-16 | 美商基利科學股份有限公司 | Phosphatidylinositol 3-kinase inhibitors |
| TW201815787A (en) | 2016-09-23 | 2018-05-01 | 美商基利科學股份有限公司 | Phosphatidylinositol 3-kinase inhibitors |
| TW201813963A (en) | 2016-09-23 | 2018-04-16 | 美商基利科學股份有限公司 | Phosphatidylinositol 3-kinase inhibitors |
| WO2023203254A2 (en) * | 2022-04-22 | 2023-10-26 | Fundamental Pharma Gmbh | Effective means to modulate nmda receptor-mediated toxicity |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0056637A1 (en) * | 1981-01-16 | 1982-07-28 | Masayuki Ishikawa | 4(3H)-quinazolinone derivatives, process for production thereof and pharmaceutical compositions comprising said compounds |
| EP0884310A1 (en) * | 1997-06-09 | 1998-12-16 | Pfizer Products Inc. | Quinazolin-4-one ampa antagonists |
| WO2001016114A2 (en) * | 1999-08-27 | 2001-03-08 | Chemocentryx, Inc. | Heterocyclic compounds and methods for modulating cxcr3 function |
| WO2001030768A1 (en) * | 1999-10-27 | 2001-05-03 | Cytokinetics, Inc. | Methods and compositions utilizing quinazolinones |
Family Cites Families (134)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1249281B (en) | 1963-05-18 | |||
| US3320124A (en) | 1964-07-20 | 1967-05-16 | American Cyanamid Co | Method for treating coccidiosis with quinazolinones |
| US3846549A (en) | 1966-09-03 | 1974-11-05 | Boehringer Sohn Ingelheim | Pharmaceutical compositions containing an n-(1-bicyclic aryl-propyl -2)-n-phenyl-piperazine |
| BG16042A3 (en) | 1968-11-12 | 1972-05-20 | Nans Ott | METHOD FOR OBTAINING NEW 2(1H)-QUINAZOLINONES |
| US3962244A (en) | 1971-01-23 | 1976-06-08 | Hoechst Aktiengesellschaft | Benzene sulfonyl ureas |
| US3740442A (en) | 1972-01-14 | 1973-06-19 | Sandoz Ag | 2-isopropylaminobenzophenones in treating inflammation |
| US4011324A (en) * | 1976-01-20 | 1977-03-08 | Pfizer Inc. | Esters and amides of pyrimido[4,5-b]quinolin-4(3H)-one-2-carboxylic acids as antiulcer agents |
| EP0009008A3 (en) | 1978-09-08 | 1980-05-14 | Ciba-Geigy Ag | Cephalosporin derivatives, process for their preparation and pharmaceutical compositions containing them |
| US4281127A (en) | 1979-07-09 | 1981-07-28 | Hoffmann-La Roche Inc. | Trans-3-(4-oxo-4H-quinazolin-3-yl)-2-propenoic acid derivatives |
| EP0046953B1 (en) * | 1980-08-30 | 1989-12-06 | Hoechst Aktiengesellschaft | Amino acid derivatives, processes for their preparation, compositions containing them and their use |
| EP0090068A1 (en) | 1982-03-31 | 1983-10-05 | Hans Wenner | Automatic vending machine for newspapers, magazines, foodstuffs and other articles |
| GB8524663D0 (en) | 1985-10-07 | 1985-11-13 | Fujisawa Pharmaceutical Co | Quinazoline derivatives |
| DE3609598A1 (en) | 1986-03-21 | 1987-10-01 | Hoechst Ag | 2-AZOLYLMETHYL-2-ARYL-1,3-DIOXOLANE AND THE SALTS THEREOF, METHOD FOR THE PRODUCTION THEREOF, MEANS CONTAINING IT AND THEIR USE |
| US5187167A (en) | 1986-03-27 | 1993-02-16 | Imperial Chemical Industries Plc | Pharmaceutical compositions comprising quinazolin-4-one derivatives |
| GB8607683D0 (en) | 1986-03-27 | 1986-04-30 | Ici Plc | Anti-tumor agents |
| US4670560A (en) * | 1986-04-28 | 1987-06-02 | Ortho Pharmaceutical Corporation | Thienopyrimidine-2,4-dione derivatives and intermediates thereof |
| US4729996A (en) | 1986-05-29 | 1988-03-08 | Harbor Branch Oceanographic Institution, Inc. | Antitumor compositions and their methods of use |
| DE3721855A1 (en) | 1987-03-12 | 1988-09-22 | Merck Patent Gmbh | AMINO ACID DERIVATIVES |
| GB8707053D0 (en) | 1987-03-25 | 1987-04-29 | Ici Plc | Anti-tumour agents |
| US4866084A (en) | 1987-07-17 | 1989-09-12 | Harbor Branch Oceanographic Institution, Inc. | Topsentin compounds effective against viruses and certain tumors |
| US4808590A (en) | 1987-07-17 | 1989-02-28 | Harbor Branch Oceanographic Institution, Inc. | Antiviral, antitumor and antifungal compositions and their methods of use |
| JPS6430768A (en) * | 1987-07-28 | 1989-02-01 | Toshiba Corp | Thermal head |
| US5756450A (en) | 1987-09-15 | 1998-05-26 | Novartis Corporation | Water soluble monoesters as solubilisers for pharmacologically active compounds and pharmaceutical excipients and novel cyclosporin galenic forms |
| US4857530A (en) | 1987-11-03 | 1989-08-15 | Warner-Lambert Company | Substituted quinazolinones as anticancer agents |
| GB8820129D0 (en) | 1988-08-24 | 1988-09-28 | Schering Agrochemicals Ltd | Fungicides |
| GB8827820D0 (en) | 1988-11-29 | 1988-12-29 | Janssen Pharmaceutica Nv | (1h-azol-1-ylmethyl)substituted quinoline derivatives |
| GB8827988D0 (en) | 1988-11-30 | 1989-01-05 | Smith Kline French Lab | Chemical compounds |
| US4970226A (en) | 1989-10-03 | 1990-11-13 | Harbor Branch Oceanographic Institution, Inc. | Bis-indole imidazole compounds which are useful antitumor and antimicrobial agents |
| US5264439A (en) | 1990-02-13 | 1993-11-23 | Merck & Co., Inc. | Quinazolinone, triazolinone and pyrimidinone angiotensin II antagonists incorporating a substituted benzyl element |
| AU640016B2 (en) | 1990-05-30 | 1993-08-12 | Imperial Chemical Industries Plc | Hydroquinazoline derivatives |
| FR2665159B1 (en) * | 1990-07-24 | 1992-11-13 | Rhone Poulenc Sante | NEW PYRIDINE AND QUINOLEIN DERIVATIVES, THEIR PREPARATION AND THE PHARMACEUTICAL COMPOSITIONS CONTAINING THEM. |
| EP0481614A1 (en) | 1990-10-01 | 1992-04-22 | Merck & Co. Inc. | Substituted pyridopyrimidinones and related heterocycles as angiotensin II antagonists |
| US5411431A (en) * | 1991-01-09 | 1995-05-02 | Crown Gear, B.V. | Method for crown gear grinding by generation |
| GB9105771D0 (en) | 1991-03-19 | 1991-05-01 | Cancer Res Inst Royal | Anti-cancer compounds |
| US5714493A (en) | 1991-05-10 | 1998-02-03 | Rhone-Poulenc Rorer Pharmaceuticals, Inc. | Aryl and heteroaryl quinazoline compounds which inhibit CSF-1R receptor tyrosine kinase |
| US5316906A (en) | 1991-08-23 | 1994-05-31 | Molecular Probes, Inc. | Enzymatic analysis using substrates that yield fluorescent precipitates |
| US5204354A (en) | 1992-02-14 | 1993-04-20 | Merck & Co., Inc. | Substituted quinazolinones as neurotensin antagonists useful in the treatment of CNS disorders |
| MY110110A (en) | 1992-03-12 | 1998-01-27 | Smithkline Beecham Plc | Pharmaceuticals in the treatment of gastrointestinal disorders, cardiovascular disorders and cns disorders |
| GB9205907D0 (en) | 1992-03-18 | 1992-04-29 | Cancer Res Inst Royal | Anti-cancer compounds |
| US5430148A (en) | 1992-03-31 | 1995-07-04 | Agouron Pharmaceuticals, Inc. | Antiproliferative quinazolines |
| GB9220571D0 (en) | 1992-09-30 | 1992-11-11 | Ici Plc | Quinazoline derivatives |
| US5817662A (en) | 1992-11-09 | 1998-10-06 | Cell Therapeutics, Inc. | Substituted amino alkyl compounds |
| US5780476A (en) | 1992-11-16 | 1998-07-14 | Cell Therapeutics, Inc. | Hydroxyl-containing xanthine compounds |
| AU6087894A (en) | 1993-01-14 | 1994-08-15 | Cell Therapeutics, Inc. | Acetal or ketal substituted therapeutic compounds |
| WO1994016704A1 (en) | 1993-01-19 | 1994-08-04 | Cell Therapeutics, Inc. | Oxime-substituted therapeutic compounds |
| US5574057A (en) | 1993-02-03 | 1996-11-12 | University Of Utah Research Foundation | Naamidine A extracted from sea sponges and methods for its use as an anti-tumor agent |
| US5401745A (en) | 1993-03-19 | 1995-03-28 | Merck & Co., Inc. | Quinazolinones substituted with phenoxyphenylacetic acid derivatives |
| US5837703A (en) | 1993-03-31 | 1998-11-17 | Cell Therapeutics, Inc. | Amino-alcohol substituted cyclic compounds |
| US5342944A (en) | 1993-07-19 | 1994-08-30 | American Cyanamid Company | Process for the preparation of 2-alkyl-3,5,6,7- or 8-substituted-4(3H)-quinazolinones |
| US20020198326A1 (en) | 1993-11-12 | 2002-12-26 | Taizo Aoyama | Polyolefin resin composition |
| CA2113229C (en) | 1994-01-11 | 1999-04-20 | Mark Pines | Anti-fibrotic quinazolinone-containing compositions and methods for the use thereof |
| CA2192470A1 (en) | 1994-01-14 | 1995-07-20 | Paul A. Brown | Method for treating diseases mediated by cellular proliferation in response to pdgf, egf, fgf and vegf |
| JPH09511496A (en) | 1994-02-18 | 1997-11-18 | セル・セラピューティックス・インコーポレーテッド | Intracellular messenger |
| WO1995024190A2 (en) | 1994-03-07 | 1995-09-14 | Sugen, Inc. | Receptor tyrosine kinase inhibitors for inhibiting cell proliferative disorders and compositions thereof |
| GB9404485D0 (en) | 1994-03-09 | 1994-04-20 | Cancer Res Campaign Tech | Benzamide analogues |
| US5801182A (en) | 1994-03-24 | 1998-09-01 | Cell Therapeutics, Inc. | Amine substituted compounds |
| US5807861A (en) | 1994-03-24 | 1998-09-15 | Cell Therapeutics, Inc. | Amine substituted xanthinyl compounds |
| US5756502A (en) | 1994-08-08 | 1998-05-26 | Warner-Lambert Company | Quinazolinone derivatives as cholyecystokinin (CCK) ligands |
| JPH09165385A (en) * | 1994-08-26 | 1997-06-24 | Kyowa Hakko Kogyo Co Ltd | Quinazoline derivative |
| US5891879A (en) | 1994-08-31 | 1999-04-06 | Hadasit Medical Research Services & Development Co., Inc. | Quinazolinone-containing pharmaceutical compositions and methods for the use thereof |
| IL110831A (en) | 1994-08-31 | 1998-12-27 | Hadasit Med Res Service | Pharmaceutical compositions containing quinazolinone derivatives for preventing restenosis |
| IL112125A (en) | 1994-12-22 | 1998-02-08 | Hadasit Med Res Service | Quinazolinone-containing pharmaceutical compositions |
| US5753664A (en) | 1995-03-16 | 1998-05-19 | Takeda Chemical Industries, Ltd. | Heterocyclic compounds, their production and use |
| EP0735143B1 (en) * | 1995-03-28 | 1998-11-18 | Tanabe Seiyaku Co., Ltd. | Process for preparing D-amino acids |
| US6245768B1 (en) | 1995-06-05 | 2001-06-12 | Neurogen Corporation | 1-(N′-(arylalkylaminoalkyl)) aminoisoindoles; a new class of dopamine receptor subtype specific ligands |
| US5747498A (en) | 1996-05-28 | 1998-05-05 | Pfizer Inc. | Alkynyl and azido-substituted 4-anilinoquinazolines |
| CN1090621C (en) | 1995-08-30 | 2002-09-11 | 株式会社大冢制药厂 | Process for producing quinazoline-4-one derivatives |
| US5783577A (en) | 1995-09-15 | 1998-07-21 | Trega Biosciences, Inc. | Synthesis of quinazolinone libraries and derivatives thereof |
| AU4858596A (en) | 1995-09-15 | 1997-04-01 | Torrey Pines Institute For Molecular Studies | Synthesis of quinazolinone libraries |
| DE19546918A1 (en) | 1995-12-15 | 1997-06-19 | Bayer Ag | Bicyclic heterocycles |
| DE19615262A1 (en) | 1996-04-18 | 1997-10-23 | Bayer Ag | Hetero-linked phenylglycinolamides |
| PT901487E (en) | 1996-05-15 | 2003-08-29 | Pfizer | 4 (3H) -QUINAZOLINONES 2,3,6-TRISSUBSTITUIDES |
| US6008010A (en) | 1996-11-01 | 1999-12-28 | University Of Pittsburgh | Method and apparatus for holding cells |
| AU5380398A (en) * | 1996-12-17 | 1998-07-15 | E.I. Du Pont De Nemours And Company | Fungicidal quinazolinones |
| US5852024A (en) | 1997-02-11 | 1998-12-22 | Hadasit | Treatment and prevention of adhesions |
| US6028075A (en) | 1997-02-11 | 2000-02-22 | Pines; Mark | Quinazolinone containing pharmaceutical compositions for prevention of neovascularization and for treating malignancies |
| US5948775A (en) | 1997-03-19 | 1999-09-07 | American Home Products Corporation | 2- or 3-(substitutedaminoalkoxyphenyl)quinazolin-4-ones |
| US6060479A (en) | 1997-06-09 | 2000-05-09 | Pfizer Inc | Quinazoline-4-one AMPA antagonists |
| US6627755B1 (en) | 1997-06-09 | 2003-09-30 | Pfizer Inc | Quinazolin-4-one AMPA antagonists |
| US5939421A (en) | 1997-07-01 | 1999-08-17 | Signal Pharmaceuticals, Inc. | Quinazoline analogs and related compounds and methods for treating inflammatory conditions |
| US5972929A (en) | 1997-08-21 | 1999-10-26 | Shiseido Co., Ltd. | Quinazolinone derivative, hair growth promoter and external composition for skin using the same |
| JP2934628B2 (en) * | 1997-08-21 | 1999-08-16 | 株式会社資生堂 | Quinazolinone derivatives, hair nourishing agents, skin external preparations |
| US6207403B1 (en) * | 1998-01-08 | 2001-03-27 | The Regents Of The University Of California | Kinesin motor modulators derived from the marine sponge Adocia |
| IL125950A0 (en) | 1997-09-05 | 1999-04-11 | Pfizer Prod Inc | Methods of administering ampa receptor antagonists to treat dyskinesias associated with dopamine agonist therapy |
| US6214879B1 (en) * | 1998-03-24 | 2001-04-10 | Virginia Commonwealth University | Allosteric inhibitors of pyruvate kinase |
| AU5322399A (en) | 1998-07-27 | 2000-02-21 | President And Fellows Of Harvard College | Method of high-throughput screening of molecules and compounds for their effectson biological and chemical processes |
| US6596497B1 (en) * | 1999-03-17 | 2003-07-22 | New York Blood Center, Inc. | Screening of antiviral compounds targeted to the HIV-1 gp41 core structure |
| US6156758A (en) | 1999-09-08 | 2000-12-05 | Isis Pharmaceuticals, Inc. | Antibacterial quinazoline compounds |
| AU780846B2 (en) | 1999-09-16 | 2005-04-21 | Curis, Inc. | Mediators of hedgehog signaling pathways, compositions and uses related thereto |
| AU7654800A (en) | 1999-09-28 | 2001-04-30 | Merck Patent Gmbh | Quinazolinones |
| AU7419300A (en) | 1999-09-28 | 2001-04-30 | Merck Patent Gmbh | Quinazolinones |
| WO2001025234A1 (en) | 1999-10-01 | 2001-04-12 | Advanced Medicine, Inc. | Quinazolinones and analogues and their use as local anesthetics |
| US6545004B1 (en) | 1999-10-27 | 2003-04-08 | Cytokinetics, Inc. | Methods and compositions utilizing quinazolinones |
| US7230000B1 (en) * | 1999-10-27 | 2007-06-12 | Cytokinetics, Incorporated | Methods and compositions utilizing quinazolinones |
| US6495561B2 (en) * | 1999-10-29 | 2002-12-17 | Merck & Co., Inc. | 2-cyclohexyl imidazopyridine NMDA/NR2B antagonists |
| US6380205B1 (en) | 1999-10-29 | 2002-04-30 | Merck & Co., Inc. | 2-cyclohexyl quinazoline NMDA/NR2B antagonists |
| US6476041B1 (en) * | 1999-10-29 | 2002-11-05 | Merck & Co., Inc. | 1,4 substituted piperidinyl NMDA/NR2B antagonists |
| AR026748A1 (en) | 1999-12-08 | 2003-02-26 | Vertex Pharma | A CASPASE INHIBITING COMPOUND, A PHARMACEUTICAL COMPOSITION THAT INCLUDES IT, A METHOD FOR THE SYNTHESIS OF THE SAME AND AN INTERMEDIATE COMPOUND PARADICHA SYNTHESIS |
| ATE348382T1 (en) | 2000-01-13 | 2007-01-15 | Koninkl Philips Electronics Nv | METHOD FOR PROTECTING COMPRESSED CONTENT AFTER SEPARATION FROM ORIGINAL SOURCE |
| ES2159488B1 (en) | 2000-03-07 | 2002-04-16 | Uriach & Cia Sa J | PROCEDURE FOR THE PREPARATION OF PIRIMIDONE DERIVATIVES WITH ANTIFUNGIC ACTIVITY. |
| AU4966401A (en) | 2000-03-30 | 2001-10-15 | Curis Inc | Small organic molecule regulators of cell proliferation |
| US7115653B2 (en) | 2000-03-30 | 2006-10-03 | Curis, Inc. | Small organic molecule regulators of cell proliferation |
| US6613798B1 (en) | 2000-03-30 | 2003-09-02 | Curis, Inc. | Small organic molecule regulators of cell proliferation |
| GEP20084317B (en) | 2000-04-25 | 2008-02-25 | Icos Corp | Inhibitors of human phosphatidyl-inositol 3-kinase delta |
| US20020165221A1 (en) | 2000-10-13 | 2002-11-07 | Baxter Anthony David | Mediators of hedgehog signaling pathways, compositions and uses related thereto |
| HUP0500880A2 (en) | 2000-12-11 | 2006-05-29 | Tularik Inc | Cxcr3 antagonists and pharmaceutical compositions thereof |
| AUPR201600A0 (en) | 2000-12-11 | 2001-01-11 | Fujisawa Pharmaceutical Co., Ltd. | Quinazolinone derivative |
| EP1389205B1 (en) * | 2001-04-09 | 2005-12-21 | Ortho-McNeil Pharmaceutical Research Inc. | Quinazoline and quinazoline-like compounds for the treatment of integrin-mediated disorders |
| EP1383771A1 (en) * | 2001-04-20 | 2004-01-28 | Vertex Pharmaceuticals Incorporated | 9-deazaguanine derivatives as inhibitors of gsk-3 |
| US7365199B2 (en) * | 2001-04-20 | 2008-04-29 | Fujifilm Corporation | Dye-forming coupler, silver halide photographic light-sensitive material, and azomethine dye compound |
| US6794379B2 (en) | 2001-06-06 | 2004-09-21 | Tularik Inc. | CXCR3 antagonists |
| US6936842B2 (en) * | 2001-06-27 | 2005-08-30 | Applied Materials, Inc. | Method and apparatus for process monitoring |
| US6596723B1 (en) * | 2001-07-16 | 2003-07-22 | Essential Therapeutics, Inc. | Fungal efflux pump inhibitors |
| US20030220338A1 (en) * | 2001-07-16 | 2003-11-27 | Watkins Will J. | Fungal efflux pump inhibitors |
| US20030144350A1 (en) * | 2001-07-20 | 2003-07-31 | Adipogenix, Inc. | Fat accumulation-modulation compounds |
| EP2116248A1 (en) * | 2001-09-05 | 2009-11-11 | Minerva Biotechnologies Corporation | Compositions and Methods of Treatment of Cancer |
| CA2459584A1 (en) * | 2001-09-05 | 2003-03-13 | Cynthia C. Bamdad | Compositions and use thereof in the treatment of cancer |
| US7053216B2 (en) * | 2001-11-19 | 2006-05-30 | Iconix Pharmaceuticals, Inc. | Modulators of Rho C activity |
| AU2002346471A1 (en) | 2001-11-20 | 2003-06-10 | Cytokinetics, Inc. | Process for the racemization of chiral quinazolinones |
| CA2475879A1 (en) | 2002-02-15 | 2003-08-28 | Cytokinetics, Inc. | Synthesis of quinazolinones |
| US20030158188A1 (en) * | 2002-02-20 | 2003-08-21 | Chih-Hung Lee | Fused azabicyclic compounds that inhibit vanilloid receptor subtype 1 (VR1) receptor |
| US7074805B2 (en) * | 2002-02-20 | 2006-07-11 | Abbott Laboratories | Fused azabicyclic compounds that inhibit vanilloid receptor subtype 1 (VR1) receptor |
| WO2003084544A2 (en) * | 2002-04-04 | 2003-10-16 | Cv Therapeutics, Inc. | Compounds dor increasing abca-1 expression useful for treating coronary artery disease and atherosclerosis |
| RU2004135554A (en) | 2002-05-09 | 2006-01-20 | Цитокинетикс, Инк. (Us) | Pyrimidinones, compositions based on them and methods for their use |
| JP2005530785A (en) | 2002-05-09 | 2005-10-13 | サイトキネティクス・インコーポレーテッド | Compounds, compositions and methods |
| WO2003103575A2 (en) | 2002-05-23 | 2003-12-18 | Cytokinetics, Inc. | Compounds, compositions, and methods |
| WO2003106426A1 (en) | 2002-06-14 | 2003-12-24 | Cytokinetics, Inc. | Compounds, compositions, and methods |
| US20040092561A1 (en) * | 2002-11-07 | 2004-05-13 | Thomas Ruckle | Azolidinone-vinyl fused -benzene derivatives |
| US7211580B2 (en) | 2002-07-23 | 2007-05-01 | Cytokinetics, Incorporated | Compounds, compositions, and methods |
| AU2003262747A1 (en) | 2002-08-21 | 2004-03-11 | Cytokinetics, Inc. | Compounds, compositions, and methods |
| WO2005025293A2 (en) * | 2003-09-10 | 2005-03-24 | Icagen, Inc. | Fused ring heterocycles as potassium channel modulators |
| CA2546932A1 (en) * | 2003-11-25 | 2005-06-09 | Chiron Corporation | Quinazolinone compounds as anticancer agents |
| US20050152940A1 (en) * | 2003-12-23 | 2005-07-14 | Medtronic Vascular, Inc. | Medical devices to treat or inhibit restenosis |
| US20050282834A1 (en) * | 2004-06-09 | 2005-12-22 | Fady Malik | Compositions, devices and methods for treating cardiovascular disease |
-
2000
- 2000-10-24 US US09/699,047 patent/US6545004B1/en not_active Expired - Lifetime
- 2000-11-28 US US09/724,897 patent/US7105668B1/en not_active Expired - Fee Related
- 2000-11-28 US US09/724,644 patent/US6562831B1/en not_active Expired - Fee Related
- 2000-11-28 US US09/724,713 patent/US6630479B1/en not_active Expired - Lifetime
- 2000-11-28 US US09/724,941 patent/US6831085B1/en not_active Expired - Lifetime
-
2001
- 2001-04-27 MX MXPA02012627A patent/MXPA02012627A/en active IP Right Grant
- 2001-04-27 IL IL153554A patent/IL153554A/en active IP Right Grant
- 2001-04-27 DK DK01932769T patent/DK1296959T3/en active
- 2001-04-27 WO PCT/US2001/013901 patent/WO2001098278A1/en active IP Right Grant
- 2001-04-27 HU HU0301201A patent/HU229078B1/en not_active IP Right Cessation
- 2001-04-27 ES ES01932769T patent/ES2262649T3/en not_active Expired - Lifetime
- 2001-04-27 JP JP2002504234A patent/JP4854906B2/en not_active Expired - Fee Related
- 2001-04-27 NZ NZ523233A patent/NZ523233A/en not_active IP Right Cessation
- 2001-04-27 EP EP06075276A patent/EP1707563A3/en not_active Withdrawn
- 2001-04-27 PT PT01932769T patent/PT1296959E/en unknown
- 2001-04-27 KR KR1020027017358A patent/KR100847788B1/en not_active IP Right Cessation
- 2001-04-27 EP EP01932769A patent/EP1296959B1/en not_active Expired - Lifetime
- 2001-04-27 BR BR0111898-6A patent/BR0111898A/en not_active Application Discontinuation
- 2001-04-27 PL PL358412A patent/PL204525B1/en not_active IP Right Cessation
- 2001-04-27 DE DE60118921T patent/DE60118921T2/en not_active Expired - Lifetime
- 2001-04-27 US US10/312,323 patent/US20040023996A1/en not_active Abandoned
- 2001-04-27 CA CA2413426A patent/CA2413426C/en not_active Expired - Fee Related
- 2001-04-27 CZ CZ20024178A patent/CZ303007B6/en not_active IP Right Cessation
- 2001-04-27 AU AU5927001A patent/AU5927001A/en active Pending
- 2001-04-27 AT AT01932769T patent/ATE323684T1/en active
- 2001-04-27 CN CNB018115829A patent/CN1229353C/en not_active Expired - Lifetime
- 2001-04-27 SI SI200130539T patent/SI1296959T1/en unknown
- 2001-04-27 AU AU2001259270A patent/AU2001259270B2/en not_active Ceased
-
2002
- 2002-12-20 NO NO20026172A patent/NO324440B1/en not_active IP Right Cessation
-
2003
- 2003-08-26 HK HK03106128A patent/HK1053837A1/en unknown
-
2004
- 2004-07-20 US US10/893,929 patent/US7294634B2/en not_active Expired - Lifetime
-
2006
- 2006-07-07 CY CY20061100942T patent/CY1105070T1/en unknown
- 2006-11-16 AU AU2006236024A patent/AU2006236024A1/en not_active Abandoned
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0056637A1 (en) * | 1981-01-16 | 1982-07-28 | Masayuki Ishikawa | 4(3H)-quinazolinone derivatives, process for production thereof and pharmaceutical compositions comprising said compounds |
| EP0884310A1 (en) * | 1997-06-09 | 1998-12-16 | Pfizer Products Inc. | Quinazolin-4-one ampa antagonists |
| WO2001016114A2 (en) * | 1999-08-27 | 2001-03-08 | Chemocentryx, Inc. | Heterocyclic compounds and methods for modulating cxcr3 function |
| WO2001030768A1 (en) * | 1999-10-27 | 2001-05-03 | Cytokinetics, Inc. | Methods and compositions utilizing quinazolinones |
Non-Patent Citations (3)
| Title |
|---|
| A.K. DEBNATH: "STRUCTURE BASED IDENTIFICATION OF SMALL MOLECULE ANTIVIRAL COMPOUNDS" JOURNAL OF MEDICINAL CHEMISTRY., vol. 42, no. 17, 26 August 1999 (1999-08-26), pages 3203-9, XP002159846 AMERICAN CHEMICAL SOCIETY. WASHINGTON., US ISSN: 0022-2623 * |
| CHEMICAL ABSTRACTS, vol. 122, no. 26, 1995, Columbus, Ohio, US; abstract no.: 327093n, BOCAKEI,ZAOLT: "TWO ANTITHROMBOTIC QUINAZOLONE DERIVATIVES." page 944 XP002159847 & ACTA CRYSTALLOGR.SECT.C:CRYST.STRUCT.COMMUN., vol. C51, no. 4, 1995, pages 723-6, * |
| SZABO, M. ET AL: "POTENCIALIS CCK-ANTAGONISTA KINAZOLON-SZARMAZEKOK SZINTEZISE II SYNTHESIS OF POTENTIAL CCK ANTAGONIST QUINAZOLONE DERIVATIVES" ACTA PHARMACEUTICA HUNGARICA, HUNGARICAN PHARMACEUTICAL ASSOCIATION, BUDAPEST, HU, vol. 65, no. 5, September 1995 (1995-09), pages 175-181, XP001018341 ISSN: 0001-6659 * |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6630479B1 (en) | Methods and compositions utilizing quinazolinones | |
| US8658656B2 (en) | Methods and compositions utilizing quinazolinones | |
| AU774748B2 (en) | Methods and compositions utilizing quinazolinones | |
| US8008311B2 (en) | Methods and compostions utilizing quinazolinones | |
| AU2001259270A1 (en) | Methods and compositions utilizing quinazolinones | |
| ZA200210133B (en) | Methods and compositions utilizing quinazolinones. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AC | Divisional application: reference to earlier application |
Ref document number: 1296959 Country of ref document: EP Kind code of ref document: P |
|
| AK | Designated contracting states |
Kind code of ref document: A2 Designated state(s): AT BE CH CY DE DK ES FI FR GB GR IE IT LI LU MC NL PT SE TR |
|
| AX | Request for extension of the european patent |
Extension state: SI |
|
| PUAL | Search report despatched |
Free format text: ORIGINAL CODE: 0009013 |
|
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: DE Ref document number: 1096393 Country of ref document: HK |
|
| AK | Designated contracting states |
Kind code of ref document: A3 Designated state(s): AT BE CH CY DE DK ES FI FR GB GR IE IT LI LU MC NL PT SE TR |
|
| AX | Request for extension of the european patent |
Extension state: SI |
|
| 17P | Request for examination filed |
Effective date: 20070712 |
|
| 17Q | First examination report despatched |
Effective date: 20071130 |
|
| AKX | Designation fees paid |
Designated state(s): AT BE CH CY DE DK ES FI FR GB GR IE IT LI LU MC NL PT SE TR |
|
| AXX | Extension fees paid |
Extension state: SI Payment date: 20070712 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: THE APPLICATION IS DEEMED TO BE WITHDRAWN |
|
| 18D | Application deemed to be withdrawn |
Effective date: 20100529 |
|
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: WD Ref document number: 1096393 Country of ref document: HK |