-
Die
vorliegende Erfindung betrifft bestimmte Purinderivate. Insbesondere
betrifft die vorliegende Erfindung Purin-2-ylcarboxamidderivate, deren Herstellung
und Zusammensetzungen, Verwendungsmöglichkeiten und bei der Herstellung
derselben verwendete Zwischenprodukte. Diese Derivate sind selektive
funktionale Agonisten des humanen Adenosin-A2a-Rezeptors und sie können als entzündungshemmende
Mittel bei der Behandlung von u.a. Erkrankungen der Atemwege verwendet
werden.
-
Adenosin
ist ein ubiquitäres
Molekül
mit einer zentralen Rolle im Zwischenstoffwechsel von Säugern. Unabhängig davon
wirkt Adenosin auf viele Oberflächenrezeptoren
unter Hervorrufen einer Vielzahl von Reaktionen. Die Rezeptorklassifizierung
ergab das Vorhandensein von mindestens vier Subtypen: A1, A2a, A2b und
A3. Es wurde berichtet, dass die Stimulation von Adenosin-A2-Rezeptoren
auf der Oberfläche
humaner Neutrophile wirksam einen Bereich von Neutrophilenfunktionen
hemmt. Aktivierte Neutrophile können
Lungengewebe durch Freisetzung reaktiver Sauerstoffspezies, wie
Superoxidanionradikale (O2 –),
und Granulumprodukte, wie humane Neutrophilenelastase (HNE), unter
anderen Entzündungsmediatoren
schädigen.
Ferner führen
aktivierte Neutrophile sowohl eine Neusynthese als auch die Freisetzung
von Arachidonatprodukten, wie Leukotrien B4 (LTB4), durch. LTB4 ist
ein wirksames Chemokin, das weitere Neutrophile zum Entzündungsherd
rekrutiert, während
freigesetztes O2 – und
HNE die pulmonale extrazelluläre
Matrix nachteilig beeinflussen. Der A2-Rezeptorsubtyp, der viele
dieser Reaktionen (O2 –-
und LTB4/HNE-Freisetzung und Zelladhäsion) vermittelt,
ist als A2a bekannt. Der A2-Subtyp (A2a oder A2b), der die anderen
Wirkungen vermittelt, ist noch festzustellen.
-
Es
wird angenommen, dass selektive Agonistenaktivität am A2a-Rezeptor einen größeren therapeutischen
Vorteil als nichtselektive Adenosinrezeptoragonisten bietet, da
die Interaktion mit anderen Rezeptorsubtypen in Tiermodellen und
Humangewebeuntersuchungen mit schädlichen Wirkungen in der Lunge
verbunden ist. Beispielsweise erleiden Asthmatiker, jedoch nicht
Nichtasthmatiker, Bronchokonstriktion, wenn sie inhaliertem Adenosin
ausgesetzt werden. Diese Reaktion beruht mindestens teilweise auf
der Aktivierung des A1-Rezeptorsubtyps. Die Aktivierung von A1-Rezeptoren
fördert
auch Neutrophilenchemotaxis und -haftung an Endothelzellen, wodurch
eine Lungenläsion
gefördert
wird. Ferner werden vielen Patienten mit einer Atemwegserkrankung
gleichzeitig β2-Agonisten verschrieben und eine negative
Interaktion wurde in Tieruntersuchungen zwischen Isoprenalin und
Adenosinrezeptoren, die negativ an Adenylatcyclase gekoppelt sind,
gezeigt. Die Degranulation humaner Mastzellen wird durch die Aktivierung
von Adenosin-A2b-Rezeptoren gefördert,
weshalb Selektivität
gegenüber
diesem Rezeptor ebenfalls vorteilhaft ist.
-
Wir
ermittelten nun überraschenderweise,
dass die vorliegenden Purinderivate die Neutrophilenfunktion hemmen
und selektive Agonisten des Adenosin-A2a-Rezeptors sind.
-
Die
vorliegenden Verbindungen können
zur Behandlung jeder Erkrankung, für die ein Adenosin-A2a-Rezeptoragonist
indiziert ist, verwendet werden. Sie können zur Behandlung einer Erkrankung,
an der eine durch Leukocyten (beispielsweise Neutrophile, Eosinophile,
Basophile, Lymphocyten, Makrophagen) induzierte Gewebeschädigung beteiligt
ist, verwendet werden. Sie sind als entzündungshemmende Mittel bei der
Behandlung von Erkrankungen der Atemwege, wie Schocklunge (ARDS),
Bronchitis, chronischer Bronchitis, chronisch-obstruktiver Lungenerkrankung,
Mukoviszidose, Asthma, Emphysem, Bronchiektase, chronischer Sinusitis
und Rhinitis, verwendbar. Die vorliegenden Verbindungen können auch
bei der Behandlung von septischem Schock, männlicher erektiler Dysfunktion,
Hypertonie, Schlaganfall, Epilepsie, Hirnischämie, peripherer Verschlusskrankheit,
postischämischer
Reperfusionsläsion,
Diabetes, rheumatoider Arthritis, Multipler Sklerose, Psoriasis,
allergischer Kontaktdermatitisfatopischer Dermatitis, Ekzem, ulzeröser Colitis,
Morbus Crohn, entzündlicher
Darmerkrankung, Heliobacter pylori-Gastritis, nicht-Heliobacter pylori-Gastritis,
einer durch nichtsteroidale entzündungshemmende
Arzneimittel induzierten Schädigung
des Magen-Darm-Trakts oder einer psychotischen Störung oder
zur Wundheilung verwendet werden.
-
Daher
erfolgt durch die vorliegenden Erfindung in einem Aspekt die Bereitstellung
einer Verbindung der Formel:
oder eines pharmazeutisch
akzeptablen Salzes oder Solvats derselben, worin
R
1 für Wasserstoff,
C
1-C
6-Alkyl oder
C
3-C
7-Cylcoalkyl
steht, die jeweils optional mit 1 oder 2 Substituenten substituiert
sind, die jeweils unabhängig
voneinander aus Hydroxyl, Fluorenyl, Phenyl und Naphthyl ausgewählt sind,
wobei Phenyl und Naphthyl optional mit
C
1-C
6-Alkyl, C
1-C
6-Alkoxy, Halogen oder Cyano substituiert
sind;
A für
eine Bindung oder C
1-C
6-Alkylen
steht;
R
2 (i) für Wasserstoff, C
1-C
6-Alkyl, C
3-C
7-Cycloalkyl, Phenyl oder Naphthyl steht,
wobei das C
3-C
7-Cycloalkyl, Phenyl
und Naphthyl optional mit C
1-C
6-Alkyl,
Phenyl, C
1-C
6-Alkoxy-
(C
1-C
6)-alkyl, Amino-(C
1-C
6)-alkyl, Fluor-(C
1-C
6)-alkyl, Fluor-(C
1-C
6)-alkoxy, C
2-C
5-Alkanoyl, Halogen, -OR
3,
Cyano, -COOR
3, C
3-C
7-Cycloalkyl, -S(O)
mR
4, -NR
3R
3,
-SO
2NR
3R
3, -CONR
3R
3, -NR
3COR
4 oder -NR
3SO
2R
4 substituiert
sind, mit der Maßgabe, dass
R
2 nicht für Wasserstoff steht, wenn A
eine Bindung ist,
oder (ii) wenn A für C
2-C
6-Alkylen steht, für -NR
3R
3, -OR
3, -COOR
3, -OCOR
4, -SO
2R
4, -CN, -SO
2NR
3R
3, -NR
3SO
2R
4,
-NR
3COR
4 oder -CONR
3R
3 steht,
oder
(iii) für
einen C-gebundenen, 4- bis 11-gliedrigen mono- oder bicyclischen
Heterocyclus steht, der entweder 1 bis 4 Ringstickstoffatome oder
1 oder 2 Stickstoff- und 1 Sauerstoff- oder 1 Schwefelringatom(e)
aufweist, der optional mit Oxo, C
1-C
6-Alkoxy-(C
1-C
6)-alkyl, Amino-(C
1-C
6)-alkyl,
Fluor-(C
1-C
6)-alkyl,
Fluor-(C
1-C
6)-alkoxy, Fluor-(C
2-C
5)-alkanoyl, Halogen, Cyano, -OR
5,
R
6, -COR
5, -NR
5R
5, -COOR
5, -S(O)
mR
6, -SO
2NR
5R
5, -CONR
5R
5, -NR
5SO
2R
5 oder -NR
5COR
6 C-substituiert
ist und optional mit C
1-C
6-Alkoxy-(C
1-C
6)-alkyl, Amino-(C
2-C
6)-alkyl, Fluor-(C
1-C
6)-alkyl, Fluor-(C
2-C
5)-alkanoyl, R
6,
-COR
5, -COOR
6, -SO
2R
6, -SO
2NR
5R
5 oder -CONR
5R
5 N-substituiert
ist,
oder (iv), wenn A für
C
2-C
6-Alkylen steht,
für N-gebundenes
Azetidinyl, Pyrrolidinyl, Morpholinyl, Tetrahydroisochinolinyl,
Piperazinyl oder Piperazinyl steht, die jeweils optional mit C
1-C
6-Alkyl, Phenyl,
C
1-C
6-Alkoxy-(C
1-C
6)-alkyl, Amino-(C
1-C
6)-alkyl, Fluor-(C
1-C
6)-alkyl, Fluor-(C
1-C
6)-alkoxy, C
2-C
5-Alkanoyl, Halogen,
-OR
3, Cyano, -COOR
3,
C
3-C
7-Cycloalkyl, -S(O)
mR
4, -NR
3R
3, -SO
2NR
3R
3, -CONR
3R
3, -NR
3COR
4 oder -NR
3SO
2R
4 C-substituiert
sind und das Piperazinyl optional mit C
1-C
6-Alkyl, Phenyl, C
1-C
6-Alkoxy-(C
1-C
6)-alkyl, Amino-(C
2-C
6)-alkyl, Fluor-(C
1-C
6)-alkyl, C
2-C
5-Alkanoyl, -COOR
4,
C
3-C
7-Cycloalkyl,
-SO
2R
4, -SO
2NR
3R
3 oder -CONR
3R
3 N-substituiert ist;
jedes
R
3 unabhängig
voneinander aus H, C
1-C
6-Alkyl,
Phenyl oder Pyridinyl ausgewählt
ist;
R
4 für C
1-C
6-Alkyl oder Phenyl steht;
R
5 für
H, C
1-C
6-Alkyl,
C
3-C
7-Cycloalkyl,
Phenyl, Naphthyl oder Het steht;
R
6 für C
1-C
6-Alkyl, C
3-C
7-Cycloalkyl,
Phenyl, Naphthyl oder Het steht;
m 0, 1 oder 2 ist;
R
7 für
Wasserstoff, C
1-C
6-Alkyl,
C
3-C
7-Cycloalkyl,
Phenyl, Naphthyl, Azetidin-3-yl, Pyrrolidin-3-yl, Piperidin-3-yl, Piperidin-4-yl
oder Het steht, wobei das Azetidin-3-yl, Pyrrolidin-3-yl, Piperidin-3-yl
und Piperidin-4-yl optional mit C
1-C
6-Alkyl substituiert sind;
R
8 für
H oder C
1-C
6-Alkyl
steht; und
"Het", das in den Definitionen
von R
5, R
6 und R
7 verwendet wird, C-gebundenes Pyrrolyl,
Imidazolyl, Triazolyl, Thienyl, Furyl, Thiazolyl, Oxazolyl, Thiadiazolyl,
Oxadiazolyl, Pyridinyl, Pyrimidinyl, Pyridazinyl, Pyrazinyl, Chinolinyl,
Isochinolinyl, Benzimidazolyl, Chinazolinyl, Phthalazinyl, Benzoxazolyl
oder Chinoxalinyl bedeutet, die jeweils optional mit C
1-C
6-Alkyl, C
1-C
6-Alkoxy, Cyano oder Halogen substituiert
sind.
-
Vorzugsweise
ist R1 Cyclohexyl oder optional substituiertes
C1-C6-Alkyl. Noch
günstiger
ist R1 optional substituiertes C1-C5-Alkyl und noch
besser ist R1 optional substituiertes C1-C2-Alkyl.
-
Vorzugsweise
sind die C1-C6-,
C1-C5- oder C1-C2-Alkylsubstituenten
aus Benzyl, Fluorenyl, Phenyl und Hydroxyl ausgewählt. Vorzugsweise
ist, wenn R1 mit Phenyl substituiert ist,
1 oder 2 Phenylgruppen vorhanden. Vorzugsweise ist R1 aus
2,2-Diphenylethyl, Cyclohexyl, 1-Ethylpropyl, 1-Benzyl-2-hydroxyethyl, 9H-Fluorin-9-ylmethyl
und 1-Benzyl-2- phenylethyl
ausgewählt.
-
Vorzugsweise
ist R1 2,2-Diphenylethyl.
-
Vorzugsweise
ist A C1-C6-Alkylen.
-
Vorzugsweise
ist A C1-C4-Alkylen.
-
Vorzugsweise
ist A aus Methylen, 1,2-Ethylen, 1,3-Propylen und 1,4-Butylen ausgewählt.
-
Vorzugsweise
ist A 1,2-Ethylen.
-
Vorzugsweise
ist R2 aus Phenyl, Pyrrolidinyl, Pyridinyl,
optional substituiertem Piperidinyl, optional substituiertem Piperazinyl,
optional substituiertem Imidazolyl, Morpholinyl, Tetrahydroisochinolyl,
C1-C6-Alkylamino,
Di-C1-C6-alkylamino, Pyridinylamino und -NR3SO2R4 ausgewählt.
-
Vorzugsweise
ist R2 aus Phenyl, 2-Pyridinyl, 1-Piperidinyl,
4-Piperazinyl, 1-Pyrrolidinyl, 4-Morpholinyl, 3,4-Tetrahydro-2(1H)-isochinolinyl,
C1-C3-Alkylamino,
Di-C1-C3-alkylamino und substituiertem
1H-Imidazol-4-yl ausgewählt.
-
Vorzugsweise
ist R2 aus Isopropylamino, Methylamino,
Dimethylamino, Diethylamino, 1-Methyl-1H-imidazol-4-yl, 5-Methyl-1H-imidazol-4-yl,
4-Methylpiperazin-1-yl, 1-(2-Propyl)piperidin-4-yl
und 2-Pyridylamino ausgewählt.
-
Vorzugsweise
ist R2 Piperidinyl, das optional mit C1-C6-Alkyl oder Methoxy
substituiert ist.
-
Vorzugsweise
ist R2 Piperidinyl, das optional an der
1-Position oder
4-Position mit C1-C6-Alkyl
oder Methoxy substituiert ist.
-
Vorzugsweise
ist R2 N-gebundenes Piperidinyl, das mit
C1-C6-Alkyl oder Methoxy
optional C-substituiert ist.
-
Vorzugsweise
ist R2 N-gebundenes Piperidinyl, das mit
C1-C3-Alkyl oder Methoxy
optional C-substituiert ist.
-
Vorzugsweise
ist R2 N-gebundenes Piperidinyl, das mit
Methyl, Methoxy oder Propyl optional C-substituiert ist.
-
Vorzugsweise
ist R2 N-gebundenes Piperidinyl, das an
der 4-Position mit
Methyl, Methoxy oder Propyl optional substituiert ist.
-
Vorzugsweise
ist R2 Piperidin-1-yl, 4-(Methyl)piperidin-1-yl, 4-(Methoxy)piperidin-1-yl
oder 4-(Prop-2-yl)piperidin-1-yl.
-
Vorzugsweise
ist R3 Methyl.
-
Vorzugsweise
ist R4 Methyl oder Phenyl.
-
Vorzugsweise
ist R7 C1-C6-Alkyl.
-
Vorzugsweise
ist R7 C1-C4-Alkyl.
-
Vorzugsweise
ist R7 Ethyl oder n-Propyl.
-
Vorzugsweise
ist R8 H.
-
In
einem weiteren Aspekt der vorliegenden Erfindung wird eine Verbindung
der Formel (II), (III), (XI), (XIII), (XIV), (XV), (XVI), (XIX),
(XIXb), (XIXc) oder (XIXd) bereitgestellt:
worin
R
1 bis R
8, A, "Het" und m, wenn sie
vorhanden sind, wie oben definiert sind; P
1,
P
2 und P
3 Schutzgruppen sind
und Z eine Abgangsgruppe ist.
-
In
einem weiteren Aspekt der vorliegenden Erfindung erfolgt die Bereitstellung
eines Verfahrens zur Herstellung einer Verbindung der Formel (I)
oder eines pharmazeutisch akzeptablen Salzes derselben, wobei das
Verfahren die Stufe der Umsetzung eines Esters der Formel (II):
mit einem Amin der Formel
R
2-A-NHR
8 (X), worin
R
1 bis R
8, A, "Het" und m, wenn sie
vorhanden sind, wie oben definiert sind, umfasst.
-
In
einem weiteren Aspekt der vorliegenden Erfindung erfolgt die Bereitstellung
eines Verfahrens zur Herstellung einer Verbindung der Formel (II):
wobei das Verfahren die Stufe
des Entschützens
einer Verbindung der Formel (III):
worin P
1 und
P
2 Schutzgruppen sind, die gleich oder verschieden
sein können
oder Teil der gleichen Schutzgruppe sein können und worin R
1 bis
R
8, A, "Het", wenn sie vorhanden
sind, wie oben definiert sind, umfasst.
-
In
einem weiteren Aspekt der vorliegenden Erfindung erfolgt die Bereitstellung
eines Verfahrens zur Herstellung einer Verbindung der Formel (III):
wobei das Verfahren die Stufe
der Umsetzung einer Verbindung der Formel (XI):
worin P
1 und
P
2 Schutzgruppen sind, mit Trimethylsilyltrifluormethansulfonat
und einer Verbindung der Formel (IV) umfasst:
wobei die Verbindung der
Formel (IV) mit N,O-Bis(trimethylsilyl)acetamid derivatisiert und
dann mit der Verbindung der Formel (XI) umgesetzt wird, und wobei
R
1 bis R
8, A, "Het" und m, wenn sie
vorhanden sind, wie oben definiert sind.
-
In
einem weiteren Aspekt der vorliegenden Erfindung erfolgt die Bereitstellung
eines Verfahrens zur Herstellung einer Verbindung der Formel (IV):
durch Alkoholyse und anschließende Hydrolyse
eines Nitrils der Formel (V):
worin R
1 bis
R
8, A, "Het" und m, wenn sie
vorhanden sind, wie oben definiert sind.
-
In
einem weiteren Aspekt der vorliegenden Erfindung erfolgt die Bereitstellung
eines Verfahrens zur Herstellung einer Verbindung der Formel (V):
durch Entschützen einer
Verbindung der Formel (VI):
worin P
3 eine
Schutzgruppe ist und R
1 bis R
8,
A, "Het" und m, wenn sie
vorhanden sind, wie oben definiert sind.
-
In
einem weiteren Aspekt der vorliegenden Erfindung erfolgt die Bereitstellung
eines Verfahrens zur Herstellung einer Verbindung der Formel (VI):
durch Substitution der Chlorgruppe
in einer Verbindung der Formel (VII):
durch eine Cyanogruppe, wobei
R
1 bis R
8, A, "Het" und m, wenn sie
vorhanden sind, wie oben definiert sind.
-
In
einem weiteren Aspekt der vorliegenden Erfindung erfolgt die Bereitstellung
eines Verfahrens zur Herstellung einer Verbindung der Formel (VII):
durch Umsetzung einer Verbindung
der Formel (VIII):
mit einem Amin der Formel
R
1NH
2 (XII), wobei
R
1 bis R
8, A, "Het" und m, wenn sie
vorhanden sind, wie oben definiert sind.
-
In
einem weiteren Aspekt der vorliegenden Erfindung erfolgt die Bereitstellung
eines Verfahrens zur Herstellung einer Verbindung der Formel (VIII):
durch Schützen von 2,6-Dichlor-9H-purin
(IX):
-
-
In
den obigen Definitionen bedeutet Halogen Fluor, Chlor, Brom oder
Iod, und Alkyl-, Alkylen-, Alkanoyl- und Alkoxygruppen, die die
erforderliche Zahl an Kohlenstoffatomen enthalten, können eine
unverzweigte oder verzweigte Kette sein. Der im obigen R2, Teil (iii) definierte Heterocyclus kann
aromatisch oder vollständig oder
partiell gesättigt
sein. Der in den Definitionen von R2 und
Het verwendete Ausdruck "C-gebunden" bedeutet, dass die
Gruppe durch einen Ringkohlenstoff an das benachbarte Atom gebunden
ist. Der in den Definitionen von R2 verwendete
Ausdruck "N-gebunden" bedeutet, dass die
Gruppe durch einen Ringstickstoff an das benachbarte Atom gebunden
ist. Beispiele für
Alkyl umfassen Methyl, Ethyl, n-Proyl, Isopropyl, n-Butyl, Isobutyl,
sek-Butyl und tert-Butyl.
Beispiele für
Alkoxy umfassen Methoxy, Ethoxy, n-Propoxy, Isopropoxy, n-Butoxy, Isobutoxy,
sek-Butoxy und tert-Butoxy. Beispiele für Alkylen umfassen Methylen,
1,1-Ethylen, 1,2-Ethylen, 1,3-Propylen und 1,2-Propylen. Beispiele
für Cycloalkyl
umfassen Cyclopropyl, Cyclobutyl, Cyclopentyl, Cyclohexyl und Cycloheptyl.
-
Die
pharmazeutisch akzeptablen Salze der Verbindungen der Formel I umfassen
die Säureadditions- und
Basesalze derselben. Geeignete Säureadditionssalze
werden mit Säuren
gebildet, die nichttoxische Salze bilden, und Beispiele sind die
Hydrochlorid-, Hydrobromid-, Hydroiodid-, Sulfat-, Bisulfat-, Nitrat-,
Phosphat-, Hydrogenphosphat-, Acetat-, Maleat-, Fumarat-, Lactat-,
Tartrat, Citrat-, Gluconat-, Succinat-, Saccharat-, Benzoat-, Methansulfonat-,
Ethansulfonat-, Benzolsulfonat-, p-Toluolsulfonat- und Pamoatsalze.
-
Geeignete
Basesalze werden von Basen gebildet, die nichttoxische Salze bilden,
und Beispiele sind die Natrium-, Kalium-, Aluminium-, Calcium-,
Magnesium-, Zink- und Diethanolaminsalze.
-
Für eine Übersicht
geeigneter Salze siehe Berge et al., J. Pharm. Sci., 1977, 66, 1–19.
-
Die
pharmazeutisch akzeptablen Solvate der Verbindungen der Formel (I)
umfassen die Hydrate derselben.
-
Ebenfalls
umfasst vom vorliegenden Umfang der Verbindungen der Formel (I)
sind Polymorphe derselben.
-
Eine
Verbindung der Formel (I) kann ein oder mehrere zusätzliche
asymmetrische Kohlenstoffatome enthalten und daher in zwei oder
mehreren stereoisomeren Formen existieren. Die vorliegende Erfindung
umfasst die individuellen Stereoisomere der Verbindungen der Formel
(I) zusammen mit Gemischen derselben zusammen mit ggf. den individuellen
Tautomeren der Verbindungen der Formel (I) und Gemischen dersel ben.
-
Eine
Trennung von Diastereoisomeren kann durch herkömmliche Techniken, beispielsweise
durch fraktionierte Destillation, Chromatographie oder HPLC eines
Stereoisomerengemischs von einer Verbindung der Formel (I) oder
einem geeigneten Salz oder Derivat derselben erreicht werden. Ein
individuelles Enantiomer einer Verbindung der Formel (I) kann auch
aus einem entsprechenden optisch reinen Zwischenprodukt oder durch
Trennung, beispielsweise durch HPLC, des entsprechenden Racemats
unter Verwendung eines geeigneten chiralen Trägers oder durch fraktionierte
Kristallisation der durch Reaktion des entsprechenden Racemats mit
einer geeigneten optisch aktiven Säure oder ggf. Base gebildeten
Diastereoisomerensalze hergestellt werden.
-
Alle
Verbindungen der Formel (I) können
auf herkömmlichen
Wegen, beispielsweise durch die in den im folgenden angegebenen
allgemeinen Verfahren beschriebenen Verfahrensmaßnahmen oder durch die im Abschnitt
Beispiele beschriebenen speziellen Verfahren oder durch dazu ähnliche
Verfahren hergestellt werden. Die vorliegende Erfindung umfasst
auch ein beliebiges oder mehrere dieser Verfahren zur Herstellung
der Verbindungen der Formel (I) zusätzlich zu etwaigen darin verwendeten
neuen Zwischenprodukten. Bei den beschriebenen allgemeinen Verfahren
sind R1, R2, R7, R8 und A wie zuvor
für eine
Verbindung der Formel (I) definiert, falls nicht anders angegeben.
-
Alle
Verbindungen der Formel (I) können
gemäß dem in
Reaktionsschema 1 gezeigten Weg hergestellt werden, wobei R9 für
C1-C4-Alkyl, vorzugsweise
Methyl oder Ethyl, steht und P1, P2 und P3 für Schutzgruppen
stehen.
-
-
In
Reaktionsschema 1 können
die Verbindungen der Formel (I) durch Reaktion eines Esters der
Formel (II) mit einem Amin der Formel R2-A-NHR8 (X)hergestellt
werden.
-
Die
Reaktion kann bei erhöhter
Temperatur, vorzugsweise 100 bis 150°C, und optional in Gegenwart eines
geeigneten Lösemittels,
wie Ethanol, durchgeführt
werden. In einem typischen Verfahren werden die Verbindungen der
Formel (II) und das Amin der Formel (X) zusammen bei etwa 120°C erhitzt.
Verbindungen der Formel (II) können
durch Entschützen
einer Verbindung der Formel (III), worin die Schutzgruppen P
1 und P
2 gleich oder
verschieden sein können
und optional einen Teil der gleichen Schutzgruppe bilden können, hergestellt
werden. Beispiele für
geeignete Schutzgruppen sind dem Fachmann klar [siehe beispielsweise "Protecting Groups
in Organic Synthesis (Second Edition)", Theodora W. Green und Peter G. M.
Wuts, John Wiley and Sons, 1991]. Bevorzugte individuelle Schutzgruppen
sind Alkanoyl und Aroyl. Bevorzugte Schutzgruppen, wobei P
1 und P
2 einen Teil
der gleichen Schutzgruppe bilden, sind, wobei P
1 und
P
2 zusammengenommen sind, C
1-C
6-Alkylen. Besonders bevorzugte individuelle
Schutzgruppen sind Acetyl und Benzoyl. Eine besonders bevorzugte
Schutzgruppe, wobei P
1 und P
2 einen
Teil der gleichen Schutzgruppe bilden, ist, wenn P
1 und P
2 zusammengenommen werden, Dimethylmethylen.
Geeignete Bedingungen für
das Entschützen
sind einschlägig
bekannt [siehe beispielsweise "Protecting
Groups in Organic Synthesis (Second Edition)", Theodora W. Green und Peter G. M.
Wuts, John Wiley and Sons, 1991]. In einem typischen Verfahren können, wenn
P
1 und P
2 jeweils
Benzoyl sind, die Schutzgruppen durch Behandeln einer Lösung der
Verbindung der Formel (III) in einem geeigneten Lösemittel,
wie Methanol, mit einer Base, wie Kaliumcarbonat, typischerweise
bei Raumtemperatur entfernt werden. Verbindungen der Formel (III)
können
durch Reaktion einer Verbindung der Formel (XI):
(worin P
1 und
P
2 wie oben definiert sind) mit Trimethylsilyltrifluormethansulfonat
und einer Verbindung der Formel (IV), die mit N,O-Bis(trimethylsilyl)acetamid
derivatisiert wurde, hergestellt werden. In einem typischen Verfahren
wird die Verbindung der Formel (IV) in Gegenwart eines geeigneten
Lösemittels,
wie 1,1,1-Trichlorethan, und von N,O-Bis(trimethylsilyl)acetamid bei erhöhter Temperatur,
vorzugsweise unter Refluxieren erhitzt. Das Gemisch wird dann abkühlen gelassen
und vom Lösemittel
befreit. Der Rückstand
wird mit einer Lösung
der Verbindung der Formel (XI) in einem geeigneten Lösemittel,
wie Toluol, und anschließend
Trimethylsilyltrifluormethansulfonat behandelt und das Gemisch wird
unter Stickstoffatmosphäre
bei einer Temperatur zwischen Raumtemperatur und Rückflusstemperatur
des gewählten
Lösemittels
erhitzt, wobei die Verbindung der Formel (III) erhalten wird. Im
Falle von Toluol ist beispielsweise eine Temperatur von 110°C bevorzugt.
Verbindungen der Formel (IV) können
durch eine Reaktionsfolge von Alkoholyse und Hydrolyse, die auf
ein Nitril der Formel (V) angewandt wird, hergestellt werden. In
einem typischen Verfahren wird eine Lösung des Nitrils der Formel
(V) in einem alkoholischen Lösemittel
R
9OH mit einem Natriumalkoxid der Formel
R
9ONa behandelt und unter Refluxieren erhitzt
(R
9 ist wie oben definiert). Das erhaltene
Gemisch wird gekühlt,
eingedampft, in einem geeigneten Lösemittel, wie Tetrahydrofuran,
gelöst
und mit einer Säure,
wie Salzsäure,
vorzugsweise 2 N Salzsäure,
behandelt, wobei eine Verbindung der Formel (IV) erhalten wird.
Verbindungen der Formel (V) kön nen
durch Entschützen
einer Verbindung der Formel (VI), worin P
3 eine
geeignete Schutzgruppe ist, hergestellt werden. Beispiele für geeignete
Schutzgruppen sind dem Fachmann klar [siehe beispielsweise "Protecting Groups
in Organic Synthesis (Second Edition)", Theodora W. Green und Peter G. M.
Wuts, John Wiley and Sons, 1991]. Eine bevorzugte Schutzgruppe ist
diejenige, worin P
3 für Tetrahydropyran-2-yl steht. Geeignete
Bedingungen für
das Entschützen
sind einschlägig
bekannt [siehe beispielsweise "Protecting
Groups in Organic Synthesis (Second Edition)", Theodora W. Green und Peter G. M.
Wuts, John Wiley and Sons, 1991]. In einem typischen Verfahren kann,
wenn P
3 Tetrahydropyran-2-yl ist, die Schutzgruppe
durch Behandeln einer Lösung
der Verbindung der Formel (VI) in einem geeigneten Lösemittel,
wie Ethanol, mit einer Säure,
wie Salzsäure,
entfernt werden. Verbindungen der Formel (VI) können durch Substitution der
Chlorgruppe in einer Verbindung der Formel (VII) durch eine Cyanogruppe
hergestellt werden. In einem typischen Verfahren wird eine Lösung der
Verbindung der Formel (VII) in einem geeigneten Lösemittel,
wie N,N-Dimethylformamid, mit Zinkcyanid, Tetrakis(triphenylphosphin)palladium(0)
und einem Säureakzeptor,
wie Triethylamin, behandelt und unter Stickstoffatmosphäre auf zwischen
80 und 120°C,
vorzugsweise 100°C,
erhitzt. Das Produkt der Reaktion ist üblicherweise mit einer Menge
der entsprechenden Verbindung der Formel (V) kontaminiert, die durch Routinechromatographie
abgetrennt werden kann. Verbindungen der Formel (VII) können durch
Reaktion einer Verbindung der Formel (VIII) mit einem Amin der Formel
R1NH2 (XII)hergestellt
werden. In einem typischen Verfahren wird eine Lösung der Verbindung der Formel
(VIII) in einem geeigneten Lösemittel,
wie Isopropylalkohol, mit der Verbindung der Formel (XII) behandelt
und unter Refluxieren, optional in Gegenwart eines Säureakzeptors,
wie N-Ethyl-N-isopropyl-2- propylamin,
erhitzt. Verbindungen der Formel (VIII) können durch Schützen von
2,6-Dichlor-9H-purin (IX) hergestellt werden. Beispiele für geeignete
Schutzgruppen P
3 sind dem Fachmann klar
[siehe beispielsweise "Protecting
Groups in Organic Synthesis (Second Edition)", Theodora W. Green und Peter G. M.
Wuts, John Wiley and Sons, 1991]. Eine bevorzugte Schutzgruppe ist
diejenige, worin P
3 für Tetrahydropyran-2-yl steht.
Geeignete Bedingungen für das
Schützen
sind einschlägig
bekannt [siehe beispielsweise "Protecting
Groups in Organic Synthesis (Second Edition)", Theodora W. Green und Peter G. M.
Wuts, John Wiley and Sons, 1991]. In einem typischen Verfahren wird,
wenn P
3 Tetrahydropyran-2-yl ist, eine Lösung aus
2,6-Dichlor-9H-purin (IX) und einem Säurekatalysator, wie 4-Toluolsulfonsäuremonohydrat,
in einem geeigneten Lösemittel,
wie Ethylacetat, auf zwischen 30 und 70°C, vorzugsweise 50°C erhitzt
und mit einer Lösung
von 2,3-Dihydropyran in einem geeigneten Lösemittel, wie Ethylacetat,
behandelt.
-
Alle
Verbindungen der Formel (I) können
auch gemäß dem in
Reaktionsschema 2 angegebenen Weg hergestellt werden, worin R9, P1, P2 und
P3 wie oben definiert sind.
-
-
In
Reaktionsschema 2 können
die Verbindungen der Formel I durch Entschützen einer Verbindung der Formel
(XIII) hergestellt werden. Geeignete Bedingungen für das Entschützen sind
einschlägig
bekannt [siehe beispielsweise "Protecting
Groups in Organic Synthesis (Second Edition)", Theodora W. Green und Peter G. M.
Wuts, John Wiley and Sons, 1991]. In einem typischen Verfahren können, wenn
P
1 und P
2 jeweils
Benzoyl sind, die Schutzgruppen durch Behandeln einer Lösung der
Verbindung der Formel (XIII) in einem geeigneten Lösemittel,
wie Methanol, mit einer Base, wie Kaliumcarbonat, typischerweise
bei Raumtemperatur entfernt werden. Verbindungen der Formel (XIII)
können
durch Reaktion einer Verbindung der Formel (XI):
(worin P
1 und
P
2 wie oben definiert sind) mit Trimethylsilyltrifluormethansulfonat
und einer Verbindung der Formel (XIV), die mit N,O-Bis(trimethylsilyl)acetamid
derivatisiert wurde, hergestellt werden. In einem typischen Verfahren
wird die Verbindung der Formel (XIV) in Gegenwart eines geeigneten
Lösemittels,
wie 1,1,1-Trichlorethan, und von N,O-Bis(trimethylsilyl)acetamid
bei erhöhter
Temperatur, vorzugsweise unter Refluxieren erhitzt. Das Gemisch
wird dann abkühlen
gelassen und das Lösemittel
wird entfernt. Der Rückstand
wird mit einer Lösung
der Verbindung der Formel (XI) in einem geeigneten Lösemittel,
wie Toluol, und anschließend Trimethylsilyltrifluormethansulfonat
behandelt und das Gemisch wird unter Stickstoffatmosphäre bei einer Temperatur
zwischen Raumtemperatur und der Rückflusstemperatur des gewählten Lösemittels
erhitzt, wobei die Verbindung der Formel (XIII) erhalten wird. Im
Falle von Toluol ist beispielsweise eine Temperatur von 90°C bevorzugt.
Verbindungen der Formel (XIV) können
durch Entschützen
einer Verbindung der Formel (XV) hergestellt werden. Geeignete Bedingungen
für das
Entschützen
sind einschlägig
bekannt [siehe beispielsweise "Protecting
Groups in Organic Synthesis (Second Edition)", Theodora W. Green und Peter G. M.
Wuts, John Wiley and Sons, 1991]. In einem typischen Verfahren kann,
wenn P
3 Tetrahydropyran-2-yl ist, die Schutzgruppe
durch Behandeln einer Lösung
der Verbindung der Formel (XV) in einem geeigneten Lösemittel,
wie Ethanol, mit einer Säure,
wie Salzsäure,
entfernt werden. Verbindungen der Formel (XV) können durch Reaktion eines Es ters
der Formel (XVI) mit einem Amin der Formel (X):
R2-A-NHR8 (X)bei erhöhter Temperatur,
vorzugsweise 100 bis 150°C,
hergestellt werden. In einem typischen Verfahren werden die Verbindung
der Formel (XVI) und das Amin der Formel (X) zusammen bei 130°C erhitzt.
Verbindungen der Formel (XVI) können
durch eine Reaktionsfolge von Alkoholyse und Hydrolyse, die an einem
Nitril der Formel (VI) durchgeführt
wird, hergestellt werden. In einem typischen Verfahren wird eine
Lösung
des Nitrils der Formel (VI) in einem alkoholischen Lösemittel
R
9OH mit dem Natriumalkoxid der Formel R
9ONa behandelt und unter Refluxieren erhitzt.
Das gebildete Gemisch wird gekühlt,
eingedampft, in einem geeigneten Lösemittel, wie einem Gemisch
aus Tetrahydrofuran und Wasser, (vorzugsweise 3:1, bezogen auf das
Volumen) gelöst und
mit einer Säure,
wie Essigsäure,
behandelt. Das gebildete Gemisch wird bei erhöhter Temperatur, vorzugsweise
unter Refluxieren erhitzt, wobei die Verbindung der Formel (XVI)
erhalten wird.
-
Verbindungen
der Formel (XI), die in den Reaktionsschemata 1 und 2 verwendet
werden, können
wie in Reaktionsschema 3 angegeben hergestellt werden, wobei P1 und P2 wie oben
definiert sind.
-
-
Verbindungen
der Formel (XI) können
durch Behandlung einer Verbindung der Formel (XVII) mit einem Gemisch
aus Essigsäure,
Essigsäureanhydrid
und einer starken Säure,
wie Salzsäure
oder Schwefelsäure,
unter Kühlen
(typischerweise auf –10°C) hergestellt
werden. Eine Verbindung der Formel (XVII) kann aus einer Säure der
Formel (XVIII) durch Aktivierung der Säure als beispielsweise Säurechlorid
und Behandeln dieses aktivierten Zwischenprodukts mit einem Amin
der Formel (XIXa): R7NH2 (XIXa)hergestellt
werden. In einem typischen Verfahren wird eine Verbindung der Formel
(XVIII) in einem geeigneten inerten Lösemittel (beispielsweise Dichlormethan)
gelöst
und mit Oxalylchlorid und einer katalytischen Menge N,N-Dimethylformamid
behandelt. Nach Entfernen von überschüssigem Lösemittel
und Reagens durch Eindampfen unter vermindertem Druck wird der Rückstand
in wasserfreiem Dichlormethan gelöst und mit einem Amin der Formel
(XIXa) behandelt. Im Hinblick auf die in späteren Stufen verwendeten Bedingungen
kann es günstig
sein, die Schutzgruppen P1 und P2 in Verbindungen der Formel (XVII) zu ändern. Alternativ
sind geeignete Schutzgruppen dem Fachmann bekannt [beispielsweise "Protecting Groups
in Organic Synthesis (Second Edition)", Theodora W. Green und Peter G. M.
Wuts, John Wiley and Sons, 1991]. In einem typischen Fall kann eine
Lösung
der Verbindung der Formel (XVII), worin P1 und
P2 zusammengenommen Dimethylmethylen sind,
in einem geeigneten Lösemittel,
wie Methanol, mit einer Säure,
wie Pyridinium-para-toluolsulfonat, behandelt werden, wobei eine
Verbindung der Formel (XVII) erhalten wird, worin P1 und
P2 beide durch H ersetzt sind, die anschließend mit
anderer Funktionalität
erneut geschützt
werden können.
Beispielsweise kann die Verbindung der Formel (XVII), worin P1 und P2 beide durch
H ersetzt sind, in einem geeigneten Lösemittel, wie Dichlormethan,
gelöst
und die gebildete Lösung
mit einem Säureakzeptor,
wie Pyridin, und Benzoylchlorid behandelt werden, wobei eine Verbindung
der Formel (XVII) erhalten wird, worin P1 und
P2 jeweils Benzoyl sind. Verbindungen der
Formel (XVIII) sind einschlägig
bekannt, beispielsweise in J. Amer. Chem. Soc., 1958, 80, 5168,
worin P1 und P2 zusammengenommen
Dimethylmethylen sind.
-
Amine
der Formeln R7NH2 (XIXa),
R1NH2 (XII) und
R2-A-NHR8 (X) sind
entweder im Handel erhältlich oder
können
durch Fachleuten bekannte Standardtechniken hergestellt werden.
-
Alle
Verbindungen der Formel (I) können
alternativ durch Kondensation einer Säure der Formel (XIX) mit einem
Amin der Formel (X), wie in Reaktionsschema 4 angegeben, hergestellt
werden.
-
-
Die
Kondensation wird typischerweise unter herkömmlichen Peptidkopplungsbedingungen
durchgeführt.
Beispielsweise kann eine Lösung
der Säure
(XIX) in einem geeigneten Lösemittel,
wie Dichlormethan, zunächst
mit einem geeigneten Kopplungsmittel, wie Carbonyldiimidazol, und
anschließend
mit einer Verbindung der Formel (X) behandelt werden. Eine Säure der
Formel (XIX) kann durch Hydrolyse einer Verbindung der Formel (II)
hergestellt werden. Zur Durchführung
der Hydrolyse wird eine Verbindung der Formel (II) typischerweise
in einem geeigneten Lösemittel,
wie Ethanol, gelöst
und mit einer geeigneten Base, wie wässriges Natriumhydroxid, behandelt.
-
Verbindungen
der Formel (I) können
alternativ durch eine Aminocarbonylierungsreaktion einer Verbindung
der Formel (XIXb):
worin Z eine geeignete Abgangsgruppe,
wie Brom, Iod, Sn(C
1-C
12-Alkyl)
3 oder CF
3SO
2O-, vorzugsweise Iod, ist, mit einer Verbindung
der Formel R
2-A-NHR
8 (X)
in Gegenwart von Kohlenmonoxid und eines geeigneten Kopplungskatalysators
hergestellt werden. Vorzugsweise ist der Katalysator ein Palladium(II)-Katalysator, noch
besser 1,1'-Bis(diphenylphosphino)ferrocendichlorpalladium(II)
(optional als 1:1-Komplex mit Dichlormethan). Alternativ kann Palladium(II)-acetat
in Gegenwart eines geeigneten Liganden, wie 1,1'-Bis(diphenylphosphino)ferrocen, Triphenyl phosphin,
Tri(o-tolyl)phosphin oder (R)-, (S)- oder racemisches 2,2'-Bis(diphenylphosphino)-1,1'-binaphthyl, verwendet
werden.
-
In
einem typischen Verfahren wird die Reaktion in einem hermetisch
verschlossenen Gefäß in Gegenwart
von Kohlenmonoxid bei erhöhtem
Druck beispielsweise etwa 345 kPa (50 psi) bei erhöhter Temperatur, beispielsweise
etwa 60°C,
und in einem geeigneten Lösemittel,
beispielsweise Tetrahydrofuran, Methanol oder Ethanol, durchgeführt. Optional
kann eine geeignete organische Base, wie ein tertiäres Amin,
beispielsweise Triethylamin, N-Ethyldiisopropylamin oder 4-Methylmorpholin,
vorhanden sein.
-
Die
Zwischenprodukte der Formel (XIXc) können wie in Reaktionsschema
5 angegeben hergestellt werden. Reaktionsschema
5
worin Z wie zuvor für die Verbindung (XIXb) definiert
ist und Ac Acetyl ist.
-
In
einem typischen Verfahren wird eine Verbindung der Formel (XIXd)
mit einem Amin der Formel R1NH2 (XII)
in Gegenwart eines geeigneten Säureakzeptors,
beispielsweise Triethylamin, und in einem geeigneten Lösemittel,
beispielsweise Acetonitril, falls nötig, bei erhöhter Temperatur
umgesetzt. Das erhaltene Produkt der Formel (XIXc) kann durch Hydrolyse
unter Bildung einer Verbindung der Formel (XIXb) durch ein herkömmliches
Verfahren, beispielsweise durch die Verwendung einer geeigneten
anorganischen Base, beispielsweise Natriumcarbonat, Natriumhydroxid,
Kaliumhydroxid, Lithiumhydroxid, Natriumcarbonat, Kaliumcarbonat
oder Cäsiumcarbonat,
und in einem geeigneten Lösemittel,
beispielsweise Methanol, Ethanol, Isopropanol, 1,2-Dimethoxyethan,
Tetrahydrofuran, Dimethylformamid, Aceton, 2-Butanon oder 4-Methyl-2-pentanon, optional
unter wässrigen
Bedingungen bei 0°C
bis zur Rückflusstemperatur
des Lösemittels,
beispielsweise Raumtemperatur, entschützt werden. Alternativ kann
das Entschützen
unter Verwendung einer geeigneten Aminbase, wie Triethylamin, Diisopropylethylamin,
4-Methylmorpholin, Ammonium, Methylamin, Ethylamin oder Dimethylamin,
in einem geeigneten Lösemittel,
wie Methanol, Ethanol, n-Propanol, Isopropanol, Tetrahydrofuran
oder Dichlormethan, bei 0°C
bis Rückflusstemperatur
des Lösemittels
durchgeführt
werden. Verbindungen der Formel (XIXd) können durch ein herkömmliches
Verfahren hergestellt werden.
-
Ein
pharmazeutisch akzeptables Salz einer Verbindung der Formel (I)
kann durch Zusammenmischen von Lösungen
einer Verbindung der Formel (I) und der gewünschten Säure oder ggf. Base ohne weiteres
hergestellt werden. Das Salz kann aus der Lösung ausfallen und durch Filtration
gewonnen werden oder durch Abdampfen des Lösemittels gewonnen werden.
-
Die
entzündungshemmenden
Eigenschaften der Verbindungen der Formel (I) werden durch deren
Fähigkeit
zur Hemmung der Neutrophilenfunktion, die A2a-Rezeptoragonistenaktivität anzeigt,
belegt. Dies wird durch Bestimmen des Verbindungsprofils in einem
Test, bei dem die Superoxidproduktion von durch fMLP aktivierten
Neutrophilen ermittelt wurde, beurteilt. Neutrophile wurden aus
humanem peripherem Blut unter Verwendung von Dextransedimentation
und anschließende
Zentrifugation über
eine Ficoll-Hypaque-Lösung
isoliert. Etwaige kontaminierende Erythrocyten in dem Granulocyten pellet
wurden durch Lyse mit eiskaltem destilliertem Wasser entfernt. Die
Superoxidproduktion von den Neutrophilen wurde durch fMLP in Gegenwart
einer Primingkonzentration von Cytochalasin B induziert. Adenosindeaminase
wurde in den Test eingearbeitet, um etwaiges endogen produziertes
Adenosin, das die Superoxidproduktion unterdrücken könnte, zu entfernen. Die Wirkung
der Verbindung auf die fMLP-induzierte Reaktion wurde kolorimetrisch über die
Reduktion von Cytochrom C in dem Testpuffer überwacht. Die Wirksamkeit der
Verbindungen wurde durch die Konzentration, die im Vergleich zur
Reaktion der Kontrolle auf fMLP 50% Hemmung ergab (IC50),
festgestellt.
-
Bei
diesem Test weisen die Verbindungen der Erfindung IC50-Werte von weniger
als 200 nM auf. Die Verbindungen der Beispiele 7, 10, 12, 16, 20,
21, 27 und 31 sind äußerst wirksam
und weisen IC50-Werte von weniger als 40
nM auf.
-
Die
vorliegende Erfindung stellt daher eine pharmazeutische Zusammensetzung
bereit, die eine Verbindung der Formel (I) gemäß der obigen Definition oder
ein pharmazeutisch akzeptables Salz oder Solvat derselben zusammen
mit einem pharmazeutisch akzeptablen Streckmittel, Verdünnungsmittel
oder Träger
umfasst.
-
Die
vorliegende Erfindung stellt ferner eine Verbindung der Formel (I)
gemäß der obigen
Definition oder ein pharmazeutisch akzeptables Salz, Solvat oder
eine Zusammensetzung derselben zur Verwendung als Medikament bereit.
-
Die
vorliegende Erfindung stellt ferner eine Verbindung der Formel (I)
gemäß der obigen
Definition oder ein pharmazeutisch akzeptables Salz, Solvat oder
eine Zusammensetzung derselben zur Verwendung als Medikament zur
Behandlung einer Erkrankung, für
die ein A2a-Rezeptoragonist indiziert ist, bereit.
-
Die
vorliegende Erfindung stellt ferner die Verwendung einer Verbindung
der Formel (I) gemäß der obigen
Definition oder eines pharmazeutisch akzeptablen Salzes, Solvats
oder einer Zusammensetzung derselben zur Herstellung eines Medikaments
zur Behandlung einer Erkrankung, für die ein A2a-Rezeptoragonist indiziert
ist, bereit.
-
Die
vorliegende Erfindung stellt ferner eine Verbindung der Formel (I)
gemäß der obigen
Definition oder ein pharmazeutisch akzeptables Salz, Solvat oder
eine Zusammensetzung derselben zur Verwendung als entzündungshemmendes
Mittel bereit.
-
Die
vorliegende Erfindung stellt ferner die Verwendung einer Verbindung
der Formel (I) gemäß der obigen
Definition oder eines pharmazeutisch akzeptablen Salzes, Solvats
oder einer Zusammensetzung derselben zur Herstellung eines entzündungshemmenden
Mittels bereit.
-
Die
vorliegende Erfindung stellt ferner die Verwendung einer Verbindung
der Formel (I) gemäß der obigen
Definition oder eines pharmazeutisch akzeptablen Salzes, Solvats
oder einer Zusammensetzung derselben zur Herstellung eines Medikaments
zur Behandlung einer Atemwegserkrankung bereit.
-
Die
vorliegende Erfindung stellt ferner eine wie oben angegebene Verwendung
bereit, wobei die Erkrankung aus der Gruppe von Schocklunge (ARDS),
Bronchitis, chronischer Bronchitis, chronisch-obstruktiver Lungenerkrankung,
Mukoviszidose, Asthma, Emphysem, Bronchiektase, chronischer Sinusitis
und Rhinitis ausgewählt
ist.
-
Die
vorliegende Erfindung stellt ferner die Verwendung einer Verbindung
der Formel (I) gemäß der obigen
Definition oder eines pharmazeutisch akzeptablen Salzes, Solvats
oder einer Zusammensetzung derselben zur Herstellung eines Medikaments
zur Behandlung von septischem Schock, männlicher erektiler Dysfunktion,
Hypertonie, Schlaganfall, Epilepsie, Hirnischämie, peripherer Verschlusskrankheit,
postischämischer Reperfusionsläsion, Diabetes,
rheumatoider Arthritis, Multipler Sklerose, Psoriasis, allergischer
Kontaktdermatitis/atopischer Dermatitis, Ekzem, ulzeröser Colitis,
Morbus Crohn, entzündlicher
Darmerkrankung, Heliobacter pylori-Gastritis, nicht-Heliobacter pylori-Gastritis,
einer durch nichtsteroidale entzündungshemmende
Arzneimittel induzierten Schädigung
des Magen-Darm-Trakts oder einer psychotischen Störung oder
zur Wundheilung bereit.
-
Die
vorliegende Erfindung stellt ferner ein Verfahren zur Behandlung
eines Säugers
einschließlich
eines Menschen zur Behandlung einer Erkrankung, für die ein
A2a-Rezeptoragonist indiziert ist, bereit, wobei das Verfahren das
Behandeln des Säugers
mit einer wirksamen Menge einer Verbindung der Formel (I) gemäß der obigen
Definition oder mit einem pharmazeutisch akzeptablen Salz, Solvat
oder einer Zusammensetzung derselben umfasst.
-
Die
vorliegende Erfindung stellt ferner ein Verfahren zur Behandlung
eines Säugers
einschließlich
eines Menschen zur Behandlung einer entzündlichen Erkrankung bereit,
wobei das Verfahren das Behandeln des Säugers mit einer wirksamen Menge
einer Verbindung der Formel (I) gemäß der obigen Definition oder
mit einem pharmazeutisch akzeptablen Salz, Solvat oder einer Zusammensetzung
derselben umfasst.
-
Die
vorliegende Erfindung stellt ferner ein Verfahren zur Behandlung
eines Säugers
einschließlich
eines Menschen zur Behandlung einer Atemwegserkrankung bereit, wobei
das Verfahren die Behandlung des Säugers mit einer wirksamen Menge einer
Verbindung der Formel (I) gemäß der obigen
Definition oder mit einem pharmazeutisch akzeptablen Salz, Solvat
oder einer Zusammensetzung derselben umfasst. Die vorliegende Erfindung
stellt ferner ein wie oben angegebenes Verfahren bereit, wobei die
Erkrankung aus der Gruppe von Schocklunge (ARDS), Bronchitis, chronischer
Bronchitis, chronisch-obstruktiver Lungenerkrankung, Mukoviszidose,
Asthma, Emphysem, Bronchiektase, chronischer Sinusitis und Rhinitis
ausgewählt
ist.
-
Die
vorliegende Erfindung stellt ferner ein Verfahren zur Behandlung
eines Säugers
einschließlich
eines Menschen zur Behandlung von septischem Schock, männlicher
erektiler Dysfunktion, Hypertonie, Schlaganfall, Epilepsie, Hirnischämie, peripherer
Verschlusskrankheit, postischämischer
Reperfusionsläsion,
Diabetes, rheumatoider Arthritis, Multipler Sklerose, Psoriasis,
allergischer Kontaktdermatitis/atopischer Dermatitis, Ekzem, ulzeröser Colitis,
Morbus Crohn, entzündlicher
Darmerkrankung, Heliobacter pylori-Gastritis, nicht-Heliobacter pylori-Gastritis,
einer durch nichtsteroidale entzündungshemmende
Arzneimittel induzierten Schädigung
des Magen-Darm-Trakts oder einer psychotischen Störung oder
zur Wundheilung bereit, wobei das Verfahren die Behandlung des Säugers mit
einer wirksamen Menge einer Verbindung der Formel (I) gemäß der obigen
Definition oder mit einem pharmazeutisch akzeptablen Salz, Solvat
oder einer Zusammensetzung desselben umfasst.
-
Die
Verbindungen der Formel (I) können
allein verabreicht werden, werden jedoch allgemein im Gemisch mit
einem geeigneten pharmazeutisch akzeptablen Streckmittel, Verdünnungsmittel
oder Träger,
die im Hinblick auf den geplanten Verabreichungsweg und die pharmazeutische
Standardpraxis ausgewählt
sind, verabreicht.
-
Beispielsweise
können
die Verbindungen der Formel (I) oral, bukkal oder sublingual in
der Form von Tabletten, Kapseln, Ovuli, Elixieren, Lösungen oder
Suspensionen, die Aromatisierungs- oder Farbmittel enthalten können, für Anwendungen
mit unmittelbarer, verzögerter,
nachhaltiger, gepulster oder gesteuerter Freisetzung verabreicht
werden.
-
Derartige
Tabletten können
Streckmittel, wie mikrokristalline Cellulose, Lactose, Natriumcitrat,
Calciumcarbonat, dibasisches Calciumphosphat und Glycin, den Zerfall
fördernde
Mittel, wie Stärke
(vorzugsweise Mais-, Kartoffel- oder
Tapiokastärke),
Natriumstärkeglycollat,
Croscarmellosenatrium und bestimmte komplexe Silicate, und Granulationsbindemittel,
wie Polyvinylpyrrolidon, Hydroxypropylmethylcellulose (HPMC), Hydroxypropylcellulose
(HPC), Saccharose, Gelatine und Akaziengummi enthalten. Zusätzlich können Gleitmittel, wie
Magnesiumstearat, Stearinsäure,
Glycerylbehenat und Talkum, eingearbeitet werden.
-
Feste
Zusammensetzungen einer ähnlichen
Art können
auch als Füllstoffe
in Gelatinekapseln verwendet werden. Bevorzugte Streckmittel umfassen
im Hinblick darauf Lactose, Stärke,
eine Cellulose, Milchzucker oder ein Polyethylenglykol mit hohem
Molekulargewicht. Für
wässrige
Suspensionen und/oder Elixiere können die
Verbindungen der Formel (I) mit verschiedenen Süßungs- oder Aromatisierungsmitteln,
Farbmitteln oder -stoffen, mit Emulgatoren und/oder Suspendiermitteln
und mit Verdünnungsmitteln,
wie Wasser, Ethanol, Propylenglykol oder Glycerin, und Kombinationen
derselben kombiniert werden.
-
Die
Verbindungen der Formel (I) können
auch parenteral, beispielsweise intravenös, intraarteriell, intraperitoneal,
intrathekal, intraventrikulär,
intrasternal, intrakranial, intramuskulär oder subkutan verabreicht werden
oder sie können
durch Infusionstechniken verabreicht werden. Sie werden am besten
in der Form einer sterilen wässrigen
Lösung
verwendet, die andere Substanzen, beispielsweise genügend Salze
oder Glucose, um die Lösung
mit Blut isotonisch zu machen, enthalten kann. Die wässrigen
Lösungen
sollten, falls nötig,
geeignet gepuffert sein (vorzugsweise auf einen pH-Wert von 3 bis
9). Die Herstellung geeigneter parenteraler Formulierungen unter
sterilen Bedingungen wird durch dem Fachmann bekannte pharmazeutische
Standardtechniken ohne weiteres erreicht. Zur oralen und parenteralen
Verabreichung an Humanpatienten beträgt die Tagesdosierungsmenge
der Verbindungen der Formel (I) üblicherweise
0,01 bis 100 mg/kg Körpergewicht
des zu behandelnden Subjekts, vorzugsweise 0,1 bis 100 mg/kg (in
Einzeldosen oder geteilten Dosen).
-
Daher
können
Tabletten oder Kapseln der Verbindung der Formel (I) 5 bis 500 mg
aktive Verbindung zur Verabreichung einzeln oder ggf. von zwei oder
mehreren gleichzeitig enthalten. Der Arzt bestimmt in jedem Fall
die tatsächliche
Dosierung, die für
jeden individuellen Patienten am geeignetsten ist, und diese variiert
mit dem Alter, Gewicht und Ansprechen des speziellen Patienten.
Die obigen Dosierungen sind Beispiele für den durchschnittlichen Fall.
Natürlich
können
individuelle Fälle
auftreten, in denen höhere
oder niedrigere Dosierungsbereiche erforderlich sind, und diese
liegen innerhalb des Umfangs dieser Erfindung.
-
Die
Verbindungen der Formel (I) können
auch intranasal oder durch Inhalation verabreicht werden und werden
günstigerweise
in der Form einer Inhaliervorrichtung eines trockenen Pulvers oder
einer Aerosolspraypräsentation
aus einem Druckbehälter,
einer Pumpe, einem Spray, einer Zerstäubungsvorrichtung oder Vernebelungsvorrichtung
unter Verwendung eines geeigneten Treibmittels, beispielsweise Dichlor difluormethan, Trichlorfluormethan,
Dichlortetrafluorethan, ein Hydrofluoralkan, wie 1,1,1,2-Tetrafluorethan
(HFA 134A [Marke]) oder 1,1,1,2,3,3,3-Heptafluorpropan (HFA 227EA
[Marke]), Kohlendioxid oder ein anderes geeignetes Gas, zugeführt. Im
Falle eines druckbeaufschlagten Aerosols kann die Dosierungseinheit
durch Anbringen eines Ventils zur Abgabe einer abgemessenen Menge
bestimmt werden. Der Druckbehälter,
die Pumpe, das Spray, die Zerstäubungsvorrichtung
oder Vernebelungsvorrichtung können
eine Lösung
oder Suspension der aktiven Verbindung, beispielsweise unter Verwendung
eines Gemischs von Ethanol und dem Treibmittel als Lösemittel,
die zusätzlich
ein Gleitmittel, beispielsweise Sorbitantrioleat, enthalten kann,
enthalten. Kapseln oder Patronen (die beispielsweise aus Gelatine
bestehen) zur Verwendung in einer Inhaliervorrichtung oder einem
Insufflator können
so formuliert werden, dass sie ein Pulvergemisch aus einer Verbindung
der Formel (I) und einer geeigneten Pulvergrundlage, wie Lactose
oder Stärke,
enthalten.
-
Aerosol-
oder Trockenpulverformulierungen sind vorzugsweise so eingestellt,
dass jede abgemessene Dosis oder jeder "Stoß" 20 bis 4000 μg einer Verbindung
der Formel (I) zur Abgabe an den Patienten enthält. Die Gesamttagesdosis bei
einem Aerosol liegt im Bereich von 20 μg bis 20 mg, die in einer einzigen
Dosis oder üblicher
in geteilten Dosen über
den Tag verabreicht werden kann.
-
Alternativ
können
die Verbindungen der Formel (I) in der Form eines Suppositoriums
oder Zäpfchens verabreicht
werden oder sie können
topisch in der Form einer Lotion, Lösung, Creme, Salbe, eines Gels,
einer Suspension, eines stäubenden
Pulvers, eines Sprays oder einer Auflage, in die das Arzneimittel
eingearbeitet ist, (beispielsweise eine Tüllauflage, eine mit weißem weichem
Paraffin oder Polyethylen glykol imprägnierte Gazeauflage oder eine
Hydrogel-, Hydrokolloid-, Alginat- oder Filmauflage) appliziert
werden. Die Verbindungen der Formel (I) können auch transdermal, beispielsweise
durch Verwendung eines Hautpflasters, verabreicht werden.
-
Zur
topischen Applikation auf die Haut können die Verbindungen der Formel
(I) als geeignete Salbe, die die aktive Verbindung in beispielsweise
einem Gemisch mit einem oder mehreren der folgenden Bestandteile:
Mineralöl,
flüssiges
Petrolatum, weißes
Petrolatum, Propylenglykol, eine Polyoxyethylenpolyoxypropylenverbindung,
emulgierendes Wachs und Wasser, gelöst oder suspendiert enthält, formuliert
werden. Alternativ können
sie als geeignete Lotion oder Creme in beispielsweise einem Gemisch
von einem oder mehreren der folgenden: Mineralöl, Sorbitanmonostearat, ein
Polyethylenglykol, Paraffinöl,
Polysorbat 60, Cetylesterwachs, Cetearylalkohol, 2-Octyldodecanol,
Benzylalkohol und Wasser, suspendiert oder gelöst formuliert werden. Sie können auch
als Hydrogel mit Cellulose- oder Polyacrylatderivaten oder anderen
Viskositätsmodifizierungsmitteln
formuliert werden.
-
Es
ist auch klar, dass alle hier angegebenen Verweise auf eine Behandlung
eine kurative, palliative und prophylaktische Behandlung umfassen.
-
BEISPIELE
-
Die
folgenden Beispiele erläutern
die Herstellung der Verbindungen der Formel (I). 1H-Kernresonanz(NMR)spektren
waren in allen Fällen
mit den vorgeschlagenen Strukturen konsistent. Charakteristische chemische
Verschiebungen (δ)
sind in parts per million von Tetramethylsilan zu niederem Feld
unter Verwendung herkömmlicher
Abkürzungen
zur Be zeichnung der Hauptpeaks angegeben: beispielsweise s, Singulett; d,
Dublett; t, Triplett; q, Quartett; m, Multiplett; br, breit. Die
Massenspektren (m/z) wurden im Thermosprayionisationsmodus aufgezeichnet.
Die folgenden Abkürzungen
wurden für übliche Lösemittel
verwendet: CDCl3, Deuterochloroform; DMSO,
Dimethylsulfoxid; THF, Tetrahydrofuran. Wenn Dünnschichtchromatographie (DC) verwendet
wurde, bezieht sich dies auf Silicagel-DC unter Verwendung von Silicagel
60 F254-Platten, wobei Rf die
von der Verbindung zurückgelegte
Distanz, geteilt durch die von der Lösemittelfront zurückgelegte
Distanz auf einer DC-Platte ist.
-
BEISPIEL
1 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-[2-(1-piperidinyl)ethyl]-9H-purin-2-carboxamid
-
Eine
Lösung
von (2R,3R,4R,5S)-4-(benzoyloxy)-2-[6-[(2,2-diphenylethyl)amino]-2-({[2-(1-piperidinyl)ethyl]amino}carbonyl)-9H-purin-9-yl]-5-[(ethyl amino)carbonyl]tetrahydro-3-furanylbenzoat
(Herstellungsbeispiel 7) (104 mg, 0,12 mmol) in Methanol (3 ml)
wurde mit Kaliumcarbonat (100 mg, 0,72 mmol) behandelt. Das Gemisch
wurde 30 min bei Raumtemperatur gerührt. Das Lösemittel wurde unter vermindertem
Druck entfernt und der Rückstand
wurde zwischen Ethylacetat und Wasser verteilt. Die organische Schicht
wurde abgetrennt, mit wasserfreiem Magnesiumsulfat getrocknet, filtriert
und unter vermindertem Druck eingedampft. Der Rückstand wurde durch Säulenchromatographie
auf Silicagel unter Elution mit Dichlormethan:Methanol (90:10, bezogen
auf das Volumen) gereinigt, wobei die Titelverbindung als weißer Feststoff
(44 mg) erhalten wurde.
1H-NMR (400
MHz, CDCl3) δ: 8,76 (1H, s), 8,67 (1H, br
m), 7,63 (1H, br m), 7,38–7,19
(10H, m), 6,92 (1H, m), 6,07 (1H, br m), 5,10 (2H, m), 4,55 (1H,
m), 4,36 (4H, m), 3,52 (2H, m), 3,40 (2H, m), 2,60 (2H, br m), 2,40
(4H, br m), 1,37 (6H, m), 1,23 (3H, t).
LRMS (Thermospray:
m/z [MH+] 643, [MNa+]
665.
-
BEISPIEL
2 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-[2-(4-isopropyl-1-piperidinyl)ethyl]-9H-purin-2-carboxamid
-
Methyl-6-[(2,2-diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetra-hydro-2-furanyl}-9H-purin-2-carboxylat
(Herstellungsbeispiel 11) (100 mg, 0,18 mmol) und 2-(4-Isopropyl-1-piperidinyl)ethylamin
(Herstellungsbeispiel 17) (300 mg, 1,76 mmol) wurden 3 h bei 120°C unter Stickstoffatmosphäre erhitzt.
Das Reaktionsgemisch wurde auf Raumtemperatur abkühlen gelassen
und durch Säulenchromatographie
auf Silicagel unter Elution mit Dichlormethan:Methanol:konzentriertes
wässriges
Ammoniak (90:10:1, bezogen auf das Volumen) gereinigt, wobei die
Titelverbindung als weißer
Feststoff erhalten wurde (76 mg, 62%).
1H-NMR
(400 MHz, CDCl3) δ: 8,80 (1H, br s), 8,60 (1H,
br m), 7,55 (2H, br m), 7,35–7,20
(10H, m), 6,95 (1H, br m), 5,95 (1H, br m), 5,10 (2H, m), 4,50 (1H,
br m), 4,40–4,20
(5H, m), 3,60–3,30
(5H, m), 2,85 (1H, br m), 2,60 (2H, br m), 1,95 (1H, br m), 1,60–1,50 (3H,
br m), 1,30–0,90
(6H, m), 0,80 (6H, d).
LRMS (Thermospray: m/z [MH+]
686.
-
BEISPIELE 3–34
-
Die
Beispiele 3 bis 33 wurden durch ein ähnliches Verfahren wie das
für Beispiel
2 oder Beispiel 34 (für
die Beispiele 10, 11, 20 und 28) verwendete hergestellt. Massenspektroskopiedaten
wurden durch das Elektrosprayverfahren erhalten. BEISPIEL
3 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-(2-pyridinylmethyl)-9H-purin-2-carboxamid
- H-NMR (400 MHz, CDCl3) δ:
9,35
(1H, br s), 8,75 (1H, s), 8,30 (1H, br s), 7,70 (1 H, t), 7,40 (1H,
br s), 7,35–7,15
(10H, m), 6,85 (1H, m), 6,60 (1H, m), 5,00 (1H, s), 4,75 (2H, m),
4,55 4,30 (5H, m), 3,40 (2H, m), 1,20 (3H, t).
- [MH+] 623, [MNa+]
645
BEISPIEL
4 N-Benzyl-6-[(2,2-diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
8,85
(1H, s), 8,50 (1H, br s), 7,40–7,10
(15H, m), 6,95 (1H, d), 6,80 (1H, br s), 5,10 (1H, s), 4,65 (2H,
m), 4,50 (1H, m), 4,35 (2H, m), 4,25 (2H, m), 3,25 (2H, m), 1,05
(3H, t).
- [MH] 622 [MNa+] 644
BEISPIEL
5 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-(2-phenylethyl)-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 8,80 (1H,
s), 8,20 (1H, m), 7,45 (1H, m), 7,40–7,10 (15H, m), 6,95 (1H, d),
6,30 (1H, br s), 5,10 (1H, s), 4,50 (1H, m), 4,35 (2H, m), 4,20
(1H, br s), 3,70 (2H, m), 3,40 (2H, m), 2,90 (2H, t), 1,20 (3H,
t).
- [MH+] 636,[MNa+]
658
BEISPIEL
6 N-[2-(Dimethylamino)ethyl]-6-[(2,2-diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 8,80 (2H,
s), 7,70–7,45
(2H, m), 7,35–7,20
(10H, m), 6,95 (1H, d), 6,00 (1H, br s), 5,20 (1H, m), 4,50 (1H, s),
4,40–4,20
(4H, m), 3,55–3,35
(4H, m), 2,55 (2H, m), 2,10 (6H, 2), 1,20 (3H, t).
- [MH+] 604
BEISPIEL
7 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-[3-(1-pyrrolidinyl)propyl]-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 8,75 (1H,
br s), 8,40 (1H, br s), 7,35–7,20
(10H, m), 6,90 (1H, br s), 6,00 (1H, br s), 5,10 (2H, m), 4,60 (1H, br
s), 4,40–4,20
(4H, m), 3,55 (2H, m), 3,40 (2H, m), 2,60–2,40 (6H, m), 1,85 (2H, m),
1,70 (4H, br s), 1,20 (3H, t).
- [MH+] 644
BEISPIEL
8 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-[2-(1-pyridinyl)ethyl]-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 9,02 (1H,
m), 8,80 (1H, s), 8,06 (1H, br s), 7,77 (1H, s), 7,61 (1H, m), 7,50
(1H, t), 7,23–7,39
(10H, m), 7,12 (1H, d), 7,00 (1H, d), 6,92 (1H, m), 5,95 (1H, br
s), 5,18 (1H, d), 5,11 (1H, s), 4,55 (1H, d), 4,18–4,42 (4H,
m), 3,88 (2H, m), 3,43 (2H, m), 3,11 (2H, t), 1,22 (3H, t)
- [MH+] 638, [MNa+]
660
BEISPIEL
9 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-[2-(4-morpholinyl)ethyl]-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 8,80 (1H,
s), 8,65 (1H, br s), 7,60 (10H, br s), 7,50 (1H, br s), 7,20–7,40 (10H,
m), 6,95 (1H, d), 6,00 (1H, br d), 5,10 (2H, m), 4,55 (1H, d), 4,20–4,40 (4H,
m), 3,30–3,60
(8H, m), 2,60 (2H, t), 2,40 (4H, m), 1,25 (3H, t)
- [MH+] 646, [MNa+]
668
BEISPIEL
10 9-{(2R,3R,4S,5S)-5-[(Ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-6-[(1-ethylpropyl)amino]-N-[2-(1-piperidinyl)ethyl]-9H-purin-2-carboxamid - H-NMR (400 MHz, CD3OD) δ:
δ: 8,45 (1H,
s), 6,10 (1H, d), 4,85 (1H, t), 4,35–4,50 (3H, m), 3,60 (2H, t),
3,25–3,45
(3H, m), 2,40–2,70
(6H, m), 1,40–1,80
(10H, m), 1,05 (3H, t), 0,95 (6H, m)
- [MH+] 534, [MNa+]
556
BEISPIEL
11 6-{[(1S)-1-Benzyl-2-hydroxyethyl]amino}-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-[2-(1-piperidinyl)ethyl]-9H-purin-2-carboxamid - H-NMR (400 MHz, CD3OD) δ:
δ: 8,45 (1H,
s), 7,30 (2H, m), 7,15 (2H, m), 7,05 (1H, m), 6,10 (1H, d), 4,80
(1H, t), 4,45 (1H, s), 4,40 (1H, d), 3,70 (2H, m), 3,55 (2H, m),
3,20–3,45
(2H, m), 3,10 (1H, m), 2,95 (1H, m), 2,40–2,70 (7H, m), 1,65 (4H, m), 1,50
(2H, m), 1,05 (3H, t)
- [MH+] 598, [MNa+]
620
BEISPIEL
12 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-[2-(4-isopropyl-1-piperidinyl)ethyl]-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 8,80 (1H,
br s), 8,55 (1H, br s), 7,65 (2H, br m), 7,55 (1H, br s), 7,20–7,35 (10H,
m), 6,95 (1H, br d), 5,95 (1H, br s), 5,05–5,15 (2H, m), 4,55 (1H, br
s), 4,20–4,45
(4H, m), 3,30–3,60
(4H, m), 2,85 (2H, br s), 2,55 (2H, br s), 1,95 (2H, br t), 0,90–1,55 (9H,
m), 0,80 (6H, d)
- [MH+] 686, [MNa+]
708
BEISPIEL
13 N-[2-(3,4-Dihydro-2(1H)-isochinolinyl)ethyl]-6-[(2,2-diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 8,75 (1H,
br s), 8,60 (1H, br s), 7,50 (2H, br m), 6,85–7,30 (15H, m), 5,85 (1H, br
s), 5,05 (2H, br m), 4,50 (1H, br s), 4,05–4,40 (4H, m), 3,50–3,75 (4H,
m), 3,40 (2H, m), 2,60–2,90
(6H, m), 1,20 (3H, t)
- [MH+] 692, [MNa+]
714
BEISPIEL
14 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-(4-piperidinylmethyl)-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3 +
DMSO-d6)
δ:
8,45 (1H, s), 8,10 (1H, br s), 7,00–7,40 (11H, m), 6,55 (1H, br
s), 6,25 (1H, br s), 4,15–4,90
(6H, m), 3,15–3,50 (4H,
m), 3,00–3,15
(2H, m), 1,55–1,75
(3H, m), 0,90–1,40
(5H, m)
- [MH+] 630, [MNa+]
652
BEISPIEL
15 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-[4-(1-piperidinyl)butyl]-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 8,73 (1H,
m), 8,56–8,42
(1H, m), 7,52–7,40
(1H, m), 7,40–7,26
(10H, m), 6,90 (1H, m), 6,06–5,94
(1H, m), 5,05 (1H, m), 4,69–4,53
(1H, m), 4,45–4,16
(4H, m), 3,61–3,29
(4H, m), 2,77–2,60
(3H, m), 1,85–1,73
(2H, m), 1,24–1,14
(3H, m), 1,00–0,93
(6H, m)
- [MH+] 671
BEISPIEL
16 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-[3-(isopropylamino)propyl]-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: ,873 (1H,
m), 8,56–8,42
(1H, m), 7,52–7,40
(1H, m), 7,40–7,26
(10H, m), 6,90 (1H, m), 6,06–5,94
(1H, m), 5,05 (1H, m), 4,69–4,53
(1H, m), 4,45–4,16
(4H, m), 3,61–3,29
(4H, m), 2,77–2,60
(3H, m), 1,85–1,73
(2H, m), 1,24–1,14
(3H, m), 1,00–0,93
(6H, m)
- [MH+] 631, [MNa+]
653
BEISPIEL
17 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-[3-(2-pyridinylamino)propyl]-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 8,45 (2H,
m), 7,80–7,60
(1H, m), 7,40 (1H, m), 7,30–7,00
(10H, m), 6,80–6,64
(1H, m), 6,48 (1H, m), 6,35 (1H, m), 5,03–4,90 (1H, m), 4,77–4,60 (1H,
m), 4,52–4,13
(4H, m), 3,68–3,20
(8H, m), 1,97–1,85
(2H, m), 1,20–1,05
(3H, m)
- [MH+] 666, [MNa+]
688
BEISPIEL
18 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-{3-[methyl(phenylsulfonyl)amino]propyl}-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 8,95 (1H,
m), 8,76 (1H, m), 7,75 (2H, m), 7,63–7,40 (5H, m), 7,40–7,06 (10H,
m), 6,97 (1H, m), 6,00–5,87 (1H,
m), 5,16 (1H, m), 5,10 (1H, m), 4,60–4,24 (5H, m), 3,70–3,55 (2H,
m), 3,48–3,34
(2H, m), 3,22–3,00
(2H, m), 2,82–2,60
(3H, m), 1,93–1,73
(2H, m), 1,23 (2H, t)
- [MH+] 743, [MNa+]
765
BEISPIEL
19 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-{3-[methyl(methylsulfonyl)amino]propyl}-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 8,93-8,80
(1H, m), 8,77–8,68
(1H, m), 7,53–7,45
(1H, m), 7,30–7,20
(10H, m), 6,90 (1H, m), 5,97–5,87
(1H, m), 5,19–5,10
(1H, m), 5,06 (1H, m), 4,56–4,27
(5H, m), 3,64–3,48
(2H, m), 3,47–3,30
(2H, m), 3,30–3,16
(2H, m), 2,90–2,77
(3H, m), 2,60–2,43
(3H, m), 1,90–1,74
(2H, m), 1,22 (3H, t)
- [MH+] 681
BEISPIEL
20 9-{(2R,3R,4S,5S)-5-[(Ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-6-[(9H-fluoren-9-ylmethyl)amino]-N-[2-(1-piperidinyl)ethyl]-9H-purin-2-carboxamid - H-NMR (400 MHz, CD3OD)
δ: 8,45 (1H,
s), 7,80 (2H, m), 7,68 (2H, m), 7,34 (2H, m), 7,24 (2H, m), 6,13
(1H, m), 4,84 (1H, m), 4,43 (1H, d), 4,40 (2H, m), 4,27–4,18 (2H,
m), 3,53 (2H, m), 3,40–3,23
(2H, m), 2,55 (2H, m), 2,50–2,35
(4H, m), 1,55–1,42
(4H, m), 1,42–1,32
(2H, m), 1,06 (3H, t)
- [MH+] 641
BEISPIEL
21 N-[3-(Diethylamino)propyl]-6-[(2,2-diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 8,95–8,60 (2H,
m), 7,90–7,65
(1H, m), 7,65–7,50
(1H, m), 7,45–7,30
(10H, m), 7,05–6,90
(1H, m), 6,10–5,85 (1H,
m), 5,25–5,05
(2H, m), 4,60–4,50
(1H, m), 4,50–4,20
(4H, m), 3,65–3,30
(4H, m), 2,60–2,50
(2H, m), 2,50–2,30
(4H, m), 1,80–1,70
(2H, m), 1,30–1,15
(3H, m), 1,00–0,85
(1H, m).
- [MH+] 645, [MNa+]
667
BEISPIEL
22 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-[3-(4-morpholinyl)propyl)-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3 +
DMSO-d6)
δ:
8,65–8,50
(1H, m), 8,30–8,10
(1H, m), 7,60–7,45
(1H, m), 7,35–7,15
(10H, m), 7,15–7,00
(1H, m), 6,75–6,65 (1H,
m), 6,30–6,15
(1H, m), 5,00–4,85
(2H, m), 4,50–4,45
(1H, m), 4,45–4,35
(2H, m), 4,35–4,15
(2H, m), 3,65–3,55
(4H, m), 3,55–3,30
(4H, m), 2,45–2,25
(6H, m), 1,85–1,70
(2H, m), 1,15 (3H, t)
- [MH+] 659
BEISPIEL
23 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-[3-(methylamino)propyl]-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 9,10–8,95 (1H,
m), 8,80–8,70
(1H, m), 7,60–7,50
(1H, m), 7,40–7,25
(10H, m), 7,00–6,85
(1H, m), 6,10–5,90 (1H,
m), 5,10–5,05
(1H, m), 4,65–4,55
(1H, m), 4,40–4,20
(4H, m), 3,60–3,45
(2H, m), 3,45–3,30
(2H, m), 2,70–2,60
(2H, m), 2,25–2,15
(3H, m), 1,80–1,70
(2H, m), 1,25–1,10
(3H, m),
- [MH+] 603
BEISPIEL
24 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-[2-(5-methyl-1H-imidazol-4-yl))ethyl]-9H-purin-2-carboxamid - H-NMR (400 MHz, CD3OD)
δ: 8,20 (1H,
s), 7,40–7,10
(11H, m), 6,10 (1H, d), 4,90–4,70
(1H, m), 4,55–4,25
(5H, m), 3,70–3,55
(2H, m), 3,45–3,20
(2H, m), 2,90–2,80
(2H, m), 2,25–2,05
(3H, m), 1,10 (1H, t)
- [MH+] 640
BEISPIEL
25 N-[3-(Dimethylamino)butyl]-6-[(2,2-diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 8,60–4,50 (1H,
m), 8,20–8,10
(1H, m), 7,35–7,25
(10H, m), 6,80–6,60
(1H, m), 6,10–5,90
(1H, m), 4,45–4,20 (1H,
m), 3,70–3,50
(1H, m), 3,45–3,20
(3H, m), 2,60–2,35
(1H, m), 2,30–2,10
(9H, m), 1,80–1,50
(6H, m), 1,15–1,00
(3H, m)
- [MH+] 631, [MNa+]
653
BEISPIEL
26 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-[3-(4-methyl-1-piperazinyl)propyl]-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 8,90–8,70 (1H,
m), 8,35–8,20
(1H, m), 7,70–7,40
(2H, m), 7,40–7,25
(10H, m), 7,00–6,85
(1H, m), 6,10–5,90 (1H,
m), 5,20–5,00
(2H, m), 4,65–4,50
(1H, m), 4,45–4,20
(4H, m), 3,60–3,30
(4H, m), 2,55–2,20
(8H, m), 2,30–2,15
(3H, m), 1,90–1,85
(2H, m), 1,30–1,10
(3H, m)
- [MH+] 672
BEISPIEL
27 N-[3-(Dimethylamino)propyl]-6-[(2,2-diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 8,90–8,70 (1H,
m), 7,60–7,45
(1H, m), 7,45–7,25
(10H, m), 7,00–6,90
(1H, m), 6,00–5,90
(1H, m), 5,20–5,00 (2H,
m), 4,65–4,50
(1H, m), 4,45–4,20
(4H, m), 3,60–3,45
(2H, m), 3,45–3,30
(2H, m), 2,45–2,30
(2H, m), 2,15–2,00
(6H, m), 1,85–1,70
(2H, m), 1,25–1,10
(3H, m)
- [MH+] 617, [MNa+]
639
BEISPIEL
28 6-[(1-Benzyl-2-phenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-[1-(1-piperidinyl)ethyl]-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 8,40 (1H,
s), 7,30–7,25
(4H, m), 7,20–7,10
(4H, m), 7,10–7,05
(2H, m), 6,05–6,00
(1H, m), 5,10–5,00
(1H, m), 4,40 (1H, s), 4,40–4,30
(1H, m), 3,60–3,50
(2H, m), 3,40–3,20
(2H, m), 3,10–3,00
(2H, m), 3,00–2,90
(2H, m), 2,65–2,60
(2H, m), 2,60–2,50
(4H, m), 1,70–1,60
(4H, m), 1,55–1,45
(2H, m), 1,05 (3H, t)
- [MH+] 657
BEISPIEL
29 9-{(2R,3R,4S,5S)-3,4-dihydroxy-5-[(propylamino)carbonyl]tetrahydro-2-furanyl}-6-[(2,2-diphenylethyl)amino]-N-[2-(1-piperidinyl)ethyl]-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 8,75 (1H,
s), 8,65 (1H, s), 7,50 (1H, s), 7,35–7,15 (10H, m), 6,90 (1H, m),
6,10 (1H, m), 5,05 (1H, s), 4,55 (1H, s), 4,45–4,20 (4H, m), 3,60–3,40 (2H,
m), 3,40–3,20
(2H, m), 2,70–2,50
(2H, m), 2,50–2,30
(4H, m), 1,65–1,50
(2H, m), 1,50–1,20
(6H, m), 0,95 (3H, m).
BEISPIEL
30 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-[2-(1-isopropyl-4-piperidinyl)ethyl]-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 8,80–8,70 (1H,
m), 8,15–8,05
(1H, m), 7,75–7,55
(1H, m), 7,50–7,40
(1H, m), 7,40–7,20
(10H, m), 7,00–6,90 (1H,
m), 5,15–5,00
(2H, m), 4,60–4,50
(1H, m), 4,45–4,15
(4H, m), 3,55–3,30
(4H, m), 2,90–2,80
(2H, m), 2,80–2,60
(1H, m), 2,10–1,95
(2H, m), 1,75–1,50
(4H, m), 1,40–1,15
(6H, m), 1,05–0,95
(6H, m).
BEISPIEL
31 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-[2-(4-methyl-1-piperidinyl)ethyl]-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 8,75 (1H,
s), 8,60–8,50
(1H, m), 7,60–7,50
(1H, m), 7,40–7,20
(10H, m), 6,00–6,90
(1H, m), 5,20–5,00
(2H, m), 4,50–4,45
(1H, m), 4,45–4,20
(4H, m), 3,60–3,35
(4H, m), 2,80–2,70
(2H, m), 2,60–2,50
(2H, m), 2,00–1,90 (2H,
m), 1,55–1,40
(2H, m), 1,40–1,10
(5H, m), 1,10–0,95
(2H, m), 0,95–0,80
(3H, m).
- [MH+] 657
BEISPIEL
32 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-[2-(4-methoxy-1-piperidinyl)ethyl]-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 8,80 (1H,
s), 8,65–8,55
(1H, m), 7,70–7,50
(2H, m), 7,40–7,20
(10H, m), 7,00–6,95
(1H, m), 6,00–5,90
(1H, m), 5,10 (2H, s), 4,55–4,40
(1H, m), 4,40–4,20
(4H, m), 3,55–3,35
(4H, m), 3,20–3,10
(1H, m), 2,70–2,50
(4H, m), 2,25–2,15
(2H, m), 1,75–1,40
(5H, m), 1,25–1,20
(3H, t).
BEISPIEL
33 6-[(2,2-Diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-[(1-methyl-1H-imidazol-4-yl)methyl]-9H-purin-2-carboxamid - H-NMR (400 MHz, CDCl3) δ:
δ: 8,75 (1H,
s), 8,65–8,55
(1H, m), 7,55 (1H, s), 7,35–7,20
(10H, m), 6,95–6,90
(1H, m), 6,80 (1H, s), 5,05 (1H, s), 4,60–4,20 (6H, m), 3,60 (3H, s),
3,40–3,25
(2H, m), 1,20–1,10
(3H, t).
BEISPIEL
34 6-(Cyclohexylamino)-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-N-[2-(1-piperidinyl)ethyl]-9H-purin-2-carboxamid 
-
Eine
Lösung
von (2S,3S,4R,5R)-5-[6-(Cyclohexylamino)-2-iod-9H-purin-9-yl]-N-ethyl-3,4-dihydroxytetrahydro-2-furancarboxamid
(Herstellungsbeispiel 31) (125 mg, 0,24 mmol), 2-(1-Piperidinyl)ethylamine
(0,14 ml, 0,96 mmol) und Tetrakis(triphenylphosphin)palladium(0)
(20 mg, 0,024 mmol) in THF (5 ml) wurde bei 60°C und 345 kPa Kohlenmonoxid
18 h carbonyliert. Das Lösemittel
wurde unter vermindertem Druck abgedampft und der Rückstand
wurde durch Säulenchromatographie
auf Silicagel unter Elution mit Dichlormethan:Methanol:konzentriertes
wässriges
Ammoniak (95:5:1, bezogen auf das Volumen) gereinigt. Nach Abdampfen
passender Fraktionen wurde der Rückstand
mit Diethylether verrieben, filtriert und getrocknet, wobei die
Titelverbindung als beigefarbenes Pulver erhalten wurde (77 mg,
58%).
1H-NMR (400 MHz, CDCl3) δ:
8,45 (1H, s), 6,15–6,10
(1H, m), 4,90–4,85
(1H, m), 4,50–4,3
(3H, m), 3,65–3,50 (2H,
m), 3,45–3,25
(2H, m), 2,65–2,45
(6H, m), 2,10–2,00
(2H, m), 1,90–1,80
(2H, m), 1,75–1,30
(13H, m), 1,10–1,05
(3 m).
-
Die
folgenden Herstellungsbeispiele beschreiben die Herstellung von
bestimmten Zwischenprodukten, die in den vorhergehenden Beispielen
verwendet wurden.
-
HERSTELLUNGSBEISPIEL
1 2,6-Dichlor-9-tetrahydro-2H-pyran-2-yl-9H-purin
-
2,6-Dichlor-9H-purin
(20 g, 0,11 mol) und 4-Toluolsulfonsäuremonohydrat (0,2 g) wurden
in Ethylacetat (300 ml) gelöst,
das Gemisch wurde auf 50°C
erhitzt und mit einer Lösung
von 2,3-Dihydropyran (12,6 ml, 0,14 mol) in Ethylacetat (50 ml)
langsam über
30 min versetzt. Das Reaktionsgemisch wurde dann auf Raumtemperatur
gekühlt,
mit Wasser (100 ml) versetzt, und der pH-Wert der Lösung wurde
durch Zugabe einer gesättigten
wässrigen
Natriumhydrogencarbonatlösung
auf 7 eingestellt. Die organische Schicht wurde abgetrennt, nacheinander
mit Wasser und Kochsalzlösung
gewaschen, über
wasserfreiem Magnesiumsulfat getrocknet, filtriert und unter vermindertem
Druck eingedampft. Der Rückstand
wurde mit Pentan (× 2)
azeotrop behandelt, wobei die Titelverbindung als leicht unreiner
weißer
Feststoff erhalten wurde (30,9 g).
1H-NMR
(400 MHz, CDCl3) δ: 8,30 (1H, s), 5,75 (1H, dd),
4,25–4,15
(1H, m), 3,85–3,70
(1H, m), 2,20–1,60
(6H, m).
-
HERSTELLUNGSBEISPIEL
2 2-Chlor-N-(2,2-diphenylethyl)-9-tetrahydro-2H-pyran-2-yl-9H-purin-6-amin
-
Eine
Lösung
von 2,6-Dichlor-9-tetrahydro-2H-pyran-2-yl-9H-purin (Herstellungsbeispiel 1) (30,9
g, 0,11 mol) in Isopropylalkohol (600 ml) wurde mit N-Ethyl-N-isopropyl-2-propanamin (47,5
ml, 0,27 mol) und 2,2-Diphenylethylamin (24,8 g, 0,13 mol) behandelt
und das gebildete Gemisch wurde 3 h unter Refluxieren erhitzt. Das
Lösemittel
wurde unter vermindertem Druck entfernt und der Rückstand
wurde mit Ethylacetat azeotropbehandelt. Der Rückstand wurde dann durch Säulenchromatographie
auf Silicagel unter Elution mit einem Gradientensystem von Ethylacetat:Hexan
(40:60, bezogen auf das Volumen) unter allmählichem Wechsel zu Ethylacetat:Hexan
(60:40, bezogen auf das Volumen) gereinigt, wobei die Titelverbindung
als Schaum erhalten wurde (49,7 g).
1H-NMR
(400 MHz, CDCl3) δ: 7,95–7,75 (1H, br s), 7,35–7,15 (10H,
m), 5,80–5,70
(1H, br s), 5,65 (1H, d), 4,35 (1H, m), 4,30–4,18 (1H, br s), 4,10 (1H,
d), 3,70 (1H, t), 2,05–1,95
(2H, m), 1,95–1,80
(1H, m), 1,80–1,55
(3H, m).
-
HERSTELLUNGSBEISPIEL
3 6-[(2,2-Diphenylethyl)amino]-9-tetrahydro-2H-pyran-2-yl-9H-purin-2-carbonitril
-
Eine
Lösung
von 2-Chlor-N-(2,2-diphenylethyl)-9-tetrahydro-2H-pyran-2-yl-9H-purin-6-amin (Herstellungsbeispiel
2) (1,0 g, 2,31 mmol), Zinkcyanid (0,162 g, 1,38 mmol), Triethylamin
(0,28 g, 2,77 mmol) und Tetrakis(triphenylphosphin)palladium(0)
(0,133 g, 0,12 mmol) in N,N-Dimethylformamid
(3 ml) wurde 6 h unter Stickstoffatmosphäre bei 100°C erhitzt. Das Reaktionsgemisch
wurde abkühlen
gelassen und zwischen Ethylacetat (100 ml) und 2 M Natriumhydroxidlösung (100
ml) verteilt. Die organische Schicht wurde abgetrennt, über wasserfreiem
Magnesiumsulfat getrocknet und unter vermindertem Druck eingedampft.
Das gebildete 1:1-Gemisch von 6-[(2,2-Diphenylethyl)amino]-9-tetrahydro-2H-pyran-2-yl-9H-purin-2-carbonitril
und 6-[(2,2-Diphenylethyl)amino]-9H-purin-2-carbonitril
(Herstellungsbeispiel 8) wurde durch Säulenchromatographie auf Silicagel
unter Elution mit einem Gradientensystem von Ethylacetat:Hexan (40:60,
bezogen auf das Volumen) unter allmählichem Wechsel zu Ethylacetat:Hexan
(60:40, bezogen auf das Volumen) getrennt, wobei die Titelverbindung
als weißer
Feststoff erhalten wurde (0,4 g).
1H-NMR
(400 MHz, CDCl3) δ: 8,00 (1H, s), 7,40–7,20 (10H,
m), 6,00–5,75
(1H, br s), 5,70 (1H, d), 4,40–4,20 (3H,
m), 4,20–4,10
(1H, m), 3,80–3,70
(1H, m), 2,20–1,90
(3H, m), 1,90–1,60
(3H, m).
-
HERSTELLUNGSBEISPIEL
4 Methyl-6-[(2,2-diphenylethyl)amino]-9-tetrahydro-2H-pyran-2-yl-9H-purin-2-carboxylat
-
Eine
Suspension von 6-[(2,2-Diphenylethyl)amino]-9-tetrahydro-2H-pyran-2-yl-9H-purin-2-carbonitril (Herstellungsbeispiel
3) (1,00 g, 2,36 mmol) in Methanol (20 ml) wurde mit Natriummethoxid
(0,14 g, 2,59 mmol) behandelt und das gebildete Gemisch wurde 20
h unter Refluxieren unter Stickstoffatmosphäre erhitzt. DC-Analyse zeigte,
dass noch einiges Ausgangsmaterial verblieben war, weshalb weiteres
Natriummethoxid (64 mg, 1,18 mmol) zugegeben und das Gemisch eine
weitere Stunde unter Refluxieren unter Stickstoffatmosphäre erhitzt
wurde. Das Gemisch wurde dann auf Raumtemperatur gekühlt und
das Lösemittel
wurde unter vermindertem Druck entfernt. Tetrahydrofuran (30 ml)
und Wasser (10 ml) wurden zu dem Rückstand gegeben und der pH-Wert wurde durch
Zugabe von Essigsäure
(1 ml) auf 4 eingestellt. Das Gemisch wurde 1 h unter Refluxieren
erhitzt. DC-Analyse zeigte, dass etwas Ausgangsmaterial immer noch
vorhanden war und deshalb wurde weitere Essigsäure (0,5 ml) zugegeben und
das Erhitzen unter Refluxieren 18 h fortgesetzt. Das Reaktionsgemisch
wurde auf Raumtemperatur gekühlt
und zwischen Ethylacetat und einer gesättigten wässrigen Natriumhydrogencarbonatlösung verteilt.
Die organische Phase wurde abgetrennt, mit Kochsalzlösung gewaschen, über wasserfreiem
Magnesiumsulfat getrocknet, filtriert und unter vermindertem Druck
eingedampft. Der Rückstand
wurde durch Säulenchromatographie
auf Silicagel unter Elution mit Dichlormethan:Methanol (98,5:1,5,
bezogen auf das Volumen) gereinigt, wobei die Titelverbindung erhalten
wurde (521 mg).
1H-NMR (400 MHz, CDCl3) δ:
8,05 (1H, br s), 7,37–7,18
(10H, m), 5,84 (2H, m), 4,40 (3H, m), 4,14 (1H, d), 4,00 (3H, s),
3,78 (1H, t), 2,17–1,60
(6H, m).
LRMS (Thermospray): m/z [MH+]
458, [MNa+] 480.
-
HERSTELLUNGSBEISPIEL
5 6-[(2,2-Diphenylethyl)amino]-N-[2-(1-piperidinyl)ethyl]-9-tetrahydro-2H-pyran-2-yl-9H-purin-2-carboxamid
-
Methyl-6-[(2,2-diphenylethyl)amino]-9-tetrahydro-2H-pyran-2-yl-9H-purin-2-carboxylat
(Herstellungsbeispiel 4) (100 mg, 0,22 mmol) und 1-(2-Aminoethyl)piperidin
(0,31 ml, 2,19 mmol) wurden 2 h zusammen bei 130°C erhitzt. Das überschüssige Amin
wurde unter vermindertem Druck entfernt und der Rückstand
wurde durch Säulenchromatographie
auf Silicagel unter Elution mit Dichlormethan:Methanol (95:5, bezogen
auf das Volumen) gereinigt, wobei die Titelverbindung als gelber
Schaum erhalten wurde (104 mg).
1H-NMR
(400 MHz, CDCl3) δ: 8,43 (1H, br m), 8,00 (1H,
br s), 7,17–7,36
(10H, m), 5,94 (1H, d), 5,80 (1H, br m), 4,37 (3H, m), 4,33 (1H,
d), 3,78 (1H, t), 3,57 (2H, m), 2,55 (2H, m), 2,40 (4H, br m), 1,65–2,17 (6H,
m), 1,26–1,33
(6H, br m).
LRMS (Thermospray): m/z [MH+]
554.
-
HERSTELLUNGSBEISPIEL
6 6-[(2,2-Diphenylethyl)amino]-N-[2-(1-piperidinyl)ethyl]-9H-purin-2-carboxamid
-
Eine
Lösung
von 6-[(2,2-Diphenylethyl)amino]-N-[2-(1-piperidinyl)ethyl]-9-tetrahydro-2H-pyran-2-yl-9H-purin-2- carboxamid (Herstellungsbeispiel
5) (420 mg, 0,76 mmol) in Ethanol (20 ml) wurde mit Salzsäure (2 M,
0,9 ml) behandelt. Das Gemisch wurde 30 min unter Refluxieren erhitzt,
wonach sich ein weißer
Niederschlag gebildet hatte. Das Gemisch wurde auf Raumtemperatur
gekühlt
und das Lösemittel
wurde unter vermindertem Druck entfernt. Der Rückstand wurde zwischen Dichlormethan
und 10 Gewichtsprozent, bezogen auf das Volumen, von wässrigem
Ammoniak verteilt. Die organische Phase wurde abgetrennt, über wasserfreiem
Magnesiumsulfat getrocknet, filtriert und unter vermindertem Druck
abgedampft, wobei die Titelverbindung als weißer Feststoff erhalten wurde
(319 mg).
1H-NMR (400 MHz, CDCl3) δ:
8,57 (1H, br m), 8,30 (1H, s), 7,40–7,20 (10H, m), 5,93 (1H, br
s), 4,39 (3H, m), 3,62 (2H, m), 2,56 (2H, t), 2,40 (4H, br m), 1,47–1,24 (6H,
br m).
LRMS (Thermospray): m/z [MH+]
470, [MNa+] 492.
-
HERSTELLUNGSBEISPIEL
7 (2R,3R,4R,5S)-4-(Benzoyloxy)-2-[6-[(2,2-diphenylethyl)amino]-2-({[2-(1-piperidinylethyl]amino}carbonyl)-9H-purin-9-yl]-5-[(ethylamino)carbonyl]tetrahydro-3-furanylbenzoat
-
Eine
Suspension von 6-[(2,2-Diphenylethyl)amino]-N-[2-(1-piperidinyl)ethyl]-9H-purin-2-carboxamid (Herstellungsbeispiel
6) (100 mg, 0,21 mmol) in 1,1,1-Trichlorethan (2 ml) wurde mit N,O-Bis(trimethylsilyl)acetamid
(0,21 ml, 0,85 mmol) behandelt. Das Gemisch wurde 90 min unter Refluxieren
erhitzt. Die Lösung wurde
auf Raumtemperatur abkühlen
gelassen und das Lösemittel
wurde unter vermindertem Druck entfernt. Der Rückstand wurde mit einer Lösung von
(2S,3R,4R,5R)- und (2S,3R,4R,5S)-5-(Acetyloxy)-4-(benzoyloxy)-2-[(ethylamino)carbonyl]-tetrahydro-3-furanylbenzoat (Herstellungsbeispiel
15) (111 mg, 0,25 mmol) in wasserfreiem Toluol (2 ml) und Trimethylsilyltrifluormethansulfonat
(0,05 ml, 0,30 mmol) behandelt. Die gebildete Lösung wurde dann 1 h unter Stickstoffatmosphäre bei 90°C erhitzt.
DC-Analyse zeigte, dass einiges Ausgangsmaterial noch verblieben
war und deshalb wurde weiteres Trimethylsilyltrifluormethansulfonat
(0,05 ml, 0,30 mmol) zugegeben und das Erhitzen 2 h fortgesetzt.
Erneut zeigte DC-Analyse, dass etwas Ausgangsmaterial noch verblieben
war, weshalb weiteres Trimethylsilyltrifluormethansulfonat (0,025
ml, 0,15 mmol) zugegeben und das Erhitzen eine weitere Stunde fortgesetzt
wurde. Das Gemisch wurde auf Raumtemperatur gekühlt, mit Ethylacetat (20 ml)
verdünnt
und mit einer gesättigten
wässrigen
Natriumhydrogencarbonatlösung gewaschen.
Die organische Schicht wurde abgetrennt, über wasserfreiem Magnesiumsulfat
getrocknet, filtriert und unter vermindertem Druck eingedampft.
Der Rückstand
wurde durch Säulenchromatographie
auf Silicagel unter Elution mit Dichlormethan:Methanol (90:10, bezogen
auf das Volumen) gereinigt, wobei die Titelverbindung als Schaum
erhalten wurde (109 mg).
1H-NMR (400
MHz, CDCl3) δ: 8,35 (1H, br m), 8,06 (3H,
d), 7,39 (2H, d), 7,69–7,18
(15H, m), 6,44 (1H, d), 6,23 (2H, br m), 5,91 (1H, br m), 4,92 (1H,
d), 4,37 (3H, m), 3,53 (4H, m), 2,60 (2H, br m), 2,44 (2H, br m),
1,40 (6H, m), 1,13 (3H, t).
LRMS (Thermospray): m/z [MH+] 851.
-
HERSTELLUNGSBEISPIEL
8 6-[(2,2-Diphenylethyl)amino]-9H-purin-2-carbonitril
-
Eine
Lösung
von 6-[(2,2-Diphenylethyl)amino]-9-tetrahydro-2H-pyran-2-yl-9H-purin-2-carbonitril
(Herstellungsbeispiel 3) (17 g, 40,1 mmol) in Ethanol (850 ml) wurde
mit 2 M wässriger
Salzsäure
(50 ml) behandelt und das Gemisch wurde 24 h bei Raumtemperatur
gerührt.
Das Lösemittel
wurde unter vermindertem Druck entfernt, der Rückstand wurde in Ethanol gelöst und das
Lösemittel
wurde erneut unter vermindertem Druck entfernt. Der Rückstand
wurde mit Diethylether verrieben, filtriert, mit Diethylether und
Pentan gewaschen und getrocknet, wobei die Titelverbindung als Feststoff
erhalten wurde (13,6 g).
1H-NMR 400
MHz, DMSO-d6) δ: 8,30 (1H, s), 8,20–8,05 (10H,
br s), 7,40–7,10
(10H, m), 4,60–4,40
(1,4H, m), 4,20–4,00
(1,6H, m).
LRMS (Thermospray): m/z [MH+]
341.
-
HERSTELLUNGSBEISPIEL
9 Methyl-6-[(2,2-diphenylethyl)amino]-9H-purin-2-carboxylat
-
Eine
Lösung
von 6-[(2,2-Diphenylethyl)amino]-9H-purin-2-carbonitril (Herstellungsbeispiel 8)
(5,0 g, 14,7 mmol) und Natriummethoxid (4,0 g, 74,1 mmol) in Methanol
(300 ml) wurde 24 h unter Refluxieren erhitzt. Weiteres Natriummethoxid
(2,0 g, 37 mmol) und Methanol (100 ml) wurden dann zugegeben und
das Erhitzen wurde weitere 24 h fortgesetzt. Das Reaktionsgemisch
wurde abkühlen
gelassen und das Lösemittel
wurde unter vermindertem Druck entfernt. Der Rückstand wurde in THF (375 ml)
gelöst,
2 M Salzsäure
(125 ml) wurde zugegeben und das Gemisch wurde 24 h bei Raumtemperatur
gerührt.
Das THF wurde unter vermindertem Druck entfernt und die Suspension
wurde mit gesättigter
wässriger
Natriumbicarbonatlösung
auf einen pH-Wert von 7 eingestellt. Ethylacetat (100 ml) wurde
dann zugegeben und der weiße
Feststoff, der hauptsächlich
aus dem gewünschten
Produkt bestand, wurde abfiltriert, mit etwas Wasser und Ethylacetat
gewaschen und getrocknet. Reinigung durch Säulenchromatographie auf Silicagel
unter Elution mit einem Gradientensystem von Dichlormethan:Methanol
(90:10, bezogen auf das Volumen) unter allmählichem Wechsel zu Dichlormethan:Methanol
(75:25, bezogen auf das Volumen) ergab die Titelverbindung als weißen Feststoff
(1,25 g). Eindampfen des Ethylacetatfiltrats führte zur Rückgewinnung von 2,6 g des Ausgangsmaterials.
1H-NMR (400 MHz, CDCl3) δ: 12,40 (1H,
br s), 8,05 (1H, s), 7,55 (1H, s), 7,30–7,20 (10H, m), 4,80 (2H, m), 4,75
(1H, m), 3,80 (3H, s).
LRMS (Thermospray): m/z [MH+]
375.
-
HERSTELLUNGSBEISPIEL
10 Methyl-9-{(2R,3R,4R,5S)-3,4-bis(benzoyloxy)-5-[(ethylamino)carbonyl]tetrahydro-2-furanyl}-6-[(2,2-diphenylethyl)amino]-9H-purin-2-carboxylat
-
Eine
Suspension von Methyl-6-[(2,2-diphenylethyl)amino]-9H-purin-2-carboxylat
(Herstellungsbeispiel 9) (440 mg, 1,18 mmol) in 1,1,1-Trichlorethan
(25 ml) wurde mit N,O-Bis(trimethylsilyl)acetamid (1,7 ml, 6,95 mmol)
behandelt. Das Gemisch wurde 1 h unter Refluxieren erhitzt. Die
Lösung
wurde auf Raumtemperatur abkühlen
gelassen und das Lösemittel
wurde unter vermindertem Druck entfernt. Der Rückstand wurde mit einer Lösung von
(2S,3R,4R,5R)- und (2S,3R,4R,5S)-5-(Acetyloxy)-4-(benzoyloxy)-2-[(ethylamino)carbonyl]-tetrahydro-3-furanylbenzoat
(Herstellungsbeispiel 15) (620 mg, 1,4 mmol) in wasserfreiem Toluol
(25 ml) und Trimethylsilyltrifluormethansulfonat (0,26 ml, 1,42
mmol) behandelt. Die gebildete Lösung
wurde dann 2,5 h unter Stickstoffatmosphäre bei 110°C erhitzt. Das Gemisch wurde
auf Raumtemperatur gekühlt,
mit Ethylacetat (200 ml) verdünnt
und mit einer gesättigten
wässrigen
Natriumhydrogencarbonatlösung
gewaschen. Die organische Lösung
wurde abgetrennt, über
wasserfreiem Magnesiumsulfat getrocknet, filtriert und unter vermindertem
Druck eingedampft. Der Rückstand
wurde durch Säulenchromatographie
auf Silicagel unter Elution mit Dichlormethan:Ethylacetat (5:1,
bezogen auf das Volumen), dann Dichlormethan:Ethylacetat (1:1, bezogen auf
das Volumen) gereinigt, wobei die Titelverbindung als Schaum erhalten
wurde (540 mg).
1H-NMR (400 MHz, CDCl3) δ:
8,10 (3H, m), 7,80 (2H, d), 7,60 (1H, m), 7,50–7,40 (4H, m), 7,35–7,20 (16H,
m), 6,40 (1H, m), 6,20 (2H, m), 5,90 (1H, m), 4,90 (1H, d), 4,40
(3H, m), 4,00 (3H, s), 3,55 (1H, m), 3,35 (1H, m), 1,15 (3H, t).
LRMS
(Thermospray): m/z [MNa+] 777.
-
HERSTELLUNGSBEISPIEL
11 Methyl-6-[(2,2-diphenylethyl)amino]-9-{(2R,3R,4S,5S)-5-[(ethylamino)carbonyl]-3,4-dihydroxytetrahydro-2-furanyl}-9H-purin-2-carboxylat
-
Eine
Lösung
von Methyl-9-{(2R,3R,4R,5S)-3,4-bis(benzoyloxy)-5-[(ethylamino)carbonyl]tetrahydro-2-furanyl}-6-[(2,2-diphenylethyl)amino]-9H-purin-2-carboxylat (Herstellungsbeispiel
10) (3,4 g, 4,5 mmol) und Natriumcarbonat (50 mg) in trockenem Methanol
(60 ml) wurde 4 h bei Raumtemperatur gerührt. Das Lösemittel wurde unter vermindertem Druck
entfernt und der Rückstand
wurde in einem Gemisch aus Dichlormethan:Methanol (95:5, bezogen
auf das Volumen, 60 ml) aufgenommen. Anorganische Salze wurden abfiltriert
und das Filtrat wurde unter vermindertem Druck eingedampft. Der
Rückstand
wurde mit Ether verrieben, abfiltriert und getrocknet, wobei die
Titelverbindung als weißer
Feststoff erhalten wurde (2,4 g).
1H-NMR
400 MHz, DMSO-d6) δ: 8,60 (1H, m), 8,15 (1H, br
s), 7,40–7,15
(10H, m), 6,00 (1H, br m), 5,60 (1H, br s), 5,50 (1H, br s), 4,60–4,40 (3H,
m), 4,30 (1H, s), 4,10–4,05
(2H, m), 4,00–3,80
(3H, m), 3,20 (2H, m), 1,00 (3H, t).
LRMS (Thermospray): m/z
[MH+] 547.
-
HERSTELLUNGSBEISPIEL
12 (3aS,4S,6R,6aR)-N-Ethyl-6-methoxy-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-carboxamid
-
Oxalylchlorid
(14,0 ml, 160 mmol) wurde tropfenweise zu einer gerührten Lösung von (3aR,4S,6R,6aR)-6-methoxy-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-carbonsäure (J.
Am. Chem. Soc., 1958, 80, 5168) (23,30 g, 107 mmol) in wasserfreiem
Dichlormethan (120 ml) und N,N-Dimethyl formamid (2 Tropfen) gegeben
und das Gemisch wurde 3 h bei Raumtemperatur gerührt, bis die Gasentwicklung
aufgehört hatte.
DC-Analyse zeigte, dass etwas Ausgangsmaterial immer noch verblieben
war und so wurde weiteres N,N-Dimethylformamid (2 Tropfen) zugegeben
und das Rühren
1 h fortgesetzt. Das Lösemittel
wurde unter vermindertem Druck entfernt und der Rückstand
wurde mit wasserfreiem Dichlormethan (× 2) azeotropbehandelt. Der
Rückstand
wurde dann in wasserfreiem Dichlormethan (200 ml) gelöst und die
Lösung
wurde tropfenweise mit Ethylamin (2 M in Tetrahydrofuran, 140 ml,
280 mmol) behandelt. Diese Lösung
wurde 48 h bei Raumtemperatur belassen. Diethyletehr (250 ml) wurde
zugegeben und das Gemisch wurde 15 min gerührt. Das Gemisch wurde filtriert
und das Lösemittel
wurde von dem Filtrat unter vermindertem Druck entfernt. Der Rückstand
wurde durch Säulenchromatographie
auf Silicagel unter Elution mit einem Gradientensystem von Dichlormethan:Ethylacetat
(100:0, bezogen auf das Volumen) unter allmählichem Wechsel zu Dichlormethan:Ethylacetat
(44:66, bezogen auf das Volumen) gereinigt, wobei die Titelverbindung
als gelber Feststoff (24,70 g) erhalten wurde.
1H-NMR
(400 MHz, CDCl3) δ: 6,53 (1H, br m), 5,12 (1H,
dd), 5,07 (1H, d), 4,60 (1H, d), 4,54 (1H, dd), 3,46 (3H, s), 3,32
(2H, m), 1,51 (3H, s), 1,34 (3H, s), 1,15 (3H, t).
LRMS (Thermospray):
m/z [MH+] 246.
-
HERSTELLUNGSBEISPIEL
13 (2S,3S,4R,5R)-
und (2S,3S,4R,5S)-N-Ethyl-3,4-dihydroxy-5-methoxytetrahydro-2-furancarboxamid
-
Eine
Lösung
von (3aS,4S,6R,6aR)-N-Ethyl-6-methoxy-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-carboxamid
(Herstellungsbeispiel 12) (24,60 g, 100 mmol) und Pyridinium-p-toluolsulfonat (2,50
g, 10 mmol) in Methanol (500 ml) wurde 18 h unter Refluxieren erhitzt.
NMR-Analyse zeigte, dass etwas Ausgangsmaterial immer noch verblieben
war. Das Lösemittel
wurde unter vermindertem Druck entfernt. Der Rückstand wurde in Methanol (500
ml) gelöst
und 8 h unter Refluxieren erhitzt. NMR-Analyse zeigte, dass etwas
Ausgangsmaterial immer noch verblieben war, weshalb das Lösemittel
noch einmal unter vermindertem Druck entfernt wurde. Der Rückstand
wurde in Methanol (500 ml) gelöst
und die verbliebene Lösung
wurde 24 h unter Refluxieren erhitzt. Das Lösemittel wurde dann unter vermindertem
Druck entfernt und der Rückstand
wurde mit Dichlormethan (× 3)
azeotropbehandelt, wobei die Titelverbindung als Öl (20,50
g) erhalten wurde.
1H-NMR (400 MHz,
CDCl3) δ:
6,58 (1H, br m), 4,99 (0,25H, d), 4,94 (0,75H, d), 4,46 (0,25H,
d), 4,37 (1,5H, m), 4,24 (0,25H, dd), 4,05 (1H, m), 3,52 (0,75H,
s), 3,47 (2,25H, s), 3,30 (2H, m), 1,16 (3H, m).
-
HERSTELLUNGSBEISPIEL
14 (2S,3R,4R,5S)-
und (2S,3R,4R,5S)-4-(Benzoyloxy)-5-[(ethylamino)carbonyl]-2-methoxytetrahydro-3-furanylbenzoat
-
Eine
Lösung
von Benzoylchlorid (30,0 ml, 259 mmol) in Dichlormethan (100 ml)
wurde langsam zu einer Lösung
von (2S,3S,4R,5R)- und (2S,3S,4R,5S)-N-Ethyl-3,4-dihydroxy-5-methoxytetrahydro-2-furancarboxamid
(Herstellungsbeispiel 13) (20,50 g, 100 mmol) und Pyridin (33,0
ml, 409 mmol) in Dichlormethan (400 ml) gegeben und das gebildete
Gemisch wurde 18 h bei Raumtemperatur gerührt. Das Lösemittel wurde unter vermindertem
Druck entfernt und der Rückstand
wurde zwischen Diethylether und Salzsäure (1 M, 300 ml) verteilt.
Die Schichten wurden getrennt und die wässrige Schicht wurde mit Diethylether
erneut extrahiert. Die organischen Schichten wurden vereinigt, nacheinander
mit Wasser und Kochsalzlösung
gewaschen, über
wasserfreiem Magnesiumsulfat getrocknet, filtriert und unter vermindertem
Druck eingedampft. Der Rückstand wurde
durch Säulenchromatographie
auf Silicagel unter Elution mit einem Gradientensystem von Dichlormethan:Diethylether
(95:5, bezogen auf das Volumen) unter allmählichem Wechsel zu Dichlormethan:Diethylether
(80:20, bezogen auf das Volumen) gereinigt, wobei die Titelverbindung
als Öl
und als Gemisch von α-
und β-Anomeren
erhalten wurde (37,0 g).
1H-NMR (400
MHz, CDCl3) δ: 8,16 (0,5H, d), 7,95 (1,5H,
d), 7,88 (1,5H, d), 7,81 (0,5H, d), 7,66–7,25 (6H, m), 6,65 (1H, br
m), 5,88 (1H, m), 5,60 (0,75H, dd), 5,46 (0,25 H, d), 5,23 (0,75H,
d), 5,17 (0,25H, t), 4,80 (1H, m), 3,59 (2,25H, s), 3,49 (0,75H,
s), 3,39 (2H, m), 1,23 (3H, t).
-
HERSTELLUNGSBEISPIEL
15 (2S,3R,4R,5R)-
und (2S,3R,4R,5S)-5-(Acetyloxy)-4-(benzoyloxy)-2-[(ethylamino)carbonyl]-tetrahydro-3-furanylbenzoat
-
Eine
Lösung
von (2S,3R,4R,5S)- und (2R,3R,4R,5S)-4-(Benzoyloxy)-5-[(ethylamino)carbonyl]-2-methoxytetrahydro-3-furanylbenzoat
(Herstellungsbeispiel 14) (37,0 ml, 89,6 mmol) in einem Gemisch
aus Essigsäure
(330 ml, 5,77 mol) und Essigsäureanhydrid
(67 ml, 709 mmol) wurde auf –10°C gekühlt und
tropfenweise mit Salzsäure
(12 N, 7,0 ml, 132 mmol) behandelt. Das Gemisch wurde 18 h gerührt, wobei
es sich während dieses
Zeitraums auf Raumtemperatur erwärmen
gelassen wurde. Nach Kühlen
des Gemischs auf 0°C
wurde es mit Wasser (100 ml) verdünnt und dann mit Ethylacetat
(3 × 500
ml) extrahiert. Die organischen Schichten wurden vereinigt, nacheinander
mit Wasser, einer gesättigten
wässrigen
Natriumhydrogencarbonatlösung
und Kochsalzlösung
gewaschen, über
wasserfreiem Magnesiumsulfat getrocknet, filtriert und unter vermindertem Druck
eingedampft. Der Rückstand
wurde durch Säulenchromatographie
auf Silicagel unter Elution mit einem Gradientensystem von Diethylether:Pentan
(66:44, bezogen auf das Volumen) unter allmählichem Wechsel zu Diethylether:Pentan
(100:0, bezogen auf das Volumen) gereinigt. Der Rückstand
wurde ferner durch Säulenchromatographie
auf Silicagel unter Elution mit einem Gradientensystem von Dichlormethan:Diethylether (95:5,
bezogen auf das Volumen) unter allmählichem Wechsel zu Dichlormethan:Diethylether
(90:10, bezogen auf das Volumen) gereinigt, wobei die Titelverbindung
als Gemisch von α-
und β-Anomeren
erhalten wurde (15,40 g).
1H-NMR (400
MHz, CDCl3) δ: 8,12 (0,8H, d), 7,97 (1,2H,
d), 7,92 (1,2H, d), 7,79 (0,8H, d), 7,65–7,24 (6H, m), 6,73 (0,4H,
d), 6,62 (0,4H, br m), 6,46 (0,6H, br m), 6,42 (0,6H, d), 6,07 (0,4H,
dd), 5,95 (0,6H, t), 5,72 (0,6H, d), 5,44 (0,4H, t), 4,94 (0,4H,
d), 4,86 (0,6H, d), 3,36 (2H, m), 2,17 (1,8H, s), 2,10 (1,2H, s),
1,20 (3H, m).
-
(2R,3R,4R,5R)-
und (2S,3R,4R,5S)-5-(Acetyloxy)-4-(benzoyloxy)-2-[(propylamino)carbonyl]-tetrahydro-3-furanylbenzoat wurden
auf analoge Weise hergestellt.
-
HERSTELLUNGSBEISPIEL
16 2-[2-(4-Isopropyl-1-piperidinyl)ethyl]-1H-isoindol-1,3(2H)-dion
-
Eine
Lösung
von 4-Isopropylpiperidin (3,3 g, 20,2 mmol) und 2-Bromethylphthalimid
(5,4 g, 21,3 mmol) in Acetonitril (100 ml) wurde mit Kaliumcarbonat
(5,9 g, 45,4 mmol) behan delt, 2,5 h unter Refluxieren erhitzt und
dann über
Nacht bei Raumtemperatur gerührt.
Das Lösemittel
wurde unter vermindertem Druck entfernt und der Rückstand
wurde zwischen Ethylacetat (100 ml) und Wasser (100 ml) verteilt.
Die organische Schicht wurde abgetrennt und die wässrige Schicht
wurde mit weiterem Ethylacetat (100 ml) extrahiert. Die vereinigten organischen
Extrakte wurden über
Natriumsulfat getrocknet und das Lösemittel wurde unter vermindertem Druck
entfernt. Das gebildete Öl
wurde durch Säulenchromatographie
auf Siliagel unter Elution mit einem Gradientensystem von Dichlormethan:Diethylether
(50:50, bezogen auf das Volumen) unter Wechsel zu reinem Diethylether
gereinigt, wobei die Titelverbindung (3,3 g) erhalten wurde.
1H-NMR (400 MHz, CDCl3) δ: 7,80 (2H,
m), 7,70 (2H, m), 3,80 (2H, t), 3,00 (2H, m), 2,60 (2H, t), 1,95
(2H, m), 1,60 (2H, m), 1,40 (1H, m), 1,20 (2H, dq), 0,95 (1H, m),
0,80 (6H, d).
LRMS (Thermospray): m/z [MH+]
301.
-
HERSTELLUNGSBEISPIEL
17 2-(4-Isopropyl-1-piperidinyl)ethylamine
-
Eine
Lösung
von 2-[2-(4-Isopropyl-1-piperidinyl)ethyl]-1H-isoindol-1,3(2H)-dion (Herstellungsbeispiel 16)
(3,2 g, 10,6 mmol) in einer 33%-igen Lösung von Methylamin in Ethanol
(60 ml) wurde 3 h unter Refluxieren erhitzt. Das Lösemittel
wurde unter vermindertem Druck entfernt, Ethanol (60 ml) wurde des
weiteren zugegeben und das Lösemittel
wurde erneut unter vermindertem Druck entfernt. Der Rückstand
wurde in Dichlormethan (100 ml) suspendiert und der Fest- stoff wurde abfiltriert
und mit weiterem Dichlormethan (100 ml) gewaschen. Das Filtrat wurde
unter vermindertem Druck eingedampft und durch Säulenchromatographie auf Silicagel
unter Elution mit Dichlormethan:Methanol:konzentriertes wässriges
Ammoniak (90:10:1, bezogen auf das Volumen) gereinigt, wobei ein
farbloses Öl
erhalten wurde. Kolben-Kolben-Destillation (150–160°C, 3 mmHg) ergab die Titelverbindung
(1,0 g).
1H-NMR (400 MHz, CDCl3) δ:
2,90 (2H, m), 2,80 (2H, t), 2,40 (2H, t), 1,95 (2H, m), 1,65 (2H,
m), 1,40 (1H, m), 1,30–1,20
(4H, m), 1,00 (1H, m), 0,85 (6H, d).
LRMS (Thermospray): m/z
[MH+] 171.
-
HERSTELLUNGSBEISPIEL
18 (2R,3R,4S,5S)-2-(2-Amino-6-chlor-9H-purin-9-yl)-4-(benzoyloxy)-5-[(ethylamino)carbonyl]-tetrahydro-3-furanylbenzoat
-
Eine
Suspension von 2-Amino-6-chlorpurin (4,60 g, 27,13 mmol) in 1,1,1-Trichlorethan
(230 ml) wurde mit N,O-Bis(trimethylsilyl)acetamid
(20 ml, 81,4 mmol) behandelt. Das Gemisch wurde 6 h unter Refluxieren erhitzt.
Die Lösung wurde
sich auf Raumtemperatur abkühlen
gelassen und das Lösemittel
wurde unter vermindertem Druck entfernt. Der Rückstand wurde mit einer Lösung von
(2S,3R,4R,5R)- und (2S,3R,4R,5S)-5-(Acetyloxy)-4-(benzoyloxy)-2-[(ethylamino)carbonyl]-tetrahydro-3-furanylbenzoat
(Herstellungsbeispiel 15) (14,39 g, 32,6 mmol) in wasserfreiem Toluol
(230 ml) und Trimethylsilyltrifluormethansulfonat (20 ml, 108,5
mmol) behandelt. Die gebildete Lösung
wurde dann 90 min unter Stickstoffatmosphäre bei 90°C erhitzt. Das Gemisch wurde
auf Raumtemperatur gekühlt,
mit Ethylacetat (250 ml) verdünnt
und mit einer gesättigten
wässrigen
Natriumhydrogencarbonatlösung
(350 ml) und dann Kochsalzlösung
(350 ml) gewaschen. Die organische Schicht wurde abgetrennt, über wasserfreiem
Magnesiumsulfat getrocknet, filtriert und unter vermindertem Druck
eingedampft. Der Rückstand
wurde durch Säulenchromatographie
auf Silicagel unter Elution mit Dichlormethan:Methanol (98:2, bezogen
auf das Volumen) gereinigt, wobei die Titelverbindung als weißer Schaum
erhalten wurde (8,1 g).
1H-NMR (400
MHz, CDCl3) δ: 8,10–7,95 (3H, m), 7,80 (2H, m),
7,50–7,30
(6H, m), 6,90 (1H, m), 6,40–6,20
(3H, m), 5,20 (2H, br s), 4,90 (1H, m), 3,45 (1H, m), 3,30 (1H,
m), 1,15 (3H, t).
LRMS (Thermospray): m/z [MH+]
552.
-
HERSTELLUNGSBEISPIEL
19 (2R,3R,4S,5S)-4-(Benzoyloxy)-2-(6-chlor-2-iod-9H-purin-9-yl)-5-[(ethylamino)carbonyl]-tetrahydro-3-furanylbenzoat
-
n-Butylnitrit
(4,65 ml, 39,7 mmol) wurde zu einer Suspension von (2R,3R,4S,5S)-2-(2-Amino-6-chlor-9H-purin-9-yl)-4-(benzoyloxy)-5-[(ethylamino)carbonyl]-tetrahydro-3-furanylbenzoat
(Herstellungsbeispiel 18) (8,10 g, 14,7 mmol), Iod (3,73 g, 14,7
mmol), Kupfer(I)-iodid (6,16 g, 32,3 mmol) und Diiodmethan (12,55
ml, 155,8 mmol) in THF (100 ml) gegeben und das Gemisch wurde 2,5
h unter Refluxieren erhitzt. Die Lösung wurde auf Raumtemperatur
abkühlen
gelassen und das Lösemittel
wurde unter vermindertem Druck entfernt. Der Rückstand wurde zwischen wässriger
Natriummetabisulfitlösung
(5%, 100 ml) und Dichlormethan (100 ml) verteilt. Die organische
Schicht wurde abgetrennt, über
Arbacel filtriert, getrocknet (MgSO4) und
das Lösemittel
wurde unter vermindertem Druck abgedampft. Der Rückstand wurde durch Säulenchromatographie auf
Silicagel unter Elution mit Dichlormethan:Methanol (99:1, bezogen
auf das Volumen) gereinigt, wobei die Titelverbindung als gelber
Schaum erhalten wurde (7,55 g, 78%).
1H-NMR
(400 MHz, CDCl3) δ: 8,55 (1H, s), 8,05 (2H, m),
7,80 (2H, m), 7,65–7,30
(6H, m), 6,75 (1H, m), 6,50 (1H, m), 6,10–6,00 (2H, m), 4,90 (1H, m),
3,60–3,40
(2H, m), 1,25 (3H, t).
LRMS: m/z [MNa+]
684.
-
HERSTELLUNGSBEISPIEL
20 (2R,3R,4S,5S)-4-(Benzoyloxy)-2-{6-[(2-benzyl-3-phenylpropyl)amino]-2-iod-9H-purin-9-yl}-5-[(ethylamino)carbonyl]-tetrahydro-3-furanylbenzoat
-
Eine
Lösung
von (2R,3R,4S,5S)-4-(Benzoyloxy)-2-(6-chlor-2-iod-9H-purin-9-yl)-5-[(ethylamino)carbonyl]-tetrahydro-3-furanylbenzoat (Herstellungsbeispiel
19) (0,25 g, 0,38 mmol) und 2-Benzyl-3-phenylpropylamin (0,16 g,
0,76 mmol) in Isopropanol (10 ml) wurde 48 h bei Raumtemperatur
gerührt.
Lösemittel
wurde unter vermindertem Druck entfernt und der Rückstand
wurde durch Säulenchromatographie
auf Silicagel unter Elution mit Dichlormethan:Methanol (99:1, bezogen
auf das Volumen) gereinigt, wobei die Titelverbindung als gelber
Schaum erhalten wurde (0,26 g, 83%).
1H-NMR
(400 MHz, CDCl3) δ: 8,00 (2H, m), 7,80 (3H, m),
7,60–7,40
(5H, m), 7,35–7,15
(9H, m), 6,25 (1H, m), 6,15–6,05
(2H, m), 5,75 (1H, br s), 4,90–4,80
(2H, m), 3,65 (1H, m), 3,50 (1H, m), 3,00–2,85 (4H, m), 1,20 (3H, t).
LRMS:
m/z [MH+] 837.
-
HERSTELLUNGSBEISPIEL
21 (2R,3R,4S,5S)-4-(Benzoyloxy)-2-(6-{[(2,2-bis(3-phenyl)ethyl]amino}-2-iod-9H-purin-9-yl}-5-[(ethylamino)carbonyl]-tetrahydro-3-furanylbenzoat
-
Herstellung
aus (2R,3R,4S,5S)-4-(Benzoyloxy)-2-(6-chlor-2-iod-9H-purin-9-yl)-5-[(ethylamino)carbonyl]-tetrahydro-3-furanylbenzoat (Herstellungsbeispiel
19) und 2,2-Bis(phenyl)ethylamin
durch das Verfahren gemäß Herstellungsbeispiel
20. Die Titelverbindung wurde als gelber Schaum erhalten.
1H-NMR (400 MHz, CDCl3) δ: 8,05 (2H,
m), 7,80 (3H, m), 7,60–7,40
(5H, m), 7,35–7,15
(9H, m), 6,25 (1H, m), 6,15–6,05
(2H, m), 5,75 (1H, br s), 4,90 (1H, m), 4,40 (1H, m), 4,25 (1H,
br s), 3,65 (1H, m), 3,50 (1H, m), 1,25 (3H, t).
LRMS: m/z
[MH+] 823.
-
HERSTELLUNGSBEISPIEL
22 (2R,3R,4S,5S)-4-(Benzoyloxy)-5-[(ethylamino)carbonyl]-2-{6-[(1-ethylpropyl)amino]-2-iod-9H-purin-9-yl}tetrahydro-3-furanylbenzoat
-
Herstellung
aus (2R,3R,4S,5S)-4-(Benzoyloxy)-2-(6-chlor-2-iod-9H-purin-9-yl)-5-[(ethylamino)carbonyl]-tetrahydro-3-furanylbenzoat (Herstellungsbeispiel
19) und 1-Ethylpropylamin durch das Verfahren gemäß Herstellungsbeispiel
20. Die Titelverbindung wurde als gelber Schaum erhalten.
1H-NMR (400 MHz, CDCl3) δ: 8,10–8,00 (3H,
m), 7,80 (2H, m), 7,60 (1H, m), 7,50–7,40 (3H, m), 7,30 (2H, m), 6,40
(1H, m), 6,15–6,05
(2H, m), 5,75 (1H, br s), 4,90 (1H, m), 4,40 (1H, m), 4,00 (1H,
m), 3,55 (1H, m), 3,35 (1H, m), 1,75 (2H, m), 1,10 (2H, m), 1,25
(6H, t).
-
HERSTELLUNGSBEISPIEL
23 (2R,3R,4S,5S)-4-(Benzoyloxy)-2-(6-{[(1S)-1-benzyl-2-hydroxyethyl]amino}-2-iod-9H-purin-9-yl)-5-[(ethylamino)carbonyl]tetrahydro-3-furanylbenzoat
-
Herstellung
aus (2R,3R,4S,5S)-4-(Benzoyloxy)-2-(6-chlor-2-iod-9H-purin-9-yl)-5-[(ethylamino)carbonyl]-tetrahydro-3-furanylbenzoat (Herstellungsbeispiel
19) und (2S)-2-Amino-3-phenyl-1-propanol
durch das Verfahren gemäß Herstellungsbeispiel
20. Die Titelverbindung wurde als gelber Schaum erhalten.
1H-NMR (400 MHz, CDCl3) δ: 8,20 (1H,
s), 8,05 (2H, d), 7,80 (2H, d), 7,60 (1H, m), 7,50–7,35 (6H,
m), 7,30–7,20
(6H, m), 6,40 (1H, m), 6,30–6,10
(3H, m), 4,90 (1H, m), 4,40 (1H, m), 3,90 (1H, m), 3,80 (1H, m), 4,25
(1H, br s), 3,55 (1H, m), 3,40 (1H, m), 3,10 (2H, m), 1,15 (3H,
t).
-
HERSTELLUNGSBEISPIEL
24 (2R,3R,4S,5S)-4-(Benzoyloxy)-5-[(ethylamino)carbonyl]-2-{6-[(9H-fluoren-9-ylmethyl)amino]-2-iod-9H-purin-9-yl}tetrahydro-3-furanylbenzoat
-
Herstellung
aus (2R,3R,4S,5S)-4-(Benzoyloxy)-2-(6-chlor-2-iod-9H-purin-9-yl)-5-[(ethylamino)carbonyl]-tetrahydro-3-furanylbenzoat (Herstellungsbeispiel
19) und 9H-Fluoren-9-ylmethylamin
durch das Verfahren gemäß Herstellungsbeispiel
20. Die Titelverbindung wurde als gelber Schaum erhalten.
1H-NMR (400 MHz, CDCl3) δ: 8,05 (2H,
m), 7,80 (4H, m), 7,70–7,25
(13H, m), 6,25 (2H, m), 6,15–6,05
(2H, m), 5,85 (1H, br s), 4,90 (1H, m), 4,40 (1H, m), 4,15 (1H,
br s), 3,65 (1H, m), 3,50 (1H, m), 1,25 (3H, t).
LRMS: m/z
[MH+] 821.
-
HERSTELLUNGSBEISPIEL
25 (2R,3R,4S,5S)-4-(Benzoyloxy)-2-[6-(cyclohexylamino)-2-iod-9H-purin-9-yl]-5-[(ethylamino)carbonyl]tetrahydro-3-furanylbenzoat
-
Herstellung
aus (2R,3R,45,5S)-4-(Benzoyloxy)-2-(6-chlor-2-iod-9H-purin-9-yl)-5-[(ethylamino)carbonyl]-tetrahydro-3-furanylbenzoat (Herstellungsbeispiel
19) und Cyclohexylamin durch das Verfahren gemäß Herstellungsbeispiel 20.
Die Titelverbindung wurde als gelber Schaum erhalten.
1H-NMR (400 MHz, CDCl3) δ: 8,05 (2H,
m), 7,80 (3H, m), 7,60 (1H, m), 7,50–7,40 (4H, m), 7,30–7,25 (2H,
m), 6,25 (1H, m), 6,15–6,05
(2H, m), 5,70 (1H, br s), 4,90 (1H, m), 4,15 (1H, br s), 3,65 (1H,
m), 3,50 (1H, m), 2,10 (2H, m), 1,80 (2H, m), 1,65 (1H, m), 1,50–1,40 (2H,
m), 1,30–1,20
(6H, m).
LRMS: m/z [MH+] 725.
-
HERSTELLUNGSBEISPIEL
26 (2S,3S,4R,5R)-5-{6-[(2-Benzyl-3-phenylpropyl)amino]-2-iod-9H-purin-9-yl}-N-ethyl-3,4-dihydroxytetrahydro-2-furancarboxamid
-
Eine
Lösung
von (2R,3R,4S,5S)-4-(Benzoyloxy)-2-{6-[(2-benzyl-3-phenylpropyl)amino]-2-iod-9H-purin-9-yl}-5-[(ethylamino)carbonyl]-tetrahydro-3-furanylbenzoat
(Herstellungsbeispiel 20) (0,26 g, 0,31 mmol) und Natriumcarbonat
(33 mg, 0,3 mmol) in Methanol (5 ml) wurde 14 h bei Raumtemperatur
gerührt.
Lösemittel
wurde unter vermindertem Druck entfernt und der Rückstand
wurde in Dichlormethan:Methanol (99:1, bezogen auf das Volumen,
5 ml) gelöst
und filtriert. Lösemittel
wurde erneut unter vermindertem Druck abgedampft und der Rückstand
wurde mit Diethylether verrieben, wobei die Titelverbindung als
beigefarbenes Pulver erhalten wurde (0,17 g, 86%).
1H-NMR (400 MHz, CDCl3) δ: 8,10 (1H,
m), 7,30–7,10
(10H, m), 5,90 (1H, d), 4,70 (1H, m), 4,55–4,30 (2H, m), 3,55–3,40 (2H,
m), 3,30 (1H, m), 3,00–2,90
(4H, m), 1,20 (3H, t).
LRMS: m/z [MH+]
629.
-
HERSTELLUNGSBEISPIEL
27 (2S,3S,4R,5R)-5-{6-[(2,2-Diphenylethyl)amino]-2-iod-9H-purin-9-yl}-N-ethyl-3,4-dihydroxytetrahydro-2-furancarboxamid
-
Herstellung
aus (2R,3R,4S,5S)-4-(Benzoyloxy)-2-(6-{[(2,2-bis(3-phenyl)ethyl]amino}-2-iod-9H-purin-9-yl}-5-[(ethylamino)carbonyl]-tetrahydro-3-furanylbenzoat
(Herstellungsbeispiel 21) durch das Verfahren gemäß Herstellungsbeispiel 26.
Die Titelverbindung wurde als gelbes Pulver erhalten.
1H-NMR 400 MHz, CD3OD) δ: 8,10 (1H,
m), 7,35–7,15
(10H, m), 5,90 (1H, m), 4,75 (1H, m), 4,45 (1H, m), 4,40 (1H, m),
4,35 (1H, m), 4,20 (2H, m), 3,55–3,40 (2H, m), 1,20 (3H, t).
LRMS:
m/z [MH+] 615.
-
HERSTELLUNGSBEISPIEL
28 (2S,3S,4R,5R)-N-Ethyl-5-{6-[(1-ethylpropyl)amino]-2-iod-9H-purin-9-yl}-3,4-dihydroxytetrahydro-2-furancarboxamid
-
Herstellung
aus (2R,3R,4S,5S)-4-(Benzoyloxy)-5-[(ethylamino)carbonyl]-2-{6-[(1-ethylpropyl)amino]-2-iod-9H-purin-9-yl}tetrahydro-3-furanylbenzoat
(Herstellungsbeispiel 22) durch das Verfahren gemäß Herstellungsbeispiel
26. Die Titelverbindung wurde als gelbes Pulver erhalten.
1H-NMR (400 MHz, CDCl3) δ: 8,10 (1H,
m), 7,75 (1H, br s), 5,95 (1H, d), 5,70 (1H, m), 5,15 (1H, br s),
4,80 (1H, m), 4,60–4,45
(3H, m), 3,50 (1H, m), 3,25 (1H, m), 1,70 (2H, m), 1,50 (2H, m),
1,10 (3H, m), 0,90 (6H, t).
-
HERSTELLUNGSBEISPIEL
29 (2S,3S,4R,5R)-5-(6-{[(1S)-1-Benzyl-2-hydroxyethyl]amino}-2-iod-9H-purin-9-yl)-N-ethyl-3,4-dihydroxytetrahydro-2-furancarboxamid
-
Herstellung
aus (2R,3R,4S,5S)-4-(Benzoyloxy)-2-(6-{[(1S)-1-benzyl-2-hydroxyethyl]amino}-2-iod-9H-purin-9-yl)-5-[(ethylamino)carbonyl]tetrahydro-3-furanylbenzoat
(Herstellungsbeispiel 23) durch das Verfahren gemäß Herstellungsbeispiel
26. Die Titelverbindung wurde als gelbes Pulver erhalten.
1H-NMR (400 MHz, CDCl3) δ: 8,10 (1H,
br s), 7,50–7,10
(5H, m), 6,65 (1H, br s), 5,95 (1H, m), 4,80 (1H, m), 4,60–4,40 (2H,
m), 3,70 (1H, m), 3,50 (1H, m), 3,35 (1H, m), 3,10 (1H, m), 1,15
(3H, t).
-
HERSTELLUNGSBEISPIEL
30 (2S,3S,4R,5R)-N-Ethyl-5-{6-[(9H-fluoren-9-ylmethyl)amino]-2-iod-9H-purin-9-yl}-3,4-dihydroxytetrahydro-2-furancarboxamid
-
Herstellung
aus (2R,3R,4S,5S)-4-(Benzoyloxy)-5-[(ethylamino)carbonyl]-2-{6-[(9H-fluoren-9-ylmethyl)amino]-2-iod-9H-purin-9-yl}tetrahydro-3-furanylbenzoat
(Herstellungsbeispiel 24) durch das Verfahren gemäß Herstellungsbeispiel
26. Die Titelverbindung wurde als gelbes Pulver erhalten.
1H-NMR (400 MHz, CD3OD) δ: 8,10 (1H,
s), 7,80 (2H, d), 7,65 (2H, m), 7,40–7,20 (4H, m), 5,90 (1H, d),
4,75 (1H, s), 4,40 (1H, s), 4,35 (2H, br s), 4,10 (2H, m), 3,45
(2H, m), 1,20 (3H, t).
LRMS: m/z [MH+]
613.
-
HERSTELLUNGSBEISPIEL
31 (2S,3S,4R,5R)-5-[6-Cyclohexylamino)-2-iod-9H-purin-9-yl]-N-ethyl-3,4-dihydroxytetrahydro-2-furancarboxamid
-
Herstellung
aus (2R,3R,4S,5S)-4-(Benzoyloxy)-2-[6-(cyclohexylamino)-2-iod-9H-purin-9-yl]-5-[(ethylamino)carbonyl]tetrahydro-3-furanylbenzoat
(Herstellungsbeispiel 25) durch das Verfahren gemäß Herstellungsbeispiel
26. Die Titelverbindung wurde als gelbes Pulver erhalten.
1H-NMR (400 MHz, CD3OD) δ: 8,15 (1H,
s), 5,90 (1H, d), 4,70 (1H, m), 4,40 (1H, s), 4,30 (1H, d), 4,05
(1H, br s), 3,50–3,40
(2H, m), 2,00 (2H, m), 1,80 (2H, m), 1,60 (1H, m), 1,50–1,20 (6H,
m), 1,15 (3H, t).
LRMS: m/z [MH+] 517.


 worin R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie oben definiert sind; P1, P2 und P3 Schutzgruppen sind und Z eine Abgangsgruppe ist.
worin R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie oben definiert sind; P1, P2 und P3 Schutzgruppen sind und Z eine Abgangsgruppe ist.

 worin P1 und P2 Schutzgruppen sind, die gleich oder verschieden sein können oder Teil der gleichen Schutzgruppe sein können und worin R1 bis R8, A, "Het", wenn sie vorhanden sind, wie oben definiert sind, umfasst.
worin P1 und P2 Schutzgruppen sind, die gleich oder verschieden sein können oder Teil der gleichen Schutzgruppe sein können und worin R1 bis R8, A, "Het", wenn sie vorhanden sind, wie oben definiert sind, umfasst.
 worin P1 und P2 Schutzgruppen sind, mit Trimethylsilyltrifluormethansulfonat und einer Verbindung der Formel (IV) umfasst:
worin P1 und P2 Schutzgruppen sind, mit Trimethylsilyltrifluormethansulfonat und einer Verbindung der Formel (IV) umfasst: wobei die Verbindung der Formel (IV) mit N,O-Bis(trimethylsilyl)acetamid derivatisiert und dann mit der Verbindung der Formel (XI) umgesetzt wird, und wobei R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie oben definiert sind.
wobei die Verbindung der Formel (IV) mit N,O-Bis(trimethylsilyl)acetamid derivatisiert und dann mit der Verbindung der Formel (XI) umgesetzt wird, und wobei R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie oben definiert sind.
 worin R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie oben definiert sind.
worin R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie oben definiert sind.
 worin P3 eine Schutzgruppe ist und R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie oben definiert sind.
worin P3 eine Schutzgruppe ist und R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie oben definiert sind.
 durch eine Cyanogruppe, wobei R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie oben definiert sind.
durch eine Cyanogruppe, wobei R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie oben definiert sind.
 mit einem Amin der Formel R1NH2 (XII), wobei R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie oben definiert sind.
mit einem Amin der Formel R1NH2 (XII), wobei R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie oben definiert sind.










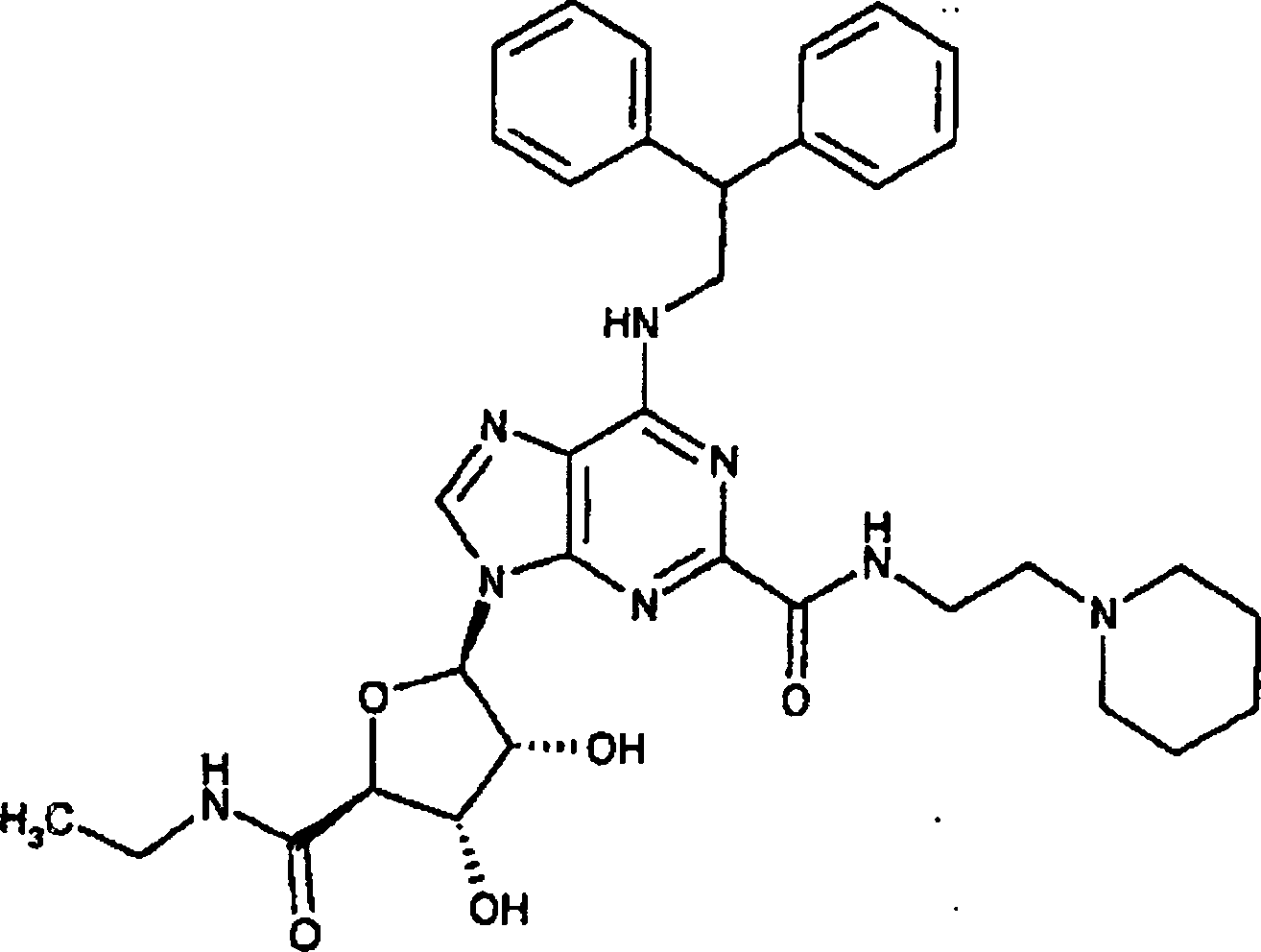

































































 worin R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie in Anspruch 1 definiert sind; worin P1 und P2 aus Alkanoyl und Aroyl ausgewählte Schutzgruppen sind und, wenn sie zusammengenommen sind, für C1-C6-Alkylen stehen, und P3 für eine Schutzgruppe steht, die Tetrahydropyran-2-yl ist; und Z eine Abgangsgruppe ist, die aus Brom, Iod, Sn(C1-C12-alkyl)3 oder CF3SO2O- ausgewählt ist.
worin R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie in Anspruch 1 definiert sind; worin P1 und P2 aus Alkanoyl und Aroyl ausgewählte Schutzgruppen sind und, wenn sie zusammengenommen sind, für C1-C6-Alkylen stehen, und P3 für eine Schutzgruppe steht, die Tetrahydropyran-2-yl ist; und Z eine Abgangsgruppe ist, die aus Brom, Iod, Sn(C1-C12-alkyl)3 oder CF3SO2O- ausgewählt ist.

 worin P1 und P2 wie in Anspruch 37 definierte Schutzgruppen sind, mit Trimethylsilyltrifluormethansulfonat und einer Verbindung der Formel (IV) umfasst:
worin P1 und P2 wie in Anspruch 37 definierte Schutzgruppen sind, mit Trimethylsilyltrifluormethansulfonat und einer Verbindung der Formel (IV) umfasst: wobei die Verbindung der Formel (IV) mit N,O-Bis(trimethylsilyl)acetamid derivatisiert und dann mit der Verbindung der Formel (XI) umgesetzt wird, und wobei R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie in Anspruch 1 definiert sind.
wobei die Verbindung der Formel (IV) mit N,O-Bis(trimethylsilyl)acetamid derivatisiert und dann mit der Verbindung der Formel (XI) umgesetzt wird, und wobei R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie in Anspruch 1 definiert sind.
 worin R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie in Anspruch 1 definiert sind.
worin R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie in Anspruch 1 definiert sind.
 durch eine Cyanogruppe, wobei R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie in Anspruch 1 definiert sind.
durch eine Cyanogruppe, wobei R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie in Anspruch 1 definiert sind.
 mit einem Amin der Formel R1NH2 (XII), wobei R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie in Anspruch 1 definiert sind.
mit einem Amin der Formel R1NH2 (XII), wobei R1 bis R8, A, "Het" und m, wenn sie vorhanden sind, wie in Anspruch 1 definiert sind.



