-
Die Erfindung betrifft einen porösen monolithischen Hybridreaktor für kontinuierliche flüssig-flüssig-Reaktionen, ein Verfahren zu dessen Herstellung sowie dessen Verwendung zur zweiphasigen Katalyse.
-
Enzyme sind Proteine, die in der Lage sind komplexe chemische Reaktionen unter milden Bedingungen und ökologisch vorteilhaft mit hohen Aktivitäten, Selektivitäten und Spezifitäten zu katalysieren (C. Mateo et al., Enzyme Microb. Technol., 2007, 40, 1451). Um biokatalytische Prozesse wirtschaftlich zu gestalten, müssen jedoch viele Reaktionszyklen mit hohen Enzymaktivitäten und -selektivitäten erreicht werden. Allerdings haben Enzyme eine sehr geringe Stabilität unter nicht-natürlichen Bedingungen (L. Batistella et al., Bioprocess and Biosyst. Eng., 2012, 35, 351, A. S. Bommarius, B. R. Riebel, Biocatalysis: Fundamentals and Applications, 2004, Wiley, Weinheim).
-
Es ist bekannt, dass ionische Flüssigkeiten (ILs) Enzyme stabilisieren können und somit eine hohe Anzahl an Reaktionszyklen und damit hohe Wechselzahlen ermöglichen. Ionische Flüssigkeiten sind organische Salze mit niedrigen Schmelzpunkten (< 100°C) und vernachlässigbaren Dampfdrücken. Häufig sind die Kationen der ILs 1,3-Dialkylimidazolium-, Tetraalkylphosphonium- oder Tetraalkylammonium-basierte Ionen und die Anionen polyatomare anorganische Ionen, wie Hexafluorophosphat, Tetrafluoroborat, Chlorid, Bromid, Trifluormethylsulfonat, Bis(trifluormethylsulfon)imid, Dicyanamid, Acetat oder Carbonat, etc. (T. Welton, Chem. Rev. 1999, 99, 2071). Auf Grund ihres sehr geringen Dampfdrucks sind ILs eine Alternative zu leicht flüchtigen organischen Lösungsmitteln (V. Plechkova et al., Chem. Soc. Rev. 2008, 37, 123). Wegen ihrer Polarität sind viele organische Moleküle sowie Enzyme oder Organometallkatalysatoren in ILs löslich.
-
Für eine kontinuierliche Prozessführung kann die Flüssigphase, in welcher der Katalysator gelöst vorliegt, auf einem Trägermaterial immobilisiert werden, so dass es zu keinem Auswaschen der stationären Phase kommt. In der sogenannten supported ionic liquid Phase (SILP) Technologie wird die ionische Flüssigkeit dabei als dünner Film auf einem porösen Trägermaterial immobilisiert, wobei der Katalysator in der IL-Schicht gelöst vorliegt. Im Vergleich zu klassischen Zweiphasenbedingungen mit einer ionischen Flüssigphase und einer weiteren mit der IL nicht mischbaren organischen oder wässrigen Flüssigphase haben SIL-Phasen den Vorteil, dass die IL und somit der darin gelöste Katalysator effizienter genutzt werden, da die Oberfläche der auf einem Träger immobilisierten ionischen Flüssigphase im Vergleich zu ihrem Volumen sehr groß ist. In SILP-Systemen können Substrate wegen des kurzen Diffusionswegs in den dünnen ionischen Flüssigkeitsfilmen leichter zum Katalysator diffundieren als unter klassischen Bedingungen, bei denen der Massentransfer stark limitiert ist. Des Weiteren werden bei SILP-Systemen nur geringe Mengen IL immobilisiert, was ökonomisch vorteilhaft ist.
-
Die Verwendung von SIL-Phasen kombiniert somit Vorteile homogener Katalyse, einer hohen Katalysatoraktivität und -selektivität mit denen der heterogenen Katalyse, wie einer großen Reaktionsfläche, an der die Katalyse stattfinden kann und der leichten Produktseparation. Die Produkte können leicht mit einer mit der IL nicht mischbaren organischen Phase abgetrennt werden, wobei der Katalysator in der immobilisierten IL zurückbleibt.
-
Mit Hilfe der SILP-Technologie (R. Fehrmann et al., Supported ionic liquids, 2014, Wiley, Weinheim) wurden bereits Hydroformylierungen (A. Riisager et al., Eur. J. Inorg. Chem. 2006, 695; C. P. Mehnert et al., Chem. Commun. 2002, 3010), Metathesereaktionen (R. Duque et al., Green Chem. 2011, 13, 1187; J. Scholz et al., Adv. Synth. Catal. 2011, 353, 2701; B. Autenrieth, W. Frey, M. R. Buchmeiser, Chem. Eur. J. 2012, 18, 14069)), Hydrierungen (C. P. Mehnert et al., Chem. Commun. 2002, 3010) sowie C-C-Kupplungsreaktionen durchgeführt (H. Hagiwara et al., Org. Lett. 2004, 6, 2325; C. P. Ferraz, B. Autenrieth, W. Frey, M. R. Buchmeiser, ChemCatChem 2014, 6, 191).
-
Bekannt sind Immobilisierungen der ionischen Flüssigkeiten auf der Oberfläche des Trägermaterials über Physisorption oder kovalente Anbindung. Die Mehrheit der für die SILP-Technologie verwendeten Trägermaterialien besteht dabei aus porösen Silikagelen, mesoporösen Silikamaterialien oder kristallinen Silika-Aluminium-Materialen (Y. Yang et al., Chem. Lett. 2005, 34, 220; A. Riisager et al., Chem. Commun. 2006, 994). Auch polymere Trägermaterialien wie Poly(diallydimethylammoniumchlorid), Polystyrol oder Chitosan (A. Wolfson et al., Tetrahedron Lett. 2004, 44, 1195; D. W. Kim et al., Angew. Chem. Int. Ed. 2004, 43, 483; J. Baudoux et al., Green Chem. 2007, 9, 1346) sowie monolithische Polymere kamen bereits zum Einsatz (P. Lozano et al., Adv. Synth. Catal. 2007, 349, 1077). Monolithische Materialien erlauben dabei durch ihren speziellen Aufbau hohe Durchflussraten bei geringen Gegendrücken (B. Autenrieth, W. Frey, M. R. Buchmeiser, Chem. Eur. J. 2012, 18, 14069).
-
Bekannte Bioreaktoren sind Festbett- oder Rieselstrom-Bioreaktoren, bei denen Enzyme an eine feste Partikeloberfläche gebunden sind und die Partikel von einer flüssigen Phase durchströmt werden (E. G. Vlakh, T. B. Tennikova, J. Sep. Sci. 2013, 36, 1149). Dies ermöglicht eine einfache Produktseparation und hohe Raum-Zeit-Ausbeuten. Nachteilig sind die häufig auftretenden inhomogenen Durchströmungen sowie das Verschließen der Hohlräume der Partikelschüttungen.
-
Kontinuierliche Prozesse mit physisorbierten SILP-Katalysatoren werden in Verbindung mit Gasphasenreaktionen oder mit überkritischen Flüssigphasen verwendet, da somit ein Auswaschen der immobilisierten IL sowie des Katalysators verhindert wird und gute Langzeitstabilitäten erreicht werden (A. Riisager et al., Chem. Commun. 2006, 9, 994; M. Jakuttis et al., Angew. Chem. Int. Ed. 2011, 19, 4492). Bei kontinuierlichen flüssig-flüssig-Systemen müssen die IL sowie der Katalysator eine sehr geringe Löslichkeit in der zweiten organischen Phase aufweisen. Zudem darf die IL nicht durch mechanische Kräfte, die bei der Verwendung einer zweiten Flüssigphase auftreten, vom Trägermaterial abgelöst werden. Folglich wird bei flüssig-flüssig-Prozessen die IL oft kovalent auf dem Trägermaterial immobilisiert (P. Lozano et al., Green Chem, 2010, 10, 1803). Dies schränkt die Wiederverwendbarkeit solcher Systeme stark ein.
-
Die Erfindung hat sich im Lichte des Standes der Technik die Aufgabe gestellt, neue vorteilhafte Trägermaterialien bzw. poröse monolithische Hybridreaktoren vorzuschlagen, welche die Immobilisierung größerer Mengen Enzym- bzw. Katalysator-haltiger ionischer Flüssigkeiten ermöglichen. Hiermit sollen längere Standzeiten der katalytischen Systeme und erhöhte Raum-Zeit-Ausbeuten ermöglicht werden. Die neuen heterogenkatalytischen Systeme sollten dabei derart beschaffen sein, dass sie leicht und bei geringen Gegendruck mit einer zweiten flüssigen Transportphase durchströmt werden können, dabei ein guter Kontakt zwischen der Transportphase und der ionischen Flüssigkeit besteht und damit ein rascher Austausch von Reaktanden und Produkten in diesen beiden flüssigen Phasen entsteht. Des Weiteren sollte ein Ausbluten der IL-Phase über lange Zeiträume hinweg unterdrückt werden können. Demzufolge ist es aus ökonomischer Sicht auch Ziel der Erfindung, dass die Wiederverwendbarkeit des bezeichneten Trägermaterials bzw. des bezeichneten Hybridreaktors vorteilhaft ermöglicht wird.
-
Erfindungsgemäß wird diese Aufgabe durch einen porösen monolithischen Hybridreaktor für kontinuierliche flüssig-flüssig-Reaktionen gelöst, der dadurch gekennzeichnet ist, dass der Hybridreaktor eine poröse Cellulose- oder Cellulosederivat-haltige polymere Matrix mit Transportporen eines Durchmessers von 1 bis 200 μm aufweist. Dabei wird bevorzugt, dass die Transportporen einen Durchmesser von 5 bis 150 μm, insbesondere 10 bis 100 μm, aufweisen.
-
Bei der Wahl der Cellulose bzw. der Cellulosederivate unterliegt man keiner kritischen Einschränkung. Die Cellulose kann beispielsweise zurückgehen auf Zellstoffe, welche bei der Papiererzeugung isoliert werden, aber auch auf andere Quellen, z. B. auf Baumwolle. Vorzugsweise handelt es sich bei den Cellulosederivaten um Celluloseacetat, Cellulosepropionat, Cellulosebutyrat und/oder Cellulosecarbamat, Methylcellulose oder Silylcellulose. Bei der Verwirklichung der Erfindung hat es sich gezeigt, dass die Cellulosederivate vorzugsweise einen Substitutionsgrad von 0 (reine Cellulose) bis 3, insbesondere von 1 bis 2,5, aufweisen. Besonders vorteilhaft ist es, wenn die Cellulose und/oder auch das Cellulosederivat als Perlmaterial, d. h. als Perlcellulose oder als Perlcellulosederivat vorliegt. Darüber hinaus ist es vorteilhaft, wenn die Cellulose und/oder die Cellulosederivate teilchenförmig, insbesondere kugelförmig sind und einen Durchmesser von 0,1 bis 100 μm, insbesondere von 0,5 bis 25 μm, aufweisen. Der Durchmesserbereich von 1 bis 10 μm ist besonders vorteilhaft.
-
Bei der Verwendung des erfindungsgemäßen Hybridreaktors hat es sich gezeigt, dass hier keine kritische Dimensionsbeschränkung vorliegt. Dennoch ist es zweckmäßig, wenn der Hybridreaktor einen Innendurchmesser von 0,1 bis 30 cm, insbesondere von 0,1 bis 10 cm, sowie eine Länge von 2 bis 200 cm, insbesondere von 5 bis 100 cm, aufweist. Besonders vorteilhaft ist es, wenn der Hybridreaktor einen Innendurchmesser von 0,1 bis 5 cm und/oder eine Länge von 10 bis 70 cm aufweist.
-
Der erfindungsgemäße Gedanke der besonderen Ausbildung eines Hybridreaktors bezieht insbesondere die vorteilhafte Weiterbildung ein, wonach sich auf der Oberfläche der Transportporen des Hybridreaktors ein Film einer ionischen Flüssigkeit mit darin gelöstem Katalysator befindet, wobei der Katalysator als Enzym oder als Organometallkatalysator, insbesondere als ionischer Organometallkatalysator, z. B. solche beschrieben in B. Autenrieth, E. B. Anderson, D. Wang, M. R. Buchmeiser, Macromol. Chem. Phys. 2013, 214, 33; B. Autenrieth, W. Frey, M. R. Buchmeiser, Chem. Eur. J. 2012, 18, 14069; B. Autenrieth, F. Willig, D. Pursley, S. Naumann, M. R. Buchmeiser, ChemCatChem 2013, 5, 3033; C. P. Ferraz, B. Autenrieth, W. Frey, M. R. Buchmeiser, ChemCatChem 2014, 6, 191; J. Zhao, D. Wang, B. Autenrieth, M. R. Buchmeiser, Macromol. Rapid Commun. 2015, 36, 190, vorliegt. Zu den bevorzugten Organometallkatalysatoren zählen ionische Rutheniumalkyliden- und Palladiumkatalysatoren.
-
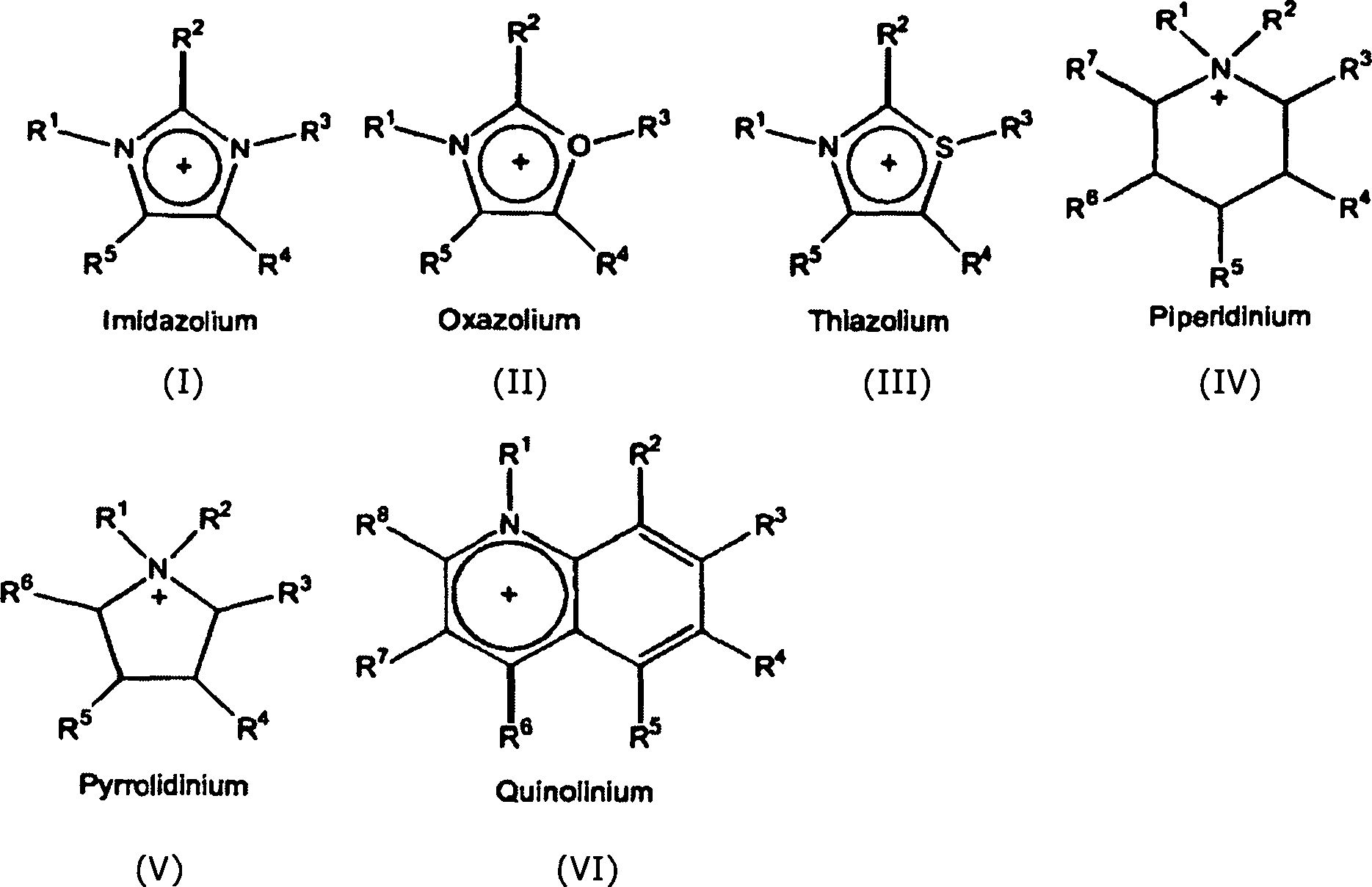
Bei der Durchführung der Erfindung gibt es keine relevante Beschränkung auf die Art der ionischen Flüssigkeit. Es ist bevorzugt, wenn ionische Flüssigkeiten gemäß der allgemeinen Formel [Q+]
n[Z]
n– eingesetzt werden, wobei das Kation [Q+]
n ein quaterniertes Ammonium-[R1R2R3R4N+], Phosphonium-[R1R2R3R4P+] oder Sulfonium-[R1R2R3S+]-Kation oder ein analoger quaternierter Stickstoff-, Phosphor- oder Schwefel-Heteroaromat der folgenden Formeln (I), (II), (III), (IV), (V) und (VI)
darstellt, wobei die Reste R1, R2, R3, R4 bzw. die Reste R1 bis R8 in den Formeln (I) bis (VI) unabhängig voneinander lineare, zyklische, verzweigte, gesättigte oder ungesättigte Alkylreste, mono- oder polycyclische, aromatische oder heteroaromatische Reste oder mit weiteren funktionellen Gruppen substituierte Derivate dieser Reste sind, wobei R1, R2, R3 und R4 untereinander verbunden sein können, wobei das Anion [Z]
n– in Form eines Halogenids, Pseudohalogenids, Amids, in Form von Phosphorverbindungen oder Nitroverbindungen vorliegt.
-
Eine Ausbildung dieser im Rahmen der Erfindung besonders vorteilhaften ionischen Flüssigkeiten ist darin zu sehen, dass die Halogenide bzw. Pseudohalogenide die Formel F–, Cl–, Br–, I–, BF4 –, PF6 –, AlCl4 –, Al2Cl7 –, Al3Cl10 –, AlBr4, FeCl4 –, BCl4 –, SbF5 –, AsF6 –, ZnCl3 –, SnCl3 –, CuCl2 –, CF3SO3 –, (CN)2N–, (CF3SO3)2N–, CF3CO2 –, CCl3CO2 –, CN–, SCN–, OCN–, die Phosphorverbindungen Phosphate der Formel PO4 3–, HPO4 2–, H2PO4 –, R1PO4 2–, HR1PO4 –, R1R2PO4 –; Phosphonate und Phosphinate der Formel: R1HPO3 –, R1R2PO2 –, R1R2PO3 –; Phosphite der Formel: PO3 3–, HPO3 2–, H2PO3 –, R1PO3 2–, R1HPO3 –, R1R2PO3 –; sowie Phosphonite und Phosphinite der Formel: R1R2PO2 –, R1HPO2 –, R1R2PO–, R1HPO– darstellen.
-
Von weitergehendem Vorteil ist es, wenn der Alkylrest in Form eines C1-C18-Alkylrestes, insbesondere eines Alkylrestes mit 1 bis 4 Kohlenstoffatomen, vorzugsweise eines Methyl-, Ethyl-, 1-Propyl-, 2-Propyl-, 1-Butyl-, oder 2-Butylrestes vorliegt, der cyclische Alkylrest in Form eines C3-10-Cycloalkylrestes, insbesondere in Form eines Cyclopropyl-, Cyclobutyl-, Cyclopentyl- oder Cyclohexyl-Restes vorliegt, der ungesättigte Alkylrest in Form eines Vinyl, 2-Propenyl, 3-Butenyl, cis-2-butenyl, trans-2-butenyl-Restes vorliegt, der aromatische Rest in Form eines Phenyl- oder Naphthyl-Restes vorliegt, der mit 1 bis 3 Halogenatomen, Alkylresten mit 1 bis 4 Kohlenstoffatomen oder Phenyl-Resten substituiert sein kann, und der heteroaromatische Rest in Form eines O-, S- oder N-enthaltenden heterocyclischen Restes mit 2 bis 5 Kohlenstoffatomen vorliegt. Als besonders vorteilhaft hat es sich erwiesen, als ionische Flüssigkeit [EMIM] [DCA], [EMIM] [Cl], [EMIM] [SCN], [EMIM] [Acetat], [EMIM] [DEP] und/oder [MMIM] [DMP] eingesetzt werden. Darin haben die Abkürzungen folgende Bedeutung: EMIM = Ethylmethylimidazolium; [MMIM] = Dimethylimidazolium; [DCA] = Dicyanamid; [DMP] = Dimethylphosphat; [DEP] = Diethylphosphat; BMIM = 1-Butyl-3-methylimidazolium; OMIM = 1-Octyl-3-methylimidazolium.
-
Folgende ionische Flüssigkeiten haben sich im Rahmen der praktischen Ausübung der Erfindung als besonders vorteilhaft erwiesen: das 1,3-Dimethylimidazolium-, 1,2,3-Trimethylimidazolium-, 1-Butyl-3-methylimidazolium-, 1-Butyl-2,3-dimethylimidazolium-, 1-Ethyl-3-methylimid-azolium- und/oder 1-Octyl-3-methylimidazoliumsalz.
-
Auch bezüglich der Menge an einbezogener ionischer Flüssigkeit unterliegt der erfindungsgemäße Hybridreaktor keiner relevanten Beschränkung. Es ist bevorzugt, dass der Hybridreaktor 5 bis 60 Gew.-%, insbesondere 20 bis 50 Gew.-% ionische Flüssigkeit, bezogen auf das Gesamtsystem, enthält. Besonders bevorzugt ist der Rahmen von 25 bis 48 Gew.-% an ionischer Flüssigkeit.
-
Weiteres zu vorteilhaften Ausbildungen des erfindungsgemäßen Hybridreaktors:
Bei diesem handelt es sich ausdrücklich um einen porösen monolithischen Hybridreaktor, wie gezeigt. Daher ist es zweckmäßig, eine vorteilhafte Porositätsangabe vorzunehmen. Die Porosität beträgt vorzugsweise 20 bis 90%, insbesondere 40 bis 80% (gemessen mittels N2-Adsorption). Die polymere Matrix besteht vorzugsweise aus Polyurethan. Daneben können mit Vorteil weitere polymere Materialien in Betracht gezogen werden, wie insbesondere Poly(norbornen)e, Poly(cycloocten)e, Poly(acrylat)e, Poly(methacrylat)e, Poly(acrylamid)e, Poly(styrol)e oder Epoxidharze. Für die quantitative Darstellung der polymeren Matrix des erfindungsgemäßen Hybridreaktors ist es eine vorteilhafte Angabe, wenn die polymere Matrix 0,1 bis 20 Gew.-%, insbesondere 1 bis 10 Gew.-% Cellulose oder Cellulosederivate enthält.
-
Gegenstand der Erfindung ist auch ein vorteilhaftes Verfahren zur Herstellung des erfindungsgemäßen Hybridreaktors, das dadurch gekennzeichnet ist, dass im Verlaufe einer Polyreaktion mit Monomeren, Initiatoren und Lösungsmitteln in Gegenwart von Cellulose oder Cellulosederivaten, insbesondere in sphärischer Teilchenform, eine poröse polymere Matrix mit Transportporen gebildet wird. Hierbei gilt es als bevorzugt, wenn in Abhängigkeit von der zu realisierenden Polymermatrix als Lösungsmittel ein porogenes Lösungsmittelgemisch in Form von Tetrahydrofuran (THF), Dichlormethan, Chloroform oder Toluol und n-Pentan eingesetzt wird. Zur Ausbildung der polymeren Matrix des erfindungsgemäßen Hybridreaktors wird vorzugsweise eine Polyreaktion als Polyaddition, Polykondensation oder als Polymerisation, in Abhängigkeit von dem zugrunde zu legenden Reaktionsmechanismus, durchgeführt.
-
Dabei unterliegt es rein fachmännischen Erwägungen, welche Monomere, Initiatoren und Lösungsmittel für die jeweilige Polyreaktion herangezogen werden. Auch lässt sich für den jeweiligen Anwendungszweck eine besonders vorteilhafte Cellulose und/oder das Cellulosederivat fachmännisch auswählen, wobei sinnvollerweise die vorstehend gegebenen technischen Ausführungen berücksichtigt werden.
-
Bei der Ausbildung der Matrix des erfindungsgemäßen Hybridreaktors auf der Basis eines Polyurethans ist es besonders bevorzugt, dass als Monomere 1,1,1-Tris(hydroxymethyl)propan oder N,N,N',N'',N'''-Pentakis(2-hydroxypropyl)-diethylentriamin und Hexamethylendiisocyanat-Trimer oder 4,4'-Methylen-bis(phenylisocyanat) sowie als Lösungsmittel n-Heptan und/oder Tetrahydrofuran, als Initiator 4-(Dimethylamino)pyridin oder Dibutylzinndilaurat verwendet werden, um durch Polyaddition ein Polyurethan zu bilden.
-
Bei der Ausbildung eines Polyurethans durch Polyaddition ist es vorteilhaft, insbesondere als Cellulosederivat Cellulose-2,5-acetat heranzuziehen. Gleichermaßen vorteilhaft ist es, wenn die Cellulose als Perlcellulose und das Cellulosederivat als Perlcellulosederivat mit den vorstehend bezeichneten Merkmalen herangezogen wird.
-
Nach Abschluss der Ausbildung der polymeren Matrix unter Einbezug der Cellulose und/oder der Cellulosederivate wird der erhaltene Hybridreaktor zur Entfernung verbliebener Reaktionspartner zweckmäßigerweise mit einem Lösungsmittel gespült und anschließend getrocknet. Es kann sich hierbei vorzugsweise handeln um Chloroform, Dichlormethan, THF, Ethylacetat, Methanol, Ethanol, 1-Propanol, 2-Propanol, Acetonitril oder Mischungen davon. Es wird zum Spülen insbesondere ein Lösungsmittelgemisch aus Ethylacetat/Tetrahydrofuran oder Chloroform/Methanol gewählt.
-
Zweckmäßigerweise erfolgt zur optimalen Vorbereitung des erfindungsgemäßen Hybridreaktors für die später geschilderten Anwendungszwecke ein mehrstündiges Trocknen, vorzugsweise bis zu 6 Stunden, insbesondere 3 bis 5 Stunden, dies bei erhöhter Temperatur, insbesondere bei 40 bis 70°C, im Hochvakuum (p < 0.1 Torr). Dieser für vorteilhafte Verwendungen vorbereitete erfindungsgemäße Hybridreaktor wird dahingehend modifiziert, dass in ihm, konkret in den Cellulose oder Cellulosederivat-Domänen, eine Katalysator-haltige ionische Flüssigkeit (IL) immobilisiert wird, wie sie vorstehend definiert wurde, indem folgende Maßnahmen durchgeführt werden: nach dem Lösen einer geeigneten Menge an Enzym oder Organometallkatalysator in einer geeigneten IL, gegebenenfalls unter Zusatz geringer Mengen an Wasser (< 10 Gew.-%), wird diese Lösung bzw. Emulsion, insbesondere mit Hilfe einer Pumpe, in den Monolithen eingebracht. Nachdem die Cellulose- oder Cellulosederivat-Domänen mit IL gequollen wurden (2–12 Stunden), wird der Überschuss an Enzym- oder Katalysator-haltiger IL durch bis zu 6-stündiges Spülen mit einem geeigneten, d. h. mit der IL nicht mischbaren Lösemittel, z. B. Methyl-t-butylether, entfernt.
-
Als Katalysator können Biokatalysatoren bzw. Enzyme, aber auch Organometallkatalysatoren, wie insbesondere ionische Rutheniumalkyliden- oder Palladiumkatalysatoren, genutzt werden.
-
Der in der oben beschriebenen Weise endgültig zur Weiterverwendung vorbereitete erfindungsgemäße Hybridreaktor, dies mit einbezogener Katalysator-haltiger ionischer Flüssigkeit, wird zur zweiphasigen Katalyse, insbesondere von kontinuierlichen Umesterungen und Amidierungen, herangezogen. Dies erfolgt vorzugsweise dadurch, dass der Hybridreaktor mit einem mit der jeweiligen in den Hybridreaktor einbezogenen ionischen Flüssigkeit nicht mischbaren Lösungsmittel, das die Reaktanden der jeweiligen Reaktion enthält, oder einem Reaktivgas oder einem Reaktivgasgemisch durchströmt wird. Bei der Wahl des nicht mischbaren Lösungsmittels unterliegt der Fachmann keiner relevanten Einschränkung. Vorzugsweise hat es eine möglichst niedere Viskosität, insbesondere von bis zu 10 mPa·s. Dabei gilt ein mit der ionischen Flüssigkeit nicht mischbares Lösungsmittel in Form von Toluol, Xylol, Methyl-t-butylether, Pentan, Hexan, Heptan, Octan, Nonan, Decan, Dodecanol, t-Butanol, Tetrahydrofuran, Wasser oder Gemische hiervon als besonders bevorzugt.
-
Die Vorteile, die mit der vorliegenden Erfindung verbunden sind, lassen sich wie folgt darstellen: Die Erfindung ermöglicht die vorteilhafte Nutzung eines porösen monolithischen Hybridreaktors, insbesondere für die kontinuierliche Biokatalyse. Ein besonderer Vorteil besteht darin, dass ionische Flüssigkeiten, integriert in den Hybridreaktor, bei einer Biokatalyse genutzt werden können, in denen geeignete Enzyme gelöst sind, wobei die ionischen Flüssigkeiten in sehr vorteilhafter Weise in den Transportporen des monolithischen Hybridträgermaterials gebunden sind. Demzufolge wird in vorteilhafter Weise ein dünner Film, der auf der ionischen Flüssigkeit mit darin dispergiertem Katalysator beruht, auf einem porösen Trägermaterial weitgehend immobilisiert. Im Anschluss besteht die Möglichkeit, dass die Reaktionsprodukte leicht mit einer mit der ionischen Flüssigkeit nicht mischbaren organischen Phase abgetrennt werden, wobei der jeweilige Katalysator in der immobilisierten ionischen Flüssigkeit zurückbleibt.
-
Die nachfolgende und auch vorausgehende Beschreibung der Erfindung, wobei nicht nur deren Vorteile, sondern auch technologische Erläuterungen herausgestellt sind, gilt nicht nur für die konkret bezeichneten Fälle, wobei vorzugsweise das Polyurethan als Matrix und Perlcellulose und Perlcellulosederivate eingesetzt werden. Es ist ohne Weiteres erkennbar, dass auch andere Polymere genutzt werden können sowie außer der Perlcellulose bzw. der Perlcellulosederivate auch andere Cellulosematerialien, sofern sie in den erfindungsgemäßen Rahmen fallen.
-
Grundsätzlich werden erfindungsgemäß polymere monolithische Hybridmaterialien, insbesondere aus Polyurethan, in Gegenwart von Perlcellulose oder Cellulosederivaten, wie z. B. Cellulose-2,5-acetat, unter Phasenseparations-bedingungen hergestellt. Die dabei entstehenden hochporösen, druckstabilen Perlcellulose-Polyurethan-Monolithe können erhebliche Mengen an ILs, in denen ein geeignetes Enzym bzw. ein geeigneter Katalysator in Form eines Organometallkatalysators in zweckmäßiger Konzentration gelöst ist, gut binden. Die mikroskalige Perlcellulose oder Perlcellulosederivate werden dabei von den Enzym/Organometallkatalysator-haltigen ILs gequollen und dienen somit als Reservoir für das Enzyme/den Katalysator. Wechselwirkungen zwischen den ILs und der Perlcellulose und/oder des Perlcellulosederivats bewirken eine sehr gute Retardierung der ILs auch unter Durchflussbedingungen und verhindern das Auswaschen der Flüssigphase und somit des Enzyms/Katalysators. Durch die große Oberfläche des vernetzten monolithischen Polymergerüsts wird eine gute Kontaktierung einer zweiten mit der IL nicht mischbaren, Substrat enthaltenden Flüssigphase mit den Enzymen erreicht, woraus ein schneller Stoffaustausch der Edukte und Produkte zwischen den beiden Phasen resultiert und hohe Raum-Zeit-Ausbeuten bei niedrigen Gegendrücken möglich sind. Die Nichtmischbarkeit der mobilen Flüssigphase mit der immobilisierten IL führt zu einer leichten Produktseparation ohne weitere Aufbereitungsschritte. Da von einer kovalenten Bindung der IL an das Trägermaterial abgesehen wird, ist ein Entfernen der IL und somit des verbrauchten Enzyms/Organometallkatalysators durch einfaches Auswaschen mit einer dafür geeigneten Waschflüssigkeit, z. B. Ethanol, möglich. Im Anschluss kann der Hybridmonolith wieder mit Enzym/Organometallkatalysator-haltiger IL beladen werden. Sowohl das Trägermaterial wie auch der Katalysator und die IL können somit für weitere Reaktionszyklen wiederverwendet werden.
-
Besondere Vorteile der Erfindung liegen auch darin, dass sie Modifizierungen zulässt. So kann es vorteilhaft sein, die Cellulose bzw. das Cellulosederivat in feinster Form, so in Form von Nanoteilchen, kontinuierlich in der polymeren Matrix des Hybridreaktors zu verteilen.
-
Von besonderem Vorteil hat sich die Erfindung bei der kontinuierlichen Umesterung erwiesen, so insbesondere bei der Umesterung von 1-Butanol mit Vinylbutyrat zu Butylbutyrat, von (R)-1-Phenylethanol mit Vinylbutyrat zu (R)-1-Phenylethylbutyrat sowie von (R)-1-Phenylethanol mit Vinylacetat zu (R)-1-Phenylethylacetat, aber auch zur Amidierung von (R)-1-Phenylethylamin mit Ethylmethoxyacetat zu (R)-2-Methoxy-N-(1-phenylethyl)acetamid.
-
Schließlich ist es in Einzelfällen denkbar, von der oben angesprochenen kontinuierlichen Reaktionsführung abzuweichen und das Verfahren chargenweise durchzuführen.
-
Beispiel 1 (Synthese von Perlcellulose)
-
Die Umwandlung von Cellulosefasern in Perlcellulose wurde nach der Vorschrift von Song et al. (sh. J. Chromatogr. A, 2010, 1217, 1298–1304) durchgeführt. Hierfür wurde eine 6,5 gew.-%-ige Lösung von Cellulose in 1-Ethyl-3-Methylimidazoliumacetat hergestellt, indem die Cellulose für eine Stunde bei 40°C in der ionischen Flüssigkeit gerührt wurde. 5,5 g der Celluloselösung wurden anschließend zu einer auf 40°C temperierten Lösung von 0,2 ml Polyethylenglycolsorbitanmonostearat (vertrieben unter dem Handelsnamen Tween 60) in 2,5 ml Cyclohexan gegeben. Die Lösung wurde daraufhin für 10 Minuten mit 1500 U/min durchmischt, wodurch eine Emulsion der organischen Phase in der ionischen Flüssigkeit entstand. Anschließend wurde eine auf 40°C temperierte Lösung von 0,9 ml Sorbitantrioleat (vertrieben unter dem Handelsnamen Span 85) in 20 ml Paraffinöl hinzugegeben. Um eine doppelte Emulsion zu erhalten, wurde die Lösung für 10 Minuten mit 800 U/min durchmischt. Eine ebenfalls auf 40°C erwärmte Lösung von 1,13 ml Sorbitantrioleat und 10 ml 0,2 M wässrige Natriumsulfatlösung in 25 ml Paraffinöl wurde daraufhin zugegeben, um die Cellulose wieder auszufällen. Hierzu wurde erneut 10 Minuten gerührt und die Lösung langsam auf Raumtemperatur abgekühlt. Die so erhaltene Perlcellulose wurde jeweils viermal mit reichlich Chloroform, Ethanol und anschließend mit demineralisiertem Wasser gewaschen und bei 60°C über Nacht im Hochvakuum getrocknet.
-
Beispiel 2 (Synthese von Perlcellulose-2,5-acetat)
-
Perlcellulose-2,5-acetat wurde nach der Vorschrift von Wagenknecht et al. (sh. ”Verfahren zur Herstellung von sphärischen Mikropartikeln auf Celluloseacetat-Basis”,
EP0750007 ) hergestellt. Hierfür wurden 3,6 g Cellulose-2,5-acetat (vertrieben von der Fa. Rhodia) in einem Lösungsmittelgemisch von 7,4 ml Methanol und 48 ml Ethylacetat gelöst. Anschließend wurde diese Lösung bei Raumtemperatur in einer Lösung aus 0,94 g Natriumacetat, 0,72 g Methylcellulose, 0,72 g Polyethylenglycol-4-tert-octylphenyl-ether (vertrieben unter dem Handelsnamen Triton X45) und 15,6 ml Ethylacetat in 120 ml demineralisiertem Wasser dispergiert, indem für 40 Minuten bei Raumtemperatur mit einem Hochleistungsdispergiergerät (Ultra-Turrax) bei 12.000 U/min kombiniert mit einem KPG-Rührer bei 250 U/min gearbeitet wurde. Nachdem der Ultra-Turrax abgeschaltet wurde, wurden die leichtflüchtigen Bestandteile mit Hilfe eines konstanten N
2-Stroms bei einer Temperatur von 30°C, die für eine Stunde gehalten wurde, entfernt. Ohne zu rühren wurde die Temperatur mit einer Heizrate von 2 K/h auf 45°C erhöht und für 12 h gehalten. Die erhaltene Perlcellulose wurde daraufhin mit reichlich demineralisiertem Wasser aufgeschlämmt und für 12 Minuten bei 4000 U/min zentrifugiert. Dieser Vorgang wurde insgesamt fünfmal durchgeführt. Zur Trocknung wurde die Cellulose über Nacht bei 80°C im Hochvakuum behandelt.
-
Beispiel 3 (Synthese der Hybridmonolithen mit den Dimensionen (0,46 × 15) cm)
-
Zur Synthese benötigtes n-Heptan wurde vor dem Gebrauch destilliert, entgast und über N2 gelagert. Tetrahydrofuran (THF) wurde dem marktüblichen Lösemittelreinigungssystem (MBraun SPS-800) entnommen, nachdem es über Aluminiumoxid getrocknet wurde, und entgast. Die Synthese der Monolithe erfolgte in (0,46 i. D. × 15) cm Stahlsäulen. Hierfür wurden 2 Lösungen vorbereitet. Die nachfolgenden prozentualen Angaben zu den Lösungen A und B beziehen sich auf die daraus hergestellte Fertigmischung. Für Lösung A wurde 1,1,1-Tris(hydroxymethyl)propan (5 Gew.-%) in den porogenen Lösungsmitteln Tetrahydrofuran (THF) (34 Gew.-%) und n-Heptan (12 Gew.-%) gelöst. Anschließend wurden 4-(Dimethylamino)pyridin (1 Gew.-%) und Cellulose-2,5-acetat (2 Gew.-%) zugegeben. Lösung B bestand aus 4,4'-Methylenbis(phenylisocyanat) (14 Gew.-%) in Tetrahydrofuran (THF) (32 Gew.-%). Beide Lösungen wurden unter Rühren vereint und die Lösung in die vertikal ausgerichtete Stahlsäule gefüllt. Die Stahlsäule wurde am oberen Ende mit einer weiteren Stahlsäule verlängert, um die Volumenabnahme der Polymerisation zu kompensieren und nach dem Befüllen verschlossen. Die Polymerisation wurde für 12 h bei 25°C durchgeführt. Anschließend wurde die Größe der monolithischen Säule an die Stahlsäule angepasst. Der Initiator sowie lösliche Komponenten wurden entfernt, indem die Säule für 4 Stunden mit einer Flussrate von 0,5 mlmin–1 mit einem Lösungsmittelgemisch aus Ethylacetat:Tetrahydrofuran (THF) (3:1, Vol:Vol) gespült wurde. Das Trocknen der Säulen erfolgte, indem sie 5 Stunden bei 50°C am Hochvakuum temperiert wurden.
-
Hybridmonolithe mit Cellulose anstatt Cellulose-2,5-acetat wurden ebenso mit 2 Gew.-% Cellulose synthetisiert. Die Synthese erfolgte analog derer für Cellulose-2,5-acetat. Lediglich das Entfernen von löslichen Komponenten erfolgte durch Spülen für 4 Stunden mit einem Lösungsmittelgemisch aus Chloroform:Methanol (1:2, Vol:Vol) bei einer Flussrate von 0,5 mlmin–1.
-
Beispiel 4 (Synthese der Hybridmonolithen mit den Dimensionen (2 × 30) cm)
-
Die Synthese der Monolithen erfolgte, indem zunächst 2 Lösungen hergestellt wurden. Für Lösung A wurde 1,1,1-Tris(hydroxymethyl)propan (5 Gew.-%) in Tetrahydrofuran (THF) (34 Gew.-%) gelöst. Um ein porogenes Lösungsmittelgemisch zu erhalten, wurden n-Heptan (15 Gew.-%), Dibutylzinndilaurat (6 Gew.-%) sowie Perlcellulose-2.5-acetat (1,7 Gew.-%) zugegeben. Lösung B bestand aus Hexamethylendiisocyanat-Trimer (19 Gew.-%) in Tetrahydrofuran (THF) (27 Gew.-%). Die Lösungen wurden vermischt und in eine vertikal ausgerichtete Stahlsäule, die am unteren Ende verschlossen wurde, gefüllt. Um die Volumenkontraktion während der Polymerisation auszugleichen, wurde die Stahlsäule am oberen Ende mit einer weiteren Stahlsäule verlängert und verschlossen, nachdem das Polymerisationsgemisch eingefüllt wurde. Die Polymerisation war nach 12 Stunden bei Raumtemperatur beendet und die monolithische Säule wurde für 5 Stunden mit einer Flussrate von 1,5 ml/min mit einem Lösungsmittelgemisch aus Ethylacetat:Tetrahydrofuran (THF) (3:1, V:V) durchspült, um lösliche Komponenten zu entfernen. Anschließend wurden die Monolithe für 5 Stunden bei 50°C im Vakuum getrocknet.
-
Beispiel 5 (Kontinuierliche Umesterung von 1-Butanol mit Vinylbutyrat zu Butylbutyrat)
-
Reaktionsgleichung 1: CALB-katalysierte Umesterung von Vinylbutyrat und 1-Butanol zu Butylbutyrat.
-
Bei der beschriebenen Umesterung wird der nach Beispiel 3 (Tabelle 1, oben) und der nach Beispiel 4 (Tabelle 1, unten) hergestellte Hybridreaktor herangezogen.
-
Es wurden diverse kontinuierlich geführte Reaktionen der Umesterung von 1-Butanol und Vinylbutyrat zu Butylbutyrat durchgeführt. Die entsprechenden Reaktionsbedingungen sind in Tabelle 1 zusammengefasst. Tabelle 1 (Reaktionsparameter sowie Produktivitäten der kontinuierlich geführten, CALB-katalysierten Umesterungen von 1-Butanol und Vinylbutyrat zu Butylbutyrat)
| IL | pH | T [°C] | linearer Fluss [mm/min] | Umsatz [%] | Produktivität [μmol/min·mgEnzym] |
| 1 | [OMIM+] [PF6 –] | 5 | 25 | 0,8 | 65 | 5 |
| 2 | | 7,5 | 25 | 5,3 | 60 | 19 |
| 3 | [OMIM+] [BF4 –] | 5 | 25 | 5,3 | 65 | 30 |
| 4 | | 7,5 | 25 | 2,3 | 83 | 27 |
| 5 | | 7,5 | 50 | 5,3 | 67 | 58 |
| 6 | | 7,5 | 25 | 23 | 45 | 19 |
| 7 | | 7,5 | 50 | 76 | 41 | 67 |
| 8 | | 7,5 | 50 | 23 | 65 | 26 |
| 9 | | 7,5 | 50 | 7,6 | 96 | 16 |
-
Anmerkungen:
-
- Einträge 1 und 2: 0,46 × 15 cm Säule; Einträge 3–9: 2 × 30 cm Säulen. Bedeutung der Abkürzung: OMIM = 1-Octyl-3-methylimidazolium
-
Beispiel 6 (Kontinuierliche Umesterung von (R)-1-Phenylethanol mit Vinylbutyrat zu (R)-1-Phenylethylbutyrat)
-
Reaktionsgleichung 2: CALB-katalysierte Bildung von (R)-1-Phenylethylbutyrat aus (R, S)-1-Phenylethanol und Vinylbutyrat.
-
Bei der beschriebenen Umesterung wird der nach Beispiel 3 hergestellte Hybridreaktor herangezogen.
-
Die CALB-katalysierte Reaktion von (R)-1-Phenylethanol und Vinylbutyrat wurde jeweils mit dem Enzym, gelöst in Phosphatpufferlösung (pH 7,5), bei 50°C und mit einer Flussrate von 2,3 mm/min durchgeführt. Die jeweils immobilisierte IL sowie die entsprechenden Umsätze und Produktivitäten sind in Tabelle 2 zusammengefasst. Tabelle 2 (Umsätze und Produktivitäten der kontinuierlich geführten, CALB-katalysierten Umesterungen von (R)-1-Phenylethanol und Vinylbutyrat zu (R)-1-Phenylethylbutyrat)
| IL | Umsatz [%] | Produktivität [μmol/min·mgEnzym] | TON1) |
| 1 | [OMIM+] [BF4 –] | 36 | 4,6 | 5,1·106 |
| 2 | [BMIM+] [PF6 –] | 60 | 7 | 7,2·106 |
| 3 | [BMIM+] [CF3SO3 –] | 31 | 4 | 4,6·106 |
-
Anmerkung zu den Abkürzungen:
-
- 1) nach 21 Tagen.
- OMIM (sh. Beispiel 5),
- BMIM = 1-Butyl-3-methylimidazolium
-
Beispiel 7 (Kontinuierliche Umesterung von (R)-1-Phenylethanol mit Vinylacetat zu (R)-1-Phenylethylacetat)
-
Reaktionsgleichung 3: CALB-katalysierte Umesterung von (R, S)-1-Phenylethanol mit Vinylacetat zu (R)-1-Phenylethylacetat.
-
Bei der beschriebenen Umesterung wird der nach Beispiel 3 hergestellte Hybridreaktor herangezogen.
-
Die CALB-katalysierte Reaktion von (R)-1-Phenylethanol und Vinylacetat wurde durchgeführt, indem das Enzym, gelöst in Phosphatpufferlösung (pH 7,5) in [OMIM
+] [BF
4 –], auf dem Hybridreaktor immobilisiert, und die Substratlösung bei 50°C und mit einer Flussrate von 2,3 mm/min durch den Monolith gepumpt wurde. Der entsprechende Umsatz sowie die Produktivität sind in Tabelle 3 zusammengefasst. Tabelle 3 (Umsatz und Produktivität der kontinuierlich geführten, CALB-katalysierten Umesterungen von (R)-1-Phenylethanol und Vinylacetat zu (R)-1-Phenylethylacetat)
| | IL | Umsatz [%] | Produktivität [μmol/min·mgEnzym] | TON1) |
| 1 | [OMIM+] [BF4 –] | 69 | 7 | 7,1·106 |
-
Anmerkung zu den Abkürzungen:
-
- 1) nach 21 Tagen.
- OMIM (s. Beispiel 5)
-
Beispiel 8 (Amidierung von (R)-1-Phenylethylamin mit Ethylmethoxyacetat zu (R)-2-Methoxy-N-(1-phenylethyl)acetamid)
-
Reaktionsgleichung 4: CALB-katalysierte Amidierung von (R, S)-Phenylethylamin mit Ethylmethoxyacetat zu (R)-2-Methoxy-N-(1-phenylethyl)acetamid.
-
- Beispiel 9 [(Ru(DMF)3(1,3-dimesitylimidazolin-2-yliden)-(=CH-2-(2-PrO)-C6H4)][(BF4)2] katalysierte Ringschlussmetathese von 1,7-Octadien zu Cyclohexen)
-
ZITATE ENTHALTEN IN DER BESCHREIBUNG
-
Diese Liste der vom Anmelder aufgeführten Dokumente wurde automatisiert erzeugt und ist ausschließlich zur besseren Information des Lesers aufgenommen. Die Liste ist nicht Bestandteil der deutschen Patent- bzw. Gebrauchsmusteranmeldung. Das DPMA übernimmt keinerlei Haftung für etwaige Fehler oder Auslassungen.
-
Zitierte Patentliteratur
-
-
Zitierte Nicht-Patentliteratur
-
- C. Mateo et al., Enzyme Microb. Technol., 2007, 40, 1451 [0002]
- L. Batistella et al., Bioprocess and Biosyst. Eng., 2012, 35, 351 [0002]
- A. S. Bommarius, B. R. Riebel, Biocatalysis: Fundamentals and Applications, 2004, Wiley, Weinheim [0002]
- T. Welton, Chem. Rev. 1999, 99, 2071 [0003]
- V. Plechkova et al., Chem. Soc. Rev. 2008, 37, 123 [0003]
- A. Riisager et al., Eur. J. Inorg. Chem. 2006, 695 [0006]
- C. P. Mehnert et al., Chem. Commun. 2002, 3010 [0006]
- R. Duque et al., Green Chem. 2011, 13, 1187 [0006]
- J. Scholz et al., Adv. Synth. Catal. 2011, 353, 2701 [0006]
- B. Autenrieth, W. Frey, M. R. Buchmeiser, Chem. Eur. J. 2012, 18, 14069 [0006]
- C. P. Mehnert et al., Chem. Commun. 2002, 3010 [0006]
- H. Hagiwara et al., Org. Lett. 2004, 6, 2325 [0006]
- C. P. Ferraz, B. Autenrieth, W. Frey, M. R. Buchmeiser, ChemCatChem 2014, 6, 191 [0006]
- Y. Yang et al., Chem. Lett. 2005, 34, 220; A. Riisager et al., Chem. Commun. 2006, 994 [0007]
- A. Wolfson et al., Tetrahedron Lett. 2004, 44, 1195 [0007]
- D. W. Kim et al., Angew. Chem. Int. Ed. 2004, 43, 483 [0007]
- J. Baudoux et al., Green Chem. 2007, 9, 1346 [0007]
- P. Lozano et al., Adv. Synth. Catal. 2007, 349, 1077 [0007]
- B. Autenrieth, W. Frey, M. R. Buchmeiser, Chem. Eur. J. 2012, 18, 14069 [0007]
- E. G. Vlakh, T. B. Tennikova, J. Sep. Sci. 2013, 36, 1149 [0008]
- A. Riisager et al., Chem. Commun. 2006, 9, 994 [0009]
- M. Jakuttis et al., Angew. Chem. Int. Ed. 2011, 19, 4492 [0009]
- P. Lozano et al., Green Chem, 2010, 10, 1803 [0009]
- B. Autenrieth, E. B. Anderson, D. Wang, M. R. Buchmeiser, Macromol. Chem. Phys. 2013, 214, 33 [0014]
- B. Autenrieth, W. Frey, M. R. Buchmeiser, Chem. Eur. J. 2012, 18, 14069 [0014]
- B. Autenrieth, F. Willig, D. Pursley, S. Naumann, M. R. Buchmeiser, ChemCatChem 2013, 5, 3033 [0014]
- C. P. Ferraz, B. Autenrieth, W. Frey, M. R. Buchmeiser, ChemCatChem 2014, 6, 191 [0014]
- J. Zhao, D. Wang, B. Autenrieth, M. R. Buchmeiser, Macromol. Rapid Commun. 2015, 36, 190 [0014]
- J. Chromatogr. A, 2010, 1217, 1298–1304 [0035]