CN105358704B - PTD-SMAD7 therapeutics - Google Patents
PTD-SMAD7 therapeutics Download PDFInfo
- Publication number
- CN105358704B CN105358704B CN201480025596.9A CN201480025596A CN105358704B CN 105358704 B CN105358704 B CN 105358704B CN 201480025596 A CN201480025596 A CN 201480025596A CN 105358704 B CN105358704 B CN 105358704B
- Authority
- CN
- China
- Prior art keywords
- smad7
- protein
- tat
- oral
- cells
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images












































































Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/18—Growth factors; Growth regulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/162—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from virus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/005—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/46—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- C07K14/47—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
- C07K14/4701—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals not used
- C07K14/4702—Regulators; Modulating activity
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/46—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- C07K14/47—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
- C07K14/4701—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals not used
- C07K14/4702—Regulators; Modulating activity
- C07K14/4703—Inhibitors; Suppressors
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/475—Growth factors; Growth regulators
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N7/00—Viruses; Bacteriophages; Compositions thereof; Preparation or purification thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/005—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'active' part of the composition delivered, i.e. the nucleic acid delivered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/01—Fusion polypeptide containing a localisation/targetting motif
- C07K2319/10—Fusion polypeptide containing a localisation/targetting motif containing a tag for extracellular membrane crossing, e.g. TAT or VP22
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/20—Fusion polypeptide containing a tag with affinity for a non-protein ligand
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/20—Fusion polypeptide containing a tag with affinity for a non-protein ligand
- C07K2319/23—Fusion polypeptide containing a tag with affinity for a non-protein ligand containing a GST-tag
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2740/00—Reverse transcribing RNA viruses
- C12N2740/00011—Details
- C12N2740/10011—Retroviridae
- C12N2740/16011—Human Immunodeficiency Virus, HIV
- C12N2740/16311—Human Immunodeficiency Virus, HIV concerning HIV regulatory proteins
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2740/00—Reverse transcribing RNA viruses
- C12N2740/00011—Details
- C12N2740/10011—Retroviridae
- C12N2740/16011—Human Immunodeficiency Virus, HIV
- C12N2740/16311—Human Immunodeficiency Virus, HIV concerning HIV regulatory proteins
- C12N2740/16322—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2800/00—Nucleic acids vectors
- C12N2800/22—Vectors comprising a coding region that has been codon optimised for expression in a respective host
Abstract
The present technology provides methods and compositions for treating inflammatory and/or tissue damage conditions. In particular, the use of Smad7 compositions for local or systemic delivery to sites of inflammation and/or tissue injury is described. Other particular embodiments relate to side effects caused by radiation and/or chemotherapy, including (but not limited to) treatment or prevention of oral and gastric mucositis. Codon-optimized nucleic acids encoding Smad7 fusion proteins are also provided.
Description
Cross Reference to Related Applications
This application claims priority from us provisional patent application USSN 61/775,252 filed on 8.3.2013, which is incorporated herein by reference in its entirety.
Statement of government interest
The invention was made with government support under the grant number AR061796 awarded by the National Institutes of Health. The government has certain rights in this invention.
Sequence listing
The present invention includes a sequence listing that has been submitted electronically in ASCII format and is incorporated by reference herein in its entirety. The ASCII copy was created on day 29 of 4 months 2014, named 089491-.
Background
Oral mucositis, a severe oral ulcer, is a common side effect of high dose radiation for bone marrow transplantation or craniofacial radiotherapy for cancer. Severe oral mucositis may require feeding tubes, management of severe pain, and premature cessation of radiation therapy. Excessive inflammation and epithelial ablation are key features of oral mucositis.
Palifermin (Palifermin), a KGF (human keratinocyte growth factor) recombinant protein, is approved for the prevention of oral mucositis in bone marrow transplant patients. Two clinical trials of palifermin in patients with head and neck cancer showed that palifermin reduced the incidence of severe oral mucositis from 67% and 69% to 51% and 54%, respectively. Other oral mucositis drugs in clinical trials or preclinical studies include growth factors, radioprotectors, anti-inflammatory agents or immunomodulators.
The modest effects of palifermin and the drugs under development in the above categories highlight the need to identify biomarkers for novel therapies. However, this effort has been hampered by the lack of routine diagnostic biopsy or waste tissue from oral mucositis patients.
Skin wound healing progresses through three overlapping phases: inflammation, tissue formation, and tissue remodeling. These are dynamic processes involving interactions between the epidermis, leukocytes, extracellular matrix (ECM), and skin fibroblasts. In response to skin injury, blood clots, infiltrating inflammatory cells, and other cell types in the wound release a variety of cytokines and chemokines. These cytokines initiate fibroblast proliferation and synthesis of ECM that fills the wound gap and causes the wound to close.
At the same time, keratinocytes at the wound edges begin to proliferate and migrate to cover the wound surface. Under the re-epithelialized epidermis, a new matrix, called granulation tissue, begins to fill the wound space, this matrix containing temporary ECM, inflammatory cells, fibroblasts, and blood vessels. Once the wound area is filled with granulation tissue and covered with newly re-epithelialized epidermis, the wound closure process is complete. Later, the wound gradually returns to normal strength and texture via tissue remodeling.
Among the many molecules known to affect wound healing, transforming growth factor beta (TGF-. beta.) has the broadest spectrum of actions, affecting all Cell types involved in all stages of wound healing (Feng et al, Annu Rev Cell DevBiol 21: 659-. The various functions of TGF- β are mediated by a number of signaling molecules, including Smad family members. TGF- β RI phosphorylates Smad2 and Smad3 when ligands bind to TGF- β type I and type II receptors (TGF β RI and TGF- β RII). Phosphorylated Smad2 and Smad3 bind co-Smad, Smad4, to form a heteromeric Smad complex and translocate into the nucleus to regulate transcription of TGF- β target genes.
TGF- β signaling has been reported to exert both positive and negative effects on wound healing (Wang et al, J Investig Dermatol Symp Proc 11: 112-. For example, Smad3 deficient mice that partially abolish TGF- β signaling exhibit accelerated wound healing (Ashcroft et al, Nat Cell Biol 1: 260-266, 1999). In contrast, introduction of exogenous Smad3 to the wound site to enhance TGF- β signaling also accelerated wound healing in a rabbit skin ulcer model (Sumiyoshi et al, J Invest Dermatol 123: 229-236, 2004). Smad 4-deficient mice have a marked increase in inflammation and angiogenesis, causing delay in wound closure and excessive scarring (Owens et al, Am J Pathol 176: 122-. Transient adenoviral gene transfer of an antagonist Smad7 of TGF-beta signaling in the corneal epithelium and stroma causes accelerated corneal wound healing with reduced inflammation (Saika et al, Am J Pathol 166: 1405-1418, 2005). Furthermore, the transfer of the Smad7 gene to the lens epithelium and stroma prevents injury-induced epithelial-mesenchymal transition of lens epithelial cells and suggests a potential role for Smad7 in the prevention of cystic fibrosis (Saika et al, Labinvest 84: 1259-. However, delivery of Smad7 to balloon lesions in rat carotid arteries via adenoviral vectors results in decreased vascular healing (Mallawaarachchi et al, Arterioscler Thromb Vasc Biol 25: 1383-1387, 2005). These studies indicate that the effects of TGF- β signalling components (such as Smad7) on wound healing are complex and highly situation specific. In addition, the effect of Smad7 may not always be explained by its role in TGF- β signaling. For example, Smad7 has also been shown to interact with components of the Wnt/β -catenin (Han et al, Dev Cell Biol 11: 301-312, 2006) and TNF β/NF- κ B (Hong et al, Nat Immunol 8: 504-513, 2007) families.
Disclosure of Invention
The present technology provides a nucleic acid molecule comprising a codon-optimized human Smad7cDNA nucleotide sequence. In some embodiments, the codon-optimized human Smad7 nucleotide sequence may include one or more arginine codons optimized for expression in one or more of bacteria or yeast, one or more serine codons optimized for expression in one or more of bacteria or yeast, and/or one or more histidine codons optimized for expression in one or more of bacteria or yeast. In some embodiments, the codon optimized human Smad7 nucleotide sequence may include 28 serine codons, 6 histidine codons, and 9 arginine codons optimized for expression in one or more of bacteria or yeast. In some embodiments, the codon optimized human Smad7 nucleotide sequence may be selected from the group consisting of SEQ ID NOs: 9. 21, 23, 24, 26, 28, 30, 32-34, 36, 38, 39, 87, 89, 91, 93, 96, 97, 99 and 100. In some embodiments, the codon optimized human Smad7 nucleotide sequence may have about 65% to 75% homology to the human Smad7cDNA, may comprise a nucleotide sequence encoding an N-terminal fragment of Smad7, may comprise a nucleotide sequence encoding a C-terminal fragment of Smad7, may comprise nucleotides encoding amino acids 2-258 of the human Smad7 protein, may comprise nucleotides encoding amino acids 259-426 of the human Smad7 protein, or may comprise nucleotides encoding amino acids 204-258 of the human Smad7 protein. In some embodiments, any of the foregoing may further comprise a nucleotide sequence encoding a protein transduction domain, such as Tat. In some embodiments, any of the foregoing may further comprise a nucleotide sequence encoding one or more of an epitope tag or a purification tag, such as V5, glutathione-S-transferase, or 6-histidine (SEQ ID NO: 40).
In some embodiments, any of the foregoing may be isolated and/or purified. In some embodiments, any of the foregoing may further encode a polypeptide having one or more biological activities selected from the group consisting of: reducing or eliminating phosphorylation of Smad2, reducing or eliminating nuclear translocation of the NF κ B p50 subunit, increasing cell proliferation, reducing apoptosis, reducing radiation-induced DNA damage, reducing inflammation, reducing angiogenesis, promoting healing of oral mucositis, promoting wound healing, and treating autoimmune diseases. In some embodiments, pharmaceutical compositions are provided comprising the nucleic acid molecules described above and one or more pharmaceutically acceptable excipients. In some embodiments, expression vectors comprising the above-described nucleic acid molecules operatively linked to a promoter are provided, as are host cells comprising such expression vectors, and pharmaceutical compositions comprising such vectors and host cells with one or more pharmaceutically acceptable excipients.
In one aspect, a protein molecule is provided comprising a human Smad7 protein having a leucine at position 216. In some embodiments, the human Smad7 protein may be truncated at the C-terminus, or truncated at the N-terminus. In some embodiments, the truncated human Smad7 protein may include about 50% of a full-length Smad7 sequence, or may include about 13% of a full-length Smad7 sequence. In some embodiments, the human Smad7 protein may comprise or consist of: amino acids 2-258, amino acids 204-258 or amino acids 259-426 of the human Smad7 protein. In some embodiments, the protein molecule may have one or more biological activities selected from the group consisting of: reducing or eliminating phosphorylation of Smad2, reducing or eliminating nuclear translocation of the NF κ B p50 subunit, increasing cell proliferation, reducing apoptosis, reducing radiation-induced DNA damage, reducing inflammation, reducing angiogenesis, promoting healing of oral mucositis, promoting wound healing, and treating autoimmune diseases. In some embodiments, any of the foregoing may further comprise a protein transduction domain, such as Tat. In some embodiments, any of the foregoing may further comprise one or more of an epitope tag or a purification tag, such as V5, glutathione-S-transferase, or 6-histidine (SEQ ID NO: 40). In some embodiments, a pharmaceutical composition is provided comprising any of the foregoing, a protein molecule, and one or more pharmaceutically acceptable excipients.
In another aspect, there is provided a method of treating or preventing an inflammatory condition in a subject comprising providing to the subject a therapeutically effective amount of a pharmaceutical composition described above. In some embodiments, the inflammatory condition may be one or more of a chronic wound, skin inflammation, psoriasis, or autoimmune disease. In some embodiments, the compositions can reduce inflammation via inhibition of TGF- β and NF- κ B signaling.
In another aspect, there is provided a method of preventing or treating a disease or disorder in a subject, comprising one or more of increasing one or more of cell proliferation or cell migration or preventing one or more of apoptosis or DNA damage in the subject, the method comprising providing to the subject a therapeutically effective amount of a pharmaceutical composition as described above, wherein one or more of increasing one or more of cell proliferation or cell migration or preventing one or more of apoptosis or DNA damage is suitable for preventing or treating the disease or disorder. In some embodiments, the disease or disorder may include one or more of a chronic wound, an acute wound, or mucositis. In some embodiments, the chronic wound may include one or more of a diabetic ulcer, a pressure ulcer, a venous ulcer, or an oral ulcer, the acute wound may include one or more of a wound-induced wound, a surgical wound, or a scar, the mucositis may include one or more of radiation-induced mucositis or chemotherapy-induced mucositis, and the mucositis may include one or more of oral mucositis or digestive tract mucositis.
It is contemplated that any method or composition described herein can be practiced with respect to any other method or composition described herein.
The use of the word "a/an" when used in conjunction with the term "comprising" in the claims and/or the specification can mean "one" but is also consistent with the meaning of "one or more", "at least one", and "one or more than one". The word "about" means plus or minus 5% of the stated number.
Other objects, features, and advantages of the present technology will be apparent from the following detailed description. It should be understood, however, that the detailed description and the specific examples, while indicating specific embodiments of the present technology, are given by way of illustration only, since various changes and modifications within the spirit and scope of the present technology will become apparent to those skilled in the art from this detailed description.
Drawings
The following drawings form part of the present specification and are included to further demonstrate certain implementations of the present technology. Implementations may be better understood by reference to one or more of these drawings in combination with specific implementations of particular implementations presented herein.
Fig. 1A-G provide illustrative embodiments of data showing resistance of k5.smad7 mice to radiation-induced oral mucositis. Fig. 1A provides illustrative embodiments of H & E staining of non-irradiated and irradiated (day 9 after initial irradiation) Wild Type (WT) and k5.smad7 tongues. Vertical lines in the image of the tongue from the WT mouse highlight the ulcer boundaries and dashed lines in the image indicate the epithelial-stromal boundaries (scale bar, 50 μm). Figure 1B provides a graphical representation of the quantification of the size of tongue ulcers (mean ± s.e.m); in 8Gy × 3 irradiation, n-8 for WT mice and n-7 for k5.smad7 mice; n-5 for WT mice and n-4 for k5.smad7 mice in 18-Gy radiation; n-5 per group for WT and k5.smad7 mice in 22-Gy irradiation. Fig. 1C provides an illustrative embodiment of the back of the human chamber of tongue (superior) and radiation-induced tongue mucositis (inferior) visualized using H & E (left) and CD45 staining (right). The solid line indicates the ulcer boundary and the dashed line indicates the basement membrane (scale bar, 25 μm). Fig. 1D provides an illustrative embodiment of immunostaining and TUNEL assay of Proliferating Cell Nuclear Antigen (PCNA) CD45 in irradiated sections adjacent to ulcers from WT mice and in injured areas from k5.smad7 mice (PI, propidium iodide). The dashed line indicates the basement membrane (scale bar, 25 μm). Figures 1E-1G provide a graphical representation of the staining quantification in figure 1D (n-3 or 4 per group). Data are expressed as mean ± s.e.m (fig. 1B) or mean ± s.d (fig. 1E-1G) and two-tailed t-test (two-tail Student t-test) is used to calculate P-values. P < 0.05, P < 0.01, P < 0.001, NS, no significance was determined by the two-tailed t-test. The dotted lines in (fig. 1A), (fig. 1C), and (fig. 1D) highlight the base film. Scale bar: 50 μm for all pictures in (FIG. 1A) and (FIG. 1C) and 25 μm for all pictures in (FIG. 1D).
Fig. 2A-G provide illustrative embodiments of data showing the attenuation of molecular changes by Smad 7. FIG. 2A provides an illustrative embodiment of immunostaining for NF- κ B p50, TGF- β 1, and pSmad 2. The irradiated tongue section of Wild Type (WT) was adjacent to the ulcer and the section of k5.smad7 was from the lesion area. Human samples were from non-irradiated oral mucosa and radiation-induced mucositis. The dashed line depicts the epithelial-stromal boundary. Scale bar, 25 μm for all pictures. FIG. 2B provides a graphical representation of the quantification of immunostaining for NF- κ B p50 and pSmad2 shown in (FIG. 2A). Fig. 2C provides illustrative embodiments of qRT-PCR of TGF- β 1 (normalized by keratin 5, n-6 per group for day 0, n-4 for days 7 and 9, and n-7 for day 10). Figure 2D provides a graphical representation of the quantification of human oral keratinocyte migration (see image in figure 8). Out-of-order, out-of-order siRNA; each group n is 3. Fig. 2E provides an illustrative embodiment of a western analysis of the knockdown efficiency at 72 hours after Smad7 knockdown for Smad7 and for siSmad7-1 and siSmad7-2 for Rac 1. M, molecular marker. Fig. 2F provides an illustrative embodiment of a western analysis of total and activated (GTP-bound) Rac1 protein. M: a molecular marker. Figure 2G provides a graphical representation of the quantification of the effect of Rac1 knockdown on Smad 7-mediated keratinocyte migration (see knockdown efficiency in figure 9A and images in figure 9D). Each group n is 3. Data are presented as mean ± s.d. and the two-tailed student's t-test is used to calculate P-values (fig. 2B-2D) and (fig. 2G). P < 0.05, P < 0.01, P < 0.001. NS, no significance.
Fig. 3A-H provide illustrative embodiments of data showing that Smad7 increases Rac1 expression by repressing the binding of individual Smad and CtBP1 to SBE of the Rac1 promoter. Fig. 3A provides a graphical representation of the quantification of Rac1mRNA in Wild Type (WT) and Smad7 transgenic keratinocytes. Each group n is 4. FIG. 3B provides an illustrative embodiment of a Western analysis of GTP-Rac1 and total Rac1 in WT and Smad7 keratinocytes. Smad7 protein levels in WT and Smad7 keratinocytes were determined by re-probing the tubulin western blot with antibodies to Smad7 (see additional western blots and quantitation in fig. 10A-B). Fig. 3C provides an illustrative embodiment of a western analysis of Rac1 protein levels after knocking off individual Smad2, Smad3, or Smad4 in human keratinocytes (for Smad knock-off efficiency, see fig. 10C-10E). FIG. 3D provides an illustrative embodiment of the ChIP assay for binding of Smad-2, Smad-3, Smad-4, and Smad-7 to the-1.5 KbSBE site of the Rac1 promoter in WT and Smad7 transgenic keratinocytes. Figure 3E provides a graphical representation of the quantification of Rac1 luciferase reporter gene assays in mouse keratinocytes. Disorder: out-of-order siRNA. n is 6. Figure 3F provides a graphical representation of the quantification of the activity of Rac1 luciferase reporter containing SBE or mutant (mut) SBE in WT or Smad7 transgenic keratinocytes. n is 6. FIG. 3G provides an illustrative embodiment of images from ChIP analysis of CtBP1 binding to the SBE-1.5Kb site of the Rac1 promoter in WT or K5.Smad7 keratinocytes. FIG. 3H provides a graphical representation of ChIP-qPCR quantitation of CtBP1 binding to SBE shown in FIG. 3G in WT and Smad7 transgenic keratinocytes. n is 4. Data are presented as mean ± s.d. and the two-tailed student's t-test was used to calculate P-values for figures 3A, 3E, 3F and 3H. P < 0.05, P < 0.01, P < 0.001.
Fig. 4A-G provide illustrative embodiments of data showing that CtBP 1-associated Rac1 repression contributes to inhibition of keratinocyte migration. Fig. 4A provides an illustrative embodiment of a western analysis of Rac1 protein after knockdown of CtBP1 in human oral keratinocytes. FIG. 4B provides a graphical representation of the quantification of the Rac1luc reporter gene activity with SBE. n is 6. Fig. 4C provides a graphical representation of the quantification of the effect of CtBP1 knockdown on human oral keratinocyte migration. Each group n is 3. Fig. 4D provides an illustrative embodiment of immunostaining for CtBP 1. The irradiated sections were adjacent to the ulcer (WT) or lesion area (k5.smad 7). The dotted line indicates the base film. Scale bar, 50 μm for all pictures. Fig. 4E provides an illustrative embodiment of immunostaining of CtBP1 in unirradiated oral mucosa and radiation-induced oral mucositis in human samples. The dotted line indicates the base film. Scale bar, 50 μm for both pictures. FIG. 4F provides a graphical representation of the quantification of CtBP1 nuclear positive cells in FIGS. 4D-E. Each group n is 3 or 4. Figure 4G provides a graphical representation of qRT-PCR quantification of CtBP1 (normalized with keratin K5). N is 6 for each group for day 0,4 for days 7 and 9, and 7 for day 10. Data are presented as mean ± s.d. and the two-tailed student's t-test was used to calculate P-values for fig. 4B, 4C, 4F and 4G. P < 0.05, P < 0.01, P < 0.001.
Fig. 5A-G provide illustrative embodiments of data showing that oral administration of Tat-Smad7 prevents radiation-induced oral mucositis in mice. Figure 5A provides a graphical representation of the quantification of oral mucositis ulcer size on day 9 after the initial 8Gy x 3 irradiation. Vehicle-saline or 50% glycerol/PBS. Fig. 5B provides an illustrative embodiment of pathological changes on day 9 of the initial 8Gy x 3 irradiation. Vehicle-saline or 50% glycerol/PBS. Scale bar, 50 μm for H & E pictures and 25 μm for the remaining pictures. The dashed line depicts the epithelial-stromal boundary; the solid line highlights the ulcer border. Fig. 5C, 5D, 5E, 5F, and 5G provide graphical representations of the quantification of immunostaining shown in fig. 5B. Each group n is 3 or 4. Data are presented as mean ± s.em (fig. 5A) or mean ± s.d. (fig. 5C-5G) and a two-tailed t-test was used to calculate P-values. P < 0.05, P < 0.01, P < 0.001. NS, no significance.
Fig. 6A-G provide illustrative embodiments of data showing Tat-Smad7 treatment for oral mucositis. Figure 6A provides a graphical representation of the quantification of ulcer size measured on day 10 after the initiation of 8Gy x 3 irradiation. Glycerol-50% glycerol/PBS. Fig. 6B provides an illustrative embodiment of H & E staining of the oral mucosa. And (4) picture uploading: open ulceration in mucosa treated with palifermin but not treated with Tat-Smad 7. The following pictures: comparison of epithelial thickness between palifermin-treated and Tat-Smad 7-treated mucosa. The dashed lines depict the basement membrane. The vertical lines highlight the ulcer boundaries. Scale bar, 50 μm for all pictures. Fig. 6C provides an illustrative embodiment of the immunostaining of Tat-Smad7 treatment after ulcer healing in 20 Gy-induced oral mucositis. Immunostaining with V5 revealed Tat-Smad7 in the oral epithelium (sections away from the lesion). K14 immunostaining was used as a contrast stain. The dashed lines depict the basement membrane. Scale bar, 25 μm for all pictures. Fig. 6D provides an illustrative embodiment of the Rac1 western analysis of the tongue of Tat-Smad7 treated mice on day 10 after the initial 8Gy x 3 irradiation. Fig. 6E provides an illustrative embodiment of the Rac1 western analysis of Tat-Smad7 treated normal human oral keratinocytes 48 hours after treatment. Fig. 6F provides an illustrative embodiment of the effect of Tat-Smad7 treatment on oral human keratinocyte migration (NOK-SI, see images in fig. 13A). Each group n is 4. FIG. 6G provides a quantitative graphical representation of the survival curves of NOK-SI keratinocytes and SCC lines (Cal27 and MSK921) with and without treatment with Tat-Smad 7. N-4 per group for each radiation dose. Data are presented as mean ± s.e.m (fig. 6A) or mean ± s.d. (fig. 6F, 6G) and the two-tailed t-test is used to calculate P-values. P < 0.05, P < 0.01, P < 0.001. NS, no significance.
Fig. 7A-E provide illustrative embodiments of data showing that k5.smad7 oral mucosal tissue is resistant to radiation-induced oral mucositis. Fig. 7A provides an illustrative embodiment of a Smad7 western ink dot: no detection in the non-irradiated Wild Type (WT) tongue and little detection after irradiation. Smad7 tongue has comparable Smad7 protein levels before and after irradiation. M: a molecular marker. Fig. 7B provides an illustrative embodiment of Smad7 immunostaining. Note that the nuclei in some irradiated epithelial cells are hypertrophied. The dashed line depicts the epithelial-stromal boundary. Figure 7C provides a graphical representation of the quantification of the reduction in the incidence of oral mucositis-induced morbidity in k5.smad7 mice. Fisher's exact test (Fisher's exact test) was used to calculate the p-value. P ═ 0.007. Fig. 7D provides an illustrative embodiment of immunostaining of k5.smad7 tongue showing reduced infiltration of neutrophils (Ly-6G), macrophages (BM8), and activated T cells (CD4) compared to WT oral mucositis. The dashed line depicts the epithelial-stromal boundary. Fig. 7E provides an illustrative embodiment of an immunostaining showing no significant difference in pSmad 1/5/8-nuclear positive cells (green) between WT and k5.smad7 oral mucosa before or after irradiation. The epithelial compartment was highlighted by the immunostaining (red) of keratin (K14). Note that the nuclei of irradiated epithelial cells are hypertrophic. The scale bar is 50 μm for all pictures.
Figures 8A-D provide illustrative embodiments of data showing that migration of spontaneously immortalized human oral epithelial cells (NOK-SI) is delayed by knocking-off Smad7 but accelerated by knocking-off TGF- β 1. Fig. 8A and 8B provide illustrative embodiments of representative images of cell migration. The dashed pair depicts a scratch wound. Quantification of cell migration and efficiency of Smad7 knockouts is presented in fig. 2D and 2E (above). Scrambled, scrambled siRNA. Figure 8C provides a graphical representation of the quantification of cell migration after TGF- β 1 knockdown from 3 independent experiments. Figure 8D provides a graphical representation of qRT-PCR showing TGF- β 1 knock-out efficiency. Data are presented as mean ± s.d. and the two-tailed student's t-test is used to calculate P-values. P < 0.05, P < 0.01. NS, no significance.
Fig. 9A-D provide illustrative embodiments of data showing that knock-out Rac1 reduces proliferation and migration of wild-type (WT) and Smad7 transgenic keratinocytes. FIG. 9A provides an illustrative embodiment of Western blot analysis of Rac1 48 hours after Rac1siRNA (siRac1-1, siRac1-2) transfection. Control, scrambled siRNA. Fig. 9B provides a graphical representation of the percentage of BrdU labeled cells in WT and Smad7 cultured cells in the BrdU incorporation assay with or without Rac1 knockdown. Data from 3 independent experiments are presented as mean ± s.d. P < 0.001. Fig. 9C provides an illustrative embodiment of representative immunofluorescence for BrdU positive cells presented in (fig. 9B). Antibodies against keratin 14(K14, red) were used for counterstaining. Fig. 9D provides an illustrative embodiment of an in vitro cell migration assay for Smad7transgene and WT keratinocytes after Rac1 knock-out. The dashed pair depicts a scratch wound. Quantification of cell migration is presented in fig. 2G.
Fig. 10A-F provide illustrative embodiments of data showing that Smad7 increases Rac1 expression by repressing the binding of Smad and CtBP1 to SBE of the Rac1 promoter. Fig. 10A provides an illustrative embodiment of western blot analysis of GTP-Rac1 and total Rac1 in Smad7 transgenic keratinocytes. Additional samples are shown in fig. 3B. M, molecular marker. Fig. 10B provides a graphical representation of the quantification of GTP-Rac1, total Rac1, and Smad7 in the WT and k5.Smad7 keratinocytes shown in fig. 10A and in fig. 3B. The protein level in the WT keratinocytes of each ink dot was normalized to "1". Data are presented as mean ± s.d. and the two-tailed student's t-test is used to calculate P-values. P < 0.01, P < 0.001. Fig. 10C and 10D provide illustrative embodiments of western blot analysis for Smad2, Smad3, and Smad4 knockouts in NOK-SI cells. Its effect on Rac1 expression is shown in fig. 3C. M, molecular marker. GAPDH, by probing the internal protein control of the same spot again. Fig. 10F provides an illustrative embodiment showing that CtBP1 knockdown promotes NOK-SI cell migration. The dashed pair depicts a scratch wound. Quantification of cell migration and efficiency of CtBP1 knockouts is shown in fig. 4A and 4C.
FIGS. 11A-G provide illustrative embodiments of data showing the purification and characterization of Tat-Smad7 and Tat-Cre protein. FIG. 11A shows an illustrative embodiment of a schematic diagram of the Tat-Smad7 protein. FIG. 11A discloses SEQ ID NO 49 and 101, respectively, in order of appearance. FIG. 11B provides an illustrative embodiment of a Western blot of purified Tat-Smad7 protein. FIG. 11C provides an illustrative embodiment of immunostaining for Tat-Smad7 protein transduction in keratinocytes. Left and middle panels: Tat-Smad7 staining with V5 antibody (green) and counterstaining with K14 antibody (red). Cells showed Tat-Smad7 in the nucleus 5 min after transduction and in both the nucleus and cytoplasm 12 hours after transduction. Right side frame: Tat-Smad7 abolished Smad2 phosphorylation (pSmad2, green). Contrasting staining with V5 (red) revealed Tat-Smad7 transduced cells. Fig. 11D provides an illustrative embodiment showing that V5 antibody staining detects Tat-Smad7 transduced immunostaining in the buccal mucosa 12 hours after topical application of Tat-Smad 7. The K14 antibody was used for counterstaining. Scale bar, 50 μm for both pictures. Fig. 11E provides an illustrative embodiment of the western ink dot of the purified Tat-Cre protein shown in fig. 11A having the same Tat and V5 tags. FIG. 11F provides an illustrative embodiment of an agarose gel showing the activity of Tat-Cre: Tat-Cre excises a 1,460bp fragment flanked by loxP (floxed) from the 7,650bp vector pLL3.7. FIG. 11G provides a graph showing that prophylactic treatment with Tat-Smad7 protein reduced 20Gy radiation-induced canker sores. Data are expressed as mean ± s.em. The two-tailed student's t-test was used to calculate P-values. P < 0.05, P < 0.001.
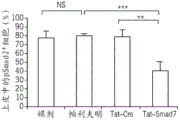
Fig. 12A-I provide illustrative embodiments of data showing the effect of Tat-Smad7 treatment on oral mucositis. FIG. 12A provides a graphical representation of the quantification of ulcer size reduction in the oral mucosa treated with Tat-Smad7 (0.8 μ g per day, day 6 to day 9). Samples were collected on day 10. Each group n is 8. Fig. 12B provides an illustrative embodiment of immunostaining for molecular markers from the sample of fig. 12A. Scale bar, 50 μm for the top two panels and 25 μm for the other panels. Propidium Iodide (PI) and K14 were used as contrast stains. Fig. 12C-G provide graphical representations of the quantification of immunostaining shown in fig. 12C. 3-4 samples were used. Figure 12H provides a graphical representation of the quantification of luciferase assay. Tat-Smad7 treatment increased the activity of the Rac1 promoter with SBE but not mutant SBE in mouse keratinocytes. FIG. 12I provides an illustrative embodiment of the ChIP assay for CtBP1 binding to SBE of the mouse Rac1 promoter in Tat-Smad7 treated mouse keratinocytes. Data are expressed as mean ± s.e.m (a) or mean ± s.d (c-h) and the two-tailed t-test is used to calculate P-values. P < 0.05, P < 0.01, P < 0.001. NS, no significance.
FIGS. 13A-H provide illustrative embodiments of data showing the effect of Tat-Smad7 treatment on migration of human keratinocytes and tumor cell lines. FIG. 13A provides an illustrative embodiment showing that Tat-Smad7 accelerates NOK-SI cell migration. Quantification from four independent experiments is shown in fig. 6F (above). The dashed pair depicts the initial wound. FIG. 13B provides an illustrative embodiment of the immunostaining of Tat-Smad7 treatment showing attenuated radiation-induced pSmad2 and NF- κ B p50 nuclear localization in NOK-SI cells. Fig. 13C provides an illustrative embodiment showing V5 staining of MSK921 cells 2 hours after Tat-Smad7 treatment. K14 staining was used as a contrast stain. Fig. 13D provides an illustrative embodiment of Rac1 western analysis in MSK921 60 hours after Tat-Smad7 treatment. M, molecular marker. Figure 13E provides a graphical representation of the quantification of MSK921 cell migration from 3 independent experiments. FIG. 13F provides an illustrative embodiment showing a representative MSK921 cell migration assay treated with Tat-Smad7 and PBS. The solid line pair depicts the initial wound. The dotted line highlights the forefront of migrating cells. Figure 13G provides a graphical representation of the quantification of Cal27 cell migration from 3 independent experiments. Fig. 13H provides an illustrative embodiment showing a representative image of fig. 13G. The solid line pair depicts the initial wound. The dotted line highlights the forefront of migrating cells. Data are expressed as mean ± s.d. and the two-tailed student's t-test is used to calculate P-values. NS, no significance.
Fig. 14A-B show a generalized illustrative schematic of the underlying mechanisms of Smad 7-mediated protection and healing of oral mucositis. FIG. 14A shows an illustrative schematic of how radiation activates NF-. kappa.B, increases TGF-. beta.1 and CtBP 1. NF-. kappa.B and TGF-. beta.1 induce inflammation. TGF-B1 induces apoptosis, growth arrest and activates Smad-2, Smad-3 and Smad-4, which recruits CtBP1 to the Rac1 promoter to repress Rac1 transcription, resulting in reduced re-epithelialization. FIG. 14B shows an illustrative schematic of how Smad7 blocks NF-. kappa.B and TGF-. beta.1-induced inflammation and blocks TGF-. beta.1-induced apoptosis and growth arrest. Smad7 derepresses Rac1 transcription by preventing TGF- β 1 mediated Smad activation (phosphorylation) or competition with the signaling Smad/CtBP1 transcription repression complex for binding to the Rac1 promoter. The increase in Rac1 induced by Smad7 contributes to keratinocyte migration during re-epithelialization.
Fig. 15 shows an illustrative schematic of Smad7 domains associated with chaperones, potential target effects, and potential physiological effects.
FIGS. 16A-B are graphs showing the ability of a truncated Smad7 protein to accelerate wound healing in a mouse wound healing model. FIG. 16A is a graph showing the effect of Tat-C-Smad7 truncated at the C-terminus (259-426aa) versus the full-length Tat-Smad7 and control (PBS) on the average percent wound healing over time. Each group n is 3. FIG. 16B is a graph illustrating the effect of Tat-N-Smad7(1-258aa) versus full-length Tat-Smad7 and control (PBS) on the average percent wound healing over time. Each group n is 6. Data are presented as mean ± s.d. and the two-tailed student's t-test is used to calculate P-values. P < 0.05, compared to control (PBS); # p < 0.05, compared to Tat-Smad 7.
Fig. 17A-C are photographs and graphs showing Smad7 accelerating wound healing in a model of impaired wound healing. Fig. 17A is a digital photograph illustrating the general appearance of wounds in diabetic (db/db) mice treated with PBS or Tat-Smad7 over a period of thirteen days. FIG. 17B is a graph showing Tat-Smad7 vsAnd control (PBS) versus average percent wound healing over time. Each group n is 6. Data are presented as mean ± s.d. and the two-tailed student's t-test is used to calculate P-values. P < 0.05, compared to control (PBS). Fig. 17C is a histological comparison of wound samples taken eight days after opening the wound. The vertical dashed line (upper panel) in the image from the control (PBS) db/db mouse highlights the wound boundaries.
Detailed Description
As further described herein, the present disclosure provides Smad7 proteins and biologically active fragments and derivatives thereof, nucleic acids encoding such proteins, vectors including such nucleic acids, and cells including vectors, nucleic acids and/or proteins, all for use in formulating a medicament and for treating and/or preventing one or more diseases or disorders. Also provided are methods of making and screening Smad7 proteins, and biologically active fragments and derivatives thereof, useful for the treatment and/or prevention of one or more diseases or disorders. Also provided are methods of using one or more markers associated with exposure to Smad7 to predict and/or evaluate a response to a treatment. Such markers may include, but are not limited to, Rac1 for cell migration, NF-. kappa.B for inflammation, and TGF-. beta.for growth arrest and inflammation.
Smad 7-treatable diseases and conditions may include those that include one or more of the following: reduced cell proliferation, reduced cell migration, increased cell death, excessive inflammation, and/or DNA damage. Smad 7-treatable diseases and conditions may include those treated with Smad7 protein and biologically active fragments and derivatives thereof having one or more of the following activities: including, but not limited to, increasing proliferation, reducing or inhibiting cell death, reducing excessive inflammation, preventing DNA damage, and/or increasing cell migration. Such diseases and/or conditions may include, but are not limited to, acute (e.g., via surgery, fight, trauma) and chronic wounds (e.g., ulcers, such as diabetic, pressure, venous), scars, fibrosis and abnormal healing, mucositis (e.g., oral and/or gastrointestinal), stomatitis, proctitis, autoimmune diseases (e.g., psoriasis, arthritis), and cancer.
It is crucial for the prevention and treatment of oral mucositis to overcome epithelial ablation due to massive apoptosis and reduced keratinocyte proliferation. The proliferative and anti-apoptotic effects of Smad7 were more pronounced in oral mucositis than in normal oral mucosa when potent growth inhibitors of epithelial cells and the apoptosis inducer TGF- β 1 were increased.
While not wishing to be bound by theory, it is believed that increased Rac1 activation is primarily responsible for Smad 7-mediated keratinocyte migration in wound closure. This finding was unexpected in view of the documented role of TGF- β signaling in Rho/Rac activation in cancer cells via a Smad-independent mechanism (Dernyck et al, Nature 415: 577-.
It is believed that Smad-dependent Rac1 repression overcomes Smad-independent Rac1 activation, if any, due to increased Smad signaling (as demonstrated by increased pSmad 2) and Smad transcription co-repressor CtBP1 during oral mucositis. When this repression is removed by Smad7, it allows for keratinocyte migration mediated by Rac1 activation. However, in oral cancer cells, loss or inactivation of signaling Smad, or other mechanisms, independently activate Rac 1. Thus, Smad 7-mediated elimination of Rac1 repression will no longer occur.
While Rac1 activation also contributed to keratinocyte proliferation, knock-out Rac1 only partially attenuated the proliferative effects of Smad 7. Thus, the contribution of Rac1 to proliferation appears to be limited, and there is also a need to block TGF- β 1-induced growth arrest to overcome radiation-induced growth inhibition.
Suppression of excessive inflammation creates a microenvironment for oral mucositis healing. Antagonism of TGF- β and NF- κ B signaling by Smad7 makes Smad 7a more potent anti-inflammatory molecule than other agents that target NF- κ B alone. Because inflammatory cells produce cytokines that further activate TGF- β and NF- κ B, the reduced TGF- β and NF- κ B signaling found in k5.Smad7 or Tat-Smad7 treated oral mucosa following irradiation reflects the direct antagonism of these two pathways by Smad7 and the resultant reduction in inflammatory cytokines from infiltrating leukocytes. However, Smad7 does not reduce NF-. kappa.B or TGF-. beta.signaling below its normal physiological state. This incomplete blocking of NF-. kappa.B or TGF-. beta.signaling may be beneficial for oral mucositis healing, as complete loss of either pathway may induce excessive inflammation.
A major obstacle to the use of growth factors to treat oral mucositis in cancer patients is the potential risk of promoting the growth of cancer cells. Most human oral cancers lose TGF- β signaling in tumor epithelial cells. Thus, anti-Smad associated cell proliferation and migration achieved by Smad7 would be ineffective in cancer cells. In tumors with intact TGF- β signaling, activation of other oncogenic pathways may suppress TGF- β induced tumor suppression. These two situations may explain why Smad7 was not observed to increase proliferation and migration of oral cancer cells with mutated or intact TGF- β signaling components.
In addition, TGF- β signaling promotes tumor invasion primarily via Smad-independent mechanisms following TGF- β induced loss of tumor inhibition. Thus, blockade of TGF- β signaling by Smad7 in cancer cells may abrogate TGF- β mediated tumor promotion, which appears similar to TGF- β inhibitors currently used in clinical trials for advanced cancer. In addition, the potent anti-inflammatory effect of Smad7 may reduce the risk of tumor progression. Thus, chronic administration of Smad7 may also be helpful in cancer therapy.
Spontaneous tumor formation in k5.smad7 mice has not been observed. Because Smad7 is not a secreted protein, local and short-term Smad7 protein delivery in the treatment of oral mucositis should have little systemic effect. In bone marrow transplant patients with oral epithelia free of cancer cells, topical application of Smad7 may be suitable for the prevention and treatment of oral mucositis.
While not wishing to be bound by any theory, Smad 7-mediated healing of oral mucositis appears to be the result of targeting multiple pathogenic processes mediated by one or more molecules (see, e.g., fig. 14A-B). It is believed that one or more of these molecules (e.g., TGF- β, NF- κ B, CtBP1, Rac1) may also be useful as a predictive and therapeutic response marker for oral mucositis in patients.
A. Nucleic acids, vectors and host cells
In another embodiment, the disclosure also provides a gene encoding Smad 7. In addition to the wild-type SMAD7 gene (SEQ ID NO: 22, 88) and various codon-optimized versions (SEQ ID NO: 9,21, 23, 24, 26, 28, 30, 32-34, 36, 38, 39, 87, 89, 91, 93, 96, 97, 99, and 100), it should be understood that the present technology is not limited to the specific nucleic acids disclosed herein. The "Smad 7 gene" as discussed below can contain a variety of different bases and still produce a corresponding polypeptide that is functionally indistinguishable from, and in some cases structurally identical to, the human genes disclosed herein.
1. Nucleic acid encoding Smad7
Nucleic acids according to the present technology may represent the entire Smad7 gene, truncated portion, and/or fragment of Smad7 that expresses a polypeptide having one or more Smad 7-related activities, such as, but not limited to, increasing proliferation, reducing or inhibiting cell death, reducing excessive inflammation, preventing DNA damage, and/or increasing cell migration, and treating or preventing one or more diseases or disorders for which such treatment would be helpful as discussed further herein. Such activity can be assessed using one or more assays, including, but not limited to, the ability to block phosphorylation of Smad2 and/or nuclear translocation of the NF- κ B p50 subunit, increase cell proliferation, decrease apoptosis and/or radiation-induced DNA damage, decrease inflammation and/or angiogenesis, promote healing of oral mucositis, surgical wounds, diabetic wounds, and/or wounds associated with chronic inflammation in mice. The nucleic acid may be derived from genomic DNA, i.e., cloned directly from the genome of a particular organism. However, in particular embodiments, the nucleic acid will comprise complementary dna (cdna). Also provided are cDNAs incorporating a native intron or an intron derived from another gene; such engineered molecules are sometimes referred to as "minigenes". At a minimum, these and other nucleic acids of the present technology can be used as molecular weight standards in, for example, gel electrophoresis.
The term "cDNA" is intended to refer to DNA prepared using messenger rna (mrna) as a template. The advantage of using cDNA, as opposed to genomic DNA or DNA polymerized from genomic, unprocessed or partially processed RNA templates, is that the cDNA contains predominantly the coding sequence for the corresponding protein. It may sometimes be preferable to have the genome sequence in whole or in part, such as where a non-coding region is required for optimal expression or where a non-coding region, such as an intron, is to be targeted in an antisense strategy.
The term "Smad 7-encoding nucleic acid" as used herein may refer to a nucleic acid molecule that has been isolated from total cellular nucleic acid and/or may refer to a cDNA encoding a Smad7 polypeptide. The term "isolated from total cellular nucleic acid" as used herein means that the nucleic acid molecule is about or at least about 75% pure, 80% pure, 85% pure, 90% pure, 95% pure, 96% pure, 97% pure, 98% pure, 99% pure, or 100% pure other cellular nucleic acid molecules as determined using standard biochemical techniques, such as (but not limited to) agarose gel electrophoresis. The term "isolated from total cellular protein" as used herein means that the protein molecule is about or at least about 75% pure, 80% pure, 85% pure, 90% pure, 95% pure, 96% pure, 97% pure, 98% pure, 99% pure, or 100% pure of other cellular nucleic acid molecules as determined using standard biochemical techniques, such as, but not limited to, western blots. In certain embodiments, the technology of the present invention relates to a polypeptide substantially as set forth in SEQ ID NO: 9. 21, 23, 24, 26, 28, 30, 32-34, 36, 38, 39, 87, 89, 91, 93, 96, 97, 99 and 100 and/or comprises any of SEQ ID NOs: 9. 21, 23, 24, 26, 28, 30, 32-34, 36, 38, 39, 87, 89, 91, 93, 96, 97, 99 and 100.
Isolated nucleic acid molecules can be produced using recombinant DNA techniques (e.g., Polymerase Chain Reaction (PCR) amplification, cloning) or chemical synthesis. Isolated nucleic acid molecules include natural nucleic acid molecules and homologs thereof, including, but not limited to, natural allelic variants and modified nucleic acid molecules in which nucleotides have been inserted, deleted, substituted, and/or inverted in a manner such that such modifications provide a desired effect (e.g., production of Smad7 protein in a non-human expression system).
The term "substantially as set forth in one or more nucleic acid sequences (e.g., SEQ ID NOS: 9-11, 21, 23-41)" means that the nucleic acid sequence substantially corresponds to at least a portion and in some cases all of one or more nucleic acid sequences (e.g., SEQ ID NOS: 9,21, 23, 24, 26, 28, 30, 32-34, 36, 38, 39, 87, 89, 91, 93, 96, 97, 99, and 100). In some embodiments, a sequence substantially corresponding to at least a portion of a nucleic acid sequence may correspond to about or at least about 50 nucleic acids, 75 nucleic acids, 150 nucleic acids, 200 nucleic acids, 250 nucleic acids, 300 nucleic acids, 350 nucleic acids, 400 nucleic acids, 450 nucleic acids, 500 nucleic acids, 550 nucleic acids, 600 nucleic acids, 650 nucleic acids, 700 nucleic acids, 750 nucleic acids, 800 nucleic acids, 900 nucleic acids, 1000 nucleic acids, 1100 nucleic acids, 1200 nucleic acids, or 1250 nucleic acids of one or more of the sequences described herein. In some embodiments, a sequence substantially corresponding to at least a portion of a nucleic acid sequence may correspond to about the following ranges: about 50-1250 nucleic acids, 75-1250 nucleic acids, 150-1250 nucleic acids, 200-1250 nucleic acids, 250-1250 nucleic acids, 300-1250 nucleic acids, 350-1250 nucleic acids, 400-1250 nucleic acids, 450-1250 nucleic acids, 500-1250 nucleic acids, 550-1250 nucleic acids, 600-1250 nucleic acids, 650-1250 nucleic acids, 700-1250 nucleic acids, 750-1250 nucleic acids, 800-1250 nucleic acids, 900-1250 nucleic acids, 1000-1250 nucleic acids, 1100-1250 nucleic acids, 1200-1250 nucleic acids, at least about 50-75 nucleic acids, 75-150 nucleic acids, 75-200 nucleic acids, 75-250 nucleic acids, 75-300 nucleic acids, 75-350 nucleic acids, 75-400 nucleic acids, 75-450 nucleic acids, 75-500 nucleic acids, 75-550 nucleic acids, 75-600 nucleic acids, 75-650 nucleic acids, 75-700 nucleic acids, 75-750 nucleic acids, 75-800 nucleic acids, 75-900 nucleic acids, 75-1000 nucleic acids, 75-1100 nucleic acids, 75-1200 nucleic acids, or 75-1250 nucleic acids or 1250 nucleic acids.
In some embodiments, a sequence substantially corresponding to at least a portion of a nucleic acid sequence includes a sequence identical to that portion of the nucleic acid sequence. In some embodiments, a sequence corresponding to substantially at least a portion of a nucleic acid sequence or all of a nucleic acid sequence may include one or more functionally equivalent codons. The term "functionally equivalent codon" is used herein to refer to one or more codons encoding the same amino acid, such as six codons for arginine or serine, and in some embodiments to codons encoding a biologically equivalent amino acid as discussed in the following page. The term "bioequivalent" amino acid is used herein to refer to one or more of the following: when changed from amino acids present in the amino acid sequence of a wild-type protein of human Smad7, one or more (or in some embodiments, any) of the biological activities of Smad7 described herein are not altered, such as, but not limited to, increasing proliferation, reducing or inhibiting cell death, reducing excessive inflammation, preventing DNA damage, and/or increasing cell migration, and treating or preventing one or more diseases or disorders for which such treatment as further discussed herein would be helpful.
In some embodiments that allow for degeneracy of the genetic code, sequences having about or at least about 60%, 70%, 80%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, and/or 99% nucleotides that are identical to the nucleotides of any of the codon-optimized nucleic acid sequences (e.g., SEQ ID NOS: 9-11, 21, 23-41) can be considered substantially corresponding nucleic acid sequences. Sequences substantially identical to those set forth in any of the nucleic acid sequences (e.g., SEQ ID NOS: 9-11, 21, 23-41) can also be defined functionally as being capable of hybridizing to a nucleic acid sequence comprising SEQ ID NO: 9-11, 21, 23-41, or a sequence that hybridizes to a nucleic acid segment of a complement.
For applications requiring high selectivity, it will generally be desirable to employ relatively high stringency conditions to form hybrids. For example, relatively low salt and/or high temperature conditions, such as provided by about 0.02M to about 0.10M NaCl at a temperature of about 50 ℃ to about 70 ℃. Such high stringency conditions tolerate little, if any, mismatch between the probe or primer and the template or target strand and would be particularly suitable for isolating a particular gene or detecting a particular mRNA transcript. It will generally be appreciated that conditions can be made more stringent by adding increasing amounts of formamide.
For certain applications, it will be appreciated that lower stringency conditions are preferred. Under these conditions, hybridization can occur even where the sequences of the hybridizing strands are not perfectly complementary, but are mismatched at one or more positions. Conditions can be made less stringent by increasing the salt concentration and/or decreasing the temperature. For example, medium stringency conditions can be provided by about 0.1 to 0.25M NaCl at a temperature of about 37 ℃ to about 55 ℃, while low stringency conditions can be provided by about 0.15M to about 0.9M salt at a temperature in the range of about 20 ℃ to about 55 ℃. Hybridization conditions can be readily manipulated depending on the desired results.
In other embodiments, hybridization can be at, e.g., 50mM Tris-HCl (pH 8.3), 75mM KCl, 3mM MgCl21.0mM dithiothreitol at a temperature of between about 20 ℃ and about 37 ℃. Other hybridization conditions utilized may include about 10mM Tris-HCl (pH 8.3), 50mM KCl, 1.5mM MgCl2At a temperature in the range of about 40 ℃ to about 72 ℃.
To determine the homology of two amino acid sequences or two nucleic acids, the sequences are aligned for optimal comparison purposes (e.g., gaps are introduced in the sequence of a first amino acid or nucleic acid sequence for optimal alignment with a second amino acid or nucleic acid sequence). The amino acid residues or nucleotides at corresponding amino acid positions or nucleotide positions can then be compared. When a position in the first sequence is occupied by the same amino acid residue or nucleotide as the corresponding position in the second sequence, then the molecules are identical at that position. The% homology between two sequences is a function of the number of identical positions shared by the sequences (identity% ═ number of identical positions/total number of positions (e.g., overlapping positions) × 100). In some embodiments, the two sequences are the same length.
To determine the% homology between two sequences, Karlin and Altschul (1990) proc.natl.acad.sci.usa 87: 2264. sup. 2268, as described in Karlin and Altschul (1993) Proc.Natl.Acad.Sci.USA 90: 5873 and 5877. Such an algorithm is incorporated into Altschul et al (1990) J.mol biol.215: 403-410 NBLAST and XBLAST programs. A BLAST nucleotide search was performed using the NBLAST program, score 100, word length 12 to obtain nucleotide sequences homologous to the nucleic acid molecules described or disclosed herein. BLAST protein searches were performed using the XBLAST program with a score of 50 and a word length of 3. To obtain gap alignments for comparison purposes, for example, Altschul et al (1997) Nucleic Acids Res.25: 3389 blank BLAST described in 3402. When utilizing BLAST and gapped BLAST programs, the default parameters of the corresponding programs (e.g., XBLAST and NBLAST) are used. For further details, see website of the National Center for biotechnology Information. Proteins suitable for use in the methods described herein also include the following: there are between 1 and 15 amino acid changes, e.g., 1,2, 3, 4, 5,6, 7, 8, 9, 10, 11, 12, 13, 14, or 15 amino acid substitutions, deletions, or additions compared to the amino acid sequence of any of the proteins described herein. In other embodiments, the altered amino acid sequence is at least 75% identical, e.g., 77%, 80%, 82%, 85%, 88%, 90%, 92%, 95%, 97%, 98%, 99%, or 100% identical to the amino acid sequence of any of the protein inhibitors described herein. Such sequence variant proteins are suitable for use in the methods described herein, so long as the altered amino acid sequence retains sufficient biological activity to function in the compositions and methods described herein. In some cases, conservative amino acid substitutions are utilized. Illustrative conservative substitutions among amino acids are within each of the following groups: (1) glycine, alanine, valine, leucine, and isoleucine, (2) phenylalanine, tyrosine, and tryptophan, (3) serine and threonine, (4) aspartic acid and glutamic acid, (5) glutamine and asparagine, and (6) lysine, arginine, and histidine. BLOSUM62 is represented by an amino acid substitution matrix derived from approximately 2,000 local multiple alignments of segments of protein sequences representing highly conserved regions of more than 500 groups of related proteins (Henikoff et al (1992), Proc. Natl Acad. Sci. USA, 89: 10915-. The frequency of BLOSUM62 substitutions may be used to define conservative amino acid substitutions that are, in some embodiments, introduced into the amino acid sequences described or disclosed herein. Although it is possible to design amino acid substitutions based solely on chemical properties (as discussed above), the language "conservative amino acid substitution" preferably refers to a substitution represented by a BLOSUM62 value greater than-1. For example, an amino acid substitution is conservative if it is characterized by a BLOSUM62 value of 0, 1,2, or 3. According to this system, preferred conservative amino acid substitutions are characterized by a BLOSUM62 value of at least 1 (e.g., 1,2, or 3), while more preferred conservative amino acid substitutions are characterized by a BLOSUM62 value of at least 2 (e.g., 2 or 3).
DNA segments of the present technology include those encoding biologically functional equivalent Smad7 proteins and peptides as described above. Such sequences may arise as a result of codon redundancy and amino acid functional equivalence that are known to occur naturally within nucleic acid sequences and proteins encoded thereby. Alternatively, functionally equivalent proteins or peptides can be created via the application of recombinant DNA techniques, wherein changes in protein structure can be engineered based on considerations of the identity of the amino acids being exchanged. As described elsewhere, the changes designed by humans can be introduced via the application of site-directed mutagenesis techniques or can be introduced randomly and screened later for the desired function.
As described in more detail below, Smad7 nucleic acid sequences have been optimized for expression in alternative host organisms (e.g., non-human). Although the genetic code is degenerate as described above, so frequently, one amino acid may be encoded by two or more nucleotide codons. Thus, multiple nucleic acid sequences may encode one amino acid sequence. Although this creates a uniform protein, the nucleic acids themselves are different and may have different properties. As described herein, one aspect of the choice of codon usage can be, but is not limited to, the ability to express a protein in a non-native cell (e.g., a human protein in bacteria or yeast), or the level of expression in such cells. Efficient protein expression in non-human systems is required in order to obtain sufficient protein for purification, testing and use in vitro assays, in animal models and ultimately in clinical development.
A series of 23 arginine amino acids in the sequence of human Smad7 protein encoded by one or more of AGG (1.7% codon usage; 9 residues), AGA (2.8% codon usage; 2 residues), CGA (3.5% codon usage; 4 residues), or CGG (5.4% codon usage; 8 residues) has been identified, and it has been determined that in order to have efficient protein expression from non-human sources, such as (but not limited to) bacteria and/or yeast, one or more, and potentially all, arginine codons should be modified to CGT (20.6% codon usage). Thus, in some embodiments, a Smad7 codon-optimized nucleic acid sequence includes at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, at least 10, at least 11, at least 12, at least 13, at least 14, at least 15, at least 16, at least 17, at least 18, at least 19, at least 20, at least 21, at least 22, or 23 arginine codons that have become CGT. In some embodiments, the Smad7 codon optimized nucleic acid sequence includes one or more or all arginine codons that become CGT at positions 7-9, 43-45, 169-171, 403-405, 490-492, 526-528, 823-825, 1057-1059, 16-18, 136-138, 199-201, 598-600, 31-33, 112-114, 316-318, 772-774, 940-942, 973-975, 1135-1137, 1276-1278, 637-639, or 814-816 of the nucleic acid sequence.
A series of 33 serine residues in the human Smad7 protein sequence encoded by TCC or TCG (9%) have been identified and it has been determined that it may be beneficial for efficient protein expression and purification from non-human sources such as, but not limited to, bacteria and/or yeast that one or more, and potentially all, serine codons are modified to AGC (15% codon usage). Thus, in some embodiments, a Smad7 codon-optimized nucleic acid sequence includes at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, at least 10, at least 11, at least 12, at least 13, at least 14, at least 15, at least 16, at least 17, at least 18, at least 19, at least 20, at least 21, at least 22, at least 23, at least 24, at least 25, at least 26, at least 27, at least 28, at least 29, at least 30, at least 31, at least 32, or 33 serine codons that have become (AGC). In some embodiments, the Smad7 codon optimized nucleic acid sequence includes one or more or all of the serine codons 1272, 133-. Among these, 23 codons (19-21, 292-.
A series of 12 histidine residues in the sequence of the human Smad7 protein encoded by CAC (9.6% codon usage) has also been identified, and it has been determined that it may be beneficial for efficient protein expression and purification from non-human sources such as (but not limited to) bacteria and/or yeast that one or more, and potentially all, serine codons are modified to CAT (optionally up to 12.6% usage). Thus, in some embodiments, a Smad7 codon-optimized nucleic acid sequence includes at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, at least 10, at least 11, or 12 histidine codons that have been changed to (CAT). In some embodiments, the Smad7 codon optimized nucleic acid sequence includes one or more or all of the serine codons at nucleic acid sequence positions 142-144, 214-216, 217-219, 220-222, 226-228, 289-291, 589-591, 778-780, 1072-1074, 1147-1149. Among these, 4 codons (nucleotides 217-219, 220-222, 589-591, 778-780) can be changed without introducing a potentially alternative open reading frame.
In some embodiments, the one or more codon-optimized nucleic acids may comprise one or more of at least one and any integer to 22 of its arginine codons modified to CGT, at least one and any integer to 28 of its serine codons modified to AGC (optionally capable of being modified by introduction of an open reading frame), or at least one and any integer to 12 of its histidine codons modified to CAT (optionally capable of being modified by introduction of an open reading frame). In some embodiments, the one or more codon-optimized nucleic acids may include at least one and any integer to 22 of its arginine codons modified to CGT, at least one and any integer to 28 of its serine codons modified to AGC (optionally capable of modification by introduction of an open reading frame), and at least one and any integer to 12 of its histidine codons modified to CAT (optionally capable of modification by introduction of an open reading frame). In some embodiments, the one or more codon-optimized nucleic acids may include 22 of its arginine codons modified to CGT, 28 of its serine codons modified to AGC (optionally capable of modification by introduction of an open reading frame), and 12 of its histidine codons modified to CAT (optionally capable of modification by introduction of an open reading frame). In some embodiments, one or more codon-optimized nucleic acids may also have nucleotide substitutions in the codon for Met216(ATG) to form the codon for Leu216 (CTG).
In some embodiments, one or more codon-optimized nucleic acids may have about 65% to 75%, about 65% to 68%, about 68% to 75%, or about 68% to 71% homology to a wild-type cDNA of human Smad7(SEQ ID NO: 12, 22). In some embodiments, one or more codon-optimized nucleic acids may have about 65%, 66%, 67%, 68%, 69%, 70%, 71%, 72%, 73%, 74%, or 75% homology to a wild-type cDNA of human Smad7(SEQ ID NO: 22, 88). In some embodiments, one or more codon-optimized nucleic acids may also have nucleotide substitutions in the codon for Met216(ATG) to form the codon for Leu216 (CTG).
Methionine codons (Met 216; ATG) have been identified that have the potential to be considered as alternative open reading frames by translation machinery, such as, but not limited to, bacteria or yeast. While not intending to be bound by theory, it is believed that the presence of the second potential open reading frame may reduce the expression of Smad7 protein. In some embodiments, one or more Smad7 nucleic acid sequences are modified at nucleotide position (646-.
It has also been found that various truncated forms and fragments of Smad7 protein retain one or more of the activities of full-length human Smad7, such as, but not limited to, increasing proliferation, reducing or inhibiting cell death, reducing excessive inflammation, preventing DNA damage and/or increasing cell migration, and treating or preventing one or more diseases or disorders for which such treatment as discussed further herein would be helpful. Such activity can be assessed using one or more assays, including, but not limited to, the ability to block phosphorylation of Smad2 and/or nuclear translocation of the NF- κ B p50 subunit, increase cell proliferation, decrease apoptosis and/or radiation-induced DNA damage, decrease inflammation and/or angiogenesis, promote healing of oral mucositis, surgical wounds, diabetic wounds, and/or wounds associated with chronic inflammation in mice.
Furthermore, in some embodiments, various truncated forms and fragments of Smad7 protein retain only a subset of one or more of the activities of full-length human Smad 7. For example, the C-terminal MH2 domain of Smad7 may primarily mediate the anti-inflammatory effects of Smad 7. Smad7 peptides having this anti-inflammatory function may be sufficient and optionally an improvement for the treatment of chronic inflammation-related conditions, such as (but not limited to) oral mucositis, stomatitis, arthritis, and psoriasis, among others. The N-terminal MH1 domain may primarily mediate cell migration and/or block TGF-beta induced growth arrest and/or fibrotic responses. Smad7 peptides with this cell migration and proliferation function may be sufficient and optionally an improvement for enhanced healing not associated with excessive inflammation. Types of wounds that may benefit from this form of treatment include, but are not limited to, surgical wounds, fibrotic scars, and diabetic wounds, defective healing, and/or scars, among others.
In some embodiments, the nucleic acid molecule (optionally, a codon-optimized nucleic acid molecule as described above and herein) encodes a fragment or truncated form of a Smad7 protein (optionally including Leu 216). In some embodiments, these fragments and/or truncated forms of Smad7 protein retain one or more or all of the activity of the full-length human Smad7 protein. In some embodiments, such truncated nucleic acid sequences encode the N-terminal portion of the Smad7 protein. In some embodiments, such truncated nucleic acid sequences encode the C-terminal portion of the Smad7 protein. In some embodiments, such truncated nucleic acid sequences (nucleotide positions 4-774) encode amino acids 2-258 of the human Smad7 protein. In some embodiments, such truncated nucleic acid sequences (nucleotide positions 775-1278) encode amino acids 259-426 of the human Smad7 protein. In some embodiments, such fragments of the nucleic acid sequence (nucleotide positions 610-774) encode amino acids 204-258 of the human Smad7 protein.
The term "truncated" as used herein with respect to a nucleic acid molecule refers to a molecule that contains a native N-terminal nucleotide sequence encoding the corresponding protein (with or without a cleaved leader sequence), but lacks one or more nucleotides from the C-terminal coding region of the molecule, or a molecule that contains a native C-terminal nucleotide sequence encoding the corresponding protein (with or without a cleaved leader sequence), but lacks one or more nucleotides from the N-terminal coding region of the molecule. In some embodiments, molecules lacking nucleotides encoding at least about 25, at least about 50, at least about 75, at least about 100, at least about 125, at least about 150, at least about 200, at least about 250, at least about 300, or at least about 350, or at least about 400 amino acids from one end or the other are specifically provided. Similarly, the term "truncated" may also be used in relation to protein molecules encoded by truncated nucleic acid molecules. In some embodiments, a "truncated" molecule is biologically active, having one or more of the Smad7 activities described herein (or encoding a polypeptide having these activities).
The term "fragment" as used herein with respect to a nucleic acid molecule refers to a molecule that contains contiguous residues of the full-length sequence, but lacks some of the 5 'and/or 3' sequence of the full-length sequence. In some embodiments, a "fragment" includes a portion of one or more of the full-length sequences described herein. In some embodiments, a "fragment" does not include sequences encoding the N-terminus or C-terminus, but only internal fragments. In some embodiments, a "fragment" encodes a biologically active polypeptide having one or more of the Smad7 activities described herein. In some embodiments, a nucleic acid fragment may encode a protein having at least about 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80,85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, 1000, 1050, 1100, 1150 amino acids. Similarly, "fragments" may also be used with respect to protein molecules encoded by Smad7 nucleic acid fragments.
The term "N-terminal portion" as used herein refers to a fragment of the corresponding protein that contains all of the sequence of the N-terminus of the protein, but lacks the C-terminus of internal residues.
The term "C-terminal portion" as used herein refers to a fragment of the corresponding protein that contains all of the sequence of the C-terminus of the protein, but lacks the N-terminus of internal residues.
While not intending to be bound by theory, it is generally believed that Smad7 protein activity is the result of interactions in the cytoplasm and nucleus of the cell. For that reason in particular, Smad7 protein is generally believed not to be a candidate for therapeutic effect. However, it was decided to pursue the development of Smad7 as a protein therapeutic, and to modify the Smad7 nucleic acid sequence to encode a Protein Transduction Domain (PTD) in frame with a Smad7 nucleic acid sequence (e.g., optionally any of the nucleic acid sequences described herein that encode the Smad7 protein, including human wild-type and codon-optimized sequences, full-length and biologically active fragments or truncated portions). In some embodiments, the PTD is located at the 3 'terminus of the Smad7 nucleic acid sequence, and in some embodiments, the PTD is located at the 5' terminus of the Smad7 nucleic acid sequence. In some embodiments, there is a linker sequence encoding 1,2, 3, 4, 5, or 6 amino acids linking the PTD and Smad7 nucleic acid sequences.
In some embodiments, the PTD nucleic acid sequence is a Tat nucleic acid sequence. Ggccgtaaaaaacgccgtcaacgccgccgt (SEQ ID NO: 1) encoding GRKKRRQRRR (SEQ ID NO: 2), tatggccgtaaaaaacgccgtcaacgccgccgt (SEQ ID NO: 3) encoding YGRKKRRQRRR (SEQ ID NO: 4), or ggccgtaaaaaacgccgtcaa (SEQ ID NO: 5) encoding GRKKRRQ (SEQ ID NO: 6).
In some embodiments, the nucleic acid sequence further comprises a nucleotide sequence encoding one or more of an epitope tag or a purification tag. In some embodiments, the epitope tag is V5. In some embodiments, the purification tag is one or more of glutathione-S-transferase (GST) or 6-histidine (H6) (SEQ ID NO: 40).
The term "epitope tag" as used herein with respect to a nucleic acid molecule refers to a nucleotide encoding a peptide sequence that is recognized and bound by the variable region of an antibody or fragment. In some embodiments, the epitope tag is not part of the native protein. In some embodiments, the epitope tag is removable. In some embodiments, the epitope tag is not inherent to the native biological activity of the protein. Examples of epitope tags include, but are not limited to, V5.
The term "purification tag" as used herein with respect to a nucleic acid molecule refers to a nucleotide that encodes a peptide sequence that facilitates purification of a protein, but is not generally necessary for the biological activity of the protein. In some embodiments, the purification tag may be removed after protein purification. Examples of purification tags include, but are not limited to, GST and H-6(SEQ ID NO: 40).
2. Vectors for cloning, gene transfer and expression
In certain embodiments, the Smad7 polypeptide product is expressed using an expression vector, and this product can then be purified for various uses. In other embodiments, the expression vector is used in gene therapy. Expression requires the provision of appropriate signals in vectors, and these vectors include various regulatory elements (such as enhancers/promoters) from viral and mammalian sources that drive expression of the gene of interest in the host cell. Elements designed to optimize messenger RNA stability and translatability in host cells are also defined. Also provided are conditions for use of a plurality of dominant drug selection markers for establishing permanent stable cell clones expressing the product, and elements linking expression of the drug selection markers to expression of the polypeptide.
Throughout this application, the term "expression construct" is intended to include any type of genetic construct that contains a nucleic acid that encodes a gene product in which a portion or all of the nucleic acid coding sequence is capable of being transcribed. The transcript may be translated into a protein, but need not be. In certain embodiments, expression comprises gene transcription and translation of mRNA into a gene product. In other embodiments, expression includes transcription of only the nucleic acid encoding the gene of interest.
The term "vector" is used to refer to a cargo nucleic acid molecule into which a nucleic acid sequence can be inserted for introduction into a cell that can replicate this nucleic acid sequence. The nucleic acid sequence may be "foreign," meaning that it is foreign to the cell into which the vector is introduced, or that the sequence is homologous to sequences in the cell but in a position within the host cell nucleic acid where it is not normally found. Vectors include plasmids, cosmids, viruses (phage, animal, and plant viruses), and artificial chromosomes (e.g., YACs). Those skilled in the art will be well equipped to construct vectors via standard recombinant techniques described, for example, in Sambrook et al, Molecular Cloning (Cold Spring Harbor Lab Press, 1989) and Ausubel et al, Current Protocols in Molecular Biology (Wiley, 1994), both of which are incorporated herein by reference.
The term "expression vector" refers to a vector containing a nucleic acid sequence encoding at least a portion of a gene product capable of being transcribed. In some cases, the RNA molecule is then translated into a protein, polypeptide, or peptide. In other cases, for example, in the production of antisense molecules or ribozymes, these sequences are not translated. Expression vectors may contain a variety of "control sequences," which refer to nucleic acid sequences, including promoters and enhancers, necessary for the transcription and possibly translation of an operably linked coding sequence in a particular host organism. In addition to control sequences that govern transcription and translation, vectors and expression vectors may contain nucleic acid sequences that serve other functions, such as transcription termination signals and polyadenylation sites.
The ability of certain viral vectors to efficiently infect or enter cells for integration into the host cell genome and stable expression of viral genes has prompted the development and application of many different viral vector systems. Robbins et al, Pharmacol, Ther.80: 35-47(1998). Viral systems are currently used as vectors for ex vivo and in vivo gene transfer. For example, adenoviruses, herpes simplex viruses, lentiviruses, retroviruses, and adeno-associated viruses are currently being evaluated for the treatment of diseases such as cancer, cystic fibrosis, Gaucher disease, kidney disease, and arthritis. Robbins et al, Pharmacol, Ther.80: 35-47 (1998); imai et al, neuroprogeries 19: 379-402 (1998); U.S. Pat. No. 5,670,488. Depending on the particular gene therapy application, various viral vectors exhibit particular advantages and disadvantages.
Non-viral methods of nucleic acid delivery suitable for use in transformation of organelles, cells, tissues, or organisms that are believed to be useful in the present techniques include virtually any method by which a nucleic acid (e.g., DNA) can be introduced into an organelle, cell, tissue, or organism, as described herein or as will be known to one of ordinary skill in the art. Such methods include, but are not limited to, direct delivery of DNA, such as by injection (U.S. Pat. nos. 5,994,624, 5,981,274, 5,945,100, 5,780,448, 5,736,524, 5,702,932, 5,656,610, 5,589,466, and 5,580,859, each of which is incorporated herein by reference), including microinjection (Harland and Weintraub, 1985; U.S. Pat. No. 5,789,215, incorporated herein by reference); by electroporation (U.S. Pat. No. 5,384,253, incorporated herein by reference); by calcium phosphate precipitation (Graham et al, Virology 52: 456-467 (1973); Chen et al, mol.cell biol.7: 2745-2752 (1987); Rippe et al, mol.cell biol.10: 689-695 (1990)); by using DEAE-dextran followed by polyethylene glycol (Gopal, mol. cell biol. 5: 1188-1190 (1985)); by direct acoustic loading (Fechheimer et al, PNAS 84: 8463-; by liposome-mediated transfection (Nicolau et al, Biochim. Biophys. acta 721: 185-190 (1982); Fraley et al, PNAS 76: 3348-3352 (1979); Nicolau et al, Methods enzymol.149: 157-176 (1987); Wong et al, Gene 10: 87-94 (1980); Kaneda et al, J.biol.chem.264: 12126-12129 (1989); Kato et al, J.biol.chem.266: 3361-3364 (1991)); by microprojectile bombardment (PCT application Nos. WO 94/09699 and 95/06128; U.S. Pat. Nos. 5,610,042, 5,322,783, 5,563,055, 5,550,318, 5,538,877, and 5,538,880, each of which is incorporated herein by reference); by agitation with silicon carbide fibers (Kaeppler et al, plant cell Rep.9: 415-418 (1990); U.S. Pat. Nos. 5,302,523 and 5,464,765, each incorporated herein by reference); or by PEG-mediated protoplast transformation (Omirulleh et al, Plant mol. biol. 21: 415-428 (1993); U.S. Pat. Nos. 4,684,611 and 4,952,500, each incorporated herein by reference); DNA uptake mediated by drying/inhibition (Potrykus et al, mol. Gen. Genet. 199: 169-177 (1985)). By applying such techniques, organelles, cells, tissues, or organisms can be stably or transiently transformed.
3. Expression system
There are numerous expression systems that include at least a portion or all of the compositions discussed above. Prokaryote-and/or eukaryote-based systems can be employed in the present technology to produce nucleic acid sequences or their cognate polypeptides, proteins, and peptides. Many such systems are commercially and widely available.
Insect cell/baculovirus systems can produce high levels of protein expression of heterologous nucleic acid segments, such as described in U.S. Pat. Nos. 5,871,986 and 4,879,236, both of which are incorporated herein by reference, and such systems can be, for example, by name 2.0 from
2.0 from And BacpackTMBaculovirus expression system from
And BacpackTMBaculovirus expression system from And (6) obtaining.
And (6) obtaining.
Other examples of expression systems include COMPLETE CONTROL (C)TMAn inducible mammalian expression system, which relates to the synthetic ecdysone-inducible receptor or its pET expression system, an E.coli expression system. Another example of an inducible expression system is available from
COMPLETE CONTROL (C)TMAn inducible mammalian expression system, which relates to the synthetic ecdysone-inducible receptor or its pET expression system, an E.coli expression system. Another example of an inducible expression system is available from It carries T-REXTM(tetracycline regulated expression) system, an inducible mammalian expression system using the full-length CMV promoter.
It carries T-REXTM(tetracycline regulated expression) system, an inducible mammalian expression system using the full-length CMV promoter. Also provided are yeast expression systems, referred to as Pichia methanolica (Pichia methanolica) expression systems, designed for high level production of recombinant proteins in the methylotrophic yeast Pichia methanolica. One skilled in the art will know how to express a vector, such as an expression construct, to produce a nucleic acid sequence or its cognate polypeptide, protein, or peptide.
Also provided are yeast expression systems, referred to as Pichia methanolica (Pichia methanolica) expression systems, designed for high level production of recombinant proteins in the methylotrophic yeast Pichia methanolica. One skilled in the art will know how to express a vector, such as an expression construct, to produce a nucleic acid sequence or its cognate polypeptide, protein, or peptide.
The primary mammalian cell culture can be prepared in various ways. In order for the cells to remain viable in vitro and when contacted with the expression construct, it must be ensured that the cells remain in contact with the correct ratio of oxygen and carbon dioxide and nutrients, but are protected from microbial contamination. Cell culture techniques are well documented.
One embodiment of the foregoing relates to the use of gene transfer to immortalize cells for the production of proteins. The gene for the protein of interest can be transferred into an appropriate host cell as described above, followed by culturing the cell under appropriate conditions. Genes for almost any polypeptide can be used in this manner. The production of recombinant expression vectors and the elements included therein are discussed above. Alternatively, the protein to be produced may be an endogenous protein which is normally synthesized by the cell in question.
Examples of suitable mammalian host cell lines are Vero and HeLa cells and Chinese hamster ovary cell lines, W138, BHK, COS-7, 293, HepG2, NIH3T3, RIN and MDCK cells. In addition, host cell strains may be selected which regulate the expression of the inserted sequences or modify and process the gene product in a desired manner. Such modifications (e.g., glycosylation) and processing (e.g., cleavage) of the protein product may be important for the function of the protein. Different host cells have the characteristics and specific mechanisms of post-translational processing and modification of proteins. Appropriate cell lines or host systems may be selected to ensure proper modification and processing of the expressed foreign protein.
Many selection systems can be used, including but not limited to HSV thymidine kinase, hypoxanthine-guanine phosphoribosyl transferase, and adenine phosphoribosyl transferase genes, in tk cells, hgprt cells, or aprt cells, respectively. Furthermore, antimetabolite resistance can be used as a basis for the selection of: dhfr conferring resistance; gpt conferring resistance to mycophenolic acid; neo conferring resistance to the aminoglycoside G418; and hygro, which confers resistance to hygromycin.
The terms "cell," "cell line," and "cell culture" as used herein may be used interchangeably. All of these terms also include their progeny, i.e., any and all progeny. It will be appreciated that all progeny may not be identical due to deliberate or inadvertent mutations. In the context of expressing a heterologous nucleic acid sequence, a "host cell" refers to a prokaryotic or eukaryotic cell, and includes any transformable organism capable of replicating a vector and/or expressing a heterologous gene encoded by the vector. Host cells can and have been used as recipients of vectors. A host cell can be "transfected" or "transformed," which refers to the process of transferring or introducing an exogenous nucleic acid into the host cell. Transformed cells include the primary subject cell and its progeny.
The host cell may be of prokaryotic or eukaryotic origin (e.g., bacterial or yeast), depending on whether the desired result is replication of the vector or expression of a portion or all of the nucleic acid sequence encoded by the vector. Numerous cell lines and cultures are available for use as host cells, and are available via the American Type Culture Collection (ATCC), an organization that serves as an archive for live cultures and genetic material (ATCC. Suitable hosts can be determined by those skilled in the art based on the vector backbone and the desired results. For example, plasmids or cosmids can be introduced into prokaryotic host cells for the replication of many vectors. Bacterial cells useful as host cells for vector replication and/or expression include DH5 alpha, JM109, and KC8, as well as a number of commercially available bacterial hosts, such as Competent cells and SolopackTMGold cell (A)La Jolla). Alternatively, bacterial cells such as E.coli LE392 can be used as host cells for phage viruses.
Competent cells and SolopackTMGold cell (A)La Jolla). Alternatively, bacterial cells such as E.coli LE392 can be used as host cells for phage viruses.
Examples of eukaryotic host cells for replication and/or expression of the vector include HeLa, NIH3T3, Jurkat, 293, Cos, CHO, Saos and PC 12. Many host cells from a variety of cell types and organisms are available and will be known to those skilled in the art. Similarly, viral vectors may be used in conjunction with eukaryotic or prokaryotic host cells, particularly host cells that allow for replication or expression of the vector.
Some vectors may employ control sequences that allow them to replicate and/or be expressed in prokaryotic and eukaryotic cells. One skilled in the art will further understand the conditions under which all of the above-described host cells are incubated to maintain them and allow the vector to replicate. Techniques and conditions are also known and known that will allow large scale production of vectors, as well as the production of nucleic acids encoded by the vectors and their homologous polypeptides, proteins or peptides.
Smad7 proteins and protein fragments
Maternal anti-pentadecaplegic homolog 7(mothers against decapentaplegic homolog 7, Smad7) was previously identified as an antagonist of TGF- β signaling via several mechanisms including: (a) blockade of TGF- β receptor mediated signaling Smad phosphorylation and nuclear translocation; (b) TGF- β receptor and signaling Smad degradation increased via specific ubiquitin-proteasome pathways; and (c) inhibition of signaling Smad for its binding to a Smad Binding Element (SBE). Smad7 also antagonizes other signaling pathways, such as the NF-. kappa.B pathway.
The Smad7 protein is encoded by the Smad7 gene discussed above. Smad7 is involved in cell signaling as are many other TGF- β family members. Which are TGF-beta type 1 receptor antagonists. It blocks TGF- β 1 and receptor-associated activin (activin), thereby blocking access to Smad 2. It is inhibitory Smad (I-Smad) and is enhanced by SMURF 2. Smad7 also enhances muscle differentiation.
The terms "polypeptide," "peptide," and "protein" are used interchangeably herein to refer to a polymer of amino acid residues. These terms apply to naturally occurring amino acid polymers as well as amino acid polymers in which one or more amino acid residues are a non-naturally occurring amino acid (e.g., an amino acid analog). These terms as used herein encompass amino acid chains of any length, including full length proteins, in which the amino acid residues are linked by covalent peptide bonds.
In one embodiment, the present technology relates to Smad7 protein compositions. In addition to the entire Smad7 molecule, the present technology relates to truncated portions and fragments of polypeptides that retain one or more activities associated with Smad7, such as (but not limited to) increasing proliferation, reducing or inhibiting cell death, reducing excessive inflammation, preventing DNA damage, and/or increasing cell migration, and treating or preventing one or more diseases or disorders for which such treatment would be helpful as discussed further herein. Such activity can be assessed using one or more assays, including, but not limited to, the ability to block phosphorylation of Smad2 and/or nuclear translocation of the NF- κ B p50 subunit, increase cell proliferation, decrease apoptosis and/or radiation-induced DNA damage, decrease inflammation and/or angiogenesis, promote healing of oral mucositis, surgical wounds, diabetic wounds, and/or wounds associated with chronic inflammation in mice.
Protein fragments can be generated by genetic engineering (discussed below) of translation termination sites within the coding region. Alternatively, treatment of Smad7 molecules with proteolytic enzymes (known as proteases) can produce a variety of N-terminal, C-terminal, and internal fragments. These fragments can be purified according to known methods, such as precipitation (e.g., ammonium sulfate), HPLC, ion exchange chromatography, affinity chromatography (including immunoaffinity chromatography), or various size separations (sedimentation, gel electrophoresis, gel filtration).
As used herein, reference to an isolated protein or polypeptide in embodiments of the invention includes a full-length protein, a fusion protein, a chimeric protein, or any fragment (truncated form, portion) or homolog of such protein. More specifically, an isolated protein may be a protein (including a polypeptide or peptide) that has been removed from its natural environment (i.e., has been subjected to human manipulation), and may include, but is not limited to, purified proteins, partially purified proteins, recombinantly produced proteins, proteins complexed with lipids, soluble proteins, synthetically produced proteins, and isolated proteins related to other proteins. As such, "isolated" does not reflect the extent to which the protein has been purified. Preferably, the isolated protein is recombinantly produced.
Also provided are variants of Smad 7-these may be substituted, inserted or deleted variants. Deletion variants lack one or more residues of the native protein that are not essential for activity, including truncation mutants described above and herein. Substitution variants typically contain exchanges of one amino acid for another at one or more sites within the protein, and may be designed to modulate one or more properties of the polypeptide, such as stability to proteolytic cleavage and/or translation and/or transcription (protein expression), without loss of other function or property. Substitutions of this kind are preferably conservative, i.e. one amino acid is replaced by an amino acid of similar shape and charge. Conservative substitutions are well known in the art and include, for example, the replacement or substitution of each amino acid with a different amino acid. In making substitution variants, hydropathic index, hydrophilicity, charge, and size are generally considered.
Specifically contemplated deletion variants of Smad7 include truncations and fragments, e.g., including polypeptide molecules having an N-terminal sequence without a C-terminal sequence, having a C-terminal sequence without an N-terminal sequence, or having internal sequences without an N-terminal or C-terminal sequence. Specifically contemplated truncations or fragments of Smad7 polypeptides include, but are not limited to, molecules comprising amino acid residues 2-258, 259-426, 204-258 corresponding to the native human Smad7 protein sequence.
The term "truncated" as used herein with respect to a protein sequence refers to a molecule that contains the native N-terminus of the corresponding protein (with or without a cleaved leader sequence), but lacks one or more amino acids from the C-terminus of the molecule, or a molecule that contains the native C-terminus of the corresponding protein (with or without a cleaved leader sequence), but lacks one or more amino acids from the N-terminus of the molecule. In some embodiments, molecules lacking at least about 25, at least about 50, at least about 75, at least about 100, at least about 125, at least about 150, at least about 200, at least about 250, at least about 300, or at least about 350, or at least about 400 amino acids from one end or the other are specifically provided. In some embodiments, a "truncated" molecule is biologically active, having one or more of the Smad7 activities described herein.
The term "fragment" as used herein with respect to a polypeptide sequence refers to a molecule that contains contiguous residues of the full-length sequence, but lacks some of the N-terminal and/or C-terminal residues of the full-length sequence. In some embodiments, a "fragment" includes a portion of one or more of the full-length sequences described herein. In some embodiments, a "fragment" does not include sequences encoding the N-terminus or C-terminus, but only internal fragments. In some embodiments, a "fragment" encodes a biologically active polypeptide having one or more of the Smad7 activities described herein. In some embodiments, a polypeptide fragment encodes at least about 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80,85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, 1000, 1050, 1100, 1150 amino acids.
One particular class of variants is fusion proteins. Such molecules typically have all or a substantial portion of the native molecule attached at the N-or C-terminus to all or a portion of the second polypeptide. However, in some embodiments, the fusion protein may include any of the fragments and/or truncated (N-terminal, C-terminal) Smad7 proteins described throughout the present disclosure. For example, fusion may employ leader sequences from other species to allow recombinant expression of the protein in a heterologous host. Another useful fusion includes the addition of optional functionally active domains such as, but not limited to, antibody epitopes and/or purification tags (e.g., V5: GKPIPNPLLGLDST (SEQ ID NO: 41); Flag: KYKDDDDK (SEQ ID NO: 42); HA: YPYDVPDYA (SEQ ID NO: 43)). Another type of fusion involves attaching a domain that can serve as a target for activating or inactivating the ligand, allowing control of the function of the fusion protein upon delivery to a subject. Such domains include, for example, steroid ligand binding (e.g., ER, PR, GR), which may be activated by small molecules, such as 4-hydroxyttamoxifen (4-hydroxytamoxifen) or RU486, which are uniquely capable of activating those steroid ligand binding domains and/or do not exist in nature and would therefore be able to fully control Smad7 function through the presence of these small molecules.
Another particular form of fusion protein that has particular utility in the present technology is a fusion comprising a Protein Transduction Domain (PTD), also known as a cell delivery domain or cell transduction domain. Such domains have been described in the art and are generally characterized as short amphiphilic or cationic peptides and peptide derivatives, often containing multiple lysine and arginine residues (Fischer, med. res. rev. 27: 755-795 (2007)). In some embodiments, the PTD is one or more variants of the TAT protein from HIV (GRKKRRQRRR (SEQ ID NO: 2), YGRKKRRQRRR (SEQ ID NO: 4), or GRKKRRQ (SEQ ID NO: 6)) or alternatively HSV VP 16. Alternative forms of Tat may be used. In some embodiments, a linker may be used to link one or more PTDs and SMad 7. In some embodiments, the PTD (optionally Tat) is fused or linked in frame to the N-terminus and/or C-terminus of any of the Smad7 full-length, fragment, and/or truncated (N-terminus, C-terminus) proteins described throughout the present disclosure. Other examples of PTDs provided by the present technology are shown in table 1.


In certain embodiments, the present technology provides sequence variants of Smad7 in which one or more residues have been altered. For example, in one embodiment, the methionine residue found at position 216 of the human Smad7 sequence is modified to a leucine residue (ATG to CTG).
C. Method of treatment
Smad 7-treatable diseases and conditions may include those that include one or more of the following: reduced cell proliferation, reduced cell migration, increased cell death, excessive inflammation, and/or DNA damage. Smad 7-associated diseases and disorders may include those that would be therapeutically helpful with Smad7 proteins and biologically active fragments and derivatives thereof having one or more of the following activities: including, but not limited to, increasing proliferation, reducing or inhibiting cell death, reducing excessive inflammation, preventing DNA damage, and/or increasing cell migration. Such diseases and/or conditions may include, but are not limited to, acute (e.g., via surgery, fight, trauma) and chronic wounds (e.g., ulcers, such as diabetic, pressure, venous), scars, fibrosis and abnormal healing, mucositis (e.g., oral and/or gastrointestinal), stomatitis, proctitis, autoimmune diseases (e.g., psoriasis, arthritis), and cancer.
In some embodiments, one or more of the diseases and/or disorders described herein can be prevented, treated, and/or ameliorated by providing to a subject in need of such treatment a therapeutically effective amount of one or more of a Smad7 protein (e.g., a full-length or biologically active truncation (e.g., N-or C-terminal) or fragment thereof) described in the present disclosure. In some embodiments, the one or more Smad7 proteins are fusion proteins comprising a PTD domain. In some embodiments, the one or more Smad7 proteins include Leu 216. In some embodiments, the Smad7 protein forms part of a pharmaceutical composition that includes one or more pharmaceutically acceptable excipients.
In some embodiments, one or more of the diseases and/or disorders described herein can be prevented, treated, and/or ameliorated by providing to a subject in need of such treatment a therapeutically effective amount of one or more of a nucleic acid molecule encoding one or more Smad7 proteins (e.g., a full-length or biologically active truncation (e.g., N-or C-terminal) or fragment thereof described in this disclosure). In some embodiments, the one or more nucleic acid molecules comprise a codon optimized nucleotide sequence and/or a sequence encoding Leu 216. In some embodiments, one or more Smad7 nucleic acid molecules are provided to a subject in a construct that includes an expression vector. In some embodiments, the Smad7 nucleic acid molecule (optionally, part of an expression vector) forms part of a pharmaceutical composition that includes one or more pharmaceutically acceptable excipients.
The term "subject" or "patient" as used herein refers to a human or non-human animal in need of treatment and/or prevention using one or more of the treatments described herein. In some embodiments, non-human animals include experimental animals such as monkeys, mice, rats, and rabbits; domestic pets such as dogs and cats; and livestock such as cattle, horses, pigs, goats, and sheep.
1. Chronic wound
A chronic wound is a wound that does not heal in an orderly set of stages and in a predictable amount of time in the manner that most wounds heal; wounds that do not heal within three months are often considered chronic. Chronic wounds appear to be retained during one or more wound healing periods. For example, chronic wounds often remain too long in the inflammatory phase. In acute wounds, there is a precise balance between the production and degradation of molecules such as collagen; in chronic wounds, this balance is lost and degradation plays a major role.
As described in more detail elsewhere herein, PTD-Smad7 has been shown to enhance wound healing in mouse skin and mucosal models. The application of PTD-Smad7 was effective via a topical route, which was required for wound treatment. While not intending to be bound by theory, it is believed that PTD-Smad7 may be used to treat or ameliorate chronic wounds via a variety of routes, and may include, inter alia, one or more of: decreasing inflammation, increasing cell proliferation (e.g., keratinocytes), increasing cell migration (e.g., keratinocytes), or decreasing fibrosis (e.g., via regulation of collagen).
Chronic wounds may never heal or may take years to heal. These wounds cause severe emotional and physical stress to the patient and a significant financial burden on the patient and the overall health care system. Acute and chronic wounds are on opposite ends of a series of wound healing types that progress toward healing at different rates. The vast majority of chronic wounds can be divided into three categories: venous ulcers, diabetic and pressure ulcers. A small number of wounds that do not fall into these categories may be due to causes such as radiation poisoning or ischemia.
Venous ulcers, which typically appear in the legs, account for about 70% to 90% of chronic wounds and affect mainly the elderly. It is believed to be venous hypertension due to improper function of the valve present in the vein to prevent backflow of blood. Ischemia is caused by dysfunction and, in combination with reperfusion injury, causes tissue damage that creates the wound.
Diabetic ulcers another major cause of chronic wounds, diabetes, is becoming increasingly prevalent. The risk of amputation due to chronic ulcers in diabetic patients is 15% higher than in the general population. Diabetes results in neuropathy, which inhibits nociception and pain perception. Thus, the patient may not initially be aware of small wounds in the legs and feet, and may therefore be unable to prevent infection or repeated injury. In addition, diabetes causes immune damage and small blood vessel damage, preventing adequate oxygenation of the tissue, which can cause chronic wounds. Pressure also plays a role in the formation of diabetic ulcers.
Another major type of chronic wound is a pressure ulcer, which commonly occurs in people with pathologies (such as paralysis) that inhibit movement of body parts that are typically subject to pressure (such as the heel, shoulder blades, and sacrum). Pressure ulcers are caused by ischemia, which occurs when the pressure on the tissue is greater than the pressure in the capillaries and thus restricts the flow of blood into this area. Muscle tissue that requires more oxygen and nutrients than skin shows the worst effects due to long-term stress. As in other chronic ulcers, reperfusion injury damages the tissue.
A chronic wound may affect only the epidermis and dermis, or it may affect tissue up to the fascia. It may have previously developed from the same thing that caused the acute wound, such as surgery or accidental trauma, or it may have developed as a result of a systemic infection, vascular, immune or neurological insufficiency, or co-morbidities such as neoplasia or metabolic disorders. While not intending to be bound by theory, the reason that a wound becomes chronic is that the body's ability to manage the injury is overwhelmed by the following factors: such as repeated trauma, sustained pressure, ischemia, or disease. Some of the major factors that cause chronic wounds include, but are not limited to, ischemia, reperfusion injury, and bacterial colonization.
Ischemia is an important factor in the formation and maintenance of wounds, especially when it occurs repeatedly (as usual) or when combined with the advanced age of the patient. Ischemia causes tissues to become inflamed and cells release factors that attract neutrophils, such as interleukins, chemokines, leukotrienes, and complement factors.
While it is resistant to pathogens, neutrophils also release inflammatory cytokines and enzymes that destroy cells. One of its important functions is the production of Reactive Oxygen Species (ROS) to kill bacteria, for which purpose it uses an enzyme called myeloperoxidase. Enzymes and ROS produced by neutrophils and other leukocytes destroy cells and prevent cell proliferation and wound closure by destroying DNA, lipids, proteins, ECM, and cytokines that accelerate healing. Neutrophils remain longer in chronic wounds than in acute wounds and contribute to the fact that chronic wounds have higher levels of inflammatory cytokines and ROS. Because wound fluid from chronic wounds has excess proteases and ROS, the fluid itself can inhibit healing by inhibiting cell growth and breaking down growth factors and proteins in the ECM.
Patients with insufficient tissue oxygenation, such as those undergoing hypothermia during surgery, are at a higher risk of infection because more oxygen in the wound environment allows leukocytes to generate ROS to kill the bacteria. The host's immune response to the presence of bacteria prolongs inflammation, delays healing, and destroys tissue. Infection can lead not only to chronic wounds, but also to gangrene, loss of infected limbs, and death of the patient.
As with ischemia, bacterial colonization and infection destroys tissue by allowing a greater number of neutrophils to enter the wound site. In patients with chronic wounds, bacteria that are resistant to antibiotics may have time to develop. In addition, patients carrying drug resistant bacterial strains, such as methicillin-resistant staphylococcus aureus (MRSA), have more chronic wounds.
Chronic wounds are also compositionally different from acute wounds in that their proteolytic enzymes, such as elastase and Matrix Metalloproteinase (MMP), are present at higher levels, while their growth factors, such as platelet-derived growth factor and keratinocyte growth factor, are present at lower concentrations.
Since Growth Factor (GF) is essential in timely wound healing, insufficient GF levels may be an important factor in chronic wound formation. In chronic wounds, the formation and release of growth factors may be hindered, these factors may be sequestered and unable to carry out their metabolic actions, or excessively degraded by cellular or bacterial proteases.
Chronic wounds such as diabetic and venous ulcers are also caused by the inability of fibroblasts to produce sufficient ECM proteins and the epithelialization of the wound by keratinocytes. Fibroblast gene expression in chronic wounds is different from that in acute wounds.
While all wounds require certain levels of elastase and protease for proper healing, too high a concentration is destructive. Leukocytes in the wound area release elastase, which increases inflammation, destroys tissue, proteoglycans, and collagen, and destroys growth factors, fibronectin, and factors that inhibit proteases. Elastase activity is increased by human serum albumin, which is the most abundant protein found in chronic wounds. However, chronic wounds with insufficient albumin are particularly unlikely to heal, and therefore the level of that protein which regulates the wound may prove helpful in the future for healing chronic wounds.
Excess matrix metalloproteases released by leukocytes may also make the wound chronic. MMPs break down ECM molecules, growth factors, and protease inhibitors, and thus increase degradation while decreasing building, thereby balancing the delicate tradeoff between production and degradation.
Mouth ulcers (also known as canker sores or mucosal ulcers) are ulcers that appear on the mucosa of the mouth. More simply stated, mouth ulcers are sores or open lesions in the mouth. Mouth ulcers are very common, associated with many diseases and occur through many different mechanisms, but there are usually no serious underlying causes. The two most common causes of canker sores are local trauma (e.g., due to sharp edge rubbing on pie fillings) and aphthous stomatitis ("aphtha"), a condition characterized by recurrent formation of canker sores for a number of unknown reasons. Some consider ulcers on the skin on the lips or around the mouth to be included under the general term mouth ulcer (e.g., an ulcer left by the rupture of a blister caused by a cold sore, a cold sore). Mouth ulcers often cause pain and discomfort, and may alter a person's choice of food (e.g., avoid sour or spicy foods and beverages) while healing occurs. Mouth ulcers may appear singly or multiple ulcers may appear simultaneously (a "batch" of ulcers). Once formed, the ulcer can be maintained by inflammation and/or secondary infection. Rarely, canker sores that do not heal for many weeks may be a sign of oral cancer. Other causes include burns, chemical injury, or infection.
Mucosal ulcers are ulcers that appear specifically on the mucosa. Ulcers are tissue defects that have penetrated the epithelial-connective tissue boundary with the deep basement in the submucosa or even within the muscle or periosteum. Ulcers are deeper epithelial breaches than erosions or desquamation and involve damage to the epithelium and lamina propria. Erosion is a superficial breach of the epithelium with little damage to the lamina propria of the deep layers. Mucosal erosion is erosion that occurs specifically on the mucosa. Only the superficial epithelial cells of the epidermis or mucosa are lost and the lesions can reach the depth of the basement membrane. The erosion heals without scarring. Desquamation is a term sometimes used to describe epithelial tears that are deeper than erosions but shallower than ulcers. This type of lesion is tangent to the spiked process and shows punctate (small needle point) bleeding caused by the exposed capillary circuit.
2. Acute wound/trauma
Physical trauma is severe and alters physical damage of the body, such as limb removal. Blunt force trauma is the type of physical trauma resulting from an impact or other force applied from or with a blunt object, while penetrating trauma is the type of physical trauma in which skin or tissue is penetrated by an object. The wound may also be described as unplanned, such as unexpected, or planned in the case of surgery. Both can be characterized as mild to severe tissue damage, blood loss, and/or shock, and both can lead to subsequent infections, including sepsis. The present technology provides for the treatment of wounds, including pretreatment (in the case of medical procedures) and treatment after traumatic injury has occurred.
As described in more detail elsewhere herein (and briefly mentioned above), PTD-Smad7 has been shown to enhance wound healing in mouse skin and mucosal models. The application of PTD-Smad7 was effective via a topical route, which was required for wound treatment. While not intending to be bound by theory, it is believed that PTD-Smad7 may be used to treat or improve wounds via a variety of routes, and may include, inter alia, one or more of: decreasing inflammation, increasing cell proliferation (e.g., keratinocytes), increasing cell migration (e.g., keratinocytes), or decreasing fibrosis (e.g., via regulation of collagen). As briefly described below, reduced inflammation may significantly contribute to accelerated wound healing, optionally via reduced angiogenesis and collagen production and/or reduced leukocyte infiltration, resulting in a reduction in cytokines and chemokines, normally released by leukocytes, which are pro-angiogenic and pro-fibrogenic. Temporary treatment with Smad7 may allow early angiogenesis and collagen production required for wound repair while preventing long-term angiogenesis and collagen production. These changes can potentially accelerate wound matrix remodeling and prevent excessive scarring due to prolonged inflammation or overproduction of collagen. For surgical procedures (as well as routine injury), treatment with Smad7 may be beneficial, particularly where the potential for scarring is problematic.
Surgery uses operational manual and instrumental techniques on a patient to study and/or treat pathological conditions such as disease or injury, to help improve bodily function or appearance, or sometimes for some other reason. As further defined below, the present techniques can treat trauma caused by surgery.
Generally, when a procedure involves the cutting of a patient's tissue or the closure of a previously-sustained wound, the procedure is considered surgical. Other procedures not necessarily belonging to this class of purposes, such as angioplasty or endoscopy, may be considered surgical if they involve common surgical procedures or settings, such as the use of sterile environments, anesthesia, sterile conditions, typical surgical instruments, and suturing or sutures. All forms of surgery are considered invasive procedures; so-called non-invasive surgery generally refers to ablation (e.g., laser ablation of the cornea) without penetrating the structure being treated or to radiological procedures (e.g., tumor irradiation). The surgery may last from minutes to hours.
Surgical procedures are generally classified by urgency, type of procedure, the body system involved, degree of invasiveness, and specialized instrumentation. Elective surgery is done to correct non-life threatening conditions and is performed at the request of the patient, subject to the availability of surgeons and surgical facilities. Emergency surgery is surgery that must be completed quickly to save lives, limbs, or functional abilities. Exploratory surgery is performed to aid or confirm the diagnosis. Therapeutic surgery treats previously diagnosed conditions.
Amputation involves cutting away a body part, typically a limb or digit. Replantation involves reattaching the severed body part. Reconstructive surgery involves the reconstruction of damaged, deformity, or deformed portions of the body. Cosmetic surgery is performed to improve the appearance of otherwise normal structures. Resection is the cutting of an organ, tissue, or other body part from a patient. Transplant surgery is the replacement of an organ or body part by inserting another organ or body part from a different human (or animal) into the patient. The removal of organs or body parts from living humans or animals for transplantation is also a type of surgery.
When an organ system or structure is operated on, it may be classified by the organ, organ system or tissue involved. Examples include cardiac surgery (performed on the heart), gastrointestinal surgery (performed within the digestive tract and its appendages), and orthopedic surgery (performed on bones and/or muscles).
Minimally invasive procedures involve a small external incision to insert miniaturized instrumentation into a body cavity or structure, as in laparoscopic surgery or angioplasty. In contrast, open surgical procedures require a large incision to access the area of interest. Laser surgery involves the use of a laser to cut tissue in place of a scalpel or similar surgical instrument. Microsurgery involves the use of a surgical microscope to allow the surgeon to view small structures. Robotic surgery utilizes a surgical robot, such as a Da Vinci or Zeus surgical system, to control instruments under the direction of a surgeon.
3. Autoimmune/inflammatory diseases
The present technology encompasses the treatment of a variety of autoimmune and/or inflammatory disease conditions, such as spondyloarthropathies, ankylosing spondylitis, psoriatic arthritis, reactive arthritis, enteropathic arthritis, ulcerative colitis, Crohn's disease, irritable bowel disease, inflammatory bowel disease, rheumatoid arthritis, juvenile rheumatoid arthritis, familial mediterranean fever, amyotrophic lateral sclerosis, Sjogren's syndrome, early arthritis, viral arthritis, multiple sclerosis, or psoriasis. The diagnosis and treatment of these diseases is well documented in the literature.
In general, autoimmune diseases are associated with an overactive immune response of the body to substances and tissues normally present in the body, and are generally not the focus of the immune response. There are more than 80 types of autoimmune diseases, some of which have similar symptoms, and which can be caused by similar deep causes. A typical sign of autoimmune disease is inflammation, which can be treated with a composition of Smad7 (optionally, PTD-Smad7), as disclosed herein.
4. Toxicity of chemotherapy, radiation therapy and cytokine therapy
Various forms of cancer therapy, including chemotherapy, radiation, and cytokines, are associated with toxicity, sometimes severe, in cancer patients. The present technology seeks to reduce such toxicity using the pharmaceutical compositions of the present technology, thereby alleviating or alleviating discomfort in the part of the patient, as well as allowing higher dose therapy.
As described in detail throughout this disclosure, PTD-Smad7 has been found to heal and prevent oral mucositis in a mouse model. PTD-Smad7 was shown to be more effective in direct comparison than palifermin, an existing drug approved for the prevention of oral mucositis.
Oral cancer, the 6 th most common cancer in the world, is a subtype of head and neck cancer and includes any cancerous tissue growth located in the oral cavity. It may be produced as a primary lesion originating in any of the oral tissues, by metastasis from a distal site of origin or by extension from adjacent anatomical structures (such as the nasal cavity), or oral cancer may originate in any tissue in the mouth and may have varying histological types: teratomas, adenocarcinomas from the large or small salivary glands, lymphomas from tonsils or other lymphoid tissues, or melanomas from the pigment-producing cells of the oral mucosa. There are several types of oral cancer, but about 90% originate from squamous cell carcinoma that is the tissue inside the mouth and lips. Oral or mouth cancer most commonly involves the tongue. It may also occur on the floor of the mouth, inside the cheeks, gums (gingiva), lips or palate (roof of mouth). Most oral cancers look very similar under a microscope and are called squamous cell carcinomas. These are malignant and tend to spread rapidly.
More than 80% of oral cancer patients are treated with radiation therapy and at least 75% of these individuals will develop oral mucositis. Oral mucositis is a chronic oral ulcer. This disease frequently occurs in radiation-treated patients of all cancer types, including, but not limited to, patients who are radiation-treated for organ transplants (to eliminate graft rejection), and patients who undergo conventional chemotherapy. Severe oral mucositis is extremely painful and impairs food/fluid intake and is therefore often the most serious complication of cancer therapy. Oral mucositis is a major factor in determining the maximum dose possible for radiation and chemotherapy in the head and neck region; it can significantly complicate cancer treatment, prolong hospitalization, reduce quality of life and increase costs.
Currently, there is no established therapy to effectively treat severe oral mucositis. To date, the recombinant protein palifermin of human Keratinocyte Growth Factor (KGF)Is the only drug approved by the FDA for intravenous (i.v.) injection of severe oral mucositis in bone marrow transplant patients, and its use in cancer therapy remains to be determined. It is also used for preventing oral mucositis. Thus, this drug is only available to 4% of the population at risk. It is due to the intravenous route of administration that a healthcare provider is also required. Other potential therapies include topical rinses such as a viscous 2% lidocaine (1idocaine) rinse, or baking soda and saline solution, or cocktail solutions such as BAX (lidocaine, diphenhydramine), sorbitol, and ). Other investigational or mucoprotective adjunctive therapies include, but are not limited to, beta carotene, tocopherol, laser irradiation, prophylactic brushing of the oral mucosa with silver nitrate, misoprostol (misoprostol), folinic acid, systemic KGF, pentoxifylline (pentoxifylline), allopurinol mouthwash, systemic sucralfate, chlorhexidine gluconate (chlorexidine gluconate), and cryotherapy.
). Other investigational or mucoprotective adjunctive therapies include, but are not limited to, beta carotene, tocopherol, laser irradiation, prophylactic brushing of the oral mucosa with silver nitrate, misoprostol (misoprostol), folinic acid, systemic KGF, pentoxifylline (pentoxifylline), allopurinol mouthwash, systemic sucralfate, chlorhexidine gluconate (chlorexidine gluconate), and cryotherapy.
Chemotherapy-induced and radiation-induced mucositis of the digestive tract is an inflammatory condition that develops as a result of acute death of rapidly differentiated intestinal epithelial cells. Most chemotherapeutic drugs used to treat solid tumors alone, in combination with drugs, or with radiation will result in the death of a large number of intestinal epithelial cells. The consequent clinical manifestations of mucositis include digestive tract symptoms such as nausea and vomiting, severe diarrhea, acute weight loss, and wasting. This soon became one of the limiting factors in the administration of chemotherapy for many cancer patients. The ability of Tat-Smad7 to protect intestinal epithelial cells from chemotherapeutic agents, radiation, or a combination of those would significantly reduce the undesirable side effects of cancer therapy and enable the treatment of disease in a more invasive manner with existing tools.
Bone marrow failure syndrome is a group of conditions that develop when the hematopoietic stem cell compartment is damaged and fails to produce normal cell types. Bone marrow failure occurs as a result of genetic abnormalities, exposure to toxic substances (such as toxins, chemicals or viruses). Although the nature and identity of the environmental factors that can contribute to the development of acquired bone marrow failure are still not fully understood, a few factors have been linked to the development of acquired bone marrow failure among military personnel, including exposure to mustard gas, ionizing radiation, and infectious agents such as visceral leishmaniasis or taeniasis. The best approach for managing the bone marrow failure syndrome remains transplantation of Hematopoietic Stem Cells (HSCs) unless a sufficient number of remaining resident bone marrow HSCs can escape these pressures and encourage re-engraftment of the hematopoietic compartment. Modulation of Smad7 as described herein should be able to deliberately protect remaining resident HSCs in patients exhibiting clinical signs consistent with bone marrow failure.
5. Cancer treatment
TGF-. beta.and NF-. kappa.B activation are known to promote cancer invasion and metastasis. Currently, TGF- β inhibitors are used in clinical trials for the treatment of metastatic cancer and NF- κ B inhibitors are used for cancer prevention. The demonstrated effect of Smad7 on blocking TGF- β and NF- κ B signaling presents the potential for it to be an even stronger anti-cancer/anti-metastatic agent than other inhibitors that inhibit only one of these two pathways. Smad7 has been shown to prevent angiogenesis and fibrogenesis and may therefore be particularly useful in situations where tumors require development of a blood supply and/or matrix.
The cancer may be selected from the group consisting of: brain, lung, liver, spleen, kidney, lymph node, small intestine, pancreas, blood cells, colon, stomach, breast, endometrium, prostate, testis, cervix, uterus, ovary, skin, head and neck, esophagus, bone marrow, and blood. Cancer may be metastatic or primary, recurrent or multi-drug resistant. In some embodiments, the cancer is a solid tumor (organ tumor). Solid tumors refer to cell masses that grow in the organ system and can occur anywhere in the body. Two types of solid tumors include epithelial tumors (carcinomas) that occur in epithelial tissues inside or outside an organ, and sarcomas (connective tissue tumors) that occur in connective tissues such as, but not limited to, muscles, tendons, fat, nerves, and other connective tissues that support, surround, or connect structures and organs within the body. In some embodiments, the cancer is a liquid tumor or cancer of the blood, bone marrow, or lymph nodes. These tumors include, but are not limited to, leukemia, lymphoma, and myeloma.
6. Scar formation, fibrosis and abnormal healing
In addition to accelerated re-epithelialization (e.g., via increased cell proliferation and/or increased cell migration), the effects of Smad7 on the wound matrix include, inter alia, a reduction in one or more of inflammation, angiogenesis, or collagen production. While not intending to be bound by theory, these effects may be mediated via a reduction in NF-. kappa.B signaling (as demonstrated by reduced p 50) and blocking TGF-. beta.signaling (as demonstrated by reduced pSmad 2). Thus, reduced inflammation may significantly contribute to accelerated wound healing, optionally via reduced angiogenesis and collagen production and/or reduced leukocyte infiltration, resulting in a reduction of cytokines and chemokines, normally released by leukocytes, which are pro-angiogenic and pro-fibrogenic. Temporary treatment with Smad7 may allow early angiogenesis and collagen production required for wound repair while preventing long-term angiogenesis and collagen production. These changes can potentially accelerate wound matrix remodeling and prevent excessive scarring due to prolonged inflammation or overproduction of collagen.
7. Stomatitis (stomatitis)
Stomatitis is an inflammation of the mucous lining of any structure in the mouth that may involve the cheek, gums, tongue, lips, throat, and the top or bottom of the mouth. Inflammation may be caused by conditions in the mouth itself, such as poor oral hygiene, dietary protein deficiency, poorly installed dentures, or mouth burns caused by hot food or beverages, toxic plants, or by conditions affecting the whole body, such as medications, allergies, radiation therapy, or infections. Severe iron deficiency anemia can lead to stomatitis. Iron is required for the upregulation of transcription elements for cell replication and repair. Iron deficiency can cause gene down-regulation of these elements, leading to inefficient repair and regeneration of epithelial cells, particularly in the mouth and lips. This condition is also found in vitamin B2(Riboflavin), B3(nicotinic acid), B6(pyridoxine), B9(Folic acid) or B12(cobalamin) deficiency is prevalent in the population. When stomatitis also involves inflammation of the gums (gingiva), it is called gingivitis. Stomatitis can also be seen in riboflavin deficiency (riboflavin deficiency) or neutropenia.
Irritation and dehiscence at the labial angle is known as angular cheilitis or cheilitis. In children, angular stomatitis is a common cause of repeated licking of the lips, and in adults it can be a deep iron deficiency anemia or vitamin B deficiency (e.g., B2Riboflavin, B9Folate or B12Cobalamin), which in turn may prove poor diet or malnutrition (e.g. celiac disease). Moreover, cheilitis can be caused by "over-closure" of the patient's jaw by missing teeth or tooth wear when static, which causes the jaw to be closer together than when there is a full/unaffected dentition. This causes skin folds around the corners of the mouth, which remain moist with saliva, and are then prone to infection; is mainly infected by Candida albicans (Candida albicans) or similar species. The treatment is usuallyInvolves the administration of topical nystatin or similar antifungal agents. Another treatment may be to correct jaw position using a dental treatment (e.g., denture or bite adjustment).
Migratory stomatitis is a condition in which a wide area in the oral mucosa is affected by a ring-shaped atrophic red focus surrounded by a thin white border. This is a relatively uncommon form of topography-like tongue pathology, which is limited to the ridge and lateral orientation of the tongue mucosa, as opposed to migratory stomatitis.
8. Inflammation of the rectum
Proctitis is an inflammation of the rectum, the lining of the lower end of the large intestine leading to the anus. Proctitis, inflammation of the lining of the rectum, known as the rectal mucosa, is uncomfortable and sometimes painful. This condition may lead to bleeding from the rectum or mucosal drainage, among other symptoms. Some causes of proctitis include (but are not limited to): sexually Transmitted Diseases (STDs), such as those that can be transmitted during anal intercourse (e.g., gonorrhea, chlamydia, syphilis, and herpes); non-STD infections due to, for example, food-borne bacteria (e.g., Salmonella (Salmonella) and Shigella (Shigella)); anorectal trauma resulting from, for example, anal intercourse or insertion of an object or substance into the rectum (e.g., chemicals from enemas); ulcerative colitis and crohn's disease or other inflammatory bowel disease, may cause ulcers (e.g., sores) in the lining of the colon and rectum; radiation therapy, particularly of the pelvic region (e.g., rectal, ovarian or prostate cancer), which may lead to rectal bleeding; antibiotics, which cause symbiotic loss, allowing harmful bacteria (e.g., Clostridium difficile) to cause disease.
9. Formulations and routes of administration
Where clinical use is contemplated, it will be necessary to prepare pharmaceutical compositions-proteins, expression vectors, viral stocks, proteins and drugs-in a form suitable for the intended use. Generally, this will require the preparation of a composition that is substantially free of pyrogens and other impurities that may be harmful to humans or animals.
PTD-Smad7 (and truncated variants) was extensively purified prior to use in animal models. PTD-Smad7 (and truncated versions) were prepared using a mixture of glycerol and PBS for topical and transmucosal application.
We will generally need to employ appropriate salts and buffers to render the delivery vehicle stable and to allow uptake by the target cells. When recombinant cells are introduced into a patient, buffers will also be employed. Aqueous compositions of the present technology comprise an effective amount of a carrier to cells, dissolved or dispersed in a pharmaceutically acceptable carrier or aqueous medium. Such compositions are also known as inoculants. The phrase "pharmaceutically or pharmacologically acceptable" refers to molecular entities and compositions that do not produce adverse, allergic, or other untoward reactions when administered to an animal or human. As used herein, "pharmaceutically acceptable carrier" includes any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents, and the like. The use of such media and agents for pharmaceutically active substances is well known in the art. Unless any conventional media or agent is incompatible with the carrier or cell of the present technology, its use in therapeutic compositions is contemplated. Supplemental active ingredients may also be incorporated into the composition.
The active compositions of the present technology may include typical pharmaceutical preparations. Administration of these compositions according to the present technology will be via any of the usual routes, so long as the target tissue is available via that route. Such routes of administration may include oral, parenteral (including intravenous, intramuscular, subcutaneous, intradermal, intraarticular, intrasynovial, intrathecal, intraarterial, intracardiac, subcutaneous, intraorbital, intracapsular, intraspinal, intrasternal, and transdermal), nasal, buccal, transurethral, rectal, vaginal, transmucosal, transdermal, or topical (including transdermal, buccal, and sublingual). Alternatively, administration may be by in situ, intradermal, subcutaneous, intramuscular, intraperitoneal or intravenous injection. Such compositions will generally be administered in the pharmaceutically acceptable compositions described above. Of particular interest are direct intratumoral administration, tumor perfusion, or local or regional administration to a tumor, for example, in the local or regional vascular structure or lymphatic system, or in a resected tumor bed. Administration can also be via nasal spray, surgical implant, internal surgical paint (internal surgical paint), infusion pump, or via catheter, stent, balloon, or other delivery device. The mode of administration that is most suitable and/or beneficial may vary, depending, inter alia, on the condition of the recipient and the condition being treated.
Solutions of the active compounds in free base or pharmacologically acceptable salt form can be prepared in water suitably mixed with a surfactant, such as hydroxypropylcellulose. Dispersions can also be prepared in glycerol, liquid polyethylene glycols, and mixtures thereof, and in oils. Under ordinary conditions of storage and use, these preparations contain a preservative to prevent the growth of microorganisms.
Pharmaceutical forms suitable for injectable use include sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions. In all cases, this form must be sterile and must be fluid to the extent that it is easy to inject. It must be stable under the conditions of manufacture and storage and must be preserved against the contaminating action of microorganisms such as bacteria and fungi. The carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (for example, glycerol, propylene glycol, and liquid polyethylene glycol, and the like), suitable mixtures thereof, and vegetable oils. Proper fluidity can be maintained, for example, by the use of a coating such as lecithin, by the maintenance of the required particle size in the case of dispersions, and by the use of surfactants. Prevention of the action of microorganisms can be achieved by various antibacterial and antifungal agents, for example, parabens, chlorobutanol, phenol, sorbic acid, thimerosal (thimerosal), and the like. In many cases, it will be preferable to include isotonic agents, for example, sugars or sodium chloride. Prolonged absorption of the injectable compositions can be brought about by the use in the compositions of agents delaying absorption, for example, aluminum monostearate and gelatin.
Sterile injectable solutions are prepared by incorporating the active compound in the required amount in the appropriate solvent with various other ingredients enumerated above, as required, followed by filtered sterilization. Generally, dispersions are prepared by incorporating the various sterilized active ingredients into a sterile vehicle which contains the basic dispersion medium and the required other ingredients from those enumerated above. In the case of sterile powders for the preparation of sterile injectable solutions, the preferred methods of preparation are vacuum drying and the freeze-drying technique which yield a powder of the active ingredient plus any additional desired ingredient from a previously sterile-filtered solution thereof.
As used herein, "pharmaceutically acceptable carrier" includes any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents, and the like. The use of such media and agents for pharmaceutically active substances is well known in the art. Except insofar as any conventional media or agent is incompatible with the active ingredient, its use in the therapeutic compositions is contemplated. Supplemental active ingredients may also be incorporated into the composition.
The compositions of the present technology may be formulated in neutral or salt form. Pharmaceutically acceptable salts include the acid addition salts (formed with the free amino groups of the protein) and are formed with the following acids: inorganic acids such as hydrochloric acid or phosphoric acid, or organic acids such as acetic acid, oxalic acid, tartaric acid, mandelic acid, and the like. Salts formed with the free carboxyl groups can also be derived from inorganic bases such as sodium hydroxide, potassium hydroxide, ammonium hydroxide, calcium hydroxide, or iron hydroxide, and organic bases such as isopropylamine, trimethylamine, histidine, procaine (procaine), and the like.
The formulations are readily administered in a variety of dosage forms. Some variation in dosage will necessarily occur depending on the condition of the subject being treated. The person responsible for administration will in any case decide the appropriate dose for the individual subject. In addition, for human administration, the formulations should meet sterility, pyrogenicity, general safety, and purity standards as required by the FDA Office of Biologics standards.
For oral administration, the polypeptides of the present technology can be combined with excipients and used in the form of non-ingestible mouthwashes and dentifrices. It is contemplated that virtually any pill or capsule form known to those skilled in the art, including, for example, over-coating and time-delayed, slow-release, etc., may be used in the present techniques. Mouthwashes may be prepared by incorporating the active ingredient in the required amount in an appropriate solvent, such as a sodium borate Solution (Dobell's Solution). Alternatively, the active ingredient may be incorporated into an anti-bacterial lotion comprising sodium borate, glycerin, and potassium bicarbonate. Active ingredients may also be dispersed in the dentifrice, including: gels, pastes, creams, powders, and slurries. The active ingredient may be added to a paste dentifrice in a therapeutically effective amount, which may include water, binders, abrasives, flavoring agents, foaming agents, and humectants.
Pharmaceutical compositions suitable for oral dosage can take a variety of forms, such as tablets, capsules, caplets, and wafers (including fast dissolving or effervescent), each containing a predetermined amount of active agent. The compositions may also be in the form of powders or granules, solutions or suspensions in aqueous or non-aqueous liquids, and liquid emulsions (oil-in-water and water-in-oil). The active agent may also be delivered as a bolus, electuary or paste. It will be generally understood that the methods of making the dosage forms described above are generally known in the art, and that any such method would be suitable for making corresponding dosage forms for delivery of the compositions.
In one embodiment, the active agent compound may be administered orally in combination with a pharmaceutically acceptable vehicle, such as an inert diluent or an edible carrier. The oral compositions may be enclosed in hard or soft shell gelatin capsules, may be compressed into tablets, or may be combined directly with the diet of the patient. The percentages of the compositions and formulations may vary; however, the amount of material in such therapeutically useful compositions will preferably achieve an effective dosage level.
Hard capsules containing the active agent compounds can be made using physiologically degradable compositions such as gelatin. Such hard capsules comprise the compound and may further comprise additional ingredients including, for example, inert solid diluents such as calcium carbonate, calcium phosphate or kaolin. Soft gelatin capsules containing the compounds may be made using physiologically degradable compositions such as gelatin. Such soft capsules comprise a compound which may be mixed with water or an oil medium, such as peanut oil, liquid paraffin, or olive oil.
Sublingual tablets are designed to dissolve very rapidly. Examples of such compositions include ergotamine tartrate, isosorbide dinitrate, and isoproterenol hydrochloride. In addition to the drug, the composition of these tablets contains various soluble excipients such as lactose, powdered sucrose, glucose, and mannitol. The solid dosage forms of the present technology may optionally be coated, and examples of suitable coating materials include, but are not limited to, cellulosic polymers (such as cellulose acetate phthalate, hydroxypropyl cellulose, hydroxypropyl methylcellulose phthalate, and hydroxypropyl methylcellulose acetate succinate), polyvinyl acetate phthalate, acrylic polymers and copolymers, and methacrylic resins (such as those under the trade name hydroxypropyl methylcellulose acetate succinate), and the like Those commercially available), zein, shellac, and polysaccharides.
Those commercially available), zein, shellac, and polysaccharides.
Powdered and granular compositions of pharmaceutical preparations can be prepared using known methods. Such compositions may be administered directly to a patient or used in the further preparation of dosage forms to form tablets, fill capsules, or prepare aqueous or oily suspensions or solutions by adding thereto an aqueous or oily vehicle. Each of these compositions may further comprise one or more additives such as dispersing or wetting agents, suspending agents, and preservatives. Additional excipients (e.g., fillers, sweeteners, flavoring or coloring agents) may also be included in these compositions.
Liquid compositions of pharmaceutical compositions suitable for oral administration may be prepared, packaged and sold in liquid form or in the form of a dry product intended to be reconstituted with water or another suitable vehicle prior to use.
Tablets containing one or more active agent compounds described herein may be manufactured by any standard process readily known to those skilled in the art, such as by compression or molding, optionally with one or more adjuvants or accessory ingredients. The tablets may optionally be coated or scored and may be formulated to provide slow or controlled release of the active agent.
Solid dosage forms may be formulated to provide delayed release of the active agent, such as by application of a coating. Delayed release coatings are known in the art, and dosage forms containing the same may be prepared by any known suitable method. Such methods generally include applying a delayed release coating composition after preparing the solid dosage form (e.g., tablet or caplet). Application may be by methods such as airless spraying, fluid bed coating, use of a pan coater, and the like. Materials used as delayed release coatings may be polymeric in nature, such as cellulosic materials (e.g., cellulose butyrate phthalate, hydroxypropyl methylcellulose phthalate, and carboxymethyl ethylcellulose), and polymers and copolymers of acrylic acid, methacrylic acid, and esters thereof.
Solid dosage forms according to the present technology may also be sustained release (i.e., release of the active agent over an extended period of time) and may or may not also be delayed release. Sustained release compositions are known in the art and are generally prepared by dispersing the drug within a matrix of progressively degradable or hydrolysable material such as an insoluble plastic, hydrophilic polymer or fatty compound. Alternatively, the solid dosage form may be coated with such a material.
Compositions for parenteral administration include aqueous and non-aqueous sterile injection solutions, which may further contain additional agents such as antioxidants, buffers, bacteriostats, and solutes that render the composition isotonic with the blood of the intended recipient. The compositions may include aqueous and non-aqueous sterile suspensions containing suspending agents and thickening agents. Such compositions for parenteral administration may be presented in unit-dose or multi-dose containers, such as sealed ampoules and vials, and may be stored in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example water (for injections), immediately prior to use. Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets of the kind previously described.
Compositions for rectal delivery include rectal suppositories, creams, ointments, and liquids. Suppositories may be presented as the active agent in combination with carriers generally known in the art, such as polyethylene glycol. Such dosage forms may be designed to disintegrate rapidly or over an extended period of time, and the time to complete disintegration may range from a short period of time, such as about 10 minutes, to an extended period of time, such as about 6 hours.
The topical composition may be in any form suitable and readily known in the art for delivering an active agent to a body surface, including transdermal, buccal, and sublingual. Typical examples of topical compositions include ointments, creams, gels, pastes, and solutions. Compositions for oral administration include lozenges.
According to these embodiments, the oral (topical, transmucosal, and/or transdermal) delivery materials may also include creams, ointments, salves, patches, liposomes, nanoparticles, microparticles, timed release formulations, and other materials known in the art for delivery to the oral cavity, mucosa, and/or skin of a subject for the treatment and/or prevention of the conditions disclosed herein. Certain embodiments relate to the use of biodegradable oral (topical, transmucosal, and/or transdermal) patch delivery systems or gelling materials. These compositions may be liquid formulations or pharmaceutically acceptable delivery systems treated with formulations of these compositions, and may also include an activating/inducing agent.
The compositions used in the methods of the present technology may also be administered transdermally, by incorporating the active agent into a layered structure (commonly referred to as a "patch") suitable for maintaining intimate contact with the epidermis of the recipient for an extended period of time. Typically, such patches are used as a single layer "medicated adhesive" patch or a multi-layer patch in which the active agent is contained in a layer separate from the adhesive layer. Both types of patches also typically contain a backing layer and a liner that is removed prior to attachment to the recipient's skin. Transdermal drug delivery patches may also consist of a reservoir beneath the backing layer that is separated from the recipient's skin by a semi-permeable membrane and an adhesive layer. Transdermal drug delivery can occur via passive diffusion, electrotransport, or iontophoresis.
In certain embodiments, a patch contemplated herein may be a slow dissolving or time release patch. According to these embodiments, the slow dissolving patch may be an alginate patch. In certain examples, the patch may contain a detectable indicator dye or agent, such as a fluorescent agent. In other embodiments, a tag (e.g., a detectable tag, such as biotin or a fluorescently tagged reagent) can be associated with a therapeutic molecule to detect the molecule after delivery to the subject. In certain embodiments, one or more oral delivery patches or other therapies contemplated herein may be administered to a subject three times daily, twice daily, once daily, every other day, weekly, etc., depending on the needs of the subject as assessed by a health care professional. The patches contemplated herein may be orally biodegradable patches or external patches that may or may not be degraded. The patch contemplated herein may be 1mm, 2mm, 3mm, 4mm to 5mm in size or greater, as desired. Additionally, skin patches for subjects suffering from psoriasis, for example, are contemplated herein. In the treatment of psoriasis and chronic wounds, Smad7 may be delivered topically using vehicles such as glycerin, carboxymethyl cellulose. Transdermal systems (e.g., commercially available from 3M) may also be used for delivery. Subcutaneous injection into the lesion (in saline or PBS) may also be used.
In some embodiments, the compositions may include short-term, rapid-onset, controlled-release, sustained-release, delayed-release, and pulsed-release compositions, provided that such compositions achieve administration of the compounds as described herein. See Remtngton's Pharmaceutical Sciences (18 th edition; Mack Publishing Company, Eaton, Pennsylvania, 1990), incorporated herein by reference in its entirety.
In certain embodiments, the compounds and compositions disclosed herein can be delivered via a medical device. Such delivery may generally be via any insertable or implantable medical device, including but not limited to a stent, catheter, balloon catheter, shunt, or coil. In one embodiment, the present technology provides a medical device, such as a stent, having a surface coated with a compound or composition as described herein. The medical devices of this technology can be used, for example, in any application that treats, prevents, or otherwise affects the progress of a disease or condition, such as those disclosed herein.
It is contemplated that any molecular biological, cell biological, or biochemical technique known in the art may be used to generate and/or test the therapeutics provided herein. In addition, protein chemistry techniques are contemplated for assessing the utility of treatment in the model systems disclosed herein (e.g., mouse model systems).
10. Combination therapy
It is common in many medical fields to treat diseases with multiple treatment modalities, commonly referred to as "combination therapy". Many diseases described herein (e.g., inflammatory diseases and cancer) are without exception. In some embodiments, to treat an inflammatory disorder using the methods and compositions of the present technology, we will contact a target cell, organ or subject with a Smad7 protein, expression construct or activator, and at least one other therapy. These therapies will be provided in a combined amount effective to achieve a reduction in one or more disease parameters. This process may involve contacting the cell/subject with two agents/therapies simultaneously, for example using a single composition or pharmacological formulation that includes both agents, or by contacting the cell/subject with two different compositions or formulations simultaneously, wherein one composition includes a Smad7 agent and the other includes the other agent.
Alternatively, the Smad7 agent may be present before or after other treatments at intervals ranging from minutes to weeks. We will generally ensure that there is no interruption for a significant period of time between the time of each delivery so that the therapy will still be able to exert a beneficial combined effect on the cells/subject. In such cases, it is expected that we will contact the cells with both modalities within about 12-24 hours of each other, within about 6-12 hours of each other, or with a delay time of only about 12 hours. In some cases, it may be desirable to significantly extend the period of treatment; however, several days (2, 3, 4, 5,6 or 7) to several weeks (1, 2, 3, 4, 5,6, 7 or 8) lapse between the respective administrations.
It is also contemplated that more than one administration of Smad7 agent or other therapy will be required. Various combinations may be employed wherein the Smad7 agent is "a" and the other therapy is "B", as exemplified below:
other combinations are provided. Other agents suitable for use in combination therapy for inflammatory conditions include steroids, glucocorticoids, nonsteroidal anti-inflammatory drugs (NSAIDs; including COX-1 and COX-2 inhibitors), aspirin (aspirin), ibuprofen (ibuprofen) and naproxen (naproxen). Analgesics are usually associated with anti-inflammatory drugs, but they have no anti-inflammatory effect. One example is paracetamol (paracetamol), known in the united states as acetaminophen and sold under the trade name Tylenol. In contrast to NSAIDs, which relieve pain and inflammation by inhibiting COX enzymes, paracetamol has recently been shown to block the resorption of the endocannabinoid (endocannabinoid), which only relieves pain, possibly explaining why it has minimal effect on inflammation. A particular agent for combined use is an anti-TGF- β antibody.
The skilled person is guided by Remington's Pharmaceutical Sciences, 15 th edition, chapter 33, in particular page 624-. Some variation in dosage will necessarily occur depending on the condition of the subject being treated. The person responsible for administration will in any case decide the appropriate dose for the individual subject. In addition, for human administration, the formulations should meet sterility, pyrogenicity, general safety, and purity standards as required by FDA office of biologies standards.
It should also be noted that any of the foregoing therapies may prove themselves suitable for treating inflammation.
As discussed above, the present technology is particularly concerned with the treatment of DNA damage and/or inflammation resulting from certain anti-cancer therapies, as well as for the treatment of cancer. Thus, in particular, the present technology may be applied as a combination with cancer therapy. This process may involve contacting the cell, organ or patient with an agent/therapy simultaneously, including by contacting the cell, organ or patient with a single composition or pharmacological formulation that includes two agents, or with two different compositions or formulations simultaneously, wherein one composition includes a Smad7 agent and the other includes an additional agent. Alternatively, similar to the schedule set forth above, the compositions may be delivered at different times, including repeated doses of one or both agents.
Agents or factors suitable for use in combination therapy include any compound or method of treatment that induces DNA damage when applied to a cell. Such agents and factors include radiation and waves that induce DNA damage, such as irradiation, microwaves, electron emission, and the like. A variety of compounds, also described as "chemotherapeutic agents" or "genotoxic agents," are intended for use in the combination therapy methods disclosed herein. In treating cancer according to the present technology, we will contact tumor cells with an agent other than an expression construct. This can be achieved by irradiating a localised tumour site; alternatively, the tumor cells can be contacted with the agent by administering a therapeutically effective amount of the pharmaceutical composition to the subject.
Various classes of chemotherapeutic agents are provided for use in combination with the peptides of the present technology. For example, selective estrogen receptor antagonists ("SERMs"), such as tamoxifen, 4-hydroxy tamoxifen (Afimoxfene), fulvestrant (falsdex), Raloxifene (Raloxifene), Bazedoxifene (Bazedoxifene), clomiphene (Clomifene), flonicate (Femarelle), Lasofoxifene (Lasofoxifene), olmexifene (Ormeloxifene), and Toremifene (Toremifene).
Chemotherapeutic agents contemplated to be useful include, for example, camptothecin, actinomycin D, mitomycin C. The present technology also encompasses the use of a combination of one or more DNA damaging agents (whether radiation-based or actual compounds), such as the use of X-rays with cisplatin (cissplatin) or cisplatin with etoposide (etoposide). As described above, the agents may be prepared and used in a combined therapeutic composition or kit by combining them with the MUCl peptide.
Agents that directly cross-link DNA or form adducts are also contemplated. Agents such as cisplatin, and other DNA alkylating agents can be used. Cisplatin has been widely used in the treatment of cancer, and its effective dose in clinical application is 20mg/m2Lasting for 5 days, once every three weeks for three treatment courses. Cisplatin is not absorbed orally and therefore must be delivered via intravenous, subcutaneous, intratumoral or intraperitoneal injection.
Agents that damage DNA also include compounds that interfere with DNA replication, mitosis, and chromosome segregation. Such chemotherapeutic compounds include doxorubicin (also known as doxorubicin (doxorubicin)), etoposide, verapamil (verapamil), podophyllotoxin (podophyllotoxin), and the like. These compounds, widely used in clinical settings for the treatment of neoplasms, are injected via intravenous bolus injection at 25-75mg/m2(21 days time interval for doxorubicin) to 35-50mg/m2(for etoposide) or orally doubling the intravenous dose. Microtubule inhibitors such as taxanes (taxanes) are also contemplated. These molecules are diterpenes produced by plants of the genus taxus and include paclitaxel (paclitaxel) and docetaxel (docetaxel).
Epidermal growth factor receptor inhibitors, such as Iressa, the mammalian target of rapamycin mTOR, also known as FK 506-binding protein 12-rapamycin associated protein 1(FRAP1), are serine/threonine protein kinases that regulate cell growth, cell proliferation, cell motility, cell survival, protein synthesis, and transcription. Thus, rapamycin and its analogs ("rapalogs") are provided for use in combination with cancer therapy in accordance with the present technology.
Another possible combination therapy with the peptides claimed herein is TNF- α (tumor necrosis factor- α), a member of the group of cytokines involved in systemic inflammation and the cytokines that stimulate the acute phase response. The main role of TNF is in the regulation of immune cells. TNF also induces apoptotic cell death, induces inflammation, and inhibits tumorigenesis and viral replication.
Agents that disrupt the synthesis and fidelity of nucleic acid precursors and subunits also cause DNA damage. In this regard, a number of nucleic acid precursors have been developed. Particularly useful are agents that have undergone extensive testing and are readily available. As such, agents such as 5-fluorouracil (5-FU) are preferentially used by neoplastic tissue, making such agents particularly useful for targeting neoplastic cells. Although quite toxic, 5-FU is suitable for use in a wide range of vehicles, including topical, however, intravenous administration at doses ranging from 3 to 15 mg/kg/day is typically used.
Other factors that cause DNA damage and have been widely used include those commonly referred to as gamma rays, x-rays, and/or targeted delivery of radioisotopes to tumor cells. Other forms of DNA damaging factors are also contemplated, such as microwaves and UV irradiation. It is most likely that all of these factors effect a wide range of damage to DNA, precursors of DNA, replication and repair of DNA, and assembly and maintenance of chromosomes. The dose of x-rays ranges from a daily dose of 50 to 200 roentgens for an extended period of time (3 to 4 weeks) to a single dose of 2000 to 6000 roentgens. The dosage of radioisotopes varies widely and depends on the half-life of the isotope, the intensity and type of radiation emitted, and the uptake by neoplastic cells.
The skilled person is guided by Remington's Pharmaceutical Sciences, 15 th edition, chapter 33, in particular page 624-. Some variation in dosage will necessarily occur depending on the condition of the subject being treated. The person responsible for administration will in any case decide the appropriate dose for the individual subject. In addition, for human administration, the formulations should meet sterility, pyrogenicity, general safety, and purity standards as required by FDA office of biologies standards.
Except that Smad7 therapy was combined with chemotherapy and radiation therapyIn addition, combination with immunotherapy, hormonal therapy, toxin therapy, and surgery is also contemplated. In particular, we can employ targeted therapies, such asAnd
in other embodiments, to assess the role and mechanism of Smad7 in the context of oral mucositis, a "gene switch" transgenic mouse model was developed to allow control of the level and duration of Smad7transgene expression specifically in the oral epithelium. According to these embodiments, these models can be used to test the effect of other genes or downstream molecules on the oral epithelium and oral mucosa. Thus, these models can be used for (but not limited to) further analysis of oral wound healing biology and testing treatment methods for oral wound healing. The molecular Smad7 targets identified in these studies can provide additional therapeutic targets to subjects suffering from oral mucositis. The models and resources developed herein may provide unique tools for analytical studies to identify biomarkers and therapeutic targets associated with Smad7 overexpression and control, e.g., downstream molecules turned on or bound by Smad7 may be identified as, e.g., additional therapeutic targets to treat oral mucositis, psoriasis, and other conditions exacerbated by TGF- β activity and NF- κ B activity.
D. Reagent kit
In certain embodiments, a kit provided herein may include a composition as discussed above for treating a subject having a condition provided herein, such as (but not limited to) oral mucositis, psoriasis, or wound healing. Kits may include one or more containers containing a therapeutic Smad7 composition of the technology. Any of these kits will generally include at least one vial, test tube, flask, jar, syringe, or other container into which the composition may preferably and/or suitably be aliquoted. The kits herein can also include kits for assessing biological targets contributing to the conditions provided herein.
E. Methods of predicting or evaluating a response
Also provided are methods of predicting and/or assessing response to treatment with Smad7 by assessing the expression level of one or more markers associated with exposure to Smad 7. Such markers may include, but are not limited to, Rac1 for cell migration, NF-. kappa.B for inflammation, and TGF-. beta.for growth arrest and inflammation. As discussed in the examples, methods and/or variations of detecting the level of one or more markers associated with Smad7 activity are provided and/or known in the art. In some embodiments, the expression level of one or more of the Smad7 markers in a subject can be assessed, and based on the detected level, treatment with Smad7 (or decision to continue or discontinue treatment) can be decided, or alternative treatment employed.
The term "detecting" as used herein refers to the ability to measure the presence or absence of a marker at a certain repeatable and controllable level. Typically, detection is performed on background values, which may include noise (or detection limits) inherent to the test system. As such, there is typically a "lower limit" of detection associated with the assay, and for detection, the change may need to be above, for example, a certain cut-off level. The determination of such limits is well known in the art.
In some embodiments, the detection is compared to a control, which can include, but is not limited to, comparison to data from a normal subject and/or comparable normal tissue (in the same or different subject) lacking the disease or disorder present in the subject (or the particular tissue of the subject being tested). In some embodiments, the comparison may be made between levels detected at various time intervals (and/or locations) in the patient. In some embodiments, the detection needs to be statistically significant compared to background or control levels; the ability to assess significance is well known in the art and is exemplified in the examples.
The term "change in level" as used herein refers to a detectable change from a control or background level and/or a previously detected level. In some embodiments, the change is an increase as compared to another level, and in some embodiments, the change is a decrease as compared to another level. In some embodiments, the detectable change (increase or decrease) is statistically significant. In some embodiments, such changes can be quantitatively evaluated as at least about 5%, 10%, 25%, 50%, 100%, 200%, 500% or more changes, and/or about 5-10%, 10-25%, 10-50%, 25-50%, 50-75%, 50-100%, 100-150%, 100-200%, 200-300%, 300-500%, or 500-1000% changes.
F. Method for screening additional biologically active fragments
In another aspect, methods of screening for additional biologically active fragments of Smad7, including but not limited to truncations, are contemplated. In some embodiments, biological activity can be assessed using one of the methods described herein, including those described in examples 5 and 8 below. Some biological activities that can be evaluated include, but are not limited to, increasing cell proliferation, reducing or inhibiting cell death, alleviating excessive inflammation, preventing DNA damage, and/or increasing cell migration, as well as animal models that treat or prevent one or more diseases or disorders for which such treatment would be helpful as discussed further herein. Such activity can be assessed using one or more assays, including, but not limited to, the ability to block phosphorylation of Smad2 and/or nuclear translocation of the NF- κ B p50 subunit, increase cell proliferation, decrease apoptosis and/or radiation-induced DNA damage, decrease inflammation and/or angiogenesis, promote healing of oral mucositis, surgical wounds, diabetic wounds, and/or wounds associated with chronic inflammation in mouse and other laboratory models. Some specific examples include, but are not limited to, Immunofluorescence (IF), Immunohistochemistry (IHC), and TUNEL assays for apoptosis.
In some embodiments, biologically active fragments are those selected to include one or more or all of the activities described herein. In some embodiments, a biologically active fragment is selected to include only or predominantly 1, only or predominantly 2, only or predominantly 3, only or predominantly 4, or only or predominantly 5 of the activities described herein. In some embodiments, a biologically active fragment is selected to exclude only or predominantly 1, only or predominantly 2, only or predominantly 3, only or predominantly 4, or only or predominantly 5 of the activities described herein. In some embodiments, a biologically active fragment is selected to include or exclude a particular subset of the activities described herein. For example, increased proliferation and migration may be sufficient to treat diabetic wounds, where anti-inflammation is required. Reduced apoptotic and DNA damaging activity is desirable for the treatment of oral mucositis, but not for the treatment of surgical wounds.
The term "consisting essentially of as used herein refers to the following fragments: while some level of other biological activity may be retained, that activity is reduced compared to the full-length fragment, while the activity considered "primary" remains at about the same or an increased level as observed in the full-length native protein. Similarly, the term "predominantly excluded" as used herein refers to the following fragments: while a certain level of a particular biological activity can be retained, the level of that particular activity is reduced (optionally, significantly and/or statistically significantly reduced) compared to the full-length fragment, while one or more other biological activities remain at about the same or increased levels as observed in the full-length native protein.
In some embodiments involving selection of biologically active fragments, the methods comprise assessing changes in the expression level of one or more biological activities, including increases and decreases in one or more activities in the selected fragment, as assessed with respect to changes in activity observed in the full-length protein. In some embodiments, one or more biological activities are selected to remain the same as observed in a full-length fragment, while other activities may be increased or decreased or even eliminated (e.g., such fragments would lack the activity or activities discussed). In some embodiments, the change is an increase as compared to another level, and in some embodiments, the change is a decrease as compared to another level. In some embodiments, the detectable change (increase or decrease) is statistically significant. In some embodiments, such changes can be quantitatively evaluated as at least about 5%, 10%, 25%, 50%, 100%, 200%, 500% or more changes, and/or about 5-10%, 10-25%, 10-50%, 25-50%, 50-75%, 50-100%, 100-150%, 100-200%, 200-300%, 300-500%, or 500-1000% changes. In some embodiments, some variation in activity that is "remains the same" as compared to the activity of the full-length protein may still be observed, but such variation may be limited to, for example, about 1%, 2%, 5%, 10%, or 20% variation or less.
In one non-limiting example, fragments of interest can include those that primarily mediate the anti-inflammatory effects of Smad 7. Smad7 peptides having this anti-inflammatory function may be sufficient and optionally an improvement for the treatment of chronic inflammation-related conditions, such as (but not limited to) oral mucositis, stomatitis, and psoriasis, among others. In another non-limiting example, fragments of interest may include those that primarily mediate cell migration and/or block TGF-beta induced growth arrest and/or fibrotic responses. Smad7 peptides with this cell migration and proliferation function may be sufficient and optionally an improvement for enhanced healing not associated with excessive inflammation. Types of wounds that may benefit from this form of treatment include, but are not limited to, surgical wounds, fibrotic scars, and diabetic wounds, defective healing, and/or scars, among others.
G. Method for producing Smad7 protein
In another aspect, methods of producing Smad7 proteins, including any of the Smad7 variants, fragments, truncations, fusion proteins (e.g., PTD-Smad7) described herein, are contemplated. The present inventors have discovered methods of producing Smad7 protein, including nucleic acid codon optimization, at levels and purities sufficient for research, development or commercialization. Thus, methods of generating Smad7 are specifically contemplated, including using one or more of the codon-optimized Smad7 nucleic acid molecules described herein (e.g., in the examples).
Examples
The following examples are included to illustrate various embodiments. It will be appreciated by those of skill in the art that the techniques disclosed in the examples which follow represent techniques discovered to function well in the practice of the claimed methods, compositions, and apparatus. However, those of skill in the art should, in light of the present disclosure, appreciate that many changes can be made in the specific embodiments which are disclosed and still obtain a like or similar result without departing from the spirit and scope of the present technology.
Example 1: smad7 mice are resistant to oral mucositis
A transgenic mouse model expressing human Smad7 protein in keratinocytes was generated as previously described (Han et al, Dev. cell, 11: 301-312, 2006) (K5.Smad 7). Transgene expression in the oral epithelium was confirmed (fig. 7A-B). Mice were bred to a C57BL/6 background, and 8-10 week old male and female transgenic mice as well as wild type littermates were used in the study. These mice showed improved healing of excised skin wounds (Han et al, am. J. Pathol., 179: 1768-.
K5.smad7 mice and wild-type littermates were exposed to cranial radiation to determine the bioequivalent dose (BED) required to induce oral mucositis in the mice. 8Gy × 3(BED ═ 43.2), a clinically relevant protocol to low fraction radiotherapy, was determined to be the minimum dose required to induce oral mucositis (fig. 1A-B). To evaluate the efficacy of Smad7 action, single doses of cranial radiation were also tested and the severity of oral mucositis was found to correlate with BED values between 18Gy (BED 50.4) and 22Gy (BED 70.4) (fig. 1A-B, fig. 7C). On day 9 after the initial radiation was cut off, wild type mice developed oral ulcers (fig. 1A-B).
Smad7 oral mucosa before irradiation had a morphology similar to wild-type mice, but exhibited resistance to radiation-induced oral mucositis (fig. 1A-B). Histological analysis revealed that wild-type mice developed oral mucositis (fig. 1A), which is similar to oral mucositis in humans (fig. 1C). The first subsidiary hospital of the university of Kunming medical, China, provided archived paraffin sections of human tissue to be de-identified and approved for exemption of the study by human subjects. The oral mucositis lesion is from the mucosa of the tongue, cheek, or oropharynx adjacent to recurrent oral cancer that has undergone radiation therapy. Unirradiated oral mucosal sections are from surgically removed sleep apnea oral tissue and tongue biopsies adjacent to cysts (mucus cysts).
Smad7 oral epithelium generally shows radiation dose-dependent damage, i.e., epithelium thinning and tongue papilla flattening after 8Gy x 3 radiation, and epithelial cells more damaged (dysplasia or hypertrophy) after 18Gy and 22Gy radiation (fig. 1A). Consistent with increased leukocyte infiltration in human oral mucositis lesions (fig. 1C), lesions in wild-type mice had numerous infiltrating leukocytes (fig. 1D-E), consisting of neutrophils, macrophages, and lymphocytes (fig. 7D); all cells were substantially reduced in the k5.smad7 oral mucosa (fig. 1D-E and 7D).
Because it is difficult to capture human oral mucositis disease states in the acute phase, a mouse model is used to assess proliferation and apoptosis when ulcers are forming. Similar to previous reports, proliferating cells were rare in irradiated wild-type oral epithelium, but more was observed in irradiated k5.smad7 oral epithelium (fig. 1D and 1F). In contrast, apoptotic cells were significantly reduced in the irradiated k5.smad7 oral mucosa compared to wild type mice (fig. 1D and 1G).
As expected, cells with a nuclear NF- κ B p50 subunit were significantly increased in oral mucositis compared to non-irradiated wild-type oral mucosa (fig. 2A-B). Interestingly, TGF- β 1, which is an immunosuppressant in the visceral organs, but pro-inflammatory in the oral mucosa, together with its activated signaling mediator, phosphorylated (p) Smad2, was also increased in oral mucositis compared to the unirradiated oral mucosa in wild-type mice (fig. 2A-B). Similar changes were also detected in human oral mucositis lesions (fig. 2A-B).
Irradiated K5.Smad7 oral epithelium significantly reduced cells positive for nuclear NF-. kappa. B p50 and pSmad2, even though they still had abundant TGF-. beta.1 protein (FIGS. 2A-B). TGF- β 1mRNA in irradiated wild-type oral mucosa was significantly increased on days 9 and 10 (fig. 2C). TGF- β 1mRNA levels in smad7 mucosa resembled wild-type mucosa at earlier time points, but returned to normal by day 10 (fig. 2C). While not wishing to be bound by any theory, these data suggest that TGF- β 1 transcription is not inhibited by Smad7, and its more rapid decline in the k5.Smad7 mucosa is likely the result of accelerated healing.
The activated marker for BMP signaling phosphorylating Smad1/5/8 was not affected by Smad7 before and after irradiation (fig. 7E). This result is consistent with the ability of Smad7 to preferentially inhibit TGF- β signaling.
Example 2: rac1 promotes Smad 7-mediated keratinocyte migration
To determine whether Smad7 contributed to healing of human oral keratinocytes, Smad7 in spontaneously immortalized human oral keratinocytes (NOK-SI) was knocked out. Smad7 knockdown attenuated keratinocyte migration after opening the wound (fig. 2D and fig. 8A). In contrast, knocking-off TGF-. beta.1 accelerated keratinocyte migration (FIGS. 8B-8D), consistent with the accelerated wound healing observed in mice in the absence of TGF-. beta.1 or Smad 3.
To investigate the molecular mechanisms associated with Smad 7-mediated keratinocyte migration, the protein Rac1 essential for oral wound healing was examined. Rac1 decreased after Smad7 knock-off (fig. 2E). TGF- β 1 overexpression in oral mucositis is expected to activate Rac1 via a Smad-independent mechanism. However, although the total Rac1 protein increased 2-fold after irradiation, the Rac1 protein activated in the wild-type tongue did not change significantly (fig. 2F).
Smad7 oral mucosa, total Rac1 and activated Rac1 were significantly increased 4-fold and 8-fold, respectively, compared to wild-type oral mucosa in k5.smad7 oral mucosa (fig. 2F). To determine the functional significance of Smad 7-induced Rac1 activation, Rac1 was knocked out in primary keratinocytes isolated from wild-type and Smad7 transgenic neonatal skin and an assay for cell proliferation and migration was performed. Rac1 knockdown showed a modest decrease in proliferation of wild-type and Smad7 keratinocytes (fig. 9A-9C), but Smad 7-induced migration was almost completely abolished (fig. 2G and fig. 9D), indicating that increased Rac1 contributes to Smad 7-mediated cell migration.
Increased Rac1mRNA levels in Smad7 transgenic keratinocytes were observed to correlate with total and active Rac1 protein levels (fig. 3A-B and fig. 10A-B), indicating that increased Rac1 activation in Smad7 keratinocytes is at least in part a result of increased Rac1 transcripts. Furthermore, Rac1 protein increased approximately 3-fold after knocking off individual smads in NOK-SI cells (fig. 10C-10E) (fig. 3C). These data indicate that normal Smad signaling represses Rac1 transcription.
Among the two putative Smad Binding Elements (SBEs) (coding sequence upstream-2.1 Kb and-1.5 Kb) in the mouse Rac1 promoter, which in a similar region of the human Rac1 promoter, chromatin immunoprecipitation (ChIP) identified Smad-2, Smad-3, Smad-4 and Smad-7 bound to the-1.5 Kb site (fig. 3D), but not to the-2.1 Kb site in wild-type keratinocytes; binding of Smad-2, Smad-3 and Smad-4 was significantly reduced in Smad7 transgenic keratinocytes (FIG. 3D).
Luciferase reporter gene analysis using the Rac1-Luc construct containing SBE showed that knock-off of Smad7 in wild-type keratinocytes significantly reduced luciferase activity (fig. 3E). In contrast, Smad7 transgenic cells had increased luciferase activity compared to wild-type cells, and mutations in SBE attenuated this increase (fig. 3F). Thus, Smad7 binding to SBE appears necessary to repel signaling Smad to eliminate Rac1 thwarting.
Among the known Smad transcriptional co-repressors, CtBP1 was found to bind to the Rac1 promoter SBE-1.5Kb site in wild-type keratinocytes (fig. 3G), and Smad7transgene expression significantly reduced CtBP1 binding to SBE (fig. 3G-H). When CtBP1 was knocked out in NOK-SI cells, the Rac1 protein and Rac1-Luc activity were increased compared to keratinocytes transfected with scrambled siRNAs (FIGS. 4A-B), indicating that CtBP1 binds to SBE-1.5Kb to repress Rac1 expression. Furthermore, CtBP1 in knock-out NOK-SI cells increased their migration (fig. 4C and 10F).
When examined for CtBP1 protein in radiation-induced oral mucositis, CtBP1 was found to be rarely detected in non-irradiated mouse and human oral mucosa (fig. 4D-4F); however, CtBP1 positive cells were significantly increased in wild-type and irradiated oral mucosa in k5.smad7 mice as well as in human oral mucositis (fig. 4D-4F). In addition, CtBP1mRNA in irradiated wild-type oral mucosa increased significantly on days 9 and 10 (fig. 4G). CtBP1mRNA levels in smad7 mucosa were similar to wild-type mucosa at earlier time points, but declined to normal by day 10 (fig. 4G). These results indicate that Smad7 does not reduce CtBP1mRNA, but rather inhibits binding of CtBP1 to the Rac1 promoter by excluding the Smad/CtBP1 complex from the SBE binding site; in addition, a more rapid decrease in CtBP1 in k5.smad7 mucosa served as a marker for healing.
Example 3: Tat-Smad7 alleviates radiation-induced oral mucositis
The ability of the Smad7transgene to block multiple pathological processes of oral mucositis prompted us to explore whether localized Smad7 delivery could be used to prevent and treat oral mucositis. Because Smad7 is a nucleoprotein, local Smad7 delivery needs to allow rapid entry of Smad7 into the cell before saliva washes away proteins. Thus, a recombinant human Smad7 with an N-terminal Tat-tag was generated, which allows the protein to rapidly penetrate the cell membrane and enter the nucleus. The V5 epitope was added to the C-terminal end of the Tat-Smad7 protein to follow the penetration of Tat-Smad7 cells (FIGS. 11A-11D).
Using its ability to block phosphorylation of Smad2, Tat-Smad7 biological activity was tested (FIG. 11C). A Tat-Cre recombinant protein with the same tag as the control was generated (FIGS. 11E-F) and cloned into pET101-Topo protein expression vector (Invitrogen) containing the sequence encoding C-terminal 6XHis (SEQ ID NO: 40). Transformation of Tat-Cre to BL-21StarTMColi (Invitrogen) to produce Tat-Cre protein and purified using a Ni-NTA column.
SDS-PAGE electrophoresis was used to verify the purity and size of both proteins. To evaluate the transduction and activity of the Tat-Smad7 protein in vitro, Tat-Smad7 was added to primary mouse keratinocytes. Slides were fixed in cold methanol for 5 min and stained for V5 and pSmad 2. Tat-Cre activity was verified by digestion of a 1,460bp loxP-flanked fragment from the 7,650bp vector pLL3.7 (Addgene). For in vivo treatment, 30 μ L of 50% glycerol/PBS was used as vehicle control and Tat-Cre as related protein control. Tat-Smad7 or Tat-Cre (in 30. mu.L of 50% glycerol/PBS, the doses and schedules are specified in each figure) was topically applied to the oral cavity of the mice and the mice were restricted from oral feeding for 1 hour.
For oral mucositis prevention, Tat-Smad7 and Tat-Cre (in 50% glycerol/PBS) were topically applied daily to the oral cavity of 8-10 week old C3H females (Jackson laboratory) or C57BL/6 mice from the 24 hour prior to irradiation to day 8 after the initiation of irradiation. The treated tissue was examined on day 9. Mouse tongues were collected, fixed in 10% formalin, embedded in paraffin, and cut into 5 μm sections. Analysis of histological changes and use of H&E stained slides for ulcers. Another group received palifermin treatment using a clinical protocol, i.e., 6.25mg kg per day prior to irradiation-1(intraperitoneal) for 3 days, and 24 hours daily for 3 days after the last dose of radiation.
Tat-Cre showed no effect compared to vehicle control (FIGS. 5A-B). Tat-Smad7 treatment showed a similar prophylactic effect on ulcer formation as palifermin (FIG. 5A). The dose-dependent effect of Tat-Smad7 was more pronounced when used to administer animals irradiated with a single dose of 20Gy (BED 60) that induced oral ulceration that was greater than fractionated irradiation (fig. 11G). Under the microscope, palifermin and Tat-Smad7 treated oral mucosa prevented open ulceration in most cases (FIG. 5B). Palifermin-treated mucosa exhibited more keratinocyte growth decline than did Tat-Smad 7-treated mucosa, and more damaged keratinocytes (coagulated or charcoal-like nuclei, swollen mononuclear or multinucleated cells, and fragmented nuclear fragments in the stratum corneum) (fig. 5B). Immunostaining revealed that palifermin increased proliferation more significantly than Tat-Smad 7. Tat-Smad7 reduced apoptosis, leukocyte infiltration, nuclear pSmad2, and NF- κ B p50, but not palifermin (FIGS. 5B-5G).
To test whether Tat-Smad7 could be used to treat existing oral mucositis, mice were exposed to fractionated (8Gy X3) cranial radiation and either Tat-Smad7 (topical) or palifermin (6.25mg kg) was applied daily from day 6 after the initial radiation (when mucosal damage was visibly evident) until day 9-1Intraperitoneal). The treated tissue was examined on day 10. Albeit at an earlier point in time than the current schemeAdministration of palifermin after the initial irradiation reduced oral mucositis in mice, but palifermin administration using the current protocol did not accelerate ulcer closure (fig. 6A), independent of its hyperproliferative effect on the entire oral mucosa (fig. 6B). This is not surprising, as palifermin is approved for the prevention of, rather than the treatment of, oral mucositis.
Tat-Smad7 treated oral mucositis reduced ulcer size and pathological changes after fractionated and single dose irradiation (FIGS. 6A-B and 12A-12G). Distant from the ulcer, the Tat-Smad7 treated oral mucosa exhibited less hyperplastic and more differentiated epithelium than the palifermin treated oral mucosa (fig. 6B). The effect of Tat-Smad7 on wound recovery after closure was more pronounced with a single dose of 20Gy radiation that caused slower healing than with fractionated radiation. When the vehicle-treated ulcer had just re-epithelialized, the Tat-Smad 7-treated mucosa had nearly returned to normal morphology (FIG. 6C).
Consistent with observations in k5.Smad7 mice, Tat-Smad7 increased Rac1 promoter activity and decreased CtBP1 binding to SBE of the mouse Rac1 promoter (fig. 12G and 12I), and increased Rac1 protein in mouse oral mucositis and human oral keratinocytes (fig. 6D-E).
Tat-Smad7 treated human oral keratinocytes after wound scarification had accelerated wound closure (fig. 6F and 13A). In addition, irradiated human oral keratinocytes increased nuclear pSmad2 and NF- κ B p50, which was attenuated by Tat-Smad7 treatment (FIG. 13B). In contrast, although Tat-Smad7 efficiently penetrated oral cancer cells (fig. 13C), it did not further increase the level of Rac1 protein already abundant in cancer cells (fig. 13D). This result may indicate that cancer cells migrated faster than normal keratinocytes (FIG. 6F and FIGS. 13A, 13E-13H), and that Tat-Smad7 had a lack of effect on the migration of two oral cancer cell lines: MSK921 without genetic loss of TGF- β signaling component; and Cal27 with a mutated Smad4 (fig. 13E-13H).
Colony assay showed that the survival of human oral keratinocytes was slightly increased by treatment with Tat-Smad7 with or without radiation (fig. 6G). Consistent with the concept that decreased survival after irradiation is more prominent in cancer cells than in normal cells, SCC cells show a substantial decrease in cell survival after irradiation. Treatment with Tat-Smad7 did not affect the survival of SCC cells with or without radiation (FIG. 6G).
Example 4: engineering cell-penetrating Smad7 proteins
It is hypothesized that in order to be effective as a therapeutic agent, SMAD7 needs to be able to penetrate cells efficiently. To accomplish this, the Smad7 sequence was modified to include a protein transduction domain.
Tat sequences from HIV were selected for testing with Smad7 as the protein transduction domain. The nucleotide and protein sequences of Tat used in fusion proteins with Smad7 and Smad7 fragments were derived from Cardarelli et al, trafficApr 9 (4): 528-39(2008). The Tat nucleotide and amino acid sequences are provided below:
ggccgtaaaaaacgccgtcaacgccgccgt(SEQ ID NO:1)
G R K K R R Q R R R(SEQ ID NO:2)
preparing a fusion protein having Tat linked in-frame directly to human Smad7 complementary dna (cdna) at the 5 'or 3' end of Smad7 as shown below:
5′Tat:Ggccgtaaaaaacgccgtcaacgccgccgt(SEQ ID NO:7)-Smad7
3′Tat:Smad7-Ggccgtaaaaaacgccgtcaacgccgccgt(SEQ ID NO:8)
the 5 'Tat-Smad 7 construct included a 3' V5 tag sequence and was cloned into a pGEX-6p-1 protein expression vector (New England Biolabs) to make a GST-Tat-Smad7 fusion protein. The Tat-Smad7 gene was transformed into BL-21Star E.coli (Invitrogen) to produce the Tat-Smad7 protein. Proteins were purified by glutathione column purification and elution using enzymatic cleavage by Glutathione S Transferase (GST) fusion (Precision enzyme, GE Life Sciences).
At the time of creation of the PTD-Smad7 fusion protein, a V5 tag was included at the 3' end to monitor penetration of Tat-Smad7 into cells by immunostaining using V5 antibody. This epitope tag may be deleted for clinical use (e.g., by recloning the sequence in the absence of the V5 tag), as appropriate.
A PTD-Smad7 fusion protein ("6H" as disclosed in SEQ ID NO: 40) with a 6-histidine (6-H) tag (SEQ ID NO: 40) for protein purification was also created (Tat-Smad7-V5-6H) (as "6H" as disclosed in SEQ ID NO: 40) and is shown below. Tat-Smad7-V5-6H ("6H" as disclosed in SEQ ID NO: 40) has the following nucleotide sequence: 1-53 includes the 5' sequence of pET-TOPO; 54-1365 includes Tat-Smad 7; 1366-1497 comprises 3' pET-TOPO containing the V5 epitope and the 6XHis tag (SEQ ID NO: 40) (V5 comprises 1393-1434, the His tag comprises 1444-1461, and the stop comprises 1462-1464).
The Tat-human Smad7 codon optimized for protein production cloned into the pET101/D-Topo vector is as follows:

a comparison of the protein sequences of Tat-Smad7-v5 and Smad7 is provided below. The first amino acid of Smad7 in Tat-Smad7 is not M (unlike Smad7) because Tat-Smad7 is designed to form a GST fusion protein with Tat and/or GST in-frame. Then, Tat-Smad7 was cleaved from the GST fusion protein after purification. The upper case nucleotides identify the V5 tag. Underlined italics indicate amino acids from an optional pET101-Topo backbone vector.
A comparison of Tat-Smad7-v5 with Smad7 is presented below:

example 5: for PTD-Smad7 eggsAdditional assays for white matter activity
Immunofluorescence (IF), Immunohistochemistry (IHC), and TUNEL assays for apoptosis IF and IHC were performed as previously described (Han, G., Li, F., Ten Dijke, P. and Wang, X.J. transient smad7transgene in the epidermis of mice induces accelerated skin wound healing (Temporal small 7transgene induction in mouse epididymis infected wound healing). Am J Pathol 179, 1768-1779 (2011)). The primary antibodies used were guinea pig antibody against K14 (1: 400, Fitzgerald, 20R-CP200), rat antibody against CD4 (1: 20, BD Bioscience, 550278), Ly-6G (1: 20, BD Bioscience, 550291), BM8 (antibody against F4/80, 1: 20, Invitrogen, MF48000), FITC-labeled antibody against BrdU (BD Bioscience, 347583), rat antibody against CD45 for mouse samples (1: 50, BD Bioscience, 550539), mouse antibody against CD45 for human samples (1: 50, Abcam, Ab781), chicken antibody against TGF-. beta.1 (1: 50, R-B-S.B.B.B.B.B.B.B.&D, AF-101-NA), rabbit antibody against CtBP1 (1: 100, Millipore, 07-306), rabbit antibody against NF-. kappa.B 3p50 (1: 200, Santa Cruz Biotechnology, SC-7178), rabbit antibody against PCNA (1: 200, Santa Cruz Biotechnology, SC-7907), rabbit antibody against pSmad2 (1: 100, Cell Signaling Technology, 3101), and mouse antibody against V5 (1: 500, Invitrogen, 460705). For IF, the secondary antibody against a different class of IgG is Alexa 594 (red) or 488 (green) conjugated (all 1: 200, Invitrogen) o for IHC, secondary biotinylated antibodies against different classes of IgG (1: 300, Vector Labs) were used and developed using the Vector Abtain ABC kit (Vector Labs). Apoptotic cells were detected on formalin-fixed tissue sections using the terminal deoxynucleotidyl transferase uridine nick end labeling (TUNEL, G3250) kit (Promega). By intraperitoneal injection of 0.125
594 (red) or 488 (green) conjugated (all 1: 200, Invitrogen) o for IHC, secondary biotinylated antibodies against different classes of IgG (1: 300, Vector Labs) were used and developed using the Vector Abtain ABC kit (Vector Labs). Apoptotic cells were detected on formalin-fixed tissue sections using the terminal deoxynucleotidyl transferase uridine nick end labeling (TUNEL, G3250) kit (Promega). By intraperitoneal injection of 0.125mg g 1 hour before euthanasia-1BrdU labeling was performed in vivo. Quantification of PCNA or BrdU as cells of epithelial length per mm, TUNEL or CD45 positive cells, including all epithelial cellsCells were quantified for epithelial length per mm including all epithelial layers above the muscle layer and stroma, and nuclear pSmad2 or NF- κ B p50 positive cells were quantified as the number of positive cells/total remaining epithelial cells present (i.e., excluding exfoliated epithelial cells induced by irradiation). The continuous field of view of the slide was used to count BrdU labeled cells using metamorphh software.
Cell culture.a Smad7transgene and wild-type primary keratinocytes were prepared from neonatal mouse skin and cultured in PCT medium (lnceltec) as previously described (transient Smad7transgene induction in Han, g., Li, f, Ten Dijke, p, and Wang, x.j. mouse epidermis accelerated skin wound healing (Temporal small 7transgene inducing skin healing): Am J path 179, 1768-. Spontaneous immortalized normal oral keratinocytes (NOK-SI) derived from gingival tissue of healthy volunteers were cultured and maintained in defined keratinocyte medium (Castilho, r.m. et al Rac 1is required for epithelial stem cell function during healing of skin and oral mucosal wounds and not for tissue homeostasis in mice (Rac1 isolated for epithelial stem cell function and oral mucosal wound healing for tissue hostasis in mice) — PloS one 5, e10503 (2010)). In the presence of 10% fetal bovine serum ( (ii) a Invitrogen) of oral Cancer cells Cal27(ATCC) and MSK921(d. raben laboratory, fingerprint collected by University of Colorado Cancer center tissue Culture Core (University of Colorado Cancer center tissue Culture Core). To evaluate the effect of Tat-Smad7 in irradiated cells, the above human cell lines were cultured in chamber slides (BD Bioscience, 354108), irradiated with 3Gy, and Tat-Smad7 (1. mu. gmL) immediately after irradiation-1) Added to the culture medium. Cells were fixed in 100% cold methanol for immunostaining of pSmad2, NF-. kappa. B p50 and
(ii) a Invitrogen) of oral Cancer cells Cal27(ATCC) and MSK921(d. raben laboratory, fingerprint collected by University of Colorado Cancer center tissue Culture Core (University of Colorado Cancer center tissue Culture Core). To evaluate the effect of Tat-Smad7 in irradiated cells, the above human cell lines were cultured in chamber slides (BD Bioscience, 354108), irradiated with 3Gy, and Tat-Smad7 (1. mu. gmL) immediately after irradiation-1) Added to the culture medium. Cells were fixed in 100% cold methanol for immunostaining of pSmad2, NF-. kappa. B p50 and V5 4 hours after Tat-Smad7 treatment.
Transfection with siRNA when subjected toWhen cultured keratinocytes reach 70% confluence, use 2000(Invitrogen) transfected with 100nM target siRNA or scrambled siRNA (Dharmacon). Cells were collected 48-72 hours after transfection and subjected to western analysis to determine knockdown efficiency. For the migration assay, siRNA was transfected when the cells were plated. The target sirnas included in this study were: mouse siRac1-1(Invitrogen, MSS237708) and siRac1-2(IDT, mmc. rnai. nw 009007.12.3); human siSmad2(Dharmacon, L-003561-00-0005), siSmad3(Invitrogen, HSS106252) and siSmad4(Invitrogen, HSS 118066); human siCtBP1-1 and siCtBP 1-2; human siSmad7-1 and siSmad 7-2; human TGF-. beta.1 (Dharmacon, J-012562-08-0005); mouse siSmad 7.
2000(Invitrogen) transfected with 100nM target siRNA or scrambled siRNA (Dharmacon). Cells were collected 48-72 hours after transfection and subjected to western analysis to determine knockdown efficiency. For the migration assay, siRNA was transfected when the cells were plated. The target sirnas included in this study were: mouse siRac1-1(Invitrogen, MSS237708) and siRac1-2(IDT, mmc. rnai. nw 009007.12.3); human siSmad2(Dharmacon, L-003561-00-0005), siSmad3(Invitrogen, HSS106252) and siSmad4(Invitrogen, HSS 118066); human siCtBP1-1 and siCtBP 1-2; human siSmad7-1 and siSmad 7-2; human TGF-. beta.1 (Dharmacon, J-012562-08-0005); mouse siSmad 7.
In vitro keratinocyte proliferation assay in vitro keratinocyte proliferation was determined by BrdU incorporation in wild-type and Smad7 transgenic keratinocytes. Cells at 70% confluence were transfected with Rac1siRNA and replaced with conventional media after 24 hours. In situ cell proliferation kit (Roche Applied Science) was used for in vitro BrdU labeling and detection, and MetaMorph software was used to count BrdU labeled cells.
In vitro cell migration assay when cells reached 100% confluence, 10. mu.g mL was used-1Mitomycin c (sigma) for 2 hours to inhibit cell proliferation and a Fisherbrand pipette tip was used to introduce the scratch wound. The cell migration was photographed daily. Migration analysis was performed when cells reached confluence 24 to 36 hours after siRNA transfection, and cell migration was recorded using Image-J software with the wound area occupied by the migrating cells. For the Tat-Smad7 treatment, cells were exposed to 1 μ gmL in culture medium after wound scarification-1Tat-Smad7 protein or vehicle control (PBS), and medium was replaced every other day with freshly added Tat-Smad7 until migrating cells completely covered the scarred wound.
Cell survival assay Methods in mo were as previously described (Munshi, a., Hobbs, m. and Meyn, r.e. Clonogenic cell survival assay)Molecular medicine110, 21-28(2005)), and cell survival assay was performed with slight modification. Briefly, cells were plated at 500 cells/well (for unirradiated cells) in 12-well plates and increased to 1, 500 cells/well with increasing radiation dose. Cells were irradiated for 24 hours after plating the cells. 1. mu.g mL of-1Tat-Smad7 or the same volume of PBS used to dissolve Tat-Smad7 (control) was added to the culture medium of both irradiated and non-irradiated cells. The medium was replaced every other day with fresh addition of Tat-Smad7 or PBS for 10 to 14 days. Colonies were fixed in methanol, stained in 0.5% crystal violet solution (containing 25% methanol), counted and the average was calculated from 4 wells in each experiment. Two to three independent experiments were performed for each cell line. Relative survival scores were calculated as previously described, i.e., the absolute survival score (number of colonies/total plated cells) at each radiation dose divided by the absolute survival score of unirradiated cells.
Western analysis protein extraction and western analysis were performed as previously described (Li, a.g., Lu, s.l., Zhang, m.x., ding, c. and Wang, x.j.smad 3knockout mice exhibited resistance to the development of skin chemical cancers (Smad3knockout micro external existence to skin chemical carcinogenesis) Cancer Res 64, 7836-7845 (2004)). Antibodies used in this study included rabbit antibody to Smad7 (1: 500), rabbit antibody to Smad2 (1: 300, Zymed, 51-1300) and rabbit antibody to Smad4 (1: 300, Epitomics, 1676-1), rabbit antibody to Smad3 (1: 300, Cell Signaling Technology, 9513), mouse antibody to Rac1 (1: 500, BD Biosciences, 610651), rabbit antibody to CtBP1 (1: 500, Millipore, 07-306), mouse antibody to tubulin (1: 3000, Sigma, T5168), mouse antibody to GAPDH (1: 5000, Abcam, Ab8245), and mouse antibody to actin (1: 1000, Santa Cruz Biotechnology, SC 1616). Use of Version 1.2 software (LI-COR Biosciences) acquired grayscale images.
Version 1.2 software (LI-COR Biosciences) acquired grayscale images.
Rac1 activation divisionUse of BIOCHEM for Rac1 activationTMThe kit (Cytoskeleton Inc, BK035) examined active GTP-bound Rac 1. Wild-type and Smad7 transgenic keratinocytes were cultured in 15cm diameter tissue culture plates and protein lysates were prepared using the lysis buffer provided. To analyze Rac1 activity, 1mg of cell lysate was used. To examine the total Rac1 and Smad7 proteins, 50 μ g of lysate was used. To measure GTP-bound Rac1 in the mouse tongue, half of the tongue was ground in liquid nitrogen and lysed with lysis buffer to extract proteins, GTP-bound Rac1 was analyzed in 2mg of protein lysate per sample, and 50 μ g of protein lysate was loaded for the total Rac1 protein western blot method.
ChIP assay As described previously (HGF upregulation of proteins in microwith Keratinocyte-specific Smad2deletion) in mice with Keratinocyte-specific Smad2deletion J Clin Invest 120, 3606 3616 (2010); Hort, K.E. Keratinocyte-specific Smad2ablation increases epithelial-mesenchymal transition during skin carcinogenesis and progression (Keratino-specific Smad2 inhibition responses in capillary-mesenchymal protein formation and promotion) Owens et al J.Clin.118, Smat 2. 2732 (Biocore 4-dependent protein expression cassette 19-19), expression of protein in follicle-19 expression kit (ChIP 2008. 19-19) using the Chot, K.E. HGF upregulation contributes to microwith Keratinocyte-specific Smad2deletion) in mice with Keratinocyte-specific Smad2deletion, J.Clin.118, Smat 2. 2732 (Biofine-dependent protein expression cassette) expression kit ( Chip 35, 322, 19, 2, 3. Isolating the DNA-protein complex from primary mouse keratinocytes. For ChIP, 6.3 μ G of cleaved chromatin was incubated with protein G magnetic beads and 2 μ G of each of the following: rabbit antibody against Smad2 (Cell Signaling Technology, 3122), rabbit antibody against Smad3 (Cell Signaling Technology, 9523), rabbit antibody against Smad4 (Cell Signaling Technology, 9515), Smad7 antibody (Santa Cruz Biotechnology, SC-11392), CtBP1(Millipore), or negative control rabbit IgG (Santa Cruz Biotechnology, SC-2027). DNA eluted from the protein-DNA complexes was used for PCR analysis and CtBP1 binding to Rac1 promoter was compared in wild type and Smad7 transgenic keratinocytes by ChIP band intensity on gel images or by quantitative PCR using Power SYBR Green Master Mix (applied biosystems). Primers for amplifying the Rac1SBE-1.5Kb promoter region:
5'-TGGAATTCCTGGTCTGGTTT-3' (sense) (SEQ ID NO: 13)
5'-GCCAAGCTGCTCTTCCAGTA-3' (antisense) (SEQ ID NO: 14)
5'-TCTCAGGGGGCCAAAGGTGTT-3' (sense) (SEQ ID NO: 15)
5'-TCCCAGCACCTGAATCACATGG-3' (antisense) (SEQ ID NO: 16)
An 883bp fragment of-1671 bp to-789 bp of Rac1 promoter encompassing SBE-1.5Kb site was amplified from wild-type mouse DNA using 5 'XhoI and 3' HindIII labelled primers and this Rac1 promoter fragment was cloned into pgl4.26 vector (Promega) to make Rac1 promoter-pgl 4.26 luciferase reporter (Rac1-Luc) constructs. For site-directed mutagenesis, SBE sequence 5'-TGTCTGTGCT-3' (SEQ ID NO: 17) was mutated to 5'-TGATAGAGCT-3' (SEQ ID NO: 18). Rac1-Luc and pGL4.74 (1: 20) were co-transfected with Smad7siRNA, CtBP1siRNA or scrambled siRNA to primary mouse keratinocytes using Lipofectamine 2000(Invitrogen) or with Tat-Smad7 (1. mu.gmL)-1) Treated primary mouse keratinocytes. 48 hours after transfection or Tat-Smad7 treatment, use Reporter gene assay kit (Promega), following the manufacturer's instructions, cell lysates were collected and subjected to luciferase assay. Rac 1-luciferase activity was measured using the Glomax machine (Promega) and is expressed as the ratio of firefly activity to Renilla activity. Primers used to amplify the Rac1 promoter sequence were:
Reporter gene assay kit (Promega), following the manufacturer's instructions, cell lysates were collected and subjected to luciferase assay. Rac 1-luciferase activity was measured using the Glomax machine (Promega) and is expressed as the ratio of firefly activity to Renilla activity. Primers used to amplify the Rac1 promoter sequence were:
5′-ATCCTCGAG-TATCCTCCAGGTCTGGG-3′(SEQ ID NO:19)
5′-GCCAAGCTT-AGCGTCCAGCGTTAACCTG-3′(SEQ ID NO:20)
statistical analysis statistical differences in molecular analysis and size of oral mucositis ulcers were analyzed using the saunders t-test (Student's t-test), and all data except for ulcer size are presented as mean ± s.d. and ulcer size is presented as mean ± s.e.m. The incidence of oral mucositis was analyzed by a fisher's exact test.
Example 6: codon optimization for Smad7 protein production in E.coli or yeast
Although many mammalian proteins can be produced in bacteria in the absence of nucleotide sequence modifications, analysis indicates that modifications to the Smad7 nucleotide sequence will be required to allow protein expression in bacteria.
Analysis of mammalian codon usage of Smad7cDNA revealed nine arginine amino acids encoded by the following nucleotides: 7-9, 43-45, 169-171, 403-405, 490-492, 526-528, 823-825, 1057-1059 are rare codons (AGG, codon usage 1.7%). Because these codons are rare codons in bacteria, it is expected that they can stop or reduce protein translation and/or production in bacteria. The amino acids encoded by the rare arginine codons are indicated in the illustrated human Smad7 protein in bold capital letters below, including arginine at positions 3, 15, 57, 135, 164, 169, 176, 275, and 353. In addition, the following arginine codons also have low frequency of use. CGA (3.5% codon usage): nucleotides 16-18, 136-; CGG (5.4% codon usage): nucleotides 31-33, 112-; AGA (2.8% codon usage): nucleotides 637-639, 814-816 which encode arginines at positions 213, 272. These arginine residues are highlighted in bold uppercase R below and become CGC (20.6% codon usage) in at least one of the codon optimized nucleic acid sequences:

based on this analysis, it was decided to optimize the Smad7 nucleotide sequence to a codon believed to allow increased production of the Tat-Smad7 protein in E.coli or yeast. The optimized nucleic acid codon sequence made by Genscript is provided below. Briefly, the sequence has the following composition: nucleotides 1-6 include a restriction recognition site for BamHI; nucleotides 7 to 36 include the Tat sequence; nucleotides 37-1314 include codon-optimized human Smad7 cDNA; nucleotide 1342-1383 includes the V5 epitope; nucleotide 1384-1386 is a stop codon; and nucleotides 1387 and 1392 include a restriction recognition site for SalI. In this sequence, the ATG is removed for use with GST. The entire designed sequence was transformed into E.coli codons based on the "codon usage database". The initial optimized Smad7 sequence (SEQ ID NO: 23) is shown below:


a nucleotide sequence comparison between the Tat-Smad7-V5(SEQ ID NO: 23) and human Smad7(SEQ ID NO: 22) cDNAs is provided below. Human Smad7 shares 68% codon homology with codon-optimized Tat-Smad 7-V5. Human Smad7 and codon-optimized Tat-Smad7 share 71% codon homology. Human Smad7 and codon optimized Smad7 share 73% codon homology.
And (3) comparison: global DNA alignment against reference molecules
Parameters are as follows: the scoring matrix is: linearity (mismatch 2, open gap 4, extended gap 1)
Reference molecule: human Smad7mRNA, region 1-1281
Number of aligned sequences: 2
Setting: similarity significance cutoff value: not less than 90%
Summary of match percentage:
reference: human Smad7mRNA 1-1281(1281bp) -
Sequence 2: Tat-Smad 7-V51-1392 (1392bp) 68%

In this optimization, Met216, which could form an alternative open reading frame, was not altered if possible, as it was required to preserve the amino acid sequence of Smad 7. In future codon optimization, Met216 will be mutated to Leu216 to improve protein production without affecting in vitro and in vivo function.
Example 7: production of truncated Smad7 protein
Smad7 is believed to have several activities in vivo, including (but not limited to) one or more of the following: enhanced cell proliferation, enhanced cell migration, reduced DNA damage, reduced apoptosis, and reduced inflammation. The effect of Smad7 on these processes is due to one or more of the following: block TGF- β signaling, block NF- κ B signaling, block CtBP1 activity, and/or increase Rac1 expression and/or activity. It is believed that the smaller functional domain of Ptd-Smad7 may be sufficient to deliver a therapeutic effect (see, e.g., fig. 15). In addition, the resulting shorter protein sequences are expected to enhance protein production. In addition, it is believed that different truncated Tat-Smad7 proteins comprising a partial Smad7 sequence may be useful in different therapies.
For example, it is believed that the C-terminal MH2 domain of Smad7 (about half the length of Smad7 protein, e.g., 208-. Smad7 peptides having this anti-inflammatory function may be sufficient and optionally an improvement for the treatment of chronic inflammation-related conditions, such as (but not limited to) oral mucositis, stomatitis, and psoriasis, among others.
The N-terminal MH1 domain plus linker region (about half of the protein, e.g., 2-208aa) of Smad7 is known to activate MAPK and bind to ubiquitin E3 ligase Smurf to degrade the TGF- β receptor (Aragon et al, Structure 20: 1726-1736 (2012)). It is believed that it may primarily mediate cell migration and/or block TGF-beta induced growth arrest and/or fibrotic responses. Smad7 peptides with this cell migration and proliferation function may be sufficient and optionally an improvement for enhanced healing not associated with excessive inflammation. Types of wounds that may benefit from this form of treatment include, but are not limited to, surgical wounds, fibrotic scars, and diabetic wounds, defective healing, and/or scars, among others.
Truncated Smad 7N-and C-terminal PTD-fusion proteins were designed. An example of a codon optimized nucleotide and protein sequence for the Tat-Smad7-C terminus is provided below. In the nucleic acid sequence, nucleotides 1-6 include a restriction recognition site for BamHI; nucleotides 7-36 include the Tat PTD sequence; nucleotides 37-810 include codon optimization for the C-terminal amino acids 258 to 426 of human Smad 7; nucleotide 568-609 includes the V5 epitope sequence; nucleotides 610-612 include a termination sequence; and nucleotide 613-618 included a restriction recognition site for SalI:


in the nucleic acid sequence, nucleotides 1-6 include a restriction recognition site for BamHI; nucleotides 7-36 include the Tat PTD sequence; nucleotides 37-810 include codon optimization for the N-terminal amino acids 1-258 of human Smad 7; nucleotide 811-852 includes the V5 epitope sequence (corresponding amino acid sequence bold); nucleotide 853-855 includes a termination sequence; and nucleotides 856-861 comprise a SalI restriction recognition site. Removal of ATG to allow fusion with GST:


example 8: testing of truncated Smad7 proteins
The activity of the truncated Smad7 protein was tested using the in vitro and in vivo assays described above for testing full-length Smad7, as well as other assays. Such assays include, but are not limited to, blocking phosphorylation of Smad2 and/or nuclear translocation of the NF- κ Bp50 subunit, increasing cell proliferation, reducing apoptosis and/or radiation-induced DNA damage, reducing inflammation and/or angiogenesis, promoting the healing of oral mucositis, surgical wounds, diabetic wounds, and/or wounds associated with chronic inflammation in mice.
In the wound healing assay, a 6-mm punch biopsy was taken from wild type mice followed by daily topical application of either C-or N-terminal Tat-Smad 7. By measuring gross wound closure, the two truncated Smad7 proteins described above (e.g., Tat-Smad7C terminal and N-terminal proteins) were found to promote wound healing similar to full-length Tat-Smad 7.
Example 9: truncated Smad7 protein to accelerate wound healing
The ability of the truncated Smad7 protein to accelerate wound healing was examined in wild-type mice. FIG. 16A shows the effect of Tat-C-Smad7 truncated at the C-terminus (259-426aa) on a mouse wound healing model. Wild type C57BL/6 mice were anesthetized, biopsies were taken by 6-mm skin punch to open an incision on the dorsal side, and treated every other day by topical application of PBS (control, 3 mice, 4 wounds per mouse), full length Tat-Smad7(0.4 μ g/10 μ L PBS/wound, 3 mice, 4 wounds per mouse), or Tat-C-Smad7(0.4 μ g/10 μ L PBS/wound, 3 mice, 4 wounds per mouse, treatment of a total of 12 wounds). The wound area was photographed with a canon digital camera and measured by imaging the analysis with a 6-mm circle inside the photograph for normalization using Image J software on days 1,2, 4 and 5 after opening the wound and calculating the average wound area percentage remaining for each treatment group. The ability of Tat-C-Smad7 to accelerate wound healing was similar to full-length Smad7 (FIG. 16A).
FIG. 16B illustrates the effect of N-terminally truncated (1-258aa) Tat-N-Smad7 on wound healing. Wild type C57BL/6 mice were anesthetized, biopsies were taken through a 6-mm skin drill to open an opening in the dorsal side, and treated every other day by topical application of PBS (control, 6 mice, 4 wounds/mouse), full length Tat-Smad7(0.4 μ g/10 μ L PBS, 6 mice, 4 wounds/mouse), or Tat-N-Smad7(0.4 μ g/10 μ L PBS, 6 mice, 4 wounds/mouse). The wound area was measured by imaging the analysis with a 6-mm circle inside the photograph for normalization using Image J software on days 1,2, and 4 after opening the wound, and the average wound area percentage remaining was calculated for each treatment group. There was a significant difference in the rate of wound healing after two days between control and Tat-Smad7 treated mice, the latter healing faster (fig. 16B, p < 0.05). Although Tat-N-Smad7 was tested as a negative control, it unexpectedly promoted significantly accelerated wound healing relative to control mice only after one day, and two days later relative to full-length Tat-Smad7 treated mice (fig. 16B, p < 0.05). Tat-Cre had no effect on wound closure relative to control treatment (data not shown).
These results show that both full-length Tat-Smad7 and truncated Tat-Smad7 proteins (Tat-C-Smad7 and Tat-N-Smad7) promote wound healing. Furthermore, these results show that certain truncated Tat-Smad7 proteins are more effective at accelerating wound healing than full-length Tat-Smad 7.
Thus, compositions comprising a truncated Tat-Smad7 protein are useful for treating wounds and for accelerating wound healing.
Example 10: smad7 accelerated wound healing in a model of impaired wound healing
Diabetic (db/db) mice were opened with an incision on the dorsal side by 6-mm skin punch biopsy as described in example 9 above, and PBS (control, 6 mice, 4 wounds per mouse), full-length Tat-Smad7(0.4 μ g/10 μ L PBS, 6 mice, 4 wounds per mouse) or every other day when the wound was not completely covered with scabs by 8 days ago Cream (swab applied, 6 mice, 4 wounds/mouse) officeIs applied to the wound for treatment. After 10 days, a treatment was applied locally to the gap between the scab and the wound periphery to avoid the blockage of the hard scab and the uninjured stratum corneum. Recombinant human platelet-derived growth factor (PDGF) -containing compounds
Cream (swab applied, 6 mice, 4 wounds/mouse) officeIs applied to the wound for treatment. After 10 days, a treatment was applied locally to the gap between the scab and the wound periphery to avoid the blockage of the hard scab and the uninjured stratum corneum. Recombinant human platelet-derived growth factor (PDGF) -containing compounds Approved for topical administration to diabetic ulcers.
Approved for topical administration to diabetic ulcers.
Wound closure was visually assessed on days 1,2, 4,6, 8, 9, 10, 11, 12, and 13 after opening the wound (fig. 17A). At each time point, the wound was photographed with a canon digital camera. After 8 days, there was a clear visual improvement in oral closure in Tat-Smad7 treated mice compared to controls (fig. 17A).
Wound size was measured by using Image J software for normalization at 6-mm circles inside the photographs on days 1,2, 3, 4,7, 9, and 11 after opening the wound and calculating the average percent wound area remaining for each treatment group. The ability of Tat-Smad7 to accelerate wound healing is similar to (FIG. 17B). On day 7, image analysis results indicated that wound closure was significantly accelerated in Tat-Smad7 treated mice relative to control (fig. 17B, p < 0.05). These results are similar to the use
(FIG. 17B). On day 7, image analysis results indicated that wound closure was significantly accelerated in Tat-Smad7 treated mice relative to control (fig. 17B, p < 0.05). These results are similar to the use Those achieved.
Those achieved.
Formalin-fixed paraffin-backed wound sections (1mm) from day 8 wound samples were stained with hematoxylin and eosin (H & E). Histological comparison of wound samples on day 8 revealed complete re-epithelialization and accelerated wound closure in Smad 7-treated db/db mice relative to control (fig. 17C).
Taken together, these results demonstrate that Tat-Smad7 is useful for accelerating wound closure in poorly healing diabetic wounds and providing for Alternative regimens of treatment.
Alternative regimens of treatment.
Example 11: additional codon optimization for Smad7 protein production
Smad7 nucleic acid molecules were designed with selected additional nucleotide changes to increase protein production. For example, the utilization of codons encoding the amino acids Ser and His will be manipulated. In the codon-optimized human Smad7 in the above examples, the Ser codon (TCC or TCG) has an amino acid frequency of about 9% codon usage. It is believed that changing the codon for Ser to AGC will increase Smad7 protein production, at least in part because it can optionally increase codon usage to 15%. 33 Ser amino acids (nucleotides at positions 19-21, 46-48, 133-, 212. 231, 232, 247, 249, 259, 283, 303, 307, 315, 336, 337, 344, 352, 365, 376, 398, 413, 414, 425). Among these, 23 can be altered without introducing a potentially alternative open reading frame (nucleotides 19-21, 292-.
Similarly, in the codon-optimized human Smad7 in the above example, the His codon (CAC) had a codon usage of 9.6%. It is believed that changing the His codon to CAT (optionally to 12.6% usage) will increase Smad7 protein production. 12 His (nucleotides 142-. Of these, 4 (nucleotides 217-219, 220-222, 589-591, 778-780, histidine residues 73, 76, 197, 260) can be varied without introducing a potential alternative open reading frame.
In addition, wild-type human Smad7 includes the Met amino acid as amino acid 216(Met 216). This can be considered an alternative open reading frame by, for example, bacterial agencies, and reduces protein production. It is believed that changing Met216 to Leu216(ATG to CTG), an amino acid with biochemical properties closest to Met and therefore not expected to change the 3D structure of the protein, will increase protein production.
A comparison between the original codon-optimized Tat-Smad7-V5 and further changes is provided below. Winding: Tat-Smad7-V5(SEQ ID NO: 23): and (3) chain descending: after optimized Ser, His and M216L mutations (SEQ ID No: 30).
And (3) comparison: local DNA homology
Parameters are as follows: the double-stranded method comprises the following steps: FastScan-Max Score

Number of aligned sequences: 2
Setting: similarity significance cutoff value: not less than 60 percent
A homologous block: match percentage 94 score 1227 length 1392

The nucleic acid sequences and their corresponding amino acid sequences that will include all of these variations are provided below. The amino acid sequence includes the V5 epitope, indicated in bold, and in italicsAnd the pET101-Topo backbone, indicated underlined. Tat-Smad7M216LThe fully optimized full-length nucleotide and protein sequences are shown below:


the optimized nucleotide and amino acid sequences will also be used to make a variety of N-terminal and C-terminal Tat-Smad7 fragments. Representative examples are provided below.
Optimized nucleotide and amino acid sequences of Tat-N-Smad7-V5 are provided. The protein sequence includes the V5 epitope indicated in bold capital letters.


Optimized nucleotide and amino acid sequences for Tat-C-Smad7-V5 are provided. The protein sequence includes the V5 epitope (indicated in bold capital letters), and the pET101-Topo backbone (indicated in underlined italics).

A comparison of before and after the above optimization is provided below. C-terminal optimization (top chain) (alignment discloses SEQ ID NOs 34 and 24, respectively, in order of appearance):
and (3) comparison: local DNA homology
Parameters are as follows: the double-stranded method comprises the following steps: FastScan-Max Score
Mol 1Tat-C-Smad7-ser-his optimization (1-618) Mol 2Tat-C end Smad7-V5(1-618)
Number of aligned sequences: 2
Setting: similarity significance cutoff value: not less than 60 percent
A homologous block: match percentage 93 score 541 length 618
N-terminal optimization (top chain) (alignment discloses SEQ ID NOs 32 and 26, respectively, in order of appearance):
and (3) comparison: local DNA homology
Parameters are as follows: the double-stranded method comprises the following steps: FastScan-Max Score
Mol 1Tat-N-Smad7-V5-Ser-His optimized (1861) Mol 2Tat-N-Smad7-V5(1-861)
Number of aligned sequences: 2
Setting: similarity significance cutoff value: not less than 60 percent
A homologous block: percent match 95 score 781 length 861

In addition, other codon-optimized nucleic acids will also be evaluated for their ability to produce Smad7 protein in one or more expression systems. Another example of such a sequence is provided below.
Tat-Smad7 optimized by Optimizer programM7216L-V5:



The nucleotide sequence is as follows:
1-6: BamHI; 7-36: tat; 37-1314: codon-optimized human Smad 7; 1315-1356: v5; 137-1359: terminating; 1360 + 1365SalI
Removal of ATG for use with GST; 682ATG to CTG (M216 to L)


Comparison with the sequence of Tat-Smad7M7216L-V5 described in example 4 (alignment discloses SEQ ID NO 36 and 30, respectively, in order of appearance):
and (3) comparison: global DNA alignment against reference molecules
Parameters are as follows: the scoring matrix is: linearity (mismatch 2, open gap 4, extended gap 1)
Reference molecule: Tat-Smad 7-216L-V5-optimizer, region 1-1365
Number of aligned sequences: 2
Setting: similarity significance cutoff value: not less than 60 percent
A summary of the match percentage;
reference: Tat-Smad 7-216L-V5-optimizer 1-1365(1365bp) -
Sequence 2: tatsmad7Ser-His optimized-682 mutant 1-1392(1392bp) 79%

The following sequences were used to produce proteins by transfecting mammalian expression vectors (e.g., pCMV-6-Entry) into HEK293 or CHO cells.
An optimized GST-Tat-Smad7-myc-flag nucleotide sequence cloned in pCMV6-Entry plasmid:


use of OPTIMUMGENE from GenScriptTMThe program was codon optimized for mammalian expression for the entire sequence (up to the NotI site).
1790-1792: changing GAT to GAC (Asp Retention) to avoid alternative ORFs (highlighted)
1-82: cloning site + kozac sequence in pCMV6-Entry
83-777: GST + precision enzyme site; 776-805: tat;
806-2086: full-length human Smad7
2087-2107:MluI、NotI、XhoI
2108-2182: myc-flag from pCMV6-Entry
Bold underline: unique NotI site
The above nucleotide sequence has 79% homology with native human Smad 7.
Number of aligned sequences: 2
Setting: sorting results listed by Score



Optimized GST-Tat-Smad7-myc-flag protein sequence from the above nucleotide sequence:

1-229: GST + precision site
232-668Tat-Smad7
671-699: restriction site + myc-Flag
An optimized GST-Tat-N-Smad7-myc-flag nucleotide sequence cloned in pCMV6-Entry plasmid:

the entire sequence (up to the NotI site) was codon optimized for mammalian expression using the optimummenetm program from GenScript.
1-50: cloning site + kozac sequence in pCMV6-Entry
51-743: GST + precision enzyme site
744-773-:Tat
774-1549-: encoding human Smad7 of N-terminal 1-258aa
1550-1563: NotI, XhoI, bold underlined: unique NotI site
1564-1637: myc-flag from pCMV6-Entry
Optimized GST-Tat-N-Smad7-myc-flag protein sequence from the above nucleotide sequence:

1-241:GST-Tat
244-500:N-Smad72-258
501-528: restriction site + myc-Flag
An optimized GST-Tat-C-Smad7-myc-flag nucleotide sequence cloned in pCMV6-Entry plasmid:


the entire sequence (up to the NotI site) was codon optimized for mammalian expression using the optimummenetm program from GenScript.
1-50: cloning site + kozac sequence in pCMV6-Entry
51-743: GST + precision enzyme site
744-773:Tat
774-1274: c terminal Smad7
1275:Mlu
1283:NotI
1275-1370:mluI、NotI、XhoI、myc+flag
Optimized GST-Tat-C-Smad7-myc-flag protein sequence from the above nucleotide sequence:
1-241: GST + Precist + Tat
242-409:c-Smad7(259-426aa)
410-440: mlu + Not + myc + flag: restriction site + myc-Flag
Based on the Smad7 structure, it was expected that the PY domain (203-258aa) containing linker (203-258aa) would have therapeutic effect by blocking TGF- β induced inflammation or growth inhibition (FIG. 15).
The amino acid sequence of the Tat-Smad 7-linker peptide (203-258aa) is:
GRKKRRQRRR-ELESPPPPYSRYPMDFLKPTADCPDAVPSSAETGGTNYLAPGGLSDSQLLLEPGDR(SEQ ID NO:95)
1-10:Tat
the codon-optimized nucleotide sequences for the above peptides in bacterial production are:

1-30:Tat
31-198: smad7 linker
199-249:V5
The codon-optimized nucleotide sequences of the above peptides in mammalian cell production are:

1-30: mammalian codon optimized Tat
31-198: smad7 linker
199-285: NotI, Myc tag, Flag tag
The amino acid sequence of the Tat-Smad7-PY peptide (203-217aa) is:
GRKKRRQRRR-ELESPPPPYSRYPMD(SEQ ID NO:98)
1-10:Tat
the codon-optimized nucleotide sequences for the above peptides in bacterial production are:

1-30:Tat
31-75:Smad7-PY
76-126:V5
the codon-optimized nucleotide sequences of the above peptides in mammalian cell production are:

1-30: mammalian codon optimized Tat
31-75:Smad7PY
76-162: NotI, Myc tag, Flag tag
The foregoing discussion of the present technology has been presented for purposes of illustration and description. The foregoing is not intended to limit the present technology to the form disclosed herein. Although the description of the present technology has included description of one or more embodiments and certain variations and modifications, other variations and modifications are within the scope of the present technology, e.g., as may be within the skill and knowledge of those in the art, after understanding the present disclosure. It is intended to obtain rights which include alternative embodiments to the extent permitted, including alternate, interchangeable and/or equivalent structures, functions, ranges or steps to those claimed, whether or not such alternate, interchangeable and/or equivalent structures, functions, ranges or steps are disclosed herein, and without intending to publicly dedicate any patentable subject matter.









































Claims (7)
1. Use of a pharmaceutical composition in the manufacture of a medicament for treating a wound or promoting wound healing, wherein the pharmaceutical composition comprises a protein molecule comprising a protein transduction domain and a human Smad7 protein fragment consisting of amino acids 2-258 of the human Smad7 protein, and one or more pharmaceutically acceptable excipients, wherein the Smad7 protein fragment retains one or more of the biological activities of a functional full-length Smad7 protein, and wherein the protein molecule does not comprise the C-terminus of the Smad7 protein.
2. Use of a pharmaceutical composition in the manufacture of a medicament for treating or promoting wound healing, wherein the pharmaceutical composition comprises a protein molecule comprising a protein transduction domain and a fragment of human Smad7 protein consisting of amino acid 259-426 of the human Smad7 protein, the fragment of Smad7 protein retaining one or more of the biological activities of a functional full-length Smad7 protein, and one or more pharmaceutically acceptable excipients, and wherein the protein molecule does not comprise the MH1 domain of the Smad7 protein.
3. The use of claim 2, wherein the protein molecule does not comprise the N-terminus of a Smad7 protein molecule.
4. The use of any one of claims 1 to 3, wherein the protein transduction domain is Tat.
5. Use of a pharmaceutical composition in the manufacture of a medicament for treating or promoting healing of a wound, wherein the pharmaceutical composition comprises a nucleic acid molecule comprising a fragment of a human Smad7cDNA nucleotide sequence, wherein the nucleotide sequence encodes a protein molecule of any one of claims 1 to 3 and does not encode a full length Smad7 protein.
6. The use of claim 5, wherein the nucleotide sequence further comprises a nucleotide sequence encoding a protein transduction domain.
7. The use of claim 6, wherein the protein transduction domain is Tat.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361775252P | 2013-03-08 | 2013-03-08 | |
| US61/775,252 | 2013-03-08 | ||
| PCT/US2014/022052 WO2014138670A1 (en) | 2013-03-08 | 2014-03-07 | Ptd-smad7 therapeutics |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN105358704A CN105358704A (en) | 2016-02-24 |
| CN105358704B true CN105358704B (en) | 2020-01-03 |
Family
ID=51492023
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201480025596.9A Active CN105358704B (en) | 2013-03-08 | 2014-03-07 | PTD-SMAD7 therapeutics |
Country Status (9)
| Country | Link |
|---|---|
| US (5) | US10456448B2 (en) |
| EP (1) | EP2964774B1 (en) |
| JP (1) | JP6576251B2 (en) |
| CN (1) | CN105358704B (en) |
| CA (1) | CA2904329C (en) |
| HK (1) | HK1219986A1 (en) |
| IL (1) | IL241190B (en) |
| SG (1) | SG11201507045TA (en) |
| WO (1) | WO2014138670A1 (en) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9084746B2 (en) | 2010-09-22 | 2015-07-21 | The Regents Of The University Of Colorado, A Body Corporate | Therapeutic applications of SMAD7 |
| EP2964774B1 (en) | 2013-03-08 | 2020-05-06 | The Regents of The University of Colorado, A Body Corporate | Ptd-smad7 therapeutics |
| KR101853923B1 (en) * | 2016-01-25 | 2018-05-02 | 연세대학교 산학협력단 | A composition containing smad protein for treatment of autoimmune diseases, a fusion protein comprising smad protein, a expression vector and a method for preparing the same |
| US20200332302A1 (en) * | 2017-12-30 | 2020-10-22 | The Regents Of The University Of Colorado, A Body Corporate | Therapeutics of ptd-smad7 fusion proteins |
| WO2023044277A1 (en) * | 2021-09-15 | 2023-03-23 | The Regents Of The University Of Colorado, A Body Corporate | Smad7 polypeptide formulations |
| WO2023150743A2 (en) * | 2022-02-07 | 2023-08-10 | Aavogen Inc. | Codon-optimized smad7 gene therapy to treat and prevent muscle wasting and to enhance muscle mass |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6251628B1 (en) * | 1997-05-20 | 2001-06-26 | Ludwig Institute For Cancer Research | Isolated nucleic acid molecules encoding Smad7 |
| CN101238215A (en) * | 2005-04-05 | 2008-08-06 | 先锋高级育种国际公司 | Methods and compositions for designing nucleic acid molecules for polypeptide expression in plants using plant virus codon-bias |
| WO2012040295A2 (en) * | 2010-09-22 | 2012-03-29 | The Regents Of The University Of Colorado | Therapeutic applications of smad7 |
Family Cites Families (45)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NL8200523A (en) | 1982-02-11 | 1983-09-01 | Univ Leiden | METHOD FOR TRANSFORMING IN VITRO PLANT PROTOPLASTS WITH PLASMIDE DNA. |
| US4879236A (en) | 1984-05-16 | 1989-11-07 | The Texas A&M University System | Method for producing a recombinant baculovirus expression vector |
| US4952500A (en) | 1988-02-01 | 1990-08-28 | University Of Georgia Research Foundation, Inc. | Cloning systems for Rhodococcus and related bacteria |
| US5703055A (en) | 1989-03-21 | 1997-12-30 | Wisconsin Alumni Research Foundation | Generation of antibodies through lipid mediated DNA delivery |
| US5302523A (en) | 1989-06-21 | 1994-04-12 | Zeneca Limited | Transformation of plant cells |
| US5550318A (en) | 1990-04-17 | 1996-08-27 | Dekalb Genetics Corporation | Methods and compositions for the production of stably transformed, fertile monocot plants and cells thereof |
| US7705215B1 (en) | 1990-04-17 | 2010-04-27 | Dekalb Genetics Corporation | Methods and compositions for the production of stably transformed, fertile monocot plants and cells thereof |
| US5322783A (en) | 1989-10-17 | 1994-06-21 | Pioneer Hi-Bred International, Inc. | Soybean transformation by microparticle bombardment |
| JPH05505102A (en) | 1989-12-21 | 1993-08-05 | ホワイトヘツド・インスチチユート・フオー・バイオメデイカル・リサーチ | How to deliver molecules into eukaryotic cells |
| US5484956A (en) | 1990-01-22 | 1996-01-16 | Dekalb Genetics Corporation | Fertile transgenic Zea mays plant comprising heterologous DNA encoding Bacillus thuringiensis endotoxin |
| US5670488A (en) | 1992-12-03 | 1997-09-23 | Genzyme Corporation | Adenovirus vector for gene therapy |
| US5384253A (en) | 1990-12-28 | 1995-01-24 | Dekalb Genetics Corporation | Genetic transformation of maize cells by electroporation of cells pretreated with pectin degrading enzymes |
| AU2515992A (en) | 1991-08-20 | 1993-03-16 | Genpharm International, Inc. | Gene targeting in animal cells using isogenic dna constructs |
| US5610042A (en) | 1991-10-07 | 1997-03-11 | Ciba-Geigy Corporation | Methods for stable transformation of wheat |
| US5702932A (en) | 1992-07-20 | 1997-12-30 | University Of Florida | Microinjection methods to transform arthropods with exogenous DNA |
| JP2952041B2 (en) | 1992-07-27 | 1999-09-20 | パイオニア ハイ−ブレッド インターナショナル,インコーポレイテッド | Improved method for AGROBACTERIUM-mediated transformation of cultured soybean cells |
| EP0656950B1 (en) | 1992-08-21 | 1998-11-04 | Biogen, Inc. | Tat-derived transport polypeptides |
| GB9222888D0 (en) | 1992-10-30 | 1992-12-16 | British Tech Group | Tomography |
| US5436222A (en) | 1993-03-15 | 1995-07-25 | The Research Foundation Of State University Of New York | Use of platelet factor 4 to treat inflammatory diseases |
| US5656610A (en) | 1994-06-21 | 1997-08-12 | University Of Southern California | Producing a protein in a mammal by injection of a DNA-sequence into the tongue |
| US5871986A (en) | 1994-09-23 | 1999-02-16 | The General Hospital Corporation | Use of a baculovirus to express and exogenous gene in a mammalian cell |
| US5736524A (en) | 1994-11-14 | 1998-04-07 | Merck & Co.,. Inc. | Polynucleotide tuberculosis vaccine |
| US5780448A (en) | 1995-11-07 | 1998-07-14 | Ottawa Civic Hospital Loeb Research | DNA-based vaccination of fish |
| US5945100A (en) | 1996-07-31 | 1999-08-31 | Fbp Corporation | Tumor delivery vehicles |
| US5981274A (en) | 1996-09-18 | 1999-11-09 | Tyrrell; D. Lorne J. | Recombinant hepatitis virus vectors |
| GB2320431B (en) * | 1996-12-20 | 2000-08-30 | Johnson & Johnson Medical | Compositions for the treatment of chronic wounds |
| US6891082B2 (en) | 1997-08-01 | 2005-05-10 | The Johns Hopkins University School Of Medicine | Transgenic non-human animals expressing a truncated activintype II receptor |
| US5994624A (en) | 1997-10-20 | 1999-11-30 | Cotton Incorporated | In planta method for the production of transgenic plants |
| JP2002509711A (en) | 1998-03-27 | 2002-04-02 | イーライ・リリー・アンド・カンパニー | Treatment and prevention of vascular disease |
| US8466134B1 (en) | 1998-06-26 | 2013-06-18 | Athena Neurosciences, Inc. | Aqueous compositions containing corticosteroids for nasal and pulmonary delivery |
| US6998240B2 (en) | 2001-06-11 | 2006-02-14 | Wisconsin Alumni Research Foundation | Screen for selective inhibitors or activators of Smad protein function |
| AUPR638101A0 (en) | 2001-07-13 | 2001-08-09 | Bioa Pty Limited | Composition and method for treatment of disease |
| US7299801B2 (en) | 2002-09-06 | 2007-11-27 | 3M Innovative Properties Company | Metering valve for a metered dose inhaler providing consistent delivery |
| KR100472938B1 (en) | 2002-10-31 | 2005-03-11 | 학교법인 한림대학교 | Advanced cell-transducing transport domain-target protein-transport domain fusion protein and thereof uses |
| AU2004281309A1 (en) | 2003-10-16 | 2005-04-28 | The University Court Of The University Of Edinburgh | Control of ES cell self-renewal and lineage specification, and medium therefor |
| ES2343641T3 (en) | 2005-01-14 | 2010-08-05 | Camurus Ab | TOPIC BIOADHESIVE FORMULATIONS. |
| EP2664337B1 (en) | 2005-09-27 | 2019-08-14 | TissueTech, Inc. | Amniotic membrane preparations and purified compositions and methods of use |
| WO2008105925A2 (en) | 2006-09-01 | 2008-09-04 | Oregon Health & Science University | COMPOUNDS AND METHODS FOR MODULATING β-CATENIN/SMAD-7 INTERACTIONS |
| US20080114287A1 (en) | 2006-11-14 | 2008-05-15 | Kar Neng Lai | Ultrasound Microbubble Mediated Genes Delivery System |
| CA2718904C (en) | 2008-03-17 | 2017-01-03 | The Scripps Research Institute | Combined chemical and genetic approaches for generation of induced pluripotent stem cells |
| US8591961B2 (en) | 2008-09-04 | 2013-11-26 | Allergan, Inc. | Aesthetic treatment of scars and aging skin |
| US20110152352A1 (en) | 2008-06-10 | 2011-06-23 | Tufts University | Smad proteins control drosha-mediated mirna maturation |
| US20100184033A1 (en) | 2008-07-16 | 2010-07-22 | West Michael D | Methods to accelerate the isolation of novel cell strains from pluripotent stem cells and cells obtained thereby |
| AU2013243951A1 (en) * | 2012-04-02 | 2014-10-30 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of secreted proteins |
| EP2964774B1 (en) | 2013-03-08 | 2020-05-06 | The Regents of The University of Colorado, A Body Corporate | Ptd-smad7 therapeutics |
-
2014
- 2014-03-07 EP EP14761076.0A patent/EP2964774B1/en active Active
- 2014-03-07 SG SG11201507045TA patent/SG11201507045TA/en unknown
- 2014-03-07 CA CA2904329A patent/CA2904329C/en active Active
- 2014-03-07 WO PCT/US2014/022052 patent/WO2014138670A1/en active Application Filing
- 2014-03-07 JP JP2015561737A patent/JP6576251B2/en active Active
- 2014-03-07 US US14/773,167 patent/US10456448B2/en active Active
- 2014-03-07 CN CN201480025596.9A patent/CN105358704B/en active Active
- 2014-03-07 US US14/201,488 patent/US9422352B2/en active Active
-
2015
- 2015-09-06 IL IL241190A patent/IL241190B/en active IP Right Grant
-
2016
- 2016-06-23 HK HK16107300.2A patent/HK1219986A1/en unknown
- 2016-07-12 US US15/208,424 patent/US20170000851A1/en not_active Abandoned
-
2019
- 2019-09-26 US US16/584,608 patent/US20200023037A1/en not_active Abandoned
-
2022
- 2022-04-22 US US17/727,579 patent/US20220370560A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6251628B1 (en) * | 1997-05-20 | 2001-06-26 | Ludwig Institute For Cancer Research | Isolated nucleic acid molecules encoding Smad7 |
| CN101238215A (en) * | 2005-04-05 | 2008-08-06 | 先锋高级育种国际公司 | Methods and compositions for designing nucleic acid molecules for polypeptide expression in plants using plant virus codon-bias |
| WO2012040295A2 (en) * | 2010-09-22 | 2012-03-29 | The Regents Of The University Of Colorado | Therapeutic applications of smad7 |
Non-Patent Citations (3)
| Title |
|---|
| An ExpandedWWDomain Recognition Motif Revealed by the Interaction between Smad7 and the E3 Ubiquitin Ligase Smurf2;P. Andrew Chong等;《The Journal of Biological Chemistry》;20060623;第281卷(第25期);第17069–17075页 * |
| Smad7 binds to the adaptors TAB2 and TAB3 to block recruitment of the kinase TAK1 to the adaptor TRAF2;Suntaek Hong等;《Nature Immunology》;20070531;第8卷(第5期);第504-513页 * |
| 转化生长因子-β与软组织创伤愈合的研究进展;鄢飞等;《中国民康医学》;20080430;第20卷(第8期);第827-828页 * |
Also Published As
| Publication number | Publication date |
|---|---|
| US10456448B2 (en) | 2019-10-29 |
| US20170000851A1 (en) | 2017-01-05 |
| US20200023037A1 (en) | 2020-01-23 |
| EP2964774A1 (en) | 2016-01-13 |
| WO2014138670A1 (en) | 2014-09-12 |
| CA2904329A1 (en) | 2014-09-12 |
| JP2016510596A (en) | 2016-04-11 |
| JP6576251B2 (en) | 2019-09-18 |
| IL241190B (en) | 2019-09-26 |
| US20140288006A1 (en) | 2014-09-25 |
| CA2904329C (en) | 2021-11-30 |
| EP2964774A4 (en) | 2016-08-10 |
| CN105358704A (en) | 2016-02-24 |
| US20220370560A1 (en) | 2022-11-24 |
| US20160039894A1 (en) | 2016-02-11 |
| IL241190A0 (en) | 2015-11-30 |
| HK1219986A1 (en) | 2017-04-21 |
| EP2964774B1 (en) | 2020-05-06 |
| SG11201507045TA (en) | 2015-10-29 |
| US9422352B2 (en) | 2016-08-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20220370560A1 (en) | Ptd-smad7 therapeutics | |
| ES2281529T3 (en) | EFFECTIVE PEPTIDES IN THE TREATMENT OF TUMORS AND OTHER CONDITIONS THAT REQUIRE THE ELIMINATION OR DESTRUCTION OF CELLS. | |
| ES2924479T3 (en) | Compositions for rejuvenating skeletal muscle stem cells | |
| US10350265B2 (en) | Therapeutic applications of Smad7 | |
| JP6921755B2 (en) | Methods and Compositions to Reduce Brain Tumor Stem Cell Growth, Migration and Infiltration to Improve Survival in Brain Tumor Patients | |
| JP5749154B2 (en) | Peptide composition for cancer treatment by inhibiting TRPV6 calcium channel activity | |
| CN113209303B (en) | WWP1 degradation oncoprotein MUC1 inhibition tumor through lysosome approach and application thereof | |
| CN112004546A (en) | PTD-SMAD7 fusion protein therapeutics | |
| JP2016509590A (en) | Administration of recombinant type 7 collagen for the treatment of aging disorders |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: DE Ref document number: 1219986 Country of ref document: HK |
|
| GR01 | Patent grant | ||
| GR01 | Patent grant |