CN103086938A - Ezetimibe synthesis method - Google Patents
Ezetimibe synthesis method Download PDFInfo
- Publication number
- CN103086938A CN103086938A CN2011103327223A CN201110332722A CN103086938A CN 103086938 A CN103086938 A CN 103086938A CN 2011103327223 A CN2011103327223 A CN 2011103327223A CN 201110332722 A CN201110332722 A CN 201110332722A CN 103086938 A CN103086938 A CN 103086938A
- Authority
- CN
- China
- Prior art keywords
- reaction
- ezetimibe
- synthetic method
- add
- tetrahydrofuran
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 *[C@](CC1)CC=C1N Chemical compound *[C@](CC1)CC=C1N 0.000 description 6
- YQJQOHZYTWPQLA-UHFFFAOYSA-N CCCc1ccc(C2CC2)cc1 Chemical compound CCCc1ccc(C2CC2)cc1 YQJQOHZYTWPQLA-UHFFFAOYSA-N 0.000 description 1
- XDKLCMCQENOGDN-VWIUQNHWSA-N C[C@@H](CCC(CC/C=C/c(cc1)ccc1F)=O)C(CO)c1ccccc1 Chemical compound C[C@@H](CCC(CC/C=C/c(cc1)ccc1F)=O)C(CO)c1ccccc1 XDKLCMCQENOGDN-VWIUQNHWSA-N 0.000 description 1
- OLNTVTPDXPETLC-XPWALMASSA-N O[C@@H](CC[C@H]([C@@H](c(cc1)ccc1O)N1c(cc2)ccc2F)C1=O)c(cc1)ccc1F Chemical compound O[C@@H](CC[C@H]([C@@H](c(cc1)ccc1O)N1c(cc2)ccc2F)C1=O)c(cc1)ccc1F OLNTVTPDXPETLC-XPWALMASSA-N 0.000 description 1
Abstract
The invention belongs to the technical field of medicine, and relates to an ezetimibe synthesis method, particularly to a method for synthesizing ezetimibe by using an intermediate. According to the method, p-hydroxybenzaldehyde and R-fluoromandelic acid are adopted as starting raw materials, and protection, condensation, cyclization, reduction, bromination, protection, docking, deprotection and other conventional methods are adopted to synthesize the ezetimibe under an effect of a catalyst, wherein the method has characteristics of high yield, less steps, significantly reduced cost and less side effects, and is suitable for industrial production.
Description
Technical field:
The invention belongs to medical technical field, relate to the synthetic method of ezetimibe, be specifically related to a kind of method of utilizing the synthetic ezetimibe of intermediate.
Background technology:
Cholesterol acyltransferase (acyl CoA:cholesterol acyltransferase, ACAT) inhibitor is a novel blood lipid-lowering medicine of class of researching and developing at present.By suppressing the ACAT enzyme, can block enteron aisle to the absorption of cholesterol in food, be conducive to the reduction of blood plasma cholesterol level.In addition, the cholesterol major part that is piled up in artery plaque exists with the form of ester, can also suppress the deposition of cholesterol on atherosclerotic plaque therefore suppress the ACAT enzyme.Merck (Merck) and Schering Plough (Schering-Plough) company when this type of inhibitor of research, joint development this novel cholesterol absorption inhibitor of ezetimibe (ezetimibe).In November, 2002, went on the market in the U.S. same period in German Initial Public Offering.
Ezetimibe (ezetimibe) chemical name is 1-(4-fluorophenyl)-3 (R)-[3-(4-fluorophenyl)-3 (S)-hydroxypropyls]-4 (S)-(4-hydroxyphenyl)-2-azetidinone.Ezetimibe belongs to the beta-lactam compounds of group, is first monobactams cholesterol absorption inhibitor of develop, has structure as follows:

Ezetimibe is white crystalline powder, very easily is dissolved in ethanol, methyl alcohol and acetone, and is water insoluble, and fusing point is about 163 ℃, stable at normal temperatures.Ezetimibe only acts on small intestine, reduces intestinal cholesterol by the absorption that suppresses cholesterol and is transported to liver, reduces its storage; Can strengthen the removing of Blood Cholesterol, thereby reduce blood plasma cholesterol level.After the ezetimibe oral administration absorbs, generate active substance with glucuronidation, ezetimibe-glucuronide is through bile and renal excretion.Reach the blood peak concentration of drug after oral in 4~12h, Cmax is 3.4~5.5mg/mL, bioavailability between 35%~60%, t
1/2Be about 22h.The ezetimibe Main Function is in exogenous cholesterol, and the statins Main Function is the synthetic of cholesterol in the minimizing liver, thereby two medical instruments have synergy.Ezetimibe and statins antilipemic drugs combined utilization are 8 times of alone statins deposits yields decreasing cholesterol effect.The main antihyperlipidemic product market scale report of world market in 2003 shows that ezetimibe/Simvastatin global marketing volume of 2003 is 4.71 hundred million dollars.
In prior art, the synthetic route of ezetimibe has a variety of, but all exists many deficiencies:
Patent WO2000/34240(U.S. Schering Plough company) method of the synthetic ezetimibe of a kind of improved chirality is disclosed; at first make (S)-3-oxy-compound by the chiral reduction agent in the method; then connect side chain (E)-N-(4-fluorophenyl by single step reaction in carbonyl α position)-two hydroxyls in product structure are protected by trimethylsilyl in 4-hydroxybenzene methylene amine; then Cheng Huan, remove blocking group, obtain ezetimibe.This synthetic method is when the carbonyl of chiral reduction, and because the group with chiral structure is far away apart from carbonyl, the content of the activity chiral carbon that obtains after reduction is low, and the amount ratio of chiral reduction agent is larger; And the yield that step that connects side chain reacts is very low, so this synthetic route cost is high, is not suitable for suitability for industrialized production.
Patent WO2007072088 discloses the synthetic route of another kind of preparation ezetimibe, and this synthetic method reactions steps is few, and concrete synthetic route is as follows:
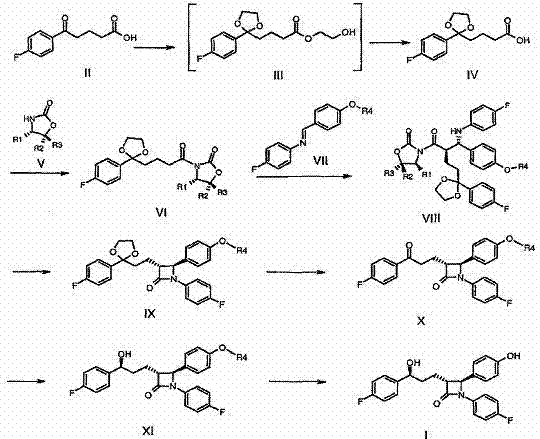
in patent WO2007072088 in the route of synthetic ezetimibe the hydroxyl on the chiral carbon of 3 side chains be to be got by the carbonyl chiral reduction, before chiral reduction not, this carbonyl makes spent glycol protection in order to it is destroyed when carrying out other chemical reactions, the midbody compound IV of gained, compound VI is thinner solid matter, in last handling process, more difficult crystallization, and because its granularity is very thin, extremely easily there is impurity to separate out in crystallisation process, in filtration procedure, the small part product can flow out along with filtrate, cause the second-rate of product, subsequent reactions is more difficult to carry out fully, whole yield is lower, increased synthetic cost.
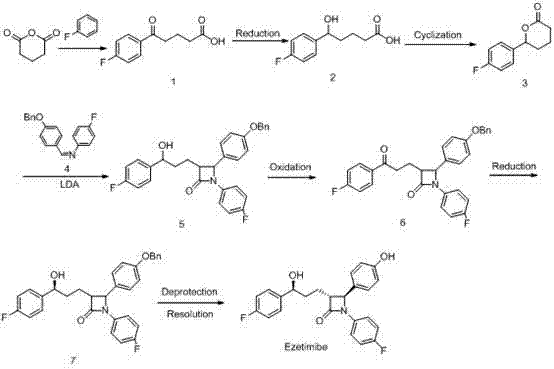
Another US:5739321; US:5886171 has reported route 1: prepare ezetimibe take (4S)-hydroxyl tetrahydrofuran-2-ketone and N-(4-fluorophenyl)-4-benzyloxy benzene methylene amine as starting raw material, its reaction scheme is as follows:
This synthetic method is: N-(4-fluorophenyl)-4-benzyloxy benzene methylene amine (1) is with (4S)-hydroxyl tetrahydrofuran-2-ketone (2) at low temperatures, (LDA) carries out cyclization with lithium diisopropylamine, reacts to carry out recrystallization after complete and can obtain compound (3).Compound (3) obtains product (4) after the oxidation of NaIO, obtain compound (6) with 4-fluoro acetophenone and trimethysilyl chloride (TMS-Cl) reaction, uses TiCl
4Carry out condensation and generate compound (7), obtain compound (8) after the TsOH catalytic dehydration.After the Pd/C shortening, reduction is sloughed simultaneously benzyl and is carried out asymmetric reduction with chiral catalyst CBS and reductive agent boronation hydrogen at low temperatures at last, obtains target product again.This reaction process can produce cis-isomeride, and content can reach 5% left and right, can remove with recrystallization in subsequent disposal.Making target product by compound (8) reduction also can be through chlorinated triphenyl〔Zeng Suji〕 phosphorus base rhodium [(PPh
3)
3RhCl] the two keys of catalytic hydrogenation, then through BH
3/ CBS asymmetric reduction, catalytic hydrogenation debenzylation obtain compound (9).This method is except cyclization reaction yield 60%, and other respectively goes on foot yield all more than 70%.In route 1, (4S)-hydroxyl tetrahydrofuran-2-ketone cost is higher, and character is unstable, and certain difficulty is arranged in production.
Another report route 2: prepare ezetimibe take (5S)-acetoxyl group-5-(4-fluorophenyl) valeric acid as reaction intermediate, its synthesis technique is as follows:

At first with (5S)-acetoxyl group-5-(4-fluorophenyl) valeric acid (27) and (4S)-4-phenyl-oxazolidones (13) carry out condensation, obtain compound (28).Product is at TiCl
4With obtain compound (30) with N-(4-fluorophenyl) 4-acetyloxy phenyl methylene amine (29) through condensation in the dichloromethane solution of DIPEA, this step reaction yield can reach 51%.Compound (30) carries out cyclization in the toluene solution that adds BSA and catalyzer TBAF, generate compound (31), and reaction yield can reach 91%.Compound (31) obtains target product after being hydrolyzed ethanoyl via lithium hydroxide.The raw material of route 2 is all more expensive, and the technique more complicated, and reaction conditions is harsher, and uses column chromatography in the fractionation to isomer, and industrialization is very difficult.
US:5767115 has reported route 3: prepare ezetimibe take Pyroglutaric acid and N-(4-fluorophenyl)-4-benzyloxy benzene methylene amine as starting raw material; it prepares by following reaction: Pyroglutaric acid and methyl alcohol effect generate monomethyl glutarate, generate 4-(chloroformyl) methyl-butyrate (21) after the sulfur oxychloride reaction.amidate action occurs with (4S)-4-phenyl-oxazolidones (13) in compound (21) in the dichloromethane solution of triethylamine and DMAP, obtain (4S)-3-(4-methyl-formiate base-1-oxo butyl)-4-phenyl-2-oxazolidone (22), at alkaline condition and titanium tetrachloride, under the effect of titanium tetraisopropylate and DIPEA with N-(4-fluorophenyl)-4-benzyloxy benzene methylene amine condensation, obtain compound (24) by BSA/TBAF (tetrabutyl ammonium fluoride) effect closed loop, obtain compound (25) after the LiOH hydrolysis.Compound (25) is prepared into acyl chlorides (26) by oxalyl chloride, through to fluorophenyl magnesium bromide (27) at ZnCl
2And Pd (PPh
3)
4Effect is lower generates compound (17), then through CBS/BH
3The asymmetric reduction carbonyl, Pd/C and H
2Remove benzyl and obtain target product.Its reaction formula is as follows:
The raw material of route 3 is cheap and easy to get, and the intermediate productive rate is relatively all higher, is fit to suitability for industrialized production, but it uses column chromatography in the fractionation of optical isomer, it is had a greatly reduced quality in practical.
US:5856473 has reported route 4: prepare ezetimibe take 5-(4-fluorophenyl)-4-pentenoic acid as reaction intermediate, it prepares by the following method: 5-(4-fluorophenyl)-4-pentenoic acid is after oxalyl chloride is prepared into acyl chlorides, carry out amidate action with chiral reagent (4S)-4-phenyl-2-oxazolidone under DMAP catalysis, product obtains compound (A) through recrystallization.With TiC1
4, DPEA and imines add in dichloromethane solution, drips at low temperatures compound (A), keeps low temperature to reaction to finish, and with acetic acid quencher reaction, it is processed rear crude product carries out recrystallization and obtain compound (B).Compound (B) cyclization in the toluene solution of TBAF obtains compound (C), and it is splashed into Pd (OAc)
2, HClO
4In the acetonitrile solution of benzoquinones, after reaction finishes dilute with water, then use organic solvent extraction, remove catalyzer, after the separating filtrate evaporate to dryness, use the column chromatography separating optical isomers to obtain compound (D), then obtain target compound through front method operation.Its reaction formula is as follows:

Route 4 is identical with 2, the somewhat expensive complex process of raw material, and industrialization is more difficult.
US:627882B; WO:2005-IB2393 has reported route 5: prepare ezetimibe take 5-(4-fluorophenyl)-5-oxopentanoic acid as reaction intermediate, this route is take fluorobenzene as starting raw material, use Using Aluminium Trichloride as Catalyst with Pyroglutaric acid under ice-water bath, Friedel-Crafts reaction occurs, and generates 5-(4-fluorophenyl)-5-oxopentanoic acid (33).5-(4-fluorophenyl)-5-oxopentanoic acid (33) adds the dichloromethane solution that is dissolved with triethylamine, is added dropwise to the synthetic mixed acid anhydride (34) of pivaloyl chloride retrude of equivalent.Amidate action occurs with (4S)-4-phenyl-2-oxazolidone (13) in mixed acid anhydride (34) under the catalysis of DMAP, thick product obtains 4 (S)-3-[5-(4-fluorophenyl)-1,5-dioxo amyl group after the Virahol recrystallization purifying]-4-phenyl-2-oxazolidone (35).Compound (35) take [(R)-MeCBS)] as catalyzer generation asymmetric reduction reaction, reacts the processing of complete rear use hydrogen peroxide and sulphuric acid soln and makes (4S)-3-[(5S)-(4-fluorophenyl)-5-hydroxyl-1-oxo amyl group]-4-phenyl-2-oxazolidone (36) in the low temperature tetrahydrofuran solution.Compound (36) adds and contains DIPEA, TMS-Cl and TiCl
4Dichloromethane solution in, at low temperatures, slowly add 4-(4-fluorophenyl imido grpup) phenol (37), after finishing, condensation reaction processes with glacial acetic acid, 7% tartaric acid solution and 20% sodium sulfite solution, add again the abundant back flow reaction of BSA, then the concentrated recrystallization of product is obtained compound (38), compound (38) carries out cyclization under BSA and TBAF effect, and removes (4S)-4-phenyl-2-oxazolidone.With the desiliconization alkane protection in Virahol and H2SO4 mixing solutions of cyclization product, namely obtain target product (10).Its reaction formula is as follows:

Route 5 is the patent of 2005, owing to relating to patent protection, its research is restricted.
CN:1805926 (A) has reported route 6: by Pyroglutaric acid (19) solid and fluorobenzene (32), in dichloromethane solution, use the Using Aluminium Trichloride as Catalyst Friedel-Crafts reaction, obtain 4-(4-fluorobenzoyl) butyric acid (33).4-(4-fluorobenzoyl) butyric acid (33) is used the hydrochloric acid methanol catalytic esterification in methanol solution, obtain 4-(4-fluorobenzoyl) methyl-butyrate (39).4-(4-fluorobenzoyl) methyl-butyrate (39) is in the tetrahydrofuran solution of-25 ℃~-35 ℃, with (-)-β-chloro diisopinocampheylchloroborane base borine (DIP-Cl), the carbonyl at benzene ortho position is carried out asymmetric reduction, obtain compound (40).Compound (40) obtains compound (41) after the sodium hydroxide solution hydrolysis, then with the para-methylbenzenepyridinsulfonate sulfonate catalyzing lactone, reaction obtains (6S)-6-(4-fluorophenyl) tetrahydrochysene-2H-pyran-2-one (42) to compound (41) in toluene solution.(6S)-6-(4-fluorophenyl) tetrahydrochysene-2H-pyran-2-one (42) is in the tetrahydrofuran solution of-50 ℃, add lithium diisopropylamine (LDA) alkalization, again N-(4-fluorophenyl)-4-benzyloxy benzene methylene amine (1) is added with after DMF dissolving, react and obtain anti-1-(4-fluorophenyl)-3-[3 (S)-(4-fluorophenyl-3-hydroxypropyl)-4-(4-benzyloxy phenyl) 2-azetidinone (43) after complete.Anti-1-(4-fluorophenyl)-3-[3 (S)-(4-fluorophenyl-3-hydroxypropyl)-4-(4-benzyloxy phenyl) 2-azetidinone (43) is dissolved in after methyl alcohol degassed, with the catalysis of palladium carbon, formic acid and ammonium formiate carry out debenzylation under 55 ℃, obtain compound (44).Compound (44) is dissolved in ethyl acetate, dripping methyl tertiary butyl ether splits two optical isomers, insolubles is filtered, after getting that filtrate is concentrated and doing, carry out recrystallization with ethyl acetate/normal hexane, obtain (3R, 4S)-1-(4-fluorophenyl)-3-[(3S)-3-(4-fluorophenyl)-3-hydroxypropyl]-4-(4-hydroxy phenyl)-2-azetidinone (10).

The problem that all exists in the synthetic method of above-mentioned ezetimibe is that synthetic route is long; Perhaps yield, purity are low; The amount of the chiral reduction agent of perhaps using is many, solvent toxicity is large, and in sum, in prior art, the cost compare of the synthetic method of ezetimibe is high, brings very large inconvenience for its suitability for industrialized production.
Summary of the invention:
Technical problem to be solved by this invention is to provide a kind of synthetic method of new ezetimibe, thereby it is high to overcome the synthetic method cost that exists in prior art, is not suitable for the shortcomings such as industrialization.
The present invention is achieved through the following technical solutions:
The synthetic route of ezetimibe of the present invention is:

The synthetic method of ezetimibe is characterized in that, by the following method preparation:
(1) Pyroglutaric acid and fluorobenzene in solvent, under Louis acid catalysis, obtain intermediate 1 by the Fu Ke acylation reaction;
(2) in protic solvent, under alkaline condition, add intermediate 1, add reductive agent, generate intermediate 2;
(3) intermediate 2 in inert solvent, makes acyl chlorides with chlorinating agent, and molecule inner ring condensation occurs, and obtains intermediate 3;
(4) in inert solvent, under the highly basic condition, low-temp reaction obtains intermediate 5 to intermediate 3 with intermediate 4;
(5) intermediate 5 obtains intermediate 6 through oxidation;
(6) intermediate 6 obtains intermediate 7 through asymmetric reduction;
(7) intermediate 7 obtains the target product ezetimibe through deprotection, recrystallization;
Solvent described in step (1) be in methylene dichloride, fluorobenzene any one; Lewis acid is selected from tin tetrachloride, aluminum chloride, and titanium tetrachloride, temperature of reaction is room temperature.
Protic solvent described in step (2) is selected from methyl alcohol, ethanol, water; Alkali is selected from sodium carbonate, salt of wormwood, sodium hydroxide, reductive agent be in lithium borohydride, sodium borohydride, POTASSIUM BOROHYDRIDE any one, temperature of reaction is that zero degree is to room temperature.
Inert solvent in step (3) is selected from methylene dichloride, ethyl acetate, tetrahydrofuran (THF), and halogenating agent is sulfur oxychloride, phosphorus oxychloride.
Inert solvent is selected from tetrahydrofuran (THF), anhydrous diethyl ether in step (4), and alkali is selected from diisopropylamine lithium, n-Butyl Lithium, and temperature of reaction is subzero 50 to spend to room temperature.
Described intermediate 6 prepares by the following method:
Intermediate 5 is dissolved in methylene dichloride, adds PCC, PDC, MnO
2In any one, stirring at room, stopped reaction after 2-4 hour adds entry, separatory, a small amount of dichloromethane extraction merges organic phase, the saturated sodium bicarbonate washing, the saturated common salt washing, drying, concentrated.
Described intermediate 7 prepares by the following method:
Getting intermediate 6 is dissolved in anhydrous diethyl ether or tetrahydrofuran (THF), add latent chiral reagent under ice bath, chiral reagent is selected from (-)-two different ancient name for Chinese cabbage base pinane or R-CBS and borine tetrahydrofuran (THF), stirring at room 8-14h, stopped reaction, add diethanolamine cancellation reaction, after removing by filter precipitation, add the methylene dichloride dissolving after steaming desolventizes, the saturated ammonium chloride washing, the saturated common salt water washing, drying, concentrated.
Intermediate 7 is dissolved in any one solvent of methyl alcohol, ethanol, Virahol, adds ammonium formiate, formic acid, catalytic amount palladium carbon, reaction 3-6h, reaction finishes.Suction filtration, filtrate is concentrated, silica gel column chromatography, obtain the compound trans ezetimibe, after be dissolved in ethyl acetate or methylene dichloride, drip that in dioxane, tetrahydrofuran (THF), methyl tertiary butyl ether, anhydrous diethyl ether, any one two optical isomers split, insolubles is filtered, after getting that filtrate is concentrated and doing, carry out recrystallization with ethyl acetate/petroleum ether, obtain ezetimibe.
The invention provides a kind of new synthetic method of ezetimibe.Present method take p-Hydroxybenzaldehyde, R-to the fluorine amygdalic acid as starting raw material, under the effect of catalyzer, through the synthetic ezetimibe of the ordinary methods such as protection, condensation, cyclization, reduction, bromo, protection, docking, deprotection.The method yield is high, and step is few, reduces costs on a large scale, and side reaction is few, is fit to industrialized production.
Embodiment
Embodiment one
under the ice-water bath condition to aluminum chloride (103.0g, 0.77mol) and methylene dichloride (320mL) in be added dropwise to Pyroglutaric acid (40.0g, 0.35mol) dichloromethane solution, slowly drip fluorobenzene (48mL after stirring 30min, 0.52mol), stir 3h under 0-5 ℃, TLC monitoring reaction is complete, in reaction solution impouring frozen water, filter after stirring 30min, filter cake is dissolved in 3% aqueous sodium hydroxide solution, use washed with dichloromethane, add concentrated hydrochloric acid and transfer to pH=2, filter, filter cake is washed till neutrality with frozen water, dry, get white solid 1 65.6g, yield 89.2%.1H-NMR(CDCl3)δ:12.1(1H)、8.0(2H)、7.3(2H)、3.1(2H)、2.3(2H)、1.8(2H)。
Embodiment two
With 1(55g, 0.26mol) be dissolved in 15% aqueous sodium hydroxide solution, add lithium borohydride (6.24g, 0.52mol) under room temperature condition in batches, after stirring at room 3h, the TLC detection reaction is completed.Reaction solution ethyl acetate extraction three times, saturated common salt water washing three times, drying.Remove solvent under reduced pressure, get white solid 2 50.1g, yield 89.7%.1H-NMR(CDCl3)δ:11.95(s,1H)、7.36-7.31(t,2H)、7.15-7.10(t,
2H)、5.20(s,1H)、4.52(s,1H)、2.21-2.17(t,2H)、1.56-1.43(m,4H)。
Embodiment three
2 (47.3g, 0.22mol) are dissolved in the 500mL methylene dichloride, slowly drip sulfur oxychloride (33mL, 0.44mmol), reflux, the TLC detection reaction is completed.Steaming desolventizes, and thinks to add in residual solvent methylene dichloride 200mL, saturated sodium bicarbonate washing, saturated common salt water washing three times, drying.Remove solvent under reduced pressure and obtain white solid 3 35g, yield 80%.1H-NMR(CDCl3)δ:?7.35-7.29(m,?2H)、7.09-7.02(m,?2H)、?5.32(dd,?1H)、?2.74-2.53(m,?2H)、?2.17-2.12?(m,?1H)、?2.06-1.92?(m,?2H)、1.85-1.82?(m,?1H)。
Embodiment four
Under-30 ℃, 3 (33g, 0.17mol) are dissolved in the 40mL tetrahydrofuran (THF), add lithium diisopropylamine (170mL, 0.34mol), stir 20min, slowly add 4-benzyloxy α-tolylene-4-fluoroaniline (52g, 0.17mol) in the reaction solution at tetrahydrofuran (THF) and
N,NSolution in-dimethyl formamide.Keep-30 ℃ to stir 45min, TLC shows that reaction adds glacial acetic acid quencher reaction after complete.Stir 15min, reaction solution is poured in the mixing solutions of hydrochloric acid and ethyl acetate into vigorous stirring.Separate water layer, with ethyl acetate, water layer is extracted again.Merge organic layer, use the saturated common salt water washing, anhydrous sodium sulfate drying filters, and filtrate is concentrated obtains white solid 5 61.2g, yield 72.1% by column chromatography purification.1H-NMR?(CDCl3)?δ:?7.44-7.20?(m,?11H)、7.05-6.89?(m,?6H)、5.06?(s,?2H)、4.71?(t,?J=5,1,?1H)、4.57?(dd,?J=2.4,?J=2.4,?1H)、3.15-3.08?(m?,1H)、2.27?(brs,?1H)、2.01-1.84?(m,?4H)。
Embodiment five
With 5(50g, 0.1mol) be dissolved in the methylene dichloride of 600mL drying, add pyridinium chloro-chromate (43g, 0.2mol) under ice bath in batches, finish, stirring at room 2h, the TLC detection reaction is completed.Organic phase washing twice, saturated sodium bicarbonate washed twice, saturated common salt water washing, drying.Remove solvent under reduced pressure, get white solid 6 49.2g, yield 100%.1H-NMR?(CDCl3)?δ:?8.03-8.01?(2H)、8.00-7.43?(2H)、7.40-7.33?(7H)、7.24-7.23(2H)、7.22-7.14?(2H)、7.01-7.00?(2H)、5.07(2H)、4.99-4.98(1H)、3.24-3.46(3H)、2.16-2.12?(2H)。
Embodiment six
With 6(25g, 0.05mol) be dissolved in the dichloromethane solution of 500mL drying, add the R-CBS toluene solution of 25mL 1mol/L, system is cooled to 10 ℃, slowly drip the dichloromethane solution of 10mL 1mol/L borine tetrahydrofuran (THF) in about 5h, finish insulation reaction 8h.After reaction finishes, add 50mL methyl alcohol, the hydrogen peroxide of 20mL 5%, the sulphuric acid soln of 400mL 2mol/L stirs layering after half an hour, and organic layer is with the sulphuric acid soln washing of 200mL 2mol/L, the saturated common salt water washing, anhydrous sodium sulfate drying spends the night.Remove solvent under reduced pressure, concentrate to get white solid 7 23g, yield 92%.1H-NMR?(CDCl3)?δ:?7.44-7.20?(m,?11H)、7.05-6.89?(m,?6H)、5.06?(s,?2H)、4.71?(t,?J=5,1,?1H)、4.57?(dd,?J=2.4,?J=2.4,?1H)、3.15-3.08?(m?,1H)、2.27?(brs,?1H)、2.01-1.84?(m,?4H)。
Embodiment seven
7 (20g, 0.04mol) are dissolved in methyl alcohol (250mL), add ammonium formiate (25g, 0.4mol), 10% palladium/carbon (1g) and formic acid (2mL, 0.04mol), stirring at room 20min, filtering palladium/carbon, filtrate is concentrated into dried.Residuum is dissolved in ethyl acetate, uses the saturated common salt water washing, drying.Organic phase is concentrated into approximately 40mL, slowly adds wherein methyl tertiary butyl ether under room temperature, stir 1h, the filtering floss is concentrated into filtrate dried.Residuum is dissolved in ethyl acetate, adds sherwood oil, and stirring at room 2h filters, and gets white solid Ezetimibe 6.4g, yield 38.9%, [α] after drying
20 D=-23.7.1H-NMR(DMSO-d6)?δ:?9.51?(s,?1H)、7.32-7.08?(m,?10H)、6.75?(d,?J=8.4,?2H)、?5.27?(d,?J=4.5,?1H)、4.80?(d,?J=2.1,?1H)、4.49?(m,?1H)、3.08?(m,?1H)、1.68-1.82?(m,?4H)。
Claims (8)
1. the synthetic method of ezetimibe, is characterized in that, by the following method preparation:
(1) Pyroglutaric acid and fluorobenzene in solvent, under the Lewis acid effect, obtain intermediate 1 by friedel-crafts acylation;
(2) in protic solvent, under alkaline condition, add intermediate 1, add reductive agent, generate intermediate 2;
(3) intermediate 2 in inert solvent, makes acyl chlorides with halogenating agent, and molecule inner ring condensation occurs, and obtains intermediate 3;
(4) in inert solvent, under the highly basic condition, low-temp reaction obtains intermediate 5 to intermediate 3 with intermediate 4;
(5) intermediate 5 obtains intermediate 6 through oxidation;
(6) intermediate 6 obtains intermediate 7 through asymmetric reduction;
(7) intermediate 7 obtains the target product ezetimibe through deprotection, recrystallization;
Its reaction formula is as follows:
2. synthetic method according to claim 1, is characterized in that, the solvent described in step (1) be in methylene dichloride, fluorobenzene any one; Lewis acid is selected from tin tetrachloride, aluminum chloride, and titanium tetrachloride, temperature of reaction is room temperature.
3. synthetic method according to claim 1, is characterized in that, the protic solvent described in step (2) is selected from methyl alcohol, ethanol, water; Alkali is selected from sodium carbonate, salt of wormwood, sodium hydroxide, reductive agent be in lithium borohydride, sodium borohydride, POTASSIUM BOROHYDRIDE any one, temperature of reaction is that zero degree is to room temperature.
4. synthetic method according to claim 1, is characterized in that, the inert solvent in step (3) is selected from methylene dichloride, ethyl acetate, tetrahydrofuran (THF), and halogenating agent is sulfur oxychloride, phosphorus oxychloride.
5. synthetic method according to claim 1, is characterized in that, inert solvent is selected from tetrahydrofuran (THF), anhydrous diethyl ether in step (4), and alkali is selected from diisopropylamine lithium, n-Butyl Lithium, and temperature of reaction is subzero 50 to spend to room temperature.
6. synthetic method according to claim 1, is characterized in that, described intermediate 6 prepares by the following method:
Intermediate 5 is dissolved in methylene dichloride, adds PCC, PDC, MnO
2In any one, stirring at room, stopped reaction after 2-4 hour adds entry, separatory, a small amount of dichloromethane extraction merges organic phase, the saturated sodium bicarbonate washing, the saturated common salt washing, drying, concentrated.
7. synthetic method according to claim 1, is characterized in that, described intermediate 7 prepares by the following method:
Getting intermediate 6 is dissolved in anhydrous diethyl ether or tetrahydrofuran (THF), add different basic pinane or the known material of R-CBS(of sending of latent chirality (-)-two under ice bath, needn't translate) and the borine tetrahydrofuran (THF), stirring at room 8-14h, stopped reaction, add diethanolamine cancellation reaction, after removing by filter precipitation, add the methylene dichloride dissolving after steaming desolventizes, the saturated ammonium chloride washing, the saturated common salt water washing, drying, concentrated.
8. synthetic method according to claim 1, it is characterized in that, intermediate 7 is dissolved in methyl alcohol, ethanol, in any one solvent of Virahol, add ammonium formiate, formic acid, catalytic amount palladium carbon, reaction 3-6h, reaction finishes, suction filtration, filtrate is concentrated, silica gel column chromatography, obtain the compound trans ezetimibe, after be dissolved in ethyl acetate or methylene dichloride, drip dioxane, tetrahydrofuran (THF), methyl tertiary butyl ether, in anhydrous diethyl ether, any one two optical isomers split, insolubles is filtered, after getting that filtrate is concentrated and doing, carry out recrystallization with ethyl acetate/petroleum ether, obtain ezetimibe.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2011103327223A CN103086938A (en) | 2011-10-28 | 2011-10-28 | Ezetimibe synthesis method |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2011103327223A CN103086938A (en) | 2011-10-28 | 2011-10-28 | Ezetimibe synthesis method |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN103086938A true CN103086938A (en) | 2013-05-08 |
Family
ID=48200125
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN2011103327223A Pending CN103086938A (en) | 2011-10-28 | 2011-10-28 | Ezetimibe synthesis method |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN103086938A (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104447473A (en) * | 2014-11-06 | 2015-03-25 | 成都森科制药有限公司 | Preparation method of Ezetimibe intermediate |
| WO2015179293A1 (en) * | 2014-05-18 | 2015-11-26 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Small molecule activators and inhibitors of lecithin: cholesterol acyltransferase |
| CN105272852A (en) * | 2014-07-16 | 2016-01-27 | 浙江九洲药物科技有限公司 | Ezetimibe intermediate and preparation method |
| CN106966942A (en) * | 2017-04-07 | 2017-07-21 | 四川智强医药科技开发有限公司 | A kind of preparation method of Ezetimibe |
| CN107176920A (en) * | 2017-04-19 | 2017-09-19 | 上海恒晟药业有限公司 | A kind of new technique for synthesizing of ezetimibe |
| CN111138276A (en) * | 2019-12-27 | 2020-05-12 | 郑州手性药物研究院有限公司 | Synthesis method of chiral 5- (4-fluorophenyl) -5-hydroxypentanoate |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5739321A (en) * | 1996-05-31 | 1998-04-14 | Schering Corporation | 3-hydroxy γ-lactone based enantionselective synthesis of azetidinones |
| CN1805926A (en) * | 2003-05-05 | 2006-07-19 | 兰贝克赛实验室有限公司 | Process for the preparation of trans-isomers of diphenylazetidinone derivatives |
| CN1931838A (en) * | 2006-10-20 | 2007-03-21 | 屠勇军 | Azetidinone derivative and synthetic method thereof |
| CN101297969A (en) * | 2001-01-26 | 2008-11-05 | 先灵公司 | Combinations of the peroxisome proliferator-activated receptor (PPAR) activator fenofibrate with sterol absorption inhibitor ezetimibe for vascular indications |
| CN101423515A (en) * | 2007-10-30 | 2009-05-06 | 杜焕达 | Novel preparation method of Ezetimibe |
| WO2009067960A2 (en) * | 2007-11-30 | 2009-06-04 | Zentiva, A.S. | A method of manufacturing (3r,4s)-l-(4-fluorophenyl)-3-[(3s)-3-(4-fluorophenyl)-3- hydroxypropyl)]-4-(4-hydroxyphenyl)-2-azetidinone and its intermediates |
| WO2009157019A2 (en) * | 2008-06-23 | 2009-12-30 | Ind-Swift Laboratories Limited | Process for preparing ezetimibe using novel allyl intermediates |
| WO2010071358A2 (en) * | 2008-12-17 | 2010-06-24 | Hanmi Pharm. Co., Ltd. | Method of preparing ezetimibe and intermediates used therein |
-
2011
- 2011-10-28 CN CN2011103327223A patent/CN103086938A/en active Pending
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5739321A (en) * | 1996-05-31 | 1998-04-14 | Schering Corporation | 3-hydroxy γ-lactone based enantionselective synthesis of azetidinones |
| CN101297969A (en) * | 2001-01-26 | 2008-11-05 | 先灵公司 | Combinations of the peroxisome proliferator-activated receptor (PPAR) activator fenofibrate with sterol absorption inhibitor ezetimibe for vascular indications |
| CN1805926A (en) * | 2003-05-05 | 2006-07-19 | 兰贝克赛实验室有限公司 | Process for the preparation of trans-isomers of diphenylazetidinone derivatives |
| CN1931838A (en) * | 2006-10-20 | 2007-03-21 | 屠勇军 | Azetidinone derivative and synthetic method thereof |
| CN101423515A (en) * | 2007-10-30 | 2009-05-06 | 杜焕达 | Novel preparation method of Ezetimibe |
| WO2009067960A2 (en) * | 2007-11-30 | 2009-06-04 | Zentiva, A.S. | A method of manufacturing (3r,4s)-l-(4-fluorophenyl)-3-[(3s)-3-(4-fluorophenyl)-3- hydroxypropyl)]-4-(4-hydroxyphenyl)-2-azetidinone and its intermediates |
| WO2009157019A2 (en) * | 2008-06-23 | 2009-12-30 | Ind-Swift Laboratories Limited | Process for preparing ezetimibe using novel allyl intermediates |
| WO2010071358A2 (en) * | 2008-12-17 | 2010-06-24 | Hanmi Pharm. Co., Ltd. | Method of preparing ezetimibe and intermediates used therein |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015179293A1 (en) * | 2014-05-18 | 2015-11-26 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Small molecule activators and inhibitors of lecithin: cholesterol acyltransferase |
| CN105272852A (en) * | 2014-07-16 | 2016-01-27 | 浙江九洲药物科技有限公司 | Ezetimibe intermediate and preparation method |
| CN105272852B (en) * | 2014-07-16 | 2019-04-23 | 浙江九洲药物科技有限公司 | A kind of Ezetimible intermediate and preparation method thereof |
| CN104447473A (en) * | 2014-11-06 | 2015-03-25 | 成都森科制药有限公司 | Preparation method of Ezetimibe intermediate |
| CN106966942A (en) * | 2017-04-07 | 2017-07-21 | 四川智强医药科技开发有限公司 | A kind of preparation method of Ezetimibe |
| CN107176920A (en) * | 2017-04-19 | 2017-09-19 | 上海恒晟药业有限公司 | A kind of new technique for synthesizing of ezetimibe |
| CN107176920B (en) * | 2017-04-19 | 2019-09-20 | 江苏恒盛药业有限公司 | A kind of new technique for synthesizing of ezetimibe |
| CN111138276A (en) * | 2019-12-27 | 2020-05-12 | 郑州手性药物研究院有限公司 | Synthesis method of chiral 5- (4-fluorophenyl) -5-hydroxypentanoate |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN102753537B (en) | Prepare the method for razaxaban | |
| CN103086938A (en) | Ezetimibe synthesis method | |
| CN103508899B (en) | Method for preparing ticagrelor key intermediate and racemate thereof and special intermediate for implementing method | |
| CN101423511A (en) | Ezetimible intermediate and synthetic method of ezetimible | |
| KR20040077883A (en) | Process for the Manufacture of Organic Compounds | |
| CN101423515A (en) | Novel preparation method of Ezetimibe | |
| CN105294426B (en) | Azetidinone compounds Preparation Method And Their Intermediate | |
| CN101265271B (en) | Method for synthesizing penem-like pharmaceutical intermediate 4AA | |
| CN101367746B (en) | Novel method for synthesis of (S)-propisochlor | |
| JP5108383B2 (en) | Process for producing optically active monosulfonate compound | |
| CN114105860A (en) | Catalytic asymmetric synthesis method and application of chiral oxindole spiro-analogue | |
| CN103254107B (en) | A kind of Ezetimibe analogue and preparation method thereof | |
| CN112154140B (en) | Compound and application thereof in synthesizing Brivaracetam (Brivaracetam) bulk drug | |
| CN102477008B (en) | Method for synthesizing ezetimibe | |
| CN105017048A (en) | Alpha-fluoroalkyl-alpha-amino acid compound containing tetrasubstituted carbon chiral center and preparation method thereof | |
| ZA200403722B (en) | Process for producing optically active oxoheptenoic acid ester. | |
| CN106397292A (en) | Ezetimibe intermediate, synthesis method of intermediate and synthesis method of ezetimibe | |
| CN105732648A (en) | Nitrogen heterocyclic ring compound of pyrrolofuran and synthetic method | |
| CN101348475A (en) | Novel method for synthesizing orlistat, intermediate compound and preparation thereof | |
| CN111333560B (en) | Method for preparing spiro beta-lactam | |
| CN101955482B (en) | Method for preparing protected meropenem | |
| CN113004296A (en) | General synthetic method for preparing chiral oxygen heterocyclic compound by novel [4+1] and [5+1] cyclization strategies | |
| CN101857602B (en) | Preparation method for Prulifloxacin | |
| CN111777554A (en) | Method for synthesizing cisatracurium besilate | |
| CN107629039A (en) | The preparation method and intermediate of deuterated acrylamide |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Application publication date: 20130508 |