WO2013019635A1 - Compounds and methods - Google Patents
Compounds and methods Download PDFInfo
- Publication number
- WO2013019635A1 WO2013019635A1 PCT/US2012/048588 US2012048588W WO2013019635A1 WO 2013019635 A1 WO2013019635 A1 WO 2013019635A1 US 2012048588 W US2012048588 W US 2012048588W WO 2013019635 A1 WO2013019635 A1 WO 2013019635A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- alkoxy
- amino
- compound
- hydrogen
- Prior art date
Links
- 0 CCC[C@](C1)C1C(C)C1*=C(C2)C2CC1 Chemical compound CCC[C@](C1)C1C(C)C1*=C(C2)C2CC1 0.000 description 5
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/30—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D263/32—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/06—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members
- C07D261/08—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/06—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members
- C07D261/10—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D261/18—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/06—1,2,4-Oxadiazoles; Hydrogenated 1,2,4-oxadiazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/06—1,2,4-Oxadiazoles; Hydrogenated 1,2,4-oxadiazoles
- C07D271/07—1,2,4-Oxadiazoles; Hydrogenated 1,2,4-oxadiazoles with oxygen, sulfur or nitrogen atoms, directly attached to ring carbon atoms, the nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/10—1,3,4-Oxadiazoles; Hydrogenated 1,3,4-oxadiazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D275/00—Heterocyclic compounds containing 1,2-thiazole or hydrogenated 1,2-thiazole rings
- C07D275/02—Heterocyclic compounds containing 1,2-thiazole or hydrogenated 1,2-thiazole rings not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/22—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D277/24—Radicals substituted by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/22—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D277/30—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention relates to no vel retinoid-related orphan receptor gamma (RORy) modulators and their use in the treatment of diseases mediated by RORy.
- RORy vel retinoid-related orphan receptor gamma
- RORs Retinoid-related orphan receptors
- the ROR. family consists of three members, ROR. alpha (RORa), ROR beta (RORp), and ROR gamma (RORy), each encoded by a separate gene (RORA, RORB, and RORC, respectively).
- RORs contain four principal domains shared by the majorit '- of nuclear receptors: an N-terminal A/B domain, a DNA-binding domain, a hinge domain, and a ligand binding domain.
- RORyl and RORyt are two isoforms of RORy which differ only in their N-terminal A/B domain.
- RORyl and RORyt also known as RORy2
- RORy is a term used to describe both RORyl and/or RORyt.
- RORyl While RORyl is expressed in a variety of tissues including thymus, muscle, kidney and li v er, RORyt is exclusi v ely expressed in the cells of the immune system. RORyt has been identified as a key regulator of Thl 7 cell differentiation. Thl 7 cells are a subset of T helper cells which produce IL- 17 and other proinflammatory cytokines. Thl 7 cells have been shown to have key functions in several mouse autoimmune disease models including experimental autoimmune encephalomyelitis (EAE) and collagen-induced arthritis (CIA).
- EAE experimental autoimmune encephalomyelitis
- CIA collagen-induced arthritis
- Thl 7 cells or their products have been shown to be associated with the pathology of a variety of human inflammatory and autoimmune disorders including multiple sclerosis, rheumatoid arthritis, psoriasis, Crohn's disease and asthma (Jetten (2009) Nucl. Recept. Signal. 7:e003; Manel et ai. (2008) Nat. Immunol 9:641 -649).
- the pathogenesis of chronic autoimmune diseases including multiple sclerosis and rheumatoid arthritis arises from the break in tolerance towards self-antigens and the development of auto-aggressive effector T cells infiltrating the target tissues.
- Thl 7 cells are one of the important drivers of the inflammatory process in tissue-specific autoimmunity (Steinman (2008) J. Exp. Med. 205: 1517- 1522; Leung et ai. (2010) Cell. Mol. Immunol. 7: 182- 189). There is evidence that Thl 7 cells are activated during the disease process and are responsible for recruiting other inflammatory cells types, especially neutrophils, to mediate pathology in the target tissues (Korn et ai. (2009) Aram. Rev. Immunol. 27:485-517).
- RORyt plays a critical role in the pathogenic responses of Thl 7 cells (Ivanov et al. (2006) Cell 126: 1 121 -1 13 ). RORyt deficient mice produce few Thl 7 cells, in addition, RORyt deficiency resulted in amelioration of EAE. Further support for the role of RORyt in the pathogenesis of autoimmune or inflammatory diseases can be found in the following references: Jetteii & .Too (2006) Adv. Dev. Biol. 16:313-355; Meier et al. (2007) Immunity 26:643-654; Aloisi & Pujol-Borrell (2006) Nat. Rev. Immunol. 6:205-217; Jager et al. (2009) J. Immunol.
- the invention is directed to novel RORy modulators and their use in the treatment of diseases mediated by RORy. Specifically, the invention is directed to compounds according to Formula I):
- n 0, 1, or 2;
- n 0, 1, 2, or 3;
- X 1 , X " , X', X 4 , and J are each independently selected from N, N + -0 ⁇ , CH, and CR ' ⁇ wherein 0-3 of X 1 , X 2 , X 3 , X 4 , and X s are N or N + -0 " and 1-3 of X 1 , X 2 , X 3 , X 4 , and X 3 are CR 5 ; provided that when zero of X 1 , X 2 , X 4 , and X 3 are N or N "f -0 ⁇ and X ""1 is CR 3 , 1-2 of X 1 , X , X 4 , and X s are CR 5 ;
- Y 7" is O or NR 8 and the other is a bond
- X 1 is CR 5
- Y 1 is NR 8
- Y 2 is a bond
- R 5 and R 8 taken together with the atoms to which they are attached form a five to seven membered ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by (Ci-C 4 )alkyl;
- Cy is (CrCgkycloalkyl, heterocycloalkyl, phenyl, or 5- or 6-membered heteroaryl, each of which is optionally substttiEted one, two, or three times, independently, by (Ci -Ce kyl,
- a 1 , A-, A 3 , and A 4 are each independently selected from N, R b , O, S, CH, and CR t0 , wherein one of A 1 , A 2 , A 3 , and A 4 is NR 6 , O, or S, 0-2 of A 1 , A 2 , A 3 , and A 4 are CR 10 , and 0-3 of A 1 , A 2 , A " , and A 4 are CH or N;
- R l is (C 3 » C 6 )alkyl, (CrC 6 )haloalkyi, (C 3 -C 3 )cyeloalkyl, (C 3 -C 6 )alkoxy,
- R 2 is hydrogen, (Ci"C 6 )aikyl, or (Ci -Cejhaloalkyl;
- R 2 taken together with the carbon atom to which they are attached form a three to eight membered ring, optionally containing a heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted one, two, or three times, independently, by R 5 ;
- R' and R' a are each independently hydrogen, hydroxy ⁇ , (Ci-C6)alkyl, (Ci -C6)haloalkyl, halogen, (C- ; -C6)aikoxy, amino, (Ci-C 4 )alkyj.ammo, or ((Ci-C4)alky])((Ci -C 4 )a]kyi)amino;
- each R 4 is independently selected from hydrogen, halogen, (Ci-Ceja!kyf, (Ci-C6)haloalkyl, -COjR 7 , -CONR 7 R 8 , -OR 9 , and -NR 8 R 9 , wherein said (C, -C 6 )alkyl or (C x -C 6 )haloalkyl is optionally substituted by hydroxy ⁇ , -OR 9 , -C0 2 R 7 , -CO R 7 R 8 , or -NR S R 9 ;
- each R 4a is independently selected from hydrogen, halogen, hydroxy!, amino, and
- R " and R 4a taken together with the carbon atom to which they are attached form a three to eight membered ring, optionally containing a heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by cyano, (Ci-C4)alkyl, (Ci-C 4 )haloalkyl,
- each R 5 is independently selected from (Ci-Cejalkyl, (Ci-Cejhaloalkyl, (C 3 -C,s)cyeloalkyl, halogen, cyano, hydroxy!, hydroxy(C] -C 6 )alkyl, (Ci-C 6 )alkoxy, (Ci-C 4 )alkoxy(Ci-C6)alkyl, amino, (Ci-C 4 )alkylamino, ((Ci-C 4 )alkyl)((Ci-C4)alkyl)amino, aryl, heteroaryl, aryl(Ci-C 6 )alkyl, heteroaryi(Ci-C6)alkyl, and heterocycloalkyl; R 6 is hydrogen, (Ci-C 6 )alkyl, (Ci-C 6 ) aloalkyl, (C ⁇ -C ⁇ jcycloalky!, hydroxy(Ci-C 6 )alkyl
- R 7 is hydrogen, (C C 6 )aikyl, (C[-C 6 )haloalkyl, (C C 6 )cycloaikyl,
- aryl aryl, heteroaryl, arylCCi-CgJalkyl, heteroaryl(Ci -C6)alkyl, or heterocycloalkyl;
- R 8 is hydrogen, (Ci-C 6 )alkyl, or (Ci -Cg)haloalkyl;
- R' and R 8 taken together with the nitrogen atom to which they are attached form a four to eight membered ring, opiionaiiy containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by (Ci-G alkyi, (Cj-C 4 )haioaiky1, (C 3 -C 6 )cycloalkyl, -C0 2 H, -C0 2 (Ci-C 4 )alkyl, hydroxyl, hydroxy(Ci-C 6 )alkyl, (C C 4 )alkoxy, (Ci-C 4 )alkoxy(C i-C6)alk l, amino, (Ci-C 4 )alkylamino, or ((C[-C 4 )alkyl)((Ci-C 4 )alkyl)amino;
- R 9 is -C(0)R 7 , ⁇ ( ' (>. -R . -C(0)NR 7 R 8 , (C r C 6 )alkyl, (CrC ⁇ haioaikyl, (C 3 -C6)cycloalkyl, aryl, heteroaryl, heteroaryl(Ci-Cg)alkyi, or heterocycloalkyl, wherein said (Ci -Cg)alkyl, (Ci-C6)haloalky , (C 3 -C6)cycloalkyl, aryl, heteroaryl, aryl(Ci -Cg)alkyl,
- heteroaryi(Ci-Cg)alkyl, or heterocycloalkyl is optionally substituted by -C0 2 R ', -CONH 2 , -CONH(Ci-C 4 )alkyl, -CON((C t -C 4 )alkyl)((Ci - C 4 )alkyl), hydroxyl, ⁇ ( . -C, yM o ⁇ .
- R 6 and R 9 taken together with the nitrogen atom to which they are attached form a four to eight membered ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by cyano, (Ci-C 4 )alkyl, (Ci-C 4 )haloalkyl, (C 3 -C 6 )cycloalkyl, -C0 2 H, -C0 2 (Ci-C 4 )alkyl, -CONR 7 R 8 , hydroxyl, hydroxy(C r C 6 )alkyl, (CrCi)alkoxy, (Ci-C 4 )alkoxy(Ci-C6)alkyl, amino, (Ci-C 4 )alkylamino,
- R 10 is (Ci-Ce)alkyl, (Ci-Cr hafoafkyl, (Qj-Cejcycloalkyl, halogen, cyano, hydroxyl, hydroxy(Ci-C 6 )alkyl, (C C 6 )alkoxy, (Ci-C 4 )alkoxy(C r C 6 )alkyl, -((C 0 -C 3 )alkyl)CO 2 R 7 ,
- the compound of Formula I does not include (2-(4- ((3,5-dimethylisoxazol-4-yl)memoxy)ph
- this invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula (1), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient.
- this invention provides for the use of a compound of Formula (I) or a pharmaceutically acceptable salt thereof for the treatment of diseases mediated by RORy.
- the invention further provides for the use of a compound of of Formula (I) or a pharmaceutically acceptable salt thereof as an active therapeutic substance in the treatment of a disease mediated by RORy.
- the invention provides a compound of Formula (I) or a pharmaceutically acceptable salt thereof for use in therapy.
- the invention provides the use of a compound of Fonnula (I) or a pharmaceutically acceptable salt thereof in the manufacture of a medicament for the treatment of diseases mediated by RORy.
- diseases for which compounds of Formula (I) may be used include autoimmune or inflammatory diseases such as multiple sclerosis, rheumatoid arthritis, psoriasis, uveitis, dry eye, glomerulonephritis, Crohn's disease and asthma, especially psoriasis
- the invention is directed to meihods of treating such diseases for example by administering to a patient (e.g. human) in need thereof an effective amount of a compound of Formula (I) or a pharmaceutically acceptable salt thereof, Detailed Description of the invention
- alkyl represents a saturated, straight, or branched hydrocarbon moiety.
- the term refers to an alkyl moiety containing from 1 to 6 carbon atoms.
- Exemplary alkyls include, but are not limited to methyl, ethyl, n-propyl, isopropyl, w-butyl, isobutyl, s-butyi, /-butyl, pentyl, and hexyl.
- C 0 aikyl means that no alkyl group is present in the moiety.
- -((Co)alkyl)CONH 2 is equivalent to -CONH 2 .
- alkyl When the term “alkyl” is used in combination with other substituent groups, such as “haloalkyl”, “hydroxyalkyl”, “alkoxyalkyl”, “arylalkyl”, or “heteroarylalkyl”, the term “alkyl” is intended to encompass a divalent straight or branched-chain hydrocarbon radical.
- arylalkyl is intended to mean the radical -alkylaryl, wherein the alkyl moiety thereof is a divalent straight or branched-chain carbon radical and the aryl moiety thereof is as defsned herein, and is represented by, for example, the bonding arrangement present in a benzyl group (-CH 2 -phenyl);
- )alkyi is intended to mean a radical having one or more halogen atoms, which may be the same or different, at one or more carbon atoms of an alkyl moiety containing from 1 to 4 carbon atoms, which is a straight or branched-chain carbon radical, and is represented by, for example, a trifiuoromethyl group (-CF 3 ).
- cycloalkyl refers to a non-aromatic, saturated, cyclic hydrocarbon ring.
- (C3-Cg)cycloalkyl refers to a non-aromatic cyclic hydrocarbon ring having from three to eight ring carbon atoms.
- Exemplary "(Cs-C ⁇ cycloalkyi” groups useful in the present invention include eyclopropyf, cyclobutyf, cyclopentyl, cycfohexyl, cyciobeptyl, and cyclooctyl.
- Alkoxy means an alkyi radical containing the specified number of carbon atoms attached through an oxygen linking atom.
- (Ci-C4)alkoxy refers to a straight- or branched-chain hydrocarbon radical having at least 1 and up to 4 carbon atoms attached through an oxygen linking atom.
- Exemplar '- "(Ci-C 4 )alkoxy” groups useful in the present invention include, but are not limited to, niethoxy, ethoxy, w-propoxy, isopropoxy, H-butoxy, s-butoxy, and /-butoxy.
- Aryl represents a group or moiety comprising an aromatic, monovalent monocyclic or bicyclic hydrocarbon radical containing from 6 to 10 carbon ring atoms, to which may be fused one or more cycloalkyl rings.
- aryl is phenyl
- Heterocyclic groups may be heteroaryl or heterocycloalkyl groups.
- Heteroaryl represents a group or moiety comprising an aromatic monovalent monocyclic or bicyclic radical, containing 5 to 10 ring atoms, including 1 to 4 heteroatoms independently selected from nitrogen, oxygen and sulfur. This term also encompasses bicyclic heterocyclic-aryl compounds containing an aryl ring moiety fused to a heterocycloalkyl ring moiety, containing 5 to 10 ring atoms, including 1 to 4 heteroatoms independently selected from nitrogen, oxygen and sulfur, illustrative examples of heteroary Is useful in the present invention include, but are not limited to, furanyl, thienyi, pyrrolyl, imidazolyl, pvrazolyl, triazolyl, tetrazolyl, thiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiadiazolyl, isothiazoiy i, pyridinyl, pyridazinyl, pyrazin
- benzimidazolyl dihydrobenzimidazolyl, henzoxazoiyi, dihydrobenzoxazolyl, benzihiazoiyi, benzoisothiazolyl, dihydrobenzoisothiazolyl, indazolyl, imidazopyridinyl, pyrazolopyridinyl, benzotriazolyi, rriazolopyridinyl, purinyJ, quinolinyi, tetrahydroquinoiinyl, isoquinolinyi, tetrahydroisoquinolinyl, quinoxaiinyl, cinnofinyf, phthalazinyl, quinazo!iny!, 1 ,5-napbthyridinyl, 1 ,6-naphthyridinyl, 1,7-naphthyridinyl, 1,8-naphthyridmyl, and pteridiny
- heteroaryl groups present in the compounds of this invention are
- Selected 5-membered and/or 6-memebred monocyclic heteroaryl groups contain one nitrogen, oxygen, or sulfur ring heteroatom, and optionally contain 1 , 2, or 3 additional nitrogen ring atoms.
- Selected 6-membered heteroaryl groups contain 1 , 2, or 3 nitrogen ring heteroatoms.
- 5- or 6-membered heteroaryl groups useful in the present invention include, but are not limited to furanyi, thienyl, pyrrolyl, imidazolyl, pyrazoiyi, triazolyl, tetrazolyl, thiazolyl, oxazoiyl, isoxazolyl, oxadiazolyl, thiadiazolyl, isothiazoiyl, pyridinyi, pyridazinyl, pyrazinyl, pyrimidinyl, and triazinyi.
- Heterocycloalkyl represents a group or moiety comprising a on-aromatic, monovalent monocyclic or bicyclic radical, which is saturated or partially unsaturated, containing 3 to 10 ring atoms, which includes 1 to 3 heteroatoms independently selected from nitrogen, oxygen and sulfur.
- heterocycloalkyls useful in the present invention include, but are not limited to, azetidinyl, pyrrolidmyl, pyrazolidinyl, pyrazolinyl, imidazolidinyi, imidazolinyl, oxazofinyf, thiazolinyl, tetrahydrofuranyl, dihydrofuranvi, 1 ,3-dioxofanyl, piperidinyl, piperazmyl, morpholinyl, thiomorpholinyl, tetrahydropyranyl, dihydropyranyl, 1 ,3-dioxanyl, 1 ,4-dioxanyl, 1 ,3- oxat iolanyl, 1 ,3 -oxathianyi, 1 ,3-dithianyl, hexahydro- 1H- 1 ,4-diazepinyl, azabicyl
- heterocycloalkyl groups are 5-7 membered heterocycloalkyl groups, such as pyrrolidmyl, pyrazolidinyl, pyrazolinyl, imidazolidinyl, imidazolinyl, oxazolinyl, thiazolinyl, tetrahydrofuranyl, dihydrofuranyl, 1 ,3-dioxolanyl, piperidinyl, piperazmyl, ⁇ >4, thiomorpholinyl, tetrahydropyranyl, dihydropyranyl, and hexahydro- I/f- I,4-diazepinyl.
- heterocycloalkyl groups are 5-7 membered heterocycloalkyl groups, such as pyrrolidmyl, pyrazolidinyl, pyrazolinyl, imidazolidinyl, imidazolinyl, oxazolinyl, thiazolin
- halogen and "halo” represent chloro, fluoro, bromo, or iodo substifuents, "Hydroxy” or "hydroxyl” is intended to mean the radical -OH.
- RORy refers to all isoforms encoded by the RORC gene which include RORy l and RORyt.
- RORy modulator refers to a chemical compound that inhibits, either directly or indirectly, the activity of RORy.
- RORy modulators include antagonists and inverse agonists of RORy.
- “Pharmaceutically acceptable” refers to those compounds, materials, compositions, and dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicit '-, irritation, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- pharmaceutically acceptable salts refers to salts that retain the desired biological activity of the subject compound and exhibit minimal undesired toxicological effects. These pharmaceutically acceptable salts may be prepared in situ during the final isolation and purification of the compound, or by separately reacting the purified compound in its free acid or free base form with a suitable base or acid, respectively.
- the term "compound(s) of the mvention” means a compound of Formula (I) (as defined above) in any form, i.e., any salt or non-salt form (e.g., as a free acid or base form, or as a pharmaceutically acceptable salt thereof) and any physical form thereof (e.g., including non-solid forms (e.g., liquid or semi-solid forms), and solid forms (e.g., amorphous or crystalline forms, specific polymorphic forms, solvates, including hydrates (e.g., mono-, di- and hemi- hydrates)), and mixtures of various forms.
- any salt or non-salt form e.g., as a free acid or base form, or as a pharmaceutically acceptable salt thereof
- any physical form thereof e.g., including non-solid forms (e.g., liquid or semi-solid forms), and solid forms (e.g., amorphous or crystalline forms, specific polymorphic forms, solvates, including
- the term "optionally substituted” indicates that a group, such as aikyl, eycloalkyl, alkoxy, heterocycloalkyl, aryi, or heteroaryi, may be unsubstituted, or the group may be substituted with one or more substituent(s) as defined. In the case where groups may be selected from a number of alternative groups the selected groups may be the same or different.
- n is 0, 1 , or 2. In a specific embodiment of this invention, m is 1.
- n is 0, 1 , 2, or 3. In another embodiment of this invention, n is 1 or 2.
- X 1 , X 2 , X', X 4 , and X 3 are each independently selected from N, ⁇ - ⁇ " (i.e. iV- oxide), CH, and CR 5 , wherein 0-3 of X ⁇ X 2 , X 3 , X 4 , and X 5 are N or lST-O " and 1 -3 of X 1 , X 2 , X 3 , X 4 , and X 5 are CR 5 ; provided that when zero of X 1 , X ⁇ , X ⁇ and X 5 are N or N + -0 " and X 3 is CR 5 , 1 -2 of X 1 , ⁇ ⁇ , X 4 , and X 5 are CR 3 .
- X 1 , X , X ⁇ X " , and X 5 are each independently selected from N, N + -Q " , CH, and CR 5 , wherein 0-2 of X 1 , X 2 , X 3 , X 4 , and X 5 are N or N + -0 ⁇ and 1 -3 of X 1 , X 2 , X 3 , X 4 , and X 5 are CR 5 ; provided that when zero of X 1 , X 2 , X 4 , and X 5 are N or N " -0 " and X 3 is CR 5 , 1 -2 of X 1 , X 2 , X", and X 5 are CR 3 .
- X 1 and X 3 are each independently selected from N, IST-O " , CH, and CR 5
- X X', and X 4 are each independently selected from CH and CR 5
- at least one of X 1 and X 3 is N or r -0 " and 0-3 of X 1 , X 2 , X X 4 , and X 5 are CR 3 .
- X 1 and X 3 are each independently selected from N, N + -0 " , and a carbon atom substituted by hydrogen, halogen, cyano, (Ci -C 4 )alkyl, (CrC ⁇ haloalkyL (Ci -G alkoxy, or
- X 2 is N or N " '-0 "
- X 1 , X 3 , X 4 , and X 5 are each independently a carbon atom substituted by hydrogen, halogen, cyano, (Ci-C 4 )alkyL (Ci-C )haloalkyl, (Ci-C 4 )alkoxy, or ((Ci-C 4 )aIkyl)((Ci-C )alkyl)amino, wherein 2-4 of X 1 , X 3 , X 4 , and X 3 are a carbon atom substituted by hy drogen.
- X 1 , X , X 3 , X " , and X s are each independently selected from CH and CR 3 , wherein 0-3 of X 1 , X 2 , X', X 4 , and X 3 are CR 3
- X 1 , X 2 , X 3 , X 4 , and X 5 are each independently a carbon atom substituted by hydrogen, halogen, cyano, (Ci-C 4 )alkyl, (CrC 4 )haloalkyl, (CrC 4 )alkoxy, or ((C[-C 4 )alkyl)((Ci-C4)alkyl)amino, wherein 2-5 of X 1 , X , X J , X * , and X 5 are a carbon atom substituted by hydrogen.
- X ' is a carbon atom substituted by halogen, (Ci-C 4 )alkyi., (Ci-C 4 )ha]oa]kyl, cyano, (Ci-C 4 )alkoxy, or
- X 2 , X 3 , X 4 , and X s are each independently a carbon atom substituted by hydrogen, halogen, (Ci-C 4 )alkyl, (Ci-C 4 )haloalkyL cyano, (Ci-C 4 )alkoxy, or ((Ci-C 4 )alkyl)((C t -C 4 )alkyl)amino, wherein 2-4 of X 2 , X 3 , X 4 , and X 3 are a carbon atom substituted by hydrogen.
- one of Y 1 and Y 2 is O or NR S and the other is a bond.
- one of Y 1 and Y ⁇ is O, NH, or N((Ci-C 4 )alkyl) and the other is a bond.
- Y l is NH or NCH 3 and Y * is a bond.
- Y 1 is NH and Y" is a bond.
- Y 1 is a bond and ⁇ ⁇ is NH.
- X 1 is CR 3 , Y 1 is NR.”, Y is a bond, and R 3 and R" taken together with the atoms to which they are attached form a five to seven membered ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by (CrC 4 )alkyl.
- X 1 is CR 5 , Y 1 is R* Y' is a bond, and R 3 and R 8 taken together represent -CH ? -, -CH 2 CH 2 -, or
- Cy is (Cs-CgjcycJoaikyl, lieterocycloalkyl, phenyl, or 5- or 6-membered heteroaryl, each of which is optionally substituted one, two, or three times, independently, by (Ci-Cg)alkyl, (CrC 6 )haloalkyL iC 3 -C 6 )cycloalkyl, halogen, oxo, cyano, hydroxy!,
- Cy is heterocycloalkyl, phenyl, or 5- or 6-membered heteroaryl, each of which is optionally substituted one or two times, independently, by (Ci ⁇ C 6 )alkyl, (Ci-Cg)haloalkyl, halogen, cyano, (Ci-C 4 )alkoxy, ((Ci-C 4 )alkyl)ammo ((C 1 -C4)alkyl)((C C 4 )alkyi)a-mmo, -((C 0 -C 3 )alkyl)CO 2 R 7 ,
- Cy is (C 3 -C 6 )cycloalkyl, azetidinyl, pyrrolidinyl, pyrazolidinyi, pyrazolinyi, imidazolidinyl, imidazolinyl, oxazolinyl, thiazolinyl, tetrahydrofuranyi, dihydrofuranyl, piperidinyi, piperazinyl, morpholinyi,
- thiomorphofinyl tetrahydropyranyl, dihydropyranyl, dioxanyf, oxathianyf, phenyl, furanyl, thieiiyl, pyrrolyl, imidazolyl, pyrazolyl, triazolyl, tetrazoiyl, thiazoiyl, oxazolyi, isoxazolyl, oxadiazolyi, thiadiazolyl, isothiazolyl, pyridinyl, pyridazinyl, pyrazinyl, pyrimidinyl, or triazinyl, each of which is optionally substituted one, two, or three times, mdependently, by (C Cyaikyi, (CrCsjhaloalkyl, (Cs-Cejcycloalliyl, halogen, oxo, cyano, hydroxy!, hydroxy(CrC 6 )a1ky
- (Ci-C4)alkylamino ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, aryl, heteroaryl, aryl(Ci-C6)alkyl, heteroaryi(Ci - C6)alkyi, or heterocycloalkyl.
- Cy is piperidinyi, piperazinyl, phenyl, pyridinyl, py ridazinyl, pyrazinyl, or pyrimidinyl, each of which is optionally substituted one, two, or three times, independently, by (Ci-Cejalkyl, (Ci-C6)haIoaikyl, (C -C'6)cycloalkyl, halogen, oxo, cyano, hydroxy 1, hydroxy(C; -C6)alkyl,
- Cy is piperidinyi, piperazinyl, phenyl, pyridinyl, pyridazinyl, pyrazinyl, or pyrimidinyl, each of which is optionally substituted one or two times, independently, by (Ci-C 6 )alkyl, (Ci -Ce haloalkyl, halogen, cyano, (Q -C 4 )alkoxy, (C r -C4)alkyl)((C l --C4)alkyi)amino, -((C 0 -C 3 )alkyl)CO 2 H,
- Cy is phenyl, which is optionally substituted one, two, or three times, independently, by (Ci ⁇ C 6 )alkyl, (Cj-C 6 )haloalky], (CVCe ⁇ ycioalkyl, halogen, oxo, cyano, hydroxy!,
- (Ci-C 4 )alkylamino ((Ci-C4)alkyl)((Ci-C-4)alkyl)animo, aryl, heteroaiyl, aryl(Ci-C 6 )aikyl, heteroaryi(Ci-C6)alkyl, or heterocycloalkyl.
- Cy is phenyl, which is optionally substituted one or two times, independently, by halogen, (d -C 4 )alkyi, (Ci-C 4 )ha1oa1kyl, cyano, (Ci -C 4 )alkoxy, • i i i " 1 r ( " : ⁇ aik> !)( "( ) R . or -((C 0 -C 3 )aUcy-)CONR 7 R 8 or
- Cy is phenyl, which is optionally substituted one or two times, independently, by halogen, (C-i-C 4 )alkyl,
- Cy is phenyl
- Z is O, NR°, or a bond. In another embodiment of this invention, Z is O, NH,
- Z is a bond, O, or NH. In another embodiment of this invention, Z. is O or NH. In a specific embodiment of this invention, Z is O.
- a 1 , A 2 , A 3 , and A 4 are each independently selected from , NR 6 , O, S, CH, and
- a 1 , A 2 , A 3 , and A 4 are NR 6 , O, or S
- 0-2 of A 1 , A 2 , A 3 , and A 4 are CR 10
- 0-3 of A 1 , A " , A', and A 4 are CH or N.
- a 1 , A , A 3 , and A 4 are each independently selected from N, N((Ci-C 4 )alkyl), O, S, CH, and C((Ci-C 4 )alkyl), wherein one of A 1 , A 2 , A 3 , and A 4 is N((C C 4 )alkyl), O, or S, 0-2 of A 1 , A 2 , A 3 , and A 4 are
- a 1 and A 4 are each independently selected from CH and CR t0 , and one of A 2 and A' is NR 6 , O, or S and the other is N or CH.
- a 1 and A are each independently selected from CH and C((Ci-C4)aIkyl), and one of ⁇ ⁇ and A J is N((Ci-C-4)alkyl), O, or S and the other is N or CH.
- a ' and A 4 are each independently selected from CH and C((Ci-C 4 )alkyJ), and one of A 2 and A' is O or S and the other is N.
- R ! is (C C 6 )alkyl, (C 3 -C 6 )haloaIkyl, (C 3 -C 8 )cycloalkyL (C 3 -C 6 )alkoxy,
- R 1 is (C 3 -C6)alkyl, (Cj-Cgjcycfoafkyl, (Cj-C6) ⁇ koxy(Ci -C2) lk ⁇ , aryl, or heteroaryl, each of which is optionally substituted one, two, or three times, independently, by R ' ⁇
- R l is (C-3-C6)aikyl, (CVC 6 )cyck)alkyl, (Ci-C 6 )alkOxy(Ci-C 2 )alkyl, phenyl, furanyl, thienyl, pyrrolyl, imidazolyi, pyrazoiyi, triazolyl, tetrazolyl, thiazolyl, oxazoiyl isoxazolyl, oxadiazolyl, thiadiazolvl, isothiazolyl, pyridinyi, pyrid
- 3 is halogen, (Ci -C 4 )alkyl, (Ci-C 4 )haloalkyl, cyano, (CrC 4 )alkoxy, or ((Ci-C 4 )alkyl)((CrC 4 )alkyl)araino).
- R 1 is (C ⁇ -Cejalkyf, (CrCekycioalkyl, phenyl, furanyl, thienyl, pyrrolyl, imidazolyi, pyrazoiyi, triazolyl, tetrazolyl, thiazolyl, oxazoiyl, isoxazolyl, oxadiazolyl, thiadiazolyl, isothiazolyl, pyridinyi, pyridazinyl, pyrazinyl, pyrimidinyl, or triazinyl, wherein said phenyl, furanyl, thienyl, pyrrolyl, imidazolyi, pyrazoiyi, triazolyl, tetrazolyl, thiazolyl, oxazoiyl, isoxazolyl, oxadiazolyl, thiadiazolyl, isothiazolyl,
- R' is (Cs-C ⁇ alkyl.
- R 1 is (Cs-Cejalkyl,
- R 1 is phenyl or pyridinyi, each of which is optionally substituted one or two times, independently, by halogen, (C[-C 4 )alkyl, ( -C 4 )haloalkyl, cyano, (Ci-C 4 )alkoxy, or ((Ci-C 4 )a.lkyl)((Ci -C 4 ) lky i) amino.
- R 1 is phenyl or pyridinyi, each of which is optionally substituted one or two times, independently, by halogen, (Ci-Ci)alkyl, (CrC 4 )alkoxy, or ((Ci-C 4 )alkyl)((Ci-C )alkyl)amino.
- R 1 is phenyl optionally substituted one or two times, independently, by halogen,
- R l is phenyl or pyridinyi. In another specific embodiment of this invention, R 1 is phenyl.
- R ⁇ is hydrogen, (C ' CeJalkyl, or (Ci-C6)haloalkyl. In another embodiment of this invention, hydrogen or (Ci-C 4 )alkyl. In another embodiment of this invention, R 2 is hy drogen or methyl. In a specific embodiment of this invention, R 2 is hydrogen.
- R 1 and R 7" taken together represent -C! i ( ' ! ! ( ! ! -. or -CH 2 CH 2 CH 2 CH 2 CH 2 -.
- R ;' and R 3a are each independently hydrogen, hydroxy!, (Ci-C 6 )a ⁇ kyl
- R and R 3a are each independently hydrogen or methy l.
- R J and R Ja are each independently hydrogen.
- each R 4 is independently selected from hydrogen, halogen, (Ci ⁇ C 6 )alkyl,
- each R is independently selected from hydrogen
- each R 4 is independently selected from hydrogen, halogen, (Ci-C 4 )alkyl,
- each R 4 is independently selected from hydrogen, halogen, (CrCYiaJkyi, (Ci-C4)alkylamino, ((Ci-C4)a3kyl)C(Ci-C4)alkyl)ami.no,
- each IT is independently selected from hydrogen, (Ci-C )alkyl, (CrC 4 )alkoxy,
- each R 4 is independently selected from (C 3 -C 4 )alkoxy, hydroxy(C 2 -C4)alkoxy,
- each R 4 is independently selected from (Ci-C 4 )alkoxy, -0((Ci-C 3 )alkyl)C0 2 H,
- each R" is independently selected from (Ci-C 4 )alkyl and (Ci-C 4 )alkoxy. In a specific embodiment of this invention, each R 4 is hydrogen.
- each R 4a is independently selected from hydrogen, halogen, hydroxy!, amino, and (Ci-C6 lkyl.
- each R 4a is independently selected from hydrogen, halogen, and
- each R 4a is independently selected from is hydrogen, fluorine, and methyl.
- each R a is independently selected from is hydrogen and methyl , in a specific embodiment of this invention, each R 4a is hydrogen.
- each "a is methyl.
- R * and R 4a taken together with the carbon atom to which they are attached form a three to eight nienibered ring, optionally containing a heieroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by cyano, (Ci-C 4 )alkyl, (C C 4 ) oalkyl, (C 3 -C 6 )cycloaIkyl, -C0 2 R 7 , -CONR 7 R 8 , hydroxy!,
- R 4 and R 43 taken together represent -CH 2 CH 2 -, -C! i ( i i ( ⁇ -. -CH 2 CH 2 CH 2 CH 2 -, or -CH 2 CH 2 CH 2 CH 2 CH 2 ⁇ .
- One particular embodiment of the invention is a com ound of Formula (la):
- m 1 ;
- n 1 or 2;
- X 1 , X " , X', and X 4 are each independently selected from N, N + -0 ⁇ , CH, and CR ' , wherein 0-2 of X 1 , X 2 , X 3 , and X 4 are N or N + ⁇ (T and 0-2 X 1 , X 2 , X 3 , and X 4 are CR 5 ;
- Y 1 is NH or NCH 3 and Y 2 is a bond
- K 1 , K “ , K 3 , and K 4 are each independently selected from N, N + -0 " , CH, and CR'°, wherein 0-2 of K 1 , . K 3 , and K 4 are N or N + -0 " and 0-2 of K l , K 2 , K 3 , and K 4 are CR 10 ;
- Z is O, NR 6 , or a bond
- a 1 , A “ , A J , and A 4 are each independently selected from N, NR 6 , O, S, CH, and CR l0 , wherein one of A 1 , A 2 , A 3 , and A 4 is NR 6 , O, or S, 0-2 of A 1 , A 2 , A 3 , and A 4 are CR 10 , and 0-3 of A 1 , A 2 , A 3 , and A 4 are CH or N; R !
- R 2 is hydrogen, (Ci-C 6 )aikyl, or (d -d)haioaikyl;
- R J and R 3a are each independently hydrogen, hydroxy!, (d ⁇ d)aikyl, (d-d haloalkyl, halogen, (d. -G alkoxy, amino, (Ci -C 4 )alkyiamino, or ((Ci-C4)alkyl)((Ci -C ⁇ alky amino;
- each R 4 is independently selected from hydrogen, halogen, (d ⁇ di)a!kyl, (CrC 4 )haloalkyl, -OR 9 , and -NR 8 R 9 , wherein said (Ci-C4)alkyl or (C] -C4)haloalkyl is optionally substituted by hydroxy!, -OR 9 , -CO.
- each R 4a is independently selected from hydrogen, halogen, hydroxy!, amino, and
- each R 3 is independently selected from (d -C6)alkyl, (Ci-G haloalkyl, (CrdOcycloalkyl, halogen, cyano, hydroxy!, hydroxy(d-C6)alkyl, (G -GOalkoxy, (Ci-C4)alkoxy(Ci -C6)alkyl, amino, (CrG)alky!amino, ((CrC 4 )alkyl)((Ci-C4)alkyl)ammo, and, heteroaryl, ary ⁇ (Ci-C6)alkyl, heteroaryl(d ⁇ d)alkyl, and heterocycloalkyl;
- R 6 is hydrogen, (Ci-C 6 )alkyl, (Ci-C 6 )haloalkyl, (C 3 -C 6 )eycloalkyl, hydroxy(Ci-C 6 )alkyl,
- R ' is hydrogen, (Ci-Cejalkyl, (Ci -Ce ⁇ haloalkyl, (d-djcyc!oa!kyl,
- R s is hydrogen, or (Ci-CejhaloalkyJ ;
- R ' and R 8 taken together with the nitrogen atom to which they are attached form a four to eight membered ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by (Ci -G alkyi, (d--d)haloalkyl, (C 3 -d)cycloaJkyl, -C0 2 H, -CQ 2 (d -C 4 )alkyL hydroxy!, hydroxy(Ci-d)alkyl, (Ci-C 4 )alkoxy, (Ci-d)a!koxy(Ci-Q)alky!, amino, (G-C 4 )alkylammo, or ((Ci-C4)alkyl)((G-C 4 )alkyl)ammo;
- R 9 is -C(0)R 7 , -C0 2 R 7 , -C(0) R 7 R 8 , (C r C 6 )alkyl, (C C 6 )haioaikyl, (C 3 -C 6 )cycloalkyl, aryl, heteroaryl, aryl(Ci - C 6 )alkyi, heteroaryl(Ci-C6)alkyl, or heterocycloalkyl, wherein said (d-d)aJky!, (d-C ⁇ haloalkyl, (d-djcyeloaJky!, aryl, heteroaryl, aiyl(d-d)alky!,
- heteiOar d(Ci-C 6 )alkyl, or heterocycloalkyl is optionally substituted by -C0 2 R 7 , -CONH 2 , -CONH(Ci-C 4 )alkyl, -CON((C C 4 )alkyl)((Ci-C 4 )alky ⁇ ), hydroxy!, (C r C )alkoxy, amino,
- R 6 and R 9 taken together with the nitrogen ato n to which they are attached form a four to eight membered ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by cyano, (Ci ⁇ C 4 )alkyl, (C]-C 4 )haloalky1, (C 3 -C 6 )cycloalkyl, -C0 2 H, -C0 2 (C 1 -C 4 )alkyl, -CONR 7 R 8 , hydroxy!, hydroxy(C r C 6 )alkyl,
- R 10 is (C-i-C6)alkyl, (Ci-Cr haloalky!, (C 3 -C-6)cyc3oa3kyl, halogen, cyano, hydroxy!, hydroxy(Ci-C 6 )alkyL (C C 6 )alkoxy, (Ci-C 4 )alkoxy(C r C 6 )alkyl, -((C 0 -C 3 )alkyl)CO 2 R 7 ,
- Another particular embodiment of the in v ention is a compound of Formula (la.) wherein: m is 1 ;
- n 1 or 2;
- X 1 , X X 3 , and X 4 are each independently a carbon atom substituted by hydrogen, halogen, cyano, (Ci-C 4 )alkyL (Ci-C 4 ) aloalkyl, (C; -C 4 )aikoxy, or ((Ct-C 4 )alJcyl)((Ci-C 4 )aikyl)aiiiino, wherein 2-4 of X 1 , X 2 , X', and X 4 are a carbon atom substituted by hydrogen;
- Y 1 is NH or NC! h and Y 2 is a bond
- K 1 , K 2 , K s , and K " are each independently a carbon atom substituted by hydrogen, halogen, (CrC 4 )aIkyL (Ci-C 4 )aikoxy, or ((Ci-C 4 )alkyl)((Ct-C )alkyl)amino, wherein 2-4 of K 1 , K " , K', and K 4 are a carbon atom substituted by hydrogen;
- Z is 0, NH, -N(CrC 4 )aJkyl, -N((C 0 -C 3 )alkyl)CO 2 R 7 , -N((C 0 -C 3 )alkyl)CONR 7 R 8 , or a bond;
- a 1 and A 4 are each independently selected from CH and CR 10 , and one of A" and A ' is NR", O, or S and the other is N or CH;
- R. 1 is (C 3 -C6)alkyl, (C 3 -C6)haloalkyl, (C 3 -Cg)cycloalky3, (GVCejaikoxy,
- heterocycloalkyl each of which is optionally substituted one, two, or three times, independently, by R 5 ;
- R ⁇ is hydrogen
- R 3 and R 3a are each independently hydrogen or methyl; each R 4 is independently selected from hydrogen, (Q -Chalky!, (CVG alkoxy, bydiOxy(C2-C 4 )afkoxy, (Ci -C 4 )alkylammo, ((Ci-C-4)aikyl)((Ci-C4)alkyl)amino,
- each R 4a is independently selected from hydrogen, hydroxyl, amino, and (CrC 4 )alkyl; each R 5 is independently selected from (C r C 6 )alkyL (Ci-C 6 )haloalkyl, (C 3 -C 6 )cycloalkyi, halogen, eyano, hydroxyl, hyciroxy(C rC 6 )alkyl, (C; -C 6 )alkoxy, (Ci-C4)alkoxy(C; -C 6 )alkyl, amino, (Ci-C4)alkylamino, ((Ci -C4)alkyl)((Ci -C4)alkyl)amino, aryl, heteroaryl, aryl(Ct-C6)aIkyl, heteroaryl(Ci-C6)alkyl, and heterocyeloalkyi;
- R ' is hydrogen, (Ci-C 6 )alkyl, (Ci -C ⁇ haloalkyf, (CrC 6 )cycloalky],
- R 8 is hydrogen, (C CeWkyl, or (Cj-Cejhaloalkyl
- R 10 is (Ci-Cfijalkyl, (Ci-CejhaloalkyL (C 3 -C6)cycloalkyl, halogen, eyano, hydroxyl, hydroxy(C r C 6 )aikyl, (C, -C 6 )alkoxy, (C x -C4)alkoxy(Ci-C 6 )alkyl, -((C 0 -C 3 )alkyl)CO 2 R 7 ,
- Another particular embodim ent of the invention is a compound of Formul a (la) wherein: m is 1 ;
- X 1 , X 2 , X', and X 4 are each independently a carbon atom substituted by hydrogen, halogen, eyano, (Ci -C 4 )alkyl, (C-i-C4)haloalkyl, (C-. -C4)aikoxy, or ((Ci -C 4 )alkyl)((Ci-C4)a]kyl)amino, wherein 2-4 of X 1 , X " , X 3 , and X are a carbon atom substituted by hydrogen;
- Y 1 is NH and Y 2 is a bond
- K l , K , K 3 , and K 4 are each independently a carbon atom substituted by hydrogen, halogen, (Cj-C 4 )aiky1, (d-C4)a]koxy, or ((Ci-C 4 )alkyl)((Ci-C 4 )alkyl)amino, wherein 2-4 of K l , K 7" , K 3 , and K 4 are a carbon atom substituted by hydrogen;
- Z is O, NH, -N(Ci-C 4 )alkyL or a bond:
- a 1 and A 4 are each independently selected from CH and C((CV C 4 )alkyl), and one of A 2 and A 3 is O or S and the other is N;
- R ! is phenyl optionally substituted one or two times, independently, by halogen, (Ci-C 4 )alkyl, (Ci ⁇ C 4 )haloalkyi, cyano, (Ci-C 4 )alkoxy, or ((Ci-C 4 )alkyl)((Ci-C 4 )a1kyl)amino;
- R 2 is hydrogen
- R ""1 and 3 ⁇ 4 are each independently hydrogen or methyl
- each R 4 is independently selected from hydrogen, (Ci-C )alkyl, (Cj -C 4 )alkyiamino,
- each R 4a is independently selected from hydrogen, hydroxyl, amino, and (Ci-C 4 )alkyl; or a pharmaceutically acceptable salt thereof.
- Specific compounds of this invention include:
- the compounds according to Formula (I) may contain one or more asymmetric centers (also referred to as a chiral center) and may, therefore, exist as individual enantiomers, diastereomers, or other stereoisomeric forms, or as mixtures thereof.
- Chiral centers such as chiral carbon atoms, may also be present in a substituent such as an alkyl group.
- Individual stereoisomers of a compound according to Formula (T) which contain one or more asymmetric centers may be resolved by methods known to those skilled in the art. For example, such resolution may be carried out (1) by formation of diastereoisomeric salts, complexes or other derivatives; (2.) by selective reaction with a stereoisomer-specific reagent, for example by enzymatic oxidation or reduction: or (3) by gas-liquid or liquid chromatography in a chiral environment, for example, on a chiral support such as silica with a bound chiral ligand or in the presence of a chiral solvent.
- stereoisomers may be synthesized by asymmetric s nthesis using optically active reagents, substrates, catalysts or solvents, or by converting one enantiomer to the other by asymmetric transformation.
- Enantiomerically enriched refers to products whose enantiomeric excess is greater than zero.
- enantiomerically enriched refers to products whose enantiomeric excess is greater than 50% ee, greater than 75% ee, and greater than 90% ee.
- Enantiomeric excess or "ee” is the excess of one enantiomer over the other expressed as a percentage. As a result, since both enantiomers are present in equal amounts in a racemic mixture, the enantiomeric excess is zero (0% ee). However, if one enantiomer was enriched such that it constitutes 95% of the product, then the enantiomeric excess would be 90% ee (the amount of the enriched enantiomer, 95%, minus the amount of the other enantiomer, 5%).
- Enantiomerically pure means products whose enantiomeric excess is 99% ee or greater.
- the compound or salt including solvates (particularly, hydrates) thereof, may exist in crystalline forms, non-crystalline forms or a mixture thereof.
- the compound or salt, or solvates (particularly, hydrates) thereof may also exhibit polymorphism (i.e. the capacity to occur in different crystalline forms). These different crystalline forms are typically known as
- polymorphs It is to be understood that when named or depicted by structure, the disclosed compound, or solvates (particularly, hydrates) thereof, also include all polymorphs thereof.
- Polymorphs have the same chemical composition but differ in packing, geometrical arrangement, and other descriptive properties of the crystalline solid state. Polymorphs, therefore, may have different physical properties such as shape, density, hardness, deform ability, stability, and dissolution properties. Polymorphs typically exhibit different melting points, 1R spectra, and X-ray powder diffraction patterns, which may be used for identification. One of ordinary skill in the art will appreciate that different polymorphs may be produced, for example, by changing or adjusting the conditions used in crystallizing/recrystallizing the compound.
- solvates of the compounds of Formula (1), or salts thereof, that are in crystalline form may be formed wherein solvent molecules are incorporated into the crystalline lattice during crystallization.
- Solvates may- involve nonaqueous solvents such as ethanol, isopropanol, DMSO, acetic acid, ethanolamine, and ethyl acetate, or they may involve water as the solvent that is incorporated into the crystalline lattice.
- Solvates wherein water is the solvent that is incorporated into the crystalline lattice are typically referred to as "hydrates.” Hydrates include stoichiometric hydrates as well as compositions containing variable amounts of water. The invention includes all such solvates.
- salts of the compounds of Formula (I) are preferably pharmaceutically acceptable. Suitable pharmaceutically acceptable salts include those described by Berge, Bighley and Monkhouse J.Pharm.Sci (1977) 66, pp 1 -19. Salts encompassed within the term "pharmaceutically acceptable salts" refer to non-toxic salts of the compounds of Formula (1).
- Salts of the compounds of Formul (!) containing a basic amine or other basic functional group may be prepared by any suitable method known in the art, including treatment of the free base with an inorganic acid, such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like, or with an organic acid, such as acetic acid, trifluoroacetic acid, maJeic acid, succinic acid, mandelic acid, fumaric acid, malonic acid, pyruvic acid, oxalic acid, glycolic acid, salicylic acid, pyranosidyl acid, such as glucuronic acid or galacturonic acid, alpha- hydroxy acid, such as citric acid or tartaric acid, amino acid, such as aspartic acid or glutamic acid, aromatic acid, such as benzoic acid or cinnamic acid, sulfonic acid, such as p-toluenesulfonic acid, methanesulfonic acid,
- Examples of pharmaceutically acceptable salts include sulfates, pyrosulfates, bisulfates, sulfites, bisulfites, phosphates, chlorides, bromides, iodides, acetates, propionates, decanoates, caprylates, acrylates, formates, isobutyrates, caproates, lieptanoates, propiolates, oxalates, malonates succinates, suberaies, sebacates, fimiaraies, maleates, buryne- 1 ,4-dioates, hexyne- 1 ,6-dioates, benzoates, chlorobenzoates, methyibenzoates,
- Salts of the compounds of Formula (1) containing a carboxyiic acid or other acidic functional group can be prepared by reacting with a suitable base.
- Such a pharmaceutically acceptable salt may be made with a base which affords a pharmaceutically acceptable cation, which includes alkali metal salts (especially sodium and potassium), alkaline earth metal salts (especially calcium and magnesium), aluminum salts and ammonium salts, as well as salts made from physiologically acceptable organic bases such as trimethylamine, triethyl mine, morpholine, pyridine, piperidine, picoline, dicyclohexylamine, N,N-dibenzylethylenediamine, 2- hydroxyethylamine, te-(2-hydroxyemyl)amine, tri-(2-hydroxyethyl)amine, procaine,
- a base which affords a pharmaceutically acceptable cation, which includes alkali metal salts (especially sodium and potassium), alkaline earth metal salts (especially calcium and magnesium), aluminum salts and ammonium salts, as well as salts made from physiologically acceptable organic bases such as trimethylamine, triethyl mine, morpholine, pyridine,
- dibenzylpiperidine dehydroabietylamine, ⁇ , ⁇ -bisdshy droabietylamine, giucamine, N- methylglucamine, collidine, quinine, quinoline, and basic amino acid such as lysine and arginine.
- non-pharmaceutically acceptable salts e.g. triffuoroacetate
- triffuoroacetate may be used, for example in the isolation of compounds of the invention, and are included within the scope of this invention.
- the invention includes within its scope all possible stoichiometric and non-stoichiometric forms of the salts of the compounds of Formul (I).
- a compound of Formula (I) containing a basic amine or other basic functional group is isolated as a salt, (he corresponding free base form of that compound may be prepared by any suitable method known to the art, including treatment of the salt with an inorganic or organic base, suitably an inorganic or organic base having a higher pK a than the free base form of the compound, Similarly, if a compound of Formula (I) containing a carboxyiic acid or other acidic functional group is isolated as a salt, the corresponding free acid form of that compound may be prepared by any suitable method known to the art, including treatment of the salt with an inorganic or organic acid, suitably an inorganic or organic acid having a lower pK a than the free acid form of the compound.
- the invention also includes various deuterated forms of the compounds of Formula (I).
- Each available hydrogen atom attached to a carbon atom may be independently replaced with a deuterium atom, A person of ordinary skill in the art will know how to synthesize deuterated forms of the compounds of Formula (I).
- Commercially available deuterated starting materials may be employed in the preparation of deuterated forms of the compounds of Formula (I), or they may be synthesized using conventional techniques employing deuterated reagents (e.g. lithium aluminum deuteride or sodium borodeuteride).
- Modulators of RORy can be useful in the treatment of diseases mediated by RORy, particularly autoimmune or inflammatory diseases and cancer.
- inflammatory or autoimmune diseases include multiple sclerosis, rheumatoid arthritis, psoriasis, Crohn's disease, inflammatory bowel disease, graft-versus-host disease (GVHD), Sjorgen's syndrome, optic neuritis, chronic obstructive pulmonary disease, asthma, type I diabetes, neuromyelitis optica, myasthenia gravis, uveitis, Behcets disease, Guillain-Barre syndrome, psoriatic arthritis, Graves' disease, allergic contact dermatitis, systemic lupus erythematosus, cutaneous lupus erythematos s, ankylosing spondylitis, Hashimoto Thyroiditis, dry eye and glomerulonephritis, myocarditis, especially psoriasis
- Such cancers include multiple mye
- the inv ention is directed to methods of treating such diseases using a compound of Formula (I) or a pharmaceutically acceptable salt thereof.
- the methods of treatment of the invention comprise administering an effective amount of a compound according to Formula (I) or a pharmaceutically acceptable salt thereof to a patient (particularly a human) in need thereof.
- the invention is directed to a compound of Formula (I) or a
- the invention is directed to the use of a compound of Formula (I) or a phannaceuticaliy acceptable salt thereof in the manufacture of a medicament for the treatment of diseases mediated by RORy, particularly autoimmune or inflammatory diseases and cancer, such as those disclosed above.
- treatment in reference to a condition means: (1 ) the amelioration or prevention of the condition being treated or one or more of the biological manifestations of the condition being treated, (2) the interference with (a) one or more points in the biological cascade that leads to or is responsible for the condition being treated or (b) one or more of the biological manifestations of the condition being treated, or (3) the alleviation of one or more of the symptoms or effects associated with the condition being treated.
- prevention of a condition includes prevention of the condition.
- prevention is not an absolute term. In medicine, “prevention” is understood to refer to the prophylactic administration of a drug to substantially diminish the likelihood or severity of a condition or biological manifestation thereof, or to delay the onset of such condition or biological m nifestation thereof.
- an “effective amount” means that amount of a drug or pharmaceutical agent that will elicit the biological or medical response of a tissue, system, animal or human that is being sought, for instance, by a researcher or clinician.
- therapeutically effective amount means any amount which, as compared to a corresponding subject who has not received such amount, results in improved treatment, healing, prevention, or amelioration of a disease, disorder, or side effect, or a decrease in the rate of advancement of a disease or disorder.
- the term also includes within its scope amounts effective to enhance normal physiological function,
- patient refers to a human or a mammal, especially a human.
- the compounds of the invention may be administered by any suitable route of administration, including both systemic administration and topical administration.
- Systemic administration includes oral administration, parenteral adminisiration, transdermal administration, rectal administration, and administration by inhalation.
- Parenteral administration refers to routes of administration other than enteral, transdermal, or by inhalation, and is typically by injection or infusion.
- Parenteral administration includes intravenous, intramuscular, and subcutaneous injection or infusion, inhalation refers to administration into the patient's lungs whether inhaled through the mouth or through the nasal passages.
- Topical administration includes application to the skin as well as intraocular, otic, intravaginal, and intranasal administration,
- the compounds of the invention may be administered once or according to a dosing regimen wherein a number of doses are administered at vary ing intervals of time for a given period of time. For example, doses may be administered one, two, three, or four times per day. Doses may be administered until the desired therapeutic effect is achieved or indefinitely to maintain the desired therapeutic effect.
- Suitable dosing regimens for a compound of the invention depend on the pharmacokinetic properties of that compound, such as absorption, distribution, and half-life, which can be determined by the skilled artisan, in addition, suitable dosing regimens, including the amount administered and the duration such regimens are administered, for a compound of the inven tion depend on the condition being treated, the severity of the condition being treated, the age and physical condition of the patient being treaied, the medical history of the patient to be treaied, the nature of concurrent therapy, the particular route of administration chosen, the desired therapeutic effect, and like factors within the knowledge and expertise of the skilled artisan.
- suitable dosing regimens may require adjustment given an individual patient's response to the dosing regimen or over time as individual patient needs change.
- Typical daily dosages range from 1 mg to 1000 mg.
- certain protected derivatives of compounds of Formula (I) which may be made prior to a final deprotection stage, may not possess pharmacological activity as such, but may, in certain instances, be administered orally or paremerally and thereafter metabolized in the body to form compounds of the invention which are pharmacologically active.
- Such derivatives may therefore be described as "prodrugs”.
- certain compounds of the invention may act as prodrugs of other compounds of the invention. All protected derivatives and prodrugs of compounds of the invention are included within the scope of the invention.
- pro-drags examples of suitable pro-drags for the compounds of the present invention are described in Drags of Today, Volume 19, Number 9, 1983, pp 499 - 538 and in Topics in Chemistry, Chapter 31, pp 306 - 316 and in "Design of Prodrugs" by H. Bundgaard, Elsevier, 1985, Chapter 1 (the disclosures in which documents are incorporated herein by reference). It will further be appreciated by those skilled in the art, that certain moieties, known to those skilled in the art as “pro-moieties”, for example as described by H. Bundgaard in “Design of Prodrugs” (the disclosure in which document is incorporated herein by reference) may be placed on appropriate
- Preferred "pro-moieties" for compounds of the invention include: ester, carbonate ester, hemi- ester, phosphate ester, nitro ester, sulfate ester, sulfoxide, amide, carbamate, azo-, phosphatide, glycoside, ether, acetal, and ketaf derivatives of the compounds of Formula (I).
- Administration of a compound of the invention as a prodrug may enable the skilled artisan to do one or more of the following: (a) modify the onset of the compound in vivo; (b) modify the duration of action of the compound in vivo; (c) modify the transportation or distribution of the compound in vivo; (d) modify the solubility of the compound in vivo; and (e) overcome or overcome a side effect or other difficulty encountered with the compound.
- the invention further includes the use of compounds of the invention as an active therapeutic substance, in particular in the treatment of diseases mediated by RORy.
- the invention relates to the use of compounds of the invention in the preparation of a medicament for the treatment of diseases mediated by RORy.
- diseases include autoimmune or inflammatory diseases such as multiple sclerosis, rheumatoid arthritis, psoriasis, Crohn's disease, inflammatory bowel disease, Sjorgen's syndrome, optic neuritis, chronic obstructive pulmonary disease, asthma, type I diabetes, neuromyelitis optica.
- autoimmune or inflammatory diseases such as multiple sclerosis, rheumatoid arthritis, psoriasis, Crohn's disease, inflammatory bowel disease, Sjorgen's syndrome, optic neuritis, chronic obstructive pulmonary disease, asthma, type I diabetes, neuromyelitis optica.
- Myasthenia Gravis uveitis, Guillain-Barre syndrome, psoriatic arthritis, Graves' disease, allergic contact dermatitis, systemic lupus erythematosus, cutaneous lupus erythematosus, ankylosing spondylitis, Hashimoto Thyroiditis, Dry Eye, glomerulonephritis, myocarditis and cancer diseases including multiple myeloma and lytic bone disease associated with multiple myeloma, acute myelogenous leukemia (AML), bead and neck squamous cell carcinoma, bladder carcinoma, gastric cancer, hepatocellular carcinoma, melanoma, medulloblastoma and colon cancer.
- AML acute myelogenous leukemia
- the invention includes the use of compounds of the invention for the preparation of a composition for treating or ameliorating diseases mediated by RQRy in a subject in need thereof, wherein the composition comprises a mixture of one or more of the compounds of the invention and an optional pharmaceutically acceptable excipient.
- the compounds of the invention may be used alone or in combination with one or more other therapeutic agents. Accordingly the present invention provides a combination comprising a compound of Formula (I) or a pharmaceutically acceptable salt thereof and one or more other therapeutic agents. Such combinations may be presented individually (wherein each active is in separate composition) or the actives are presented in a combined composition.
- This invention provides a combination of a compound of Formula (I), or a
- an inflammator '' disease and/or an autoimmune disease for example, a TNF-a inhibitor; a nonselective COX-l/COX-2 inhibitor; a selective COX-2 inhibitor, such as celecoxib; agents including methotrexate, leflunomide, sulfasalazine, azathioprine, penicillamine, bucillamine, actarit, mizorihine, lobenzarit, hydroxychloroquine, d-penicillamme, aurothiom.ai.ate, auranofin, parenteral and/or oral gold, cyclophosphamide, a BAFF/ APRIL inhibitor, CTLA-4-Ig, or a mimetic of CTLA-4-Ig; 5-lipoxygenase (5-LO) inhibitor, or a 5 -lipoxygenase activating protein (FLAP) antagonist; a leukotriene
- a TNF-a inhibitor for example, a TNF
- PDE-TV phosphodiesterase type IV
- ciioniiiast ariflo
- roilumilast an antihistamine HI receptor antagonist
- anticholinergic agents such as muscarinic antagonists (ipratropium bromide and tiotropium bromide), as well as selective muscarinic M3 antagonists
- ⁇ -adrenoceptor agonists such as salmeteroi, formoterol, arformoteroi, terbutaline, metaproterenol, albuterol and the like
- a DP receptor antagonist such as S-5751 and laropiprant
- TP receptor antagonists such as seratrodast
- VLA-4 antagonists a corticosteroid, such as triamcinolone acetonide, budesonide, beclomethasone, fluticasone and mometasone
- insulin-like growth factor type I I
- This invention further provides a combination of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, and one or more therapeutic agents for the treatment of multiple myeloma, for example, Boriezomib-dexamethasone, Bortezomib-dexamethasone- cyclophosphamide, Bortezomib-dexamethasone-lenalidoniide, Lenalidomide-dexamethasone, Melphalan-prednisone-thaiidoniide, Melphalan-prednisone-bortezo ib, Melphalan-prednisone- lenalidomide, Lenaiidomide- dexamethasone- clarithromycin and any of the above combinations plus agents used to treat bone disease in multiple myeloma including bisphosponates, RA K-L inhibitors such as Denusomab and anabolic bone building drugs such as parathyroid hormone (PTH).
- PTH par
- This invention also provides a combination of a compound of Formula (I), or a
- FOLFOX® leucovorin [folinic acid], 5-Fluoruracil, and oxaliplatin
- FOLF1R1® leucovorin, 5-Fluoruraeii, and irinotecan
- CapeOX® capecitabine and oxaliplatin
- 5- Fluoruraeil and leucovorin with or without bevacizumab, Capecitabine, with or without bevacizumab
- FOLFOXIRI® leucovorin, 5-Fluoruracil, oxaliplatin, and irinotecan
- Irinotecan with or without cetuximab, Cetuximab alone, and Panitumumab alone.
- the compounds of the invention will normally, but not necessarily, be formulated into pharmaceutical compositions prior to administration to a patient. Accordingly, in another aspect the invention is directed to pharmaceutical compositions comprising a compound of the invention and one or more pharmaceutically acceptable excipieni(s).
- compositions of the invention may be prepared and packaged in bulk form wherein an effective amount of a compound of the invention can be extracted and then given to the patient such as with powders, syrups, and solutions for injection.
- the pharmaceutical compositions of the invention may be prepared and packaged in unit dosage form.
- a dose of the pharmaceutical composition contains at least a therapeutically effective amount of a compound of this invention (i.e., a compound of Formula I or a salt, particularly a pharmaceutically acceptable salt, thereof).
- the pharmaceutical compositions may contain from 1 mg to 1000 mg of a compound of this invention.
- the pharmaceuiicai compositions of the invention typically contain one compound of the invention. However, in certain embodiments, the pharmaceutical compositions of the invention contain more than one compound of the invention. For example, in certain embodiments the pharmaceutical compositions of the invention contain two compounds of the invention. In addition, the pharmaceutical compositions of the invention may optionally further comprise one or more additional therapeutically active compounds.
- pharmaceutically acceptable excipient means a pharmaceutically acceptable material, composition, or vehicle involved in giving form or consistency to the pharmaceutical composition.
- Each excipient must be compatible with the other ingredients of the pharmaceutical composition when commingled such that interactions which would substantially reduce the efficacy of the compound of the invention when administered to a patient and interactions which would result in pharmaceutical compositions that are not pharmaceutically acceptable are avoided.
- each excipient must of course be of sufficiently high purity to render it pharmaceutically acceptable.
- dosage forms include those adapted for (1 ) oral administration such as tablets, capsules, caplets, pills, troches, powders, syrups, elixers, suspensions, solutions, emulsions, sachets, and cachets: (2) parenteral administration such as sterile solutions, suspensions, and powders for reconstitution; (3) transdermal administration such as transdermal patches; (4) rectal administration such as suppositories; (5) inhalation such as dry- powders, aerosols, suspensions, and solutions; and (6) topical administration such as creams, ointments, lotions, solutions, pastes, sprays, foams, and gels.
- oral administration such as tablets, capsules, caplets, pills, troches, powders, syrups, elixers, suspensions, solutions, emulsions, sachets, and cachets
- parenteral administration such as sterile solutions, suspensions, and powders for reconstitution
- transdermal administration such as transdermal patches
- Suitable pharmaceutically acceptable excipients will vary depending upon the particular dosage form chosen.
- suitable pharmaceutically acceptable excipients may be chosen for a particular function that they may serve in the composition. For example, certain
- pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of uniform dosage forms. Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of stable dosage forms. Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the carrying or transporting of the compound or compounds of the invention once administered to the patient from one organ, or portion of the body, to another organ, or portion of the body. Certain pharmaceutically acceptable excipients may be chosen for their ability to enhance patient compliance.
- Suitable pharmaceutically acceptable excipients include the following types of excipients: diluents, fillers, binders, disintegrants, lubricants, glidants, granulating agents, coating agents, wetting agents, solvents, co-solvents, suspending agents, emulsifiers, sweeteners, flavoring agents, flavor masking agents, coloring agents, anticaking agents, hemectants, chelating agents, plasticizers, viscosity increasing agents, antioxidants, preservatives, stabilizers, surfactants, and buffering agents.
- excipients may serve more than one function and may serve alternative functions depending on how much of the excipient is present in the formulation and what other ingredients are present in the formulation.
- Skilled artisans possess the knowledge and skill in the art to enable the to select suitable pharmaceutically acceptable excipients in appropri te amounts for use in the invention.
- compositions of the invention are prepared using techniques and methods known to those skilled in the art. Some of the methods commonly used in the art are described in Remington's Pharmaceutical Sciences (Mack Publishing Company).
- the invention is directed to a solid oral dosage form such as a tablet or capsule comprising a safe and effective amount of a compound of the invention and a diluent or filler.
- Suitable diluents and fillers include lactose, sucrose, dextrose, mannitol, sorbitol, starch (e.g. corn starch, potato starch, and pre-gelatinized starch), cellulose and its derivatives (e.g.
- the oral solid dosage form may further comprise a binder. Suitable binders include starch (e.g. corn starch, potato starch, and pre-gelatinized starch), gelatin, acacia, sodium alginate, alginic acid, tragacanth, guar gum, povidone, and cellulose and its derivatives (e.g. microcrystaliine cellulose).
- the oral solid dosage form may further comprise a disintegrant. Suitable disintegrants include crospovidone, sodium starch glycolate, croscamielose, alginic acid, and sodium carboxymethyl cellulose.
- the oral solid dosage form may further comprise a lubricant. Suitable lubricants include stearic acid, magnesium stearate, calcium stearate, and talc.
- the compounds of Formula (I) may be obtained by using synthetic procedures illustrated in the Schemes below or by drawing on the knowledge of a skilled organic chemist.
- the reaction sequences provided in these Schemes are applicable for producing compounds of the invention having a variety of different X'-X 3 , R 1 , R 3 , R 3a , R*, R" a , K'-K 4 , and A'-A 4 groups, as defined above, employing appropriate precursors.
- the skilled artisan will appreciate that if a substituent described herein is not compatible with the synthetic methods described herein, the substituent may be protected with a suitable protecting group that is stable to the reaction conditions. The protecting group may be removed at a suitable point in the reaction sequence to provide a desired intermediate or target compound.
- Suitable protecting groups and the methods for protecting and de-protecting different substituents using such suitable protecting groups are well known to those skilled in the art; examples of which may be found in T. Greene and P. Wuts, Protecting Groups in Chemical Synthesis (3rd ed.), John Wiley & Sons, NY (1999).
- a substituent may be specifically selected to be reactive under the reaction conditions used. Under these circumstances, the reaction conditions convert the selected substituent into another substituent that is either useful as an intermediate compound or is a desired substituent in a target compound.
- LCMS-P1 Column: Waters Sunfire C I 8, 3.5 ⁇ , 50 x 4.6 mm: Temperature: 50 °C;
- LCMS-G7 Column: XBridge CI 8, 3.6 ⁇ , 50 x 4.6 mm; Temperature: 50 °C;
- Mobile Phase A: water (0.1% formic acid) B: methanol; Gradient: 10% B for 0.1 min, increase to 95% B within 2,5 min, 95% B for 2.5 min, return to 10% B within 0.1 min, 10% B for 2 min.;
- LCMS-G12 Column: Sunfire CI 8, 5 ⁇ , 50 x 4.6 mm; Temperature: 50 °C; Mobile
- Phase A: water (0.1% formic acid) B: methanol; Gradient: 30% B for 0.1 min, increase to 90% B within 4 min, 99% B for 4 min, return to 30% B within 0.1 min, 10% B for 2 min.; Flow Rate:
- This compound was synthesized from 2-(4-((3,5-dimethylisoxazol-4- yl)methoxy)phenyl)acetic acid and (4-chloro-2-methylphenyl)(4-chlorophenyl)methanamine essentially as described in example 1 (f), except the title compound was isolated as follows: after completion of the reaction, water ( 1 0 niL) was added into the reaction mixture very slowly with cooling and the obtained white solid was filtered off and washed with water (20 mL) and hexanes (20 mL) and dried under reduced pressure to obtain the title compound (240 nig, 55.98%).
- This compound was synthesized from 2,4-dimethylhenzomtriie and 4-chloro
- This compound was synthesized from 2-(4-hydroxyphenyl)acetic acid and (4- chlorophenyi)(phenyl)methananiine essentially as described in example 1 (f) and the product was purified by silica gel column chromatography (40% EtOAc/hexanes) to provide the title compound (4.5 g, 65.2%).
- This compound was synthesized from 2-( ' 4-(((3,5-dimethylisoxazol-4- yl)methyl)amino)pheny])acetic acid and heny3(p-tolyl)methanamii e essentially as described in example 4 (b) and purified by silica gel column chromatography using 0- 100% ethyl
- This compound was synthesized from (3,5-diisopropylisoxazol-4-yl)methan.ol essentially as described in example 10(d). (1.0 g, 76.33%), 3 ⁇ 4 NMR (400 MHz, DMSO-de) ⁇ ppm 4.45 (s, 2 H), 3.16-3.22, (m, 1 IT), 3.03 3.10 (m, 1 H), 1.29-1.39 (m, 12 IT).
- This compound was synthesized from methyl 2-(4-hydroxyphenyl)acetate and 4- (chloromethyl)-3,5-diisopropylisoxazole essentially as described in example 1 1(a). (0.300 g, 60.4%).
- reaction mixiure was extracted with 8% aqueous Na 2 C0 3 (10 x 50 mL) and the combined aqueous layer was then acidified using 6 N HCl (20 mL) and the solid obtained was filtered and dried.
- the crude solid product was purified using silica gel column chromatography using 20%
- This compound was prepared from methyl 2-(4-(2-(3,5-dimethylisoxazol-4-yl)-2- hydroxyethoxy)phenyl)acetate essentially as described in example 24 (d) (99 mg, 100%).
- This compound was prepared from 2-(4 ⁇ (2-(3,5-dimethylisoxazol ⁇ 4-yl) ⁇ 2 ⁇
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention relates to novel retinoid-reiated orphan receptor gamma (RORy) modulators and their use in the treatment of diseases mediated by RORy.
Description
COMPOUNDS AND METHODS
The present invention relates to no vel retinoid-related orphan receptor gamma (RORy) modulators and their use in the treatment of diseases mediated by RORy.
Background of the Invention Retinoid-related orphan receptors (RORs) are transcription factors which belong to the steroid hormone nuclear receptor superfamily (Jetten & Joo (2006) Adv. Dev. Biol. 16:313-355). The ROR. family consists of three members, ROR. alpha (RORa), ROR beta (RORp), and ROR gamma (RORy), each encoded by a separate gene (RORA, RORB, and RORC, respectively). RORs contain four principal domains shared by the majorit '- of nuclear receptors: an N-terminal A/B domain, a DNA-binding domain, a hinge domain, and a ligand binding domain. Each ROR gene generates several isoforms which differ only in their N-terminal A/B domain. Two isoforms of RORy have been identified: RORyl and RORyt (also known as RORy2). RORy is a term used to describe both RORyl and/or RORyt.
While RORyl is expressed in a variety of tissues including thymus, muscle, kidney and li v er, RORyt is exclusi v ely expressed in the cells of the immune system. RORyt has been identified as a key regulator of Thl 7 cell differentiation. Thl 7 cells are a subset of T helper cells which produce IL- 17 and other proinflammatory cytokines. Thl 7 cells have been shown to have key functions in several mouse autoimmune disease models including experimental autoimmune encephalomyelitis (EAE) and collagen-induced arthritis (CIA). In addition, Thl 7 cells or their products have been shown to be associated with the pathology of a variety of human inflammatory and autoimmune disorders including multiple sclerosis, rheumatoid arthritis, psoriasis, Crohn's disease and asthma (Jetten (2009) Nucl. Recept. Signal. 7:e003; Manel et ai. (2008) Nat. Immunol 9:641 -649). The pathogenesis of chronic autoimmune diseases including multiple sclerosis and rheumatoid arthritis arises from the break in tolerance towards self-antigens and the development of auto-aggressive effector T cells infiltrating the target tissues. Studies have shown that Thl 7 cells are one of the important drivers of the inflammatory process in tissue-specific autoimmunity (Steinman (2008) J. Exp. Med. 205: 1517- 1522; Leung et ai. (2010) Cell. Mol. Immunol. 7: 182- 189). There is evidence that Thl 7 cells are activated during the disease process and are responsible for recruiting other inflammatory cells types, especially neutrophils, to mediate pathology in the target tissues (Korn et ai. (2009) Aram. Rev. Immunol. 27:485-517).
RORyt plays a critical role in the pathogenic responses of Thl 7 cells (Ivanov et al. (2006) Cell 126: 1 121 -1 13 ). RORyt deficient mice produce few Thl 7 cells, in addition, RORyt deficiency resulted in amelioration of EAE. Further support for the role of RORyt in the
pathogenesis of autoimmune or inflammatory diseases can be found in the following references: Jetteii & .Too (2006) Adv. Dev. Biol. 16:313-355; Meier et al. (2007) Immunity 26:643-654; Aloisi & Pujol-Borrell (2006) Nat. Rev. Immunol. 6:205-217; Jager et al. (2009) J. Immunol. 183:7169- 7177; Serafmi et al (2004) Brain Pathol. l 4: 164- 174; Magliozzi et al. (2007) Brain 130: 1089-1 104 Barnes (2008) Nat. Rev. Immunol. 8: 1 83-192.
in light of the role RORy plays in the pathogenesis of diseases, it is desirable to prepare compounds that modulate RORy activity, which can be used in the treatment of diseases mediated by RORy.
Summary of the Invention
The invention is directed to novel RORy modulators and their use in the treatment of diseases mediated by RORy. Specifically, the invention is directed to compounds according to Formula I):

wherein:
m is 0, 1, or 2;
n is 0, 1, 2, or 3;
X1 , X", X', X4, and J are each independently selected from N, N+-0~, CH, and CR'\ wherein 0-3 of X1, X2, X3, X4, and Xs are N or N+-0" and 1-3 of X1, X2, X3, X4, and X3 are CR5; provided that when zero of X1, X2, X4, and X3 are N or N"f-0~ and X""1 is CR3, 1-2 of X1, X , X4, and Xs are CR5;
one of Y ; and Y7" is O or NR8 and the other is a bond;
or X1 is CR5, Y1 is NR8, Y2 is a bond, and R5 and R8 taken together with the atoms to which they are attached form a five to seven membered ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by (Ci-C4)alkyl;
Cy is (CrCgkycloalkyl, heterocycloalkyl, phenyl, or 5- or 6-membered heteroaryl, each of which is optionally substttiEted one, two, or three times, independently, by (Ci -Ce kyl,
·: ( ·ί'.,;·!·ΐϋ!οϋ!1·. ν!. (CVCe cycioaikyl, halogen, oxo, cyano, hydroxy!, hydroxy(Ci-C6)alkyl, (Ci-C6)alkoxy, -(( i',;-C'. !ulkvi

-((Co-C3)alkyl)NHC(0)R7, -((Co-C3)alkyl)N((Ci-C4)alkyl)C(0)R7, ~((Co-C3)alkyl)C02R7,
-(((Y.-( alky! )C NR ' \ -iiC0-C3)alky!)C(O)R7, (Ci-C4)a1koxy(Ci-C6)alky1, amino(C C6)alkyi, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino(Ci-C6)alkyl, (Ci-C4)alkylamino( -C6)alkyl, amino,
(Ci-C4)alkylamino, ((Ci -C4)alkyl)((Q-C4)alkyl)amino, aryl, heteroaryl, aryl(Ci-C6)alkyL heteroaryl(C] -C6)alkyl, or beterocycloalkyl;
Z is O, S, S02, C=0, N 6, or a bond;
A1, A-, A3, and A4 are each independently selected from N, Rb, O, S, CH, and CRt0, wherein one of A1, A2, A3, and A4 is NR6, O, or S, 0-2 of A1, A2, A3, and A4 are CR10, and 0-3 of A1, A2, A", and A4 are CH or N;
Rl is (C3 »C6)alkyl, (CrC6)haloalkyi, (C3-C3)cyeloalkyl, (C3-C6)alkoxy,
(Ci-C6)a{koxy(Ci-C2)alky{, aryl, heteroaryl, aryl(Ci-C6)alkyl, heteroaryl(Ci-C6)alkyl, or beterocycloalkyl, each of which is optionally substituted one, two, or three times, independently, by
R5;
R2 is hydrogen, (Ci"C6)aikyl, or (Ci -Cejhaloalkyl;
or R; and R2 taken together with the carbon atom to which they are attached form a three to eight membered ring, optionally containing a heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted one, two, or three times, independently, by R5;
R' and R'a are each independently hydrogen, hydroxy}, (Ci-C6)alkyl, (Ci -C6)haloalkyl, halogen, (C-;-C6)aikoxy, amino, (Ci-C4)alkyj.ammo, or ((Ci-C4)alky])((Ci -C4)a]kyi)amino;
each R4 is independently selected from hydrogen, halogen, (Ci-Ceja!kyf, (Ci-C6)haloalkyl, -COjR7, -CONR7R8, -OR9, and -NR8R9, wherein said (C, -C6)alkyl or (Cx-C6)haloalkyl is optionally substituted by hydroxy}, -OR9, -C02R7, -CO R7R8, or -NRSR9;
each R4a is independently selected from hydrogen, halogen, hydroxy!, amino, and
(Q-Qjaikyl;
or R" and R4a taken together with the carbon atom to which they are attached form a three to eight membered ring, optionally containing a heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by cyano, (Ci-C4)alkyl, (Ci-C4)haloalkyl,
(C3-C6)cycloalkyl, ~C02R7, -CONR7R8, bydroxyl, hydroxy(Ci-C6)alkyl, (Ci-C4)alkoxy,
(Ci-C4)alkoxy(C[-C6)alkyl, amino, (Ci-C4)alkylamino, ((C[-C4)alkyl)((Ci-C4)alkyl)amino, -NHCO2R7, -N((Ci-C4)alkyl)C02R7, -NHC(0)R7, or -N((CrC4)alkyl)C(0)R7;
each R5 is independently selected from (Ci-Cejalkyl, (Ci-Cejhaloalkyl, (C3-C,s)cyeloalkyl, halogen, cyano, hydroxy!, hydroxy(C] -C6)alkyl, (Ci-C6)alkoxy, (Ci-C4)alkoxy(Ci-C6)alkyl, amino, (Ci-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, aryl, heteroaryl, aryl(Ci-C6)alkyl, heteroaryi(Ci-C6)alkyl, and heterocycloalkyl;
R6 is hydrogen, (Ci-C6)alkyl, (Ci-C6) aloalkyl, (C^-C^jcycloalky!, hydroxy(Ci-C6)alkyl, (CrC4)alkoxy(Ci-C6)alkyl, -((C0-C3)alkyi)CO2R7, -((Q,-C3)alkyl)CO R7R8, aryl, heteroaryl, aiyl(CrC6)alliyl, heteroaryl(Ci-Cg)alkyl, or heterocycloalkyl;
R7 is hydrogen, (C C6)aikyl, (C[-C6)haloalkyl, (C C6)cycloaikyl,
R8 is hydrogen, (Ci-C6)alkyl, or (Ci -Cg)haloalkyl;
or R' and R8 taken together with the nitrogen atom to which they are attached form a four to eight membered ring, opiionaiiy containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by (Ci-G alkyi, (Cj-C4)haioaiky1, (C3-C6)cycloalkyl, -C02H, -C02(Ci-C4)alkyl, hydroxyl, hydroxy(Ci-C6)alkyl, (C C4)alkoxy, (Ci-C4)alkoxy(C i-C6)alk l, amino, (Ci-C4)alkylamino, or ((C[-C4)alkyl)((Ci-C4)alkyl)amino;
R9 is -C(0)R7, ·( '(>. -R . -C(0)NR7R8, (CrC6)alkyl, (CrC^haioaikyl, (C3-C6)cycloalkyl, aryl, heteroaryl,
 heteroaryl(Ci-Cg)alkyi, or heterocycloalkyl, wherein said (Ci -Cg)alkyl, (Ci-C6)haloalky , (C3-C6)cycloalkyl, aryl, heteroaryl, aryl(Ci -Cg)alkyl,
heteroaryl(Ci-Cg)alkyi, or heterocycloalkyl, wherein said (Ci -Cg)alkyl, (Ci-C6)haloalky , (C3-C6)cycloalkyl, aryl, heteroaryl, aryl(Ci -Cg)alkyl,
heteroaryi(Ci-Cg)alkyl, or heterocycloalkyl is optionally substituted by -C02R ', -CONH2, -CONH(Ci-C4)alkyl, -CON((Ct-C4)alkyl)((Ci - C4)alkyl), hydroxyl, \ (. -C, yM o\ . amino, (Ci-C4)a]kylammo, ((C C4)alky1)((Ci-C4)a1kyl)ammo, -NHC02R7, -N((CrC4)alkyl)C02R7, -NHC(0)R7, or -\ i.{C -C . m!k i K K)} :
or R6 and R9 taken together with the nitrogen atom to which they are attached form a four to eight membered ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by cyano, (Ci-C4)alkyl, (Ci-C4)haloalkyl, (C3-C6)cycloalkyl, -C02H, -C02(Ci-C4)alkyl, -CONR7R8, hydroxyl, hydroxy(CrC6)alkyl, (CrCi)alkoxy, (Ci-C4)alkoxy(Ci-C6)alkyl, amino, (Ci-C4)alkylamino,
((Ci-C4)alkyl)((Ci-C4)alkyl)amino, -N HCO.-R . -N((Ci-C4)alkyl)C02R7, -NHC(0)R7, or
-N((Ci-C4)alkyl)C(0)R7; and
R10 is (Ci-Ce)alkyl, (Ci-Cr hafoafkyl, (Qj-Cejcycloalkyl, halogen, cyano, hydroxyl, hydroxy(Ci-C6)alkyl, (C C6)alkoxy, (Ci-C4)alkoxy(CrC6)alkyl, -((C0-C3)alkyl)CO2R7,
-((Co-C3)alkyl)CONR?R8, amino(Cx-C6)alkyl, ((Cr-C4)alkyl)((C1 - C4)arkyl)amino(Cr-C6)alkyl, (Ci-C4)alky1amino(Ci-C6)alkyl, amino, (Ci-C4)aUkylamino, ((Ci-C4)alkyl)((Ci-C4)alky{)amino, aryL heteroaryl, aryl(Ci -Cg)alkyl, heteroaryl(C-;-C6)aikyl, or heterocycloalkyl;
or a salt thereof, particularly, a pharmaceutically acceptable salt thereof.
In one embodiment of this invention, the compound of Formula I does not include (2-(4- ((3,5-dimethylisoxazol-4-yl)memoxy)ph
yl)m ethyl) acetam de) .
In another aspect, this invention provides a pharmaceutical composition comprising a compound of Formula (1), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient.
In another aspect, this invention provides for the use of a compound of Formula (I) or a pharmaceutically acceptable salt thereof for the treatment of diseases mediated by RORy. The invention further provides for the use of a compound of of Formula (I) or a pharmaceutically acceptable salt thereof as an active therapeutic substance in the treatment of a disease mediated by RORy.
In another aspect, the invention provides a compound of Formula (I) or a pharmaceutically acceptable salt thereof for use in therapy.
In another aspect, the invention provides the use of a compound of Fonnula (I) or a pharmaceutically acceptable salt thereof in the manufacture of a medicament for the treatment of diseases mediated by RORy.
Examples of such diseases for which compounds of Formula (I) may be used include autoimmune or inflammatory diseases such as multiple sclerosis, rheumatoid arthritis, psoriasis, uveitis, dry eye, glomerulonephritis, Crohn's disease and asthma, especially psoriasis
In yet another aspect, the invention is directed to meihods of treating such diseases for example by administering to a patient (e.g. human) in need thereof an effective amount of a compound of Formula (I) or a pharmaceutically acceptable salt thereof, Detailed Description of the invention
As used herein, the term "alkyl" represents a saturated, straight, or branched hydrocarbon moiety. The term
 refers to an alkyl moiety containing from 1 to 6 carbon atoms. Exemplary alkyls include, but are not limited to methyl, ethyl, n-propyl, isopropyl, w-butyl, isobutyl, s-butyi, /-butyl, pentyl, and hexyl. C0aikyl means that no alkyl group is present in the moiety. Thus, -((Co)alkyl)CONH2 is equivalent to -CONH2.
refers to an alkyl moiety containing from 1 to 6 carbon atoms. Exemplary alkyls include, but are not limited to methyl, ethyl, n-propyl, isopropyl, w-butyl, isobutyl, s-butyi, /-butyl, pentyl, and hexyl. C0aikyl means that no alkyl group is present in the moiety. Thus, -((Co)alkyl)CONH2 is equivalent to -CONH2.
When the term "alkyl" is used in combination with other substituent groups, such as "haloalkyl", "hydroxyalkyl", "alkoxyalkyl", "arylalkyl", or "heteroarylalkyl", the term "alkyl" is intended to encompass a divalent straight or branched-chain hydrocarbon radical. For example, "arylalkyl" is intended to mean the radical -alkylaryl, wherein the alkyl moiety thereof is a divalent straight or branched-chain carbon radical and the aryl moiety thereof is as defsned herein, and is represented by, for example, the bonding arrangement present in a benzyl group (-CH2-phenyl); "halo(CrC |)alkyi" is intended to mean a radical having one or more halogen atoms, which may be the same or different, at one or more carbon atoms of an alkyl moiety containing from 1 to 4 carbon
atoms, which is a straight or branched-chain carbon radical, and is represented by, for example, a trifiuoromethyl group (-CF3).
As used herein, the term "cycloalkyl" refers to a non-aromatic, saturated, cyclic hydrocarbon ring. The term "(C3-Cg)cycloalkyl" refers to a non-aromatic cyclic hydrocarbon ring having from three to eight ring carbon atoms. Exemplary "(Cs-C^cycloalkyi" groups useful in the present invention include eyclopropyf, cyclobutyf, cyclopentyl, cycfohexyl, cyciobeptyl, and cyclooctyl.
"Alkoxy" means an alkyi radical containing the specified number of carbon atoms attached through an oxygen linking atom. The term "(Ci-C4)alkoxy" refers to a straight- or branched-chain hydrocarbon radical having at least 1 and up to 4 carbon atoms attached through an oxygen linking atom. Exemplar '- "(Ci-C4)alkoxy" groups useful in the present invention include, but are not limited to, niethoxy, ethoxy, w-propoxy, isopropoxy, H-butoxy, s-butoxy, and /-butoxy.
"Aryl" represents a group or moiety comprising an aromatic, monovalent monocyclic or bicyclic hydrocarbon radical containing from 6 to 10 carbon ring atoms, to which may be fused one or more cycloalkyl rings.
Generally, in the compounds of this invention, aryl is phenyl.
Heterocyclic groups may be heteroaryl or heterocycloalkyl groups.
"Heteroaryl" represents a group or moiety comprising an aromatic monovalent monocyclic or bicyclic radical, containing 5 to 10 ring atoms, including 1 to 4 heteroatoms independently selected from nitrogen, oxygen and sulfur. This term also encompasses bicyclic heterocyclic-aryl compounds containing an aryl ring moiety fused to a heterocycloalkyl ring moiety, containing 5 to 10 ring atoms, including 1 to 4 heteroatoms independently selected from nitrogen, oxygen and sulfur, illustrative examples of heteroary Is useful in the present invention include, but are not limited to, furanyl, thienyi, pyrrolyl, imidazolyl, pvrazolyl, triazolyl, tetrazolyl, thiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiadiazolyl, isothiazoiy i, pyridinyl, pyridazinyl, pyrazinyl, pyrimidinyl, triazinyJ, benzofuranyl, isobenzofuryl, 2,3-dibydrobenzofuryl, 1 ,3-benzodioxolyl,
dihydrobenzodioxinyl, benzothienyl, indolizinyl, indolyl, isoindofyl, d hydroindolyl,
benzimidazolyl, dihydrobenzimidazolyl, henzoxazoiyi, dihydrobenzoxazolyl, benzihiazoiyi, benzoisothiazolyl, dihydrobenzoisothiazolyl, indazolyl, imidazopyridinyl, pyrazolopyridinyl, benzotriazolyi, rriazolopyridinyl, purinyJ, quinolinyi, tetrahydroquinoiinyl, isoquinolinyi, tetrahydroisoquinolinyl, quinoxaiinyl, cinnofinyf, phthalazinyl, quinazo!iny!, 1 ,5-napbthyridinyl, 1 ,6-naphthyridinyl, 1,7-naphthyridinyl, 1,8-naphthyridmyl, and pteridinyl.
Generally, the heteroaryl groups present in the compounds of this invention are
5-membered and/or 6-memebred monocyclic heteroaryl groups. Selected 5-membered heteroaryl groups contain one nitrogen, oxygen, or sulfur ring heteroatom, and optionally contain 1 , 2, or 3
additional nitrogen ring atoms. Selected 6-membered heteroaryl groups contain 1 , 2, or 3 nitrogen ring heteroatoms. Illustrative examples of 5- or 6-membered heteroaryl groups useful in the present invention include, but are not limited to furanyi, thienyl, pyrrolyl, imidazolyl, pyrazoiyi, triazolyl, tetrazolyl, thiazolyl, oxazoiyl, isoxazolyl, oxadiazolyl, thiadiazolyl, isothiazoiyl, pyridinyi, pyridazinyl, pyrazinyl, pyrimidinyl, and triazinyi.
"Heterocycloalkyl" represents a group or moiety comprising a on-aromatic, monovalent monocyclic or bicyclic radical, which is saturated or partially unsaturated, containing 3 to 10 ring atoms, which includes 1 to 3 heteroatoms independently selected from nitrogen, oxygen and sulfur. Illustrative examples of heterocycloalkyls useful in the present invention include, but are not limited to, azetidinyl, pyrrolidmyl, pyrazolidinyl, pyrazolinyl, imidazolidinyi, imidazolinyl, oxazofinyf, thiazolinyl, tetrahydrofuranyl, dihydrofuranvi, 1 ,3-dioxofanyl, piperidinyl, piperazmyl, morpholinyl, thiomorpholinyl, tetrahydropyranyl, dihydropyranyl, 1 ,3-dioxanyl, 1 ,4-dioxanyl, 1 ,3- oxat iolanyl, 1 ,3 -oxathianyi, 1 ,3-dithianyl, hexahydro- 1H- 1 ,4-diazepinyl, azabicylo[3.2. ljoctyl, azabicylo[3.3.1 ]nonyl, azabieylo[4.3.Q]nonyl, oxabicylo[2.2.1 ]hepty] and 1 ,5,9-triazacyclododecyl.
Generally, in the compounds of this invention, heterocycloalkyl groups are 5-7 membered heterocycloalkyl groups, such as pyrrolidmyl, pyrazolidinyl, pyrazolinyl, imidazolidinyl, imidazolinyl, oxazolinyl, thiazolinyl, tetrahydrofuranyl, dihydrofuranyl, 1 ,3-dioxolanyl, piperidinyl, piperazmyl, ηιοφηοίϊη>4, thiomorpholinyl, tetrahydropyranyl, dihydropyranyl, and hexahydro- I/f- I,4-diazepinyl.
"Oxo" represents a double-bonded oxygen moiety; for example, if attached directly to a carbon atom forms a carbonyi moiety (C=0).
The terms "halogen" and "halo" represent chloro, fluoro, bromo, or iodo substifuents, "Hydroxy" or "hydroxyl" is intended to mean the radical -OH.
"RORy" refers to all isoforms encoded by the RORC gene which include RORy l and RORyt.
"RORy modulator" refers to a chemical compound that inhibits, either directly or indirectly, the activity of RORy. RORy modulators include antagonists and inverse agonists of RORy.
"Pharmaceutically acceptable" refers to those compounds, materials, compositions, and dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicit '-, irritation, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
As used herein, the term "pharmaceutically acceptable salts" refers to salts that retain the desired biological activity of the subject compound and exhibit minimal undesired toxicological effects. These pharmaceutically acceptable salts may be prepared in situ during the final isolation
and purification of the compound, or by separately reacting the purified compound in its free acid or free base form with a suitable base or acid, respectively.
As used herein, the term "compound(s) of the mvention" means a compound of Formula (I) (as defined above) in any form, i.e., any salt or non-salt form (e.g., as a free acid or base form, or as a pharmaceutically acceptable salt thereof) and any physical form thereof (e.g., including non-solid forms (e.g., liquid or semi-solid forms), and solid forms (e.g., amorphous or crystalline forms, specific polymorphic forms, solvates, including hydrates (e.g., mono-, di- and hemi- hydrates)), and mixtures of various forms.
As used herein, the term "optionally substituted" indicates that a group, such as aikyl, eycloalkyl, alkoxy, heterocycloalkyl, aryi, or heteroaryi, may be unsubstituted, or the group may be substituted with one or more substituent(s) as defined. In the case where groups may be selected from a number of alternative groups the selected groups may be the same or different.
The term "independently" means that where more than one substituent is selected from a number of possible substituents, those substituents may be the same or different. The alternative definitions for the various groups and substituent groups of Fonnuia (T) provided throughout the specification are intended to particularly describe each compound species disclosed herein, individually, as well as groups of one or more compound species. The scope of this invention includes any combination of these group and substituent group definitions.
Suitably, m is 0, 1 , or 2. In a specific embodiment of this invention, m is 1.
Suitably , n is 0, 1 , 2, or 3. In another embodiment of this invention, n is 1 or 2.
Suitably, X1 , X2, X', X4, and X3 are each independently selected from N, ΊΝΓ-Ο" (i.e. iV- oxide), CH, and CR5, wherein 0-3 of X\ X2, X3, X4, and X5 are N or lST-O" and 1 -3 of X1, X2, X3, X4, and X5 are CR5; provided that when zero of X1, X^, X\ and X5 are N or N+-0" and X3 is CR5, 1 -2 of X1, ΧΔ, X4, and X5 are CR3. In another embodiment of this invention, X1, X , X\ X", and X5 are each independently selected from N, N+-Q", CH, and CR5, wherein 0-2 of X1, X2, X3, X4, and X5 are N or N+-0~ and 1 -3 of X1, X2, X3, X4, and X5 are CR5; provided that when zero of X1 , X2, X4, and X5 are N or N"-0" and X3 is CR5, 1 -2 of X1, X2, X", and X5 are CR3. In another embodiment of this invention, X1 and X3 are each independently selected from N, IST-O", CH, and CR5, and X X', and X4 are each independently selected from CH and CR5, wherein at least one of X1 and X3 is N or r-0" and 0-3 of X1, X2, X X4, and X5 are CR3. In another embodiment of this invention, X1 and X3 are each independently selected from N, N+-0", and a carbon atom substituted by hydrogen, halogen, cyano, (Ci -C4)alkyl, (CrC^haloalkyL (Ci -G alkoxy, or
((Ci-C4)alkyl)((C[-C4)alkyl)amino (i.e. N, N -Q", CH, and CR3, wherein R5 is halogen, cyano, (CrCYiaikyl, (Ci-C4)haloalkyl, (Q-Cijalkoxy, or ((Ci-C4)alkyl)((Ci-C4)alkyl)amino), and X2, X"', and X4 are each independently a carbon atom substituted by hydrogen, halogen, cyano,
(C-.-C4)ajkyl, (Ci-C4)haloalkyL (Ci -C4)alkoxy, or ((Ci-C4)alkyl)((Ci-C4)alkyl)amino (i.e. CH or CR5, wherein R5 is halogen, cyano, (Ci-C4)alkyl, (Ci-C )haloalkyl, (Ci -C4)alkoxy, or
((Ci-C4)alkyl)((Ci-C4)alkyl)amino), wherein at least one of X1 and X5 is N or 1ΝΓ-0" and 2-4 of X1, X2, XJ, X4, and X5 are a carbon atom substituted by hydrogen (i.e. CH). In another embodiment of this invention, X2 is N or N"'-0", and X1, X3, X4, and X5 are each independently a carbon atom substituted by hydrogen, halogen, cyano, (Ci-C4)alkyL (Ci-C )haloalkyl, (Ci-C4)alkoxy, or ((Ci-C4)aIkyl)((Ci-C )alkyl)amino, wherein 2-4 of X1, X3, X4, and X3 are a carbon atom substituted by hy drogen. In another embodiment of this in v ention, X1, X , X3, X", and Xs are each independently selected from CH and CR3, wherein 0-3 of X1 , X2, X', X4, and X3 are CR3, In another embodiment of this invention, X1, X2, X3, X4, and X5 are each independently a carbon atom substituted by hydrogen, halogen, cyano, (Ci-C4)alkyl, (CrC4)haloalkyl, (CrC4)alkoxy, or ((C[-C4)alkyl)((Ci-C4)alkyl)amino, wherein 2-5 of X1, X , XJ, X*, and X5 are a carbon atom substituted by hydrogen. In another embodiment of this invention, X ' is a carbon atom substituted by halogen, (Ci-C4)alkyi., (Ci-C4)ha]oa]kyl, cyano, (Ci-C4)alkoxy, or
((Ci-C4)alkyl)((Ci-C4)alkyl)amino, and X2, X3, X4, and Xs are each independently a carbon atom substituted by hydrogen, halogen, (Ci-C4)alkyl, (Ci-C4)haloalkyL cyano, (Ci-C4)alkoxy, or ((Ci-C4)alkyl)((Ct-C4)alkyl)amino, wherein 2-4 of X2, X3, X4, and X3 are a carbon atom substituted by hydrogen.
Suitably, one of Y1 and Y2 is O or NRS and the other is a bond. In another embodiment of this invention, one of Y1 and Y~ is O, NH, or N((Ci-C4)alkyl) and the other is a bond. In a specific embodiment of this invention, Yl is NH or NCH3 and Y* is a bond. In another specific embodiment of this invention, Y1 is NH and Y" is a bond. In another specific embodiment of this invention, Y1 is a bond and ΎΔ is NH.
In another embodiment of this invention, X1 is CR3, Y1 is NR.", Y is a bond, and R3 and R" taken together with the atoms to which they are attached form a five to seven membered ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by (CrC4)alkyl. In another embodiment of this invention, X1 is CR5, Y1 is R* Y' is a bond, and R3 and R8 taken together represent -CH?-, -CH2CH2-, or
-CH2CH2CH2-.
Suitably, Cy is (Cs-CgjcycJoaikyl, lieterocycloalkyl, phenyl, or 5- or 6-membered heteroaryl, each of which is optionally substituted one, two, or three times, independently, by (Ci-Cg)alkyl, (CrC6)haloalkyL iC3-C6)cycloalkyl, halogen, oxo, cyano, hydroxy!,
hydroxy(Cl-C6)alkyl, (CrC6)alkoxy, -((C0-C3)alkyl)NHCO2R7,
-((Co-C3)a1kyl)N((Ci-C4)alkyl)C02R7, -((Co-C3)alkyl)NHC(0)R7,
-((Co-C3)a1kyl)N((Ci-C4)alkyl)C(0)R7, -((Co-C3)alkyl)C02R7, -((C0-C3)alkyl)CONR7R8,
-((Co-C3)a1kjd)C(0)R7, (C C4)alkoxy(Ci-C6)alkyl, amino(Ci -C6)alkyl,
((Ci-C4)alkyl)((Ci-C4)alkyl)amino(Ci-C6)alkyl, (Ci-C4)alkylamino( -C6)alkyl, amino,
(Ci-C4)alkylamino, ((Ci-C4)alkyl)((CrC4)alkyl)amino, aryl, heteroaryl, aryl(Ci-C6)alkyL heteroaryl(Ci -C6)alkyl, or heterocycloalkyl. In another embodiment of this invention, Cy is heterocycloalkyl, phenyl, or 5- or 6-membered heteroaryl, each of which is optionally substituted one or two times, independently, by (Ci~C6)alkyl, (Ci-Cg)haloalkyl, halogen, cyano, (Ci-C4)alkoxy, ((Ci-C4)alkyl)ammo ((C1-C4)alkyl)((C C4)alkyi)a-mmo, -((C0-C3)alkyl)CO2R7,
or -((Co-C3)alkyl)CO >!R'R8. In another embodiment of this invention, Cy is (C3 -C6)cycloalkyl, azetidinyl, pyrrolidinyl, pyrazolidinyi, pyrazolinyi, imidazolidinyl, imidazolinyl, oxazolinyl, thiazolinyl, tetrahydrofuranyi, dihydrofuranyl, piperidinyi, piperazinyl, morpholinyi,
thiomorphofinyl, tetrahydropyranyl, dihydropyranyl, dioxanyf, oxathianyf, phenyl, furanyl, thieiiyl, pyrrolyl, imidazolyl, pyrazolyl, triazolyl, tetrazoiyl, thiazoiyl, oxazolyi, isoxazolyl, oxadiazolyi, thiadiazolyl, isothiazolyl, pyridinyl, pyridazinyl, pyrazinyl, pyrimidinyl, or triazinyl, each of which is optionally substituted one, two, or three times, mdependently, by (C Cyaikyi, (CrCsjhaloalkyl, (Cs-Cejcycloalliyl, halogen, oxo, cyano, hydroxy!, hydroxy(CrC6)a1kyl,
(CrC6)alkoxy, -((Co-C3)alkyl)NHC02R7, -((C0-C3)alkyl)N((Ci-C4)alkyl)CO2R7,
-((Co-C3)alkyl)NHC(0)R7, -((Co-C3)alkyl)N((Ci-C4)alkyl)C(0)R7. -((Co-C3)alkyl)C02R7,
-((Co-C3)al l)CONR7R8, -((Co~C3)alkyl)C(0)R7, (C - ( " i ia!koxyi C -(Vjniky! . amino(CrC6)aIkyl, ((Ci-C4)alkyl)((Ci-C4)alkyl)ammo(CrC6)a1kyl, (Ci-C4)alkylamino(Ci-C6)alkyl, amino,
(Ci-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, aryl, heteroaryl, aryl(Ci-C6)alkyl, heteroaryi(Ci - C6)alkyi, or heterocycloalkyl. In another embodiment of this invention, Cy is piperidinyi, piperazinyl, phenyl, pyridinyl, py ridazinyl, pyrazinyl, or pyrimidinyl, each of which is optionally substituted one, two, or three times, independently, by (Ci-Cejalkyl, (Ci-C6)haIoaikyl, (C -C'6)cycloalkyl, halogen, oxo, cyano, hydroxy 1, hydroxy(C; -C6)alkyl,
(Ci-C6)alkoxy, - u i ,:-C )alk l ;N UC(}; . (Co-C3)alkyl)N((Cr-C4)allvyl)C02R7,
-((Co-C3)alkyd)NHC(0)R7, -((Co-C3)alky1)N((Ci-C4)alkyl)C(0)R7, -((Co~C3)aikyl)C02R7,
-((Co-C3)a1kyl)CONR7R8, -(( ,-C3)alkyl)C(0)R7, (Ci-C4)a1koxy(Ci-C6)alky1, ammo(Ci-C6)afkyl, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino(Ci-C6)alkyl, (Ci-C4)alkylamino(Ci-C6)alkyl, amino,
(Cj-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, aryl, heteroaryl, aryl(Ci -Ce alkyl, heteroaryl(Ci-C6)alkyl, or heterocyci oalkyl. In another embodiment of this invention, Cy is piperidinyi, piperazinyl, phenyl, pyridinyl, pyridazinyl, pyrazinyl, or pyrimidinyl, each of which is optionally substituted one or two times, independently, by (Ci-C6)alkyl, (Ci -Ce haloalkyl, halogen, cyano, (Q -C4)alkoxy, (Cr-C4)alkyl)((Cl--C4)alkyi)amino, -((C0-C3)alkyl)CO2H,
-((Co-C3)alk d)C02(Ci-C6)aikyi, or -((Co-C-3)alkyl)C NH(Ci-C6)a]kyl. In another embodiment of this invention, Cy is phenyl, which is optionally substituted one, two, or three times, independently,
by (Ci~C6)alkyl, (Cj-C6)haloalky], (CVCe^ycioalkyl, halogen, oxo, cyano, hydroxy!,
hydroxy(Ci-C6)alkyl, (C -( Jalkoxy. -! ! ('.r(' . ;alkyi }\l iC'O R .
-((C¾-C3)alkyl)N((Ci-C4)alkyl)C02R7, -((Co-C3)alkyl) HC(0)R7,
-((Co-C3)alkyi)N((CrC4^
-((C0-C3)alkyl)C(O)R7, ((" -C iaikoxyii' -C6)alkyJ, ammo(Ct-C6)alkyl,
((Ci-C-4)alkyl)((Ci -C4)alkyl)amino(Ci~C6)alkyl, (Cj -C4)alkyIamino(Ci-Cg)alky3, amino,
(Ci-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C-4)alkyl)animo, aryl, heteroaiyl, aryl(Ci-C6)aikyl, heteroaryi(Ci-C6)alkyl, or heterocycloalkyl. In another embodiment of this inv ention, Cy is phenyl, which is optionally substituted one or two times, independently, by halogen, (d -C4)alkyi, (Ci-C4)ha1oa1kyl, cyano, (Ci -C4)alkoxy,•i i i" 1r (" : }aik> !)("( ) R . or -((C0-C3)aUcy-)CONR7R8 or
((Ci-C4)alkyr)((Ci-C4)alkyl)ammo. In another embodiment of this invention, Cy is phenyl, which is optionally substituted one or two times, independently, by halogen, (C-i-C4)alkyl,
(Cj-C4)haloaikyl, cyano, (Ci-C4)alkoxy, or ((C) -C4)alkyl)((Ci-C4)alJ{yI)aniino. In a specific embodiment of this invention, Cy is phenyl,
Suitably, Z is O, S, S02, C=0, NR'', or a bond. In another embodiment of this invention,
Z is O, NR°, or a bond. In another embodiment of this invention, Z is O, NH,
-N(C C4)alkyl, - Ni ( ( ,:-C ;)ali, I ;C(); . -N((Co-C3)alkyl)CONR7R8 or a bond. In another embodiment of this invention, Z is a bond, O, or NH. In another embodiment of this invention, Z. is O or NH. In a specific embodiment of this invention, Z is O.
Suitably, A1, A2, A3, and A4 are each independently selected from , NR6, O, S, CH, and
CRi0, wherein one of A1, A2, A3, and A4 is NR6, O, or S, 0-2 of A1, A2, A3, and A4 are CR10, and 0-3 of A1 , A", A', and A4 are CH or N. In another embodiment of this invention, A1 , A , A3, and A4 are each independently selected from N, N((Ci-C4)alkyl), O, S, CH, and C((Ci-C4)alkyl), wherein one of A1, A2, A3, and A4 is N((C C4)alkyl), O, or S, 0-2 of A1, A2, A3, and A4 are
C((C] -C4)alkyi), and 0-3 of A1 , A"', A3, and A4 are CH or N. In another embodiment of this invention, A1 and A4 are each independently selected from CH and CRt0, and one of A2 and A' is NR6, O, or S and the other is N or CH. In another embodiment of this invention. A1 and A are each independently selected from CH and C((Ci-C4)aIkyl), and one of ΑΔ and AJ is N((Ci-C-4)alkyl), O, or S and the other is N or CH. In another embodiment of this invention. A ' and A4 are each independently selected from CH and C((Ci-C4)alkyJ), and one of A2 and A' is O or S and the other is N.
Suitably, R! is (C C6)alkyl, (C3-C6)haloaIkyl, (C3-C8)cycloalkyL (C3-C6)alkoxy,
(Ci-C6)alkoxy(Ci - C2)alk>rl, aryl, heteroaryi, aryi(Ci -C6)alkyi, heteroaryI(C[-C6)alkyl, or heterocycloalkyl, each of which is optionally substituted one, two, or three times, independently, by R5. In another embodiment of this invention, R1 is (C3-C6)alkyl, (Cj-Cgjcycfoafkyl,
(Cj-C6) {koxy(Ci -C2) lk {, aryl, or heteroaryl, each of which is optionally substituted one, two, or three times, independently, by R'\ In another embodiment of this invention, Rl is (C-3-C6)aikyl, (CVC6)cyck)alkyl, (Ci-C6)alkOxy(Ci-C2)alkyl, phenyl, furanyl, thienyl, pyrrolyl, imidazolyi, pyrazoiyi, triazolyl, tetrazolyl, thiazolyl, oxazoiyl isoxazolyl, oxadiazolyl, thiadiazolvl, isothiazolyl, pyridinyi, pyridazinyl, pyrazinyl, pyrimidinyl, or triazinyl, wherein said phenyl, furanyl, thienyl, pyrrolyl, imidazolyi, pyrazoiyi, triazolyl, tetrazolyl, thiazolyl, oxazoiyl, isoxazolyl, oxadiazolyl, thiadiazolvl, isothiazolyl, pyridinyi, pyridazinyl, pyrazinyl, pyrimidinyl, or triazinyl is optionally substituted one or two times, independently, by halogen, (Ci-C4)alkyl, (Ci-Q)haloalkyl, cyano, (Ci-C4)alkoxy, or ((Ci -C4)alkyl)((Ci -C4)alkyl)amino (i.e. wherein 3 is halogen, (Ci -C4)alkyl, (Ci-C4)haloalkyl, cyano, (CrC4)alkoxy, or ((Ci-C4)alkyl)((CrC4)alkyl)araino). In another embodiment of this invention, R1 is (C^-Cejalkyf, (CrCekycioalkyl, phenyl, furanyl, thienyl, pyrrolyl, imidazolyi, pyrazoiyi, triazolyl, tetrazolyl, thiazolyl, oxazoiyl, isoxazolyl, oxadiazolyl, thiadiazolyl, isothiazolyl, pyridinyi, pyridazinyl, pyrazinyl, pyrimidinyl, or triazinyl, wherein said phenyl, furanyl, thienyl, pyrrolyl, imidazolyi, pyrazoiyi, triazolyl, tetrazolyl, thiazolyl, oxazoiyl, isoxazolyl, oxadiazolyl, thiadiazolyl, isothiazolyl, pyridinyi, pyridazinyl, pyrazinyl, pyrimidinyl, or triazinyl is optionally substituted one or two times, independently, by halogen, (d-C4)alkyl, (CrC4)alkoxy, or ((Cj -C4)alkyl)((Ci -C4)alkyl)amino. In another embodiment of this invention, R' is (Cs-C^alkyl. In another embodiment of this invention, R1 is (Cs-Cejalkyl, In another embodiment of this invention, R1 is phenyl or pyridinyi, each of which is optionally substituted one or two times, independently, by halogen, (C[-C4)alkyl, ( -C4)haloalkyl, cyano, (Ci-C4)alkoxy, or ((Ci-C4)a.lkyl)((Ci -C4) lky i) amino. In another embodiment of this invention, R1 is phenyl or pyridinyi, each of which is optionally substituted one or two times, independently, by halogen, (Ci-Ci)alkyl, (CrC4)alkoxy, or ((Ci-C4)alkyl)((Ci-C )alkyl)amino. In another embodiment of this invention, R1 is phenyl optionally substituted one or two times, independently, by halogen,
(Ci-C4)alkyl, (CrC4)haloalkyl, cyano, (Ci-C4)aikoxy, or ((Ci -C4)alkyl)((Ci-C4)alkyl)amino. In a specific embodiment of this invention, Rl is phenyl or pyridinyi. In another specific embodiment of this invention, R1 is phenyl.
Suitably, R~ is hydrogen, (C' CeJalkyl, or (Ci-C6)haloalkyl. In another embodiment of this invention, hydrogen or (Ci-C4)alkyl. In another embodiment of this invention, R2 is hy drogen or methyl. In a specific embodiment of this invention, R2 is hydrogen.
In another embodiment of this invention, R1 and R" taken together with the carbon atom to which they are attached form a three to eight membered ring, optionally containing a heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted one, two, or three times, independently, by R\ In another embodiment of this invention, R1 and R7" taken together represent -C! i ('! ! ( ! ! -. or -CH2CH2CH2CH2CH2-.
Suitably, R ;' and R3a are each independently hydrogen, hydroxy!, (Ci-C6)a{kyl,
(C-. -C6)ha3oa3kyl, halogen, (Ci~C6)a3koxy, amino, (Ci-C4)a3kylamino, or
((Ci-C4)alkyl)((Ci-C4)alkyl)amino. In another embodiment of this invention, R and R3a are each independently hydrogen or methy l. In a specific embodiment of this invention, RJ and RJa are each independently hydrogen.
Suitably, each R4 is independently selected from hydrogen, halogen, (Ci~C6)alkyl,
(d-Cyiialoalkyi -C02R7, · ( ON R R\ -OR9, and -NR8R9, wherein said (CrC6)alkyl or
(CrC6)haloalkyl is optionally substituted by hydroxy!, -OR9, -C02R7, - ( ON R R \ or - R8R9. In another embodiment of this invention, each R is independently selected from hydrogen,
(C-;-C4)alkyi, (Ci-C4)haloalkyl, -OR9, and -NR¾9, wherein said (Ci-C4)alky1 or (Ci-C4)iialoalky3 is optionally substituted by hydroxyl, -OR9, -C02R', -CONR7Rs, or ~NRSR9, In another embodiment of this invention, each R4 is independently selected from hydrogen, halogen, (Ci-C4)alkyl,
(Ci -C4)alkylammo, ((Ci-C4)aikyl)((CrC4)alkyl)amino, (Ci-C4)alkoxy(Ci-C4)alkylamino,
-MHC02(Ci-C4)alkyl, (Ci-C4)alkoxy, hydroxy(C2-C4)alkoxy, (Ci-C4)a3koxy(C2-C4)alkoxy, amino(C2-C4)alkoxy, -0((Ci-C4)alkyl)C02R7, -Ot t C -C : lalky! iCON j I.·.
·( )(( (· -< . !alky DC ON i li C •-('•ja ikyl. -Oi( C -< , sulkyi }C ONi i C••Cjaiky i K i C -i iky! s. and CO R . In another embodiment of this invention, each R4 is independently selected from hydrogen, halogen, (CrCYiaJkyi, (Ci-C4)alkylamino, ((Ci-C4)a3kyl)C(Ci-C4)alkyl)ami.no,
(Ci-C4)alkoxy(Ci-C4)alkylamino, (Ci-C4)alkoxy, hydroxy(C2-C4)alkoxy,
(Ci-C4)alkoxy(C2-C4)alkoxy, amino(C2-C4)alkoxy, -0((Ci-C3)alkyl)C02H,
-0((Ci-C3)alkyl)C02(Ci -C4)alkyl, -0((Ci-C3)alkyl)CONH2, ((CT-C3)alJcyl)CONH(Ci -C4)aJj£yl, and -OC(Ci-C3)alkyl)CON((Ci -C4)alky3)((Ci-C4)a3kyl). In another embodiment of this invention, each IT is independently selected from hydrogen, (Ci-C )alkyl, (CrC4)alkoxy,
hydroxy(C2-C4)alkoxy, (Ci-C4)alkoxy(C2-C4)alkoxy, amino(C2-C4)alkoxy, -0((Ci-C3)alkyl)C02H, -0((Ci-C3)aU£yl)C02(CT-C4)alJcyl, -0((Q^
and -0((Ci-C3)alkyl)CON((Ci-C4)a{kyl)((Ci-C4)alkyl). In another embodiment of this invention, each R4 is independently selected from (C3-C4)alkoxy, hydroxy(C2-C4)alkoxy,
(Ci-C4)alkoxy(C2-C4)alkoxy, amino(C2-C4)alkoxy, -0((Ci-C3)alkyl)C02H,
•Oi i ( •C ; );!ikyi !( · ) ·; (■ -C
 ((CT-C3)alJcyl)CONH(Ci -C4)aJj£yl, and -OC(Ci-C3)alkyl)CON((Ci -C4)alky3)((Ci-C4)a3kyl). In another embodiment of this invention, each R4 is independently selected from (Ci-C4)alkoxy, -0((Ci-C3)alkyl)C02H,
((CT-C3)alJcyl)CONH(Ci -C4)aJj£yl, and -OC(Ci-C3)alkyl)CON((Ci -C4)alky3)((Ci-C4)a3kyl). In another embodiment of this invention, each R4 is independently selected from (Ci-C4)alkoxy, -0((Ci-C3)alkyl)C02H,
-0((C1-C3)alkyl)C02(C1-C4)alkyl, -0((CrC3)alkyl)CONH2, -0((C C3)alkyl)CONH(C1-C4)alkyl, and -0((C] -C3)a)-kyl)CON((Ci-C4)aJiiyl)((Ci-C4)alkyl). In another embodiment of this invention, each R" is independently selected from (Ci-C4)alkyl and (Ci-C4)alkoxy. In a specific embodiment of this invention, each R4 is hydrogen.
Suitably, each R4a is independently selected from hydrogen, halogen, hydroxy!, amino, and (Ci-C6 lkyl. In another embodiment of this invention, each R4a is independently selected from hydrogen, halogen, and
 In another embodiment of this invention, each R4a is independently selected from is hydrogen, fluorine, and methyl. In another embodiment of this invention, each R a is independently selected from is hydrogen and methyl , in a specific embodiment of this invention, each R4a is hydrogen. In a specific embodiment of this invention, each "a is methyl.
In another embodiment of this invention, each R4a is independently selected from is hydrogen, fluorine, and methyl. In another embodiment of this invention, each R a is independently selected from is hydrogen and methyl , in a specific embodiment of this invention, each R4a is hydrogen. In a specific embodiment of this invention, each "a is methyl.
In another embodiment of this invention, R* and R4a taken together with the carbon atom to which they are attached form a three to eight nienibered ring, optionally containing a heieroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by cyano, (Ci-C4)alkyl, (C C4) oalkyl, (C3-C6)cycloaIkyl, -C02R7, -CONR7R8, hydroxy!,
hydroxy(Ci-C6)alkyl, (Ci -C-i alkoxy, (C-i-C4)alkoxy(Ci-C6)alkyl, amino, (Ci-C4)alkylamino, ((Ci-C4)alkyl)((Ci -C4)alkyl)amino, - N I K O R '. - ((C, -C4)alkyl)C02R7, -NHC(0)R7, or
-M((Ci-C4)aIky1)C(0) '. In another embodiment of this invention, R4 and R43 taken together represent -CH2CH2-, -C! i ( i i ( Ή -. -CH2CH2CH2CH2-, or -CH2CH2CH2CH2CH2~.
One particular embodiment of the invention is a com ound of Formula (la):

wherein:
m is 1 ;
n is 1 or 2;
X1 , X", X', and X4 are each independently selected from N, N+-0~, CH, and CR' , wherein 0-2 of X1, X2, X3, and X4are N or N+~(T and 0-2 X1, X2, X3, and X4 are CR5;
Y1 is NH or NCH3 and Y2 is a bond;
K1 , K" , K3, and K4 are each independently selected from N, N+-0", CH, and CR'°, wherein 0-2 of K1 , . K3, and K4 are N or N+-0" and 0-2 of Kl, K2, K3, and K4 are CR10;
Z is O, NR6, or a bond;
A1, A", AJ, and A4 are each independently selected from N, NR6, O, S, CH, and CRl0, wherein one of A1 , A2, A3, and A4 is NR6, O, or S, 0-2 of A1 , A2, A3, and A4 are CR10, and 0-3 of A1, A2, A3, and A4 are CH or N;
R! is (CVQjalky!, (QrC^haloaJkyl, (d-d)cyeloalkyJ, (QrCeiaJ oxy, (Ci-C6)alkoxy(Ci -C2)alkyl, aiyl, heteroaryl, aryl(C-i -C6)alkyi, heteroaryi(d-C6)alkyl, or heterocycloalkyl, each of which is optionally substituted one, two, or three times, independently, by
R5;
R2 is hydrogen, (Ci-C6)aikyl, or (d -d)haioaikyl;
or R1 and R" taken together with the carbon atom to which they are attached form a three to eight membered ring, optionally containing a heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted one, two, or three times, independently, by R5;
RJ and R3a are each independently hydrogen, hydroxy!, (d~d)aikyl, (d-d haloalkyl, halogen, (d. -G alkoxy, amino, (Ci -C4)alkyiamino, or ((Ci-C4)alkyl)((Ci -C^alky amino;
each R4 is independently selected from hydrogen, halogen, (d ~di)a!kyl, (CrC4)haloalkyl, -OR9, and -NR8R9, wherein said (Ci-C4)alkyl or (C] -C4)haloalkyl is optionally substituted by hydroxy!, -OR9, -CO. R '. -COX R F; . or -NR8R9;
each R4a is independently selected from hydrogen, halogen, hydroxy!, amino, and
(d -djalkyl;
each R3 is independently selected from (d -C6)alkyl, (Ci-G haloalkyl, (CrdOcycloalkyl, halogen, cyano, hydroxy!, hydroxy(d-C6)alkyl, (G -GOalkoxy, (Ci-C4)alkoxy(Ci -C6)alkyl, amino, (CrG)alky!amino, ((CrC4)alkyl)((Ci-C4)alkyl)ammo, and, heteroaryl, ary{(Ci-C6)alkyl, heteroaryl(d~d)alkyl, and heterocycloalkyl;
R6 is hydrogen, (Ci-C6)alkyl, (Ci-C6)haloalkyl, (C3-C6)eycloalkyl, hydroxy(Ci-C6)alkyl,
(Ci-C4)alkoxy(Ci-C6)alkyl, -((Co-C3)alkyl)C02 V((Co-C3)alkyl)CO RV^ aryl, heteroaryl, arylCd -Gjaikyl, heteroaiyliCi -G ky!, or heterocycloalkyl;
R ' is hydrogen, (Ci-Cejalkyl, (Ci -Ce^haloalkyl, (d-djcyc!oa!kyl,
(CrG)alkoxy d-d kyl, aryl, heteroaryl, aryl(Ci -C6)alkyl, heteroaryl(C[-C6)alkyl, or heterocycloalkyl;
R s is hydrogen,
 or (Ci-CejhaloalkyJ ;
or (Ci-CejhaloalkyJ ;
or R ' and R8 taken together with the nitrogen atom to which they are attached form a four to eight membered ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by (Ci -G alkyi, (d--d)haloalkyl, (C3-d)cycloaJkyl, -C02H, -CQ2(d -C4)alkyL hydroxy!, hydroxy(Ci-d)alkyl, (Ci-C4)alkoxy, (Ci-d)a!koxy(Ci-Q)alky!, amino, (G-C4)alkylammo, or ((Ci-C4)alkyl)((G-C4)alkyl)ammo;
R9 is -C(0)R7, -C02R7, -C(0) R7R8, (CrC6)alkyl, (C C6)haioaikyl, (C3-C6)cycloalkyl, aryl, heteroaryl, aryl(Ci - C6)alkyi, heteroaryl(Ci-C6)alkyl, or heterocycloalkyl, wherein said (d-d)aJky!, (d-C^haloalkyl, (d-djcyeloaJky!, aryl, heteroaryl, aiyl(d-d)alky!,
heteiOar d(Ci-C6)alkyl, or heterocycloalkyl is optionally substituted by -C02R7, -CONH2,
-CONH(Ci-C4)alkyl, -CON((C C4)alkyl)((Ci-C4)alky{), hydroxy!, (CrC )alkoxy, amino,
(C C4)alkylammo, ((C C4)alkyl)((Ci-C4)alkyl)amino, -NHC02R7, -N((Ci-C4)alkyl)C02R7, -NHC(0)R7, or -N((C1-C4)aikyl)C(0)R7;
or R6 and R9 taken together with the nitrogen ato n to which they are attached form a four to eight membered ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by cyano, (Ci~C4)alkyl, (C]-C4)haloalky1, (C3-C6)cycloalkyl, -C02H, -C02(C1-C4)alkyl, -CONR7R8, hydroxy!, hydroxy(CrC6)alkyl,
(C;-C4)alkoxy, (Ci-C4)alkoxy(Ci-C6)aikyl, amino, (C; -C )aUcylamino,
((Ci-C4)alkyl)((Ci-C4)alkyl)amino, -N HCO.-R . -N((Ci-C4)alkyl)C02R7, -NHC(0)R7, or
-N((Ci-C4)alkyl)C(0)R7; and
R10 is (C-i-C6)alkyl, (Ci-Cr haloalky!, (C3-C-6)cyc3oa3kyl, halogen, cyano, hydroxy!, hydroxy(Ci-C6)alkyL (C C6)alkoxy, (Ci-C4)alkoxy(CrC6)alkyl, -((C0-C3)alkyl)CO2R7,
-((Co-C3)alkyl)CONR?R8, amino(C C6)alkyl, ((CrC )alkyi)((C) -C4)alkyl)amino(CrC6)alkyL (Ci-C4)alky1araino(Ci-C6)alkyl, amino, (Ci-C4)alkylammo, ((Ci-C4)alkyl)((Ci-C4)alky{)araino, aryJ, heteroaryl, ary!(Ci~Ci,)a!ky3, heteroarylfCi-Celaiky!, or lieterocyc!oalkyl;
or a pharmaceutically acceptable salt thereof.
Another particular embodiment of the in v ention is a compound of Formula (la.) wherein: m is 1 ;
n is 1 or 2;
X1, X X3, and X4 are each independently a carbon atom substituted by hydrogen, halogen, cyano, (Ci-C4)alkyL (Ci-C4) aloalkyl, (C; -C4)aikoxy, or ((Ct-C4)alJcyl)((Ci-C4)aikyl)aiiiino, wherein 2-4 of X1, X2, X', and X4 are a carbon atom substituted by hydrogen;
Y1 is NH or NC! h and Y2 is a bond;
K1, K2, Ks, and K" are each independently a carbon atom substituted by hydrogen, halogen, (CrC4)aIkyL (Ci-C4)aikoxy, or ((Ci-C4)alkyl)((Ct-C )alkyl)amino, wherein 2-4 of K1 , K", K', and K4 are a carbon atom substituted by hydrogen;
Z is 0, NH, -N(CrC4)aJkyl, -N((C0-C3)alkyl)CO2R7, -N((C0-C3)alkyl)CONR7R8, or a bond;
A1 and A4 are each independently selected from CH and CR10, and one of A" and A ' is NR", O, or S and the other is N or CH;
R.1 is (C3-C6)alkyl, (C3-C6)haloalkyl, (C3-Cg)cycloalky3, (GVCejaikoxy,
(C!-Q)alkoxy(Ci-C2)alkyl, aryl, heteroaryl, aryl(Ci~C6)alkyl, heteroaryl(Ci -Ce)alkyl, or
heterocycloalkyl, each of which is optionally substituted one, two, or three times, independently, by R5;
R ~ is hydrogen;
R3 and R3a are each independently hydrogen or methyl;
each R4 is independently selected from hydrogen, (Q -Chalky!, (CVG alkoxy, bydiOxy(C2-C4)afkoxy, (Ci -C4)alkylammo, ((Ci-C-4)aikyl)((Ci-C4)alkyl)amino,
(Ci-C4)alkoxy(Ci-Cj)alkylamino, (Ci-C4)alkoxy(C-2-C4)alkoxy, anrino(C2-C4)alkoxy,
-0((Ci-C3)alkyl)C02H, -0((Ci-C3)alkyl)C02(Ci -C4)alkyl, -0((Ci-C3)alkyl)CONH2,
··(); ( (" -C niky COX l !i C C . fa ik L and -0< ( C ^ } ik ! )(X)Xi ((^ -i :; ) iky! H ( ( ' -( ' l ia!ky h:
each R4a is independently selected from hydrogen, hydroxyl, amino, and (CrC4)alkyl; each R5 is independently selected from (CrC6)alkyL (Ci-C6)haloalkyl, (C3-C6)cycloalkyi, halogen, eyano, hydroxyl, hyciroxy(C rC6)alkyl, (C; -C6)alkoxy, (Ci-C4)alkoxy(C; -C6)alkyl, amino, (Ci-C4)alkylamino, ((Ci -C4)alkyl)((Ci -C4)alkyl)amino, aryl, heteroaryl, aryl(Ct-C6)aIkyl, heteroaryl(Ci-C6)alkyl, and heterocyeloalkyi;
R ' is hydrogen, (Ci-C6)alkyl, (Ci -C^haloalkyf, (CrC6)cycloalky],
(Ci-C4)alkoxy(C [-C6)alkyl, aryl, heteroaryl, a yiiCi-Cyalk L heteroaryl(Ci -C6)alkyl, or heterocycloalkyl;
R8 is hydrogen, (C CeWkyl, or (Cj-Cejhaloalkyl; and
R10 is (Ci-Cfijalkyl, (Ci-CejhaloalkyL (C3-C6)cycloalkyl, halogen, eyano, hydroxyl, hydroxy(C rC6)aikyl, (C, -C6)alkoxy, (Cx-C4)alkoxy(Ci-C6)alkyl, -((C0-C3)alkyl)CO2R7,
-((C0-C3)alkyl)CONR7R8, amino(C, -C6)alkyl, ((CrC4)alkyl)( Ci-C4)alkyl)amino(Ci-C6)alkyl, (Ci-C4)a{kylamino(Ci-C6)aUkyl, amino, (Ci-C4)alkylamino, ((Ci-C4)a1kyl)((Ci-C4)alkyl)amino, aryl, heteroaryl, ary!(Ci-C6)alkyl, heteroaryl(Ci-C6)alkyJ, or heterocycloalkyl;
or a pharmaceutically acceptable salt thereof.
Another particular embodim ent of the invention is a compound of Formul a (la) wherein: m is 1 ;
is 1 or 2;
X1 , X2, X', and X4 are each independently a carbon atom substituted by hydrogen, halogen, eyano, (Ci -C4)alkyl, (C-i-C4)haloalkyl, (C-. -C4)aikoxy, or ((Ci -C4)alkyl)((Ci-C4)a]kyl)amino, wherein 2-4 of X1, X", X3, and X are a carbon atom substituted by hydrogen;
Y1 is NH and Y2 is a bond;
Kl, K , K3, and K4 are each independently a carbon atom substituted by hydrogen, halogen, (Cj-C4)aiky1, (d-C4)a]koxy, or ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, wherein 2-4 of Kl, K7", K3, and K4 are a carbon atom substituted by hydrogen;
Z is O, NH, -N(Ci-C4)alkyL or a bond:
A1 and A4 are each independently selected from CH and C((CV C4)alkyl), and one of A2 and A3 is O or S and the other is N;
R! is phenyl optionally substituted one or two times, independently, by halogen, (Ci-C4)alkyl, (Ci~C4)haloalkyi, cyano, (Ci-C4)alkoxy, or ((Ci-C4)alkyl)((Ci-C4)a1kyl)amino;
R2 is hydrogen;
R""1 and ¾ are each independently hydrogen or methyl;
each R4 is independently selected from hydrogen, (Ci-C )alkyl, (Cj -C4)alkyiamino,
((Ci-C4)alkyl)((C]-C4)alkyl)amino, and (Ci-C4)alkoxy; and
each R4a is independently selected from hydrogen, hydroxyl, amino, and (Ci-C4)alkyl; or a pharmaceutically acceptable salt thereof. Specific compounds of this invention include:
N-((4-chloro-2-methylphenyi)(/ ^
acetamide ;
N-((4-chloro-2-methylphenyl)( -chiorophenyl)methyl)-2-(4-((3,5-dimethyli
yl)m ethoxyjpheny 1) acetami de;
N-(0fi(2-chlorophei yl)methyl)-2-(4-((3,5-dimethy3isoxazol-4-yi)methoxy)ph
N-(di- ?-tolylmethyl)-2-(4-((3,5-dimethylisoxazol-4-yl)methoxy)phenyl)acetaim
2-(4-((3,5-dimethylisoxazol-4-yl)methoxy)phenyl)-N-(l -phenyl- l-(p-tolyl)emyl)acetam N-((4-chlorophenyl)(2,4-dimethylphenyl)methyl)-2-(4-(2-(3,5-dimethyfe
yi)ethoxy)phenyi)acetam de ;
A-((4-chlorophenyl)(phenyl)methyl)-2-(4-(((3,5-dirnethylisoxazol-4- yl)methyl)(methyi)amino)phenyl)acetamide:
A'-((4~chlorophenyl)(phenyi)methyl)-2-(4-(thiazol-4~y
A'-((4-chlorophenyl)(2,4-dimethyfphenyl)methyl)-2-(4-((3
yi)methoxy)pheny i) acetami de;
N"((4-chlorophenyl)iphenyl)methyi)-2--(4-((3-(hydroxymethyl)--5-me
yl)methoxy)phenyl)acetamide;
N-((4-chlorophenyl)(pheny3)methyl)-2-^
N-((4-chlorophenyl)(phenyl)mdhyl)-2-(4-((3>5-dimethylisoxazol-4-yl)methoxy)ph
meihyiacetamide;
2.-{4-((3,5~dimethylisoxazol-4-yi)methoxy)phenyl)~A'-(phenyl(o-tolyl^
A'-((4-chlorophenyl)(phenyr)methyf)-2-(4-(oxazol-5-ylmethoxy)pheny
2-(((3,5-dimethylisoxazol-4-yl)methyl)(4-(2-oxo-2-((phenyl p- tolyi)methyl)amino)ethyl)phenyl)ammo)acetic acid;
N-(6?s(4-fluorophenyl)methy^
2-methylpropanamide;
N-((4-chiorophenyl)( heny])methy^
methylpTopanamide;
2-(4-((3,5-diisopropylisoxazol-4-yl)methoxy)phenyl)-N-((2,4- dimethylphenyl)(phe"nyl)methyl)acetamide;
N-(4-((3,5-dimethylisoxazol-4-yl)metho
pheiiylacetamide;
N-((4-chlorophenyl)(phenyl)me1hyl)-2-(5-((3,5-dimethyiisoxazol-4-yl)methoxy)pyri
yi)acetamide;
N~((4-chio"rophenyl)(phenyl)niethyl)-2-(4-((3,5-dimeihyiisoxazoi-4- y{)methy1thio)phenyl)acetamide;
N-((4-chloro-2-methylphenyl)(phenyl)methyl)-2-(4-(2-(3,5-dimethylisoxazol-4-y
l-yl)acetamide;
methyl 2~((3,5-dimethylisoxazol-4~yl)methoxy)-5-(2-(((2,4- dimethylphenyl)(phenyl)meth.yl)amino)-2-oxoet yl)benzoate;
2-(4-(2-(3,5-dimethylisoxazol-4-yl)-2-hydroxyethoxy)phenyl)-N-((2,4- dimethylphenyl)(phenyl)methyl)acetamide;
2-amino-2~(4-((3,5-dimethylisoxazol-4-yl)methoxy)phenyl)-N-((2,4- dimethylpbenyl)(phenyl)methy])acetamide;
Ar-((4-chloro-2-methylphenyi)(4-chloropheny3)methyl)-2-(4-((3,5-dimetbylisoxazol-4- yi)methoxy)phei yl)acetamide ;
N-((4-chloro-2-methylphenyl)(phe"nyl)methyl)-2-(4-((3,5-dimethylisoxazol-4- yl)methoxy)phenyl)acetamide ;
2-(4-((3,5-dimeihyiisoxazol-4-yl)methoxy)phenyl)-jV-((4- methoxyphenyl)(phenyl)meihyl)acetamide;
N-(bis(4-methoxyphenyl)methyl)-2-^ ; 2-(4-((3,5-dimethyiisoxazol-4-yr)methoxy)phenyl)-AL((2- methoxyphenyl)(phenyl)methyl)acetamide ;
N-((4-(dimethylamino)phenyl) (phenyl)methyl)-2-(4-((3,5-dimethylisoxazol-4-yl)
methoxy)phenyl)acetamide ;
N~((2-(dimethylamino)phenyl) (phenyl)methyl)-2-(4-((3,5-dimethylisoxazol-4- yi)methoxy)phenyl)aceiainide ;
2-(4-((3,5-dimethylisoxazol-4-yl)methoxy)phenyty^ : N~(6z'i(4-fluorophenyl)methyl)-2-(4~((3,5-dimethylisoxazol-4-yl)methoxy)phenyl)acetamide ; A'-((4~cliloiO-2-methylphenyl)(pyridin-4~y{)methy1)~
yl)methoxy)phenyi)acetamide ;
N-((2-chloro-4-methylphenyl)(4^
yi)methoxy)phenyl)acetamide ;
N-((4-chloropheny3)(o-tolyl)methy3)-2-(4-((3,5-diTnethylisoxazol-4-yl)methoxy) phenyl)acetamide 2-(4-((3,5-dimethylisoxazol-4-yl)methoxy)phenyl)-N-((2,4- dimethylphenyi)(phenyl)methyl)a.cetamide;
Ar-((2-chloro-4-methy3phenyl)(pheny3 met3iyl)-2-(4-((3,5-dimethylisoxazol-4- yl)meth.oxy)phenyl)acetamide;
2-(4-((3,5-dimethylisoxazol-4-y3)methoxy)^
2-(4-((3,5-dimethyiisoxazQl-4-yl)meth^
acetamide;
A-((4-chloro-2-methylphenyl)(cyclopropyl)niethyl)-2-(4-((3,5-dimethyiisoxazoi-4- yl)methoxy)phenyl)acetamide ;
N~(cyciohexyl(2,4-dimeihylphenyl)methyl)-2-(4-(('3,5-dimethylisoxazol-4- yi)methoxy)phenyl)aceiainide;
2-C4-((3,5-dimethylisoxazol-4-yl)methoxy)phenyl)-A'-(l -(2,4-dimethylpheny3)-2- methylpropyl)acetamide;
2-(4-((3,5-dimethylisoxazol-4-yl)methoxy)phenyl)-N-(l -( ?-tolyl)cyclohexyl)acetaraide;
2-(4-((3,5-dimethylisoxazol-4-yl)methoxy)^^
Ar-((3,4-dichlorophenyi)(phei yl)methyl)-2-(4-((3,5-dimethylisoxazol-4- yi)methoxy)phei yl)acetamide;
jV-((4-chlorophenyl)(2,4-dimethyiphenyi)methyi)-2-(4~((
yl)methoxy)phen.yl)acetatnide;
2-(4-((3,5-dimcthyiisoxazol-4-y3)metho
f3uoropheny3)methy3)acetamide;
2-(4-((3,5-dimethylisoxazol-4-yl)methoxy^
methoxypbenyl)methyl)acetarmde;
N-(di-o Qlylmethyl)-2-(4-(2-(3,5-dime^
2-(4^2-(3,5-dimethylisoxazo1-4-yl)ethoxy acetamide; 2-(4-(2-(3,5-dimethylisoxazol-4-yi)ethoxy)phenyl)-N-((2-isopropylphenyl)(o-tolyl)me
acetamide;
2-(4-((3,5-dimethylisoxazol-4-y3)methoxy)phenyl)-N-(3 -(3,5-dimethylpyridm-2-yl)-4- methylpentyl)acetamide;
N-((4-clilorophenyl)(2,4-dimethyiphenyl)methyl)-2-(4-(2-(3,5-dimethylisoxa^
yl)ethoxy)phenyl)acetainide ;
N-((4-chlorophenyl)(o-tolyl)raethyl)-2-(4-(2-(3,5-dimethyUsoxazol-4-yl)ethoxy) phenyl)aeetamide
N-((4-chloro-2-methylphei yl)(phenyl)methyl)-2-(4-(2-(3,5-dimethylisoxazoi-4- yl)ethoxy)pbenyl)acetamide ;
2-(4-(2-(3,5-dimetbylisoxazol-4-yl)e1hoxy)ph^ ;
A'-(di- ? ,olylniet yl)-2-(4-(2-(3,5-dimethylisoxazol-4-yl)et oxy)
Λ - ((4 - chloro-2 - methylphe nyl) (p -tolyl) methyl) -2 - (4 - (2 - (3 , 5 -dimethyHsoxazol-4-yl)ethoxy)phenyl)acetamide; A'-((2-chloro-4-meihylphenyI)(4-chiorophenyl)meihyl)
yl)ethoxy)phenyl)acetamide ;
N-((2-(dimethylamko)phenyl) (phenyi)methyl)-2-(4-(2-(3,5-dimetliylisoxazol-4- yl)ethoxy)phenyl)acetamide;
N~((4-chlorophenyl)(phenyl)metliyl)-2-(4-((3,5-diineihyiisoxazol-4-yl)inethoxy)-3 - fluorophenyl)aceta.mide;
Ar-C(4-chlorophenyj)(phenyl)met yl)-2-(4-((3,5-dimethylisoxazol-4-y])methoxy)-2- fiuorophenyl)acetamide ;
N-((4-cWotophmyl)(phenyl)methyl)-2-(4-((2-methylfliiazoM ;
N-((4-chlorophmyl)(phenyl)methyl)-2-(4-(^^ ; 2-(4-((3,5-diraetliylisothiazo1~4-yl)t)iethoxy)phenyl)- ;
2.-{4-((5-iT!eihyl-l,2,4~oxadiazo1-3~yl)t)iedioxy)phenyl)- 2~(4-((5-etbyl-l,2,4~oxadiazo1-3~yl)t)iethoxy)phenyl)-/V-(ph
2.-{4-((3-iT!eihyl-l,2,4~oxadiazo1-5~yl)t)iedioxy)phenyl)- ; 2-(4-((5-methy]- l ,3,4-oxadiazol-2-yl)me†n^ ; 2-(4-((3,5-dirnethyiisoxazol-4-yr)methoxy)phenyl)-AL((4-methoxy-2- methyiphei yl)(phenyi)methy3)acetamide ;
N-((2-chloro-4-methylphenyl)(phe^^
yl)methoxy)phenyl)acetamide ;
N-((2,4-dimethylphenyl)(phenyl)methyl)-2-(4-((3-(hydroxyTOethyl)-5-m
yi)methoxy)phenyl)aceiainide ;
2-C4-((3,5-dimethylisoxazol-4-yl)methoxy)phenyl)-A'-(pheny](4- Ctrifiuoromethyl)phenyl)methyl)acetamide;
N-((4-chlorophenyl)(phenyl)meth.yl)-2-(4-((3,5-dirnethylisoxazol-4- yi)methoxy)phei yl)pTopanamide;
N-((4-chlorophenyl)(phenyl)methyl)-2-(4-(2-(3,5-dimethylisoxazol-4-yl)ethoxy)phenyl)acetamid ; 2-(4-((3-ethyl-5-methylisoxazol-4-yl)methoxy)phenyl)-N-{phenyl(p olyl)methyi)acetamide;
N-(di-p-tolylmethyl)-2-(4-(l-(3,5-dimeth^
A/'-C(4-chlorophenyj.)(phenyl)met yl)-2-(4-((3-ethyl-5-methylisoxazol-4- yl)meth.oxy)phenyl)acetamide ;
N-((4-chlorophenyl)(pheny])methyl)-2-^ ; A'-(i4-elilorQphenyl)(phenyl)m ;
N-((4-chiorophenyl)(pheny])methyl)-2-(4-^ ;
2-(4-chlorophenyl)-N-(4-((3,5-dimethylisoxazol-4-yl)methoxy)benzyl)-2-pheny{acetam
2-(l -(3,5-DimethylisQxazoi-4-yl)-2-(4-(2- oxoethyl)phenoxy)ethoxy)-2-methylpropanoic acid;
fS^-N-((4-chloro-2~methylphenyi)(4-chlorophenyl)methyl)~2-(4-((3,5-diinethyli^
yl)meth.oxy)phenyl)acetamide;
(7?)-N-((4-chJoro-2-methylphenylX
yl)methoxy)phenyl)acetamide;
4-((4-(2-(((4-chloiO-2-methylphenyi)(phenyl)methyl)amino)-2-oxoethyl)phenoxy)metliyl)
methylisoxazole-3-carboxyiic acid;
2-((3,5-dimethyHsoxazol-4-yl)methoxy)-5-(2-(((2,4-dimethylphenyl)(phOTy
oxoethyl)benzoic acid;
2-(4-((3,5-dimethyiisoxazoi-4-yl)methoxy)phenyl)~ l -( l ^henyl-3,4~dihydroisoquinolin-2
yl)ethan.one;
AL((4-cliloro-2-methylphenyl)(4-chforophenyl)methyf)- yl)ethoxy)phenyl)acetamide;
2-(4-(2-(3,5-dimethylisoxazol-4-yl)ethoxy)phenyl)-N-((2,4- dimethylpbenyl)(phenyj.)methy])acetamide;
N-(¾is(2-chlorophenyl)methyl)-2-(4-((3,5-dimethylisoxazol-4-yl)methoxy
2-(4-((3-ethyl-5-methylisoxazol-4-yl)methoxy)phenyl)-N-{phenyl(o-tolyl)methyl)acetamide;
and pharmaceutically acceptable salts thereof.
The meaning of any functional group or substituent thereon at any one occurrence in Formula (I), or any sub formula thereof, is independent of its meaning, or any other functional group's or substituent's meaning, at any other occurrence, unless stated otherwise.
The compounds according to Formula (I) may contain one or more asymmetric centers (also referred to as a chiral center) and may, therefore, exist as individual enantiomers, diastereomers, or other stereoisomeric forms, or as mixtures thereof. Chiral centers, such as chiral carbon atoms, may also be present in a substituent such as an alkyl group. Where the
stereochemistry of a chiral center present in Formula (I), or in any chemical structure illustrated
'5
herein, is not specified the stracture is intended to encompass all individual stereoisomers and all mixtures thereof. Thus, compounds according to Formula (I) containing one or more chiral center may be used as racemic mixtitres, enantiomerically enriched mixtures, or as enantiomerically pure indi vidua! s tereoi somers .
Individual stereoisomers of a compound according to Formula (T) which contain one or more asymmetric centers may be resolved by methods known to those skilled in the art. For example, such resolution may be carried out (1) by formation of diastereoisomeric salts, complexes or other derivatives; (2.) by selective reaction with a stereoisomer-specific reagent, for example by enzymatic oxidation or reduction: or (3) by gas-liquid or liquid chromatography in a chiral environment, for example, on a chiral support such as silica with a bound chiral ligand or in the presence of a chiral solvent. The skilled artisan will appreciate that where the desired stereoisomer is converted into another chemical entity by one of the separation procedures described above, a further step is required to liberate the desired form. Alternatively, specific stereoisomers may be synthesized by asymmetric s nthesis using optically active reagents, substrates, catalysts or solvents, or by converting one enantiomer to the other by asymmetric transformation.
"Enantiomerically enriched" refers to products whose enantiomeric excess is greater than zero. For example, enantiomerically enriched refers to products whose enantiomeric excess is greater than 50% ee, greater than 75% ee, and greater than 90% ee.
"Enantiomeric excess" or "ee" is the excess of one enantiomer over the other expressed as a percentage. As a result, since both enantiomers are present in equal amounts in a racemic mixture, the enantiomeric excess is zero (0% ee). However, if one enantiomer was enriched such that it constitutes 95% of the product, then the enantiomeric excess would be 90% ee (the amount of the enriched enantiomer, 95%, minus the amount of the other enantiomer, 5%).
"Enantiomerically pure" means products whose enantiomeric excess is 99% ee or greater.
When a disclosed compound or its salt is named or depicted by stracture, it is to be understood that the compound or salt, including solvates (particularly, hydrates) thereof, may exist in crystalline forms, non-crystalline forms or a mixture thereof. The compound or salt, or solvates (particularly, hydrates) thereof, may also exhibit polymorphism (i.e. the capacity to occur in different crystalline forms). These different crystalline forms are typically known as
"polymorphs." It is to be understood that when named or depicted by structure, the disclosed compound, or solvates (particularly, hydrates) thereof, also include all polymorphs thereof.
Polymorphs have the same chemical composition but differ in packing, geometrical arrangement, and other descriptive properties of the crystalline solid state. Polymorphs, therefore, may have different physical properties such as shape, density, hardness, deform ability, stability, and
dissolution properties. Polymorphs typically exhibit different melting points, 1R spectra, and X-ray powder diffraction patterns, which may be used for identification. One of ordinary skill in the art will appreciate that different polymorphs may be produced, for example, by changing or adjusting the conditions used in crystallizing/recrystallizing the compound.
For solvates of the compounds of Formula (1), or salts thereof, that are in crystalline form, the skilled artisan will appreciate that pharmaceutically acceptable solvates may be formed wherein solvent molecules are incorporated into the crystalline lattice during crystallization. Solvates may- involve nonaqueous solvents such as ethanol, isopropanol, DMSO, acetic acid, ethanolamine, and ethyl acetate, or they may involve water as the solvent that is incorporated into the crystalline lattice. Solvates wherein water is the solvent that is incorporated into the crystalline lattice are typically referred to as "hydrates." Hydrates include stoichiometric hydrates as well as compositions containing variable amounts of water. The invention includes all such solvates.
Because of their potential use in medicine, the salts of the compounds of Formula (I) are preferably pharmaceutically acceptable. Suitable pharmaceutically acceptable salts include those described by Berge, Bighley and Monkhouse J.Pharm.Sci (1977) 66, pp 1 -19. Salts encompassed within the term "pharmaceutically acceptable salts" refer to non-toxic salts of the compounds of Formula (1).
Salts of the compounds of Formul (!) containing a basic amine or other basic functional group may be prepared by any suitable method known in the art, including treatment of the free base with an inorganic acid, such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like, or with an organic acid, such as acetic acid, trifluoroacetic acid, maJeic acid, succinic acid, mandelic acid, fumaric acid, malonic acid, pyruvic acid, oxalic acid, glycolic acid, salicylic acid, pyranosidyl acid, such as glucuronic acid or galacturonic acid, alpha- hydroxy acid, such as citric acid or tartaric acid, amino acid, such as aspartic acid or glutamic acid, aromatic acid, such as benzoic acid or cinnamic acid, sulfonic acid, such as p-toluenesulfonic acid, methanesulfonic acid, ethanesulfonic acid or the like. Examples of pharmaceutically acceptable salts include sulfates, pyrosulfates, bisulfates, sulfites, bisulfites, phosphates, chlorides, bromides, iodides, acetates, propionates, decanoates, caprylates, acrylates, formates, isobutyrates, caproates, lieptanoates, propiolates, oxalates, malonates succinates, suberaies, sebacates, fimiaraies, maleates, buryne- 1 ,4-dioates, hexyne- 1 ,6-dioates, benzoates, chlorobenzoates, methyibenzoates,
dinitrobenzoates, hydroxybenzoates, methoxybenzoates, phthalates, phenylacetates,
phenylpropionates, phenylbutrates, citrates, lactates, γ-hydroxybutyrates, glycolates, tartrates mandelates, and sulfonates, such as xylenesulfonates, methanesulfonates, propanesulfonates, naphthalene- 1 -sulfonates and naphihalene-2-suifonates.
Salts of the compounds of Formula (1) containing a carboxyiic acid or other acidic functional group can be prepared by reacting with a suitable base. Such a pharmaceutically acceptable salt may be made with a base which affords a pharmaceutically acceptable cation, which includes alkali metal salts (especially sodium and potassium), alkaline earth metal salts (especially calcium and magnesium), aluminum salts and ammonium salts, as well as salts made from physiologically acceptable organic bases such as trimethylamine, triethyl mine, morpholine, pyridine, piperidine, picoline, dicyclohexylamine, N,N-dibenzylethylenediamine, 2- hydroxyethylamine, te-(2-hydroxyemyl)amine, tri-(2-hydroxyethyl)amine, procaine,
dibenzylpiperidine, dehydroabietylamine, Ν,λΓ -bisdshy droabietylamine, giucamine, N- methylglucamine, collidine, quinine, quinoline, and basic amino acid such as lysine and arginine.
Other non-pharmaceutically acceptable salts, e.g. triffuoroacetate, may be used, for example in the isolation of compounds of the invention, and are included within the scope of this invention.
The invention includes within its scope all possible stoichiometric and non-stoichiometric forms of the salts of the compounds of Formul (I).
If a compound of Formula (I) containing a basic amine or other basic functional group is isolated as a salt, (he corresponding free base form of that compound may be prepared by any suitable method known to the art, including treatment of the salt with an inorganic or organic base, suitably an inorganic or organic base having a higher pKa than the free base form of the compound, Similarly, if a compound of Formula (I) containing a carboxyiic acid or other acidic functional group is isolated as a salt, the corresponding free acid form of that compound may be prepared by any suitable method known to the art, including treatment of the salt with an inorganic or organic acid, suitably an inorganic or organic acid having a lower pKa than the free acid form of the compound.
The invention also includes various deuterated forms of the compounds of Formula (I).
Each available hydrogen atom attached to a carbon atom may be independently replaced with a deuterium atom, A person of ordinary skill in the art will know how to synthesize deuterated forms of the compounds of Formula (I). Commercially available deuterated starting materials may be employed in the preparation of deuterated forms of the compounds of Formula (I), or they may be synthesized using conventional techniques employing deuterated reagents (e.g. lithium aluminum deuteride or sodium borodeuteride). See for example the compound of Example 5, N-((4-chlorophenyl)(2,4-dimethylphenyl)methyl)-2-(4-((3,5-d"irnethylisoxazol-4- yi)me thoxy)pheny 1) ace tamide -dj .
Methods of Use
Modulators of RORy can be useful in the treatment of diseases mediated by RORy, particularly autoimmune or inflammatory diseases and cancer. Such inflammatory or autoimmune diseases include multiple sclerosis, rheumatoid arthritis, psoriasis, Crohn's disease, inflammatory bowel disease, graft-versus-host disease (GVHD), Sjorgen's syndrome, optic neuritis, chronic obstructive pulmonary disease, asthma, type I diabetes, neuromyelitis optica, myasthenia gravis, uveitis, Behcets disease, Guillain-Barre syndrome, psoriatic arthritis, Graves' disease, allergic contact dermatitis, systemic lupus erythematosus, cutaneous lupus erythematos s, ankylosing spondylitis, Hashimoto Thyroiditis, dry eye and glomerulonephritis, myocarditis, especially psoriasis Such cancers include multiple myeloma and lytic bone disease associated with multiple myeloma, acute myelogenous leukemia (AML), head and neck squamous cell carcinoma, bladder carcinoma, gastric cancer, hepatocellular carcinoma, melanoma, medulloblastoma and colon cancer. Accordingly, in another aspect the inv ention is directed to methods of treating such diseases using a compound of Formula (I) or a pharmaceutically acceptable salt thereof. The methods of treatment of the invention comprise administering an effective amount of a compound according to Formula (I) or a pharmaceutically acceptable salt thereof to a patient (particularly a human) in need thereof.
in a further aspect, the invention is directed to a compound of Formula (I) or a
pharmaceutically acceptable salt thereof for use in therapy. In particular, for use in the treatment of diseases mediated by RORy, particularly autoimmune or inflammatory diseases and cancer, such as those disclosed above.
In a further aspect, the invention is directed to the use of a compound of Formula (I) or a phannaceuticaliy acceptable salt thereof in the manufacture of a medicament for the treatment of diseases mediated by RORy, particularly autoimmune or inflammatory diseases and cancer, such as those disclosed above.
As used herein, "treatment" in reference to a condition means: (1 ) the amelioration or prevention of the condition being treated or one or more of the biological manifestations of the condition being treated, (2) the interference with (a) one or more points in the biological cascade that leads to or is responsible for the condition being treated or (b) one or more of the biological manifestations of the condition being treated, or (3) the alleviation of one or more of the symptoms or effects associated with the condition being treated.
As indicated above, "treatment" of a condition includes prevention of the condition. The skilled artisan will appreciate that "prevention" is not an absolute term. In medicine, "prevention" is understood to refer to the prophylactic administration of a drug to substantially diminish the
likelihood or severity of a condition or biological manifestation thereof, or to delay the onset of such condition or biological m nifestation thereof.
An "effective amount" means that amount of a drug or pharmaceutical agent that will elicit the biological or medical response of a tissue, system, animal or human that is being sought, for instance, by a researcher or clinician. Furthermore, the term "therapeutically effective amount" means any amount which, as compared to a corresponding subject who has not received such amount, results in improved treatment, healing, prevention, or amelioration of a disease, disorder, or side effect, or a decrease in the rate of advancement of a disease or disorder. The term also includes within its scope amounts effective to enhance normal physiological function,
As used herein, "patient" refers to a human or a mammal, especially a human.
The compounds of the invention may be administered by any suitable route of administration, including both systemic administration and topical administration. Systemic administration includes oral administration, parenteral adminisiration, transdermal administration, rectal administration, and administration by inhalation. Parenteral administration refers to routes of administration other than enteral, transdermal, or by inhalation, and is typically by injection or infusion. Parenteral administration includes intravenous, intramuscular, and subcutaneous injection or infusion, inhalation refers to administration into the patient's lungs whether inhaled through the mouth or through the nasal passages. Topical administration includes application to the skin as well as intraocular, otic, intravaginal, and intranasal administration,
The compounds of the invention may be administered once or according to a dosing regimen wherein a number of doses are administered at vary ing intervals of time for a given period of time. For example, doses may be administered one, two, three, or four times per day. Doses may be administered until the desired therapeutic effect is achieved or indefinitely to maintain the desired therapeutic effect. Suitable dosing regimens for a compound of the invention depend on the pharmacokinetic properties of that compound, such as absorption, distribution, and half-life, which can be determined by the skilled artisan, in addition, suitable dosing regimens, including the amount administered and the duration such regimens are administered, for a compound of the inven tion depend on the condition being treated, the severity of the condition being treated, the age and physical condition of the patient being treaied, the medical history of the patient to be treaied, the nature of concurrent therapy, the particular route of administration chosen, the desired therapeutic effect, and like factors within the knowledge and expertise of the skilled artisan. It will be further understood by such skilled artisans that suitable dosing regimens may require adjustment given an individual patient's response to the dosing regimen or over time as individual patient needs change. Typical daily dosages range from 1 mg to 1000 mg.
it will be appreciated by those skilled in the art that certain protected derivatives of compounds of Formula (I), which may be made prior to a final deprotection stage, may not possess pharmacological activity as such, but may, in certain instances, be administered orally or paremerally and thereafter metabolized in the body to form compounds of the invention which are pharmacologically active. Such derivatives may therefore be described as "prodrugs". Further, certain compounds of the invention may act as prodrugs of other compounds of the invention. All protected derivatives and prodrugs of compounds of the invention are included within the scope of the invention.
Examples of suitable pro-drags for the compounds of the present invention are described in Drags of Today, Volume 19, Number 9, 1983, pp 499 - 538 and in Topics in Chemistry, Chapter 31, pp 306 - 316 and in "Design of Prodrugs" by H. Bundgaard, Elsevier, 1985, Chapter 1 (the disclosures in which documents are incorporated herein by reference). It will further be appreciated by those skilled in the art, that certain moieties, known to those skilled in the art as "pro-moieties", for example as described by H. Bundgaard in "Design of Prodrugs" (the disclosure in which document is incorporated herein by reference) may be placed on appropriate
functionalities when such functionalities are present within compounds of the invention.
Preferred "pro-moieties" for compounds of the invention include: ester, carbonate ester, hemi- ester, phosphate ester, nitro ester, sulfate ester, sulfoxide, amide, carbamate, azo-, phosphatide, glycoside, ether, acetal, and ketaf derivatives of the compounds of Formula (I).
Administration of a compound of the invention as a prodrug may enable the skilled artisan to do one or more of the following: (a) modify the onset of the compound in vivo; (b) modify the duration of action of the compound in vivo; (c) modify the transportation or distribution of the compound in vivo; (d) modify the solubility of the compound in vivo; and (e) overcome or overcome a side effect or other difficulty encountered with the compound.
The invention further includes the use of compounds of the invention as an active therapeutic substance, in particular in the treatment of diseases mediated by RORy. In another embodiment, the invention relates to the use of compounds of the invention in the preparation of a medicament for the treatment of diseases mediated by RORy.
Examples of such diseases include autoimmune or inflammatory diseases such as multiple sclerosis, rheumatoid arthritis, psoriasis, Crohn's disease, inflammatory bowel disease, Sjorgen's syndrome, optic neuritis, chronic obstructive pulmonary disease, asthma, type I diabetes, neuromyelitis optica. Myasthenia Gravis, uveitis, Guillain-Barre syndrome, psoriatic arthritis, Graves' disease, allergic contact dermatitis, systemic lupus erythematosus, cutaneous lupus erythematosus, ankylosing spondylitis, Hashimoto Thyroiditis, Dry Eye, glomerulonephritis, myocarditis and cancer diseases including multiple myeloma and lytic bone disease associated with
multiple myeloma, acute myelogenous leukemia (AML), bead and neck squamous cell carcinoma, bladder carcinoma, gastric cancer, hepatocellular carcinoma, melanoma, medulloblastoma and colon cancer.
The invention includes the use of compounds of the invention for the preparation of a composition for treating or ameliorating diseases mediated by RQRy in a subject in need thereof, wherein the composition comprises a mixture of one or more of the compounds of the invention and an optional pharmaceutically acceptable excipient.
The compounds of the invention may be used alone or in combination with one or more other therapeutic agents. Accordingly the present invention provides a combination comprising a compound of Formula (I) or a pharmaceutically acceptable salt thereof and one or more other therapeutic agents. Such combinations may be presented individually (wherein each active is in separate composition) or the actives are presented in a combined composition.
This invention provides a combination of a compound of Formula (I), or a
pharmaceutically acceptable salt thereof, and one or more therapeutic agents for the treatment of an inflammator '' disease and/or an autoimmune disease, for example, a TNF-a inhibitor; a nonselective COX-l/COX-2 inhibitor; a selective COX-2 inhibitor, such as celecoxib; agents including methotrexate, leflunomide, sulfasalazine, azathioprine, penicillamine, bucillamine, actarit, mizorihine, lobenzarit, hydroxychloroquine, d-penicillamme, aurothiom.ai.ate, auranofin, parenteral and/or oral gold, cyclophosphamide, a BAFF/ APRIL inhibitor, CTLA-4-Ig, or a mimetic of CTLA-4-Ig; 5-lipoxygenase (5-LO) inhibitor, or a 5 -lipoxygenase activating protein (FLAP) antagonist; a leukotriene modifier, including a leukotriene receptor antagonist, such as montelukast, zafirlukast, pranlukast; a. phosphodiesterase type IV (PDE-TV) inhibitor, such as ciioniiiast (ariflo) or roilumilast; an antihistamine HI receptor antagonist; anticholinergic agents such as muscarinic antagonists (ipratropium bromide and tiotropium bromide), as well as selective muscarinic M3 antagonists; β-adrenoceptor agonists such as salmeteroi, formoterol, arformoteroi, terbutaline, metaproterenol, albuterol and the like; a DP receptor antagonist, such as S-5751 and laropiprant; TP receptor antagonists such as seratrodast; neurokinin antagonists ( 1 K2); VLA-4 antagonists; a corticosteroid, such as triamcinolone acetonide, budesonide, beclomethasone, fluticasone and mometasone; insulin-like growth factor type I (IGF- 1) mimetic; kinase inhibitors including Janus Kinase inhibitors (e.g., JAK 1 and/or JAK2 and/or JAK 3 and/or TYK2), p38 MAPK, Syk or
IKK2; rituximab; selective co-stimulation modulator such as abatacept; IL-1 inhibitor anakinra, IL- 6 inhibitor tocilizumab, and IL12/1L-23 inhibitor ustekimumab; anti-IL17 antibody, anti-IL17R antibody, anti-IL21 antibody, or anti-lL22 antibody, SlPl agonists including iingolimod; interferon beta 1 ; natalizumab; a niTOR inhibitor such as rapamycin, cyeiosporine, tacrolimus; non-steroidal antiinflammatory agent (NSAID), including alminoprofen, benoxaprofen, bucloxic acid, carprofen,
fenbufen, fenoprofen, fluprofen, flurbiprofen, ibuprofen, indoprofen, ketoprofen, miroprofen, naproxen, oxaprozin, pirprofen, pranoprofen, suprofen, tiaprofenic acid, and tioxaprofen, indomethacm, acemetacin, alclofenac, clidanac, diclofenac, fenclotenac, fenclozic acid, tentiazac, furofenac, ibufenac, isoxepac, oxpinac, sulindac, tiopinac, tolmetin, zidometacin, and zomepirac, flufenamic acid, meclofenamic acid, mefenamic acid, nifiumic acid, tolfenamic acid, difiunisal and flufenisal, isoxicam, piroxicam, sudoxicam, tenoxican, acetyl salicylic acid, apazone, bezpiperylon, feprazone, mofebutazone, oxyphenbutazone, phenylbutazone; fumaric acid derivative, BG-12; chemokine or chemokine receptor inhibitor, such as a CC - 1, CCR-2, CCR-3 and CCR-9 antagonist.
This invention further provides a combination of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, and one or more therapeutic agents for the treatment of multiple myeloma, for example, Boriezomib-dexamethasone, Bortezomib-dexamethasone- cyclophosphamide, Bortezomib-dexamethasone-lenalidoniide, Lenalidomide-dexamethasone, Melphalan-prednisone-thaiidoniide, Melphalan-prednisone-bortezo ib, Melphalan-prednisone- lenalidomide, Lenaiidomide- dexamethasone- clarithromycin and any of the above combinations plus agents used to treat bone disease in multiple myeloma including bisphosponates, RA K-L inhibitors such as Denusomab and anabolic bone building drugs such as parathyroid hormone (PTH).
This invention also provides a combination of a compound of Formula (I), or a
pharmaceutically acceptable salt thereof, and one or more therapeutic agents for the treatment of colon and/or rectal cancer, for example FOLFOX® (leucovorin [folinic acid], 5-Fluoruracil, and oxaliplatin), FOLF1R1® (leucovorin, 5-Fluoruraeii, and irinotecan), CapeOX® (capecitabine and oxaliplatin), any of the above combinations plus either bevacizumab or cetuximab (but not both), 5- Fluoruraeil and leucovorin, with or without bevacizumab, Capecitabine, with or without bevacizumab, FOLFOXIRI® (leucovorin, 5-Fluoruracil, oxaliplatin, and irinotecan), Irinotecan, with or without cetuximab, Cetuximab alone, and Panitumumab alone.
Compositions
The compounds of the invention will normally, but not necessarily, be formulated into pharmaceutical compositions prior to administration to a patient. Accordingly, in another aspect the invention is directed to pharmaceutical compositions comprising a compound of the invention and one or more pharmaceutically acceptable excipieni(s).
The pharmaceutical compositions of the invention may be prepared and packaged in bulk form wherein an effective amount of a compound of the invention can be extracted and then given to the patient such as with powders, syrups, and solutions for injection. Alternatively, the
pharmaceutical compositions of the invention may be prepared and packaged in unit dosage form. For oral application, for example, one or more tablets or capsules may be administered. A dose of the pharmaceutical composition contains at least a therapeutically effective amount of a compound of this invention (i.e., a compound of Formula I or a salt, particularly a pharmaceutically acceptable salt, thereof). When prepared in unit dosage form, the pharmaceutical compositions may contain from 1 mg to 1000 mg of a compound of this invention.
The pharmaceuiicai compositions of the invention typically contain one compound of the invention. However, in certain embodiments, the pharmaceutical compositions of the invention contain more than one compound of the invention. For example, in certain embodiments the pharmaceutical compositions of the invention contain two compounds of the invention. In addition, the pharmaceutical compositions of the invention may optionally further comprise one or more additional therapeutically active compounds.
As used herein, "pharmaceutically acceptable excipient" means a pharmaceutically acceptable material, composition, or vehicle involved in giving form or consistency to the pharmaceutical composition. Each excipient must be compatible with the other ingredients of the pharmaceutical composition when commingled such that interactions which would substantially reduce the efficacy of the compound of the invention when administered to a patient and interactions which would result in pharmaceutical compositions that are not pharmaceutically acceptable are avoided. In addition, each excipient must of course be of sufficiently high purity to render it pharmaceutically acceptable.
The compounds of the invention and the pharmaceutically acceptable excipient or excipients will typically be formulated into a dosage form adapted for administration to the patient by the desired route of administration. For example, dosage forms include those adapted for (1 ) oral administration such as tablets, capsules, caplets, pills, troches, powders, syrups, elixers, suspensions, solutions, emulsions, sachets, and cachets: (2) parenteral administration such as sterile solutions, suspensions, and powders for reconstitution; (3) transdermal administration such as transdermal patches; (4) rectal administration such as suppositories; (5) inhalation such as dry- powders, aerosols, suspensions, and solutions; and (6) topical administration such as creams, ointments, lotions, solutions, pastes, sprays, foams, and gels.
Suitable pharmaceutically acceptable excipients will vary depending upon the particular dosage form chosen. In addition, suitable pharmaceutically acceptable excipients may be chosen for a particular function that they may serve in the composition. For example, certain
pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of uniform dosage forms. Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of stable dosage forms. Certain pharmaceutically acceptable
excipients may be chosen for their ability to facilitate the carrying or transporting of the compound or compounds of the invention once administered to the patient from one organ, or portion of the body, to another organ, or portion of the body. Certain pharmaceutically acceptable excipients may be chosen for their ability to enhance patient compliance.
Suitable pharmaceutically acceptable excipients include the following types of excipients: diluents, fillers, binders, disintegrants, lubricants, glidants, granulating agents, coating agents, wetting agents, solvents, co-solvents, suspending agents, emulsifiers, sweeteners, flavoring agents, flavor masking agents, coloring agents, anticaking agents, hemectants, chelating agents, plasticizers, viscosity increasing agents, antioxidants, preservatives, stabilizers, surfactants, and buffering agents. The skilled artisan will appreciate that certain pharmaceutically acceptable excipients may serve more than one function and may serve alternative functions depending on how much of the excipient is present in the formulation and what other ingredients are present in the formulation.
Skilled artisans possess the knowledge and skill in the art to enable the to select suitable pharmaceutically acceptable excipients in appropri te amounts for use in the invention. In addition, there are a nitmber of resources that are available to the skilled artisan which describe pharmaceutically acceptable excipients and may be useful in selecting suitable pharmaceutically acceptable excipients. Examples include Remington's Pharmaceutical Sciences (Mack Publishing Company), The Handbook of Pharmaceutical Additives (Gower Publishing Limited), and The Handbook of Pharmaceutical Excipients (the American Pharmaceutical Association and the Pharmaceutical Press).
The pharmaceutical compositions of the invention are prepared using techniques and methods known to those skilled in the art. Some of the methods commonly used in the art are described in Remington's Pharmaceutical Sciences (Mack Publishing Company).
In one aspect, the invention is directed to a solid oral dosage form such as a tablet or capsule comprising a safe and effective amount of a compound of the invention and a diluent or filler. Suitable diluents and fillers include lactose, sucrose, dextrose, mannitol, sorbitol, starch (e.g. corn starch, potato starch, and pre-gelatinized starch), cellulose and its derivatives (e.g.
microciystalline cellulose), calcium sulfate, and dibasic calcium phosphate. The oral solid dosage form may further comprise a binder. Suitable binders include starch (e.g. corn starch, potato starch, and pre-gelatinized starch), gelatin, acacia, sodium alginate, alginic acid, tragacanth, guar gum, povidone, and cellulose and its derivatives (e.g. microcrystaliine cellulose). The oral solid dosage form may further comprise a disintegrant. Suitable disintegrants include crospovidone, sodium starch glycolate, croscamielose, alginic acid, and sodium carboxymethyl cellulose. The
oral solid dosage form may further comprise a lubricant. Suitable lubricants include stearic acid, magnesium stearate, calcium stearate, and talc.
Compound Preparation
The compounds of Formula (I) may be obtained by using synthetic procedures illustrated in the Schemes below or by drawing on the knowledge of a skilled organic chemist. The reaction sequences provided in these Schemes are applicable for producing compounds of the invention having a variety of different X'-X3, R1, R3, R3a, R*, R"a, K'-K4, and A'-A4 groups, as defined above, employing appropriate precursors. The skilled artisan will appreciate that if a substituent described herein is not compatible with the synthetic methods described herein, the substituent may be protected with a suitable protecting group that is stable to the reaction conditions. The protecting group may be removed at a suitable point in the reaction sequence to provide a desired intermediate or target compound. Suitable protecting groups and the methods for protecting and de-protecting different substituents using such suitable protecting groups are well known to those skilled in the art; examples of which may be found in T. Greene and P. Wuts, Protecting Groups in Chemical Synthesis (3rd ed.), John Wiley & Sons, NY (1999). In some instances, a substituent may be specifically selected to be reactive under the reaction conditions used. Under these circumstances, the reaction conditions convert the selected substituent into another substituent that is either useful as an intermediate compound or is a desired substituent in a target compound. Scheme I

Conditions: a) R Br or R l . i. THF, XaB! L. MeOH; b) NH2OH>HCl, pyridine; c) Zn, EtOFi, NH4OH, N i l O.Ac .

Conditions: a) K2C03 or Cs C0 or NaH, DMF or acetone or CH3CN; b) NaOH, M.eOH, H20; c) (II), EDC, l-IOBt, Et3N or DIPEA, DMF or THF or C'H Π : or (II), HAT LI, NMM, CU -CI .
. or OH

Conditions: a) (IT), EDC, HOBt, Et3N or DIPEA, DMF or THF or Cl !.·('! ·; or (II), HATU, NMM, CH2Ci2; b) K2C03 or Cs C03 or NaH, DMF or acetone or C¾CN (X - Cl or Br); or PPh3, DIAD. THF (X = QH).
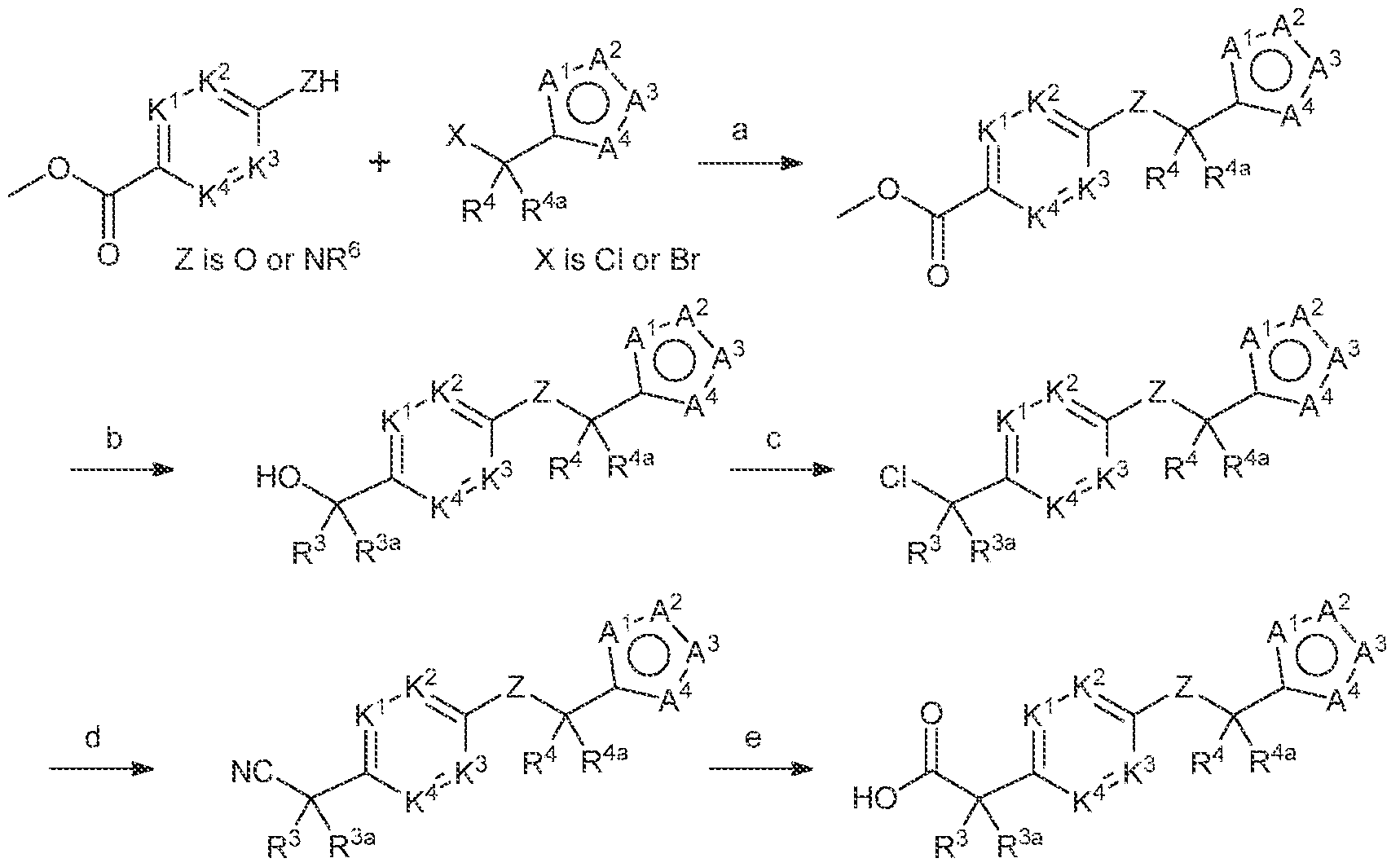
Conditions: a) K2C03 or Cs2C03 or NaH, DMF or acetone or CH3CN; b) NaBH4, MeOH; c) SOCl2, i l l (Ί : d) NaCN, i. MeOH; e) NaOH, MeOH, U O.
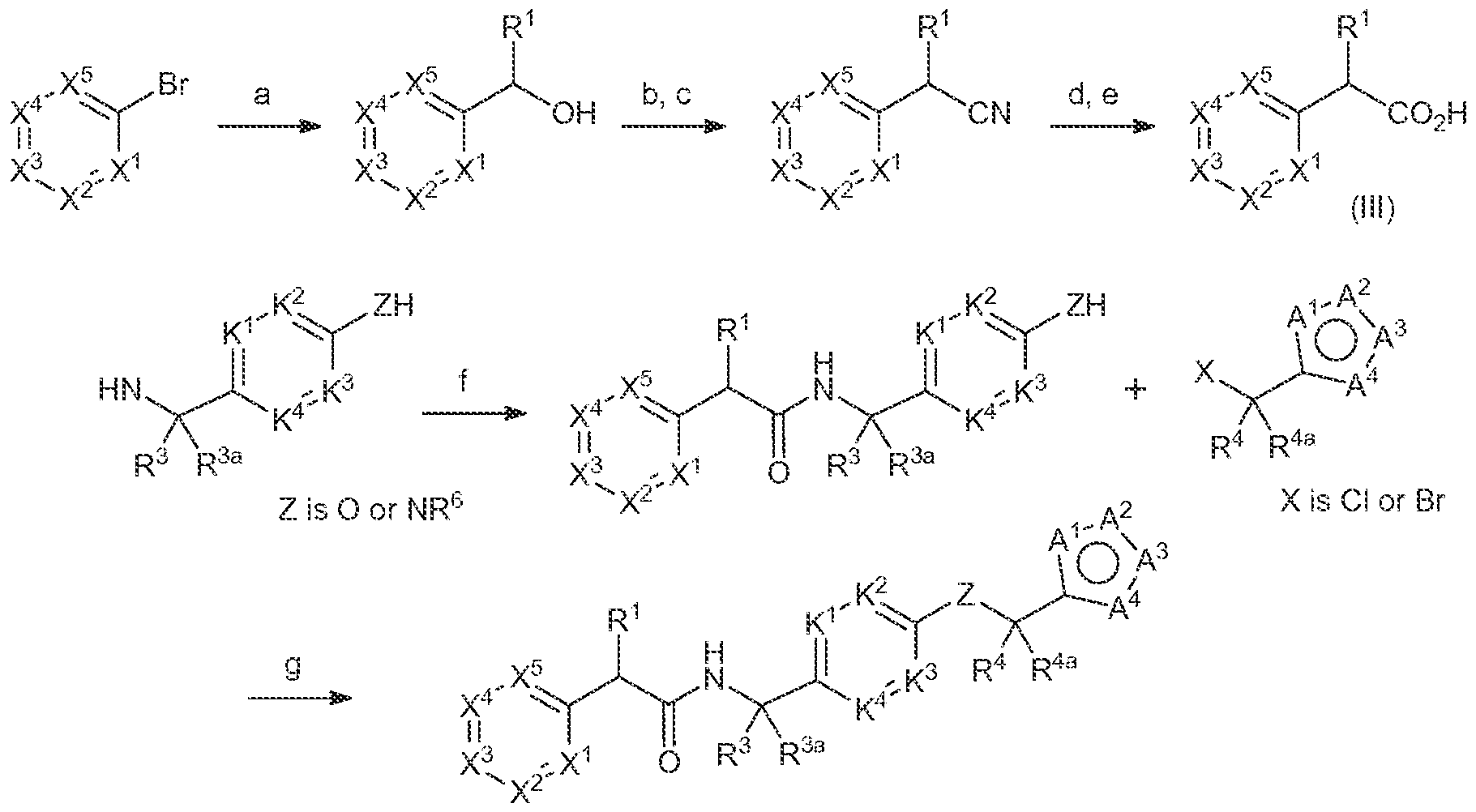
Conditions: a) n-BuLi, R:CF10, THF, -78 °C-0 °C; b) SOCi2, CFI,C12, 0 °C-rt; c) NaCN, K2C03, DMF, 60 °C: d) NaOFI, EtOFI, FLO, reilux: e) H2S04, AcOH, H20, reilux; i) (III), EDC, PIOBt, Et3N or DIPEA, DMF or TFIF or CFI2C12 or (III), HATU, NMM, CFI2C12; g) K2C03 or Cs2C03 or NaH, DMF or acetone or ( i hCX.

Conditions: a) KHCO¾ KI, CH3CN; b) HC1, Et20, MeOH; c) K2C03, BrCR3R3aC02CH3, DMF; d) LiOH, THF, H20; ε) (II), EDC, HOBt, Et3N or D1PEA, DMF or THF or CI i ( ! ·.
Examples
The following examples illustrate the invention. These examples are not intended to limit the scope of the present invention, but rather to provide guidance to the skilled artisan to prepare and use the compounds, compositions, and methods of the present invention. While particular embodiments of ihe present invention are described, the skilled artisan will appreciate that various changes and modifications can be made without departing from the spirit and scope of the invention.
Compounds names were generated using the software program ChemBioDraw Ultra VI 2.0 available from CambridgeSoft Corporation, 100 CambridgePark Drive, Cambridge, MA 02140 LISA. (http:/7 www.cambridgesoft.com).
Abbreviations
AcOH acetic acid
AJBN azob i siso butyronitril e
AiCl3 aluminum trichloride
aq. aqueous
Ar argon gas
Br2 bromine
CBr4 carbon tetrabromide
CCL carbon tetrachloride
CU -Ci dichloromethane
CH3CN acetonitrile
σ¾ΐ methyl iodide
(CH20)n paraformaldehyde
C¾S03H methanesuJfonic acid
cone. concentrated
Cs2C03 cesium carbonate
CuBr copper(I) bromide
CuCN copper(I) cyanide
Cul copper(l) iodide
(COCi)2 oxalyl chloride
DIPEA N, N-di isopr opy lethy lamine
DMAP 4-(dimethy3ammo)pyridme
DME 1 ,2-dimet oxy ethane
DMF N,N- dim ethylformamid e
DMSO dimethyls ulfoxide
EtOAc ethyl acetate
EDC A-(3-dimethyiaminopropyl)-N-ethylcarbodiimide hydrochloride
E†3N triethylamine
Et20 diethyl ether
EtOH ethanol
FeS04 iron(II) sulfate
h hour(s)
H2 hydrogen gas
HATU 0-(7-azabenzotriazol- 1 -yl)-N,N,N',N'-tetramethy1uronium hexafluoropliosphate
HBr hydrobromic acid
HCi hydrochloric acid
¾0 water
HN03 nitric acid
HOBt hydroxyhenzotriazole
HPLC high-performance liquid chromatography- H2SO4 sulfuric acid
h iodine
2-PrMgCl isopropylmagnesium chloride
K2CO3 potassium carbonate
K3Fe(C )6 potassium ferricyanide
KO/-Bu potassium /eri-butoxide
K3PO4 potassium phosphate tribasic
LCMS liquid chromatography mass spectrometry
L1AIH4 lithium aluminum hy dride
LiOH lithium hydroxide
m-CPBA me/a-chloroperbenzoic acid
MeMgBr methyl magnesium bromide
MeOH methanol
Mg magnesium
MgCl2 magnesium chloride
min minute(s)
M11O2 manganese dioxide
N2 nitrogen gas
NaBH4 sodium borohydride
NaCN sodium cyanide
Na2C03 sodium carbonate
Naii sodium hydride
NaHC03 sodium bicarbonate
Xai iSO : sodium bisulfite
NaN3 sodium azide
NaOH sodium hydroxide
Na2S04 sodium sulfate
«-BuLi H-butyllithium
NH4CI ammonium chloride
NMM A;~methylmorpholine
PCC pyridinium chlorochromate
Pd/C palladium on carbon
Pd(dppf)Cl2 [l ,l ''-bis(diphenylphosphino)ferrocene]dichloropalladiuni(ll)
Pd(PPh3)4 tetrak.is(triphenylphosphine)palladium(0)
PI1 O2 nitrobenzene
POCI3 phosphoryl chloride
PPh3 triphenylphosphine
/ TsGH para -toluene sulfonic acid
Rf retention factor
rt room temperature
t retention time
SOCl; thionyi chloride
TFA trifiuoroacetic acid
THF tetrahydrofuran
TLC thin layer chromatography
2,4,6- tripropyl- 1 ,3,5,2,4,6-trioxatriphosphorinane 2,4,6-trioxide
Zn zinc powder
LCMS Conditions
LCMS-P1 : Column: Waters Sunfire C I 8, 3.5 μηι, 50 x 4.6 mm: Temperature: 50 °C;
Mobile Phase: A: water (0.05% TFA.) B: acetonitrile (0.05% TFA); Gradient: 5% B for 0.2. min, increase to 95% B within 1.2 min, 95% B for 1.6 min, return to 5% B within 0.0 ! min.; Flow
Rate: 1.8 mL/min; Detection: PDA 190-400 nm
LCMS-G7: Column: XBridge CI 8, 3.6 μιη, 50 x 4.6 mm; Temperature: 50 °C;
Mobile Phase: A: water (0.1% formic acid) B: methanol; Gradient: 10% B for 0.1 min, increase to 95% B within 2,5 min, 95% B for 2.5 min, return to 10% B within 0.1 min, 10% B for 2 min.;
Flow Rate: 1.0 mL/min; Detection: PDA 190-400 nm
LCMS-G9: Column: XBridge C18, 3,6 μτη, 50 x 4.6 mm: Temperature: 50 °C:
Mobile Phase: A: water (0.1% ammonium acetate) B: methanol; Gradient: 10% B for 0.2 min, increase to 95% B within 5 min, 95% B for 2 min, return to 10% B within 0.1 min, 10% B for 2 min.; Flow Rate: 0.8 mL/min; Detection: PDA 190-400 nm
LCMS-G12: Column: Sunfire CI 8, 5 μηι, 50 x 4.6 mm; Temperature: 50 °C; Mobile
Phase: A: water (0.1% formic acid) B: methanol; Gradient: 30% B for 0.1 min, increase to 90% B within 4 min, 99% B for 4 min, return to 30% B within 0.1 min, 10% B for 2 min.; Flow Rate:
0.8 mL/min; Detection: PDA 190-400 nm
LCMS-G30: Column: Eclipse XDB CI 8, 5 μιη, 250 x 4.6 mm; Temperature: 50 °C:
Mobile Phase: A: water (0.05% TFA) B: acetonitrile (0.05% TFA); Gradient: 30% B for 0.2 min, increase to 95% B within 15 min, 95% B for 5 min, return to 30% B within 3 min 30% B for 5 min.; Flow Rate: 0.8 mL/min; Detection: PDA 190-400 nm
LCMS-X: Column: Eclipse XDB CI 8, 5 μηι, 150 x 4.6 mm; Temperature: 50 °C; Mobile Phase: A: water (0.1% formic acid) B: acetonitrile (0.1 % formic acid); Gradient: 10% B for 0.1 min, increase to 90% B within 5 min, 100% within 2 min, 100% B for 4 min, return to 10% B within 0.01 min, 10% B for 1 min.: Flow Rate: 1.0 mL/min; Detection: PDA 190-400 nm LCMS-Tl : Column: Eclipse XDB C I 8, 5 ,um, 150 x 4.6 mm; Temperature: 50 °C;
Mobile Phase: water (0.05% TFA) B: acetonitrile (0.05% TEA); Gradient: 5% B for 0.1 min, increase to 95% B within 7 min, 100% within 2 min, return to 5% B within 0.1 min, 5% B for 3 min.; Flow Rate: 1.0 mL/min; Detection: PDA 190-400 nm
Example 1
N~((4-chloro-2-memylphenyi)(^~tolyi)methyl)-2~(4-((3,5-dimemylisoxazol-4-yl^
acetamide

(a) methyl 2-(4-hydroxyphenyl)acetate
To a solution of 2-(4-hydroxyphenyl)acetic acid (5.0 g, 32,89 mmol) in methanol (75 mL), HQ gas was purged for 2 hours at 0 °C. After completion of the reaction, the reaction mixture was cooled to rt and methanol was distilled out under reduced pressure. The crude obtained was dissolved in water and neutralized to pH=7 using sodium bi carbonate solution. The aqueous layer was extracted with ethyl acetate (2 x 50 mL), The combined organic layers were dried over Na2S04 and distilled under reduced pressure to provide the title compound. LCMS-P1 : 167.2 [M+Fif; Rt: 1.276 min. (b) methyl 2~(4-((3,5-dimethylisoxazol-4-yl)methoxy)phenyi)aceta.te
To a solution of methyl 2-(4-hydroxyphenyl)acetate (3.0 g, 18.0 mmol) in
dimethylforrn amide (35 mL) was added potassium carbonate (3.7 g, 27.1 mmol), and the reaction mixture was stirred at rt for 30 min.4-(chloromethyl)-3, 5-dimethylisoxazole(2.62 g, 21.6 mmol) was then added and the resulting mixture was stirred at 80 °C for 6 h. After completion of the reaction, water (30 mL) was added and the reaction mixture was extracted with ethyl acetate (2 x 50 mL). The organic layer was dried over Na2S04 and concentrated to obtain a crude product which was purified by silica gel column chromatography using (30% EtOAc/hexanes) to provide
the title compound (3.3 g, 67%). U NMR (400 MHz, DMSO-d6) δ ppm 7.17-7.20 (d, 2 i n. 6.94-6.96 (d, 2 H), 4.88 (s, 2 H), 3.60 ($, 3 H), 3.41 (s, 2 H), 2.39 (s, 3 H), 2.20 (s, 3 H). MS (ES1+) === 276.12 (M · i l ). (c) 2-(4-((3,5-diniethy1isoxazo1-4-yl)niethoxy)phenyl)acetic acid
To a solution of methyl 2-(4-((3,5-dimethylisoxazof-4-yi)methoxy)phenyl)acetate (3.2 g, 1 1.6 mmoi) in methanol (35 mL), sodium hydroxide (0.93 g, 23.2 mmol) in 50 mL of water was added dropwise and the reaction mixture was stirred at rt for 2 h. After completion of the reaction, (he methanol was distilled off at reduced pressure and water (50 mL) was added to the reaction mixture. The aqueous layer was washed with ethyl acetate and discarded. Then, the aqueous layer was acidified to pH=3 using 10% Hydrochloric acid and extracted with ethyl acetate (2 X 50 mL). The organic layer was dried over Na2S0 and concentrated to provide the title compound (2.9 g, 95.70%). 1 H NMR (400 MHz, DMSO-d6) δ ppm 7.03-7.16 (d, 2 i l l. 6.91 -6.93 (d, 2 H), 4.84 (s, 2 H), 3.46 (s, 2 H), 2.35 (s, 3 H), 2.17 (s, 3 H). MS (ESI+) 262.06 (M + H).
(d) p-tolylmagnesium bromide
Magnesium turnings (0.224 g, 9.35 mmoi) were added to a three neck RBF equipped with nitrogen flow and cold water condenser. Anhydrous THF (25 mL) was added to the reaction mixture and heated at 70-80 °C. A solution of 1 -bromo-4-methy3benzene (0.80 g, 4,67 mmol) in THF (10 mL) was added over 20 minutes dropwise to the reaction mixture and the resulting mixture was retluxed at 80 °C for 2 h. The resulting gray mixture was cooled to 10 °C and used in next step without further purification.
(e) (4-chloro-2-methylphenyl)(p-tolyl)methanamme
To a solution of 4-chloro-2~methylbenzonitrile (0.50 g, 3.31 mmoi) in anhydrous THF (15 mL) at 0 °C was added solution of >-tolylmagnesium bromide in THF (15 mL) slowly over 10 minutes and the resulting mixture was allowed to warm to rt. The reaction mixture was stirred at rt for 5 h and heated to 60 °C where it was further stirred for 2 h. After completion of the imine formation, the reaction mixture was cooled to 0 °C and 5 mL of methanol was added very slowly followed by sodium borohydride (0.244 g, 6.62 mmol). The resulting mixture was warmed to rt and stirred overnight. After completion of the reaction, water (10 mL) was added in to the reaction mixture and the mixture was extracted with ethyl acetate (2 X 40 mL). The combined organic layers were washed with brine (25 mL), dried over Na2804, and evaporated to obtained a crude product which was purified using silica gel column chromatography using 10% ethyl acetate in hexanes to obtain the title compound (0.40 g, 40.0%) as a light brown oil. ]H NMR (400 MHz,
DMSO-de) δ ppm 7.54-7.56 (d, 1 H), 7.22-7.28 (d, 1 H), 7.15-7.21 (m, 3 H), 7.1 1 -7.13 (m, 2 H), 5.16 (s, 1 IT), 2.24 (s, 3 H), 2.21 (s, 3 IT).
(f) N-((4-chloro-2-memylphenyl)( olyl)methyl)-2-(4-^^
phenyi) acetamide
To a solution of 2-(4-((3,5-dimethylisoxazol-4-y3)methoxy)phenyl)acetic acid (0.20 g 0.766 mmol) in tetrahydrofuran (25 mL), EDC (0.20 g, 1.149 mmol) was added portion wise and the reaction was stirred at ri for 2. h.(4-chloro-2-niethylphenyl)(p-tolyl)methanamine (0.206 g, 0.842 mmol), HOBt (0.1 17 g, 0.766 mmol), and triethylamine (0.318 mL, 2.29 mmol) were added and the reaction mixture was stirred for 24 h at rt. After completion of the reaction, water (50 mL) was added to the reaction mixture and the mixture was extracted with ethyl acetate (2 X 50 mL). The organic layer was dried over Na2S0 and concentrated to obtain a cmde product which was purified by silica gel column chromatography using 30% EtQAc/hexane as mobile phase to provide the title compound (0.1 15 g, 30.74%). LCMS-X1 : 489.3 ; M i i f : Rt = 5.89 min. ! i MR (400 MHz, DMSO-d6) δ ppm 8.85-8.87 (d, I H), 7.12-7.29 (m, 6 IT), 7.01-7.09 (m, 2 IT), 6.91 -6.99 (d, 2 IT), 6.1 1-6.13 (d, 1 IT), 4.88 (s, 2 IT), 3.43 (s, 2 H), 2.38 (s, 3 IT), 2.27 (s, 3 IT), 2.20 (s, 3 H), 2.17 (s, 3 H).
Example 2
A-((4-chloro-2-methylphenyl)(4-chlorophenyl)methyl)-2-(4-((3,5-dimethylisoxazol-4- yl)methoxy)phenyl)aeetamide

(a) (4-chloro-2-methylphenyl)(4-chlorophenyj)methanamine
This compound was synthesized from 4-chloro-2-methy1benzonitrile and (4- chlorophenyl)magnesium bromide essentially as described in example 1 (e) to give the title compound (0.31 1 g, 35.42%) out of which 2.24 mg was used in the next step without further purification. lH NMR (400 MHz, DMSO-d6) δ ppm 7.49-7.51 (d, 1 H), 7.37-7.40 (d, 2 H), 7.26- 7.35 (m, 3 H), 7.21 -7.22 (d, 1 IT), 5.29 (s, 1 H), 2.08 (s, 3 IT).
(b) N-((4-chloro-2-methylphenyl)(4-chlorophenyj)methyl)-2-(4-((3,5-dimethylisoxazol-4- yl)methoxy)pheny3)acetamide
This compound was synthesized from 2-(4-((3,5-dimethylisoxazol-4- yl)methoxy)phenyl)acetic acid and (4-chloro-2-methylphenyl)(4-chlorophenyl)methanamine essentially as described in example 1 (f), except the title compound was isolated as follows: after completion of the reaction, water ( 1 0 niL) was added into the reaction mixture very slowly with cooling and the obtained white solid was filtered off and washed with water (20 mL) and hexanes (20 mL) and dried under reduced pressure to obtain the title compound (240 nig, 55.98%).
LCMS-X1 : 509.2 j YM I j : R- = 6.03 min. Ή MMR (400 MHz, DMSG-d6) δ ppm 8.92-8.94 (d, 1 H), 7.39-7.4 i (d, 2 H), 7.28 (s, 1 H), 7.17-7.25 (m, 5 H), 7.04-7.06 (d, 1 H), 6.92-6.94 (d, 2 H), 6.17-6.19 (d, i H), 4.88 (s, 2 ! ! }. 3.45 (s, 2 R), 2.39 (s, 3 H), 2.20 (s, 6 H),
Example 3
A'-(6 s(2-chiorophenyl)methyl)- -(4-((3,5-dimethylisoxazoi-4-y{)

(a) ??',v(2-chiorophenyl)methanone oxime
To a solution of .fe(2~chlorophenyf)methanone (1 .0 g, 3.98 mmol) in pyridine (10,0 mL) was added hydroxylamine hydrochloride (1.10 g, 15.90 mmol), and the reaction was heated at reflux temperature overnight. After completion of the reaction, the reaction mixture was concentrated, diluted with EtOAc (50 mL), and washed with water (25 mL) and 2M HCl solution. The combined organic layers were dried over Na2S04 and concentrated. The the title compound was purified by triturating with diethyl ether ( 10 mL) to provide (0.80 g, 75.54%). Ή NMR (400 MHz, DMSO-de) δ ppm 1 1.84 (s, 1 H), 7.44-7.51 , (m, 3 H), 7.40-7.44 (m, 2 H), 7.36-7.40 (m, 2 H), 7.27-7.29 (dd 1 H).
(b) v(2-chlorophenyl)methanamine
To a soiution of A«(2-chlorophenyl)methanone oxime (0.800 g, 3.00 mmol) in ethanol (4,0 mL) was added concentrated ammonia soiution (20 mL), ammonium acetate (0, 1 15 g, 1 ,50 mmol), and zinc powder (1 .05 g, 16.23 mmol). The mixture was heated at reflux temperature for 4 h. After completion of the reaction, the reaction mixture was cooled to rt and filtered through Celite. A 10% NaOH solution was added and then the mixture was washed with ethyl acetate (100 mL).
The organic layer was dried over Na2S04 and concentrated. Title compound was obtained (0.80 g, 75.54%) and was used in the next step without any further purification. Ή NMR (400 MHz, DMSO-ds) δ ppm 7.38-7.41, fm, 4 H), 7.32-7.35 (m, 2 H), 7.25-7.30 (m,2 H), 5.66 (s 1 H),2.33 (br, 2 H).
(c) N-(¾i5(2-chforophenyl)metbyi)-2-(4-((3,5-dimethy1isoxazo]-4-
This compound was synthesized from te(2-chlorophenyl)methanamine and 2-(4-((3, 5- dimethylisoxazol-4-yl) methoxy) phenyl) acetic acid essentially as described in example 1 (f) except the reaction was carried out in DMF.(87 mg, 22.19%). LCMS-X1 : 495.2 [M+Hj+; ¾ = 6.81 min. lH NMR (400 MHz, DMSG-d6) δ ppm 8.90-8.92 (d, 1 H), 7.48-7.49 (d, 2 H), 7.32-7.41 (m, 4 H), 7.18-7.20 (d, 2 H), 7.10-7.12 (dd, 1 H), 6.92-6.94 (d, 2 H), 6.57-6.59 (d, i H), 4.88 (s, 2 IT), 3.46 (s, 2 m. 2.39 fs, 3 IT), 2.20 (s, 3 i n.
Example 4
N-(di-/j-tolylmethyl)-2-(4-((3,5-dimeth^

(a) di-p-tolylmethanamine
This compound was synihesized from 4-niethylbenzonitrile and -torylmagnesiurn broniid essentially as described in example 1 (e). LCMS-P1 : 195 [M- H2] +; Rt: 1.217 min. Ή NMR (400 MHz, CDCU) δ ppm 7.24 (d, J= 7.6 Hz, 4H), 7.1 1 (d, J= 8 Hz, 4H), 5.15 (s, lH), 2.31 (s, 611), 1.82 (br, 2H).
(b) A'-(di- 7-toly]methyl)-2-(4-((3,5-dimethylisoxazol-4-yl)methoxy)pheny])acetamide
To a stirred solution of 2-(4-((3, 5-dimethylisoxazol-4-yl)methoxy)phenyl)acetic acid(261 mg, 1 mmol) in dichloromethane (10 mL) was added di-p-tolylmethanamine (21 1 mg, 1 mmol), DIPEA (258 mg, 2. mmol), EDC (230 mg, 1.2 mmol) and HOBt (162 mg, 1.2. mmol) successively. The resulting mixture was stirred at rt overnight. Additional dichloromethane (20 mL) was added, the resulting mixture was washed with (10 mL 3), 1% HCl (10 mL,x3), and the organic layer was dried over Na2S04. After removal of the organic solvent, the crude product was purified by preparator HPLC using 10- 100% water/acetonitrile with 0.1% TFA to give the title compound (275 mg, 61%), LCMS-P1 : 455 [M+H]÷; Rt: 1.753 min. lH NMR (500 MHz, CDC13) δ ppm 7.14 (d, J- 8.0 Hz, 2H), 7.02 (d, 8.0 Hz, 4H), 6.92 (d, j= 8.2 Hz, 4H), 6.85 (d, J= 8.5 Hz,
2H), 6. 10 (d, J = 8.5 Hz, 1 H), 5.90 (d, J= 8.5 Hz, IH), 4.71 (s, 2H), 3.51 (s, 2H), 2.33 (s, 3H), 2.24 (s, 6H), 2.22 (s, 3H).
Example 5
Ar-C(4-chloropheny])(2,4-dimethylphenyl)methyl)-2-(4-((3,5-dimethylisoxazol-4- y3)methoxy)phenyl)acetamide-i /

(a) (4-chlorophenyl)(2,4-diinethylphe"nyl)methanamme-£/;
This compound was synthesized from 2,4-dimethylbenzonitriie and (4- ch]orophenyl)magnes un bromide essentially as described in example 1 (e) except NaBD4 was used. LCMS-T1 : 230.1 ΓΜ- NH2]+; Rt - 4.50 mm.
(b) A-((4-chlorophenyi)(2,4-dimethylphenyl)methyl)-2-(4-((3,5-dnnethylisox
y])methoxy)phenyl)acetami.de-i//
This compound was synthesized from (4-chforophenyl)(2,4-dimethylpher!yf)methanamiiie- di and 2-(4-((3,5-dimethylisoxazol-4-yi)methoxy)phenyl)acetic acid essentially as described in example 4 (b). LCMS-T1 491.2 [M+H ]. lH NMR (DMSO~d6) δ 8.81 (IH, d), 7.35 (2R d), 7.17■■ 7.14 ( -H i . m), 6.96■■ 6.88 (5H, m), 4.85 (2H, s), 3.41 (2H, s), 2.36 (3H, s), 2.21 (3H, s), 2.17 (3H, s), 2.12 (3H, s).
Example 6
2-(4-((3,5-dimethylisoxazol-4 olyl)ethyl)acetamide

2-(4-((3,5-dimethylisoxazol-4-yl)methoxy)phenyl)acetic acid (60 mg, 0.2 mmol) and 1 - phenyl- 1 -(p-tolyl)ethanamine (24 mg, 0.1 mmoi) were dissolved in dichloromethane (15 mL). ! i ATI, (349 mg, 0.9 mmol) was added followed by N-methyl morpholine (200 mg, 1.6 mmol) and the reaction was stirred overnight. Solvent was removed and the title compound was purified by
reverse phase chromatography using aeetonitrile/water with 0.05% TFA. (2 mg, 2%). LCMS-T1 : 454.5 i Vi - i l ) ; Rt - 5.74 mm. Ή NMR (400 MHz, CDC13) δ ppm 7.28 (m, 2H), 7.15 (d, 2H), 7.09 (d, 2H), 6.89 (t, 2H), 6.83 (d, 2H) 6.1 8 (d, 1H), 5.91 (d, 2H), 3.99 (s, 3H), 3.70 (s, 2H), 3.55 (s, 21 n. 2.31 (s, M l ). 2.09 (s, 3H), 2.07 (s, M l ).
Example 7
N~((4-c orophenyl)(2,4-ditnethylphenyl)methyI)-2-(4-(2-(3,5-dimethylisoxazol- yi)ethoxy)phenyi)aeetamide

This compound was synthesized from 2,4-dimethylhenzomtriie and 4-chloro
phenylmagnesiumbromide essentially as described in example 1 (e) and was purified by silica gel column chromatography (20% ethyl acetate in hexanes) to provide the title compound ( 1.20 g, 29.62%). !H NMR (400 MHz, DMSO-de) δ ppm 7.27-7.36 (m, 5 H), 6.97-6.99 (d, 1 H), 6.91 (d, i H), 5.18 (s, 1 H), 2.17-2.22 (d, 8 H).
(b) methyl 2-(4-(2-(3,5-dimethylisoxazol-4-yl)ethoxy)phenyl)acetate
To 2-(3,5-dimethylisoxazoI-4-yl)etlianoI (100 mg, 0.7 mmol) and triefhylamine (0.12 mL, 0.8 mmol) in dichioromethane (5 mL) at 0 °C, raesyl chloride (97 mg, 0.8 mmol) was added dropwise. The reaction was stirred for 1 h at 0 °C and then for 30 minutes at rt. When the reaction was complete, water(5 mL) was added and the mixture was extracted with
dichioromethane. The organic layer was washed with saturated sodium bicarbonate and brine and dried over sodium sulfate. Solvent was removed and 2~(3,5~dimethyJisoxazoJ-4~y{)ethyl methanesulfonate was dissolved in DMF (1 mL) and used crude. To methyl 2~(4- hydroxyphenyl)aeetate ( 166 mg, 0.6 mmol) in DMF (2 mL), was added to sodium hydride (60%, 32. mg, 0.7 mmol) in DMF (2 mL) at 0 °C. The reaction was stirred for 30 min at 0 °C and then for 30 minutes at rt. 2-(3,5-dimethylisoxazol-4-yl)ethyl methanesulfonate in DMF was added and the reaction was stirred for 30 min at rt and then at 90 °C overnight. Water (10 mL) was added and the mixture was extracted with ethyl acetate (2X15 mL), dried over sodium sulfate, and solvent was removed. Title compound was purified by silica gel column chromatography (0- 18% ethyl acetate in hexanes) to provide (40 mg, 24%).
(c) 2-(4-(2-(3,5-dimethyiisoxazol-4-yl)ethoxy)phenyl)acetic acid
This compound was synthesized from methyl 2-(4-(2-(3,5-dimethylisoxazol-4- yl)ethoxy)phenyl)acetate essentially as described in example 1 (c). (800 mg, 80%). MS 275.9 I NT H i ;
(d) N-((4-chlorophenyl)(2,4-dimethylphenyl)methyl)-2-(4-(2-(3,5-dimethylisoxazol-4- yl)ethoxy)phenyl)acetamide
This compound was synthesized from 2-(4-(2-(3,5-dimethylisoxazol-4- yl)ethoxy)phenyl)acetic acid and (4-chlorophenyl)(2,4-dimethy1pheiiyl)raethanamine essentially as described in example 1 (f) (0.023 g, 25.27%). LCMS-X1 : 503.5 [M+H]+; R, - 5.52 min. Ή NMR (400 MHz, DMSO-de) δ ppm 8.80-8.82 (d, 1 H), 7.36-7.38 (d, 2 H), 7.14-7.18 (1 4 IT), 6.90- 6.98 (m, 2 H), 6.81 -6.83 (d, 2 H), 6.14-6.17 (d, 1 H), 3.97-4.00 (t, 2 H), 3.39-3.42 (d, 2 H), 2.72- 2.75 (t, 2H), 2.32 (s, 3 H), 2.23 (s, 3 H), 2.20 (s, 3 H), 2.14 (s, 3 H)
Example 8
N~((4-chlorophenyl)(phenyl)nietliyl)~2-(4-(((3,5-dimeihyiisoxazoi-4- yl)methyl)(methyj.)amino)ph

To a solution of N-((4-chlorophenyl)(phenyj.)methyl)-2-(4-(((3,5-dimethylisoxazol-4- yl)methyl)amino)phenyl)acetamide (0.200 g, 0.435 mmol) in acetone (10 mL) at 25 °C was added potassium carbonate (0.151 g, 1.08 mmol) followed by addition of iodomethane (0.123 g, 0.87 mmol) at Q°C and the reaction was allowed to stir for 24 h. Then the solvent was evaporated under reduced pressure and waf er (10 mL) was added. The solution was extracted with ethyl acetate (3 x 25 mL) and the combined organic layers were washed with water (20 mL), brine (25 mL), dried over Na2S04, and evaporated under reduced pressure to obtain a crude product which was purified using preparative TLC on silica gel using 40% ethyl acetate in hexanes to provide the title compound was isolated. (22 mg, 10.67%). LCMS-X 1 : 474.6 [M+H]+; Rt - 6.78 min. Ή NMR (400 MHz, DMSO-d6) δ ppm 8.92-8.94 (d, 1 H), 7.34-7.39 (d, 2 H), 7.24-7.32 (m, 7 H),
7.09-7.1 1 (d, 2 H), 6.76-6.79 (d, 2 H), 6.07-6.09 (d, 1 IT), 4.19 (d, 2 IT), 3.36 (s, 2 IT), 2.69 (s, 3 H), 2.33 (s, M l ). 2.05 (s, 3 IT).
Example 9
A-((4-chlorophenyl)(phenyl)m acet amide

(a) Ar-((4-chlorophenyl)(phenyi)methyl)-2-(4-hydroxypheny3)acetamide
This compound was synthesized from 2-(4-hydroxyphenyl)acetic acid and (4- chlorophenyi)(phenyl)methananiine essentially as described in example 1 (f) and the product was purified by silica gel column chromatography (40% EtOAc/hexanes) to provide the title compound (4.5 g, 65.2%). Ή NMR (400 MHz, DMSO-d6) δ ppm 9.25 (s, 1 H), 8.94-8.96, (d, 1 H), 3.37-3.39 id, 2 H), 7.28-7.34 (m,3 H), 7.24-7.26 (m 3 H),7.04-7.06 (d, 2 H), 6.65-6.67 (d, 2 H), 6.07-6.09 id, 1 H), 4.08 (s, 2 H MS (ESI+) 352.0 (M + H).
(b) AL((4-chloroplienyl)(phenyl)methy
To a solution of N-((4-chlorophenyl)(phenyl)methyl)-2-(4-hydroxyphenyl)acetamide
(0.300 g, 0.85 mmol) in DMF (4.0 mL) was added potassium carbonate (0.353 g, 0.2.55 mmol) and 4-(chloromethyl) thiazole (0.136 g, 1.02 mmol), and the reaction was stirred for 20 h. After completion of the reaction, the reaction mixture was poured into water (20 mL) and extracted with ethyl acetate (100 mL). The organic layer was dried over Na2S04 and concentrated. The compound was purified by silica gel column chromatography (32% EtOAc/hexanes) to provide the title compound (0.185 g, 48.42%). LCMS-X1 : 449.1 [M+Hf ; Rt = 5.58 min. lH NMR (400 MHz, DMSO-d6) δ ppm 9.12-9.13 (d, 1 H), 9.00-9.02 (d, 1 H), 7.77 (s, 1 H), 7.37-7.40 (d, 2 H), 7.25-7.35 (m, 7 H), 6.95-6.97 (d, 2 H), 6.07-6.09 (s, 1 i n. 5.18 (s, 2 H), 3.47 (s, 2 H).
Example Id
Ai-((4-chforophenyl)(2,4-dimetliylphenyi)methyf)-2-(4-((3-ethyl-5-methylisoxazof'
yl)methoxy)pheny3)acetamide

This compound was synthesized from 2,4-dimethylbenzonitrile and p-chloromagnesium bromide essentially as described in example 1 (e). ( 1.20 g, 29.62%). Ή NMR (400 MHz, DMSO- d6) δ ppm 7.27-7.36 (m, 5 H), 6.97-6.99 (d, 1 H), 6.91 (d, 1 H), 5.18 (s, 1 H), 2.17-2.22 (d, 8 H).
(b) N-((4-chlorophenyl)(2,4-dimethylphenyl)methyl)-2-(4-hydroxyphenyl)acetamide
This compound was synthesized from 2-(4-hydroxyphenyi)acetic acid and (4- ehlorophenyl)(2,4-dimethylphenyl)methanamine essentially as described in example 4 (b). The product was purified by silica gel column chromatography (25% ethyl acetate/he anes) to provide the title compound (0.81 g, 43.78%) MS (ESI+) 380.1 (M + H). ]H NMR (400 MHz, DMSO~d6) δ ppm 9.23 (s, 1 H), 8.79-8.81 (d, 1 H), 7.37-7.39 fd, 2 H), 7.16-7.18 (d, 2 H), 7.03-7.05 (d, 2 H), 6.97-6.99 (d, 1 H), 6.91-6.95 (t, 2 H), 6.65-6.67 (d, 2 H), 6.17-6.15 (d, 1 H), 3.33-3.37 (t 2 H), 2.14-2.23 (m, 7 H).
(c) (3-ethyl-5-methylisoxazol-4-yl)methanol

To a solution ethyl 3-ethy1-5-methylisoxazole-4-carboxy1ate (0.50 g, 2.72 mmol) in anhydrous THF (7 mL) at -78 °C was added 1 .0 M solution of LiAlH4 (4.00 mL, 4.08 mmol) dropwise. After completion of the addition, the reaction mixture was warmed to -40 °C and stirred for 1 h. After completion of the reaction, the reaction mixture was warmed to -10 °C and water (5 mL) was added and the mixture was extracted with ethyl acetate (2 X 15 mL). The combined organic layers were dried over Na2S04 and concentrated to provide the title compound (0.325 g, 84.63%)' MS (ESI+) 142.1 (M + H). Ή NMR (400 MHz, DMSO-d6) δ ppm 4.91 (s, 1 H i. 4.26 (s, 2 H), 2.58-2.67 fm, 2 H), 2.32 (s, 3 H), 1.17-1.20 (t, 3 H).
(d) 4-(chloroniethyl)-3-ethy1-5-methyiiso

To a solution (3-ethyl-5-methylisoxazol-4-yl)methanol (0.10 g, 0.85 mmol) in
dicbloromethane (4 mL) at 0 °C was added SOCL (0.09 mL, 0.12 mmol) dropwise. After completion of the addition, the reaction mixture was warmed to rt and stirred for 2 h. After completion of the reaction, solvent was evaporated and azeotroped by dicbloromethane (2 X 10
mL) to provide the title compound (0.1 1 g, 97.34%), Ή NMR (400 MHz, DMSO-d6) δ ppm 4.68 (s, 2 H), 2.64-2.69 (q, 2 I I I 2.40 (s, 3 H), 1.20- 1.23 (t, 3 H). MS 160.1 (M + H).
(e) Ar-((4-chlorophenyl)(2,4-dimethylphenyl)methyl)-2-(4-((3-ethyl-5-methylisoxazol-4- y1)metlioxy)pherjyl)acetamide
To a solution of N-((4-chlorophenyl)(2,4-d methylphenyl)methyi)-2-(4- hydroxyphenyl)acelamide (0.20 g, 0.52 mmol) in anhydrous DMF (5 mL) at 25 °C was added cesium carbonate (0.51 g, 1.57 mmol) and 4-(chloromethyl)-3-ethyl-5-methyiisoxazoie (0,085 g, 0.52 mmol). The reaction was stirred for 4 h at 90-100 °C under argon atmosphere. After completion of the reaction, water (20 mL) was added and extracted with ethyl acetate (2 x 35 mL). The combined organic layers were dried over a2S04 and concentrated and the product was purified by silica gel column chromatography (18% ethyl acetate/hexanes) to provide the title compound (0.095 g, 35.98%). LCMS-X1 : 504.1 [M+H]+: t = 5.61 min. lH NMR (400 MHz, DMSO-de) δ ppm 8.82-8.84 (d, 1 H), 7.36-7.39 (d, 2 H), 7, 16-7, 19 (q, 4 H), 6.91 -6.99 (m, 5 H), 6.16-6.1 8 (d, 1 H), 4.90 (s, 2H), 3.44 (s, 2 H), 2.60-2.66 (m, 2 H), 2.39 (s, 3 H), 2.23 (s, 3 H), 2.15 (s, 3 H), 1.16-1.20 (t, 3 H).
Example 11
Ar-((4-chlorophenyl)(pheny3)methyl)-2-(4-((3-(1 ydroxyTnethyl)-5-methy3iso^

To a solution of A'-((4-chloropheny3.)(phenyl)methyl)-2-(4-3iydroxyp3ienyl)acetamide (0.259 g, 0.73 mmol) in dimetbylformamide (5 mL) was added ethyl 4-(chloromethy3)-5- methylisoxazole-3-carboxylate (0.150 g, 0.73 mmol) and anhydrous potassium carbonate (0.304 g,
2.20 mmol), and the reaction was ref!uxed for 2. h. A fter completion of the reaction, water (30 mL) was added into the reaction mixture and the mixture was extracted with ethyl acetate (125 mL). The organic layer was washed with brine (25 mL), dried over Na2S(¼. and evaporated to obtain a crude product which was purified using silica gel column chromatography using 32% EtOAc: hexanes to obtain the title compound (0.310 g, 81.3%) as a light yellow semi solid. MS (EST+) 5.19.4 (M + H),
(b) N-((4-cUorophenyl)(phenyl)methyl)~2-(4-((3-(¾ydroxymethyl)-5-meihylisoxa
yl)me thoxy)pheny 1) ace tamide

To a solution ethyl 4-((4-(2-((4-ch]oroplienyf)(phenyl)methylamino)-2- oxoethyl)phenoxy)methyl)-5-methylisoxazole-3-carboxylate (0.300 g, 0.579 mmol) in THF (5 mL) at 0 °C was added 1 M lithium aluminum hydride solution in THF (0.57 mL, 0.579 mmol) and the reaction was stirred at 0 °C for 1 h. After completion of the reaction, crashed ice was added to the reaction mixture and extracted with ethyl acetate (300 mL). The combined organic layers were dried over Na2S04 and concentrated to obtain a crude product which was puiified by preparative TLC on silica gel using (50% EtOAc /hexanes) as mobile phase to provide the title compound (0.075 g, 51.7%). LCMS-X1 : 477.3 [M+H]+; t = 6.32 min. Ή NMR (400 MHz, DM80-d6) δ ppm 8.99-9.01 (d, 1 H), 7.38-7.40 (d, 2 H), 7.24-7.35 (m, 7 H), 7.17-7.20 (m, 2 H), 6.91-6.93 (d, 2 H), 6.07-6.10 (d, 1 H), 5.42-5.45 (t, 1 H), 4.93 (s, 2 H), 4.51-4.52 (d, 2 H), 3.47 (s, 2 H), 2.40 (s, 3H).
Example 12
A'-((4-cliloiOphenyl)(phenyl)methyl)-2-(4-((5-methy]isoxazol-3-yf)methoxy

To a. solution of A-((4-chlorophenyl)(phenyl)methyl)-2-(4-hydroxyphenyI)acetaniide (0.250 g, 0.710 mmol) in DMF (2.0 mL) was added 60% sodium hydride (0.034g, 0.85 mmol) at 0
°C and stirred for 20 min, 3-(chloromethyl)-5-methylisoxazole (0.1 12 g, 0.852 mmol) was added and stirred for lh. After completion of the reaction, the reaction mixture was poured into crushed ice and extracted with ethyl acetate (100 mL). The organic layer was dried over Na2S04 and concentrated and product was purified by silica gel column chromatography (40% EtOAc/hexanes) to provide the title compound (0.080 g, 25.23%). LCMS-Xl : 447.1 [M+H]+; R, = 5.60 min. !H MR (400 MHz, DMSO-d6) δ ppm 9.00-9.02 (d, 1 H), 7.38-7.40 (d, 2 H), 7.33-7.34 (d, 2 H), 7.24- 7.33 (m, 4 H), 7.1 8-7.20 (d, 2 H), 6.92-6.94 (d, 2 H), 6.31 (s, 1 H), 6.07-6.09 (d, 1 H), 5.10 (s, 2 H), 3.47 is, 2 H), 2.40 (s, 3 H). Example 13
N-((4-chlorophOTyl)(phenyJ)methy
methylacetamide

(a) 1 -(4-chlorophenyl)-N-methyl- 1 -phenyimethanamine
This compound was synthesized from (4-chloropheny3)(phenyl)methanamine
hydrochloride essentially as described in example 8 and was purified by silica gel column chromatography ( 10% EtOAc/hexane) to provide the title compound (0.1 1 g, 40.44%). ¾ NMR (400 MHz, DMSO-de) δ ppm 7.39-7.43 (t, 3 H), 7.32-7.37 (t, 4 H), 7.26-7.30 (t, 2 H), 7.1 6-7.22 (t, 1 H), 4.65 (s, 1 H), 2.12 (s, 3 H i.
(b) N-((4-chlorophenyl)(phenyl)methyl)~2-(4-((3,5-dimethylisoxazol-4-yl)methoxy)phenyl)-N- methylacetamide
This compound was synthesized from 2~(4-((3,5-dimetliylisoxazol~4- yl)methoxy)phenyl)acetic acid and 1 -(4-chlorophen l)-N-methyl- 1 -phenyimethanamine essentially as described in example 3 (e). (0.184 g, 50.68%). LCMS-Xl : 475.2 [M+H]+; Rt = 5.95 min. ¾ NMR (400 MHz, DMSO-d6) δ ppm 7.32-7.44 (m 6H), 7.04-7.17 (m, 6H), 6.91 -6.96 (m, 3H), 4.89 (s 2H), 3.77 (s, 2H), 2.75 (s, 2H), 2.39 (s, 3H) 2.20 (s 3H).
Example 14
2-(4-((3,5-dimethylisoxazol-4-yl)methoxy)phenyl)^

This compound was synthesized from 2-niethylbenzonitrile and phenylniagnesium chloride essentially as described in example 1 (e). (360 mg, 43%). LCMS-P1 : 1 81 [M-NH2] +; Rt: 1.604 mm. ιΉ NMR (500 MHz, CDC ) δ ppm 7.59 -7.18 (m, 9H), 5.41 (s, IH), 2.30 (s, M l ). 1.84 (s, 2H).
(b) 2-(4-hydroxyphenyl)-A -(phenyl(o-tolyl)methyl)acetamide
This compound was synthesized from 2-(4-hydroxyphenyl)acetic acid and phenyl(o- tolyl)methanamine essentially as described in example 4 (b) and this the title compound was used directly without further purification. LCMS-P 1 : 332 [M+H] ' ; Rt: 1.544 min.
(c) 2-(4-((3,5-dimethylisoxazol-4-yl)methoxy)phenyl)-N-(phenyl(o-tolyl)methyl)acetamide
To a stirred solution of 2-(4-hydroxyphenyl)-N-(phenyl(o-tolyl)methyl)acetamide (400 mg, 1.21 mmol) in 10 mL of DMF was added 4-(chloromethyl)-3,5-dimethylisoxazole (176 mg, 1.21 mmol), K2CO3 (334 mg, 2.42 mmol), and tetrabutylammonium iodide successively. The reaction mixture was stirred at rt overnight. Water (20 mL) was added and the reaction was extracted with ethyl acetate (30 mL><3). The combined organics were washed with brine and dried over a2S04. After removal of the organic solvent, the crude product was purified by silica gel column chromotography (petroleum ether/EtOAc = 3/1) to give 262 mg product (49%). The product was repurified by preparatory HPLC using 10- 100% water/acetonitrile with 0.1 % TFA to obtain the title compound (156 mg, 29%). LCMS-Pl : 441 [M+H]+; Rt: 1.696 min. XH NMR (500 MHz, CDCI3) δ ppm 7.29-6.91 (m, 1 1 i s. 6.40 (d, ./ 10.0 Hz, i ! Π. 5.96 i d. ./ 10.0 Hz, 1H), 4.78 (s, 2H), 3.59 (s, 2H), 2.40 (s, 3H), 2.29 (s, 3H), 2.24 (s, 3H).
Example 15
Ar-((4-chlorophenyl)(pheny3) l)acetarr)ide

(0.423 g, 1.20 mmol), in THF ( 1.0 mL) was added oxazol-5-ylmethanol (0.150 g, 1.50 mmol) and iriphenyiphosplime (0.409 g, 1.56 mmol) and the reaction was sonicated in a 33 -KHz sonicating bath at 0 °C until a clear solution was obtained. To the reaction mixture, di-isopropyl azodicarboxylate (0.315 g, 1 .56 mmol) was added dropwise over a period of 5 minutes at 0 °C and sonicated for 15 min. The THF was removed under vacumn and then remaining mixture was poured into water (20 mL) and extracted with diehloromethane ( 100 mL), The organic layer was dried over Na2S04 and concentrated and the product was purified by silica gel column
chromatography (40% EtOAc/hexanes) and again repurified by preparative TLC o silica gel (80% EtOAc/hexanes) to provide the title compound. (0.128 g, 24.6%). LCMS-X1 : 433.1 [M+I-I] ; Rt - 5.01 min. Π NMR (400 MHz, DMSO-d6) δ ppm 8.99-9.02 (d, 1 I I I 8.41 (s, 1 H), 7.38-7.40 (d, 2 H), 7.29-7.35 (m, 3 H), 7.24-7.27 (m, 5 H), 7.18-7.20 (d, 2 H), 6.94-6.96 (d, 2 H), 6.08-6.10 (d, 1 H), 5.14 (s, 2 H), 3.47 (s, 2 H).
Example 16
2-(((3,5-dimethylisoxazol-4-yl)methyl)(4-(2-oxo-2-((phenyl(j!?- toly])methyl)ammo)ethyl)phenyl)amino)acettc acid

(a) 2-(4-(((3,5-dimethylisoxazol-4-yl)methy^^
This compound was synthesized from 2-('4-(((3,5-dimethylisoxazol-4- yl)methyl)amino)pheny])acetic acid and heny3(p-tolyl)methanamii e essentially as described in example 4 (b) and purified by silica gel column chromatography using 0- 100% ethyl
acetate hexane (1 18 mg, 12%). LCMS-T 1 : 440.3 [M+H] "; ¾: 5.53 min.
(b) methyl 2-(((3,5-diraethylisoxazol-4-yl)raethyl)(4-(2-oxo-2-((phenyl( ?- to]yl)methyl)amino)ethyl)phenyr)amino)acetate

2-(4-(((3,5-dimethylisoxazol-4-yl)methyl)amino)phenyl)-N-(phenyl(p- tolyl)niethyl)acetamide ( 1 18 mg, 0.3 mmol), methyl 2-bromoacetate (41 mg, 0.3 mmol), tetrabutylammomum iodide (198 mg, 0.5 mmol), arid cesium carbonate (174 mg, 0.5 mmol) in DMF (lmL) were stirred overnight. The reaction was filtered to remove solids. Water was added and the solution was purified by reverse phase using 10-90% acetonitrile/water with 0.05% TFA to give the title compound (130 mg, 94%). LCMS-T1 : 512.2. [M+Hj+; Rt: 6.30 min.
(c) 2-(((3,5-dimethylisoxazol-4-y3)methyl)(4-(2-oxo-2-((phenyl( 7- tolyl)methyl)amino)ethyl)phenyl)amino)acetic acid
Methyl 2-(((3,5-dimethyiisoxazoi-4-yl)meihyi)(4-(2-oxo-2-((phenyi(p- toly])methyl)ammo)ethyl)phenyl)amino)acetate ( 130 mg, 0.3 mmol) was stirred for 3 hours in aqueous 1 M LiOH (1.5 mL) and dioxane (6 mL). The reaction was acidified and the solution was purified by reversed phase chromatography using 10-90% acetonitrile/water with 0.05% TFA to give the title compound (130 mg, 94%). LCMS-T1 : 498.3 [M+H ; R;: 5.74 min. Ή NMR (400 MHz, MeOH-d4) δ ppm 7.30-7.05 (m, 1 1H), 6.80 (d, 2H), 6.10 (t, 1H), 4.38 (s, 2H), 3.97 (s, 2H), 3.58 (s, 2H), 2.31 (s, 6H), 2.17 (s, 3H).
Example 17
N-(0 s(4-fluorophenyl)methyl)~2-(4-((3-(hydroxymethyl)-5-methyiisoxazoi-4- 2-methylpropanamide

This compound was synthesized from 2~(4-methoxyphenyI)acetic acid essentially as described in example 1 (a) (4.0 g, 92.37%). ¾ NMR (400 MHz, CDCL) δ ppm 7.17-7.19 (d, H), 6.87-6.89 (d, 2 H), 3.73 (s, 3 H), 3.60 (s, 5 H).
(b) methyl 2-(4-methoxypheny3)- -methylpropanoate

To a solution of methyl 2-(4-methoxyphenyl)acetate (4.0 g, 22.22 mmol) in THF (25 mL), was added iodomethane (9.45 g, 66.66 mmol) and the reaction mixture was cooled to -60 °C, followed by addition of potassium tert-butoxide (7.48 g, 66.66 mmol) portion wise. The reaction mixture was stirred for 30 minutes at the same temperature. After completion of the reaction, water (30 mL) was added into the reaction mixture at -60 °C and extracted with ethyl acetate (2 x 250 mL). The combined organic layers were dried over NaiSC and concentrated to obtain a crude product which was purified by silica gel column chromatography using 2.5% EtOAc/hexanes to provide the title compound (3,52 g, 75,59%).
(c) 2-(4-hydroxyphenyl)-2-met

A solution of methyl 2-(4-methoxyphenyl)-2-methylpropanoate (3.50 g, 6.80 mmol) in dichloromethane (20 mL) was cooled to -78 °C and boron tribromide was added (1.0 M solution in dichloromethane, 50.4 mL, 50.40 mmol) over 30 minutes. After completion of addition, the temperature was raised to 0 °C and the reaction mixture was stirred for 30 minutes. After completion of the reaction, water was slowly added to the reaction mixture. The organic layer was separated, dried over Na2S04j and concentrated to provide the title compound (1.4 g, 77.40%). Ί-Ι NMR (400 MHz, DMSO-d6) 8 ppm 12.10 (br, 1 i n. 9.35 (s, 1 H), 7.08-7.15 (dd, 2 H), 6.68-6.71 (dd, 2 H), 1.41 (s, 6 H).
(d) 6 s(4-fluorophenyj.)methanamine
This compound was synthesized from 4~fluorobenzoiiitrile and (4-fluorophenyl)magnesium bromide essentially as described in example 1 (e). (0.450 g, 12.46%). !H NMR (400 MHz, DMSO-d6) δ ppm 7.30-7.46 (m, 4 H), 7.03-7.16 (m, 4 H), 5.15 (s, 1 H).
(e) N-(& 5(4-fluorophenyl)methyl)-2-(4-hydroxyphenyl)-2-methylpropanam e
To a solution of 2-(4-hydroxyphenyl)-2-methylpropanoic acid (0. 8 g, 0.19 mmol) in dhnethylformamide (2 mL), EDC (0.228 g, 1.18 mmol), HOBt (0.18 Ig, 1.18 mmol), DMAP (0.242 g, 1.98 mmol) and 6 s(4-fluorophenyl)methanamine (0.2.18 g, 0.021 mmol) was added and the reaction mixture was stirred overnight at rt. After completion of the reaction, water (50 mL) was added to the reaction mixture and extracted with ethyl acetate ( 100 mL). The organic layer was dried over Na2S04 and concentrated io obtain a crude product which was purified by silica gel column chromatography ( 1.5% methanol in dichloromethane) to provide the title compound (200 mg, 52.49%). Ή ΜΙΙ (400 MHz, DMSO-d6) δ ppm 9.24-9.26 (d, 1 H), 7.99-8.02 (d, 1 H), 7.16- 7.19 (m, 4 i i ). 7.06-7.18 (m, 5 H), 6.66-6.68 (d, 2 H), 6.18-6.20 (d, 1 H), 1.43 (s, 6 H).
(f) ethyl 4-((4■( 1 ~(bis(4 -fluorophenyl)methylamino)-2 -methyl- 1 -oxopropan-2 -yl)phenoxy)methyl)- 5-methylisoxazole-3-carboxylate
This compound was synthesized from N-(Ws(4-fluorophenyl)methyl)-2-(4- hydroxyphenyl)-2-methylpropanamide and ethyl 4-(chloromethyl)-5-methylisoxazole-3- carboxylate essentially as described in example 11(a) (0.200 g, 69.7%) as a light yellow semi solid,
(g) N-(6w(4-fluorophenyl)methyl)-2- yl)methoxy)phenyl)-2-methylpropanamide
This compound was synthesized from ethyl 4-((4-(l-(6/s(4-fluorophenyl)methylamino)-2- methyi- 1 -oxopropan-2-yl)phenoxy)methyi)-5-methylisoxazole-3-carboxylate essentially as described in example 1 1 (b). (44 mg, 23.8%). LCMS-X1 : 507.6 [M+Hf; Rt = 6.59 rain. !H NMR (400 MHz, DMSO-d6) δ ppm 8.07-8.10 (d, 1 H), 7.15-7.21 (m, 6 H), 7.09-7.13 (m, 4 H), 6.91 -6.93 (d, 2 H), 6.18-6.20 (d, 1 H), 5.43-5.45 (t, 1 H), 4.96 (s, 2 H), 4.52-4.53 id, 2 H), 2.40 (s, 3 H), 1.46 (s, 6 H).
Example 18
N-((4-clilorophenyl)(phenyl)methyi)-2-(4-((3,5-dimethylisoxazol-4-yl)methoxy)phenyl)-2- methylpropanamide


To a solution of methyl 2-(4-((3, 5~dimethylisoxazol-4~y{)methoxy)phenyl)acetate(0.5 g, 1.818 mmol) in THF (25 mL) at -78 °C, iodomethane (0.34 mL, 5.454 mmol) was added dropwise. To this solution, potassium fcrt-butoxide (0.627 g, 5.454 mmol) was added portion wise over 30 min and the reaction was stirred at -78 °C for Ih followed by rt for another one hour. After completion of the reaction, the reaction mixture was quenched by addition of ammonium chloride solution in water (25 mL) and extracted with (2 X 50 mL) ethyl acetate. The organic layers were dried (Na2SC>4) and concentrated to obtain crude the title compound (0.60 g, 91.66%). MS (ESI+) 304.2 (M + H),
(b) 2-(4-((3,5-dimethylisoxazol-4-yl)methoxy)phenyl)-2-methy3propanoic acid
This compound was synthesized from methyl 2-(4-((3,5-dimethylisoxazol-4- yl)methoxy)phenyl)-2-methyrpropanoate essentially as described in example 1 (c). (0.350 g, 61.2%). MS (ESI+) 290.2 (M + H). lH NMR (400 MHz, DMSO-d6) δ ppm 12.33 (s, 1H), 7.26- 7.28 (d, 2H), 6.94-6.97 (d, 2H), 4.88 (s, 2H), 2.39 (s, 3 H), 2.20 (s, 3 H), 1.42 (s, 6 H).
(c) N-((4-chlorophenyl)(phenyl)methyl)-2-(4-((3,5-dimethylisoxazol-4-yl)methoxy)phenyl)-2 methy Ipr opanamide
To a solution of 2-(4-((3,5-dimet ylisoxazol-4-yl)methoxy)pheny])-2-methylpropanoic acid (0.20 g, 0.692 mmol) in tetrahydrofuran (35 mL), EDC (0.2 g, 1.41 mmol) was added portion wise and the reaction was stirred at rt for 2 h. (4-chlorophenyl)(phenyl)methanamine
hydrochloride (0.21 g, 0,826 mmol), HOBt (0.105 g, 0.686 mmol), and triethylaniine (0.287 mL, 2.074 mmol) were then added, and the reaction mixture was stirred for 24 h at rt. After completion of the reaction, the reaction mixture was quenched by addition of water (50 ml.) and extracted with 2 X 50 mL ethyl acetate. The organic layers were dried (Na2S04) and concentrated to obtain a crude product which was purified by silica gel column chromatography (35%
EtOAc/hexane) to provide the title compound (0.074 g, 21.95%). MS (ES1+) 489.3 (M + H). Ή NMR (400 MHz, CDC13) δ ppm 7.25-7.31 (m, 8 H), 7.03-7.04 (m, 4 H), 6.92-6.99(m, 2 H), 6.16- 6.18 (d, 1 H), 5.72-5.74 (d, 1 H), 4.80 is, 2 H), 2.43 (s, 3 H), 2.31 (s, 3 H), 1.59- 1.63 (d 6H).
Example 1
2-(4-((3,5-diisopropylisoxazol-4-yl)rr)ethoxy)phenyl)-N-((2,4- dim ethylpheny l)(phenyl)me

(a) ethyl 2-isobutvT d-4-methyl-3-oxopeiitanoate

A solution of ethyl 4-methyl-3-oxopentatioate (5.00 g, 31.6 mmol) in dry benzene (5.0 niL) was added to a mixture of a-metal (0.80 g, 34.7 mmol) in dry benzene (50 mL) dropwise within 10 minutes at rt. After completion of the addition, the reaction mixture was stirred at rt until Na metal was dissolved and the reaction was refluxed for 1 h. After 1 h, the reaction mixture was cooled to 0 °C and isobutyryl chloride (3.70 g 34.7 mmol) was added dropwise at 0 °C. The reaction mixture was stirred at 0 °C for 5 min. and then heated to reflux and stirred at reflux for 3 h. The reaction mixture was then cooled to rt and stirred overnight. After completion of the reaction, water was added and the reaction was extracted with ethyl acetate (2 X 250 mL). The combined organic layers were dried using ajSC and concentrated. The compound was purified by silica gel column chromatography using 100% hexanes to provide the title compound (3.52 g 48.88%).
(b) ethyl 3, 5-diisopropylisoxazoie-4-c

To a solution of ethyl 2-isobutyryl-4-methyj-3-oxopentanoate (2.80 g, 12.28 mmol) in Ethaiiol (10 mL) at 25 °C was added hydroxy] amine hydrochloride (1.49 g, 21.49 mmol) in water (5 mL) and the resulting mixture was refluxed for 1 h. After i h, water (10 mL) was added into the reaction mixture and the reaction mixture was extracted with diethyl ether (3 X 50 mL). The combined organic layers were washed with 1 NaOH solution (3 X 10 mL). The organic layer was dried using Na2S04 and concentrated. The compound was purified by silica gel column
chromatography eiuting 2% EtOAC:hexanes to provide the title compound (1,80 g 65.21%), Ή NMR (400 MHz, CDC13) δ ppm 4.31 -4.36 (q, 2 H), 3.73-3.80 (m, 1 H), 3.42-3.49 (m, 1 H), 1.27- 1.38 (m, 15 H). (c) (3,5-diisopropylisoxazol-4-yl)methan

To a solution of ethyl 3, 5-diisopropylisoxazole-4-carboxylate ( 1.7 g 7.55 mmol) in anhydrous tetrahydrofuran ( 15 mL) was added lithium aluminum hydride (1.0 M solution in THF, 7.5 ml., 7.55 mmol) at 0 °C over 30 minutes, and the reaction mixture was stirred at 0 °C for 90 minutes. The reaction mixture was quenched with water, and extracted with ethyl acetate (2 X 50 mL). The combined organic layers were dried using Na2SC>4 and concentrated. The compound was purified by silica gel column chromatography eiuting 15% EtOAc/hexanes to provide the title compound (1.2 g, 86.95%), TH NMR (400 MHz, DMSO-d6) δ ppm 4.90-4.93 (t, 1 H), 4.29-4.30 (d, 2 H), 3.18-3.25, (m, 1 H), 3.00 3.07 (m, 1 H), 1.21 -1.25 (m, 12 H).
(d) 4-(chloromethyl)-3,5-diisopropylisoxazole
This compound was synthesized from (3,5-diisopropylisoxazol-4-yl)methan.ol essentially as described in example 10(d). (1.0 g, 76.33%), ¾ NMR (400 MHz, DMSO-de) δ ppm 4.45 (s, 2 H), 3.16-3.22, (m, 1 IT), 3.03 3.10 (m, 1 H), 1.29-1.39 (m, 12 IT).
(e) methyl 2-(4-((3,5-diisopropylisoxazol-4-yl)methoxy)phenyl)acetate
This compound was synthesized from methyl 2-(4-hydroxyphenyl)acetate and 4- (chloromethyl)-3,5-diisopropylisoxazole essentially as described in example 1 1(a). (0.300 g, 60.4%).
(f) 2-(4-((3,5-diisopropylisoxazol-4-yl)methoxy)phenyl)acetic acid
This compound was synthesized from methyl 2-(4-((3,5-diisopropylisoxazol-4- yl methoxy )phenyl)acetate essentially as described in ex ample 1 (c). (0.215 g, 74.91%), 1 ! \ M R (400 MHz, DMSO-de) δ ppm 12.25 (br, 1 H), 7.18-7.20 (d, 2 H), 6.95-6.97 (d, 2 IT), 4.98 (s, 2 H), 3.49 (s, 2 H), 3.25-3.32 (m, 1 H), 3.01-3.08 (m, 1 H), 1 .21- 1.30(m, 12 H).
(g) 2-(4-((3,5-diisopropylisoxazol-4-yl)methoxy)pheny{)-N-((2,4- dimethylpheny3)(phenyl)methyi)acetamide
This compound was synthesized from 2-(4-((3,5-diisopropyrisoxazol-4- yl)methoxy)phenyl)acetic acid and (2,4-dimethylphenyl)(phenyi)methanamine essentially as described in example 3 (c). (56 mg, 35,00%), LCMS-X1 : 51 1.4 [M+H]+; t = 5,99 min. Ή MR (400 MHz, DMSO-d6) δ ppm 8.83-8.85 (d, 1 H), 7.29.-733 (m, 2 H), 7.15-7.26 (m, 5 H), 6.92-6.99 l i!i. 5 H), 6.17-6.19 i d. 1 H), 4.88 (s, 2 U s. 3.35 (s, 2 H i. 3.24-3.33 (m, 1 H), 3.00-3.07 (m, 1 H), 2.15 (s, 3 H), 2,20 (s, 3 H), 1.20- 1.24 (m, 12 H). Example 20
N-(4-((3,5-dimethylisQxazol-4-y!)m
phenylacetamide
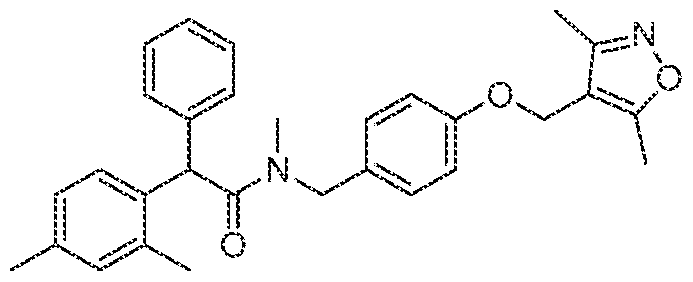

The solution of mandelic acid (10.0 g, 65.7 mmoJ) in w-xylene (56.79 mL, 460 mmol) was heated to 60-70 °C followed by addition of SnCl4 (1 1.5 mL, 98,6 mmol) over 2 h. The reaction mixture was then cooled to rt and stirred for 6 h at rt. The completion of the reaction was monitored by TLC on silica gel using exanes: EtOAc (1 : 1). After completion of the reaction, iced-water (100 mL) was added into the reaction mixture, and the mixiure was extracted with diethyl ether (3 x 250 mL). The combined organic layers were discarded. The remaining reaction mixiure was extracted with 8% aqueous Na2C03 (10 x 50 mL) and the combined aqueous layer was then acidified using 6 N HCl (20 mL) and the solid obtained was filtered and dried. The crude solid product was purified using silica gel column chromatography using 20%
EtOAc/hexanes to obtain the title compound (6.25 g, 39.63%) as a solid. XH NMR (400 MHz, DMSO-de) δ ppm 12.6 (s, 1 H), 7.21-7.33 (m, 6 H), 7.09-7.12 (m, 1 H), 6.97-7.06 (m, 3 H), 5.12 i H), 2.23 (s, 3 H), 2.17-2.18 (d, 4 H).
(b) 2-(2,4-dimethylphenyl)-A'-C4-hydroxybenzyl)-2-phenylacetamide.
To a solution of 2-(2,4-dimethylphenyl)-2-phenylacetic acid(0.25 g, 1.04 mmol) in anhydrous DMF (10 mL) at 25 °C was added EDC (0.237 g, 1.24 mmol) and the reaction mixture was stirred for 30 min at it under argon atmosphere followed by addition of HOBi (0.19 g, 1.24 mmol), 4-dimethylamino pyridine (0. 9 g, 1.56 mmol) and 4-(ammomethyl)p enol (0.154 g, 1 .24 mmol), and the reaction mixture was stirred overnight under argon atmosphere. The raction mixture was cooled to 0 °C. Water ( 10 mL) was added and the solid precipitate was filtered, washed with hexanes (2 x 10 mL), and dried under vacuum to provide ihe title compound (0.065 g,
18.10%) 'H NMR (400 MHz, DMSO-d6) δ ppm 9.29 (s, 1 H), 8.55-8.58 (t, 1 H), 7.26-7.30 (t 2 H), 7.16-7.20 (m, 4 H), 7.00-7.02 (d, 2 H), 6.93-6.95 (d, 2 H), 6.65-6.68 (d, 2 H), 5.07 (s, i H), 4.1 1- 4.22 (m, 2 H), 2.22 (s, 3 H), 2.15 (s, 3 H).
(c) N-(4-((3,5-dimethylisoxazol- -yi)methoxy)^^

To a solution of 2-(2,4-dimeihyiphenyl)-A'-(4-hydroxybenzyl)-2-phenylaceiamide (0.06 g,
0.17 mmol) in acetonitrile (3 mL) was added cesium carbonate (0. 7 g, 0.52 mmol) and 4- (chloromethy])-3,5-dimethyfisoxazofe (0,03 g, 0.21 mmol). The resulting mixture was heated at 70 °C and stirred for 2 h in a sealed-tube. After completion of the reaction, water ( 10 mL) was added, and ihe mixture was extracted with ethyl acetate (2 x 15 mL). The combined organic layers were dried over a2S04 and evaporated under vacuum to obtain crade product which was purified by preparatory TLC on silica gel to provide the title compound (50, 1 mg, 63.57%) ]H NMR (400 MHz, DMSO-d6) δ ppm 8.61-8.64 (s, 1 H), 7.26-7.30 (t, 2 H), 7.14-7.23 (m, 5 H), 5.08 is, 1 H), 4.88 (s, 2 H), 4.21-4.24 (t, 2 H), 2.38 (s, 3 H), 2.15-2.22 (i, 9 H). (d) N-(4-((3,5-dimethylisoxazol-4-yl)m
phenylace tami de

Example 21
N-((4-chlorophenyl)(phenyl)me^
yl)acetamide

(a) methyl 5-hydroxypicolinate

To a solution of 5-hydroxypieolinic acid (4.0 g, 28.77 mmol) in methanol (75 mL), cone. H2SO4 (5 mL) was added dropwise at 0 °C and the reaction mixture was ref!uxed at 80 °C for 6 h. Methanol was removed under reduced pressure. The residue was dissolved in water and neutralized by sodium bicarbonate solution. Product was extracted with (3 X 50 mL) ethyl acetate. The combined organic layers were dried over Na2S04 and concentrated to provide the title compound (2.10 g, 47.72%). MS (ESI+) 1 54.08 (M + H). i l NMR (400 MHz, DMSO-d6) δ ppm 10.86 (s, 1 H), 8.20 (s, i H), 7.93-7.98 (d, 1 H), 7.25-7.28 (s, 1 H), 3.81 (s, 3 H).
(b) methyl 5-((3,5-dimethylisoxazol-4-yl)methoxy)picolinate
To a solution of methyl 5-hydroxypicoiinate (3.0 g, 19.60 mmol) in dimethyiformamide (35 mL) was added potassium carbonate (4.05 g, 29.41 mmol) and the reaction was stirred at rt for 30 min. 4-(chloromethyl)-3,5-dimethylisoxazole (3.41 g, 23.52 mmol) was added and the reaction mixture was stirred at 80 °C for 6 h. After completion of the reaction, the reaction mixture was quenched by addition of water (50 mL) and the mixture was extracted with 2 X 50 mL ethyl acetate. The organic layers were dried (Na2Si¾) and concentrated to obtain a crude product which was purified by silica gel column chromatography (30% EtOAc/hexanes) to provide the title compound (3.9 g, 76.47%). MS (ESI+) 263.00 (M + H). ]H NMR (400 MHz, DM80-d6) δ ppm 8.43 (s, 1 H), 8.07-8.09 (d, 1 H), 7.60-7.63 (d, 1 H), 5.1 1 (s, 2 ! i ) 3.85 (s, 3 H), 2.43 (s, 3 H), 2.23 (s, 3 l i s.
(c) (5-((3,5-dimeihyIisoxazol-4-yl)inethoxy)pyridin-2-yl)methanoI
To a solution of methyl 5-((3,5-dimemyiisoxazol-4-yl)methoxy) picoiinate(3.80 g, 14.50 mmol) in methanol (150 mL), sodium borohydride (10.74 g, 290.07 mmol) was added portion wise at 0 °C and stirred at rt for 3 h. After completion of the reaction, methanol was evaporated and diluted with water. The product was extracted with 2 X 100 mL ethyl acetate. The combined organic layers were dried over Na2SG4 and concentrated to obtain the title compound (3.1 g, 91.44%). f i NMR (400 MHz, DMSO-d6) δ ppm 8.23 (s, 1 H i. 7.46-7.48 (s, 1 H), 7.38-7.40 (s, 1 H), 5.31-5.34 (t, 1 i n. 4.99 (s, 2 H), 4.48 (s, 2 I D. 2.40 (s 3! i s. 2.21 (s, 3H).
(d) 4-(((6-(chloromethyl)pyridin-3-yl)oxy)methyl)-3,5-dimethylisoxazole
This compound was synthesized from (5-((3,5-dimethylisoxazol-4-yl)methoxy)pyridin-2- yl)methanol essentially as described in example 10(d). (3.5 g, 94.59%). MS (ESI+) 253.00 [M + H], Ή NMR (400 MHz, DMSO-d6) δ ppm 8.40 (s, 1 H), 7.61-7.67 (m, 2 H), 5.06 (s, 2 H), 4.81 (s, 2 H), 2.42 (s, 3 H), 2.22 (s, 3 H).
(e) 2-(5-((3,5-dimethylisoxazol-4-yl)methoxy)pyridin-2-yl)acetonitrile
To a solution of 4-(((6-(chloromethyl)pyridin-3-yl)oxy)methyl)-3,5-dimethylisoxazole (1.0 g, 3.968 mmol) in methanol (35 mL) and water mixture was added potassium iodide (0,05 g, 0.30 mmol) and sodium cyanide (0.388 g, 7.936 mmol). Reaction was refSuxed at 80°C for 4 h. After completion of the reaction, the methanol was evaporated. The reaction mixture was quenched by addition of water (50 mL) and extracted it with ethyl acetate (2 X 25 mL). The combined organic layers were dried over Na2S04and concentrated to provide the title compound which was used as crude in the next step, (0.90 g, 93.75%). MS (ES1+) 244.10 (M + H),
(f) 2-(5-((3,5-dimethylisoxazol-4-y{)methoxy)pyridin-2-yl)acetic acid
To a solution of 2-(5-((3,5-dimethyiisoxazol-4-yl)methoxy)pyrid n-2-yl)acetonitrile (0.90 g, 3.70 mmol) in methanol (35 mL), sodium hydroxide (0.444 g, 1 1.1 mmol) in water (50 mL) was added dropwise and the reaction was refluxed at 80 °C for 6 h. After completion of the reaction, the methanol was distilled off at reduced pressure and water (50 ml.) was added to the reaction mixture. The aqueous layer was washed with ethyl acetate and then the aqueous layer was acidified to pH=3 using 10% Hydrochloric acid and extracted with ethyl acetate (2 X 50 mL). The combined organic layers were dried over NaiSC and concentrated to provide the title compound (0.160 g, 16.49%) MS (ESI+) 263.1 (M + H).
(g) N-((4-chlorophenyl)(phenyl)methy1)-2-(5-((3,5-dimethyli
yljacetamide
To a solution of 2-(5-((3,5-diniethylisoxazol-4-yl)niethoxy)pyridin-2-yl)a.cetic acid (0, 150 g 0.572 mmol) in tetrahydrofuran (25 mL), EDC (0.164 g, 0.858 mmol) was added portion wise and the reaction was stirred at rt for 2 h. (4-chforophenyl) (phenyl) methanamine hydrochloride (0.200 g, 0.858 mmol), HOBt (0.100 g, 0.572 mmol), and triethylamine (0.237 mL, 1.751 mmol) were then added and the reaction mixture was stirred for 24 h at rt. After completion of the reaction, water (50 mL) was added to the reaction mixture and extracted with ethyl acetate (2 X 50 mL). The combined organic layers were dried over 2S04 and concentrated to obtain a crude product which was purified by silica gel column chromatography using (30% EtOAc/hexanes) as mobile phase to provide the title compound (0.130 g, 49.24%). MS (ESI+) 462.34 [M + ITj. Ή NMR (400 MHz, DMSO-d6) δ ppm 9.05 (d, 1 H), 8.24 (s, 1 H), 7.33-7.42 (m, 3 H), 7.24-7.32 (m, 7 H), 6.10-6.12 (d, 1 H), 4.98 (s, 2 H), 3.69 (s, 2 H), 2.40 (s, 3 H), 2.18 (s, 3 H). Example 22
N-((4-chlorophenyl)(phenyl)methyl)-2-(4-((3,5-dimethylisoxazol-4- yl)methylthio)phenyl)acetami


To a solution of 2-(4-mercaptophenyl)acetic acid (0.25 g, 1,40 mmol) in DMF (3 mL) at 25 °C was added 4-(chlorophenyl)-3,5-dimethylisoxazole (0.227 g, 1.5 mmol) followed by addition of potassium hydroxide (0.208 g, 3.7 mmol) in two drops of water. After completion of the addition, the reaction mixture was heated to 120 "C for 2 h. After completion of the reaction, the reaction mixture was cooled to rt, water (10 mL) was added and the mixture was acidified to pH=2 using 10% aqueous HCJ, The aqueous layer then was extracted with ethyl acetate (2 x 25 mL). The combine organic layers were washed with brine (25 mL), dried over (N^SC , and evaporated to obtain the title compound as a light yellow oil (0.46 g) which was used in the next step without further purification. Ή NMR (400 MHz, DM80-ci6) δ ppm 12.45 (br, 1 H), 7.19-7.29 (m, 4 H), 3.93 (s, 2 H), 3.55 (s, 2 H), 2.14 (s, 3 H), 2.04 (s, 3 H). (b) N-((4-chlorophenyl)(phenyl)methyl)-2-(4-((3,5-dimethylisoxazol-4- yl)methylthio)phenyi)acetamide
To a solution of 2-(4-((3,5-dimethylisoxazol-4-y])methylthio)phenyl)acetic acid (0.460 g, 1.6 mmol) in DMF (5 mL) at 25 °C was added HOBt (0.304 g, 1.8 mmol), EDC (0.382 g, 1.9 mmol), and DMAP (0.405 g, 3.2 mmol) and the resulting mixture was stirred at rt for 10 minutes. (4-chlorophenyl)(phenyl)methanamine hydrochloride (0.466 g, 1.8 mmol) in DMF ( 1 mL) was added slowly and the stirring at rt was continued overnight. After completion of the reaction, water (10 mL) was added into the reaction mixture and extracted with ethyl acetate (2 X 25 mL). The combined organic layers were washed with brine (10 mL), dried over(Na2S04), and evaporated to obtain a crude product which was purified using silica gel column chromatography using 24% ethyl acetate :hexanes and then purified using prep TLC on silica gel (50% ethyl acetate in hexanes -r 5 drops of acetic acid) to obtain the title compound (66 mg, 16,68%). LCMS-X1 : 477,2
[M+H]+; R = 3.61 min. !H NMR (400 MHz, CDC13) δ ppm 7.28-7.35 (m, 6 H), 7,21-7,23 (d, 2 i i ). 7.06-7.1 i (m, 4 H), 6.21-6.23 (d, 1 H), 5.93-5.94 (d, i H), 3.76 (s, 2 H), 3.63 (s, 2 H), 2.24 (s, 3 H), 2.01 (s, 3 H).
Example 23
N-((4-chloro-2-methylplienyi)(phenyl)meth
i-yl)acet amide

(a) tert-butyl 4-(2~(3,5~dimethylisoxazol-4-yl)etliyl)piperazine- 1 -carboxviate
To the solution of 4-(2-e3i3oroemyi)-3,5-dimethylisoxazole (159 mg, I mmol) in MeCN (5 mL) was added tert-butyl piperazine- 1 -carboxviate (205 mg, 1 . Immol), then potassium bicarbonate (200 mg, 2 mmol) and potassium iodide (34mg, 0.2mmol). Then the resulting mixture was heated to reflux overnight. After cooling to rt, the mixture was extracted with EtOAc. The combined organic layers were washed with brine and dried over Na2S04. A fter removal of solvent under reduced pressure, the residue was purified by silica gel column chromotography with
EtOAc:petroleum ether=2: l to afford the title compound as a yellow solid ( 189 mg, 61%).
LCMS-P1 : 310, I m · ί 11 .
(b) 3,5-dimethy]-4-(2-(piperazm- 1 -y3)ethy3)isoxazo3e hydrochloride
To the solution of tert-butyl 4-(2-(3,5-dimethylisoxazol-4-yl)ethyl)piperazine- l- carboxylate (189 mg, Immol) in MeOH (5mL) was added HCT/Et20 (1M, 2.5mL, 2.5mmol), then the mixture was stirred at rt overnight. The reaction mixture was evaporated under reduced pressure, the crude the title compound (140mg, yield 97.5%) was used directly to the next step without further purification. LCMS-P 1 : 210 [M- H]+;
(c) methyl 2-(4-(2-(3,5-dimethyiisoxazol-4-yl)etliyl)piperazin- 1 -yl)acetate
the mixture of 3,5-dhnethyl-4-(2-(piperazin-l -y3)ethyl)isoxazole liydrochloride (140m 0.57mmol) in DMF (3mL) was added K2CO3 (236mg, 1.7 Immol) and methyl 2-bromoacetate ( 105 mg, 0,68mmol), then the mixture was heated at 60 °C overnight under argon. The reaction mixture was cooled to rt and poured into water. The mixture was extracted with EtOAc. The combined organic layers were washed with brine and dried over Na2S04. After evaporation of solvent under reduced pressure, the residue was purified by flash chromotography to give the title compound as light-yellow oil (97nig, yield 60%). LCMS-P1 : 282 [M+H]+;
86
(d) 2-(4-(2-(3,5-dimethylisoxazol-4-yl)ethyl)piperazin- -yl)aeetic acid
To the suspension of methyl 2-(4-(2-(3,5-dimethylisoxazol-4-yl)ethyf)piperazin-l- yliacetate (97mg, 0.345mmol) in THF/Water (4mL, 1 : 1) was added LiOH (29 mg, 0.69 mmol), and the resulting mixture was stirred at rt for 30 min. The reaction mixture was neutralized by HC1 (1M). After removal of all the solvent, the crude the title compound was used directly to the next step without further purification (1 10 mg). LCMS-P1 : 268 [ M i l !
(e) N-((4-chloro-2-memylphenyl)(phenyl)methyI)-2-(4-(2-(3,5-dimethylisoxazol-4- yl)ethyl)piperazin- 1 -yl)acetamide
To the solution of 2-(4-(2-(3,5-dimethylisoxazol-4-yl)ethy{)piperazin- l-yl)acetic acid (1 10 mg, 0,345 mmol) in DMF (3 mL) was added (4-chloiO-2-methylphenyi)(pheiiyl)methanamine (1 1 1 mg, 0.414 mmol), EDC (79 mg, 0.414 mmol), HOBt (56 mg, 0.414 mmol), then DIPEA (89 mg, 0.69 mmol). After stirring at 45 "C overnight, the mixture was poured into water, and extracted with EtOAc (15 mL X 2). The combined organic layers were washed with brine and dried over Na2Si)4. After removal of solvent, the residue was purified by preparatory HPLC using 10- 100% water/acetonitrile with 0.1% TFA to afford the the title compound as a white solid (20 mg, yield 12%). LCMS-P1 : 4S I | \1 - I I | . i i NMR (400 MHz, DMSO-d6) δ ppm 8.48 i d. ./ 8Hz, IH), 7.34 (t, J= 0.76 Hz, 2H), 7.29-7.25 (m, 3H), 7.20-7.15 (m, 3H), 6.23 (d, J = 8Hz, 1H), 3.02 (s, 2H), 2.43-2.30 (m 12H), 2.27 (s, 3H), 2.23 (s, 3H), 2.14 (s 3H).
Example 24
methyl 2-((3,5-dimethylisoxazol-4-yl)methoxy)-5-(2.-(((2,4- dimethylphenyl)(pheiiyl)methy
(a) benzyl 2-(4-hydroxyphenyl

Sodium hydride was suspended in 100 ml of DMF and 2-(4-hydroxyphenyi)acetic acid (9 g, 39.4 mmol) was added potionwise to the stirred mixture at 0 °C. After 30 minutes, benzyl bromide (30 ml) was added dropwise in 1 h. The mixture was diluted with water (100 mL) and
extracted with EtOAc (100 mL¾ 3). The combined organic solvents were dried over Isla^SO^ After removal of solvent, the crude compound was purified by column chromatography on silica gel (EtOAc: petroleum ether = 1 : 5) to provide the title compound (8.7 g, 91.2%). LCMS-P1: 243.0 ί 1 i 1 1 : R- 1.85 min,
(b) benzyl 2-(3-fonnyl-4

To a solution of benzyl 2-(4-hydroxyphenyl)acetate (4.84 g, 20 mmof) in CH3CN (150 mL) was added MgC (3.8 g, 40 mmoi) and triethylamine (8.08g, 80 mmol) under nitrogen and the mixture was refluxed for lh. Then (O (·() · . was added and the reaction was refiuxeci overnight. After cooling to rt, ether (200 mL) and ΓΜ HCl (100 mL) were added. The organic layer was separated and washed with 1 M HCl (100 mL X 3), and dried over Na2SC)4. After removal of solvent, the residue was purified by silica gel column chromotography (petroleum
ether/EtOAc=5/l) to obtain the title compound (2.71 g, 50%). LCMS-P1 : 271.1 ; \1 i i ! : Rt : 1.93 min
(c) benzyl 2-(4-((3,5- enyl)acetate

To a solution of benzyl 2-(3-formyl-4-hydroxyphenyl)acetate (L35g, 10 mmol) in MeCN (50mL) were added 4-(chloromethyl)-3,5-dimethylisoxazole (0.73g,10 mmol), K3CO3 (0,74 g, 15 mmol) and potassium iodide (0.41g, 5 mmol). The reaction mixture was refluxed overnight. After the completion of the reaction, the mixture was diluted with water (30 mL) and extracted with EtOAc (20 mL* 3). The combined organic layers were dried over Na2S04. After removal of the solvent, the crude product was purified by column chromatography on silica gel (EtOAc: petroleum ether = 2 : 1) to provide the title compound (1.5 g, 41%). LCMS-Pl : 380.0 [M+H]+; R. = 1.72 min,
(d) 5-(2-(benzyloxy)-2-o ethoxy)beiizoic acid

To a solution of benzyl 2-(4-((3 ,5-dimet y]isoxazoj-4-yl)met oxy)-3-formylphenyl)acetate (0.6 g, 1.56 mmol) in DMSO (15 mL) were added sodium dibydrogen phosphate (0.48 g, 3.96 mmol, dissolved in 4 mL water) and Sodium chlorite (0.29 g, 3.12 mmol in 8 ml water). The reaction mixture was stirred at rt overnight. Then sat. aq. Na2CO;, (20 ml) was added in 5 min. The mixture was diluted with water (30 mL) and extracted with EtOAe (20 mL* 3). The combined organic solvents were dried over Na2S04. After removal of solvent, the crude compound was purified by column chromatography on silica gel (EtOAe: petroleum ether = 1 : 1) to provide the title compound (430mg, 69.8%). LCMS-P1 : 396.1 [M+H] "; Rt - 1.57 min
(e) methyl 5-(2-(benzylo -2-oxoethy])-2-((3,5-dimethylisoxazol-4-y])methoxy)beiizoate

To a solution of 5-(2-(benzyloxy)-2-oxoethyl)-2-((3,5-dimethylisoxazol-4- yl)metboxy)benzoic acid (0.2 g, 0.51 mmol) in DMF (1 5 ml.) were added iodomethane (72 mg,
0.51 mmol) and KJCOJ (140 mg, 1 .02 mmol). The reaction mixture was stirred at rt overnight.
Then sat, aq. Na2C03 (20 ml) was added over 5 min. The mixture was diluted with water (30 mL) and extracted with EtOAe (2.0 mL* 3). The combined organic solvents were dried over Na2S04.
After removal of solvent, the crude compound was purified by column chromatography on silica gel (EtOAe: petroleum ether = i : 3) to provide the title compound (1 60 mg, 76.7%). LCMS-P1:
410.1 [M+H] 4'; Rt = 1.99 min
(f) 2-(4-((3,5-dimethylisoxazol-4-yl)methoxy)-3-(methoxycarbonyl)ph.enyl)acetic acid

(g) methyl 2-((3,5-dimethylisoxazol-4-yl)methoxy)-5-(2-(((2,4- dimethylpheny])(p enyl)methyl)amino)-2-oxoethy])benzoate
This compound was synthesized from 2-(4-((3,5-dimethylisoxazol-4-y3)methoxy)-3- (methoxycarbonyl)phenyl)acetic acid and (2,4-dimethyiphenyl)(phenyi)methanamine essentially as described in example 4 (b) and was purified by column chromatography on silica gel (EtOAc: petroleum ether = 1 : 3) to provide the title compound (25mg, 40.7%). LCMS-P1 : 535.0
[M+Na]+; ¾ - 1.83 min. ]H NMR (400 MHz, DMSO-d6) δ ppm 7.65 (d, J= 2.0 Hz, 1 H), 7.36 (dd, .1 2.4 Hz, i l l ). 7.20 fs, 2H), 7.18 (s, 1H), 7.03 (d, ,/ 6.8 Hz, 2H), 6.93 ft, J= 6.0 Hz, 2H), 6.87 (d, J = 8.0 Hz, IH), 6.76 (d, J= 8.0 Hz, 1H), 6.30 (d, . 8.0 Hz, U I s . 5.85 i d. ./ 8.0 Hz, 1H), 4.80 (s, 2H), 3.77 (3, 3H), 3.51 (s, 2H), 2.32 (s, 3H), 2.25 (s, 3H), 2.22 (s, 3H), 2.15 (s, 3H). Example 25
2-(4-(2-(3,5-dimethylisoxazol-4-yl)-2-hydroxyethoxy)phenyl)-N-((2,4- dimethylphenyl)(phenyl)m
(a) methyl 3,5-dimethyliso

To a solution of 3,5-dimethylisoxazole-4-carboxy3ic acid (9.2 g, 65.2 mmol) in MeOH (50 mL) was added SOCl2 (15.3 g, 130.4 mmol) very slowly. The reaction mixture was heated to 70 °C overnight. The reaction mixture was then cooled to rt, concentrated, and purified by column
chromatography (10% EtO Ac/petroleum ether) to afford the title compound (9.0 g, 89%), LCMS- Pl : 156 | \] i l l : R:: 1.404 min.
(b)(3,5-dimethylisoxazol-4-yl)methanol

To a stirred solution of methyl 3,5-dimethylisoxazole-4-carboxylate (9.0 g, 58 mmol) in THF (200 mL) at 0 °C was added LiAH¾ (2.42 g, 63.8 mmol) portionwise. After addition, the reaciion mixture was allowed to warm up to rt and stirred overnight. The reaction mixture was quenched by the successive addition of water (2.5 mL), 10% aq. NaOH (5 mL), and water (7,5 mL). The organic layer was separated, dried over Na2S04, filtered, and concentrated to afford the title compound (5.0 g, 68%). LCMS-P1 : 128 [M+H ; Rt: 0.963 min. lH NMR (400 MHz, CDCL) δ ppm 4.37 (s, 2H), 2.30 (s, 3H), 2.20 (s, 3H).
(c) 3,5-dimethyiisoxazole-4-carbaldebyd

To a solution of (3,5-dimethylisoxazol-4-yl)methano3 (1.00 g, 7.86 mmol) in CH2CI2 (20 mL) at 0 °C was added Dess-Martin periodinane (4.17 g, 9.83 mmol) over 10 min and the reaction mixture was warmed to rt. The reaction mixture was stirred at rt for 60 min and then filtered through CeHte* and washed through with 0¾ί¾. The organic layer was dried over N82SO4, concentrated, and purified by column chromatography (15% EtO Ac/hex anes) to provide the title compound (0.450 g, 46%). T NMR (400 MHz, DM80-d6) δ ppm 9.92 (s, 1H), 2.68 (s, 3H), 2.3' (s, 3H).
(d) 1 -(3,5-dimethylisoxazol-4-y3)ethanol

To a solution of 3,5-dimethylisoxazole-4-carbaldehyde (0.450 g, 0.36 mmol) in dry THF (3 mL) at 0 °C was added MeMgBr (3.0 M solution in Et20) (1.2 mL, 3.60 mmol) dropwise over 10 min and the reaciion mixture was warmed to rt. The reaction mixture was stirred at rt for 90 min. Water ( 10 mL) was added very slowly and ihe reaciion mixture was extracted wiih EtO Ac ( 100
rnL). The organic layer was dried over Na2SQ4, concentrated, and purified by column chromatography (27% EtOAc/hexanes) to provide the title compound (0,480 g, 95%). Ή NMR (400 MHz, DMSO-de) δ ppm 5.07 (d, 1H), 4.65 - 4.70 (m, IH), 2.33 (s, 3H), 2.20 (s, 3H), 1.32 (d, 3H).
(e) 1 -i'3,5-dimethylisoxazol-4-yl)ethanon

To a solution of l-(3,5-dimethyiisoxazol-4-yl)etha.nol (0.30 g 2.12 mmol) in CH2C12 (8 ml,) at 0 °C was added PCC (0.068 g, 3.19 mmol) and reaction was stirred for 1 h at rt. Water (25 ml,) was added slowly and the reaction mixture was extracted with (¾(¾ (3 x 10 raL). The organic layer was dried over Na¾S04, filtered, concentrated, and purified by column
chromatography (10% EtOAc/hexane) to provide the title compound (0.10 g, 34%). Ή NMR (400 MHz, CDCI3) δ ppm 2.68 (s, 3H), 2.44 (s, 51 1 :·. 2.24 (s, I H). (f) 2-bromo- 1 -(3,5-dimethylisoxazol-4-yl)ethanone

To a solution of l -(3,5-dimethylisoxazol-4-yl)ethanone (1.0 g, 7, 13 mmol) in CCI4 (40 mL) was added AcOH (1.0 g, 1.142 mmol). The reaction mixture was stirred at 48 °C, followed by dropwise addition of Br2 (0.37 mL, 7.19 mmol) in CCI4 (30 mL), The reaction mixture was stirred at 48 °C for an additional 20 mm. Tee cold water (50 mL) was added and the reaction mixture was extracted with CH2CI2 (3 x 25 mL). The organic layer was dried over Na2SC)4, filtered, concentrated, and purified by column chromatography (5% EtOAc/hexane) to provide the title compound (0.80 g, 51%). Ή NMR (400 MHz, CDC13) δ ppm 4.18 (s, 2H), 2,74 (s, 3H), 2.52 (s, 3H).
(g) methyl 2-(4-(2-(3,5-dimethyl )acetate

A mixture of 2-bromo- 1 -(3,5-dimethylisoxazol-4-yl)ethanone (193 mg, 0.89 mmol), methyl 2-(4-hydroxyphenyl)acetate (147 mg, 0.89 mmol) and potassium carbonate (246 mg, 1 ,78
mol) in acetomtrile (10 mL) was stirred at ri for 1 .5 h. After filtration, the filtrate was concentrated to obtain the title compound (270 mg, 99%) as a yellow oil. LCMS-P l : 304.1 [M+H]+; ¾ = 1.82 min. (h) methyl 2-(4-(2-(3,5-dimethylisoxazol-4-yl)-2-hydroxyethoxy)phenyl)acetate
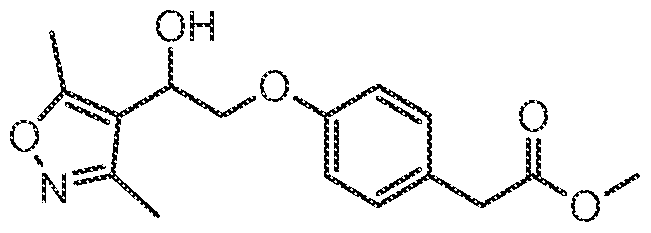
To a cooled (0 °C) solution of methyl 2-(4-(2-(3,5-diniethylisoxazol-4-yl)-2- oxoethoxy)phenyl)acetate ( 120 mg, 0.4 mmol) in MeOH (10 mL) was added NaB¾ (76 mg, 2 mniol). After the addition, the reaction mixture was stirred at rt for l . Then solvent was removed and water (10 mL) was added to the mixture. The mixture was extracted with EtOAc
(15ml x 3). The organic layers were dried over anhydrous a2S04 and filtered. The solvent was removed to obtain the title compound (96 mg, 79%) as colorless oil. The the title compound was used in the next step without any further purification. LCMS-Pl : 306.1 [M+H]+; Rt = 1.73 min.
(i) 2-(4-(2-(3,5-diinethyiisoxazoi-4-yl)-2-hydroxyethoxy)phenyi)acetic acid
This compound was prepared from methyl 2-(4-(2-(3,5-dimethylisoxazol-4-yl)-2- hydroxyethoxy)phenyl)acetate essentially as described in example 24 (d) (99 mg, 100%).
PI : 292.0 [M+Hf; Rt - 1.32 min. (j) 2-(4-(2-(3,5-dimethyiisoxazoi-4-yl)-2-hydroxyethoxy)phenyi)-N-((2,4- dim ethylphenyl ) (phenyl) methy 1) acetami de
This compound was prepared from 2-(4~(2-(3,5-dimethylisoxazol~4-yl)~2~
hydroxyethoxy)phenyi)acetic acid and (2,4-dimethylphenyl)(phenyl)methanamine essentially as described in example 4 (b)(46 mg, 56%) as a white solid. LCMS-Pl : 485.0 [M+H ; Rt - 1.75 min. Ή NMR (400 MHz, CDC13) δ ppm 7.23-7.16 (m, J = 7.6Hz, 5H), 7.08 (d, J = 6.4 Hz, 2H), 6.99 (s, IH), 6.93-6.86 (m, 3H), 6.77 (d, J = 8.0 Hz, IH), 6.37 i d. ./ 8.0Hz, 1H), 5.89 ((!. ./ 8.4 ! ! /. Hi), 5.03-5.06 (m, I H), 4.1 1 -4.06 (m, IH), 3.99 aid. ./ 9.6Hz, 3.6! iz. IH), 3.59 (s, 21 ! }. 2.46 (s, 3H), 2.34 (s, 3H), 2.29 (s, 3H), 2.22 (s,3H).
Example 26
2-amino-2-(4-((3,5-dimethylisoxazol-4-yl)methoxy)phenyl)-N-((2,4- diniethylphenyl)(phenyl)methyl)acet amide

(a) lert-butyl ( 1 -(4-((3,5-dimethylisoxazol-4-yl)methoxy)phenyl)-2-(((2,4- dimethylphenyl)(phenyl)niethyl)atnino)-2-oxoethyl)carbaniate
This compound was synthesized essentially as described in example 1 starting from methyl
2-((teri-butoxycarbonyl)amino)-2-(4-hydroxyphenyl)acetate.
(b) 2-amino-2-(4-((3,5-dimethylisoxazol-4-yl)methoxy)phenyl)-N-((2,4- diinethylphenyl)(phenyl)methyi)acetaniide
tert-B tyl (1 -(4-((3,5-dimethylisoxazol-4-yl)methoxy)pheny])-2-(((2,4- dirriethylpheny3)(phenyr)methyi)amino)-2-oxoethy3)carbamate (85 mg, 0.16 mmo3) in
dichloromethane (2 mL) and trifluorocetic acid (1 mL) were stirred for 15 minutes. Solvent was removed and product was purified by re versed phase HPLC column chromotography using 10-90% aeetomtrile/water with 0.05% TFA to give the title compound (130 mg, 94%). LCMS-Tl : 470.2 I M - I ! ; : Rt: 5.75 min. ¾ NMR (400 MHz, (TX'i .) δ ppm 7.34-7.19 (m, 5H), 7.10 (m, IH), 6.97 (m, I H), 6.93-6.87 (m, 2H), 6.81 (m, IH), 6.60 (m, IH), 6.19 (m, IH), 5.10 (d, 1H), 4.78 (d, 2H), 2.39 (s, 3H), 2.27 (s, 3H), 2.25 (s, 3H), 1.98 (s, 3H).
Using essentially the same procedure as described in example 2, the following compounds in table 1 were made.
Table 1



y l)me ihoxy)pheny 1) ace !amide
H NMR (400 MHz, DMSO-de) δ
ppm 8.97-8.99 id, 1H), 7.38-7.40
(d, 2 H), 7.25-7.28 (ra, 2 H), 7.15- LCMS-Xl :
37 " 7.19 (m, 5 H), 6.91-6.94 (d, 2 H), 509.2 [M-i-Hf;
N-((2-chloro-4-methylphenyl)(4- 6.32-6.34 (d, 1 H), 4.87 (s, 2 H), R = 5.47 rain chlorophenyl)methyl)-2-(4-((3,5- 3.45 (s, 2 H), 2.38 (s, 3H), 2.28 (s,
dimethyiisoxazol-4- 3 I I I. 2.20 (s, 3 H
yl)methoxy)phenyl)acetamide
! ! NMR (400 MHz, DMSO~d6) δ
ppm 8.90-8.92 (d, 1H), 7.36-7.40
(t, 2 H), 7.1 1 -7.33 (m, 7 H), 7.04- LCMS-Xl :
38 Uv H 7.06 (d, 1 H), 6.91-6.93 (d, 2 H), 475.5 [M+H]+;
N-((4-chlorophenyl)(o- 6.20-6.22 (d, 1 H), 4.78 (s, 2 H), Rt - 7.04 min tolyl)methyl)-2-(4-((3,5- 3.45 (s, 2 H), 2.38 (s, 3 H), 2.22 (s,
dimethyiisoxazol-4-yl)methoxy) 6 H).
pheny])acetamide
! ! NMR (400 MHz, DMSO-d6) δ
ppm 8.82-8.84 (d,l H), 7.29-7.33
(m, 2 H), 7.22-7.26 (ra, 1 H), 7, 15-
LCMS-Xl : 7.20 (ni, 4 H), 6.91-6.99 (m, 5 H),
39 4^ 3 [Μ+Η ;
2-(4-((3,5-dimethylisoxazoi-4- 6.16-6.18 (d, 1 H), 4.87 (s, 2 H),
Rt - 7.02 mm yl)methoxy)phenyl)-A'-((2,4- 3.41 -3.46 (s, 2 H), 2.38 (s, 3 H),
diniethylphenyl)(phenyl)methyi)ae 2.37 fs, 3H), 2,20 (s, 3 H), 2.15 (s,
etamide 3 H).
Ή NMR (400 MHz, DMSO-d6) δ
ppm 8.96-8.98 (s, 1 H), 7.24-7.34
(m, 5 H), 7.13-7.20 (ra, 5 H), 6.91 - LCMS-Xl :
40 6.93 (d, 2 H), 6.33-6.35 (d, 2 H), 475.3 [M+H]+;
N-((2-chioro-4- 4.87 (s, 2 H), 3.42-3.49 (m, 2 H), R. = 6.95 min methyiphenyl)(phenyl)methyl)-2~ 2.38 (s, 3 H), 2.33 (s, 3 H), 2.19 (s,
(4-((3,5-dimethylisoxazol-4- 3 H).
ylimethoxy )pheny 1) acetamide
Using essentially the same procedure as described in example 4, the following compounds in table 2 were made.
Table 2.


yl)methoxy)pbenyl)acetamide

δ ppm 7.29 (1H, d), 7.24 (IH, d),
7.16 - 7.12 (4H, m), 7.08 (lH, t),
7.02 ( 11-1, i), 6.89 (IH, d), 6.80 -
MS 497.3
54 6.76 (3H, m), 6.56 ( IH, d), 5.81
I M · H 1 .
2-(4-(2-(3,5-dimethylisoxazol-4- (IH, d), 3.96 (2H, t), 3.53 (2H,
yl)ethoxy)phenyl)-N-((2- s),3.05 (IH, m), 2.76 (2H, t), 2.34
isopropylphenyi)(o-iolyl)memy1) (3H, s), 2.25 (3H, s), 2.16 (3H, s),
acetatnide 1.20 (3H, d), 1.06 (3H, d).
LCMS-A024:
55 1 ! NMR (CDC , 400MHz): δ
9.38 (s, IH), 8.36 (s, IH), 7.81 (s, 450.7 ; M · i 11 : IH), 7.24 (d, .1 8.0 Hz, 2H), 6.85 Rt : 1.44 min. ·: d. ./ 8.0 Hz, 2H), 5.19 (s, i l l ).
4.75 (s, 2H), 3.51 (s, 2H), 2.61 (s,
2-(4-((3,5-dimethylisoxazol-4- 3H), 2.45 (s, 3H), 2.38 (s, 3H),
yl)methoxy)phenyl)-A-(l-(3,5- 2.26 (s, 3H), 2.06-1.93 (m, IH),
dimet y]pyridin-2-yl)-4- 1.83- 1.72 (m, IH), 1.54-1.51 (m,
methylpentyl)acetamide IH), 1.33- 1.25 (m, IH), 1.13- 1.04
(m, H), 0.85 (s, 3H), 0.82 (s, 3H).
Using essentially the same procedure as described in example 7, the following compounds in table 3 were made.
Table 3

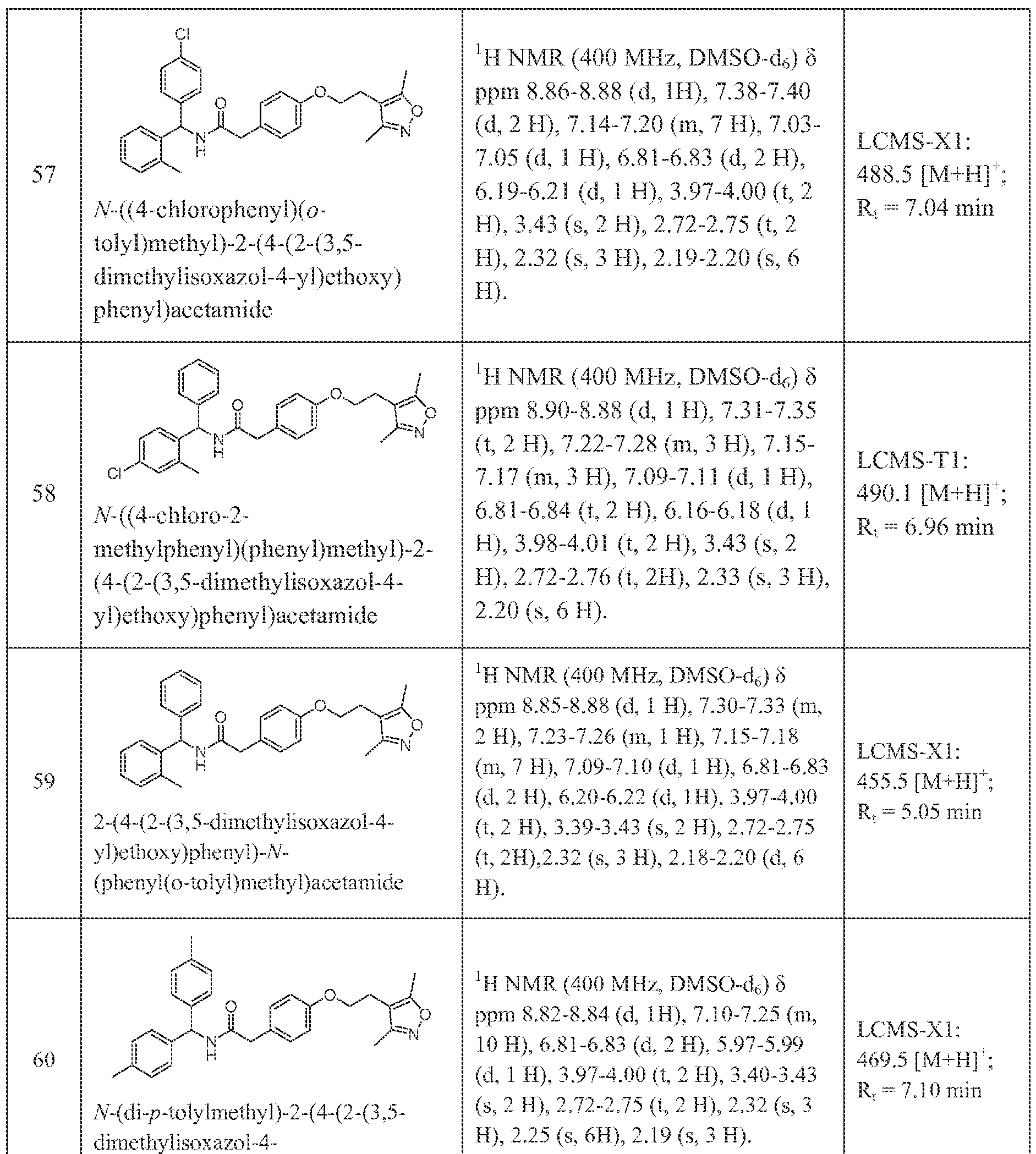
y])ethoxy)phenyl)acetamide
NMR (400 MHz, DMSO-d6) δ
ppm 8.82-8.84 (d. 1 H). 7.19-7.28 (m,
3 H), 7.08-7.16 (m, 4 H), 7.03-7.05 (d,
LCMS-X1 : 2 H), 6.81-6.83(d, 2 H), 6.10-6.13 (d,
503.4 ( V! \ \ \ ■
1 H), 3.97-4.00 (t, 2 H), 3.38-3.41 (s,
N- ((4■■ chlo i'o- 2 -methy iphenyl) (p - R; = 5.56 min
2 H), 2.72-2.75 (t, 2H), 2.23 (s, 3 H),
toly])methyl)-2-(4-(2-(3,5- 2.20 (s, 3 H), 2.14 (s, 3H), 2.08 (s,
dimethylisoxazoi-4- 3H).
yl)ethoxy)plienyi)aceiamide
C!
Ή NMR (400 MHz, DMSO-d6) δ
ppm 8.95-8.97 (d, 1 H), 7.38-7.40 (d,
!j ! H 2 H), 7.24-7.27 (t, 2 H), 7.13-7.16 (m, LCMS-X1 :
4 H), 6.81 -6.83(d, 2 H), 6.30-6.32 (d, 523.4 1 Vi · ! 1 i : 1 H), 3.97-4.00 (t, 2 H), 3.41 -3.46 (t, 2 R. = 5.51 min
Λ' - ((2 - chioro-4 -methy Iphenyl) (4 - H), 2.72-2.75 (m, 2H),2.28-2.32 (d, 6
ch iorophenyl)methy 1) - 2 - (4 - (2 - (3,5- H), 2.19 (s, 3 H).
dimethylisoxazol-4- yl)e†hoxy)phenyl)acetamide
Ή NMR (400 MHz, DMSO-d6) δ
ppm 8.80-8.82 (d. Iff), 7.16-7.28 (m,
8 H), 7.04-7.12 (m, 3 H), 6.81-6.83 (d, LCMS-Xi :
 2 H), 6.61 -6.63 (d, 1 H), 3.97-4.00 (t, 484.6 [M÷Hf;V- ((2 - (dimethyl am i no) phenyl) 2 H), 3.39 (s, 2 H), 2.72-2.75 (i, 2 H), R;— 6.43 min (phenyi)methyl)~2-(4-(2-(3,5- 2.53 (s, 6H), 2.32 (s, 3 H), 2.19 (s, 3
2 H), 6.61 -6.63 (d, 1 H), 3.97-4.00 (t, 484.6 [M÷Hf;V- ((2 - (dimethyl am i no) phenyl) 2 H), 3.39 (s, 2 H), 2.72-2.75 (i, 2 H), R;— 6.43 min (phenyi)methyl)~2-(4-(2-(3,5- 2.53 (s, 6H), 2.32 (s, 3 H), 2.19 (s, 3
dimethylisoxazol-4- H).
y])ethoxy)phenyI)acetamide
1 ! NMR (400 MHz, DMSO-d6) δ
ppm 9.00-9.02 (d, 1H), 7.17-7.40
LCMS-X1 : (m, 10 H), 7.09-7.12 (dd, 1 H),
479.2 [M÷Hj+;
N-((4- 7.02-7.04 (d, i H), 6.08-6.10 (d, 1
chlorophenyl)(pheny3)methyl)-2- R-t = 6.88 min
H), 4.96 (s, 2 H), 3.49 (s, 2 H),
(4-((3,5-dimethylisoxazol-4- 2.36 (s, 3 H), 2.20 (s, 3H).
yl)methoxy)-3- fluorophenyl)acetamide

flu orophenyl )acetamide
Using essentially the same procedure as described in example 9, the following compounds 4 were made.
Table 4


tolyl)methyl)acetamide 2.26 (s, 3 Us.
Ή NMR (400 MHz, DM80-d6) δ
ppm 8.80-8.82 (d, 1 H), 7.24-7.31
LCMS-T1 : (m, 2 H), 7.1 5-7.22 (m, 5 H), 6.91 -
493.1
73 2-(4-((3,5-dimethylisoxazol-4- 6.97 (m, 3 H), 6.70-6.76 (m, 2 H),
yl)methoxy)pbenyl)-N-((4- 6.14-6.17 (d, i H), 4.87 (s, 2 H),
6.4 ! min methoxy-2- 3.71 (s, 3 H), 3.44 (s, 2 H), 2.33 (s,
methylpheny!){phenyl)methyl)acet 3 H), 2.19 (s, 3 H), 2.17 (s, 3 H).
amide
Using essentially the same procedure as described in example 1 1 , the following compounds in table 5 were made.
Table 5
Ex. Structure/Name NMR LCMS
U NMR (400 MHz, DMSO-d6) δ
ppm 8.95-8.93 id, 1 H), 7.27-7.19
(m, 9 H), 6.91 -6.93 (d, 2 H), 6.33- LCMS-X1 :
74
N-((2-chloro-4- 6.36 (d, 1 H), 5.41 -5.44 (t, 1 H), 490.9 [M+Hf ; methyiphenyl)(phenyl)methyl)-2- 4.95 (s, 2 H), 4.51 -4.53 (d, 2 1 11 Rt = 3.77 min
(4-((3-(hydroxymethyl)-5- 3.39-3.45 (s, 2 i n. 2.40 (s, 3 H),
methylisoxazol-4- 2.28 (s, 3 H).
y!) methoxy)ph eny 1) ace tamide
Ή NMR (400 MHz, DMSO-d6) δ
ppm 8.82-8.84 (d, 1 H), 7.29-7.33
(m, 2 H), 7.72-7.76 (d, 1 H), 7.15-
 7.19 (m, 3H), 6.97 (s, 3 H), 6.91 - LCMS-X1 :
7.19 (m, 3H), 6.97 (s, 3 H), 6.91 - LCMS-X1 :
75 N-((2,4- 6.93 (d, 2 1 1 :·. 6.16-6.18 (d, 1 H), 470.0 [M-i-Hf; dimethylphenyl)(phenyl)methyl)- 5.41 -5.44 (t, 1 H), 4.94 (s, 2 H), R, = 3.80 2-(4-((3-(hydroxymethyl)-5- 4.51 -4.53 (d, 2 H), 3.44(s, 2 H),
methylisoxazol-4- 2.40 (s, 3 H), 2.23 (s, 3 H),2.15 (s,
yl)methoxy)ph eny 1) ace tamide 3 H).
Using essentially ihe same procedure as described in example 14, the following compounds in table 6 were made.
Table 6

ppm 8,91 -8.93 (d, l H), 7.29-7.32
(m, 2 H), 7.1 5-7.24 (m, 5 H), 7.10-
LCMS-X1 : 7.13 (ni, 4 H), 6.91 -6.93 (d, 2 H),
79 4^ 3 [M÷Hf;
6.02-6.04 (d, 1 i n. 4.87 (s, 2 i n.
2-(4-((3-ethyl-5-methylisoxazol-4- Rt = 6.94 min
3.42-3.46 (s, 2 H), 2.60-2.67 (d, 2
yl)memoxy)phenyl)-N-(phenyl( .?- H), 2.38 (s, 3 H), 2.26 (s, 3 H),
tolyl)methyl)acetamide
1.15- 1.19 (s, 3 H). i i NMR (400 MHz, DMSO-d6) δ
ppm 8.82-8.85 (d, 1 H), 7.12. -
7.14 (d, 2 H), 7.08-7.10 (m,7 H), LCMS-X1 :
80 6.80-6.82 (d, 2 H), 5.96-5.98 (d, 1 469.3 [M-i-Hf ;
H), 5.43-5.48 (q, 1 H), 3.37 (s, 2 Rt = 7.16 mm
N-(di-p-tolylmethyl)-2-(4-(l -(3,5- H), 2.50 (s, 3 H), 2.37 (s, 6 H),
dimethylisoxazol-4-
2.15 (s, 3 H), 1 .52- 1 .53 (d, 3 H).
yl)ethoxy)phenyl)acetamide if Vq ! ! NMR (400 MHz, DMSO-d6) δ
ppm 9.00-9.25 (d, 1 H), 8.90 (s,
1 H), 7.38-7.40, (d, 2 H), 7.33-7.35
LCMS-X1 : (d,2 H), 7.18-7.31 (m, 5 H),6.91 -
81 475.1 [M÷H]+;
N-((4- 6.9.3 (d, 2 H), 6.07-6.09 (d, 1 H),
Rt = 3.43 min chlorophenyl) (phenyl )methy 1) -2 - 5.87 (s, 2 H), 3.45-3.50 (1 2 H),
(4-((3-ethyl-5-meihyiisoxazoi-4- 2.59-2.65 (m, 2 H), 2.50-2.51 ( 3
ylimethoxy )pheny 1) acetamide H), 1.15- 1.19 (t, 3 H).
lH NMR (400 MHz, DM80-d6) δ
ppm 8.99-9.01 (d, 1 H), 8.9 l (s, 1
LCMS-X1 : H), 7.38-7.40 (d, 2 i n. 7.24-7.35
82 447.2 [M+H]+;
N-((4- (m,8 H), 7.18-7.20 (d, 2 H),6.93- , 7. 1 min chlorophenyl)(phenyl)methyl)-2- 6.95 (d, 1 H 14.96 (s, 2 H), 3.47 (s,
(4■ ((3 -methylisoxazol-4- 2. H), 2.33 (s, 3 H).
yl)memoxy)phenyl)acetamide

yl)methoxy)phenyl)acetamide
Using essentially the same procedure as described in example 20, the following compound in iabie 8 was made.
Table 8

phenylacetamide
Example 86
2-(l -(3,5-DimethylisQxazoi-4-yl)-2-(4-(^
oxoethyl)phenoxy)ethox -2-methylpropanoic acid

(a) 2-(4-(2-(3,5-Dimethylisoxazol-4-y3)-2-oxoethoxy)phenyi
dimethylphenyl)(pheny

2-Bromo-l -(3,5-dimethylisoxazol-4-yl)ethanone (1.3g,0.006mmol) and K2CO3 (l . lg, O.OOSmoi) were added to a solution of .N-((2,4-dimethylphenyl)(phenyl)methy3)-2-(4- hydroxyphenyl)acetamide (1 .5g, 0.004mmol) in MeCN (25mL) under N2. The mixture was stirred at 70 °C for 8 h. Water (50 mL) was then added to reaction mixture, and the mixture was extracted with ethyl acetate (3 x 50 ml,). The combined extracts were washed with brine (3 x 15 mL), dried over Na2S0 , and concentrated under reduced pressure to yield a yellow oil. The oil was purified by silica gel chromatography using 50% EtOAc in PE to give white solid (1.2g, yiled 60%). LC- MSA022: 505.1 [M+H] ; RT =1.84 min, purity 96.54% (254nm).
(b) 2-(4-(2-(3,5-dimethylisoxazol-4-y3)-2-3iyclTOxyethoxy)phenyl)-A'-(C2,4- dimethylphenyl)

NaBH (105mg, 2.75mmol) was added to a solution of 2-(4-(2-(3,5-dimethylisoxazol-4-yl)- 2-oxoethoxy)pheny3)-A-((2,4-dimet3iylpheny3)(pheiiyl)methyl)acetamide (1.2g, 2.5mmo3) in MeOH (18mL). The mixture was stiiTed at rt for 0.5 h. After completion of the reaction, water (30 mL) was added and the mixture was extracted with ethyl acetate (3 x 30 mL), The combined extracts were washed with brine (3 x 15 mL), dried over Na2S04, and concentrated under reduced pressure
to yield a white solid (680mg, yield 56%), which was used in the next step without further purification. LC-MSAO l 1 : 485.2 [M+H]+; RT - 2.00 mm, purity 94.20% (254nm).
(c) Methyl 2-(l -(3,5-dimethylisoxazol-4-yl)-2-(4-(2-(((2,4- dimethylphenyl)(phenyl)methyl)amino)-2-oxoethyl)phenoxy)ethoxy)-2-methylpropanoate:

Methyl 2-bromo-2-methylpropanoate (42mg, 0.23mmol) and Cs2C03 (147mg, 0.45mmol) were added to a solution of 2-(4-(2~(3,5-dimethylisoxazol-4-yi)-2-hy^Oxyethoxy)phenyl)-N-((2,4- dimethylphenyl)(pheny])methyl)acetamide (75mg, 0.15mmoi) in CH3C (1 5mL). The mixture was stirred at 70 °C for 8 h. Water (30 mL) was added to reaction mixture and the mixture was extracted with ethyl acetate (3 x 30 mL). The combined extracts were washed with brine (3 x 15 mL), dried over a2S04, and concentrated under reduced pressure to give a yellow oil. The oil was purified by silica gel Prep-TLC using 50% EtOAc in petroleum ether to give methyl 2-(l-(3,5- dimethyl soxazol-4-yl)-2-(4~(2-(((2^
oxoethyl)phenoxy)ethoxy)-2-methylpropanoate (lOOmg). LCMS-A026: 585.3.2[M+H]+ ; Rt =2.0Gmin.
(d) 2-(l -(3,5-Dimethylisoxazol-4-yl)-2-(4-(2-(((2,4- dimethylphenyi)(phenyl)metliyl)amm^ acid:

A solution of LiOFi (6mg, 0.24mmoi) in water (3mL) was added to the suspension of methyl 2-(l -(3,5-dimethylisoxazol-4-yl)-2-(4-(2-(((2,4-dim
oxoethyl)phenoxy)ethoxy)-2-methylpropanoate (68mg, Q.12mmoi) in THF (5 mL). After stirring at rt for 2 li, the reaction mixture was evaporated under reduced pressure to remove the excess THF. The reaction was acidified to pH ~ 7 with 1 M FiCi solution and extracted with ethyl acetate (3 x 50 mL), and dried over NasSO After removal of the solvent, the residue was purified by reverse
phase HPLC using water/acetonitrile with 0.05% TFA to obtain 2-(l-(3,5-dimethylisoxazol-4-yl)-
2-(4-(2-(((2,4-dimethyfpheiiyl)(phenyf)methyl)aiT!iiio
methylpropanoic acid (1 1 mg, yield: 16%). LCMS-A024: 571.3 [M+Hf; Rt = 1 .74min. !H NMR (MeOD, 400MHz): δ 7.31 -7.20 (in, H i. 7.13 i d. 7.2 Hz, 2l h. 6.99 (s, 1 H), 6.94 (s, 2H), 6.84 (d, J = 8.8 Hz, 2H), 6.29-6.27 (m, ! ! ! }. 4.97-4.89 (m, 1 H), 4.36-4.31 (ra, 1H), 4.08-4.05 (m, 1H), 3.50 (s, 2H), 2.35 (s, M l ). 2.29 (s, 3H), 2.28 (s, 3H), 2.17 (s, 3H), 1 .46 (s, 3H), 1 .35 (s, 3H).
Examples 87 and 88
(^-N-((4-chloro-2-methylphenyl)(4-chloro^
yl)methoxy)phenyl)acetamide and R -N-((4~chloro-2-niethylphenyl)(4-chlorophenyl)methyl)-2~(4- ((3,5-dimethylisoxazol-4-yl)met oxy)pheny])acetamide

N~((4-chloro-2-meth.ylphenyl)(4-chloropheny])methyl)-2-(4-((3,5- d methylisoxazo]-4-yl)methoxy)phenyl)acetamide (180 mg) was resolved using the following method:
Instrument: Thar SFC Prep 80 (Thar Technologies, Waters); Column: ChiralCell OJ-H, 20 mm LD. x 250 mm Length, 5 μιη (Daicel Chemical Industries Co., Ltd); Column Temperature: 35 °C; Mobile Phase: C02/MeOH7DEA -70/30/0.1 ; Flow rate: 50 g/min; Back Pressure: 100 Bar;
Wavelength: 214 mn; Cycle time: 3.0 min; Injection Volume: 0.6 mL; Load per injection: 6 mg; Feed solution: 180 mg dissolved in 18 mL MeOH
Said resolution yielded 2 enantiomers:
-peak 1 , 75 mg, RT 6.4 min, 95.0 % de, 97.5 % purity
-peak 2, 58 mg, RT 7.7 min, 100% de, 100% purity
Example 89
4-((4-(2-(((4-chloro-2-methylphenyl)(ph^
methylisoxazole-3-carbo

(a) (4-ch3oro-2-methy3phenyl)magnesium bromide: This compound was synthesized from l -bromo-4-chloro-2-methylbenzene essentially as described in example 1 (d) to give the title compound which was used in the next step without further purification,
(b) (4-chloro-2-methylphenyl)(pheny])methanamme: This compound was synthesized from benzomtrile and (4-chioro-2-methylpheny3)magnesium bromide essentially as described in example i (e) to give the title compound (17.1 g, 64% yield), which was used in the next step without further purification. LC-MS : 215.0 [M-NH2]+; Rt = 1.261 min.
(c) N-((4-chloro-2-methylphenyl)(phenyl)methyl)-2-(4-3iydroxyp3ienyl)acetamide: This compound was synthesized from (4-cl loro-2-methy3phenyl)(phenyl)methanamine hydrochloride and 2-(4-hydrox phenyl)acetic acid essentially as described in example 4 (b) to give the title compound (650mg, Yield: 63.6%). LC-MS: m/z 366 [M+H]+; Rt - 1.93 min.
(d) methyl 4-((4-(2-(((4-c oro-2-methylphenyl)(phenyl)methyl)ainino)-2- oxoethyl)phenoxy)methyl)-5-methy3isoxazo3e-3-carboxylate: This compound was synthesized from A-((4-chloro-2-methylphenyl)(phenyl)methyl)-2-(4-hydroxyphenyl)acetamide and methyl 4- (chloromethyl)-5-methylisoxazole-3-carboxylate essentially as described in example 1 (b) to give the title compound (21 Omg, Yield: 70%). LC-MS: m/z 519 [M+H]+; Rt = 2.06 min.
(e) 4-((4-(2-(((4-chloro-2-met3iylpheny3)(phenyl)methyl)amino)-2- oxoethyl)phenoxy)methyl)-5-methylisoxazole-3-carboxylic acid: NaOH (33 mg, 0.82 mmol) was added to a solution of methyl 4-((4-(2-(((4-c oro-2-methylphenyl)^ enyl)methyl)aniino)-2- oxoethy{)phenoxy)methyl)-5-xnethylisoxazo{e-3-carboxylate (210 mg, 0.41 mmol) in THF (8 mL) and water (2 mL). The mixture was stirred at rt overnight. Then water (10 ml.) was added to the mixture, and then AcOH was used to adjust the aqueous phase to pH ~ 6. The mixture was then extracted with ethyl acetate (3 x 30 mL). The combined extracts were washed with brine (3 x 15 mL), dried over Na2S04, and concentrated under reduced pressure to give a yellow oil The residue was purified by column chromatography on silica gel (EA: PE = 1 : 2) to provide 4-((4-(2-(((4- chloro-2-methylphenyl)(phenyi)methyl)amino)-2-oxoethyl)phenoxy)methyl)-5-methylisoxazole-3- carboxylic acid (30mg, Yield: 14.6%). LC-MS: 505[M+H]+; Rt = 1.71 min. ¾ NMR (MEOD,
400MHz): 7,30-7,33 (m, 3H), 7.13-7.30 (m, 6H), 7.03-7.29 (m, 5H), 7.03 (m, 3H), 6.94 (m, 3H), 6.27 (s, 1H), 5.18 (s, 2H). 3.51 (s, 2H). 2.45 (s, 3H). 2.20 (s, 3H).
Example 90
2-((3,5-dimethylisoxazol-4-yi)methoxy)-5-(2-(((2,4-dimethylphenyi)(phenyl)methyl)
oxoethyl)benzoic acid

This compound was synthesized from methyl 2-((3,5-dimethylisoxazol-4-yl)methoxy)-5- (2-(((2,4-dimethylphenyl)(phenyl)methyl)amino)-2-oxoethyl)benzoate essentially as described in example 1 (c) (18 mg, 78%). LCMS-P1 : 521.2 [M+Naf; R, = 1.676 min. !H NMR (400 MHz, DMSO-de) δ ppm 12.69 (s, 1H), 8.93 (d, J= 8.4 Hz, 1H), 7.64 (d, J= 2.0 Hz, 1H), 7.46-7.25 (m, 7H), 7.05-7.00 (m, 3H), 6.23 (d, J= 8.4 Hz, 1H), 5.00 (s, 2H), 3.55 (s, 2H), 2.45 (s, 3H), 2.30 (s, 3H), 2,29 (s, 3H), 2.21 (s, 3H).
Example 91
2-(4-((3 ,5 -dimethylisoxazol-4-yl)methoxy)phenyl)- 1 -( 1 -phenyl-3 ,4-dihydroisoquinolin-2( 1 H)- yl)ethanone

This compound was synthesized from 1 -phenyl- 1 ,2, 3, 4-tetraliydroisoquinoline hydrochloride and 2-(4-((3,5-dimethylisoxazol-4-y3)methoxy)phenyl)acetic acid essentially as described in example 4 (b) ( 136 mg, 63% yield). LCMS: 453.2 [M+H]+; Rt = 6.795 min
Example 92
Ar-((4-chloro-2-methylphenyi)(4-chloropheny3)methyl)-2-(4-(2-(3,5-dimethylisoxazol-4- yl)ethoxy)phenyl)acetamide

This compound was synthesized from (4-chioro-2-methylpheny3)(4- chlorophenyl)methanamine and 2-(4-((3,5-dimethylisoxazol-4-yl)methoxy)phenyl)acetic acid essentially as described in example 20 (b) (0.017 g, 17.89%) Ή NMR (400 MHz, DMSO) δ 8.88- 8.90 (d, 1 H), 7.34-7.41 (m, 2 H), 7.24-7.27 (t 2 H), 7.13 -7.28 (m, 6 H), 7.03-7.05 (d, 1 H), 6.81 - 6.84 (d, 2 H), 6.15-6.17 (d, 1 H), 3.97-4.01 (t, 2 H), 3.33-3.38 (d, 1 H), 2.72-2.76 (m, 2 H), 2.32- 2.36 (d, 3 IT), 2.1 8-2.20 (d, 3 H), LCMS purity: 94.52%.
Example 93
2-(4-(2-(3,5-dimethylisoxazol-4-y{)ethoxy)phenyl)-N-((2,4- dimethylphenyl)(phenyl)methyl)acetamide

This compound was synthesized from (2,4-diniethylphenyl)(phenyl)methanamine and 2-(4- (2-(3,5-dimethylisoxazol-4-yl)ethoxy)phenyl)acetic acid essentially as described in example 20 (b) (7.0 mg, 8.23 %). ¾ NMR (400 MHz, DMSO) δ 8.83 -8.80 i d, 1 H), 7.29-7.32 ( 2 H), 7.22-7.25 (t, 1 l ! ). 7.15-7.17 (m, 3 H), 6.94-6.97 (t, 2 I D. 6.81 -6.83 (d, 2 H), 6.16-6.18 (d, 1 H i. 3.97-4.00 (t, 2 H), 3.39 (s, 2 H), 2.72-2.76 (t, 2H), 2.32 (s, 3 IT), 2.20 (s, 6 H), 2.14 (s, 3 H), LCMS purity: 97.58%.

This compound was synthesized from bis(2-chiorophenyl)methanamine and 2-(4-((3,5- dimethylisoxazo1~4~yl)methoxy)phenyl)acetic acid essentially as described in example 2.0 (h) (40.2 mg, 21.72 % ). ¾ NMR (400 MHz, DMSO) δ 7.44-7.45 (d, 2 H), 7.33-7.35 (d, 1 H), 7.31 - 7.35 (d, i H), 7.21-7.30 (m, 4 H), 7.07-7.09 (dd, 2 H), 6.84-6.86 (d, 2 H), 6.72-6.73 (d, 1 H), 4.05- 4.08 (t, 2 H), 3.53 (s, 2 H), 2.85-2.88 (t, 2 H), 2.38 (s, 3 H), 2.28 (s, 3 H), LCMS purity 97.72%. Example 95
2-(4-((3-ethyl-5-methyli

Triphenylphosphine (0.189 g, 0.725 mmol) and (3-etliyl-5-methyfisoxazol-4-yl)methanol (0. 02 g, 0.725 mmol) were added to a solution of 2-(4-hydroxyphenyl)-A7-(phenyl(o- toiyl)methyl)aceta:mide (0.20 g, 0.604 mmol) in dry THF (1.0 mL). The reaction mixture was then cooled in a 33 -KHz sonicating hath and sonicated it for 3 min giving a clear solution .While sonicating, diisopropylazod carboxyl te (0.146 mL, 0,725 mmol) was added drop-wise to the reaction mixture during 2 min. The reaction mixture was then sonicated for an additional 15 min, and men THF was removed under reduced pressure. The resulting residue was purified by silica gel chromatography (0.2% methanol/ CH2C12) and then re-purified with prep HPLC to provide 2- (4-((3~ethyf-5~memy1isoxazo!~4-yl)met!io (95 mg,
34.67 %). Π N M R (400 MHz, DMSO) δ 8.89-8.91 (d, l H), 7.30-7.34 id, 2 H), 7.23-7.27 (m, 1 H), 7.18-7.20 (m, 6 H), 7.10-7.17 (m, 1 i n. 6.91-6.93 (d, 2 H), 6.21 -6.23 (d, 1 H), 4.87 is, 2 H), 3.45 (s, 2 H), 2.60-2.64 (q, 2 H), 2.39 (s, 3H), 2.19 (s, 3H), 1.15-1.19(t, 3H). MS (ESH-) m/z 455.30 (M + H), LCMS purity: 99.93%.
Biological Date
As stated above, the compounds according to Formula (I) are RORy modulators, and are useful in the treatment of diseases mediated by RORy. The biological activities of the compounds according to Formula 0) can be determined using any suitable assay for determining the activity of a candidate compound as a RORy modulator, as well as tissue and in vivo models.
Dual Fluorescence Energy Transfer (FRET) Assay
This assay is based on the knowledge that nuclear receptors interact with cofactors (transcription factors) in a ligand dependent manner. RORy is a typical nuclear receptor in that it has an AF2 domain in the ligand binding domain (LBD) which interacts with co-activators. The sites of interaction have been mapped to the LXXLL motifs in the co-activator SRC 1(2) sequences. Short peptide sequences containing the LXXLL motif mimic the behavior of full-length co- activator.
The assay measures ligand-mediated interaction of the co-activator peptide with the purified bacterial-expressed RORy ligand binding domain (RORy-LBD) to indirectly assess ligand binding, RORy has a basal level of interaction with the co-activator SRC 1 (2) in the absence of ligand, thus it is possible to find ligands that inhibit or enhance the RORy/SRCl(2) interaction.
Materials
Generation of RORy-LBD bacterial expression piasmid
Human RORy Ligand Binding Domain (RORy-LBD) was expressed in E.coli strain BL21(DE3) as an amino -terminal polyhistidine tagged fusion protein. DNA encoding this recombinant protein was sub-cloned into a modified pET21 expression vector (Novagen). A modified polyhistidine tag (MKKHHHHHHLVPRGS) (SEQ ID No: 1) was fused in frame to residues 263-518 of the human RORy sequence.
Protein Purification
Approximately 50 g E.coli cell pellet was resuspended in 300 mL of lysis buffer (30 mM imidazole pH 7.0 and 150 mM NaCl). Cells were lysed by sonication and cell debris was removed by centrifugation for 30 minutes at 20,000 g at 4 °C. The cleared supernatant was filtered through a 0.45 μΜ cellulose acetate membrane filter. The clarified iysate was loaded onto a column (XK-26) packed with ProBond Nickel Chelating resin (InVitrogen), pre-equiiibrated with 30 mM imidazole pH 7.0 and 150 mM NaCl. After washing to baseline absorbance with the equilibration buffer, the column was developed with a gradient from 30 to 500 mM imidazole pH 7.0. Column fractions containing the RORy-LBD protein were pooled and concentrated to a volume of 5 mL. The concentrated protein was loaded onto a Superdex 200 column pre-
equilibrated with 20 mM Tris-Ci pH 7.2 and 200 mM NaCl. T he fractions containing the desired RORy-LBD protein were pooled together.
Protein Biotinylation
Purified RORy-LBD was buffer exchanged by exhaustive dialysis [3 changes of at least 20 volumes (>8000x)] against PBS [100 mM NaPhosphate, pH 8 and 150 mM aCll The concentration of RORy-LBD was approximately 30 μΜ in PBS. Five-fold molar excess of NHS- LC-Biotin (Pierce) was added in a minimal volume of PBS. This solution was incubated with occasional gentle mixing for 60 minutes at ambient rt. The modified RORy-LBD was dialyzed against 2 buffer changes - TBS pH 8.0 containing 5 mM DTT, 2 mM EDTA and 2% sucrose - each at least 20 times of the volume. The modified protein was distributed inf o aliquots, frozen on dry ice and stored at -80 °C, The biotinylated RORy-LBD was subjected to mass spectrometric analysis to reveal the extent of modification by the biotinylation reagent, in general,
approximately 95% of the protein had at least a single site of biotinylation and the overall extent of biotinylation followed a normal distribution of multiple sites ranged from one to five.
A biotinylated peptide corresponding to amino acid 676 to 700
(CPS SHS SLTERHKILHRLLQEG SPS) (SEQ ID No: 2) of the co-activator steroid receptor coactivator SRC 1(2) was generated using similar method.
j sa
Preparation of Europium labeled SRC 1(2) peptide: biotinylated SRC 1(2) solution was prepared by adding an appropriate amount of biotinylated SRC 1 (2) from the 100 μΜ stock solution to a buffer containing 10 mM of freshly added DTT from solid to give a final concentration of 40 nM. An appropriate amount of Europium labeled Streptavidin was then added to the biotinylated SRC 1(2) solution in a tube to give a final concentration of 10 nM. The tube was inverted gently and incubated for 15 minutes at rt. Twenty-fold excess biotin from the 10 mM stock solution was added and the tube was inverted gently and incubated for 10 minutes at rt.
Preparation of APC labeled RORy-LBD: biotinylated RORy-LBD solution was prepared by adding an appropriate amount of biotinylated RORy-LBD from the stock solution to a buffer containing 10 mM of freshly added DTT from solid to give a final concentration of 40 nM. An appropriate amount of APC labeled Streptavidin was then added to the biotinylated RORy-LBD solution in a tube to give a final concentration of 20 nM. The tube was inverted gently and incubated for 15 minutes at rt. Twenty-fold excess biotin from the 10 mM stock solution was then added and the tube was inverted gently and incubated for 10 minutes at rt.
Equal volumes of the above-described Europium labeled SRC 1 (2) peptide and the APC labeled RORy-LBD were gently mixed together to give 20 nM RORy-LBD, 10 nM APC- Strepavidin, 20 nM SRC 1(2) and 5 nM Europium- Streptavidin. The reaction mixtures were
incubated for 5 minutes. Using a Thermo Corabi Multidrop 384 stacker unit, 25 \iL of the reaction mixtures per well was added to the 384-well assay plates containing 1 jxL of test compound per well in 100% DMSO. The plates were incubated for 1 hour and then read on ViewLux in Lance mode for EU/APC.
Results
The compounds of Examples 1 -95 were tested in the dual FRET assay described abov and were found to have a pIC50 between 4.4 and 9.
Claims
1. A compound according to Formula (I):

wherein:
m is 0, 1 , or 2;
n is 0, i, 2, or 3;
X1, X , X3, X4, and X5 are each independently selected from N, N f -Q~, CH, and C 5, wherein 0-3 of X!, X2, X3, X4, and X5 are N or +-0" and 1 -3 ofX1, X2, X3, X4, and Xs are CR5; provided that when zero of X!, X2, X , and X5 are N or W-O" and X3 is CR5, 1-2 of X , X", X4, and X5 are CR5;
one of Y1 and Y' is O or NR8 and the other is a bond;
or X1 is CR'\ Y1 is NR8, Y7" is a bond, and R5 and R8 taken together with the atoms to which they are attached form a five to seven membered ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by (Cr-C4)alkyl;
Cy is (CVCsjcycloaikyi, lieterocycloaikyl, phenyl, or 5- or 6-membered heteroaryl, each of which is optionally substituted one, two, or three times, independently, by (d-Cejalkyl,
(Cj-Cyhaloalkyl, (C3-C6)cycloalkyl, halogen, oxo, cyano, hydroxy!, hydroxy(C; -C6)alkyl, (CT-C6)alkoxy, · ! ( ( ,: ·( ' )alk i ;N UC(};R . · H Cn ' : ialk ! }N; i C -C : C -(>.- .
(Co-C3)alkyl)NHCiO)R7, ·-{ ( ' ^ !n!ky 1 1% ( ί " - C- : laiky l)C" ) sii . -{ ((VC alkyhCO -R .
-((Co-C3)all<yl)CONR7R8, -((Co-C3)alkyl)C(0)R7, (Ci-C4)alkoxy(Ci-C6)alkyl, amino(Ci-C6)alkyl, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino(Ci-C6)alkyl, (Ci-C4)alkyiamino(Ci-C6)alkyl, amino,
(Cj-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, aryl, heteroaryl, aryl(Cj -C6)alkyl,
heteroaryl(C] -C6)alkyl, or lieterocycloaikyl;
Z is O, S, S02, C>0, NR6, or a bond;
A1, A2, A3, and A4 are each independently selected from N, NR°, O, S, CH, and CRl!), wherein one of A1, A2, A3, and A4 is NR6, O, or S, 0-2 of A1, A2, A3, and A4 are CR10, and 0-3 of A.1, A2, A3, and A4 are CH or N;
Rl is (C3-C6)aikyl, (C3~Gs)!ialoalkyl, (C3-C3)cycloalkyl, (C3-C6)alkoxy,
(CrC6)alkoxy(Ci-C2)aikyl, aryl, heteroaryl, aryi(Ci-C6)alkyL heteiOaryi(CrC6)alkyi, or heterocycloalkyl, each of which is optionally substituted one, two, or three times, independently, by R5;
R2 is hydrogen, (Ci -C^alky!, or iCi-C6)haloalkyl;
or R1 and taken together with the carbon atom to which they are attached form a three to eight membered ring, optionally containing a heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted one, two, or three times, independently, by ';
R3 and R3a are each independently hydrogen, hydroxyi, (CrC6)alkyl, (Q-C/jhaloalkyl, halogen, (C rC6)alkoxy, amino, (Ci -C4)alkylamino, or ((C; -C4)alkyl)((CVC4)alkyl)amino;
each R4 is independently selected from hydrogen, halogen, (Ci -C6)alk l, (CV-CV^haloalkyl, -C02R7, -CONR7R8, -OR9, and -NR8R9, wherein said (Ci-C6)alkyl or (Ci-Csjlialoalkyl is optionally substituted by hydroxy!, -OR9, -C02R7, -CONR7R8, or ~NR8R9;
each R4"* is independently selected from hydrogen, halogen, hydroxyi, amino, and i ( -C, S ikyi :
or R4 and R4a taken together with the carbon atom to which they are attached form a three to eight membered ring, optionally containing a heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by cyano, (C rC- alkyl, (C; -C4)haloalkyl,
(C C6)eycloalkyl, -C02R7, -C \ R R \ hydroxy!, hydroxy(Ci -C6)alkyi, (C C4)alkoxy,
(C;-C4)alkoxy(Ci-C6)aikyl, amino, (Ci-C4)a{ky!amino, ((Ci -C4)alky{)((Ci-C4)alky'!)anisno, -NHCO2R7, - ((C1-C4)alkyl)C02R7, -NHC(0)R7, or -N((Ci-C4)alkyl)C(0)R7;
each R5 is independently selected from (Ci -Chalky 1, (CVCyhaloalkyl, (CrC^cyeloalkyl, halogen, cyano, hydroxy!, hydroxy(Ci -C6)alkyl, (CV-Celalkoxy, (Ci-C4)alkoxy(Ci-C6)aIkyl, amino, (Ci-C4)a!kylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)ammo, aryl, heteroaryl, aryl(Ci-C6)alkyl, heteroaiyl(Ci-C6)alky!, and heterocycloalkyl;
R6 is hydrogen, (C C6)alkyL (C; -C6)haloalliyi, (C C6)cycloalkyl, hydroxy(C1-C6)alkyl, (Ci-C4)alkoxy(C, -C6)a!kyl, ·((( ',;· ( ; mlkyhCO . - n CVC )a!k> ! )( N R R ". aryl, heteroaryl, aiyl(Ci-C6)aJkyl, heteroary Ci -Cejaikyl, or heterocycloalkyl;
R ' is hydrogen, (Ci-C6)alkyL (Ci -C6)haloalky!, (CrC6)cyc!oa!kyl,
(Ci-C4)alkoxy(C [-C6)alkyl, aryl, heteroaryl, a yliCrCyalkyi, heteroaryl(Ci -C6)alkyl, or heterocycloalkyl;
R8 is hydrogen, (C CeWkyi, or (Ci -Cejhaloalkyl;
or R' and 8 taken together with the nitrogen atom to which they are attached form a four to eight membered ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by (Ci-C4)alkyl, (Ci -C4)haloalkyl, (CVCyeycioaikyl, -C02H, -C02(C-;-C4)alkyi, hydroxy!, hydroxyfC; -Chalky!, (Ci-C4)alkoxy, (Ci-C4)alkoxy(Ci-C6)alkyl, amino, (Ci-C4)alkylamino, or ((Ci-C4)alkyl)((Ci-C4)alkyl)ammo; R" is -C(0)R7, -CO R . -C(0)NR7R8, (CrC6)alkyl, (C C6)haloalkyl, (C3-C6)cycloaikyl, aiy], heteroaryl, aiyl(Ci -C6)alkyi, he†eraaryl(Ci~C6)alkyl, or heterocycloalkyl, wherein said (Ci -C6)alkyl, (Q-C^haloalkyl, (C3-C6)cycloalkyl, aryl, heteroaryl, aryl(Ci -C6)alkyl,
heteroaryl(Ci -C6)alkyl, or heterocycloalkyl is optionally substituted by -C02R?, -CONH ,
-COMH(C] -C4)alkyl, -i "i)\( (C -C4)a]kyl)((CrC4)aIkyl), hydroxyl, (C - C - ia!koxy. amino,
(C C4)alky]arnino, ((CI-C4)alkyl)((C1-C4)alkyl)amino, -NHC02R7, -N((C1-C4)alkyl)C02R7, -NHC(0)R7, or -N((CrC4)alkyl)C(0)R7;
or R8 and R1' taken together with the nitrogen atom to which they are attached form a four to eight niembered ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by cyano, (Ci-C4)a1kyl, (Ci-C4)haioalkyl, (C3-C6)cycloalkyl, ~C02H, -C02(CrC4)alkyl, -CONR7R8, hydroxy!, hydroxy(Ci-C6)alkyl,
(Ci-C4)alkoxy, (CrC4)alkoxy(CrC6)aikyL amino, (CrCyalkylamino,
i i ( •C : H! ii, l !( ; C -C4)alkyl)amino, - N I K O R '. - ((C, -C4)alkyl)C02R?, -NHC(0)R7, or
-N((Ci-C4)alky1)C(0)R7; and
R10 is (Ci-Cfijalkyl, (Ci-Cejhaloalkyl, (C3-C6)cycloalkyl, halogen, cyano, hydroxyl, hydroxy(C rC6)alkyl, (C, -C6)alkoxy, (Cx-C4)alkoxy(Ci-C6)alkyl, -((C0-C3)alkyl)CO2R7,
-((Co-C3)alkyl)CONR7R8, amino(C, -C6)alkyl, ((CrC4)alkyl)( Ci-C )alkyl)amino(Ci-C6)alkyl, (Ci-C4)a{kylamino(Ci-C6)aUkyl, amino, (Ci-C4)alkylamino, ((Ci-C4)a1kyl)((Ci-C4)alkyl)amino, aryl, heteroaryl, aryl(Ci-C6)alkyl, heteroaryl(Ci-C6)alkyJ, or heterocycloalkyl;
provided that the compound is not (2-(4-((3,5-dimethylisoxazol-4-yl)methoxy)phenyl)-Ar-
((2-methoxyphenyl)(l -methyl- lH-imidazol-2~yi)methyl)acetamide);
or a salt thereof.
2. The compound or salt according to claim 1 , wherein m is 1 and n is 1 or 2.
3. The compound or salt according to claim 1 or claim 2, wherein X1 and XJ are each independently selected from N, N"-0", CH, and CR5, and X", X3, and X4 are each independently selected from CH and CR5, wherein at least one of X1 and X3 is N or N+-0" and 0-3 of X1, X XJ, X4, and X5 are CR5.
4. The compound or salt according to claim 1 or claim 2, wherein X1 is a carbon atom substituted by halogen, (CrC4)alkyl, (Ci-C4)haloalkyl, cyano, (CrC4)alkoxy, or
((Ci-C4)alkyl)((Ci-C4)alkyl)amino, and X2, XJ, X4, and X5 are each independently a carbon atom substituted by hydrogen, halogen, (d-C4)a]kyl, (Ci-C4)haioaIkyl, cyano, (CrC4)aikoxy, or ((Ci-C4)alkyl){(Ci~C4)alkyl)amino, wherein 2-4 of X2, X3, X4, and Xs are a carbon atom substituted by hydrogen.
5. The compound or salt according to any one of claims 1-4, wherein Y1 is NH or NCH3 and Y2 is a bond.
6. The compound or salt according to any one of claims 1 -5, wherein Cy is heterocycfoafkyl, phenyl, or 5- or 6-membered heteroaryl, each of which is optionally substituted one or two times, independently, by (C; -C6)alkyL (C) -C6)haloalkyL halogen, cya.no, 
((CrC4)alkyl)aniino ((C C4)alkyl)( Cx-C )alkyl)amino, -((C0 3)alkyl)CO2R?,
or -((Co-C3)alkyl)CONR7R8.
7. The compound or salt according to any one of claims 1-5, wherein Cy is piperidinyl, piperazinyi, phenyi, pyridinyl, pyridazinyl, pyrazinyl, or pyrimidinyi, each of which is optionally substituted one or two times, independently, by (Ci -Cejalkyl, (Ci-Cejhaloalkyl, halogen, cya.no, (d-C4)alkoxy, (Ci-C4)alkyl)((Ci-C4)alkyl)amino, ~((Co-C3)alkyl)C02H,
-((Co-C3)alkyl)C02(Ci-C6)alkyl, or -((Co-C3)alkyl)CONH(Ci-C6)alkyl.
8. The compound or salt according to any one of claims 1-5, wherein Cy is phenyi, which is optionally substituted one or two times, independently, by halogen, (Ci -Gi)alkyl, (Ci -C haloalkyf, cyano, (C ··( '.. !askoxy. or ((Ci-C4)alkyl)((Ci-C )alkyl)ammo.
9. The compound or salt according to any one of claims 1 -8, wherein Z is a bond, O, or NH.
10. The compound or salt according to any one of claims 1 -9, wherein R1 is (CrCejalkyl, (CrCekycloalkyl, phenyl, furanyi, thienyl, pyrrolyl, imidazoiyi, pyrazoiyl, triazolyl, tetrazolyl, thiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiadiazoly!, isothiazolyl, pyridinyl, pyridazinyl, pyrazinyl, pyrimidinyi, or triazinyl, wherein said phenyl, furanyi, thienyl, pyrrolyl, imidazoiyi, pyrazoiyl, triazolyl, tetrazolyl, thiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiadiazolyl, isothiazolyl, pyridinyl, pyridazinyl, pyrazinyl, pyrimidinyi, or triazinyl is optionally substituted one or two times, independently, by halogen, (Ci -C^alky.!, (Ci-C4)alkoxy, or ((Ci-C4)a]kyl)C(Ci-C4)alkyl)ammo.
1 1. The compound or salt according to any one of claims 1 -9, wherein R: is phenyl or pyridinyl, each of which is optionally substituted one or two times, independently, by halogen, (Ci-C4)aikyl, (Ci-C4)aikoxy, or ((Ci-C )a]kyl)((Ci-C4)alkyl)ammo.
12. The compound or salt according to any one of claims 1-1 1 , wherein R is hydrogen or methyl.
13. The compound or salt according to any one of claims 1-12, wherein R3 and RJa are each independently hydrogen or methyl.
14. The compound or salt according to any one of claims 1 -13, wherein each R4 is independently selected from hydrogen, (Q -C4)alkyl, (Ci-C4)haloalkyl, -OR9, and -NR8R9, wherein said (Ci -C4)alkyl or (Ci -C4)haloalkyl is optionally substituted by hydroxy!, -OR9, -CO2R', -CONR7Rs, or - RSR9.
15. The compound or salt accordmg to any one of claims 1-13, wherem each R4 is independently selected from hydrogen, ( ( -Oaik i. (d -C4)alkoxy, hydroxy(C2-C4)alkoxy, (CrC4)alkoxy(C2-C4)alkoxy, amino(C2-C4)a1koxy, -0((Ci-C3)alk l)C02H,
-0((Ci-C3)alky1)C02(C1-C4)alkyI, -0((Ci-C3)a1kyl)CONH2, -©((Ci-Csialky CO Hid-C^alkyl, and -0((Ci-C3)alkyl)CON((Ci-C4)alkyl)((Ci-C,)alkyl).
16. The compound or salt according to any one of claims 1-15, wherein each R4a is independently selected from hydrogen and methyl,
17. The compound or salt according to any one of claims 1-16, wherein A1 and A4 are each independently selected from CH and CR10, and one of A" and A3 is NR.", O, or S and the other is N or CH.
18. The compound or salt according to any one of claims 1- 16, wherein A1 and A4 are each independently selected from CH and C((Ci-C4)alkyl), and one of A" and A3 is N((CrC4)alkyi), O, or S and the other is or CH.
19. A compound according to Formula (la):

ra is 1 ;
n is 1 or 2;
X1, X , X', and X4 are each independently selected from N, N";-0\ CH, and CR3, wherein 0-2 of X1 , X2, X3, and X4are N or N"h-0" and 0-2 X1 , X2, X3, and X4 are CR5;
Y1 is NH or NCH3 and Y2 is a bond;
K1, K2, KJ, and K" are each independently selected from N, N+-0~, CH, and CRl0, wherein 0-2 of K1, K2, '. and K4 are N or IST-O" and 0-2 of K1, K2, K3, and K4 are CR10;
Z is O, NR6, or a. bond:
A1 , A", A3, and A4 are each independently selected from N, NR.6, O, S, CH, and C.R!", wherein one of A1 , A2, A3, and A4 is NR6, O, or S, 0-2 of A1 , A2, A3, and A4 are CR10, and 0-3 of
A1, A2, A3, and A4 are CH or N;
R! is (C3-C6)alkyl, (C3-C6)haloalkyl, (C3-Cs)cycloalkyl, (C3-C6)alkoxy,
(Ci -C6)alkoxy(Ci-C2)alkyl, aryl, heteroaryl, aryl(Ci-C6)alkyl, heteroaryl(Ci -C6)alkyl, or heterocycloalkyl, each of which is optionally substituted one, two, or three times, independently, by
R5;
R2 is hydrogen, (G -Cejaikyl, or (Ci-C6)haloalkyl;
or Rl and R- taken together with the carbon atom to which they are attached form a three to eight membered ring, optionally containing a heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted one, two, or three times, independently, by Rs:
R3 and R'a are each independently hydrogen, hydroxy!, (Cj -C4)alkyl, (Ci-C4)haloalkyl, halogen, (Ci-C4)alkoxy, amino, (Ci-C4)alkylamin.o, or ((Ci-C4)ajkyl)CCCi-C4)alkyl)ammo;
each R4 is independently selected from hydrogen, halogen, (CrC4)alkyl, (Ci-C4)haloalkyl, -OR9, and -NRSR9, wherein said (C C^alkyl or (C rC^haloalkyl is optionally substituted by hydroxy!, -OR9, -C02R?, - CONR'R8, or -NRSR9:
each R4a is independently selected from hydrogen, halogen, hydroxyi, amino, and
(C;-C4)alkyl;
each R5 is independently selected from (Ci -Ce kyl, (Q -C^haloalkyl, (C3-C6)cycloalkyl, halogen, cyano, hydroxy!, hydroxy(Ci -C6)alkyl, (Cr-Celalkoxy, (Ci-C4)alkoxy(Ci-C6)alkyl, amino, (Ci-C4)alky1araino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, aryl, heteroaryl, aryl(Ci-C6)alkyl, heteroaiyl(Ci -C6)alkyl, and heterocycloalkyl;
R6 is hydrogen, (Ci-C6)alkyl, (Ci -Cf^haloalkyl, (C3-C6)cycloalkyL hydroxy(Ci-C6)alkyl, (Ci-C4)alkoxy(Ci -C6)a!kyl, ·((( '«;· ( ; mlkyhCO . - i f Co -C )aik> UC 'ON R \ aryl, heteroaryl, aiyl(Ci-C6)aikyl, heteroary Ci -Cejaikyl, or heterocycloalkyl; R' is hydrogen, (Ci~C6)alkyl, (C; -C3)ha!oa!ky1, (C^-Cejeycloalkyl,
(Ci-C4)alkoxy(Ci-C6)alkyl, aryl, heteroaryl, aryl(C-i -C6)alkyi, heteroaryf(Ci~C6)alkyl, or heterocycloalkyl;
R8 is hydrogen, (d -Chalky!, or (Ci-C6)haioaikyl;
or R / and Rs taken together with the nitrogen atom to which they are attached form a four to eight membered ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by (C-i-Q)alkyl, (CrC^haloalkyl, (C C6)cycloalkyl, -C02H, -C02(CrC4)alkyL hydroxyl, hydroxy(C C6)alkyl, (CrC4)alkoxy, (Ci-C4)alkoxy(C] -Ce alkyl, amino, (Ci-C4)alkylamino, or ((Ci-C4)alkyl)((Ci-C4)alkyl)amino;
R9 is -C(0)R7, -C02R7, -C(0)NR7R8, (C •C. i iky! (CrC6)haloalkyl, (Cs- cycloalkyl, aryl, heteroaryl, aryl(CrC6)alkyl, heter arylfCi-Cr alkyl, or heterocycloalkyl, wherein said (CrCe kyl, (Ci -C6)haloalkyl, (C;,-C6)cycloalkyl, aryl, heteroaryl, aiylfCpCe kyl,
heteroaryi(Ci - C6)alkyi, or heterocycloalkyl is optionally substituted by -C02R'', -CONH2,
-CONH(Ci-C4)alkyJ, -CON((C-i-C4)a]kyl)((Ci-C4)alkyl), hydroxyl, (Ci-C4)alkoxy, amino,
(Ci-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, -NHC02R7, -N((Ci-C4)alkyl)C02R7, -NHC(0)R7, or -N((Ci-C4)alkyl)C(0)R7;
or Rs and R"" taken together with the nitrogen atom (0 which they are attached form a four to eight membered ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, which ring is optionally substituted by cyano, (Ci-C4)alkyl, (Ci-C4)haloalkyl, (C3-C6)cycloalkyl, -CO2H, -C02(C1-C4)alkyl> -CO R'R8, hydroxy!, hydroxy(C C6)alkyl,
·; ( ·(' : S iko . (Ci-C4)alkoxy(Ci -Cejaikyl, amino, (Ci-C4)alkylamino,
((Ci-C4)alkyl)((Ci -C4)alkyl)amino, - N l ·:("() R -N((Ci -C4)alkyl)C02R7, -NHC(0)R7, or
-N((Ci-C4)alkyl)C(0)R7; and
R10 is (Ci-Ce)alkyl, (Ci-C6)haloalkyl, (C3-C6)cycloalkyl, halogen, cyano, hydroxyl, hyciroxy(Cr-C6)aikyl, (Cr-C6)alkoxy, (C · ( i !a lkoAVi i -C s ikyi . -((C(r C3)aikyl)C02R7,
-((Co-C3)alk d)CONR7R8, amino(Ci -C6)alkyl, t i C - C ^ i ik ! )(( ('••( )a!k ! ;nn .ino( i ' - (" il )aiky!. (Ci-C4)alkylamino(Ci-C6)alkyf, amino, (Ci-C4)alkylamino, ((CrC4)al3 yl)((Ci-C4)alkyl)amino, aryl, heteroaryl, aryl(Ci-C6)alkyl, heteroaryl(C;-C6)alkyl, or heterocycloalkyl;
or a pharmaceutically acceptable salt thereof,
20. The compound or salt according to claim 1 9, wherein:
X1, X', X3, and X4 are each independently a carbon atom substituted by hydrogen, halogen, cyano, (Ci -C4)alkyl, (C] -C )haloalkyi, (Ci-C4)alkoxy, or ((Ci -C4)alkyl)((Ci-C )alkyl)aniino, wherein 2-4 of X\ X", X and X4 are a carbon atom substituted by hydrogen; K1 , 2, Κ', and " are each independently a carbon atom substituted by hydrogen, halogen, (Ci-C4)alkyl, (Ci-C4)alkoxy, or (( -C )alkyl)((Ci-C4)alkyl)ammo, wherein 2-4 of K1 , K2, K', and K4 are a carbon atom substituted by hydrogen;
Z is O, NH, -N(Ci-C )alkyl, -N((Q-C3)alkyl)C02R7, -N((Co-C3)alkyl)CONR7R8, or a bond; A1 and A4 are each independently selected from CH and CR10, and one of A' and A3 is NR6,
O, or S and the other is N or CH;
Rz is hydrogen;
R3 and R"' are each independently hydrogen or methyl;
each R4 is independently selected from hydrogen, (C] -C4)alkyi, (Ci-C4)aikoxy,
hydroxy(C2-C4)alkoxy, (Ci -C4)alkyiamino, C(Ci-C4)alkyl)((Ci-C4)alkyl)ammo,
(Ci-C4)alkoxy(Ci-C4)alkylamino, (Ci-C4)alkoxy(C2-C4)a1koxy, amino(C2-C4)alkoxy,
-0((Ci-C3)alkyl)C02H, -0((Ci-C3)alkyl)C02(Ci -C4)alkyl, -0((C C3)alkyl)CONH2,
•Oi i ( •(•; );!ik i !C( )\ U( C ··( ·.. >aik> I. and -OH C - : ialk ! }<; ON; -C \: }a II. ! >( ( " - C' : Uuk> 1 >:
each R4a is independently selected from hydrogen, hydroxyl, amino, and (Ci-C4)a{kyl; R7 is hydrogen, (C C6)aikyl, (Ci-Cejhaloalkyl, (C3-C6)cycloalkyl,
(C C4)alkoxy(Ci-C6) lkyL aryl, heteroaryl, aryl(C] -C6)alkyl, heteroaryl(C[-C6)alkyl, or heterocycloalkyi; and
Rs is hydrogen, (Ci -Cejalkyl, or (C-. -C^haloaJkyl.
21. The compound or salt according to claim 20, wherein:
Z is O, NH, -N(C; -C4)aikyL or a bond;
A1 and A4 are each independently selected from CH and C((Ci-C4)alkyl), and one of A" and A" is O or S and the other is N;
R1 is phenyl optionally substituted one or two times, independently, by halogen,
(CrC4)alkyl, (Ct-C4)kaloalkyl, cyano, (Ci-C4)alkoxy, or ((Ci -C4)aIkyl)((CrC4)aikyl)aniino; and each R4 is independently selected from hydrogen, (CrC4)alky1, (Ci-C4)a1kylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, and (Ci-C4)alkoxy.
22. The compound or salt according to any one of claims 1 - 18, wherein the salt is a
pharmaceutically acceptable salt of said compound.
23. A compound of any one of Examples 1 -95, or a pharmaceutically acceptable salt thereof.
24. A pharmaceutical composition comprising the compound, or pharmaceutically acceptable salt thereof, according to one of claims 19-23 and a pharmaceutically acceptable excipient.
25. A method of treatment of a disease mediated by RORy which comprises administering to a human in need thereof an effective amount of the compound, or pharmaceutically acceptable salt thereof, according to any one of claims 19-23, or the pharmaceutical composition according to claim 17 or claim 18.
26. The method according to claim 25, wherein said disease is an inflammatory or autoimmune disease.
27. The method according to claim 26, wherein said inflammatory or autoimmune disease is selected from the group consisting of multiple sclerosis, rheumatoid arthritis, psoriasis, u v eitis, dry eye, glomerulonephritis, and Crohn's disease.
28. The method according to claim 25, wherein said disease is colon cancer, multiple myeloma, or bone disease associated with multiple myeloma.
29. Use of the compound, or pharmaceutically acceptable salt thereof, according to any of claims 19-23 for the treatment of diseases mediated by RORy.
30. Use of the compound, or pharmaceutically acceptable salt thereof, according to any of claims 19-23 as an active therapeutic substance in the treatment of a disease mediated by RORy.
31. A compound or pharmaceutically acceptable salt thereof according to any of claims 19-23 for use in therapy.
32. Use of the compound, or pharmaceutically acceptable salt thereof, according to any of claims 19-23 in the manufacture of a medicament for the treatment of diseases mediated by RORy.
33. The use according to any of claims 29-32, wherein said disease is an inflammatory or autoimmune disease.
34. The use according to claim 33, wherein said inflammatory or autoimmune disease is selected from the group consisting of multiple sclerosis, rheumatoid arthritis, psoriasis, uveitis, dry- eye, glomerulonephritis, and Crohn's disease.
35. The use according to any of claims 29-32, wherein said disease is colon cancer, multiple myeloma, or bone disease associated with multiple myeloma.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/235,489 US20140256740A1 (en) | 2011-07-29 | 2012-07-27 | Compounds and methods |
| EP12820262.9A EP2747560A4 (en) | 2011-07-29 | 2012-07-27 | Compounds and methods |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161513121P | 2011-07-29 | 2011-07-29 | |
| US61/513,121 | 2011-07-29 | ||
| US201161521838P | 2011-08-10 | 2011-08-10 | |
| US61/521,838 | 2011-08-10 | ||
| US201161533913P | 2011-09-13 | 2011-09-13 | |
| US61/533,913 | 2011-09-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013019635A1 true WO2013019635A1 (en) | 2013-02-07 |
Family
ID=47629617
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2012/048588 WO2013019635A1 (en) | 2011-07-29 | 2012-07-27 | Compounds and methods |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US20140256740A1 (en) |
| EP (1) | EP2747560A4 (en) |
| WO (1) | WO2013019635A1 (en) |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013178362A1 (en) | 2012-05-31 | 2013-12-05 | Phenex Pharmaceuticals Ag | Carboxamide or sulfonamide substituted thiazoles and related derivatives as modulators for the orphan nuclear receptor ror[gamma] |
| WO2014023367A1 (en) | 2012-08-09 | 2014-02-13 | Phenex Pharmaceuticals Ag | Carboxamide or sulfonamide substituted nitrogen-containing 5-membered heterocycles as modulators for the orphan nuclear receptor ror gamma |
| WO2015095265A1 (en) * | 2013-12-19 | 2015-06-25 | Merck Sharp & Dohme Corp. | Hiv protease inhibitors |
| US9067898B1 (en) * | 2014-03-07 | 2015-06-30 | Janssen Pharmaceutica Nv | Isothiazole derivatives as GPR120 agonists for the treatment of type II diabetes |
| WO2015145371A1 (en) * | 2014-03-27 | 2015-10-01 | Piramal Enterprises Limited | Ror-gamma modulators and uses thereof |
| US9266886B2 (en) | 2014-02-03 | 2016-02-23 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US9481674B1 (en) | 2016-06-10 | 2016-11-01 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US9663515B2 (en) | 2014-11-05 | 2017-05-30 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US9796710B2 (en) | 2014-10-14 | 2017-10-24 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US9845308B2 (en) | 2014-11-05 | 2017-12-19 | Vitae Pharmaceuticals, Inc. | Isoindoline inhibitors of ROR-gamma |
| WO2018011746A1 (en) * | 2016-07-14 | 2018-01-18 | Cadila Healthcare Limited | Cyclopropyl derivatives as ror-gamma modulators |
| WO2018138362A1 (en) | 2017-01-27 | 2018-08-02 | Genfit | N-{[2-(piperidin-1-yl)phenyl](phenyl)methyl}-2-(3-oxo-3,4-dihydro-2h-1,4-benzoxa zin-7-yl)acetamide derivatives and related compounds as ror-gamma modulators for treating autoimmune diseases |
| US10155737B2 (en) | 2013-03-14 | 2018-12-18 | Janssen Pharmaceutica Nv | Benzo-fused heterocyclic derivatives useful as agonists of GPR120 |
| CN109574825A (en) * | 2018-12-27 | 2019-04-05 | 江南大学 | A kind of synthetic method of phenylacetic acid |
| US10301261B2 (en) | 2015-08-05 | 2019-05-28 | Vitae Pharmaceuticals, Llc | Substituted indoles as modulators of ROR-gamma |
| US10829481B2 (en) | 2016-01-29 | 2020-11-10 | Vitae Pharmaceuticals, Llc | Benzimidazole derivatives as modulators of ROR-gamma |
| US10913739B2 (en) | 2017-07-24 | 2021-02-09 | Vitae Pharmaceuticals, LLC (121374) | Inhibitors of RORγ |
| US10975056B2 (en) | 2016-06-13 | 2021-04-13 | Glaxosmithkline Intellectual Property Development Limited | Substituted pyridines as inhibitors of DNMT1 |
| US11008340B2 (en) | 2015-11-20 | 2021-05-18 | Vitae Pharmaceuticals, Llc | Modulators of ROR-gamma |
| US11186573B2 (en) | 2017-07-24 | 2021-11-30 | Vitae Pharmaceuticals, Llc | Inhibitors of ROR gamma |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2982162C (en) | 2015-04-10 | 2023-10-10 | Bioresponse, L.L.C. | Self-emulsifying formulations of dim-related indoles |
| CN114716389A (en) * | 2021-12-31 | 2022-07-08 | 北京岳达生物科技有限公司 | Synthetic method of 3, 5-diethylisoxazole-4-carboxylic acid |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6297375B1 (en) * | 1999-02-24 | 2001-10-02 | Hoffmann-La Roche Inc. | 4-phenyl-pyridine derivatives |
| US7015246B2 (en) * | 2001-03-29 | 2006-03-21 | Bayer Aktiengesellschaft | Benzofuran derivatives |
| US20090149514A1 (en) * | 2003-02-21 | 2009-06-11 | Intrexon Corporation | Oxadiazoline ligands for modulating the expression of exogenous genes via an ecdysone receptor complex |
| US20100216816A1 (en) * | 2007-10-24 | 2010-08-26 | Barrow James C | Pyrazinyl amide-t type calcium channel antagonists |
| US20100249176A1 (en) * | 2007-10-24 | 2010-09-30 | Barrow James C | Heterocycle amide t-type calcium channel antagonists |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2875805B1 (en) * | 2004-09-27 | 2006-12-29 | Genfit S A | SUBSTITUTED N- (BENZYL) PHENYLACETAMIDE DERIVATIVE COMPOUNDS, PREPARATION AND USES |
| JP4675801B2 (en) * | 2006-03-06 | 2011-04-27 | 日本メナード化粧品株式会社 | ROR activator |
-
2012
- 2012-07-27 WO PCT/US2012/048588 patent/WO2013019635A1/en active Application Filing
- 2012-07-27 EP EP12820262.9A patent/EP2747560A4/en not_active Withdrawn
- 2012-07-27 US US14/235,489 patent/US20140256740A1/en not_active Abandoned
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6297375B1 (en) * | 1999-02-24 | 2001-10-02 | Hoffmann-La Roche Inc. | 4-phenyl-pyridine derivatives |
| US7015246B2 (en) * | 2001-03-29 | 2006-03-21 | Bayer Aktiengesellschaft | Benzofuran derivatives |
| US20090149514A1 (en) * | 2003-02-21 | 2009-06-11 | Intrexon Corporation | Oxadiazoline ligands for modulating the expression of exogenous genes via an ecdysone receptor complex |
| US20100216816A1 (en) * | 2007-10-24 | 2010-08-26 | Barrow James C | Pyrazinyl amide-t type calcium channel antagonists |
| US20100249176A1 (en) * | 2007-10-24 | 2010-09-30 | Barrow James C | Heterocycle amide t-type calcium channel antagonists |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2747560A4 * |
Cited By (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013178362A1 (en) | 2012-05-31 | 2013-12-05 | Phenex Pharmaceuticals Ag | Carboxamide or sulfonamide substituted thiazoles and related derivatives as modulators for the orphan nuclear receptor ror[gamma] |
| US10301272B2 (en) | 2012-05-31 | 2019-05-28 | Phenex Pharmaceuticals Ag | Carboxamide or sulfonamide substituted thiazoles and related derivatives as modulators for the orphan nuclear receptor ROR[γ] |
| WO2014023367A1 (en) | 2012-08-09 | 2014-02-13 | Phenex Pharmaceuticals Ag | Carboxamide or sulfonamide substituted nitrogen-containing 5-membered heterocycles as modulators for the orphan nuclear receptor ror gamma |
| US9458104B2 (en) | 2012-08-09 | 2016-10-04 | Phenex Pharmaceuticals Ag | Carboxamide or sulfonamide substituted nitrogen-containing 5-membered heterocycles as modulators for the orphan nuclear receptor RORγ |
| EP3118189A1 (en) | 2012-08-09 | 2017-01-18 | Phenex Pharmaceuticals AG | Carboxamide or sulfonamide substituted nitrogen-containing 5-membered heterocycles as modulators for the orphan nuclear receptor ror gamma |
| US11254647B2 (en) | 2013-03-14 | 2022-02-22 | Janssen Pharmaceutica Nv | Benzo-fused heterocyclic derivatives useful as agonists of GPR120 |
| US10155737B2 (en) | 2013-03-14 | 2018-12-18 | Janssen Pharmaceutica Nv | Benzo-fused heterocyclic derivatives useful as agonists of GPR120 |
| US10730847B2 (en) | 2013-03-14 | 2020-08-04 | Janssen Pharmaceutica Nv | Benzo-fused heterocyclic derivatives useful as agonists of GPR120 |
| WO2015095265A1 (en) * | 2013-12-19 | 2015-06-25 | Merck Sharp & Dohme Corp. | Hiv protease inhibitors |
| US9737545B2 (en) | 2013-12-19 | 2017-08-22 | Merck Sharp & Dohme Corp. | HIV protease inhibitors |
| US9266886B2 (en) | 2014-02-03 | 2016-02-23 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US9624217B2 (en) | 2014-02-03 | 2017-04-18 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US10807980B2 (en) | 2014-02-03 | 2020-10-20 | Vitae Pharmaceuticals, Llc | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US11535614B2 (en) | 2014-02-03 | 2022-12-27 | Vitae Pharmaceuticals, Llc | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US10047085B2 (en) | 2014-02-03 | 2018-08-14 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US10399976B2 (en) | 2014-02-03 | 2019-09-03 | Vitae Pharmaceuticals, Llc | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US9067898B1 (en) * | 2014-03-07 | 2015-06-30 | Janssen Pharmaceutica Nv | Isothiazole derivatives as GPR120 agonists for the treatment of type II diabetes |
| US11084784B2 (en) | 2014-03-27 | 2021-08-10 | Piramal Enterprises Limited | ROR-gamma modulators and uses thereof |
| AU2015237788B2 (en) * | 2014-03-27 | 2021-03-11 | Piramal Enterprises Limited | ROR-gamma modulators and uses thereof |
| EP3122721A4 (en) * | 2014-03-27 | 2018-01-10 | Piramal Enterprises Limited | Ror-gamma modulators and uses thereof |
| WO2015145371A1 (en) * | 2014-03-27 | 2015-10-01 | Piramal Enterprises Limited | Ror-gamma modulators and uses thereof |
| US10087184B2 (en) | 2014-10-14 | 2018-10-02 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of RORγ |
| US9796710B2 (en) | 2014-10-14 | 2017-10-24 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US9845308B2 (en) | 2014-11-05 | 2017-12-19 | Vitae Pharmaceuticals, Inc. | Isoindoline inhibitors of ROR-gamma |
| US11001583B2 (en) | 2014-11-05 | 2021-05-11 | Vitae Pharmaceuticals, Llc | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US9663515B2 (en) | 2014-11-05 | 2017-05-30 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US10301261B2 (en) | 2015-08-05 | 2019-05-28 | Vitae Pharmaceuticals, Llc | Substituted indoles as modulators of ROR-gamma |
| US10829448B2 (en) | 2015-08-05 | 2020-11-10 | Vitae Pharmaceuticals, Llc | Substituted benzoimidazoles as modulators of ROR-γ |
| US11008340B2 (en) | 2015-11-20 | 2021-05-18 | Vitae Pharmaceuticals, Llc | Modulators of ROR-gamma |
| US10829481B2 (en) | 2016-01-29 | 2020-11-10 | Vitae Pharmaceuticals, Llc | Benzimidazole derivatives as modulators of ROR-gamma |
| US9481674B1 (en) | 2016-06-10 | 2016-11-01 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US10975056B2 (en) | 2016-06-13 | 2021-04-13 | Glaxosmithkline Intellectual Property Development Limited | Substituted pyridines as inhibitors of DNMT1 |
| WO2018011746A1 (en) * | 2016-07-14 | 2018-01-18 | Cadila Healthcare Limited | Cyclopropyl derivatives as ror-gamma modulators |
| JP2019520400A (en) * | 2016-07-14 | 2019-07-18 | カディラ ヘルスケア リミティド | Novel cyclopropyl derivative |
| WO2018138362A1 (en) | 2017-01-27 | 2018-08-02 | Genfit | N-{[2-(piperidin-1-yl)phenyl](phenyl)methyl}-2-(3-oxo-3,4-dihydro-2h-1,4-benzoxa zin-7-yl)acetamide derivatives and related compounds as ror-gamma modulators for treating autoimmune diseases |
| US10913739B2 (en) | 2017-07-24 | 2021-02-09 | Vitae Pharmaceuticals, LLC (121374) | Inhibitors of RORγ |
| US11186573B2 (en) | 2017-07-24 | 2021-11-30 | Vitae Pharmaceuticals, Llc | Inhibitors of ROR gamma |
| CN109574825A (en) * | 2018-12-27 | 2019-04-05 | 江南大学 | A kind of synthetic method of phenylacetic acid |
Also Published As
| Publication number | Publication date |
|---|---|
| US20140256740A1 (en) | 2014-09-11 |
| EP2747560A4 (en) | 2015-02-25 |
| EP2747560A1 (en) | 2014-07-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2747560A1 (en) | Compounds and methods | |
| KR102203552B1 (en) | Allosteric modulator of nicotinic acid acetylcholine receptor | |
| US20140155381A1 (en) | Compounds and methods | |
| WO2013019621A1 (en) | Compounds and methods | |
| ES2346335T3 (en) | DERIVATIVES OF (4- (HETEROARIL) PIPERACIN-1-IL) - (PHENYLL 2,5-REPLACED) METANONE AS INHIBITORS OF THE GLICINE 1 CARRIER (GLYT-1) FOR THE TREATMENT OF NEUROLOGICAL AND NEUROPSYCHIATRIC DISORDERS. | |
| WO2013019682A1 (en) | Compounds and methods | |
| CA2787018A1 (en) | Inhibitors of histone deacetylase (hdac) enzymes | |
| WO2013085890A1 (en) | Therapeutic methods | |
| WO2011088192A1 (en) | Compounds and methods | |
| EP2611772B1 (en) | 2-(benzyloxy)benzamides as lrrk2 kinase inhibitors | |
| CA2985542A1 (en) | Triazole agonists of the apj receptor | |
| AU2005230847A1 (en) | Anaplastic lymphoma kinase modulators and methods of use | |
| TW201018677A (en) | Compounds which selectively modulate the CB2 receptor | |
| US5622953A (en) | 1-amino-3-phenoxy propane derivatives as modulator agents and their applications | |
| US20120322827A1 (en) | Compounds and methods | |
| CA2510793A1 (en) | Indane acetic acid derivatives and their use as pharmaceutical agents, intermediates, and method of preparation | |
| WO2003095441A1 (en) | Inhibitors of hepatitis c virus rna-dependent rna polymerase, and compositions and treatments using the same | |
| CA2702976A1 (en) | 2-aminooxazole derivatives as trpv1 antagonists useful for treating pain | |
| WO2011056126A1 (en) | Novel 1,3-oxazolidine compounds and their use as renin inhibitors | |
| AU2020204341A1 (en) | Naphthyridinone derivatives and their use in the treatment of arrhythmia | |
| JP2014510145A (en) | Benzodioxepin and benzodioxin compounds for the treatment of diabetes that interact with glucokinase regulatory proteins | |
| AU2014246609A1 (en) | Compounds and methods |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12820262 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14235489 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012820262 Country of ref document: EP |