WO2005073224A2 - Quinoline quinazoline pyridine and pyrimidine counds and their use in the treatment of inflammation angiogenesis and cancer - Google Patents
Quinoline quinazoline pyridine and pyrimidine counds and their use in the treatment of inflammation angiogenesis and cancer Download PDFInfo
- Publication number
- WO2005073224A2 WO2005073224A2 PCT/US2005/002304 US2005002304W WO2005073224A2 WO 2005073224 A2 WO2005073224 A2 WO 2005073224A2 US 2005002304 W US2005002304 W US 2005002304W WO 2005073224 A2 WO2005073224 A2 WO 2005073224A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- methoxy
- thienyl
- pyridyl
- chloro
- Prior art date
Links
- 0 CC(*)(CCCCCC1C(C)=S=CN=C1)*=* Chemical compound CC(*)(CCCCCC1C(C)=S=CN=C1)*=* 0.000 description 5
- MHXVTWXNTMJEOR-UHFFFAOYSA-N CCC(C)c1cnc2NC(C)Sc2n1 Chemical compound CCC(C)c1cnc2NC(C)Sc2n1 MHXVTWXNTMJEOR-UHFFFAOYSA-N 0.000 description 1
- UMOVOQPXTOLOHZ-UHFFFAOYSA-N Cc([s]c1c2)nc1ccc2[IH]C Chemical compound Cc([s]c1c2)nc1ccc2[IH]C UMOVOQPXTOLOHZ-UHFFFAOYSA-N 0.000 description 1
- VIJYHDMLYXAHEV-UHFFFAOYSA-N Cc1nc2ccc(C)cc2[o]1 Chemical compound Cc1nc2ccc(C)cc2[o]1 VIJYHDMLYXAHEV-UHFFFAOYSA-N 0.000 description 1
- JEKCSLMWKCKDCC-UHFFFAOYSA-N Cc1nc2ccc(C)cc2[s]1 Chemical compound Cc1nc2ccc(C)cc2[s]1 JEKCSLMWKCKDCC-UHFFFAOYSA-N 0.000 description 1
- ONMCRIXONKHAPC-UHFFFAOYSA-N Cc1nc2ccc(C)nc2[o]1 Chemical compound Cc1nc2ccc(C)nc2[o]1 ONMCRIXONKHAPC-UHFFFAOYSA-N 0.000 description 1
- YKFLBOGLUHQJAH-UHFFFAOYSA-N Cc1nc2cnc(C)nc2[s]1 Chemical compound Cc1nc2cnc(C)nc2[s]1 YKFLBOGLUHQJAH-UHFFFAOYSA-N 0.000 description 1
- PNYSLKIJTVYMDI-UHFFFAOYSA-N Cc1nc2ncc(C)cc2[o]1 Chemical compound Cc1nc2ncc(C)cc2[o]1 PNYSLKIJTVYMDI-UHFFFAOYSA-N 0.000 description 1
- BTCHKDMPQAWXEY-UHFFFAOYSA-N Cc1nc2ncc(C)nc2[o]1 Chemical compound Cc1nc2ncc(C)nc2[o]1 BTCHKDMPQAWXEY-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- FIELD OF THE INVENTION This invention is in the field of pharmaceutical agents and specifically relates to compounds, compositions, uses and methods for treating inflammation, angiogenesis and cancer.
- BACKGROUND OF THE INVENTION Protein kinases represent a large family of proteins which play a central role in the regulation of a wide variety of cellular processes, maintaining control over cellular function.
- a partial list of such kinases includes abl, Akt, bcr-abl, Blk, Brk, Btk, c-kit, c-Met, c-src, c-fms, CDK1, CDK2, CDK3, CDK4, CDK5, CDK6, CDK7, CDK8, CDK9, CDK10, cRafl, CSF1R, CSK, EGFR, ErbB2, ErbB3, ErbB4, Erk, Fak, fes, FGFRl, FGFR2, FGFR3, FGFR4, FGFR5, Fgr, flt-1, Fps, Frk, Fyn, Hck, IGF-IR, INS-R, Jak, KDR, ck, Lyn, MEK, p38, PDGFR, PI , PKC, PYK2, ros, tie, tie2, TRK, Yes, and Zap70.
- c-Met The hepatocyte growth factor receptor
- c-Met is a unique receptor tyrosine kinase shown to be overexpressed in a variety of malignancies.
- c-Met typically comprises, in its native form, a 190-kDa heterodimeric (a disulfide-linked 50-kDa ⁇ -chain and a 145-kDa ⁇ -chain) membrane-spanning tyrosine kinase protein (Proc. Natl. Acad. Sci. USA, 84:6379-6383 (1987)).
- c-Met is mainly expressed in epithelial cells and stimulation of c-Met leads to scattering, angiogenesis, proliferation and metastasis.
- HGF hepatocyte growth factor
- HGF-SF Hepatocyte Growth Factor- Scatter Factor
- HGF/SF The biological effect of HGF/SF may depend in part on the target cell.
- HGF induces a spectrum of biological activities in epithelial cells, including mitogenesis, stimulation of cell motility and promotion of matrix invasion (Biochem. Biophys. Res. Comm., 122:1450- 1459 (1984); Proc. Natl. Acad. Sci. USA, 88:415-419 (1991)). It stimulates the motility and invasiveness of carcinoma cells, the former having been implicated in the migration of cells required for metastasis.
- HGF can also act as a "scatter factor", an activity that promotes the dissociation of epithelial and vascular endothelial cells (Nature, 327:239-242 (1987); J.
- HGF Hepatocyte Growth Factor-Scatter Factor
- C-Met Receptor Goldberg and Rosen, eds., Birkhauser Verlag-Basel, 131-165 (1993)
- HGF and c-Met are expressed at abnormally high levels in a large variety of solid tumors.
- HGF and/or c-Met have been observed in liver, breast, pancreas, lung, kidney, bladder, ovary, brain, prostate, gallbladder and myeloma tumors in addition to many others.
- the role of HGF/c-Met in metastasis has been investigated in mice using cell lines transformed with HGF/c-Met (J. Mol. Med., 74:505-513 (1996)).
- Overexpression of the c-Met oncogene has also been suggested to play a role in the pathogenesis and progression of thyroid tumors derived from follicular epithelium (Oncogene, 7:2549- 2553 (1992)).
- HGF is a morphogen (Development, 110:1271-1284 (1990); Cell, 66:697-71 1 (1991)) and a potent angiogenic factor (J. Cell BioL, 119:629-641 (1992)).
- a morphogen Development, 110:1271-1284 (1990); Cell, 66:697-71 1 (1991)
- a potent angiogenic factor J. Cell BioL, 119:629-641 (1992)
- Recent work on the relationship between inhibition of angiogenesis and the suppression or reversion of tumor progression shows great promise in the treatment of cancer (Nature, 390 :404-407
- Angiogenesis can be stimulated by HGF, as well as vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF).
- VEGF vascular endothelial growth factor
- bFGF basic fibroblast growth factor
- Angiogenesis the process of sprouting new blood vessels from existing vasculature and arteriogenesis, the remodeling of small vessels into larger conduit vessels are both physiologically important aspects of vascular growth in adult tissues. These processes of vascular growth are required for beneficial processes such as tissue repair, wound healing, recovery from tissue ischemia and menstrual cycling.
- vascular growth in these contexts has also shown beneficial effects in preclinical animal models. For example, inhibition of angiogenesis by olocking vascular endothelial growth factor or its receptor has resulted in inhibition of tumor growth, and in retinopathy. Also, the development of pathological pannus tissue in rheumatoid arthritis involves angiogenesis and might be blocked by inhibitors of angiogenesis.
- the ability to stimulate vascular growth has potential utility for treatment of ischemia-induced pathologies such as myocardial infarction, coronary artery disease, peripheral vascular disease, and stroke.
- ischemia-induced pathologies such as myocardial infarction, coronary artery disease, peripheral vascular disease, and stroke.
- the sprouting of new vessels and/or the expansion of small vessels in ischemic tissues prevents ischemic tissue death and induces tissue repair.
- Certain diseases are known to be associated with deregulated angiogenesis, for example ocular neovascularization, such as retinopathies (including diabetic retinopathy), age-related macular degeneration, psoriasis, hemangioblastoma, hemangioma, arteriosclerosis, inflammatory disease, such as a rheumatoid or rheumatic inflammatory disease, especially arthritis (including rheumatoid arthritis), or other chronic inflammatory disorders, such as chronic asthma, arterial or post-transplantational atherosclerosis, endometriosis, and neoplastic diseases, for example so-called solid tumors and liquid tumors (such as leukemias).
- retinopathies including diabetic retinopathy
- age-related macular degeneration psoriasis
- hemangioblastoma hemangioblastoma
- arteriosclerosis arteriosclerosis
- inflammatory disease such as a rheumatoid
- HGF and cMet Treatment of malaria and related viral diseases may also be mediated by HGF and cMet. Elevated levels of HGF and c-Met have also been observed in non-oncological settings, such as hypertension, myocardial infarction and rheumatoid arthritis. It has been observed that levels of HGF increase in the plasma of patients with hepatic failure (Gohda et al., supra) and in the plasma (HepatoL, 13:734-750 (1991)) or serum (J. Biochem., 109:8-13 (1991)) of animals with experimentally induced liver damage.
- HGF has also been shown to be a mitogen for certain cell types, including melanocytes, renal tubular cells, keratinocytes, certain endothelial cells and cells of epithelial origin (Biochem. Biophys. Res. Commun., 176:45-51 (1991); Biochem. Biophys. Res. Commun., 174:831-838 (1991); Biochem., 30:9768-9780 (1991); Proc. Natl. Acad. Sci. USA, 88:415-419 (1991)). Both HGF and the c-Met protooncogene have been postulated to play a role in microglial reactions to CNS injuries (Oncogene, 8:219-222 (1993)).
- T cells play a pivotal role in the regulation of immune responses and are important for establishing immunity to pathogens.
- T cells are often activated during inflammatory autoimmune diseases, such as rheumatoid arthritis, inflammatory bowel disease, type I diabetes, multiple sclerosis, Sjogren's disease, myasthenia gravis, psoriasis, and lupus.
- T cell activation is also an important component of transplant rejection, allergic reactions, and asthma.
- T cells are activated by specific antigens through the T cell receptor (TCR) which is expressed on the cell surface.
- TCR T cell receptor
- This activation triggers a series of intracellular signaling cascades mediated by enzymes expressed within the cell (Kane, LP et al. Cunent Opinion in Immunol. 200, 12, 242). These cascades lead to gene regulation events that result in the production of cytokines, like interleukin-2 (IL-2).
- IL-2 is a critical cytokine in T cell activation, leading to proliferation and amplification of specific immune responses.
- kinase enzymes Members of the Src-family of tyrosine kinases include, for example: Lck, Fyn(B), Fyn(T), Lyn, Src, Yes, Hck, Fgr and Blk (for review see: Bolen, JB, and Brugge, JS Annu. Rev. Immunol 1997, 15, 371).
- Gene disruption studies suggest that inhibition of some members of the src family of kinases would potentially lead to therapeutic benefit.
- Src(-/-) mice have abnormalities in bone remodeling or osteopetrosis (Soriano, P.
- the compounds disclosed in the present invention possess pharmacological activity only by virtue of an effect on a single biological process, it is believed that the compounds modulate T cell activation by way of inhibition of one or more of the multiple protein tyrosine kinases involved in early signal transduction steps leading to T cell activation, for example by way of inhibition of Lck kinase.
- Src-family kinases are also important for signaling downstream of other immune cell receptors.
- Fyn like Lck, is involved in TCR signaling in T cells (Appleby, MW et al. Cell 1992, 70, 751).
- Hck and Fgr are involved in Fc ⁇ receptor signaling leading to neutrophil activation (Vicentini, L. et al. J.
- a class of compounds useful in treating cancer and angiogenesis is defined by Formula I
- the invention also relates to compounds of Formula I R is selected from H, 6-10 membered aryl, 4-10 membered heterocyclyl, 4-6 membered cycloalkyl, C ⁇ -6 -alkyl, C 2-6 -alkenyl and C 2 . 6 -aLkynyl; wherein R is substituted or unsubstituted; in conjunction with any of the above or below embodiments.
- R is phenyl or naphthyl; in conjunction with any of the above or below embodiments.
- the invention also relates to compounds of Formula I R is a substituted or unsubstituted heterocyclyl ring selected from pyrrolidinyl, pyrrolyl, imidazolyl, pyrazolyl, pyrazinyl, pyrimidinyl, pyridyl, quinolinyl, isoquinolinyl, 2,3-dihydrobenzofuryl, 2,3-dihydro-l,4-benzodioxinyl, 1,3- benzodioxolyl, isoxazolyl, isothiazolyl, oxazolyl, thiazolyl, furanyl, and thienyl; in conjunction with any of the above or below embodiments.
- the invention also relates to compounds of Formula I wherein R is 4-6 membered cycloalkyl selected from cyclobutyl, cyclopentyl and cyclohexyl; in conjunction with any of the above or below embodiments.
- R is selected from methyl, ethyl, propyl, butyl and pentyl; in conjunction with any of the above or below embodiments.
- R is selected from ethenyl and propenyl; in conjunction with any of the above or below embodiments.
- the invention also relates to compounds of Formula I wherein A is selected from
- R 7 is selected from H, halo and methyl; and pharmaceutically acceptable derivatives thereof; in conjunction with any of the above or below embodiments.
- the invention also relates to compounds of Formula I wherein A is benzothiazole; and pharmaceutically acceptable derivatives thereof; in conjunction with any of the above or below embodiments.
- the invention also relates to compounds of Formula I wherein R 1 is selected from 9-10- membered bicyclic heterocyclyl; in conjunction with any of the above or below embodiments.
- R 1 is selected from 9-10- membered bicyclic heteroaryl; in conjunction with any of the above or below embodiments.
- the invention also relates to compounds of Formula I wherein R 1 is selected from I
- R U - wherein ring T is selected from phenyl and 5-6-membered heteroaryl; wherein Z is selected from N or CH; wherein R 10 is one or more substituents selected from R 5 O-; and wherein R 5 is selected from . ⁇ -alkyl, C ⁇ -6 -haloalkyl, C ⁇ -6 -alkylamino-C 1-6 -alkyl, aryl-C ⁇ -6 -alkyl, heterocyclyl-C 1-6 - alkyl, cycloalkyl-C ⁇ -6 -alkyl, aryl, heterocyclyl, and cycloalkyl; in conjunction with any of the above or below embodiments.
- the invention also relates to compounds of Formula I wherein R 1 is selected from
- R 10 is selected from C ⁇ -3 -alkoxy, C ⁇ -3 -alkylamino-C ⁇ -3 -alkoxy, phenyl-C ⁇ -3 -alkoxy, 5-6 membered heterocyclyl-C, -3 -alkoxy and C 4-6 -cycloalkyl-C ⁇ -3 -alkoxy; in conjunction with any of the above or below embodiments.
- the invention also relates to compounds of Formula I wherein R 1 is selected from 6,7-dimethoxy- 4-quinolinyl, 6-methoxy-7-(dimethylaminopropoxy)-4-quinolinyl, 6,7-dimethoxy-4-quinazolinyl, and 6- methoxy-7-(dimethylaminopropoxy)-4-quinazolinyl; in conjunction with any of the above or below embodiments.
- the invention also relates to compounds of Formula I wherein R is selected from ethyl, isopropyl, (CH 3 ) 3 CCH 2 -, ethenyl, and an unsubstituted or substituted ring selected from phenyl, cyclobutyl, cyclopentyl, cyclohexyl, 2-pynolidinyl, 2-pyrrolyl, 5-imidazolyl, 5-pyrazolyl, 2-pyrazinyl, 4-pyrimidinyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 8-quinolinyl, 2,3-dihydrobenzofur-7-yl, 2,3-dihydro-l,4-benzodioxin-5-yl, l,3-benzodioxol-4-yl, 4-isoxazolyl, 3 -isothiazolyl, 5-oxazolyl, 4-thiazolyl, 5-thiazolyl, 2-
- the invention also relates to compounds of Formula F
- R A' R wherein R is selected from a) substituted or unsubstituted aryl, b) substituted or unsubstituted heterocyclyl, c) substituted or unsubstituted cycloalkyl, d) substituted or unsubstituted cycloalkenyl, e) H, f) substituted or unsubstituted alkyl, g) substituted or unsubstituted alkenyl, h) substituted or unsubstituted alkynyl, i) alkylaminocarbonyl, j) aminocarbonyl, and k) cyano; wherein R 1 is selected from a) substituted or unsubstituted aryl, b) substituted or unsubstituted heterocyclyl, c) substituted or unsubstituted cycloalkyl, d) substituted or unsubstituted cycloalkenyl, e) H, f) substitute
- ring T is selected from phenyl and 5-6-membered heteroaryl; wherein Z is selected from N or CR X ; wherein R x is selected from H, CN, NH 2 , F, alkylcarbonylamino, and alkylaminocarbonyl; wherein R 10 is one or more substituents selected from Ci- ⁇ -alkoxy, Ci- ⁇ - haloalkoxy, C ⁇ -6 -alkylamino-C ⁇ -6 -alkoxy, aryl-C ⁇ - 6 -alkoxy, heterocyclyl-C ⁇ . 6 -alkoxy, cycloalkyl- C ⁇ -6 -alkoxy, heterocyclyl-C].
- the invention also relates to compounds of Formula F wherein R is selected from H, 6-10 membered aryl, 4-10 membered heterocyclyl, 3-6 membered cycloalkyl, C ⁇ . 6 -alkyl, C 2-6 -alkenyl and C 2-6 - alkynyl; wherein R is substituted or unsubstituted.
- R is optionally substituted phenyl or optionally substituted naphthyl.
- the invention also relates to compounds of Formula I' wherein R is a substituted or unsubstituted heterocyclyl ring selected from pynolidinyl, pynolyl, imidazolyl, pyrazolyl, pyrazinyl, pyrimidinyl, pyridyl, quinolinyl, isoquinolinyl, tefrahydrofuryl, 2,3-dihydrothiazolyl, 2,3-dihydrobenzofuryl, 2,3- dihydro-l,4-benzodioxinyl, 1,3-benzodioxolyl, benzisoxazolyl, benzthiazolyl, benzimidazolyl, benzothiadiazolyl, indolinyl, imidazo[l,2-a]pyridyl, isoxazolyl, isothiazolyl, oxazolyl, thiazolyl, thiadiazolyl, fur
- R is an unsubstituted or substituted ring selected from 2-pyrrolidinyl, 2-pyrrolyl, 5-imidazolyl, 5 -pyrazolyl, 2-pyrazinyl, 4- pyrimidinyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 8-quinolinyl, 2,3-dihydrobenzofur-7-yl, 2,3-dihydro-l,4- benzodioxin-5-yl, l,3-benzodioxol-4-yl, 4-isoxazolyl, 3-isothiazolyl, 5-oxazolyl, 4-thiazolyl, 5-thiazolyl, 2-furanyl, 3-furanyl, 3-thienyl and 2-thienyl.
- the invention also relates to compounds of Formula I' wherein R is selected from 1-methyl- cyclopropyl, cyclopropyl, 2-fluorocyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
- R is selected from 1-methyl- cyclopropyl, cyclopropyl, 2-fluorocyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
- R also relates to compounds of Formula F wherein R is selected from methyl, trifluoromethyl, ethyl, propyl, butyl and pentyl.
- R also relates to compounds of Formula F wherein R is selected from cyclohexenyl, ethenyl and propenyl.
- R also relates to compounds of Formula I' wherein R is H.
- R also relates to compounds of Formula F wherein R is dimethylamino.
- A is selected from
- R 7 is selected from H, halo and methyl.
- the invention also relates to compounds of Formula I' wherein A is
- the invention also relates to compounds of Formula F wherein A is
- the invention also relates to compounds of Formula F wherein
- the invention also relates to compounds of Formula F wherein R is
- ring T is selected from phenyl and 5-6-membered heteroaryl
- Z is selected from N or CH; wherein R 10 is one or more substituents selected from R 8 0-; and wherein R 8 is selected from C ⁇ -6 -alkyl, C ⁇ -6 -haloalkyl, C ⁇ -6 -alkylamino-C ⁇ . 6 -alkyl, aryl-C ⁇ _ 6 -alkyl, heterocyclyl-C ⁇ .
- R is selected from C 1-3 -alkoxy, C ⁇ -3 -alkylamino-C ⁇ . 3 -alkoxy, 5-6 membered heterocyclyl-Ci. 3 -alkoxy, C . 6 -cycloalkyl-C ⁇ -3 -alkoxy, 5-6 membered heterocyclyl-C ⁇ -3 -(hydroxyalkoxy), C 3-6 -cycloalkyl- C ⁇ -3 -(hydroxyalkoxy), phenyl-C ⁇ -3 -(hydroxyalkoxy), C ⁇ -2 -alkoxy-C ⁇ -3 -alkoxy, phenyloxy-C ⁇ .
- the invention also relates to compounds of Formula I' wherein R 1 is selected from 6,7- dimethoxy-4-quinolinyl, 6-methoxy-7-(dimethylaminopropoxy)-4-quinolinyl, 6-methoxy-7-(4- morpholinylpropoxy)-4-quinolinyl, 6,7-dimethoxy-4-quinazolinyl, and 6-methoxy-7- (dimethylaminopropoxy)-4-quinazolinyl.
- the invention also relates to compounds of Formula I' wherein R is selected from cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, phenyl, 4-methylphenyl, 3-methylphenyl, 2-methylphenyl, 2- ethylphenyl, 4-tert-butyl-phenyl, 2,3-dimethylphenyl, 4-fluorophenyl, 3 -fluorophenyl, 2-fluorophenyl, 2,4-difluorophenyl, 3,4-difluorophenyl, 4-bromophenyl, 4-chlorophenyl, 3-chlorophenyl, 2-chlorophenyl, 2,4-dichlorophenyl, 3-chloro-4-fluorophenyl, 2-methoxyphenyl, 3-methoxyphenyl, 4-methoxyphenyl, 4-methoxyphenyl, 4-methoxyphenyl, 4-methoxyphenyl
- the invention also relates to compounds of Formula I' wherein X is O.
- the invention also relates to compounds of Formula F selected from N-[6-(6,7-Dimethoxy-quinolin-4-yloxy)-benzothiazol-2-yl]-3-methyl-benzamide; Thiophene-3-carboxylic acid [6-(6,7-dimethoxy-quinolin-4-yloxy)-benzothiazol-2-yl]-amide; 2-Phenyl-N-[6-(7-trifluoromethyl-quinolin-4-yloxy)-benzothiazol-2-yl]-acetamide; N-[6-(2-Methyl-pyridin-4-yloxy)-benzothiazol-2-yl]-2-phenyl-acetamide;
- the invention also relates to compounds of Formula JJ wherein R' is an unsubstituted or substituted ring selected from phenyl, cyclobutyl, cyclopentyl, cyclohexyl, 2-pyrrolidinyl, 2-pyrrolyl, 5- imidazolyl, 5-pyrazolyl, 2-pyrazinyl, 4-pyrimidinyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 8-quinolinyl, 2,3- dihydrobenzofur-7-yl, 2,3-dihydro-l,4-benzodioxin-5-yl, l,3-benzodioxol-4-yl, 4-isoxazolyl, 3- isothiazolyl, 5-oxazolyl, 4-thiazolyl, 5-thiazolyl, 2-furanyl, 3-furanyl, 3-thienyl and 2-thienyl; and pharmaceutically acceptable derivatives thereof; in conjunction with any of the above
- the invention also relates to compounds of Formula II wherein R' is a ring selected from phenyl, cyclobutyl, cyclopentyl, cyclohexyl, 2-pyrrolidinyl, 2-pyrrolyl, 5-imidazolyl, 5-pyrazolyl, 2-pyrazinyl, 4- pyrimidinyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 8-quinolinyl, 2,3-dihydrobenzofur-7-yl, 2,3-dihydro-l,4- benzodioxin-5-yl, l,3-benzodioxol-4-yl, 4-isoxazolyl, 3-isothiazolyl, 5-oxazolyl, 4-thiazolyl, 5-thiazolyl, 2-furanyl, 3-furanyl, 3-thienyl and 2-thienyl; wherein the ring is substituted with or or two substituents selected from methoxymethylthi
- 3 -alkyl and an unsubstituted or substituted ring selected from phenyl, naphthyl, 1,3-benzodioxolyl, 2,3-dihydro-l,4-benzodioxinyl, C 3-6 -cycloalkyl, C 5-6 - cycloalkenyl, pyrrolidinyl, pyrrolyl, imidazolyl, pyrazolyl, pyrazinyl, pyrimidinyl, pyridyl, quinolinyl, dihydrothiazolyl, 2,3-dihydrobenzofuryl, piperidinyl, 1-methyl-oxopyridyl, tetrahydropyran-4-yl, indolinyl, imidazo[l,2-a]pyridinyl, quinolinyl, benzofuryl, benzo[l,2,5]thiadiazolyl, benzothiazolyl, benzimidazoly
- the invention also relates to compounds of wherein R' is selected from H, methyl, ethyl, n-butyl, isobutyl, tert-butyl, isopropyl, propyl, cyanomethyl, aminocarbonylmethyl, dimethylaminocarbonylmethyl, dimethylaminoethyl, 2-methoxy-l -methylethyl, methoxycarbonylmethyl, methoxyethyl, methoxypropyl, methylsulfonylethyl, dimethylaminoethyl, methoxycarbonylmethyl, ethenyl, thiazol-2-yl-CH(CH 3 )-, phenyl-CH(CH 3 )-, 5-methylisoxazol-3-ylmethyl, pynolidin-1-ylethyl, tetrahydrofur-2-ylmethyl, 4-methyl-2-oxo-oxazolidin-5-yl, pyrid
- R 10a is methoxy; and wherein R 10b is selected from 4-morpholinopropoxy, 2-hydroxy-3- morpholin-4-yl-propoxy, pyrrolidin-1-ylpropoxy, 1-pyreolidinylethoxy, 4-piperidinyloxypropoxy, (4- methylpiperazin- 1 -yl)propoxy, 3 -(4-methylpiperazin- 1 -yl)propoxy, 3 -( 1 ,2,4-triazol- 1 -yl)propoxy, triazinylpropoxy, 3-(piperidin-4-yl)propoxy, dimethylaminoethoxy, dimethylaminopropoxy and methoxy.
- the invention also relates to compounds of wherein R 8 is H; and wherein R 9 is H, methyl or fluoro.
- the invention also relates to compounds of wherein X is O.
- the invention also relates to compounds of wherein R' is selected from cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, phenyl, 4-methylphenyl, 3-methylphenyl, 2-methylphenyl, 2-ethylphenyl, 3- isopropylphenyl, 4-tert-butyl-phenyl, 2,3-dimethylphenyl, 4-isopropyl-3-methylphenyl, 3-chloro-4- methylphenyl, 4-fluorophenyl, 3 -fluorophenyl, 2-fluorophenyl, 2,4-difluorophenyl, 3,4-difluorophenyl, 4- bromophenyl, 4-chlorophenyl, 3-chlorophenyl, 2-chlorophenyl, 2,4-dichlorophenyl, 3,4-dichlorophenyl, 3-chloro-4-fluorophenyl, 2-methoxy
- the invention also relates to compounds of wherein Z is CH; wherein R 10a is methoxy; and wherein R 10b is selected from 4-morpholinopropoxy, 2-hydroxy-3-morpholin-4-yl-propoxy, pynolidin-1- ylpropoxy, l-pyrrolidinylethoxy, 4-piperidinyloxypropoxy, (4-methylpiperazin-l-yl)propoxy, 3-(4- methylpiperazin-l-yl)propoxy, 3-(l,2,4-friazol-l-yl)propoxy, friazinylpropoxy, 3-(piperidin-4-yl)propoxy, dimethylaminoethoxy, dimethylaminopropoxy and methoxy.
- the invention also relates to compounds of wherein R' is selected from H, methyl, ethyl, n-butyl, isobutyl, tert-butyl, isopropyl, propyl, cyanomethyl, aminocarbonylmethyl, dimethylaminocarbonylmethyl, dimethylaminoethyl, 2-methoxy- 1 -methylethyl, methoxycarbonylmethyl, methoxyethyl, methoxypropyl, methylsulfonylethyl, dimethylaminoethyl, methoxycarbonylmethyl, ethenyl, thiazol-2-yl-CH(CH 3 )-, phenyl-CH(CH 3 )-, 5-methylisoxazol-3-ylmethyl, pyrrolidin-1-ylethyl, tetrahydrofur-2-ylmethyl, 4-methyl-2-oxo-oxazolidin-5-yl, pyrid-4
- the invention also relates to compounds of wherein R 8 and R 9 is H.
- the invention also relates to compounds of wherein R 10b is methoxy; and pharmaceutically acceptable salts thereof.
- the invention also relates to compounds of wherein R a is methyl.
- the invention also relates to compounds of wherein Z is CH.
- the invention also relates to compounds selected from N-(6-((6,7-bis(methyloxy)-4-quinolinyl)oxy)-l-methyl-lH-indazol-3-yl)-4-chlorobenzamide; N-(6-((6,7-bis(methyloxy)-4-quinolinyl)oxy)- 1 -methyl- 1 H-indazol-3 -yl)-4- (trifluoromethyl)benzamide; N-(6-((6,7-bis(methyloxy)-4-quinolinyl)oxy)-l-methyl-lH-indazol-3-yl)-4-(methyloxy)benzamide; N-(6-((6,7-bis(methyloxy)-4-quinolinyl)oxy)-l-methyl-lH-indazol-3-yl)-3-chloro-4-fluorobenzamide; N-(6-((6,7-bis(methyloxy)-4-quinolinyl)oxy)-l -methyl- 1 H
- compounds of the present invention would be useful for, but not limited to, the prevention or treatment of angiogenesis related diseases.
- the compounds of the invention have kinase inhibitory activity, such as NEGFR/KDR inhibitory activity.
- the compounds of the invention are useful in therapy as antineoplasia agents or to minimize deleterious effects of VEGF.
- neoplasia including cancer and metastasis, including, but not limited to: carcinoma such as cancer of the bladder, breast, colon, kidney, liver, lung (including small cell lung cancer), esophagus, gall-bladder, ovary, pancreas, stomach, cervix, thyroid, prostate, and skin (including squamous cell carcinoma); hematopoietic tumors of lymphoid lineage (including leukemia, acute lymphocitic leukemia, acute lymphoblastic leukemia, B-cell lymphoma, T-cell-lymphoma, Hodgkin's lymphoma, non-Hodgkin's lymphoma, hairy cell lymphoma and Burkett's lymphoma); hematopoietic tumors of myeloid lineage (including acute and chronic myelogenous leukemias, myelodysplastic syndrome and promyelocytic leukemia); tumors of me
- the compounds are useful for the treatment of neoplasia selected from lung cancer, colon cancer and breast cancer.
- the compounds of the present invention are also useful in the treatment of cancer related indications such as solid tumors, sarcomas (especially Ewing's sarcoma and osteosarcoma), retinoblastoma, rhabdomyosarcomas, neuroblastoma, hematopoietic malignancies, including leukemia and lymphoma, tumor- induced pleural or pericardial effusions, and malignant ascites.
- the compounds of this invention may also act as inhibitors of other protein kinases, e.g. tie-2, lck, src, fgf, KDR, ron, ckit and ret, and thus be effective in the treatment of diseases associated with other protein kinases.
- these compounds are also useful for veterinary treatment of companion animals, exotic animals and farm animals, including mammals, rodents, and the like. More preferred animals include horses, dogs, and cats.
- the compounds of the present invention include the pharmaceutically acceptable derivatives thereof. Where the plural form is used for compounds, salts, and the like, this is taken to mean also a Sindle compound, salt and the like.
- Angiogenesis is defined as any alteration of an existing vascular bed or the formation of new vasculature which benefits tissue perfasion. This includes the formation of new vessels by sprouting of endothelial cells from existing blood vessels or the remodeling of existing vessels to alter size, maturity, direction or flow properties to improve blood perfusion of tissue.
- HGF refers to hepatocyte growth factor/scatter factor.
- HGF hepatocyte growth factor/scatter factor
- fragments of hepatocyte growth factor/scatter factor chemically synthesized fragments of hepatocyte growth factor/scatter factor, derivatives or mutated versions of hepatocyte growth factor/scatter factor
- fusion proteins comprising hepatocyte growth factor/scatter factor and another protein.
- HGF as used herein also includes hepatocyte growth factor/scatter factor isolated from species other than humans.
- c-Met refers to the receptor for HGF. This includes purified receptor, fragments of receptor, chemically synthesized fragments of receptor, derivatives or mutated versions of receptor, and fusion proteins comprising the receptor and another protein.
- c-Met as used herein also includes the HGF receptor isolated from a species other than humans.
- HGF refers to hepatocyte growth factor/scatter factor. This includes purified hepatocyte growth factor/scatter factor, fragments of hepatocyte growth factor/scatter factor, chemically synthesized fragments of hepatocyte growth factor/scatter factor, derivatives or mutated versions of hepatocyte growth factor/scatter factor, and fusion proteins comprising hepatocyte growth factor/scatter factor and another protein.
- HGF as used herein also includes hepatocyte growth factor/scatter factor isolated from species other than humans.
- c-Met refers to the receptor for HGF.
- c-Met as used herein also includes the HGF receptor isolated from a species other than humans.
- hepatocyte growth factor and "HGF” refer to a growth factor typically having a structure with six domains (finger, Kringle 1, Kringle 2, Kringle 3, Kringle 4 and serine protease domains). Fragments of HGF constitute HGF with fewer domains and variants of HGF may have some of the domains of HGF repeated; both are included if they still retain their respective ability to bind a HGF receptor.
- hepatocyte growth factor and HGF include hepatocyte growth factor from humans (“huHGF”) and any non-human mammalian species, and in particular rat HGF.
- the terms as used herein include mature, pre, pre-pro, and pro forms, purified from a natural source, chemically synthesized or recombinantly produced.
- Human HGF is encoded by the cDNA sequence published by Miyazawa et al. (1989), supra, or Nakamura et al. (1989), supra.
- the sequences reported by Miyazawa et al. and Nakamura et al. differ in 14 amino acids. The reason for the differences is not entirely clear; polymorphism or cloning artifacts are among the possibilities.
- HGF hepatocyte growth factor
- HGF receptor and "c-Met” include the polypeptide molecule that comprises the full-length, native amino acid sequence encoded by the gene variously known as pl90.sup.MET.
- the present definition specifically encompasses soluble forms of HGF receptor, and HGF receptor from natural sources, synthetically produced in vitro or obtained by genetic manipulation including methods of recombinant DNA technology.
- the HGF receptor variants or fragments preferably share at least about 65% sequence homology, and more preferably at least about 75% sequence homology with any domain of the human c-Met amino acid sequence published in Rodrigues et al., Mol. Cell. BioL, 11 :2962-2970 (1991); Park et al., Proc. Natl. Acad. Sci., 84:6379-
- agonist and agonistic when used herein refer to or describe a molecule which is capable of, directly or indirectly, substantially inducing, promoting or enhancing HGF biological activity or HGF receptor activation.
- cancer and “cancerous” when used herein refer to or describe the physiological condition in mammals that is typically characterized by unregulated cell growth. Examples of cancer include but are not limited to, carcinoma, lymphoma, sarcoma, blastoma and leukemia.
- cancers include squamous cell carcinoma, lung cancer, pancreatic cancer, cervical cancer, bladder cancer, hepatoma, breast cancer, colon carcinoma, and head and neck cancer.
- cancer as used herein is not limited to any one specific form of the disease, it is believed that the methods of the invention will be particularly effective for cancers which are found to be accompanied by increased levels of HGF or expression of c-Met in the mammal.
- treating,” “treatment,” and “therapy” as used herein refer to curative therapy, prophylactic therapy, and preventative therapy.
- mammal refers to any mammal classified as a mammal, including humans, cows, horses, dogs and cats.
- the mammal is a human.
- treatment includes therapeutic treatment as well as prophylactic treatment (either preventing the onset of disorders altogether or delaying the onset of a pre-clinically evident stage of disorders in individuals).
- a “pharmaceutically-acceptable derivative” denotes any salt, ester of a compound of this invention, or any other compound which upon administration to a patient is capable of providing (directly or indirectly) a compound of this invention, or a metabolite or residue thereof, characterized by the ability to inhibit angiogenesis.
- the phrase "therapeutically-effective" is intended to qualify the amount of each agent, which will achieve the goal of improvement in disorder severity and the frequency of incidence over treatment of each agent by itself, while avoiding adverse side effects typically associated with alternative therapies.
- H denotes a single hydrogen atom.
- This radical may be attached, for example, to an oxygen atom to form a hydroxyl radical.
- alkyl is used, either alone or within other terms such as “haloalkyl” and “alkylamino", it embraces linear or branched radicals having one to about twelve carbon atoms. More prefened alkyl radicals are "lower alkyl” radicals having one to about six carbon atoms.
- radicals examples include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, pentyl, isoamyl, hexyl and the like. Even more prefened are lower alkyl radicals having one or two carbon atoms.
- alkylenyl embraces bridging divalent alkyl radicals such as methylenyl and ethylenyl.
- lower alkyl substituted with R 2 does not include an acetal moiety.
- alkenyl embraces linear or branched radicals having at least one carbon-carbon double bond of two to about twelve carbon atoms. More prefened alkenyl radicals are “lower alkenyl” radicals having two to about six carbon atoms. Most prefened lower alkenyl radicals are radicals having two to about four carbon atoms. Examples of alkenyl radicals include ethenyl, propenyl, allyl, propenyl, butenyl and 4-methylbutenyl.
- alkenyl and “lower alkenyl” embrace radicals having "cis” and “trans” orientations, or alternatively, "E” and "Z” orientations.
- alkynyl denotes linear or branched radicals having at least one carbon-carbon triple bond and having two to about twelve carbon atoms. More prefened alkynyl radicals are "lower alkynyl” radicals having two to about six carbon atoms. Most prefened are lower alkynyl radicals having two to about four carbon atoms. Examples of such radicals include propargyl, butynyl, and the like.
- halo means halogens such as fluorine, chlorine, bromine or iodine atoms.
- haloalkyl embraces radicals wherein any one or more of the alkyl carbon atoms is substituted with halo as defined above.
- monohaloalkyl, dihaloalkyl and polyhaloalkyl radicals including perhaloalkyl may have either an iodo, bromo, chloro or fluoro atom within the radical.
- Dihalo and polyhaloalkyl radicals may have two or more of the same halo atoms or a combination of different halo radicals.
- “Lower haloalkyl” embraces radicals having 1-6 carbon atoms. Even more prefened are lower haloalkyl radicals having one to three carbon atoms.
- haloalkyl radicals include fluoromethyl, difluoromethyl, trifluoromethyl, chloromethyl, dichloromethyl, trichloromethyl, pentafluoroethyl, heptafluoropropyl, difluorochloromethyl, dichlorofluoromethyl, difluoroethyl, difluoropropyl, dichloroethyl and dichloropropyl.
- Perfluoroalkyl means alkyl radicals having all hydrogen atoms replaced with fluoro atoms. Examples include trifluoromethyl and pentafluoroethyl.
- hydroxyalkyl embraces linear or branched alkyl radicals having one to about ten carbon atoms any one of which may be substituted with one or more hydroxyl radicals. More prefened hydroxyalkyl radicals are "lower hydroxyalkyl” radicals having one to six carbon atoms and one or more hydroxyl radicals. Examples of such radicals include hydroxymethyl, hydroxyethyl, hydroxypropyl, hydroxybutyl and hydroxyhexyl. Even more prefened are lower hydroxyalkyl radicals having one to three carbon atoms.
- alkoxy embrace linear or branched oxy-containing radicals each having alkyl portions of one to about ten carbon atoms.
- More prefened alkoxy radicals are "lower alkoxy" radicals having one to six carbon atoms. Examples of such radicals include methoxy, ethoxy, propoxy, butoxy and tert- butoxy. Even more prefened are lower alkoxy radicals having one to three carbon atoms. Alkoxy radicals may be further substituted with one or more halo atoms, such as fluoro, chloro or bromo, to provide "haloalkoxy" radicals. Even more prefened are lower haloalkoxy radicals having one to three carbon atoms. Examples of such radicals include fluoromethoxy, chloromethoxy, trifluoromethoxy, trifluoroethoxy, fluoroethoxy and fluoropropoxy.
- aryl alone or in combination, means a carbocyclic aromatic system containing one or two rings wherein such rings may be attached together in a fused manner.
- aryl embraces aromatic radicals such as phenyl, naphthyl, indenyl, tetrahydronaphthyl, and indanyl. More prefened aryl is phenyl.
- Said "aryl” group may have 1 to 3 substituents such as lower alkyl, hydroxyl, halo, haloalkyl, nitro, cyano, alkoxy and lower alkylamino. Phenyl substituted with -O-CH 2 -O- forms the aryl benzodioxolyl substituent.
- heterocyclyl embraces saturated, partially saturated and unsaturated heteroatom- containing ring radicals, where the heteroatoms may be selected from nitrogen, sulfur and oxygen. It does not include rings containing -O-O-,-O-S- or -S-S- portions.
- Said "heterocyclyl” group may have 1 to 3 substituents such as hydroxyl, Boc, halo, haloalkyl, cyano, lower alkyl, lower aralkyl, oxo, lower alkoxy, amino and lower alkylamino.
- saturated heterocyclic radicals include saturated 3 to 6-membered heteromonocyclic groups containing 1 to 4 nitrogen atoms [e.g.
- pynolidinyl imidazolidinyl, piperidinyl, pyrrolinyl, piperazinyl]; saturated 3 to 6-membered heteromonocyclic group containing 1 to 2 oxygen atoms and 1 to 3 nitrogen atoms [e.g. morpholinyl]; saturated 3 to 6-membered heteromonocyclic group containing 1 to 2 sulfur atoms and 1 to 3 nifrogen atoms [e.g., thiazolidinyl].
- partially saturated heterocyclyl radicals include dihydrothienyl, dihydropyranyl, dihydrofuryl and dihydrothiazolyl.
- unsaturated aromatic heterocyclic radicals also termed "heteroaryl” radicals
- unsaturated aromatic heterocyclic radicals include unsaturated 5 to 6 membered heteromonocyclyl group containing 1 to 4 nitrogen atoms, for example, pynolyl, imidazolyl, pyrazolyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, pyrimidyl, pyrazinyl, pyridazinyl, friazolyl [e.g., 4H-l,2,4-triazolyl, lH-l,2,3-triazolyl, 2H-l,2,3-triazolyl]; unsaturated 5- to 6-membered heteromonocyclic group containing an oxygen atom, for example, pyranyl, 2-furyl, 3-furyl, etc.; unsaturated 5 to 6-membered heteromonocyclic group containing a sulfur atom, for example, 2-thienyl, 3-
- heterocyclyl also embraces radicals where heterocyclic radicals are fused/condensed with aryl radicals: unsaturated condensed heterocyclic group containing 1 to 5 nitrogen atoms, for example, indolyl, isoindolyl, indolizinyl, benzimidazolyl, quinolyl, isoquinolyl, indazolyl, benzotriazolyl, tetrazolopyridazinyl [e.g., tetrazolo [l,5-b]pyridazinyl]; unsaturated condensed heterocyclic group containing 1 to 2 oxygen atoms and 1 to 3 nifrogen atoms [e.g.
- Prefened heterocyclic radicals include five to ten membered fused or unfused radicals.
- heteroaryl radicals include quinolyl, isoquinolyl, imidazolyl, pyridyl, thienyl, thiazolyl, oxazolyl, furyl, and pyrazinyl.
- Other prefened heteroaryl radicals are 5- or 6- membered heteroaryl, containing one or two heteroatoms selected from sulfur, nitrogen and oxygen, selected from thienyl, furyl, pynolyl, indazolyl, pyrazolyl, oxazolyl, friazolyl, imidazolyl, pyrazolyl, isoxazolyl, isothiazolyl, pyridyl, piperidinyl and pyrazinyl.
- non-nitrogen containing heteroaryl include pyranyl, 2-furyl, 3-furyl, 2- thienyl, 3-thienyl, benzofuryl, benzothienyl, and the like.
- partially saturated and saturated heterocyclyl include pyrrolidinyl, imidazolidinyl, piperidinyl, pynolinyl, pyrazolidinyl, piperazinyl, morpholinyl, tetrahydropyranyl, thiazolidinyl, dihydrothienyl, 2,3-dihydro-benzo[l,4]dioxanyl, indolinyl, isoindolinyl, dihydrobenzothienyl, dihydrobenzofuryl, isochromanyl, chromanyl, 1 ,2-dihydroquinolyl, 1,2,3,4- tetrahydro-isoquinolyl, 1,2,3,4-tetrahydro-quino
- alkylsulfonyl denotes respectively divalent radicals -SO 2 -.
- sulfamyl denotes a sulfonyl radical substituted with an amine radical, forming a sulfonamide (-S0 2 NH 2 ).
- alkylaminosulfonyl includes "N-alkylaminosulfonyl” where sulfamyl radicals are independently substituted with one or two alkyl radical(s).
- More prefened alkylaminosulfonyl radicals are "lower alkylaminosulfonyl” radicals having one to six carbon atoms. Even more prefened are lower alkylaminosulfonyl radicals having one to three carbon atoms. Examples of such lower alkylaminosulfonyl radicals include N-methylaminosulfonyl, and N-ethylaminosulfonyl.
- the tenri "carbonyl”, whether used alone or with other terms, such as “aminocarbonyl”, denotes - (C 0)-.
- N-alkylaminocarbonyl and N,N-dialkylaminocarbonyl denote aminocarbonyl radicals independently substituted with one or two alkyl radicals, respectively. More prefened are "lower alkylaminocarbonyl” having lower alkyl radicals as described above attached to an aminocarbonyl radical.
- N-arylaminocarbonyl and “N-alkyl-N-arylaminocarbonyl” denote aminocarbonyl radicals substituted, respectively, with one aryl radical, or one alkyl and one aryl radical.
- heterocyclylalkylenyl and “heterocyclylalkyl” embrace heterocyclic-substituted alkyl radicals. More prefened heterocyclylalkyl radicals are "5- or 6-membered heteroarylalkyl” radicals having alkyl portions of one to six carbon atoms and a 5- or 6-membered heteroaryl radical.
- Even more prefened are lower heteroarylalkylenyl radicals having alkyl portions of one to three carbon atoms. Examples include such radicals as pyridylmethyl and thienylmethyl.
- arylalkyl and aralkyl embraces aryl-substituted alkyl radicals.
- Preferable aralkyl radicals are "lower aralkyl” radicals having aryl radicals attached to alkyl radicals having one to six carbon atoms.
- Even more prefened are "phenylalkylenyl” attached to alkyl portions having one to three carbon atoms. Examples of such radicals include benzyl, diphenylmethyl and phenylethyl.
- alkylthio embraces radicals containing a linear or branched alkyl radical, of one to ten carbon atoms, attached to a divalent sulfur atom. Even more prefened are lower alkylthio radicals having one to three carbon atoms.
- alkylthio is methylthio, (CH 3 S-).
- haloalkylthio embraces radicals containing a haloalkyl radical, of one to ten carbon atoms, attached to a divalent sulfur atom.
- alkylamino embraces "N-alkylamino" and "N,N-dialkylamino" where amino groups are independently substituted with one alkyl radical and with two alkyl radicals, respectively. More prefened alkylamino radicals are "lower alkylamino” radicals having one or two alkyl radicals of one to six carbon atoms, attached to a nitrogen atom. Even more prefened are lower alkylamino radicals having one to three carbon atoms.
- Suitable alkylamino radicals may be mono or dialkylamino such as N- methylamino, N-ethylamino, N,N-dimethylamino, N,N-diethylamino and the like.
- arylamino denotes amino groups which have been substituted with one or two aryl radicals, such as N-phenylamino.
- the arylamino radicals may be further substituted on the aryl ring portion of the radical.
- heteroarylamino denotes amino groups which have been substituted with one or two heteroaryl radicals, such as N-thienylamino.
- heteroarylamino radicals may be further substituted on the heteroaryl ring portion of the radical.
- aralkylamino denotes amino groups which have been substituted with one or two aralkyl radicals. More prefened are phenyl-C ⁇ -C 3 -alkylamino radicals, such as N-benzylamino.
- the aralkylamino radicals may be further substituted on the aryl ring portion.
- N-alkyl-N-arylamino and “N-aralkyl-N-alkylamino” denote amino groups which have been independently substituted with one aralkyl and one alkyl radical, or one aryl and one alkyl radical, respectively, to an amino group.
- aminoalkyl embraces linear or branched alkyl radicals having one to about ten carbon atoms any one of which may be substituted with one or more amino radicals. More prefened aminoalkyl radicals are "lower aminoalkyl” radicals having one to six carbon atoms and one or more amino radicals. Examples of such radicals include aminomethyl, aminoethyl, aminopropyl, aminobutyl and aminohexyl.
- alkylaminoalkyl embraces alkyl radicals substituted with alkylamino radicals. More prefened alkylaminoalkyl radicals are "lower alkylaminoalkyl” radicals having alkyl radicals of one to six carbon atoms. Even more prefened are lower alkylaminoalkyl radicals having alkyl radicals of one to three carbon atoms.
- Suitable all ylaminoalkyl radicals may be mono or dialkyl substituted, such as N- methylaminomethyl, N,N-dimethyl-aminoethyl, N,N-diethylaminomethyl and the like.
- alkylaminoalkoxy embraces alkoxy radicals substituted with alkylamino radicals. More prefened alkylaminoalkoxy radicals are "lower alkylaminoalkoxy" radicals having alkoxy radicals of one to six carbon atoms. Even more prefened are lower alkylaminoalkoxy radicals having alkyl radicals of one to three carbon atoms.
- alkylaminoalkoxy radicals may be mono or dialkyl substituted, such as N-methylaminoethoxy, N,N-dimethylaminoethoxy, N,N-diethylaminoethoxy and the like.
- alkylaminoalkoxyalkoxy embraces alkoxy radicals substituted with alkylaminoalkoxy radicals. More prefened alkylaminoalkoxyalkoxy radicals are "lower alkylaminoalkoxyalkoxy" radicals having alkoxy radicals of one to six carbon atoms. Even more prefened are lower alkylaminoalkoxyalkoxy radicals having alkyl radicals of one to three carbon atoms.
- Suitable alkylaminoalkoxyalkoxy radicals may be mono or dialkyl substituted, such as N- methylaminomethoxyethoxy, N-methylaminoethoxyethoxy, N,N-dimethylaminoethoxyethoxy, N,N- diethylaminomethoxymethoxy and the like.
- the term "carboxyalkyl” embraces linear or branched alkyl radicals having one to about ten carbon atoms any one of which may be substituted with one or more carboxy radicals. More prefened carboxyalkyl radicals are "lower carboxyalkyl" radicals having one to six carbon atoms and one carboxy radical.
- radicals examples include carboxymethyl, carboxypropyl, and the like. Even more prefened are lower carboxyalkyl radicals having one to three CH 2 groups.
- halosulfonyl embraces sulfonyl radicals substituted with a halogen radical. Examples of such halosulfonyl radicals include chlorosulfonyl and fluorosulfonyl.
- arylthio embraces aryl radicals of six to ten carbon atoms, attached to a divalent sulfur atom. An example of “arylthio” is phenylthio.
- aralkylthio embraces aralkyl radicals as described above, attached to a divalent sulfur atom.
- More prefened are phenyl-C ⁇ -C 3 -alkylthio radicals.
- An example of “aralkylthio” is benzylthio.
- aryloxy embraces optionally substituted aryl radicals, as defined above, attached to an oxygen atom. Examples of such radicals include phenoxy.
- aralkoxy embraces oxy-containing aralkyl radicals attached through an oxygen atom to other radicals. More prefened aralkoxy radicals are "lower aralkoxy” radicals having optionally substituted phenyl radicals attached to lower alkoxy radical as described above.
- heteroaryloxy embraces optionally substituted heteroaryl radicals, as defined above, attached to an oxygen atom.
- heteroarylalkoxy embraces oxy-containing heteroarylalkyl radicals attached through an oxygen atom to other radicals. More prefened heteroarylalkoxy radicals are "lower heteroarylalkoxy” radicals having optionally substituted heteroaryl radicals attached to lower alkoxy radical as described above.
- cycloalkyl includes saturated carbocyclic groups. Prefened cycloalkyl groups include C 3 -C 6 rings. More prefened compounds include, cyclopentyl, cyclopropyl, and cyclohexyl.
- cycloalkylalkyl embraces cycloalkyl-substituted alkyl radicals.
- Preferable cycloalkylalkyl radicals are "lower cycloalkylalkyl” radicals having cycloalkyl radicals attached to alkyl radicals having one to six carbon atoms. Even more prefened are "5-6-membered cycloalkylalkyl” attached to alkyl portions having one to three carbon atoms. Examples of such radicals include cyclohexylmethyl.
- the cycloalkyl in said radicals may be additionally substituted with halo, alkyl, alkoxy and hydroxy.
- cycloalkenyl includes carbocyclic groups having one or more carbon-carbon double bonds including “cycloalkyldienyl” compounds.
- Prefened cycloalkenyl groups include C 3 -C 6 rings. More prefened compounds include, for example, cyclopentenyl, cyclopentadienyl, cyclohexenyl and cycloheptadienyl .
- the term “comprising” is meant to be open ended, including the indicated component but not excluding other elements.
- Formmulas I-ffl includes any sub formulas.
- the compounds of the invention are endowed with kinase inhibitory activity, such as c-Met inhibitory activity.
- the present invention also comprises the use of a compound of the invention, or pharmaceutically acceptable salt thereof, in the manufacture of a medicament for the treatment either acutely or chronically of an angiogenesis mediated disease state, including those described previously.
- the compounds of the present invention are useful in the manufacture of an anti-cancer medicament.
- the compounds of the present invention are also useful in the manufacture of a medicament to attenuate or prevent disorders through inhibition of c-Met.
- the present invention comprises a pharmaceutical composition comprising a therapeutically- effective amount of a compound of Formulas I-TTI in association with a least one pharmaceutically- acceptable canier, adjuvant or diluent.
- the present invention also comprises a method of treating angiogenesis related disorders in a subject having or susceptible to such disorder, the method comprising treating the subject with a therapeutically-effective amount of a compound of Formula I-III.
- a compound of Formula I-III can be administered as the sole active pharmaceutical agent, they can also be used in combination with one or more compounds of the invention or other agents.
- the therapeutic agents can be formulated as separate compositions that are administered at the same time or sequentially at different times, or the therapeutic agents can be given as a single composition.
- co-therapy in defining use of a compound of the present invention and another pharmaceutical agent, is intended to embrace administration of each agent in a sequential manner in a regimen that will provide beneficial effects of the drug combination, and is intended as well to embrace co-administration of these agents in a substantially simultaneous manner, such as in a single capsule having a fixed ratio of these active agents or in multiple, separate capsules for each agent.
- administration of compounds of the present invention may be in conjunction with additional therapies known to those skilled in the art in the prevention or treatment of neoplasia, such as with radiation therapy or with cytostatic or cytotoxic agents. If formulated as a fixed dose, such combination products employ the compounds of this invention within the accepted dosage ranges.
- Compounds of Formula I may also be administered sequentially with known anticancer or cytotoxic agents when a combination formulation is inappropriate.
- the invention is not limited in the sequence of administration; compounds of the invention may be administered either prior to, simultaneous with or after administration of the known anticancer or cytotoxic agent.
- standard treatment of primary tumors consists of surgical excision followed by either radiation or IN administered chemotherapy.
- the typical chemotherapy regime consists of either D ⁇ A alkylating agents, D ⁇ A intercalating agents, CDK inhibitors, or microtubule poisons.
- the chemotherapy doses used are just below the maximal tolerated dose and therefore dose limiting toxicities typically include, nausea, vomiting, dianhea, hair loss, neutropenia and the like.
- antineoplastic agents available in commercial use, in clinical evaluation and in pre-clinical development, which would be selected for treatment of neoplasia by combination drug chemotherapy.
- Such antineoplastic agents fall into several major categories, namely, antibiotic-type agents, alkylating agents, antimetabolite agents, hormonal agents, immunological agents, interferon-type agents and a category of miscellaneous agents.
- a first family of antineoplastic agents which may be used in combination with compounds of the present invention consists of antimetabolite-type/thymidilate synthase inhibitor antineoplastic agents.
- Suitable antimetabolite antineoplastic agents may be selected from but not limited to the group consisting of 5-FU-fibrinogen, acanthifolic acid, aminothiadiazole, brequinar sodium, carmofur, Ciba-Geigy CGP- 30694, cyclopentyl cytosine, cytarabine phosphate stearate, cytarabine conjugates, Lilly DATHF, Menel Dow DDFC, dezaguanine, dideoxycytidine, dideoxyguanosine, didox, Yoshitomi DMDC, doxifluridine, Wellcome EH ⁇ A, Merck & Co.
- EX-015 isopropyl pynolizine, Lilly LY-188011, Lilly LY-264618, methobenzaprim, methotrexate, Wellcome MZPES, norspermidine, NCI NSC- 127716, NCI NSC-264880, NCI NSC-39661, NCI NSC-612567, Warner-Lambert PALA, pentostatin, piritrexim, plicamycin, Asahi Chemical PL- AC, Takeda TAC-788, thioguanine, tiazofurin, Erbamont TIF, trimetrexate, tyrosine kinase inhibitors, Taiho UFT and uricytin.
- a second family of antineoplastic agents which may be used in combination with compounds of the present invention consists of alkylating-type antineoplastic agents.
- Suitable alkylating-type antineoplastic agents may be selected from but not limited to the group consisting of Shionogi 254-S, aldo-phosphamide analogues, altretamine, anaxirone, Boehringer Mannheim BBR-2207, bestrabucil, budotitane, Wakunaga CA-102, carboplatin, carmustine, Chinoin-139, Chinoin-153, chlorambucil, cisplatin, cyclophosphamide, American Cyanamid CL-286558, Sanofi CY-233, cyplatate, Degussa D-19- 384, Sumimoto DACHP(Myr)2, diphenylspiromustine, diplatinum cytostatic, Erba distamycin derivatives, Chugai DWA-2114R, ITI
- a third family of antineoplastic agents which may be used in combination with compounds of the present invention consists of antibiotic-type antineoplastic agents.
- Suitable antibiotic-type antineoplastic agents may be selected from but not limited to the group consisting of Taiho 4181 -A, aclarubicin, actinomycin D, actinoplanone, Erbamont ADR-456, aeroplysinin derivative, Ajinomoto AN-201-II, Ajinomoto AN-3, Nippon Soda anisomycins, anthracycline, azino-mycin-A, bisucaberin, Bristol-Myers BL-6859, Bristol-Myers BMY-25067, Bristol-Myers BMY-25551, Bristol-Myers BMY-26605, Bristol- Myers BMY-27557, Bristol-Myers BMY-28438, bleomycin sulfate, bryostatin-1, Taiho C-1027, calichemycin, chrom
- a fourth family of antineoplastic agents which may be used in combination with compounds of the present invention consists of a miscellaneous family of antineoplastic agents, including tubulin interacting agents, topoisomerase JJ inhibitors, topoisomerase I inhibitors and hormonal agents, selected from but not limited to the group consisting of ⁇ -carotene, ⁇ -difluoromethyl-arginine, acitretin, Biotec AD-5, Kyorin AHC-52, alstonine, amonafide, amphethinile, amsacrine, Angiostat, ankinomycin, anti- neoplaston A10, antineoplaston A2, antineoplaston A3, antineoplaston A5, antineoplaston AS2-1, Henkel APD, aphidicolin glycinate, asparaginase, Avarol, baccharin, batracylin, benfluron, benzofript, Ipsen- Beaufour BLM-23015,
- the present compounds may also be used in co-therapies with other anti-neoplastic agents, such as acemannan, aclarubicin, aldesleukin, alemtuzumab, alitretinoin, altretamine, amifostine, aminolevulinic acid, amrubicin, amsacrine, anagrelide, anastrozole, ANCER, ancestim, ARGLABIN, arsenic trioxide, BAM 002 (Novelos), bexarotene, bicalutamide, broxuridine, capecitabine, celmoleukin, cetrorelix, cladribine, clotrimazole, cytarabine ocfosfate, DA 3030 (Dong-A), daclizumab, denileukin diftitox, deslorelin, dexrazoxane, dilazep, docetaxel, docosanol,
- the present compounds may also be used in co-therapies with other anti-neoplastic agents, such as other kinase inhibitors including p38 inhibitors and CDK inhibitors, TNF inhibitors, metallomatrix proteases inhibitors (MMP), COX-2 inhibitors including celecoxib, rofecoxib, parecoxib, valdecoxib, and etoricoxib, NSAJD's, SOD mimics or ⁇ v ⁇ 3 inhibitors.
- MMP metallomatrix proteases inhibitors
- COX-2 inhibitors including celecoxib, rofecoxib, parecoxib, valdecoxib, and etoricoxib
- NSAJD's SOD mimics or ⁇ v ⁇ 3 inhibitors.
- pharmaceutically-acceptable salts embraces salts commonly used to form alkali metal salts and to form addition salts of free acids or free bases.
- the nature of the salt is not critical, provided that it is pharmaceutically-acceptable.
- Suitable pharmaceutically-acceptable acid addition salts of compounds of Formula I-III may be prepared from an inorganic acid or from an organic acid. Examples of such inorganic acids are hydrochloric, hydrobromic, hydroiodic, nitric, carbonic, sulfuric and phosphoric acid.
- organic acids may be selected from aliphatic, cycloaliphatic, aromatic, arylaliphatic, heterocyclic, carboxylic and sulfonic classes of organic acids, example of which are formic, acetic, adipic, butyric, propionic, succinic, glycolic, gluconic, lactic, malic, tartaric, citric, ascorbic, glucuronic, maleic, fumaric, pyruvic, aspartic, glutamic, benzoic, anthranilic, mesylic, 4-hydroxybenzoic, phenylacetic, mandelic, embonic (pamoic), methanesulfonic, ethanesulfonic, ethanedisulfonic, benzenesulfonic, pantothenic, 2-hydroxyethanesulfonic, toluenesulfonic, sulfanilic, cyclohexylaminosulfonic, camphoric, camphorsulfonic,
- Suitable pharmaceutically-acceptable base addition salts of compounds of Formula I-III include metallic salts, such as salts made from aluminum, calcium, lithium, magnesium, potassium, sodium and zinc, or salts made from organic bases including primary, secondary and tertiary amines, substituted amines including cyclic amines, such as caffeine, arginine, diethylamine, N-ethyl piperidine, aistidine, glucamine, isopropylamine, lysine, morpholine, N-ethyl morpholine, piperazine, piperidine, triethylamine, trimethylamine.
- metallic salts such as salts made from aluminum, calcium, lithium, magnesium, potassium, sodium and zinc
- organic bases including primary, secondary and tertiary amines, substituted amines including cyclic amines, such as caffeine, arginine, diethylamine, N-ethyl piperidine, aistidine, glucamine, isopropyl
- Substituted bicyclic compounds 4 [where Y a is substituted amines or amides] can be prepared by the process outlined in Scheme 1.
- the amine 2 [where X is O] is coupled with a quinoline derivative 1 [where LG is halo, and the like] such as in the presence of KOH and Cu, in a solvent such as dry CH 2 C1 2 at a temperature above RT, preferably above about 70 °C, more preferably at a temperature of about 100 °C, to form the linked compound 3.
- a solvent such as dry CH 2 C1 2 at a temperature above RT, preferably above about 70 °C, more preferably at a temperature of about 100 °C, to form the linked compound 3.
- Such heating is preferably heated by microwave.
- the amine 3 can be reductively aminated with aldehydes, such as in the presence of NaBH(OAc) 3 , in a solvent such as dry CH 2 C1 2 at a temperature at about RT, to form amides 4 of the present invention.
- aldehydes such as in the presence of NaBH(OAc) 3
- a solvent such as dry CH 2 C1 2 at a temperature at about RT
- the amine 3 can be coupled with compounds having an active acyl moiety, such as acid chlorides and carboxylic acids, such as in the presence of PyBOP and a base such as K 2 CO 3 , to form amides 4 of the present invention.
- amines 3 can be prepared via amination of a bromo-derivative 5 e.g. a halo derivative more preferably a bromo-derivative such as in the presence of Pd and a strong base, e.g. LiHMDS.
- a bromo-derivative 5 e.g. a halo derivative more preferably a bromo-derivative such as in the presence of Pd and a strong base, e.g. LiHMDS.
- Pd 2 (dba) 3 in the presence of P(t-Bu) 3 is used.
- the reaction is kept at about RT.
- compounds where Y is -CO 2 - 6 can be prepared as described in Scheme 3.
- a substituted carboxylic acid ester 6 is treated with strong base, such as NaH, preferably in a solvent such as DMF, to form the anion.
- the reaction temperature is at about RT.
- Substituted nitrogen containing heteroaryl compounds 1, such as substituted quinazolines or quinolines, are coupled to the anion to form the compounds of the present invention 7.
- the reaction temperature is above RT, preferably above about 50 °C, more preferably at about 60 °C.
- a substituted carboxylic acid 8 can be coupled with an amine under standard coupling chemistry, such as with EDC in the presence of a solvent like DMF, to form amides 9.
- the reaction temperature is preferably kept at about RT.
- a substituted amine 11 can be coupled with an active carbonyl compound (Y"-R) as described in Scheme 1, to form amides 12.
- Coupling the amides 12 with nitrogen containing heterocyclic compounds, such as quinolines and quinazolines 1 by the method described in Scheme 4, provides compounds of the present invention 10 [where Y is -NHC( O)-] .
- Bromonaphthyl intermediates 14 can be provided by the method described in Scheme 6.
- Ureas of the present invention 18 are prepared by the method outlined in Scheme 8.
- Amines 11 are treated with isocyantates, preferably an excess of isocyanate, in the presence of base, preferably an excess of base, in a solvent such as DMF to form the ureas 18.
- the reaction temperature is maintained at around RT.
- Scheme 9 R ⁇ xylation -A-Br R D-l-rC'tHr - _A ⁇ _- ⁇ Nv ⁇ H ⁇ _-rC' I( 24
- Substituted bicyclic compounds 24 [where Y is an amide] can be prepared by the process outlined in Scheme 9.
- 6-Bromo-2 -hydroxymethyl compounds 21 are prepared such as by the coupling of 6- carbaldehydes 20 and activated R 1 containing compounds 19, such as phenylsulfinyl substituted compounds.
- the coupling occurs in the presence of a Grignard reagent, such as phenylmagnesium bromide, in an appropriate protic solvent such as THF.
- the temperature is preferably maintained at about RT.
- the Grignard is first added to the R 1 containing compound 19 prior to the addition of the carbaldehyde 20.
- the resulting hydroxymethyl compound 21 is dehydroxylated, such as in the presence of Zn and formic acid.
- the dehydroxylation preferably occurs at a temperature above RT, more preferably above about 50 °C, and most preferably at about reflux temperature.
- the resulting 6- bromonaphtyl compound 22 is animated similar to that described in Scheme 2 to form naphthyl amine 23 and the amides 24 are consequently formed similar to that described in Scheme 1.
- Activated R 1 containing compounds 19 can be prepared such as by the method identified in Scheme 10.
- Halosubstituted compounds 25 are dehalogenated, such as with aqueous base, e.g. KOH, then treated with a thiol compound, such as thiophenol, at a temperature above RT, preferably above 75 °C, more preferably at about 100 °C.
- the thio compound 26 is oxidized, such as with mCPBA, at a temperature below RT, preferably below -23 °C, more preferably at about -78 °C, to form the sulfinyl compounds 19.
- Scheme 11 einreb reagent Br-A-C0 2 H Br-A-CON (CH 3 ) OCH 3 Br-A-COH DIBAL 20 27 28
- 6-Bromonaphthyl-2-carbaldehydes 20 are prepared from the carboxylic acid 27 via reduction of the amide intermediate 28.
- the amide 28 is formed via peptide type coupling, such as in the presence of EDC, HOBt and base, of a substituted hydroxylamine, at a temperature preferably at about RT.
- Reduction of the amide 28, such as with DIBAL, in a solvent such as THF, at a temperature between -78 °C and RT, preferably at about RT provides the desired 6-bromonaphthalene-2 -carbaldehyde 20.
- Other benzothiazoles and benzoxazoles can be prepared by methods described in the literature (e.g. J. Heterocycl. Chem., 17(4):817 (1980); Tetrahedron, 42(20):5739 (1986); and Chem. Pharm. Bull., 43(10):1614 (1995)).
- compounds (where A is indazoles) can be prepared as described in Scheme 12.
- a solution of 6-fluoro-4-hydroxy benzonitrile is reacted with a hydrazine, such as methyl hydrazine at a temperature above RT, preferably above about 50 °C and more preferably at about 80 °C, to provide the lH-indazol-6-ol 30.
- the alcohol 30 is coupled with the appropriately substituted quinolines or quinazolines (where LG is halo, and the like) in the presence of a base such as cesium carbonate, to form the ethers 31.
- the reaction temperature is above RT, preferably above about 50 °C, more preferably at about 100 °C.
- Other compounds of the invention 32 can be prepared by substituting the amine 31 using chemistry such as reductive amination of aldehydes such as using NaBH(OAc) 3 at a temperature of about RT.
- chemistry such as reductive amination of aldehydes such as using NaBH(OAc) 3 at a temperature of about RT.
- a solution/suspension of Pd 2 dba 3 , 2-(dicyclohexylphosphino)-2'-4'-6'-fri-/-propyl- l,l '-biphenyl, a base such as sodium tert-butoxide, the 1 H-indazol-3 -amine 31 and a halo compound, such as an aryl halide, in a solvent such as toluene can be used to form substituted amines 32.
- the reaction temperature is above RT, preferably above about 50 °C, more preferably at about 100 °C.
- a solution of 1 H-indazol-3 -amine 31 in a solvent such as pyridine is treated with an substituted acid chloride such as p-anisoyl chloride to form the substituted amides 32.
- the reaction is preferably maintained at a temperature of about RT.
- a solution of 1 H-indazol-3 -amine in a solvent such as benzene is treated with a substituted isocyanate to form the substituted ureas 32.
- the reaction is preferably maintained at a temperature of about RT.
- Dimethoxy-quinolines 35 can be prepared from the conesponding nifro 33 compounds via the method described in Scheme 13. Reduction of the nitro compound 33 to the amine 34, such as with H 2 in the presence of a catalyst, such as Pd, e.g. Pd/C, followed by treatment with base and dimethyl ether, yields the desired quinolines 35.
- a catalyst such as Pd, e.g. Pd/C
- Various substituted quinolines and quinazolines can be prepared by the methods described in WO 98/13350.
- the starting compounds defined in Schemes 1-13 may also be present with functional groups in protected form if necessary and/or in the form of salts, provided a salt-forming group is present and the reaction in salt form is possible.
- one compound of Formula I can be converted into another compound of Formula I or a N-oxide thereof; a compound of Formula I can be converted into a salt; a salt of a compound of Formula I can be converted into the free compound or another salt; and/or a mixture of isomeric compounds of Formula I can be separated into the individual isomers.
- N-Oxides can be obtained in a known matter by reacting a compound of Formula I with H 2 O 2 , Oxone, or a peracid, e.g. mCPBA, in an inert solvent, e.g.
- the protecting groups may already be present in precursors and should protect the functional groups concerned against unwanted secondary reactions, such as acylations, etherifications, esterifications, oxidations, solvolysis, and similar reactions. It is a characteristic of protecting groups that they lend themselves readily, i.e. without undesired secondary reactions, to removal, typically by solvolysis, reduction, photolysis or also by enzyme activity, for example under conditions analogous to physiological conditions, and that they are not present in the end-products.
- the specialist knows, or can easily establish, which protecting groups are suitable with the reactions mentioned above and hereinafter. The protection of such functional groups by such protecting groups, the protecting groups themselves, and their removal reactions are described for example in standard reference works, such as J.F.W.
- Salts of a compound of Formula I with a salt-forming group may be prepared in a manner known per se. Acid addition salts of compounds of Formula I may thus be obtained by treatment with an acid or with a suitable anion exchange reagent.
- a salt with two acid molecules for example a dihalogenide of a compound of Formula I
- Salts may be present in all starting compounds and transients, if these contain salt-forming groups. Salts may also be present during the reaction of such compounds, provided the reaction is not thereby disturbed. In certain cases, typically in hydrogenation processes, it is possible to achieve stereoselective reactions, allowing for example easier recovery of individual isomers.
- the solvents from which those can be selected which are suitable for the reaction in question include for example water, esters, typically lower alkyl-lower alkanoates, e.g., EtOAc, ethers, typically aliphatic ethers, e.g., Et 2 O, or cyclic ethers, e.g., THF, liquid aromatic hydrocarbons, typically benzene or toluene, alcohols, typically MeOH, EtOH or 1-propanol, IPOH, nifriles, typically CH 3 CN, halogenated hydrocarbons, typically CH 2 C1 2 , acid amides, typically DMF, bases, typically heterocyclic nitrogen bases, e.g.
- esters typically lower alkyl-lower alkanoates
- ethers typically aliphatic ethers, e.g., Et 2 O
- cyclic ethers e.g., THF
- liquid aromatic hydrocarbons typically benzene or toluene
- alcohols typically
- carboxylic acids typically lower alkanecarboxylic acids, e.g., AcOH
- carboxylic acid anhydrides typically lower alkane acid anhydrides, e.g., acetic anhydride
- cyclic linear, or branched hydrocarbons, typically cyclohexane, hexane, or isopentane, or mixtures of these solvents, e.g., aqueous solutions, unless otherwise stated in the description of the process.
- solvent mixtures may also be used in processing, for example in chromatography.
- the invention relates also to those forms of the process in which one starts from a compound obtainable at any stage as a transient and canies out the missing steps, or breaks off the process at any stage, or forms a starting material under the reaction conditions, or uses said starting material in the form of a reactive derivative or salt, or produces a compound obtainable by means of the process according to the invention and processes the said compound in situ.
- a compound obtainable at any stage as a transient canies out the missing steps, or breaks off the process at any stage, or forms a starting material under the reaction conditions, or uses said starting material in the form of a reactive derivative or salt, or produces a compound obtainable by means of the process according to the invention and processes the said compound in situ.
- the prefened embodiment one starts from those starting materials which lead to the compounds described above as prefened.
- the compounds of Formula I, including their salts are also obtainable in the form of hydrates, or their crystals can include for example the solvent used for crystallization (present as solvates).
- New starting materials and/or intermediates, as well as processes for the preparation thereof, are likewise the subject of this invention.
- such starting materials are used and reaction conditions so selected as to enable the prefened compounds to be obtained.
- Starting materials of the invention are known, are commercially available, or can be synthesized in analogy to or according to methods that are known in the art.
- existing functional groups which do not participate in the reaction should, if necessary, be protected.
- Prefened protecting groups, their introduction and their removal are described above or in the examples. All remaining starting materials are known, capable of being prepared according to known processes, or commercially obtainable; in particular, they can be prepared using processes as described in the examples.
- optical isomers can be obtained by resolution of the racemic mixtures according to conventional processes, e.g., by formation of diastereoisomeric salts, by treatment with an optically active acid or base.
- appropriate acids are tartaric, diacetyltartaric, dibenzoyltartaric, ditoluoyltartaric, and camphorsulfonic acid and then separation of the mixture of diastereoisomers by crystallization followed by liberation of the optically active bases from these salts.
- a different process for separation of optical isomers involves the use of a chiral chromatography column optimally chosen to maximize the separation of the enantiomers.
- Still another available method involves synthesis of covalent diastereoisomeric molecules by reacting compounds of the invention with an optically pure acid in an activated form or an optically pure isocyanate.
- the synthesized diastereoisomers can be separated by conventional means such as chromatography, distillation, crystallization or sublimation, and then hydrolyzed to deliver the enantiomerically pure compound.
- the optically active compounds of the invention can likewise be obtained by using optically active starting materials. These isomers may be in the form of a free acid, a free base, an ester or a salt.
- the compounds of this invention may contain one or more asymmetric centers and thus occur as racemates and racemic mixtures, scalemic mixtures, single enantiomers, individual diastereomers and diastereomeric mixtures. All such isomeric forms of these compounds are expressly included in the present invention.
- the compounds of this invention may also be represented in multiple tautomeric forms, for example, as illustrated below:
- the invention expressly includes all tautomeric forms of the compounds described herein.
- the compounds may also occur in cis- or trans- or E- or Z- double bond isomeric forms. All such isomeric forms of such compounds are expressly included in the present invention. All crystal forms of the compounds described herein are expressly included in the present invention.
- Substituents on ring moieties e.g., phenyl, thienyl, etc.
- the compounds of this invention may contain heterocyclic ring systems attached to another ring system. Such heterocyclic ring systems may be attached through a carbon atom or a heteroatom in the ring system.
- a compound of any of the formulas delineated herein may be synthesized according to any of the processes delineated herein. In the processes delineated herein, the steps may be performed in an alternate order and may be preceded, or followed, by additional protection/deprotection steps as necessary.
- the processes may further comprise use of appropriate reaction conditions, including inert solvents, additional reagents, such as bases (e.g., LDA, DIEA, pyridine, K 2 CO 3 , and the like), catalysts, and salt forms of the above.
- the intermediates may be isolated or carried on in situ, with or without purification.
- Purification methods are known in the art and include, for example, crystallization, chromatography (liquid and gas phase, and the like), extraction, distillation, trituration, reverse phase HPLC and the like. Reactions conditions such as temperature, duration, pressure, and atmosphere (inert gas, ambient) are known in the art and may be adjusted as appropriate for the reaction.
- the above synthetic schemes are not intended to comprise a comprehensive list of all means by which the compounds described and claimed in this application may be synthesized. Further methods will be evident to those of ordinary skill in the art. Additionally, the various synthetic steps described above may be performed in an alternate sequence or order to give the desired compounds.
- Synthetic chemistry transformations and protecting group methodologies useful in synthesizing the inhibitor compounds described herein are known in the art and include, for example, those such as described in R. Larock, “Comprehensive Organic Transformations", VCH Publishers (1989); T.W. Greene and P.G.M. Wuts, "Protective Groups in Organic Synthesis", 3 rd edition, John Wiley and Sons (1999); L. Fieser and M. Fieser, “Fieser and Fieser's Reagents for Organic Synthesis", John Wiley and Sons (1994); A. Kafritzky and A. Pozharski, "Handbook of Heterocyclic Chemistry", 2 nd edition (2001); M.
- Bodanszky A. Bodanszky, "The Practice of Peptide Synthesis", Springer-Verlag, Berlin Heidelberg (1984); J. Seyden- Penne, “Reductions by the Alumino- and Borohydrides in Organic Synthesis", 2 nd edition, Wiley- VCH, (1997); and L. Paquette, editor, “Encyclopedia of Reagents for Organic Synthesis", John Wiley and Sons (1995).
- the compounds of this invention may be modified by appending appropriate functionalities to enhance selective biological properties. Such modifications are known in the art and include those which increase biological penetration into a given biological compartment (e.g., blood, lymphatic system, central nervous system), increase oral availability, increase solubility to allow administration by injection, alter metabolism and alter rate of excretion.
- Example 105 The following Examples were prepared similar to that described in Example 105.
- Example 106 The following Examples were prepared similar to that described in Example 106.
- Step (a) Preparation of 3-amino-l-methyl-lH-indazol-6-ol
- a solution of 6-fluoro-4-hydroxy benzonitrile (Aldrich, 10.1 g, 73.9 mmol) in methyl hydrazine (Aldrich, 20.0 mL, 379 mmol) was stined at 80 °C for 16 h. After cooling to RT, the volatile portion was removed in vacuo. The residue was triturated with DCM and MeOH. The title compound was obtained by filtration as an off-white solid. The filtrate was concenfrated in vacuo and purified by silica gel chromatography (DCM/MeOH: 100/0 to 95/5) to give an additional amount of the title compound as a white solid.
- DCM/MeOH silica gel chromatography
- Step (b) Preparation of 6-((6,7-bis(methyloxy)-4-quinolinyl)oxy)-l-methyl-lH-indazol-3-amine 4-Chloro-6,7-dimethoxyquinoline (2.60 g, 11.7 mmol), 3 -amino- 1 -methyl- lH-indazol-6-ol (Step a, 2.72 g, 16.7 mmol) and cesium carbonate (Aldrich, 16.3 g, 50.1 mmol) in DMSO (16.7 mL, Aldrich) were stined at 100 °C for 16h. After cooling to RT, water was added. The aqueous layer was extracted with DCM.
- Step (a) Preparation of 5-hydroxybenzofuran-2-carboxylic acid
- Step (b) Preparation of 5-(6,7-dimethoxyquinolin-4-yloxy)benzofuran-2-carboxylic acid
- a stined solution of 5-hydroxybenzofuran-2 -carboxylic acid (Step a, 2.00 g, 11.23 mmol) in DMSO (23 mL) at RT was added cesium carbonate (11.0 g, 33.7 mmol).
- the reaction was stined for 10 min followed by the addition of 4-chloro-6,7-dimethoxyquinoline (prepared by the method described in WO 03/33472) (1.75 g, 7.86 mmol).
- the reaction was stined at 100 °C for 16h. After cooling to RT, water was added.
- Step (b) Preparation of 6-methoxybenzofuran-3-yl trifluoromethane sulfonate.
- 6-Methoxybenzofuran-3(2H)-one (5.6 g, 34.15 mmol) was dissolved in dichloromethane (100 mL) then cooled to —65 C.
- N,N-diisopropylethylamine (6.84 mL, 39.27 mmol) was added dropwise via syringe, followed by addition of trifluoromethane sulfonic anhydride (6.88 mL, 40.97 mmol). The mixture was slowly warmed from -65C to 0 C over 2.5 h.
- Step (c) Preparation of methyl 6-methoxybenzofuran-3-carboxylate.
- 6-Methoxybenzofuran-3-yl trifluoromethane sulfonate 500 mg, 1.69 mmol was added to a stainless steel high-pressure reaction vessel then dissolved in N,N-dimethylformamide (6.5 mL). Carbon monoxide gas was bubbled through the solution for 10 minutes.
- Step (d) Preparation of methyl 6-hydroxybenzofuran-3-carboxylate.
- Methyl 6-methoxybenzofuran-3-carboxylate (498 mg, 2.42 mmol) was dissolved in dichloromethane (100 mL) then cooled to -10 C.
- a solution of IM boron tribromide in dichloromethane (9.67 mL, 9.67 mmol) was added slowly via syringe.
- the dark purple reaction mixture was warmed to RT over 3 h then continued stirring at RT for an additional 4 h.
- the mixture was slowly quenched with aqueous IN hydrochloric acid until it became colorless.
- the mixture was diluted with dichloromethane and washed with water (2x) and brine.
- Step (e) Preparation of methyl 6-(6,7-dimethoxyquinolin-4-yloxy)benzofuran-3-carboxylate.
- Methyl 6-hydroxybenzofuran-3-carboxylate (312 mg, 1.62 mmol) was dissolved inN,N- dimethylformamide (5 mL) then added 4-chloro-6,7-dimethoxyquinoline (451 mg, 2.02 mmol), 2- (dicyclohexylphosphino)-2',4',6'-friisopropyl-l,r-biphenyl (77 mg, 0.162 mmol), palladium acetate (36 mg, 0.162 mmol) and potassium phosphate (687 mg, 3.24 mmol).
- the reaction mixture was stined at 100 °C overnight. Additional palladium acetate (36 mg, 0.162 mmol) was added to the mixture and the heating was continued at 100 °C for 4 h. Additional 4-chloro-6,7-dimethoxyquinoline (181 mg, 0.812 mmol) and 2-(dicyclohexylphosphino)-2',4',6'-friisopropyl-l, -biphenyl (77 mg, 0.162 mmol) were added to the mixture and heating at 100 °C was continued for 2 days. The reaction mixture was filtered and the filtrate was concentrated in vacuo.
- Step (f) Preparation of 6-(6,7-dimethoxyquinoIin-4-yloxy)benzofuran-3-carboxyIic acid.
- Methyl 6-(6,7-dimethoxyquinolin-4-yloxy)benzofuran-3-carboxylate (102 mg, 0.269 mmol) was dissolved in dioxane (1.6 mL) and water (0.5 mL), then added 2N aqueous sodium hydroxide (141 DL). The reaction mixture was stined at room temperature overnight. The mixture was concenfrated in vacuo then diluted with water. The aqueous layer was extracted with ethyl acetate.
- Step (g) Preparation of ⁇ (4-chlorophenyl)-6-(6,7-dimethoxyquinolin-4-yloxy)benzofuran-3- carboxamide. 6-(6,7-Dimethoxyquinolin-4-yloxy)benzofuran-3-carboxylic acid (31 mg, 0.085 mmol) and 2-
- GST-c-Met kinase domain fusion (GST-Met) gene is transposed into full-length baculovirus DNA using the BacToBacTM system (Invitrogen). High5 cells are infected with the recombinant baculovirus for 72 h at 27 °C. The infected cells are harvested by centrifugation and the pellet is stored at -80 °C.
- the pellet is resuspended in buffer A (50 mM HEPES, pH 8.0, 0.25 M NaCl, 10 mM 2-mercaptoethanol, 10% (w/v) glycerol, 0.5 % (v/v) protease inhibitor cocktail (Sigma P8340), stined at 4 °C to homogeneity, and the cells are disrupted by microfluidization (Microfluidics) at 10,000 psi. The resulting lysate is centrifuged at 50,000 x g for 90 min at 4 °C, and the supernatant is adsorbed onto 10 mL of glutathione sepharoseTM 4B (Amersham) by batch method.
- buffer A 50 mM HEPES, pH 8.0, 0.25 M NaCl, 10 mM 2-mercaptoethanol, 10% (w/v) glycerol, 0.5 % (v/v) protease inhibitor cocktail (Sigma P8340), stined
- the slurry is rocked gently overnight at 4 °C.
- the glutathione resin is harvested by centrifugation and washed three times with 40 mL buffer A by batch method.
- the resin is washed three times with buffer B (buffer A adjusted to 0.1 M NaCl, less protease inhibitors).
- the protein is eluted with buffer B containing 25 mM reduced glutathione. Eluted fractions are analyzed by SDS- PAGE and concentrated to ⁇ 10 mL ( ⁇ 10 mg/mL total protein).
- the concentrated protein is separated by SuperdexTM 200 (Amersham) size exclusion chromatography in buffer C (25 mM Tris, pH 7.5, 0.1 M NaCl, 10 mM 2-mercaptoethanol, 10% glycerol). The fractions are analyzed by SDS-PAGE and the appropriate fractions are pooled and concentrated to ⁇ 1 mg/mL. The protein is aliquotted and stored at - 80 °C.
- the peak tubes with GST-cMET are pooled and the QD 2S0 is read with the column buffer listed above as the blank buffer.
- Phosphorylation of the purified GST-cMET is performed by incubating the protein for 3 h at RT with the following: Final concentration a) 100 mM ATP (Sigma #A7699) 25 mM b) 1.0 M MgCl 2 (Sigma #M-O250) 100 mM c) 200 mM Sodium Orthovanadate (Sigma #S-6508) 15 mM d) 1.0 M Tris-HCl, pH 7.00 (in house) 50 mM e) H 2 0 f) GST-cMET 0.2 - 0.5 mg/mL
- a Kinase reaction Buffer is prepared as follows: Per l L
- the HTRF buffer contains:
- K M ATP, K M gastrin for various enzymes were determined by HTRF/ 33 P labeling and HTRF methods.
- Examples 1, 2, 4-31, 90-93 and 96 exhibited activity with IC 50 values less than 0.5 ⁇ M.
- c-Met cell-based autophosphorylation assay Human PC3 and mouse CT26 cells are available obtained from ATCC. The cells were cultured in a growth medium containing RPMI 1640 ,penicillin/streptomycin/ glutamine (IX) and 5% FBS. 2xl0 4 cells in medium were plated per well in a 96 well plate and incubated at 37 °C overnight.
- the cells were serum-starved by replacing the growth media with basic medium (DMEM low glucose + 0.1 BSA, 120 ⁇ L per well) at 37 °C for 16 h.
- basic medium DMEM low glucose + 0.1 BSA, 120 ⁇ L per well
- Compounds either 1 mM and 0.2 mM in 100% DMSO were serially diluted (1:3) 3333 fold on a 96 well plate, diluting 1:3 with DMSO from column 1 to 11 (columns 6 and 12 receive no compound).
- Compound samples (2.4 ⁇ L per well) were diluted with basic medium (240 ⁇ L) in a 96 well plate.
- the cells were washed once with basic medium (GIBCO, DMEM 11885-076) then compound solution was added (100 ⁇ L).
- the cells were incubated at 37 °C for 1 h.
- a (2 mg/mL) solution of CHO-HGF (7.5 ⁇ L) was diluted with 30 mL basic medium to provide a final concentration of 500 ng/mL.
- This HGF-containing media 120 ⁇ L was transfened to a 96 well plate.
- Compounds 1.2 ⁇ L was added to the HGF-containing media and mixed well.
- the mixture of media/HGF/compound (100 ⁇ L) was added to the cells (final HGF concentration - 250 ng/mL) then incubated at 37 °C for 10 min.
- a cell lysate buffer (20 mL) was prepared containing 1% Triton X-100, 50 mM Tris pH 8.0, 100 mM NaCl, Protease inhibitor (Sigma, #P-8340) 200 ⁇ L, Roche Protease inhibitor (Complete, # 1-697- 498 ) 2 tablets, Phosphatase Inhibitor II (Sigma, #P-5726) 200 ⁇ L, and a sodium vanadate solution
- the IGEN assay was performed as follows: Dynabeads M-280 sfreptavidin beads were pre- incubated with biotinylated anti-human HGFR (240 ⁇ L anti-human-HGFR (R&D system, BAF527 or BAF328) @ 100 ⁇ g/mL + 360 ⁇ L Beads (IGEN #10029 + 5.4 ⁇ L buffer - PBS/1% BSA/0.1% Tween20) by rotating for 30 min at RT. Antibody beads (25 ⁇ L) were transfened to a 96 well plate. Cell lysate solution (25 ⁇ L) was transfened added and the plate was shaken at RT for 1 h.
- biotinylated anti-human HGFR 240 ⁇ L anti-human-HGFR (R&D system, BAF527 or BAF328) @ 100 ⁇ g/mL + 360 ⁇ L Beads (IGEN #10029 + 5.4 ⁇ L buffer - PBS/1%
- Anti-phosphotyrosine 4G10 (Upstate 05-321) (19.7 ⁇ L antibody + 6 mL IX PBS) (12.5 ⁇ L) was added to each well, then incubated for 1 h at RT.
- Anti-mouse IgG ORI-Tag (ORIGEN #110087) (24 ⁇ L Antibody + 6 mL buffer) (12.5 ⁇ L) was added to each well, then incubated at RT for 30 min.
- IX PBS (175 ⁇ L) was added to each well and the electrochemiluminescence was read by an IGEN M8. Raw data was analyzed using a 4- parameter fit equation in XLFit. IC 50 values are then determined using Grafit software.
- Examples 46, 82, 96, 102 and 104 exhibited activity in PC3 cells with IC 50 values less than 1.0 ⁇ M.
- Examples 36, 46, 71, 82, 89, 96, 100-102 and 104 exhibited activity in CT26 cells with IC 50 values less than 1.0 ⁇ M.
- HUVEC Proliferation Assay Human Umbilical Vein Endothelial cells are purchased from Clonetics, Inc., as cryopreserved cells harvested from a pool of donors. These cells, at passage 1, are thawed and expanded in EBM-2 complete medium, until passage 2 or 3. The cells are trypsinized, washed in DMEM + 10% FBS + antibiotics, and spun at 1000 rpm for 10 min. Prior to centrifugation of the cells, a small amount is collected for a cell count. After centrifugation, the medium is discarded, and the cells are resuspended in the appropriate volume of DMEM + 10% FBS + antibiotics to achieve a concenfration of 3xl0 5 cells/mL.
- the cells are diluted to 3xl0 4 cells/mL in DMEM + 10% FBS + antibiotics, and 100 ⁇ L of cells are added to a 96-well plate. The cells are incubated at 37 °C for 22 h. Prior to the completion of the incubation period, compound dilutions are prepared. Five-point, five-fold serial dilutions are prepared in DMSO, at concentrations 400-fold greater than the final concentrations desired. 2.5 ⁇ L of each compound dilution are diluted further in a total of 1 mL DMEM + 10% FBS + antibiotics (400x dilution). Medium containing 0.25%) DMSO is also prepared for the 10 ⁇ M compound sample.
- the medium is removed from the cells, and 100 ⁇ L of each compound dilution is added.
- the cells are incubated at 37 °C for 2-3 h.
- the growth factors are diluted to the appropriate concentrations. Solutions of DMEM + 10% FBS + antibiotics, containing either VEGF or bFGF at the following concentrations: 50, 10, 2, 0.4, 0.08, and 0 ng/mL are prepared.
- VEGF vascular endothelial growth factor
- bFGF vascular endothelial growth factor
- the medium is removed, and the cells are washed twice with PBS. After the second wash with PBS, the plates are tapped gently to remove excess PBS, and the cells are placed at —70 °C for at least 30 min. The cells are thawed and analyzed using the CyQuant fluorescent dye (Molecular Probes C-7026), following the manufacturer's recommendations. The plates are read on a Victor/Wallac 1420 workstation at 485 nm/530 nm (excitation/emission). Raw data are collected and analyzed using a 4-parameter fit equation in XLFit. IC 50 values are then determined.
- Examples 132, 134-140, 142 144 and 145 inhibited VEGF-stimulated HUVEC proliferation at a level below 500 nM.
- compositions comprising the active compounds of Formula I-II in association with one or more non-toxic, pharmaceutically-acceptable caniers and/or diluents and/or adjuvants (collectively refened to herein as "carrier” materials) and, if desired, other active ingredients.
- carrier non-toxic, pharmaceutically-acceptable caniers and/or diluents and/or adjuvants
- the active compounds of the present invention may be administered by any suitable route, preferably in the form of a pharmaceutical composition adapted to such a route, and in a dose effective for the treatment intended.
- the compounds and compositions of the present invention may, for example, be administered orally, mucosally, topically, rectally, pulmonarily such as by inhalation spray, or parentally including intravascularly, intravenously, intraperitoneally, subcutaneously, intramuscularly intrasternaHy and infusion techniques, in dosage unit formulations containing conventional pharmaceutically acceptable caniers, adjuvants, and vehicles.
- the pharmaceutically active compounds of this invention can be processed in accordance with conventional methods of pharmacy to produce medicinal agents for administration to patients, including humans and other mammals.
- the pharmaceutical composition may be in the form of, for example, a tablet, capsule, suspension or liquid.
- the pharmaceutical composition is preferably made in the form of a dosage unit containing a particular amount of the active ingredient.
- dosage units are tablets or capsules.
- these may contain an amount of active ingredient from about 1 to 2000 mg, preferably from about 1 to 500 mg.
- a suitable daily dose for a human or other mammal may vary widely depending on the condition of the patient and other factors, but, once again, can be determined using routine methods.
- the amount of compounds which are administered and the dosage regimen for treating a disease condition with the compounds and/or compositions of this invention depends on a variety of factors, including the age, weight, sex and medical condition of the subject, the type of disease, the severity of the disease, the route and frequency of administration, and the particular compound employed. Thus, the dosage regimen may vary widely, but can be determined routinely using standard methods.
- a daily dose of about 0.01 to 500 mg/kg, preferably between about 0.01 and about 50 mg/kg, and more preferably about 0.01 and about 30 mg/kg body weight may be appropriate.
- the daily dose can be administered in one to four doses per day.
- the active compounds of this invention are ordinarily combined with one or more adjuvants appropriate to the indicated route of administration.
- the compounds may be admixed with lactose, sucrose, starch powder, cellulose esters of alkanoic acids, cellulose alkyl esters, talc, stearic acid, magnesium stearate, magnesium oxide, sodium and calcium salts of phosphoric and sulfuric acids, gelatin, acacia gum, sodium alginate, polyvinylpyrrolidone, and/or polyvinyl alcohol, and then tableted or encapsulated for convenient administration.
- Such capsules or tablets may contain a controlled-release formulation as may be provided in a dispersion of active compound in hydroxypropylmethyl cellulose.
- Topical preparation of compounds of this invention In the case of psoriasis and other skin conditions, it may be preferable to apply a topical preparation of compounds of this invention to the affected area two to four times a day.
- Formulations suitable for topical administration include liquid or semi-liquid preparations suitable for penetration through the skin (e.g. , liniments, lotions, ointments, creams, or pastes) and drops suitable for administration to the eye, ear, or nose.
- a suitable topical dose of active ingredient of a compound of the invention is 0.1 mg to 150 mg administered one to four, preferably one or two times daily.
- the active ingredient may comprise from 0.001% to 10% w/w, e.g., from 1% to 2% by weight of the formulation, although it may comprise as much as 10% w/w, but preferably not more than 5% w/w, and more preferably from 0.1 % to 1 % of the formulation.
- the active ingredients may be employed with either paraffinic or a water-miscible ointment base.
- the active ingredients may be formulated in a cream with an oil-in-water cream base.
- the aqueous phase of the cream base may include, for example at least 30% w/w of a polyhydric alcohol such as propylene glycol, butane-l,3-diol, mannitol, sorbitol, glycerol, polyethylene glycol and mixtures thereof.
- the topical formulation may desirably include a compound which enhances absorption or penetration of the active ingredient through the skin or other affected areas. Examples of such dermal penetration enhancers include DMSO and related analogs.
- the compounds of this invention can also be administered by a transdermal device. Preferably fransdermal administration will be accomplished using a patch either of the reservoir and porous membrane type or of a solid matrix variety.
- the active agent is delivered continuously from the reservoir or microcapsules through a membrane into the active agent permeable adhesive, which is in contact with the skin or mucosa of the recipient. If the active agent is absorbed through the skin, a controlled and predetermined flow of the active agent is administered to the recipient.
- the encapsulating agent may also function as the membrane.
- the oily phase of the emulsions of this invention may be constituted from known ingredients in a known manner. While the phase may comprise merely an emulsifier, it may comprise a mixture of at least one emulsifier with a fat or an oil or with both a fat and an oil.
- a hydrophilic emulsifier is included together with a lipophilic emulsifier which acts as a stabilizer. It is also prefened to include both an oil and a fat. Together, the emulsifier(s) with or without stabilizer(s) make-up the so-called emulsifying wax, and the wax together with the oil and fat make up the so-called emulsifying ointment base which forms the oily dispersed phase of the cream formulations.
- Emulsif ⁇ ers and emulsion stabilizers suitable for use in the formulation of the present invention include Tween 60, Span 80, cetostearyl alcohol, myristyl alcohol, glyceryl monostearate, sodium lauryl sulfate, glyceryl distearate alone or with a wax, or other materials well known in the art.
- the choice of suitable oils or fats for the formulation is based on achieving the desired cosmetic properties, since the solubility of the active compound in most oils likely to be used in pharmaceutical emulsion formulations is very low.
- the cream should preferably be a non-greasy, non-staining and washable product with suitable consistency to avoid leakage from tubes or other containers.
- Sfraight or branched chain, mono- or dibasic alkyl esters such as di-isoadipate, isocetyl stearate, propylene glycol diester of coconut fatty acids, isopropyl myristate, decyl oleate, isopropyl palmitate, butyl stearate, 2- ethylhexyl palmitate or a blend of branched chain esters may be used. These may be used alone or in combination depending on the properties required. Alternatively, high melting point lipids such as white soft paraffin and/or liquid paraffin or other mineral oils can be used.
- Formulations suitable for topical administration to the eye also include eye drops wherein the active ingredients are dissolved or suspended in suitable canier, especially an aqueous solvent for the active ingredients.
- the active ingredients are preferably present in such formulations in a concenfration of 0.5 to 20%, advantageously 0.5 to 10% and particularly about 1.5%) w/w.
- Formulations for parenteral administration may be in the form of aqueous or non-aqueous isotonic sterile injection solutions or suspensions. These solutions and suspensions may be prepared from sterile powders or granules using one or more of the caniers or diluents mentioned for use in the formulations for oral administration or by using other suitable dispersing or wetting agents and suspending agents.
- the compounds may be dissolved in water, polyethylene glycol, propylene glycol, ethanol, corn oil, cottonseed oil, peanut oil, sesame oil, benzyl alcohol, sodium chloride, tragacanth gum, and/or various buffers.
- Other adjuvants and modes of administration are well and widely known in the pharmaceutical art.
- the active ingredient may also be administered by injection as a composition with suitable caniers including saline, dextrose, or water, or with cyclodextrin (ie. Captisol), cosolvent solubilization (ie. propylene glycol) or micellar solubilization (ie. Tween 80).
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non- toxic parenterally acceptable diluent or solvent, for example as a solution in 1,3-butanediol.
- a non- toxic parenterally acceptable diluent or solvent for example as a solution in 1,3-butanediol.
- acceptable vehicles and solvents that may be employed are water, Ringer's solution, and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil may be employed, including synthetic mono- or diglycerides.
- fatty acids such as oleic acid find use in the preparation of injectables.
- the pharmaceutical composition may be administered in the form of an aerosol or with an inhaler including dry powder aerosol.
- Suppositories for rectal administration of the drug can be prepared by mixing the drug with a suitable non-irritating excipient such as cocoa butter and polyethylene glycols that are solid at ordinary temperatures but liquid at the rectal temperature and will therefore melt in the rectum and release the drug.
- a suitable non-irritating excipient such as cocoa butter and polyethylene glycols that are solid at ordinary temperatures but liquid at the rectal temperature and will therefore melt in the rectum and release the drug.
- the pharmaceutical compositions may be subjected to conventional pharmaceutical operations such as sterilization and/or may contain conventional adjuvants, such as preservatives, stabilizers, wetting agents, emulsif ⁇ ers, buffers etc. Tablets and pills can additionally be prepared with enteric coatings.
- Such compositions may also comprise adjuvants, such as wetting, sweetening, flavoring, and perfuming agents.
Abstract
Description










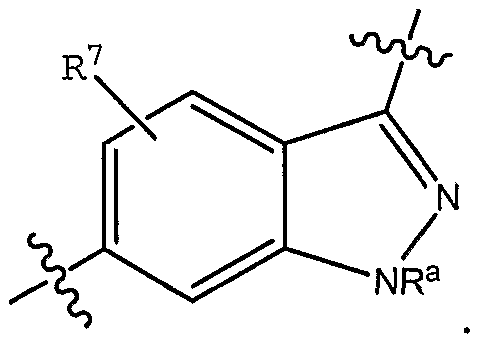
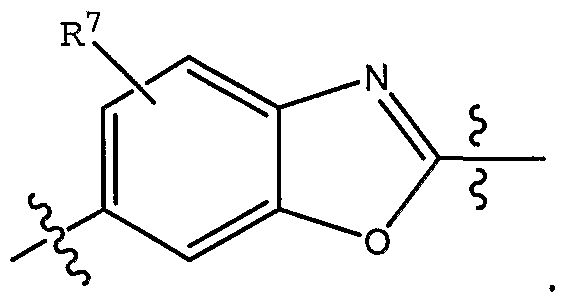












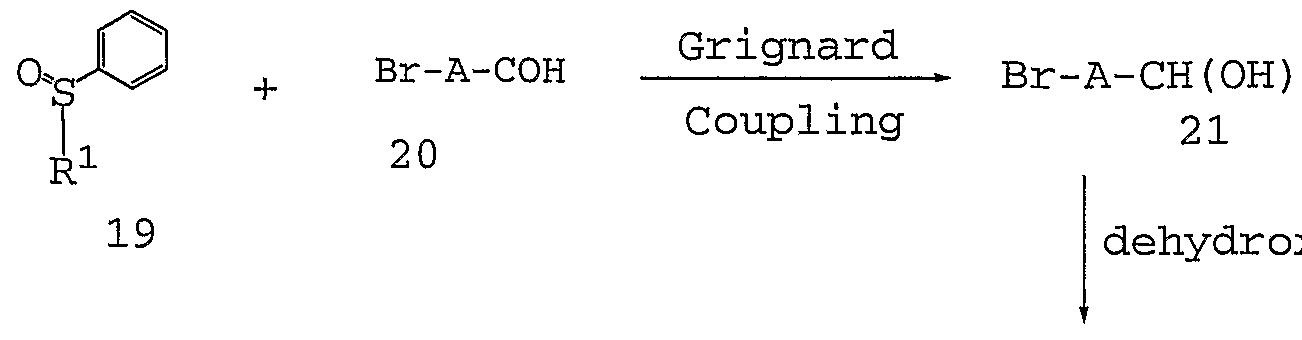









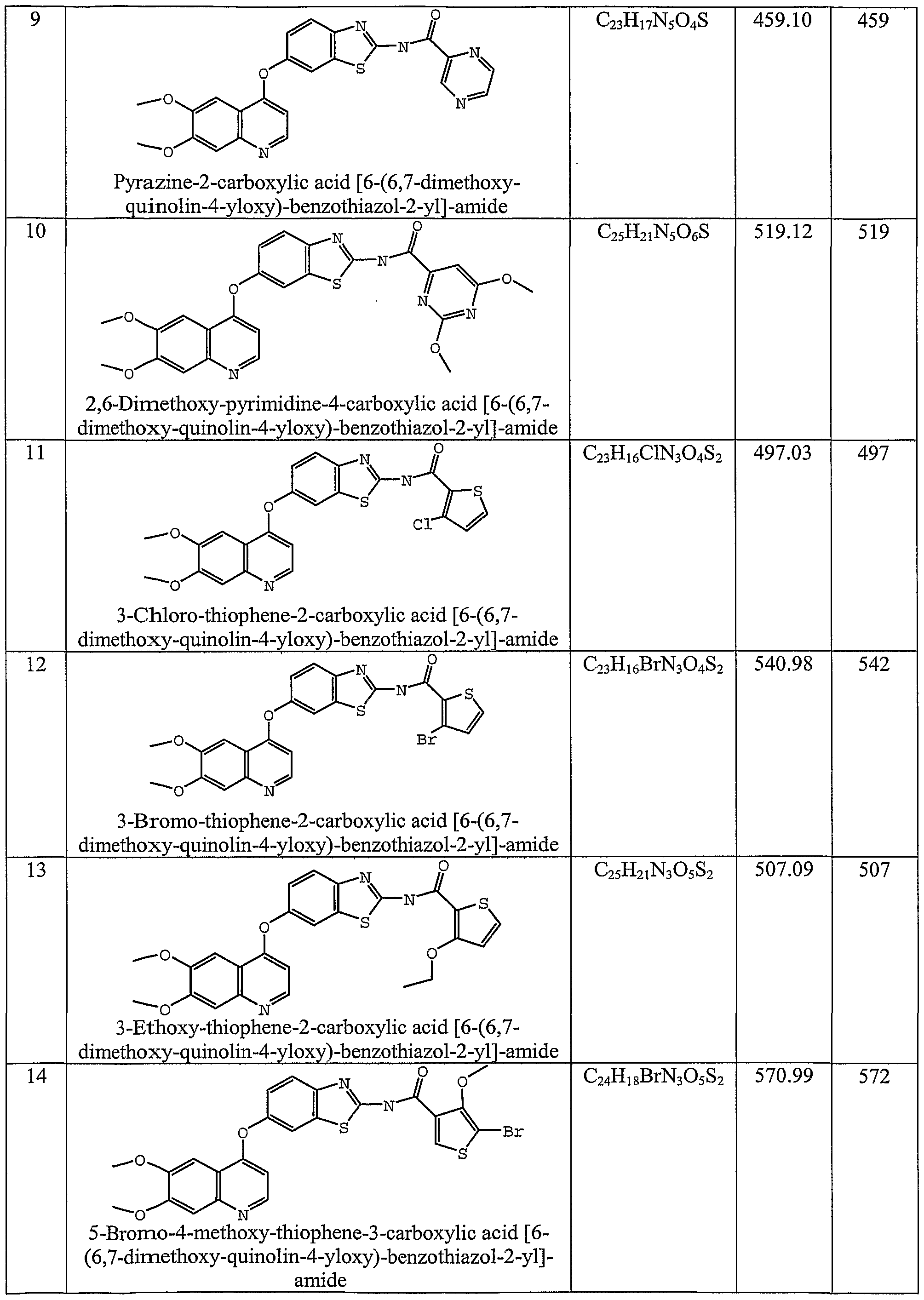






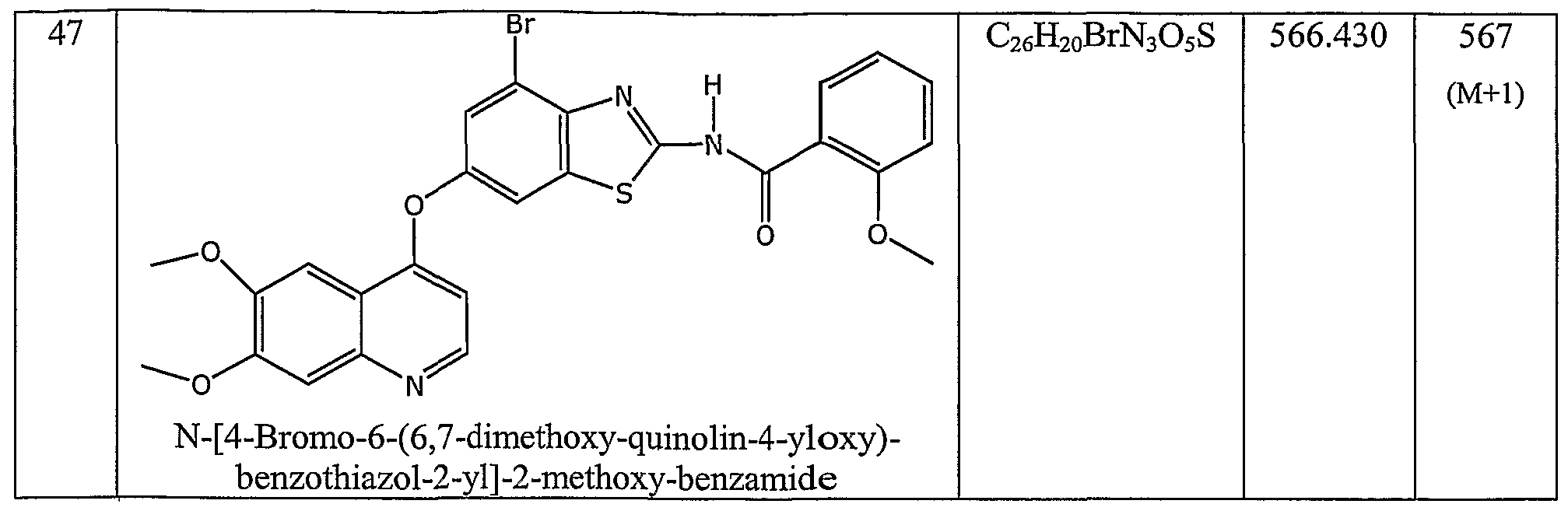




























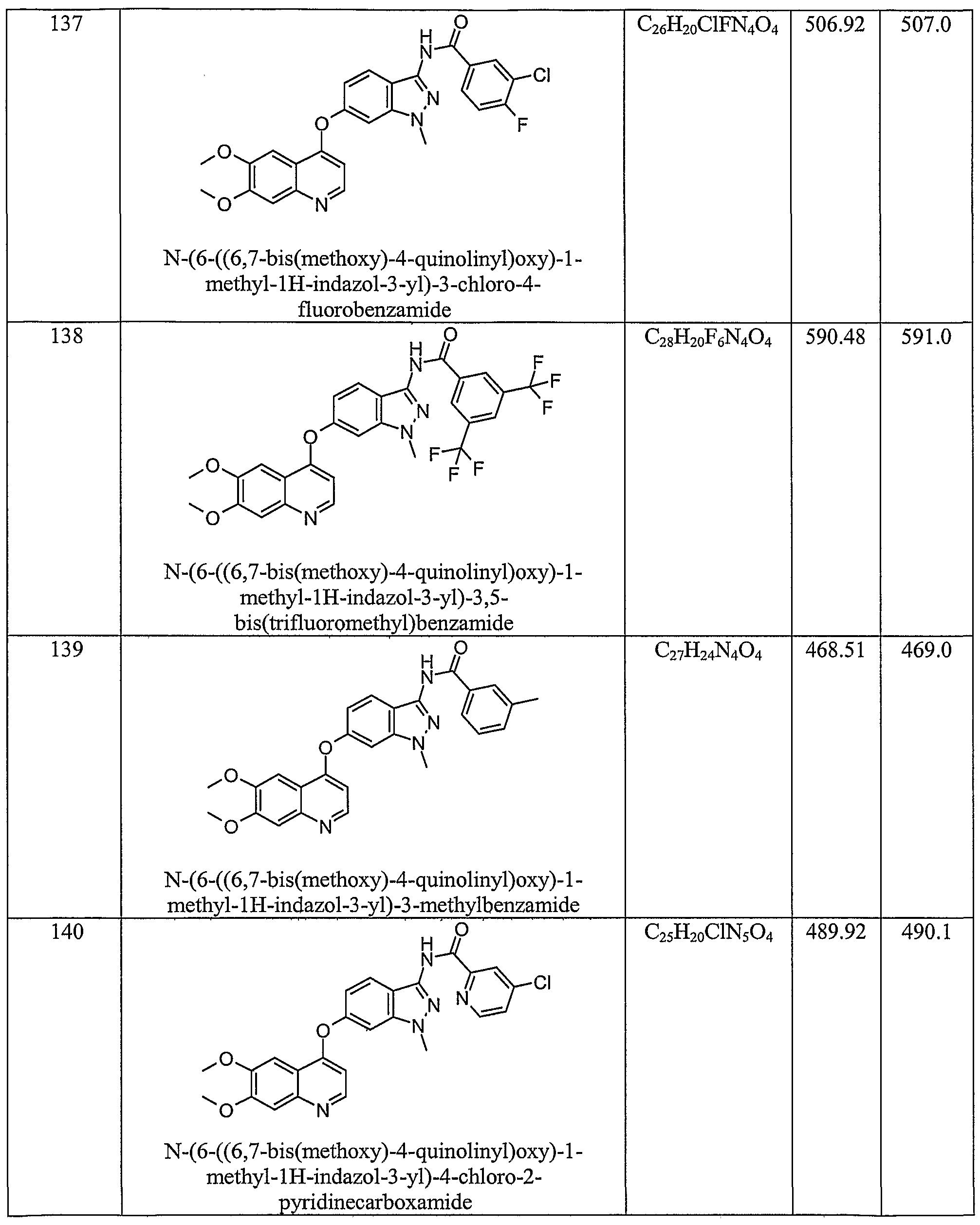











Claims











Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP05711976A EP1711495A2 (en) | 2004-01-23 | 2005-01-24 | Quinoline, quinazoline, pyridine and pyrimidine counds and their use in the treatment of inflammation, angiogenesis and cancer |
| AU2005207946A AU2005207946A1 (en) | 2004-01-23 | 2005-01-24 | Quinoline quinazoline pyridine and pyrimidine counds and their use in the treatment of inflammation angiogenesis and cancer |
| CA002553433A CA2553433A1 (en) | 2004-01-23 | 2005-01-24 | Quinoline quinazoline pyridine and pyrimidine compounds and their use in the treatment of inflammation angiogenesis and cancer |
| JP2006551396A JP2007518823A (en) | 2004-01-23 | 2005-01-24 | Quinoline, quinazoline, pyridine, and pyrimidine compounds and their use in the treatment of inflammation, angiogenesis, and cancer |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US53893504P | 2004-01-23 | 2004-01-23 | |
| US60/538,935 | 2004-01-23 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2005073224A2 true WO2005073224A2 (en) | 2005-08-11 |
| WO2005073224A3 WO2005073224A3 (en) | 2005-09-29 |
Family
ID=34826028
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2005/002304 WO2005073224A2 (en) | 2004-01-23 | 2005-01-24 | Quinoline quinazoline pyridine and pyrimidine counds and their use in the treatment of inflammation angiogenesis and cancer |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US7626030B2 (en) |
| EP (1) | EP1711495A2 (en) |
| JP (1) | JP2007518823A (en) |
| AU (1) | AU2005207946A1 (en) |
| CA (1) | CA2553433A1 (en) |
| WO (1) | WO2005073224A2 (en) |
Cited By (57)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007121484A2 (en) * | 2006-04-19 | 2007-10-25 | Novartis Ag | 6-o-substituted benzoxazole and benzothiazole compounds and methods of inhibiting csf-1r signaling |
| WO2008089307A2 (en) * | 2007-01-18 | 2008-07-24 | Lexicon Pharmaceuticals, Inc. | Delta 5 desaturase inhibitors for the treatment of pain, inflammation and cancer |
| WO2008112407A1 (en) | 2007-03-14 | 2008-09-18 | Advenchen Laboratories, Llc | Spiro substituted compounds as angiogenesis inhibitors |
| WO2008116919A1 (en) * | 2007-03-27 | 2008-10-02 | 4Sc Ag | Pharmaceutical composition comprising a cytokine |
| EP1977759A1 (en) * | 2007-03-27 | 2008-10-08 | 4Sc Ag | Pharmaceutical composition comprising a cytokine |
| WO2008144062A1 (en) | 2007-05-21 | 2008-11-27 | Novartis Ag | Csf-1r inhibitors, compositions, and methods of use |
| JP2009532333A (en) * | 2006-03-10 | 2009-09-10 | ノバルティス アクチエンゲゼルシャフト | Heterobicyclic carboxamides as inhibitors of kinases |
| EP2125777A1 (en) * | 2007-03-14 | 2009-12-02 | Advenchen Laboratories, LLC | Spiro substituted compounds as angiogenesis inhibitors |
| WO2010021918A1 (en) | 2008-08-19 | 2010-02-25 | Advenchen Laboratories, Llc | Compounds as kinase inhibitors |
| US7683060B2 (en) | 2006-08-07 | 2010-03-23 | Incyte Corporation | Triazolotriazines as kinase inhibitors |
| US7687522B2 (en) | 2006-12-20 | 2010-03-30 | Amgen Inc. | Substituted pyridines and pyrimidines and their use in treatment of cancer |
| US7732465B2 (en) | 2005-08-30 | 2010-06-08 | Novartis Vaccines And Diagnostics, Inc. | Substituted benzimidazoles and methods of their use |
| US7767675B2 (en) | 2006-11-22 | 2010-08-03 | Incyte Corporation | Imidazotriazines and imidazopyrimidines as kinase inhibitors |
| US7772247B2 (en) | 2004-07-30 | 2010-08-10 | Methylgene Inc. | Substituted thieno[3,2-d]pyridines as inhibitors of the VEGF receptor and HGF receptor |
| US7790729B2 (en) | 2005-05-20 | 2010-09-07 | Methylgene Inc. | Inhibitors of VEGF receptor and HGF receptor signaling |
| EP2339918A1 (en) * | 2008-09-26 | 2011-07-06 | GlaxoSmithKline LLC | Preparation of a quinolinyloxydiphenylcyclopropanedicarboxamide |
| AU2005321517B2 (en) * | 2004-12-27 | 2011-09-29 | 4Sc Ag | 2, 5 and 2, 6-disubstituted benzazole analogues useful as protein kinase inhibitors |
| WO2011162835A1 (en) | 2010-02-03 | 2011-12-29 | Incyte Corporation | Imidazo[1,2-b][1,2,4]triazines as c-met inhibitors |
| US8093264B2 (en) | 2005-05-20 | 2012-01-10 | Methylgene Inc. | Fused heterocycles as inhibitors of VEGF receptor and HGF receptor signaling |
| WO2012152763A1 (en) * | 2011-05-12 | 2012-11-15 | Nerviano Medical Sciences S.R.L. | Substituted indazole derivatives active as kinase inhibitors |
| US8338455B2 (en) | 2006-12-20 | 2012-12-25 | Amgen Inc. | Compounds and methods of use |
| US8389526B2 (en) | 2009-08-07 | 2013-03-05 | Novartis Ag | 3-heteroarylmethyl-imidazo[1,2-b]pyridazin-6-yl derivatives |
| US8420645B2 (en) | 2008-05-21 | 2013-04-16 | Incyte Corporation | Salts of 2-fluoro-N-methyl-4-[7-(quinolin-6-yl-methyl)-imidazo[1,2-b][1,2,4]triazin-2-yl]benzamide and processes related to preparing the same |
| CN101432281B (en) * | 2006-04-19 | 2013-08-28 | 诺瓦提斯公司 | 6-o-substituted benzoxazole and benzothiazole compounds and methods of inhibiting CSF-1R signaling |
| US8729268B2 (en) | 2008-03-05 | 2014-05-20 | Methylgene Inc. | Inhibitors of protein tyrosine kinase activity |
| US8822468B2 (en) | 2008-02-28 | 2014-09-02 | Novartis Ag | 3-Methyl-imidazo[1,2-b]pyridazine derivatives |
| WO2015138920A1 (en) | 2014-03-14 | 2015-09-17 | Novartis Ag | Antibody molecules to lag-3 and uses thereof |
| WO2016057841A1 (en) | 2014-10-08 | 2016-04-14 | Novartis Ag | Compositions and methods of use for augmented immune response and cancer therapy |
| WO2016061142A1 (en) | 2014-10-14 | 2016-04-21 | Novartis Ag | Antibody molecules to pd-l1 and uses thereof |
| WO2016091167A1 (en) * | 2014-12-09 | 2016-06-16 | 正大天晴药业集团股份有限公司 | Quinoline derivative against squamous cell carcinoma of lung |
| WO2016096709A1 (en) * | 2014-12-16 | 2016-06-23 | Eudendron S.R.L. | Heterocyclic derivatives modulating activity of certain protein kinases |
| WO2016100882A1 (en) | 2014-12-19 | 2016-06-23 | Novartis Ag | Combination therapies |
| WO2016145102A1 (en) | 2015-03-10 | 2016-09-15 | Aduro Biotech, Inc. | Compositions and methods for activating "stimulator of interferon gene" -dependent signalling |
| WO2016161952A1 (en) * | 2015-04-07 | 2016-10-13 | 广东众生药业股份有限公司 | Tyrosine kinase inhibitor and pharmaceutical composition comprising same |
| WO2017019894A1 (en) | 2015-07-29 | 2017-02-02 | Novartis Ag | Combination therapies comprising antibody molecules to lag-3 |
| WO2017019897A1 (en) | 2015-07-29 | 2017-02-02 | Novartis Ag | Combination therapies comprising antibody molecules to tim-3 |
| CN106543145A (en) * | 2016-10-28 | 2017-03-29 | 山西医科大学 | C Met kinases suppression 5 benzoic acid amides derivant of agent 3 (4 fluorophenyl) pyrimidone, preparation method and application |
| CN106543143A (en) * | 2015-09-22 | 2017-03-29 | 合肥中科普瑞昇生物医药科技有限公司 | New FLT3 kinase inhibitors of one class and application thereof |
| WO2017106656A1 (en) | 2015-12-17 | 2017-06-22 | Novartis Ag | Antibody molecules to pd-1 and uses thereof |
| US9809549B2 (en) | 2009-01-16 | 2017-11-07 | Exelixis, Inc. | Malate salt of N-(4-{[6,7-bis(methyloxy)quinolin-4-yl]oxy}phenyl)-N′(4-fluorophenyl)cyclopropane-1,1-dicarboxamide, and crystalline forms therof for the treatment of cancer |
| KR20180006167A (en) * | 2016-07-08 | 2018-01-17 | 주식회사유한양행 | Benzo[d]thiazole derivative or its salt and pharmaceutical composition comprising the same |
| AU2014219024B2 (en) * | 2013-02-20 | 2018-04-05 | KALA BIO, Inc. | Therapeutic compounds and uses thereof |
| US10016448B2 (en) | 2011-06-10 | 2018-07-10 | Merck Patent Gmbh | Compositions and methods for the production of pyrimidine and pyridine compounds with BTK inhibitory activity |
| WO2018237173A1 (en) | 2017-06-22 | 2018-12-27 | Novartis Ag | Antibody molecules to cd73 and uses thereof |
| WO2018237157A1 (en) | 2017-06-22 | 2018-12-27 | Novartis Ag | Antibody molecules to cd73 and uses thereof |
| EP3514179A1 (en) | 2014-01-24 | 2019-07-24 | Dana-Farber Cancer Institute, Inc. | Antibody molecules to pd-1 and uses thereof |
| WO2019218928A1 (en) * | 2018-05-15 | 2019-11-21 | 北京诺诚健华医药科技有限公司 | Indoline-1-formamide compound, preparation method therefor, and medical use thereof |
| WO2019232244A2 (en) | 2018-05-31 | 2019-12-05 | Novartis Ag | Antibody molecules to cd73 and uses thereof |
| WO2019229658A1 (en) | 2018-05-30 | 2019-12-05 | Novartis Ag | Entpd2 antibodies, combination therapies, and methods of using the antibodies and combination therapies |
| US10736886B2 (en) | 2009-08-07 | 2020-08-11 | Exelixis, Inc. | Methods of using c-Met modulators |
| US10894830B2 (en) | 2015-11-03 | 2021-01-19 | Janssen Biotech, Inc. | Antibodies specifically binding PD-1, TIM-3 or PD-1 and TIM-3 and their uses |
| WO2021053559A1 (en) | 2019-09-18 | 2021-03-25 | Novartis Ag | Entpd2 antibodies, combination therapies, and methods of using the antibodies and combination therapies |
| WO2021067801A1 (en) * | 2019-10-03 | 2021-04-08 | Ifm Due, Inc. | Compounds and compositions for treating conditions associated with sting activity |
| US11098077B2 (en) | 2016-07-05 | 2021-08-24 | Chinook Therapeutics, Inc. | Locked nucleic acid cyclic dinucleotide compounds and uses thereof |
| US11618749B2 (en) | 2018-07-03 | 2023-04-04 | Ifm Due, Inc. | Compounds and compositions for treating conditions associated with STING activity |
| EP4324518A2 (en) | 2014-01-31 | 2024-02-21 | Novartis AG | Antibody molecules to tim-3 and uses thereof |
| US11970480B2 (en) | 2022-05-17 | 2024-04-30 | Glaxosmithkline Intellectual Property Development Limited | Heterocyclic amides useful as protein modulators |
Families Citing this family (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003078448A1 (en) | 2002-03-13 | 2003-09-25 | Signum Biosciences, Inc. | Modulation of protein methylation and phosphoprotein phosphate |
| US7531553B2 (en) * | 2003-03-21 | 2009-05-12 | Amgen Inc. | Heterocyclic compounds and methods of use |
| EP2609919A3 (en) | 2003-09-26 | 2014-02-26 | Exelixis, Inc. | c-Met modulators and methods of use |
| ES2450566T3 (en) * | 2004-11-30 | 2014-03-25 | Amgen Inc. | Quinazoline and quinoline analogues and their use as medicines to treat cancer |
| US8221804B2 (en) | 2005-02-03 | 2012-07-17 | Signum Biosciences, Inc. | Compositions and methods for enhancing cognitive function |
| US7923041B2 (en) | 2005-02-03 | 2011-04-12 | Signum Biosciences, Inc. | Compositions and methods for enhancing cognitive function |
| JO2787B1 (en) * | 2005-04-27 | 2014-03-15 | امجين إنك, | Substituted Amid derivatives & methods of use |
| WO2007107005A1 (en) * | 2006-03-22 | 2007-09-27 | Methylgene, Inc. | Inhibitors of protein tyrosine kinase activity |
| US20110053931A1 (en) * | 2006-06-08 | 2011-03-03 | John Gaudino | Quinoline compounds and methods of use |
| CN102014897B (en) | 2008-04-21 | 2015-08-05 | 西格纳姆生物科学公司 | Compound, compositions and its preparation method |
| FR2933982A1 (en) * | 2008-07-18 | 2010-01-22 | Sanofi Aventis | NOVEL IMIDAZO-1,2-A! PYRIMIDINE DERIVATIVES, PROCESS FOR THEIR PREPARATION, THEIR USE AS MEDICAMENTS, PHARMACEUTICAL COMPOSITIONS AND NOVEL USE IN PARTICULAR AS MET INHIBITORS |
| FR2933981A1 (en) * | 2008-07-18 | 2010-01-22 | Sanofi Aventis | New imidazo(1,2-a)pyridine derivatives are mesenchymal-epithelial transition protein kinase inhibitors useful in a pharmaceutical composition for preparing a medicament to treat/prevent e.g. fibrotic disorders, allergies and asthma |
| FR2933980B1 (en) * | 2008-07-18 | 2011-07-29 | Sanofi Aventis | NOVEL TRIAZOLO®4,3-A! PYRIDINE DERIVATIVES, PROCESS FOR THEIR PREPARATION, THEIR USE AS MEDICAMENTS, PHARMACEUTICAL COMPOSITIONS AND NOVEL USE IN PARTICULAR AS MET INHIBITORS |
| AR073231A1 (en) * | 2008-08-29 | 2010-10-20 | Genentech Inc | DIAGNOSTIC METHODS AND TREATMENTS FOR VEGF INDEPENDENT TUMORS (VASCULAR ENDOTELIAL GROWTH FACTOR) |
| JP5734193B2 (en) * | 2008-10-14 | 2015-06-17 | クイ ニング | Compounds and methods of use |
| US20100136096A1 (en) * | 2008-12-02 | 2010-06-03 | Searete Llc, A Limited Liability Corporation Of The State Of Delaware | Systems for modulating inflammation |
| US20100136095A1 (en) * | 2008-12-02 | 2010-06-03 | Searete Llc, A Limited Liability Corporation Of The State Of Delaware | Systems for modulating inflammation |
| US20100135984A1 (en) * | 2008-12-02 | 2010-06-03 | Searete Llc, A Limited Liability Corporation Of The State Of Delaware | Anti-inflammatory compositions and methods |
| US20100135908A1 (en) * | 2008-12-02 | 2010-06-03 | Searete Llc, A Limited Liability Corporation Of The State Of Delaware | Delivery devices for modulating inflammation |
| US20100136094A1 (en) * | 2008-12-02 | 2010-06-03 | Searete Llc, A Limited Liability Corporation Of The State Of Delaware | Systems for modulating inflammation |
| US20100137246A1 (en) * | 2008-12-02 | 2010-06-03 | Searete Llc, A Limited Liability Corporation Of The State Of Delaware | Anti-inflammatory compositions and methods |
| RU2011142597A (en) * | 2009-03-21 | 2013-04-27 | Саншайн Лейк Фарма Ко., Лтд. | DERIVATIVES OF COMPOUND ETHERS OF AMINO ACIDS, THEIR SALTS AND METHODS OF APPLICATION |
| IN2012DN01983A (en) * | 2009-08-24 | 2015-07-24 | Ascepion Pharmaceuticals Inc | |
| KR20130108543A (en) | 2010-08-05 | 2013-10-04 | 암젠 인코포레이션 | Benzimidazole and azabenzimidazole compounds that inhibit anaplastic lymphoma kinase |
| WO2013152252A1 (en) | 2012-04-06 | 2013-10-10 | OSI Pharmaceuticals, LLC | Combination anti-cancer therapy |
| US9353122B2 (en) | 2013-02-15 | 2016-05-31 | Kala Pharmaceuticals, Inc. | Therapeutic compounds and uses thereof |
| US9688688B2 (en) | 2013-02-20 | 2017-06-27 | Kala Pharmaceuticals, Inc. | Crystalline forms of 4-((4-((4-fluoro-2-methyl-1H-indol-5-yl)oxy)-6-methoxyquinazolin-7-yl)oxy)-1-(2-oxa-7-azaspiro[3.5]nonan-7-yl)butan-1-one and uses thereof |
| AU2014342042B2 (en) | 2013-11-01 | 2017-08-17 | KALA BIO, Inc. | Crystalline forms of therapeutic compounds and uses thereof |
| US9890173B2 (en) | 2013-11-01 | 2018-02-13 | Kala Pharmaceuticals, Inc. | Crystalline forms of therapeutic compounds and uses thereof |
| MA39776A (en) | 2014-03-24 | 2017-02-01 | Hoffmann La Roche | Cancer treatment with c-met antagonists and correlation of the latter with hgf expression |
| WO2017151409A1 (en) | 2016-02-29 | 2017-09-08 | University Of Florida Research Foundation, Incorporated | Chemotherapeutic methods |
| EP3509421A4 (en) | 2016-09-08 | 2020-05-20 | Kala Pharmaceuticals, Inc. | Crystalline forms of therapeutic compounds and uses thereof |
| US10253036B2 (en) | 2016-09-08 | 2019-04-09 | Kala Pharmaceuticals, Inc. | Crystalline forms of therapeutic compounds and uses thereof |
| US10392399B2 (en) | 2016-09-08 | 2019-08-27 | Kala Pharmaceuticals, Inc. | Crystalline forms of therapeutic compounds and uses thereof |
| CN106478621B (en) * | 2016-09-30 | 2018-12-25 | 遵义医学院 | Quinoline or quinazoline derivative, preparation method and applications |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0602851A1 (en) * | 1992-12-10 | 1994-06-22 | Zeneca Limited | Quinazoline derivatives |
| WO1997003069A1 (en) * | 1995-07-13 | 1997-01-30 | Glaxo Group Limited | Heterocyclic compounds and pharmaceutical compositions containing them |
| WO2000018761A1 (en) * | 1998-09-29 | 2000-04-06 | American Cyanamid Company | Substituted 3-cyanoquinolines as protein tyrosine kinases inhibitors |
| WO2000047212A1 (en) * | 1999-02-10 | 2000-08-17 | Astrazeneca Ab | Quinazoline derivatives as angiogenesis inhibitors |
| US6288082B1 (en) * | 1998-09-29 | 2001-09-11 | American Cyanamid Company | Substituted 3-cyanoquinolines |
| WO2002092571A1 (en) * | 2001-05-11 | 2002-11-21 | Astrazeneca Ab | Novel 4-anilinoquinoline-3-carboxamides |
| WO2004085425A1 (en) * | 2003-03-21 | 2004-10-07 | Amgen Inc | Fused azoles such as 2,5-disubstituted benzimidazoles, benzoxazoles and benzothiazoles as kinase inhibitors |
| EP1548008A1 (en) * | 2002-08-23 | 2005-06-29 | Kirin Beer Kabushiki Kaisha | Compound having tgf-beta inhibitory activity and medicinal composition containing the same |
Family Cites Families (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3755332A (en) | 1971-07-01 | 1973-08-28 | Ciba Geigy Corp | Substituted 4 indazolaminoquinolines |
| US4916135A (en) | 1989-05-08 | 1990-04-10 | Hoechst Roussel Pharmaceuticals Inc. | N-heteroaryl-4-quinolinamines |
| AU4547396A (en) | 1995-01-31 | 1996-08-21 | Zenyaku Kogyo Kabushiki Kaisha | Thioquinoline derivatives |
| GB9505702D0 (en) | 1995-03-21 | 1995-05-10 | Agrevo Uk Ltd | Fungicidal compounds |
| GB9505651D0 (en) | 1995-03-21 | 1995-05-10 | Agrevo Uk Ltd | AgrEvo UK Limited |
| WO1997017329A1 (en) | 1995-11-07 | 1997-05-15 | Kirin Beer Kabushiki Kaisha | Quinoline derivatives and quinazoline derivatives inhibiting autophosphorylation of growth factor receptor originating in platelet and pharmaceutical compositions containing the same |
| US5965563A (en) | 1995-11-14 | 1999-10-12 | Pharmacia & Upjohn S.P.A. | Aryl and heteroaryl purine compounds |
| GB9603095D0 (en) | 1996-02-14 | 1996-04-10 | Zeneca Ltd | Quinazoline derivatives |
| DE19614718A1 (en) | 1996-04-15 | 1997-10-16 | Hoechst Schering Agrevo Gmbh | Substituted pyridines / pyrimidines, processes for their preparation and their use as pesticides |
| GB9800575D0 (en) | 1998-01-12 | 1998-03-11 | Glaxo Group Ltd | Heterocyclic compounds |
| ATE293102T1 (en) | 1998-04-23 | 2005-04-15 | Takeda Pharmaceutical | NAPHTHALENE DERIVATIVES, THEIR PRODUCTION AND USE |
| MXPA00011773A (en) | 1998-05-28 | 2002-06-04 | Parker Hughes Inst | Quinazolines for treating brain tumor. |
| JP2002523403A (en) | 1998-08-21 | 2002-07-30 | パーカー ヒューズ インスティテュート | Quinazoline derivatives |
| US6258820B1 (en) | 1999-03-19 | 2001-07-10 | Parker Hughes Institute | Synthesis and anti-tumor activity of 6,7-dialkoxy-4-phenylamino-quinazolines |
| ES2231179T3 (en) | 1999-04-12 | 2005-05-16 | Aventis Pharma Limited | SUBSTITUTED BICYCLE HETEROARILO COMPOUNDS AS INTEGRINE ANTAGONISTS. |
| CN1422262A (en) * | 2000-02-07 | 2003-06-04 | 艾博特股份有限两合公司 | 2-benzothiazolyl urea derivatives and their use as protein kinase inhibitors |
| DE60102137T2 (en) | 2000-03-17 | 2004-10-21 | Bristol Myers Squibb Pharma Co | BETA-AMINIC ACID DERIVATIVES FOR USE AS MATRIX METALLOPROTEASES AND TNA ALPHA INHIBITORS |
| CN1481377A (en) | 2000-03-17 | 2004-03-10 | ����˹�ж�-����˹˹����ҩƷ��˾ | Cyclic Beta-amino acid derivatives as inhibitors of matrix metalloproteases and TNF alhpa |
| EP1317449B1 (en) | 2000-09-15 | 2006-05-31 | Vertex Pharmaceuticals Incorporated | Pyrazole compounds useful as protein kinase inhibitors |
| JP2004511479A (en) | 2000-10-13 | 2004-04-15 | アストラゼネカ アクチボラグ | Quinazoline derivatives |
| TWI304061B (en) * | 2000-10-20 | 2008-12-11 | Eisai R&D Man Co Ltd | Nitrogen-containing aromatic ring derivatives |
| WO2002070662A2 (en) | 2001-03-02 | 2002-09-12 | Gpc Biotech Ag | Three hybrid assay system |
| US20030125344A1 (en) | 2001-03-23 | 2003-07-03 | Bayer Corporation | Rho-kinase inhibitors |
| EP1382604B1 (en) * | 2001-04-27 | 2005-12-28 | Kirin Beer Kabushiki Kaisha | Quinoline derivative having azolyl group and quinazoline derivative |
| CN100415720C (en) | 2001-06-22 | 2008-09-03 | 麒麟医药株式会社 | Quinoline derivative and quinazoline derivative inhibiting self-phosphorylation of hepatocytus proliferator receptor and medicinal composition containing the same |
| GB0126433D0 (en) | 2001-11-03 | 2002-01-02 | Astrazeneca Ab | Compounds |
| CN100343238C (en) | 2001-11-03 | 2007-10-17 | 阿斯特拉曾尼卡有限公司 | Quinazoline derivatives as antitumor agents |
| PL371486A1 (en) | 2002-02-01 | 2005-06-13 | Astrazeneca Ab | Quinazoline compounds |
| US7645878B2 (en) | 2002-03-22 | 2010-01-12 | Bayer Healthcare Llc | Process for preparing quinazoline Rho-kinase inhibitors and intermediates thereof |
| CA2480638C (en) | 2002-03-29 | 2013-02-12 | Chiron Corporation | Substituted benzazoles and use thereof as raf kinase inhibitors |
| UA77303C2 (en) * | 2002-06-14 | 2006-11-15 | Pfizer | Derivatives of thienopyridines substituted by benzocondensed heteroarylamide useful as therapeutic agents, pharmaceutical compositions and methods for their use |
| WO2004078114A2 (en) | 2003-02-28 | 2004-09-16 | Encysive Pharmaceuticals Inc. | Pyridine, pyrimidine, quinoline, quinazoline, and naphthalene urotensin-ii receptor antagonists. |
| GB0310401D0 (en) | 2003-05-07 | 2003-06-11 | Astrazeneca Ab | Therapeutic agent |
| EP2609919A3 (en) * | 2003-09-26 | 2014-02-26 | Exelixis, Inc. | c-Met modulators and methods of use |
| UA82577C2 (en) * | 2003-12-23 | 2008-04-25 | Пфайзер Инк. | Quinoline derivatives |
| US7576090B2 (en) * | 2004-12-27 | 2009-08-18 | 4Sc Ag | Benzazole analogues and uses thereof |
-
2005
- 2005-01-24 AU AU2005207946A patent/AU2005207946A1/en not_active Abandoned
- 2005-01-24 EP EP05711976A patent/EP1711495A2/en not_active Withdrawn
- 2005-01-24 US US11/042,398 patent/US7626030B2/en active Active
- 2005-01-24 JP JP2006551396A patent/JP2007518823A/en active Pending
- 2005-01-24 CA CA002553433A patent/CA2553433A1/en not_active Abandoned
- 2005-01-24 WO PCT/US2005/002304 patent/WO2005073224A2/en active Application Filing
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0602851A1 (en) * | 1992-12-10 | 1994-06-22 | Zeneca Limited | Quinazoline derivatives |
| WO1997003069A1 (en) * | 1995-07-13 | 1997-01-30 | Glaxo Group Limited | Heterocyclic compounds and pharmaceutical compositions containing them |
| WO2000018761A1 (en) * | 1998-09-29 | 2000-04-06 | American Cyanamid Company | Substituted 3-cyanoquinolines as protein tyrosine kinases inhibitors |
| US6288082B1 (en) * | 1998-09-29 | 2001-09-11 | American Cyanamid Company | Substituted 3-cyanoquinolines |
| WO2000047212A1 (en) * | 1999-02-10 | 2000-08-17 | Astrazeneca Ab | Quinazoline derivatives as angiogenesis inhibitors |
| WO2002092571A1 (en) * | 2001-05-11 | 2002-11-21 | Astrazeneca Ab | Novel 4-anilinoquinoline-3-carboxamides |
| EP1548008A1 (en) * | 2002-08-23 | 2005-06-29 | Kirin Beer Kabushiki Kaisha | Compound having tgf-beta inhibitory activity and medicinal composition containing the same |
| WO2004085425A1 (en) * | 2003-03-21 | 2004-10-07 | Amgen Inc | Fused azoles such as 2,5-disubstituted benzimidazoles, benzoxazoles and benzothiazoles as kinase inhibitors |
Cited By (124)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8470850B2 (en) | 2004-07-30 | 2013-06-25 | Methylgene Inc. | Inhibitors of VEGF receptor and HGF receptor signalling |
| US7772247B2 (en) | 2004-07-30 | 2010-08-10 | Methylgene Inc. | Substituted thieno[3,2-d]pyridines as inhibitors of the VEGF receptor and HGF receptor |
| AU2005321517B2 (en) * | 2004-12-27 | 2011-09-29 | 4Sc Ag | 2, 5 and 2, 6-disubstituted benzazole analogues useful as protein kinase inhibitors |
| US8093264B2 (en) | 2005-05-20 | 2012-01-10 | Methylgene Inc. | Fused heterocycles as inhibitors of VEGF receptor and HGF receptor signaling |
| US8329726B2 (en) | 2005-05-20 | 2012-12-11 | Methylgene Inc. | Inhibitors of VEGF receptor and HGF receptor signaling |
| US7790729B2 (en) | 2005-05-20 | 2010-09-07 | Methylgene Inc. | Inhibitors of VEGF receptor and HGF receptor signaling |
| US8592459B2 (en) | 2005-08-30 | 2013-11-26 | Novartis Ag | Substituted benzimidazoles and methods of their use |
| US7767820B2 (en) | 2005-08-30 | 2010-08-03 | Novartis Vaccines And Diagnostics, Inc. | Substituted benzimidazoles and methods of preparation |
| US7732465B2 (en) | 2005-08-30 | 2010-06-08 | Novartis Vaccines And Diagnostics, Inc. | Substituted benzimidazoles and methods of their use |
| JP2009532333A (en) * | 2006-03-10 | 2009-09-10 | ノバルティス アクチエンゲゼルシャフト | Heterobicyclic carboxamides as inhibitors of kinases |
| JP2009534410A (en) * | 2006-04-19 | 2009-09-24 | ノバルティス アクチエンゲゼルシャフト | 6-O-substituted benzoxazole and benzothiazole compounds and methods for inhibiting CSF-1R signaling |
| KR101464385B1 (en) * | 2006-04-19 | 2014-11-21 | 노파르티스 아게 | 6-o-substituted benzoxazole and benzothiazole compounds and methods of inhibiting csf-1r signaling |
| EA018917B1 (en) * | 2006-04-19 | 2013-11-29 | Новартис Аг | 6-o-substituted benzoxazole and benzothiazole compounds and methods of inhibiting csf-1r signaling |
| AU2007237904B2 (en) * | 2006-04-19 | 2011-03-03 | Novartis Ag | 6-O-substituted benzoxazole and benzothiazole compounds and methods of inhibiting CSF-1R signaling |
| CN101432281B (en) * | 2006-04-19 | 2013-08-28 | 诺瓦提斯公司 | 6-o-substituted benzoxazole and benzothiazole compounds and methods of inhibiting CSF-1R signaling |
| WO2007121484A2 (en) * | 2006-04-19 | 2007-10-25 | Novartis Ag | 6-o-substituted benzoxazole and benzothiazole compounds and methods of inhibiting csf-1r signaling |
| US8143251B2 (en) | 2006-08-07 | 2012-03-27 | Incyte Corporation | Triazolotriazines as kinase inhibitors |
| US7683060B2 (en) | 2006-08-07 | 2010-03-23 | Incyte Corporation | Triazolotriazines as kinase inhibitors |
| US7915408B2 (en) | 2006-08-07 | 2011-03-29 | Incyte Corporation | Triazolotriazines as kinase inhibitors |
| US11261191B2 (en) | 2006-11-22 | 2022-03-01 | Incyte Holdings Corporation | Imidazotriaines and imidazopyrimidines as kinase inhibitors |
| US10738052B2 (en) | 2006-11-22 | 2020-08-11 | Incyte Holdings Corporation | Imidazotriaines and imidazopyrimidines as kinase inhibitors |
| EP3443958A1 (en) | 2006-11-22 | 2019-02-20 | Incyte Holdings Corporation | Imidazotriazines and imidazopyrimidines as kinase inhibitors |
| EP2497470A1 (en) | 2006-11-22 | 2012-09-12 | Incyte Corporation | Imidazotriazines and imidazopyrimidines as kinase inhibitors |
| EP3034075A1 (en) | 2006-11-22 | 2016-06-22 | Incyte Holdings Corporation | Imidazotriazines and imidazopyrimidines as kinase inhibitors |
| US7767675B2 (en) | 2006-11-22 | 2010-08-03 | Incyte Corporation | Imidazotriazines and imidazopyrimidines as kinase inhibitors |
| US8461330B2 (en) | 2006-11-22 | 2013-06-11 | Incyte Corporation | Imidazotriazines and imidazopyrimidines as kinase inhibitors |
| US9944645B2 (en) | 2006-11-22 | 2018-04-17 | Incyte Corporation | Imidazotriazines and imidazopyrimidines as kinase inhibitors |
| US7687522B2 (en) | 2006-12-20 | 2010-03-30 | Amgen Inc. | Substituted pyridines and pyrimidines and their use in treatment of cancer |
| US8338455B2 (en) | 2006-12-20 | 2012-12-25 | Amgen Inc. | Compounds and methods of use |
| WO2008089307A3 (en) * | 2007-01-18 | 2008-11-06 | Lexicon Pharmaceuticals Inc | Delta 5 desaturase inhibitors for the treatment of pain, inflammation and cancer |
| WO2008089307A2 (en) * | 2007-01-18 | 2008-07-24 | Lexicon Pharmaceuticals, Inc. | Delta 5 desaturase inhibitors for the treatment of pain, inflammation and cancer |
| EP2125777A1 (en) * | 2007-03-14 | 2009-12-02 | Advenchen Laboratories, LLC | Spiro substituted compounds as angiogenesis inhibitors |
| US8163923B2 (en) | 2007-03-14 | 2012-04-24 | Advenchen Laboratories, Llc | Spiro substituted compounds as angiogenesis inhibitors |
| EP2125776A1 (en) * | 2007-03-14 | 2009-12-02 | Advenchen Laboratories, LLC | Spiro substituted compounds as angiogenesis inhibitors |
| EP2125776A4 (en) * | 2007-03-14 | 2011-06-22 | Advenchen Lab Llc | Spiro substituted compounds as angiogenesis inhibitors |
| US8513283B2 (en) | 2007-03-14 | 2013-08-20 | Advenchen Laboratories, Llc | Spiro substituted compounds as angiogenesis inhibitors |
| WO2008112407A1 (en) | 2007-03-14 | 2008-09-18 | Advenchen Laboratories, Llc | Spiro substituted compounds as angiogenesis inhibitors |
| EP2125777A4 (en) * | 2007-03-14 | 2011-06-22 | Advenchen Lab Llc | Spiro substituted compounds as angiogenesis inhibitors |
| EP1977759A1 (en) * | 2007-03-27 | 2008-10-08 | 4Sc Ag | Pharmaceutical composition comprising a cytokine |
| WO2008116919A1 (en) * | 2007-03-27 | 2008-10-02 | 4Sc Ag | Pharmaceutical composition comprising a cytokine |
| US8293769B2 (en) | 2007-05-21 | 2012-10-23 | Novartis Ag | CSF-1R inhibitors, compositions, and methods of use |
| JP2010528009A (en) * | 2007-05-21 | 2010-08-19 | ノバルティス アーゲー | CSF-1R inhibitors, compositions and methods of use |
| WO2008144062A1 (en) | 2007-05-21 | 2008-11-27 | Novartis Ag | Csf-1r inhibitors, compositions, and methods of use |
| US20100130490A1 (en) * | 2007-05-21 | 2010-05-27 | Ng Simon C | Csf-1r inhibitors,compositions, and methods of use |
| US8822468B2 (en) | 2008-02-28 | 2014-09-02 | Novartis Ag | 3-Methyl-imidazo[1,2-b]pyridazine derivatives |
| US8729268B2 (en) | 2008-03-05 | 2014-05-20 | Methylgene Inc. | Inhibitors of protein tyrosine kinase activity |
| US8759522B2 (en) | 2008-03-05 | 2014-06-24 | Methylgene Inc. | Inhibitors of protein tyrosine kinase activity |
| US11452726B2 (en) | 2008-05-21 | 2022-09-27 | Incyte Corporation | Salts of 2-fluoro-N-methyl-4-[7-(quinolin-6-yl-methyl)-imidazo[1,2-b][1,2,4]triazin-2-yl]benzamide and processes related to preparing the same |
| US10245265B2 (en) | 2008-05-21 | 2019-04-02 | Incyte Incorporation | Salts of 2-fluoro-N-methyl-4-[7-(quinolin-6-yl-methyl)-imidazo[1,2-B][1,2,4]triazin-2-yl]benzamide and processes related to preparing the same |
| US8901123B2 (en) | 2008-05-21 | 2014-12-02 | Incyte Corporation | Salts of 2-fluoro-N-methyl-4-[7-(quinolin-6-yl-methyl)-imidazo[1,2-B][1,2,4]triazin-2-yl]benzamide and processes related to preparing the same |
| US8420645B2 (en) | 2008-05-21 | 2013-04-16 | Incyte Corporation | Salts of 2-fluoro-N-methyl-4-[7-(quinolin-6-yl-methyl)-imidazo[1,2-b][1,2,4]triazin-2-yl]benzamide and processes related to preparing the same |
| EP3287449A2 (en) | 2008-05-21 | 2018-02-28 | Incyte Holdings Corporation | Salts of 2-fluoro-n-methyl-4-[7-(quinolin-6-yl-methyl)- imidazo[1,2-b][1,2,4]triazin-2-yl]benzamide and processes related to preparing the same |
| US10799509B2 (en) | 2008-05-21 | 2020-10-13 | Incyte Corporation | Salts of 2-fluoro-N-methyl-4-[7-(quinolin-6-yl-methyl)-imidazo[1,2-B][1,2,4]triazin-2-yl]benzamide and processes related to preparing the same |
| WO2010021918A1 (en) | 2008-08-19 | 2010-02-25 | Advenchen Laboratories, Llc | Compounds as kinase inhibitors |
| JP2012503671A (en) * | 2008-09-26 | 2012-02-09 | グラクソスミスクライン・リミテッド・ライアビリティ・カンパニー | Preparation of quinolinyloxydiphenylcyclopropanedicarboxamide |
| EP2339918A4 (en) * | 2008-09-26 | 2012-06-06 | Glaxosmithkline Llc | Preparation of a quinolinyloxydiphenylcyclopropanedicarboxamide |
| EP2339918A1 (en) * | 2008-09-26 | 2011-07-06 | GlaxoSmithKline LLC | Preparation of a quinolinyloxydiphenylcyclopropanedicarboxamide |
| US8314232B2 (en) | 2008-09-26 | 2012-11-20 | Glaxosmithkline Llc | Preparation of a quinolinyloxydiphenylcyclopropanedicarboxamide |
| US11098015B2 (en) | 2009-01-16 | 2021-08-24 | Exelixis, Inc. | Malate salt of N-(4-{[6,7-bis(methyloxy) quinolin-4-yl]oxy}phenyl)-N′-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide, and crystalline forms thereof for the treatment of cancer |
| US11091439B2 (en) | 2009-01-16 | 2021-08-17 | Exelixis, Inc. | Malate salt of N-(4-{[6,7-bis(methyloxy) quinolin-4-yl]oxy}phenyl)-N′-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide, and crystalline forms therof for the treatment of cancer |
| US11091440B2 (en) | 2009-01-16 | 2021-08-17 | Exelixis, Inc. | Malate salt of N-(4-{[6,7-bis(methyloxy) quinolin-4-yl]oxy}phenyl)- N′-(4-fluorophenyl)cyclopropane-1,1 -dicarboxamide, and crystalline forms thereof for the treatment of cancer |
| US9809549B2 (en) | 2009-01-16 | 2017-11-07 | Exelixis, Inc. | Malate salt of N-(4-{[6,7-bis(methyloxy)quinolin-4-yl]oxy}phenyl)-N′(4-fluorophenyl)cyclopropane-1,1-dicarboxamide, and crystalline forms therof for the treatment of cancer |
| US10736886B2 (en) | 2009-08-07 | 2020-08-11 | Exelixis, Inc. | Methods of using c-Met modulators |
| US11433064B2 (en) | 2009-08-07 | 2022-09-06 | Exelixis, Inc. | Methods of using c-Met modulators |
| US8389526B2 (en) | 2009-08-07 | 2013-03-05 | Novartis Ag | 3-heteroarylmethyl-imidazo[1,2-b]pyridazin-6-yl derivatives |
| US10919901B2 (en) | 2010-02-03 | 2021-02-16 | Incyte Holdings Corporation | Imidazo[1,2-B][1,2,4]triazines as c-Met inhibitors |
| US9221824B2 (en) | 2010-02-03 | 2015-12-29 | Incyte Holdings Corporation | Imidazo[1,2-B][1,2,4]triazines as c-Met inhibitors |
| WO2011162835A1 (en) | 2010-02-03 | 2011-12-29 | Incyte Corporation | Imidazo[1,2-b][1,2,4]triazines as c-met inhibitors |
| US10472367B2 (en) | 2010-02-03 | 2019-11-12 | Incyte Incorporation | Imidazo[1,2-B][1,2,4]triazines as c-Met inhibitors |
| US9988387B2 (en) | 2010-02-03 | 2018-06-05 | Incyte Holdings Corporation | Imidazo[1,2-B][1,2,4]triazines as c-Met inhibitors |
| US8487096B2 (en) | 2010-02-03 | 2013-07-16 | Incyte Corporation | Imidazo[1,2-B][1,2,4]triazines as C-MET inhibitors |
| US10478423B2 (en) | 2011-05-12 | 2019-11-19 | Nerviano Medical Sciences S.R.L. | Substituted indazole derivatives active as kinase inhibitiors |
| KR101953272B1 (en) * | 2011-05-12 | 2019-02-28 | 네르비아노 메디칼 사이언시스 에스.알.엘. | Substituted indazole derivatives active as kinase inhibitors |
| EA023579B1 (en) * | 2011-05-12 | 2016-06-30 | НЕРВИАНО МЕДИКАЛ САЙЕНСИЗ С.р.л. | Substituted indazole derivatives active as kinase inhibitors |
| US9408850B2 (en) | 2011-05-12 | 2016-08-09 | Nerviano Medical Sciences S.R.L. | Substituted indazole derivatives active as kinase inhibitors |
| WO2012152763A1 (en) * | 2011-05-12 | 2012-11-15 | Nerviano Medical Sciences S.R.L. | Substituted indazole derivatives active as kinase inhibitors |
| KR20140037871A (en) * | 2011-05-12 | 2014-03-27 | 네르비아노 메디칼 사이언시스 에스.알.엘. | Substituted indazole derivatives active as kinase inhibitors |
| US9597317B2 (en) | 2011-05-12 | 2017-03-21 | Nerviano Medical Sciences S.R.L. | Substituted indazole derivatives active as kinase inhibitiors |
| CN103534239A (en) * | 2011-05-12 | 2014-01-22 | 内尔维阿诺医学科学有限公司 | Substituted indazole derivatives active as kinase inhibitors |
| US10028934B2 (en) | 2011-05-12 | 2018-07-24 | Nerviano Medical Sciences S.R.L. | Substituted indazole derivatives active as kinase inhibitiors |
| US10016448B2 (en) | 2011-06-10 | 2018-07-10 | Merck Patent Gmbh | Compositions and methods for the production of pyrimidine and pyridine compounds with BTK inhibitory activity |
| US10413562B2 (en) | 2011-06-10 | 2019-09-17 | Merck Patent Gmbh | Compositions and methods for the production of pyrimidine and pyridine compounds with BTK inhibitory activity |
| AU2014219024B2 (en) * | 2013-02-20 | 2018-04-05 | KALA BIO, Inc. | Therapeutic compounds and uses thereof |
| US10285991B2 (en) | 2013-02-20 | 2019-05-14 | Kala Pharmaceuticals, Inc. | Therapeutic compounds and uses thereof |
| EP3514179A1 (en) | 2014-01-24 | 2019-07-24 | Dana-Farber Cancer Institute, Inc. | Antibody molecules to pd-1 and uses thereof |
| EP4324518A2 (en) | 2014-01-31 | 2024-02-21 | Novartis AG | Antibody molecules to tim-3 and uses thereof |
| WO2015138920A1 (en) | 2014-03-14 | 2015-09-17 | Novartis Ag | Antibody molecules to lag-3 and uses thereof |
| EP3660050A1 (en) | 2014-03-14 | 2020-06-03 | Novartis AG | Antibody molecules to lag-3 and uses thereof |
| WO2016057841A1 (en) | 2014-10-08 | 2016-04-14 | Novartis Ag | Compositions and methods of use for augmented immune response and cancer therapy |
| EP4245376A2 (en) | 2014-10-14 | 2023-09-20 | Novartis AG | Antibody molecules to pd-l1 and uses thereof |
| WO2016061142A1 (en) | 2014-10-14 | 2016-04-21 | Novartis Ag | Antibody molecules to pd-l1 and uses thereof |
| WO2016091167A1 (en) * | 2014-12-09 | 2016-06-16 | 正大天晴药业集团股份有限公司 | Quinoline derivative against squamous cell carcinoma of lung |
| US10336707B2 (en) | 2014-12-16 | 2019-07-02 | Eudendron S.R.L. | Heterocyclic derivatives modulating activity of certain protein kinases |
| WO2016096709A1 (en) * | 2014-12-16 | 2016-06-23 | Eudendron S.R.L. | Heterocyclic derivatives modulating activity of certain protein kinases |
| WO2016100882A1 (en) | 2014-12-19 | 2016-06-23 | Novartis Ag | Combination therapies |
| WO2016145102A1 (en) | 2015-03-10 | 2016-09-15 | Aduro Biotech, Inc. | Compositions and methods for activating "stimulator of interferon gene" -dependent signalling |
| US10449211B2 (en) | 2015-03-10 | 2019-10-22 | Aduro Biotech, Inc. | Compositions and methods for activating “stimulator of interferon gene”—dependent signalling |
| US11040053B2 (en) | 2015-03-10 | 2021-06-22 | Chinook Therapeutics, Inc. | Compositions and methods for activating “stimulator of interferon gene”13 dependent signalling |
| WO2016161952A1 (en) * | 2015-04-07 | 2016-10-13 | 广东众生药业股份有限公司 | Tyrosine kinase inhibitor and pharmaceutical composition comprising same |
| US10654808B2 (en) | 2015-04-07 | 2020-05-19 | Guangdong Raynovent Biotech Co., Ltd. | Tyrosine kinase inhibitor and pharmaceutical composition comprising same |
| EP3964528A1 (en) | 2015-07-29 | 2022-03-09 | Novartis AG | Combination therapies comprising antibody molecules to lag-3 |
| WO2017019894A1 (en) | 2015-07-29 | 2017-02-02 | Novartis Ag | Combination therapies comprising antibody molecules to lag-3 |
| EP3878465A1 (en) | 2015-07-29 | 2021-09-15 | Novartis AG | Combination therapies comprising antibody molecules to tim-3 |
| WO2017019897A1 (en) | 2015-07-29 | 2017-02-02 | Novartis Ag | Combination therapies comprising antibody molecules to tim-3 |
| CN106543143A (en) * | 2015-09-22 | 2017-03-29 | 合肥中科普瑞昇生物医药科技有限公司 | New FLT3 kinase inhibitors of one class and application thereof |
| CN106543143B (en) * | 2015-09-22 | 2019-03-22 | 合肥中科普瑞昇生物医药科技有限公司 | A kind of novel FLT3 kinase inhibitor and application thereof |
| US10894830B2 (en) | 2015-11-03 | 2021-01-19 | Janssen Biotech, Inc. | Antibodies specifically binding PD-1, TIM-3 or PD-1 and TIM-3 and their uses |
| WO2017106656A1 (en) | 2015-12-17 | 2017-06-22 | Novartis Ag | Antibody molecules to pd-1 and uses thereof |
| US11098077B2 (en) | 2016-07-05 | 2021-08-24 | Chinook Therapeutics, Inc. | Locked nucleic acid cyclic dinucleotide compounds and uses thereof |
| KR102556214B1 (en) | 2016-07-08 | 2023-07-19 | 주식회사유한양행 | Benzo[d]thiazole derivative or its salt and pharmaceutical composition comprising the same |
| KR20180006167A (en) * | 2016-07-08 | 2018-01-17 | 주식회사유한양행 | Benzo[d]thiazole derivative or its salt and pharmaceutical composition comprising the same |
| EP3483149A4 (en) * | 2016-07-08 | 2019-12-25 | Yuhan Corporation | Benzo[d]thiazole derivative or salt thereof, and pharmaceutical composition including same |
| CN106543145A (en) * | 2016-10-28 | 2017-03-29 | 山西医科大学 | C Met kinases suppression 5 benzoic acid amides derivant of agent 3 (4 fluorophenyl) pyrimidone, preparation method and application |
| WO2018237157A1 (en) | 2017-06-22 | 2018-12-27 | Novartis Ag | Antibody molecules to cd73 and uses thereof |
| WO2018237173A1 (en) | 2017-06-22 | 2018-12-27 | Novartis Ag | Antibody molecules to cd73 and uses thereof |
| CN112384506A (en) * | 2018-05-15 | 2021-02-19 | 北京诺诚健华医药科技有限公司 | Indoline-1-formamide compound, preparation method and application thereof in medicine and pharmacology |
| CN112384506B (en) * | 2018-05-15 | 2023-01-24 | 北京诺诚健华医药科技有限公司 | Indoline-1-formamide compound, preparation method and application thereof in medicine and pharmacology |
| WO2019218928A1 (en) * | 2018-05-15 | 2019-11-21 | 北京诺诚健华医药科技有限公司 | Indoline-1-formamide compound, preparation method therefor, and medical use thereof |
| WO2019229658A1 (en) | 2018-05-30 | 2019-12-05 | Novartis Ag | Entpd2 antibodies, combination therapies, and methods of using the antibodies and combination therapies |
| WO2019232244A2 (en) | 2018-05-31 | 2019-12-05 | Novartis Ag | Antibody molecules to cd73 and uses thereof |
| US11618749B2 (en) | 2018-07-03 | 2023-04-04 | Ifm Due, Inc. | Compounds and compositions for treating conditions associated with STING activity |
| WO2021053559A1 (en) | 2019-09-18 | 2021-03-25 | Novartis Ag | Entpd2 antibodies, combination therapies, and methods of using the antibodies and combination therapies |
| WO2021067801A1 (en) * | 2019-10-03 | 2021-04-08 | Ifm Due, Inc. | Compounds and compositions for treating conditions associated with sting activity |
| US11970480B2 (en) | 2022-05-17 | 2024-04-30 | Glaxosmithkline Intellectual Property Development Limited | Heterocyclic amides useful as protein modulators |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2005073224A3 (en) | 2005-09-29 |
| EP1711495A2 (en) | 2006-10-18 |
| US20050245547A1 (en) | 2005-11-03 |
| AU2005207946A1 (en) | 2005-08-11 |
| US7626030B2 (en) | 2009-12-01 |
| JP2007518823A (en) | 2007-07-12 |
| CA2553433A1 (en) | 2005-08-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7626030B2 (en) | Compounds and methods of use | |
| EP1664023B1 (en) | Sustituted isoquinoline derivatives and methods of use | |
| EP1664027B1 (en) | Substituted 2,3-dihydro-1h-isoindol-1-one derivatives and methods of use | |
| US8017601B2 (en) | Bis-aryl kinase inhibitors and method | |
| US6822097B1 (en) | Compounds and methods of uses | |
| CA2434178C (en) | Substituted amine derivatives and methods of use | |
| CA2434274A1 (en) | N-pyridyl carboxamide derivatives and pharmaceutical compositions containing them | |
| US20060241151A1 (en) | Pyrid-2-one derivatives and methods of use | |
| AU2002248339A1 (en) | Substituted Arylamine Derivatives and Methods of Use | |
| CA2489166A1 (en) | Substituted anthranilic amide derivatives and methods of use | |
| AU2002253890A1 (en) | Substituted Amine Derivatives and Methods of Use | |
| JP2004531484A (en) | Substituted alkylamine derivatives and methods of using the same | |
| WO2005070891A2 (en) | Compounds and methods of use | |
| EP1583744A2 (en) | Substituted benzylamine derivatives and methods of use | |
| EP2118069A2 (en) | Bis-aryl amide derivatives useful for the treatment of cancer | |
| MXPA06008314A (en) | Quinoline quinazoline pyridine and pyrimidine counds and their use in the treatment of inflammation angiogenesis and cancer |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2553433 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006551396 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2006/008314 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005711976 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005207946 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2005207946 Country of ref document: AU Date of ref document: 20050124 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005207946 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005711976 Country of ref document: EP |